
ANEXO I RESUMO DAS CARACTERÍSTICAS DO MEDICAMENTO · alternativa sem ou com potencial mínimo de...
75
1 ANEXO I RESUMO DAS CARACTERÍSTICAS DO MEDICAMENTO
Transcript of ANEXO I RESUMO DAS CARACTERÍSTICAS DO MEDICAMENTO · alternativa sem ou com potencial mínimo de...

1
ANEXO I
RESUMO DAS CARACTERÍSTICAS DO MEDICAMENTO

2
1. DENOMINAÇÃO DO MEDICAMENTO SUTENT 12,5 mg cápsulas 2. COMPOSIÇÃO QUALITATIVA E QUANTITATIVA Cada cápsula contém malato de sunitinib equivalente a 12,5 mg de sunitinib. Excipiente(s): 80,0 mg de manitol. Lista completa de excipientes, ver secção 6.1. 3. FORMA FARMACÊUTICA Cápsulas. Cápsulas de gelatina com a cabeça e o corpo cor de laranja, com “Pfizer” impresso a tinta branca na cabeça, “STN 12,5 mg” no corpo e contendo grânulos amarelo alaranjados. 4. INFORMAÇÕES CLÍNICAS 4.1 Indicações terapêuticas O SUTENT está indicado para o tratamento de tumores malignos do estroma gastrointestinal irressecáveis e/ou metastáticos (GIST) após insucesso do tratamento com mesilato de imatinib, devida a resistência ou intolerância. O SUTENT está indicado para o tratamento do carcinoma de células renais avançado e/ou metastático (MRCC) após insucesso da terapêutica com interferão-alfa ou interleucina-2. A eficácia é baseada no tempo até progressão tumoral e num aumento de sobrevivência no GIST e nas taxas de resposta objectivas para o MRCC (ver secção 5.1). 4.2 Posologia e modo de administração A terapêutica deve ser iniciada por um médico experiente no tratamento do carcinoma de células renais ou de GIST. A posologia recomendada de SUTENT é uma dose oral diária de 50 mg tomada durante 4 semanas consecutivas, a que se segue um período de repouso de 2 semanas (esquema de tratamento 4/2), completando um ciclo de 6 semanas. Podem ser aplicados ajustes de dose em intervalos de 12,5 mg com base na segurança e tolerabilidade individuais. A dose diária não deve exceder os 87,5 mg nem ser reduzida abaixo dos 37,5 mg. A co-administração de indutores potentes da CYP3A4, tal como a rifampicina, deverá ser evitada (ver secções 4.4 e 4.5). Caso tal não seja possível, poderá ser necessário aumentar a dose de SUTENT com pequenos incrementos de 12,5 mg (até 87,5 mg por dia), com base na monitorização cuidadosa da tolerabilidade. A co-administração de SUTENT com inibidores potentes da CYP3A4, tal como o cetoconazol, deverá ser evitada (ver secções 4.4 e 4.5). Caso tal não seja possível, poderá ser necessário reduzir a dose de SUTENT para um mínimo de 37,5 mg diários, com base na monitorização cuidadosa da tolerabilidade. Deverá ser considerada a selecção de medicação concomitante alternativa sem ou com potencial mínimo de inibição ou indução da CYP3A4.

3
Utilização pediátrica: A segurança e eficácia de SUTENT em doentes pediátricos não foram estabelecidas. SUTENT não deverá ser utilizado na população pediátrica até estarem disponíveis mais dados. Utilização em doentes idosos: Aproximadamente 25% dos indivíduos que participaram nos estudos clínicos de SUTENT tinham 65 ou mais anos. Não se observaram diferenças significativas relativas à segurança ou efectividade entre os doentes mais novos e os doentes mais velhos. Insuficiência hepática: Não foram realizados estudos clínicos em doentes com diminuição da função hepática (ver secção 5.2). Insuficiência renal: Não foram realizados estudos clínicos em doentes com diminuição da função renal (ver secção 5.2). SUTENT pode ser tomado com ou sem alimentos. Se o doente não tomar uma dose, não deverá compensar com uma dose adicional. O doente deverá tomar a dose recomendada no dia seguinte, da forma habitual. 4.3 Contra-indicações Hipersensibilidade ao malato de sunitinib ou a qualquer um dos excipientes. 4.4 Advertências e precauções especiais de utilização A co-administração de indutores potentes da CYP3A4, tal como a rifampicina, poderá diminuir a concentração plasmática de sunitinib. Como tal, a combinação com indutores deverá ser evitada. Se tal não for possível, poderá ser necessário aumentar a dose de SUTENT (ver secções 4.2 e 4.5). A co-administração de inibidores potentes da CYP3A4, como o cetoconazol, poderá aumentar as concentrações plasmáticas de sunitinib. Recomenda-se a selecção de medicação concomitante alternativa sem ou com potencial mínimo de inibição enzimática. Se não for possível, poderá ser necessário reduzir a dose de SUTENT (ver secções 4.2 e 4.5). Pele e tecidos A alteração da coloração cutânea, possivelmente devida à cor da substância activa (amarela) é um acontecimento adverso comum relacionado com o tratamento, ocorrendo em aproximadamente 30% dos doentes. Os doentes devem ser avisados que a despigmentação do cabelo ou da pele poderá também ocorrer durante o tratamento com SUTENT. Outros efeitos dermatológicos possíveis podem incluir secura, espessamento ou fissuras na pele, bolhas ou erupção cutânea ocasional nas palmas das mãos e nas plantas dos pés. Foi notificada dor/irritação na cavidade oral em aproximadamente 14% dos doentes. Foi reportada disgeusia (alterações do paladar) em aproximadamente 28% dos doentes. Os acontecimentos acima descritos não foram cumulativos, foram tipicamente reversíveis e geralmente não resultaram em descontinuação do tratamento. Acontecimentos gastrointestinais Os acontecimentos gastrointestinais mais frequentemente relatados foram náuseas, diarreia, estomatite, dispepsia e vómitos. As medidas de suporte para os acontecimentos gastrointestinais que requerem tratamento podem incluir administração de medicamentos anti-eméticos ou anti-diarreicos. Hemorragia Ocorreu hemorragia tumoral relacionada com o tratamento em aproximadamente 2% dos doentes com GIST. Estes acontecimentos poderão ocorrer subitamente, e no caso de tumores pulmonares podem surgir como hemoptises graves ou hemorragias pulmonares graves e potencialmente fatais. Num ensaio clínico conduzido em doentes com carcinoma do pulmão de não-pequenas células (NSCLC)

4
metastizado, ocorreu hemorragia pulmonar fatal em 2 doentes que estavam a receber SUTENT. Ambos os doentes apresentavam histologia celular escamosa. O SUTENT não está aprovado para a utilização em doentes com carcinoma do pulmão de não-pequenas células. A avaliação de rotina deste acontecimento deve incluir contagem completa das células sanguíneas e exame físico. A epistaxis foi o acontecimento adverso hemorrágico relacionado com o tratamento mais frequente, tendo sido relatado para aproximadamente metade dos doentes com tumores sólidos que apresentaram acontecimentos hemorrágicos. Nenhum destes acontecimentos foi grave. Tracto gastrointestinal Raramente, ocorreram complicações gastrointestinais graves e por vezes fatais, incluindo perfuração gastrointestinal, em doentes com doença maligna intra-abdominal tratados com SUTENT. Hipertensão Foi notificada hipertensão relacionada com o tratamento em aproximadamente 16% dos doentes com tumores sólidos. A posologia de SUTENT foi reduzida ou temporariamente adiada em 2,7% desta população de doentes. Nenhum destes doentes descontinuou o tratamento com SUTENT. Ocorreu hipertensão grave (sistólica >200 mmHg ou diastólica de 110 mmHg) em 4,7% desta população de doentes. Os doentes devem ser monitorizados para a hipertensão e apropriadamente controlados. É recomendada a suspensão temporária em doentes com hipertensão grave que não se encontra controlada com o acompanhamento médico. O tratamento poderá ser retomado assim que a hipertensão estiver apropriadamente controlada. Hematológicos Foram relatados decréscimos na contagem absoluta de neutrófilos de gravidade grau 3 e 4 em 13,1% e 0,9% dos doentes, respectivamente. Foram relatados decréscimos na contagem de plaquetas de gravidade grau 3 e 4 em 4% e 0,5% dos doentes, respectivamente. Os acontecimentos adversos acima descritos não foram cumulativos, foram tipicamente reversíveis e geralmente não resultaram na descontinuação do tratamento. Deve ser realizada uma contagem completa das células sanguíneas no início de cada ciclo de tratamento, nos doentes a receber tratamento com SUTENT.

5
Cardiovascular Ocorreram diminuições na fracção de ejecção ventricular esquerda (FEVE) ≥ 20% e abaixo do limite inferior do normal, em aproximadamente 2% dos doentes com GIST tratados com SUTENT, em 4% dos doentes com carcinoma de células renais metastático e em 2% dos doentes tratados com placebo. Estas diminuições do FEVE não aparentam ter sido progressivas e melhoraram frequentemente com a continuação do tratamento. Foram relatados acontecimentos adversos relacionados com o tratamento de “insuficiência cardíaca”, “insuficiência cardíaca congestiva” ou “insuficiência ventricular esquerda” em 0,7% dos doentes com tumores sólidos e em 1% dos doentes tratados com placebo. Todos os doentes tinham GIST. A relação entre a inibição dos receptores da tirosina-cinase e a função cardíaca, se existir, permanece por esclarecer. Os doentes que apresentaram acontecimentos cardíacos nos 12 meses anteriores à administração de SUTENT, como enfarte do miocárdio (incluindo angina instável/grave), bypass coronário/arterial periférico, insuficiência cardíaca congestiva (ICC) sintomática, acidente vascular cerebral ou isquémico transitório ou embolia pulmonar foram excluídos dos ensaios clínicos com SUTENT. Não se sabe se os doentes com estas condições concomitantes podem apresentar um risco superior de desenvolver disfunção ventricular esquerda relacionada com o fármaco. Recomenda-se que os médicos avaliem estes riscos versus os potenciais benefícios do medicamento. Estes doentes deverão ser cuidadosamente monitorizados quanto a sinais e sintomas de ICC enquanto estiverem a receber SUTENT. Deverão ser igualmente consideradas avaliações de base e periódicas da FEVE enquanto o doente estiver a receber SUTENT. Em doentes sem factores de risco cardíacos, deverá ser considerada uma avaliação de base da fracção de ejecção. Na presença de manifestações clínicas de ICC, é recomendada a descontinuação de SUTENT. A dose de SUTENT deve ser interrompida e/ou reduzida em doentes sem evidência clínica de ICC, mas que apresentem uma fracção de ejecção <50% e > 20% abaixo do valor de base. Prolongamento do intervalo QT O prolongamento do intervalo QT foi investigado num ensaio com 24 doentes, com idades entre os 20-87 anos, com doença maligna avançada. Em concentrações de aproximadamente o dobro das concentrações terapêuticas, o SUTENT demonstrou prolongar o intervalo QTcF (correcção de Frederica). Não existiram doentes com prolongamento do intervalo QT/QTc superior a grau 2 (CTCAE v3.0) e nenhum doente apresentou arritmia cardíaca. A relevância clínica dos efeitos observados é pouco clara e dependerá dos factores de risco individuais e das susceptibilidades que o doente apresentar. O SUTENT deve ser usado com precaução em doentes com uma história conhecida de prolongamento do intervalo QT, em doentes que se encontrem a tomar antiarrítmicos ou em doentes com doença cardíaca pré-existente relevante, bradicardia ou perturbações electrolíticas. O tratamento concomitante com inibidores potentes da CYP3A4, que podem aumentar as concentrações plasmáticas de sunitinib, deve ser administrado com precaução e a dose de SUTENT reduzida (ver secção 4.5). Acontecimentos tromboembólicos venosos Foram notificados acontecimentos tromboembólicos venosos em quatro doentes (2%) dos dois estudos conduzidos para o carcinoma de células renais metastático; dois doentes com embolia pulmonar (ambas grau 4) e dois doentes com trombose venosa profunda (ambas grau 3). Ocorreu interrupção da administração num destes casos. No estudo pivot de GIST, sete doentes (3%) a receber SUTENT e nenhum dos doentes em placebo apresentaram acontecimentos tromboembólicos venosos; cinco dos sete foram tromboses venosas profundas de grau 3 e dois foram grau 1 ou 2. Quatro destes sete doentes com GIST descontinuaram o tratamento após a primeira observação de trombose venosa profunda. Embolia pulmonar Foi reportada embolia pulmonar relacionada com o tratamento em aproximadamente 1,1% dos doentes com tumores sólidos que receberam SUTENT. Nenhum destes acontecimentos resultou na descontinuação do tratamento com SUTENT; contudo, ocorreu, num número reduzido de casos, uma redução da dose ou um adiamento temporário do tratamento. Não houve mais ocorrências de embolia pulmonar nestes doentes após o tratamento ter sido retomado.

6
Hipotiroidismo O hipotiroidismo foi reportado como acontecimento adverso em 7 doentes (4%) dos dois estudos realizados no carcinoma de células renais metastático. Adicionalmente, registaram-se aumentos da TSH em 4 doentes (2%). Globalmente, 7% da população com carcinoma de células renais metastático apresentaram evidência clínica ou laboratorial de hipotiroidismo emergente do tratamento. Foi observado hipotiroidismo adquirido emergente do tratamento em 8 (4%) doentes com GIST a receber SUTENT versus 1 (1%) a receber placebo. Os doentes com sintomas sugestivos de hipotiroidismo devem realizar monitorização laboratorial da função tiroideia e devem ser tratados de acordo com a prática clínica considerada padrão. Função pancreática Foram observados aumentos nas actividades da lipase e da amilase séricas em doentes com diversos tumores sólidos medicados com SUTENT. Os aumentos das actividades da lipase, em doentes com diversos tipos de tumores sólidos, foram transitórios e geralmente não foram acompanhados por sinais ou sintomas de pancreatite. Foi observada pancreatite em 0,4% dos doentes com tumores sólidos. Se estiverem presentes sintomas de pancreatite, os doentes devem ter acompanhamento médico adequado. Convulsões Foram observadas convulsões nos estudos clínicos com SUTENT, em indivíduos com evidência radiológica de metástases cerebrais. Adicionalmente, existiram notificações raras (<1%) de indivíduos que apresentaram convulsões e evidência radiológica da síndrome de leocoencefalopatia posterior reversível. Em nenhum dos indivíduos este acontecimento foi fatal. Os doentes com convulsões e sinais/sintomas consistentes com síndrome de leucoencefalopatia posterior reversível, tais como hipertensão, cefaleias, diminuição do estado de alerta, alterações da função mental e perda visual, incluindo cegueira cortical, deverão ser controlados através de acompanhamento médico, que inclua o controlo da hipertensão. Recomenda-se a suspensão temporária de SUTENT; o tratamento pode ser retomado após resolução, de acordo com o critério do médico. 4.5 Interacções medicamentosas e outras formas de interacção Medicamentos que podem aumentar as concentrações plasmáticas de sunitinib: A administração concomitante de malato de sunitinib com o inibidor potente da CYP3A4, cetoconazol, em voluntários saudáveis resultou em aumentos de, respectivamente, 49% e 51% nos valores de Cmáx e AUC0-∞ do complexo [sunitinib+metabolito principal], após uma dose única de malato de sunitinib. A administração de SUTENT com outros inibidores potentes da família da CYP3A4 (ex., ritonavir, itraconazol, eritromicina, claritromicina e sumo de toranja) poderá aumentar as concentrações de sunitinib. Assim, deverá ser evitada a administração de SUTENT com os inibidores ou deverá ser considerada a escolha de medicação concomitante alternativa sem ou com potencial mínimo de inibição da CYP3A4. Se tal não for possível, poderá ser necessário reduzir a dose de SUTENT até um mínimo de 37,5 mg diários, com base na monitorização cuidadosa da tolerabilidade (ver secção 4.2). Medicamentos que podem diminuir as concentrações plasmáticas de sunitinib: A administração concomitante de SUTENT com o indutor da CYP3A4, rifampicina, em voluntários saudáveis resultou em reduções de, respectivamente, 23% e 46% nos valores de Cmáx e AUC0-∞ do complexo [sunitinib+metabolito principal] após uma dose única de SUTENT. A administração de SUTENT com indutores potentes da família da CYP3A4 (ex., dexametasona, fenitoína, carbamazepina, rifampicina, fenobarbital ou Hypericum perforatum, também conhecido por hipericão) poderá diminuir as concentrações de sunitinib. Como tal, a administração concomitante de indutores deverá ser evitada ou deverá ser considerada a selecção de medicação concomitante alternativa sem ou com potencial mínimo de inibição ou indução da CYP3A4. Se tal não for possível, poderá ser necessário aumentar a dose de SUTENT em incrementos de 12,5 mg (até 87,5 mg por dia), com base na monitorização cuidadosa da tolerabilidade (ver secção 4.2).

7
Para manter as concentrações alvo de sunitinib, deve ser considerada a selecção de medicação concomitante com menor potencial de indução enzimática. Se tal não for possível, podem ser necessários ajustes de dose de SUTENT (ver secção 4.2). Em doentes tratados com SUTENT raramente foi observada hemorragia (ver secção 4.4). Os doentes a receber tratamento concomitante com anti-coagulantes (ex., varfarina, acenocumarol) podem ser monitorizados periodicamente por contagem completa das células sanguíneas (plaquetas), factores de coagulação (tempo de protrombina/Relação Internacional Normalizada (INR)) e exame físico. 4.6 Gravidez e aleitamento Gravidez Não existem estudos em mulheres grávidas a utilizar SUTENT. Estudos em animais demonstraram toxicidade reprodutiva, incluindo malformações fetais (ver secção 5.3). O SUTENT não deve ser utilizado durante a gravidez nem em mulheres em idade fértil que não utilizem um método contraceptivo adequado, excepto se o potencial benefício justificar o potencial risco para o feto. Se o medicamento for administrado durante a gravidez ou se a doente ficar grávida enquanto estiver a receber este fármaco, deverá ser informada dos potenciais riscos para o feto. As mulheres em período fértil devem ser informadas de que deverão evitar engravidar durante o tratamento com SUTENT. Com base em achados não clínicos, a fertilidade masculina e feminina poderão ser comprometidas pelo tratamento com SUTENT (ver secção 5.3). Aleitamento Em ratos, o sunitinib e/ou os seus metabolitos são excretados pelo leite. Não se sabe se o sunitinib ou o seu principal metabolito activo são excretados no leite humano. Uma vez que os medicamentos são frequentemente excretados no leite humano e devido à possibilidade de reacções adversas graves em crianças que estão a ser amamentadas, as mulheres não deverão amamentar enquanto estiverem a tomar SUTENT. 4.7 Efeitos sobre a capacidade de conduzir e utilizar máquinas Não foram realizados estudos para avaliar os efeitos sobre a capacidade de conduzir e utilizar máquinas. Os doentes devem ser advertidos para o facto de poderem sentir tonturas durante o tratamento com SUTENT. 4.8 Efeitos indesejáveis Os acontecimentos adversos graves mais importantes relacionados com o tratamento, associados ao tratamento com SUTENT de doentes com tumores sólidos foram: embolia pulmonar (1%), trombocitopenia (1%), hemorragia tumoral (0,9%), neutropenia febril (0,4%) e hipertensão (0,4%). Os acontecimentos adversos mais frequentes, de qualquer grau, relacionados com o tratamento (ocorridos em pelo menos 20% dos doentes), incluíram: fadiga, doenças gastrointestinais, tais como diarreia, náuseas, estomatite, dispepsia e vómitos; alteração da coloração cutânea; disgeusia e anorexia. Os acontecimentos adversos relacionados com o tratamento mais frequentes, de gravidade máxima grau 3, foram a fadiga, a hipertensão e a neutropenia, e o aumento da lipase foi o acontecimento adverso relacionado com o tratamento mais frequente, de gravidade máxima grau 4, em doentes com tumores sólidos. As reacções adversas relacionadas com o tratamento que foram notificadas em >5% dos doentes com tumores sólidos encontram-se abaixo descritas. Os efeitos indesejáveis são apresentados por ordem decrescente de gravidade dentro de cada classe de frequência. As frequências são definidas por: muito frequentes (>1/10), frequentes (>1/100 ou <1/10), pouco frequentes (>1/1000 a <1/100), raros (>1/10000 a 1/1000), muito raros (<1/10000).

