
1. INTRODUÇÃO - USP · minimizar os efeitos deletérios do diabetes em seus principais órgãos...
84
1 1. INTRODUÇÃO O Diabetes Mellitus (DM) é uma patologia que vem preocupando cada vez mais os órgãos de saúde mundial em função do contínuo aumento da prevalência e incidência nas últimas décadas. Atualmente, existem 285 milhões de pessoas com DM no mundo, e a expectativa é que em 2030 sejam 438 milhões (INTERNATIONAL DIABETES FEDERATION, 2010). Nos Estados Unidos, 18 milhões de americanos com idade acima de 20 anos tinham DM em 2002, enquanto que em 2007, os números subiram para 23,5 milhões (CENTER for DISEASE CONTROL and PREVENTION, 2007). No Brasil não existem estudos comparativos recentes, entretanto, a Sociedade Brasileira de Diabetes (SBD) estima uma população aproximada de 11 milhões de pessoas portadoras de DM, representando 5,9% da população total (SBD, 2010). De acordo com a American Diabetes Association (ADA, 2008), o custo anual com os cuidados relacionados ao DM é em torno de 116 bilhões de dólares. O DM é um distúrbio que consiste na resposta secretória deficiente ou ação defeituosa de insulina, manifestando-se pela utilização inadequada de glicose pelos tecidos com consequente hiperglicemia (OKOSHI, GUIMARAES, DI MUZIO, FERNANDES & OKOSHI, 2007). O quadro crônico de hiperglicemia pode causar complicações que envolvem doenças cardiovasculares, incluindo o desenvolvimento de hipertensão arterial sistêmica (PETI-PETERDI, KANG & TOMA, 2008) e de cardiomiopatia diabética (RUBERG, 2007), contribuindo significativamente para o aumento da morbidade e mortalidade relacionada à doença (KIVELA, SILVENNOINEN, TOUVRA, LEHTI, KAINULAINEN & VIHKO, 2006). No DM classificado como tipo 1, os prejuízos metabólicos são decorrentes da falência das células ß das ilhotas endócrinas pancreáticas (POORNIMA, PARIKH &
Transcript of 1. INTRODUÇÃO - USP · minimizar os efeitos deletérios do diabetes em seus principais órgãos...

1
1. INTRODUÇÃO
O Diabetes Mellitus (DM) é uma patologia que vem preocupando cada vez mais
os órgãos de saúde mundial em função do contínuo aumento da prevalência e incidência
nas últimas décadas. Atualmente, existem 285 milhões de pessoas com DM no mundo,
e a expectativa é que em 2030 sejam 438 milhões (INTERNATIONAL DIABETES
FEDERATION, 2010). Nos Estados Unidos, 18 milhões de americanos com idade
acima de 20 anos tinham DM em 2002, enquanto que em 2007, os números subiram
para 23,5 milhões (CENTER for DISEASE CONTROL and PREVENTION, 2007). No
Brasil não existem estudos comparativos recentes, entretanto, a Sociedade Brasileira de
Diabetes (SBD) estima uma população aproximada de 11 milhões de pessoas portadoras
de DM, representando 5,9% da população total (SBD, 2010). De acordo com a
American Diabetes Association (ADA, 2008), o custo anual com os cuidados
relacionados ao DM é em torno de 116 bilhões de dólares.
O DM é um distúrbio que consiste na resposta secretória deficiente ou ação
defeituosa de insulina, manifestando-se pela utilização inadequada de glicose pelos
tecidos com consequente hiperglicemia (OKOSHI, GUIMARAES, DI MUZIO,
FERNANDES & OKOSHI, 2007). O quadro crônico de hiperglicemia pode causar
complicações que envolvem doenças cardiovasculares, incluindo o desenvolvimento de
hipertensão arterial sistêmica (PETI-PETERDI, KANG & TOMA, 2008) e de
cardiomiopatia diabética (RUBERG, 2007), contribuindo significativamente para o
aumento da morbidade e mortalidade relacionada à doença (KIVELA,
SILVENNOINEN, TOUVRA, LEHTI, KAINULAINEN & VIHKO, 2 006).
No DM classificado como tipo 1, os prejuízos metabólicos são decorrentes da
falência das células ß das ilhotas endócrinas pancreáticas (POORNIMA, PARIKH &

2
SHANNON, 2006), onde além da hiperglicemia, observa-se também aumento do
metabolismo energético basal e maior catabolismo protéico (HEBERT & NAIR, 2009),
poliúria, polidpsia e perda de peso corporal, apesar do aumento do apetite (HEIMANN,
2003). Alterações ocorrem também no fenótipo do músculo esquelético, tais como
redução da capacidade oxidativa e de capilarização (KRAUSE, RIDDELL, GORDON,
IMAM, CAFARELLI & HAWKE, 2009), atrofia muscular, diminuição da quantidade
de fibras musculares tipo 1 e intolerância aos exercícios físicos (ZOLL, MONASSIER,
GARNIER, N'GUESSAN, METTAUER, VEKSLER, PIQUARD, VENTURA-
CLAPIER & GENY, 2006), caracterizando, assim, o quadro de miopatia diabética
(AUGHSTEEN, KHAIR & SULEIMAN, 2006; KRAUSE et al., 2009). Observa-se
também elevados níveis de ácidos graxos livres e triglicerídeos oriundos da maior
atividade lipolítica do tecido adiposo (AN & RODRIGUES, 2006), o que coincide com
a redução considerável do tecido adiposo (GHORBANI, VAREDI, HADJZADEH &
OMRANI, 2010), perda de peso corporal (BLAAK, 2003) e prejuízo no metabolismo de
glicose (TAKADA, MACHADO, PERES, BRITO, BORGES-SILVA, COSTA,
FONSECA-ALANIZ, ANDREOTTI & LIMA, 2007).
Já no DM classificado como tipo 2, a hiperglicemia está diretamente relacionada
com prejuízos na ação da insulina. Distúrbios no metabolismo de glicose e de lipídios
são observados frequentemente e, no músculo-esquelético observa-se menor síntese de
glicogênio muscular e menor utilização da glicose como substrato energético (RABOL,
BOUSHEL, ALMDAL, HANSEN, PLOUG, HAUGAARD, PRATS, MADSBAD &
DELA, 2010; SCHRAUWEN-HINDERLING, KOOI, HESSELINK, JENESON,
BACKES, VAN ECHTELD, VAN ENGELSHOVEN, MENSINK & SCHRAUWEN,
2007), acúmulo de ácido graxo decorrente da menor taxa de oxidação associada à
disfunção mitocondrial, o que resulta em menor capacidade da musculatura esquelética

3
em produzir energia em indivíduos diabéticos (ZELEZNIAK, PERS, SOARES, PATTI
& PATIL, 2010).
Os mecanismos envolvidos na progressão do diabetes que diferenciam um
indivíduo do outro até seu estágio final tornaram-se alvos de estudos para o tratamento
dessa patologia. Fatores humorais parecem interferir diretamente nas complicações
metabólicas induzidas pelo diabetes, e um exemplo que nos últimos anos vem sendo
bastante investigado é o peptídio angiotensina II (Ang II), produto final do sistema
renina angiotensina (SRA) (RIBEIRO-OLIVEIRA, NOGUEIRA, PEREIRA, BOAS,
DOS SANTOS & SIMOES E SILVA, 2008). Estudos na literatura demonstraram a
influência negativa da Ang II na progressão do diabetes até o seu desfecho final
(PERKINS, AIELLO & KROLEWSKI, 2009; SAIKI, OHIRA, ENDO, KOIDE,
OYAMA, MURANO, WATANABE, MIYASHITA & SHIRAI, 2009).
TIKELLIS; COOPER & THOMAS (2006), relataram que tanto no DM1 quanto
no DM2 pode-se observar ativação do SRA associada ao aumento do estresse oxidativo,
inflamação e aumento dos níveis de ácidos graxos livres, contribuindo diretamente para
a disfunção das células β pancreáticas. Esses dados são reforçados em estudos com
modelos experimentais de diabetes, nos quais o bloqueio do SRA via inibidor da ECA
ou bloqueador do receptor AT1 resulta em melhora na estrutura e função das ilhotas, e
na redução das complicações associadas ao diabetes (AROZAL, WATANABE,
VEERAVEEDU, MA, THANDAVARAYAN, SUZUKI, TACHIKAWA, K ODAMA &
AIZAWA, 2009; LEUNG, 2007; OLTMAN, DAVIDSON, COPPEY,
KLEINSCHMIDT, DAKE & YOREK, 2010). De fato, o uso de inibidores da ECA
(CLERK, VINCENT, BARRETT, LANKFORD & LINDNER, 2007; PERKINS,
AIELLO & KROLEWSKI, 2009) e bloqueadores do receptor AT1 (BILOUS, 2008;
DALLA VESTRA, SIMIONI & MASIERO, 2009; MAUER, ZINMAN, GARDINER,

4
SUISSA, SINAIKO, STRAND, DRUMMOND, DONNELLY, GOODYER, GUBLER
& KLEIN, 2009) tem sido fortemente recomendados como estratégias terapêuticas para
minimizar os efeitos deletérios do diabetes em seus principais órgãos alvo, tais como
vasos, olhos, rins e coração.
O treinamento físico tem sido indicado para o controle do diabetes por promover
benefícios na captação de glicose, melhora da sensibilidade à ação da insulina no
músculo esquelético e no tecido adiposo (KIVELA et al., 2006). Melhoras significantes
nos níveis de insulina foram observadas em ratos diabéticos por estreptozotocina
treinados durante dez semanas (DE ANGELIS, OLIVEIRA, DALL'AGO, PEIXOTO,
GADONSKI, LACCHINI, FERNANDES & IRIGOYEN, 2000). Aumento na
expressão e atividade de enzimas oxidativas (EVANGELISTA & KRIEGER, 2006;
LEICK, HELLSTEN, FENTZ, LYNGBY, WOJTASZEWSKI, HIDALGO &
PILEGAARD, 2009), aumento de proteínas transportadoras de triglicerídeos e ácidos
graxos no músculo esquelético (LIMA-SILVA, ADAMI, NAKAMURA, DE
OLIVEIRA & GEVAERD, 2006), biogênese mitocondrial (HOOD, 2009; LJUBICIC,
JOSEPH, SALEEM, UGUCCIONI, COLLU-MARCHESE, LAI, NGUYEN & HOOD,
2009), e aumento no predomínio de fibras musculares oxidativas tipo 1
(DASTMALCHI, ALEXANDERSON, LOELL, STAHLBERG, BORG, LUNDBERG
& ESBJORNSSON, 2007) são alguns dos benefícios promovidos pelo treinamento
físico que podem auxiliar no melhor controle do diabetes.
Esses dados ressaltam a potencialidade do treinamento físico aeróbio para
promover adaptações no metabolismo energético que poderiam contribuir efetivamente
para o tratamento do diabetes. No entanto, é interessante observar que a maioria dos
estudos que investigou o potencial terapêutico do treinamento físico para o diabetes,
introduziu o treinamento físico na fase inicial da doença. Mas, ainda não está muito

5
claro na literatura se os benefícios promovidos pelo treinamento físico também podem
ser obtidos quando este for iniciado somente após um período crônico de hiperglicemia.
Embora a literatura demonstre a associação do genótipo da ECA com o pior
prognóstico do diabetes e ressalte os benefícios do treinamento físico para o tratamento
do diabetes, até o presente momento pouco se sabe sobre os efeitos do treinamento
físico sobre o metabolismo energético de animais diabéticos induzidos
experimentalmente por STZ com diferentes níveis de expressão do gene da ECA. Sendo
assim, considerando a importância dos ajustes metabólicos associados ao diabetes, o
papel da Ang II na progressão da patologia e a aplicabilidade do treinamento físico
como importante tratamento não farmacológico, o presente estudo testou a hipótese de
que a expressão de ECA aumentada induz maior prejuízo metabólico no diabetes
experimental e, que o treinamento físico é capaz de atenuar essa resposta.

6
2. OBJETIVOS
2.1 Objetivo Geral
Estudar os efeitos do treinamento físico aeróbio sobre as alterações do
metabolismo energético induzidas pelo diabetes em camundongos com diferentes
dosagens de ECA (1 cópia vs. 3 cópias).
2.2 Objetivos Específicos
Estudar em camundongos diabéticos com 1 e 3 cópias do gene da ECA,
sedentários e treinados:
- a evolução do peso corporal;
- a ingestão de ração e a taxa metabólica de repouso;
- a capacidade de realização de exercício físico;
- a tolerância à glicose;
- o perfil hormonal através da dosagem de leptina e insulina;
- a atividade lipolítica do tecido adiposo;
- a função lipogênica através da dosagem da enzima ácido graxo sintase no
tecido adiposo e no tecido hepático;
- a atividade máxima da enzima citrato sintase no músculo esquelético, a
tipagem de fibras musculares e capilarização.

7
3. REVISÃO DE LITERATURA
3.1 Diabetes
O DM1 é causado pela destruição das células β pancreáticas consequente de um
processo auto-imune, e geralmente provoca deficiência absoluta de insulina
(WELTMAN, SALIBA, BARRETT & WELTMAN, 2009). Alguns indivíduos com
DM1, principalmente adultos, conseguem manter o funcionamento de algumas células β
por alguns anos, entretanto, a maioria acaba se tornando dependente de insulina exógena
(ADA, 2008b). Diversos distúrbios metabólicos são evidentes no DM1, tais como
hiperglicemia, excreção de glicose na urina (glicosúria), aumento na produção de urina
(poliúria), aumento da sede (polidipsia), aumento do apetite (polifagia) e redução no
peso corporal (TAKADA, FONSECA-ALANIZ, DE CAMPOS, ANDREOTTI,
CAMPANA, OKAMOTO, BORGES-SILVA, MACHADO & LIMA, 2008).
No DM1, a hiperglicemia crônica provoca alterações importantes em toda a rede
metabólica. A indisponibilidade de glicose em quantidade suficiente para ser utilizada
como substrato energético influencia os sítios metabólicos envolvidos no fornecimento
de energia, especialmente aqueles relacionados com o metabolismo de lipídios e
proteínas (HEBERT & NAIR, 2009; VIRTUE & VIDAL-PUIG, 2010). De fato, o
aumento do catabolismo lipídico e protéico observado em portadores de DM1 contribui
ainda mais para o prejuízo metabólico e, consequentemente, para o pior prognóstico da
patologia (VAN HERPEN & SCHRAUWEN-HINDERLING, 2008).
Segundo PIGHIN, KARABATAS, PASTORALE, DASCAL, CARBONE,
CHICCO, LOMBARDO & BASABE (2005), a redução significativa da massa adiposa
e do peso corporal estão diretamente relacionados com o aumento da atividade lipolítica
do tecido adiposo. Como consequência, há uma grande disponibilidade de ácido graxo

8
para ser utilizado como fonte de energia, no entanto, o ácido graxo, na forma de Acetil-
CoA no fígado, não consegue se unir ao oxalacetato e formar citrato para entrar no ciclo
de Krebs, uma vez que o oxalacetato é utilizado na gliconeogênese. Então a Acetil-CoA
acumulada na célula sofre ação das tiolases, formando corpos cetônicos que serão
liberados na corrente sanguínea, resultando em um quadro de acidose metabólica
(BARONE, RODACKI, CENCI, ZAJDENVERG, MILECH & OLIVEIRA, 2007).
Paralelo ao aumento da lipólise, a ausência ou decréscimo na quantidade de
insulina circulante associada à elevação dos hormônios contra-regulatórios, incluindo
glucagon, catecolaminas, cortisol e hormônio do crescimento (GH), resulta em aumento
da produção hepática e renal de glicose, diminuição da utilização periférica da mesma,
hiperglicemia e hiperosmolaridade. A associação entre hiperglicemia e acidose causa
diurese osmótica, com consequente desidratação e desequilíbrio eletrolítico. O estágio
mais avançado é de extrema desidratação celular, contração do volume plasmático,
hipoperfusão cerebral e alteração progressiva do estado de consciência (AXELROD &
LEVINE, 1982).
Apesar de os tecidos metabolicamente ativos aumentarem a oxidação de ácido
graxo, ainda observa-se predomínio da produção, resultando em acúmulo de ácido
graxo e outros produtos tóxicos do seu metabolismo denominado por lipotoxicidade
(AN & RODRIGUES, 2006). No coração, essas alterações precedem a cardiomiopatia
diabética, independentemente de hipertensão ou doença coronariana prévia (RUBERG,
2007).
No músculo esquelético, o aumento do conteúdo de triglicerídeos provoca
grande prejuízo na utilização de substratos energéticos (STUMP, SHORT, BIGELOW,
SCHIMKE & NAIR, 2003) em decorrência da diminuição da atividade do complexo
piruvato desidrogenase, fundamental para o controle da oxidação de glicose, e aumento

9
da piruvato desidrogenase quinase, que por fosforilação, inativa a enzima piruvato
desidrogenase provocando diminuição da oxidação de piruvato na mitocôndria e
aumento da conversão de piruvato em lactato no citosol (PIGHIN et al., 2005).
Ainda, alterações no fenótipo do músculo esquelético, tais como diminuição da
contratilidade (ANDERSEN, SCHMITZ & NIELSEN, 2005), redução da capacidade
oxidativa e capilarização (KRAUSE et al., 2009), atrofia muscular, redução de fibras
musculares tipo 1 e intolerância aos esforços físicos também são observados em
pacientes com DM1 (ZOLL et al., 2006), caracterizando o quadro de miopatia diabética
(AUGHSTEEN, KHAIR & SULEIMAN, 2006).
Entre os diversos problemas associados ao diabetes, as complicações
cardiovasculares têm aumentado significativamente as taxas de morbidade e
mortalidade (PERKINS, AIELLO & KROLEWSKI, 2009). De fato, cerca de 80% das
mortes associadas ao diabetes são devidas às complicações cardiovasculares (HAYAT,
PATEL, KHATTAR & MALIK, 2004) como obesidade, hipertensão arterial,
aterosclerose, nefropatia, isquemia coronariana, doença vascular periférica e acidente
vascular cerebral (ALVAREZ-AGUILAR, ENRIQUEZ-RAMIREZ, FIGUEROA-
NUNEZ, GOMEZ-GARCIA, RODRIGUEZ-AYALA, MORAN-MOGUEL, FARIAS-
RODRIGUEZ, MINO-LEON & LOPEZ-MEZA, 2007; BOUDINA & ABEL, 2010;
LAWN, 1992; RUBERG, 2007; VIRTUE & VIDAL-PUIG, 2010). Quanto mais longo
o tempo de diabetes, maiores são as complicações associadas à patologia e o
comprometimento dos pacientes, mesmo com intenso tratamento insulínico e controle
metabólico (NORDWALL, HYLLIENMARK & LUDVIGSSON, 2006). Desta forma,
reduzir o período de exposição à hiperglicemia pode significar reduzir os riscos de
complicações cardiovasculares no paciente diabético (KUSMINSKI, SHETTY, ORCI,
UNGER & SCHERER, 2009).

10
Diferentes modelos experimentais têm sido utilizados na literatura para o estudo
do diabetes. Entre os vários agentes químicos citotóxicos para as células β do pâncreas,
a estreptozotocina (STZ), um derivado N-nitroso de glucosamina, tem sido
sistematicamente empregada para induzir diabetes nos animais (SAITO, KINOSHITA,
SATOH, SHINBORI, KONO, HANADA, UEMASU, SUZUKI, YAMADA &
SATOH, 2006). A ação da STZ é seletivamente direcionada para as células produtoras
de insulina devido à especial capacidade dessas células em reconhecer e metabolizar
glicose rapidamente, causando necrose de forma rápida e irreversível (JUNOD,
LAMBERT, ORCI, PICTET, GONET & RENOLD, 1967). Evidências de danos pela
STZ aparecem entre 30 minutos e 2 horas (SHARMA & THOMAS, 1999) e são
dependentes da dose utilizada. Doses 4 ou 5 vezes menores que a dose letal são
suficientes para gerar hipoinsulinemia, hiperglicemia, hiperfagia, polidipsia, perda de
peso corporal, aumento de lípides séricos e estresse oxidativo (OKOSHI et al., 2007;
PIGHIN et al., 2005).
Interessante observar que uma única injeção intraperitoneal ou intravenosa de
STZ induz diabetes experimental que pode ser mantido sem tratamento com insulina
por um ou dois anos (SHARMA & THOMAS, 1999). Isso porque, embora ocorra
depleção de células β pela STZ, a indução de deficiência de insulina é incompleta
quando doses abaixo de 65 mg/Kg são utilizadas, haja vista a ausência de cetose e a
persistência de pequena quantidade de insulina próxima de valores de jejum encontrada
no soro dos animais (JUNOD, LAMBERT, STAUFFACHER & RENOLD, 1969).
Dessa forma, apesar de o diabetes induzido pela STZ apresentar alterações metabólicas
que mimetizam o DM1, as particularidades observadas no modelo são suficientes para
que seja definido como diabetes experimental.

11
3.2 Sistema renina angiotensina
Classicamente, o SRA é visto como um sistema hormonal que envolve uma
cascata proteolítica conectada a um sistema de transdução, onde a renina, uma enzima
proteolítica sintetizada e estocada nas células justaglomerulares da arteríola eferente
renal, cliva o angiotensinogênio, seu substrato que é produzido no fígado, na porção N-
terminal em um decapeptídeo, a angiotensina I (Ang I). Esta é convertida
subsequentemente em Ang II pela ECA primariamente na circulação pulmonar. A ECA,
uma dipeptidil-carboxipeptidade, por sua vez é predominantemente uma enzima ligada
à membrana que é responsável pela produção de Ang II a partir do precursor inativo
Ang I e também pela degradação da bradicinina em fragmentos inativos (JOHNSTON,
1990).
A interação da Ang II extracelular com receptores de membrana AT1 acoplados
à proteína G estimula primariamente o aumento de inositol fosfato e da atividade de
proteína quinase C (MORGAN & BAKER, 1991), bem como as vias de sinalização
Ras, Raf-1, MAP quinases, fosfolipase A, Jak-Stat e jun quinase (PELLIEUX,
SAUTHIER, AUBERT, BRUNNER & PEDRAZZINI, 2000; SUGDEN & CLERK,
1998; THORBURN, 1994). Apesar de a Ang II gerada através do SRA ter sido
considerada classicamente como um hormônio, outras evidências indicam a síntese de
todos os componentes do SRA, ou pelo menos parte deles em diversos órgãos e
sistemas, tais como coração, rim, pâncreas, tecido adiposo, tecido muscular, vasos,
sistema nervoso, sistema digestivo e sistema reprodutor (PAUL, POYAN MEHR &
KREUTZ, 2006).
O SRA tecidual exerce múltiplos efeitos fisiológicos em nível celular tais como
regulação do crescimento celular, diferenciação, proliferação e apoptose, geração de

12
espécies reativas de oxigênio, inflamação, fibrose e secreção hormonal. No pâncreas, o
SRA influencia a regulação do fluxo sanguíneo local, secreção de enzimas digestivas,
biossíntese de pró-insulina, proliferação e diferenciação de células pancreáticas. Além
disso, pode mediar a inflamação induzida por estresse oxidativo, apoptose, fibrose e
necrose (LEUNG, 2007). Os efeitos negativos do SRA sobre o pâncreas estão
associados à disfunção das células β pancreáticas, e consequentemente, ao
desenvolvimento e pior prognóstico do DM (LEUNG, 2007). De fato, YUAN, LI, XU
& QI (2010) mostraram que o bloqueio farmacológico do SRA resultou em melhora na
função das células β pancreáticas em ratos diabéticos devido à redução do estresse
oxidativo, fibrose e apoptose.
O tecido adiposo é um importante órgão que sintetiza a Ang II independente dos
níveis circulantes, e a ação da Ang II no metabolismo das células adiposas tem sido
bastante estudada nos últimos anos. Evidências na literatura mostraram que a Ang II
pode estar envolvida na modulação da massa adiposa por influenciar a conversão de
pré-adipócito em adipócito (CASSIS, POLICE, YIANNIKOURIS & THATCHER,
2008), aumentando a síntese de lipídios e o crescimento dos adipócitos (KARLSSON,
LINDELL, OTTOSSON, SJOSTROM, CARLSSON & CARLSSON, 1998; ZORAD,
DOU, BENICKY, HUTANU, TYBITANCLOVA, ZHOU & SAAVEDRA, 2006).
Dados obtidos em nosso grupo sugeriram fortemente a participação da Ang II na
modulação da massa adiposa e do metabolismo do tecido adiposo ao confirmarem que
animais com aumento na expressão do gene da ECA (3 cópias) ganham mais massa
adiposa quando submetidos a uma dieta hiperlipídica comparados ao animais com
menor expressão de ECA (1 cópia) (EVANGELISTA, BERGAMO, FONSECA-
ALANIZ, NAKAMUTA & KRIEGER, 2008).

