
universidade federal da bahia saulo de tarso figueiredo grecco
146
UNIVERSIDADE FEDERAL DA BAHIA INSTITUTO DE QUÍMICA PROGRAMA DE PÓS-GRADUAÇÃO EM QUÍMICA SAULO DE TARSO FIGUEIREDO GRECCO “PREPARAÇÃO DE ZEÓLITAS MORDENITA COM ESTRUTURA HIERÁRQUICA DE POROS” Salvador 2013
Transcript of universidade federal da bahia saulo de tarso figueiredo grecco

UNI VERS ID ADE FEDER AL DA BAH IA
I N S T I T U T O D E Q U Í M I C A P R O G R A M A D E P Ó S - G R A D U A Ç Ã O E M Q U Í M I C A
SAULO DE TARSO FIGUEIREDO GRECCO
“PREPARAÇÃO DE ZEÓLITAS MORDENITA COM
ESTRUTURA HIERÁRQUICA DE POROS”
Salvador
2013

SAULO DE TARSO FIGUEIREDO GRECCO
“PREPARAÇÃO DE ZEÓLITAS MORDENITA COM
ESTRUTURA HIERÁRQUICA DE POROS”
Tese apresentada ao Programa de Pós-
Graduação em Química da Universidade
Federal da Bahia como parte dos requisitos
necessários à obtenção do grau de Doutor
em Química.
Orientadora: Profa. Dra. Maria do Carmo Rangel
Salvador
2013

Sistema de Bibliotecas – IQ/UFBA
Grecco, Saulo de Tarso Figueiredo
Preparação de zeólitas mordenita com estrutura hierárquica de poros. / Saulo de Tarso Figueiredo Grecco. - 2014.
144 f. : il. Orientadora: Profª. Drª. Maria do Carmo Rangel Santos Valera. Tese (doutorado) - Universidade Federal da Bahia, Instituto de Química, Salvador, 2013.
1. Catalise. 2. Catalisadores. 3. Zeolitos. I. Varela, Maria do Carmo Rangel Santos. II. Universidade Federal da Bahia. Instituto de Química. III. Título.
CDD – 660.2 CDU – 544.47


Aos meus pais, Marcus e Ana Cristina,
ao meu filho William e aos meus irmãos.

AGRADECIMENTOS
À Professora Dra. Maria do Carmo Rangel pela orientação e confiança, durante o
desenvolvimento deste trabalho e pela oportunidade de crescimento profissional.
Ao Professor Dr. Ernesto Antonio Urquieta-González pela colaboração e pelos
conselhos, principalmente, durante a etapa de preparação dos primeiros materiais com
estrutura hierárquica de poros.
Aos Professores que participaram da banca examinadora.
Ao Professor Patricio Reyes e ao Marcelo Oportus pelas medidas de acidez realizadas
na Facultad de Ciências Químicas da Universidad de Concepción.
Ao Professor Luciano Lião pelas análises de ressonância magnética nuclear de 27
Al e 29
Si realizadas no Instituto de Química da Universidade Federal de Goiás.
Ao Professor Marcus e a Jessica pelas análises de microscopia eletrônica de varredura
realizadas no Instituto de Física da Universidade Federal de Bahia.
À Professora Zênis pelas análises de espectroscopia no infravermelho com
Transformada de Fourier realizadas no Instituto de Química da Universidade Federal
de Bahia.
À Professora Heloysa e ao Raildo pelas análises de termogravimetria realizadas no
Instituto de Química da Universidade Federal de Bahia.
A todos os meus amigos do GECCAT, principalmente, Sirlene, Sarah, Hilma, Olivia,
Márcia, Karla, Hadma, Lindaura, Jessília, Ivoneide, Henrique, Tarcisio, Caio, Igor,
Peterson, André, Leonardo, Jadosn, Guillermo e tantos outros que, de contribuíram de
alguma forma para a realização deste trabalho.
Aos colegas da UFSCar, especialmente Edilene Deise, pelo apoio na preparação dos
primeiros materiais com estrutura hierárquica de poros e pelas análises no
equipamento de difração de raios X do Laboratório de Catálise da Universidade
Federal de São Carlos.
Aos meus amigos Paulo e Diego por estarem sempre dispostos a ajudar no que for
preciso.
A toda minha família pelos exemplos de vida e por tudo mais.
A minha namorada pela compreensão, paciência e amor.
Ao CNPq pela bolsa concedida.
À FINEP e ao CNPq pelo apoio financeiro.
A todos que contribuíram de alguma forma para a realização deste trabalho.

“Quando tudo nos parece dar errado, acontecem coisas boas, que não teriam
acontecido, se tudo tivesse dado certo”.
Renato Russo

RESUMO
As restrições difusionais aos reagentes, causadas pelos microporos, limitam o
uso das zeólitas no processamento de moléculas pesadas. Isto demanda o
desenvolvimento de materiais que combinem as propriedades de zeólitas com as de
materiais mesoporosos. Um número significativo de procedimentos experimentais,
pré ou pós síntese, vem sendo sugerido para a obtenção de zeólitas hierarquicamente
estruturadas. As metodologias de síntese mais bem sucedidas envolvem o uso de
agentes geradores de mesoporosidade (agentes orgânicos e nanopartículas) ou
nanomoldes (moldagem em nanoespaços), que geram sólidos com mesoporosidade
intracristalina com uma estreita distribuição de tamanho de poros; isto resulta em
sólidos contendo mesoporos, além dos microporos intrínsecos das zeólitas. Entretanto,
ainda não existem estudos sistemáticos, que permitam estabelecer o efeito das
variáveis de preparação sobre as características dos sólidos finais. A fim de superar
essa dificuldade, neste trabalho foi estudado o efeito do tempo e da temperatura de
cristalização do gel de síntese sobre as características de materiais baseados em
mordenita com estrutura hierárquica de poros. Na preparação das amostras,
adicionou-se um organossilano gerador de mesoporosidade (TPOAC, cloreto de [3-
(trimetoxissilil)propil]octadecildimetilamônio), ao gel de síntese da mordenita, que foi
cristalizado por diferentes períodos e em distintas temperaturas. Os sólidos obtidos
foram submetidos à troca iônica com cloreto de amônio e posterior calcinação, de
modo a obter a forma ácida do material. As amostras foram caracterizadas por
termogravimetria, espectroscopia no infravermelho com transformada de Fourier,
difração de raios X, análise textural por adsorção de nitrogênio, ressonância
magnética nuclear de 29
Si e de 27
Al, microscopia eletrônica de varredura e medidas de
acidez por dessorção de amônia à temperatura programada. Observou-se que a
formação da mordenita contendo mesoporos é influenciada pelo tempo e temperatura
de cristalização do gel da zeólita. O emprego de tempos relativamente curtos ou
baixas temperaturas favorece a formação de um sólido amorfo, enquanto longos
tempos ou elevadas temperaturas favorecem a formação de mesoporos intracristalinos
na mordenita. Por outro lado, tempos e temperaturas intermediárias favoreceram a
formação da mordenita com uma estrutura hierárquica de poros e mesoporos
desordenados. O aumento da cristalinidade da mordenita acarreta uma diminuição na
área e no volume de mesoporos, mas promove um acréscimo na área e no volume de
microporos. A área externa também tende a diminuir devido ao aumento do tamanho
do cristal da mordenita em função da cristalinidade. Os sólidos obtidos foram
susceptíveis à desaluminação durante a etapa de calcinação. A extensão da
desaluminação diminuiu com o aumento do tempo ou da temperatura de cristalização,
devido à inserção dos átomos de alumínio na rede da zeólita em formação. Porém, em
tempos de cristalização longos e temperaturas altas, pode ocorrer a redispersão dos
átomos de alumínio. Todos os sólidos apresentaram elevada acidez que aumentou
com a cristalinidade. Entretanto, nas amostras preparadas em tempos curtos e
temperaturas baixas, a maioria dos sítios apresentou força ácida moderada, enquanto
aquelas obtidas em tempos longos e temperaturas altas apresentaram maior
quantidade de sítios ácidos fortes.
Palavras-chave: zeólita hierarquicamente estruturada, mordenita, TPOAC,
microporos, mesoporos.

ABSTRACT
The diffusion restrictions of the reactants caused by the micropores limit the
use of zeolites for processing heavy molecules. This demands for the development of
materials that can combine the properties of zeolites and of mesoporous materials. A
significant number of experimental procedures, pre or post synthesis, has been
suggested for obtaining hierarchically structured zeolites. The most successful
synthesis involve the use of mesoporosity generating agents (nanoparticles and
organic agents) or nanotemplates (templating in nanospaces), which generate solids
with intracristaline mesoporosity with a narrow pore size distribution. This results in
solids containing mesoporous besides the intrinsic zeolite micropores. However,
there is not any systematic study which allows to state the effect of crystallization
time and temperature of the synthesis gel on the properties of the final solid. In order
to overcome this difficulty, the effect of time and temperature of the synthesis gel on
the properties of mordenite-based materials with hierarchical pore structure was
studied in this work. In the samples preparation a mesoporosity generating
organosilane (TPOAC, [3-(trimethoxysilyl) propyl] octadecyldimethylammonium
chloride) was added to the synthesis gel of mordenite, which was crystallized for
different times and temperatures. The solids were then submitted to ion exchange
with ammonium chloride and further calcination to obtain the acidic form of the
zeolite. The samples were characterized by thermogravimetry, Fourier transformed
infrared spectroscopy, X-ray diffraction, textural analysis by nitrogen adsorption, 29
Si
and 27
Al NMR, scanning electron microscopy and acidity measurements by ammonia
desorption. It was observed that the formation of mordenite containing mesoporous is
affected by the time and temperature of crystallization of the zeolite gel. The use of
relatively short times and low temperatures favors the formation of an amorphous
solid, while long times or high temperatures favor the formation of intracristaline
mesoporosity in the mordenite. On the other hand, intermediate times and
temperatures favor the formation of mordenite with hierarchical pore structure and
disordered mesopores. The increase in mordenite crystallinity leads to a decrease in
mesopore area and volume but promotes an increase in micropore area and volume.
The external area also tends to decrease due to the increased crystal size as a function
of mordenite crystallinity. The solids obtained were susceptible to dealumination
during the calcination step. The degree of dealumination decreased with the
increasing crystallization time or temperature due to the insertion of aluminum atoms
in the zeolite lattice. However, at long crystallization times and high crystallization
temperatures the redispersion of aluminum atoms can occur. All solids showed high
acidity which increased as a function of crystallinity. However, the samples prepared
at short times and low temperatures showed the majority of moderate acid sites of
medium strength, whereas those obtained at long times and high temperatures have
more strong acid sites. Thus, intermediate times and temperatures favor the formation
of solids having zeolitic characteristics and high mesoporosity.
Keywords: hierarchically structured zeolite, mordenita, TPOAC, microporous,
mesoporous.

LISTA DE FIGURAS
Figura 2.1 Esquema das reações químicas envolvidas na geração de sítios ácidos de
Brönsted e de Lewis em zeólitas.
06
Figura 2.2 Ilustração esquemática da (a) unidade de doze átomos T composta por
duas unidades 5-1 ao longo do eixo c (visão superior) e ao longo do eixo
a (visão inferior), (b) unidade de construção periódica bidimensional da
mordenita projetada ao longo do eixo b (uma unidade de doze átomos T
em negrito) e (c) unidade de construção periódica bidimensional da
mordenita projetada ao longo do eixo c.
07
Figura 2.3 Ilustração esquemática da estrutura da mordenita ao longo do plano
[001].
08
Figura 2.4 Ilustração esquemática do mecanismo de síntese das zeólitas. 10
Figura 2.5 Esquema mostrando os caminhos mecanísticos possíveis de formação da
MCM-41: (1) inicialmente, forma-se a fase cristal líquido; (2) os ânions
silicato interagem inicialmente com as micelas do surfactante.
12
Figura 2.6 Ilustração esquemática das diferentes estruturas do surfactante em uma
solução aquosa em função da concentração.
13
Figura 2.7 Ilustração da estrutura, (a) MCM-41, MCM-48 e MCM-50 e (b) SBA-
15. POP= Polímero do óxido de propileno; POE= polímero do óxido de
etileno.
14
Figura 2.8 Padrão de difração de raios X, (a) MCM-41 e (b) SBA-15. 16
Figura 2.9 Ilustração dos cristais e poros de materiais zeolíticos com estrutura
hierárquica de poros.
19
Figura 2.10 Ilustração (a) das etapas de síntese de cristais zeolíticos nanométricos
em espaço confinado em um suporte poroso e (b) do crescimento dos
cristais zeolíticos em torno das nanopartículas de carbono.
23
Figura 2.11 Ilustração esquemática do crescimento dos cristais zeolíticos em torno
dos nanotubos de carbono.
24
Figura 2.12 Ilustração esquemática do mecanismo hipotético da formação de zeólitas
mesoporosas hierárquicas através da moldagem com peneiras
moleculares mesoporosas de carbono.
26
Figura 2.13 Ilustração esquemática da síntese de zeólitas com mesoporos
intracristalinos usando polímeros silanizados como molde.
27

Figura 2.14 Ilustração esquemática da rota de síntese de cristais de zeólitas
mesoporosas usando sacarose como molde de carbono.
28
Figura 2.15 Ilustração de possíveis organizações de precursores zeolíticos com uma
fase micelar do surfactante. (a) Organização das nanopartículas do
precursor zeolítico coloidal; (b) organização de agregados amorfos
mediado pelo direcionador.
30
Figura 2.16 Ilustração esquemática da preparação de zeólitas deslaminadas. 31
Figura 2.17 (a) Ilustração do mecanismo proposto para a síntese de cristais de
aluminofosfatos hierárquicos, empregando organossilanos anfifílicos
como molde supramolecular. (b) Ilustração esquemática da estrutura
hierárquica obtida usando surfactantes bifuncionais.
32
Figura 2.18 Ilustração das etapas de síntese de nanocristais de zeólita a partir da
silanização de sementes.
35
Figura 4.1 Curvas de perda de massa e de derivada da perda de massa das amostras
obtidas durante o estudo do tempo de cristalização a 140 ºC. O primeiro
e o segundo números representam a temperatura e o tempo de
cristalização, respectivamente. A letra P indica os precursores dos
materiais hierárquicos baseados na mordenita.
44
Figura 4.2 Espectros de FTIR das amostras obtidas durante o estudo do tempo de
cristalização a 140 °C. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra P indica
os precursores dos materiais hierárquicos baseados na mordenita. As
linhas tracejadas na figura indicam a região das bandas de estiramento e
de deformação angular de grupos CH2 e CH3.
47
Figura 4.3 Espectros de FTIR das amostras na forma sódica obtidas durante o
estudo do tempo de cristalização a 140 °C, ampliados na região do
infravermelho médio de 1300 a 400 cm-1
. O primeiro e o segundo
números representam a temperatura e o tempo de cristalização,
respectivamente.
48
Figura 4.4 Espectros de FTIR das amostras na forma protônica obtidas durante o
estudo do tempo de cristalização a 140 °C, ampliados na região do
infravermelho médio de 1300 a 400 cm-1
. O primeiro e o segundo
números representam a temperatura e o tempo de cristalização,
respectivamente. A letra H indica as amostras na forma protônica.
50
Figura 4.5 Difratogramas de raios X na região de altos ângulos das amostras na
forma sódica obtidas durante o estudo do tempo de cristalização a 140
°C. O primeiro e o segundo números representam a temperatura e o
tempo de cristalização, respectivamente.
51
Figura 4.6 Difratogramas de raios X na região de altos ângulos das amostras na
forma protônica obtidas durante o estudo do tempo de cristalização a
52

140 °C. O primeiro e o segundo números representam a temperatura e o
tempo de cristalização, respectivamente. A letra H indica as amostras na
protônica.
Figura 4.7 Difratogramas de raios X na região de baixos ângulos das amostras na
forma sódica obtidas durante o estudo do tempo de cristalização a 140
°C. O primeiro e o segundo números representam a temperatura e o
tempo de cristalização, respectivamente.
53
Figura 4.8 Isotermas de adsorção (-•-) e dessorção (-o-) de nitrogênio das amostras
na forma sódica obtidas durante o estudo do tempo de cristalização a
140 °C. O primeiro e o segundo números representam a temperatura e o
tempo de cristalização, respectivamente.
55
Figura 4.9 Isotermas de adsorção (-•-) e dessorção (-o-) de nitrogênio das amostras
na forma protônica obtidas durante o estudo do tempo de cristalização a
140 °C. O primeiro e o segundo números representam a temperatura e o
tempo de cristalização, respectivamente. A letra H indica as amostras na
forma protônica.
56
Figura 4.10 Distribuição do tamanho de poro das amostras na forma sódica obtidas
durante o estudo do tempo de cristalização a 140 °C. O primeiro e o
segundo números representam a temperatura e o tempo de cristalização,
respectivamente.
57
Figura 4.11 Deslocamento químico devido à substituição de átomos de silício por
átomos de alumínio nas zeólitas.
58
Figura 4.12 Espectros de RMN de 29
Si das amostras na forma sódica obtidas durante
o estudo do tempo de cristalização a 140 °C. O primeiro e o segundo
números representam a temperatura e o tempo de cristalização,
respectivamente.
59
Figura 4.13 Espectros de RMN de 29
Si das amostras na forma sódica obtidas durante
o estudo do tempo de cristalização a 140 °C. O primeiro e o segundo
números representam a temperatura e o tempo de cristalização,
respectivamente. A letra P indica os precursores dos materiais
hierárquicos baseados na mordenita.
61
Figura 4.14 Espectros de RMN de 27
Al das amostras na forma sódica obtidas durante
o estudo do tempo de cristalização a 140 °C. O primeiro e o segundo
números representam a temperatura e o tempo de cristalização,
respectivamente. A letra P indica os precursores dos materiais
hierárquicos baseados na mordenita.
62
Figura 4.15 Micrografias eletrônicas de varredura da (a) Amostra MT-140/24, (b)
Amostra MT-140/48, (c) Amostra MT-140/96 e (d) Amostra MT-
140/192. O primeiro e o segundo números representam a temperatura e
o tempo de cristalização, respectivamente.
64

Figura 4.16 Número de moléculas de amônia dessorvidas por grama de sólido em
função da temperatura. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra H
indica as amostras na forma protônica.
66
Figura 4.17 Curvas de perda de massa e da derivada da perda de massa das amostras
obtidas durante o estudo da temperatura de cristalização. O primeiro e o
segundo números representam a temperatura e o tempo de cristalização,
respectivamente. A letra P indica os precursores dos materiais
hierárquicos baseados na mordenita.
67
Figura 4.18 Espectros de FTIR das amostras na obtidas durante o estudo da
temperatura de cristalização, ampliados na região do infravermelho
médio de 1300 a 400 cm-1
. O primeiro e o segundo números
representam a temperatura e o tempo de cristalização, respectivamente.
A letra H indica as amostras na forma protônica.
69
Figura 4.19 Difratogramas de raios X na região de altos ângulos das amostras
obtidas durante o estudo da temperatura de cristalização. O primeiro e o
segundo números representam a temperatura e o tempo de cristalização,
respectivamente. As curvas em preto representam as amostras após a
troca iônica e as curvas em vermelho representam as amostras não
trocadas.
70
Figura 4.20 Difratogramas de raios X na região de baixos ângulos das amostras na
forma sódica obtidas durante o estudo da temperatura de cristalização. O
primeiro e o segundo números representam a temperatura e o tempo de
cristalização, respectivamente.
71
Figura 4.21 Isotermas de adsorção (-•-) e dessorção (-o-) de nitrogênio das amostras
na forma sódica obtidas durante o estudo da temperatura de
cristalização. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente.
72
Figura 4.22 Distribuição do tamanho de poro das amostras na forma sódica obtidas
durante o estudo da temperatura de cristalização. O primeiro e o
segundo números representam a temperatura e o tempo de cristalização,
respectivamente.
74
Figura 4.23 Espectros de RMN de 29
Si das amostras na forma sódica obtidas durante
o estudo da temperatura de cristalização. O primeiro e o segundo
números representam a temperatura e o tempo de cristalização,
respectivamente. A letra P indica os precursores dos materiais
hierárquicos baseados na mordenita.
75
Figura 4.24 Espectros de RMN de 27
Al das amostras na forma sódica obtidas durante
o estudo da temperatura de cristalização. O primeiro e o segundo
números representam a temperatura e o tempo de cristalização,
respectivamente. A letra P indica os precursores dos materiais
hierárquicos baseados na mordenita.
77

Figura 4.25 Micrografias eletrônicas de varredura da (a) Amostra MT-120/96, (b)
Amostra MT-140/96, (c) Amostra MT-170/96 e (d) ampliação da
micrografia da Amostra MT-170/96. O primeiro e o segundo números
representam a temperatura e o tempo de cristalização, respectivamente.
78
Figura 4.26 Número de moléculas de amônia dessorvidas por grama de sólido em
função da temperatura. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra H
indica as amostras na forma protônica.
80

LISTA DE TABELAS
Tabela 3.1 Características e procedência dos reagentes empregados na preparação
dos materiais com estrutura hierárquica de poros baseados na mordenita.
36
Tabela 3.2 Nomenclatura e condições de preparação dos materiais com estrutura
hierárquica de poros baseados na mordenita. Tabela 3.2. Nomenclatura e
condições de preparação dos materiais com estrutura hierárquica de
poros baseados na mordenita. A Amostra HM-170/96 refere-se à
amostra obtida na ausência do organossilano, na forma protônica.
37
Tabela 4.1 Perda de massa total das amostras obtidas no estudo do tempo de
cristalização a 140 °C. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra P
indica os precursores dos materiais hierárquicos baseados na mordenita.
46
Tabela 4.2 Cristalinidade (C), diâmetro médio do cristal () e relação entre as
intensidades de picos (R), das amostras obtidas durante o estudo do
tempo de cristalização a 140 °C. O primeiro e o segundo números
representam a temperatura e o tempo de cristalização, respectivamente.
A letra H indica as amostras na forma protônica.
52
Tabela 4.3 Área de microporos (Sgm), área de externa (Sgext), área de mesoporos
(Sgmp), volume de microporos (Vm), volume de mesoporos (Vmp) e
diâmetro de poros (dp) das amostras em função do tempo de
cristalização. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra H
indica as amostras na forma protônica.
56
Tabela 4.4 Quantidade de alumínio tetraédrico (Altetra) e quantidade de alumínio
octaédrico (Alocta) das amostras obtidas durante o estudo do tempo de
cristalização a 140 °C. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente.
63
Tabela 4.5 Quantidade (em número de mols) de amônia dessorvida por massa (em
gramas) de sólido em função da temperatura. Os números entre
parênteses representam a quantidade de sítios expressos em
percentagem. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra H
indica as amostras na forma protônica.
66

Tabela 4.6 Perda de massa total das amostras obtidas no estudo da temperatura de
cristalização. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra P
indica os precursores dos materiais hierárquicos baseados na mordenita.
68
Tabela 4.7 Cristalinidade (C), diâmetro médio do cristal () e relação entre as
intensidades de picos (R) das amostras obtidas durante o estudo da
temperatura de cristalização. O primeiro e o segundo números
representam a temperatura e o tempo de cristalização, respectivamente.
A letra H indica as amostras na forma protônica.
70
Tabela 4.8 Área de microporos (Sgm), área de externa (Sgext), área de mesoporos
(Sgmp), volume de microporos (Vm) volume de mesoporos (Vmp) e
diâmetro de poros (dp) das amostras em função da temperatura de
cristalização. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra H
indica as amostras na forma protônica.
73
Tabela 4.9 Quantidade de alumínio tetraédrico (Altetra) e quantidade de alumínio
octaédrico (Alocta) das amostras obtidas durante o estudo da temperatura
de cristalização. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente.
76
Tabela 4.10 Quantidade (em número de mols) de amônia dessorvida por massa (em
gramas) de sólido em função da temperatura. Os números entre
parênteses representam as quantidades dessorvidas de amônia expressas
em percentagem. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra H
indica as amostras na forma protônica.
80

LISTA DE ABREVIATURAS E SIGLAS
Al-MSU-S Sílica mesoporosa hexagonal desenvolvida na Michigan State University
contendo alumínio e mesoestruturada
APTMS 3-aminopropil-trimetoxissilano
BJH Barret, Joyner e Halenda
CMK Carbon Mesoestructured from Kaist
CTAB Brometo de cetiltrimetilamônio
C (%) Cristalinidade da estrutura microporosa da mordenita
dp Diâmetro de poros obtido através do método de BJH
DRX Difração de raios X
DTA Análise térmica diferencial
DTG Termogravimetria derivada
FAU Fórmula estrutural da zeólita faujasita
FTIR Espectrofotometria no infravermelho com transformada de Fourier
HMS Sílica mesoporosa hexagonal
IBTES Isobutil-trietoxissilano
ITQ-2 Zeólita deslaminada desenvolvida no Instituto de Tecnología Química da
Universidad Politécnica de Valencia
KIT-1 Sílica mesoporosa desordenada desenvolvida no Korea Advanced
Institute of Science and Technology Number 1
LCT Liquid Crystal Templating
LTA Linde Type A
MCM-n Mobil Composition Matter de número n
MEV Microscopia eletrônica de varredura
MFI Código da International Zeolite Association para representar a
topologia MFI
MSU Sílica mesoporosa hexagonal desenvolvida na Michigan State University
M41S Família de materiais mesoporosos, que inclui a MCM-41, MCM-48 e

MCM-50
ODTMS Octadecil-trimetoxissilano
PHAPTMS Fenilaminopropil-trimetoxissilano
R Relação entre as intensidades dos picos dos planos (111) e (310)
RMN Ressonância Magnética Nuclear
SBA-15 Sílica mesoporosa hexagonal descoberta por pesquisadores da
Universidade de Santa Bárbara
Sgext Área externa obtida através do método t-plot
Sgm Área microporos obtido através do método t-plot
Sgmp Área de mesoporos obtido através do método de BJH
S+I
- Rota de síntese empregando surfactante catiônico e precursores
inorgânicos aniônicos
S-I
+ Rota de síntese empregando surfactante aniônicos e precursores
inorgânicos catiônico
S-M
+I
- Rota de síntese empregando surfactante e precursores inorgânicos
aniônicos com contra-íons catiônicos
S+X
-I
+ Rota de síntese empregando surfactante e precursores inorgânicos
catiônicos com contra-íons aniônicos
S0I
0 Rota de síntese empregando surfactante neutro e precursores
inorgânicos neutros
TEOS Tetraetilortossilicato
TG Termogravimetria
TPAOH Hidróxido de tetrapropilamônio
TPD-NH3 Dessorção de amônia à temperatura programada
TPOAC Cloreto de [3-(trimetoxissilil)propil]octadecildimetilamônio
TS-1 Titanossilicalita-1
Vm Volume de microporos obtido através do método t-plot
Vmp Volume de mesoporos obtido através do método de BJH
ZSM-5 Zeolite Socony Móbil
λ Comprimento de onda da radiação

SUMÁRIO
1. INTRODUÇÃO E OBJETIVOS...........................................................................01
1.1. Introdução......................................................................................................01
1.2. Objetivos.........................................................................................................02
1.2.1. Objetivo Geral......................................................................................02
1.2.2. Objetivos Específicos............................................................................03
2. REVISÃO BIBLIOGRÁFICA..............................................................................04
2.1. Zeólitas: Estrutura e Métodos de Preparação............................................04
2.1.1 Estrutura das Zeólitas e Propriedades Gerais....................................04
2.1.2. Estrutura da Mordenita.......................................................................06
2.1.3. Métodos de Preparação das Zeólitas..................................................08
2.2. Propriedades e Obtenção dos Materiais Mesoporosos Ordenados...........10
2.2.1. A Família M41S: Estruturas e Métodos de Preparação...................14
2.2.2. A Família SBA: Estruturas e Métodos de Preparação.....................16
2.3. Materiais Zeolíticos com Estrutura Hierárquica de Poros.......................18
2.3.1. Zeólitas com Estrutura Hierárquica de Poros Preparadas por
Moldagem Sólida............................................................................................21
2.3.2. Zeólitas com Estrutura Hierárquica de Poros Preparadas por
Moldagem Supramolecular...........................................................................28
3. PARTE EXPERIMENTAL...................................................................................36
3.1. Reagentes Utilizados......................................................................................36
3.2. Nomenclatura das Amostras........................................................................36
3.3. Preparação dos Materiais com Estrutura Hierárquica de Poros baseados
na Mordenita.........................................................................................................37
3.3.1. Preparação do Gel de Síntese da Mordenita......................................37
3.3.2. Cristalização do Gel de Síntese da Mordenita...................................38
3.3.3. Remoção do Agente Gerador de Mesoporosidade............................38
3.3.4. Troca Iônica dos Cátions Na+ por Cátions H
+..................................38
3.4. Preparação da Mordenita na Ausência do Organossilano........................39
3.5. Caracterização dos Materiais.......................................................................39

3.5.1. Termogravimetria................................................................................39
3.5.2. Espectroscopia no Infravermelho com Transformada de
Fourier.............................................................................................................40
3.5.3. Difração de Raios X..............................................................................40
3.5.4. Análise Textural a Partir de Dados de Adsorção/Dessorção de
Nitrogênio........................................................................................................41
3.5.5. Ressonância Magnética Nuclear de 29
Si e de 27
Al .............................42
3.5.6. Microscopia Eletrônica de Varredura................................................42
3.5.7. Medidas de Acidez por Dessorção de Amônia à Temperatura
Programada....................................................................................................43
4. RESULTADOS E DISCUSSÃO...........................................................................44
4.1. Estudo do Tempo de Cristalização..............................................................44
4.1.1. Termogravimetria................................................................................44
4.1.2. Espectroscopia no Infravermelho com Transformada de
Fourier.............................................................................................................46
4.1.3. Difração de Raios X..............................................................................50
4.1.4. Análise Textural a Partir de Dados de Adsorção/Dessorção de
Nitrogênio........................................................................................................54
4.1.5. Ressonância Magnética Nuclear de 29
Si e de 27
Al .............................57
4.1.6. Microscopia Eletrônica de Varredura................................................63
4.1.7. Medidas de Acidez por Dessorção de Amônia à Temperatura
Programada....................................................................................................65
4.2. Estudo da Temperatura de Cristalização...................................................66
4.2.1. Termogravimetria................................................................................66
4.2.2. Espectroscopia no Infravermelho com Transformada de
Fourier.............................................................................................................68
4.2.3. Difração de Raios X..............................................................................69
4.2.4. Análise Textural a Partir de Dados de Adsorção/Dessorção de
Nitrogênio........................................................................................................72
4.2.5. Ressonância Magnética Nuclear de 29
Si e de 27
Al .............................74
4.2.6. Microscopia Eletrônica de Varredura................................................78
4.2.7. Medidas de Acidez por Dessorção de Amônia à Temperatura
Programada....................................................................................................79

4.3. Efeito do tempo e da temperatura nas propriedades dos materiais
hierárquicos baseados em mordenita.................................................................80
5. CONCLUSÕES......................................................................................................82
6. PERSPECTIVAS....................................................................................................84
7. REFERÊNCIAS.....................................................................................................85
8. PUBLICAÇÕES NO TEMA DA TESE...............................................................97

1 – INTRODUÇÃO E OBJETIVOS 1
1. INTRODUÇÃO E OBJETIVOS
1.1. Introdução
As zeólitas, materiais cristalinos microporosos, têm se consolidado ao longo da segunda
metade do século XX, e nestas primeiras décadas do século XXI, como importantes catalisadores
em processos da indústria de refino de petróleo e da indústria química e petroquímica, assim
como no controle ambiental (DIMITROVA, 2004; ESCOBAR, 2000; FONSECA, 2010;
GRECCO, 2005; PEREIRA, 2011; RAMOS, 2005). Esse amplo espectro de aplicações está
relacionado às propriedades físicas e químicas desses materiais, que podem ser controladas
durante a sua preparação, visando a uma aplicação específica. Além de seu particular sistema de
microporos, as zeólitas apresentam propriedades específicas, que as diferenciam de outros
materiais, tais como sua acidez ou basicidade superficial e sua capacidade de troca iônica
(CORMA, 1997). Entretanto, apesar dessas propriedades desejáveis, a presença apenas de
microporos nas zeólitas impõe limitações à difusão de reagentes ou produtos volumosos. Embora
a limitação difusional seja usada, em alguns casos, para controlar beneficamente a seletividade a
um determinado produto da reação catalítica, a difusividade relativamente baixa de moléculas
volumosas, nos microporos da zeólita, limita a taxa de reação, devido ao transporte mais lento
dos reagentes e produtos, ocasionando um maior tempo de residência e, como consequência,
favorecendo a ocorrência de reações indesejáveis (MAJANO, 2005). Em processos envolvendo
hidrocarbonetos, a limitação difusional contribui para a formação do coque, que provoca a
desativação da zeólita por obstrução dos canais ou envenenamentos dos sítios ativos (GUISNET,
1989; RANGEL, 2003).
Com a expectativa de superar as limitações difusionais, no fim década de 80 se deu início
à busca por materiais contendo mesoporos, que resultou na síntese de materiais mesoporosos
ordenados no início dos anos 90 (BECK, 1992; KRESGE, 1992; TANEV, 1994; ZHAO, 1998 a;
ZHAO, 1998 b). Diversas peneiras moleculares mesoporosas com tamanho de poros ajustável
têm sido desenvolvidas, apresentando potencial de emprego em reações catalíticas. Entretanto,
comparado às zeólitas, esses materiais mesoporosos possuem acidez e estabilidade hidrotérmica

1 – INTRODUÇÃO E OBJETIVOS 2
mais baixas, o que limita as suas aplicações catalíticas. Assim, consideráveis esforços têm sido
dedicados ao desenvolvimento de materiais zeolíticos com estrutura hierárquica de poros, que
combinam as propriedades intrínsecas das zeólitas com a facilidade de difusão resultante da
geração de mesoporosidade.
Um número significativo de procedimentos experimentais, pré ou pós-síntese, vem sendo
sugerido para a obtenção de zeólitas hierarquicamente estruturadas (BORGES, 2012; CHOI,
2006 a; CORMA, 1998; JANSSEN, 2003; LIU, 2000; MADSEN, 1999; SAKTHIVEL, 2004;
SCHMIDT, 2001; TAO, 2003 a; WANG, 2006; XIAO, 2006; YANG, 2004). As metodologias de
síntese mais bem sucedidas envolvem o uso de agentes geradores de mesoporosidade (agentes
orgânicos e nanopartículas) ou nanomoldes (moldagem em nanoespaços), que geram sólidos com
mesoporosidade intracristalina com uma estreita distribuição de tamanho de poros; isto resulta
em sólidos contendo mesoporos, além dos microporos intrínsecos das zeólitas. Além disso, pode
ser gerada uma mesoporosidade intercristalina, resultante da aglomeração dos cristais de zeólita
com tamanhos nanométricos.
Nesse contexto, no presente trabalho foram preparadas zeólitas mordenita com estrutura
hierárquica de poros empregando um organossilano como agente gerador de mesoporosidade. Foi
estudado o efeito do tempo e da temperatura de cristalização sobre as características e
propriedades desse material. O efeito dessas variáveis de síntese sobre as propriedades de zeólitas
mordenita ainda não foi estudado
1.2. Objetivos
1.2.1. Objetivo Geral
Desenvolver sólidos baseados na zeólita mordenita, com estrutura hierárquica de poros,
com propriedades adequadas ao seu emprego como catalisadores no processamento de moléculas
volumosas, estabelecendo-se o papel de algumas variáveis de síntese, de modo a obter um
material otimizado.

1 – INTRODUÇÃO E OBJETIVOS 3
1.2.2. Objetivos Específicos
1.2.2.1. Estudar o efeito do tempo e da temperatura de cristalização nas características
estruturais de zeólitas mordenita com estrutura hierárquica de poros.
1.2.2.2. Avaliar a influência do tempo e da temperatura de cristalização nas propriedades
texturais e ácidas de catalisadores com estrutura hierárquica de poros baseados na mordenita.

2. REVISÃO BIBLIOGRÁFICA 4
2. REVISÃO BIBLIOGRÁFICA
2.1. Zeólitas: Estrutura e Métodos de Preparação
As zeólitas são materiais complexos, consistindo no maior grupo de silicatos com
estrutura aberta (BRECK, 1984; KLEIN, 1977). A primeira zeólita, hoje identificada como
estilbita, foi descoberta em uma mina de cobre na Suécia em 1756, pelo químico e
mineralogista Axel Crönstedt (PÉREZ-PARIENTE, 2002). Esses minerais foram
denominados zeólitas, nome de origem grega, composto pelas palavras zeo (ferver) e lithos
(pedra), devido à sua capacidade de liberar vapor d’água sob aquecimento. Até o presente
momento, foram descobertas sessenta e sete zeólitas naturais, sendo algumas delas
encontradas em abundância na natureza (IZA, 2013). Apesar disso, as zeólitas sintéticas são
mais empregadas comercialmente, devido à sua maior uniformidade em composição, sua
pureza elevada e a possibilidade de modelar as suas propriedades, de modo a otimizá-las para
aplicações industriais específicas (BRAGA, 2007).
2.1.1. Estrutura das Zeólitas e Propriedades Gerais
As zeólitas são aluminossilicatos cristalinos hidratados de estrutura aberta, geralmente
contendo metais alcalinos ou alcalinos terrosos como contra-íons. Estruturalmente, estes
materiais são formados por uma rede tridimensional de tetraedros interligados, contendo
canais e cavidades de dimensões moleculares. Os tetraedros são constituídos por unidades do
tipo [SiO4] ou [AlO4]- que se ligam entre si, através do compartilhamento de átomos de
oxigênio para formar as unidades secundárias de construção (BRAGA, 2007; BRECK, 1984).
A diversidade e a complexidade dos materiais zeolíticos se devem às diferentes maneiras,
pelas quais essas unidades secundárias de construção podem se ligar para formar uma
estrutura tridimensional (IZA, 2013).
A estrutura resultante possui cargas negativas, que são geradas durante a substituição
de átomos de silício da rede por átomos de alumínio. Estas cargas são compensadas por íons
positivos, chamados de cátions de compensação (WRIGTH, 2011), que se distribuem de
forma a minimizar a energia livre do sistema; a sua distribuição, na estrutura, depende da

2. REVISÃO BIBLIOGRÁFICA 5
temperatura do tratamento térmico, das espécies catiônicas e do grau de hidratação da zeólita
(YANG, 2003). Na estrutura das zeólitas, a quantidade de átomos de silício e de alumínio
presentes na rede pode variar em uma ampla faixa, desde a razão Si/Al unitária até um valor
tendendo ao infinito, que corresponde a materiais contendo apenas átomos silício, tais como
os polimorfos da sílica (SiO2). Cabe ressaltar que a maioria das zeólitas só pode ser obtida em
uma faixa de razão Si/Al limitada, que depende da estrutura. De acordo com a regra de
Loewenstein, a razão Si/Al não pode ser inferior a 1, uma vez que a existência de tetraedros
AlO4- adjacentes não é possível, devido à repulsão entre as cargas negativas (PAYRA, 2003).
Devido às suas características estruturais, as zeólitas possuem algumas propriedades
únicas que as tornam úteis em diversas aplicações industriais, especialmente em catálise, tais
como (CORMA, 2003; LUNA, 2001): (i) área superficial específica elevada; (ii) dimensões
moleculares dos poros, canais e cavidades, que lhes confere diferentes tipos de seletividade de
forma; (iii) capacidade de adsorção elevada; (iv) facilidade na separação de reagentes e
produtos; (v) possibilidade de modelar as propriedades eletrônicas dos sítios ativos; (vi)
possibilidade de pré-ativar as moléculas dentro dos poros, pela existência de campos elétricos
elevados e do confinamento molecular e (vii) propriedades ácidas e básicas.
A última propriedade é especialmente importante do ponto de vista de aplicação, uma
vez que a maioria das reações de hidrocarbonetos e muitas das reações dos compostos
orgânicos são catalisadas por zeólitas ácidas. Esta propriedade é gerada pela substituição dos
cátions alcalinos e alcalinos terrosos por prótons, que se ligam fracamente aos átomos de
oxigênio (ligados aos átomos de silício e alumínio), gerando grupos hidroxila ligados em
ponte (sítios ácidos de Brönsted), como mostrado na Figura 2.1 (SOUSA-AGUIAR, 2002).
Por outro lado, o aquecimento desses sólidos leva à formação de sítios ácidos de Lewis, como
consequência da saída desses grupos na forma de água (Figura 2.1) (MORENO, 2009). A
força ácida de um centro protônico, bem como a sua atividade catalítica, depende de vários
parâmetros, tais como: o ângulo da ligação Al-(OH)-Si, a proximidade entre os centros
protônicos, a velocidade de troca iônica e a interação com sítios ácidos de Lewis. Diversos
estudos (GUISNET, 2004; XU, 2007; NUR, 2011), abordando importantes processos do
refino do petróleo e da petroquímica conduzidos sobre catalisadores zeolíticos, mostraram que
os centros de acidez protônica (sítios ácidos de Brönsted) são altamente ativos nessas reações;
quando os sítios ácidos de Lewis estão presentes, eles promovem um aumento da força ácida
dos centros protônicos. Nesses casos, a atividade catalítica depende também da acessibilidade
dos reagentes ao centro protônico (GUISNET, 2004).