8
Reacções adversas reportadas nos estudos de GIST Classes de sistemas de órgãos
Frequência Reacções adversas
Todos os Graus n (%)
Grau 3 n (%)
Grau 4 n (%)
Muito frequentes
Anemia 33 (12,8%) 13 (5,1%) 1 (0,4%)
Frequentes Neutropenia 24 (9,3%) 15 (5,8%) 1 (0,4%)
Doenças do sangue e do sistema linfático
Frequentes Trombocitopenia 23 (8,9%) 6 (2,3%) 1 (0,4%) Doenças endócrinas Frequentes Hipotiroidismo 15 (5,8%) 0 (0,0%) 1 (0,4%) Doenças do metabolismo e da nutrição
Frequentes Anorexia 44 (7,1%) 1 (0,4%) 0 (0,0%)
Muito frequentes
Disgeusia 48 (18,7%) 0 (0,0%) 0 (0,0%) Doenças do sistema nervoso
Muito frequentes
Cefaleias 27 (10,5%) 2 (0,8%) 0 (0,0%)
Vasculopatias Muito frequentes
Hipertensão 43 (16,7%) 18 (7,0%) 0 (0,0%)
Doenças respiratórias, torácicas e do mediastino
Frequentes Epistaxis 17 (6,6%) 0 (0,0%) 0 (0,0%)
Doenças renais e urinárias
Frequentes Cromatúria 13 (5,1%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Diarreia 90 (35,0%) 13 (5,1%) 0 (0,0%)
Muito frequentes
Náuseas 69 (26,8%) 2 (0,8%) 0 (0,0%)
Muito frequentes
Estomatite 49 (19,1%) 2 (0,8%) 0 (0,0%)
Muito frequentes
Vómitos 46 (17,9%) 1 (0,4%) 0 (0,0%)
Muito frequentes
Dispepsia 32 (12,5%) 2 (0,8%) 0 (0,0%)
Frequentes Glossodinia 17 (6,6%) 0 (0,0%) 0 (0,0%) Frequentes Obstipação 13 (5,1%) 1 (0,4%) 0 (0,0%) Muito frequentes
Dor abdominal* 30 (11,7%) 5 (1,9%) 1 (0,4%)
Frequentes Dor na cavidade oral
16 (6,2%) 0 (0,0%) 0 (0,0%)
Frequentes Flatulência 15 (5,8%) 0 (0,0%) 0 (0,0%) Frequentes Xerostomia 15 (5,8%) 0 (0,0%) 0 (0,0%)
Doenças gastrointestinais
Frequentes Refluxo gastroesofágico
15 (5,8%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Alteração da coloração cutânea
65 (25,3%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Síndrome da Eritrodisestesia palmo-plantar
55 (21,4%) 14 (5,4%) 0 (0,0%)
Afecções dos tecidos cutâneos e subcutâneas
Muito frequentes
Erupção cutânea***
39 (15,2%) 2 (0,8%) 0 (0,0%)
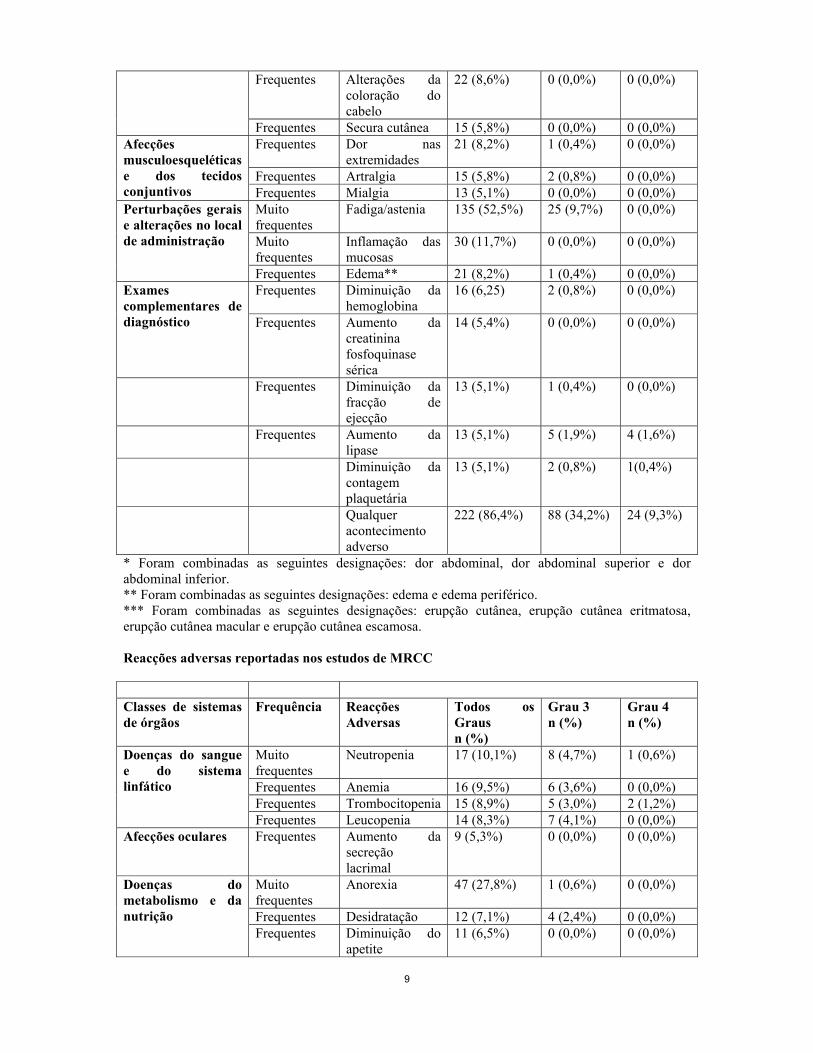
9
Frequentes Alterações da coloração do cabelo
22 (8,6%) 0 (0,0%) 0 (0,0%)
Frequentes Secura cutânea 15 (5,8%) 0 (0,0%) 0 (0,0%) Frequentes Dor nas
extremidades 21 (8,2%) 1 (0,4%) 0 (0,0%)
Frequentes Artralgia 15 (5,8%) 2 (0,8%) 0 (0,0%)
Afecções musculoesqueléticas e dos tecidos conjuntivos Frequentes Mialgia 13 (5,1%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Fadiga/astenia 135 (52,5%) 25 (9,7%) 0 (0,0%)
Muito frequentes
Inflamação das mucosas
30 (11,7%) 0 (0,0%) 0 (0,0%)
Perturbações gerais e alterações no local de administração
Frequentes Edema** 21 (8,2%) 1 (0,4%) 0 (0,0%) Frequentes Diminuição da
hemoglobina 16 (6,25) 2 (0,8%) 0 (0,0%) Exames
complementares de diagnóstico Frequentes Aumento da
creatinina fosfoquinase sérica
14 (5,4%) 0 (0,0%) 0 (0,0%)
Frequentes Diminuição da fracção de ejecção
13 (5,1%) 1 (0,4%) 0 (0,0%)
Frequentes Aumento da lipase
13 (5,1%) 5 (1,9%) 4 (1,6%)
Diminuição da contagem plaquetária
13 (5,1%) 2 (0,8%) 1(0,4%)
Qualquer acontecimento adverso
222 (86,4%) 88 (34,2%) 24 (9,3%)
* Foram combinadas as seguintes designações: dor abdominal, dor abdominal superior e dor abdominal inferior. ** Foram combinadas as seguintes designações: edema e edema periférico. *** Foram combinadas as seguintes designações: erupção cutânea, erupção cutânea eritmatosa, erupção cutânea macular e erupção cutânea escamosa. Reacções adversas reportadas nos estudos de MRCC Classes de sistemas de órgãos
Frequência Reacções Adversas
Todos os Graus n (%)
Grau 3 n (%)
Grau 4 n (%)
Muito frequentes
Neutropenia 17 (10,1%) 8 (4,7%) 1 (0,6%)
Frequentes Anemia 16 (9,5%) 6 (3,6%) 0 (0,0%) Frequentes Trombocitopenia 15 (8,9%) 5 (3,0%) 2 (1,2%)
Doenças do sangue e do sistema linfático
Frequentes Leucopenia 14 (8,3%) 7 (4,1%) 0 (0,0%) Afecções oculares Frequentes Aumento da
secreção lacrimal
9 (5,3%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Anorexia 47 (27,8%) 1 (0,6%) 0 (0,0%)
Frequentes Desidratação 12 (7,1%) 4 (2,4%) 0 (0,0%)
Doenças do metabolismo e da nutrição
Frequentes Diminuição do apetite
11 (6,5%) 0 (0,0%) 0 (0,0%)

10
Muito frequentes
Disgeusia 71 (42%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Cefaleias 25 (14,8%) 1 (0,6%) 0 (0,0%)
Frequentes Tonturas 13 (7,7%) 2 (1,2%) 0 (0,0%)
Doenças do sistema nervoso
Frequentes Parestesia 9 (5,3%) 0 (0,0%) 0 (0,0%) Vasculopatias Muito
frequentes Hipertensão 28 (16,6%) 7 (4,1%) 0 (0,0%)
Frequentes Epistaxis 16 (9,5%) 0 (0,0%) 0 (0,0%) Doenças respiratórias, torácicas e do mediastino
Frequentes Dispneia 9 (5,3%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Diarreia 83 (49,1%) 5 (3,0%) 0 (0,0%)
Muito frequentes
Náuseas 84 (49,7%) 2 (1,2%) 0 (0,0%)
Muito frequentes
Estomatite 70 (41,4%) 6 (3,6%) 0 (0,0%)
Muito frequentes
Dispepsia 69 (40,8%) 1 (0,6%) 0 (0,0%)
Muito frequentes
Vómitos 52 (30,8%) 2 (1,2%) 0 (0,0%)
Muito frequentes
Obstipação 34 (20,1%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Glossodinia 25 (14,8%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Dor abdominal* 17 (10,1%) 2 (1,2%) 0 (0,0%)
Frequentes Flatulência 16 (9,5%) 0 (0,0%) 0 (0,0%) Frequentes Distensão
abdominal 9 (5,3%) 0 (0,0%) 0 (0,0%)
Doenças gastrointestinais
Frequentes Xerostomia 9 (5,3%) 0 (0,0%) 0 (0,0%) Muito frequentes
Alteração da coloração cutânea
54 (32,0%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Erupção cutânea**
46 (27,2%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Alteração da coloração do cabelo
24 (14,2%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Síndrome da Eritrodisestesia palmo-plantar
21 (12,4%) 6 (3,6%) 0 (0,0%)
Frequentes Alopécia 13 (7,7%) 0 (0,0%) 0 (0,0%) Frequentes Dermatite
esfoliativa 10 (5,9%) 2 (1,2%) 0 (0,0%)
Frequentes Edema periorbitário
9 (5,3%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Secura cutânea 22 (13,0%) 0 (0,0%) 0 (0,0%)
Afecções dos tecidos cutâneos e subcutâneas
Muito frequentes
Eritema 20 (11,8%) 0 (0,0%) 0 (0,0%)
Afecções Muito frequente
Dor nas extremidades
21 (12,4%) 1 (0,6%) 0 (0,0%)

11
musculoesqueléticas e dos tecidos conjuntivos
Frequentes Mialgia 15 (8,9%) 1 (0,6%) 0 (0,0%)
Muito frequentes
Fadiga/astenia 108 (63,9%) 19 (11,2%) 0 (0,0%) Perturbações gerais e alterações no local de administração Muito
frequentes Inflamação das mucosas
30 (17,8%) 1 (0,6%) 0 (0,0%)
Complicações de intervenções relacionadas com lesões e intoxicações
Muito frequentes
Bolhas 7 (11,1%) 2 (3,2%) 0 (0,0%)
Muito frequentes
Aumento da lipase
17 (10,1%) 12 (7,1%) 3 (1,8%)
Frequentes Fracção de ejecção alterada
16 (9,5%) 1 (0,6%) 0 (0,0%)
Exames complementares de diagnóstico
Frequentes Aumento da amilase sérica
9 (5,3%) 6 (3,6%) 0 (0,0%)
Frequentes Diminuição de peso
11 (6,5%) 0 (0,0%) 0 (0,0%)
Frequentes Diminuição da contagem dos glóbulos brancos
10 (5,9%) 3 (1,8%) 0 (0,0%)
Frequentes Diminuição da contagem plaquetária
9 (5,3%) 3 (1,8%) 2 (1,2%)
Qualquer acontecimento adverso
166 (98,2%) 77 (45,6%) 14 (8,3%)
* Foram combinadas as seguintes designações: dor abdominal, dor abdominal superior e dor abdominal inferior. ** Foram combinadas as seguintes designações: erupção cutânea, erupção cutânea eritmatosa, erupção cutânea folicular, erupção cutânea generalizada, erupção cutânea papular e erupção cutânea associada a prurido. 4.9 Sobredosagem Não existe experiência de sobredosagem aguda com SUTENT. Não existe antídoto específico para a sobredosagem com SUTENT, devendo o tratamento destes casos consistir em medidas gerais de suporte. Se estiver indicado, a eliminação do medicamento não absorvido poderá ser conseguida por indução do vómito ou lavagem gástrica. 5. PROPRIEDADES FARMACOLÓGICAS 5.1 Propriedades farmacodinâmicas Grupo farmacoterapêutico: Agente antineoplásico - Inibidor da proteína tirosina-cinase; Código ATC: L01XE04. O malato de sunitinib inibe múltiplos receptores da tirosina-cinase que estão implicados no crescimento tumoral, na angiogénese patológica e na progressão metastática do cancro. O sunitinib foi identificado como inibidor dos receptores do factor de crescimento derivado das plaquetas (PDGFRα e PDGFRβ), receptores do factor de crescimento endotelial vascular (VEGFR1, VEGFR2 e VEGFR3), receptor do factor das células estaminais (KIT), receptor Fms-like da tirosina-cinase-3 (FLT3), receptor do factor estimulador de colónias (CSF-1R) e o receptor do factor neutrotrófico derivado de

12
células gliais (RET). O metabolito principal apresenta potência similar ao sunitinib em ensaios bioquímicos e celulares. ESTUDOS CLÍNICOS A segurança e eficácia clínica do SUTENT foi estudada no tratamento de doentes com tumores malignos do estroma gastrointestinal resistentes ao imatinib (isto é, doentes em que a doença evoluiu durante ou após o tratamento com imatinib) ou intolerantes ao imatinib (isto é, doentes que sofreram toxicidade significativa durante o tratamento com imatinib, que impediu a continuação tratamento), e no tratamento de doentes com carcinoma de células renais metastático após falha de terapêutica à base de citocinas. A eficácia é baseada no tempo até progressão tumoral e no aumento da sobrevivência no GIST e nas taxas de resposta objectiva no carcinoma das células renais metastático. Tumores do estroma gastrointestinal Foi conduzido um estudo inicial de desenho aberto para determinação de dose em doentes com GIST após insucesso da terapêutica com imatinib devida a resistência ou intolerância (dose máxima diária mediana de 800 mg). Foram incluídos noventa e sete doentes em várias doses e regimes; 55 doentes receberam 50 mg na posologia recomendada de tratamento, em ciclos de 4 semanas de tratamento/2 semanas em repouso (ciclos 4/2). Neste estudo, o tempo mediano até progressão tumoral (TTP) foi de 34,0 semanas (IC 95% = 22,0 – 46,0 semanas). Foi realizado um estudo de fase 3 de SUTENT, aleatorizado, em dupla ocultação e controlado com placebo, em doentes com tumores do estroma gastrointestinal que apresentaram intolerância ou progressão da doença durante ou após o tratamento com imatinib (dose máxima mediana diária de 800 mg). Neste estudo, foram aleatorizados 312 doentes (2:1) para receber 50 mg de SUTENT ou placebo, por via oral, com o esquema de tratamento 4/2, até progressão da doença ou saída do estudo por outra razão (207 doentes receberam SUTENT e 105 doentes receberam placebo). O objectivo primário de avaliação da eficácia foi o tempo até progressão do tumor, definido como o período de tempo entre a aleatorização e a primeira documentação objectiva de progressão do tumor. O tempo mediano até progressão do tumor com SUTENT foi de 28,9 semanas (IC de 95% = 21,3–34,1 semanas), mais longo que o tempo até progressão do tumor de 5,1 semanas (IC de 95% = 4,4–10,1 semanas) registado com o placebo, com diferença estatisticamente significativa. A diferença na sobrevivência global foi estatisticamente favorável a SUTENT [risco relativo: 0,491 (IC de 95%, 0,290-0,0831)]; o risco de morte foi duas vezes superior para os doentes no braço de placebo comparativamente ao braço de SUTENT. As percentagens de mortes foram de 14% para o SUTENT versus 25% para o placebo. No momento da análise, ainda não tinha sido alcançada a sobrevivência global mediana em ambos os braços do tratamento. Carcinoma das células renais metastático refractário às citocinas (MRCC) Foi realizado um estudo de fase 2 com o SUTENT em doentes refractários a um tratamento anterior com citocinas, com interleucina-2 ou interferão-α. Sessenta e três doentes receberam uma dose inicial diária de 50 mg de SUTENT, por via oral, durante 4 semanas consecutivas, seguida de um período de repouso de 2 semanas, de forma a cumprir um ciclo completo de 6 semanas (esquema de tratamento 4/2). O objectivo primário de avaliação da eficácia foi a taxa de resposta objectiva (ORR), baseada nos critérios de avaliação de resposta em tumores sólidos (Response Evaluation Criteria in Solid Tumors, RECIST). Neste estudo, a taxa de resposta objectiva foi de 36,5% (IC 95%, 24,7% – 49,6%) e o tempo mediano até progressão (TTP) foi de 37,7 semanas (IC de 95%, 24,0- 46,4 semanas). Foi realizado um estudo confirmatório de desenho aberto, de braço único e multicêntrico para avaliação da eficácia e segurança do SUTENT em doentes com carcinoma das células renais metastático refractário a um tratamento prévio com citocinas. Cento e seis doentes receberam pelo menos uma dose de 50 mg de SUTENT com o esquema de tratamento 4/2.

13
O objectivo primário de avaliação da eficácia deste estudo foi a taxa de resposta objectiva (ORR). Os objectivos de avaliação secundários incluíram o tempo até progressão do tumor (TTP), a duração da resposta e a sobrevivência global (OS). Neste estudo, a taxa de resposta objectiva foi de 38% (IC 95%, 26,8% – 47,5%). A duração da resposta e sobrevivência global medianas ainda não tinham sido alcançadas. Este medicamento foi autorizado sujeito a uma Autorização de Introdução no Mercado condicionada. Isto significa que se aguardam evidências adicionaissobre este medicamento, em particular sobre o efeito de SUTENT em termos de sobrevivência livre de progressão em doentes com MRCC. Está a ser conduzido um estudo para avaliar esta questão. A Agência Europeia de Medicamentos (EMEA) irá rever anualmente nova informação sobre o medicamento, e este RCM será actualizado se necessário. 5.2 Propriedades farmacocinéticas A farmacocinética do sunitinib e do malato de sunitinib foi avaliada em 135 voluntários saudáveis e em 266 doentes com tumores sólidos. Absorção Após administração oral, as concentrações máximas (Cmáx) de sunitinib são geralmente observadas entre 6 a 12 horas (Tmáx) . Os alimentos não afectam a biodisponibilidade de sunitinib. Distribuição Em ensaios in vitro, a ligação de sunitinib e do seu principal metabolito activo às proteínas plasmáticas humanas foi de, respectivamente, 95% e 90%, aparentemente não dependente da concentração. O volume de distribuição aparente de sunitinib (V/F) foi elevado – 2230l -, indicando distribuição tecidular. Metabolismo Os valores Ki calculados in vitro para todas as isoformas de CYP testadas (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 e CYP4A9/11) indicam que é improvável que o sunitinib e o seu metabolito activo principal inibam o metabolismo, em qualquer extensão clinicamente relevante, dos fármacos que possam ser metabolizados por estas enzimas. Estudos in vitro indicam que o SUTENT não induz nem inibe as principais enzimas CYP, incluindo a CYP3A4. Biotransformação O sunitinib é metabolizado principalmente pela enzima do citocromo P450, CYP3A4, que produz o seu principal metabolito activo, que por sua vez é ainda metabolizado pela CYP3A4. A administração concomitante de SUTENT com o indutor potente da CYP3A4, a rifampicina, resultou na redução de, aproximadamente, 56% e 78% dos valores de Cmáx e de AUC0-∞ de sunitinib, respectivamente, após a administração única de SUTENT em voluntários saudáveis. A administração de SUTENT com outros indutores da família da CYP3A4 (ex.: dexametasona, fenitoína, carbamazepina, fenobarbital ou Hypericum perfuratum, também conhecido por hipericão) pode diminuir as concentrações de sunitinib. Eliminação A principal via de excreção é a fecal (61%) e a via renal é responsável pela eliminação de 16% da dose administrada do medicamento e dos seus metabolitos. Os principais compostos identificados no plasma, urina e fezes relacionados com o fármaco foram o sunitinib e o seu principal metabolito, representando 91,5%, 86,4% e 73,8%, respectivamente, da reactividade em amostras agregadas. Foram identificados metabolitos secundários na urina e fezes, mas não foram, geralmente, encontrados no plasma. A depuração oral total (CL/F) foi de 34-62 l/hora. Insuficiência de funções orgânicas

14
Insuficiência hepática: Não foram realizados estudos clínicos em doentes com alteração da função hepática. Os estudos excluíram os doentes com ALT ou AST> 2,5 vezes o limite superior do normal ou >5,0 vezes o limite superior do normal, se devido a patologia subjacente. Insuficiência renal: Não foram realizados estudos clínicos em doentes com alteração da função renal. Os estudos excluíram doentes com creatinina sérica> 2,0 vezes o limite superior do normal. Análises populacionais farmacocinéticas indicam que a depuração aparente de sunitinib (CL/F) não foi afectada pela depuração da creatinina dentro da gama de valores avaliada (42-347 ml/min). Farmacocinética plasmática Após a administração oral em voluntários saudáveis, as semi-vidas de eliminação de sunitinib e do seu principal metabolito activo, desetil, foram, aproximadamente, 40 a 60 horas, e 80-110 horas, respectivamente. A área sob a curva (AUC) e a Cmáx aumentaram proporcionalmente com a dose, no intervalo de dose de 25-100 mg. Com a administração diária reiterada, o sunitinib acumula-se 3 a 4 vezes e o seu principal metabolito activo acumula-se 7 a 10 vezes. As concentrações de sunitinib e do seu principal metabolito activo em estado estacionário foram atingidas em 10 a 14 dias. Por volta do dia 14, as concentrações plasmáticas combinadas de sunitinib e do seu metabolito activo são de 62,9-101 ng/ml, que correspondem às concentrações alvo que se prevê inibirem, através de dados pré-clínicos, a fosforilação in vitro do receptor e resultarem em estabilização/redução do crescimento tumoral in vivo. O principal metabolito activo representa 23 a 37% da exposição total. Não foram observadas alterações significativas na farmacocinética do sunitinib ou do principal metabolito activo após administração diária reiterada ou com repetição dos ciclos nos regimes de dose testados. A farmacocinética foi similar em todas as populações com tumores sólidos testadas e nos voluntários saudáveis. A análise farmacocinética de dados demográficos populacionais indica que não é necessário efectuar ajuste de dose com base no peso ou score ECOG. Os dados disponíveis indicam que as mulheres podem apresentar uma depuração aparente (CL/F) de sunitinib cerca de 30% inferior à dos homens: contudo, esta diferença não requer ajustes de dose. 5.3 Dados de segurança pré-clínica Em estudos de toxicidade reiterada realizados em ratos e macacos, com duração até 9 meses, os principais efeitos em órgãos alvo foram identificados no tracto gastrointestinal (emése e diarreia nos macacos), glândulas supra-renais (congestão cortical e/ou hemorragia em ratos e macacos, com necrose seguida de fibrose em ratos), sistema hematopoiético e linfático (hipocelularidade da medula óssea e deplecção linfóide do timo, baço e gânglios linfáticos), pâncreas exócrino (desgranulação das células acinares com necrose celular), glândula salivar (hipertrofia acinar), articulações (espessamento da placa de crescimento), útero (atrofia) e ovários (diminuição do desenvolvimento folicular). Todos os achados ocorreram com níveis de exposição plasmática de sunitinib clinicamente relevantes. Foram observados efeitos adicionais noutros estudos, incluindo prolongamento do intervalo QTc, redução do FEVE, hipertrofia pituitária, atrofia tubular testicular, aumento das células mesangiais no rim, hemorragia gastrointestinal e da mucosa oral e hipertrofia das células pituitárias anteriores. Pensa-se que as alterações no útero (atrofia do endométrio) e na placa óssea de crescimento (espessamento fisário ou displasia da cartilagem) estejam relacionadas com a acção farmacológica do sunitinib. A maioria destes efeitos foi reversível após 2 a 6 semanas sem tratamento. Genotoxicidade O potencial genotóxico do sunitinib foi avaliado in vitro e in vivo. O sunitinib não foi mutagénico em bactérias com activação metabólica efectuada por fígado de rato. O sunitinib não induziu aberrações cromossómicas estruturais in vitro nos linfócitos periféricos humanos. Foi observada poliploidia in vitro (aberrações cromossómicas numéricas), em linfócitos periféricos, tanto na presença como na ausência de activação metabólica. O sunitinib não foi clastogénico in vivo na medula óssea de rato. O potencial de toxicidade genética não foi avaliado para o principal metabolito activo.