13
A influência do SRA no controle do metabolismo e do peso corporal pode estar
ocorrendo também em nível central além do nível do tecido adiposo, no entanto, o efeito
da Ang II acontece de forma antagônica. Nesse sentido, a Ang II pode aumentar a
lipólise via estimulação da liberação de noradrenalina pelo sistema nervoso simpático,
contribuindo para a redução de peso corporal. De fato, CASSIS et. al., (2008),
observaram redução de peso corporal e da ingestão de alimentos induzidas por infusão
central crônica de Ang II.
No tecido muscular, a Ang II com seu efeito vasoconstritor é responsável pela
redução da perfusão sanguínea e aporte de nutrientes energéticos para o músculo
esquelético (ZOLL et al., 2006), redução da capilarização no diabetes (CLERK et al.,
2007), redução da eficiência da musculatura periférica no trabalho aeróbio (ZHANG,
YAN, ZHOU, HUANG, YANG, LI, LIN & ZHOU, 2008) e influência negativa sobre o
metabolismo glicêmico (MACKNIGHT, MISTRY, PASTORS, HOLMES &
RYNDERS, 2009).
O estudo da participação do SRA em mecanismos fisiopatológicos específicos
tornaram-se mais evidentes na literatura através da identificação do polimorfismo do
gene da ECA, que é definido pela presença ou ausência de uma sequência de 287 pares
de bases não codificantes inserida (I) ou deletada (D) do íntron 16 do gene da ECA, o
que confere níveis mais elevados de ECA sérica e tecidual em indivíduos homozigotos
DD (VAN BERLO & PINTO, 2003). Aumento nos níveis de ECA resulta em maior
formação de Ang II, e esta resposta está associada com aumento da suscetibilidade ao
infarto do miocárdio, aterosclerose, desenvolvimento de DM2 e maior mortalidade de
pacientes diabéticos, doenças coronárias, insuficiência cardíaca e hipertrofia cardíaca
(ALVAREZ-AGUILAR et al., 2007; BACKLUND, LAKKISTO, PALOJOKI,
GRONHOLM, SARASTE, FINCKENBERG, MERVAALA, TIKKANEN & LAINE,

14
2007; BOZKURT, VERSCHUREN, VAN WIEREN-DE WIJER, KNOL, DE BOER,
GROBBEE, GEERLINGS, HEERDINK & KLUNGEL, 2008; MAUER et al., 2009;
OAK & CAI, 2007; PEDERSEN-BJERGAARD, DHAMRAIT, SETHI, FRANDSEN,
NORDESTGAARD, MONTGOMERY, PRAMMING, HOUGAARD &
THORSTEINSSON, 2008).
Experimentalmente, a criação de modelos transgênicos tem oferecido inúmeras
oportunidades para o estudo da participação de genes específicos no desenvolvimento e
evolução de doenças complexas no contexto do animal intacto. Nesse sentido,
camundongos modificados geneticamente com 1, 2, 3 e 4 cópias funcionais do gene da
ECA foram produzidos pelo Dr. John Krege no laboratório do Dr. Oliver Smithies
(University of North Carolina, Chapel Hill) através de recombinação homóloga de
células tronco embrionárias, que foram posteriormente introduzidas em zigotos. Pode-se
observar um aumento progressivo da atividade da ECA sérica e tecidual conforme
aumento do número de cópias do gene, ainda que os níveis de pressão arterial
permaneçam inalterados (KREGE, MOYER, LANGENBACH, PENG, ZHANG,
MAEDA, REDDICK & SMITHIES, 1997). Este modelo tem permitido reproduzir as
características observadas no polimorfismo da ECA presentes em humanos, onde os
níveis de ECA do genótipo II, ID e DD correlacionam-se com os níveis de ECA dos
animais com 1, 2 e 3 cópias, respectivamente.
3.3 Diabetes e Sistema Renina Angiotensina
A associação do SRA com o DM tem sido amplamente demonstrada na
literatura, especialmente em função do papel da Ang II para o desenvolvimento e
progressão do DM (TIKELLIS, COOPER & THOMAS, 2006). Ensaios clínicos

15
confirmaram que a inibição do SRA resulta em proteção do músculo esquelético e
eficácia no transporte de glicose (FROSSARD, JOUKHADAR, STEFFEN, SCHMID,
EICHLER & MULLER, 2000), proteção estrutural e funcional as ilhotas pancreáticas
(YUAN et al., 2010), reduzindo significativamente a incidência de complicações
vasculares em pacientes diabéticos (OAK & CAI, 2007; PARVING & ROSSING,
2001). De fato, a utilização de inibidores da ECA (iECA) e/ou bloqueadores do receptor
AT1 da ECA têm sido benéfico para a redução da disfunção metabólica provocada pelo
DM (CLERK et al., 2007), restauração da biodisponibilidade de óxido nítrico (NO) e da
enzima óxido nítrico sintetase (CARVALHO-PINTO, GARCIA, GOMEZ,
BALLESTEROS, ZABALLOS, FLORES, MELLADO, RODRIGUEZ-FRADE,
BALOMENOS & MARTINEZ) no endotélio (OAK & CAI, 2007), e
consequentemente, redução na disfunção endotelial.
A Ang II exerce ação potencializadora para o desenvolvimento de
microalbuminúria (MAUER et al., 2009), lesão tubular, glomerular, albuminúria
(NIELSEN, SUGAYA, TARNOW, LAJER, SCHJOEDT, ASTRUP, BABA,
PARVING & ROSSING, 2009) e proteinúria (COHEN & TOWNSEND, 2009). No
DM observa-se aumento da Ang II tecidual, e nos rins, isso está diretamente associado
ao desenvolvimento de nefropatia diabética (GIACCHETTI, SECHI, RILLI & CAREY,
2005). De fato, animais diabéticos quando tratados com bloqueadores do SRA
apresentaram redução de proteinúria, demonstrando que inibir a ação do SRA é
fundamental para a proteção e prevenção da nefropatia diabética (KAMAL,
YANAKIEVA-GEORGIEVA, PIAO, MORIOKA & OITE, 2010; ZOIA &
ARGYROPOULOS, 2010; ZOJA, CORNA, GAGLIARDINI, CONTI, ARNABOLDI,
BENIGNI & REMUZZI, 2010).

16
Outros estudos na literatura envolvendo modelos experimentais dão suporte à
associação entre SRA e DM. YUAN et al., (2010) observaram que animais diabéticos
por STZ quando tratados com bloqueadores do SRA, apresentavam aumento na
expressão de mRNA da insulina, redução da apoptose de ilhotas e redução do estresse
oxidativo. AROZAL et. al., (2009) demonstraram nesse mesmo modelo experimental
que o bloqueio do SRA reduziu o conteúdo de fibrose cardíaca e renal, reduziu a
proteinúria e o estresse oxidativo. Em síntese, o conjunto de achados confirma que
reduzir a ação do SRA no DM é fundamental para a proteção contra o desenvolvimento
de complicações renais e cardiovasculares associadas à patologia.
3.4 Adaptações metabólicas ao treinamento físico
A prática regular de exercícios físicos tem mostrado diminuição no risco de
desenvolvimento de DM, obesidade, hipertensão, entre outras doenças de risco
cardiovascular (FALLUCCA & POZZILLI, 2009). Estudos do nosso laboratório
mostraram que o treinamento físico aeróbio tem efeito positivo na função cardíaca
(EVANGELISTA, BRUM & KRIEGER, 2003) e no metabolismo energético
(EVANGELISTA & KRIEGER, 2006).
O treinamento físico aeróbio tem trazido muitos benefícios para indivíduos
portadores de DM e tem sido amplamente recomendado como uma estratégia não
farmacológica para o tratamento do DM (APOR, 2009; WELTMAN et al., 2009).
ERIKSEN; DAHL-PETERSEN; HAUGAARD; DELA (2007) observaram melhora na
glicemia de jejum e na tolerância à glicose em indivíduos que treinaram 3 sessões de 10
minutos por dia, com intensidade moderada a alta durante 4 semanas. SOUZA et. al.,
(2009), mostraram que oito semanas de treinamento físico aeróbio foram capazes de

17
reduzir a taxa de mortalidade em ratas diabéticas induzidas por STZ. Sabendo que a
redução do período de exposição à hiperglicemia está diretamente relacionada com a
redução do risco de complicações cardiovasculares no paciente diabético (KUSMINSKI
et al., 2009), o treinamento físico assume um papel importante no tratamento do DM.
A sensibilidade à ação da insulina é aumentada após um período de treinamento
físico através da melhora na sinalização do receptor de insulina e translocação do
GLUT4 (NAKAI, MIYAZAKI, SATO, OSHIDA, NAGASAKI, TAN AKA,
NAKASHIMA & SHIMOMURA, 2002; SHERMAN, FRIEDMAN, GAO, REED,
ELTON & DOHM, 1993). Enquanto isso, o triglicerídeo plasmático, aumentado no
DM1, diminui após o treinamento físico (LEME, GOMES, DE MELLO & LUCIANO,
2008), possivelmente associado à atividade da lipase lipoprotéica (HAMILTON,
AREIQAT, HAMILTON & BEY, 2001; KRAUSS, 2002).
Classicamente, a melhora do perfil glicêmico associado ao treinamento físico
está diretamente relacionada com as mudanças na sensibilidade da via insulínica
(HOWLETT, SAKAMOTO, HIRSHMAN, ASCHENBACH, DOW, WHITE &
GOODYEAR, 2002). No entanto, MACKNIGHT et al., (2009) observaram que a
contração muscular realizada durante o treinamento físico é capaz de aumentar o
transporte de glicose no músculo esquelético por mecanismos independentes da insulina
moduladas pela proteína quinase ativada por AMP (AMPK) e pelo cálcio (JESSEN &
GOODYEAR, 2005).
Quando fosforilada, a AMPK ativa vias que aumentam a síntese de ATP e
inativa as vias que consomem ATP (TAYLOR, ELLINGSON, LAMB, CHESSER,
COMPTON & WINDER, 2006). Durante o treinamento físico, devido à necessidade de
síntese de ATP, a AMPK atua nas vias de captação de glicose e oxidação de ácidos
graxos (MUSI, HIRSHMAN, ARAD, XING, FUJII, POMERLEAU, AHMAD,

18
BERUL, SEIDMAN, TIAN & GOODYEAR, 2005; STEPHENS, CHEN, CANNY,
MICHELL, KEMP & MCCONELL, 2002). De fato, MUSI et al., (2001), demonstraram
a relação entre o aumento da atividade da AMPK e a redução da glicemia durante o
treinamento físico em indivíduos diabéticos.
Diversas pesquisas têm procurado investigar a importância do polimorfismo do
gene da ECA na resposta ao treinamento físico. Estudos mostraram que a capacidade
máxima de esforço foi significativamente maior em indivíduos com genótipo II
comparados aos que apresentavam genótipo ID ou DD (MONTGOMERY,
CLARKSON, BARNARD, BELL, BRYNES, DOLLERY, HAJNAL, HEMINGWAY,
MERCER, JARMAN, MARSHALL, PRASAD, RAYSON, SAEED, TALMUD,
THOMAS, JUBB, WORLD & HUMPHRIES, 1999; ZHANG, WANG, DAI, LIN &
ZHANG, 2008). WILLIAMS, ANDERSON, SELIG, CAREY, FEBBRAIO, HAYES,
TOIA, HARRAP & HARE (2010) observaram que indivíduos que apresentaram
genótipo II tiveram melhor VO2 pico que seus pares homozigotos DD antes de iniciar
um período de treinamento físico, entretanto, essa diferença não se modificou após o
período de treino. Um dos possíveis mecanismos que poderia explicar essa diferença foi
a maior quantidade de fibras musculares tipo 1 e menor quantidade de fibras musculares
tipo 2 observadas em indivíduos homozigotos II (ZHANG, TANAKA, SHONO,
MIURA, KIYONAGA, SHINDO & SAKU, 2003).
Em contrapartida, EVANGELISTA & KRIEGER (2006), não encontraram
diferença nos ajustes metabólicos promovidos pelo treinamento físico de natação em
animais com 1 e 3 cópias do gene da ECA, cujos níveis diferenciais de ECA mimetizam
o polimorfismo do gene no ser humano. Esses dados demonstram as limitações que os
estudos com polimorfismo podem oferecer, tais como seleção e quantidade da amostra,
heterogeneidade genética das populações estudadas, dificuldade de controle dos fatores

19
ambientais como, por exemplo, a dieta das populações estudadas. Assim, a associação
do SRA com as respostas ao treinamento físico, especialmente no DM, ainda precisa ser
mais investigada.
3.5 O modelo experimental
A dificuldade em definir a relação entre genótipo e fenótipo de maneira
controlada tem sido um dos principais fatores limitantes nos estudos sobre a interação
gene-meio ambiente para o desenvolvimento de doenças complexas. Isso se torna
particularmente importante quando se considera que o DM é um fenótipo complexo
determinado por influências genéticas e ambientais. Supõe-se nestas condições que, a
contribuição individual de um fator genético seja modesta, impondo restrições
experimentais importantes à sua demonstração.
A possibilidade de modificar precisamente o conteúdo genético de camundongos
vem oferecendo inúmeras oportunidades para sobrepor essas limitações e possibilitar o
entendimento da participação individual de um determinado fator genético sobre
processos complexos no contexto do animal intacto (XIAO, ZHANG, CHAKIR,
AVDONIN, ZHU, BOND, BALKE, LAKATTA & CHENG, 2003).
No presente estudo, utilizamos camundongos modificados geneticamente com 1
e 3 cópias funcionais do gene da ECA, o que nos possibilitou efetivamente testar a
nossa hipótese de trabalho, já que os níveis de ECA circulante e tecidual são
significativamente superiores no animal com 3 cópias comparado ao animal com 1
cópia (EVANGELISTA & KRIEGER, 2006). Além disso, esses animais foram
submetidos à injeção de STZ para o desenvolvimento de diabetes experimental, e
posteriormente, submetidos ao treinamento físico aeróbio.

20
É importante salientar que, a utilização preferencial de camundongos para
manipulações genéticas é decorrente da disponibilidade de células tronco embrionárias
pluripotentes para esta espécie e do custo relativamente baixo para criação e
manutenção (RAO & VERKMAN, 2000). No mais, o modelo transgênico da ECA tem
sido frequentemente utilizado em estudos experimentais que visam investigar a
interação gene-meio ambiente para determinação de fenótipos complexos
(EVANGELISTA & KRIEGER, 2006; HUANG, GALLOIS, BOUBY, BRUNEVAL,
HEUDES, BELAIR, KREGE, MENETON, MARRE, SMITHIES & ALHENC-
GELAS, 2001; MESSADI, VINCENT, GRIOL-CHARHBILI, MANDET, COLUCCI,
KREGE, BRUNEVAL, BOUBY, SMITHIES, ALHENC-GELAS & RICHER, 2010)

21
4. MATERIAIS E MÉTODOS
4.1 Amostra
Foram utilizados camundongos machos C57BL/6 modificados geneticamente,
com 1 e 3 cópias do gene da ECA, apresentando idade entre 7 e 8 semanas, com peso
inicial variando entre 15 e 25 gramas, provenientes do Biotério Central da Faculdade de
Medicina da USP. Todos os animais foram submetidos ao diabetes, e posteriormente,
distribuídos em grupos de acordo com o número de cópias do gene da ECA, sendo que
parte dos animais de cada grupo foi mantido sedentário (S) e a outra parte foi submetida
ao protocolo de treinamento físico (T), conforme demonstrado na Tabela 1. Os animais
foram mantidos em caixas com água e comida administradas ad libitum, em ambiente
com temperatura controlada entre 22ºC e 24ºC e ciclo invertido claro-escuro (7am –
7pm).
TABELA 1: Separação dos grupos de animais submetidos ao protocolo experimental.
Grupos Sub-grupos Identificação (n)
Sedentário 1S 14
1 cópia
Treinado 1T 15
Sedentário 3S 10
3 cópias
Treinado 3T 11

22
Os procedimentos experimentais foram aprovados pelo Comitê de Ética em
Pesquisa da Escola de Educação Física e Esporte da Universidade de São Paulo
(protocolo n. 2008/38).
4.2 Modelo de camundongos geneticamente modificados
O modelo de animais geneticamente modificados foi desenvolvido pelo grupo
do Dr. Oliver Smithies através da técnica de recombinação gênica (KREGE et al., 1997;
KUNJATHOOR, WILSON & LEBOEUF, 1996). Esta técnica consiste em inserir por
recombinação uma sequência de DNA no lugar do gene a ser desativado, ou inserir uma
cópia extra de um gene ao “lado” do gene existente. Os nossos animais possuem
inserido o gene que confere resistência à neomicina entre o exon 14 e o íntron 13 da
ECA (animais “knockout” – gene da ECA modificado), ou inserida uma cópia extra do
gene da ECA (animais “knockin” – gene da ECA modificado) contendo pelo menos
3Kbs de seqüência a montante (5´) e a juzante (3´) das sequências codificadoras.
4.3 Planejamento dos cruzamentos dos camundongos com 1 a 4 cópias do
gene da ECA
A produção dos camundongos transgênicos é feita seguindo um padrão
específico de acasalamentos. Animais heterozigotos para a deleção (genótipo 1/0) e
duplicação (genótipo 1/2) são acasalados com os respectivos pares de tal sorte que em
cada geração nascem em média para o acasalamento deleção 25% de animais com os
genótipos homozigoto 1/1 e 0/0 e 50% de animais com o genótipo heterozigoto 1/0. Da
mesma forma, em cada geração nascem em média para o acasalamento duplicação 25%
de animais com genótipos homozigoto 1/1 e 2/2 e 50% de animais com o genótipo

23
heterozigoto 1/2. Este esquema de acasalamento permite que o ambiente entre os vários
grupos permaneça semelhante, pois os diferentes genótipos utilizados no estudo – 1 e 3
cópias do gene da ECA - são originados de apenas 2 acasalamentos.
4.4 Identificação dos animais
Após o nascimento, os animais foram identificados com um dispositivo
eletrônico introduzido subcutaneamente no dorso durante anestesia inalatória. Cada
dispositivo contém um código com letras e números, que pode ser identificado por
telemetria através de um leitor automático. O procedimento é simples, pois não
necessita sutura e em poucos minutos o animal estava totalmente recuperado.
4.5 Genotipagem
A genotipagem dos camundongos com 1 e 3 cópias do gene da ECA foi
realizada previamente ao início do protocolo. Após a identificação dos animais, biópsias
da orelha direita foram retiradas e o DNA genômico foi extraído a partir da digestão
com proteinase K e precipitação do DNA com isopropanol. A genotipagem foi realizada
através da técnica de PCR, determinando assim o número de cópias do gene da ECA.
No caso dos animais “knockout” foram utilizados três “primers”:
A (TAATTCCTTGGGAGGCAGCACT)
B (AGTGGAGGGTATTTGTCAGGGC)
C (TAAAGCGCATGCTCCAGACTGC).
Para os animais "knockin" foram utilizados dois "primers":
D11M+ (AAACAGAGATAAACCACGGGG)

24
D11M- (TGTGGAACTAACTCTCAGAAGGC)
Todos os experimentos foram realizados cegamente quanto ao genótipo dos
animais, de forma que a identificação dos grupos ocorreu somente após o término
completo das análises.
4.6 Indução do Diabetes Experimental
Na sétima semana de vida dos animais foi realizada a indução do diabetes
experimental através de injeção intraperitonial de estreptozotocina (STZ) (125 mg/kg de
peso corporal) (KUNJATHOOR, WILSON & LEBOEUF, 1996). Após sete dias da
administração de STZ, os animais foram mantidos em jejum por 6 horas e, após coleta
de sangue na cauda, a glicemia foi determinada utilizando-se fitas reativas para medida
de glicose (fitas reativas Advantage – Roche). Esse procedimento se faz necessário em
função do tempo de ação da STZ, pois apesar de causar destruição importante das
células β pacreáticas poucas horas após a injeção, a hiperglicemia resultante do
procedimento pode ser observada somente após alguns dias (JUNOD et al., 1969). O
critério de inclusão da amostra foi definido para a glicemia de jejum maior ou igual a
200mg/dl. Caso os níveis de glicose estivessem abaixo de 200mg/dl, uma dose adicional
de STZ com a mesma concentração de 125mg/kg de peso corporal seria administrada, e
novamente foram aguardados mais sete dias para a avaliação da glicemia de jejum
(KUNJATHOOR el al, 1996). De acordo com JUNOD et al. (1967), uma segunda dose
de STZ tem maior potencial para causar danos às células β pancreáticas devido ao
aumento da sensibilidade à ação da STZ provocado pela primeira dose.

25
4.7 Treinamento Físico
O início do treinamento físico aconteceu após 7 semanas de diabetes confirmado
pela verificação da glicemia de jejum acima de 200mg/dl. Buscamos com isso, avaliar o
efeito do treinamento físico iniciado em uma fase da patologia cuja hiperglicemia
estivesse cronicamente instalada. Além disso, utilizamos os dados prévios que
demonstraram aumento da mortalidade dos animais diabéticos com 3 cópias do gene da
ECA na 14ª semana após indução do diabetes por estreptozotocina para definir o curso
temporal das complicações associadas ao diabetes e seu desfecho final. Assim, sabendo
que o treinamento físico duraria 8 semanas, o início do protocolo de natação foi
programado para a 7ª semana após indução do diabetes, encerrando-se na 15ª semana de
diabetes (HEIMANN, 2003).
O treinamento físico aeróbio foi realizado com natação adaptado do estudo
realizado por EVANGELISTA et al. (2003). Após uma semana de adaptação com 10
minutos diários, iniciou-se o treinamento com duração de 20 minutos, aumentando
diariamente 10 minutos até atingir-se 90 minutos na segunda semana, uma vez ao dia,
cinco dias por semana, durante oito semanas. Os dias de descanso dos animais
ocorreram apenas às quartas-feiras e domingos.
Para minimizar a influência do efeito do estresse exercido pelo meio aquoso, os
grupos sedentários foram colocados no sistema de natação 2 vezes por semana durante 5
minutos.
4.8 Avaliação da capacidade de esforço físico
A capacidade de realização de esforço físico foi avaliada nos grupos estudados
através de um teste progressivo até a exaustão em esteira rolante (FERREIRA; ROLIM;

26
GRANÁ; BARTHOLOMEU & BRUM, 2004). O teste iniciou com velocidade de
6m/min e teve incremento de 3m/min a cada 3 minutos até a exaustão do animal. Este
teste foi realizado antes e após o período de treinamento físico, e foram quantificados o
tempo máximo de realização do teste, a velocidade atingida no pico do esforço e a
distância total percorrida. Embora o teste em esteira não seja específico ao tipo de
treinamento físico realizado no presente estudo, utilizamos esse teste apenas para
indicar a capacidade de execução de exercício físico.
4.9 Avaliações durante o protocolo experimental in vivo
4.9.1 Fatores determinantes do balanço energético
Ao longo das 15 semanas de protocolo experimental, o peso corporal foi
acompanhado semanalmente em balança digital (Gehaka, modelo BG4001), sempre no
mesmo dia e horário.
O consumo de ração foi avaliado em grupos de animais com genótipos
semelhantes mantidos na mesma gaiola ao longo de todo o período experimental,
durante períodos de 24 horas, por 3 dias consecutivos em cada semana. Foi utilizada a
média dos 3 dias avaliados para determinar o consumo de ração diário.
A taxa metabólica de repouso dos animais foi determinada através da avaliação
do consumo de oxigênio por calorimetria indireta antes e após o período de treinamento
físico. Para isso, os animais permaneceram 24 horas sem realização de exercício físico e
foram mantidos por 6 horas em jejum. O tempo de permanência dos animais dentro da
caixa metabólica foi de 30 minutos, sendo que o repouso foi considerado o menor valor
de oxigênio consumido observado. Para o cálculo de consumo de oxigênio, foi utilizada
a seguinte fórmula: VO2 = (Fi O2 -Fe O2) x 1.165 / peso corporal, onde: Fi O2 = fração

27
inspirada de oxigênio; Fe O2 = fração expirada de oxigênio; 1.165 = fluxo de ar na
bomba de ar.
4.9.2 Teste de tolerância à glicose intraperitoneal (TTGI)
A tolerância à glicose foi avaliada antes e após o período de treinamento físico,
através da determinação da curva glicêmica após administração intraperitoneal de
glicose (2 g/kg p.c.) nos animais previamente submetidos ao jejum de 6h. A glicemia foi
determinada por meio de glicosímetro (Accu-Chek Advantage Roche Diagnostics) em
amostras de sangue retiradas da cauda nos tempos 0 (basal), 15, 30, 60, 90 e 120
minutos após a administração de glicose.
4.10 Sacrifício e coleta de tecidos e sangue
Após 24 horas da última sessão de treinamento físico, os camundongos foram
anestesiados por injeção i.p. de Pentobarbital Sódico. Em seguida, foram pesados em
balança digital, e posicionados sob uma régua para a medida do comprimento naso-anal
que serviu para a determinação do Índice de Lee através da fórmula: (³√peso
corporal/comprimento naso-anal) (BERNARDIS & PATTERSON, 1968). No momento
em que o animal não mais demonstrou sinais de reflexo nas patas traseiras, a cavidade
abdominal foi exposta e a coleta de sangue foi realizada através de punção na veia cava
inferior. Em seguida, foram coletados e pesados os depósitos de gordura branca (tecido
adiposo periepididimal e retroperitoneal), os músculos esqueléticos das patas
(gastrocnêmio e sóleos) e os órgãos (coração, pulmão, rins, fígado, glândulas adrenais e
baço).