2. REVISÃO BIBLIOGRÁFICA 6
Figura 2.1. Esquema das reações químicas envolvidas na geração de sítios ácidos de Brönsted e de Lewis em
zeólitas (MISTRY, 2012).
2.1.2. Estrutura da Mordenita
O mineral, conhecido como mordenita, foi descoberto ao longo da costa da Baía de
Fundy no Canadá (IZA; 2013). A mordenita é uma zeólita com composição química igual a
Na8Al8Si40O96.24H2O e possui cela unitária ortorrômbica com as seguintes dimensões: a =
18,1 Å, b = 20,5 Å e c = 7,5 Å e simetria do grupo espacial Cmcm.
A estrutura do cristal da mordenita foi determinada por Meier em 1961 e refinada por
Gramlich em 1971, usando cristais naturais trocados com sódio (IZA, 2013). A estrutura
forma-se a partir de unidades de construção de doze átomos T (em que T pode ser Al ou Si)
compostas por duas unidades 5-1, como mostrado na Figura 2.2a. A unidade de construção
periódica bidimensional é obtida através da translação destas unidades de doze átomos T ao
longo do eixo c ou por uma rotação em 180º, acompanhada por um deslocamento de ½ c
(Figura 2.2b). Infinitas cadeias ao longo do eixo c são formadas (Figura 2.2c).

2. REVISÃO BIBLIOGRÁFICA 7
Figura 2.2. Ilustração esquemática da (a) unidade de doze átomos T composta por duas unidades 5-1 ao longo do
eixo c (visão superior) e ao longo do eixo a (visão inferior), (b) unidade de construção periódica bidimensional
da mordenita projetada ao longo do eixo b (uma unidade de doze átomos T em negrito) e (c) unidade de
construção periódica bidimensional da mordenita projetada ao longo do eixo c (IZA, 2013).
A mordenita é uma zeólita com estrutura de poros bidimensional e o seu sistema de
poros consiste em dois canais interconectados (IZA, 2013): um canal elíptico paralelo ao eixo
c (medindo 6,5 x 7,0 Å) e outro canal paralelo ao eixo b (medindo 2,6 x 5,7 Å), como
mostrado na Figura 2.3. Uma vez que este último é tão pequeno para ser acessado pelas
moléculas, a mordenita é considerada como uma zeólita unidimensional.
Devido à sua elevada acidez e estabilidade térmica, a mordenita tem sido empregada
na catálise de diversas reações, tais como hidrocraqueamento, hidroisomerização, alquilação e
reforma, entre outros processos (ALY, 2012). Por outro lado, esta zeólita apresenta como
desvantagem o tamanho limitado dos seus canais e a falta de interconectividade, o que impõe
limitações à difusão de reagentes ou produtos volumosos; isto limita sua atividade,
seletividade e estabilidade e, portanto, as suas aplicações catalíticas.

2. REVISÃO BIBLIOGRÁFICA 8
Figura 2.3. Ilustração esquemática da estrutura da mordenita ao longo do plano [001] (IZA, 2013).
2.1.3. Métodos de Preparação das Zeólitas
Por muitos anos as zeólitas naturais, encontradas em abundância na natureza, foram
estudadas e consideradas atrativas para uso industrial. Entretanto, a baixa pureza desses
sólidos, bem como a sua ampla variedade de composições, tornou-os inviáveis para uma
aplicação comercial em larga escala. Dessa forma, consideráveis esforços têm sido realizados
para obter esses materiais em laboratório, através de metodologias que possam produzir
materiais reprodutíveis em larga escala e com características pré-determinadas.
Os trabalhos de Richard Barrer e Robert Milton foram os pioneiros na preparação de
zeólitas sintéticas (BARRER, 1948; MILTON, 1959 a). A metodologia desenvolvida por
Richard Barrer foi baseada na conversão de fases minerais sob ação de soluções salinas
concentradas, na faixa de 180 a 270 °C. Robert Milton, por sua vez, empregou espécies mais
reativas, por exemplo, sílica gel ou silicato de sódio como fontes de silício e alumina ou
aluminato de sódio como fontes de alumínio. Também foi usado hidróxido de sódio como
fonte de sódio e meio mineralizante. Isto permitiu o emprego de condições mais brandas, tais
como temperaturas e pressões mais baixas. A partir desses trabalhos, muitos outros foram
conduzidos (MILTON, 1959 b; RABO, 2001) levando a avanços significativos, tanto em
termos da descoberta de novas estruturas zeolíticas quanto na elucidação do mecanismo da
síntese.

2. REVISÃO BIBLIOGRÁFICA 9
As zeólitas são, em geral, preparadas sob condições hidrotérmicas porque a estrutura
aberta desses aluminossilicatos deve ser estabilizada durante o seu crescimento, através da
ocupação dos canais e das cavidades formadas por moléculas hospedeiras (BARRER, 1982;
CUNDY, 2003). A síntese hidrotérmica de uma zeólita consiste na mistura das fontes
contendo silício e alumínio, geralmente em meio básico (meio mineralizante), com uma fonte
do cátion de compensação. Em seguida, a mistura reacional é aquecida em uma autoclave sob
pressão autógena (no caso de temperaturas de reação acima de 100 °C). Após o período de
indução, pode-se detectar cristais do produto zeolítico, que são recuperados por filtração,
lavagem e secagem (CUNDY, 2005).
Muitas zeólitas podem ser preparadas empregando-se apenas reagentes inorgânicos.
Entretanto, nos anos 60, introduziu-se o uso de compostos orgânicos na síntese de zeólitas,
denominados agentes direcionadores de estrutura, que levaram a uma melhoria significativa
na qualidade dos produtos. Os agentes direcionadores de estrutura mais utilizados são os sais
de amônio quaternário. Devido a restrições estéricas, que limitam a quantidade de espécies
orgânicas envolvidas na formação da estrutura, e devido à função de cátion compensador do
íon amônio quaternário, são impostas restrições na densidade da carga, o que resulta na
produção de estruturas com razões Si/Al elevadas (CUNDY, 2003). A primeira estrutura
obtida utilizando um agente orgânico foi a zeólita beta, cuja síntese foi desenvolvida em 1967
por pesquisadores da Mobil (WADLINGER, 1967). Um dos prováveis mecanismos da síntese
hidrotérmica, empregando direcionadores de estrutura, é apresentado na Figura 2.4 (DAVIS,
1996). O mecanismo consiste na formação de um compósito inorgânico-orgânico, no meio
reacional, através da substituição parcial ou completa da esfera de hidratação do íon amônio
por espécies de silicato. As interações de van der Waals, entre os íons amônio e as espécies de
silicato, fornecem a entalpia requerida para a formação da estrutura zeolítica, enquanto a
liberação das moléculas de água fornece uma entropia adicional para compensar o processo de
auto-organização. A agregação destes compósitos inorgânicos-orgânicos é responsável pela
etapa de nucleação.
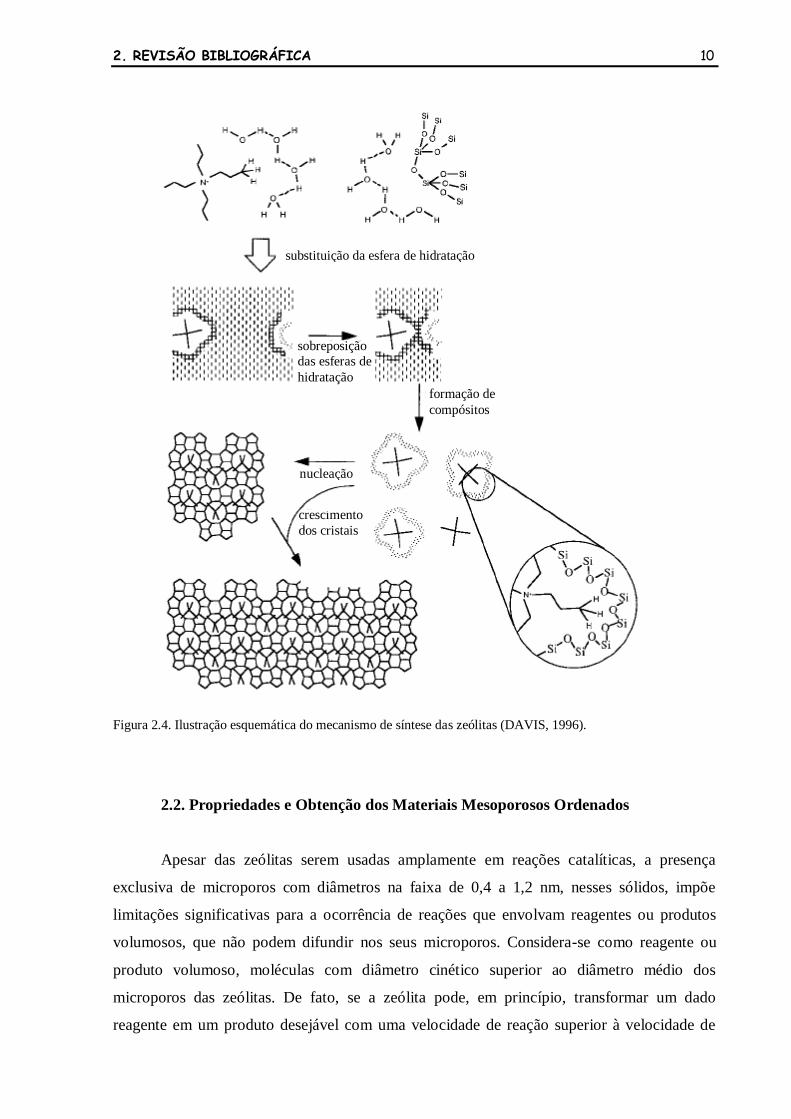
2. REVISÃO BIBLIOGRÁFICA 10
substituição da esfera de hidratação
sobreposição
das esferas de
hidratação
formação de
compósitos
crescimento
dos cristais
nucleação
Figura 2.4. Ilustração esquemática do mecanismo de síntese das zeólitas (DAVIS, 1996).
2.2. Propriedades e Obtenção dos Materiais Mesoporosos Ordenados
Apesar das zeólitas serem usadas amplamente em reações catalíticas, a presença
exclusiva de microporos com diâmetros na faixa de 0,4 a 1,2 nm, nesses sólidos, impõe
limitações significativas para a ocorrência de reações que envolvam reagentes ou produtos
volumosos, que não podem difundir nos seus microporos. Considera-se como reagente ou
produto volumoso, moléculas com diâmetro cinético superior ao diâmetro médio dos
microporos das zeólitas. De fato, se a zeólita pode, em princípio, transformar um dado
reagente em um produto desejável com uma velocidade de reação superior à velocidade de

2. REVISÃO BIBLIOGRÁFICA 11
difusão dos reagentes e produtos nos seus poros, a velocidade da reação global será limitada
pela taxa de difusão. Portanto, a reação ocorrerá sob regime de controle de difusão. Devido a
essa limitação, consideráveis esforços vêm sendo realizados na tentativa de desenvolver
materiais com diâmetros de poros na região dos mesoporos (TRONG ON, 2003).
Desde a descoberta da família de peneiras moleculares mesoporosas denominada
M41S, em 1992, por pesquisadores da Mobil Corporation (BECK, 1992; KRESGE, 1992),
diversos materiais mesoporosos têm sido preparados, usando o mesmo mecanismo de
modelagem via a formação de um cristal líquido, do inglês Liquid Crystal Templating (LCT).
No processo LCT, as espécies orgânicas atuam como estrutura central, em torno da qual as
espécies óxido são organizadas (VARTULI, 1998). O processo de formação da estrutura
mesoporosa tem sido explicado através de dois mecanismos (BECK, 1992), como mostrado
na Figura 2.5. O primeiro deles reflete o significado literal do mecanismo LCT, isto é,
envolve a pré-existência de agregados do surfactante (fase cristal líquido) na solução
precursora. A subsequente formação da estrutura mesoporosa ocorre através da migração e
polimerização das espécies de silicato na fase aquosa. No entanto, essa ideia parece ter apenas
um significado conceitual, uma vez que a concentração do surfactante está abaixo da
necessária para formar cristais líquidos; de fato, nenhuma fase cristal líquido foi observada
em estudos de RMN e espalhamento de nêutrons a baixos ângulos (VARTULI, 1998). O
segundo mecanismo considera a auto-organização das espécies de silicato, através da
interação mútua entre eles e as espécies surfactantes (mecanismo cooperativo) (BECK, 1992).
Entretanto, esses dois modelos são insuficientes para o entendimento mecanístico de
formação da estrutura mesoporosa (VARTULI, 1998).
Posteriormente a Beck (1992), Monnier e colaboradores (1993) propuseram um
modelo baseado no mecanismo cooperativo, que explica detalhadamente a formação da
mesofase. De acordo com esse modelo, a formação da estrutura mesoporosa ocorre através de
três etapas: (i) interação entre as espécies silicato e as espécies surfactantes na fase micelar;
(ii) polimerização preferencial de espécies silicato na região interfacial silicato/surfactante em
detrimento da polimerização em solução e (iii) equilíbrio entre as densidades de carga do
silicato e do surfactante. A compensação da carga superficial das micelas por anions silicato é
o parâmetro mais importante na formação das diferentes fases mesoporosas, uma vez que
variando algumas condições de síntese, tais como pH, temperatura, etc, pode-se afetar o grau
de polimerização e, consequentemente, a densidade de carga do silicato (REY, 2002). Em
presença de espécies silicato muito despolimerizadas, há a tendência de formação de
estruturas lamelares ou cúbicas, que possuem um menor raio de curvatura e um fator de

2. REVISÃO BIBLIOGRÁFICA 12
Figura 2.5. Esquema mostrando os caminhos mecanísticos possíveis de formação da MCM-41: (1) inicialmente,
forma-se a fase cristal líquido; (2) os ânions silicato interagem inicialmente com as micelas do surfactante
(BECK, 1992).
empacotamento mais elevado, resultando em uma maior densidade de carga positiva. Desta
forma, estes arranjos são capazes de compensar a elevada densidade de carga negativa das
espécies de silicato mais despolimerizadas. Por outro lado, as espécies mais polimerizadas
tendem a formar estruturas hexagonais com menor densidade de carga.
A natureza da mesofase obtida também depende da concentração do surfactante
(VARTULI, 1998; ZHAO, 1996), como mostrado na Figura 2.6. Em concentrações mais
baixas, as moléculas de surfactante se encontram isoladas enquanto que, aumentando-se a
concentração, as moléculas do surfactante se agregam formando micelas. Se a concentração
continua aumentando, são formados arranjos hexagonais empacotados dando origem à fase
hexagonal. A próxima etapa, nesse processo de formação micelar, é a coalescência dos
cilindros adjacentes para formar a fase lamelar. Em alguns casos, forma-se previamente a fase
cúbica. A natureza da fase presente na solução aquosa do surfactante depende, não apenas da
concentração, mas também da natureza do surfactante, bem como dos parâmetros de síntese
(ZHAO, 1996).
Arranjo Hexagonal
MCM-41
Cal
cinaç
ão
Silicato
Micela do
Surfactante
Cilindro Micelar
Surfactante

2. REVISÃO BIBLIOGRÁFICA 13
Figura 2.6. Ilustração esquemática das diferentes estruturas do surfactante em uma solução aquosa em função da
concentração (BALMBRA, 1969).
Existem três tipos principais de peneiras moleculares mesoporosas, que são obtidos
através de diferentes rotas de formação da mesofase. O primeiro tipo é a família M41S de
silicatos ou aluminossilicatos mesoporosos, que inclui a fase hexagonal (MCM-41), a fase
cúbica (MCM-48) e a fase lamelar (MCM-50) (VARTULI, 1998; ZHAO, 1996), que são
mostradas na Figura 2.7a. A preparação destes materiais envolve o uso de surfactantes
iônicos, como um molde (template) e um mecanismo de organização iônica, representado por
S+I
-. De acordo com este mecanismo, ocorre a formação de um par iônico entre as espécies
inorgânicas aniônicas e o surfactante catiônico. Este método foi estendido a uma série de
mecanismos de organização eletrostática, incluindo a rota reversa S-I
+, em que as espécies
inorgânicas são catiônicas e as espécies surfactantes são aniônicas. Outro mecanismo refere-
se às rotas auxiliadas por contra-íons, representadas por S+X
-I
+ e S
-M
+I
-, em que as espécies
inorgânicas interagem com as espécies do surfactante por intermédio de um contra-íon (HUO,
1996). O segundo tipo de peneiras moleculares mesoporosas envolve uma interação entre
espécies surfactantes e espécies inorgânicas neutras (S0I
0) (TANEV, 1994). Entretanto, as
sílicas mesoporosas hexagonais (HMS e MSU), obtidas através desta rota, são menos
ordenadas do que as peneiras moleculares mesoporosas preparadas via o mecanismo iônico
(TRONG ON, 2003). O último tipo de peneiras moleculares mesoporosas, que inclui a SBA-
15 (Figura 2.7b), é obtido através de uma nova rota de síntese, empregando copolímeros di e
triblocos anfifílicos, como moldes (templates) (ZHAO, 1998 a; ZHAO, 1998 b).
Cristais em solução
Cúbica
Lamelar
Solução
ideal
Hexagonal
cmc2
cmc1
Percentagem em massa de CTAB (%)
Tem
per
atura
(ºC
)
Fase micelar

2. REVISÃO BIBLIOGRÁFICA 14
Figura 2.7. Ilustração da estrutura, (a) MCM-41, MCM-48 e MCM-50 (VARTULI, 1998) e (b) SBA-15. POP= Polímero do óxido de propileno; POE= polímero do óxido de etileno (MEYNEN, 2009).
A principal diferença entre os materiais mesoporosos e as zeólitas consiste no
ordenamento da estrutura resultante. Nas estruturas zeolíticas, os tetraedros TO4 (T = Si, Al)
estão espacialmente ordenados e, portanto, é possível criar uma estrutura cristalina a partir da
repetição no espaço de uma unidade elementar, denominada célula unitária. Nos materiais
mesoporosos, não é possível definir uma célula unitária e nem as posições cristalográficas. A
única organização é a geometria tetraédrica dos átomos T. A partir da unidade TO4 não existe
um arranjo definido de átomos. Esses materiais, portanto, possuem paredes amorfas (BECK,
1992). Desta forma, os materiais mesoporosos apresentam apenas um ordenamento dos
mesoporos de longo alcance, enquanto as zeólitas possuem uma organização dos átomos em
longo alcance, que é característica de materiais cristalinos.
2.2.1. A Família M41S: Estruturas e Métodos de Preparação
A família M41S inclui as estruturas MCM-41, MCM-48 e MCM-50 (Figura 2.7a). A
estrutura MCM-41 é formada por um arranjo hexagonal de mesoporos uniformes e
unidimensionais, enquanto a MCM-48 possui um arranjo cúbico de mesoporos
interconectados, resultando em um sistema de poros tridimensional. Por outro lado, a
estrutura MCM-50 possui um arranjo lamelar constituído por uma camada dupla do
(b)
MCM-50
MCM-41 MCM-48
(a)
microporos
Calcinação
POE
POP
mesoporos
(4-14 nm)

2. REVISÃO BIBLIOGRÁFICA 15
surfactante alternada por camadas de sílica; após a remoção do surfactante, se formará um
sistema de poros bidimensional, cuidando-se para que a estrutura seja devidamente
estabilizada, por exemplo, via o processo de pilarização.
Entre as estruturas da família M41S, aquela da MCM-41 é a que tem sido mais
investigada, uma vez que as demais estruturas são instáveis termicamente ou difíceis de
sintetizar (MEYNEN, 2009; SAYARI, 1996). Este material pode apresentar área superficial
específica superior a 1000 m2.g
-1 e diâmetro de mesoporos na faixa de 1,5 a 10 nm
(MEYNEN, 2009; ZHAO, 1996), que podem ser controlados através da escolha cuidadosa do
molde (template), da adição de compostos orgânicos auxiliares ou modificando-se os
parâmetros de síntese (VARTULI, 1998). Além disso, seus mesoporos são uniformes, o que
resulta em uma distribuição estreita de diâmetros (MEYNEN, 2009). Entretanto, as paredes
dos mesoporos são finas, com uma espessura que varia de 1 a 1,5 nm, o que impõe limitações
na estabilidade térmica e hidrotérmica do material.
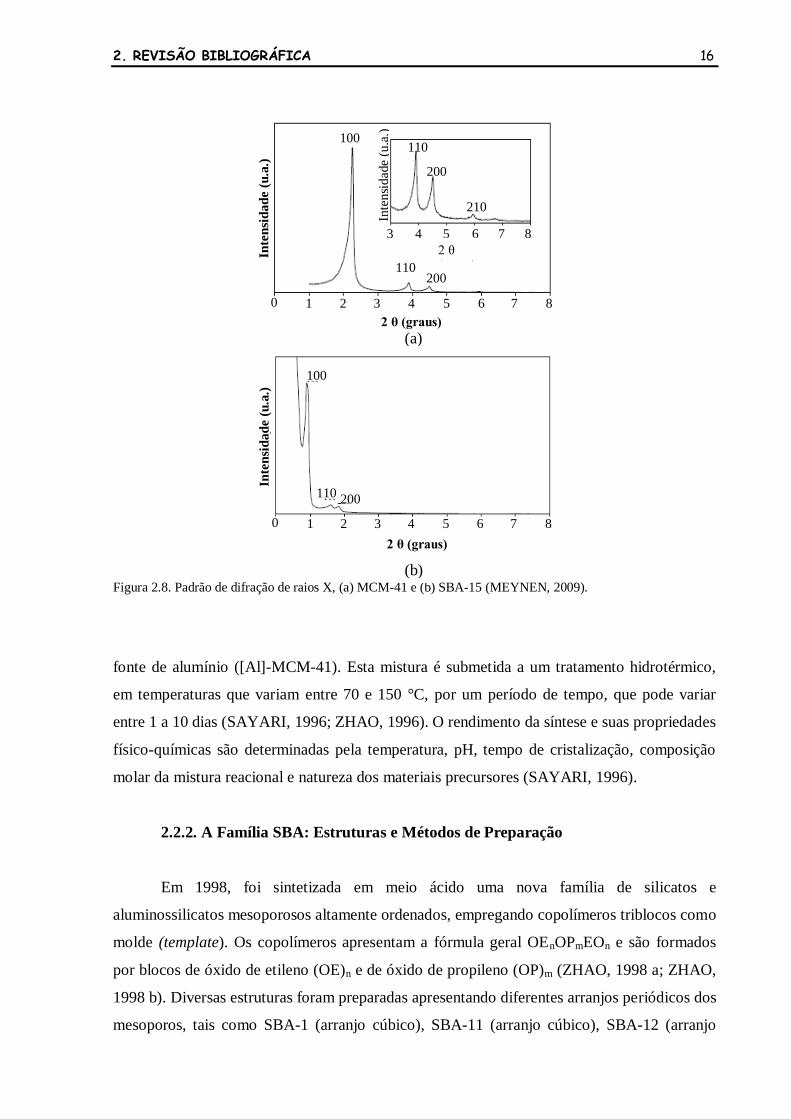
O padrão de difração da MCM-41 (Figura 2.8a) apresenta apenas reflexões na região
de baixos ângulos e, em casos de baixo ordenamento, observa-se apenas um pico de difração
em 2 igual a 2° (ZHAO, 1996). Diferentemente das zeólitas, estas reflexões são provenientes
do ordenamento dos mesoporos, que resulta em uma organização de longo alcance nestes
materiais amorfos.
A MCM-41 apresenta um grande potencial de aplicação como catalisador ou suporte
catalítico, especialmente em reações de conversão de moléculas volumosas, devido às suas
características, principalmente a área superficial específica elevada e a estrutura de poros com
canais de tamanho e forma definidos. Entretanto, os materiais mesoporosos contendo apenas
silício apresentam acidez baixa, atribuída aos grupos silanóis, presentes na superfície
(TRONG ON, 2003).
Como nas zeólitas, a substituição de átomos de silício por átomos de alumínio na
estrutura da MCM-41 cria cargas negativas que, ao serem compensadas por prótons, geram os
sítios ácidos de Brönsted. Comparada às zeólitas, a MCM-41 contendo alumínio exibe uma
forte tendência à desaluminação durante a remoção do surfactante. Este processo ocorre,
principalmente, devido à hidrólise do alumínio da rede pelo vapor d’água gerado durante a
combustão do surfactante (ZHAO, 1996). Além disso, independentemente da quantidade de
alumínio existente na estrutura, os materiais obtidos apresentam acidez de Brönsted mais
baixa quando comparado às zeólitas (CORMA, 1994).
Assim como as zeólitas, a MCM-41 é obtida a partir de uma mistura reacional
contendo o surfactante e uma fonte de silício ([Si]-MCM-41), podendo também conter uma
2. REVISÃO BIBLIOGRÁFICA 16
Figura 2.8. Padrão de difração de raios X, (a) MCM-41 e (b) SBA-15 (MEYNEN, 2009).
fonte de alumínio ([Al]-MCM-41). Esta mistura é submetida a um tratamento hidrotérmico,
em temperaturas que variam entre 70 e 150 °C, por um período de tempo, que pode variar
entre 1 a 10 dias (SAYARI, 1996; ZHAO, 1996). O rendimento da síntese e suas propriedades
físico-químicas são determinadas pela temperatura, pH, tempo de cristalização, composição
molar da mistura reacional e natureza dos materiais precursores (SAYARI, 1996).
2.2.2. A Família SBA: Estruturas e Métodos de Preparação
Em 1998, foi sintetizada em meio ácido uma nova família de silicatos e
aluminossilicatos mesoporosos altamente ordenados, empregando copolímeros triblocos como
molde (template). Os copolímeros apresentam a fórmula geral OEnOPmEOn e são formados
por blocos de óxido de etileno (OE)n e de óxido de propileno (OP)m (ZHAO, 1998 a; ZHAO,
1998 b). Diversas estruturas foram preparadas apresentando diferentes arranjos periódicos dos
mesoporos, tais como SBA-1 (arranjo cúbico), SBA-11 (arranjo cúbico), SBA-12 (arranjo
0 1 2 3 4 5 6 7 8
100
110 200
2 θ (graus)
Inte
nsi
dad
e (
u.a
.)
(a)
In
ten
sid
ad
e (
u.a
.)
110
200
100
110
200
210
3 4 5 6 7 8
2 θ
(graus)
Inte
nsi
dad
e (u
.a.)
0 1 2 3 4 5 6 7 8
2 θ (graus)
Inte
nsi
dad
e (
u.a
.)
(b)

2. REVISÃO BIBLIOGRÁFICA 17
hexagonal com sistema de poros tridimensional), SBA-14 (arranjo lamelar), SBA-15 (arranjo
hexagonal com sistema de poros bidimensional) e SBA-16 (arranjo cúbico centrado em uma
cavidade mesoporosa) (MEYNEN, 2009). Entre elas, a SBA-15 é a estrutura que tem atraído
mais atenção, devido às suas propriedades, tais com estabilidade térmica e hidrotérmica mais
elevadas quando comparada aos outros materiais mesoporosos (MEYNEN, 2009).
A estrutura SBA-15 é formada por um arranjo hexagonal de mesoporos similar à
estrutura MCM-41, porém com espessuras da parede dos mesoporos superiores (entre 3 e 6
nm) e diâmetros de poros na faixa de 4 a 14 nm (MEYNEN, 2009). Devido à maior espessura
da parede, a SBA-15 possui uma estabilidade térmica e hidrotérmica maior do que a MCM-
41, o que aumenta o seu potencial de aplicação em processos catalíticos industriais. Essas
paredes espessas são formadas por uma matriz de sílica microporosa. Assim, a SBA-15 é um
material composto por microporos e mesoporos, como mostrado na Figura 2.7b. Essa
estrutura apresenta um arranjo mesoporoso altamente ordenado e o diâmetro dos microporos
pode variar entre 0,5 e 3 nm, dependendo das condições empregadas na síntese (MEYNEN,
2009).
Os copolímeros di e triblocos proporcionam a formação da estrutura da SBA-15, uma
vez que as cadeias do polímero de óxido de etileno são hidrofílicas, enquanto as cadeias do
polímero de óxido de propileno são hidrofóbicas; isto conduz à formação de micelas
cilíndricas, com as cadeias do polímero de óxido de etileno localizadas no lado externo das
micelas. Desta forma, as cadeias do polímero de óxido de etileno podem ser ocluídas nas
paredes do silicato, como mostrado na Figura 2.7b. Após a calcinação, a estrutura resultante
possui mesoporos e microporos, formados durante a remoção do polímero de óxido de etileno
e do polímero de óxido de propileno, respectivamente. Entretanto, Garlaneau e colaboradores
(2001) observaram que a formação de uma estrutura de poros tridimensional, na qual os
mesoporos estão interconectados através dos microporos, só é possível quando se utiliza
temperaturas inferiores a 130 °C. Acima desta temperatura, o óxido de etileno não permanece
ocluído nas paredes do silicato.
A SBA-15 apresenta apenas reflexões na região de baixos ângulos no seu padrão de
difração de raios X (Figura 2.8b), o que evidencia apenas uma organização de longo alcance
dos mesoporos. Nenhuma reflexão pode ser observada na região de altos ângulos, devido à
natureza amorfa das paredes dos poros. Pelo fato da SBA-15 ser obtida em meio fortemente
ácido, a incorporação de alumínio nas suas paredes é dificultada, o que prejudica a
possibilidade de se introduzir prótons como cátions de compensação, inviabilizando a sua
aplicação como catalisador ácido. As dificuldades de incorporação de alumínio nestes

2. REVISÃO BIBLIOGRÁFICA 18
materiais se devem à fácil dissociação da ligação Al-O-Si sob condições hidrotérmicas ácidas
e à diferença entre as taxas de hidrólise dos precursores de silício e de alumínio. Várias
estratégias foram empregadas para solucionar estes problemas, tais como a pré-hidrólise dos
alcoxissilanos antes da adição do alcóxido de alumínio (YOLDAS, 1998) ou a diminuição da
taxa de hidrólise dos precursores de alumínio por complexação com agentes quelantes, tais
como o acetoacetato de etila (PIERRE, 1998). O problema também pode ser resolvido
adicionando alguns catalisadores, tais como aqueles baseados em fluoreto, para acelerar as
taxas de hidrólise do precursor de silício (LI, 2004; ZHANG, 2002). Apesar destes esforços,
estes materiais também possuem acidez mais baixa que as zeólitas.
Na primeira vez em que a SBA-15 foi sintetizada, Zhao e colaboradores (1998)
empregaram um copolímero tribloco, conhecido como Pluronic P123, que apresenta a fórmula
geral OE20OP70OE20. Foram empregadas baixas concentrações do Pluronic P123 e meio ácido
(pH<1), variando-se a concentração do copolímero e a temperatura de mesoestruturação.
Observou-se que, usando concentrações do copolímero superiores a 6 % ou inferiores a 0,5 %,
formava-se apenas um silicato não ordenado. Por outro lado, em temperaturas inferiores a 35
°C e superiores a 80 °C era produzido, também, um silicato pouco ordenado. A partir desses
resultados, concluiu-se que as condições mais adequadas para a formação de uma estrutura
mesoporosa ordenada eram concentrações do copolímero na faixa de 0,5 a 6 % e temperatura
de mesoestruturação entre 35 e 80 °C.
Outra importante variável estudada, na formação de sólidos do tipo SBA, foi a relação
entre o número de unidades do óxido de etileno e do óxido de propileno, que controla o tipo
da estrutura mesoporosa que será obtida (fase lamelar, cúbica, hexagonal e outras). Quando
são empregadas relações óxido de etileno/óxido de propileno mais baixas, a formação da fase
hexagonal é favorecida (IMPÉROR-CLERC, 2000; KIPKEMBOI, 2001). Por outro lado,
alterando o comprimento da cadeia do polímero de óxido de propileno, pode-se controlar o
diâmetro dos mesoporos (KIPKEMBOI, 2001). O diâmetro dos mesoporos também pode ser
controlado através da adição de aditivos, tais como co-surfactantes, agentes expansivos,
eletrólitos e sais (MEYNEN, 2009).
2.3. Materiais Zeolíticos com Estrutura Hierárquica de Poros
Comparados às zeólitas, os aluminossilicatos mesoporosos apresentam acidez e
estabilidade hidrotérmica e mecânica mais baixas, o que limita as suas aplicações industriais,
especialmente em catálise (TRONG ON, 2003).

2. REVISÃO BIBLIOGRÁFICA 19
Uma vez que a estabilidade térmica, hidrotérmica e mecânica, assim como a acidez
elevada, são parâmetros fundamentais para as aplicações catalíticas industriais, muitos
esforços têm sido realizados com o intuito de obter novos materiais que combinem as
vantagens das zeólitas e dos materiais mesoporosos, principalmente em relação à elevada
acidez e mesoporosidade. Dessa forma, diversas metodologias de síntese têm sido
desenvolvidas para aumentar a acessibilidade aos sítios ácidos das zeólitas. As estratégias
envolvem a geração de mesoporos dentro do cristal da zeólita ou a síntese de nanocristais. Os
materiais resultantes, que constituem as zeólitas com estrutura hierárquica de poros, foram
classificados em três tipos diferentes (EGEBLAD, 2008; GRECCO, 2013) como mostrado na
Figura 2.9, que podem ser denominados cristais hierárquicos, cristais nanométricos e cristais
suportados de zeólitas.
Figura 2.9. Ilustração dos cristais e poros de materiais zeolíticos com estrutura hierárquica de poros (GRECCO,
2013).
O primeiro tipo de material, os cristais hierárquicos de zeólita, possui um sistema de
mesoporos intracristalinos e um sistema de macroporos intercristalinos (porosidade gerada
Mesoporos
Intracristalino
Macroporos
Intercristalino
Microporos
da Zeólita
Cristais Nanométricos de Zeólitas
Microporos da
Zeólita Meso-macroporos
Intercristalino
Cristais Suportados de Zeólitas
Meso-macroporos
Intercristalino Microporos da Zeólita
Suporte
Cristais Hierárquicos de Zeólita

2. REVISÃO BIBLIOGRÁFICA 20
pela aglomeração de cristais de zeólita), além da microporosidade intrínseca das zeólitas. O
segundo tipo, os cristais nanométricos de zeólita, por definição, inclui todos os cristais de
zeólitas com tamanhos de até 1000 nm (TOSHEVA, 2005). Entretanto, na maioria dos
trabalhos, têm sido obtidos cristais de zeólitas com tamanhos inferiores a 100 nm, que são
muito menores do que aqueles apresentados pelas zeólitas convencionais. Esses materiais
apresentam um sistema de microporos bem definido e, adicionalmente, um sistema de
mesoporos intercristalinos como resultado do empacotamento dos cristais nanométricos. No
último tipo de materiais, os cristais suportados de zeólita, os cristais da zeólita estão dispersos
ou suportados no sistema de poros de outro material. Neste caso, o sólido resultante não é
uma zeólita pura, mas um sólido constituído pelo sistema de microporos da zeólita e um
sistema de mesoporos e macroporos intercristalinos, cujo diâmetro de mesoporos é,
principalmente, determinado pelo suporte (material não zeolítico). Esses sólidos têm
apresentado uma acidez relativamente mais baixa quando comparada com a zeólita pura, o
que limita sua aplicação como catalisador em processos da indústria de refino de petróleo,
petroquímica e outras potenciais aplicações na indústria química.
Estes sólidos podem ser obtidos através de diferentes métodos de síntese, usando a
técnica de nanomoldagem ou não; alguns deles podem ser usados para produzir diferentes
tipos de materiais. Geralmente, os métodos que empregam a técnica de nanomoldagem
tornam possível ajustar o tamanho dos poros através do uso de um molde de mesoporos com
um tamanho característico que, ao ser removido, permite a formação de poros com o mesmo
tamanho e forma do molde. Estes métodos podem ser classificados como moldagem sólida,
moldagem supramolecular e moldagem indireta (EGEBLAD, 2008).
No método de moldagem sólida, a cristalização controlada da fase zeolítica ocorre em
presença de um material sólido, que é posteriormente removido para gerar a porosidade.
Portanto, o sólido atua como um molde dos mesoporos e pode ser removido por combustão ou
por dissolução. Por outro lado, na moldagem supramolecular, emprega-se um agregado
supramolecular de surfactantes como molde dos mesoporos, que pode ser removido por
combustão ou por extração com solventes. No caso da moldagem indireta, ocorre a
transformação parcial de um material mesoporoso não zeolítico, previamente sintetizado em
uma estrutura zeolítica hierárquica mesoporosa. Uma vez que o molde não está presente
quando a zeólita cristaliza, o efeito da moldagem é considerado como indireto. A deposição
controlada de uma zeólita, no sistema de poros de um material mesoporoso, também pode ser
considerada como um método de moldagem indireta. A maioria das abordagens está
relacionada com uma cristalização (secundária) parcial dos materiais mesoporosos ordenados

2. REVISÃO BIBLIOGRÁFICA 21
em uma estrutura zeolítica (CAMPOS, 2004; CAMPOS, 2005; EGEBLAD, 2008).
Geralmente, os materiais obtidos através da moldagem indireta podem ser classificados como
cristais suportados de zeólitas e, portanto, não são objeto desta revisão.
As zeólitas mesoporosas hierárquicas também podem ser obtidas na ausência de um
molde, através da desmetalização ou de uma cristalização controlada. Na desmetalização,
geralmente, a zeólita é submetida a um tratamento pós-síntese para a remoção de um elemento
metálico específico da estrutura. Quando uma zeólita é submetida a um tratamento térmico
em presença de vapor d’água ou a um lixiviamento ácido, ocorre a remoção preferencial de
átomos de alumínio da rede (desaluminização). Embora os tratamentos térmicos sem vapor
d´água também possam criar defeitos na estrutura de zeólita, o uso do vapor d´água aumenta
muito a mobilidade das espécies de alumínio e silício. Entretanto, esse procedimento
apresenta alguns problemas, tal como o fato de que a remoção seletiva do alumínio da rede
altera a razão Si/Al e, consequentemente, modifica as propriedades ácidas e de troca iônica da
zeólita. Além disso, nas estruturas zeolíticas ricas em silício, o baixo conteúdo de alumínio
limita a geração de mesoporos, uma vez que são criados poucos sítios de defeitos. Desta
forma, a extração seletiva do silício da rede através de um tratamento alcalino (dessilicação)
constitui um método mais conveniente para gerar mesoporos nas zeólitas ricas em silício, tais
como ZSM-5 e zeólita beta. Porém, a extração do metal, independente do método, conduz à
formação de mesoporos intracristalinos não uniformes e com uma larga distribuição de
diâmetros.
Os cristais nanométricos zeolíticos também podem ser obtidos na ausência de moldes,
através do controle das condições de cristalização da zeólita. Entretanto, os materiais obtidos
apresentam apenas mesoporos interparticulares (TOSHEVA, 2005).
2.3.1. Zeólitas com Estrutura Hierárquica de Poros Preparadas por Moldagem
Sólida
Diversos materiais sólidos podem ser empregados como molde de mesoporos, o que
torna este método muito versátil na síntese dos diferentes tipos de materiais zeolíticos
mesoporosos hierárquicos. Entre eles, podem ser destacados os seguintes materiais:
nanopartículas de carbono (MADSEN, 1999), nanotubos de carbono (BORGES, 2012;
SCHMIDT, 2001), nanofibras de carbono (JANSSEN, 2003), aerogéis de carbono (TAO,
2003 a), peneiras moleculares de carbono (SAKTHIVEL, 2004; YANG, 2004), polímeros
catiônicos (XIAO, 2006 a) e polímeros silanizados (WANG, 2006).