15
Carcinogénese Não foram realizados estudos de carcinogénese com malato de sunitinib. Toxicidade reprodutiva e de desenvolvimento Não foram observados efeitos na fertilidade dos ratos machos ou fêmea nos estudos de toxicidade reprodutiva. No entanto, foram observados efeitos na fertilidade feminina em estudos de toxicidade reiterada realizados em ratos e macacos, sob a forma de atresia folicular, degeneração do corpo lúteo, alterações no endométrio uterino e diminuição do peso do útero e dos ovários, com níveis de exposição sistémica clinicamente relevantes. Foram observados efeitos na fertilidade masculina em ratos, sob a forma de atrofia tubular testicular, redução dos espermatozóides nos epididimos e deplecção colóide na próstata e vesículas seminais, a níveis plasmáticos de exposição 18 vezes superiores aos clinicamente observados. A mortalidade embriofetal foi evidente em ratos, sob a forma de redução significativa do número de fetos vivos, aumento do número de reabsorções, aumento da perda pós implantação e perda total de ninhadas em 8 de 28 fêmeas grávidas, para níveis de exposição plasmática 5,5 vezes superiores aos observados na clínica. As reduções do peso do útero de coelhos fêmeas grávidas e no número de fetos vivos foram devidas ao aumento do número de reabsorções, aumento da perda pós-implantação e perda de ninhadas completas em 4 de 6 fêmeas grávidas, para níveis de exposição plasmática 3 vezes superiores aos observados na clínica. O tratamento de ratos com sunitinib durante a organogénese resultou em efeitos no desenvolvimento, consistindo no aumento da incidência de malformações do esqueleto dos fetos, predominantemente caracterizadas por ossificação tardia das vértebras torácicas/lombares e ocorreram a níveis de exposição plasmática 6 vezes superiores aos observados na clínica. Os efeitos no desenvolvimento dos coelhos consistiram no aumento da incidência de fenda labial para níveis de concentração plasmática aproximadamente iguais aos observados na clínica e fenda labial e fenda palatina, para níveis de exposição plasmática 2,7 vezes superiores aos observados na clínica. 6. INFORMAÇÕES FARMACÊUTICAS 6.1 Lista dos excipientes Conteúdo da cápsula Manitol Croscarmelose sódica Povidona Estereato de magnésio Cápsula Gelatina Dióxido de titânio (E171) Óxido de ferro vermelho (E172) Tinta de impressão: Goma laca Propilenoglicol Hidróxido de sódio Povidona Dióxido de titânio (E171)

16
6.2 Incompatibilidades Não aplicável. 6.3 Prazo de validade 2 anos. 6.4 Precauções especiais de conservação O medicamento não necessita de quaisquer precauções especiais de conservação. 6.5 Natureza e conteúdo do recipiente Frascos de polietileno de alta densidade (HDPE) com sistema de fecho de polipropileno, contendo 30 cápsulas. 6.6 Precauções especiais de eliminação Não existem requisitos especiais. 7. TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Pfizer Ltd Ramsgate Road Sandwich, Kent CT13 9NJ United Kingdom 8. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO 9. DATA DA PRIMEIRA AUTORIZAÇÃO/RENOVAÇÃO DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Data da primeira autorização: Data da próxima renovação: 10. DATA DA REVISÃO DO TEXTO

17
1. DENOMINAÇÃO DO MEDICAMENTO SUTENT 25 mg cápsulas 2. COMPOSIÇÃO QUALITATIVA E QUANTITATIVA Cada cápsula contém malato de sunitinib, equivalente a 25,0 mg de sunitinib. Excipiente(s): 39,663 mg de manitol. Lista completa de excipientes, ver secção 6.1. 3. FORMA FARMACÊUTICA Cápsulas. Cápsulas de gelatina com a cabeça cor de caramelo e o corpo cor de laranja, com “Pfizer” impresso a tinta branca na cabeça, “STN 25 mg” impresso no corpo e contendo grânulos amarelo alaranjados. 4. INFORMAÇÕES CLÍNICAS 4.1 Indicações terapêuticas O SUTENT está indicado para o tratamento de tumores malignos do estroma gastrointestinal irressecáveis e/ou metastáticos (GIST) após insucesso do tratamento com mesilato de imatinib, devida a resistência ou intolerância. O SUTENT está indicado para o tratamento do carcinoma de células renais avançado e/ou metastático (MRCC) após insucesso da terapêutica com interferão-alfa ou interleucina-2. A eficácia é baseada no tempo até progressão tumoral e num aumento de sobrevivência no GIST e nas taxas de resposta objectivas para o MRCC (ver secção 5.1). 4.2 Posologia e modo de administração A terapêutica deve ser iniciada por um médico experiente no tratamento do carcinoma de células renais ou de GIST. A posologia recomendada de SUTENT é uma dose oral diária de 50 mg tomada durante 4 semanas consecutivas, a que se segue um período de repouso de 2 semanas (esquema de tratamento 4/2), completando um ciclo de 6 semanas. Podem ser aplicados ajustes de dose em intervalos de 12,5 mg com base na segurança e tolerabilidade individuais. A dose diária não deve exceder os 87,5 mg nem ser reduzida abaixo dos 37,5 mg. A co-administração de indutores potentes da CYP3A4, tal como a rifampicina, deverá ser evitada (ver secções 4.4 e 4.5). Caso tal não seja possível, poderá ser necessário aumentar a dose de SUTENT com pequenos incrementos de 12,5 mg (até 87,5 mg por dia), com base na monitorização cuidadosa da tolerabilidade. A co-administração de SUTENT com inibidores potentes da CYP3A4, tal como o cetoconazol, deverá ser evitada (ver secções 4.4 e 4.5). Caso tal não seja possível, poderá ser necessário reduzir a dose de SUTENT para um mínimo de 37,5 mg diários, com base na monitorização cuidadosa da tolerabilidade. Deverá ser considerada a selecção de medicação concomitante alternativa sem ou com potencial mínimo de inibição ou indução da CYP3A4.

18
Utilização pediátrica: A segurança e eficácia de SUTENT em doentes pediátricos não foram estabelecidas. SUTENT não deverá ser utilizado na população pediátrica até estarem disponíveis mais dados. Utilização em doentes idosos: Aproximadamente 25% dos indivíduos que participaram nos estudos clínicos de SUTENT tinham 65 ou mais anos. Não se observaram diferenças significativas relativas à segurança ou efectividade entre os doentes mais novos e os doentes mais velhos. Insuficiência hepática: Não foram realizados estudos clínicos em doentes com diminuição da função hepática (ver secção 5.2). Insuficiência renal: Não foram realizados estudos clínicos em doentes com diminuição da função renal (ver secção 5.2). SUTENT pode ser tomado com ou sem alimentos. Se o doente não tomar uma dose, não deverá compensar com uma dose adicional. O doente deverá tomar a dose recomendada no dia seguinte, da forma habitual. 4.3 Contra-indicações Hipersensibilidade ao malato de sunitinib ou a qualquer um dos excipientes. 4.4 Advertências e precauções especiais de utilização A co-administração de indutores potentes da CYP3A4, tal como a rifampicina, poderá diminuir a concentração plasmática de sunitinib. Como tal, a combinação com indutores deverá ser evitada. Se tal não for possível, poderá ser necessário aumentar a dose de SUTENT (ver secções 4.2 e 4.5). A co-administração de inibidores potentes da CYP3A4, como o cetoconazol, poderá aumentar as concentrações plasmáticas de sunitinib. Recomenda-se a selecção de medicação concomitante alternativa sem ou com potencial mínimo de inibição enzimática. Se não for possível, poderá ser necessário reduzir a dose de SUTENT (ver secções 4.2 e 4.5). Pele e tecidos A alteração da coloração cutânea, possivelmente devida à cor da substância activa (amarela) é um acontecimento adverso comum relacionado com o tratamento, ocorrendo em aproximadamente 30% dos doentes. Os doentes devem ser avisados que a despigmentação do cabelo ou da pele poderá também ocorrer durante o tratamento com SUTENT. Outros efeitos dermatológicos possíveis podem incluir secura, espessamento ou fissuras na pele, bolhas ou erupção cutânea ocasional nas palmas das mãos e nas plantas dos pés. Foi notificada dor/irritação na cavidade oral em aproximadamente 14% dos doentes. Foi reportada disgeusia (alterações do paladar) em aproximadamente 28% dos doentes. Os acontecimentos acima descritos não foram cumulativos, foram tipicamente reversíveis e geralmente não resultaram em descontinuação do tratamento. Acontecimentos gastrointestinais Os acontecimentos gastrointestinais mais frequentemente relatados foram náuseas, diarreia, estomatite, dispepsia e vómitos. As medidas de suporte para os acontecimentos gastrointestinais que requerem tratamento podem incluir administração de medicamentos anti-eméticos ou anti-diarreicos. Hemorragia Ocorreu hemorragia tumoral relacionada com o tratamento em aproximadamente 2% dos doentes com GIST. Estes acontecimentos poderão ocorrer subitamente, e no caso de tumores pulmonares podem surgir como hemoptises graves ou hemorragias pulmonares graves e potencialmente fatais. Num

19
ensaio clínico conduzido em doentes com carcinoma do pulmão de não-pequenas células (NSCLC) metastizado, ocorreu hemorragia pulmonar fatal em 2 doentes que estavam a receber SUTENT. Ambos os doentes apresentavam histologia celular escamosa. O SUTENT não está aprovado para a utilização em doentes com carcinoma do pulmão de não-pequenas células. A avaliação de rotina deste acontecimento deve incluir contagem completa das células sanguíneas e exame físico. A epistaxis foi o acontecimento adverso hemorrágico relacionado com o tratamento mais frequente, tendo sido relatado para aproximadamente metade dos doentes com tumores sólidos que apresentaram acontecimentos hemorrágicos. Nenhum destes acontecimentos foi grave. Tracto gastrointestinal Raramente, ocorreram complicações gastrointestinais graves e por vezes fatais, incluindo perfuração gastrointestinal, em doentes com doença maligna intra-abdominal tratados com SUTENT. Hipertensão Foi notificada hipertensão relacionada com o tratamento em aproximadamente 16% dos doentes com tumores sólidos. A posologia de SUTENT foi reduzida ou temporariamente adiada em 2,7% desta população de doentes. Nenhum destes doentes descontinuou o tratamento com SUTENT. Ocorreu hipertensão grave (sistólica >200 mmHg ou diastólica de 110 mmHg) em 4,7% desta população de doentes. Os doentes devem ser monitorizados para a hipertensão e apropriadamente controlados. É recomendada a suspensão temporária em doentes com hipertensão grave que não se encontra controlada com o acompanhamento médico. O tratamento poderá ser retomado assim que a hipertensão estiver apropriadamente controlada. Hematológicos Foram relatados decréscimos na contagem absoluta de neutrófilos de gravidade grau 3 e 4 em 13,1% e 0,9% dos doentes, respectivamente. Foram relatados decréscimos na contagem de plaquetas de gravidade grau 3 e 4 em 4% e 0,5% dos doentes, respectivamente. Os acontecimentos adversos acima descritos não foram cumulativos, foram tipicamente reversíveis e geralmente não resultaram na descontinuação do tratamento. Deve ser realizada uma contagem completa das células sanguíneas no início de cada ciclo de tratamento, nos doentes a receber tratamento com SUTENT.

20
Cardiovascular Ocorreram diminuições na fracção de ejecção ventricular esquerda (FEVE) ≥ 20% e abaixo do limite inferior do normal, em aproximadamente 2% dos doentes com GIST tratados com SUTENT, em 4% dos doentes com carcinoma de células renais metastático e em 2% dos doentes tratados com placebo. Estas diminuições do FEVE não aparentam ter sido progressivas e melhoraram frequentemente com a continuação do tratamento. Foram relatados acontecimentos adversos relacionados com o tratamento de “insuficiência cardíaca”, “insuficiência cardíaca congestiva” ou “insuficiência ventricular esquerda” em 0,7% dos doentes com tumores sólidos e em 1% dos doentes tratados com placebo. Todos os doentes tinham GIST. A relação entre a inibição dos receptores da tirosina-cinase e a função cardíaca, se existir, permanece por esclarecer. Os doentes que apresentaram acontecimentos cardíacos nos 12 meses anteriores à administração de SUTENT, como enfarte do miocárdio (incluindo angina instável/grave), bypass coronário/arterial periférico, insuficiência cardíaca congestiva (ICC) sintomática, acidente vascular cerebral ou isquémico transitório ou embolia pulmonar foram excluídos dos ensaios clínicos com SUTENT. Não se sabe se os doentes com estas condições concomitantes podem apresentar um risco superior de desenvolver disfunção ventricular esquerda relacionada com o fármaco. Recomenda-se que os médicos avaliem estes riscos versus os potenciais benefícios do medicamento. Estes doentes deverão ser cuidadosamente monitorizados quanto a sinais e sintomas de ICC enquanto estiverem a receber SUTENT. Deverão ser igualmente consideradas avaliações de base, e periódicas, da FEVE enquanto o doente estiver a receber SUTENT. Em doentes sem factores de risco cardíacos, deverá ser considerada uma avaliação de base da fracção de ejecção. Na presença de manifestações clínicas de ICC, é recomendada a descontinuação de SUTENT. A dose de SUTENT deve ser interrompida e/ou reduzida em doentes sem evidência clínica de ICC, mas que apresentem uma fracção de ejecção < 50% e > 20% abaixo do valor de base. Prolongamento do intervalo QT O prolongamento do intervalo QT foi investigado num ensaio com 24 doentes, com idades entre os 20-87 anos, com doença maligna avançada. Em concentrações de aproximadamente o dobro das concentrações terapêuticas, o SUTENT demonstrou prolongar o intervalo QTcF (correcção de Frederica). Não existiram doentes com prolongamento do intervalo QT/QTc superior a grau 2 (CTCAE v3.0) e nenhum doente apresentou arritmia cardíaca. A relevância clínica dos efeitos observados é pouco clara e dependerá dos factores de risco individuais e das susceptibilidades que o doente apresentar. O SUTENT deve ser usado com precaução em doentes com uma história conhecida de prolongamento do intervalo QT, em doentes que se encontrem a tomar antiarrítmicos ou em doentes com doença cardíaca pré-existente relevante, bradicardia ou perturbações electrolíticas. O tratamento concomitante com inibidores potentes da CYP3A4, que podem aumentar as concentrações plasmáticas de sunitinib, deve ser administrado com precaução e a dose de SUTENT reduzida (ver secção 4.5). Acontecimentos tromboembólicos venosos Foram notificados acontecimentos tromboembólicos venosos em quatro doentes (2%) dos dois estudos conduzidos para o carcinoma de células renais metastático; dois doentes com embolia pulmonar (ambas grau 4) e dois doentes com trombose venosa profunda (ambas grau 3). Ocorreu interrupção da administração num destes casos. No estudo pivot de GIST, sete doentes (3%) a receber SUTENT e nenhum dos doentes em placebo apresentaram acontecimentos tromboembólicos venosos; cinco dos sete foram tromboses venosas profundas de grau 3 e dois foram grau 1 ou 2. Quatro destes sete doentes com GIST descontinuaram o tratamento após a primeira observação de trombose venosa profunda. Embolia pulmonar Foi reportada embolia pulmonar relacionada com o tratamento em aproximadamente 1,1% dos doentes com tumores sólidos que receberam SUTENT. Nenhum destes acontecimentos resultou na descontinuação do tratamento com SUTENT; contudo, ocorreu, num número reduzido de casos, uma

21
redução da dose ou um adiamento temporário do tratamento. Não houve mais ocorrências de embolia pulmonar nestes doentes após o tratamento ter sido retomado. Hipotiroidismo O hipotiroidismo foi reportado como acontecimento adverso em 7 doentes (4%) dos dois estudos realizados no carcinoma de células renais metastático. Adicionalmente, registaram-se aumentos da TSH em 4 doentes (2%). Globalmente, 7% da população com carcinoma de células renais metastático apresentaram evidência clínica ou laboratorial de hipotiroidismo emergente do tratamento. Foi observado hipotiroidismo adquirido emergente do tratamento em 8 (4%) doentes com GIST a receber SUTENT versus 1 (1%) a receber placebo. Os doentes com sintomas sugestivos de hipotiroidismo devem realizar monitorização laboratorial da função tiroideia e devem ser tratados de acordo com a prática clínica considerada padrão. Função pancreática Foram observados aumentos nas actividades da lipase e da amilase séricas em doentes com diversos tumores sólidos medicados com SUTENT. Os aumentos nas actividades da lipase, em doentes com diversos tipos de tumores sólidos, foram transitórios e geralmente não foram acompanhados por sinais ou sintomas de pancreatite. Foi observada pancreatite em 0,4% dos doentes com tumores sólidos. Se estiverem presentes sintomas de pancreatite, os doentes devem ter acompanhamento médico adequado. Convulsões Foram observadas convulsões nos estudos clínicos com SUTENT, em indivíduos com evidência radiológica de metástases cerebrais. Adicionalmente, existiram notificações raras (<1%) de indivíduos que apresentaram convulsões e evidência radiológica da síndrome de leucoencefalopatia posterior reversível. Em nenhum dos indivíduos este acontecimento foi fatal. Os doentes com convulsões e sinais/sintomas consistentes com síndrome de leucoencefalopatia posterior reversível, tais como hipertensão, cefaleias, diminuição do estado de alerta, alterações da função mental e perda visual, incluindo cegueira cortical, deverão ser controlados com seguimento médico, que inclua o controlo da hipertensão. Recomenda-se a suspensão temporária de SUTENT; o tratamento pode ser retomado após resolução, de acordo com o critério do médico. 4.5 Interacções medicamentosas e outras formas de interacção Medicamentos que podem aumentar as concentrações plasmáticas de sunitinib: A administração concomitante de malato de sunitinib com o inibidor potente da CYP3A4, cetoconazol, em voluntários saudáveis resultou em aumentos de, respectivamente, 49% e 51% nos valores de Cmáx e AUC0-∞ do complexo [sunitinib+metabolito principal], após uma dose única de malato de sunitinib. A administração de SUTENT com outros inibidores potentes da família da CYP3A4 (ex., ritonavir, itraconazol, eritromicina, claritromicina e sumo de toranja) poderá aumentar as concentrações de sunitinib. Assim, deverá ser evitada a administração de SUTENT com os inibidores ou deverá ser considerada a escolha de medicação concomitante alternativa sem ou com potencial mínimo de inibição da CYP3A4. Se tal não for possível, poderá ser necessário reduzir a dose de SUTENT até um mínimo de 37,5 mg diários, com base na monitorização cuidadosa da tolerabilidade (ver secção 4.2). Medicamentos que podem diminuir as concentrações plasmáticas de sunitinib: A administração concomitante de SUTENT com o indutor da CYP3A4, rifampicina, em voluntários saudáveis resultou em reduções de, respectivamente, 23% e 46% nos valores de Cmáx e AUC0-∞ do complexo [sunitinib+metabolito principal] após uma dose única de SUTENT. A administração de SUTENT com indutores potentes da família da CYP3A4 (ex., dexametasona, fenitoína, carbamazepina, rifampicina, fenobarbital ou Hypericum perforatum, também conhecido por hipericão) poderá diminuir as concentrações de sunitinib. Como tal, a administração concomitante de indutores deverá ser evitada ou deverá ser considerada a selecção de medicação concomitante alternativa sem ou com potencial mínimo de inibição ou indução da CYP3A4. Se tal não for possível,

22
poderá ser necessário aumentar a dose de SUTENT em incrementos de 12,5 mg (até 87,5 mg por dia), com base na monitorização cuidadosa da tolerabilidade (ver secção 4.2). Para manter as concentrações alvo de sunitinib, deve ser considerada a selecção de medicação concomitante com menor potencial de indução enzimática. Se tal não for possível, podem ser necessários ajustes de dose de SUTENT (ver secção 4.2). Para manter as concentrações alvo de sunitinib, deve ser considerada a selecção de medicação concomitante com menor potencial de indução enzimática. Se tal não for possível, podem ser necessários ajustes de dose de SUTENT (ver secção 4.2). Em doentes tratados com SUTENT raramente foi observada hemorragia (ver secção 4.4). Os doentes a receber tratamento concomitante com anti-coagulantes (ex., varfarina; acenocumarol) podem ser monitorizados periodicamente por contagem completa das células sanguíneas (plaquetas), factores de coagulação (tempo de protrombina/ Relação Internacional Normalizada (INR)) e exame físico. 4.6 Gravidez e aleitamento Gravidez Não existem estudos em mulheres grávidas a utilizar SUTENT. Estudos em animais demonstraram toxicidade reprodutiva, incluindo malformações fetais (ver secção 5.3). O SUTENT não deve ser utilizado durante a gravidez nem em mulheres em idade fértil que não utilizem um método contraceptivo adequado, excepto se o potencial benefício justificar o potencial risco para o feto. Se o medicamento for administrado durante a gravidez ou se a doente ficar grávida enquanto estiver a receber este fármaco, deverá ser informada dos potenciais riscos para o feto. As mulheres em período fértil devem ser informadas de que deverão evitar engravidar durante o tratamento com SUTENT. Com base em achados não clínicos, a fertilidade masculina e feminina poderão ser comprometidas pelo tratamento com SUTENT (ver secção 5.3). Aleitamento Em ratos, o sunitinib e/ou os seus metabolitos são excretados pelo leite. Não se sabe se o sunitinib ou o seu principal metabolito activo são excretados no leite humano. Uma vez que os medicamentos são frequentemente excretados no leite humano e devido à possibilidade de reacções adversas graves em crianças que estão a ser amamentadas, as mulheres não deverão amamentar enquanto estiverem a tomar SUTENT. 4.7 Efeitos sobre a capacidade de conduzir e utilizar máquinas Não foram realizados estudos para avaliar os efeitos sobre a capacidade de conduzir e utilizar máquinas. Os doentes devem ser advertidos para o facto de poderem sentir tonturas durante o tratamento com SUTENT. 4.8 Efeitos indesejáveis Os acontecimentos adversos graves mais importantes relacionados com o tratamento, associados ao tratamento com SUTENT de doentes com tumores sólidos foram: embolia pulmonar (1%), trombocitopenia (1%), hemorragia tumoral (0,9%), neutropenia febril (0,4%) e hipertensão (0,4%). Os acontecimentos adversos mais frequentes, de qualquer grau, relacionados com o tratamento (ocorridos em pelo menos 20% dos doentes), incluíram: fadiga, doenças gastrointestinais, tais como diarreia, náuseas, estomatite, dispepsia e vómitos; alteração da coloração cutânea; disgeusia e anorexia. Os acontecimentos adversos relacionados com o tratamento mais frequentes, de gravidade máxima grau 3, foram a fadiga, a hipertensão e a neutropenia, e o aumento da lipase foi o acontecimento adverso relacionado com o tratamento mais frequente, de gravidade máxima grau 4, em doentes com tumores sólidos.