28
O sangue foi centrifugado a 40C, 12000 rpm por 10 min, e o plasma armazenado
em freezer a –800C para determinação da concentração de leptina por radioimunoensaio
(RIA) utilizando anticorpo espécie-específico (kit da Linco Research Inc.®) e da
insulina através de kit (Sensitive rat insulin RIA Kit - Linco research, Inc.). O tecido
adiposo periepididimal foi destinado para a avaliação de lipólise e medida do diâmetro
dos adipócitos, o tecido adiposo retroperitoneal e hepático foram utilizados para a
dosagem da atividade da enzima ácido graxo sintase (FAS), o músculo sóleo de uma das
patas foi retirado para a dosagem da atividade da enzima citrato sintase, além da loja
muscular da outra pata, contendo os músculos gastrocnênio, sóleo e plantar para
quantificação da tipagem de fibra muscular e razão capilar/fibra muscular.
4.11 Estudo do tecido adiposo branco
4.11.1 Extração dos adipócitos e análise morfométrica
Os adipócitos foram isolados do tecido adiposo periepididimal mediante a
técnica de digestão de tecido pela colagenase, descrita por RODBELL (1964), com
algumas modificações para adaptar o método às nossas condições laboratoriais. Em
resumo, o depósito adiposo periepididimal foi retirado, picado com tesoura em finos
fragmentos e incubado em 4,0 mL de tampão digestivo [DMEM, HEPES 25 mM, BSA
4 %, colagenase tipo II (Sigma Chemical, St. Louis, MO, Estados Unidos) 1,25 mg/mL,
pH 7,45] por cerca de 30 min a 37 oC em banho-maria com agitação orbital (150 rpm).
Em seguida, a amostra foi filtrada em peneira plástica com malha fina (que retém restos
teciduais e vasos não digeridos) e lavada por três vezes com 25 mL de tampão EHB
(sais de EARLE, HEPES 25 mM, BSA 1 %, piruvato de sódio 1 mM, sem glicose, pH
7,45) mantido a 37 ºC. Após a segunda lavagem, a suspensão celular em 25 mL de

29
tampão EHB foi deixada 30 min em repouso em estufa a 37 °C, com a finalidade de
atenuar os efeitos da insulina e de outros hormônios endógenos. Para a determinação do
lipócrito (porcentagem de adipócitos contidos na suspensão celular total), 40 µL da
suspensão celular em tampão EHB foram colocados em capilar de vidro e submetidos à
centrifugação (2000 rpm por 1 min). O volume total da suspensão corresponde a 100 %
e o volume de adipócitos obtido após a centrifugação, nos fornece o lipócrito da
amostra.
Para a análise morfométrica, alíquotas de suspensão celular foram avaliadas em
microscópio óptico com o programa Leica Quantimet 500. Em cada preparação foram
medidas 100 células. A partir do diâmetro celular médio e admitindo-se que o adipócito
isolado é esférico, o volume e o número de células foram calculados de acordo com as
seguintes fórmulas:
(a) V= [(π/6) x D3]/1000, (b) N= (lipócrito x 107)/ V
Onde:
D é o diâmetro médio de 50 adipócitos (µm), N é o número de células e V é o
volume médio (DI GIROLAMO, MENDLINGER & FERTIG, 1971). A divisão por
1000 em (a) visa expressar o volume em picolitros (pL).
4.11.2 Avaliação da atividade lipolítica frente ao estímulo com isoproterenol
As taxas de lipólise basal e estimulada pelo agonista β-adrenérgico isoproterenol
(Sigma®) foram mensuradas em adipócitos isolados do tecido adiposo periepididimal
conforme o seguinte protocolo: 40 µL de suspensão celular (em tampão EHB) foram
transferidos para microtubos (1,5 mL) e incubados durante 5 min a 37 °C em presença

30
de 20 µL de adenosina deaminase (ADA, Sigma, 0,2 U/mL em tampão EHB, pH 7,45)
para permitir a degradação da adenosina, um metabólito com ação anti-lipolítica,
liberada no meio pelos adipócitos (HONNOR, DHILLON & LONDOS, 1985). Após
este período, as células foram incubadas por 60 min a 37 °C com ou sem 10 µL de
isoproterenol (10-5 M) totalizando um volume de 200 µL. Ao final da incubação, a
mistura de reação foi bloqueada pela transferência dos tubos para o gelo seguido por
centrifugação a 12000 rpm por 10 min a 4 °C, para separar as células do meio de reação.
Após a centrifugação, alíquotas de aproximadamente 120 µL do meio de incubação
foram retiradas e armazenadas em freezer a –20 °C para posterior mensuração do
glicerol nele contido. A quantidade de glicerol liberada pelos adipócitos para o meio de
incubação foi determinada pelo método enzimático-colorimétrico (Sigma®). Os
resultados foram expressos em nmol.10-6 cels.h-1.
4.11.3 Dosagem da atividade da enzima ácido graxo sintase (FAS)
A FAS é a principal enzima envolvida no processo de lipogênese, a qual é
definida por processos metabólicos que resultam na biossíntese e incorporação de
triglicerídeos nas gotículas de gordura do adipócito (FONSECA-ALANIZ, TAKADA,
ALONSO-VALE & LIMA, 2006). Os principais locais de ação da FAS são no tecido
adiposo e no tecido hepático. Para que ocorra a reação da lipogênese, além da enzima
FAS é necessária a disponibilidade do substrato (carboidrato) e de cofatores (NADPH),
este último produzido pela via da pentose fosfato (FOUFELLE, GIRARD & FERRE,
1996). A ação catalisadora da FAS acontece na formação de acilCoA à partir do
malonil-CoA, que será utilizado para a esterificação com glicerol-3-P, completando a

31
síntese de triacilglicerol e posterior incorporação a gotícula de lipídio (FONSECA-
ALANIZ et. al., 2006).
Para dosagem da atividade da FAS, amostras do tecido adiposo RP e fígado
(aproximadamente 0,2 g) foram suspensas em tampão de extração (na proporção de 1:2
e 1:3 peso/volume, respectivamente) contendo sacarose (250 mM), EDTA (1 mM),
DTT (1 mM), leupeptina (50 µM) e aprotinina (5 µM) pH=7.4. O material mantido em
gelo foi homogeneizado em Polytron (PT 3100, Kinematica AG, Littau-Lucene, Suíça)
por 10 segundos em velocidade máxima, centrifugado (20800 g, 15 minutos, 0 °C em
Centrífuga 5417 C/R- Eppendorf) para separação dos restos celulares e lipídicos.
Volumes de 20 µL do infranadante para o tecido adiposo RP e 15 µL do sobrenadante
para o fígado desta última centrifugação foram utilizados para a análise indireta da
atividade enzimática da FAS, como medida da oxidação (consumo) total de NADPH
(BAZIN & FERRE, 2001).
O tampão de ensaio utilizado (volume de 270 µL para tecido adiposo RP e 275
µL para fígado) consistiu de KH2PO4 (100 mM), acetilCoA (100 µM) e NADPH (200
µM), pH=6.5. A reação foi iniciada com a adição de 10 µL de malonilCoA (600 µM)
ao extrato enzimático, e acompanhada por 5 minutos (37 °C). A absorbância foi
monitorada a 340 nm, sendo o coeficiente de extinção para este comprimento de onda
igual a 6,22. As proteínas foram quantificadas pelo método de BRADFORD (1976). Os
resultados foram expressos em nmol.min-1.mg-1 de proteína presente no extrato.
4.12 Estudo do músculo esquelético
4.12.1 Dosagem da atividade máxima da enzima citrato sintase

32
A citrato sintase é uma importante enzima oxidativa, pois cataliza a primeira
reação do ciclo de Krebs, onde ocorre a condensação do Acetil coenzima A (Acetil-
CoA) com o oxalacetato para formar citrato e coenzima A (COATE & HUGGINS,
2010). É uma enzima reguladora, e em muitos tipos de células, a reação por ela
catalizada é o passo limitante na velocidade de todo o ciclo de Krebs e
consequentemente na formação de energia via metabolismo aeróbio (LEHNINGER,
1991).
Apenas uma amostra do músculo sóleo foi suspensa em tampão de extração (300
µL), contendo Tris Base (50mM), EDTA (1mM), pH 7,4. O material mantido em gelo
foi homogeneizado em Polytron (PT 3100, Kinematica AG, Littau-Lucene, Suíça) por
10 segundos, em velocidade máxima, centrifugado (3000 rpm, 15 minutos, 4 °C em
Centrífuga 5417 C/R- Eppendorf) e um volume de 15 µL do sobrenadante dessa
centrifugação foi utilizado para determinação da atividade da enzima citrato sintase
segundo ALP e cols. (1976).
A mistura de ensaio utilizada continha Tris Base (100mM), 5,5´ditio-bis– (2–
nitrobenzoic acid (DTNB, 0,2 mM), Triton 0,1%, acetil-CoA 1mg, pH 8,1 . Para análise
da cinética enzimática, foram utilizados 15 uL do homogenato das amostras e 270 uL de
mistura de ensaio. Após estabilização por 5 min, a reação foi iniciada com a adição de
15 µL de ácido oxaloacético (0,5 mM) ao extrato enzimático, e acompanhada por 10
minutos (25 °C). A absorbância foi monitorada a 412 nm, sendo o coeficiente de
extinção para este comprimento de onda igual a 13,6.
As proteínas foram quantificadas pelo método de BRADFORD (1976). Neste

33
ensaio, as proteínas da amostra são complexadas com o corante azul de coomassie
brilhante G-250. A reação foi colorimétrica e a absorbância foi medida a 595 nm. Os
resultados de absorbância obtidos foram lançados na equação da reta de uma curva
padrão de albumina sérica bovina (1 a 10 g/ml), repetida a cada determinação
enzimática. A concentração da solução padrão de albumina foi aferida antes da
preparação de cada curva, dividindo-se o valor de absorbância obtido em 280 nm por
0,66. Os resultados foram expressos em nmol.min-1.mg-1 de proteína presente no
extrato.
4.12.2 Reação histoquímica para Miosina ATPase e determinação da
tipagem de fibras musculares
Após a retirada da loja posterior composta pelos músculos gastrocnêmio, sóleo e
plantar, as amostras foram mergulhadas em isopentano durante 10 segundos (FIGURA
1A), que tem a ação crioprotetora, evitando assim artefatos nas amostras, e
posteriormente congeladas em nitrogênio líquido (FIGURA 1B), até que os cortes
fossem realizados.
FIGURA 1. Loja muscular posterior após mergulhada no isopentano (A) e
Congelamento das amostras em nitrogênio líquido (B).

34
Os cortes seriados dos músculos com espessuras de 10µm foram montados em
lâminas para as reações histoquímicas. As lâminas foram dispostas em cubetas
diferentes para os pHs 4.6 e 10.3 e nelas realizadas as reações com formol de cálcio
(CaCl e formalina 10%) seguido de tampão glicina (glicina + NaCl + CaCl . 2H2O +
NaOH 1M) para o pH 10.3 ou tampão acetato (Na-acetato + H2O destilada + Ácido
acético glacial + CaCl . 2H2O) para o pH 4.6. As lâminas das duas cubetas foram
passadas para uma única cubeta contendo solução ATP (ATP + tampão glicina, pH 9.5),
seguido de banho com cloreto de cobalto 2% (CoCl6H2O), e posteriormente solução
0,5% de sulfeto de amônio por 3 minutos. Finalmente, os cortes foram lavados em água
destilada e montados com gelatina-glicerina. A captura das imagens foi realizada
através do sistema de vídeo (LEICA QUANTIMET 500) com magnificação de 200x e
objetiva de 20x acoplado ao computador.
Os músculos sóleo e plantar foram identificados conforme descrito por
(SPURWAY, 1981). Não foram atribuídos número de campos para análise, pois a
avaliação foi realizada no músculo sóleo inteiro. A quantificação dos diferentes tipos de
fibras musculares no sóleo foi realizada por um único observador por meio do programa
Image-Pro Plus.
Para a identificação dos subtipos de fibras, em cortes seriados processados em
diferentes pHs 4,3 e 10,3, foi realizada a comparação entre as colorações obtidas nesses
ensaio segundo as observações de HAMALAINEN & PETTE (1993). A classificação
dos tipos de fibras atribuída pela coloração está exemplificada na FIGURA 2.

35
FIGURA 2. Classificação dos tipos de fibras no músculo sóleo no pH 10,3.
4.12.3 Quantificação de capilares no músculo esquelético
Por meio da reação histoquímica para miosina ATPase no pH 10,3, em uma
magnificação de 400x foi quantificado o número de capilares localizados em torno das
fibras do músculo sóleo inteiro, bem como a quantidade de fibras musculares. A
contagem dos capilares e das fibras foi realizada em sistema computadorizado (LEICA
QUANTIMET 500). Para a determinação da razão número de capilares/fibra foi
dividido o número de capilares pelo número de fibras encontrados no músculo sóleo.

36
FIGURA 3. Identificação dos capilares no músculo sóleo.
4.13 Desenho dos procedimentos experimentais

37
4.14 Análise Estatística
Os dados foram apresentados na forma de média ± erro padrão da média (EPM).
Com o propósito de avaliar o efeito do genótipo da ECA sobre os distúrbios
metabólicos associados ao diabetes, foi utilizado o Teste t de Student para dados não
pareados. Para o estudo do efeito do genótipo da ECA e do treinamento físico (fatores
independentes), foi utilizada análise de variância (ANOVA) de dois caminhos para
dados repetidos para as seguintes variáveis: evolução do peso corporal, consumo de
oxigênio e teste de esforço. As demais variáveis foram avaliadas através de análise de
variância (ANOVA) de dois caminhos (genótipo e treinamento físico como fatores
independentes). Na presença de diferenças estatísticas, foi utilizado o teste post-hoc de
Bonferroni. Em todos os casos foi adotado nível de significância de 5% (p<0,05).
38
5. RESULTADOS
5.1 Composição corporal
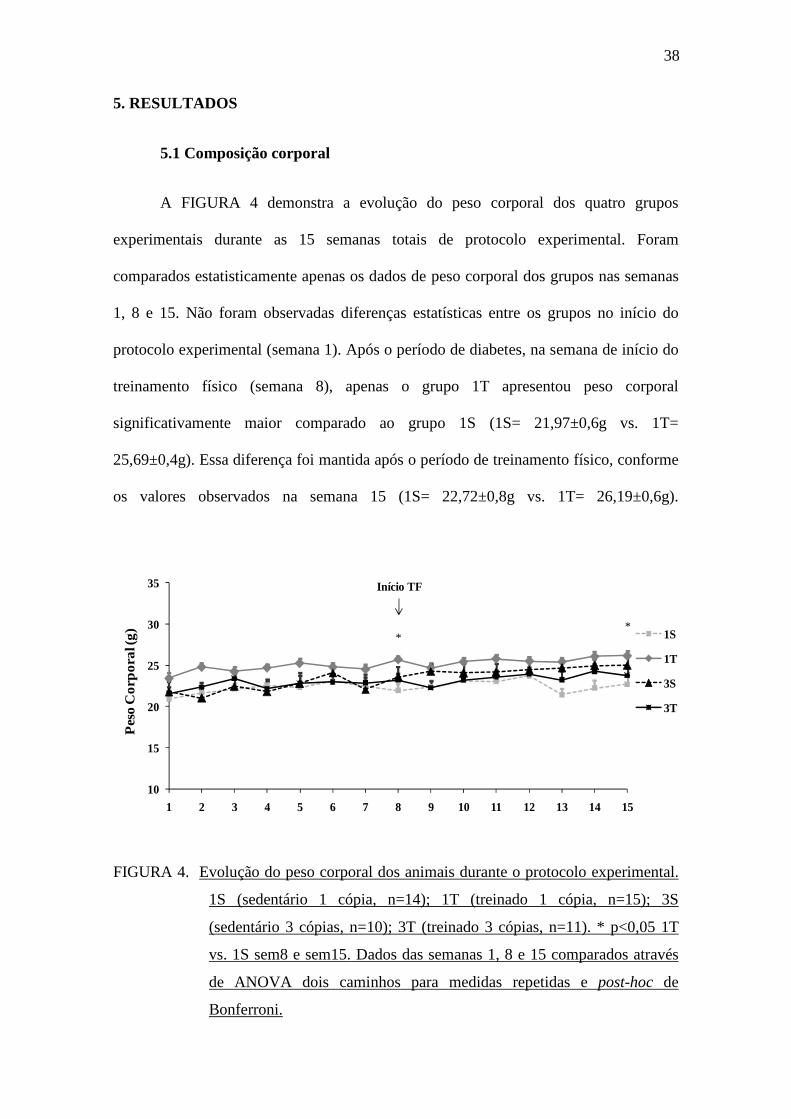
A FIGURA 4 demonstra a evolução do peso corporal dos quatro grupos
experimentais durante as 15 semanas totais de protocolo experimental. Foram
comparados estatisticamente apenas os dados de peso corporal dos grupos nas semanas
1, 8 e 15. Não foram observadas diferenças estatísticas entre os grupos no início do
protocolo experimental (semana 1). Após o período de diabetes, na semana de início do
treinamento físico (semana 8), apenas o grupo 1T apresentou peso corporal
significativamente maior comparado ao grupo 1S (1S= 21,97±0,6g vs. 1T=
25,69±0,4g). Essa diferença foi mantida após o período de treinamento físico, conforme
os valores observados na semana 15 (1S= 22,72±0,8g vs. 1T= 26,19±0,6g).
10
15
20
25
30
35
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
Pes
o C
orp
ora
l (g
)
1S
1T
3S
3T
Início TF
**
FIGURA 4. Evolução do peso corporal dos animais durante o protocolo experimental.
1S (sedentário 1 cópia, n=14); 1T (treinado 1 cópia, n=15); 3S
(sedentário 3 cópias, n=10); 3T (treinado 3 cópias, n=11). * p<0,05 1T
vs. 1S sem8 e sem15. Dados das semanas 1, 8 e 15 comparados através
de ANOVA dois caminhos para medidas repetidas e post-hoc de
Bonferroni.
39
Ainda na FIGURA 4, pode-se observar que não houve diferença estatística no
peso corporal dos grupos no término do protocolo (semana 15) comparado aos
respectivos pesos corporais avaliados no início do protocolo (semana 1).
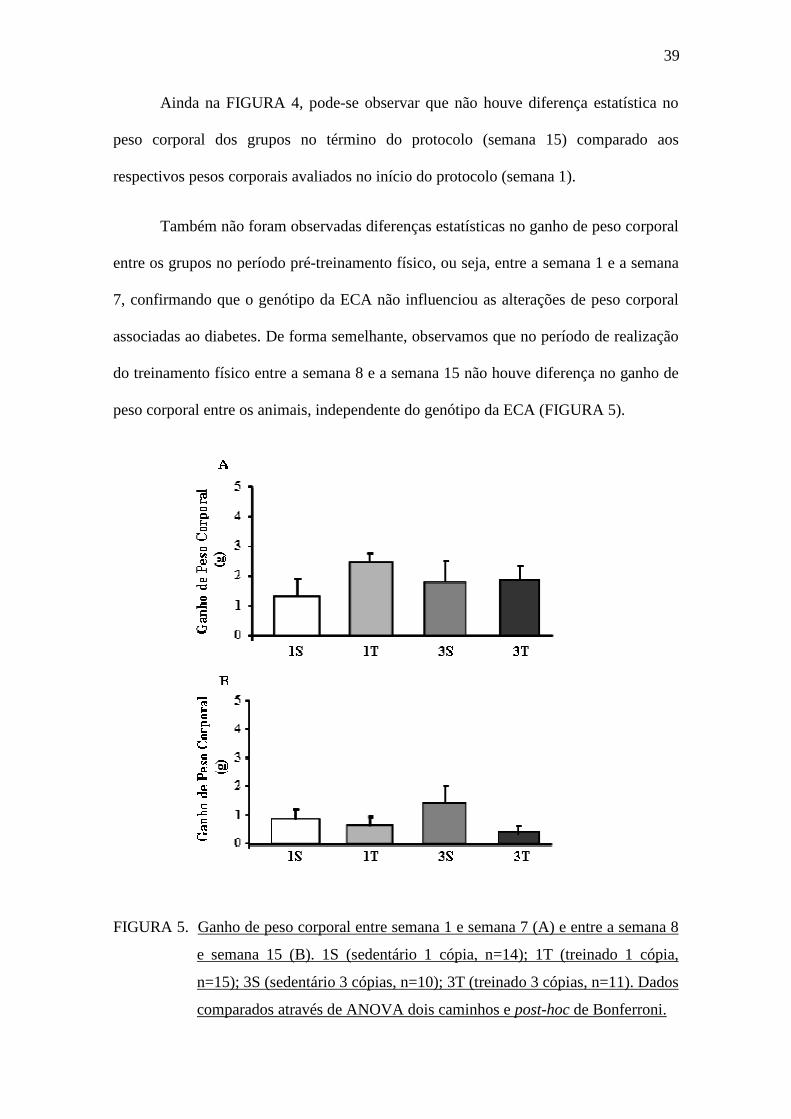
Também não foram observadas diferenças estatísticas no ganho de peso corporal
entre os grupos no período pré-treinamento físico, ou seja, entre a semana 1 e a semana
7, confirmando que o genótipo da ECA não influenciou as alterações de peso corporal
associadas ao diabetes. De forma semelhante, observamos que no período de realização
do treinamento físico entre a semana 8 e a semana 15 não houve diferença no ganho de
peso corporal entre os animais, independente do genótipo da ECA (FIGURA 5).
FIGURA 5. Ganho de peso corporal entre semana 1 e semana 7 (A) e entre a semana 8
e semana 15 (B). 1S (sedentário 1 cópia, n=14); 1T (treinado 1 cópia,
n=15); 3S (sedentário 3 cópias, n=10); 3T (treinado 3 cópias, n=11). Dados
comparados através de ANOVA dois caminhos e post-hoc de Bonferroni.
40
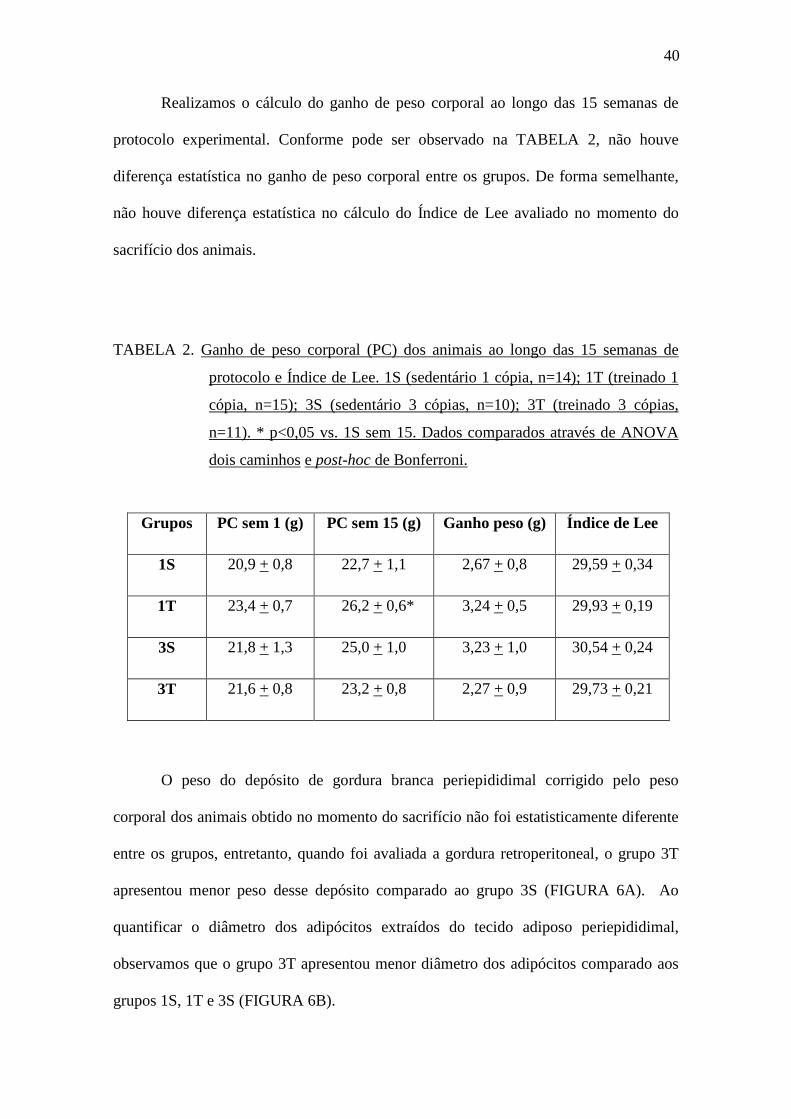
Realizamos o cálculo do ganho de peso corporal ao longo das 15 semanas de
protocolo experimental. Conforme pode ser observado na TABELA 2, não houve
diferença estatística no ganho de peso corporal entre os grupos. De forma semelhante,
não houve diferença estatística no cálculo do Índice de Lee avaliado no momento do
sacrifício dos animais.
TABELA 2. Ganho de peso corporal (PC) dos animais ao longo das 15 semanas de
protocolo e Índice de Lee. 1S (sedentário 1 cópia, n=14); 1T (treinado 1
cópia, n=15); 3S (sedentário 3 cópias, n=10); 3T (treinado 3 cópias,
n=11). * p<0,05 vs. 1S sem 15. Dados comparados através de ANOVA
dois caminhos e post-hoc de Bonferroni.
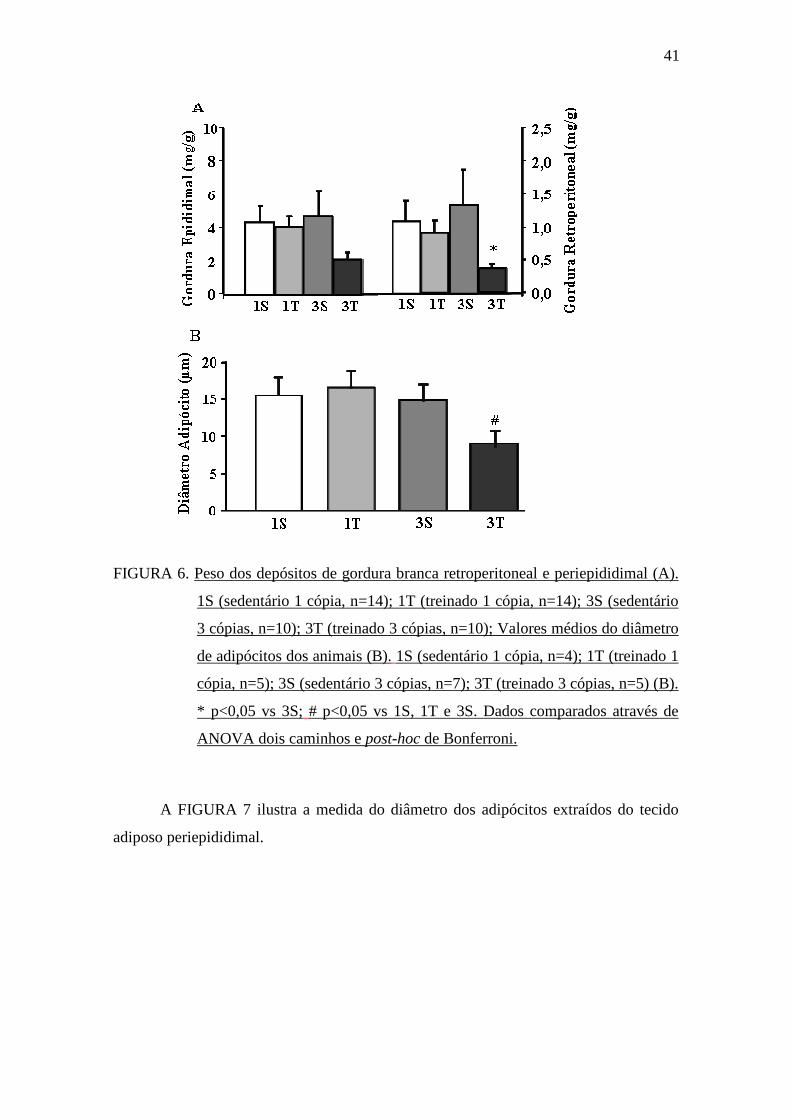
O peso do depósito de gordura branca periepididimal corrigido pelo peso
corporal dos animais obtido no momento do sacrifício não foi estatisticamente diferente
entre os grupos, entretanto, quando foi avaliada a gordura retroperitoneal, o grupo 3T
apresentou menor peso desse depósito comparado ao grupo 3S (FIGURA 6A). Ao
quantificar o diâmetro dos adipócitos extraídos do tecido adiposo periepididimal,
observamos que o grupo 3T apresentou menor diâmetro dos adipócitos comparado aos
grupos 1S, 1T e 3S (FIGURA 6B).
Grupos PC sem 1 (g) PC sem 15 (g) Ganho peso (g) Índice de Lee
1S 20,9 + 0,8 22,7 + 1,1 2,67 + 0,8 29,59 + 0,34
1T 23,4 + 0,7 26,2 + 0,6* 3,24 + 0,5 29,93 + 0,19
3S 21,8 + 1,3 25,0 + 1,0 3,23 + 1,0 30,54 + 0,24
3T 21,6 + 0,8 23,2 + 0,8 2,27 + 0,9 29,73 + 0,21
41
FIGURA 6. Peso dos depósitos de gordura branca retroperitoneal e periepididimal (A).
1S (sedentário 1 cópia, n=14); 1T (treinado 1 cópia, n=14); 3S (sedentário
3 cópias, n=10); 3T (treinado 3 cópias, n=10); Valores médios do diâmetro
de adipócitos dos animais (B). 1S (sedentário 1 cópia, n=4); 1T (treinado 1
cópia, n=5); 3S (sedentário 3 cópias, n=7); 3T (treinado 3 cópias, n=5) (B).
* p<0,05 vs 3S; # p<0,05 vs 1S, 1T e 3S. Dados comparados através de
ANOVA dois caminhos e post-hoc de Bonferroni.
A FIGURA 7 ilustra a medida do diâmetro dos adipócitos extraídos do tecido
adiposo periepididimal.