2. REVISÃO BIBLIOGRÁFICA 22
A primeira metodologia para a obtenção desses materiais foi desenvolvida em 1999
por pesquisadores da Haldor Topsøe (MADSEN, 1999) e consistiu na preparação de cristais
nanométricos de ZSM-5, através da cristalização da zeólita dentro do sistema de poros de uma
matriz de carbono, como mostrado na Figura 2.10a. O procedimento consistiu na impregnação
úmida incipiente de um carvão ativo, com uma solução contendo hidróxido de
tetrapropilamônio (TPAOH), etanol, isopropóxido de alumínio e água destilada. Após a
evaporação do etanol à temperatura ambiente, o carvão foi impregnado com
tetraetilortossilicato (TEOS) e, posteriormente, submetido a um tratamento hidrotérmico com
vapor d´água saturado, a 180 °C. A matriz de carbono foi removida por combustão a 550 °C,
por 6 h, obtendo-se cristais de ZSM-5 com diâmetros médios na faixa de 20 a 75 nm. Uma
vez que esses cristais crescem dentro da estrutura porosa das partículas de carbono, a
distribuição de tamanho do cristal é governada pelo tamanho dos poros do carvão. Desta
forma, é possível controlar o tamanho dos cristais da zeólita pela escolha adequada de um
molde de carbono. Diferentes zeólitas têm sido preparadas em escala nanométrica, a partir da
síntese em espaço confinado, tais como zeólita beta (7-30 nm), zeólita X (22–60 nm), zeólita
A (25–37 nm) (SCHMIDT, 2000). As etapas fundamentais na síntese em espaço confinado
consistem em (TOSHEVA, 2005): (i) restringir a cristalização da zeólita dentro do sistema de
poros da matriz de carbono, o que pode ser alcançada através da impregnação úmida
incipiente do carbono com o gel de síntese da zeólita e (ii) evitar a difusão das espécies
silicatos/aluminossilicatos presentes no gel de síntese para o exterior dos poros do molde, o
que pode ser obtido impedindo o contato direto entre a matriz de carbono impregnada e a
água, durante o tratamento hidrotérmico.
Os moldes de carbono também foram empregados na síntese de cristais hierárquicos
contendo mesoporosidade intrazeolítica (JACOBSEN, 2000). Utilizando um excesso do gel
de síntese da zeólita, observou-se que os cristais da zeólita cresciam em torno das partículas
do carvão ativo, resultando em cristais de zeólita embebidos com um material de carbono,
como mostrado na Figura 2.10b. Isso significa que os cristais da zeólita são nucleados dentro
do sistema de poros da matriz de carbono e, posteriormente, devido ao excesso do gel de
síntese, esses cristais crescem o suficiente para encapsular as partículas de carbono. A
remoção das partículas de carbono, por combustão, conduz à formação de cristais
hierárquicos zeolíticos contendo um sistema de mesoporosos intracristalinos na faixa de 10 a
100 nm. Portanto, através da escolha adequada do molde de carbono, é possível controlar o
sistema mesoporoso do sólido.

2. REVISÃO BIBLIOGRÁFICA 23
(a)
(b)
Matriz de carbono
impregnada com
precursor de alumínio
Nanocristais da
zeólita dentro dos
poros do carbono
Nanocristais
da zeólita
Precursor
de silício
Calcinação
ou
dissolução
Poros gerados durante a
combustão das partículas
de carbono
Partículas de
carbono (12 nm)
Cristal da zeólita (12
µm) crescendo no
sistema de poros da
partícula de carbono
O2
500 °C + CO2
Cristal da zeólita
contendo
mesoporos
Figura 2.10. Ilustração (a) das etapas de síntese de cristais zeolíticos nanométricos em espaço confinado em um
suporte poroso (TOSHEVA, 2005) e (b) do crescimento de cristais zeolíticos em torno das nanopartículas de
carbono (JACOBSEN, 2000).
A formação de cristais nanométricos ou de cristais zeolíticos mesoporosos é
determinada pelos mesmos fatores experimentais, que influenciam as taxas de nucleação e de
crescimento do cristal. Os cristais nanométricos são obtidos quando as velocidades de
nucleação são mais elevadas que as velocidades de crescimento. Os fatores chave para a
formação dos nanocristais não agregados são a elevada supersaturação e a estabilidade
estérica dos núcleos. Estas condições, geralmente, são obtidas através da utilização de
quantidades abundantes do direcionador de estrutura, além de uma quantidade reduzida dos
cátions alcalinos para limitar a agregação das partículas. Além disso, devem ser empregadas

2. REVISÃO BIBLIOGRÁFICA 24
temperaturas relativamente moderadas. Por outro lado, a formação de cristais zeolíticos
mesoporosos é favorecida, empregando velocidades de crescimento mais elevadas
(JACOBSEN, 2001).
Em diversos trabalhos, os pesquisadores da Haldor Topsøe empregaram nanotubos de
carbono e nanofibras de carbono como moldes, na síntese de zeólitas mesoporosas (BOISEN,
2003; JANSSEN, 2003; SCHMIDT, 2001). Entretanto, os nanotubos e as nanofibras de
carbono foram encapsulados apenas parcialmente durante o crescimento dos cristais
zeolíticos, como mostrado na Figura 2.11. Desta forma, a remoção seletiva destes materiais
conduziu à formação de mesoporos intracristalinos. Quando se empregou nanotubos de
carbono, foram obtidos cristais com tamanhos de 100 a 500 nm, contendo mesoporos
uniformes e retos, com diâmetros de 12 a 30 nm. Observou-se que a quantidade de
mesoporos, nos cristais da zeólita, é determinada pela quantidade de nanotubos de carbono em
relação à quantidade do gel de síntese da zeólita. Por outro lado, podem ser obtidos cristais
zeolíticos mesoporosos mais econômicos, usando nanofibras de carbono, que são de custo
muito mais baixo que os nanotubos de carbono; entretanto, os mesoporos do sólido obtido
podem variar em uma faixa mais ampla de tamanhos.
Figura 2.11. Ilustração esquemática do crescimento dos cristais zeolíticos em torno dos nanotubos de carbono (SCHMIDT, 2001).
Posteriormente, Tao e colaboradores (2003) empregaram, pela primeira vez, aerogéis
de carbono ou aerogéis orgânicos como moldes de carbono, na síntese de zeólitas
Cristal único de zeólita (0,1-10 µm)
Nanotubos de carbono (1-20 nm)

2. REVISÃO BIBLIOGRÁFICA 25
mesoporosas. Os aerogéis de carbono podem ser obtidos, principalmente, por pirólise de
aerogéis orgânicos em temperaturas acima de 500 °C (PIERRE, 2002), tais como aerogéis de
resorcinol-formaldeído e de melamina-formaldeído. Durante a pirólise, os aerogéis de
carbono, obtidos a partir dos aerogéis de resorcinol-formaldeído, mantiveram as áreas
superficiais específicas elevadas, assim como o elevado volume de mesoporos dos materiais
precursores. Uma vez que os aerogéis de carbono possuem mesoporos uniformes que são
facilmente ajustáveis, por exemplo, variando a razão molar dos materiais de partida
(resorcinol e formaldeído), eles constituem importantes moldes na preparação de zeólitas com
porosidade hierárquica.
Tao e colaboradores (2005) também empregaram um aerogel de resorcinol-
formaldeído na preparação de zeólitas mesoporosos, obtendo cristais de ZSM-5 e de zeólita A
nanométricos, com uma estrutura de poros bimodal compreendendo mesoporos de tamanhos
uniformes. Durante a etapa de cristalização, os cristais da zeólita cresceram no espaço
confinado do aerogel de carbono, resultando na formação de cristais nanométricos. As
distribuições de diâmetro de mesoporos foram bastante estreitas, entre 10 e 15 nm, que
correspondem à espessura da parede dos poros do aerogel de carbono usado (cerca de 10 nm).
Portanto, variando a estrutura zeolítica e a porosidade do aerogel de carbono, é possível obter
diferentes materiais zeolíticos com mesoporosidade controlada.
As peneiras moleculares mesoporosas de carbono também foram empregadas, de
modo independente, por dois grupos de pesquisa diferentes, como moldes na formação de
zeólitas mesoporosas hierárquicas (SAKTHIVEL, 2004; YANG, 2004). Estes sólidos
carbonáceos mesoporosos, denominados materiais CMK, podem ser sintetizados através da
carbonização de diferentes moléculas orgânicas nos poros de materiais mesoporosos
ordenados (VELOSO, 2009; WALLAU, 2006). Em seguida, os poros do carbono são
preenchidos com um gel de síntese da zeólita e o sistema é submetido à cristalização
hidrotérmica e à remoção da matriz de carbono por combustão. Se o gel precursor for
convertido em uma fase zeolítica contínua dentro dos poros do carbono mesoporoso, podem
ser obtidos cristais nanométricos com mesoporosidade interzeolítica, como mostrado na
Figura 2.12.
Entretanto, os canais mesoporosos são muito estreitos, o que dificulta uma
cristalização controlada no espaço confinado do carbono mesoporoso e, portanto, o gel tende
a migrar, parcial ou completamente, para a superfície externa encapsulando as partículas do
carbono mesoporoso, durante a cristalização (CHO, 2012). Após a remoção do molde, são
obtidos materiais zeolíticos com mesoporos intracristalinos desordenados, em forma de

2. REVISÃO BIBLIOGRÁFICA 26
Figura 2.12. Ilustração esquemática do mecanismo hipotético da formação de zeólitas mesoporosas hierárquicas
através da moldagem com peneiras moleculares mesoporosas de carbono (WALLAU, 2006).
caverna, ao invés de ocorrer uma replicação da forma dos poros do carbono. Estes resultados
sugerem que os carbonos mesoporosos não atuam como um verdadeiro molde (LI, 2007).
De modo a controlar a cristalização dentro dos poros do carbono, investigou-se o
método de conversão do gel seco na moldagem com carbonos mesoporosos (CHO, 2012;
ZHANG, 2005). Observou-se que, quando a umidade relativa foi muito elevada (> 95 %), o
gel precursor migrou para a superfície externa formando uma fase zeolítica. Por outro lado,
empregando valores de umidade entre 80 e 85 %, formou-se uma zeólita com mesoporos
altamente ordenados. Além disso, observou-se que a formação de uma estrutura zeolítica com
mesoporos ordenados não dependia apenas da umidade, mas também do diâmetro de poros e
da rigidez das paredes do carbono mesoporoso (CHO, 2012).
Por outro lado, Xiao e colaboradores (2006) empregaram polímeros catiônicos em
mesoescala, como molde de mesoporos, na síntese de zeólitas mesoporosas hierárquicas. O
procedimento consistiu na adição de um polímero catiônico ao gel de síntese da zeólita beta.
Após a cristalização hidrotérmica e calcinação, foram obtidas zeólitas hierárquicas com uma
distribuição de diâmetro de mesoporos na faixa de 5 a 40 nm, que corresponde ao tamanho do
polímero. A mesoporosidade intrazeolítica obtida pode ser ajustada através da quantidade
adicionada do polímero catiônico. Outros materiais zeolíticos, ou com características

2. REVISÃO BIBLIOGRÁFICA 27
zeólita
nucleação
compósito
zeólita-polímero
nucleado
formação da rede
polimérica
intracristalina
polímero
silanizado
crescimento do
cristal
Sementes de zeólitas
zeolíticas, foram preparados através desta metodologia, tais como a ZSM-5, zeólita Y e TS-1
(titanossilicalita) (XIAO, 2006; LIU, 2008). No mesmo período, Wang e colaboradores
(2006) empregaram polímeros funcionalizados com grupos silanos, na preparação das zeólitas
hierárquicas, como mostrado na Figura 2.13. A principal vantagem desse método consiste na
obtenção de cristais zeolíticos com mesoporos intracristalinos com diâmetros pequenos e
uniformes, na faixa de 2,0 a 3,0 nm.
Figura 2.13. Ilustração esquemática da síntese de zeólitas com mesoporos intracristalinos usando polímeros
silanizados como molde (WANG, 2006).
Outras matrizes de carbono também têm sido estudadas, porém em menor extensão.
Por exemplo, em 2005, Katsuki e colaboradores (2005) empregaram casca de arroz
carbonizada como molde de carbono. Estes materiais apresentam um elevado teor de silício e,
por esse motivo, foram também utilizados como fonte de sílica na preparação dos cristais de
zeólitas com mesoporos intracristalinos. Por outro lado, em 2007, Kustova e colaboradores
desenvolveram um método de nanomoldagem com materiais de carbono preparados in situ,
através da decomposição de um carboidrato, diretamente, na sílica usada como material de
partida na obtenção da zeólita (Figura 2.14). Na preparação de zeólitas hierárquicas com
morfologias diferentes, alguns materiais biológicos também têm sido usados como moldes de
carbono (DAVIS, 1997; DAVIS, 1998; ZHANG, 2000). Eles representam uma alternativa
atrativa em relação aos demais moldes, uma vez que são abundantes e frequentemente de
baixo custo.

2. REVISÃO BIBLIOGRÁFICA 28
Figura 2.14. Ilustração esquemática da rota de síntese de cristais de zeólitas mesoporosas usando sacarose como
molde de carbono (KUSTOVA, 2007).
2.3.2. Zeólitas com Estrutura Hierárquica de Poros Preparadas por Moldagem
Supramolecular
A primeira metodologia relacionada à obtenção de zeólitas com estrutura hierárquica
de poros, por moldagem supramolecular, foi desenvolvida por Kloetstra e colaboradores
(1996), que prepararam peneiras moleculares com distribuição de poros bimodal, combinando
microporos e mesoporos. O método consistiu na cristalização simultânea da zeólita FAU e da
MCM-41 ou através da deposição de MCM-41 sobre cristais da zeólita FAU. Os resultados de
microscopia eletrônica de transmissão mostraram que os materiais consistiam de uma fina
camada de MCM-41 cobrindo a superfície dos cristais da zeólita FAU.
Posteriormente, Karlsson e colaboradores (1999) desenvolveram uma nova
metodologia de preparação de aluminossilicatos mesoporosos, que consistiu na adição
simultânea dos agentes direcionadores das estruturas microporosa (molde molecular) e
mesoporosa (molde supramolecular). Esse método é baseado na ideia de que um molde
molecular direciona a cristalização da estrutura da zeólita, enquanto que, simultaneamente,
um agregado supramolecular de surfactantes (template) conduz à formação da estrutura
mesoporosa. Entretanto, observou-se que os moldes atuavam de modo competitivo,
resultando na formação de uma fase zeolítica pura, uma fase mesoporosa pura ou uma mistura
física do material mesoporoso com a zeólita.
Uma nova metodologia de preparação de zeólitas com estrutura hierárquica de poros
foi desenvolvida em 2000, pelo grupo de Pinnavaia (LIU, 2000), que consistiu na organização
em ar
Calcinação
2) Cristalização
em ar
1) Molde
OH-, Al
3+
Açúcar/Água
SiO2
Calcinação
em ar

2. REVISÃO BIBLIOGRÁFICA 29
dos precursores zeolíticos que normalmente nucleiam a cristalização de zeólitas microporosas.
Estes precursores, também conhecidos como sementes de zeólitas, se assemelham às unidades
secundárias de construção da estrutura zeolítica. O acoplamento destes precursores em uma
estrutura mesoporosa hexagonal do tipo MCM-41, utilizando o brometo de
cetiltrimetilamônio (CTAB), confere estabilidade hidrotérmica e acidez à estrutura
mesoporosa. O material, denominado [Al]-MSU-S (MSU significa Michigan State
University), foi submetido a um tratamento com vapor d’água a 800 °C e manteve a sua
estrutura mesoporosa hexagonal ordenada. O método não se limita apenas a sementes de
zeólita Y; diversos trabalhos foram desenvolvidos, empregando a mesoestruturação de
sementes de zeólitas para obter novos materiais hierárquicos zeolíticos, tais como os
compósitos beta/MCM-41 (GRECCO, 2008; GUO, 2001 a; GUO, 2001 b; LIU, 2001;
ZHANG, 2001), beta/MCM-48 (SHIH, 2004), ZSM-5/MCM-41 (GONÇALVES, 2006;
GONÇALVES, 2008), ZSM-5/MCM-48 (XIA, 2004), mordenita/MCM-41 (WANG, 2004),
mordenita/MCM-48 (DIMITROV, 2005). Outros materiais também foram obtidos, através do
acoplamento de precursores zeolíticos em uma estrutura mesoporosa hexagonal do tipo SBA-
15, tais como os compósitos Y/SBA-15, ZSM-5/ SBA-15 e beta/SBA-15 (LIU, 2002).
Diversas estratégias de síntese foram empregadas na obtenção de zeólitas mesoporosas
hierárquicas através da moldagem supramolecular (PÉREZ-PARIENTE, 2005). As
observações experimentais mostraram que o emprego de precursores zeolíticos, como blocos
de construção da rede mesoporosa, produzia os materiais mais estáveis. Entretanto, foi
observado que a curvatura da micela do surfactante não favorece o empacotamento dessas
nanopartículas de construção. Por exemplo, em estruturas do tipo MCM-41 que possuem
espessura da parede mesoporosa na ordem de 1 nm, seria muito difícil acomodar essas
nanopartículas para formar paredes cristalinas altamente condensadas (Figura 2.15a). Por
outro lado, pode-se propor um agrupamento de agregados de sílica/direcionador, que conduz a
unidades primárias amorfas na ordem de 1 a 3 nm que melhor se ajustam à parede
mesoporosa, como mostrado na Figura 2.15b.
Entretanto, os materiais obtidos através da mesoestruturação de precursores zeolíticos
são, provavelmente, compostos por uma matriz de sílica/alumina amorfa contendo domínios
zeolíticos na superfície ou dentro dos poros, ou seja, são materiais híbridos das estruturas
mesoporosas ordenada e zeolítica (GRECCO, 2010).

2. REVISÃO BIBLIOGRÁFICA 30
Figura 2.15. Ilustração de possíveis organizações de precursores zeolíticos com uma fase micelar do surfactante.
(a) Organização das nanopartículas do precursor zeolítico coloidal; (b) organização de agregados amorfos
mediado pelo direcionador (PÉREZ-PARIENTE, 2005).
Com a expectativa de melhorar a acessibilidade em zeólitas lamelares, Corma e
colaboradores (1998) desenvolveram um método de preparação que consistiu na
deslaminação de seus precursores, de maneira similar ao que é realizado com uma estrutura de
camadas de argila. Inicialmente, as lamelas de um precursor zeolítico, MCM-22 (P), foram
expandidas pela adição de CTAB. Esse processo de expansão pode ser monitorado através do
aumento da distância entre as lâminas, observado por difração de raios X. Nesse processo, as
lamelas do precursor zeolítico são separadas, através de sonicação, mas, a estrutura zeolítica é
preservada nas camadas individuais. Após a remoção do agente de expansão, as lâminas são
reempilhadas, de modo desorganizado, resultando em um aluminossilicato com elevada área
superficial específica (ITQ-2), como ilustrado na Figura 2.16.
Os mesoporos e macroporos formados entre as lamelas desorganizadas proporcionam
maior acessibilidade aos sítios ativos que estão localizados dentro das lâminas finas de
zeólita. Após a deslaminação, a estrutura resultante apresenta uma importante perda de
microporosidade. Por outro lado, ocorre um aumento da área externa e, consequentemente, a
formação de superfícies fortemente hidroxiladas. Durante o processo de deslaminação,
também ocorre desaluminização conduzindo a uma diminuição da acidez do sólido.
Solução
coloidal de
precursores
zeolíticos
2,8 nm
4 nm
(a)
(b)

2. REVISÃO BIBLIOGRÁFICA 31
Figura 2.16. Ilustração esquemática da preparação de zeólitas deslaminadas (EGEBLAD, 2008).
Observou-se que as zeólitas deslaminadas adsorveram uma menor quantidade de piridina
quando comparadas a seu precursor. Por outro lado, as medidas de acidez por adsorção de
2,6-di-terc-butilpiridina mostraram um comportamento inverso, revelando que o processo de
deslaminação permite o acesso de moléculas volumosas aos sítios ativos. A restrição desta
metodologia, entretanto, é que sua aplicabilidade se limita a estruturas zeolíticas lamelares
(CORMA, 2002).
De modo a obter materiais com mesoporos bem definidos, em 2006, Ryoo e
colaboradores (CHOI, 2006 a) desenvolveram uma estratégia para a síntese direta de zeólitas,
usando organossilanos anfifílicos, [(CH3O)3SiC3H6N(CH3)2CnH2n+1]Cl, como molde
supramolecular. Estes compostos são formados por um grupo amônio quaternário,
responsável pelo ordenamento da estrutura zeolítica, uma fração de organossilano ligada ao
grupo amônio e uma cadeia alquila. Esperava-se que o organossilano anfifílico pudesse
interagir fortemente com a estrutura zeolítica durante a cristalização, através da formação de
ligações covalentes entre as espécies silicato ou aluminato com a fração de organossilano.
Dessa maneira, as moléculas de surfactante não seriam removidas da esfera de
aluminossilicato ou do aluminofosfato, durante o processo de cristalização, o que tornaria
possível produzir cristais com uma estrutura mesoporosa intracristalina desordenada (Figura
2.17a). O comprimento da cadeia alquila promoveria a formação de mesoporos de tamanho
relativamente uniforme. Desta forma, diferentes estruturas zeolíticas com mesoporos
ajustáveis poderiam ser obtidas variando-se o comprimento da cadeia alquila.
No trabalho pioneiro de Ryoo e colaboradores (CHOI, 2006 a), os organossilanos
anfifílicos foram adicionados diretamente ao gel de síntese da zeólita MFI, contendo o
direcionador de estrutura. Os cristais obtidos apresentaram diferentes diâmetros médios de
mesoporos, que variaram em função do comprimento da cadeia alquila do organossilano ou
Deslaminação CTAB
ITQ-2
Deslaminada MCM-22 (P)
Expandida MCM-22 (P)

2. REVISÃO BIBLIOGRÁFICA 32
Figura 2.17. (a) Ilustração do mecanismo proposto para a síntese de cristais de aluminofosfatos hierárquicos,
empregando organossilanos anfifílicos como molde supramolecular (CHOI, 2006 b). (b) Ilustração esquemática
da estrutura hierárquica obtida usando surfactantes bifuncionais (NA, 2011; MÖLLER, 2011).
da temperatura da síntese hidrotérmica. Por exemplo, as zeólitas mesoporosas apresentaram
diâmetros médio de poros de 2,1, 3,1 e 3,9 nm, quando o número de átomos de carbono da
cadeia alquila foi 12, 16 e 18, respectivamente. Entretanto, podem ser obtidos sólidos com
diâmetros de mesoporos acima de 20 nm sob condições de síntese especificas, tais como a
adição de copolímeros triblocos ao gel de síntese ou de agentes de expansão dos poros
(CHO,2009). Usando organossilanos anfifílicos, diversas outras estruturas contendo
mesoporosidade intracristalina foram preparadas, dentre as quais pode-se citar as zeólitas
(b)
(a)
Cristalização
Calcinação
mesoporos
Cristal nanométrico
de AlPO4-n
camada orgânica
Gel de
síntese do
AlPO4-n
Nanocristais
Uniformes
Arranjo Hexagonal dos
Mesoporos nos Nanocristais
MFI

2. REVISÃO BIBLIOGRÁFICA 33
LTA (CHO, 2011; CHOI, 2006 a), a sodalita (SHANBHAG, 2009) e a mordenita (LI, 2009)
e os aluminofosfatos (CHOI, 2006 b), titanossilicatos (CHENEVIERE, 2010) e
zirconossilicatos (ZHAO, 2009). A presença de mesoporos usando organossilanos anfifílicos
foi investigada através de microscopia eletrônica de varredura de alta resolução e por
simulação de Monte Carlo de crescimento do cristal (CHO, 2011). Os cristais da zeólita LTA
apresentaram morfologia cúbica, típica da zeólita, que se tornou progressivamente
arredondada à medida que a concentração do organossilano foi aumentada. Além disso, a
estrutura mesoporosa foi preservada durante um período de cristalização de seis dias. Estes
resultados sugerem um mecanismo de geração de mesoporos intracristalinos, em que as
micelas do organossilano anfifílico permanecem ocluídas no interior do cristal da zeólita.
Em outros trabalhos, Ryoo e colaboradores (CHOI, 2009; NA, 2010; NA, 2011)
desenvolveram novas metodologias empregando surfactantes bifuncionais, que podem
direcionar a formação de ambas as estruturas, microporosa e mesoporosa, da zeólita
hierárquica. No primeiro caso (CHOI, 2009), eles empregaram um surfactante (C22H45-
N+(CH3)2-C6H12-N
+(CH3)2-C6H13) contendo um grupo alquil de cadeia longa (C22H45) e dois
grupos amônio quaternário separados por dois grupos alquílicos (CHOI, 2009). O grupo
diamônio foi responsável pelo ordenamento da estrutura zeolítica do tipo MFI, enquanto a
interação hidrofóbica das cadeias longas (C22H45) induziu a formação de uma mesoestrutura
micelar. As cadeias hidrofóbicas restringiram o crescimento da estrutura zeolítica na parte
hidrofílica das micelas, resultando na formação de lamelas de zeólita MFI em escala
nanométrica (espessura de 2 nm). Dependendo das condições de síntese, as nanolamelas
podem se auto-organizar em um arranjo unilamelar desordenado ou um arranjo multilamelar
ordenado bidimensional (CHOI, 2009; NA, 2010). As camadas lamelares também podem ser
submetidas a um processo de pilarização (NA, 2010), no qual as lamelas são separadas por
pilares de sílica que, posteriormente, são convertidos em uma parte da estrutura zeolítica;
portanto, a mesoporosidade interlamelar é preservada, após a remoção do surfactante. Em
outro trabalho, Ryoo e colaboradores (NA, 2011) empregaram uma série de surfactantes de
poliamônio quaternário, que podem atuar como direcionador de diferentes estruturas
hierárquicas ordenadas. Um membro especialmente importante desta série é o surfactante
C18H37-N+(CH3)2-C6H12-N
+(CH3)2-C6H12-N
+(CH3)2-C18H37(Br
-)3, responsável pela formação
de uma peneira molecular mesoporosa ordenada hexagonal, que possui paredes microporosas
cristalinas com características zeolíticas, como mostrado na Figura 2.17b. Os mesoporos em
arranjo hexagonal são gerados pelos agregados do surfactante, enquanto os diferentes grupos
poliamônio quaternário atuam como direcionador das diferentes estruturas zeolíticas. A

2. REVISÃO BIBLIOGRÁFICA 34
espessura da parede, o tipo de estrutura zeolítica e o diâmetro de mesoporos podem ser
controlados usando surfactantes com diferentes grupos, envolvendo um número maior de
grupos amônio quaternário e a presença de anéis fenil. Embora normalmente o ordenamento
dos mesoporos diminua com o aumento da espessura da parede, observou-se que a espessura
da parede foi uniforme, variando na faixa de 2,3 a 5,1 nm. O diâmetro de mesoporos (3,8 a 21
nm) pode ser controlado através da adição de agentes de expansão dos poros (NA, 2011).
Uma nova metodologia de preparação de cristais nanométricos foi proposta por
Serrano e colaboradores em 2006. Ela consistiu na perturbação do crescimento dos cristais da
zeólita através da silanização de sementes, de modo a impedir uma posterior agregação destes
precursores. A síntese de zeólitas a partir da silanização de sementes consiste de três etapas,
como mostrado na Figura 2.18 (SERRANO, 2006; 2011): (i) formação dos núcleos da
zeólita, que ocorre durante uma etapa de pré-cristalização. Esses núcleos devem ser
considerados como entidades pseudo-cristalinas, uma vez que apresentam propriedades
intermediárias entre os aluminossilicatos amorfos e as zeólitas. Eles são obtidos nos primeiros
estágios de cristalização (na etapa de pré-cristalização) da zeólita e são amorfos aos raios X.
Por outro lado, foram observadas vibrações da estrutura zeolítica através de bandas no
espectro de infravermelho; (ii) acoplamento do agente de silanização, em presença ou
ausência de alcoóis, na superfície externa dessas entidades, que ocorre durante a etapa de
silanização, com um agente direcionador de estrutura ocupando os microporos e (iii)
cristalização hidrotérmica das sementes funcionalizadas, obtendo-se materiais com
características cristalinas e com porosidade hierárquica, uma vez que a presença do agente de
silanização impede a agregação das entidades nanométricas.
Esta estratégia foi bem sucedida na preparação das zeólitas ZSM-5 e beta
nanométricas, usando fenilaminopropil-trimetoxissilano como agente de funcionalização
(SERRANO, 2006; 2008; 2011). Por outro lado, essa metodologia pode ser também aplicada
na preparação de outras nanozeólitas, tais como mordenita, faujasita e materiais com
características zeolíticas, como os titanossilicatos (AGUADO, 2009 a; VUONGA, 2010;
SANZA, 2011). No caso da ZSM-5 nanométrica, foram obtidas partículas de 300 a 400 nm,
que são formadas por aglomerados de cristais pequenos da ZSM-5 (10 - 20 nm) (SERRANO,
2006; SERRANO, 2008). Por outro lado, a zeólita beta consistiu de macro-partículas (500 a
600 nm), formadas por aglomerados de nanocristais de cerca de 15 nm (AGUADO, 2008). O
efeito da natureza do agente de silanização sobre as características da ZSM-5 também foi
estudado por esses pesquisadores (SERRANO, 2008), empregando quatro organossilanos
diferentes: 3-aminopropil-trimetoxissilano (APTMS), isobutil-trietoxissilano (IBTES),

2. REVISÃO BIBLIOGRÁFICA 35
Sementes da zeólita
a) Silanização
PHAPTHMS
b) Silanização e alcoxilação
PHAPTHMS +
R-OH
Interação entre o
organossilano e álcool
Figura 2.18. Ilustração das etapas de síntese de nanocristais de zeólita a partir da silanização de sementes (SERRANO, 2011).
fenilaminopropil-trimetoxissilano (PHAPTMS) e octadecil-trimetoxissilano (ODTMS). O
PHAPTMS foi o agente de silanização mais eficiente, tanto no seu acoplamento à superfície
externa dos núcleos quanto na perturbação do crescimento do cristal da ZSM-5.Foi observado
que as propriedades das nanozeólitas obtidas através da silanização dependem não apenas da
natureza do agente de silanização, mas também da concentração do agente de silanização, das
condições de pré-cristalização e da adição de alcoóis na etapa de silanização (SERRANO,
2009; SERRANO, 2011). Em outro trabalho, Carvalho e colaboradores (2011) estudaram a
influencia dos tempos de pré-cristalização de sementes e de cristalização da zeólita, nas
propriedades de zeólitas beta. Foi observado que o tempo de pré-cristalização das sementes e
de cristalização da zeólita não afetou significativamente o tamanho do cristal. Por outro lado,
o tempo de pré-cristalização das sementes mais curto levou à formação de sólidos com as
áreas superficiais específicas e áreas de microporos mais elevadas, mas com a menor
quantidade de alumínio. Na amostra preparada com o tempo de cristalização mais longo, não
foi notada a adsorção do organossilano na zeólita. A funcionalização das sementes com
organossilanos levou à produção de um sólido com menor tamanho de partícula e mais
alumínio na rede zeolítica, em comparação com as amostras não funcionalizadas. A amostra
mais promissora foi sintetizada com o tempo de pré-cristalização de um dia e um período de
cristalização de dois dias utilizando o fenil-aminopropil-trimetoxissilano, apresentando um
menor tamanho de cristal e área de microporos e área externa mais elevadas.

3 – PARTE EXPERIMENTAL 36
3. PARTE EXPERIMENTAL
3.1. Reagentes Utilizados
As características e a procedência dos reagentes, empregados na preparação e
caracterização das zeólitas mordenita com estrutura hierárquica de poros, são descritas na Tabela
3.1.
Tabela 3.1. Características e procedência dos reagentes empregados na preparação dos materiais com estrutura
hierárquica de poros baseados na mordenita.
Nome Fórmula Fabrica
nte
Pureza
(%) Sílica (Aerosil 200)* SiO2 Deguss
a
100
Sulfato de alumínio Al2(SO4)3·18
H2O
Aldrich 99,5
Hidróxido de sódio NaOH Aldrich ≥98,0
Ácido ortofosfórico** H3PO4 Aldrich -
Cloreto de amônio NH4Cl Aldrich 99
Cloreto de [3-(trimetoxissilil)propil]octadecildimetilamônio*** C19H42NCl Aldrich 72
Ar sintético**** O2 AGA -
Nitrogênio N2 AGA 99,999
*Aerosil 200 com 200 m2g-1.
**em solução aquosa (85% em massa).
***impurezas: 13% de metanol e 15% de 3-(cloropropil)-trimetoxissilano.
****em gás nitrogênio como balanço (20%).
3.2. Nomenclatura das Amostras
As amostras obtidas neste trabalho foram identificadas como MT-x/y, em que M
representa a mordenita, T representa o cloreto de [3-(trimetoxissilil)propil]
octadecildimetilamônio (TPOAC) usado como agente gerador de mesoporosidade, x e y são a
temperatura e o tempo de cristalização, respectivamente. As amostras identificadas como HMT-
x/y referem-se às amostras na forma protônica. A nomenclatura e as condições de preparação das

3 – PARTE EXPERIMENTAL 37
zeólitas mordenita com estrutura hierárquica de poros na forma sódica são apresentadas na
Tabela 3.2.
Tabela 3.2. Nomenclatura e condições de preparação dos materiais com estrutura hierárquica de poros baseados na
mordenita. A Amostra HM-170/96 refere-se à amostra obtida na ausência do organossilano, na forma protônica.
3.3. Preparação dos Materiais com Estrutura Hierárquica de Poros baseados na
Mordenita
As zeólitas mordenita com estrutura hierárquica de poros foram preparadas através da
adição de um organossilano (TPOAC), usado como agente gerador de mesoporosidade, ao gel de
síntese da mordenita.
3.3.1. Preparação do Gel de Síntese da Mordenita
O gel de síntese da mordenita foi preparado utilizando a metodologia descrita por
Samanta e colaboradores (2004), com algumas modificações. O gel de composição molar igual a
16 Na2O:1 Al2O3:30 SiO2: 3,33 H3PO4:1,6 TPOAC:1000 H2O foi obtido através da mistura de
duas soluções. A primeira foi obtida através da dissolução de 1,44 g de sulfato de alumínio
hidratado em 11 mL de água desionizada. Na preparação da segunda solução, 4,0 g de sílica
aerosil foram dissolvidos em 18 mL de água junto com 2,82 g de hidróxido de sódio, sob
aquecimento brando. A seguir, foram acrescentados 11 mL de água e 0,5 mL de ácido
Nomenclatura Tempo de cristalização Temperatura de cristalização
MT-140/24 24 h 140 °C
MT-140/48 48 h 140 °C
MT-140/96 96 h 140 °C
MT-140/192 192 h 140 °C
MT-120/96 96 h 120 °C
MT-170/96 96 h 170 °C
HM-170/96 96 h 170 °C

3 – PARTE EXPERIMENTAL 38
ortofosfórico, sob agitação mecânica. A mistura permaneceu sob agitação, por 1 h, à temperatura
ambiente. Posteriormente, 2,44 g de cloreto de [3-(trimetoxissilil)propil]octadecildimetilamônio
de foram adicionados ao gel de síntese da mordenita.
3.3.2. Cristalização do Gel de Síntese da Mordenita
Após a homogeneização, o gel foi transferido para uma autoclave e submetido a
tratamento hidrotérmico a 120, 140 e 170 °C, em diferentes intervalos de tempo. Após a
cristalização, as amostras foram filtradas, lavadas com água desionizada e secas em estufa a 60
°C, por 24 h. A secagem foi realizada nessa temperatura com o intuito de preservar a estrutura
micro-mesoporosa, não submetendo o sólido a condições hidrotérmicas que promoveriam a
hidrólise das ligações Si-O-Si.
3.3.3. Remoção do Agente Gerador de Mesoporosidade
Os materiais zeolíticos foram preparados utilizando o cloreto de [3-(trimetoxissilil)
propil] octadecildimetilamônio, como agente gerador de mesoporosidade. Para serem utilizados
como catalisadores, é necessário remover esse composto orgânico que está ocluído no interior do
sistema micro-mesoporoso, obstruindo os canais destes sólidos. A remoção do organossilano da
água de hidratação deve ser efetuada cuidadosamente, de modo a evitar o colapso da estrutura do
sólido. Dessa forma, as amostras foram aquecidas (2 °C.min-1
) sob fluxo de ar sintético (100
mL.min-1
) até 550 °C, permanecendo nesta temperatura por 6 h, obtendo-se os materiais com
estrutura hierárquica de poros baseados na mordenita forma sódica (Tabela 3.2).
3.3.4. Troca Iônica dos Cátions Na+ por Cátions H
+
Como o material com estrutura hierárquica de poros baseado na mordenita estava
inicialmente, na forma sódica, realizou-se a troca iônica dos íons Na+ por íons H
+, com o objetivo
de criar os sítios ácidos de Brønsted nos catalisadores zeolíticos. Os materiais foram submetidos a
três trocas sucessivas com uma solução aquosa de cloreto de amônio 1 mol.L-1
(50 mL/g de
sólido) a 80 °C, por 3 h. Em seguida, a mistura foi filtrada e o sólido lavado com água

3 – PARTE EXPERIMENTAL 39
desionizada até à completa remoção dos íons cloreto, o que foi verificado através de testes com
uma solução de nitrato de prata 1 mol.L-1
. Posteriormente, o sólido foi seco a 60 °C, por 24 h.
Após a secagem, a amostra foi aquecida (2 °C.min-1
) sob fluxo de ar (100 mL.min-1
) até 550 °C,
permanecendo nesta temperatura por 3 h e obtendo-se o material com estrutura hierárquica de
poros baseado na mordenita. Para garantir a eficiência, o processo de troca iônica foi repetido três
vezes.
3.4. Preparação da Mordenita na Ausência do Organossilano
A zeólita mordenita, obtida na ausência do organossilano (Amostra HM-170/96), foi
preparada usando um gel de síntese obtido nas mesmas condições da zeólita mordenita com
estrutura hierárquica de poros (item 3.3). O gel foi submetido a tratamento hidrotérmico a 170
°C, por 96 h. Após a cristalização, o sólido foi separado por filtração, lavado com água
desionizada e seco a 60 °C, por 24 h. Em seguida, a amostra foi submetida à troca iônica dos íons
Na+ por íons H
+, nas mesmas condições descritas no item 3.3.4.
3.5. Caracterização dos Materiais
As amostras obtidas durante o estudo do tempo e da temperatura de cristalização foram
caracterizadas por termogravimetria, espectroscopia no infravermelho com transformada de
Fourier, difração de raios X, análise textural por medidas de adsorção de nitrogênio, ressonância
magnética nuclear de 29
Si e de 27
Al, microscopia eletrônica de varredura e medidas de acidez por
dessorção de amônia à temperatura programada.
3.5.1. Termogravimetria
A remoção do agente direcionador de estrutura foi acompanhada através da
termogravimetria (TG). Estas análises foram realizadas em um equipamento da Shimadzu,
modelo TGA-50, usando aproximadamente 5 mg da amostra, que foi colocada em um porta-
amostra de alumina e submetida a aquecimento desde a temperatura ambiente até 1000 ºC, sob
fluxo de ar sintético (50 ml.min-1
) e utilizando uma taxa de aquecimento de 10 ºC.min-1
.