23
As reacções adversas relacionadas com o tratamento que foram notificadas em >5% dos doentes com tumores sólidos encontram-se abaixo descritas. Os efeitos indesejáveis são apresentados por ordem decrescente de gravidade dentro de cada classe de frequência. As frequências são definidas por: muito frequentes (>1/10), frequentes (>1/100 ou <1/10), pouco frequentes (>1/1000 a <1/100), raros (>1/10000 a 1/1000), muito raros (<1/10000). Reacções adversas reportadas nos estudos de GIST Classes de sistemas de órgãos
Frequência Reacções adversas
Todos os Graus n (%)
Grau 3 n (%)
Grau 4 n (%)
Muito frequentes
Anemia 33 (12,8%) 13 (5,1%) 1 (0,4%)
Frequentes Neutropenia 24 (9,3%) 15 (5,8%) 1 (0,4%)
Doenças do sangue e do sistema linfático
Frequentes Trombocitopenia 23 (8,9%) 6 (2,3%) 1 (0,4%) Doenças endócrinas Frequentes Hipotiroidismo 15 (5,8%) 0 (0,0%) 1 (0,4%) Doenças do metabolismo e da nutrição
Frequentes Anorexia 44 (7,1%) 1 (0,4%) 0 (0,0%)
Muito frequentes
Disgeusia 48 (18,7%) 0 (0,0%) 0 (0,0%) Doenças do sistema nervoso
Muito frequentes
Cefaleias 27 (10,5%) 2 (0,8%) 0 (0,0%)
Vasculopatias Muito frequentes
Hipertensão 43 (16,7%) 18 (7,0%) 0 (0,0%)
Doenças respiratórias, torácicas e do mediastino
Frequentes Epistaxis 17 (6,6%) 0 (0,0%) 0 (0,0%)
Doenças renais e urinárias
Frequentes Cromatúria 13 (5,1%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Diarreia 90 (35,0%) 13 (5,1%) 0 (0,0%)
Muito frequentes
Náuseas 69 (26,8%) 2 (0,8%) 0 (0,0%)
Muito frequentes
Estomatite 49 (19,1%) 2 (0,8%) 0 (0,0%)
Muito frequentes
Vómitos 46 (17,9%) 1 (0,4%) 0 (0,0%)
Muito frequentes
Dispepsia 32 (12,5%) 2 (0,8%) 0 (0,0%)
Frequentes Glossodinia 17 (6,6%) 0 (0,0%) 0 (0,0%) Frequentes Obstipação 13 (5,1%) 1 (0,4%) 0 (0,0%) Muito frequentes
Dor abdominal* 30 (11,7%) 5 (1,9%) 1 (0,4%)
Frequentes Dor na cavidade oral
16 (6,2%) 0 (0,0%) 0 (0,0%)
Frequentes Flatulência 15 (5,8%) 0 (0,0%) 0 (0,0%) Frequentes Xerostomia 15 (5,8%) 0 (0,0%) 0 (0,0%)
Doenças gastrointestinais
Frequentes Refluxo gastroesofágico
15 (5,8%) 0 (0,0%) 0 (0,0%)
Afecções dos tecidos cutâneos e subcutâneas
Muito frequentes
Alteração da coloração cutânea
65 (25,3%) 0 (0,0%) 0 (0,0%)
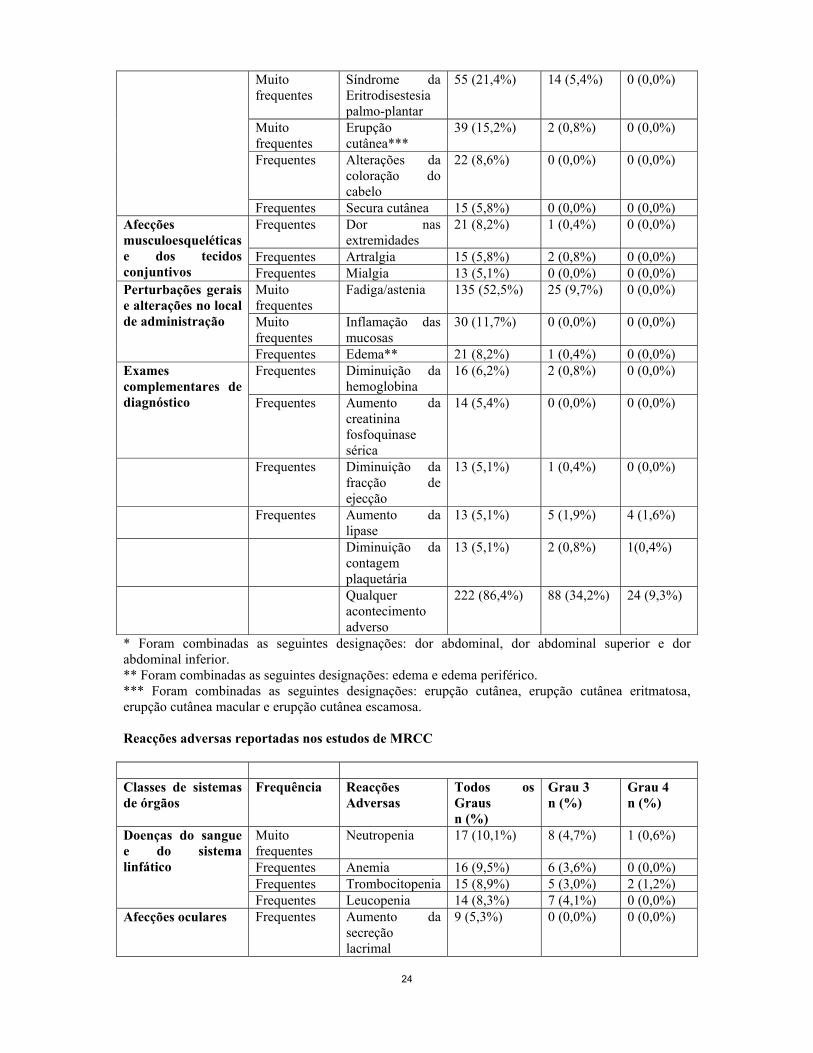
24
Muito frequentes
Síndrome da Eritrodisestesia palmo-plantar
55 (21,4%) 14 (5,4%) 0 (0,0%)
Muito frequentes
Erupção cutânea***
39 (15,2%) 2 (0,8%) 0 (0,0%)
Frequentes Alterações da coloração do cabelo
22 (8,6%) 0 (0,0%) 0 (0,0%)
Frequentes Secura cutânea 15 (5,8%) 0 (0,0%) 0 (0,0%) Frequentes Dor nas
extremidades 21 (8,2%) 1 (0,4%) 0 (0,0%)
Frequentes Artralgia 15 (5,8%) 2 (0,8%) 0 (0,0%)
Afecções musculoesqueléticas e dos tecidos conjuntivos Frequentes Mialgia 13 (5,1%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Fadiga/astenia 135 (52,5%) 25 (9,7%) 0 (0,0%)
Muito frequentes
Inflamação das mucosas
30 (11,7%) 0 (0,0%) 0 (0,0%)
Perturbações gerais e alterações no local de administração
Frequentes Edema** 21 (8,2%) 1 (0,4%) 0 (0,0%) Frequentes Diminuição da
hemoglobina 16 (6,2%) 2 (0,8%) 0 (0,0%) Exames
complementares de diagnóstico Frequentes Aumento da
creatinina fosfoquinase sérica
14 (5,4%) 0 (0,0%) 0 (0,0%)
Frequentes Diminuição da fracção de ejecção
13 (5,1%) 1 (0,4%) 0 (0,0%)
Frequentes Aumento da lipase
13 (5,1%) 5 (1,9%) 4 (1,6%)
Diminuição da contagem plaquetária
13 (5,1%) 2 (0,8%) 1(0,4%)
Qualquer acontecimento adverso
222 (86,4%) 88 (34,2%) 24 (9,3%)
* Foram combinadas as seguintes designações: dor abdominal, dor abdominal superior e dor abdominal inferior. ** Foram combinadas as seguintes designações: edema e edema periférico. *** Foram combinadas as seguintes designações: erupção cutânea, erupção cutânea eritmatosa, erupção cutânea macular e erupção cutânea escamosa. Reacções adversas reportadas nos estudos de MRCC Classes de sistemas de órgãos
Frequência Reacções Adversas
Todos os Graus n (%)
Grau 3 n (%)
Grau 4 n (%)
Muito frequentes
Neutropenia 17 (10,1%) 8 (4,7%) 1 (0,6%)
Frequentes Anemia 16 (9,5%) 6 (3,6%) 0 (0,0%) Frequentes Trombocitopenia 15 (8,9%) 5 (3,0%) 2 (1,2%)
Doenças do sangue e do sistema linfático
Frequentes Leucopenia 14 (8,3%) 7 (4,1%) 0 (0,0%) Afecções oculares Frequentes Aumento da
secreção lacrimal
9 (5,3%) 0 (0,0%) 0 (0,0%)

25
Muito frequentes
Anorexia 47 (27,8%) 1 (0,6%) 0 (0,0%)
Frequentes Desidratação 12 (7,1%) 4 (2,4%) 0 (0,0%)
Doenças do metabolismo e da nutrição
Frequentes Diminuição do apetite
11 (6,5%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Disgeusia 71 (42%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Cefaleias 25 (14,8%) 1 (0,6%) 0 (0,0%)
Frequentes Tonturas 13 (7,7%) 2 (1,2%) 0 (0,0%)
Doenças do sistema nervoso
Frequentes Parestesia 9 (5,3%) 0 (0,0%) 0 (0,0%) Vasculopatias Muito
frequentes Hipertensão 28 (16,6%) 7 (4,1%) 0 (0,0%)
Frequentes Epistaxis 16 (9,5%) 0 (0,0%) 0 (0,0%) Doenças respiratórias, torácicas e do mediastino
Frequentes Dispneia 9 (5,3%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Diarreia 83 (49,1%) 5 (3,0%) 0 (0,0%)
Muito frequentes
Náuseas 84 (49,7%) 2 (1,2%) 0 (0,0%)
Muito frequentes
Estomatite 70 (41,4%) 6 (3,6%) 0 (0,0%)
Muito frequentes
Dispepsia 69 (40,8%) 1 (0,6%) 0 (0,0%)
Muito frequentes
Vómitos 52 (30,8%) 2 (1,2%) 0 (0,0%)
Muito frequentes
Obstipação 34 (20,1%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Glossodinia 25 (14,8%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Dor abdominal* 17 (10,1%) 2 (1,2%) 0 (0,0%)
Frequentes Flatulência 16 (9,5%) 0 (0,0%) 0 (0,0%) Frequentes Distensão
abdominal 9 (5,3%) 0 (0,0%) 0 (0,0%)
Doenças gastrointestinais
Frequentes Xerostomia 9 (5,3%) 0 (0,0%) 0 (0,0%) Muito frequentes
Alteração da coloração cutânea
54 (32,0%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Erupção cutânea**
46 (27,2%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Alteração da coloração do cabelo
24 (14,2%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Síndrome da Eritrodisestesia palmo-plantar
21 (12,4%) 6 (3,6%) 0 (0,0%)
Frequentes Alopécia 13 (7,7%) 0 (0,0%) 0 (0,0%) Frequentes Dermatite
esfoliativa 10 (5,9%) 2 (1,2%) 0 (0,0%)
Frequentes Edema periorbitário
9 (5,3%) 0 (0,0%) 0 (0,0%)
Afecções dos tecidos cutâneos e subcutâneas
Muito frequentes
Secura cutânea 22 (13,0%) 0 (0,0%) 0 (0,0%)

26
Muito frequentes
Eritema 20 (11,8%) 0 (0,0%) 0 (0,0%)
Muito frequente
Dor nas extremidades
21 (12,4%) 1 (0,6%) 0 (0,0%) Afecções musculoesqueléticas e dos tecidos conjuntivos
Frequentes Mialgia 15 (8,9%) 1 (0,6%) 0 (0,0%)
Muito frequentes
Fadiga/astenia 108 (63,9%) 19 (11,2%) 0 (0,0%) Perturbações gerais e alterações no local de administração Muito
frequentes Inflamação das mucosas
30 (17,8%) 1 (0,6%) 0 (0,0%)
Complicações de intervenções relacionadas com lesões e intoxicações
Muito frequentes
Bolhas 7 (11,1%) 2 (3,2%) 0 (0,0%)
Muito frequentes
Aumento da lipase
17 (10,1%) 12 (7,1%) 3 (1,8%)
Frequentes Fracção de ejecção alterada
16 (9,5%) 1 (0,6%) 0 (0,0%)
Exames complementares de diagnóstico
Frequentes Aumento da amilase sérica
9 (5,3%) 6 (3,6%) 0 (0,0%)
Frequentes Diminuição de peso
11 (6,5%) 0 (0,0%) 0 (0,0%)
Frequentes Diminuição da contagem dos glóbulos brancos
10 (5,9%) 3 (1,8%) 0 (0,0%)
Frequentes Diminuição da contagem plaquetária
9 (5,3%) 3 (1,8%) 2 (1,2%)
Qualquer acontecimento adverso
166 (98,2%) 77 (45,6%) 14 (8,3%)
* Foram combinadas as seguintes designações: dor abdominal, dor abdominal superior e dor abdominal inferior. ** Foram combinadas as seguintes designações: erupção cutânea, erupção cutânea eritmatosa, erupção cutânea folicular, erupção cutânea generalizada, erupção cutânea papular e erupção cutânea associada a prurido. 4.9 Sobredosagem Não existe experiência de sobredosagem aguda com SUTENT. Não existe antídoto específico para a sobredosagem com SUTENT, devendo o tratamento destes casos consistir em medidas gerais de suporte. Se estiver indicado, a eliminação do medicamento não absorvido poderá ser conseguida por indução do vómito ou lavagem gástrica. 5. PROPRIEDADES FARMACOLÓGICAS 5.1 Propriedades farmacodinâmicas Grupo farmacoterapêutico: Agente antineoplásico - Inibidor da proteína tirosina-cinase; Código ATC: L01XE04. O malato de sunitinib inibe múltiplos receptores da tirosina-cinase que estão implicados no crescimento tumoral, na angiogénese patológica e na progressão metastática do cancro. O sunitinib foi identificado como inibidor dos receptores do factor de crescimento derivado das plaquetas (PDGFRα e PDGFRβ), receptores do factor de crescimento endotelial vascular (VEGFR1, VEGFR2 e VEGFR3),

27
receptor do factor das células estaminais (KIT), receptor Fms-like da tirosina-cinase-3 (FLT3), receptor do factor estimulador de colónias (CSF-1R) e o receptor do factor neutrotrófico derivado de células gliais (RET). O metabolito principal apresenta potência similar ao sunitinib em ensaios bioquímicos e celulares. ESTUDOS CLÍNICOS A segurança e eficácia clínica do SUTENT foi estudada no tratamento de doentes com tumores malignos do estroma gastrointestinal resistentes ao imatinib (isto é, doentes em que a doença evoluiu durante ou após o tratamento com imatinib) ou intolerantes ao imatinib (isto é, doentes que sofreram toxicidade significativa durante o tratamento com imatinib, que impediu a continuação tratamento), e no tratamento de doentes com carcinoma de células renais metastático após falha de terapêutica à base de citocinas. A eficácia é baseada no tempo até progressão tumoral e no aumento da sobrevivência no GIST e nas taxas de resposta objectiva no carcinoma das células renais metastático. Tumores do estroma gastrointestinal Foi conduzido um estudo inicial de desenho aberto para determinação de dose em doentes com GIST após insucesso da terapêutica com imatinib devida a resistência ou intolerância (dose máxima diária mediana de 800 mg). Foram incluídos noventa e sete doentes em várias doses e regimes; 55 doentes receberam 50 mg na posologia recomendada de tratamento, em ciclos de 4 semanas de tratamento/2 semanas em repouso (ciclos 4/2). Neste estudo, o tempo mediano até progressão tumoral foi de 34,0 semanas (IC 95% = 22,0 – 46,0 semanas). Foi realizado um estudo de fase 3 de SUTENT, aleatorizado, em dupla ocultação e controlado com placebo, em doentes com tumores do estroma gastrointestinal que apresentaram intolerância ou progressão da doença durante ou após o tratamento com imatinib (dose máxima diária mediana de 800 mg). Neste estudo, foram aleatorizados 312 doentes (2:1) para receber 50 mg de SUTENT ou placebo, por via oral, com o esquema de tratamento 4/2, até progressão da doença ou saída do estudo por outra razão (207 doentes receberam SUTENT e 105 doentes receberam placebo). O objectivo primário de avaliação da eficácia foi o tempo até progressão do tumor, definido como o período de tempo entre a aleatorização e a primeira documentação objectiva de progressão do tumor. O tempo mediano até progressão do tumor com SUTENT foi de 28,9 semanas (IC de 95% = 21,3–34,1 semanas), mais longo que o tempo até progressão do tumor de 5,1 semanas (IC de 95% = 4,4–10,1 semanas) registado com o placebo, com diferença estatisticamente significativa. A diferença na sobrevivência global foi estatisticamente favorável a SUTENT [risco relativo: 0,491 (IC de 95%, 0,290-0,0831)]; o risco de morte foi duas vezes superior para os doentes no braço de placebo comparativamente ao braço de SUTENT. As percentagens de mortes foram de 14% para o SUTENT versus 25% para o placebo. No momento da análise, ainda não tinha sido alcançada a sobrevivência global mediana em ambos os braços do tratamento. Carcinoma das células renais metastático refractário às citocinas (MRCC) Foi realizado um estudo de fase 2 com o SUTENT em doentes refractários a um tratamento anterior com citocinas, com interleucina-2 ou interferão-α. Sessenta e três doentes receberam uma dose inicial diária de 50 mg de SUTENT, por via oral, durante 4 semanas consecutivas, seguida de um período de repouso de 2 semanas, de forma a cumprir um ciclo completo de 6 semanas (esquema de tratamento 4/2). O objectivo primário de avaliação da eficácia foi a taxa de resposta objectiva (ORR), baseada nos critérios de avaliação de resposta em tumores sólidos (Response Evaluation Criteria in Solid Tumors, RECIST). Neste estudo, a taxa de resposta objectiva foi de 36,5% (IC 95%, 24,7% – 49,6%) e o tempo mediano até progressão (TTP) foi de 37,7 semanas (IC de 95%, 24,0- 46,4 semanas). Foi realizado um estudo confirmatório de desenho aberto, de braço único e multicêntrico para avaliação da eficácia e segurança do SUTENT em doentes com carcinoma das células renais

28
metastático refractário a um tratamento prévio com citocinas. Cento e seis doentes receberam pelo menos uma dose de 50 mg de SUTENT com o esquema de tratamento 4/2. O objectivo primário de avaliação da eficácia deste estudo foi a taxa de resposta objectiva (ORR). Os objectivos de avaliação secundários incluíram o tempo até progressão do tumor (TTP), a duração da resposta e a sobrevivência global (OS). Neste estudo, a taxa de resposta objectiva foi de 38% (IC 95%, 26,8% – 47,5%). A duração da resposta e sobrevivência global medianas ainda não tinham sido alcançadas. Este medicamento foi sujeito a uma Autorização de Introdução no Mercado condicionada. Isto significa que se aguardadam evidências adicionais sobre este medicamento, em particular sobre o efeito de SUTENT em termos de sobrevivência livre de progressão em doentes com MRCC. Está a ser conduzido um estudo para avaliar esta questão. A Agência Europeia de Medicamentos (EMEA) irá rever anualmente novaa informação sobre o medicamento, e este RCM será actualizado se necessário. 5.2 Propriedades farmacocinéticas A farmacocinética do sunitinib e do malato de sunitinib foi avaliada em 135 voluntários saudáveis e em 266 doentes com tumores sólidos. Absorção Após administração oral, as concentrações máximas (Cmáx) de sunitinib são geralmente observadas entre 6 a 12 horas (Tmáx) . Os alimentos não afectam a biodisponibilidade de sunitinib. Distribuição Em ensaios in vitro, a ligação de sunitinib e do seu principal metabolito activo às proteínas plasmáticas humanas foi de, respectivamente, 95% e 90%, aparentemente não dependente da concentração. O volume de distribuição aparente de sunitinib (V/F) foi elevado – 2230L - , indicando distribuição tecidular. Metabolismo Os valores Ki calculados in vitro para todas as isoformas de CYP testadas (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 e CYP4A9/11) indicam que é improvável que o sunitinib e o seu metabolito activo principal inibam o metabolismo, em qualquer extensão clinicamente relevante, dos fármacos que possam ser metabolizados por estas enzimas. Estudos in vitro indicam que o SUTENT não induz nem inibe as principais enzimas CYP, incluindo a CYP3A4. Biotransformação O sunitinib é metabolizado principalmente pela enzima do citocromo P450, CYP3A4, que produz o seu principal metabolito activo, que por sua vez é ainda metabolizado pela CYP3A4. A administração concomitante de SUTENT com o indutor potente da CYP3A4, a rifampicina, resultou na redução de, aproximadamente, 56% e 78% dos valores de Cmáx e de AUC0-∞ de sunitinib, respectivamente, após a administração única de SUTENT em voluntários saudáveis. A administração de SUTENT com outros indutores da família da CYP3A4 (ex.: dexametasona, fenitoína, carbamazepina, fenobarbital ou Hypericum perfuratum, também conhecido por hipericão) pode diminuir as concentrações de sunitinib. Eliminação A principal via de excreção é a fecal (61%) e a via renal é responsável pela eliminação de 16% da dose administrada do medicamento e dos seus metabolitos. Os principais compostos identificados no plasma, urina e fezes relacionados com o fármaco foram o sunitinib e o seu principal metabolito, representando 91,5%, 86,4% e 73,8%, respectivamente, da reactividade em amostras agregadas. Foram identificados metabolitos secundários na urina e fezes, mas não foram, geralmente, encontrados no plasma. A depuração oral total (CL/F) foi de 34-62 L/hora.