42
FIGURA 7. Imagens ilustrativas da quantificação do diâmetro dos adipócitos extraídos
do tecido adiposo periepididimal. 1S (sedentário 1 cópia); 1T (treinado 1
cópia); 3S (sedentário 3 cópias); 3T (treinado 3 cópias).
Os pesos dos órgãos corrigidos pelo peso corporal avaliado no momento do
sacrifício estão demonstrados na FIGURA 8A. Conforme pode ser observado, não
houve diferença estatística no peso dos órgãos entre os grupos, exceto para o peso dos
pulmões, onde o grupo 3S apresentou menor peso comparado aos grupos 1T e 3T. Não
foram observadas diferenças estatísticas no peso do músculo sóleo e no peso do
músculo gastrocnêmio dos animais submetidos ao protocolo experimental (FIGURA
8B).
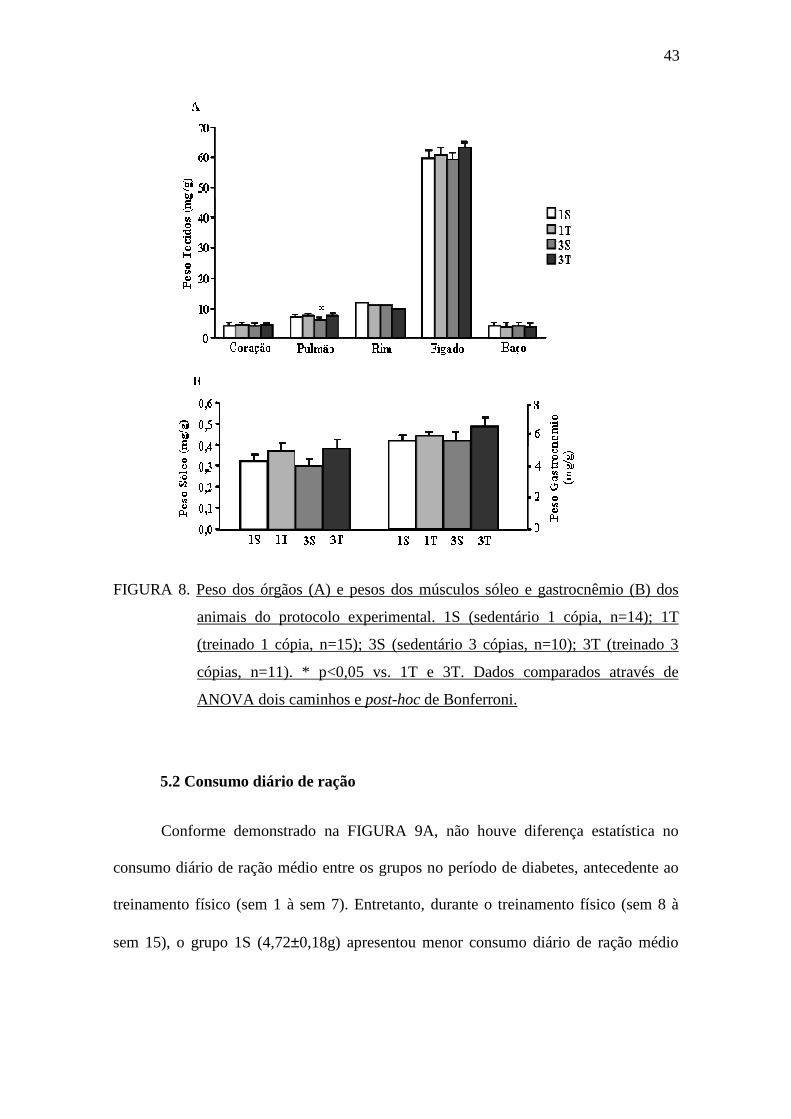
43
FIGURA 8. Peso dos órgãos (A) e pesos dos músculos sóleo e gastrocnêmio (B) dos
animais do protocolo experimental. 1S (sedentário 1 cópia, n=14); 1T
(treinado 1 cópia, n=15); 3S (sedentário 3 cópias, n=10); 3T (treinado 3
cópias, n=11). * p<0,05 vs. 1T e 3T. Dados comparados através de
ANOVA dois caminhos e post-hoc de Bonferroni.
5.2 Consumo diário de ração
Conforme demonstrado na FIGURA 9A, não houve diferença estatística no
consumo diário de ração médio entre os grupos no período de diabetes, antecedente ao
treinamento físico (sem 1 à sem 7). Entretanto, durante o treinamento físico (sem 8 à
sem 15), o grupo 1S (4,72±0,18g) apresentou menor consumo diário de ração médio
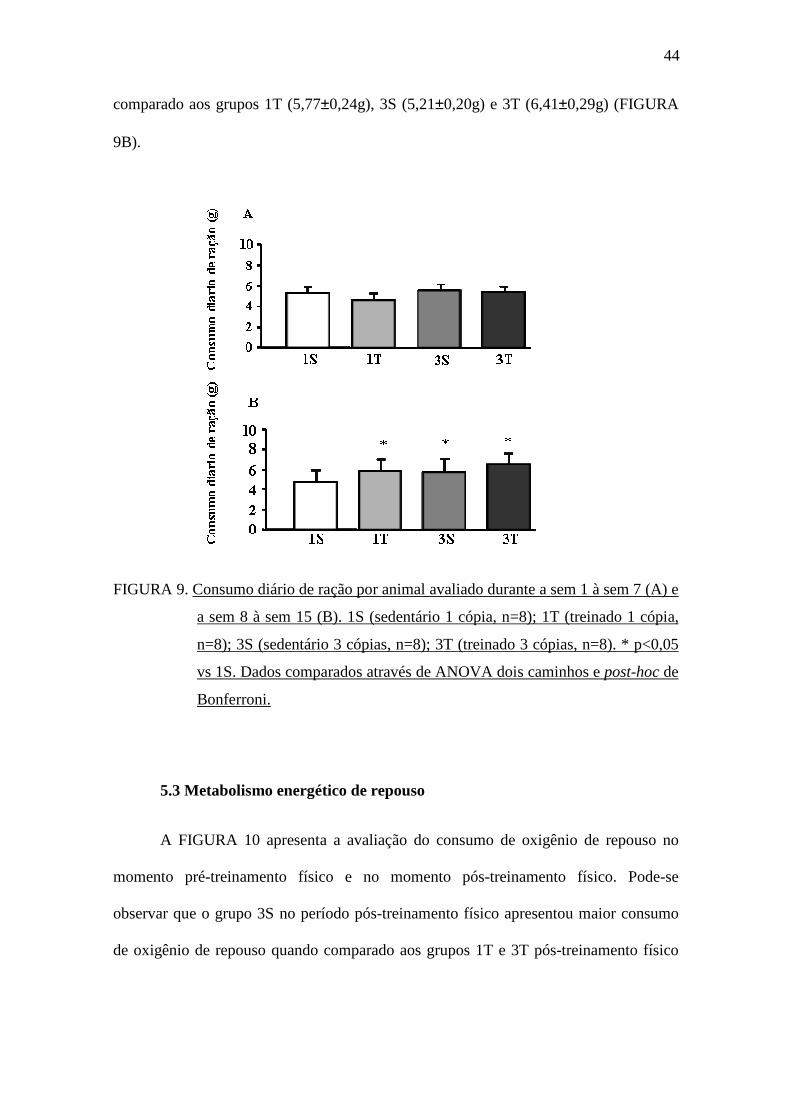
44
comparado aos grupos 1T (5,77±0,24g), 3S (5,21±0,20g) e 3T (6,41±0,29g) (FIGURA
9B).
FIGURA 9. Consumo diário de ração por animal avaliado durante a sem 1 à sem 7 (A) e
a sem 8 à sem 15 (B). 1S (sedentário 1 cópia, n=8); 1T (treinado 1 cópia,
n=8); 3S (sedentário 3 cópias, n=8); 3T (treinado 3 cópias, n=8). * p<0,05
vs 1S. Dados comparados através de ANOVA dois caminhos e post-hoc de
Bonferroni.
5.3 Metabolismo energético de repouso
A FIGURA 10 apresenta a avaliação do consumo de oxigênio de repouso no
momento pré-treinamento físico e no momento pós-treinamento físico. Pode-se
observar que o grupo 3S no período pós-treinamento físico apresentou maior consumo
de oxigênio de repouso quando comparado aos grupos 1T e 3T pós-treinamento físico
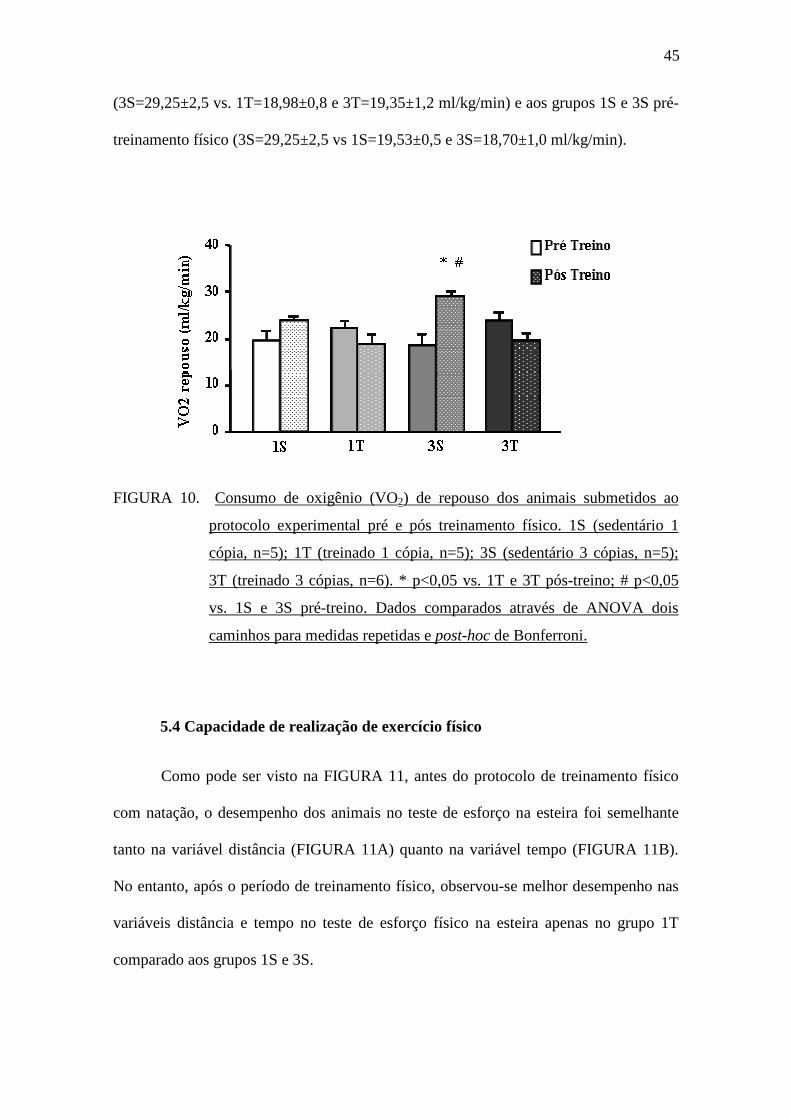
45
(3S=29,25±2,5 vs. 1T=18,98±0,8 e 3T=19,35±1,2 ml/kg/min) e aos grupos 1S e 3S pré-
treinamento físico (3S=29,25±2,5 vs 1S=19,53±0,5 e 3S=18,70±1,0 ml/kg/min).
FIGURA 10. Consumo de oxigênio (VO2) de repouso dos animais submetidos ao
protocolo experimental pré e pós treinamento físico. 1S (sedentário 1
cópia, n=5); 1T (treinado 1 cópia, n=5); 3S (sedentário 3 cópias, n=5);
3T (treinado 3 cópias, n=6). * p<0,05 vs. 1T e 3T pós-treino; # p<0,05
vs. 1S e 3S pré-treino. Dados comparados através de ANOVA dois
caminhos para medidas repetidas e post-hoc de Bonferroni.
5.4 Capacidade de realização de exercício físico
Como pode ser visto na FIGURA 11, antes do protocolo de treinamento físico
com natação, o desempenho dos animais no teste de esforço na esteira foi semelhante
tanto na variável distância (FIGURA 11A) quanto na variável tempo (FIGURA 11B).
No entanto, após o período de treinamento físico, observou-se melhor desempenho nas
variáveis distância e tempo no teste de esforço físico na esteira apenas no grupo 1T
comparado aos grupos 1S e 3S.
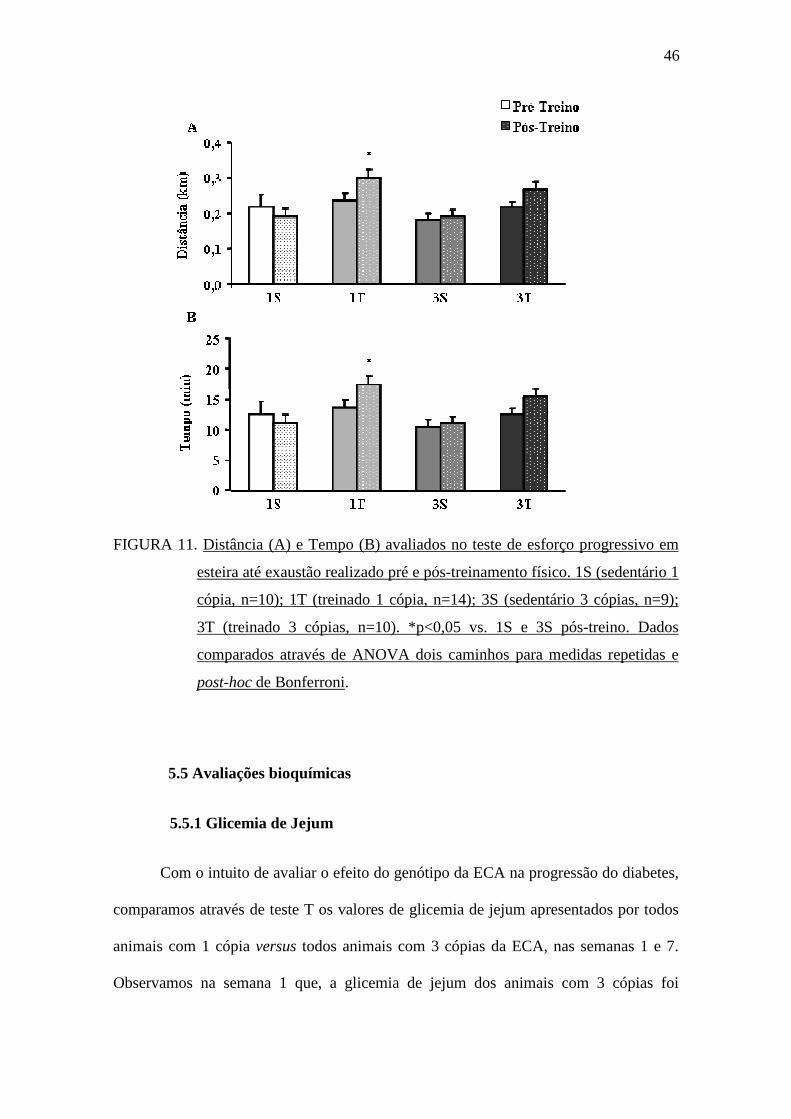
46
FIGURA 11. Distância (A) e Tempo (B) avaliados no teste de esforço progressivo em
esteira até exaustão realizado pré e pós-treinamento físico. 1S (sedentário 1
cópia, n=10); 1T (treinado 1 cópia, n=14); 3S (sedentário 3 cópias, n=9);
3T (treinado 3 cópias, n=10). *p<0,05 vs. 1S e 3S pós-treino. Dados
comparados através de ANOVA dois caminhos para medidas repetidas e
post-hoc de Bonferroni.
5.5 Avaliações bioquímicas
5.5.1 Glicemia de Jejum
Com o intuito de avaliar o efeito do genótipo da ECA na progressão do diabetes,
comparamos através de teste T os valores de glicemia de jejum apresentados por todos
animais com 1 cópia versus todos animais com 3 cópias da ECA, nas semanas 1 e 7.
Observamos na semana 1 que, a glicemia de jejum dos animais com 3 cópias foi
47
significativamente maior que dos animais com 1 cópia (313,4±21,2 vs. 263,2±14,4
mg/dl, p=0,04). Já na semana 7, antecedente ao início do treinamento físico, não
observamos diferença estatística na glicemia de jejum entre os animais com 1
(366,8±21,6 mg/dl) e 3 cópias (387±21,1 mg/dl) (p=0,512).
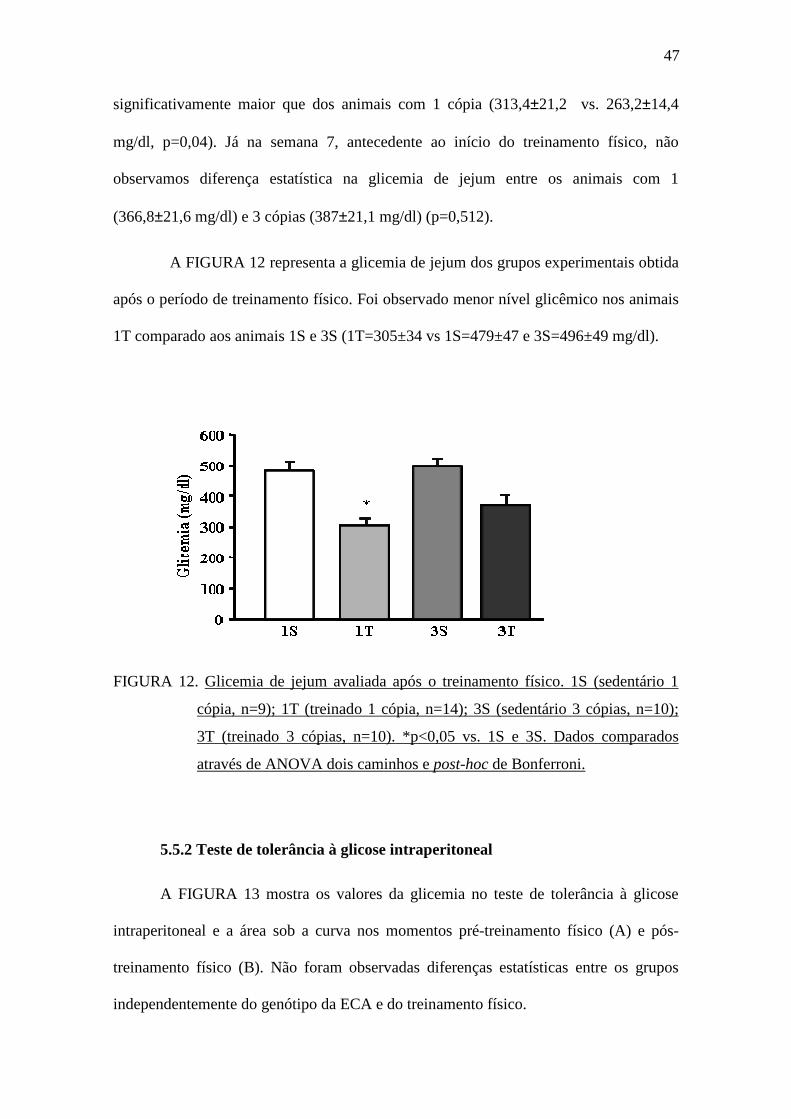
A FIGURA 12 representa a glicemia de jejum dos grupos experimentais obtida
após o período de treinamento físico. Foi observado menor nível glicêmico nos animais
1T comparado aos animais 1S e 3S (1T=305±34 vs 1S=479±47 e 3S=496±49 mg/dl).
FIGURA 12. Glicemia de jejum avaliada após o treinamento físico. 1S (sedentário 1
cópia, n=9); 1T (treinado 1 cópia, n=14); 3S (sedentário 3 cópias, n=10);
3T (treinado 3 cópias, n=10). *p<0,05 vs. 1S e 3S. Dados comparados
através de ANOVA dois caminhos e post-hoc de Bonferroni.
5.5.2 Teste de tolerância à glicose intraperitoneal
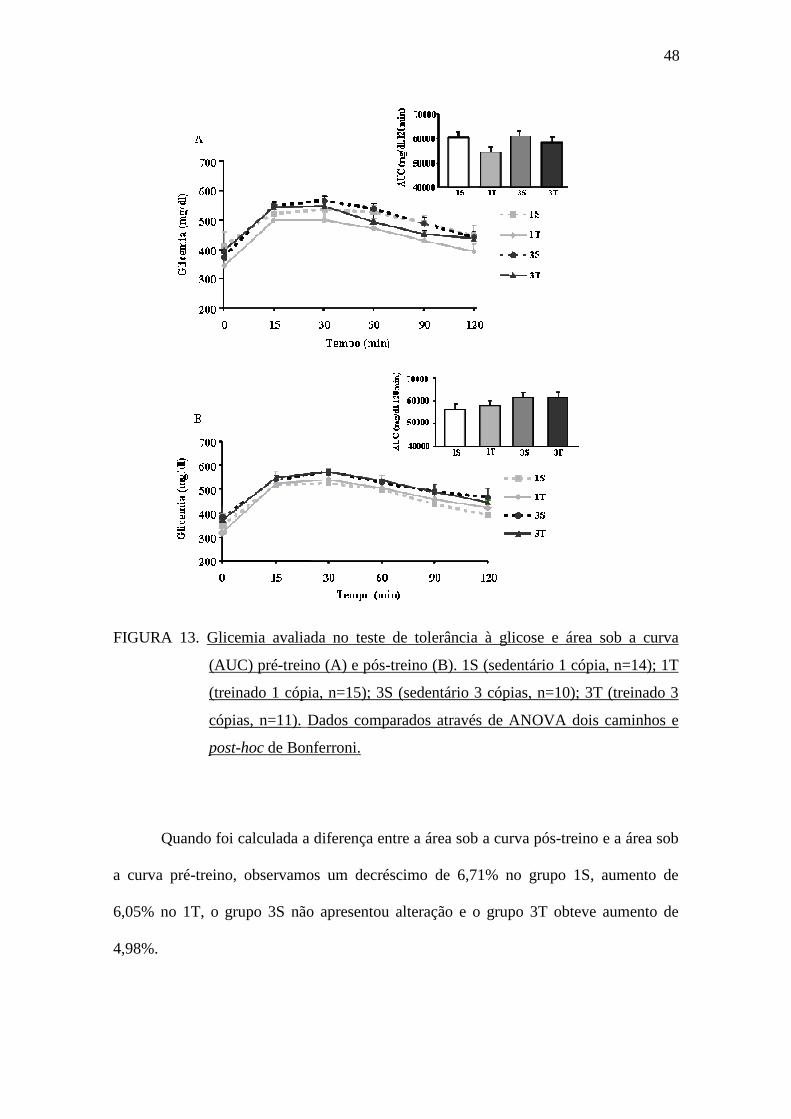
A FIGURA 13 mostra os valores da glicemia no teste de tolerância à glicose
intraperitoneal e a área sob a curva nos momentos pré-treinamento físico (A) e pós-
treinamento físico (B). Não foram observadas diferenças estatísticas entre os grupos
independentemente do genótipo da ECA e do treinamento físico.
48
FIGURA 13. Glicemia avaliada no teste de tolerância à glicose e área sob a curva
(AUC) pré-treino (A) e pós-treino (B). 1S (sedentário 1 cópia, n=14); 1T
(treinado 1 cópia, n=15); 3S (sedentário 3 cópias, n=10); 3T (treinado 3
cópias, n=11). Dados comparados através de ANOVA dois caminhos e
post-hoc de Bonferroni.
Quando foi calculada a diferença entre a área sob a curva pós-treino e a área sob
a curva pré-treino, observamos um decréscimo de 6,71% no grupo 1S, aumento de
6,05% no 1T, o grupo 3S não apresentou alteração e o grupo 3T obteve aumento de
4,98%.
49
Com o intuito de avaliar o efeito do genótipo da ECA na progressão do diabetes,
comparamos através de teste T os valores de área sob a curva apresentados por todos os
animais com 1 cópia versus todos os animais com 3 cópias da ECA na semana 7. Não
foram observadas diferenças estatísticas entre os animais (p= 0,357).
5.5.3 Insulina
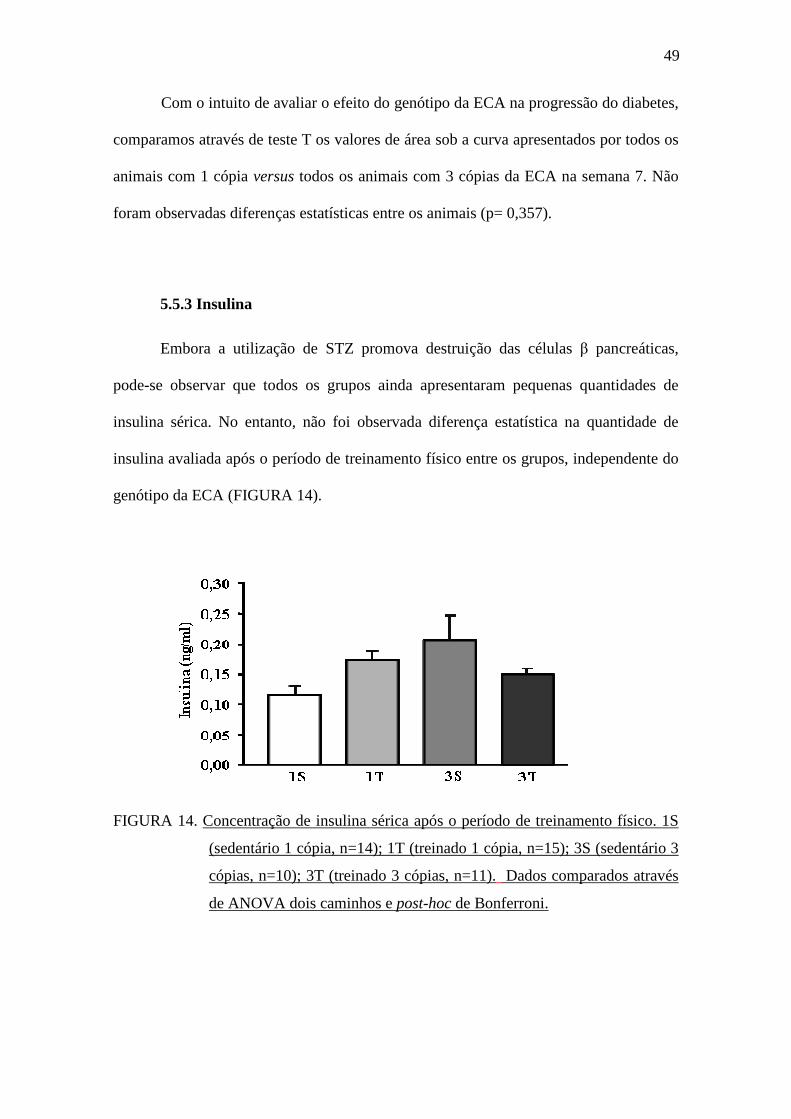
Embora a utilização de STZ promova destruição das células β pancreáticas,
pode-se observar que todos os grupos ainda apresentaram pequenas quantidades de
insulina sérica. No entanto, não foi observada diferença estatística na quantidade de
insulina avaliada após o período de treinamento físico entre os grupos, independente do
genótipo da ECA (FIGURA 14).
FIGURA 14. Concentração de insulina sérica após o período de treinamento físico. 1S
(sedentário 1 cópia, n=14); 1T (treinado 1 cópia, n=15); 3S (sedentário 3
cópias, n=10); 3T (treinado 3 cópias, n=11). Dados comparados através
de ANOVA dois caminhos e post-hoc de Bonferroni.
50
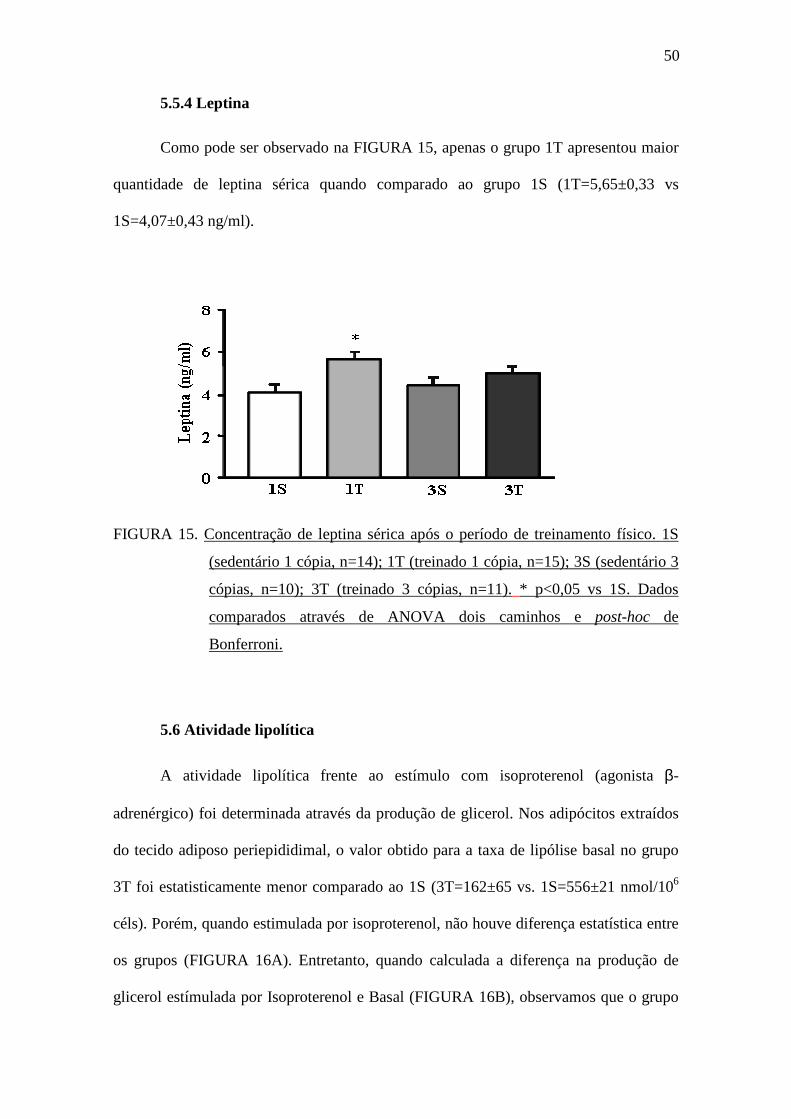
5.5.4 Leptina
Como pode ser observado na FIGURA 15, apenas o grupo 1T apresentou maior
quantidade de leptina sérica quando comparado ao grupo 1S (1T=5,65±0,33 vs
1S=4,07±0,43 ng/ml).
FIGURA 15. Concentração de leptina sérica após o período de treinamento físico. 1S
(sedentário 1 cópia, n=14); 1T (treinado 1 cópia, n=15); 3S (sedentário 3
cópias, n=10); 3T (treinado 3 cópias, n=11). * p<0,05 vs 1S. Dados
comparados através de ANOVA dois caminhos e post-hoc de
Bonferroni.
5.6 Atividade lipolítica
A atividade lipolítica frente ao estímulo com isoproterenol (agonista β-
adrenérgico) foi determinada através da produção de glicerol. Nos adipócitos extraídos
do tecido adiposo periepididimal, o valor obtido para a taxa de lipólise basal no grupo
3T foi estatisticamente menor comparado ao 1S (3T=162±65 vs. 1S=556±21 nmol/106
céls). Porém, quando estimulada por isoproterenol, não houve diferença estatística entre
os grupos (FIGURA 16A). Entretanto, quando calculada a diferença na produção de
glicerol estímulada por Isoproterenol e Basal (FIGURA 16B), observamos que o grupo
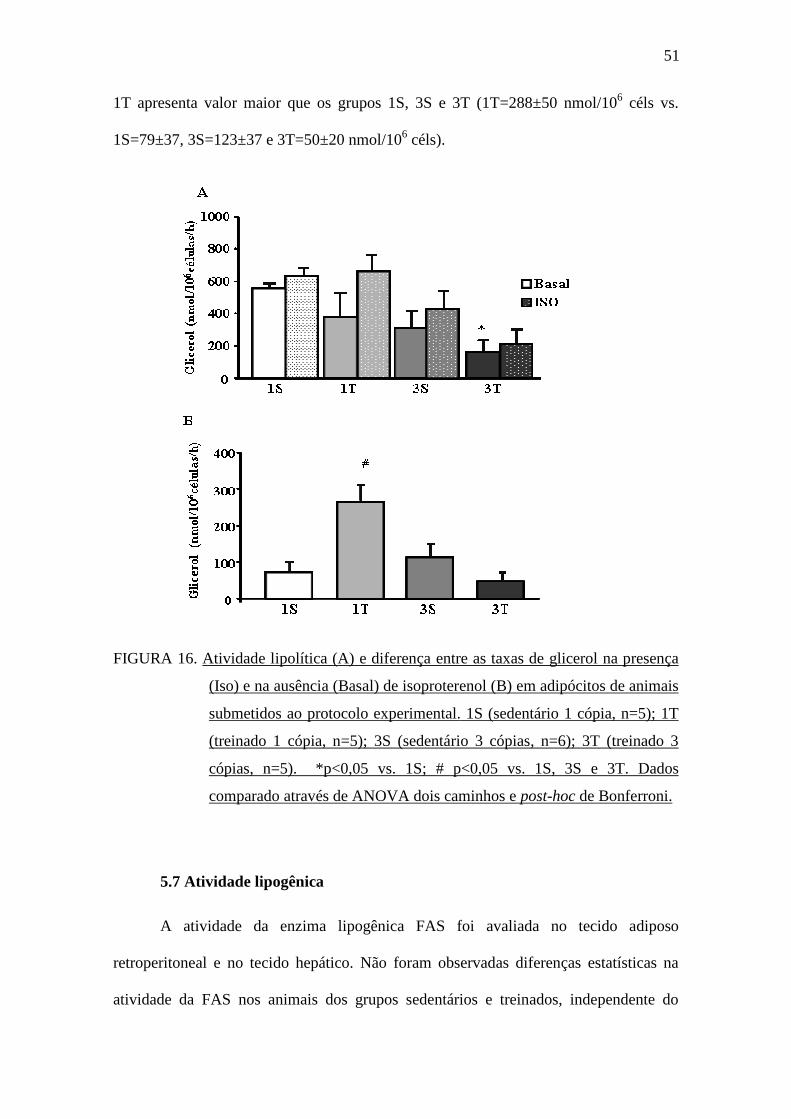
51
1T apresenta valor maior que os grupos 1S, 3S e 3T (1T=288±50 nmol/106 céls vs.
1S=79±37, 3S=123±37 e 3T=50±20 nmol/106 céls).
FIGURA 16. Atividade lipolítica (A) e diferença entre as taxas de glicerol na presença
(Iso) e na ausência (Basal) de isoproterenol (B) em adipócitos de animais
submetidos ao protocolo experimental. 1S (sedentário 1 cópia, n=5); 1T
(treinado 1 cópia, n=5); 3S (sedentário 3 cópias, n=6); 3T (treinado 3
cópias, n=5). *p<0,05 vs. 1S; # p<0,05 vs. 1S, 3S e 3T. Dados
comparado através de ANOVA dois caminhos e post-hoc de Bonferroni.
5.7 Atividade lipogênica
A atividade da enzima lipogênica FAS foi avaliada no tecido adiposo
retroperitoneal e no tecido hepático. Não foram observadas diferenças estatísticas na
atividade da FAS nos animais dos grupos sedentários e treinados, independente do
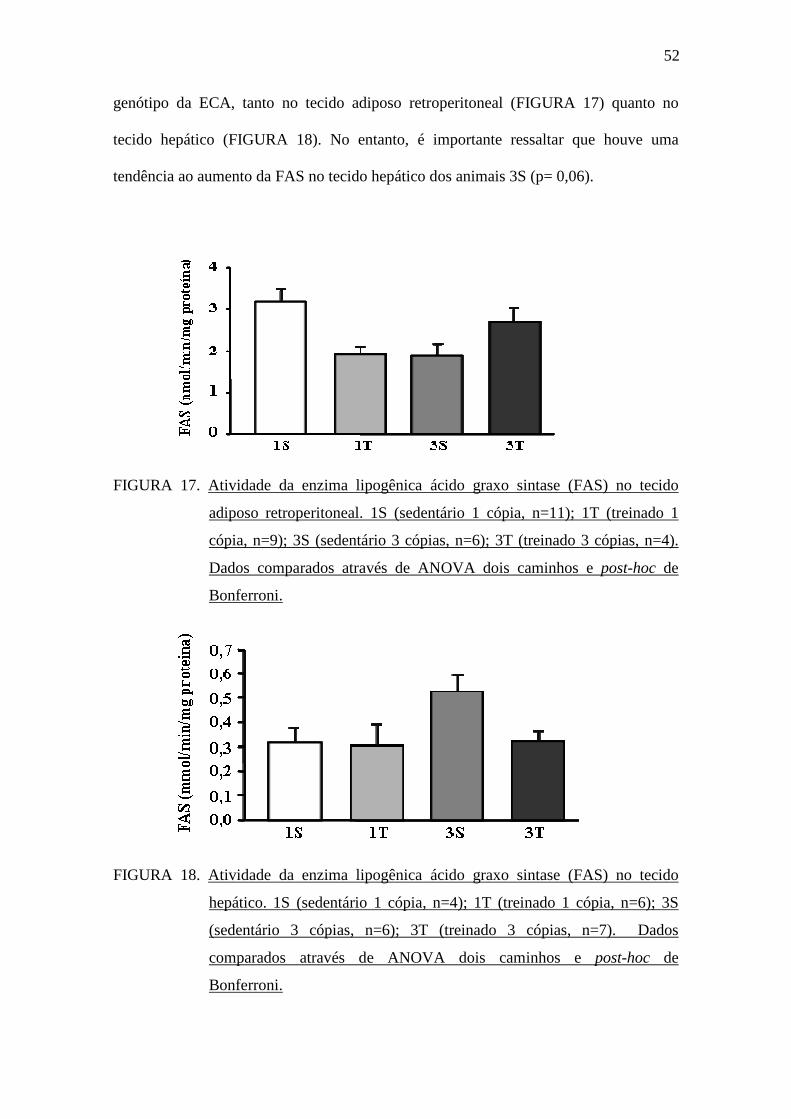
52
genótipo da ECA, tanto no tecido adiposo retroperitoneal (FIGURA 17) quanto no
tecido hepático (FIGURA 18). No entanto, é importante ressaltar que houve uma
tendência ao aumento da FAS no tecido hepático dos animais 3S (p= 0,06).
FIGURA 17. Atividade da enzima lipogênica ácido graxo sintase (FAS) no tecido
adiposo retroperitoneal. 1S (sedentário 1 cópia, n=11); 1T (treinado 1
cópia, n=9); 3S (sedentário 3 cópias, n=6); 3T (treinado 3 cópias, n=4).
Dados comparados através de ANOVA dois caminhos e post-hoc de
Bonferroni.
FIGURA 18. Atividade da enzima lipogênica ácido graxo sintase (FAS) no tecido
hepático. 1S (sedentário 1 cópia, n=4); 1T (treinado 1 cópia, n=6); 3S
(sedentário 3 cópias, n=6); 3T (treinado 3 cópias, n=7). Dados
comparados através de ANOVA dois caminhos e post-hoc de
Bonferroni.
53
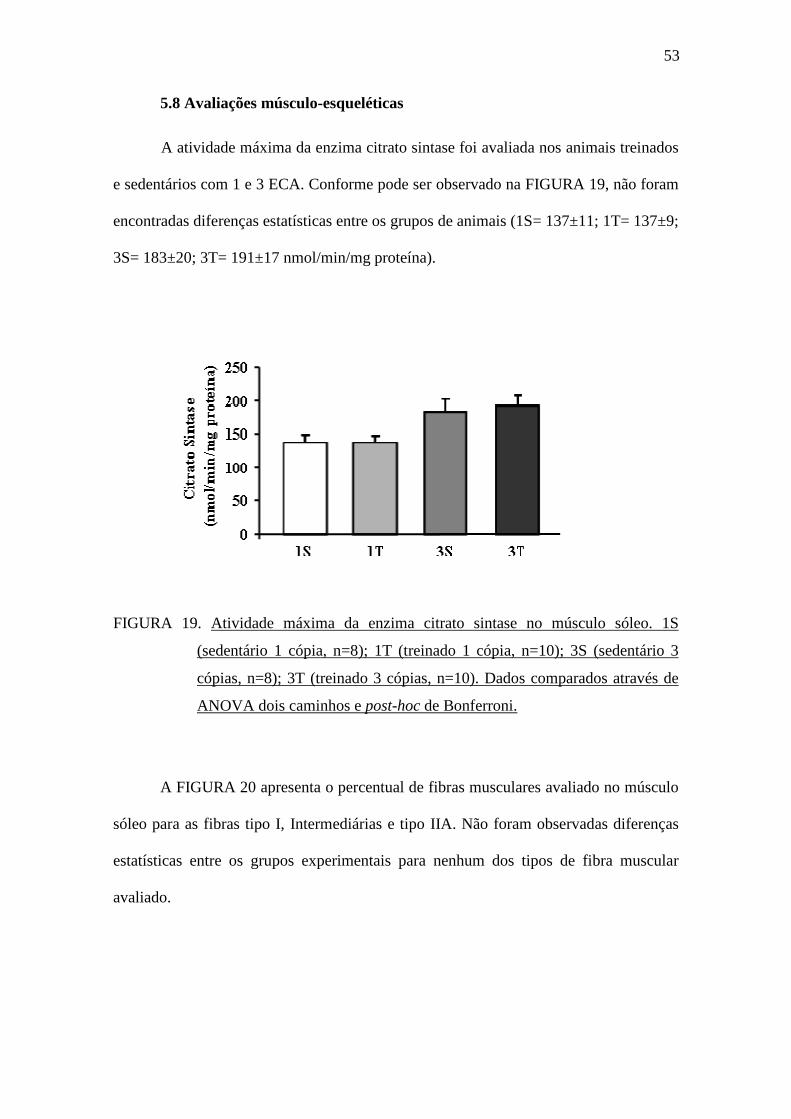
5.8 Avaliações músculo-esqueléticas
A atividade máxima da enzima citrato sintase foi avaliada nos animais treinados
e sedentários com 1 e 3 ECA. Conforme pode ser observado na FIGURA 19, não foram
encontradas diferenças estatísticas entre os grupos de animais (1S= 137±11; 1T= 137±9;
3S= 183±20; 3T= 191±17 nmol/min/mg proteína).
FIGURA 19. Atividade máxima da enzima citrato sintase no músculo sóleo. 1S
(sedentário 1 cópia, n=8); 1T (treinado 1 cópia, n=10); 3S (sedentário 3
cópias, n=8); 3T (treinado 3 cópias, n=10). Dados comparados através de
ANOVA dois caminhos e post-hoc de Bonferroni.
A FIGURA 20 apresenta o percentual de fibras musculares avaliado no músculo
sóleo para as fibras tipo I, Intermediárias e tipo IIA. Não foram observadas diferenças
estatísticas entre os grupos experimentais para nenhum dos tipos de fibra muscular
avaliado.
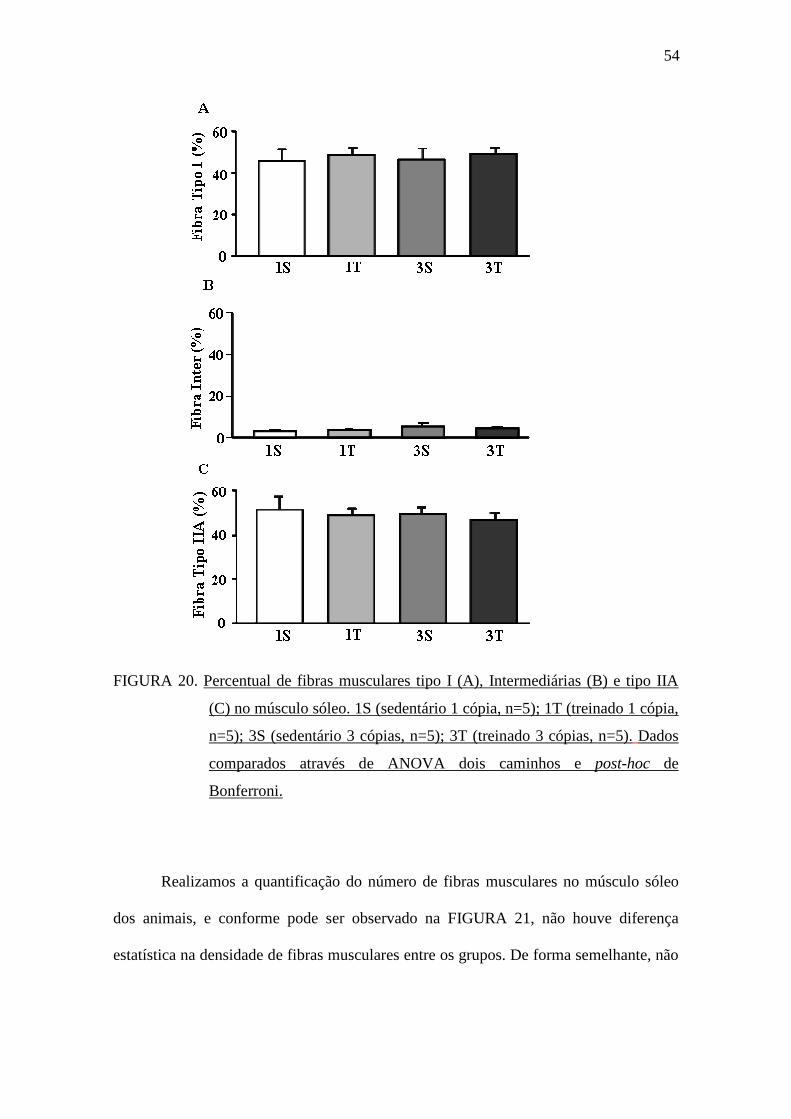
54
FIGURA 20. Percentual de fibras musculares tipo I (A), Intermediárias (B) e tipo IIA
(C) no músculo sóleo. 1S (sedentário 1 cópia, n=5); 1T (treinado 1 cópia,
n=5); 3S (sedentário 3 cópias, n=5); 3T (treinado 3 cópias, n=5). Dados
comparados através de ANOVA dois caminhos e post-hoc de
Bonferroni.
Realizamos a quantificação do número de fibras musculares no músculo sóleo
dos animais, e conforme pode ser observado na FIGURA 21, não houve diferença
estatística na densidade de fibras musculares entre os grupos. De forma semelhante, não
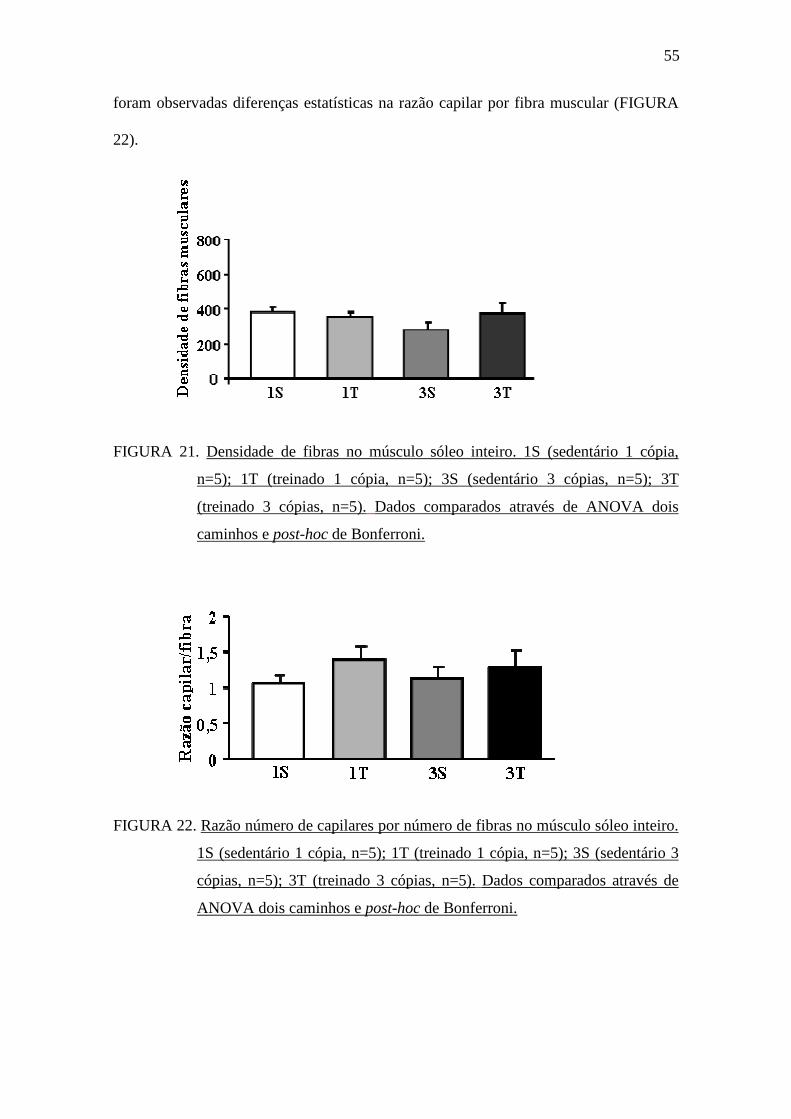
55
foram observadas diferenças estatísticas na razão capilar por fibra muscular (FIGURA
22).
FIGURA 21. Densidade de fibras no músculo sóleo inteiro. 1S (sedentário 1 cópia,
n=5); 1T (treinado 1 cópia, n=5); 3S (sedentário 3 cópias, n=5); 3T
(treinado 3 cópias, n=5). Dados comparados através de ANOVA dois
caminhos e post-hoc de Bonferroni.
FIGURA 22. Razão número de capilares por número de fibras no músculo sóleo inteiro.
1S (sedentário 1 cópia, n=5); 1T (treinado 1 cópia, n=5); 3S (sedentário 3
cópias, n=5); 3T (treinado 3 cópias, n=5). Dados comparados através de
ANOVA dois caminhos e post-hoc de Bonferroni.