3 – PARTE EXPERIMENTAL 40
3.5.2. Espectroscopia no Infravermelho com Transformada de Fourier
A remoção do agente gerador de mesoporosidade também foi acompanhada através da
espectroscopia no infravermelho com transformada de Fourier (FTIR). Os experimentos foram
conduzidos em um espectrofotômetro ABB Bomem Inc, modelo MB 102. Em cada espectro
obtido, foram realizadas 32 varreduras, na região entre 4000 e 400 cm-1
, com uma resolução de
4,0 cm-1
. As amostras analisadas, cerca de 50 mg, foram misturadas com brometo de potássio,
submetidas à pressão para formar pastilhas, que foram colocadas em um porta-amostra de metal.
Todas as análises foram conduzidas à temperatura ambiente.
Além da confirmação da remoção do direcionador de estrutura, estes experimentos foram
conduzidos com o objetivo de identificar os modos vibracionais da estrutura das zeólitas por
meio das bandas resultantes e, assim, verificar se houve mudança de estrutura após os diversos
tratamentos.
3.5.3. Difração de Raios X
A formação da estrutura mesoporosa e microporosa nas amostras obtidas foi investigada,
através dos experimentos de difração de raios X (DRX), realizados em um equipamento da
Rigaku, modelo Multiflex, utilizando radiação de CuKα gerada a 40 kV, 30 mA e com filtro de
níquel. O espectrômetro pertence à Universidade Federal de São Carlos (UFSCar).
Na identificação da estrutura mesoporosa, a análise foi conduzida na faixa de varredura 2θ
de 1 a 10 °, com a velocidade do goniômetro de 0,5 °.min-1
. Na verificação da estrutura
microporosa, o equipamento foi operado na faixa de varredura 2θ de 5 a 50 ° e com a velocidade
do goniômetro de 2,0 °.min-1
.
A cristalinidade da estrutura microporosa (C) da mordenita foi determinada como a
relação entre as intensidades dos picos de difração em 2θ igual a 19,68°, 22,4°, 25,74°, 26,4° e
27,64° da amostra analisada e as intensidades destes picos de difração para a amostra considerada
como padrão, representada na Equação 3.1 (RAYALU, 2005).
(3.1)
100
(amostra) I (%) x C
(padrão) I

3 – PARTE EXPERIMENTAL 41
em que:
I(amostra) é o somatório das intensidades dos picos de difração em 2θ igual a 19,68°, 22,4°,
25,74°, 26,4° e 27,64° da amostra analisada e
I(padrão) é o somatório das intensidades dos picos de difração em 2θ igual a 19,68°, 22,4°,
25,74°, 26,4° e 27,64° da Amostra HMT-170/96 considerada como padrão, que foi aquela que
apresentou o valor de intensidade mais elevada.
Além disso, foi determinado o diâmetro médio () dos cristais da mordenita a partir da
equação de Scherrer (GUISNET, 2004), mostrada na Equação 3.2.
(3.2)
em que:
é o diâmetro médio do cristal da mordenita;
k é a constante de proporcionalidade dependente da forma das partículas (geralmente assumem-se
as partículas esféricas, com valor da constante igual a 0,94);
λ é o comprimento de onda da radiação (nm) e
é a média da soma das larguras à meia altura dos picos de difração em 2θ igual a 19,68°, 22,4°,
25,74° e 26,4°, característico da mordenita (rad).
3.5.4. Análise Textural a Partir de Dados de Adsorção/Dessorção de Nitrogênio
As propriedades texturais dos materiais zeolíticos foram determinadas através de
experimentos de adsorção de nitrogênio, conduzidos a 77 K, em um equipamento da
Micromeritics, modelo ASAP 2020. Antes da análise, aproximadamente 0,15 g da amostra foi
acondicionada em uma cela de vidro, previamente pesada, a qual foi acoplada ao sistema de pré-
tratamento do equipamento. Nesta etapa, a amostra foi aquecida a 250 °C por 4 h, sob vácuo e
x cos(θ)
k x λ

3 – PARTE EXPERIMENTAL 42
taxa de aquecimento de 10 °C.min-1
e submetida a um aumento máximo de pressão de 50 µmHg,
para a remoção da água adsorvida na zeólita. Em seguida, a amostra foi resfriada e a cela foi
novamente pesada e acoplada ao sistema de análise do equipamento. Durante a análise, a amostra
foi submetida a pulsos de nitrogênio até um aumento máximo de pressão de 925 mmHg.
A área e o volume de microporos foram calculados pelo método t-plot, usando a Equação
de Harkins e Jura (BARRET, 1951). A determinação da área e o volume de mesoporos e a
distribuição de tamanho de poros foram calculadas utilizando-se o método BJH, seguindo o ramo
de dessorção (GREGG, 1982).
3.5.5. Ressonância Magnética Nuclear de 29
Si e de 27
Al
Os espectros de ressonância magnética nuclear de silício e alumínio (RMN 29
Si e 27
Al)
foram adquiridos a 25 ºC em um espectrômetro Bruker Avance III 500 (frequência de 99,3 MHz
para 29
Si e 130,3 MHz para 27
Al), utilizando-se uma sonda CPMAS de 4 mm e um rotor de
zircônio, girando a 7 kHz. Os espectros foram adquiridos com uma média de 1024 varreduras,
número de pontos igual a 8 e 16k, janela espectral de 5000 e 10000 Hz, tempo de aquisição de
0,082 s e d1 de 15 e 0,5 s, respectivamente para o 29
Si e 27
Al. O espectrômetro pertence à
Universidade Federal de Goiás (UFG).
3.5.6. Microscopia Eletrônica de Varredura
As imagens dos materiais zeolíticos sintetizados foram obtidas por microscopia eletrônica
de varredura (MEV) em um equipamento Jeol, modelo JSM-6610LV. O microscópio operou com
uma voltagem de aceleração de 26 KV e a imagem foi gerada por elétrons secundários. Na
preparação das amostras, os sólidos foram dispersos em acetona e uma gota da suspensão foi
colocada em um porta-amostra de alumínio e, em seguida, coberta com uma fina camada de ouro.
3.5.7. Medidas de Acidez por Dessorção de Amônia à Temperatura Programada
A quantidade e a força dos sítios ácidos das zeólitas na forma protônica foram avaliadas
através de dessorção de amônia à temperatura programada (TPD-NH3), em um equipamento
Micromeritics, modelo TPD/TPR 2900, pertencente à Facultad de Ciências Químicas da

3 – PARTE EXPERIMENTAL 43
Universidad de Concepción (Chile). Inicialmente, uma massa de 0,3 g de cada amostra foi seca a
100 °C, por 2 h, sob fluxo de ar (50 mL.min-1
). Nessa temperatura, injetaram-se pulsos de amônia
até atingir a saturação. Então, a amostra foi resfriada à temperatura ambiente e a dessorção da
amônia conduzida aquecendo-a (10 °C.min-1
) até 800 °C.

4 – RESULTADOS E DISCUSSÃO 44
4. RESULTADOS E DISCUSSÃO
4.1. Estudos do Tempo de Cristalização
4.1.1. Termogravimetria
As curvas de perda de massa dos materiais hierárquicos baseados na mordenita, como
sintetizados, são mostradas na Figura 4.1.
Figura 4.1. Curvas de perda de massa e de derivada da perda de massa das amostras obtidas durante o estudo do
tempo de cristalização a 140 ºC. O primeiro e o segundo números representam a temperatura e o tempo de
cristalização, respectivamente. A letra P indica os precursores dos materiais hierárquicos baseados na mordenita.
200 400 600 800 1000
2,8
3,2
3,6
4,0
4,4
Temperatura (ºC)
Mas
sa
(mg)
-0,002
-0,001
0,000
0,001
MT-140/192-P DT
G
(dm
/dT
)
220
70
325 180 290
-0,002
-0,001
0,000
0,001
MT-140/96-P
DT
G
(dm
/dT
)
230 65 415
200 400 600 800 1000
2,8
3,2
3,6
4,0
Mas
sa (
mg)
Temperatura (ºC)
220
-0,002
-0,001
0,000
0,001
DT
G (d
m/d
T)
200 HMT-140/24
600 800 1000
3,6
4,0
4,4
4,8
5,2
Mas
sa
(mg)
Temperatura (ºC)
60
330
590
HMT-140/192

4 – RESULTADOS E DISCUSSÃO 45
De modo geral, as curvas apresentaram quatro estágios de perda de massa, em
concordância com os trabalhos de Kleitz e colaboradores (2003). Isto era esperado, uma vez
que o agente gerador de mesoporos (TPOAC) é estruturalmente similar ao brometo de
cetiltrimetilamônio (CTAB) usado como direcionador de estrutura de materiais mesoporosos
ordenados. O primeiro estágio ocorre em temperaturas inferiores a 100 °C e é relativo à
dessorção de materiais voláteis e de água, enquanto que o segundo e terceiro estágios, entre
150 e 500 °C, se referem, principalmente, à decomposição e combustão do TPOAC presente
na estrutura da mordenita. O quarto estágio de perda de massa ocorre em temperaturas
elevadas, próximas a 600 °C e é atribuído à dehidroxilação de grupos silanol e à combustão
do carbono residual (KLEITZ, 2003).
Esses eventos podem ser melhor visualizados nas curvas da derivada da perda de
massa em relação à temperatura (DTG), exibidas na Figura 4.1. O DTG da Amostra MT-
140/48 apresentou um pico em 220 °C, relacionado à eliminação da trimetilamina através da
degradação de Hofmann (decomposição do TPOAC), o que conduz à formação de
hidrocarbonetos. Posteriormente, ocorre a sua fragmentação em compostos orgânicos de peso
molecular mais baixos e combustão destes últimos, entre 250 e 400 °C. O aumento do tempo
de cristalização conduz a uma maior interação entre o agente gerador de mesoporosidade e as
espécies aluminossilicatos, o que dificulta a decomposição deste composto orgânico. Dessa
forma, os picos relacionados à eliminação da trimetilamina e combustão dos compostos
orgânicos são deslocados para regiões de temperatura mais elevadas, no caso da Amostra MT-
140/96. Se este tempo for suficientemente elevado, parte do TPOAC se decompõe durante a
etapa de cristalização do gel. Como se pode observar na Figura 4.1, o DTG da Amostra MT-
140/192 apresentou dois picos entre 150 e 250 ºC que provavelmente se referem à eliminação
da trimetilamina e outros dois picos entre 250 e 350 ºC relacionados à fragmentação e
combustão dos hidrocarbonetos resultantes. Isto sugere que os picos podem estar associados à
eliminação da trimetilamina e à combustão de compostos orgânicos em diferentes interações
com a estrutura zeolítica.
Pode-se observar que a perda de massa variou em função do tempo de cristalização,
como mostrado na Tabela 4.1. Do ponto de vista termodinâmico e cinético, as forças que
atuam no sistema gel são resultantes da interação entre as espécies do surfactante e as espécies
inorgânicas (SI), entre as espécies do surfactante (SS) e entre as espécies inorgânicas (II)
(HUANG, 2000). Espera-se que a ordem das forças de interação necessárias para a formação
da fase mesoporosa seja SI > SS > II. Isto indica que as fortes interações entre os cátions
TPOA+ com as espécies aluminossilicatos são importantes na formação da estrutura

4 – RESULTADOS E DISCUSSÃO 46
Tabela 4.1. Perda de massa total das amostras obtidas no estudo do tempo de cristalização a 140 °C. O primeiro e o segundo números representam a temperatura e o tempo de cristalização, respectivamente. A letra P indica os precursores dos materiais hierárquicos baseados na mordenita.
Amostras Perda de massa (%)
MT-140/48-P 33
MT-140/96-P 31
MT-140/192-P 37
mesoporosa. Por outro lado, a força de interação entre as espécies inorgânicas aumenta em
função do tempo de cristalização, o que reduz a densidade de carga da superfície e,
consequentemente, diminui a força de interação dos cátions TPOA+ com as espécies
aluminossilicatos. Assim, as espécies TPOA+ que não estão interagindo são facilmente
eliminadas durante a lavagem do sólido. Como resultado, há uma ligeira diminuição da perda
de massa em função do aumento da cristalinidade. Quando este tempo é suficientemente
elevado, uma parte do TPOAC pode ser encapsulada durante o crescimento dos cristais e,
portanto, não é facilmente eliminada na lavagem. Por essa razão, há um aumento da perda de
massa total no termograma da Amostra MT-140/192.
4.1.2. Espectroscopia no Infravermelho com Transformada de Fourier
A decomposição do agente gerador de mesoporosidade, durante a calcinação das
amostras, também foi acompanhada através da espectroscopia no infravermelho com
transformada de Fourier (FTIR) dos materiais, obtidos antes e após a calcinação dos sólidos,
mostrados na Figura 4.2. Podem ser observadas bandas de estiramento simétrico e assimétrico
da ligação C-H próximas a 2923 e 2853 cm-1
, respectivamente. Estes ligações são
características de grupos CH2 e CH3 do composto orgânico. Também foram observadas as
banda de deformação angular das ligações C-H em 1454 cm-1
(δCH3) e 1394 cm-1
(δCH2), em
concordância com outros trabalhos (RANGEL, 2013; SELVARAJ, 2003). Após a calcinação,
estas bandas desapareceram, indicando que o processo de calcinação foi eficiente na remoção
destes compostos, exceto no caso da Amostra MT-140/192. Estes resultados sugerem que
ocorre um encapsulamento dos compostos orgânicos gerados pela decomposição do TPOAC
durante a etapa de cristalização do gel, o que dificulta a combustão destes compostos como
observado nos termogramas desta amostra.
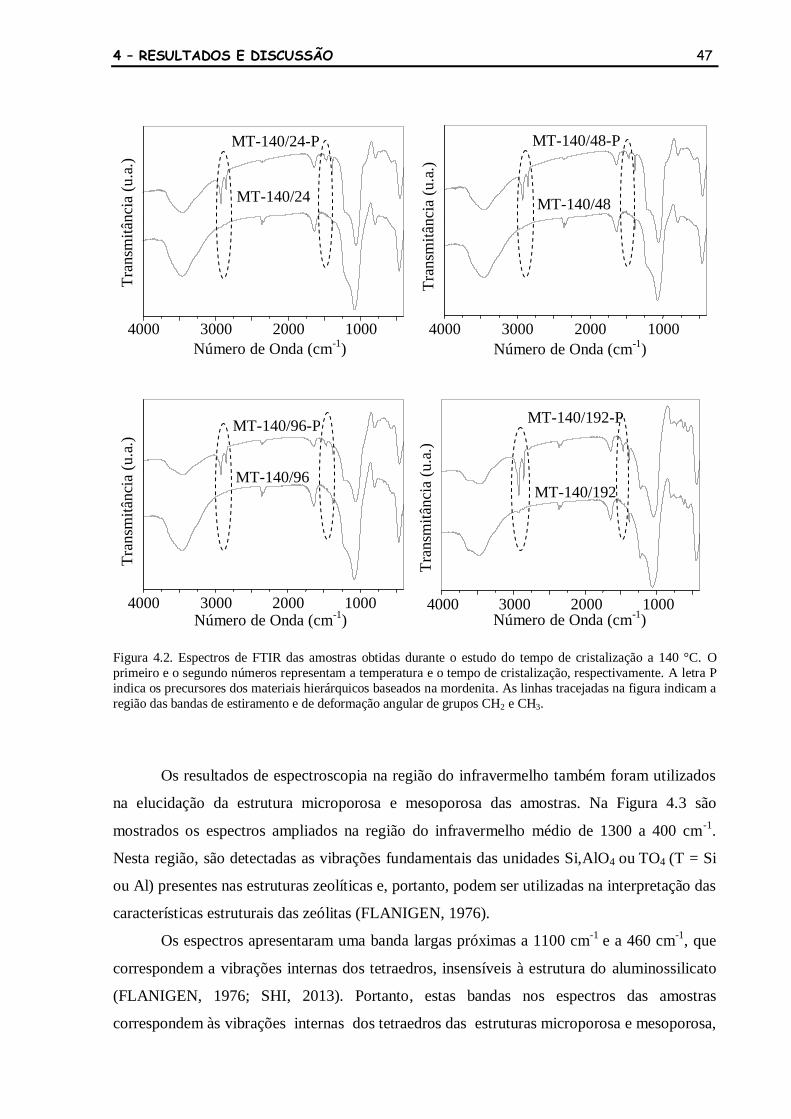
4 – RESULTADOS E DISCUSSÃO 47
Figura 4.2. Espectros de FTIR das amostras obtidas durante o estudo do tempo de cristalização a 140 °C. O
primeiro e o segundo números representam a temperatura e o tempo de cristalização, respectivamente. A letra P
indica os precursores dos materiais hierárquicos baseados na mordenita. As linhas tracejadas na figura indicam a
região das bandas de estiramento e de deformação angular de grupos CH2 e CH3.
Os resultados de espectroscopia na região do infravermelho também foram utilizados
na elucidação da estrutura microporosa e mesoporosa das amostras. Na Figura 4.3 são
mostrados os espectros ampliados na região do infravermelho médio de 1300 a 400 cm-1
.
Nesta região, são detectadas as vibrações fundamentais das unidades Si,AlO4 ou TO4 (T = Si
ou Al) presentes nas estruturas zeolíticas e, portanto, podem ser utilizadas na interpretação das
características estruturais das zeólitas (FLANIGEN, 1976).
Os espectros apresentaram uma banda largas próximas a 1100 cm-1
e a 460 cm-1
, que
correspondem a vibrações internas dos tetraedros, insensíveis à estrutura do aluminossilicato
(FLANIGEN, 1976; SHI, 2013). Portanto, estas bandas nos espectros das amostras
correspondem às vibrações internas dos tetraedros das estruturas microporosa e mesoporosa,
4000 3000 2000 1000
Tra
nsm
itân
cia
(u.a
.)
Número de Onda (cm-1
)
MT-140/96
MT-140/96-P
4000 3000 2000 1000
Tra
nsm
itân
cia
(u.a
.)
Número de Onda (cm-1
)
MT-140/24-P
MT-140/24
4000 3000 2000 1000
MT-140/48-P
Tra
nsm
itân
cia
(u.a
.)
Número de Onda (cm-1
)
MT-140/48
4000 3000 2000 1000
Tra
nsm
itân
cia
(u.a
.)
Número de Onda (cm-1
)
MT-140/192
MT-140/192-P
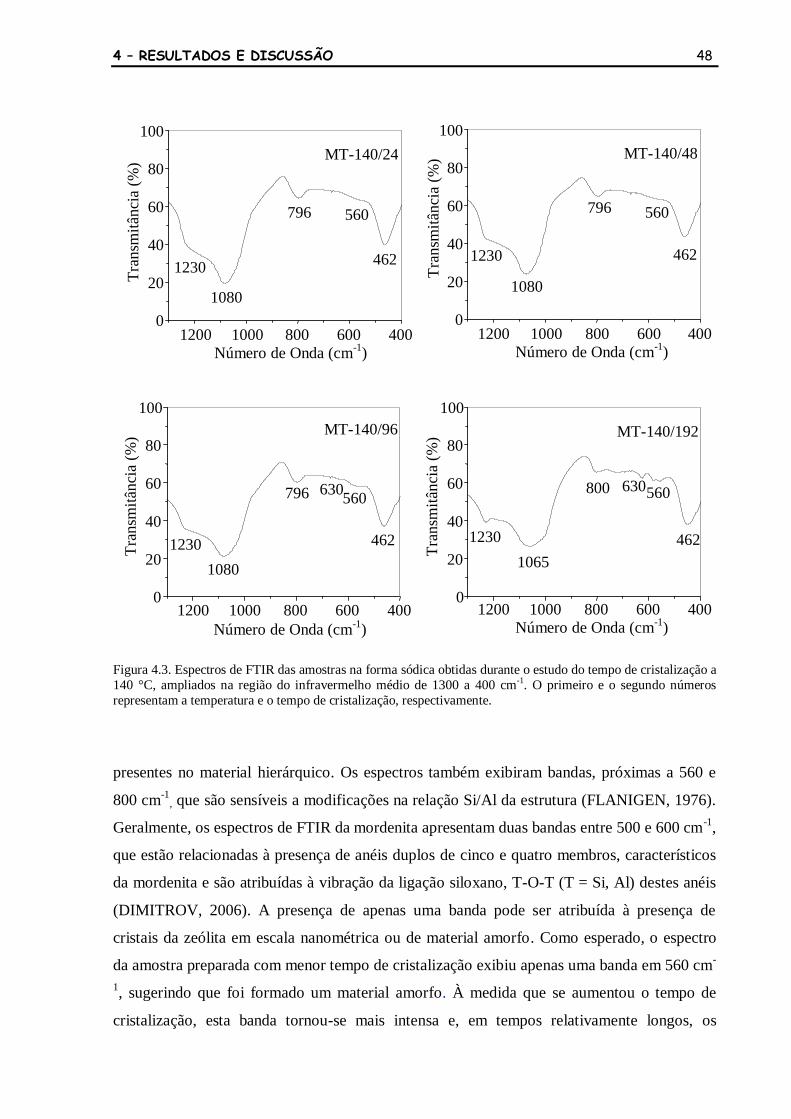
4 – RESULTADOS E DISCUSSÃO 48
Figura 4.3. Espectros de FTIR das amostras na forma sódica obtidas durante o estudo do tempo de cristalização a
140 °C, ampliados na região do infravermelho médio de 1300 a 400 cm-1. O primeiro e o segundo números
representam a temperatura e o tempo de cristalização, respectivamente.
presentes no material hierárquico. Os espectros também exibiram bandas, próximas a 560 e
800 cm-1
, que são sensíveis a modificações na relação Si/Al da estrutura (FLANIGEN, 1976).
Geralmente, os espectros de FTIR da mordenita apresentam duas bandas entre 500 e 600 cm-1
,
que estão relacionadas à presença de anéis duplos de cinco e quatro membros, característicos
da mordenita e são atribuídas à vibração da ligação siloxano, T-O-T (T = Si, Al) destes anéis
(DIMITROV, 2006). A presença de apenas uma banda pode ser atribuída à presença de
cristais da zeólita em escala nanométrica ou de material amorfo. Como esperado, o espectro
da amostra preparada com menor tempo de cristalização exibiu apenas uma banda em 560 cm-
1, sugerindo que foi formado um material amorfo. À medida que se aumentou o tempo de
cristalização, esta banda tornou-se mais intensa e, em tempos relativamente longos, os
1080
796 560
462
1200 1000 800 600 400 0
20
40
60
80
100
MT-140/48
Número de Onda (cm-1
)
Tra
nsm
itân
cia
(%)
1230
1080
796 560
462
1200 1000 800 600 400 0
20
40
60
80
100
MT-140/24
Número de Onda (cm-1
)
Tra
nsm
itân
cia
(%)
1230
1200 1000 800 600 400 0
20
40
60
80
100
MT-140/192
Número de Onda (cm-1
)
Tra
nsm
itân
cia
(%)
800 560
462
1065
1230
630
1080
796 560
462
1200 1000 800 600 400 0
20
40
60
80
100
MT-140/96
Tra
nsm
itân
cia
(%)
Número de Onda (cm-1
)
1230
630

4 – RESULTADOS E DISCUSSÃO 49
espectros exibiram duas bandas características da mordenita. O aumento da cristalinidade da
zeólita também pode ser confirmado através do aparecimento de uma banda em 630 cm-1
,
relacionada à presença de anéis duplos de seis membros, característicos da mordenita
(DIMITROV, 2006).
A banda próxima a 1200 cm-1
é atribuída à vibração assimétrica intertetraedros,
enquanto que a banda exibida em torno de 800 cm-1
pode ser atribuída ao estiramento
simétrico da ligação T-O (T = Si, Al). Esta banda está relacionada às vibrações que dependem
da estrutura e, portanto, podem ser deslocadas para frequências mais baixas com o aumento
do número de átomos de alumínio tetraédricos na rede (BRECK, 1984; FLANIGEN, 1976;
GUISNET, 2004). Este efeito está relacionado com a variação no comprimento da ligação T-
O devido à diminuição da constante de força média das ligações T-O. Uma vez que o íon
silício apresenta maior relação carga/raio quando comparado ao íon alumínio, os
comprimentos da ligação T-O aumentam em função do teor de alumínio (FLANIGEN, 1976;
GUISNET, 2004). Pode-se observar que todas as amostras apresentaram uma banda próxima
a 800 cm-1
, sugerindo que não houve alteração significativa na concentração de átomos de
alumínio na rede da zeólita, após as etapas de preparação.
Como a maioria das reações de transformação de hidrocarbonetos (craqueamento,
isomerização e transalquilação) é catalisada por sítios ácidos de Brønsted e os materiais
hierárquicos encontravam-se, inicialmente, na forma sódica, realizou-se a troca iônica dos
íons Na+ por íons NH4
+, seguido de calcinação, obtendo-se os materiais na forma protônica.
Comparando os espectros das amostras na forma protônica (Figura 4.4) com aqueles das
amostras na forma sódica (Figura 4.3), pode-se observar que não houve alterações
significativas na estrutura dos materiais hierárquicos baseados na mordenita, após o processo
de troca iônica.
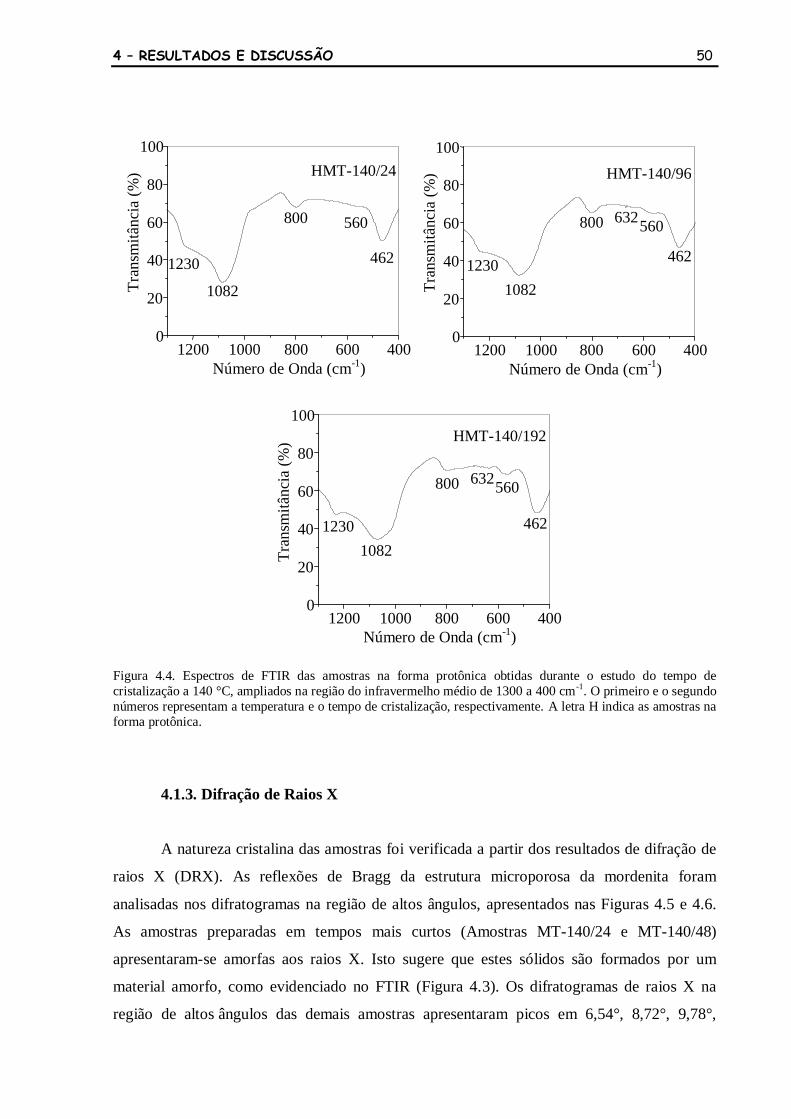
4 – RESULTADOS E DISCUSSÃO 50
Figura 4.4. Espectros de FTIR das amostras na forma protônica obtidas durante o estudo do tempo de
cristalização a 140 °C, ampliados na região do infravermelho médio de 1300 a 400 cm-1. O primeiro e o segundo
números representam a temperatura e o tempo de cristalização, respectivamente. A letra H indica as amostras na
forma protônica.
4.1.3. Difração de Raios X
A natureza cristalina das amostras foi verificada a partir dos resultados de difração de
raios X (DRX). As reflexões de Bragg da estrutura microporosa da mordenita foram
analisadas nos difratogramas na região de altos ângulos, apresentados nas Figuras 4.5 e 4.6.
As amostras preparadas em tempos mais curtos (Amostras MT-140/24 e MT-140/48)
apresentaram-se amorfas aos raios X. Isto sugere que estes sólidos são formados por um
material amorfo, como evidenciado no FTIR (Figura 4.3). Os difratogramas de raios X na
região de altos ângulos das demais amostras apresentaram picos em 6,54°, 8,72°, 9,78°,
800 560
462
1082
1230
632
1200 1000 800 600 400 0
20
40
60
80
100
Tra
nsm
itân
cia
(%)
Número de Onda (cm-1
)
HMT-140/96
800 560
462
1082
1230
632
1200 1000 800 600 400 0
20
40
60
80
100
Tra
nsm
itân
cia
(%)
Número de Onda (cm-1
)
HMT-140/192
800 560
462
1082
1230
1200 1000 800 600 400 0
20
40
60
80
100
Número de Onda (cm-1
)
Tra
nsm
itân
cia
(%) HMT-140/24
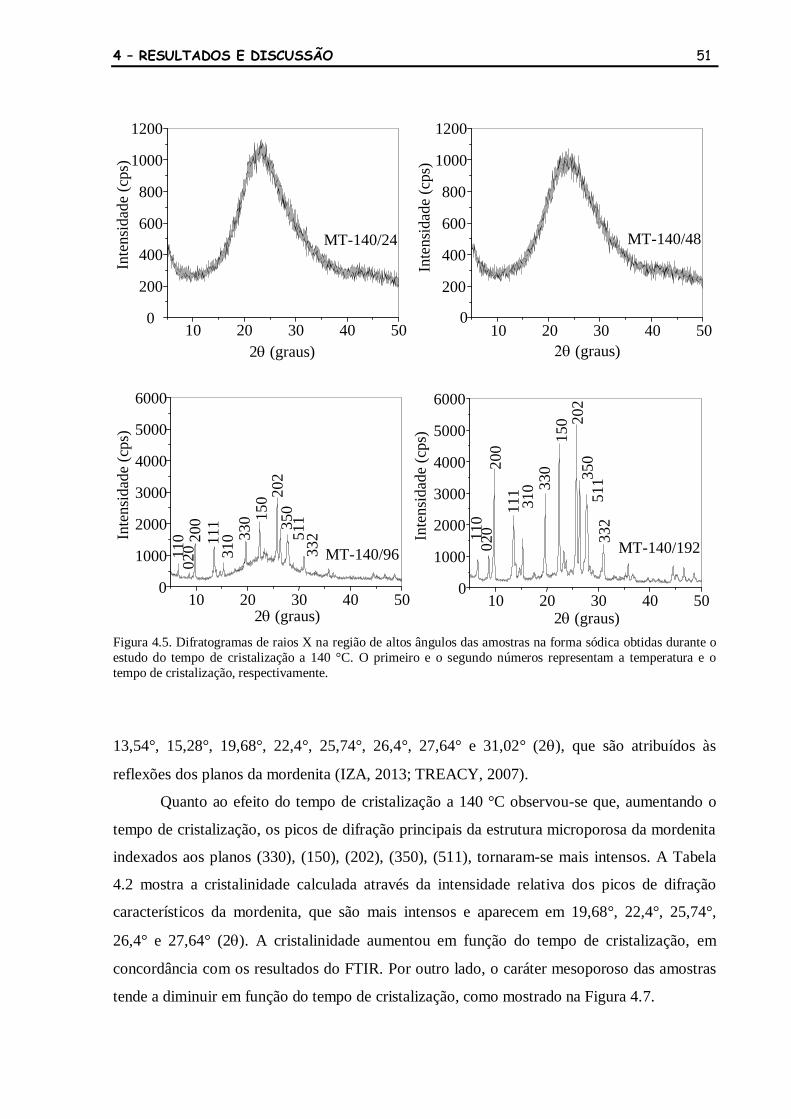
4 – RESULTADOS E DISCUSSÃO 51
Figura 4.5. Difratogramas de raios X na região de altos ângulos das amostras na forma sódica obtidas durante o
estudo do tempo de cristalização a 140 °C. O primeiro e o segundo números representam a temperatura e o
tempo de cristalização, respectivamente.
13,54°, 15,28°, 19,68°, 22,4°, 25,74°, 26,4°, 27,64° e 31,02° (2), que são atribuídos às
reflexões dos planos da mordenita (IZA, 2013; TREACY, 2007).
Quanto ao efeito do tempo de cristalização a 140 °C observou-se que, aumentando o
tempo de cristalização, os picos de difração principais da estrutura microporosa da mordenita
indexados aos planos (330), (150), (202), (350), (511), tornaram-se mais intensos. A Tabela
4.2 mostra a cristalinidade calculada através da intensidade relativa dos picos de difração
característicos da mordenita, que são mais intensos e aparecem em 19,68°, 22,4°, 25,74°,
26,4° e 27,64° (2). A cristalinidade aumentou em função do tempo de cristalização, em
concordância com os resultados do FTIR. Por outro lado, o caráter mesoporoso das amostras
tende a diminuir em função do tempo de cristalização, como mostrado na Figura 4.7.
10 20 30 40 50 0
200
400
600
800
1000
1200 In
tensi
dad
e (c
ps)
2 (graus)
MT-140/24
10 20 30 40 50 0
200
400
600
800
1000
1200
(graus)
Inte
nsi
dad
e (c
ps)
MT-140/48
10 20 30 40 50 0
1000
2000
3000
4000
5000
6000
2 (graus)
MT-140/96
Inte
nsi
dad
e (c
ps)
110
020 2
00
111
310 330
150 202
350
511
332
11
0
020
200
111
310 330
150 202
350
511
332
10 20 30 40 50
0
1000
2000
3000
4000
5000
6000
Inte
nsi
dad
e (c
ps)
2 (graus)
MT-140/192
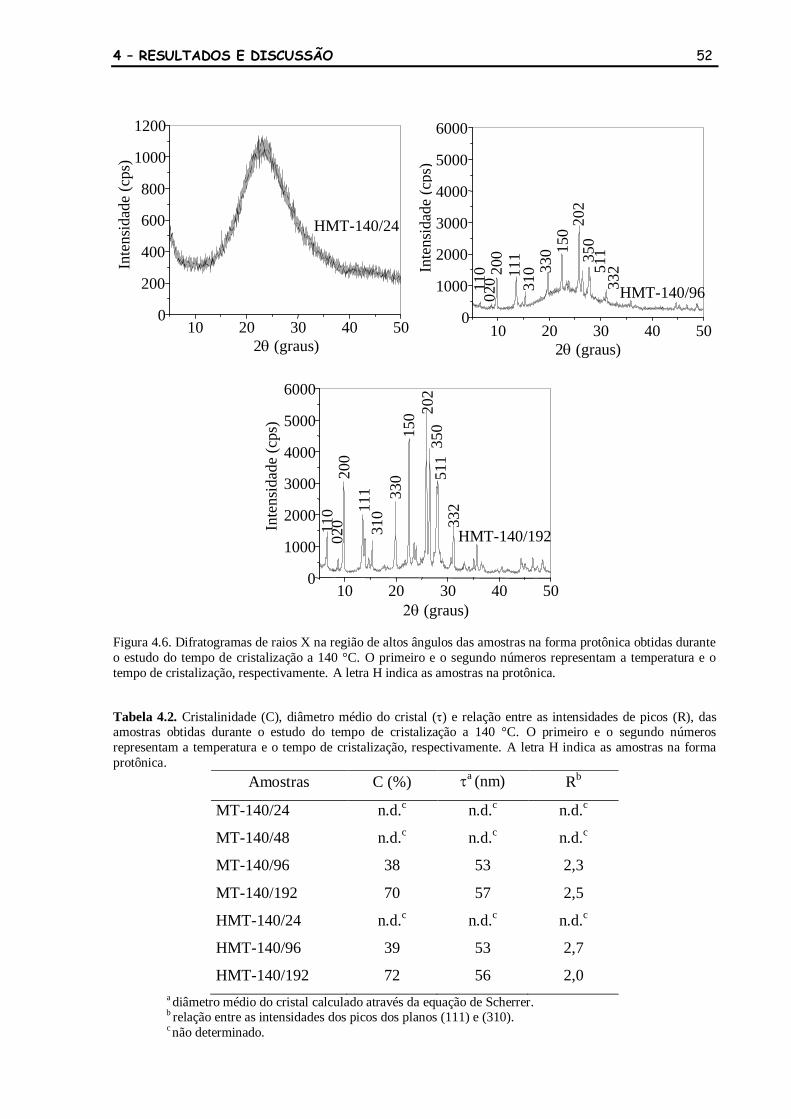
4 – RESULTADOS E DISCUSSÃO 52
Figura 4.6. Difratogramas de raios X na região de altos ângulos das amostras na forma protônica obtidas durante
o estudo do tempo de cristalização a 140 °C. O primeiro e o segundo números representam a temperatura e o
tempo de cristalização, respectivamente. A letra H indica as amostras na protônica.
Tabela 4.2. Cristalinidade (C), diâmetro médio do cristal () e relação entre as intensidades de picos (R), das amostras obtidas durante o estudo do tempo de cristalização a 140 °C. O primeiro e o segundo números
representam a temperatura e o tempo de cristalização, respectivamente. A letra H indica as amostras na forma
protônica.
Amostras C (%) a (nm) R
b
MT-140/24 n.d.c n.d.
c n.d.
c
MT-140/48 n.d.c n.d.
c n.d.
c
MT-140/96 38 53 2,3
MT-140/192 70 57 2,5
HMT-140/24 n.d.c n.d.
c n.d.
c
HMT-140/96 39 53 2,7
HMT-140/192 72 56 2,0
a diâmetro médio do cristal calculado através da equação de Scherrer. b relação entre as intensidades dos picos dos planos (111) e (310).
c não determinado.
10 20 30 40 50 0
200
400
600
800
1000
1200 In
tensi
dad
e (c
ps)
2 (graus)
HMT-140/24
11
0
020
200
111
310
330 150 2
02
350
511
332
10 20 30 40 50 0
1000
2000
3000
4000
5000
6000
Inte
nsi
dad
e (c
ps)
2 (graus)
HMT-140/96
11
0
020
200
111
310
330
150 202
35
0
511
332
10 20 30 40 50 0
1000
2000
3000
4000
5000
6000
Inte
nsi
dad
e (c
ps)
(graus)
HMT-140/192
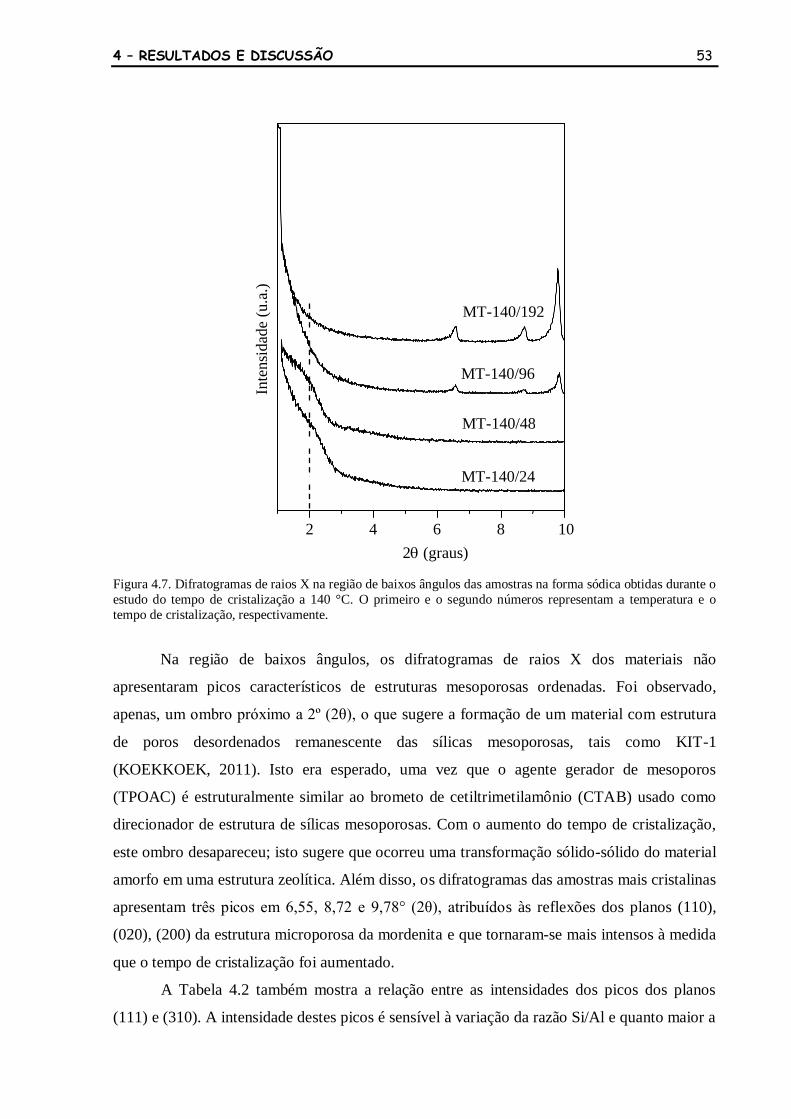
4 – RESULTADOS E DISCUSSÃO 53
Figura 4.7. Difratogramas de raios X na região de baixos ângulos das amostras na forma sódica obtidas durante o
estudo do tempo de cristalização a 140 °C. O primeiro e o segundo números representam a temperatura e o
tempo de cristalização, respectivamente.
Na região de baixos ângulos, os difratogramas de raios X dos materiais não
apresentaram picos característicos de estruturas mesoporosas ordenadas. Foi observado,
apenas, um ombro próximo a 2º (2θ), o que sugere a formação de um material com estrutura
de poros desordenados remanescente das sílicas mesoporosas, tais como KIT-1
(KOEKKOEK, 2011). Isto era esperado, uma vez que o agente gerador de mesoporos
(TPOAC) é estruturalmente similar ao brometo de cetiltrimetilamônio (CTAB) usado como
direcionador de estrutura de sílicas mesoporosas. Com o aumento do tempo de cristalização,
este ombro desapareceu; isto sugere que ocorreu uma transformação sólido-sólido do material
amorfo em uma estrutura zeolítica. Além disso, os difratogramas das amostras mais cristalinas
apresentam três picos em 6,55, 8,72 e 9,78° (2θ), atribuídos às reflexões dos planos (110),
(020), (200) da estrutura microporosa da mordenita e que tornaram-se mais intensos à medida
que o tempo de cristalização foi aumentado.
A Tabela 4.2 também mostra a relação entre as intensidades dos picos dos planos
(111) e (310). A intensidade destes picos é sensível à variação da razão Si/Al e quanto maior a
2 4 6 8 10
MT-140/192
MT-140/24
MT-140/48
MT-140/96
Inte
nsi
dad
e (u
.a.)
2 (graus)