29
Insuficiência de funções orgânicas Insuficiência hepática: Não foram realizados estudos clínicos em doentes com alteração da função hepática. Os estudos excluíram os doentes com ALT ou AST >2,5 vezes o limite superior do normal ou, >5,0 vezes o limite superior do normal, se devido a patologia subjacente. Insuficiência renal: Não foram realizados estudos clínicos em doentes com alteração da função renal. Os estudos excluíram doentes com creatinina sérica >2,0 vezes o limite superior do normal. Análises populacionais farmacocinéticas indicam que a depuração aparente de sunitinib (CL/F) não foi afectada pela depuração da creatinina dentro da gama de valores avaliada (42-347 ml/min). Farmacocinética plasmática Após a administração oral em voluntários saudáveis, as semi-vidas de eliminação de sunitinib e do seu principal metabolito activo, desetil, foram, aproximadamente, 40 a 60 horas, e 80-110 horas, respectivamente. A área sob a curva (AUC) e a Cmáx aumentaram proporcionalmente com a dose, no intervalo de dose de 25-100 mg. Com a administração diária reiterada, o sunitinib acumula-se 3 a 4 vezes e o seu principal metabolito activo acumula-se 7 a 10 vezes. As concentrações de sunitinib e do seu principal metabolito activo em estado estacionário foram atingidas em 10 a 14 dias. Por volta do dia 14, as concentrações plasmáticas combinadas de sunitinib e do seu metabolito activo são de 62,9-101 ng/mL, que correspondem às concentrações alvo que se prevê inibirem, através de dados pré-clínicos, a fosforilação in vitro do receptor e resultarem em estabilização/redução do crescimento tumoral in vivo. O principal metabolito activo representa 23 a 37% da exposição total. Não foram observadas alterações significativas na farmacocinética do sunitinib ou do principal metabolito activo após administração diária reiterada ou com repetição dos ciclos nos regimes de dose testados. A farmacocinética foi similar em todas as populações com tumores sólidos testadas e nos voluntários saudáveis. A análise farmacocinética de dados demográficos populacionais indica que não é necessário efectuar ajuste de dose com base no peso ou score ECOG. Os dados disponíveis indicam que as mulheres podem apresentar uma depuração aparente (CL/F) de sunitinib cerca de 30% inferior à dos homens: contudo, esta diferença não requer ajustes de dose. 5.3 Dados de segurança pré-clínica Em estudos de toxicidade reiterada realizados em ratos e macacos, com duração até 9 meses, os principais efeitos em órgãos alvo foram identificados no tracto gastrointestinal (emése e diarreia nos macacos), glândulas supra-renais (congestão cortical e/ou hemorragia em ratos e macacos, com necrose seguida de fibrose em ratos), sistema hematopoiético e linfático (hipocelularidade da medula óssea e deplecção linfóide do timo, baço e gânglios linfáticos), pâncreas exócrino (desgranulação das células acinares com necrose celular), glândula salivar (hipertrofia acinar), articulações (espessamento da placa de crescimento), útero (atrofia) e ovários (diminuição do desenvolvimento folicular). Todos os achados ocorreram com níveis de exposição plasmática de sunitinib clinicamente relevantes. Foram observados efeitos adicionais noutros estudos, incluindo prolongamento do intervalo QTc, redução do FEVE, hipertrofia pituitária, atrofia tubular testicular, aumento das células mesangiais no rim, hemorragia gastrointestinal e da mucosa oral e hipertrofia das células pituitárias anteriores. Pensa-se que as alterações no útero (atrofia do endométrio) e na placa óssea de crescimento (espessamento fisário ou displasia da cartilagem) estejam relacionadas com a acção farmacológica do sunitinib. A maioria destes efeitos foi reversível após 2 a 6 semanas sem tratamento. Genotoxicidade O potencial genotóxico do sunitinib foi avaliado in vitro e in vivo. O sunitinib não foi mutagénico em bactérias com activação metabólica efectuada por fígado de rato. O sunitinib não induziu aberrações cromossómicas estruturais in vitro nos linfócitos periféricos humanos. Foi observada poliploidia in vitro (aberrações cromossómicas numéricas), em linfócitos periféricos, tanto na presença como na ausência de activação metabólica. O sunitinib não foi clastogénico in vivo na

30
medula óssea de rato. O potencial de toxicidade genética não foi avaliado para o principal metabolito activo. Carcinogénese Não foram realizados estudos de carcinogénese com malato de sunitinib. Toxicidade reprodutiva e de desenvolvimento Não foram observados efeitos na fertilidade dos ratos machos ou fêmeas em estudos de toxicidade reprodutiva. No entanto, foram observados efeitos na fertilidade feminina em estudos de toxicidade reiterada realizados em ratos e macacos, sob a forma de atresia folicular, degeneração do corpo lúteo, alterações no endométrio uterino e diminuição do peso do útero e dos ovários, com níveis de exposição sistémica clinicamente relevantes. Foram observados efeitos na fertilidade masculina em ratos, sob a forma de atrofia tubular testicular, redução dos espermatozóides nos epididimos e deplecção colóide na próstata e vesículas seminais, a níveis plasmáticos de exposição 18 vezes superiores aos clinicamente observados. A mortalidade embriofetal foi evidente em ratos, sob a forma de redução significativa do número de fetos vivos, aumento do número de reabsorções, aumento da perda pós implantação e perda total de ninhadas em 8 de 28 fêmeas grávidas, para níveis de exposição plasmática 5,5 vezes superiores aos observados na clínica. As reduções do peso do útero de coelhos fêmeas grávidas e no número de fetos vivos foram devidas ao aumento do número de reabsorções, aumento da perda pós-implantação e perda de ninhadas completas em 4 de 6 fêmeas grávidas, para níveis de exposição plasmática 3 vezes superiores aos observados na clínica. O tratamento de ratos com sunitinib durante a organogénese resultou em efeitos no desenvolvimento, consistindo no aumento da incidência de malformações do esqueleto dos fetos, predominantemente caracterizadas por ossificação tardia das vértebras torácicas/lombares e ocorreram a níveis de exposição plasmática 6 vezes superiores aos observados na clínica. Os efeitos no desenvolvimento dos coelhos consistiram no aumento da incidência de fenda labial para níveis de concentração plasmática aproximadamente iguais aos observados na clínica e fenda labial e fenda palatina, para níveis de exposição plasmática 2,7 vezes superiores aos observados na clínica. 6. INFORMAÇÕES FARMACÊUTICAS 6.1 Lista dos excipientes Conteúdo da cápsula Manitol Croscarmelose sódica Povidona Estereato de magnésio Invólucro da cápsula laranja Gelatina Dióxido de titânio (E171) Óxido de ferro vermelho (E172) Invólucro da cápsula caramelo Gelatina Dióxido de titânio (E171) Óxido de ferro amarelo (E172) Óxido de ferro vermelho (E172) Óxido de ferro negro (E172) Tinta de impressão:

31
Goma laca Propilenoglicol Hidróxido de sódio Povidona Dióxido de titânio (E171) 6.2 Incompatibilidades Não aplicável. 6.3 Prazo de validade 2 anos. 6.4 Precauções especiais de conservação O medicamento não necessita de quaisquer precauções especiais de conservação. 6.5 Natureza e conteúdo do recipiente Frascos de polietileno de alta densidade (HDPE) com sistema de fecho de polipropileno, contendo 30 cápsulas. 6.6 Precauções especiais de eliminação Não existem requisitos especiais. 7. TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Pfizer Ltd Ramsgate Road Sandwich, Kent CT13 9NJ United Kingdom 8. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO 9. DATA DA PRIMEIRA AUTORIZAÇÃO/RENOVAÇÃO DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Data da primeira autorização: Data da próxima renovação: 10. DATA DA REVISÃO DO TEXTO

32
1. DENOMINAÇÃO DO MEDICAMENTO SUTENT 50 mg cápsulas 2. COMPOSIÇÃO QUALITATIVA E QUANTITATIVA Cada cápsula contém malato de sunitinib equivalente a 50 mg de sunitinib. Excipiente(s): 79,326 mg de manitol. Lista completa de excipientes, ver secção 6.1. 3. FORMA FARMACÊUTICA Cápsulas. Cápsulas de gelatina com a cabeça e o corpo cor de caramelo, com “Pfizer” impresso a tinta branca na cabeça, “STN 50 mg” no corpo e contendo grânulos amarelo alaranjados. 4. INFORMAÇÕES CLÍNICAS 4.1 Indicações terapêuticas O SUTENT está indicado para o tratamento de tumores malignos do estroma gastrointestinal irressecáveis e/ou metastáticos (GIST) após insucesso do tratamento com mesilato de imatinib, devida a resistência ou intolerância. O SUTENT está indicado para o tratamento do carcinoma de células renais metastático (MRCC) após insucesso da terapêutica com interferão-alfa e/ou interleucina-2. A eficácia é baseada no tempo até progressão tumoral e num aumento de sobrevivência no GIST e nas taxas de resposta objectivas para o MRCC (ver secção 5.1). 4.2 Posologia e modo de administração A terapêutica deve ser iniciada por um médico experiente no tratamento do carcinoma de células renais ou de GIST. A posologia recomendada de SUTENT é uma dose oral diária de 50 mg tomada durante 4 semanas consecutivas, a que se segue um período de repouso de 2 semanas (esquema de tratamento 4/2), completando um ciclo de 6 semanas. Podem ser aplicados ajustes de dose em intervalos de 12,5 mg com base na segurança e tolerabilidade individuais. A dose diária não deve exceder os 87,5 mg nem ser reduzida abaixo dos 37,5 mg. A co-administração de indutores potentes da CYP3A4, tal como a rifampicina, deverá ser evitada (ver secções 4.4 e 4.5). Caso tal não seja possível, poderá ser necessário aumentar a dose de SUTENT com pequenos incrementos de 12,5 mg (até 87,5 mg por dia), com base na monitorização cuidadosa da tolerabilidade. A co-administração de SUTENT com inibidores potentes da CYP3A4, tal como o cetoconazol, deverá ser evitada (ver secções 4.4 e 4.5). Caso tal não seja possível, poderá ser necessário reduzir a dose de SUTENT para um mínimo de 37,5 mg diários, com base na monitorização cuidadosa da tolerabilidade. Deverá ser considerada a selecção de medicação concomitante alternativa sem ou com potencial mínimo de inibição ou indução da CYP3A4.

33
Utilização pediátrica: A segurança e eficácia de SUTENT em doentes pediátricos não foram estabelecidas. SUTENT não deverá ser utilizado na população pediátrica até estarem disponíveis mais dados. Utilização em doentes idosos: Aproximadamente 25% dos indivíduos que participaram nos estudos clínicos de SUTENT tinham 65 ou mais anos. Não se observaram diferenças significativas relativas à segurança ou efectividade entre os doentes mais novos e os doentes mais velhos. Insuficiência hepática: Não foram realizados estudos clínicos em doentes com diminuição da função hepática (ver secção 5.2). Insuficiência renal: Não foram realizados estudos clínicos em doentes com diminuição da função renal (ver secção 5.2). SUTENT pode ser tomado com ou sem alimentos. Se o doente não tomar uma dose, não deverá compensar com uma dose adicional. O doente deverá tomar a dose recomendada no dia seguinte, da forma habitual. 4.3 Contra-indicações Hipersensibilidade ao malato de sunitinib ou a qualquer um dos excipientes. 4.4 Advertências e precauções especiais de utilização A co-administração de indutores potentes da CYP3A4, tal como a rifampicina, poderá diminuir a concentração plasmática de sunitinib. Como tal, a combinação com indutores deverá ser evitada. Se tal não for possível, poderá ser necessário aumentar a dose de SUTENT (ver secções 4.2 e 4.5). A co-administração de inibidores potentes da CYP3A4, como o cetoconazol, poderá aumentar as concentrações plasmáticas de sunitinib. Recomenda-se a selecção de medicação concomitante alternativa sem ou com potencial mínimo de inibição enzimática. Se não for possível, poderá ser necessário reduzir a dose de SUTENT (ver secções 4.2 e 4.5). Pele e tecidos A alteração da coloração cutânea, possivelmente devida à cor da substância activa (amarela) é um acontecimento adverso comum relacionado com o tratamento, ocorrendo em aproximadamente 30% dos doentes. Os doentes devem ser avisados que a despigmentação do cabelo ou da pele poderá também ocorrer durante o tratamento com SUTENT. Outros efeitos dermatológicos possíveis podem incluir secura, espessamento ou fissuras na pele, bolhas ou erupção cutânea ocasional nas palmas das mãos e nas plantas dos pés. Foi notificada dor/irritação na cavidade oral em aproximadamente 14% dos doentes. Foi reportada disgeusia (alterações do paladar) em aproximadamente 28% dos doentes. Os acontecimentos acima descritos não foram cumulativos, foram tipicamente reversíveis e geralmente não resultaram em descontinuação do tratamento. Acontecimentos gastrointestinais Os acontecimentos gastrointestinais mais frequentemente relatados foram náuseas, diarreia, estomatite, dispepsia e vómitos. As medidas de suporte para os acontecimentos gastrointestinais que requerem tratamento podem incluir administração de medicamentos anti-eméticos ou anti-diarreicos. Hemorragia Ocorreu hemorragia tumoral relacionada com o tratamento em aproximadamente 2% dos doentes com GIST. Estes acontecimentos poderão ocorrer subitamente, e no caso de tumores pulmonares, podem surgir como hemoptises graves ou hemorragias pulmonares graves e potencialmente fatais. Num ensaio clínico conduzido em doentes com carcinoma do pulmão de não-pequenas células (NSCLC)

34
metastizado, ocorreu hemorragia pulmonar fatal em 2 doentes que estavam a receber SUTENT. Ambos os doentes apresentavam histologia celular escamosa. O SUTENT não está aprovado para a utilização em doentes com carcinoma do pulmão de não-pequenas células. A avaliação de rotina deste acontecimento deve incluir contagem completa das células sanguíneas e exame físico. A epistaxis foi o acontecimento adverso hemorrágico relacionado com o tratamento mais frequente, tendo sido relatado para aproximadamente metade dos doentes com tumores sólidos que apresentaram acontecimentos hemorrágicos. Nenhum destes acontecimentos foi grave. Tracto gastrointestinal Raramente, ocorreram complicações gastrointestinais graves e por vezes fatais, incluindo perfuração gastrointestinal, em doentes com doença maligna intra-abdominal tratados com SUTENT. Hipertensão Foi notificada hipertensão relacionada com o tratamento em aproximadamente 16% dos doentes com tumores sólidos. A posologia de SUTENT foi reduzida ou temporariamente adiada em 2,7% desta população de doentes. Nenhum destes doentes descontinuou o tratamento com SUTENT. Ocorreu hipertensão grave (sistólica >200 mmHg ou diastólica de 110 mmHg) em 4,7% desta população de doentes. Os doentes devem ser monitorizados para a hipertensão e apropriadamente controlados. É recomendada a suspensão temporária em doentes com hipertensão grave que não se encontra controlada com o acompanhamento médico. O tratamento poderá ser retomado assim que a hipertensão estiver apropriadamente controlada. Hematológicos Foram relatados decréscimos na contagem absoluta de neutrófilos de gravidade grau 3 e 4 em 13,1% e 0,9% dos doentes, respectivamente. Foram relatados decréscimos na contagem de plaquetas de gravidade grau 3 e 4 em 4% e 0,5% dos doentes, respectivamente. Os acontecimentos adversos acima descritos não foram cumulativos, foram tipicamente reversíveis e geralmente não resultaram na descontinuação do tratamento. Deve ser realizada uma contagem completa das células sanguíneas no início de cada ciclo de tratamento, nos doentes a receber tratamento com SUTENT.

35
Cardiovascular Ocorreram diminuições na fracção de ejecção ventricular esquerda (FEVE) ≥ 20% e abaixo do limite inferior do normal, em aproximadamente 2% dos doentes com GIST tratados com SUTENT, em 4% dos doentes com carcinoma de células renais metastático e em 2% dos doentes tratados com placebo. Estas diminuições do FEVE não aparentam ter sido progressivas e melhoraram frequentemente com a continuação do tratamento. Foram relatados acontecimentos adversos relacionados com o tratamento de “insuficiência cardíaca”, “insuficiência cardíaca congestiva” ou “insuficiência ventricular esquerda” em 0,7% dos doentes com tumores sólidos e em 1% dos doentes tratados com placebo. Todos os doentes tinham GIST. A relação entre a inibição dos receptores da tirosina-cinase e a função cardíaca, se existir, permanece por esclarecer. Os doentes que apresentaram acontecimentos cardíacos nos 12 meses anteriores à administração de SUTENT, como enfarte do miocárdio (incluindo angina instável/grave), bypass coronário/arterial periférico, insuficiência cardíaca congestiva (ICC) sintomática, acidente vascular cerebral ou isquémico transitório ou embolia pulmonar foram excluídos dos ensaios clínicos com SUTENT. Não se sabe se os doentes com estas condições concomitantes podem apresentar um risco superior de desenvolver disfunção ventricular esquerda relacionada com o fármaco. Recomenda-se que os médicos avaliem estes riscos versus os potenciais benefícios do medicamento. Estes doentes deverão ser cuidadosamente monitorizados quanto a sinais e sintomas de ICC enquanto estiverem a receber SUTENT. Deverão ser igualmente consideradas avaliações de base, e periódicas, da FEVE enquanto o doente estiver a receber SUTENT. Em doentes sem factores de risco cardíacos, deverá ser considerada uma avaliação de base da fracção de ejecção. Na presença de manifestações clínicas de ICC, é recomendada a descontinuação de SUTENT. A dose de SUTENT deve ser interrompida e/ou reduzida em doentes sem evidência clínica de ICC, mas que apresentem uma fracção de ejecção < 50% e > 20% abaixo do valor de base. Prolongamento do intervalo QT O prolongamento do intervalo QT foi investigado num ensaio com 24 doentes, com idades entre os 20-87 anos, com doença maligna avançada. Em concentrações de aproximadamente o dobro das concentrações terapêuticas, o SUTENT demonstrou prolongar o intervalo QTcF (correcção de Frederica). Não existiram doentes com prolongamento do intervalo QT/QTc superior a grau 2 (CTCAE v3.0) e nenhum doente apresentou arritmia cardíaca. A relevância clínica dos efeitos observados é pouco clara e dependerá dos factores de risco individuais e das susceptibilidades que o doente apresentar. O SUTENT deve ser usado com precaução em doentes com uma história conhecida de prolongamento do intervalo QT, em doentes que se encontrem a tomar antiarrítmicos ou em doentes com doença cardíaca pré-existente relevante, bradicardia ou perturbações electrolíticas. O tratamento concomitante com inibidores potentes da CYP3A4, que podem aumentar as concentrações plasmáticas de sunitinib, deve ser administrado com precaução e a dose de SUTENT reduzida (ver secção 4.5). Acontecimentos tromboembólicos venosos Foram notificados acontecimentos tromboembólicos venosos em quatro doentes (2%) dos dois estudos conduzidos para o carcinoma de células renais metastático; dois doentes com embolia pulmonar (ambas grau 4) e dois doentes com trombose venosa profunda (ambas grau 3). Ocorreu interrupção da administração num destes casos. No estudo pivot de GIST, sete doentes (3%) a receber SUTENT e nenhum dos doentes em placebo apresentaram acontecimentos tromboembólicos venosos; cinco dos sete foram tromboses venosas profundas de grau 3 e dois foram grau 1 ou 2. Quatro destes sete doentes com GIST descontinuaram o tratamento após a primeira observação de trombose venosa profunda. Embolia pulmonar Foi reportada embolia pulmonar relacionada com o tratamento em aproximadamente 1,1% dos doentes com tumores sólidos que receberam SUTENT. Nenhum destes acontecimentos resultou na descontinuação do tratamento com SUTENT; contudo, ocorreu, num número reduzido de casos, uma redução da dose ou um adiamento temporário do tratamento. Não houve mais ocorrências de embolia pulmonar nestes doentes após o tratamento ter sido retomado.

36
Hipotiroidismo O hipotiroidismo foi reportado como acontecimento adverso em 7 doentes (4%) dos dois estudos realizados no carcinoma de células renais metastático. Adicionalmente, registaram-se aumentos da TSH em 4 doentes (2%). Globalmente, 7% da população com carcinoma de células renais metastático apresentaram evidência clínica ou laboratorial de hipotiroidismo emergente do tratamento. Foi observado hipotiroidismo adquirido emergente do tratamento em 8 (4%) doentes com GIST a receber SUTENT versus 1 (1%) a receber placebo. Os doentes com sintomas sugestivos de hipotiroidismo devem realizar monitorização laboratorial da função tiroideia e devem ser tratados de acordo com a prática clínica considerada padrão. Função pancreática Foram observados aumentos nas actividades da lipase e da amilase séricas em doentes com diversos tumores sólidos medicados com SUTENT. Os aumentos nas actividades da lipase, em doentes com diversos tipos de tumores sólidos, foram transitórios e geralmente não foram acompanhados por sinais ou sintomas de pancreatite. Foi observada pancreatite em 0,4% dos doentes com tumores sólidos. Se estiverem presentes sintomas de pancreatite, os doentes devem ter acompanhamento médico adequado. Convulsões Foram observadas convulsões nos estudos clínicos com SUTENT, em indivíduos com evidência radiológica de metástases cerebrais. Adicionalmente, existiram notificações raras (<1%) de indivíduos que apresentaram convulsões e evidência radiológica da síndrome de leucoencefalopatia posterior reversível. Em nenhum dos indivíduos este acontecimento foi fatal. Os doentes com convulsões e sinais/sintomas consistentes com síndrome de leucoencefalopatia posterior reversível, tais como hipertensão, cefaleias, diminuição do estado de alerta, alterações da função mental e perda visual, incluindo cegueira cortical, deverão ser controlados através de acompanhamento médico, que inclua o controlo da hipertensão. Recomenda-se a suspensão temporária de SUTENT; o tratamento pode ser retomado após resolução, de acordo com o critério do médico. 4.5 Interacções medicamentosas e outras formas de interacção Medicamentos que podem aumentar as concentrações plasmáticas de sunitinib: A administração concomitante de malato de sunitinib com o inibidor potente da CYP3A4, cetoconazol, em voluntários saudáveis resultou em aumentos de, respectivamente, 49% e 51% nos valores de Cmáx e AUC0-∞ do complexo [sunitinib+metabolito principal], após uma dose única de malato de sunitinib. A administração de SUTENT com outros inibidores potentes da família da CYP3A4 (ex., ritonavir, itraconazol, eritromicina, claritromicina e sumo de toranja) poderá aumentar as concentrações de sunitinib. Assim, deverá ser evitada a administração de SUTENT com os inibidores ou deverá ser considerada a escolha de medicação concomitante alternativa sem ou com potencial mínimo de inibição da CYP3A4. Se tal não for possível, poderá ser necessário reduzir a dose de SUTENT até um mínimo de 37,5 mg diários, com base na monitorização cuidadosa da tolerabilidade (ver secção 4.2). Medicamentos que podem diminuir as concentrações plasmáticas de sunitinib: A administração concomitante de SUTENT com o indutor da CYP3A4, rifampicina, em voluntários saudáveis resultou em reduções de, respectivamente, 23% e 46% nos valores de Cmáx e AUC0-∞ do complexo [sunitinib+metabolito principal] após uma dose única de SUTENT. A administração de SUTENT com indutores potentes da família da CYP3A4 (ex., dexametasona, fenitoína, carbamazepina, rifampicina, fenobarbital ou Hypericum perforatum, também conhecido por hipericão) poderá diminuir as concentrações de sunitinib. Como tal, a administração concomitante de indutores deverá ser evitada ou deverá ser considerada a selecção de medicação concomitante alternativa sem ou com potencial mínimo de inibição ou indução da CYP3A4. Se tal não for possível, poderá ser necessário aumentar a dose de SUTENT em incrementos de 12,5 mg (até 87,5 mg por dia), com base na monitorização cuidadosa da tolerabilidade (ver secção 4.2).