56
6. DISCUSSÃO
6.1 Influência do genótipo da ECA no desenvolvimento do diabetes
experimental
A proposta inicial do presente estudo previa a investigação da relação do
genótipo da ECA com as respostas metabólicas induzidas pelo diabetes experimental.
Para isso, a indução do diabetes experimental foi realizada entre a 7ª e 8ª semana de
vida dos animais, e diferente da maioria dos estudos sobre o tema na literatura,
planejamos introduzir o treinamento físico somente após um período de estabelecimento
da hiperglicemia. Utilizamos como referência para a definição do momento de
introdução do treinamento físico o estudo prévio de (HEIMANN, 2003), no qual foi
demonstrado que a taxa de mortalidade dos animais diabéticos com 3 cópias do gene da
ECA aumentava significativamente na 14ª semana após indução do diabetes por
estreptozotocina, definindo o curso temporal para o agravamento das complicações
associadas ao diabetes e seu desfecho final. Assim, sabendo que o treinamento físico
duraria 8 semanas, o início do protocolo de natação foi programado para a 7ª semana
após indução do diabetes, encerrando-se na 15ª semana de diabetes.
Considerando que o SRA exerce grande influência no desenvolvimento e
progressão do diabetes (AROZAL et al., 2009; LEUNG, 2010; OLTMAN et al., 2010),
avaliamos durante as sete semanas de desenvolvimento do diabetes se a presença de
mais ou menos ECA poderia interferir em algumas variáveis metabólicas. Nossos
principais achados demonstraram que na fase inicial do desenvolvimento do diabetes
(sete semanas), o nível diferencial de expressão gênica da ECA não refletiu em
alterações no ganho de peso corporal, no consumo de ração diário, no consumo de