4 – RESULTADOS E DISCUSSÃO 54
relação entre as intensidades desse picos, maior a razão Si/Al da zeólita (ITABASHI, 1984;
RAMOS; 2013). Apesar dos valores aproximados, há uma tendência de aumento em função
do tempo de cristalização. Isto se deve à desaluminação durante a etapa de cristalização do
sólido. Na Tabela 4.2 também são mostrados os diâmetros médios dos cristais. Pode-se
observar que todas as amostras apresentaram tamanho médio dos cristais próximo a 50 nm.
Por outro lado, o diâmetro do cristal tende a aumentar em função do aumento do tempo de
cristalização.
Através das Figuras 4.5 a 4.6, também foi possível observar que a estrutura
microporosa dos materiais hierárquicos não foi afetada significativamente pela substituição do
cátion alcalino por íons H+. Esse resultado indicou que os materiais obtidos apresentam
estabilidade térmica.
4.1.4. Análise Textural a Partir de Dados de Adsorção/Dessorção de Nitrogênio
A presença de micro e mesoporos nas amostras na forma sódica e protônica foi
confirmada através de medidas de adsorção/dessorção de nitrogênio, cujos resultados
produziram isotermas intermediárias entre os tipos I e IV (Figuras 4.8 e 4.9), características de
materiais microporosos e mesoporosos (SCHNEIDER, 1995).
Os materiais cristalizados em tempos mais curtos (Amostra MT-140/24 e Amostra
MT-140/48) exibiram isotermas mais próximas do tipo IV e com um laço estreito de histerese
do tipo H4, na faixa de pressões relativas P/P0 entre 0,4 a 0,9. Este laço de histerese, com
formato retangular, está associado à condensação capilar em uma estrutura de mesoporos
secundários, proveniente do espaçamento interparticular. Estas amostras possuem
mesoporosos com diâmetros pequenos, como mostrado na Tabela 4.3. Com o aumento do
tempo de cristalização, a curva se modifica em direção a uma isoterma do tipo I com uma
histerese intermediaria entre o tipo H1 e H4, o que sugere a formação de diferentes
mesoporos. A histerese do tipo H1 está relacionada a uma estrutura de mesoporos confinados,
enquanto que a histerese do tipo H4 indica a presença de mesoporos formados irregularmente
e pelo menos uma ordem de magnitude maior do que os mesoporos confinados (BAGSHAW,
2003). Além disso, o laço de histerese torna-se mais alargado, devido ao aumento do diâmetro
dos mesoporos (Tabela 4.3).
Todas as amostras apresentaram estreitas distribuições de tamanho de poros, como
mostrado na Figura 4.10. Porém, o diâmetro médio dos mesoporos tende a aumenta em
função do tempo de cristalização. Além disso, a Amostra MT-140/192 apresentou três picos

4 – RESULTADOS E DISCUSSÃO 55
Figura 4.8. Isotermas de adsorção (--) e dessorção (-o-) de nitrogênio das amostras na forma sódica obtidas durante o estudo do tempo de cristalização a 140 °C. O primeiro e o segundo números representam a temperatura
e o tempo de cristalização, respectivamente.
na curva de distribuição de tamanho de poros, devido à formação de mesoporos secundários
com diferentes diâmetros durante a cristalização.
As amostras apresentaram elevadas áreas de microporos, áreas de mesoporos e áreas
externas como mostrado na Tabela 4.3. Como esperado, a área de microporos e mesoporos
das amostras variaram em função da cristalinidade da mordenita formada, calculada através
da intensidade relativa das amostras em relação à Amostra HMT-170/96 (considerada como
padrão). Como esperado, o aumento da cristalinidade da mordenita produziu um aumento na
área e volume de microporos e uma redução na área e volume de mesoporos das amostras.
Através da Tabela 4.3, e das Figuras 4.8 a 4.9, também foi possível observar que as
propriedades texturais dos sólidos não foram afetadas significativamente pela substituição do
cátion alcalino por íons H+. Esses resultados indicam que os materiais obtidos apresentam
estabilidade térmica.
0,0 0,2 0,4 0,6 0,8 1,0 0
100
200
300
MT-140/24
Volu
me
Adso
rvid
o (
cm3.g
-1)
Pressão Relativa (P/P0)
0,0 0,2 0,4 0,6 0,8 1,0 0
100
200
300
Pressão Relativa (P/P0)
Volu
me
Adso
rvid
o (
cm3.g
-1)
MT-140/48
0,0 0,2 0,4 0,6 0,8 1,0 0
100
200
300
MT-140/96
Pressão Relativa (P/P0)
Volu
me
Adso
rvid
o (
cm3.g
-1)
0,0 0,2 0,4 0,6 0,8 1,0 0
100
200
300
Volu
me
Adso
rvid
o (
cm3.g
-1)
Pressao Relativa (P/P0)
MT-140/192
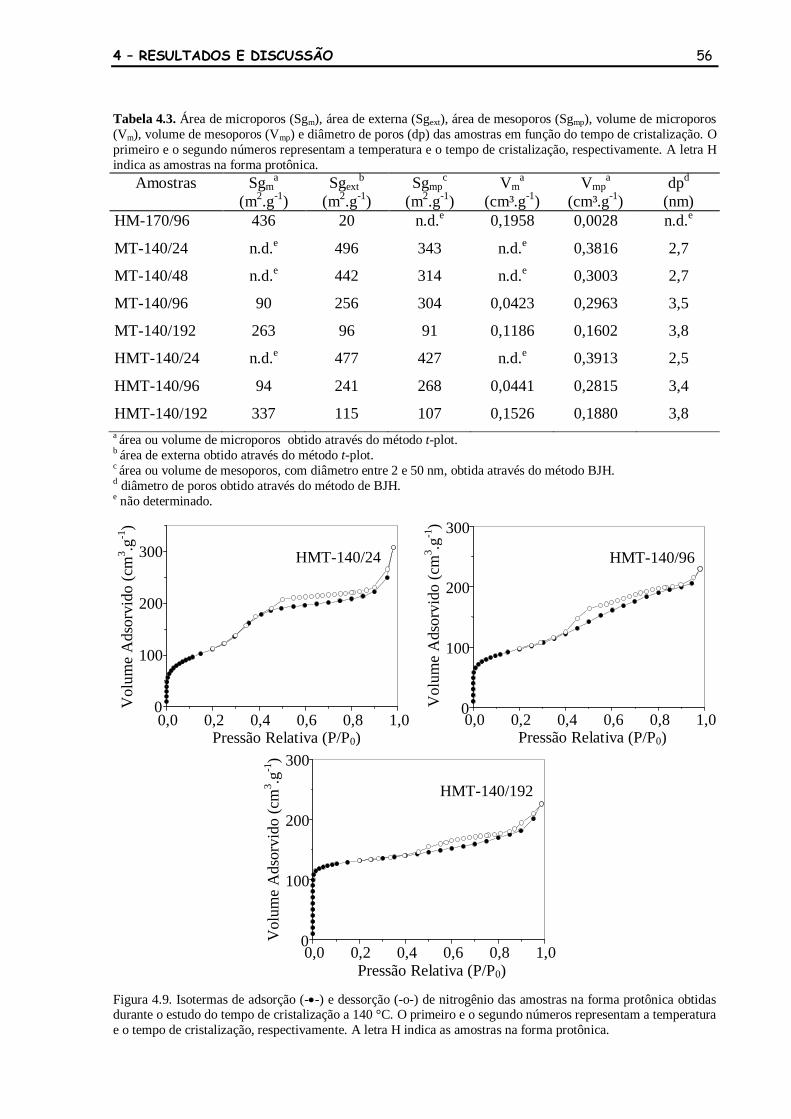
4 – RESULTADOS E DISCUSSÃO 56
Tabela 4.3. Área de microporos (Sgm), área de externa (Sgext), área de mesoporos (Sgmp), volume de microporos
(Vm), volume de mesoporos (Vmp) e diâmetro de poros (dp) das amostras em função do tempo de cristalização. O
primeiro e o segundo números representam a temperatura e o tempo de cristalização, respectivamente. A letra H
indica as amostras na forma protônica.
Amostras Sgma
(m2.g
-1)
Sgextb
(m2.g
-1)
Sgmpc
(m2.g
-1)
Vma
(cm³.g-1
)
Vmpa
(cm³.g-1
)
dpd
(nm)
HM-170/96 436 20 n.d.e 0,1958 0,0028 n.d.
e
MT-140/24 n.d.e 496 343 n.d.
e 0,3816 2,7
MT-140/48 n.d.e 442 314 n.d.
e 0,3003 2,7
MT-140/96 90 256 304 0,0423 0,2963 3,5
MT-140/192 263 96 91 0,1186 0,1602 3,8
HMT-140/24 n.d.e 477 427 n.d.
e 0,3913 2,5
HMT-140/96 94 241 268 0,0441 0,2815 3,4
HMT-140/192 337 115 107 0,1526 0,1880 3,8
a área ou volume de microporos obtido através do método t-plot. b área de externa obtido através do método t-plot. c área ou volume de mesoporos, com diâmetro entre 2 e 50 nm, obtida através do método BJH. d diâmetro de poros obtido através do método de BJH. e não determinado.
Figura 4.9. Isotermas de adsorção (--) e dessorção (-o-) de nitrogênio das amostras na forma protônica obtidas durante o estudo do tempo de cristalização a 140 °C. O primeiro e o segundo números representam a temperatura
e o tempo de cristalização, respectivamente. A letra H indica as amostras na forma protônica.
0,0 0,2 0,4 0,6 0,8 1,0 0
100
200
300
Volu
me
Adso
rvid
o (
cm3.g
-1)
Pressão Relativa (P/P0)
HMT-140/192
0,0 0,2 0,4 0,6 0,8 1,0 0
100
200
300
Volu
me
Adso
rvid
o (
cm3.g
-1)
Pressão Relativa (P/P0)
HMT-140/24
0,0 0,2 0,4 0,6 0,8 1,0 0
100
200
300
Volu
me
Adso
rvid
o (
cm3.g
-1)
Pressão Relativa (P/P0)
HMT-140/96
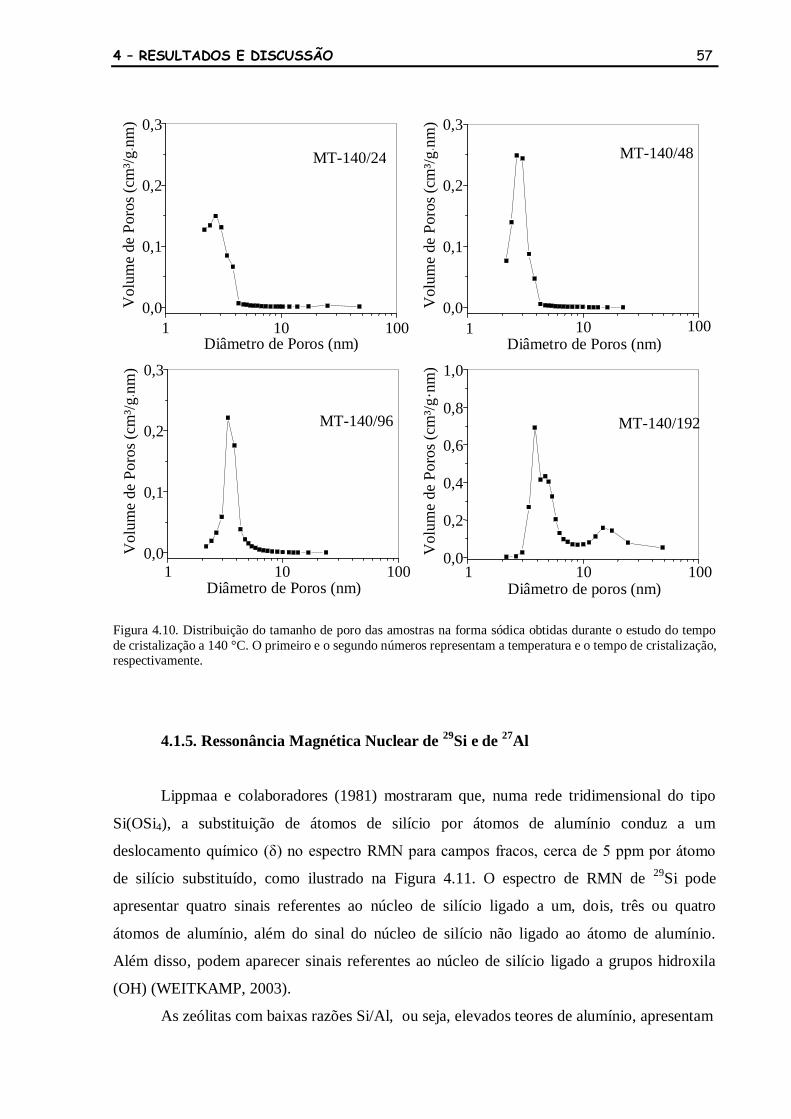
4 – RESULTADOS E DISCUSSÃO 57
Figura 4.10. Distribuição do tamanho de poro das amostras na forma sódica obtidas durante o estudo do tempo
de cristalização a 140 °C. O primeiro e o segundo números representam a temperatura e o tempo de cristalização, respectivamente.
4.1.5. Ressonância Magnética Nuclear de 29
Si e de 27
Al
Lippmaa e colaboradores (1981) mostraram que, numa rede tridimensional do tipo
Si(OSi4), a substituição de átomos de silício por átomos de alumínio conduz a um
deslocamento químico (δ) no espectro RMN para campos fracos, cerca de 5 ppm por átomo
de silício substituído, como ilustrado na Figura 4.11. O espectro de RMN de 29
Si pode
apresentar quatro sinais referentes ao núcleo de silício ligado a um, dois, três ou quatro
átomos de alumínio, além do sinal do núcleo de silício não ligado ao átomo de alumínio.
Além disso, podem aparecer sinais referentes ao núcleo de silício ligado a grupos hidroxila
(OH) (WEITKAMP, 2003).
As zeólitas com baixas razões Si/Al, ou seja, elevados teores de alumínio, apresentam
dV
/dr
(cm
3g
-1A
)
1 10 100
0,0
0,1
0,2
0,3
MT-140/24
Volu
me
de
Poro
s (c
m³/
g·n
m)
Diâmetro de Poros (nm) 1 10 100
0,0
0,1
0,2
0,3
MT-140/48
Diâmetro de Poros (nm)
Volu
me
de
Poro
s (c
m³/
g·n
m)
1 10 100
0,0
0,1
0,2
0,3
Diâmetro de Poros (nm)
Volu
me
de
Poro
s (c
m³/
g·n
m)
MT-140/96
1 10 100 0,0
0,2
0,4
0,6
0,8
1,0
MT-140/192 V
olu
me
de
Poro
s (c
m³/
g·n
m)
Diâmetro de poros (nm)

4 – RESULTADOS E DISCUSSÃO 58
Figura 4.11. Deslocamento químico devido à substituição de átomos de silício por átomos de alumínio nas
zeólitas (LIPPMA, 1981).
espectros com os sinais dos diferentes ambientes químicos do silício bem definidos. Todos os
átomos de alumínio estão conectados ao núcleo SiO4, pois não ocorrem ligações do tipo Al-
O-Al, de acordo com a regra de Loewenstein, segundo a qual “sempre que dois tetraedros
estiverem ligados por um átomo de oxigênio, se o centro de um deles for ocupado por um
átomo de alumínio, o centro do outro tetraedro deve ser ocupado por um átomo de silício ou
outro íon pequeno com valência quatro ou maior”. Nas zeólitas com elevada razão Si/Al, que
é o caso das amostras sintetizadas neste trabalho, não é possível quantificar a razão Si/Al
através do espectro de RMN de 29
Si, devido às dificuldades de integração dos picos referentes
as espécies Si(1, 2, 3 ou 4Al). Como a quantidade de alumínio nessas amostras é muito
pequena, praticamente somente o sinal da espécie Si(0Al) apareceu no espectro.
A Figura 4.12 apresenta os espectro de RMN de 29
Si das amostras calcinadas. Pode-se
observar a presença de um sinal intenso em aproximadamente -110 ppm, atribuído às espécies
Si(0Al). Além disso, a Amostra MT-140/24 apresentou dois sinais sobrepostos, em torno de -
105 e -95 ppm, relacionados às espécies Si(1Al) e Si(2Al), respectivamente. Não foram
observados os sinais referentes às espécies Si(3Al) e Si(4Al), o que já era esperado uma vez
que as amostras obtidas possuem baixas quantidades de alumínio. Devido à dificuldade de
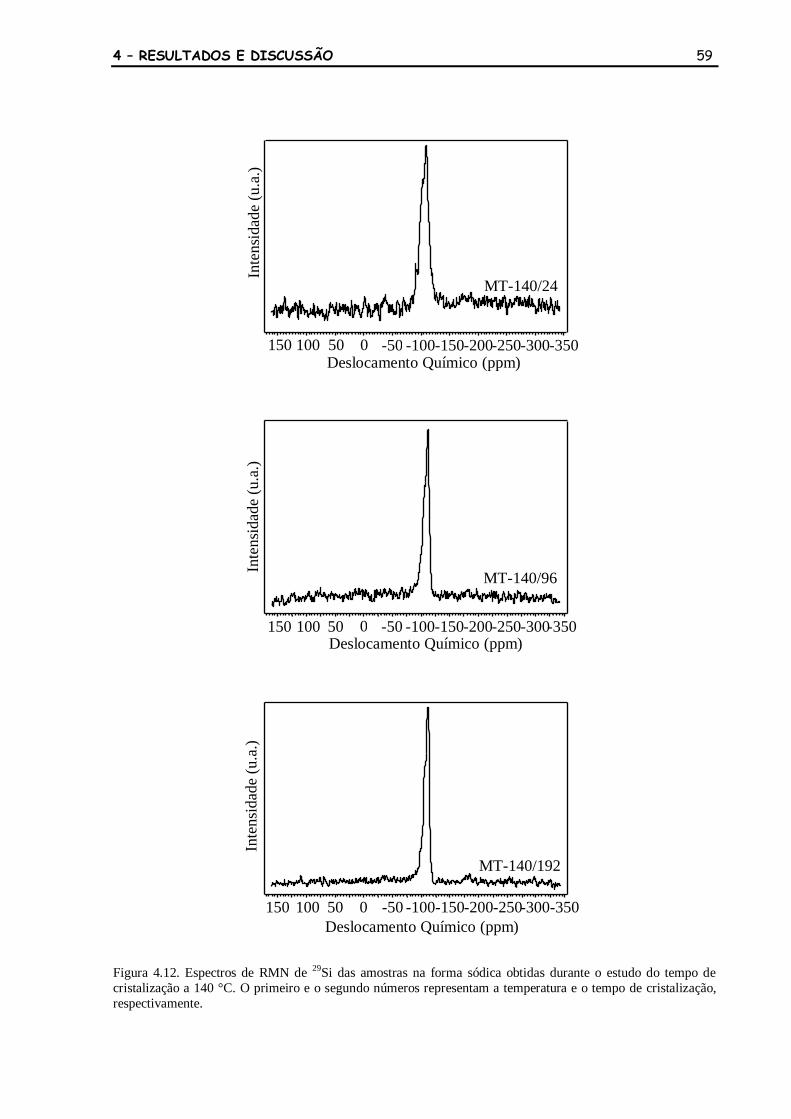
4 – RESULTADOS E DISCUSSÃO 59
Figura 4.12. Espectros de RMN de 29Si das amostras na forma sódica obtidas durante o estudo do tempo de
cristalização a 140 °C. O primeiro e o segundo números representam a temperatura e o tempo de cristalização,
respectivamente.
MT-140/96
Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm) 150 100 50 0 -50 -100 -150 -200 -250 -300 -350
MT-140/192
150 100 50 0 -50 -100 -150 -200 -250 -300 -350
Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm)
MT-140/24
Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm) 150 100 50 0 -50 -100 -150 -200 -250 -300 -350

4 – RESULTADOS E DISCUSSÃO 60
integração do sinal da espécie Si(1Al) e Si(2Al), mesmo utilizando técnicas de deconvolução,
não foi possível calcular a razão Si/Al.
Através da Figura 4.13, pode-se comparar os espectros de RMN de 29
Si das amostras,
antes e após a calcinação. Em todos os casos, ocorre uma diminuição da intensidade dos sinais
atribuídos as espécies Si(1Al) e Si(2Al), nos espectros das amostras após a calcinação; isto
indica que, durante a calcinação, está ocorrendo a desaluminação das amostras. Nos espectros
de RMN de 29
Si das amostras não calcinadas foi possível observar um sinal fraco em
aproximadamente -60 ppm, que é atribuído às espécies de silício ligadas a átomos de carbono
(CARVALHO, 2012; SERRANO, 2008), indicando que ocorreu um acoplamento do agente
gerador da mesoporosidade na superfícies da zeólita e que este composto orgânico
permaneceu ligado à superfície durante a etapa de cristalização, realizando sua função.
Após a calcinação das amostras, os espectros não apresentaram esse sinal, evidenciando que a
parte orgânica do agente funcionalizante foi eliminado durante a calcinação.
O espectro de ressonância magnética nuclear de 27
Al de zeólitas apresentou somente
um ambiente químico, correspondente ao átomo de alumínio tetraedricamente coordenado, na
estrutura zeolítica; por isso, o espectro de RMN de 27
Al das zeólitas são simples, apresentando
apenas um pico em torno de 60 ppm referente ao átomo de alumínio tetraédrico (SHI, 2013).
Entretanto, ao sofrerem processo de desaluminação, ou seja, quando ocorre a saída de átomos
de alumínio da rede zeolítica, os espectros RMN de 27
Al apresentam um pico adicional, em
aproximadamente 0 ppm, referente ao íon alumínio fora da rede, que passa a ter uma
coordenação octaédrica (GUISNET, 2004; SHI, 2013). Todas as amostras, como sintetizadas,
apresentaram espectros com apenas um pico em aproximadamente 60 ppm, atribuído ao
átomo de alumínio tetraédrico da rede da zeólita, como pode ser observado na Figura 4.14.
Através dos espectros da Figura 4.14, pode-se inferir sobre as transformações sofridas
pelas amostras durante o processo de calcinação. Antes desse processo, as amostras
apresentaram espectros com apenas o pico do átomo de alumínio tetraédrico, indicando que
todos os átomos de alumínio estão na rede da zeólita. Entretanto, após a calcinação, nota-se o
aparecimento do pico em aproximadamente 0 ppm, indicando que houve uma destruição da
estrutura ou desaluminação da amostra durante esse processo. Todas as amostras calcinadas
apresentaram espectros com os picos referentes ao átomo de alumínio tetraédrico (dentro da
rede) e do íon alumínio octaédrico (fora da rede). As percentagens dos picos dos átomos de
alumínio tetraédricos e octaédricos obtidos através dos espectros das amostras calcinadas, são
mostradas na Tabela 4.4. Observou-se que a maior parte dos átomos de alumínio tetraédricos
da Amostra MT-140/24 (menor tempo de cristalização) modificou sua coordenação para
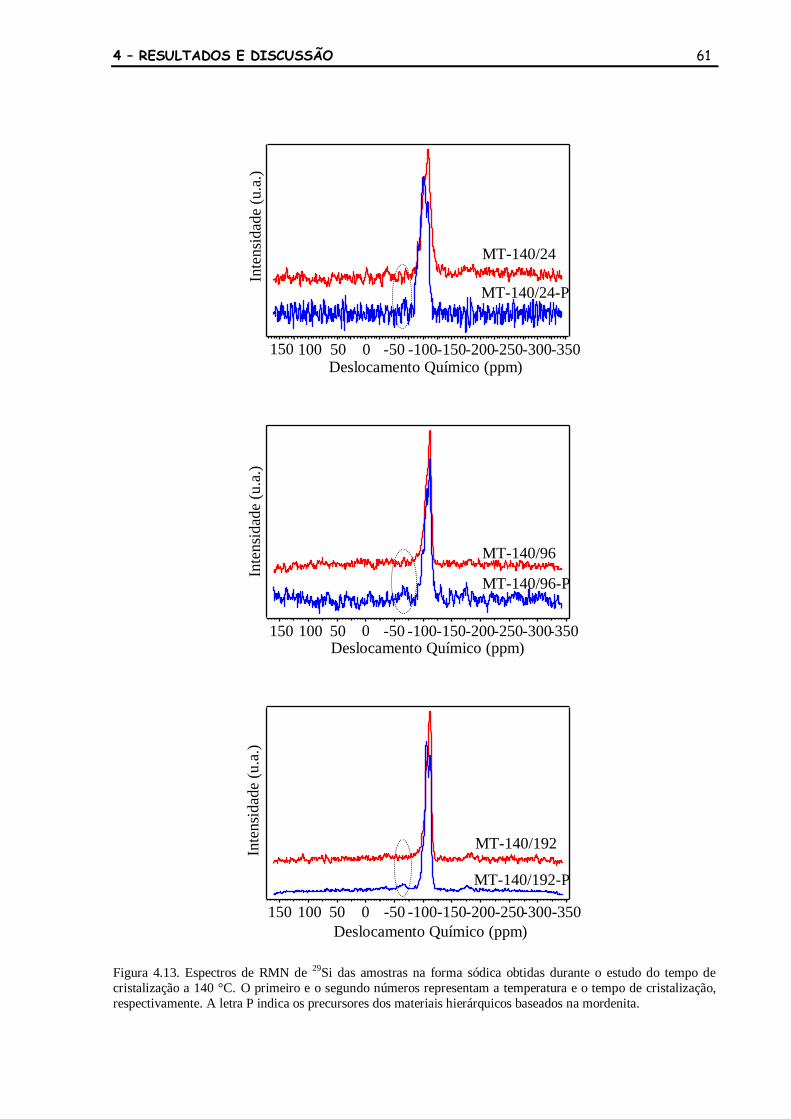
4 – RESULTADOS E DISCUSSÃO 61
Figura 4.13. Espectros de RMN de 29Si das amostras na forma sódica obtidas durante o estudo do tempo de
cristalização a 140 °C. O primeiro e o segundo números representam a temperatura e o tempo de cristalização,
respectivamente. A letra P indica os precursores dos materiais hierárquicos baseados na mordenita.
MT-140/192-P
MT-140/192
150 100 50 0 -50 -100 -150 -200 -250 -300 -350
Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm)
MT-140/24-P
MT-140/24 In
tensi
dad
e (u
.a.)
Deslocamento Químico (ppm) 150 100 50 0 -50 -100 -150 -200 -250 -300 -350
MT-140/96-P
MT-140/96
Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm) 150 100 50 0 -50 -100 -150 -200 -250 -300 -350
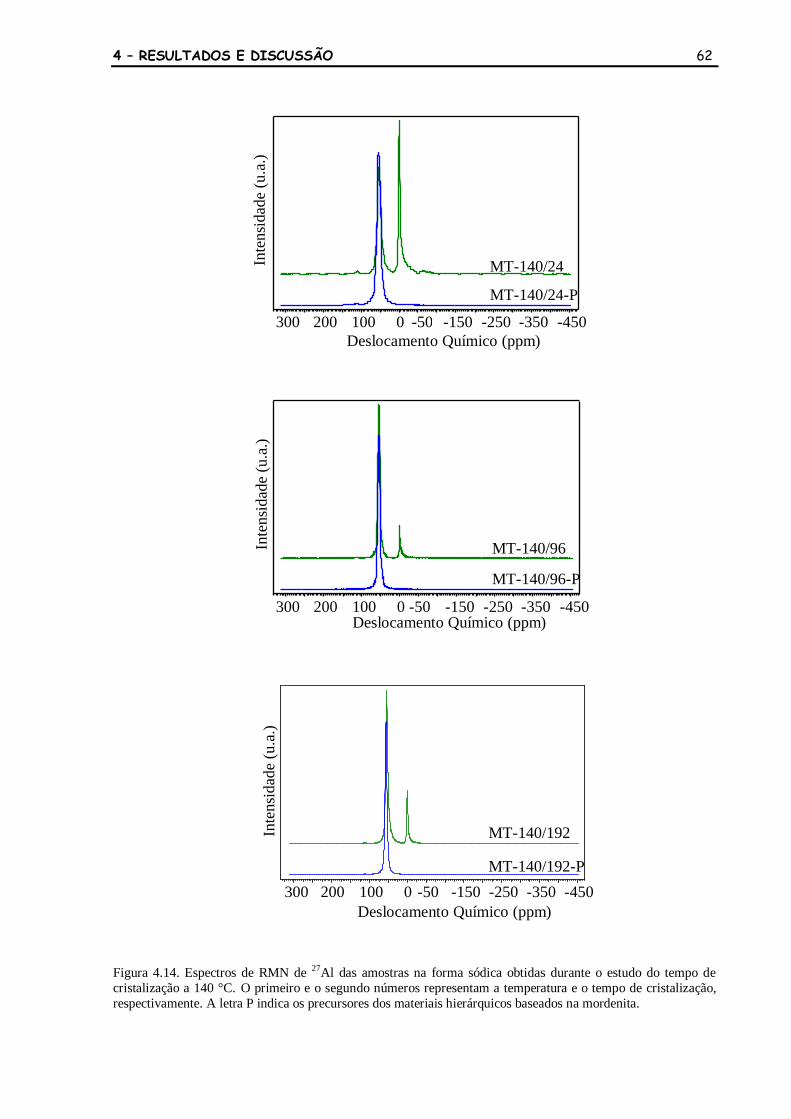
4 – RESULTADOS E DISCUSSÃO 62
Figura 4.14. Espectros de RMN de 27Al das amostras na forma sódica obtidas durante o estudo do tempo de
cristalização a 140 °C. O primeiro e o segundo números representam a temperatura e o tempo de cristalização,
respectivamente. A letra P indica os precursores dos materiais hierárquicos baseados na mordenita.
MT-140/24-P
MT-140/24 Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm)
300 200 100 0 -50 -150 -250 -350 -450
300 200 100 0 -50 -150 -250 -350 -450
MT-140/192-P
MT-140/192 Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm)
300 200 100 0 -50 -150 -250 -350 -450
MT-140/96-P
MT-140/96 Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm)

4 – RESULTADOS E DISCUSSÃO 63
Tabela 4.4. Quantidade de alumínio tetraédrico (Altetra) e quantidade de alumínio octaédrico (Alocta) das amostras
obtidas durante o estudo do tempo de cristalização a 140 °C. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente.
Amostras Altetra (%) Alocta (%)
MT-140/24 41 59
MT-140/48 n.d.a n.d.
a
MT-140/96 83 17
MT-140/192 74 26
a não determinado.
octaédrica, durante o processo de calcinação. Isto indica que o material amorfo não possui
estabilidade termica e sua estrutura mesoporosa é destruída parcialmente. Com o aumento do
tempo de cristalização, ocorre uma transformação do material amorfo em uma estrutura
zeolítica com elevada estabilidade térmica, e, portanto, os picos de ressonância em 0 ppm é
atribuído ao processo de desaluminação. Em tempos de cristalização relativamente longos,
pode ocorrer a redispersão dos átomos de alumínio e a desaluminação aumenta
(KOEKKOEK, 2011). Estes resultados estão em concordância com os valores da relação
entre as intensidades dos picos dos planos (111) e (310) nos difratogramas de raios X das
amostras (Tabela 4.2). Após a troca iônica, também ocorreu uma tendência em aumentar o
valor de R, devido à desaluminação durante este processo.
4.1.6. Microscopia Eletrônica de Varredura
As micrografias eletrônicas de varredura das amostras calcinadas, com aumentos entre
3500 a 5000 vezes, são apresentadas na Figura 4.15. Pode-se notar que as amostras tendem a
formar aglomerados. Isto já era esperado, visto que as partículas possuem uma elevada
energia superficial e, então, tendem a se aglomerar, de modo a diminuir a superfície e,
portanto, a energia.
A morfologia das amostras variou em função do tempo de cristalização. Observou-se
que os sólidos obtidos em tempos relativamente curtos apresentaram um aspecto esponjoso,
característico de partículas mesoporosas desordenadas (KOEKKOEK, 2011), como
evidenciado através da difração de raios X. O aumento do tempo de cristalização favoreceu a
transformação sólido-sólido da sílica amorfa em um material cristalino; dessa forma, a
Amostra MT-140/24 apresentou apenas aspecto esponjoso, enquanto que a Amostra

4 – RESULTADOS E DISCUSSÃO 64
Figura 4.15. Micrografias eletrônicas de varredura da (a) Amostra MT-140/24, (b) Amostra MT-140/48, (c)
Amostra MT-140/96 e (d) Amostra MT-140/192. O primeiro e o segundo números representam a temperatura e
o tempo de cristalização, respectivamente.
MT-140/192 é formada por cristais zeolíticos. Na micrografia da Amostra MT-140/96 foram
observadas partículas esféricas e partículas mesoporosas desordenadas. Isto indica que esta
amostra é formada por duas fases: uma fase cristalina referente à mordenita e uma fase
mesoporosa desordenada, como observado em trabalho anterior com a zeólita mordenita (LI,
2009). A morfologia destes materiais é similar àquela apresentada por outras zeólitas com
estrutura hierárquica de poros preparadas usando organossilanos, tais como a ZSM-5
(KOEKKOEK, 2011) e zeólita A (CHO, 2011). Quando o tempo foi suficientemente elevado
(Amostra MT-140/192), foram observados aglomerados de cristais na forma de placas,
característicos da mordenita. Porém, uma quantidade não negligenciável da fase mesoporosa
amorfa ainda pode ser detectada, como mostrado na região destacada na Figura 4.15d.
(d)
(a)
(c)
(b)