37
Para manter as concentrações alvo de sunitinib, deve ser considerada a selecção de medicação concomitante com menor potencial de indução enzimática. Se tal não for possível, podem ser necessários ajustes de dose de SUTENT (ver secção 4.2). Para manter as concentrações alvo de sunitinib, deve ser considerada a selecção de medicação concomitante com menor potencial de indução enzimática. Se tal não for possível, podem ser necessários ajustes de dose de SUTENT (ver secção 4.2). Em doentes tratados com SUTENT raramente foi observada hemorragia (ver secção 4.4). Os doentes a receber tratamento concomitante com anti-coagulantes (ex., varfarina, acenocumarole) podem ser monitorizados periodicamente por contagem completa das células sanguíneas (plaquetas), factores de coagulação (tempo de protrombina/Relação Internacional Normalizada (INR)) e exame físico. 4.6 Gravidez e aleitamento Gravidez Não existem estudos em mulheres grávidas a utilizar SUTENT. Estudos em animais demonstraram toxicidade reprodutiva, incluindo malformações fetais (ver secção 5.3). O SUTENT não deve ser utilizado durante a gravidez nem em mulheres em idade fértil que não utilizem um método contraceptivo adequado, excepto se o potencial benefício justificar o potencial risco para o feto. Se o medicamento for administrado durante a gravidez ou se a doente ficar grávida enquanto estiver a receber este fármaco, deverá ser informada dos potenciais riscos para o feto. As mulheres em período fértil devem ser informadas de que deverão evitar engravidar durante o tratamento com SUTENT. Com base em achados não clínicos, a fertilidade masculina e feminina poderão ser comprometidas pelo tratamento com SUTENT (ver secção 5.3). Aleitamento Em ratos, o sunitinib e/ou os seus metabolitos são excretados pelo leite. Não se sabe se o sunitinib ou o seu principal metabolito activo são excretados no leite humano. Uma vez que os medicamentos são frequentemente excretados no leite humano e devido à possibilidade de reacções adversas graves em crianças que estão a ser amamentadas, as mulheres não deverão amamentar enquanto estiverem a tomar SUTENT. 4.7 Efeitos sobre a capacidade de conduzir e utilizar máquinas Não foram realizados estudos para avaliar os efeitos sobre a capacidade de conduzir e utilizar máquinas. Os doentes devem ser advertidos para o facto de poderem sentir tonturas durante o tratamento com SUTENT. 4.8 Efeitos indesejáveis Os acontecimentos adversos graves mais importantes relacionados com o tratamento, associados ao tratamento com SUTENT de doentes com tumores sólidos foram: embolia pulmonar (1%), trombocitopenia (1%), hemorragia tumoral (0,9%), neutropenia febril (0,4%) e hipertensão (0,4%). Os acontecimentos adversos mais frequentes, de qualquer grau, relacionados com o tratamento (ocorridos em pelo menos 20% dos doentes), incluíram: fadiga, doenças gastrointestinais, tais como diarreia, náuseas, estomatite, dispepsia e vómitos; alteração da coloração cutânea; disgeusia e anorexia. Os acontecimentos adversos relacionados com o tratamento mais frequentes, de gravidade máxima grau 3, foram a fadiga, a hipertensão e a neutropenia, e o aumento da lipase foi o acontecimento adverso relacionado com o tratamento mais frequente, de gravidade máxima grau 4, em doentes com tumores sólidos. As reacções adversas relacionadas com o tratamento que foram notificadas em >5% dos doentes com tumores sólidos encontram-se abaixo descritas. Os efeitos indesejáveis são apresentados por ordem decrescente de gravidade dentro de cada classe de frequência.
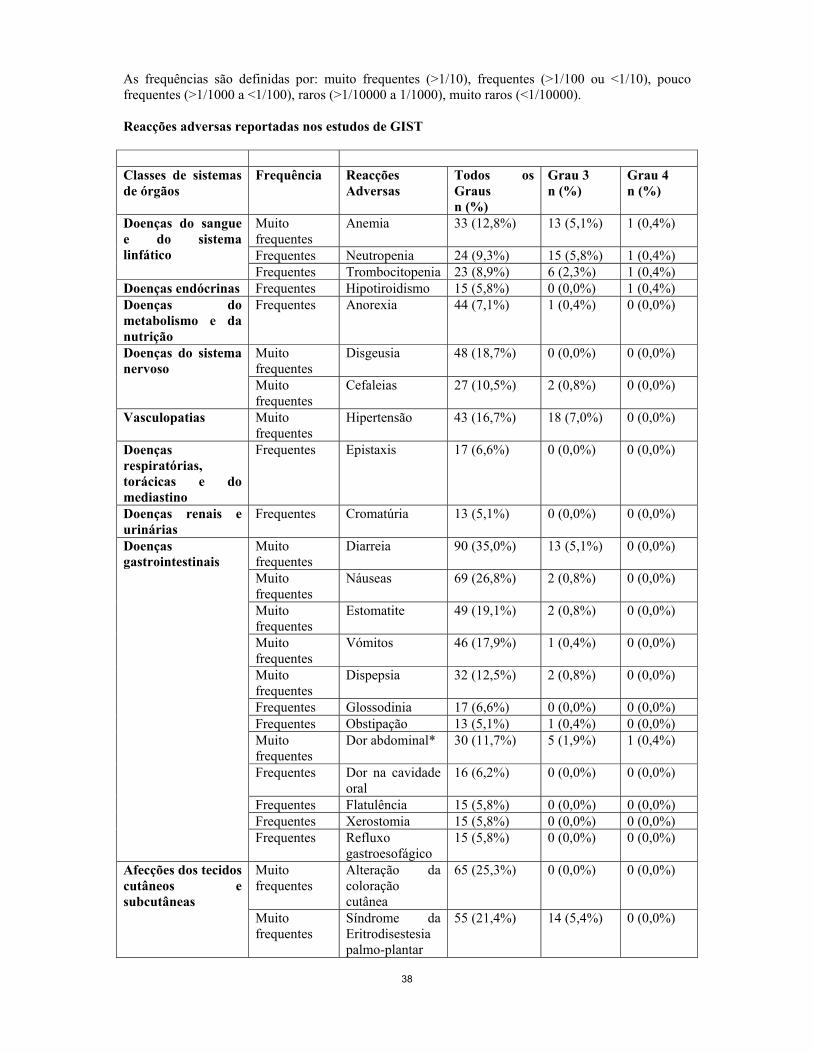
38
As frequências são definidas por: muito frequentes (>1/10), frequentes (>1/100 ou <1/10), pouco frequentes (>1/1000 a <1/100), raros (>1/10000 a 1/1000), muito raros (<1/10000). Reacções adversas reportadas nos estudos de GIST Classes de sistemas de órgãos
Frequência Reacções Adversas
Todos os Graus n (%)
Grau 3 n (%)
Grau 4 n (%)
Muito frequentes
Anemia 33 (12,8%) 13 (5,1%) 1 (0,4%)
Frequentes Neutropenia 24 (9,3%) 15 (5,8%) 1 (0,4%)
Doenças do sangue e do sistema linfático
Frequentes Trombocitopenia 23 (8,9%) 6 (2,3%) 1 (0,4%) Doenças endócrinas Frequentes Hipotiroidismo 15 (5,8%) 0 (0,0%) 1 (0,4%) Doenças do metabolismo e da nutrição
Frequentes Anorexia 44 (7,1%) 1 (0,4%) 0 (0,0%)
Muito frequentes
Disgeusia 48 (18,7%) 0 (0,0%) 0 (0,0%) Doenças do sistema nervoso
Muito frequentes
Cefaleias 27 (10,5%) 2 (0,8%) 0 (0,0%)
Vasculopatias Muito frequentes
Hipertensão 43 (16,7%) 18 (7,0%) 0 (0,0%)
Doenças respiratórias, torácicas e do mediastino
Frequentes Epistaxis 17 (6,6%) 0 (0,0%) 0 (0,0%)
Doenças renais e urinárias
Frequentes Cromatúria 13 (5,1%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Diarreia 90 (35,0%) 13 (5,1%) 0 (0,0%)
Muito frequentes
Náuseas 69 (26,8%) 2 (0,8%) 0 (0,0%)
Muito frequentes
Estomatite 49 (19,1%) 2 (0,8%) 0 (0,0%)
Muito frequentes
Vómitos 46 (17,9%) 1 (0,4%) 0 (0,0%)
Muito frequentes
Dispepsia 32 (12,5%) 2 (0,8%) 0 (0,0%)
Frequentes Glossodinia 17 (6,6%) 0 (0,0%) 0 (0,0%) Frequentes Obstipação 13 (5,1%) 1 (0,4%) 0 (0,0%) Muito frequentes
Dor abdominal* 30 (11,7%) 5 (1,9%) 1 (0,4%)
Frequentes Dor na cavidade oral
16 (6,2%) 0 (0,0%) 0 (0,0%)
Frequentes Flatulência 15 (5,8%) 0 (0,0%) 0 (0,0%) Frequentes Xerostomia 15 (5,8%) 0 (0,0%) 0 (0,0%)
Doenças gastrointestinais
Frequentes Refluxo gastroesofágico
15 (5,8%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Alteração da coloração cutânea
65 (25,3%) 0 (0,0%) 0 (0,0%) Afecções dos tecidos cutâneos e subcutâneas
Muito frequentes
Síndrome da Eritrodisestesia palmo-plantar
55 (21,4%) 14 (5,4%) 0 (0,0%)

39
Muito frequentes
Erupção cutânea***
39 (15,2%) 2 (0,8%) 0 (0,0%)
Frequentes Alterações da coloração do cabelo
22 (8,6%) 0 (0,0%) 0 (0,0%)
Frequentes Secura cutânea 15 (5,8%) 0 (0,0%) 0 (0,0%) Frequentes Dor nas
extremidades 21 (8,2%) 1 (0,4%) 0 (0,0%)
Frequentes Artralgia 15 (5,8%) 2 (0,8%) 0 (0,0%)
Afecções musculoesqueléticas e dos tecidos conjuntivos Frequentes Mialgia 13 (5,1%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Fadiga/astenia 135 (52,5%) 25 (9,7%) 0 (0,0%)
Muito frequentes
Inflamação das mucosas
30 (11,7%) 0 (0,0%) 0 (0,0%)
Perturbações gerais e alterações no local de administração
Frequentes Edema** 21 (8,2%) 1 (0,4%) 0 (0,0%) Frequentes Diminuição da
hemoglobina 16 (6,2%) 2 (0,8%) 0 (0,0%) Exames
complementares de diagnóstico Frequentes Aumento da
creatinina fosfoquinase sérica
14 (5,4%) 0 (0,0%) 0 (0,0%)
Frequentes Diminuição da fracção de ejecção
13 (5,1%) 1 (0,4%) 0 (0,0%)
Frequentes Aumento da lipase
13 (5,1%) 5 (1,9%) 4 (1,6%)
Diminuição da contagem plaquetária
13 (5,1%) 2 (0,8%) 1(0,4%)
Qualquer acontecimento adverso
222 (86,4%) 88 (34,2%) 24 (9,3%)
* Foram combinadas as seguintes designações: dor abdominal, dor abdominal superior e dor abdominal inferior. ** Foram combinadas as seguintes designações: edema e edema periférico. *** Foram combinadas as seguintes designações: erupção cutânea, erupção cutânea eritmatosa, erupção cutânea macular e erupção cutânea escamosa. Reacções adversas reportadas nos estudos de MRCC Classes de sistemas de órgãos
Frequência Reacções Adversas
Todos os Graus n (%)
Grau 3 n (%)
Grau 4 n (%)
Muito frequentes
Neutropenia 17 (10,1%) 8 (4,7%) 1 (0,6%)
Frequentes Anemia 16 (9,5%) 6 (3,6%) 0 (0,0%) Frequentes Trombocitopenia 15 (8,9%) 5 (3,0%) 2 (1,2%)
Doenças do sangue e do sistema linfático
Frequentes Leucopenia 14 (8,3%) 7 (4,1%) 0 (0,0%) Afecções oculares Frequentes Aumento da
secreção lacrimal
9 (5,3%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Anorexia 47 (27,8%) 1 (0,6%) 0 (0,0%) Doenças do metabolismo e da nutrição Frequentes Desidratação 12 (7,1%) 4 (2,4%) 0 (0,0%)

40
Frequentes Diminuição do apetite
11 (6,5%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Disgeusia 71 (42%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Cefaleias 25 (14,8%) 1 (0,6%) 0 (0,0%)
Frequentes Tonturas 13 (7,7%) 2 (1,2%) 0 (0,0%)
Doenças do sistema nervoso
Frequentes Parestesia 9 (5,3%) 0 (0,0%) 0 (0,0%) Vasculopatias Muito
frequentes Hipertensão 28 (16,6%) 7 (4,1%) 0 (0,0%)
Frequentes Epistaxis 16 (9,5%) 0 (0,0%) 0 (0,0%) Doenças respiratórias, torácicas e do mediastino
Frequentes Dispneia 9 (5,3%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Diarreia 83 (49,1%) 5 (3,0%) 0 (0,0%)
Muito frequentes
Náuseas 84 (49,7%) 2 (1,2%) 0 (0,0%)
Muito frequentes
Estomatite 70 (41,4%) 6 (3,6%) 0 (0,0%)
Muito frequentes
Dispepsia 69 (40,8%) 1 (0,6%) 0 (0,0%)
Muito frequentes
Vómitos 52 (30,8%) 2 (1,2%) 0 (0,0%)
Muito frequentes
Obstipação 34 (20,1%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Glossodinia 25 (14,8%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Dor abdominal* 17 (10,1%) 2 (1,2%) 0 (0,0%)
Frequentes Flatulência 16 (9,5%) 0 (0,0%) 0 (0,0%) Frequentes Distensão
abdominal 9 (5,3%) 0 (0,0%) 0 (0,0%)
Doenças gastrointestinais
Frequentes Xerostomia 9 (5,3%) 0 (0,0%) 0 (0,0%) Muito frequentes
Alteração da coloração cutânea
54 (32,0%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Erupção cutânea**
46 (27,2%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Alteração da coloração do cabelo
24 (14,2%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Síndrome da Eritrodisestesia palmo-plantar
21 (12,4%) 6 (3,6%) 0 (0,0%)
Frequentes Alopécia 13 (7,7%) 0 (0,0%) 0 (0,0%) Frequentes Dermatite
esfoliativa 10 (5,9%) 2 (1,2%) 0 (0,0%)
Frequentes Edema periorbitário
9 (5,3%) 0 (0,0%) 0 (0,0%)
Muito frequentes
Secura cutânea 22 (13,0%) 0 (0,0%) 0 (0,0%)
Afecções dos tecidos cutâneos e subcutâneas
Muito frequentes
Eritema 20 (11,8%) 0 (0,0%) 0 (0,0%)

41
Muito frequente
Dor nas extremidades
21 (12,4%) 1 (0,6%) 0 (0,0%) Afecções musculoesqueléticas e dos tecidos conjuntivos
Frequentes Mialgia 15 (8,9%) 1 (0,6%) 0 (0,0%)
Muito frequentes
Fadiga/astenia 108 (63,9%) 19 (11,2%) 0 (0,0%) Perturbações gerais e alterações no local de administração Muito
frequentes Inflamação das mucosas
30 (17,8%) 1 (0,6%) 0 (0,0%)
Complicações de intervenções relacionadas com lesões e intoxicações
Muito frequentes
Bolhas 7 (11,1%) 2 (3,2%) 0 (0,0%)
Muito frequentes
Aumento da lipase
17 (10,1%) 12 (7,1%) 3 (1,8%)
Frequentes Fracção de ejecção alterada
16 (9,5%) 1 (0,6%) 0 (0,0%)
Exames complementares de diagnóstico
Frequentes Aumento da amilase sérica
9 (5,3%) 6 (3,6%) 0 (0,0%)
Frequentes Diminuição de peso
11 (6,5%) 0 (0,0%) 0 (0,0%)
Frequentes Diminuição da contagem dos glóbulos brancos
10 (5,9%) 3 (1,8%) 0 (0,0%)
Frequentes Diminuição da contagem plaquetária
9 (5,3%) 3 (1,8%) 2 (1,2%)
Qualquer acontecimento adverso
166 (98,2%) 77 (45,6%) 14 (8,3%)
* Foram combinadas as seguintes designações: dor abdominal, dor abdominal superior e dor abdominal inferior. ** Foram combinadas as seguintes designações: erupção cutânea, erupção cutânea eritmatosa, erupção cutânea folicular, erupção cutânea generalizada, erupção cutânea papular e erupção cutânea associada a prurido. 4.9 Sobredosagem Não existe experiência de sobredosagem aguda com SUTENT. Não existe antídoto específico para a sobredosagem com SUTENT, devendo o tratamento destes casos consistir em medidas gerais de suporte. Se estiver indicado, a eliminação do medicamento não absorvido poderá ser conseguida por indução do vómito ou lavagem gástrica. 5. PROPRIEDADES FARMACOLÓGICAS 5.1 Propriedades farmacodinâmicas Grupo farmacoterapêutico: Agente antineoplásico - Inibidor da proteína tirosina-cinase; Código ATC: L01XE04. O malato de sunitinib inibe múltiplos receptores da tirosina-cinase que estão implicados no crescimento tumoral, na angiogénese patológica e na progressão metastática do cancro. O sunitinib foi identificado como inibidor dos receptores do factor de crescimento derivado das plaquetas (PDGFRα e PDGFRβ), receptores do factor de crescimento endotelial vascular (VEGFR1, VEGFR2 e VEGFR3), receptor do factor das células estaminais (KIT), receptor Fms-like da tirosina-cinase-3 (FLT3), receptor do factor estimulador de colónias (CSF-1R) e o receptor do factor neutrotrófico derivado de

42
células gliais (RET). O metabolito principal apresenta potência similar ao sunitinib em ensaios bioquímicos e celulares. ESTUDOS CLÍNICOS A segurança e eficácia clínica do SUTENT foi estudada no tratamento de doentes com tumores malignos do estroma gastrointestinal resistentes ao imatinib (isto é, doentes em que a doença evoluiu durante ou após o tratamento com imatinib) ou intolerantes ao imatinib (isto é, doentes que sofreram toxicidade significativa durante o tratamento com imatinib, que impediu a continuação tratamento), e no tratamento de doentes com carcinoma de células renais metastático após falha de terapêutica à base de citocinas. A eficácia é baseada no tempo até progressão tumoral e no aumento da sobrevivência no GIST e nas taxas de resposta objectiva no carcinoma das células renais metastático. Tumores do estroma gastrointestinal Foi conduzido um estudo inicial de desenho aberto para determinação de dose em doentes com GIST após insucesso da terapêutica com imatinib devida a resistência ou intolerância (dose máxima diária mediana de 800 mg). Foram incluídos noventa e sete doentes em várias doses e regimes; 55 doentes receberam 50 mg na posologia recomendada de tratamento, em ciclos de 4 semanas de tratamento/2 semanas em repouso (ciclos 4/2). Neste estudo, o tempo mediano até progressão tumoral foi de 34,0 semanas (IC 95% = 22,0 – 46,0 semanas). Foi realizado um estudo de fase 3 de SUTENT, aleatorizado, em dupla ocultação e controlado com placebo, em doentes com tumores do estroma gastrointestinal que apresentaram intolerância ou progressão da doença durante ou após o tratamento com imatinib (dose máxima diária mediana de 800 mg). Neste estudo, foram aleatorizados 312 doentes (2:1) para receber 50 mg de SUTENT ou placebo, por via oral, com o esquema de tratamento 4/2, até progressão da doença ou saída do estudo por outra razão (207 doentes receberam SUTENT e 105 doentes receberam placebo). O objectivo primário de avaliação da eficácia foi o tempo até progressão do tumor, definido como o período de tempo entre a aleatorização e a primeira documentação objectiva de progressão do tumor. O tempo mediano até progressão do tumor com SUTENT foi de 28,9 semanas (IC de 95% = 21,3–34,1 semanas), mais longo que o tempo até progressão do tumor de 5,1 semanas (IC de 95% = 4,4–10,1 semanas) registado com o placebo, com diferença estatisticamente significativa. A diferença na sobrevivência global foi estatisticamente favorável a SUTENT [risco relativo: 0,491 (IC de 95%, 0,290-0,0831)]; o risco de morte foi duas vezes superior para os doentes no braço de placebo comparativamente ao braço de SUTENT. As percentagens de mortes foram de 14% para o SUTENT versus 25% para o placebo. No momento da análise, ainda não tinha sido alcançada a sobrevivência global mediana em ambos os braços de tratamento. Carcinoma das células renais metastático refractário às citocinas Foi realizado um estudo de fase 2 com o SUTENT em doentes refractários a um tratamento anterior com citocinas, com interleucina-2 ou interferão-α. Sessenta e três doentes receberam uma dose inicial diária de 50 mg de SUTENT, por via oral, durante 4 semanas consecutivas, seguida de um período de repouso de 2 semanas, de forma a cumprir um ciclo completo de 6 semanas (esquema de tratamento 4/2). O objectivo primário de avaliação da eficácia foi a taxa de resposta objectiva (ORR), baseada nos critérios de avaliação de resposta em tumores sólidos (Response Evaluation Criteria in Solid Tumors, RECIST). Neste estudo, a taxa de resposta objectiva foi de 36,5% (IC 95%, 24,7%–49,6%) e o tempo mediano até progressão (TTP) foi de 37,7 semanas (IC de 95%, 24,0- 46,4 semanas). Foi realizado um estudo confirmatório de desenho aberto, de braço único e multicêntrico para avaliação da eficácia e segurança do SUTENT em doentes com carcinoma das células renais metastático refractário a um tratamento prévio com citocinas. Cento e seis doentes receberam pelo menos uma dose de 50 mg de SUTENT com o esquema de tratamento 4/2.

43
O objectivo primário de avaliação da eficácia deste estudo foi a taxa de resposta objectiva (ORR). Os objectivos de avaliação secundários incluíram o tempo até progressão do tumor (TTP), a duração da resposta e a sobrevivência global (OS). Neste estudo, a taxa de resposta objectiva foi de 38% (IC 95%, 26,8% – 47,5%). A duração da resposta e sobrevivência global medianas ainda não tinham sido alcançadas. Este medicamento foi sujeito a uma Autorização de Introdução no Mercado condicionada. Isto significa que se aguardam evidências adicionais sobre este medicamento, em particular sobre o efeito de SUTENT em termos de sobrevivência livre de progressão em doentes com MRCC. Está a ser conduzido um estudo para avaliar esta questão. A Agência Europeia de Medicamentos (EMEA) irá rever anualmente nova informação sobre o medicamento, e este RCM será actualizado se necessário. 5.2 Propriedades farmacocinéticas A farmacocinética do sunitinib e do malato de sunitinib foi avaliada em 135 voluntários saudáveis e em 266 doentes com tumores sólidos. Absorção Após administração oral, as concentrações máximas (Cmáx) de sunitinib são geralmente observadas entre 6 a12 horas (Tmáx) . Os alimentos não afectam a biodisponibilidade de sunitinib. Distribuição Em ensaios in vitro, a ligação de sunitinib e do seu principal metabolito activo às proteínas plasmáticas humanas foi de, respectivamente, 95% e 90%, aparentemente não dependente da concentração. O volume de distribuição aparente de sunitinib (V/F) foi elevado – 2230L - , indicando distribuição tecidular. Metabolismo Os valores Ki calculados in vitro para todas as isoformas de CYP testadas (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 e CYP4A9/11) indicam que é improvável que o sunitinib e o seu metabolito activo principal inibam o metabolismo, em qualquer extensão clinicamente relevante, dos fármacos que possam ser metabolizados por estas enzimas. Estudos in vitro indicam que o SUTENT não induz nem inibe as principais enzimas CYP, incluindo a CYP3A4. Biotransformação O sunitinib é metabolizado principalmente pela enzima do citocromo P450, CYP3A4, que produz o seu principal metabolito activo, que por sua vez é ainda metabolizado pela CYP3A4. A administração concomitante de SUTENT com o indutor potente da CYP3A4, a rifampicina, resultou na redução de, aproximadamente, 56% e 78% dos valores de Cmax e de AUC0-∞ de sunitinib, respectivamente, após a administração única de SUTENT em voluntários saudáveis. A administração de SUTENT com outros indutores da família da CYP3A4 (ex.: dexametasona, fenitoína, carbamazepina, fenobarbital ou Hypericum perfuratum, também conhecido por hipericão) pode diminuir as concentrações de sunitinib. Eliminação A principal via de excreção é a fecal (61%) e a via renal é responsável pela eliminação de 16% da dose administrada do medicamento e dos seus metabolitos. Os principais compostos identificados no plasma, urina e fezes relacionados com o fármaco foram o sunitinib e o seu principal metabolito, representando 91,5%, 86,4% e 73,8%, respectivamente, da reactividade em amostras agregadas. Foram identificados metabolitos secundários na urina e fezes, mas não foram, geralmente, encontrados no plasma. A depuração oral total (CL/F) foi de 34-62 L/hora. Insuficiência de funções orgânicas Insuficiência hepática: Não foram realizados estudos clínicos em doentes com alteração da função hepática.