57
oxigênio em repouso, na capacidade de realização de exercício físico e na tolerância à
glicose. Quanto à glicemia de jejum, observamos que o nível glicêmico dos animais 3
cópias avaliados após a indução do diabetes foi superior ao nível glicêmico dos animais
com 1 cópia do gene da ECA, porém após sete semanas de desenvolvimento do
diabetes, essa diferença já não foi mais observada.
Evidências na literatura demonstraram que a influência do SRA no controle de
peso corporal é bastante complexa, haja vista os diferentes resultados encontrados.
Enquanto alguns autores confirmaram que o aumento dos componentes do SRA,
especialmente a Ang II, associa-se ao maior ganho de peso corporal (GIACCHETTI,
FALOIA, MARINIELLO, SARDU, GATTI, CAMILLONI, GUERRIERI &
MANTERO, 2002), outros autores observaram que a atividade do SRA era
inversamente associada ao ganho de peso corporal (CASSIS, ENGLISH,
BHARADWAJ & BOUSTANY, 2004).
Para avaliar a evolução do peso corporal dos animais nas sete primeiras semanas
de protocolo, realizamos inicialmente a comparação dos dados dos animais com 1 cópia
versus animais como 3 cópias. Observamos que não havia diferença estatística entre os
grupos. No entanto, quando separamos os animais nos respectivos grupos sedentários e
treinados, observamos que o grupo 1T apresentava peso corporal superior ao grupo 1S
após sete semanas de diabetes. Essa resposta parece ser muito mais conseqüente da
distribuição dos animais nos grupos do que propriamente do genótipo da ECA, pois
embora não tenham sido estatisticamente diferentes, os valores iniciais de peso corporal
do 1S foi de 20,9 + 0,8g e do 1T foi de 23,4 + 0,7g. Além disso, quando avaliamos o
ganho de peso corporal nessa etapa, não encontramos diferença estatística entre os
grupos.

58
Apesar de os estudos na literatura demonstrarem que o animal diabético por STZ
reduz o peso corporal (TAKADA et al 2007; PINGHIN, 2005). HEIMANN et al.
(2005) utilizando o modelo animal geneticamente modificado com 1, 2 e 3 cópias do
gene da ECA alimentados com dieta padrão, não apresentaram diferença no peso
corporal avaliado durante 30 semanas. HUANG et al. (2001) estudaram animais com 1,
2 e 3 cópias do gene da ECA diabéticos por STZ e não observaram diferença no peso
corporal após 12 semanas de diabetes. Esses dados dão suporte ao nosso estudo, que
embora na presença de diabetes, o padrão de resposta de peso corporal dos animais é
mantido, mostrando que na fase inicial de desenvolvimento do diabetes, o genótipo da
ECA não influencia o peso corporal.
Embora o diabetes tenha como característica marcante o aumento do consumo
alimentar (OKOSHI et al., 2007), e que a infusão central crônica de Ang II em ratos
provoca diminuição no consumo alimentar (PORTER, ANDERSON, ROBISON &
PHILLIPS, 2003), no presente trabalho não foi encontrada diferença estatística na
média de consumo de ração diário entre os grupos com 1 e 3 cópias de ECA durante as
7 semanas de diabetes que antecederam o início do treinamento físico. Esses dados
demonstram que o modelo experimental e o procedimento metodológico influenciam
diretamente os resultados observados, e devem ser considerado quando a proposta de
estudo se volta para o entendimento da interação gene-meio ambiente para definição de
fenótipos complexos.
Os dados de metabolismo energético de repouso observados nas 7 primeiras
semanas de protocolo (avaliação pré-treino) foram relativamente inesperados, já que o
aumento da Ang II resulta em potente efeito estimulador do sistema nervoso simpático,
o que poderia contribuir diretamente para o aumento do metabolismo energético de
repouso (ENGLISH & CASSIS, 1999). No entanto, observamos que na fase inicial do

59
diabetes, não houve diferença no consumo energético de repouso dos animais associado
ao genótipo da ECA.
Considerando que a determinação do peso corporal está intimamente relacionada
com o balanço energético (MULLER, BOSY-WESTPHAL, LATER, HAAS &
HELLER, 2009), os dados de consumo de ração e de metabolismo energético de
repouso apontam para um balanço energético neutro, refletindo em inalteração no ganho
do peso corporal associado ao genótipo da ECA. Isso porque, os animais ingeriram
exatamente a mesma ração, estavam submetidos ao mesmo confinamento em caixa e
não realizavam treinamento físico, nos permitindo considerar que os principais fatores
determinantes do balanço energético foram o consumo de ração e o gasto energético de
repouso.
Considerando que o pior prognóstico do diabetes está associado ao maior
conteúdo de Ang II (TIKELLIS, COOPER & THOMAS, 2006; WEIR, 2007),
esperávamos que logo após as 7 semanas de diabetes, os animais com 3 cópias
apresentassem maiores prejuízos na glicemia de jejum e na tolerância à glicose. No
entanto, o que vimos foi apenas uma maior hiperglicemia do 3 cópias no início do
protocolo, e que depois de sete semanas foi equiparada à hiperglicemia dos animais com
1 cópia.
A literatura oferece inúmeras evidências dos prejuízos causados pelo SRA na
progressão do diabetes, no entanto, quando olhamos para os estudos experimentais, a
associação do SRA com o diabetes normalmente tem sido demonstrada através do uso
farmacológico de bloqueadores do SRA (KAMAL et al., 2010; VAISHYA, SINGH &
LAL, 2009; ZOJA et al., 2010). Por outro lado, no nosso estudo observamos que a
presença de mais ou menos ECA em níveis fisiológicos não interfere na magnitude de

60
resposta da glicemia e da tolerância à glicose na fase inicial da doença. Esses dados
fortalecem a idéia de que a modulação do SRA no diabetes parece ser não só
dependente do modelo estudado, mas também do tempo necessário para que o excesso
de Ang II reflita em complicações metabólicas associadas ao diabetes.
Um dado interessante foi a diferença estatística na glicemia de jejum entre os
animais com 1 e 3 cópias logo após a indução do diabetes, revelando que o efeito da
STZ sob as células ß pancreáticas é mais acentuado nos animais com maior conteúdo de
ECA. No entanto, esse efeito parece ser mais agudo, pois não observamos mais
diferença estatística entre os grupos após 7 semanas de diabetes. Nossos dados
corroboram o estudo de HUANG et al. (2001) no qual foi demonstrado que a glicemia
de jejum não diferiu entre os animais com 1 a 3 cópias do gene da ECA diabéticos por
STZ. Embora os mecanismos não estejam claros na literatura, uma possível explicação
para essa resposta seria a resposta compensatória de algum dos componentes do SRA
em função do elevado conteúdo de Ang II. De fato, MONOSIKOVA, ZEMAN,
VESELA & HERICHOVA (2009) observaram redução na expressão gênica do receptor
da angiotensina AT1 no pâncreas de ratos diabéticos induzido por STZ durante os
estágios iniciais do desenvolvimento do diabetes.
As adaptações encontradas no diabetes podem refletir diretamente na redução da
tolerância ao esforço físico, especialmente em função das complicações metabólicas e
de distúrbios no músculo esquelético (STUMP et al., 2003; KRAUSE et al., 2009). No
entanto, observamos que após 7 semanas de diabetes, o desempenho físico dos animais
no teste de esforço em esteira até a exaustão não foi diferente entre os genótipos da
ECA.

61
6.2 Efeitos metabólicos do treinamento físico no diabetes experimental
Após o período de 7 semanas de desenvolvimento do diabetes, os animais foram
submetidos ao treinamento físico com natação por 8 semanas ou mantidos sedentários.
Nessa etapa, avaliamos se os distúrbios metabólicos associados ao diabetes seriam
diferenciados nos animais com 1 e 3 cópias do gene da ECA, e se o treinamento físico
iria interferir nessa resposta.
Após o período de treinamento físico, observamos que o grupo 1T apresentou
peso corporal significativamente maior comparado ao grupo 1S. Essa diferença parece
ser conseqüente do diferencial de peso corporal entre esses grupos já observado no
início do treinamento físico, já que o ganho de peso corporal durante o treinamento
físico não foi estatisticamente diferente entre os grupos. Esses dados repetem
exatamente aquilo que foi observado nas 7 semanas iniciais de protocolo experimental.
No presente estudo, esperávamos redução significativa do peso corporal dos
animais treinados, especialmente com 3 cópias da ECA, pois reuníamos dois
importantes fatores determinantes para essa resposta, o diabetes por STZ (LEHTI et al.,
2007) e a maior oxidação de substratos energéticos promovida pelo treinamento físico
(LIMA-SILVA et al., 2006). No entanto, não foram observadas diferenças estatísticas
no peso corporal dos animais quando considerado o genótipo (1S vs. 3S) e o
treinamento físico (3S vs. 3T).
Apesar de o diabetes por STZ induzir respostas metabólicas mais condizentes
com o diabetes tipo 1 (OKOSHI et al., 2007), esse modelo apresenta características
peculiares, como por exemplo, pequena quantidade de insulina ainda remanescente
(JUNOD et al., 1969), que acabam refletindo diretamente no comportamento
metabólico observado. De fato, quando avaliamos a concentração de insulina nos nossos

62
animais, observamos que os quatro grupos apresentavam pequena quantidade de
insulina plasmática, porém sem diferença estatística entre eles.
Quanto ao treinamento físico, apesar de alguns estudos demonstrarem redução
de peso corporal (SNIJDERS, VERDIJK, HANSEN, DENDALE & VAN LOON,
2010) ou aumento de forma mais lenta em animais treinados (TATCHUM-TALOM,
SCHULZ, MCNEILL & KHADOUR, 2000), nossos dados corroboram os estudos que
observaram que o treinamento físico não influenciou o ganho de peso corporal de
animais. De forma semelhante ao realizado para as sete semanas iniciais de protocolo
experimental, estudamos duas possíveis variáveis determinantes da resposta de peso
corporal dos animais durante o período de treinamento físico, o consumo de ração e o
metabolismo energético de repouso.
Considerando apenas o genótipo da ECA, observamos que durante o período de
8 semanas de treinamento físico, o grupo 3S apresentou maior consumo diário de ração
comparado ao grupo 1S, porém o grupo 3S no período pós-treinamento físico
apresentou maior consumo de oxigênio comparado aos grupos 1S e 3S pré-treinamento
físico. De acordo com esses dados, o peso corporal de ambos os grupos sedentários não
diferiu porque no grupo 1S não há diferença no metabolismo energético de repouso e
também no consumo de ração, porém no grupo 3S a maior ativação metabólica em
repouso foi ajustada com o aumento no consumo de ração, garantindo assim,
manutenção do peso corporal.
Quando consideramos o treinamento físico, observamos que o grupo 1T
consumiu mais ração que o grupo 1S, porém não diferiu no metabolismo energético de
repouso. De acordo com STEVENSON, BOX, FELEKI & BEATON (1966), os animais
podem ajustar o consumo de ração equivalente ao gasto energético quando submetidos

63
ao treinamento físico regular, o que em última instância garante a manutenção do peso
corporal. Interessante observar que o consumo de ração não diferiu entre os animais 3S
e 3T, no entanto, o metabolismo energético de repouso é menor no 3T comparado ao
3S, sinalizando que a manutenção do peso corporal no grupo 3T foi decorrente do ajuste
entre o gasto energético promovido pelo treinamento físico e o gasto energético
resultante do metabolismo de repouso.
Ainda em relação à avaliação do metabolismo energético de repouso,
observamos que o grupo 3S apresentou maior consumo de oxigênio em repouso na
semana quinze (pós-treinamento físico) comparado ao seu respectivo valor obtido na
semana oito (pré-treinamento físico) e também comparado ao grupo 3T (pós-
treinamento físico). Esses dados sinalizam que o metabolismo energético de repouso
aumentou durante a progressão do diabetes e que foi genótipo-dependente, ou seja, o
maior nível de ECA resultou em maior gasto energético de repouso, e que o treinamento
físico foi capaz de atenuar essa resposta. Embora o comportamento do metabolismo
energético de repouso não tenha refletido em alteração no ganho de peso corporal
desses animais, isso pode ter sido apenas por uma questão temporal do diabetes.
Com os dados reportados até o presente momento, podemos afirmar que o
treinamento físico promoveu manutenção do peso corporal e que o genótipo da ECA
influenciou apenas os meios pelos quais essa resposta foi obtida. Na tentativa de
entender um pouco mais sobre os fatores determinantes do balanço energético,
realizamos a dosagem da concentração de leptina plasmática. Considerando que a
leptina secretada pelas células adiposas aumenta o gasto energético de repouso e reduz o
apetite (MERINO, CANO, GUZMAN, SOMOZA & RUIZ-GAYO, 2008) e a possível
influencia da Ang II na regulação da secreção de leptina (CASSIS et al., 2004),
esperávamos que os animais com 3 cópias de ECA apresentassem maior concentração

64
de leptina. No entanto, observamos que apenas o grupo 1T apresentou aumento
significante comparado ao 1S.
O aumento da secreção de leptina pelo grupo 1T parece ser mais condizente ao
efeito agudo da última sessão de treino do que propriamente uma adaptação crônica
provocada pelo treinamento físico, já que a literatura tem demonstrado que o
treinamento físico promove redução ou manutenção da concentração de leptina
(BALDUCCI, ZANUSO, NICOLUCCI, FERNANDO, CAVALLO, CARDELLI,
FALLUCCA, ALESSI, LETIZIA, JIMENEZ, FALLUCCA & PUGLIESE, 2009;
BOUASSIDA, CHAMARI, ZAOUALI, FEKI, ZBIDI & TABKA, 2 008; POLAK,
KLIMCAKOVA, MORO, VIGUERIE, BERLAN, HEJNOVA, RICHTEROVA,
KRAUS, LANGIN & STICH, 2006). Isso porque, conforme mostrado por ESSIG,
ALDERSON, FERGUSON, BARTOLI & DURSTINE (2000), a redução da
concentração de leptina foi observada apenas 48hs após a última sessão de treino,
sugerindo que o treinamento físico pode induzir mudanças na concentração de leptina,
mas que não são observadas nas primeiras horas de recuperação. De fato, a coleta de
sangue dos animais foi realizada 24 horas após a última sessão de treinamento físico.
Além disso, a inabilidade da ação periférica da leptina em alterar o metabolismo
energético de repouso e inibir o apetite dos animais 1T, reforçam ainda mais a hipótese
de que a elevação da concentração de leptina no nesse grupo tenha sido um efeito agudo
da última sessão de treinamento.
Um possível mecanismo que justifica o aumento agudo da leptina associado ao
treinamento físico é o sinergismo desta proteína com o cortisol, cuja secreção está
diretamente relacionada à intensidade do exercício. Embora não tenha sido dosada a
cortisona nos animais, possivelmente a intensidade do exercício realizado bem como o
estresse provocado pela natação seria suficiente para exacerbar a resposta da cortisona

65
e, paralelamente, elevar a concentração de leptina. De fato, foi observado aumento na
concentração de leptina durante 41 minutos de ciclismo associado ao aumento
significativo do cortisol e adrenalina (FISHER, VAN PELT, ZINDER, LANDT &
KOHRT, 2001).
O efeito agudo da última sessão de exercício também deveria ter promovido
aumento na secreção de leptina no grupo 3T, porém não visualizamos diferença
comparada ao grupo 3S. Essa resposta pode ser justificada pela redução significativa da
massa adiposa retroperitoneal e menor diâmetro de célula adiposa periepididimal
apresentada por esses animais. De acordo com GUO, HALO, LEIBEL & ZHANG
(2004), a secreção de leptina é diretamente proporcional ao conteúdo de massa adiposa
e, de fato, quando há redução na massa adiposa a secreção de leptina também é reduzida
(KONDO, KOBAYASHI & MURAKAMI, 2006). Por isso a resposta de leptina no
grupo 3T não ter sido semelhante à resposta observada no 1T.
A indisponibilidade de glicose suficiente para o fornecimento de energia causada
pelo diabetes influencia diretamente o tecido adiposo e o metabolismo de lipídios
(VIRTUE & VITAL-PUIG, 2010). Embora a combinação de diabetes, ativação do SRA
e treinamento físico não tenha refletido em alteração do peso corporal, observamos que
o grupo 3T apresentou menor peso do depósito de gordura retroperitoneal comparado ao
grupo 3S e menor diâmetro dos adipócitos comparado aos grupos 1S, 1T e 3S. Em
função das diferenças encontradas no tecido adiposo e a importância metabólica desse
tecido no diabetes, estudamos a lipólise em células adiposas estimuladas por
isoproterenol e também dosamos a enzima lipogênica FAS no tecido adiposo e também
hepático.