4 – RESULTADOS E DISCUSSÃO 65
Também foi possível observar que o diâmetro do cristal tende a aumentar em função do
aumento do tempo de cristalização, como evidenciado pela difração de raios X (Tabela 4.2).
4.1.7. Medidas de Acidez por Dessorção de Amônia à Temperatura Programada
A partir de medidas de acidez por dessorção de amônia à temperatura programada, os
sítios ácidos presentes nas zeólitas pode ser diferenciados como fraco, moderado e forte, de
acordo com a temperatura de dessorção (RAMOS, 2005). Os sítios ácidos fracos e moderados
podem estar presentes tanto em zeólitas quanto em aluminossilicatos mesoporosos (RAMOS,
2005, WANG, 1998). Por outro lado, sítios ácidos fortes são comuns somente em zeólitas.
As curvas de acidez das amostras na forma protônica estão ilustradas na Figura 4.16.
Na curva de acidez da Amostra HMT-140/24 (menor tempo de cristalização), observou-se um
ombro próximo a 200 ºC e um pico de dessorção de amônia, com máximo em 300 °C. O
ombro em baixa temperatura pode ser atribuído a sítios ácidos fracos, enquanto aquele que
ocorre em temperatura mais elevada pode ser atribuído a sítios ácidos de força média
(RAMOS, 2005). Além disso, foi observado um ombro bem suave entre 500 e 600 ºC,
relacionado à presença de sítios ácidos mais fortes (RAMOS, 2005). A intensidade desses
picos aumenta em função da cristalinidade, sendo que a curva de acidez da Amostra HMT-
140/192 (maior tempo de cristalização) apresentou dessorção de amônia mais acentuada em
temperaturas superiores a 400 °C, o que é representativo da presença de sítios ácidos mais
fortes, fato esperado em função das propriedades ácidas da mordenita [LÓNYI, 2001].
Comparando as curvas de acidez dos materiais hierárquicos com aquela da mordenita
microporosa (Amostra HM-170/96), observa-se que a curva de acidez do sólido microporoso
apresentou três picos acima de 400 ºC, relacionados a sítios ácidos fortes em diferentes
localizações da rede da zeólita microporosa.
Na Tabela 4.5 são apresentados os resultados das medidas de acidez dos sólidos na
forma protônica em função da quantidade de amônia adsorvida por massa de sólido. A
quantidade de amônia dessorvida aumentou em função da cristalinidade da zeólita, de modo
que a Amostra HMT-140/192 apresentou um valor de acidez próximo àquele da mordenita
convencional. Pode-se observar que a maioria dos sítios ácidos, presentes na amostra
preparada em tempos relativamente curtos, são sítios de força média, enquanto que, a amostra
preparada em longos tempos possui maior quantidade de sítios ácidos fortes.
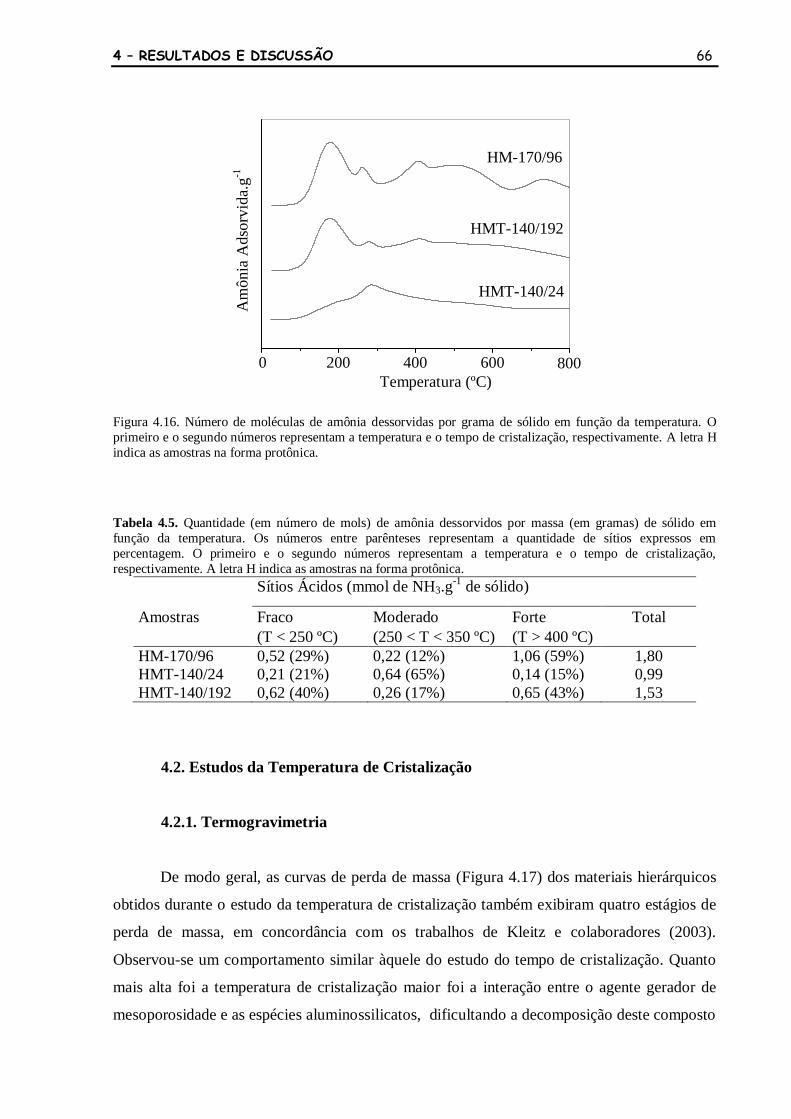
4 – RESULTADOS E DISCUSSÃO 66
Figura 4.16. Número de moléculas de amônia dessorvidas por grama de sólido em função da temperatura. O
primeiro e o segundo números representam a temperatura e o tempo de cristalização, respectivamente. A letra H
indica as amostras na forma protônica.
Tabela 4.5. Quantidade (em número de mols) de amônia dessorvidos por massa (em gramas) de sólido em
função da temperatura. Os números entre parênteses representam a quantidade de sítios expressos em
percentagem. O primeiro e o segundo números representam a temperatura e o tempo de cristalização,
respectivamente. A letra H indica as amostras na forma protônica.
Sítios Ácidos (mmol de NH3.g-1
de sólido)
Amostras Fraco
(T < 250 ºC)
Moderado
(250 < T < 350 ºC)
Forte
(T > 400 ºC)
Total
HM-170/96 0,52 (29%) 0,22 (12%) 1,06 (59%) 1,80
HMT-140/24 0,21 (21%) 0,64 (65%) 0,14 (15%) 0,99
HMT-140/192 0,62 (40%) 0,26 (17%) 0,65 (43%) 1,53
4.2. Estudos da Temperatura de Cristalização
4.2.1. Termogravimetria
De modo geral, as curvas de perda de massa (Figura 4.17) dos materiais hierárquicos
obtidos durante o estudo da temperatura de cristalização também exibiram quatro estágios de
perda de massa, em concordância com os trabalhos de Kleitz e colaboradores (2003).
Observou-se um comportamento similar àquele do estudo do tempo de cristalização. Quanto
mais alta foi a temperatura de cristalização maior foi a interação entre o agente gerador de
mesoporosidade e as espécies aluminossilicatos, dificultando a decomposição deste composto
0 200 400 600 800
A
mônia
Adso
rvid
a.g
-1
Temperatura (ºC)
HM-170/96
HMT-140/192
HMT-140/24
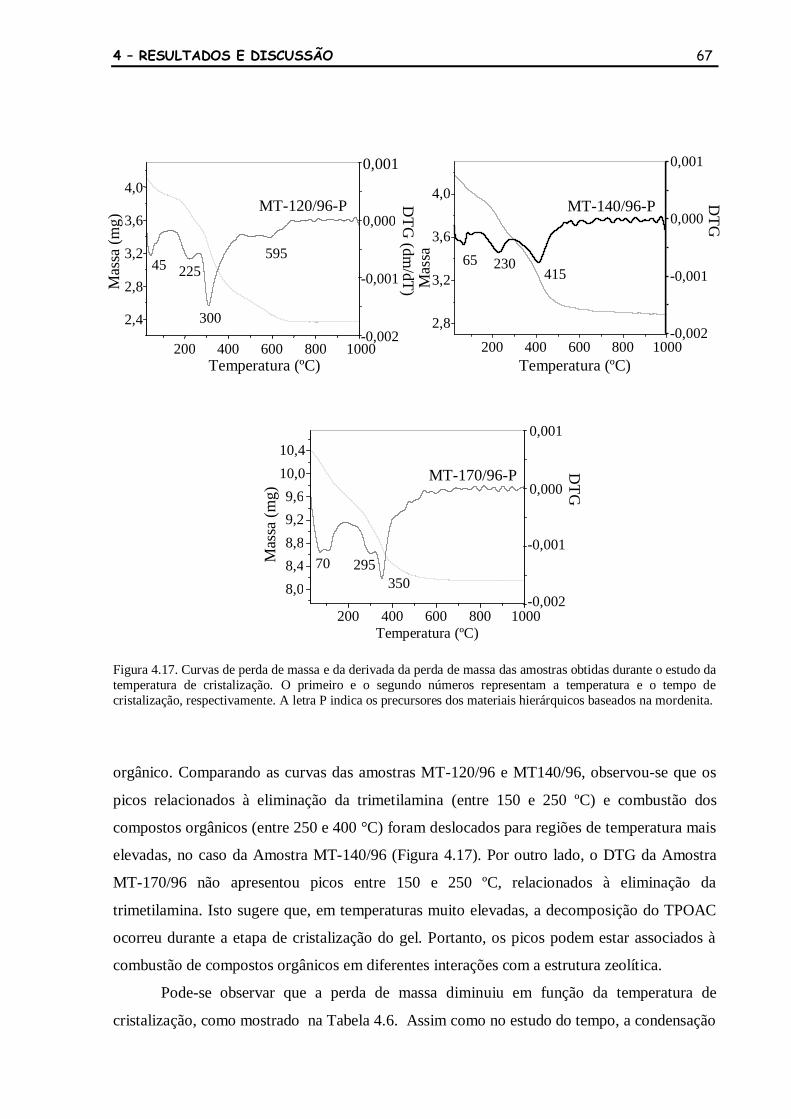
4 – RESULTADOS E DISCUSSÃO 67
Figura 4.17. Curvas de perda de massa e da derivada da perda de massa das amostras obtidas durante o estudo da
temperatura de cristalização. O primeiro e o segundo números representam a temperatura e o tempo de
cristalização, respectivamente. A letra P indica os precursores dos materiais hierárquicos baseados na mordenita.
orgânico. Comparando as curvas das amostras MT-120/96 e MT140/96, observou-se que os
picos relacionados à eliminação da trimetilamina (entre 150 e 250 ºC) e combustão dos
compostos orgânicos (entre 250 e 400 °C) foram deslocados para regiões de temperatura mais
elevadas, no caso da Amostra MT-140/96 (Figura 4.17). Por outro lado, o DTG da Amostra
MT-170/96 não apresentou picos entre 150 e 250 ºC, relacionados à eliminação da
trimetilamina. Isto sugere que, em temperaturas muito elevadas, a decomposição do TPOAC
ocorreu durante a etapa de cristalização do gel. Portanto, os picos podem estar associados à
combustão de compostos orgânicos em diferentes interações com a estrutura zeolítica.
Pode-se observar que a perda de massa diminuiu em função da temperatura de
cristalização, como mostrado na Tabela 4.6. Assim como no estudo do tempo, a condensação
-0,002
-0,001
0,000
0,001
MT-140/96-P DT
G
(dm
/dT
)
230 65 415
200 400 600 800 1000
2,8
3,2
3,6
4,0
Mas
sa
(mg)
Temperatura (ºC)
-0,002
-0,001
0,000
0,001
DT
G (d
m/d
T)
(dm
/dT
)
200 400 600 800 1000
2,4
2,8
3,2
3,6
4,0
Mas
sa (
mg)
Temperatura (ºC)
225 45
300
595
MT-120/96-P
-0,002
-0,001
0,000
0,001
DT
G
(dm
/dT
)
70
350
295
MT-170/96-P
200 400 600 800 1000
8,0
8,4
8,8
9,2
9,6
10,0
10,4
Temperatura (ºC)
Mas
sa (
mg)

4 – RESULTADOS E DISCUSSÃO 68
Tabela 4.6. Perda de massa total das amostras obtidas no estudo da temperatura de cristalização. O primeiro e o
segundo números representam a temperatura e o tempo de cristalização, respectivamente. A letra P indica os
precursores dos materiais hierárquicos baseados na mordenita.
Amostras Perda de massa (%)
MT-120/96-P 42
MT-140/96-P 31
MT-170/96-P 22
das espécies aluminossilicato aumentou com a temperatura de cristalização, reduzindo a
densidade de carga da superfície e, consequentemente, enfraquecendo as interações das
espécies TPOA+ com as espécies aluminossilicatos. Dessa forma, as espécies TPOA
+ que não
estão interagindo com as espécies aluminossilicatos são facilmente eliminadas durante a
lavagem do sólido. Como resultado, há uma diminuição da perda de massa em função do
aumento da cristalinidade. Além disso, elevadas temperaturas promovem a decomposição do
agente orgânico durante a etapa de cristalização do gel e os hidrocarbonetos resultantes são
facilmente eliminados durante a etapa de lavagem.
4.2.2. Espectroscopia no Infravermelho com Transformada de Fourier
Na Figura 4.18 são mostrados os espectros das amostras antes e após a troca iônica,
ampliados na região do infravermelho médio de 1300 a 400 cm-1
. Podem ser observadas
bandas características da zeólita mordenita em aproximadamente 460, 560, 800, 1080 e 1230
cm-1
(SHI, 2013). De modo similar ao estudo efeito do tempo de cristalização, observou-se
que a intensidade da banda entre 500 e 600 cm-1
aumentou em função da temperatura de
cristalização. Em temperaturas bastante elevadas (Amostra MT-170/96), foram observadas
duas bandas atribuídas à vibração da ligação siloxano, T-O-T (T = Si, Al) de anéis duplos de
cinco e quatro membros. Além disso, o espectro desta amostra exibiu uma banda em 630 cm-
1, relacionada à presença de anéis duplos de seis membros, característicos da mordenita
(DIMITROV, 2006). Este efeito está relacionado ao aumento da cristalinidade da zeólita,
mostrada na Tabela 4.7.
Comparando-se os espectros das amostras antes e após a troca iônica (Figura 4.18),
também é possível observar que a estrutura dos materiais hierárquicos obtidos no estudo da
temperatura não foi afetada significativamente pela substituição do cátion alcalino por íons
H+. Esse resultado indicou que os materiais obtidos apresentam estabilidade térmica.

4 – RESULTADOS E DISCUSSÃO 69
Figura 4.18. Espectros de FTIR das amostras na obtidas durante o estudo da temperatura de cristalização,
ampliados na região do infravermelho médio de 1300 a 400 cm-1. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente. A letra H indica as amostras na forma protônica.
4.2.3. Difração de Raios X
A natureza cristalina das amostras também foi investigada a partir dos resultados de
difração de raios X (Figura 4.19). Em temperatura baixa (Amostra MT-120/96), não se
observam as reflexões de Bragg da estrutura microporosa da mordenita na região de altos
ângulos (Figuras 4.19). À medida que a temperatura de cristalização foi aumentada, surgem
os picos atribuídos às reflexões de planos característicos da mordenita (IZA, 2013; TREACY,
2007). Além disso, esses picos tornam-se mais intensos com o aumento da temperatura,
devido ao aumento da cristalinidade (Tabela 4.7). A cristalinidade aumentou em função da
temperatura de cristalização, em concordância com os resultados do FTIR. Por outro lado, a
1080
796 560
462
1200 1000 800 600 400 0
20
40
60
80
100 HMT-140/96
Tra
nsm
itân
cia
(%)
Número de Onda (cm-1
)
1230
630
MT-140/96 1082
800 560
462 1230
632
1200 1000 800 600 400
Tra
nsm
itân
cia
(%)
Número de Onda (cm-1
)
MT-170/96
1080
810 560
445 1230
630
HMT-170/96
1082
810 560
455 1230
632
1080
796 560
462 1230
1200 1000 800 600 400 0
20
40
60
80
100 T
ransm
itân
cia
(%)
Número de Onda (cm-1
)
HMT-120/96
MT-120/96 1080
796 560 462
1230

4 – RESULTADOS E DISCUSSÃO 70
Figura 4.19. Difratogramas de raios X na região de altos ângulos das amostras obtidas durante o estudo da
temperatura de cristalização. O primeiro e o segundo números representam a temperatura e o tempo de
cristalização, respectivamente. As curvas em preto representam as amostras após a troca iônica e as curvas em
vermelho representam as amostras não trocadas.
Tabela 4.7. Cristalinidade (C), diâmetro médio do cristal () e relação entre as intensidades de picos (R) das amostras obtidas durante o estudo da temperatura de cristalização. O primeiro e o segundo números representam
a temperatura e o tempo de cristalização, respectivamente. A letra H indica as amostras na forma protônica.
Amostras C (%) a (nm) R
b
MT-120/96 n.d.c n.d.
c n.d.
c
MT-140/96 38 53 2,3
MT-170/96 81 61 3,5
HMT-120/96 n.d.c n.d.
c n.d.
c
HMT-140/96 39 53 2,7
HMT-170/96 100 62 5,8
a diâmetro médio do cristal calculado através da equação de Scherrer.
b relação entre as intensidades dos picos dos planos (111) e (310).
c não determinado.
Inte
nsi
dad
e (c
ps)
110
020
200
111
310 3
30 1
50
202
350
511
332
10 20 30 40 50 0
1000
2000
3000
4000
MT-140/96
Inte
nsi
dad
e (c
ps)
2 (graus) 10 20 30 40 50
0
500
1000
1500
2000
MT-120/96 Inte
nsi
dad
e (c
ps)
2 (graus)
10 20 30 40 50 0
1000
2000
3000
4000
5000
6000
7000
8000
MT-170/96
Inte
nsi
dad
e (c
ps)
2 (graus)
110
020
200
111
310
330
150
202
350 5
11
332
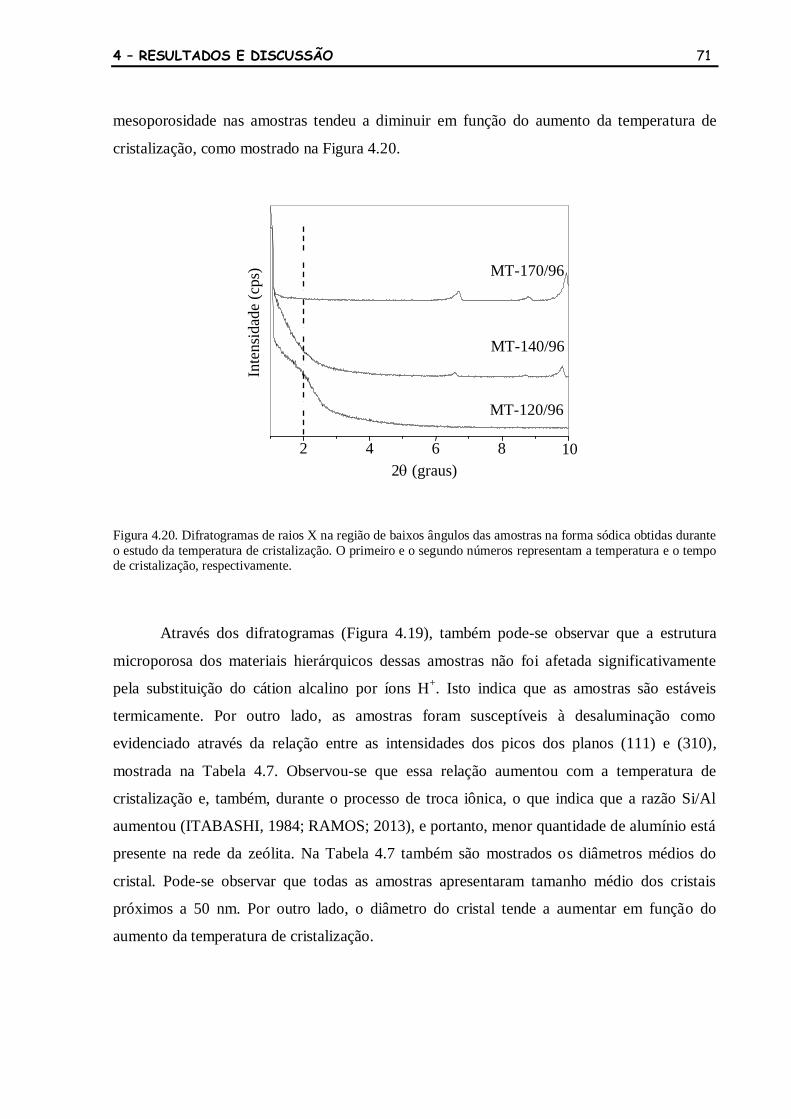
4 – RESULTADOS E DISCUSSÃO 71
mesoporosidade nas amostras tendeu a diminuir em função do aumento da temperatura de
cristalização, como mostrado na Figura 4.20.
Figura 4.20. Difratogramas de raios X na região de baixos ângulos das amostras na forma sódica obtidas durante
o estudo da temperatura de cristalização. O primeiro e o segundo números representam a temperatura e o tempo de cristalização, respectivamente.
Através dos difratogramas (Figura 4.19), também pode-se observar que a estrutura
microporosa dos materiais hierárquicos dessas amostras não foi afetada significativamente
pela substituição do cátion alcalino por íons H+. Isto indica que as amostras são estáveis
termicamente. Por outro lado, as amostras foram susceptíveis à desaluminação como
evidenciado através da relação entre as intensidades dos picos dos planos (111) e (310),
mostrada na Tabela 4.7. Observou-se que essa relação aumentou com a temperatura de
cristalização e, também, durante o processo de troca iônica, o que indica que a razão Si/Al
aumentou (ITABASHI, 1984; RAMOS; 2013), e portanto, menor quantidade de alumínio está
presente na rede da zeólita. Na Tabela 4.7 também são mostrados os diâmetros médios do
cristal. Pode-se observar que todas as amostras apresentaram tamanho médio dos cristais
próximos a 50 nm. Por outro lado, o diâmetro do cristal tende a aumentar em função do
aumento da temperatura de cristalização.
2 4 6 8 10
MT-120/96
MT-140/96
MT-170/96
Inte
nsi
dad
e (c
ps)
2 (graus)
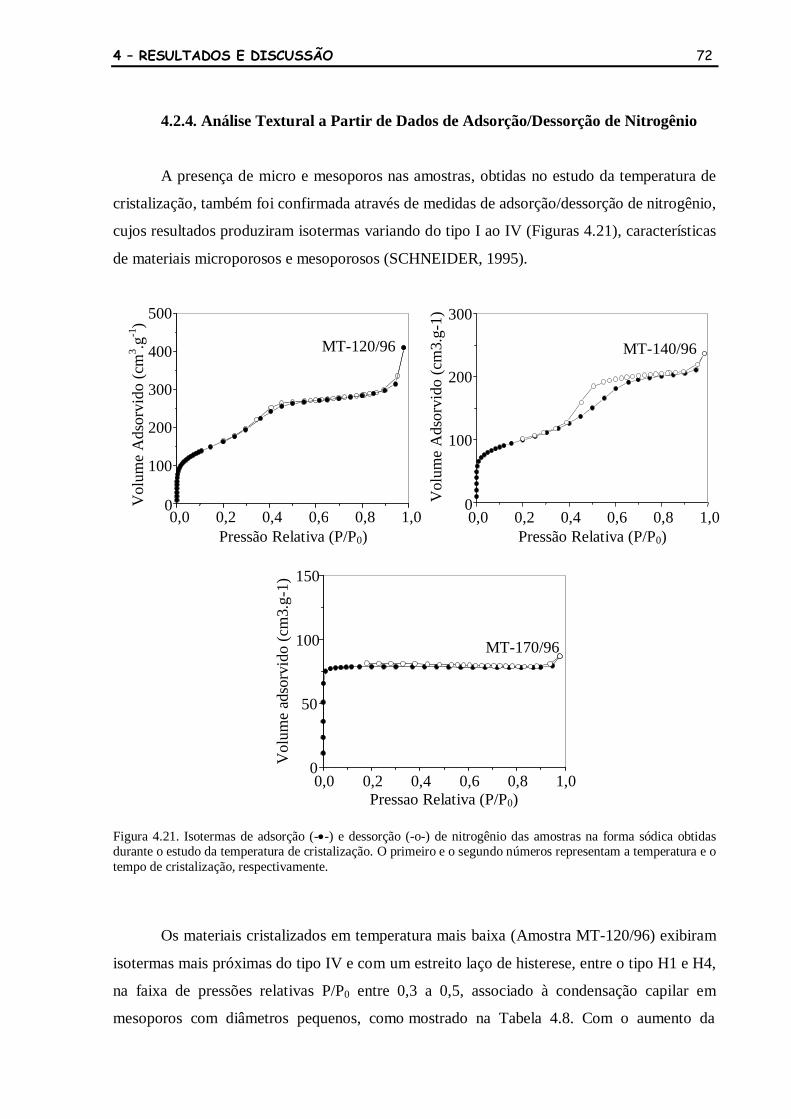
4 – RESULTADOS E DISCUSSÃO 72
4.2.4. Análise Textural a Partir de Dados de Adsorção/Dessorção de Nitrogênio
A presença de micro e mesoporos nas amostras, obtidas no estudo da temperatura de
cristalização, também foi confirmada através de medidas de adsorção/dessorção de nitrogênio,
cujos resultados produziram isotermas variando do tipo I ao IV (Figuras 4.21), características
de materiais microporosos e mesoporosos (SCHNEIDER, 1995).
Figura 4.21. Isotermas de adsorção (--) e dessorção (-o-) de nitrogênio das amostras na forma sódica obtidas durante o estudo da temperatura de cristalização. O primeiro e o segundo números representam a temperatura e o
tempo de cristalização, respectivamente.
Os materiais cristalizados em temperatura mais baixa (Amostra MT-120/96) exibiram
isotermas mais próximas do tipo IV e com um estreito laço de histerese, entre o tipo H1 e H4,
na faixa de pressões relativas P/P0 entre 0,3 a 0,5, associado à condensação capilar em
mesoporos com diâmetros pequenos, como mostrado na Tabela 4.8. Com o aumento da
0,0 0,2 0,4 0,6 0,8 1,0 0
100
200
300
MT-140/96
Pressão Relativa (P/P0)
Volu
me
Adso
rvid
o (
cm3.g
-1)
0,0 0,2 0,4 0,6 0,8 1,0 0
100
200
300
400
500
MT-120/96
Pressão Relativa (P/P0)
Volu
me
Adso
rvid
o (
cm3.g
-1)
0,0 0,2 0,4 0,6 0,8 1,0 0
50
100
150
MT-170/96
Pressao Relativa (P/P0)
Volu
me
adso
rvid
o (
cm3.g
-1)
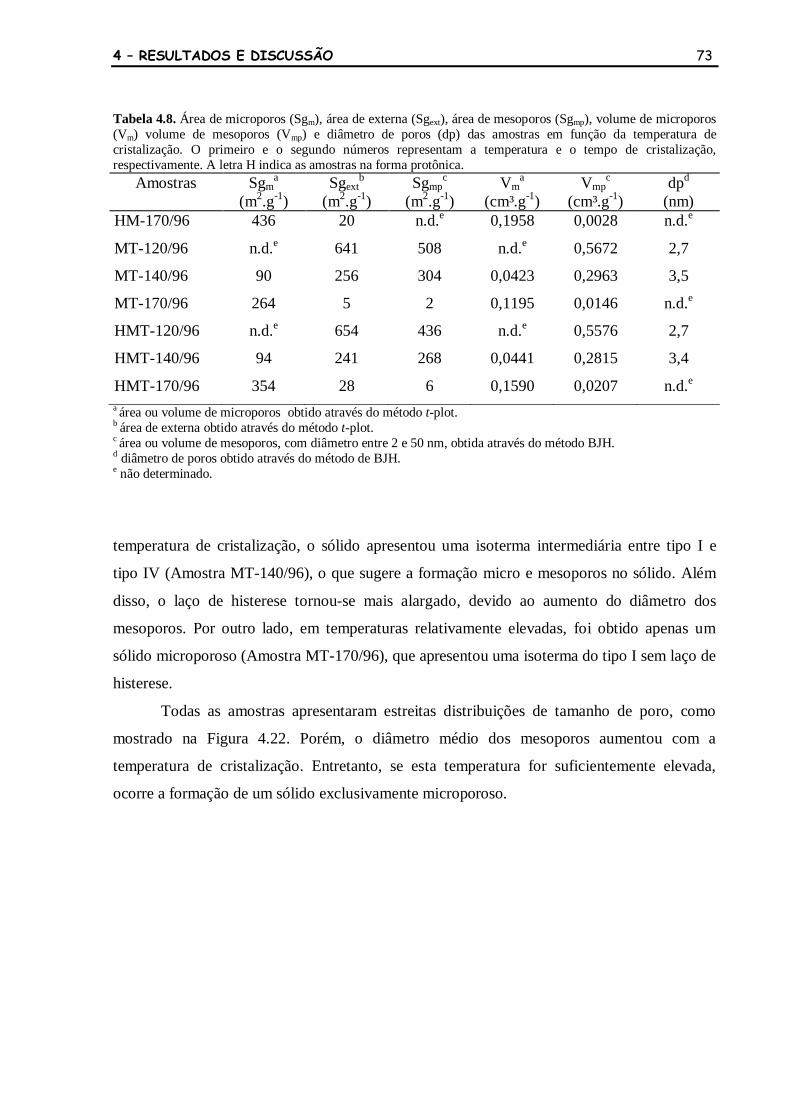
4 – RESULTADOS E DISCUSSÃO 73
Tabela 4.8. Área de microporos (Sgm), área de externa (Sgext), área de mesoporos (Sgmp), volume de microporos
(Vm) volume de mesoporos (Vmp) e diâmetro de poros (dp) das amostras em função da temperatura de
cristalização. O primeiro e o segundo números representam a temperatura e o tempo de cristalização,
respectivamente. A letra H indica as amostras na forma protônica.
Amostras Sgma
(m2.g
-1)
Sgextb
(m2.g
-1)
Sgmpc
(m2.g
-1)
Vma
(cm³.g-1
)
Vmpc
(cm³.g-1
)
dpd
(nm)
HM-170/96 436 20 n.d.e 0,1958 0,0028 n.d.
e
MT-120/96 n.d.e 641 508 n.d.
e 0,5672 2,7
MT-140/96 90 256 304 0,0423 0,2963 3,5
MT-170/96 264 5 2 0,1195 0,0146 n.d.e
HMT-120/96 n.d.e 654 436 n.d.
e 0,5576 2,7
HMT-140/96 94 241 268 0,0441 0,2815 3,4
HMT-170/96 354 28 6 0,1590 0,0207 n.d.e
a área ou volume de microporos obtido através do método t-plot. b área de externa obtido através do método t-plot. c área ou volume de mesoporos, com diâmetro entre 2 e 50 nm, obtida através do método BJH. d diâmetro de poros obtido através do método de BJH. e não determinado.
temperatura de cristalização, o sólido apresentou uma isoterma intermediária entre tipo I e
tipo IV (Amostra MT-140/96), o que sugere a formação micro e mesoporos no sólido. Além
disso, o laço de histerese tornou-se mais alargado, devido ao aumento do diâmetro dos
mesoporos. Por outro lado, em temperaturas relativamente elevadas, foi obtido apenas um
sólido microporoso (Amostra MT-170/96), que apresentou uma isoterma do tipo I sem laço de
histerese.
Todas as amostras apresentaram estreitas distribuições de tamanho de poro, como
mostrado na Figura 4.22. Porém, o diâmetro médio dos mesoporos aumentou com a
temperatura de cristalização. Entretanto, se esta temperatura for suficientemente elevada,
ocorre a formação de um sólido exclusivamente microporoso.
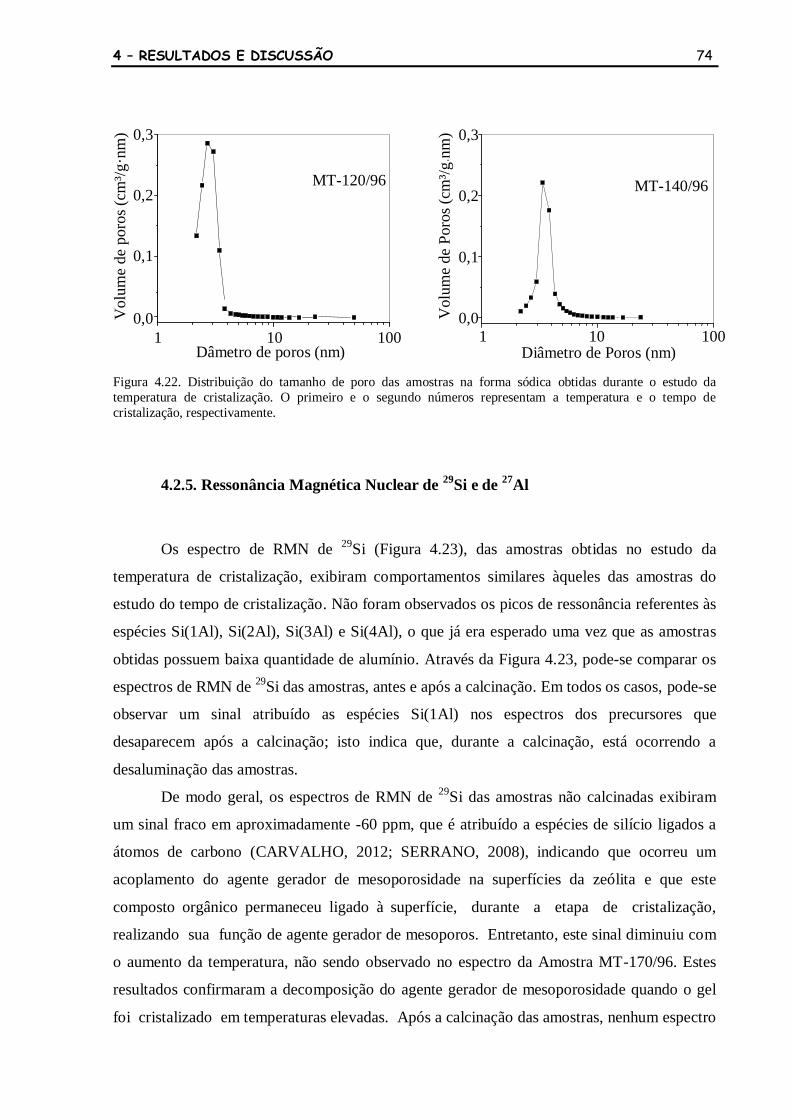
4 – RESULTADOS E DISCUSSÃO 74
Figura 4.22. Distribuição do tamanho de poro das amostras na forma sódica obtidas durante o estudo da
temperatura de cristalização. O primeiro e o segundo números representam a temperatura e o tempo de
cristalização, respectivamente.
4.2.5. Ressonância Magnética Nuclear de 29
Si e de 27
Al
Os espectro de RMN de 29
Si (Figura 4.23), das amostras obtidas no estudo da
temperatura de cristalização, exibiram comportamentos similares àqueles das amostras do
estudo do tempo de cristalização. Não foram observados os picos de ressonância referentes às
espécies Si(1Al), Si(2Al), Si(3Al) e Si(4Al), o que já era esperado uma vez que as amostras
obtidas possuem baixa quantidade de alumínio. Através da Figura 4.23, pode-se comparar os
espectros de RMN de 29
Si das amostras, antes e após a calcinação. Em todos os casos, pode-se
observar um sinal atribuído as espécies Si(1Al) nos espectros dos precursores que
desaparecem após a calcinação; isto indica que, durante a calcinação, está ocorrendo a
desaluminação das amostras.
De modo geral, os espectros de RMN de 29
Si das amostras não calcinadas exibiram
um sinal fraco em aproximadamente -60 ppm, que é atribuído a espécies de silício ligados a
átomos de carbono (CARVALHO, 2012; SERRANO, 2008), indicando que ocorreu um
acoplamento do agente gerador de mesoporosidade na superfícies da zeólita e que este
composto orgânico permaneceu ligado à superfície, durante a etapa de cristalização,
realizando sua função de agente gerador de mesoporos. Entretanto, este sinal diminuiu com
o aumento da temperatura, não sendo observado no espectro da Amostra MT-170/96. Estes
resultados confirmaram a decomposição do agente gerador de mesoporosidade quando o gel
foi cristalizado em temperaturas elevadas. Após a calcinação das amostras, nenhum espectro
1 10 100
0,0
0,1
0,2
0,3
Diâmetro de Poros (nm)
Volu
me
de
Poro
s (c
m³/
g·n
m)
MT-140/96
1 10 100
0,0
0,1
0,2
0,3
MT-120/96
Volu
me
de
poro
s (c
m³/
g·n
m)
Dâmetro de poros (nm)
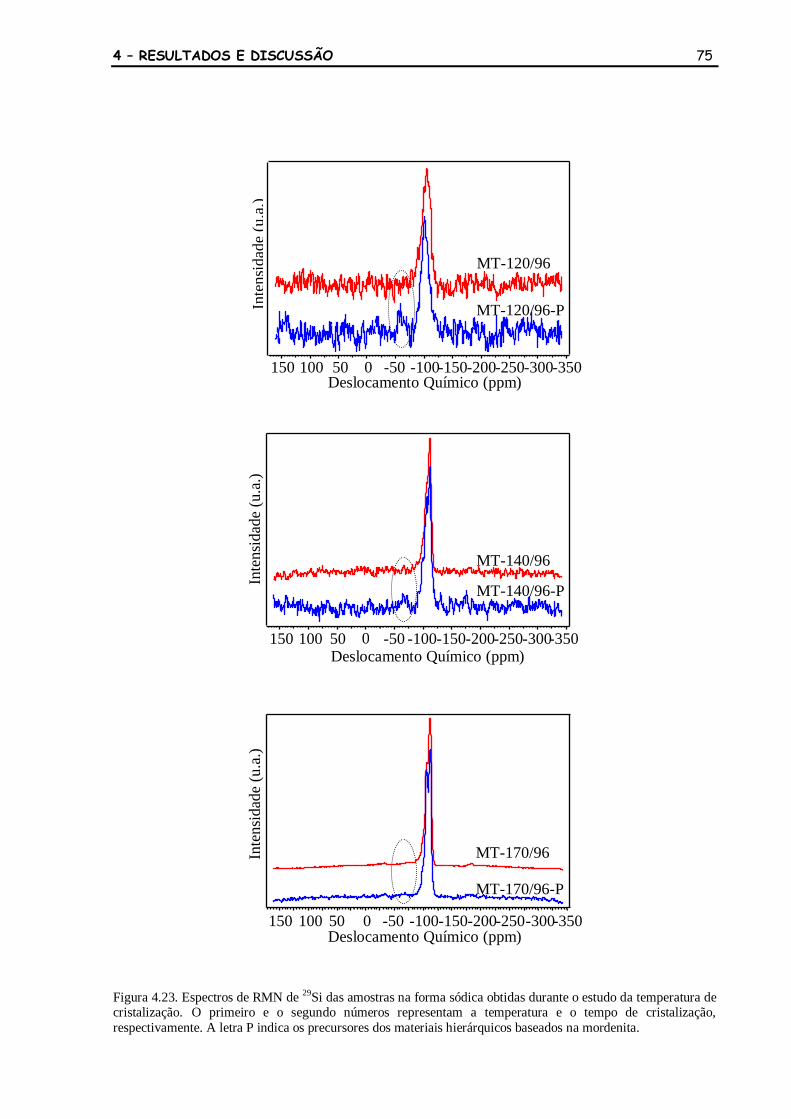
4 – RESULTADOS E DISCUSSÃO 75
Figura 4.23. Espectros de RMN de 29Si das amostras na forma sódica obtidas durante o estudo da temperatura de cristalização. O primeiro e o segundo números representam a temperatura e o tempo de cristalização,
respectivamente. A letra P indica os precursores dos materiais hierárquicos baseados na mordenita.
150 100 50 0 -50 -100 -150 -200 -250 -300 -350
MT-120/96-P
MT-120/96 In
tensi
dad
e (u
.a.)
Deslocamento Químico (ppm)
Deslocamento Químico (ppm) 150 100 50 0 -50 -100 -150 -200 -250 -300 -350
MT-170/96-P
MT-170/96 Inte
nsi
dad
e (u
.a.)
MT-140/96-P
MT-140/96
Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm) 150 100 50 0 -50 -100 -150 -200 -250 -300 -350