44
Os estudos excluíram os doentes com ALT ou AST> 2,5 vezes o limite superior do normal ou >5,0 vezes o limite superior do normal, se devido a patologia subjacente. Insuficiência renal: Não foram realizados estudos clínicos em doentes com alteração da função renal. Os estudos excluíram doentes com creatinina sérica> 2,0 vezes o limite superior do normal. Análises populacionais farmacocinéticas indicam que a depuração aparente de sunitinib (CL/F) não foi afectada pela depuração da creatinina dentro da gama de valores avaliada (42-347 ml/min). Farmacocinética plasmática Após a administração oral em voluntários saudáveis, as semi-vidas de eliminação de sunitinib e do seu principal metabolito activo, desetil, foram, aproximadamente, 40 a 60 horas, e 80-110 horas, respectivamente. A área sob a curva (AUC) e a Cmáx aumentaram proporcionalmente com a dose, no intervalo de dose de 25-100 mg. Com a administração diária reiterada, o sunitinib acumula-se 3 a 4 vezes e o seu principal metabolito activo acumula-se 7 a 10 vezes. As concentrações de sunitinib e do seu principal metabolito activo em estado estacionário foram atingidas em 10 a 14 dias. Por volta do dia 14, as concentrações plasmáticas combinadas de sunitinib e do seu metabolito activo são de 62,9-101 ng/mL, que correspondem às concentrações alvo que se prevê inibirem, através de dados pré-clínicos, a fosforilação in vitro do receptor e resultarem em estabilização/redução do crescimento tumoral in vivo. O principal metabolito activo representa 23 a 37% da exposição total. Não foram observadas alterações significativas na farmacocinética do sunitinib ou do principal metabolito activo após administração diária reiterada ou com repetição dos ciclos nos regimes de dose testados. A farmacocinética foi similar em todas as populações com tumores sólidos testadas e nos voluntários saudáveis. A análise farmacocinética de dados demográficos populacionais indica que não é necessário efectuar ajuste de dose com base no peso ou score ECOG. Os dados disponíveis indicam que as mulheres podem apresentar uma depuração aparente (CL/F) de sunitinib cerca de 30% inferior à dos homens: contudo, esta diferença não requer ajustes de dose. 5.3 Dados de segurança pré-clínica Em estudos de toxicidade reiterada realizados em ratos e macacos, com duração até 9 meses, os principais efeitos em órgãos alvo foram identificados no tracto gastrointestinal (emése e diarreia nos macacos), glândulas supra-renais (congestão cortical e/ou hemorragia em ratos e macacos, com necrose seguida de fibrose em ratos), sistema hematopoiético e linfático (hipocelularidade da medula óssea e deplecção linfóide do timo, baço e gânglios linfáticos), pâncreas exócrino (desgranulação das células acinares com necrose celular), glândula salivar (hipertrofia acinar), articulações (espessamento da placa de crescimento), útero (atrofia) e ovários (diminuição do desenvolvimento folicular). Todos os achados ocorreram com níveis de exposição plasmática de sunitinib clinicamente relevantes. Foram observados efeitos adicionais noutros estudos, incluindo prolongamento do intervalo QTc, redução do FEVE, hipertrofia pituitária, atrofia tubular testicular, aumento das células mesangiais no rim, hemorragia gastrointestinal e da mucosa oral e hipertrofia das células pituitárias anteriores. Pensa-se que as alterações no útero (atrofia do endométrio) e na placa óssea de crescimento (espessamento fisário ou displasia da cartilagem) estejam relacionadas com a acção farmacológica do sunitinib. A maioria destes efeitos foi reversível após 2 a 6 semanas sem tratamento. Genotoxicidade O potencial genotóxico do sunitinib foi avaliado in vitro e in vivo. O sunitinib não foi mutagénico em bactérias com activação metabólica efectuada por fígado de rato. O sunitinib não induziu aberrações cromossómicas estruturais in vitro nos linfócitos periféricos humanos. Foi observada poliploidia in vitro (aberrações cromossómicas numéricas), em linfócitos periféricos, tanto na presença como na ausência de activação metabólica. O sunitinib não foi clastogénico in vivo na medula óssea de rato. O potencial de toxicidade genética não foi avaliado para o principal metabolito activo. Carcinogénese Não foram realizados estudos de carcinogénese com malato de sunitinib.

45
Toxicidade reprodutiva e de desenvolvimento Não foram observados efeitos na fertilidade dos ratos machos ou fêmeas em estudos de toxicidade reprodutiva. No entanto, foram observados efeitos na fertilidade feminina em estudos de toxicidade reiterada realizados em ratos e macacos, sob a forma de atresia folicular, degeneração do corpo lúteo, alterações no endométrio uterino e diminuição do peso do útero e dos ovários, com níveis de exposição sistémica clinicamente relevantes. Foram observados efeitos na fertilidade masculina em ratos, sob a forma de atrofia tubular testicular, redução dos espermatozóides nos epididimos e deplecção colóide na próstata e vesículas seminais, a níveis plasmáticos de exposição 18 vezes superiores aos clinicamente observados. A mortalidade embriofetal foi evidente em ratos, sob a forma de redução significativa do número de fetos vivos, aumento do número de reabsorções, aumento da perda pós implantação e perda total de ninhadas em 8 de 28 fêmeas grávidas, para níveis de exposição plasmática 5,5 vezes superiores aos observados na clínica. As reduções do peso do útero de coelhos fêmeas grávidas e no número de fetos vivos foram devidas ao aumento do número de reabsorções, aumento da perda pós-implantação e perda de ninhadas completas em 4 de 6 fêmeas grávidas, para níveis de exposição plasmática 3 vezes superiores aos observados na clínica. O tratamento de ratos com sunitinib durante a organogénese resultou em efeitos no desenvolvimento, consistindo no aumento da incidência de malformações do esqueleto dos fetos, predominantemente caracterizadas por ossificação tardia das vértebras torácicas/lombares e ocorreram a níveis de exposição plasmática 6 vezes superiores aos observados na clínica. Os efeitos no desenvolvimento dos coelhos consistiram no aumento da incidência de fenda labial para níveis de concentração plasmática aproximadamente iguais aos observados na clínica e fenda labial e fenda palatina, para níveis de exposição plasmática 2,7 vezes superiores aos observados na clínica. 6. INFORMAÇÕES FARMACÊUTICAS 6.1 Lista dos excipientes Conteúdo da cápsula Manitol Croscarmelose sódica Povidona Estereato de magnésio Cápsula Gelatina Dióxido de titânio (E171) Óxido de ferro amarelo (E172) Óxido de ferro vermelho (E 172) Óxido de ferro negro (E172) Tinta de impressão: Goma laca Propilenoglicol Hidróxido de sódio Povidona Dióxido de titânio (E171) 6.2 Incompatibilidades Não aplicável. 6.3 Prazo de validade

46
2 anos. 6.4 Precauções especiais de conservação O medicamento não necessita de quaisquer precauções especiais de conservação. 6.5 Natureza e conteúdo do recipiente Frascos de polietileno de alta densidade (HDPE) com sistema de fecho de polipropileno, contendo 30 cápsulas. 6.6 Precauções especiais de eliminação Não existem requisitos especiais. 7. TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Pfizer Ltd Ramsgate Road Sandwich, Kent CT13 9NJ United Kingdom 8. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO 9. DATA DA PRIMEIRA AUTORIZAÇÃO/RENOVAÇÃO DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Data da primeira autorização: Data da próxima renovação: 10. DATA DA REVISÃO DO TEXTO

47
ANEXO II A. TITULAR(ES) DE AUTORIZAÇÃO DE FABRICO
RESPONSÁVEL(VEIS) PELA LIBERTAÇÃO DO LOTE B. CONDIÇÕES DA AUTORIZAÇÃO DE INTRODUÇÃO
NO MERCADO C. OBRIGAÇÕES ESPECÍFICAS A SEREM CUMPRIDAS PELO
TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO

48
A TITULAR(ES) DE AUTORIZAÇÃO DE FABRICO RESPONSÁVEL(VEIS) PELA
LIBERTAÇÃO DO LOTE Nome e endereço do(s) fabricante(s) responsável(veis) pela libertação do lote PFIZER Italia S.r.l. Via del Commercio Zona Industriale IT-63046 Marino del Tronto (Ascoli Piceno) Italy B. CONDIÇÕES DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO • CONDIÇÕES OU RESTRIÇÕES RELATIVAS AO FORNECIMENTO E UTILIZAÇÃO
IMPOSTAS AO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Medicamento sujeito a receita médica restrita (Ver Anexo I: Resumo das Características do Medicamento, secção 4.2). • CONDIÇÕES OU RESTRIÇÕES RELATIVAS À UTILIZAÇÃO SEGURA E EFICAZ
DO MEDICAMENTO Não aplicável. • OUTRAS CONDIÇÕES Plano de Gestão do Risco O Titular da Autorização de Introdução no Mercado compromete-se a realizar os estudos e actividades de farmacovigilância adicionais que estão detalhados no plano de Farmacovigilância. Deverá ser submetido um plano de Gestão do Risco actualizado, de acordo com a CHMP Guideline on Risk Management Systems for medicinal products for human use, em simultâneo com os RPSs, dentro de 60 dias após ter sido alcançado um marco importante (Farmacovigilância ou Minimização de Risco) ou quando os resultados de um estudo estiverem disponíveis ou a pedido da Autoridade Competente. C. OBRIGAÇÕES ESPECÍFICAS A SEREM CUMPRIDAS PELO TITULAR DA
AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO O Titular da Autorização de Introdução no Mercado deverá completar o seguinte programa de estudos dentro do intervalo de tempo especificado. Os resultados deste deverão ser tidos em consideração na relação benefício risco durante a avaliação do pedido de renovação. Aspectos clínicos: O Titular de Autorização de Introdução no Mercado compromete-se a providenciar os resultados de um estudo que se encontra a decorrer em doentes com carcinoma de células renais metastático naive para citocinas em Setembro de 2006.

49
ANEXO III ROTULAGEM E FOLHETO INFORMATIVO

50
A. ROTULAGEM

51
INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO {Cartonagem exterior – cápsulas de 12,5 mg} 1. DENOMINAÇÃO DO MEDICAMENTO SUTENT 12,5 mg cápsulas Sunitinib 2. DESCRIÇÃO DO(S) PRINCÍPIO(S) ACTIVO(S) Cada cápsula contém malato de sunitinib, equivalente a 12,5 mg de sunitinib. 3. LISTA DOS EXCIPIENTES As cápsulas contêm manitol e propilenoglicol. 4. FORMA FARMACÊUTICA E CONTEÚDO 30 cápsulas 5. MODO E VIA(S) DE ADMINISTRAÇÃO Via oral. Consultar o folheto informativo. 6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DO ALCANCE E DA VISTA DAS CRIANÇAS Manter fora do alcance e da vista das crianças. 7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO Use de acordo com as indicações do seu médico. 8. PRAZO DE VALIDADE Val.: 9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO O medicamento não necessita de quaisquer precauções especiais de conservação. 10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE FOR CASO DISSO

52
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO Pfizer Ltd Ramsgate Road Sandwich Kent CT13 9NJ United Kingdom 12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO 13. NÚMERO DO LOTE Lote 14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO Medicamento sujeito a receita médica. 15. INSTRUÇÕES DE UTILIZAÇÃO 16. INFORMAÇÃO EM BRAILLE SUTENT 12,5 mg

53
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE ACONDICIONAMENTO PRIMÁRIO {Frasco HDPE – 12,5 mg cápsulas } 1. DENOMINAÇÃO DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO SUTENT 12,5 mg cápsulas Sunitinib Via oral 2. MODO DE ADMINISTRAÇÃO Consultar o folheto informativo. 3. PRAZO DE VALIDADE Val: 4. NÚMERO DO LOTE Lote 5. CONTEÚDO EM TERMOS DE PESO, VOLUME OU UNIDADE 30 cápsulas 6. OUTRAS

54
INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO {Cartonagem exterior – cápsulas de 25 mg} 1. DENOMINAÇÃO DO MEDICAMENTO SUTENT 25 mg cápsulas Sunitinib 2. DESCRIÇÃO DO(S) PRINCÍPIO(S) ACTIVO(S) Cada cápsula contém malato de sunitinib, equivalente a 25,0 mg de sunitinib. 3. LISTA DOS EXCIPIENTES As cápsulas contêm manitol e propilenoglicol. 4. FORMA FARMACÊUTICA E CONTEÚDO 30 cápsulas 5. MODO E VIA(S) DE ADMINISTRAÇÃO Via oral. Consultar o folheto informativo. 6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DO ALCANCE E DA VISTA DAS CRIANÇAS Manter fora do alcance e da vista das crianças. 7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO Use de acordo com as indicações do seu médico. 8. PRAZO DE VALIDADE Val.: 9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO O medicamento não necessita de quaisquer precauções especiais de conservação. 10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE FOR CASO DISSO

55
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO Pfizer Ltd Ramsgate Road Sandwich Kent CT13 9NJ United Kingdom 12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO 13. NÚMERO DO LOTE Lote 14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO Medicamento sujeito a receita médica. 15. INSTRUÇÕES DE UTILIZAÇÃO 16. INFORMAÇÃO EM BRAILLE SUTENT 25 mg

56
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE ACONDICIONAMENTO PRIMÁRIO {Frasco HDPE – 25 mg cápsulas } 1. DENOMINAÇÃO DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO SUTENT 25 mg cápsulas Sunitinib Via oral 2. MODO DE ADMINISTRAÇÃO Consultar o folheto informativo. 3. PRAZO DE VALIDADE Val: 4. NÚMERO DO LOTE Lote 5. CONTEÚDO EM TERMOS DE PESO, VOLUME OU UNIDADE 30 cápsulas 6. OUTRAS

57
INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO {Cartonagem exterior – cápsulas de 50 mg} 1. DENOMINAÇÃO DO MEDICAMENTO SUTENT 50 mg cápsulas Sunitinib 2. DESCRIÇÃO DO(S) PRINCÍPIO(S) ACTIVO(S) Cada cápsula contém malato de sunitinib, equivalente a 50,0 mg de sunitinib 3. LISTA DOS EXCIPIENTES As cápsulas contêm manitol e propilenoglicol. 4. FORMA FARMACÊUTICA E CONTEÚDO 30 cápsulas 5. MODO E VIA(S) DE ADMINISTRAÇÃO Via oral. Consultar o folheto informativo. 6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DO ALCANCE E DA VISTA DAS CRIANÇAS Manter fora do alcance e da vista das crianças. 7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO Use de acordo com as indicações do seu médico. 8. PRAZO DE VALIDADE Val.: 9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO O medicamento não necessita de quaisquer precauções especiais de conservação. 10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE FOR CASO DISSO

58
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO Pfizer Ltd Ramsgate Road Sandwich Kent CT13 9NJ United Kingdom 12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO 13. NÚMERO DO LOTE Lote 14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO Medicamento sujeito a receita médica. 15. INSTRUÇÕES DE UTILIZAÇÃO 16. INFORMAÇÃO EM BRAILLE SUTENT 50 mg

59
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE ACONDICIONAMENTO PRIMÁRIO {Frasco HDPE – 50 mg cápsulas } 1. DENOMINAÇÃO DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO SUTENT 50 mg cápsulas Sunitinib Via oral 2. MODO DE ADMINISTRAÇÃO Consultar o folheto informativo. 3. PRAZO DE VALIDADE Val: 4. NÚMERO DO LOTE Lote 5. CONTEÚDO EM TERMOS DE PESO, VOLUME OU UNIDADE 30 cápsulas 6. OUTRAS

60
B. FOLHETO INFORMATIVO

61
FOLHETO INFORMATIVO: INFORMAÇÃO PARA O UTILIZADOR
SUTENT 12,5 mg cápsulas
sunitinib Leia atentamente este folheto antes de tomar o medicamento - Conserve este folheto. Pode ter necessidade de o reler. - Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico. - Este medicamento foi receitado para si. Não deve dá-lo a outros; o medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sintomas. - Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico ou farmacêutico. Neste folheto:
1. O que é SUTENT e para que é utilizado 2. Antes de tomar SUTENT 3. Como tomar SUTENT 4. Efeitos secundários possíveis 5. Como conservar SUTENT 6. Outras informações
1. O QUE É SUTENT E PARA QUE É UTILIZADO O SUTENT é um medicamento utilizado no tratamento do cancro, para prevenir a actividade de um grupo especial de proteínas que se sabe estarem envolvidas no crescimento e difusão das células cancerígenas. O SUTENT apenas lhe será prescrito por um médico com experiência no tratamento do carcinoma das células renais ou tumor do estroma gastrointestinal. O SUTENT é um medicamento utilizado no tratamento do carcinoma das células renais, uma forma de cancro dos rins que envolve alterações cancerígenas nas células do túbulo renal. O SUTENT é também utilizado no tratamento do tumor maligno do estroma gastrointestinal (GIST). O GIST é um cancro do estômago e dos intestinos. Resulta do crescimento descontrolado das células dos tecidos de suporte destes órgãos. O SUTENT inibe o crescimento destas células. Contacte o seu médico se tiver alguma questão relacionada com o modo de acção de SUTENT ou com a razão deste medicamento lhe ter sido prescrito. 2. ANTES DE TOMAR SUTENT Siga cuidadosamente todas as indicações do seu médico, mesmo que estas sejam diferentes da informação geral descrita neste Folheto. Não tome SUTENT Se tem alergia (hipersensibilidade) ao sunitinib ou a qualquer outro componente de SUTENT. Tome especial cuidado com SUTENT - Se tem ou teve problemas nos rins ou no fígado - Se tem a pressão sanguínea elevada - Se está grávida ou pensa poder estar grávida (ver descrição abaixo) - Se estiver a amamentar (ver descrição abaixo) Se algum destes se aplicar a si, informe o seu médico antes de tomar SUTENT.

62
Tomar SUTENT com outros medicamentos Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos obtidos sem receita médica, incluindo medicamentos à base de plantas (quaisquer medicamentos que possam aumentar a concentração de SUTENT, como o cetoconazol, o ritonavir, o itraconazol, a eritromicina, a claritromicina; ou quaisquer medicamentos que possam diminuir a concentração de SUTENT como a dexametasona, a fenitoína, a carbamazepina, a rifampicina, o fenobarbital ou o Hypericum perfuratum, também conhecido como hipericão). Tomar SUTENT com alimentos e bebidas O SUTENT pode ser tomado com ou sem alimentos, contudo não deverá tomar SUTENT com sumo de toranja. Gravidez e aleitamento Se estiver grávida ou pensa poder estar, informe o seu médico. O SUTENT não deverá ser utilizado durante a gravidez a menos que seja estritamente necessário. O seu médico discutirá consigo o risco potencial de tomar SUTENT durante a gravidez. Aconselham-se as mulheres em idade fértil a utilizar um método contraceptivo eficaz durante o tratamento com SUTENT. Informe o seu médico se estiver a amamentar. Não amamente durante o período de tratamento com SUTENT. Condução de veículos e utilização de máquinas Tome especial cuidado na condução ou utilização de máquinas se sentir tonturas ou se se sentir invulgarmente cansado. 3. COMO TOMAR SUTENT O seu médico irá prescrever-lhe a dose mais indicada para si. Recomenda-se que o SUTENT seja tomado durante 28 dias (4 semanas), seguido de 14 dias (2 semanas) de repouso (sem medicação), administrado em ciclos de 6 semanas. O seu médico determinará quantos ciclos de tratamento serão necessários para si.
Se tomar mais SUTENT do que deveria Informe imediatamente o seu médico se tomou acidentalmente demasiadas cápsulas. Poderá necessitar de cuidados médicos. Caso se tenha esquecido de tomar SUTENT Não tome uma dose a dobrar para compensar uma dose que se esqueceu de tomar. 4. EFEITOS SECUNDÁRIOS POSSÍVEIS Como os demais medicamentos, SUTENT pode causar efeitos secundários em algumas pessoas, no entanto, estes não se manifestam em todas as pessoas. Efeitos secundários muito frequentes, que podem afectar mais de 10 em cada 100 pessoas - dor/irritação na boca (da cavidade oral), boca (cavidade oral) sensibilizada, alterações do paladar, problemas no estômago, náuseas, vómitos, diarreia, prisão de ventre, dor abdominal, perda do apetite. - descoloração cutânea, alterações na coloração do cabelo, erupção cutânea nas palmas das mãos e nas plantas dos pés, bolhas, pele seca - cansaço, pressão arterial elevada, enxaqueca - redução do número de: glóbulos vermelhos e/ou glóbulos brancos