66
Os componentes do SRA, como angiotensinogênio, renina, ECA, Ang II e
receptor AT1 foram demonstrados no tecido adiposo (SARZANI, SALVI, DESSI-
FULGHERI & RAPPELLI, 2008). E, apesar de a Ang II aumentar a lipólise via
estimulação da liberação de noradrenalina pelo sistema nervoso simpático (BLAAK,
2003), não foram observadas diferenças estatísticas nos dados de lipólise entre os
grupos. Ainda, quando calculada a diferença na produção de glicerol estimulada por
isoproterenol e basal, observamos apenas que o 1T apresentou maior valor que os
grupos 1S, 3S e 3T. Esses dados nos revelam que o genótipo da ECA por si só não
influenciou a lipólise, pois não encontramos diferença estatística entre os grupos
sedentários. Além disso, confirma dados anteriores publicados na literatura sobre o
aumento da taxa lipolítica induzida pelo treinamento físico (ENEVOLDSEN,
STALLKNECHT, LANGFORT, PETERSEN, HOLM, PLOUG & GALBO, 2001;
SHEPHERD, NOBLE, KLUG & GOLLNICK, 1981), conforme observado no grupo
1T. Nesse sentido, era de se esperar que os animais 3T apresentassem respostas
parecidas dos animais 1T, entretanto, isso não foi observado possivelmente em função
da diferença no conteúdo de tecido adiposo periepididimal desses dois grupos, que
apesar de não ter sido estatisticamente significante, pode ter influenciado no conteúdo
de células extraídas e estimuladas. Obviamente, a quantificação do lipócrito realizada
visa normalizar o conteúdo de células adiposas estimuladas, porém não exclui
totalmente essa interferência.
O processo lipogênico, avaliado através da enzima FAS no tecido adiposo e no
tecido hepático, não foi estatisticamente diferente entre os grupos estudados, embora
tenha havido uma tendência ao aumento no tecido hepático dos animais 3S. Estudos na
literatura demonstraram que a Ang II estimula a expressão de genes lipogênicos como o
da enzima FAS e da enzima glicose-6-fosfato desidrogenase (G6PDH), resultando em

67
aumento no conteúdo de ácidos graxos e estoque de triglicerídeos (JONES,
STANDRIDGE & MOUSTAID, 1997; KIM, DUGAIL, STANDRIDGE,
CLAYCOMBE, CHUN & MOUSTAID-MOUSSA, 2001). No entanto, na presença do
diabetes e do treinamento físico, o diferencial de expressão da ECA não interferiu na
resposta enzimática.
O treinamento físico tem sido fortemente recomendado para o tratamento do
diabetes, especialmente por melhorar os mecanismos envolvidos na captação de glicose
e exercer um potente efeito hipoglicemiante (MACKNIGHT et al., 2009; WELTMAN
et al., 2009). Um dos principais resultados encontrados no nosso estudo foi a redução do
nível glicêmico demonstrado pelo grupo 1T comparado aos grupos 1S e 3S,
confirmando a eficácia do treinamento físico para atenuar a hiperglicemia induzida pelo
diabetes apenas nos animais com 1 cópia do gene da ECA. Entretanto, no grupo 3T essa
resposta não foi observada. Se considerarmos que a quantidade de insulina não diferiu
entre os grupos, e que no teste de tolerância à glicose também não observamos diferença
alguma, independente do genótipo e do treinamento físico, a melhora glicêmica pode
estar associada às adaptações intracelulares responsáveis pela captação de glicose,
principalmente no músculo esquelético e no tecido adiposo.
A associação entre o SRA e a resistência à insulina tem sido demonstrada na
literatura (MATAYOSHI, KAMIDE, TAKIUCHI, HORIO, YOSHIHARA,
NAKAMURA, NAKAHAMA & KAWANO, 2007). A maior concentração de Ang II
influencia negativamente no metabolismo glicêmico (MACKNIGHT et al., 2009; XU,
ZHANG, COHEN & DIPETRILLO, 2010), onde a infusão de Ang II induz resistência à
insulina (RICHEY, ADER, MOORE & BERGMAN, 1999) e a utilização de
bloqueadores do SRA melhora a sensibilidade à insulina (FURUHASHI, URA,
TAKIZAWA, YOSHIDA, MONIWA, MURAKAMI, HIGASHIURA &

68
SHIMAMOTO, 2004). Isso ocorre porque a Ang II pode modular negativamente as
ações da insulina através da inibição da fosforilação do receptor IRS 1 no resíduo de
tirosina e também da inibição da sinalização intracelular via IP-3 (SHINSHI,
HIGASHIURA, YOSHIDA, TOGASHI, YOSHIDA, MIYAZAKI, URA &
SHIMAMOTO, 2007). Esses achados podem contribuir para justificar o efeito do
treinamento físico sobre a glicemia observado apenas no grupo 1T, pois embora os
valores de insulina sejam semelhantes, possivelmente a presença de maior quantidade
de ECA no grupo 3T pode ter contribuído para reduzir a ação da insulina ainda existente
nesses animais. Sendo assim, a capacidade do treinamento físico em atenuar a resposta
de hiperglicemia foi genótipo-dependente, ou seja, apenas os animais com menor
expressão de ECA foram beneficiados por essa resposta.
Embora não tenhamos estudado, é sabido que durante o exercício físico, devido
à necessidade de síntese de ATP, a proteína ativada por AMP (AMPK) atua nas vias de
captação de glicose e oxidação de ácidos graxos (MUSI et al., 2005; STEPHENS et al.,
2002). Por acionar um mecanismo independente da insulina para a captação de glicose,
a ação da AMPK tem sido bastante investigada em estudos que mostram o efeito
terapêutico do treinamento físico sobre o diabetes. De fato, PÁDUA, PÁDUA, PAULI,
DE SOUZA, DA SILVA, ROPELLE, CARVALHEIRA & ROPELLE (2009),
observaram que camundongos diabéticos ob/ob submetidos à sessão aguda de exercício
físico aumentaram a atividade da AMPK em 229,4%, e os diabéticos db/db aumentaram
a atividade em 288,5%, quando comparados aos seus respectivos grupos sem exercício
físico.
Sabendo disso, uma hipótese alternativa que poderia justificar a melhora
glicêmica no grupo 1T seria a resposta mais eficaz da AMPK ao treinamento físico
associada à menor expressão de ECA. Em um elegante estudo da ação da Ang II sobre a

69
atividade da AMPK e captação de glicose, SHINSHI et al., (2007) confirmaram o efeito
inibitório que a Ang II, através do receptor AT1, exerce sobre a captação de glicose via
prejuízo da AMPK. Embora essa via não tenha sido estudada em modelo de diabetes
por STZ, os dados obtidos por SHINSHI et al., (2007) fortalecem nossa justificativa
para melhora da glicemia apenas no grupo 1T.
A melhor resposta glicêmica observada no grupo 1T corrobora o melhor
desempenho físico apresentado por esse grupo no teste de capacidade de realização de
exercício físico em esteira quando comparado com os grupos 1S e 3S. Por outro lado, o
treinamento físico não foi capaz de promover essa resposta nos animais com 3 cópias do
gene da ECA (3T).
É sabido que as alterações metabólicas encontradas no diabetes podem refletir
diretamente na redução da capacidade de realização de exercício físico, tanto em função
dos prejuízos cardiovasculares (WICHI, MALFITANO, ROSA, DE SOUZA, SALEMI,
MOSTARDA, DE ANGELIS & IRIGOYEN, 2007), quanto de alterações morfológicas
e funcionais no músculo esquelético (STUMP et al., 2003; KRAUSE et al., 2009). De
acordo com KRAUSE et al., (2009), a redução da capacidade oxidativa muscular, a
alteração de tipagem de fibras e redução da capilarização estão entre os principais
mecanismos responsáveis pela redução da capacidade de realização de exercício físico
associado ao diabetes.
Conhecendo a importância do músculo esquelético para as respostas metabólicas
ao diabetes e ao treinamento físico e, considerando os estudos na literatura que
mostraram que indivíduos com genótipo II, que dispõem de menor quantidade de ECA
local e circulante, apresentam maior capacidade máxima de realização de exercício
físico comparados com genótipos ID e DD (MONTGOMERY et al., 1999; ZANG et al.,

70
2008 e WILLIAMS et al., 2010), investigamos algumas possíveis adaptações no
músculo esquelético que pudessem esclarecer o porquê de apenas o grupo 1T melhorar
a capacidade de realização de exercício físico.
Para nossa surpresa, independente do genótipo e do treinamento físico, não
foram encontradas diferenças estatísticas no peso dos músculos sóleo e gastrocnêmio
corrigidos pelo peso corporal, na tipagem de fibras musculares, na densidade de fibras
musculares, na razão capilar por fibra muscular e, finalmente, na atividade máxima da
enzima oxidativa citrato sintase. Nossos dados obtidos em animais diabéticos não
corroboraram os achados da literatura que mostraram melhora na atividade de enzimas
oxidativas, mudanças na tipagem de fibras e aumento na capilarização associados ao
treinamento físico aeróbio (DASTMALCHI et al., 2007; EVANGELISTA et al., 2003;
EVANGELISTA & KRIEGER, 2006; LEICK et al., 2009).
Um dos possíveis fatores que poderia justificar os resultados de músculo-
esquelético do nosso estudo foi o estágio da patologia no momento em que o
treinamento físico foi iniciado. Quando buscamos na literatura informações sobre os
benefícios musculares proporcionados pelo treinamento físico que auxiliam no
tratamento de doenças metabólicas, observamos que os efeitos significativos
normalmente aconteciam quando o treinamento físico era introduzido no estágio inicial
da doença. Por outro lado, SNIJDERS et al. (2010), quando avaliaram o efeito de 6
meses de treinamento físico em pacientes obesos e com diabetes tipo 2 há mais de 12
meses, não observaram diferenças na quantidade de massa muscular na perna e nas
características das fibras musculares, fortalecendo a idéia de que quanto mais precoce a
introdução do treinamento físico para auxiliar no tratamento de doenças metabólicas,
maiores serão os benefícios gerados por ele. No entanto, uma questão que não poderá ser
respondida através do nosso estudo é se o treinamento físico introduzido precocemente

71
seria eficaz para oferecer benefícios metabólicos no músculo esquelético dos animais
diabéticos e se isso estaria relacionado ao genótipo da ECA.
Assim, os dados obtidos no presente estudo apenas sugerem que após um
período crônico de hiperglicemia, observa-se redução do potencial do treinamento físico
para promover adaptações metabólicas musculares que poderiam contribuir para atenuar
a progressão do diabetes no modelo experimental investigado. Em decorrência disso,
parece que a melhora na capacidade de realização de exercício físico apresentada pelo
grupo 1T está mais associada à melhora no metabolismo glicêmico do que propriamente
aos efeitos do treinamento físico sobre as variáveis músculo-esqueléticas estudadas.
7. CONCLUSÃO
Os resultados obtidos no presente estudo nos permitem afirmar que as respostas
metabólicas provocadas pelo diabetes não foram associadas ao aumento da expressão de
ECA e, que o treinamento físico foi capaz de atenuar parcialmente os prejuízos
metabólicos induzidos pelo diabetes apenas nos animais com 1 cópia do gene da ECA.

72
8. REFERÊNCIAS
ALVAREZ-AGUILAR, C.; ENRIQUEZ-RAMIREZ, M. L.; FIGUEROA-NUNEZ, B.; GOMEZ-GARCIA, A.; RODRIGUEZ-AYALA, E.; MORAN-MOGUEL, C.; FARIAS-RODRIGUEZ, V. M.; MINO-LEON, D.; LOPEZ-MEZA, J. E. Association between angiotensin-1 converting enzyme gene polymorphism and the metabolic syndrome in a Mexican population. Exp Mol Med, v.39, n.3, p.327-34, 2007 AMERICAN DIABETES ASSOCIATION: Economic Costs of Diabetes in the U.S. in 2007. Diabetes Care, 31: 596-615, 2008 _______:Diagnosis and Classification of Diabetes Mellitus. Diabetes Care, 31 (suppl.1): S55-60, 2008 AN, D.; RODRIGUES, B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol, v.291, n.4, p.H1489-506, 2006 ANDERSEN, H.; SCHMITZ, O.; NIELSEN, S. Decreased isometric muscle strength after acute hyperglycaemia in Type 1 diabetic patients. Diabet Med, v.22, n.10, p.1401-7, 2005 APOR, P. [Physical activity in prevention and treatment of diabetes]. Orv Hetil, v.150, n.13, p.579-87, 2009 AROZAL, W.; WATANABE, K.; VEERAVEEDU, P. T.; MA, M.; THANDAVARAYAN, R. A.; SUZUKI, K.; TACHIKAWA, H.; KO DAMA, M.; AIZAWA, Y. Effects of angiotensin receptor blocker on oxidative stress and cardio-renal function in streptozotocin-induced diabetic rats. Biol Pharm Bull, v.32, n.8, p.1411-6, 2009 AUGHSTEEN, A. A.; KHAIR, A. M.; SULEIMAN, A. A. Quantitative morphometric study of the skeletal muscles of normal and streptozotocin-diabetic rats. JOP, v.7, n.4, p.382-9, 2006 AXELROD, L.; LEVINE, L. Plasma prostaglandin levels in rats with diabetes mellitus and diabetic ketoacidosis. Diabetes, v.31, n.11, p.994-1001, 1982 BACKLUND, T.; LAKKISTO, P.; PALOJOKI, E.; GRONHOLM, T.; SARASTE, A.; FINCKENBERG, P.; MERVAALA, E.; TIKKANEN, I.; LAINE, M. Activation of protective and damaging components of the cardiac renin-angiotensin system after myocardial infarction in experimental diabetes. J Renin Angiotensin Aldosterone Syst, v.8, n.2, p.66-73, 2007 BALDUCCI, S.; ZANUSO, S.; NICOLUCCI, A.; FERNANDO, F.; CAVALLO, S.; CARDELLI, P.; FALLUCCA, S.; ALESSI, E.; LETIZIA, C.; JIMENEZ, A.; FALLUCCA, F.; PUGLIESE, G. Anti-inflammatory effect of exercise training in subjects with type 2 diabetes and the metabolic syndrome is dependent on exercise modalities and independent of weight loss. Nutr Metab Cardiovasc Dis, v.20, n.8, p.608-17, 2009

73
BARONE, B.; RODACKI, M.; CENCI, M. C.; ZAJDENVERG, L.; MILECH, A.; OLIVEIRA, J. E. [Diabetic ketoacidosis in adults--update of an old complication]. Arq Bras Endocrinol Metabol, v.51, n.9, p.1434-47, 2007 BAZIN, R.; FERRE, P. Assays of lipogenic enzymes. Methods Mol Biol, v.155, p.121-7, 2001 BERNARDIS, L. L.; PATTERSON, B. D. Correlation between 'Lee index' and carcass fat content in weanling and adult female rats with hypothalamic lesions. J Endocrinol, v.40, n.4, p.527-8, 1968 BILOUS, R. Renin angiotensin system (RAS) blockade and diabetic microvascular complications in the eye and kidney. Prim Care Diabetes, v.2, n.4, p.201-2, 2008 BLAAK, E. E. Fatty acid metabolism in obesity and type 2 diabetes mellitus. Proc Nutr Soc, v.62, n.3, p.753-60, 2003 BOUASSIDA, A.; CHAMARI, K.; ZAOUALI, M.; FEKI, Y.; ZBIDI, A.; TABKA, Z. Review on leptin and adiponectin responses and adaptations to acute and chronic exercise. Br J Sports Med, v.44, n.9, p.620-30, 2008 BOUDINA, S.; ABEL, E. D. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord, v.11, n.1, p.31-9, 2010 BOZKURT, O.; VERSCHUREN, W. M.; VAN WIEREN-DE WIJER, B. M.; KNOL, M. J.; DE BOER, A.; GROBBEE, D. E.; GEERLINGS, M. I.; HEERDINK, E. R.; KLUNGEL, O. H. Genetic variation in the renin-angiotensin system modifies the beneficial effects of ACE inhibitors on the risk of diabetes mellitus among hypertensives. J Hum Hypertens, v.22, n.11, p.774-80, 2008 BRADFORD, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, v.72, p.248-54, 1976 CARVALHO-PINTO, C.; GARCIA, M. I.; GOMEZ, L.; BALLESTEROS, A.; ZABALLOS, A.; FLORES, J. M.; MELLADO, M.; RODRIGUEZ-FRADE, J. M.; BALOMENOS, D.; MARTINEZ, A. C. Leukocyte attraction through the CCR5 receptor controls progress from insulitis to diabetes in non-obese diabetic mice. Eur J Immunol, v.34, n.2, p.548-57, 2004 CASSIS, L. A.; ENGLISH, V. L.; BHARADWAJ, K.; BOUSTANY, C. M. Differential effects of local versus systemic angiotensin II in the regulation of leptin release from adipocytes. Endocrinology, v.145, n.1, p.169-74, 2004 CASSIS, L. A.; POLICE, S. B.; YIANNIKOURIS, F.; THATCHER, S. E. Local adipose tissue renin-angiotensin system. Curr Hypertens Rep, v.10, n.2, p.93-8, 2008

74
CENTERS FOR DISEASE CONTROL AND PREVENTION: National Diabetes Fact Sheet, 2007. Available from http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2007.pdf. Acessado em Março, 2010 CLERK, L. H.; VINCENT, M. A.; BARRETT, E. J.; LANKFORD, M. F.; LINDNER, J. R. Skeletal muscle capillary responses to insulin are abnormal in late-stage diabetes and are restored by angiotensin-converting enzyme inhibition. Am J Physiol Endocrinol Metab, v.293, n.6, p.E1804-9, 2007 COATE, K. C.; HUGGINS, K. W. Consumption of a high glycemic index diet increases abdominal adiposity but does not influence adipose tissue pro-oxidant and antioxidant gene expression in C57BL/6 mice. Nutr Res, v.30, n.2, p.141-150, 2010 DALLA VESTRA, M.; SIMIONI, N.; MASIERO, A. Renal effects of dual renin-angiotensin-aldosterone system blockade in patients with diabetic nephropathy. Int Urol Nephrol, v.41, n.1, p.119-26, 2009 DASTMALCHI, M.; ALEXANDERSON, H.; LOELL, I.; STAHLBERG, M.; BORG, K.; LUNDBERG, I. E.; ESBJORNSSON, M. Effect of physical training on the proportion of slow-twitch type I muscle fibers, a novel nonimmune-mediated mechanism for muscle impairment in polymyositis or dermatomyositis. Arthritis Rheum, v.57, n.7, p.1303-10, 2007 DE ANGELIS, K. L.; OLIVEIRA, A. R.; DALL'AGO, P.; PEIXOTO, L. R.; GADONSKI, G.; LACCHINI, S.; FERNANDES, T. G.; IRIGOYEN, M. C. Effects of exercise training on autonomic and myocardial dysfunction in streptozotocin-diabetic rats. Braz J Med Biol Res, v.33, n.6, p.635-41, 2000 DI GIROLAMO, M.; MENDLINGER, S.; FERTIG, J. W. A simple method to determine fat cell size and number in four mammalian species. Am J Physiol, v.221, n.3, p.850-8, 1971 ENEVOLDSEN, L. H.; STALLKNECHT, B.; LANGFORT, J.; PETERSEN, L. N.; HOLM, C.; PLOUG, T.; GALBO, H. The effect of exercise training on hormone-sensitive lipase in rat intra-abdominal adipose tissue and muscle. J Physiol, v.536, n.Pt 3, p.871-7, 2001 ENGLISH, V.; CASSIS, L. Facilitation of sympathetic neurotransmission contributes to angiotensin regulation of body weight. J Neural Transm, v.106, n.7-8, p.631-44, 1999 ESSIG, D. A.; ALDERSON, N. L.; FERGUSON, M. A.; BARTOLI, W. P.; DURSTINE, J. L. Delayed effects of exercise on the plasma leptin concentration. Metabolism, v.49, n.3, p.395-9, 2000 EVANGELISTA, F. S.; BERGAMO, F. C.; FONSECA-ALANIZ, M. H.; NAKAMUTA, J.; KRIEGER, J. E. Body weight homeostasis associated with high fat feeding and ACE gene. Obesity Journal, v.16, n.Suppl 1, p.S260, 2008

75
EVANGELISTA, F. S.; BRUM, P. C.; KRIEGER, J. E. Duration-controlled swimming exercise training induces cardiac hypertrophy in mice. Braz J Med Biol Res, v.36, n.12, p.1751-9, 2003 EVANGELISTA, F. S.; KRIEGER, J. E. Small gene effect and exercise training-induced cardiac hypertrophy in mice: an Ace gene dosage study. Physiol Genomics, v.27, n.3, p.231-6, 2006 FALLUCCA, F.; POZZILLI, P. Physical exercise, public health and quality of life in diabetes. Diabetes Metab Res Rev, v.25 Suppl 1, p.S1-3, 2009 FISHER, J. S.; VAN PELT, R. E.; ZINDER, O.; LANDT, M.; KOHRT, W. M. Acute exercise effect on postabsorptive serum leptin. J Appl Physiol, v.91, n.2, p.680-6, 2001 FONSECA-ALANIZ, M. H.; TAKADA, J.; ALONSO-VALE, M. I.; LIMA, F. B. [The adipose tissue as a regulatory center of the metabolism]. Arq Bras Endocrinol Metabol, v.50, n.2, p.216-29, 2006 FOUFELLE, F.; GIRARD, J.; FERRE, P. Regulation of lipogenic enzyme expression by glucose in liver and adipose tissue: a review of the potential cellular and molecular mechanisms. Adv Enzyme Regul, v.36, p.199-226, 1996 FROSSARD, M.; JOUKHADAR, C.; STEFFEN, G.; SCHMID, R.; EICHLER, H. G.; MULLER, M. Paracrine effects of angiotensin-converting-enzyme- and angiotensin-II-receptor- inhibition on transcapillary glucose transport in humans. Life Sci, v.66, n.10, p.PL147-54, 2000 FURUHASHI, M.; URA, N.; TAKIZAWA, H.; YOSHIDA, D.; MONIWA, N.; MURAKAMI, H.; HIGASHIURA, K.; SHIMAMOTO, K. Blockade of the renin-angiotensin system decreases adipocyte size with improvement in insulin sensitivity. J Hypertens, v.22, n.10, p.1977-82, 2004 GHORBANI, A.; VAREDI, M.; HADJZADEH, M. A.; OMRANI, G. H. Type-1 diabetes induces depot-specific alterations in adipocyte diameter and mass of adipose tissues in the rat. Exp Clin Endocrinol Diabetes, v.118, n.7, p.442-8, 2010 GIACCHETTI, G.; FALOIA, E.; MARINIELLO, B.; SARDU, C.; GATTI, C.; CAMILLONI, M. A.; GUERRIERI, M.; MANTERO, F. Overexpression of the renin-angiotensin system in human visceral adipose tissue in normal and overweight subjects. Am J Hypertens, v.15, n.5, p.381-8, 2002 GIACCHETTI, G.; SECHI, L. A.; RILLI, S.; CAREY, R. M. The renin-angiotensin-aldosterone system, glucose metabolism and diabetes. Trends Endocrinol Metab, v.16, n.3, p.120-6, 2005 GUO, K. Y.; HALO, P.; LEIBEL, R. L.; ZHANG, Y. Effects of obesity on the relationship of leptin mRNA expression and adipocyte size in anatomically distinct fat depots in mice. Am J Physiol Regul Integr Comp Physiol, v.287, n.1, p.R112-9, 2004

76
HAMALAINEN, N.; PETTE, D. The histochemical profiles of fast fiber types IIB, IID, and IIA in skeletal muscles of mouse, rat, and rabbit. J Histochem Cytochem, v.41, n.5, p.733-43, 1993 HAMILTON, M. T.; AREIQAT, E.; HAMILTON, D. G.; BEY, L. Plasma triglyceride metabolism in humans and rats during aging and physical inactivity. Int J Sport Nutr Exerc Metab, v.11 Suppl, p.S97-104, 2001 HAYAT, S. A.; PATEL, B.; KHATTAR, R. S.; MALIK, R. A. Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci (Lond), v.107, n.6, p.539-57, 2004 HEBERT, S. L.; NAIR, K. S. Protein and energy metabolism in type 1 diabetes. Clin Nutr, v.29, n.1, p.13-7, 2009 HEIMANN, A. O gene da enzima conversora de angiotensina influencia nas alterações do diabetes. 2003. Tese de Doutorado -, Faculdade de Medicina da Universidade de São Paulo, São Paulo. HONNOR, R. C.; DHILLON, G. S.; LONDOS, C. cAMP-dependent protein kinase and lipolysis in rat adipocytes. I. Cell preparation, manipulation, and predictability in behavior. J Biol Chem, v.260, n.28, p.15122-9, 1985 HOOD, D. A. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl Physiol Nutr Metab, v.34, n.3, p.465-72, 2009 HOWLETT, K. F.; SAKAMOTO, K.; HIRSHMAN, M. F.; ASCHENBACH, W. G.; DOW, M.; WHITE, M. F.; GOODYEAR, L. J. Insulin signaling after exercise in insulin receptor substrate-2-deficient mice. Diabetes, v.51, n.2, p.479-83, 2002 HUANG, W.; GALLOIS, Y.; BOUBY, N.; BRUNEVAL, P.; HEUDES, D.; BELAIR, M. F.; KREGE, J. H.; MENETON, P.; MARRE, M.; SMITHIES, O.; ALHENC-GELAS, F. Genetically increased angiotensin I-converting enzyme level and renal complications in the diabetic mouse. Proc Natl Acad Sci U S A, v.98, n.23, p.13330-4, 2001 INTERNATIONAL DIABETES FEDERATION. Diabetes and Obesity: Urgent Action Needed. http://www.idf.org/home/index.cfm?unode_C659495D-7467-45C0-8A195B3D8EA3D172. Acessado em Março, 2010 JESSEN, N.; GOODYEAR, L. J. Contraction signaling to glucose transport in skeletal muscle. J Appl Physiol, v.99, n.1, p.330-7, 2005 JOHNSTON, C. I. Biochemistry and pharmacology of the renin-angiotensin system. Drugs, v.39 Suppl 1, p.21-31, 1990 JONES, B. H.; STANDRIDGE, M. K.; MOUSTAID, N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology, v.138, n.4, p.1512-9, 1997