4 – RESULTADOS E DISCUSSÃO 76
apresentou esse sinal, evidenciando que a parte orgânica do agente funcionalizante foi
eliminado durante a calcinação.
Os espectros de ressonância magnética nuclear de 27
Al das amostras, obtidas no estudo
da temperatura de cristalização como sintetizadas, também apresentaram somente um pico em
aproximadamente 60 ppm, atribuído ao átomo de alumínio tetraédrico da rede da zeólita,
como pode ser observado na Figura 4.24. Após a calcinação, nota-se o aparecimento do pico
em aproximadamente 0 ppm, indicando que houve uma destruição da estrutura ou
desaluminação da amostra durante esse processo. As percentagens dos picos dos átomos de
alumínio tetraédricos e octaédricos obtidas através dos espectros das amostras calcinadas, são
mostradas na Tabela 4.9. A amostra preparada em temperatura mais baixa (Amostras MT-
120/96) não possui estabilidade térmica e sua estrutura mesoporosa é destruída parcialmente.
Com o aumento da temperatura de cristalização, também ocorre uma transformação do
material amorfo em uma estrutura zeolítica com elevada estabilidade térmica, e, portanto, os
picos de ressonância em 0 ppm é atribuído ao processo de desaluminação. Em temperaturas
de cristalização elevadas, pode ocorrer a redispersão dos átomos de alumínio e a
desaluminação aumenta (KOEKKOEK, 2011). Estes resultados estão em concordância com
os resultados do DRX das amostras (Tabela 4.7).
Tabela 4.9. Quantidade de alumínio tetraédrico (Altetra) e quantidade de alumínio octaédrico (Alocta) das amostras
obtidas durante o estudo da temperatura de cristalização. O primeiro e o segundo números representam a
temperatura e o tempo de cristalização, respectivamente.
Amostras Altetra (%) Alocta (%)
MT-120/96 78 22
MT-140/96 83 17
MT-170/96 76 24

4 – RESULTADOS E DISCUSSÃO 77
Figura 4.24. Espectros de RMN de 27Al das amostras na forma sódica obtidas durante o estudo da temperatura de
cristalização. O primeiro e o segundo números representam a temperatura e o tempo de cristalização,
respectivamente. A letra P indica os precursores dos materiais hierárquicos baseados na mordenita.
300 200 100 0 -50 -150 -250 -350 -450
MT-170/96-P
MT-170/96 Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm)
300 200 100 0 -50 -150 -250 -350 -450
MT-120/96-P
MT-120/96
Inte
nsi
dad
e
(u.a
.)
Deslocamento Químico (ppm)
300 200 100 0 -50 -150 -250 -350 -450
MT-140/96-P
MT-140/96 Inte
nsi
dad
e (u
.a.)
Deslocamento Químico (ppm)

4 – RESULTADOS E DISCUSSÃO 78
4.2.6. Microscopia Eletrônica de Varredura
As micrografias eletrônicas de varredura das amostras calcinadas, com aumento entre
3500 a 5000 vezes, são apresentadas na Figura 4.25. Pode-se notar que as amostras obtidas no
estudo da temperatura de cristalização também tendem a formar aglomerados.
Figura 4.25. Micrografias eletrônicas de varredura da (a) Amostra MT-120/96, (b) Amostra MT-140/96, (c)
Amostra MT-170/96 e (d) ampliação da micrografia da Amostra MT-170/96. O primeiro e o segundo números
representam a temperatura e o tempo de cristalização, respectivamente.
A morfologia das amostras também variou em função da temperatura de cristalização.
De modo similar às amostras preparadas no estudo do efeito do tempo de cristalização, as
amostras obtidas em baixas temperaturas apresentaram um aspecto esponjoso, característico
de partículas mesoporosas desordenadas, como evidenciado através da difração de raios X.
Por outro lado, o aumento da temperatura de cristalização favoreceu a transformação sólido-
sólido da sílica amorfo em um material cristalino. Na micrografia da Amostra MT-140/96,
(c)
(a) (b)
(d)

4 – RESULTADOS E DISCUSSÃO 79
foram observadas partículas esféricas mesoporosas desordenadas. Isto indica que este sólido é
formado por duas fases: uma cristalina referente à mordenita e uma fase mesoporosa
desordenada (LI, 2009). A morfologia destes materiais é similar àquela apresentada por outras
zeólitas com estrutura hierárquica de poros preparadas usando organossilanos, tais como a
ZSM-5 (KOEKKOEK, 2011) e zeólita A (CHO, 2011). Quando a temperatura de cristalização
foi suficientemente elevada (Amostra MT-170/96), foram observados cristais da mordenita na
forma de placas, na ausência de fase mesoporosa amorfa (Figura 4.25d). Também foi possível
observar que o diâmetro do cristal tende a aumentar em função do aumento da temperatura de
cristalização, como evidenciado pela difração de raios X (Tabela 4.7).
4.2.7. Medidas de Acidez por Dessorção de Amônia à Temperatura Programada
As curvas de acidez das amostras, na forma protônica, obtidas durante o estudo da
temperatura de cristalização, estão ilustradas na Figura 4.26. Observou-se que a curva de
acidez da Amostra HMT-120/96 (temperatura de cristalização mais baixa), apresentou um
pico de dessorção de amônia bastante alargado entre 100 e 600 ºC. Este pico está relacionado,
principalmente, à presença de sítios ácidos fracos e moderados, podendo ser também
associado a poucos sítios ácidos fortes. Com o aumento da temperatura de cristalização
(Amostra HMT-170/96), pode-se observar a presença de três regiões de dessorção de amônia,
características de materiais zeolíticos (RAMOS, 2005): um pico próximo a 200 ºC, atribuído
aos sítios ácidos fracos e um ombro próximo a 300 °C, relacionado à presença de sítios ácidos
de força média. Também foram observados dois picos acima de 400 ºC, relacionado à
presença de sítios ácidos mais fortes, fato esperado em função das propriedades ácidas da
mordenita [LÓNYI, 2001].
Comparando as curvas de acidez dos materiais hierárquicos com aquela da mordenita
microporosa, observa-se que a curva de acidez do material cristalizado em elevadas
temperaturas se assemelha aquele da zeólita convencional, o que indica a formação de uma
fase microporosa possuindo principalmente sítios ácidos fortes.
Na Tabela 4.10, são apresentados os resultados das medidas de acidez dos sólidos na
forma protônica em função da quantidade de amônia adsorvida por massa de sólido. A
quantidade de amônia dessorvida aumentou em função do aumento da temperatura de
cristalização. Pode-se observar que a maioria dos sítios ácidos presentes na amostra preparada
em tempos relativamente curtos é de força média, enquanto a amostra preparada em longos
tempos possui maior quantidade de sítios ácidos fortes.
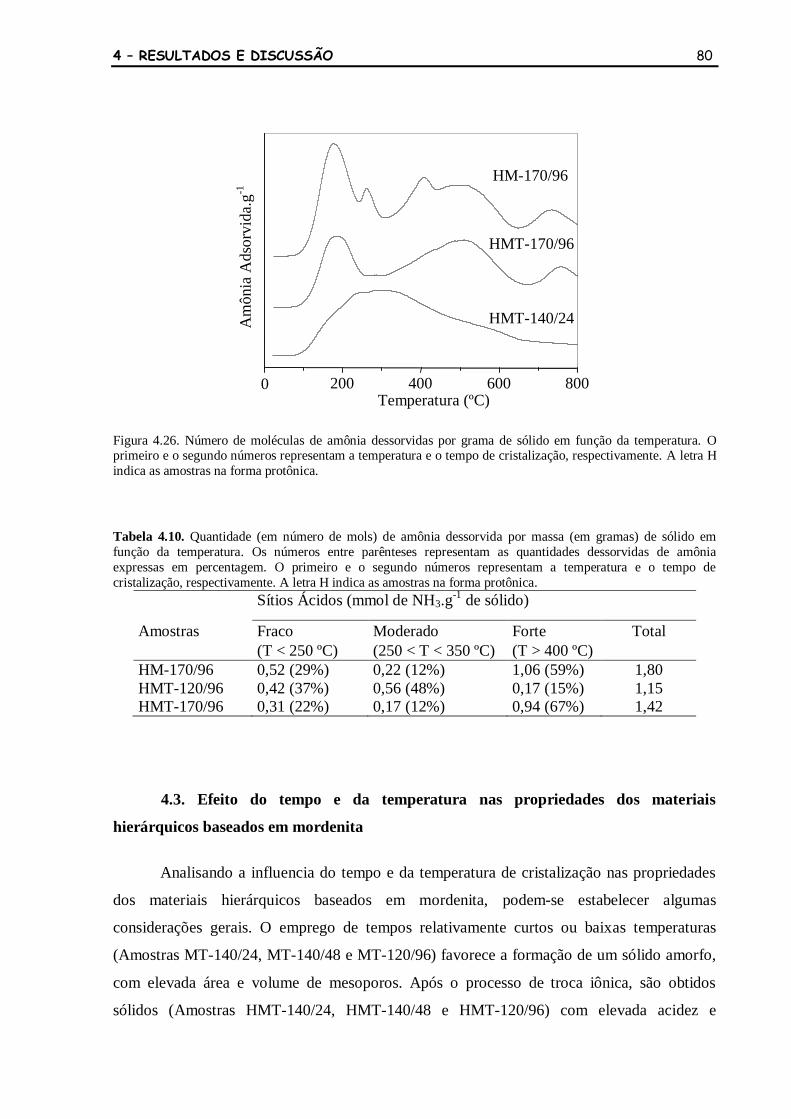
4 – RESULTADOS E DISCUSSÃO 80
Figura 4.26. Número de moléculas de amônia dessorvidas por grama de sólido em função da temperatura. O primeiro e o segundo números representam a temperatura e o tempo de cristalização, respectivamente. A letra H
indica as amostras na forma protônica.
Tabela 4.10. Quantidade (em número de mols) de amônia dessorvida por massa (em gramas) de sólido em
função da temperatura. Os números entre parênteses representam as quantidades dessorvidas de amônia
expressas em percentagem. O primeiro e o segundo números representam a temperatura e o tempo de
cristalização, respectivamente. A letra H indica as amostras na forma protônica.
Sítios Ácidos (mmol de NH3.g-1
de sólido)
Amostras Fraco
(T < 250 ºC)
Moderado
(250 < T < 350 ºC)
Forte
(T > 400 ºC)
Total
HM-170/96 0,52 (29%) 0,22 (12%) 1,06 (59%) 1,80
HMT-120/96 0,42 (37%) 0,56 (48%) 0,17 (15%) 1,15
HMT-170/96 0,31 (22%) 0,17 (12%) 0,94 (67%) 1,42
4.3. Efeito do tempo e da temperatura nas propriedades dos materiais
hierárquicos baseados em mordenita
Analisando a influencia do tempo e da temperatura de cristalização nas propriedades
dos materiais hierárquicos baseados em mordenita, podem-se estabelecer algumas
considerações gerais. O emprego de tempos relativamente curtos ou baixas temperaturas
(Amostras MT-140/24, MT-140/48 e MT-120/96) favorece a formação de um sólido amorfo,
com elevada área e volume de mesoporos. Após o processo de troca iônica, são obtidos
sólidos (Amostras HMT-140/24, HMT-140/48 e HMT-120/96) com elevada acidez e
0 200 400 600 800
HM-170/96
HMT-170/96
Am
ônia
Adso
rvid
a.g
-1
Temperatura (ºC)
HMT-140/24

4 – RESULTADOS E DISCUSSÃO 81
constituídos, principalmente, por sítios ácidos de força média. O aumento da cristalinidade da
mordenita em função do aumento do tempo ou da temperatura de cristalização, em sólidos
com estrutura hierárquica de poros, acarreta uma diminuição na área (de mesoporos) e no
volume de mesoporos. Por outro lado, este aumento da cristalinidade promove um aumento
na área (de microporos) e no volume de microporos. Entretanto, os sólidos obtidos em longos
tempos e temperaturas de cristalização (HMT-140/192 e HMT-170/96) são formados,
principalmente, por sítios ácidos fortes que estão presentes na mordenita na ausência do
organossilano (Amostra HM-170/96).
Desta forma, pode-se concluir que o aumento do tempo ou da temperatura de
cristalização favorece a taxa de crescimento do cristal em detrimento da taxa de nucleação e,
portanto, os primeiros núcleos zeolíticos tendem a crescer ao invés de novos núcleos serem
formados. Por outro lado, o sólido amorfo (Amostras MT-140/24 ou MT-120/96) é
redissolvido com o aumento do tempo e da temperatura e as espécies de silicato geradas
tendem a difundir para a superfície dos primeiros cristais formados, incorporando-se na
estrutura cristalina; em seguida, há o aumento das dimensões do cristal gerando cristais
grandes da mordenita (Amostras MT-140/96 e MT-170/96), como observado nas imagens de
MEV. Além disso, a decomposição parcial (tempos longos) ou total (elevadas temperaturas)
do TPOAC (agente gerador de mesoporosidade) conduz à formação de sólidos constituídos,
principalmente, por microporos.
Desta forma, o emprego de tempos e temperaturas intermediárias (Amostra MT-
140/96) favorece a formação de um sólido constituído por duas fases: uma fase cristalina
referente à mordenita responsável pela elevada área de microporos (90 m2.g
-1) e uma fase
mesoporosa desordenada. Espera-se que este sólido apresente melhor acessibilidade aos sítios
ácidos presentes nos seus microporos e, desta forma, sejam catalisadores mais eficientes no
processamento de moléculas volumosas.

5 – CONCLUSÕES 82
5. CONCLUSÕES
5.1. Na obtenção de materiais hierárquicos baseados na mordenita, observou-se que a
formação da fase mesoporosa desordenada e a formação da fase microporosa (mordenita) são
influenciadas pelo tempo e temperatura de cristalização do gel da zeólita. O emprego de
tempos relativamente curtos ou baixas temperaturas favorece a formação de um sólido
amorfo, enquanto longos tempos ou elevadas temperaturas favorecem a formação de um
material cristalino com características da mordenita. Por outro lado, tempos e temperaturas
intermediárias favorecem a formação de um sólido constituído por duas fases: uma fase
cristalina referente à mordenita e uma fase mesoporosa desordenada. O emprego de longos
tempos e elevadas temperaturas de cristalização conduz à decomposição prematura do
organossilano, durante a etapa de cristalização. As amostras obtidas através da adição de um
organossilano (TPOAC) no gel de síntese da mordenita apresentam elevada estabilidade
térmica e hidrotérmica.
5.2. O aumento da cristalinidade da mordenita, em sólidos com estrutura hierárquica de
poros, acarreta uma diminuição na área e no volume de mesoporos. Por outro lado, este
aumento da cristalinidade promove um aumento na área e no volume de microporos. A área
externa também tende a diminuir devido ao aumento do tamanho do cristal da mordenita em
função do aumento da cristalinidade.
5.3. Materiais hierárquicos baseados em mordenita, obtidos com TPOAC ([3-(trimetoxissilil)
propil] octadecildimetilamônio) entre 120 e 170 °C, em intervalos de tempo variando de 24 a
192 h, são susceptíveis a destruição da estrutura mesoporosa ou ao processo de desaluminação
durante a etapa de calcinação. Esta susceptibilidade aumenta com o tempo ou da temperatura
de cristalização, devido à redispersão dos átomos de alumínio.
5.4. Sólidos com estrutura hierárquica de poros baseados em mordenita, obtidos com
(TPOAC, cloreto de [3-(trimetoxissilil)propil]octadecildimetilamônio) entre 120 e 170 °C, em
intervalos de tempo variando de 24 a 192 h apresentam elevada acidez que aumenta em
função da cristalinidade. Em amostras preparadas em tempos e temperaturas relativamente

5 – CONCLUSÕES 83
curtos predominam os sítios de força média, enquanto em sólidos obtidos em longos tempos e
temperaturas possuem mais sítios ácidos fortes.
5.5. O método de adição de um organossilano (TPOAC, cloreto de [3-(trimetoxissilil)propil]
octadecildimetilamônio) ao gel de síntese da mordenita, cristalizado em temperaturas
variando de 120 a 170 °C, por 24-192 h, empregado neste trabalho, foi adequado à preparação
de materiais hierárquicos baseados na mordenita e sugere que a formação da estrutura micro-
mesoporosa ocorre através de um processo de transformação “sólido-sólido” intrapartícula, no
qual, à medida que se aumenta a cristalização, o material amorfo é convertido em um material
cristalino.
5.6. As condições experimentais mais adequadas para obter sólidos com estrutura hierárquica
de poros baseados em mordenita, empregando (TPOAC, cloreto de [3-(trimetoxissilil)propil]
octadecildimetilamônio), consistem em cristalizar o gel de síntese por 96 h a 140 ºC, que
levam à formação de sólidos mesoporosos com estrutura zeolítica e elevada acidez.

6 – PERSPECTIVAS 84
6. PERSPECTIVAS
A partir dos resultados obtidos, e para se dar continuidade à pesquisa em estudo, pode-
se propor a realização dos seguintes trabalhos:
6.1. Preparar materiais hierárquicos utilizando diferentes fontes de sílica e alumínio e avaliar a
influência das razões SiO2/Al2O3 e Na2O/Al2O3 na formação das fases zeolítica e mesoporosa.
6.2. Estudar a influência da razão TPOAC/Al2O3 na formação das estruturas mesoporosa e
microporosa da mordenita.
6.3. Realizar estudos de adição de copolímeros tribloco ou surfactantes, na presença do
organossilano, como agente de expansão dos poros na estrutura da mordenita.
6.4. Avaliar a atividade e seletividade dos materiais hierárquicos baseados na mordenita como
catalisadores no craqueamento de hidrocarbonetos de alto peso molecular.

7 – REFERÊNCIAS 85
7. REFERÊNCIAS
AGUADO, J.; SERRANO, D. P.; ESCOLA, J. M.; RODRÍGUEZ, J. M.; PERAL, A. Zeolite
Beta with Hierarchical Porosity Prepared from Organofunctionalized Seeds. Microporous
and Mesoporous Materials, v. 115, p. 504, 2008.
AGUADO, J.; SERRANO, D. P.; ESCOLA, J. M.; RODRÍGUEZ, J. M.; PERAL, A.
Catalytic Cracking of Polyethylene over Zeolite Mordenite with Enhanced Textural
Properties. Journal Analitical Applied Pyrolysis, v. 85, p. 352, 2009.
ALY, H. M.; MOUSTAFA, M. E.; ABDELRAHMAN, E. A. Synthesis of Mordenite Zeolite
in Absence of Organic Template. Advanced Powder Technology, v. 23, p. 757, 2012.
BAGSHAW, S. A.; JAENICKE, S.; KHUAN, C. G. Effect of Al Content on the Assembly of
Al-Msu-S Mesostructures: Zeolite Seed Structure Change from Zeolite LZY to LTA with
Increasing Al Content. Catalysis Communications, v. 4, p. 140, 2003.
BALMBRA, R. R.; CLUNIE, J. S.; GOODMAN, J. F. Cubic Mesomorphic Phases. Nature v.
222, p. 1159, 1969.
BARRER, R. M. Synthesis of a Zeolitic Mineral with Chabazite-Like Sorptive Properties
Journal of the Chemical Society, 1948, 127.
BARRER, R. M. Hydrothermal Chemistry of Zeolites. London: Academic Press, 1982.
BARRET, E. P.; JOYNER, L. G.; HALENDA, P. P. The Determination of Pore Volume and
Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. Journal
of the American Chemical Society, v. 73, p. 373, 1951.
BECK, J. S.; VARTULI, J. C.; ROTH, W. J.; LEONOWICZ, M. E.; KRESGE, C. T.;
SCHMITT, K. D.; CHU, C. T-W.;. OLSON, D. H.; SHEPPARD, E. W.; MCCULLEN, S. B.;
HIGGINS, J. B.; SCHLENKER, J. L. A New Family of Mesoporous Molecular Sieves
Prepared with Liquid Crystal Templates. Journal of the American Chemical Society, v.
114, p. 10834, 1992.
BOISEN, A.; SCHMIDT, I.; CARLSSON, A.; DAHL, S.; BRORSON M.; JACOBSEN, C. J.
H. TEM Stereo-Imaging of Mesoporous Zeolite Single Crystals. Chemical Communication.
p. 958, 2003.
BORGES, S. M. S.; RANGEL, M. C. Properties and Catalytic Applications of Carbon
Nanotubes. Current Topics in Catalysis, v. 10, p. 57, 2012.
BRAGA, A. A. C.; MORGON, N. H. Descrições Estruturais Cristalinas de Zeólitos. Química
Nova, v. 30, p. 178, 2007.

7 – REFERÊNCIAS 86
BRECK, D. W. Zeolite Molecular Sieves: Structure, Chemistry and Use. Malabar: Robert E.
Krieger Publishing Company, 1984.
BRUNAUER, S.; EMMETT, P. H.; TELLER, E. J. Adsorption of Gases in Multimolecular
Layers. Journal of the American Chemical Society, v. 60, p. 309, 1938. CAMPOS, A. A.; MARTINS, L.; OLIVEIRA, L. L.; SILVA, C. R.; WALLAU, M.; URQUIETA-GONZALEZ, E. A. Phase Transformation of Amorhous SBA-15 Pore Walls into Microporous MFI Structure. Studies in Surface Science and Catalysis, v. 154, p. 541, 2004. CAMPOS, A. A.; MARTINS, L.; OLIVEIRA, L. L.; SILVA, C. R.; WALLAU, M.; URQUIETA-GONZÁLEZ, E. A. Secondary Crystallization of SBA-15 Pore Walls into Microporous Material with MFI Structure. Catalysis Today, v. 107-108, p. 759, 2005.
CARVALHO, D. R. Preparação de Zeólita Beta Nanoestruturada através da Funcionalização
de Sementes com Organossilanos. 2012, Dissertação, Universidade Federal da Bahia,
Salvador, Bahia, Brasil.
CHENEVIERE, Y.; CHIEUX, F.; CAPS, V.; TUEL, A. Synthesis And Catalytic Properties of
TS-1 with Mesoporous/Microporous Hierarchical Structures Obtained in the Presence of
Amphiphilic Organosilanes. Journal of Catalysis, v. 269, p. 161, 2010.
CHO, K.; CHO, H. S.; MÉNORVAL, L. C.; RYOO, R. Generation of Mesoporosity in LTA
Zeolites by Organosilane Surfactant for Rapid Molecular Transport in Catalytic Application.
Chemistry Materials, v. 21, p.5664, 2009.
CHO, K.; RYOO, R.; ASAHINAB, S.; XIAO, C.; KLINGSTEDT, M.; UMEMURA, A.;
ANDERSON, M. W.; TERASAKI, O. Mesopore Generation by Organosilane Surfactant
During LTA Zeolite Crystallization, Investigated by High-Resolution SEM and Monte Carlo
Simulation. Solid State Sciences, v. 13, p. 750, 2011.
CHO, H. S.; RYOO, R. Synthesis of Ordered Mesoporous MFI Zeolite Using CMK Carbon
Templates. Microporous and Mesoporous Materials, v. 151, p. 107, 2012.
CHOI, M; CHO, H. S.; SRIVASTAVA, R.; VENKATESAN, C.; CHOI, D.; RYOO, R.
Amphiphilic Organosilane-Directed Synthesis of Crystalline Zeolite with Tunable
Mesoporosity. Nature Materials, v. 5, p. 718, 2006 a.
CHOI, M; SRIVASTAVA, R.; RYOO, R. Organosilane Surfactant-Directed Synthesis of
Mesoporous Aluminophosphates Constructed with Crystalline Microporous Frameworks.
Chemical Communication, p. 4380, 2006 b. CHOI, M.; NA, K.; KIM, J.; SAKAMOTO, Y.; TERASAKI, O.; RYOO, R. Stable Single-Unit-Cell Nanosheets of Zeolite MFI as Active and Long-Lived Catalysts. Nature, v. 461, p. 246, 2009. CORMA, A.; FORNÉS, V.; NAVARRO, M. T.; PÉREZ-PARIENTE, J. Acidity and Stability of MCM-41 Crystalline Aluminosilicates. Journal of Catalysis, v. 148, p. 569, 1994. CORMA, A. From Microporous to Mesoporous Molecular Sieve Materials and Their Use in
Catalysis. Chemical Reviews, v. 97, p. 2373, 1997.

7 – REFERÊNCIAS 87
CORMA, A.; FORNES, V.; PERGHER, S. B.; MAESEN, TH. L. M.; BUGLASS, J. G.
Delaminated Zeolite Precursors as Selective Acidic Catalysts. Nature, v. 396, p. 353, 1998.
CORMA, A.; Fornés, V. Zeolitas Deslaminadas: Un Nuevo Concepto de Catalizador, In:
PÉREZ-PARIENTE, J.; MARTÍNEZ, J. G. Materiales Zeolíticos: Síntesis, Propriedades y
Aplicaciones. San Vicente: Universidad Alicante, 2002. Cap. 11.
CORMA, A. State of the Art Future Challenges of Zeolites as Catalysts. Journal of
Catalysis, v. 216, p. 298, 2003.
CUNDY, C. S.; PAUL, C. The Hydrothermal Synthesis of Zeolites: History and Development
from the Earliest Days to the Present Time. Chemical Reviews, v. 103, p. 663, 2003.
CUNDY, C. S.; PAUL, C. The Hydrothermal Synthesis of Zeolites: Precursors, Intermediates
and Reaction Mechanism. Microporous and Mesoporous Materials, v. 82, p. 1, 2005.
DAVIS, M. E.; KATZ, A.; AHMAD, W. R. Rational Catalyst Design via Imprinted
Nanostructured Materials. Chemistry Materials, v. 8, p.1820, 1996.
DAVIS, S. A.; BURKETT, S. L.; MENDELSON, N. L.; MANN, S. Bacterial Templating of
Ordered Macrostructures in Silica and Silica-Surfactant Mesophases. Nature, v. 385, p. 420,
1997
DAVIS, S. A.; PATEL, H. M.; MAYES, E. L.; MENDELSON, N. L.; FRANCO, G.; MANN,
S. Bacteria: A Biomimetic Approach to the Formation of Fibrous Composite Materials.
Chemistry Materials, v. 10, p. 2516, 1998.
DIMITROV, L.; WALLAU, M.; URQUIETA-GONZÁLEZ, E. A. Mesostructuration of
Mordenite Zeolite Nanoparticles. Anais do 13° Congresso Brasileiro de Catálise / 3°
Mercosul, Foz do Iguaçu, Brasil, 2005.
DIMITROV, L. D.; WALLAU, M.; URQUIETA-GONZÁLEZ, E. A. Mordenite Seeding
Gels Mesostructured by the Nonionic Surfactant Pluronic P123. Studies in Surface Science
and Catalysis, v.162, p. 433, 2006.
DIMITROVA, R.; GUNDUZ, G.; DIMITROV, L.; TSONCHEVA, T.; YIALMAZ, S.;
URQUIETA-GONZALEZ, E. A. Acidic Sites in Beta Zeolites in Dependence of the
Preparation Methods. Journal of Molecular Catalysis A: Chemical, v. 214, p. 265, 2004.
EGEBLAD, K.; CHRISTENSEN, C. H.; KUSTOVA, M.; CHRISTENSEN, C. H.
Templating Mesoporous Zeolites. Chemistry Materials, v. 20, p. 946, 2008.
ESCOBAR, M. F. A.; BATISTA, M. S; URQUIETA-GONZÁLEZ, E. A.
Desproporcionamento de Tolueno sobre Zeólitas Tipo Mordenita - Atividade e Seletividade
na Obtenção de Xilenos. Química Nova, v. 23, p. 303, 2000.
FLANIGEN, E. M., In: RABO, J. Zeolite Chemistry and Catalysis, American Chemical
Society, Washington, DC, 1976, Cap. 2.

7 – REFERÊNCIAS 88
FONSECA, J. D. S. L.; JÚNIOR, A. D. C. F.; GRAU, J. M.; RANGEL, M. D. C.
Ethylbenzene Production over Platinum Catalysts Supported on Modified KY Zeolites.
Applied Catalysis A: General, v. 386, p. 201, 2010.
GALARNEAU, A.; CAMBON, H.; RENZO, F. D.; FAJULA, F. True Microporosity and
Surface Area of Mesoporous SBA-15 Silicas as a Function of Synthesis Temperature.
Langmuir, v. 17, p. 8328, 2001.
GONÇALVES, M. L.; DIMITROV, L. D.; WALLAU, M.; URQUIETA-GONZALEZ, E. A.
Mesoporous ZSM-5 Synthesized by Simultaneous Mesostructuring and Crystallization of
ZSM-5 Nuclei. Studies in Surface Science and Catalysis, v. 162, p. 323, 2006.
GONÇALVES, M. L.; DIMITROV,L. D.; JORDÃO, M. H.; WALLAU, M.; URQUIETA-
GONZÁLEZ, E. A. Synthesis of Mesoporous ZSM-5 by Crystallisation of Aged Gels in the
Presence of Cetyltrimethylammonium Cations. Catalysis Today, v. 133-135, 69, 2008.
GRECCO, S. T. F.; GOMES, L. P.; REYES, P.; OPORTUS, M.; RANGEL, M. C. Stud. Surf.
Sci. Catal. Effect of Platinum on the Activity of Zeolite-based Catalysts. Studies in Surface
Science and Catalysis, v. 158, p. 1937, 2005.
GRECCO, S. T. F.; NOBRE, P. S. S.; URQUIETA-GONZÁLEZ, E. A; RANGEL M. C.
Two-Step Synthesis Procedure of Beta Zeolites: Mesoporosity and Performance as Acid
Catalysts, In: SAYARI, A.; JARONIEC, M. Nanoporous Materials: Proceedings of the 5th
International Symposium, New Jersey: World Scientific Publishing Co. Pte. Ltd., 2008.
GRECCO, S. T. F.; URQUIETA-GONZÁLEZ, E. A.; REYES, P.; OPORTUS, M.; RANGEL
M. C. Influência da Temperatura e do Tempo de Envelhecimento do Gel de Sementes nas
Propriedades de Zeólitas Beta obtidas via Mesoestruturação-Cristalização. Anais do XX
Simpósio Iberoamericano de Catálise, Viña del Mar, Chile, 2010.
GRECCO, S. T. F.; RANGEL, M. C.; URQUIETA-GONZÁLEZ, E. A. Zeólitas
Hierarquicamente Estruturadas. Quimica Nova, v. 36, p. 131, 2013
GREGG, S. J.; SING, K. S. W. Adsorption. Surface Area and Porosity. 2nd Edition, London:
Academic Press, 1982.
GUISNET, M.; MAGNOUX, P. Coking and deactivation of zeolites: influence of the pore
structure. Applied Catalysis, v. 54, p. 1, 1989.
GUISNET, M.; RIBEIRO, F. R. Zeólitos: Um Nanomundo ao Serviço da Catálise. Lisboa:
Fundação Calouste Gulbenkian, 2004.
GUO, W.; HUANG, L.; DENG, P.; XUE, Z.; LI, Q. Characterization of Beta/MCM-41
Composite Molecular Sieve Compared with the Mechanical Mixture. Microporous and
Mesoporous Materials, v. 44-45, p. 427, 2001 a.
GUO, W.; XIONG, C.; HUANG, L.; LI, Q. Synthesis and Characterization of Composite
Molecular Sieves Comprising Zeolite Beta with MCM-41 Structures. Journal of Materials
Chemistry, v. 11, p. 1886, 2001 b.

7 – REFERÊNCIAS 89
HUANG, L.; GUO, W.; DENG, P.; XUE, Z.; LI, Q. Investigation of Synthesizing MCM-
41/ZSM-5 Composites. Journal of Physical Chemistry B, v. 104, p. 2817, 2000.
HUO, Q.; MARGOLESE, D. I.; STUCKY, G. D. Surfactant Control of Phases in the
Synthesis of Mesoporous Silica-Based Materials. Chemistry Materials, v. 8, p.1147, 1996.
IMPÉROR-CLERC, M.; DAVIDSON, P.; DAVIDSON, A. Existence of a Microporous
Corona around the Mesopores of Silica-Based SBA-15 Materials Templated by Triblock
Copolymers. Journal of the American Chemical Society, v. 122, 11925, 2000.
ITABASHI, K.; FUKUSHIMA, T.; IGAWA, K. Synthesis and Characteristic Properties of
Siliceous Mordenite. Zeolites, v.6, p. 30, 1986.
IZA: http://www.iza-structure.org/databases. Acessado em 20/06/2013.
JACOBSEN, C. J. H.; MADSEN, C.; HOUZVICKA, J.; SCHMIDT, I.; CARLSSON, A.
Mesoporous Zeolite Single Crystals. Journal of the American Chemical Society, v. 122, p.
7116, 2000.
JACOBSEN, C. J. H.; HOUZVICKA, J.; CARLSSON, A.; SCHMIDT, A. Mesoporous
Zeolites. Studies in Surface Science and Catalysis. v. 135, p. 167, 2001.
JANSSEN, A. H.; SCHMIDT, I.; JACOBSEN, C. J. H.; KOSTER, A. J.; DE JONG, K. P.
Exploratory Study of Mesopore Templating with Carbon During Zeolite Synthesis.
Microporous and Mesoporous Materials, v. 65, p. 59, 2003.
KARLSSON, A.; STOCKER, M.; SCHMIDT, R. Composites of Micro- and Mesoporous
Materials: Simultaneous Syntheses of MFI/MCM-41 like Phases by a Mixed Template
Approach. Microporous and Mesoporous Materials, v. 27, p. 181, 1999.
KATSUKI, H.; FURUTA S.; WATARI, T.; KOMARNENI, S. ZSM-5 Zeolite/Porous Carbon
Composite: Conventional- and Microwave-Hydrothermal Synthesis from Carbonized Rice
Husk. Microporous and Mesoporous Materials, v. 86, p. 145, 2005.
KIPKEMBOI, P.; FODGEN, A.; ALFREDSSON, V.; FLODSTRÖM, K. Triblock
Copolymers as Templates in Mesoporous Silica Formation: Structural Dependence on
Polymer Chain Length and Synthesis Temperature. Langmuir, v. 17, p. 5398, 2001.
KLEIN, C.; HURLBUT, C. S. Manual of Mineralogy. 21st Edition, New York: Jonh Wiley &
Sons, INC., 1977.
KLEITZ, F.; SCHIMIDT, W.; SCHÜTH, F. Calcination Behavior of Different Surfactant-
Templated Mesostructured Silica Materials. Microporous and Mesoporous Materials, v.
62, p.1, 2003.
KLOETSTRA, K. R.; ZANDBERGEN, H. W.; JANSEN J. C.; VAN BEKKUM H.
Overgrowth of Mesoporous MCM-41 on Faujasite. Microporous and Mesoporous
Materials, v. 6, p. 287, 1996.

7 – REFERÊNCIAS 90
KOEKKOEK, A. J. J.; TEMPELMAN, C. H. L.; DEGIRMENCI, V.; GUO, M.; FENG, Z.;
LI, C.; HENSEN, E. J. M. Hierarchical Zeolites Prepared by Organosilane Templating: A
Study of the Synthesis Mechanism and Catalytic Activity. Catalysis Today, v. 168, p. 96,
2011.
KRESGE, C. T.; LEONOWICZ, M. E.; ROTH, W. J.; VARTULI, J. C.; BECK, J. S. Ordered
Mesoporous Molecular Sieves Synthesized by a Liquid-Crystal Template Mechanism.
Nature, v. 359, p. 710, 1992.
KUSTOVA, M.; EGEBLAD, K.; ZHU, K.; CHRISTENSEN C. H. Versatile Route to Zeolite
Single Crystals with Controlled Mesoporosity: in situ Sugar Decomposition for Templating of
Hierarchical Zeolites. Chemistry Materials, v. 19, p. 2915, 2007.
LI, Y.; ZHANG, W.; ZHANG, L.; YANG, Q.; WEI, Z.; FENG, Z.; LI, C. Direct Synthesis of
Al−SBA-15 Mesoporous Materials via Hydrolysis-Controlled Approach. Journal of Physical
Chemistry B, v. 108, p. 9739, 2004.
LI, H.; SAKAMOTO, Y.; LIU, Z.; OHSUNA, T.; TERASAKI, O.; THOMMES, M.; CHE, S.
Mesoporous silicalite-1 zeolite crystals with unique pore shapes analogous to the morphology.
Microporous and Mesoporous Materials, v. 106, p. 174, 2007.
LI, X.; PRINSA, R.; VAN BOKHOVEN, J. A. Synthesis and characterization of mesoporous
mordenite. Journal of Catalysis, v. 262, p. 257, 2009.
LIPPMAA, E.; MAGI, M.; SAMOSON, A.; TARMAK, M.; ENGELHARDT, G.
Investigation of the Structure of Zeolites by Solid-state High-Resolution 29
Si NMR
Spectroscopy. Journal of the American Chemical Society, v. 103, p. 4992, 1981.
LIU, Y.; ZHANG, W.; PINNAVAIA, T. J. Steam-Stable Aluminosilicate Mesostructures
Assembled from Zeolite Type Y Seeds Journal of the American Chemical Society, v. 122,
p. 8791, 2000.
LIU, Y.; ZHANG, W.; PINNAVAIA, T. J. Steam-Stable MSU-S Aluminosilicate
Mesostructures Assembled from Zeolite ZSM-5 and Zeolite Beta Sedds. Angewandte
Chemie, v. 40, p. 1255, 2001.
LIU, Y.; PINNAVAIA, T. J. Assembly of Hydrothermally Stable Aluminosilicate Foams and
Large-Pore Hexagonal Mesostructures from Zeolite Seeds under Strongly Acidic Conditions.
Chemistry Materials, v. 14, p. 3, 2002.
LIU, S.; CAO, X.; LI, L.; LI, C.; JI, Y.; XIAO, F.-S.; Preformed zeolite precursor route for
synthesis of mesoporous X zeolite. Colloids and Surfaces A: Physicochemical and
Engineering Aspects, v. 318, 269, 2008.
LÓNYI, F.; VALYON, J. On the interpretation of the NH3-TPD patterns of H-ZSM-5 and H-
mordenite. Microporous and Mesoporous Materials, v. 47, 293, 2001.
LUNA, F. J.; SCHUCHARDT, U. Modificação de Zeólitas para Uso em Catálise. Química
Nova, v. 24, p. 885, 2001.