63
Outros efeitos secundários possíveis, que podem afectar entre 1 a 10 em cada 100 pessoas - diminuição da actividade da tiróide, fluxo sanguíneo cardíaco reduzido - hemorragia nasal, cor da urina anómala, fluxo lacrimal excessivo - dores nas articulações, dores musculares, fluidos excessivos nos tecidos incluindo a área do olho - alteração da sensação da pele, falta de ar - queda de cabelo, perda de peso Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico ou farmacêutico. 5. COMO CONSERVAR SUTENT - Manter fora do alcance e da vista das crianças. - O medicamento não necessita de precauções especiais de conservação. - Não utilize após o prazo de validade impresso na embalagem exterior e no rótulo. - Não utilize nenhuma embalagem que esteja danificada ou apresente sinais de violação. Os medicamentos não devem ser eliminados na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como eliminar os medicamentos de que já não necessita. Estas medidas irão ajudar a proteger o ambiente. 6. OUTRAS INFORMAÇÕES Qual a composição de SUTENT - A substância activa é o sunitinib (sob a forma de malato). - Os outros componentes são o manitol, a croscarmelose sódica, a povidona e o estearato de magnésio. A cápsula é composta por gelatina, óxido de ferro vermelho (E172) e dióxido de titânio (E171). A tinta de impressão contém goma laca, propilenoglicol, hidróxido de sódio, povidona e dióxido de titânio. Qual o aspecto de SUTENT e conteúdo da embalagem O SUTENT é fornecido como cápsulas de gelatina com a cabeça e o corpo cor de laranja, com “Pfizer” impresso com tinta branca na cabeça e “STN 12,5 mg” no corpo. SUTENT está disponível em embalagens de 30 cápsulas. Titular da Autorização de Introdução no Mercado Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJ United Kingdom Fabricante Pfizer Italia S.r.L Via Del Commercio – Zone Industriale 63046 Marino del Tronto (Ascoli Piceno) Italy Para quaisquer informações sobre este medicamento, queira contactar o representante local do titular da autorização de introdução no mercado.
Belgique / België /Belgien Pfizer S.A. / N.V. Tél/Tel: +32 (0)2 554 62 11
Luxembourg/Luxemburg Pfizer S.A. Tél/Tel: +32 (0)2 554 62 11

64
Česká republika Pfizer s.r.o. Tel.: +420-283-004-151
Magyarország Pfizer Kft. Tel.: +36-1-488-37-00
Danmark Pfizer ApS Tlf: +45 44 20 11 00
Malta V.J. Salomone Pharma Ltd. Tel. +356 212201
Deutschland Pfizer GmbH Tel: +49 (0)721 61 01 01
Nederland Pfizer BV Tel: +31 (0)10 406 42 00
Eesti Pfizer Luxembourg SARL, Eesti filiaal Tel.: +372 6 405 328
Norge Pfizer AS Tlf: +47 67 52 61 00
Ελλάδα Pfizer Hellas A.E. Τηλ: +30 210 7517981-3
Österreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0
España Pfizer S.A. Tél: +34 91 490 99 00
Polska Pfizer Polska Sp.zo.o Tel.:+48 22 335 61 00
France Pfizer Tél: +33 (0)1 58 07 34 40
Portugal Laboratórios Pfizer, Lda. Tel: +351 21 423 5500
Ireland Pfizer Healthcare Ireland Tel: +353 1800 633 363
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti Ljubljana Tel.: + 386 1 52 11 400
Ísland Vistor hf. Sími: +354 535 7000
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel.:+ 421 2 5941 8500
Italia Pfizer Italia S.r.l. Tel: +39 06 33 18 21
Suomi/Finland Pfizer Oy Puh./Tel: +358 (0)9 43 00 40
Kύπρος Geo. Pavlides & Araouzos Ltd. Tηλ.:+ 357 22 818087
Sverige Pfizer AB Tel: +46 (0)8 550-52000
Latvija Pfizer Luxembourg SARL filiale Latvjia Tel.: + 371 70 35 775
United Kingdom Pfizer Limited, Tel: +44 (0)1737 331111
Lietuva Pfizer Luxembourg SARL filialas Lietuvoje Tel. + 370 52 51 4000

65
Este folheto foi aprovado pela última vez em A este medicamento foi dad uma “autorização condicional”. Isto significa que se aguarda mais informação sobre este medicamento, em particular sobre o tratamento do cancro do rim. O SUTENT demonstrou diminuir o tumor. Contudo, espera-se mais informação sobre a duração deste efeito. A Agência Europeia de Medicamentos (EMEA) irá rever anualmente a nova informação sobre este medicamento e este Folheto Informativo será actualizado se necessário. Informação pormenorizada sobre este medicamento está disponível na Internet no site da Agência Europeia de Medicamentos (EMEA) http://www.emea.eu.int. Também existem links para outros sites sobre doenças raras e tratamentos.

66
FOLHETO INFORMATIVO: INFORMAÇÃO PARA O UTILIZADOR
SUTENT 25 mg cápsulas sunitinib
Leia atentamente este folheto antes de tomar o medicamento - Conserve este folheto. Pode ter necessidade de o reler. - Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico. - Este medicamento foi receitado para si. Não deve dá-lo a outros; o medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sintomas. - Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico ou farmacêutico. Neste folheto:
1. O que é SUTENT e para que é utilizado 2. Antes de tomar SUTENT 3. Como tomar SUTENT 4. Efeitos secundários possíveis 5. Como conservar SUTENT 6. Outras informações
1. O QUE É SUTENT E PARA QUE É UTILIZADO O SUTENT é um medicamento utilizado no tratamento do cancro, para prevenir a actividade de um grupo especial de proteínas que se sabe estarem envolvidas no crescimento e difusão das células cancerígenas. O SUTENT apenas lhe será prescrito por um médico com experiência no tratamento do carcinoma das células renais ou tumor do estroma gastrointestinal. O SUTENT é um medicamento utilizado no tratamento do carcinoma das células renais, uma forma de cancro dos rins que envolve alterações cancerígenas nas células do túbulo renal. O SUTENT é também utilizado no tratamento do tumor maligno do estroma gastrointestinal (GIST). O GIST é um cancro do estômago e dos intestinos. Resulta do crescimento descontrolado das células dos tecidos de suporte destes órgãos. O SUTENT inibe o crescimento destas células. Contacte o seu médico se tiver alguma questão relacionada com o modo de acção de SUTENT ou com a razão deste medicamento lhe ter sido prescrito. 2. ANTES DE TOMAR SUTENT Siga cuidadosamente todas as indicações do seu médico, mesmo que estas sejam diferentes da informação geral descrita neste Folheto. Não tome SUTENT Se tem alergia (hipersensibilidade) ao sunitinib ou a qualquer outro componente de SUTENT. Tome especial cuidado com SUTENT - Se tem ou teve problemas nos rins ou no fígado - Se tem a pressão sanguínea elevada - Se está grávida ou pensa poder estar grávida (ver descrição abaixo) - Se estiver a amamentar (ver descrição abaixo) Se algum destes se aplicar a si, informe o seu médico antes de tomar SUTENT. Tomar SUTENT com outros medicamentos

67
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos obtidos sem receita médica, incluindo medicamentos à base de plantas (quaisquer medicamentos que possam aumentar a concentração de SUTENT, como o cetoconazol, o ritonavir, o itraconazol, a eritromicina, a claritromicina; ou quaisquer medicamentos que possam diminuir a concentração de SUTENT como a dexametasona, a fenitoína, a carbamazepina, a rifampicina, o fenobarbital ou o Hypericum perfuratum, também conhecido como hipericão). Tomar SUTENT com alimentos e bebidas O SUTENT pode ser tomado com ou sem alimentos, contudo não deverá tomar SUTENT com sumo de toranja. Gravidez e aleitamento Se estiver grávida ou pensa poder estar, informe o seu médico. O SUTENT não deverá ser utilizado durante a gravidez a menos que seja estritamente necessário. O seu médico discutirá consigo o risco potencial de tomar SUTENT durante a gravidez. Aconselham-se as mulheres em idade fértil a utilizar um método contraceptivo eficaz durante o tratamento com SUTENT. Informe o seu médico se estiver a amamentar. Não amamente durante o período de tratamento com SUTENT. Condução de veículos e utilização de máquinas Tome especial cuidado na condução ou utilização de máquinas se sentir tonturas ou se se sentir invulgarmente cansado. 3. COMO TOMAR SUTENT O seu médico irá prescrever-lhe a dose mais indicada para si. Recomenda-se que o SUTENT seja tomado durante 28 dias (4 semanas), seguido de 14 dias (2 semanas) de repouso (sem medicação), administrado em ciclos de 6 semanas. O seu médico determinará quantos ciclos de tratamento serão necessários para si.
Se tomar mais SUTENT do que deveria Informe imediatamente o seu médico se tomou acidentalmente demasiadas cápsulas. Poderá necessitar de cuidados médicos. Caso se tenha esquecido de tomar SUTENT Não tome uma dose a dobrar para compensar uma dose que se esqueceu de tomar. 4.EFEITOS SECUNDÁRIOS POSSÍVEIS Como os demais medicamentos, SUTENT pode causar efeitos secundários em algumas pessoas, no entanto, estes não se manifestam em todas as pessoas. Efeitos secundários muito frequentes, que podem afectar mais de 10 em cada 100 pessoas - dor/irritação na boca (da cavidade oral), boca (cavidade oral) sensibilizada, alterações do paladar, problemas no estômago, náuseas, vómitos, diarreia, prisão de ventre, dor abdominal, perda do apetite. - descoloração cutânea, alterações na coloração do cabelo, erupção cutânea nas palmas das mãos e nas plantas dos pés, bolhas, pele seca - cansaço, pressão arterial elevada, enxaqueca - redução do número de: glóbulos vermelhos e/ou glóbulos brancos

68
Outros efeitos secundários possíveis, que podem afectar entre 1 a 10 em cada 100 pessoas - diminuição da actividade da tiróide, fluxo sanguíneo cardíaco reduzido - hemorragia nasal, cor da urina anómala, fluxo lacrimal excessivo - dores nas articulações, dores musculares, fluidos excessivos nos tecidos incluindo a área do olho - alteração da sensação da pele, falta de ar - queda de cabelo, perda de peso Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico ou farmacêutico. 5. COMO CONSERVAR SUTENT - Manter fora do alcance e da vista das crianças. - O medicamento não necessita de precauções especiais de conservação. - Não utilize após o prazo de validade impresso na embalagem exterior e no rótulo. - Não utilize nenhuma embalagem que esteja danificada ou apresente sinais de violação. Os medicamentos não devem ser eliminados na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como eliminar os medicamentos de que já não necessita. Estas medidas irão ajudar a proteger o ambiente. 6. OUTRAS INFORMAÇÕES Qual a composição de SUTENT - A substância activa é o sunitinib (sob a forma de malato). - Os outros componentes são o manitol, a croscarmelose sódica, a povidona e o estearato de magnésio. A cápsula é composta por gelatina, óxido de ferro vermelho (E172) e dióxido de titânio (E171). A tinta de impressão contém goma laca, propilenoglicol, hidróxido de sódio, povidona e dióxido de titânio. Qual o aspecto de SUTENT e conteúdo da embalagem O SUTENT é fornecido como cápsulas de gelatina com a cabeça cor de caramelo e o corpo cor de laranja, com “Pfizer” impresso com tinta branca na cabeça e “STN 25 mg” no corpo. SUTENT está disponível em embalagens de 30 cápsulas. Titular da Autorização de Introdução no Mercado Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJ United Kingdom Fabricante Pfizer Italia S.r.L Via Del Commercio – Zone Industriale 63046 Marino del Tronto (Ascoli Piceno) Italy Para quaisquer informações sobre este medicamento, queira contactar o representante local do titular da autorização de introdução no mercado.
Belgique / België /Belgien Pfizer S.A. / N.V. Tél/Tel: +32 (0)2 554 62 11
Luxembourg/Luxemburg Pfizer S.A. Tél/Tel: +32 (0)2 554 62 11

69
Česká republika Pfizer s.r.o. Tel.: +420-283-004-151
Magyarország Pfizer Kft. Tel.: +36-1-488-37-00
Danmark Pfizer ApS Tlf: +45 44 20 11 00
Malta V.J. Salomone Pharma Ltd. Tel. +356 212201
Deutschland Pfizer GmbH Tel: +49 (0)721 61 01 01
Nederland Pfizer BV Tel: +31 (0)10 406 42 00
Eesti Pfizer Luxembourg SARL, Eesti filiaal Tel.: +372 6 405 328
Norge Pfizer AS Tlf: +47 67 52 61 00
Ελλάδα Pfizer Hellas A.E. Τηλ: +30 210 7517981-3
Österreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0
España Pfizer S.A. Tél: +34 91 490 99 00
Polska Pfizer Polska Sp.zo.o Tel.:+48 22 335 61 00
France Pfizer Tél: +33 (0)1 58 07 34 40
Portugal Laboratórios Pfizer, Lda. Tel: +351 21 423 5500
Ireland Pfizer Healthcare Ireland Tel: +353 1800 633 363
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel.: + 386 1 52 11 400
Ísland Vistor hf. Sími: +354 535 7000
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel.:+ 421 2 5941 8500
Italia Pfizer Italia S.r.l. Tel: +39 06 33 18 21
Suomi/Finland Pfizer Oy Puh./Tel: +358 (0)9 43 00 40
Kύπρος Geo. Pavlides & Araouzos Ltd. Tηλ.:+ 357 22 818087
Sverige Pfizer AB Tel: +46 (0)8 550 5200
Latvija Pfizer Luxembourg SARL filiale Latvjia Tel.: + 371 70 35 775
United Kingdom Pfizer Limited, Tel: +44 (0)1737 331111
Lietuva Pfizer Luxembourg SARL filialas Lietuvoje e Tel. + 370 52 51 4000

70
Este folheto foi aprovado pela última vez em A este medicamento foi dada uma “autorização condicional”. Isto significa que se aguarda mais informação sobre este medicamento, em particular sobre o tratamento do cancro do rim. O SUTENT demonstrou diminuir o tumor. Contudo, espera-se mais informação sobre a duração deste efeito. A Agência Europeia de Medicamentos (EMEA) irá rever anualmente a nova informação sobre este medicamento e este Folheto Informativo será actualizado se necessário. Informação pormenorizada sobre este medicamento está disponível na Internet no site da Agência Europeia de Medicamentos (EMEA) http://www.emea.eu.int. Também existem links para outros sites sobre doenças raras e tratamentos.

71
FOLHETO INFORMATIVO: INFORMAÇÃO PARA O UTILIZADOR
SUTENT 50 mg cápsulas sunitinib
Leia atentamente este folheto antes de tomar o medicamento - Conserve este folheto. Pode ter necessidade de o reler. - Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico. - Este medicamento foi receitado para si. Não deve dá-lo a outros; o medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sintomas. - Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico ou farmacêutico. Neste folheto:
1. O que é SUTENT e para que é utilizado 2. Antes de tomar SUTENT 3. Como tomar SUTENT 4. Efeitos secundários possíveis 5. Como conservar SUTENT 6. Outras informações
1. O QUE É SUTENT E PARA QUE É UTILIZADO O SUTENT é um medicamento utilizado no tratamento do cancro, para prevenir a actividade de um grupo especial de proteínas que se sabe estarem envolvidas no crescimento e difusão das células cancerígenas. O SUTENT apenas lhe será prescrito por um médico com experiência no tratamento do carcinoma das células renais ou tumor do estroma gastrointestinal. O SUTENT é um medicamento utilizado no tratamento do carcinoma das células renais, uma forma de cancro dos rins que envolve alterações cancerígenas nas células do túbulo renal. O SUTENT é também utilizado no tratamento do tumor maligno do estroma gastrointestinal (GIST). O GIST é um cancro do estômago e dos intestinos. Resulta do crescimento descontrolado das células dos tecidos de suporte destes órgãos. O SUTENT inibe o crescimento destas células. Contacte o seu médico se tiver alguma questão relacionada com o modo de acção de SUTENT ou com a razão deste medicamento lhe ter sido prescrito. 2. ANTES DE TOMAR SUTENT Siga cuidadosamente todas as indicações do seu médico, mesmo que estas sejam diferentes da informação geral descrita neste Folheto. Não tome SUTENT Se tem alergia (hipersensibilidade) ao sunitinib ou a qualquer outro componente de SUTENT. Tome especial cuidado com SUTENT - Se tem ou teve problemas nos rins ou no fígado - Se tem a pressão sanguínea elevada - Se está grávida ou pensa poder estar grávida (ver descrição abaixo) - Se estiver a amamentar (ver descrição abaixo) Se algum destes se aplicar a si, informe o seu médico antes de tomar SUTENT. Tomar SUTENT com outros medicamentos

72
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos obtidos sem receita médica, incluindo medicamentos à base de plantas (quaisquer medicamentos que possam aumentar a concentração de SUTENT, como o cetoconazol, o ritonavir, o itraconazol, a eritromicina, a claritromicina; ou quaisquer medicamentos que possam diminuir a concentração de SUTENT como a dexametasona, a fenitoína, a carbamazepina, a rifampicina, o fenobarbital ou o Hypericum perfuratum, também conhecido como hipericão). Tomar SUTENT com alimentos e bebidas O SUTENT pode ser tomado com ou sem alimentos, contudo não deverá tomar SUTENT com sumo de toranja. Gravidez e aleitamento Se estiver grávida ou pensa poder estar, informe o seu médico. O SUTENT não deverá ser utilizado durante a gravidez a menos que seja estritamente necessário. O seu médico discutirá consigo o risco potencial de tomar SUTENT durante a gravidez. Aconselham-se as mulheres em idade fértil a utilizar um método contraceptivo eficaz durante o tratamento com SUTENT. Informe o seu médico se estiver a amamentar. Não amamente durante o período de tratamento com SUTENT. Condução de veículos e utilização de máquinas Tome especial cuidado na condução ou utilização de máquinas se sentir tonturas ou se se sentir invulgarmente cansado. 3. COMO TOMAR SUTENT O seu médico irá prescrever-lhe a dose mais indicada para si. Recomenda-se que o SUTENT seja tomado durante 28 dias (4 semanas), seguido de 14 dias (2 semanas) de repouso (sem medicação), administrado em ciclos de 6 semanas. O seu médico determinará quantos ciclos de tratamento serão necessários para si.
Se tomar mais SUTENT do que deveria Informe imediatamente o seu médico se tomou acidentalmente demasiadas cápsulas. Poderá necessitar de cuidados médicos. Caso se tenha esquecido de tomar SUTENT Não tome uma dose a dobrar para compensar uma dose que se esqueceu de tomar. 4. EFEITOS SECUNDÁRIOS POSSÍVEIS Como os demais medicamentos, SUTENT pode causar efeitos secundários em algumas pessoas, no entanto, estes não se manifestam em todas as pessoas. Efeitos secundários muito frequentes, que podem afectar mais de 10 em cada 100 pessoas - dor/irritação na boca (da cavidade oral), boca (cavidade oral) sensibilizada, alterações do paladar, problemas no estômago, náuseas, vómitos, diarreia, prisão de ventre, dor abdominal, perda do apetite. - descoloração cutânea, alterações na coloração do cabelo, erupção cutânea nas palmas das mãos e nas plantas dos pés, bolhas, pele seca - cansaço, pressão arterial elevada, enxaqueca - redução do número de: glóbulos vermelhos e/ou glóbulos brancos Outros efeitos secundários possíveis, que podem afectar entre 1 a 10 em cada 100 pessoas

73
- diminuição da actividade da tiróide, fluxo sanguíneo cardíaco reduzido - hemorragia nasal, cor da urina anómala, fluxo lacrimal excessivo - dores nas articulações, dores musculares, fluidos excessivos nos tecidos incluindo a área do olho - alteração da sensação da pele, falta de ar - queda de cabelo, perda de peso Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico ou farmacêutico. 5. COMO CONSERVAR SUTENT - Manter fora do alcance e da vista das crianças. - O medicamento não necessita de precauções especiais de conservação. - Não utilize após o prazo de validade impresso na embalagem exterior e no rótulo. - Não utilize nenhuma embalagem que esteja danificada ou apresente sinais de violação. Os medicamentos não devem ser eliminados na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como eliminar os medicamentos de que já não necessita. Estas medidas irão ajudar a proteger o ambiente. 6. OUTRAS INFORMAÇÕES Qual a composição de SUTENT - A substância activa é o sunitinib (sob a forma de malato). - Os outros componentes são o manitol, a croscarmelose sódica, a povidona e o estearato de magnésio. A cápsula é composta por gelatina, óxido de ferro vermelho (E172) e dióxido de titânio (E171). A tinta de impressão contém goma laca, propilenoglicol, hidróxido de sódio, povidona e dióxido de titânio. Qual o aspecto de SUTENT e conteúdo da embalagem O SUTENT é fornecido como cápsulas de gelatina com a cabeça e o corpo cor de caramelo, com “Pfizer” impresso com tinta branca na cabeça e “STN 50 mg” no corpo. SUTENT encontra-se comercializado em embalagens de 30 cápsulas. Titular da Autorização de Introdução no Mercado Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJ United Kingdom Fabricante Pfizer Italia S.r.L Via Del Commercio – Zone Industriale 63046 Marino del Tronto (Ascoli Piceno) Italy Para quaisquer informações sobre este medicamento, queira contactar o representante local do titular da autorização de introdução no mercado.
Belgique / België /Belgien Pfizer S.A. / N.V. Tél/Tel: +32 (0)2 554 62 11
Luxembourg/Luxemburg Pfizer S.A. Tél/Tel: +32 (0)2 554 62 11

74
Česká republika Pfizer s.r.o. Tel.: +420-283-004-151
Magyarország Pfizer Kft. Tel.: +36-1-488-37-00
Danmark Pfizer ApS Tlf: +45 44 20 11 00
Malta V.J. Salomone Pharma Ltd. Tel. +356 212201
Deutschland Pfizer GmbH Tel: +49 (0)721 61 01 01
Nederland Pfizer BV Tel: +31 (0)10 406 42 00
Eesti Pfizer Luxembourg SARL, Eesti filiaal Tel.: +372 6 405 328
Norge Pfizer AS Tlf: +47 67 52 61 00
Ελλάδα Pfizer Hellas A.E. Τηλ: +30 210 7517981-3
Österreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0
España Pfizer S.A. Tél: +34 91 490 99 00
Polska Pfizer Polska Sp.zo.o Tel.:+48 22 335 61 00
France Pfizer Tél: +33 (0)1 58 07 34 40
Portugal Laboratórios Pfizer, Lda. Tel: +351 21 423 5500
Ireland Pfizer Healthcare Ireland Tel: +353 1800 633 363
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, a Ljubljana Tel.: + 386 1 52 11 400
Ísland Vistor hf. Sími: +354 535 7000
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel.:+ 421 2 5941 8500
Italia Pfizer Italia S.r.l. Tel: +39 06 33 18 21
Suomi/Finland Pfizer Oy Puh./Tel: +358 (0)9 43 00 40
Kύπρος Geo. Pavlides & Araouzos Ltd. Tηλ.:+ 357 22 818087
Sverige Pfizer AB Tel: +46 (0)8 550 52000
Latvija Pfizer Luxembourg SARL filiale Latvjia Tel.: + 371 70 35 775
United Kingdom Pfizer Limited, Tel: +44 (0)1737 331111
Lietuva Pfizer Luxembourg SARL filialas Lietuvoje Tel. + 370 52 51 4000

75
Este folheto foi aprovado pela última vez em A este medicamento foi dada uma “autorização condicional”. Isto significa que se aguarda mais informação sobre este medicamento, em particular sobre o tratamento do cancro do rim. O SUTENT demonstrou diminuir o tumor. Contudo, espera-se mais informação sobre a duração deste efeito. A Agência Europeia de Medicamentos (EMEA) irá rever anualmente a nova informação sobre este medicamento e este Folheto Informativo será actualizado se necessário. Informação pormenorizada sobre este medicamento está disponível na Internet no site da Agência Europeia de Medicamentos (EMEA) http://www.emea.eu.int. Também existem links para outros sites sobre doenças raras e tratamentos.


