77
JUNOD, A.; LAMBERT, A. E.; ORCI, L.; PICTET, R.; GONET, A. E.; RENOLD, A. E. Studies of the diabetogenic action of streptozotocin. Proc Soc Exp Biol Med, v.126, n.1, p.201-5, 1967 JUNOD, A.; LAMBERT, A. E.; STAUFFACHER, W.; RENOLD, A. E. Diabetogenic action of streptozotocin: relationship of dose to metabolic response. J Clin Invest, v.48, n.11, p.2129-39, 1969 KAMAL, F.; YANAKIEVA-GEORGIEVA, N.; PIAO, H.; MORIOKA, T.; OITE, T. Local delivery of angiotensin II receptor blockers into the kidney passively attenuates inflammatory reactions during the early phases of streptozotocin-induced diabetic nephropathy through inhibition of calpain activity. Nephron Exp Nephrol, v.115, n.3, p.e69-79, 2010 KARLSSON, C.; LINDELL, K.; OTTOSSON, M.; SJOSTROM, L.; CARLSSON, B.; CARLSSON, L. M. Human adipose tissue expresses angiotensinogen and enzymes required for its conversion to angiotensin II. J Clin Endocrinol Metab, v.83, n.11, p.3925-9, 1998 KIM, S.; DUGAIL, I.; STANDRIDGE, M.; CLAYCOMBE, K.; CHUN, J.; MOUSTAID-MOUSSA, N. Angiotensin II-responsive element is the insulin-responsive element in the adipocyte fatty acid synthase gene: role of adipocyte determination and differentiation factor 1/sterol-regulatory-element-binding protein 1c. Biochem J, v.357, n.Pt 3, p.899-904, 2001 KIVELA, R.; SILVENNOINEN, M.; TOUVRA, A. M.; LEHTI, T. M.; KAINULAINEN, H.; VIHKO, V. Effects of experimental type 1 diabetes and exercise training on angiogenic gene expression and capillarization in skeletal muscle. FASEB J, v.20, n.9, p.1570-2, 2006 KONDO, T.; KOBAYASHI, I.; MURAKAMI, M. Effect of exercise on circulating adipokine levels in obese young women. Endocr J, v.53, n.2, p.189-95, 2006 KRAUSE, M. P.; RIDDELL, M. C.; GORDON, C. S.; IMAM, S. A.; CAFARELLI, E.; HAWKE, T. J. Diabetic myopathy differs between Ins2Akita+/- and streptozotocin-induced Type 1 diabetic models. J Appl Physiol, v.106, n.5, p.1650-9, 2009 KRAUSS, R. M. Is the size of low-density lipoprotein particles related to the risk of coronary heart disease? JAMA, v.287, n.6, p.712-3, 2002 KREGE, J. H.; MOYER, J. S.; LANGENBACH, L. L.; PENG, L.; ZHANG, S. H.; MAEDA, N.; REDDICK, R. L.; SMITHIES, O. Angiotensin-converting enzyme gene and atherosclerosis. Arterioscler Thromb Vasc Biol, v.17, n.7, p.1245-50, 1997 KUNJATHOOR, V. V.; WILSON, D. L.; LEBOEUF, R. C. Increased atherosclerosis in streptozotocin-induced diabetic mice. J Clin Invest, v.97, n.7, p.1767-73, 1996 KUSMINSKI, C. M.; SHETTY, S.; ORCI, L.; UNGER, R. H.; SCHERER, P. E. Diabetes and apoptosis: lipotoxicity. Apoptosis, v.14, n.12, p.1484-95, 2009

78
LAWN, R. M. Lipoprotein(a) in heart disease. Sci Am, v.266, n.6, p.54-60, 1992 LEICK, L.; HELLSTEN, Y.; FENTZ, J.; LYNGBY, S. S.; WOJTASZEWSKI, J. F.; HIDALGO, J.; PILEGAARD, H. PGC-1alpha mediates exercise-induced skeletal muscle VEGF expression in mice. Am J Physiol Endocrinol Metab, v.297, n.1, p.E92-103, 2009 LEME, J. A.; GOMES, R. J.; DE MELLO, M. A.; LUCIANO, E. Moderate physical training increases brain insulin concentrations in experimental diabetic rats. Indian J Exp Biol, v.46, n.6, p.443-6, 2008 LEUNG, P. S. Mechanisms of protective effects induced by blockade of the renin-angiotensin system: novel role of the pancreatic islet angiotensin-generating system in Type 2 diabetes. Diabet Med, v.24, n.2, p.110-6, 2007 ______. Current research of the RAS in diabetes mellitus. Adv Exp Med Biol, v.690, p.131-53, 2010 LIMA-SILVA, A. E.; ADAMI, F.; NAKAMURA, F. Y.; DE O LIVEIRA, F. R.; GEVAERD, M. S. Metabolismo de gordura durante o exercício físico: mecanismos de regulação. Rev Bras Cin Desemp Hum., v.8, n.4, p.106-111, 2006 LJUBICIC, V.; JOSEPH, A. M.; SALEEM, A.; UGUCCIONI, G.; COLLU-MARCHESE, M.; LAI, R. Y.; NGUYEN, L. M.; HOOD, D. A. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: effects of exercise and aging. Biochim Biophys Acta, v.1800, n.3, p.223-34, 2009 MACKNIGHT, J. M.; MISTRY, D. J.; PASTORS, J. G.; HOLMES, V.; RYNDERS, C. A. The daily management of athletes with diabetes. Clin Sports Med, v.28, n.3, p.479-95, 2009 MATAYOSHI, T.; KAMIDE, K.; TAKIUCHI, S.; HORIO, T.; YOSHIHARA, F.; NAKAMURA, S.; NAKAHAMA, H.; KAWANO, Y. Relationship between insulin resistance and the renin-angiotensin system: analysis for patients with essential and renovascular hypertension. Clin Exp Hypertens, v.29, n.7, p.479-87, 2007 MAUER, M.; ZINMAN, B.; GARDINER, R.; SUISSA, S.; SINAIKO, A.; STRAND, T.; DRUMMOND, K.; DONNELLY, S.; GOODYER, P.; GUBLER, M. C.; KLEIN, R. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med, v.361, n.1, p.40-51, 2009 MERINO, B.; CANO, V.; GUZMAN, R.; SOMOZA, B.; RUIZ-GAYO, M. Leptin-mediated hypothalamic pathway of cholecystokinin (CCK-8) to regulate body weight in free-feeding rats. Endocrinology, v.149, n.4, p.1994-2000, 2008 MESSADI, E.; VINCENT, M. P.; GRIOL-CHARHBILI, V.; MANDET, C.; COLUCCI, J.; KREGE, J. H.; BRUNEVAL, P.; BOUBY, N.; SMITHIES, O.; ALHENC-GELAS, F.; RICHER, C. Genetically determined angiotensin converting enzyme level and myocardial tolerance to ischemia. FASEB J, 2010

79
MONOSIKOVA, J.; ZEMAN, M.; VESELA, A.; HERICHOVA, I. Down regulation of angiotensin II receptor AT1 expression in the pancreas of diabetic rat. Exp Clin Endocrinol Diabetes, v.117, n.8, p.438-9, 2009 MONTGOMERY, H.; CLARKSON, P.; BARNARD, M.; BELL, J.; BRYNES, A.; DOLLERY, C.; HAJNAL, J.; HEMINGWAY, H.; MERCER, D.; JARMAN, P.; MARSHALL, R.; PRASAD, K.; RAYSON, M.; SAEED, N.; TALMUD, P.; THOMAS, L.; JUBB, M.; WORLD, M.; HUMPHRIES, S. Angiotensin-converting-enzyme gene insertion/deletion polymorphism and response to physical training. Lancet, v.353, n.9152, p.541-5, 1999 MORGAN, H. E.; BAKER, K. M. Cardiac hypertrophy. Mechanical, neural, and endocrine dependence. Circulation, v.83, n.1, p.13-25, 1991 MULLER, M. J.; BOSY-WESTPHAL, A.; LATER, W.; HAAS, V.; HELLER, M. Functional body composition: insights into the regulation of energy metabolism and some clinical applications. Eur J Clin Nutr, v.63, n.9, p.1045-56, 2009 MUSI, N.; HIRSHMAN, M. F.; ARAD, M.; XING, Y.; FUJII, N.; POMERLEAU, J.; AHMAD, F.; BERUL, C. I.; SEIDMAN, J. G.; TIAN, R.; GOODYEAR, L. J. Functional role of AMP-activated protein kinase in the heart during exercise. FEBS Lett, v.579, n.10, p.2045-50, 2005 NAKAI, N.; MIYAZAKI, Y.; SATO, Y.; OSHIDA, Y.; NAGA SAKI, M.; TANAKA, M.; NAKASHIMA, K.; SHIMOMURA, Y. Exercise training increases the activity of pyruvate dehydrogenase complex in skeletal muscle of diabetic rats. Endocr J, v.49, n.5, p.547-54, 2002 NIELSEN, S. E.; SUGAYA, T.; TARNOW, L.; LAJER, M.; SCHJOEDT, K. J.; ASTRUP, A. S.; BABA, T.; PARVING, H. H.; ROSSING, P. Tubular and glomerular injury in diabetes and the impact of ACE inhibition. Diabetes Care, v.32, n.9, p.1684-8, 2009 NORDWALL, M.; HYLLIENMARK, L.; LUDVIGSSON, J. Early diabetic complications in a population of young patients with type 1 diabetes mellitus despite intensive treatment. J Pediatr Endocrinol Metab, v.19, n.1, p.45-54, 2006 OAK, J. H.; CAI, H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes, v.56, n.1, p.118-26, 2007 OKOSHI, K.; GUIMARAES, J. F.; DI MUZIO, B. P.; FERNANDES, A. A.; OKOSHI, M. P. [Diabetic cardiomyopathy]. Arq Bras Endocrinol Metabol, v.51, n.2, p.160-7, 2007 OLTMAN, C. L.; DAVIDSON, E. P.; COPPEY, L. J.; KLEINSCHMIDT, T. L.; DAKE, B.; YOREK, M. A. Role of the effect of inhibition of neutral endopeptidase on vascular and neural complications in streptozotocin-induced diabetic rats. Eur J Pharmacol, 2010

80
PÁDUA, M. F.; PÁDUA, T. F.; PAULI, J. R.; DE SOUZA, C. T.; DA SILVA, A. S. R.; ROPELLE, E. C. C.; CARVALHEIRA, J. B. C.; ROPELLE, E. R. Exercício Físico Reduz a Hiperglicemia de Jejum em Camundongos Diabéticos Através da Ativação da AMPK. Rev Bras Med Esporte, v.15, p.179-184, 2009 PARVING, H. H.; ROSSING, P. Therapeutic benefits of ACE inhibitors and other antihypertensive drugs in patients with type 2 diabetes. Diabetes Care, v.24, n.1, p.177-80, 2001 PAUL, M.; POYAN MEHR, A.; KREUTZ, R. Physiology of local renin-angiotensin systems. Physiol Rev, v.86, n.3, p.747-803, 2006 PEDERSEN-BJERGAARD, U.; DHAMRAIT, S. S.; SETHI, A. A.; FRANDSEN, E.; NORDESTGAARD, B. G.; MONTGOMERY, H. E.; PRAMMING, S.; HOUGAARD, P.; THORSTEINSSON, B. Genetic variation and activity of the renin-angiotensin system and severe hypoglycemia in type 1 diabetes. Am J Med, v.121, n.3, p.246 e1-8, 2008 PELLIEUX, C.; SAUTHIER, T.; AUBERT, J. F.; BRUNNER, H. R.; PEDRAZZINI, T. Angiotensin II-induced cardiac hypertrophy is associated with different mitogen-activated protein kinase activation in normotensive and hypertensive mice. J Hypertens, v.18, n.9, p.1307-17, 2000 PERKINS, B. A.; AIELLO, L. P.; KROLEWSKI, A. S. Diabetes complications and the renin-angiotensin system. N Engl J Med, v.361, n.1, p.83-5, 2009 PETI-PETERDI, J.; KANG, J. J.; TOMA, I. Activation of the renal renin-angiotensin system in diabetes--new concepts. Nephrol Dial Transplant, v.23, n.10, p.3047-9, 2008 PIGHIN, D.; KARABATAS, L.; PASTORALE, C.; DASCAL, E.; CARBONE, C.; CHICCO, A.; LOMBARDO, Y. B.; BASABE, J. C. Role of lipids in the early developmental stages of experimental immune diabetes induced by multiple low-dose streptozotocin. J Appl Physiol, v.98, n.3, p.1064-9, 2005 POLAK, J.; KLIMCAKOVA, E.; MORO, C.; VIGUERIE, N.; BERLAN, M.; HEJNOVA, J.; RICHTEROVA, B.; KRAUS, I.; LANGIN, D.; STICH, V. Effect of aerobic training on plasma levels and subcutaneous abdominal adipose tissue gene expression of adiponectin, leptin, interleukin 6, and tumor necrosis factor alpha in obese women. Metabolism, v.55, n.10, p.1375-81, 2006 POORNIMA, I. G.; PARIKH, P.; SHANNON, R. P. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res, v.98, n.5, p.596-605, 2006 PORTER, J. P.; ANDERSON, J. M.; ROBISON, R. J.; PHILLIPS, A. C. Effect of central angiotensin II on body weight gain in young rats. Brain Res, v.959, n.1, p.20-8, 2003 RABOL, R.; BOUSHEL, R.; ALMDAL, T.; HANSEN, C. N.; PLOUG, T.; HAUGAARD, S. B.; PRATS, C.; MADSBAD, S.; DELA, F. Opposite effects of

81
pioglitazone and rosiglitazone on mitochondrial respiration in skeletal muscle of patients with type 2 diabetes. Diabetes Obes Metab, v.12, n.9, p.806-14, 2010 RAO, S.; VERKMAN, A. S. Analysis of organ physiology in transgenic mice. Am J Physiol Cell Physiol, v.279, n.1, p.C1-C18, 2000 RIBEIRO-OLIVEIRA, A., JR.; NOGUEIRA, A. I.; PEREIRA, R. M.; BOAS, W. W.; DOS SANTOS, R. A.; SIMOES E SILVA, A. C. The renin-angiotensin system and diabetes: an update. Vasc Health Risk Manag, v.4, n.4, p.787-803, 2008 RICHEY, J. M.; ADER, M.; MOORE, D.; BERGMAN, R. N. Angiotensin II induces insulin resistance independent of changes in interstitial insulin. Am J Physiol, v.277, n.5 Pt 1, p.E920-6, 1999 RODBELL, M. Metabolism of Isolated Fat Cells. I. Effects of Hormones on Glucose Metabolism and Lipolysis. J Biol Chem, v.239, p.375-80, 1964 RUBERG, F. L. Myocardial lipid accumulation in the diabetic heart. Circulation, v.116, n.10, p.1110-2, 2007 SAIKI, A.; OHIRA, M.; ENDO, K.; KOIDE, N.; OYAMA, T.; MURANO, T.; WATANABE, H.; MIYASHITA, Y.; SHIRAI, K. Circulating angiotensin II is associated with body fat accumulation and insulin resistance in obese subjects with type 2 diabetes mellitus. Metabolism, v.58, n.5, p.708-13, 2009 SAITO, M.; KINOSHITA, Y.; SATOH, I.; SHINBORI, C.; KONO, T.; HANADA, T.; UEMASU, J.; SUZUKI, H.; YAMADA, M.; SATOH, K. N-hexacosanol ameliorates streptozotocin-induced diabetic rat nephropathy. Eur J Pharmacol, v.544, n.1-3, p.132-7, 2006 SARZANI, R.; SALVI, F.; DESSI-FULGHERI, P.; RAPPELLI, A. Renin-angiotensin system, natriuretic peptides, obesity, metabolic syndrome, and hypertension: an integrated view in humans. J Hypertens, v.26, n.5, p.831-43, 2008 SCHRAUWEN-HINDERLING, V. B.; KOOI, M. E.; HESSELINK, M. K.; JENESON, J. A.; BACKES, W. H.; VAN ECHTELD, C. J.; VAN ENGELSHOVEN, J. M.; MENSINK, M.; SCHRAUWEN, P. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia, v.50, n.1, p.113-20, 2007 SHARMA, A. K.; THOMAS, P. K. Animal models: pathology and pathophysiology. In: Philadelphia (Ed.). Diabetic neurophathy: WB Saunders Company, 1999, p.237-52 SHEPHERD, R. E.; NOBLE, E. G.; KLUG, G. A.; GOLLNICK, P. D. Lipolysis and cAMP accumulation in adipocytes in response to physical training. J Appl Physiol, v.50, n.1, p.143-8, 1981 SHERMAN, W. M.; FRIEDMAN, J. E.; GAO, J. P.; REED, M. J.; ELTON, C. W.; DOHM, G. L. Glycemia and exercise training alter glucose transport and GLUT4 in the Zucker rat. Med Sci Sports Exerc, v.25, n.3, p.341-8, 1993

82
SHINSHI, Y.; HIGASHIURA, K.; YOSHIDA, D.; TOGASHI, N.; YOSHIDA, H.; MIYAZAKI, Y.; URA, N.; SHIMAMOTO, K. Angiotensin II inhibits glucose uptake of skeletal muscle via the adenosine monophosphate-activated protein kinase pathway. J Am Soc Hypertens, v.1, n.4, p.251-5, 2007 SNIJDERS, T.; VERDIJK, L. B.; HANSEN, D.; DENDALE, P.; VAN LOON, L. J. Continuous endurance-type exercise training does not modulate satellite cell content in obese type 2 diabetes patients. Muscle Nerve, 2010 SOCIEDADE BRASILEIRA DE DIABETES: Calcule o número de diabéticos na sua cidade. Disponível em http://www.diabetes.org.br/para-o-publico/calculadoras/numero-de-diabeticos. Acessado em Março, 2010 SPURWAY, N. C. Objective characterization of cells in terms of microscopical parameters: an example from muscle histochemistry. Histochem J, v.13, n.2, p.269-317, 1981 STEPHENS, T. J.; CHEN, Z. P.; CANNY, B. J.; MICHELL, B. J.; KEMP, B. E.; MCCONELL, G. K. Progressive increase in human skeletal muscle AMPKalpha2 activity and ACC phosphorylation during exercise. Am J Physiol Endocrinol Metab, v.282, n.3, p.E688-94, 2002 STEVENSON, J. A.; BOX, B. M.; FELEKI, V.; BEATON, J. R. Bouts of exercise and food intake in the rat. J Appl Physiol, v.21, n.1, p.118-22, 1966 STUMP, C. S.; SHORT, K. R.; BIGELOW, M. L.; SCHIMKE, J. M.; NAIR, K. S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci U S A, v.100, n.13, p.7996-8001, 2003 SUGDEN, P. H.; CLERK, A. Cellular mechanisms of cardiac hypertrophy. J Mol Med, v.76, n.11, p.725-46, 1998 TAKADA, J.; FONSECA-ALANIZ, M. H.; DE CAMPOS, T. B.; ANDREOTTI, S.; CAMPANA, A. B.; OKAMOTO, M.; BORGES-SILVA, C. N.; MACHADO, U. F.; LIMA, F. B. Metabolic recovery of adipose tissue is associated with improvement in insulin resistance in a model of experimental diabetes. J Endocrinol, v.198, n.1, p.51-60, 2008 TAKADA, J.; MACHADO, M. A.; PERES, S. B.; BRITO, L. C.; BORGES-SILVA, C. N.; COSTA, C. E.; FONSECA-ALANIZ, M. H.; ANDREOTTI, S.; LIMA, F. B. Neonatal streptozotocin-induced diabetes mellitus: a model of insulin resistance associated with loss of adipose mass. Metabolism, v.56, n.7, p.977-84, 2007 TATCHUM-TALOM, R.; SCHULZ, R.; MCNEILL, J. R.; KHADOUR, F. H. Upregulation of neuronal nitric oxide synthase in skeletal muscle by swim training. Am J Physiol Heart Circ Physiol, v.279, n.4, p.H1757-66, 2000

83
TAYLOR, E. B.; ELLINGSON, W. J.; LAMB, J. D.; CHESSER, D. G.; COMPTON, C. L.; WINDER, W. W. Evidence against regulation of AMP-activated protein kinase and LKB1/STRAD/MO25 activity by creatine phosphate. Am J Physiol Endocrinol Metab, v.290, n.4, p.E661-9, 2006 THORBURN, A. Ras activity is required for phenylephrine-induced activation of mitogen-activated protein kinase in cardiac muscle cells. Biochem Biophys Res Commun, v.205, n.2, p.1417-22, 1994 TIKELLIS, C.; COOPER, M. E.; THOMAS, M. C. Role of the renin-angiotensin system in the endocrine pancreas: implications for the development of diabetes. Int J Biochem Cell Biol, v.38, n.5-6, p.737-51, 2006 VAISHYA, R.; SINGH, J.; LAL, H. Biochemical effects of irbesartan in experimental diabetic nephropathy. Indian J Pharmacol, v.41, n.6, p.252-4, 2009 VAN BERLO, J. H.; PINTO, Y. M. Polymorphisms in the RAS and cardiac function. Int J Biochem Cell Biol, v.35, n.6, p.932-43, 2003 VAN HERPEN, N. A.; SCHRAUWEN-HINDERLING, V. B. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav, v.94, n.2, p.231-41, 2008 VIRTUE, S.; VIDAL-PUIG, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome--an allostatic perspective. Biochim Biophys Acta, v.1801, n.3, p.338-49, 2010 WEIR, M. R. Effects of renin-angiotensin system inhibition on end-organ protection: can we do better? Clin Ther, v.29, n.9, p.1803-24, 2007 WELTMAN, N. Y.; SALIBA, S. A.; BARRETT, E. J.; WELTMAN, A. The use of exercise in the management of type 1 and type 2 diabetes. Clin Sports Med, v.28, n.3, p.423-39, 2009 WICHI, R.; MALFITANO, C.; ROSA, K.; DE SOUZA, S. B.; SALEMI, V.; MOSTARDA, C.; DE ANGELIS, K.; IRIGOYEN, M. C. Noninvasive and invasive evaluation of cardiac dysfunction in experimental diabetes in rodents. Cardiovasc Diabetol, v.6, p.14, 2007 WILLIAMS, A. D.; ANDERSON, M. J.; SELIG, S.; CAREY, M. F.; FEBBRAIO, M. A.; HAYES, A.; TOIA, D.; HARRAP, S. B.; HARE, D. L. Differential response to resistance training in CHF according to ACE genotype. Int J Cardiol, 2010 XIAO, R. P.; ZHANG, S. J.; CHAKIR, K.; AVDONIN, P.; ZHU, W.; BOND, R. A.; BALKE, C. W.; LAKATTA, E. G.; CHENG, H. Enhanced G(i) signaling selectively negates beta2-adrenergic receptor (AR)--but not beta1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation, v.108, n.13, p.1633-9, 2003 XU, L.; ZHANG, Y.; COHEN, S. B.; DIPETRILLO, K. TrkB Agonist Antibody Dose-Dependently Raises Blood Pressure in Mice With Diet-Induced Obesity. Am J Hypertens, 2010

84
YUAN, L.; LI, X.; XU, G. L.; QI, C. J. Effects of renin-angiotensin system blockade on islet function in diabetic rats. J Endocrinol Invest, v.33, n.1, p.13-9, 2010 ZELEZNIAK, A.; PERS, T. H.; SOARES, S.; PATTI, M. E.; PATIL, K. R. Metabolic network topology reveals transcriptional regulatory signatures of type 2 diabetes. PLoS Comput Biol, v.6, n.4, p.e1000729, 2010 ZHANG, B.; TANAKA, H.; SHONO, N.; MIURA, S.; KIYONAGA, A.; SHINDO, M.; SAKU, K. The I allele of the angiotensin-converting enzyme gene is associated with an increased percentage of slow-twitch type I fibers in human skeletal muscle. Clin Genet, v.63, n.2, p.139-44, 2003 ZHANG, S.; YAN, X.; ZHOU, P. C.; HUANG, C.; YANG, L.; LI, X.; LIN, J.; ZHOU, Z. G. [Effects of pravastatin in prevention of diabetes and mechanism thereof: experiment with non-obese diabetic mice]. Zhonghua Yi Xue Za Zhi, v.88, n.8, p.568-72, 2008 ZHANG, X.; WANG, C.; DAI, H.; LIN, Y.; ZHANG, J. Association between angiotensin-converting enzyme gene polymorphisms and exercise performance in patients with COPD. Respirology, v.13, n.5, p.683-8, 2008 ZOIA, L.; ARGYROPOULOS, D. S. Characterization of free radical spin adducts of the DIPPMPO using mass spectrometry and (31)P NMR. Eur J Mass Spectrom (Chichester, Eng), v.16, n.2, p.175-85, 2010 ZOJA, C.; CORNA, D.; GAGLIARDINI, E.; CONTI, S.; ARNABOLDI, L.; BENIGNI, A.; REMUZZI, G. Adding a statin to a combination of ACE inhibitor and ARB normalizes proteinuria in experimental diabetes, which translates into full renoprotection. Am J Physiol Renal Physiol, v.299, n.5, p.F1203-11, 2010 ZOLL, J.; MONASSIER, L.; GARNIER, A.; N'GUESSAN, B.; METTAUER, B.; VEKSLER, V.; PIQUARD, F.; VENTURA-CLAPIER, R.; GENY, B. ACE inhibition prevents myocardial infarction-induced skeletal muscle mitochondrial dysfunction. J Appl Physiol, v.101, n.2, p.385-91, 2006 ZORAD, S.; DOU, J. T.; BENICKY, J.; HUTANU, D.; TYBITANCLOVA, K.; ZHOU, J.; SAAVEDRA, J. M. Long-term angiotensin II AT1 receptor inhibition produces adipose tissue hypotrophy accompanied by increased expression of adiponectin and PPARgamma. Eur J Pharmacol, v.552, n.1-3, p.112-22, 2006


