7 – REFERÊNCIAS 91
MADSEN, C.; JACOBSEN, C. J. H. Nanosized Zeolite Crystals—Convenient Control of
Crystal Size Distribution by Confined Space Synthesis. Chemical Communication, p. 673,
1999.
MAJANO G.; MINTOVA S.; OVSITSER O.; MIHAILOVA B.; BEIN T. Zeolite Beta
Nanosized Assemblies. Microporous and Mesoporous Materials, v. 80, p. 227, 2005.
MEYNEN, V.; COOL, P.; VANSANT, E. F. Verified Syntheses of Mesoporous Materials.
Microporous and Mesoporous Materials, v. 125, p. 170, 2009.
MILTON, R. M. U.S. Patent. 2.882.243, 1959 a.
MILTON, R. M. U.S. Patent 2.882.244, 1959 b.
MISTRY, S. R.; MAHERIA, K. C. Synthesis of diarylpyrimidinones (DAPMs) using large
pore zeólitas. Journal of Molecular Catalysis A: Chemical, v. 355, p. 210, 2012.
MONNIER, A.; SCHUTH, F.; HUO, Q.; KUMAR, D.; MARGOLESE, D.; STUCKY, G. D.;
KRISHNAMURTY, M.; PETROFF, P.; FIROUZI, A.; JANICKE, M.; GHMELKA, B. F.
Cooperative Formation of Inorganic-Organic Interfaces in the Synthesis of Silicate
Mesostructuers. Science, v. 261, p. 1299, 1993.
MORENO, E. L.; RAJAGOPAL, K. Desafios da acidez na catálise em estado sólido.
Química Nova, v. 32, p. 538, 2009.
NA, K.; CHOI, M.; PARK, W.; SAKAMOTO, Y.; TERASAKI, O.; RYOO, R. Pillared MFI
Zeolite Nanosheets of a Single-Unit-Cell Thickness. Journal of the American Chemical
Society, v. 132, p. 4169, 2010.
NA, K.; JO, C.; KIM, J.; CHO, K.; JUNG, J.; SEO, Y.; MESSINGER, R. J.; CHMELKA, B.
F.; RYOO, R. Directing Zeolite Structures into Hierarchically Nanoporous Architectures.
Science, v. 333, p. 328, 2011.
NUR, H.; RAMLI, Z.; EFENDI, J.; RAHMAN, A. N. A.; CHANDREN, S.; YUAN, L. S.
Synergistic role of Lewis and Brönsted acidities in Friedel–Crafts alkylation of resorcinol
over gallium-zeolite beta. Catalysis Communications, v. 12, 822, 2011.
PAYRA, P.; DUTTA, P. K. Zeolites: A Primer, In: AUERBOCH, S. M.; CARRADO, K. A.;
DUTTA, P. K. Handboook of Zeolites Science and Technology, New York: Marcel Dekker
Inc, 2003. Cap. 1.
PEREIRA, A. L. C.; GONZÁLEZ-CARBALLO, J. M.; PÉREZ-ALONSO, F. J.; ROJAS, S.;
FIERRO, J. L. G.; RANGEL, M. D. C. Effect of the Mesostructuration of the Beta Zeolite
Support on the Properties of Cobalt Catalysts for Fischer–Tropsch Synthesis. Topics in
Catalysis, v. 54, p. 179, 2011.
PÉREZ-PARIENTE, J. Química Estructural de Materiales Zeolíticos, In: PÉREZ-
PARIENTE, J.; MARTÍNEZ, J. G. Materiales Zeolíticos: Síntesis, Propriedades y
Aplicaciones. San Vicente: Universidad Alicante, 2002. Cap. 1.

7 – REFERÊNCIAS 92
PÉREZ-PARIENTE, J.; DÍAZ, I.; AGÚNDEZ, J. Organising Disordered Matter: Strategies
for Ordering the Network of Mesoporous Materials. Comptes Rendus Chimie, v. 8, p. 569,
2005.
PIERRE, A. C.; ELALOUI, E.; PAJONK, G. M. Comparison of the Structure and Porous
Texture of Alumina Gels Synthesized by Different Methods. Langmuir, v. 14, p. 66, 1998.
PIERRE, A. C.; PAJONK, G. M. Chemistry of Aerogels and Their Applications. Chemical
Reviews, v. 102, p. 4243, 2002
RABO, J. A.; SCHOONOVER, M. W. Applied Catalysis A: General, v. 222, p. 261, 2001.
RAMOS, M. S.; GRECCO, S. T.; GOMES, L. P.; OLIVEIRA, A. C.; REYES, P.;
OPORTUS, M.; RANGEL, M. C. Evaluation of MY Zeolites (M= Pt, Pd, Ni) in the
Transalkylation of Trimethylbenzene with Benzene. Studies in Surface Science and
Catalysis, v. 156, p. 809, 2005.
RAMOS, F. S. O.; MUNSIGNATTI, E. C. O.; PASTORE, H. O. 2D–3D Structures: The
Hydrothermal Transformation of a Layered Sodium Silicate, Na-RUB-18, into Mordenite
Zeolite. Microporous and Mesoporous Materials, v. 177, p. 143, 2013.
RANGEL, M. C.; VALENTINI, A.; OLIVEIRA, A. S.; DAVID, J. M.; MARQUES
BRITTO, J.; DOMINGUES, S. M.; REYES, P. Natureza do Coque Formado sobre a
Mordenita Durante a Transalquilação de Benzeno. Quimica Nova, v. 26, p. 305, 2003.
RANGEL, M. C.; GRECCO, S. T. F.; CARVALHO, D. R.; URQUIETA-GONZÁLEZ, E.
A.; GRAU, J. M.; REYES, P.; OPORTUS, M. Influence of Platinum on the Performance of
Beta Zeolite-Based Catalysts for Xylenes Production, In: Daramola, M. O. Xylenes:
Synthesis, Characterization and Physicochemical Properties, Nova Science Publishers, Inc.,
2013. Cap 4.
RAYALU, S. S.; UDHOJI, J. S.; MESHRAM, S. U.; NAIDU, R. R.; DEVOTTA, S.
Estimation of crystallinity in flyash-based zeolite-A using XRD and IR spectroscopy.
Current Science, v. 89, p. 2147, 2005.
REY, F.; JORDÁ, L. J. Síntesis y Aplicaciones de Sólidos Mesoporosos Ordenados, In:
PÉREZ-PARIENTE, J.; MARTÍNEZ, J. G. Materiales Zeolíticos: Síntesis, Propriedades y
Aplicaciones. San Vicente: Universidad Alicante, 2002. Cap. 3.
SAKTHIVEL, A.; HUANG, S. J.; CHEN, W. H.; LAN, Z. H.; CHEN, K. H.; KIM, T. W.;
RYOO, R.; CHIANG, A. S. T.; LIU, S. B. Replication of Mesoporous Aluminosilicate
Molecular Sieves (RMMs) with Zeolite Framework from Mesoporous Carbons (CMKs).
Chemistry Materials, v. 16, p. 3168, 2004.
SAMANTA, S.; MAL, N. K.; KUMAR, P.; BHAUMIK, A. Hydrothermally synthesized high
silica mordenite as an efficient catalyst in alkylation reaction under liquid phase condition.
Journal of Molecular Catalysis A: Chemical, v. 215, p. 169, 2004.

7 – REFERÊNCIAS 93
SANZA, R.; SERRANO, D. P.; PIZARRO, P.; MORENO, I. Hierarchical TS-1 Zeolite
Synthesized from SiO2 TiO2 Xerogels Imprinted with Silanized Protozeolitic Units. Chemical
Engineering Journal, v. 171, p. 1428, 2011.
SAYARI, A. Catalysis by Crystalline Mesoporous Molecular Sieves. Chemistry Materials,
v. 8, p. 1840, 1996.
SCHMIDT, I.; MADSEN, C.; JACOBSEN, C. J. H. Confined Space Synthesis. A Novel
Route to Nanosized Zeolites. Inorganic Chemistry, v. 39, p. 2279, 2000.
SCHMIDT, I.; BOISEN, A.; GUSTAVSSON, E.; STAHL, K.; PEHRSON, S.; DAHL, S.;
CARLSSON, A.; JACOBSEN, C. J. H. Carbon Nanotube Templated Growth of Mesoporous
Zeolite Single. Crystals Chemistry of Materials, v.13, p. 4416, 2001.
SELVARAJ, M.; PANDURANGAN, A.; SESHADRI, K. S.; SINHA, P. K.; LAL, K. B.
Synthesis, Characterization and Catalytic Application of MCM-41 Mesoporous Molecular
Sieves Containing Zn and Al. Applied Catalysis A: General, v. 242, p. 347, 2003.
SERRANO, D. P.; AGUADO, J.; ESCOLA, J. M.; RODRÍGUEZ, J. M.; PERAL, A.
Hierarchical Zeolites with Enhanced Textural and Catalytic Properties Synthesized from
Organofunctionalized Seeds. Chemistry Materials, v. 18, p.22462, 2006.
SERRANO, D. P.; AGUADO, J.; ESCOLA, J. M.; RODRÍGUEZ, J. M.; PERAL, A. Effect
of the Organic Moiety Nature on the Synthesis of Hierarchical ZSM-5 from Silanized
Protozeolitic Units. Journal of Materials Chemistry, v. 18, p. 4210, 2008.
SERRANO, D. P.; AGUADO, MORALES, G.; RODRÍGUEZ, J. M.; PERAL, A.;
THOMMES, M.; EPPING, J. D.; CHMELKA, B. F. Molecular and Meso- and Macroscopic
Properties of Hierarchical Nanocrystalline ZSM-5 Zeolite Prepared by Seed Silanization.
Chemistry Materials, v. 21, p. 641, 2009.
SERRANO, D. P.; AGUADO, J.; ESCOLA, J. M.; PERAL, A.; MORALES, G.; ABELLA E.
Synthesis of hierarchical ZSM-5 by silanization and alkoxylation of protozeolitic units.
Catalysis Today, v. 168, p. 86, 2011.
SCHNEIDER, P. Adsorption Isotherms of Microporous-Mesoporous Solids Revisited.
Applied Catalysis A: General, v. 129, p. 157, 1995.
SHANBHAG, G. V.CHOI, M; KIM, J.; RYOO, R. Mesoporous Sodalite: A Novel, Stable
Solid Catalyst for Base-Catalyzed Organic Transformations. Journal of Catalysis, v. 264, p.
88, 2009.
SHI, Z.; WANG, Y.; MENG, C.; LIU, X. Hydrothermal Conversion of Magadiite into
Mordenite in the Presence of Cyclohexylamine. Microporous and Mesoporous Materials,
v. 176, p. 155, 2013.
SHIH, P. C.; WANG, J. H.; MOU, C. Y. Strongly Acidic Mesoporous Aluminosilicates
Prepared from Zeolite Seeds: Acylation of Anisole with Octanyl Chloride. Catalysis Today,
v. 93, p. 365, 2004.

7 – REFERÊNCIAS 94
SOUSA-AGUIAR, E. F.; CORREA, R. J. Principios de La Catálisis por Zeolitas, In: PÉREZ-
PARIENTE, J.; MARTÍNEZ, J. G. Materiales Zeolíticos: Síntesis, Propriedades y
Aplicaciones. San Vicente: Universidad Alicante, 2002. Cap. 6.
TANEV, P. T.; CHIBWE, M.; PINNAVAIA, T. J. Titanium-Containing Mesoporous
Molecular Sieves for Catalytic Oxidation of Aromatic Compounds. Nature, v. 368, p. 321,
1994.
TAO, Y.; KANOH, H.; KANEKO, K. ZSM-5 Monolith of Uniform Mesoporous Channels.
Journal of the American Chemical Society, v. 125, p. 6044, 2003 a.
TAO, Y.; KANOH, H.; KANEKO, K. Uniform Mesopore-Donated Zeolite Y Using Carbon
Aerogel Templating. Journal of Physical Chemistry B, v. 107, p. 10974, 2003 b.
TAO, Y.; HATTORI, Y.; MATUMOTO, A.; KANOH, H.; KANEKO, K. Comparative Study
on Pore Structures of Mesoporous ZSM-5 from Resorcinol-Formaldehyde Aerogel and
Carbon Aerogel Templating. Journal of Physical Chemistry B, v. 109, p. 194, 2005 a.
TAO, Y.; KANOH, H.; KANEKO, K. Synthesis of Mesoporous Zeolite A by Resorcinol-
Formaldehyde Aerogel Templating. Langmuir, v. 21, p. 504, 2005 b.
TOSHEVA, L.; VALTCHEV V. P. Nanozeolites: Synthesis, Crystallization Mechanism, and
Applications. Chemistry of Materials, v. 17, p. 2494, 2005.
TREACY, M. M. J.; HIGGINS, J. B., Collection of Simulated XRD Powder Patterns for
Zeolites. Elsevier, 5th Revised Edition, 2007.
TRONG ON, D.; DESPLANTIER-GISCARD, D.; DANUMAH, C.; KALIAGUINE, S.
Perspectives in Catalytic Applications of Mesostructured Materials. Applied Catalysis A:
General, v. 253, p. 545, 2003.
VARTULI, J. C.; ROTH, W. J.; BECK, J. S.; MCCULLEN, S. B.; KRESGE, C. T. The
Synthesis and Properties of M41S and Related Mesoporous Materials, In: KARGE, H. G.;
WEITKAMP, J. Molecular Sieves, v. 1, Berlin: Springer-Verlag, 1998. Cap 4.
VELOSO, C. M.; RANGEL, M. C. Preparação de Carbonos Porosos por Moldagem
Sequencial. Química Nova, v. 32, p. 2133, 2009.
VUONGA, G.-T.; HOANGA, V.-T.; NGUYENB, D.-T.; DO, T.-O. Synthesis of
Nanozeolites and Nanozeolite-Based FCC Catalysts, and Their Catalytic Activity in Gas Oil
Cracking Reaction. Applied Catalysis A: General, v. 382, p. 231, 2010.
WADLINGER, R. L.; KERR, G. T.; ROSINSKI, E. J. U.S. Patent, 3.308.069, 1967.
WALLAU, M.; SILVA, C. R.; URQUIETA-GONZÁLEZ, E. A. Mesoporous ZSM-5
Prepared by Sequential Nanocasting of MCM-41 Nanospheres. Studies in Surface Science
and Catalysis, v. 162, p. 409, 2006.

7 – REFERÊNCIAS 95
WANG, J.; HUANG, L.; CHEN, H.; LI, Q. Acid Function of Al‐MCM‐41 Supported
Platinum Catalysts in Hydrogenation of Benzene, Toluene O‐Xylene. Catalysis Letters, v.
55, p. 157, 1998.
WANG, S.; DOU, T.; LI, Y.; ZHANG, Y.; LI, X.; YAN Z. Synthesis, Characterization, and
Catalytic Properties of Stable Mesoporous Molecular Sieve MCM-41 Prepared from Zeolite
Mordenite. Journal of Solid State Chemistry, v. 177 p. 4800, 2004.
WANG, H.; PINNAVAIA, T. J. MFI Zeolite with Small and Uniform Intracrystal Mesopores.
Angewandte Chemie, v. 45, p. 7603, 2006.
WEITKAMP, J. Zeolites and Catalysis. Solid State Ionics, v. 131, p. 175, 2000.
WRIGHT, P. A.; LOZINSKA, M. Structural Chemistry and Properties of Zeolites, In:
MARTÍNEZ, C.; PEREZ-PARIENTE, J. Zeolites and Ordered Porous Solids: Fundamentals
and Applications, Valencia: Universitat Politècnica de València, 2011. Cap 1.
XIA, Y.; MOKAYA, R. On the Synthesis and Characterization of ZSM-5/MCM-48
Aluminosilicate Composite Materials. Journal of Materials Chemistry, v. 14, p. 863, 2004.
XIAO, F. S.; WANG, L.; YIN, C.; LIN, K.; DI, Y.; LI, J.; XU, R.; SU, D. S.; SCHLGL, R.;
YOKOI, T.; TATSUMI, T. Catalytic Properties of Hierarchical Mesoporous Zeolites
Templated with a Mixture of Small Organic Ammonium Salts and Mesoscale Cationic
Polymer. Angewandte Chemie, v. 45, p. 3090, 2006.
XU, B.; BORDIGA, S.; PRINS, R.; VAN BOKHOVEN, J. A. Effect of Framework Si/Al
Ratio and Extra-Framework Aluminum on the Catalytic Activity of Y Zeolite. Applied
Catalysis A: General, v. 333, p. 245, 2007.
YANG, R. T. Adsorbents: Fundamentals and Applications. New Jersey: John Wiley & Sons,
2003.
YANG, Z.; XIA, Y.; MOKAYA, R. Zeolite ZSM-5 with Unique Supermicropores
Synthesized Using Mesoporous Carbon as a Template. Advanced Materials, v. 16, p. 727,
2004.
YOLDAS, B. E.; PARTLOW, D. P. J. Formation of mullite and other alumina-based
ceramics via hydrolytic polycondensation of alkoxides and resultant ultra- and microstructural
effects. Journal of Materials Science, V. 23, p. 1895, 1988.
ZHANG, B.; DAVIS, S. A.; MENDELSON, N. H.; MANN, S. Bacterial Templating of
Zeolite Fibres with Hierarchical Structure. Chemical Communication, p. 781, 2000.
ZHANG, Z.; HAN, Y.; XIAO, F. S.; QIU, S.; ZHU, L.; WANG, R.; YU, Y.; ZHANG, Z.;
ZOU, B.; WANG, Y.; SUN, H.; ZHAO, D.; WEI, Y. Mesoporous Aluminosilicates with
Ordered Hexagonal Structure, Strong Acidity, and Extraordinary Hydrothermal Stability at
High Temperatures. Journal of the American Chemical Society, v. 123, p. 5014, 2001.
ZHANG, W. H.; LU, J.; HAN, B.; LI, M.; XIU, J.; YING, P.; LI, C. Direct Synthesis and
Characterization of Titanium-Substituted Mesoporous Molecular Sieve SBA-15. Chemistry
Materials, v. 14, p. 3413, 2002.

7 – REFERÊNCIAS 96
ZHANG, Y.; OKUBO T.; OGURA, M. Synthesis of Mesoporous Aluminosilicate with
Zeolitic Characteristics Using Vapor Phase Transport. Chemical Communication, p. 2719,
2005.
ZHAO, X. S.; LU, G. K.; MILLAR, G. J. Advances in Mesoporous Molecular Sieve MCM-
41. Industrial and Engineering Chemistry Research, v. 35, p. 2075, 1996.
ZHAO, D.; FENG, J.; HUO, Q.; MELOSH, N.; FREDRICKSON, G.H.; CHMELKA, B.F.;
STUCKY, G.D. Triblock Copolymer Syntheses of Mesoporous Silica with Periodic 50 to 300
Angstrom Pores. Science, v. 279, p. 548, 1998 a.
ZHAO, D.; FENG, J.; HUO, Q.; MELOSH, N.; FREDRICKSON, G.H.; CHMELKA, B.F.;
STUCKY, G.D. Nonionic Triblock and Star Diblock Copolymer and Oligomeric Surfactant
Syntheses of Highly Ordered, Hydrothermally Stable, Mesoporous Silica Structures. Journal
of the American Chemical Society, v. 120, p. 6024, 1998 b.
ZHAO, Z.; LIU, Y.; WU, H.; LI, X.; HE, M.; WU, P. Hydrothermal Synthesis of Mesoporous
Zirconosilicate with Enhanced Textural and Catalytic Properties with the Aid of Amphiphilic
Organosilane. Microporous and Mesoporous Materials, v. 123, p. 324, 2009.

8 – PUBLICAÇÕES NO TEMA DA TESE 97
8. PUBLICAÇÕES NO TEMA DA TESE
8.1. GRECCO, S. T. F.; RANGEL, M. C.; URQUIETA-GONZÁLEZ, E. A. Zeólitas
Hierarquicamente Estruturadas. Química Nova, v. 36, p. 131, 2013.
8.2. RANGEL, M. C.; GRECCO, S. T. F.; CARVALHO, D. R.; URQUIETA-GONZÁLEZ,
E. A.; GRAU, J. M.; REYES, P.; OPORTUS, M. Influence of Platinum on the Performance
of Beta Zeolite-Based Catalysts for Xylenes Production, In: DARAMOLA, M. O. Xylenes:
Synthesis, Characterization and Physicochemical Properties, Nova Science Publishers, Inc.,
2013. Cap 4.
8.3. SILVA, L. H.; GRECCO, S. T. F.; SILVA, T. F.; ASSAF, J. M.; GRAU, J. M.;
RANGEL, M. C. Obtenção de catalisadores de platina suportada em zeólita beta para a
purificação de hidrogênio. Anais do 17º Congresso Brasileiro de Catálise e VII Congresso
de Catálise do Mercosul, Gramado, Rio Grande do Sul, Brasil, 2013.
8.4. RANGEL, M. C.; CARVALHO, D. R.; GRECCO, S. T. F.; URQUIETA-GONZÁLEZ,
E. A. Efeito do Tempo de Pré-Cristalização e de Cristalização sobre as Características da
Nanozeólita Beta obtida por Funcionalização das Sementes com Organossilanos. Anais do
XXIII Congresso Ibero-americano de Catálise, Santa Fe, Argentina, 2012.
8.5. OLIVEIRA, J. B.; D. R.; GRECCO, S. T. F.; GRAU, J. M.; RANGEL, M. C. Obtenção
de Catalisadores de Platina Suportada em MCM-41 modificada com Zircônio e/ou Alumínio
para a Reforma do n-Octano. Anais do XXIII Congresso Ibero-americano de Catálise,
Santa Fe, Argentina, 2012.
8.6. da SILVA, O. B.; GRECCO, S. T. F.; SANTOS, H. C. F, BORGES, S.; RANGEL, M. C.
Abatimento de Azul de Metileno em Efluentes Aquosos sobre Catalisadores do Tipo Fe/Sba-
15. Anais do XIX Congresso Brasileiro de Engenharia Química, Búzios, Rio de Janeiro,
Brasil, 2012.
8.7. SILVA, L. H.; GRECCO, S. T. F.; RANGEL, M. C. Efeito do teor de platina nas
propriedades texturais da nanozeólita beta. Anais do IX Encontro Norte, Nordeste e
Centro-oeste de Catálise, Maceió, Alagoas, Brasil, 2012.
8.8. GRECCO, S. T. F.; SILVA, E. D.; URQUIETA-GONZÁLEZ, E. A.; RANGEL, M. C.
Effect of Crystallization Time on the Particle Size of Beta Nanozeolites. Anais do 5th
International FEZA Conference, Valencia, Espanha, 2011.
8.9. CARVALHO, D. R.; GRECCO, S. T. F.; SILVA, E. D.; URQUIETA-GONZÁLEZ, E.
A.; RANGEL, M. C. The Effect of the Kind of Organosilane on the Textural Properties of
Beta Nanozeolite-Based Catalysts. Anais do 5th International FEZA Conference, Valencia,
Espanha, 2011.

8 – PUBLICAÇÕES NO TEMA DA TESE 98
8.10. GRECCO, S. T. F.; SILVA, E. D.; URQUIETA-GONZÁLEZ, E. A.; RANGEL, M. C.
Craqueamento de Cicloexano sobre Zeólitas Beta de Diferentes Tamanhos de Cristais. Anais
do 16º Congresso Brasileiro de Catálise, Campos do Jordão, São Paulo, Brasil, 2011.
8.11. CARVALHO, D. R.; GRECCO, S. T. F.; SILVA, L. H.; URQUIETA-GONZÁLEZ, E.
A.; RANGEL, M. C. Influência do Tempo de Envelhecimento no Tamanho do Cristal da
Zeólita Beta. Anais do 16º Congresso Brasileiro de Catálise, Campos do Jordão, São Paulo,
Brasil, 2011.
8.12. GRECCO, S. T. F.; URQUIETA-GONZÁLEZ, E. A.; REYES, P.; OPORTUS, M.;
RANGEL, M. C. Influência da Temperatura e do Tempo de Envelhecimento do Gel de
Sementes nas Propriedades de Zeólitas Beta obtidas via Mesoestruturação-Cristalização.
Anais do XXII Congresso Ibero-americano de Catálise, Viña Del Mar, Chile, 2010.
8.13. GRECCO, S. T. F.; URQUIETA-GONZÁLEZ, E. A.; RANGEL, M. C. Efeito do
Tempo de Cristalização de Sementes de Zeólita Beta na Preparação de Catalisadores Micro-
mesoporosos. Anais do 15º Congresso Brasileiro de Catálise e 5º Congresso de Catálise
do Mercosul, Armação dos Búzios, Rio de Janeiro, Brasil, 2009.
8.14. CARVALHO, D. R.; GRECCO, S. T. F.; NOBRE, P. S. S.; RANGEL, M. C. Influência
do Tempo de Cristalização e da Presença do Cátion Alcalino nas Propriedades da Nanozeólita
Beta. Anais do 15º Congresso Brasileiro de Catálise e 5º Congresso de Catálise do
Mercosul, Armação dos Búzios, Rio de Janeiro, Brasil, 2009.

Quim. Nova, Vol. 36, No. 1, 131-142, 2013
Rev
isão
*e-mail: [email protected]
ZEÓLITAS HIERARQUICAMENTE ESTRUTURADAS
Saulo de Tarso Figueiredo Grecco e Maria do Carmo Rangel* Instituto de Química. Universidade Federal da Bahia. Campus Universitário de Ondina, 40170-290 Salvador – BA, BrasilErnesto Antonio Urquieta-GonzálezDepartamento de Engenharia Química, Universidade Federal de São Carlos, 13565-905 São Carlos – SP, Brasil
Recebido em 5/3/12; aceito em 3/7/12; publicado na web em 26/11/12
HIERARCHICALLY STRUCTURED ZEOLITES. This review presents the main characteristics and properties of microporous (zeolites) and ordered mesoporous materials, focusing on structural aspects and preparation. In addition, their use as heterogeneous catalysts are also discussed, with emphasis on their advantages and disadvantages. Due to difficulty in application of zeolites in the conversion of bulky molecules, the most relevant strategies of synthesis for the preparation of zeolitic materials with hierarchical pore structure was also analyzed, which allow this limitation to be overcome.
Keywords: synthesis of zeolites; mesoporous materials; hierarchical porous structure.
INTRODUÇÃO
As zeólitas, materiais cristalinos microporosos, têm se conso-lidado ao longo da segunda metade do século XX e nesta primeira década do século XXI como importantes catalisadores em processos das indústrias de refino de petróleo, química e petroquímica, assim como no controle ambiental.1 Esse amplo espectro de aplicações está relacionado às propriedades físicas e químicas desses materiais, que podem ser controladas durante a sua preparação, visando uma aplicação específica. Além de seu particular sistema de microporos, as zeólitas apresentam propriedades específicas, que as diferenciam de outros materiais, tais como acidez ou basicidade superficial e capacidade de troca iônica.2 Entretanto, apesar dessas propriedades desejáveis, a presença apenas de microporos nas zeólitas impõe limitações à difusão de reagentes ou produtos volumosos. Embora a limitação difusional seja usada, em alguns casos, para controlar beneficamente a seletividade a um determinado produto da reação catalítica, a difusividade relativamente baixa de moléculas volumosas, nos microporos da zeólita, limita a taxa de reação, devido ao transporte mais lento dos reagentes e produtos, ocasionando um maior tempo de residência e, como consequência, favorecendo a ocorrência de reações indesejáveis.3 Em processos envolvendo hidrocarbonetos, a limitação difusional contribui para a formação do coque, que provoca a desativação da zeólita por obstrução dos canais ou envenenamentos dos sítios ativos.4
Com a expectativa de superar as limitações difusionais, no fim da década de 80 se deu início à busca por materiais contendo meso-poros, que resultou na síntese de materiais mesoporosos ordenados no início dos anos 90.5-8 Diversas peneiras moleculares mesoporosas com tamanho de poros ajustável têm sido desenvolvidas, apresentando emprego potencial em reações catalíticas. Entretanto, comparado às zeólitas, esses materiais mesoporosos possuem acidez e estabilidade hidrotérmica mais baixas, o que limita as suas aplicações catalíticas. Assim, esforços consideráveis têm sido dedicados ao desenvolvimento de materiais zeolíticos com estrutura hierárquica de poros, que com-binam as propriedades intrínsecas das zeólitas com a facilidade de difusão resultante da geração de mesoporosidade.
Um número significativo de procedimentos experimentais, pré- ou pós-síntese, vem sendo sugeridos para a obtenção de zeólitas
hierarquicamente estruturadas.9-19 As metodologias de síntese mais bem sucedidas envolvem o uso de agentes geradores de mesoporo-sidade (agentes orgânicos e nanopartículas) ou nanomoldes (mol-dagem em nanoespaços), que geram sólidos com mesoporosidade intracristalina com uma estreita distribuição de tamanho de poros; isto resulta em sólidos contendo mesoporos, além dos microporos intrínsecos das zeólitas. Além disso, é gerada uma mesoporosidade intercristalina, resultante da aglomeração dos cristais de zeólita com tamanhos nanométricos.
Nesse contexto, este artigo apresenta uma revisão crítica das principais características e propriedades de materiais microporosos (zeólitas) e mesoporosos ordenados, enfocando aspectos estruturais e de preparação. Além disso, são discutidas as vantagens e desvan-tagens do uso desses materiais como catalisadores heterogêneos. Devido à dificuldade de aplicação de zeólitas na conversão de moléculas volumosas, também são apresentadas as principais al-ternativas para superar essa limitação, descrevendo-se os métodos mais relevantes na preparação de materiais zeolíticos com estrutura hierárquica de poros.
ZEÓLITAS: ESTRUTURA E MÉTODOS DE PREPARAÇÃO
As zeólitas são materiais complexos consistindo no maior grupo de silicatos com estrutura aberta.20,21 A primeira zeólita, hoje identifi-cada como estilbita, foi descoberta em uma mina de cobre na Suécia, em 1756, pelo químico e mineralogista A. Crönstedt.22 Esses mine-rais foram denominados zeólitas, nome de origem grega, composto pelas palavras zeo (ferver) e lithos (pedra), devido à sua capacidade de liberar vapor d’água sob aquecimento. Até o presente momento, foram descobertas cerca de 60 zeólitas naturais, sendo algumas encontradas em abundância na natureza. Apesar disso, as zeólitas sintéticas são mais empregadas comercialmente, devido à sua maior uniformidade em composição, pureza elevada e possibilidade de se modelar suas propriedades, de modo a otimizá-las para aplicações industriais específicas.23
Estrutura das zeólitas e propriedades gerais
As zeólitas são aluminossilicatos cristalinos hidratados de estrutu-ra aberta, geralmente contendo metais alcalinos ou alcalinos terrosos como contra-íons. Estruturalmente, estes materiais são formados por

Grecco et al.132 Quim. Nova
uma rede tridimensional de tetraedros interligados, contendo canais e cavidades de dimensões moleculares. Os tetraedros são constituídos por unidades do tipo [SiO4] ou [AlO4]
- que se ligam entre si, através do compartilhamento de átomos de oxigênio para formar as unidades secundárias de construção.21,23 A diversidade e a complexidade dos materiais zeolíticos se devem às diferentes maneiras pelas quais essas unidades secundárias de construção podem se ligar para formar uma estrutura tridimensional.24
As cargas negativas, geradas durante a substituição de átomos de silício da rede por átomos de alumínio na estrutura da zeólita, são compensadas por íons positivos, chamados de cátions de compen-sação,21 que se distribuem de forma a minimizar a energia livre do sistema; a sua distribuição na estrutura depende da temperatura do tratamento térmico, das espécies catiônicas e do grau de hidratação da zeólita.25 Em zeólitas, a quantidade de átomos de silício e de alumínio presentes na rede pode variar em uma ampla faixa, desde a razão Si/Al unitária até o valor infinito, que corresponde a materiais contendo apenas átomos de silício, tais como os polimorfos da sílica (SiO2). Cabe ressaltar que a maioria das zeólitas só pode ser obtida em uma faixa de razão Si/Al limitada, que depende da estrutura. De acordo com a regra de Lowenstein, a razão Si/Al não pode ser inferior a 1, uma vez que a existência de tetraedros AlO4
- adjacentes não é favorável, devido à repulsão entre as cargas negativas.26
Devido a suas características estruturais, as zeólitas possuem algumas propriedades únicas que as tornam úteis em diversas apli-cações industriais, especialmente em catálise, tais como:27,28 área superficial específica elevada; dimensões moleculares dos poros, canais e cavidades, que lhes conferem diferentes tipos de seletividade de forma; capacidade de adsorção elevada; facilidade na separação de reagentes e produtos; possibilidade de modelar as propriedades eletrônicas dos sítios ativos; possibilidade de pré-ativar as moléculas dentro dos poros, pela existência de campos elétricos elevados e do confinamento molecular e, propriedades ácidas e básicas.
A última propriedade é especialmente importante do ponto de vista de aplicação, uma vez que a maioria das reações de hidrocarbo-netos e muitas das reações dos compostos orgânicos são catalisadas por zeólitas ácidas. Esta propriedade é gerada pela substituição dos cátions alcalinos e alcalinos terrosos por prótons, que se ligam fracamente aos átomos de oxigênio (ligados aos átomos de silício e alumínio), gerando grupos hidroxila ligados em ponte (sítios ácidos de Brönsted) mostrados na Figura 1.29 Por outro lado, o aquecimen-to desses sólidos leva à formação de sítios ácidos de Lewis, como consequência da saída desses grupos na forma de água (Figura 1).29 A força ácida de um centro protônico, bem como a sua atividade ca-talítica, depende de vários parâmetros, tais como, o ângulo da ligação Al-(OH)-Si, a proximidade entre os centros protônicos, a velocidade de troca iônica e a interação com sítios ácidos de Lewis. Diversos estudos, abordando importantes processos do refino do petróleo e da petroquímica conduzidos sobre catalisadores zeolíticos, mostraram que os centros de acidez protônica (sítios ácidos de Brönsted) são altamente ativos nessas reações; quando os sítios ácidos de Lewis estão presentes, promovem um aumento da força ácida dos centros protônicos.30 Nesses casos, a atividade catalítica depende também da acessibilidade dos reagentes ao centro protônico.30
Métodos de preparação de zeólitas
Por muitos anos as zeólitas naturais, encontradas em abundância na natureza, foram estudadas e consideradas atrativas para uso indus-trial. Entretanto, a baixa pureza desses sólidos, bem como a sua ampla variedade de composições, tornou-os inviáveis para uma aplicação comercial em larga escala. Dessa forma, esforços consideráveis têm sido realizados para obter esses materiais em laboratório, através de
metodologias que possam produzir materiais reprodutíveis em larga escala e com características pré-determinadas.
Os trabalhos de Barrer e Milton31 foram os pioneiros na prepara-ção de zeólitas sintéticas. A metodologia desenvolvida por Barrer foi baseada na conversão de fases minerais sob ação de soluções salinas concentradas, na faixa de 180 a 270 °C. Milton, por sua vez, empregou reagentes mais reativos, por exemplo, sílica gel ou silicato de sódio como fontes de silício e alumina ou aluminato de sódio como fontes de alumínio. Também usou hidróxido de sódio como fonte de sódio e meio mineralizante. Isto permitiu o emprego de condições mais brandas, tais como temperaturas e pressões mais baixas. A partir desses trabalhos, muitos outros foram conduzidos levando a avanços significativos, tanto em termos da descoberta de novas estruturas zeolíticas quanto na elucidação do mecanismo da síntese.
As zeólitas são, em geral, preparadas sob condições hidrotérmicas porque a estrutura aberta desses aluminossilicatos deve ser estabi-lizada durante o seu crescimento, através da ocupação dos canais e das cavidades formadas por moléculas hospedeiras.32,33 A síntese hidrotérmica de uma zeólita consiste na mistura das fontes contendo silício e alumínio, geralmente em meio básico (meio mineralizante), com uma fonte do cátion de compensação. Em seguida, a mistura reacional é aquecida em uma autoclave sob pressão autógena (no caso de temperaturas de reação acima de 100 °C). Após o período de indução, podem-se detectar cristais do produto zeolítico, que são recuperados por filtração, lavagem e secagem.34
Muitas zeólitas podem ser preparadas empregando-se apenas reagentes inorgânicos. Entretanto, nos anos 60, introduziu-se o uso de compostos orgânicos na síntese de zeólitas, denominados agentes direcionadores de estrutura, que levaram a uma melhoria significativa na qualidade dos produtos. Os agentes direcionadores de estrutura mais utilizados são os sais de amônio quaternário. Devido a res-trições estéricas, que limitam a quantidade de espécies orgânicas envolvidas na formação da estrutura, e devido à função de cátion
Figura 1. Reações químicas envolvidas na geração de sítios ácidos de Bröns-ted e de Lewis em zeólitas

Zeólitas hierarquicamente estruturadas 133Vol. 36, No. 1
compensador do íon amônio quaternário, são impostas restrições na densidade da carga, o que resulta na produção de estruturas com razões Si/Al elevadas.33 A primeira estrutura obtida utilizando um agente orgânico foi a zeólita beta, cuja síntese foi desenvolvida em 1967 por pesquisadores da Mobil.35 Um dos prováveis mecanismos da síntese hidrotérmica, empregando direcionadores de estrutura,36 consiste na formação de um compósito inorgânico-orgânico, no meio reacional, através da substituição parcial ou completa da esfera de hidratação hidrofóbica do íon amônio por espécies de silicato. As interações de van der Waals, entre os íons amônio e as espécies de silicato, fornecem a entalpia requerida para a formação da estrutura zeolítica, enquanto a liberação das moléculas de água fornece uma entropia adicional para compensar o processo de auto-organização. A agregação destes compósitos inorgânicos-orgânicos é responsável pela etapa de nucleação.
PROPRIEDADES E OBTENÇÃO DOS MATERIAIS MESOPOROSOS ORDENADOS
Apesar das zeólitas serem usadas amplamente em reações cata-líticas, a presença exclusiva de microporos com diâmetros na faixa de 0,4 a 1,2 nm, nesses sólidos, impõe limitações significativas para a ocorrência de reações que envolvam reagentes ou produtos volu-mosos, que não podem difundir nos seus microporos. Considera-se como reagente ou produto volumoso moléculas com diâmetro cinético superior ao diâmetro médio dos microporos das zeólitas. De fato, se a zeólita pode, em princípio, transformar um dado reagente em um pro-duto desejável com uma velocidade de reação superior à velocidade de difusão dos reagentes e produtos nos seus poros, então a velocidade da reação global será limitada pela taxa de difusão. Portanto, a reação ocorrerá sob regime de controle de difusão. Devido a essa limitação, consideráveis esforços têm sido feitos para tentar desenvolver mate-riais com diâmetros de poros na região dos mesoporos.
Desde a descoberta da família de peneiras moleculares meso-porosas denominada M41S em 1992, por pesquisadores da Mobil Corporation,5,6 diversos materiais mesoporosos têm sido preparados, usando o mesmo mecanismo de modelagem via a formação de um cristal líquido, liquid crystal templating (LCT). No processo LCT, as espécies orgânicas atuam como estrutura central, em torno da qual as espécies óxido são organizadas.37 O processo de formação da estrutura mesoporosa tem sido explicado através de dois mecanis-mos,5 como mostrado na Figura 1S, material suplementar. O primeiro deles reflete o significado literal do mecanismo LCT, isto é, envolve a pré-existência de agregados do surfactante (fase cristal líquido) na solução precursora. A subsequente formação da estrutura mesoporosa ocorre através da migração e polimerização das espécies de silicato na fase aquosa. No entanto, essa ideia parece ter apenas um significado conceitual, uma vez que a concentração do surfactante está abaixo da necessária para formar cristais líquidos; de fato, nenhuma fase cristal líquido foi observada em estudos de RMN e espalhamento de nêutrons a baixos ângulos.37 O segundo mecanismo considera a auto--organização das espécies de silicato, através da interação mútua entre eles e as espécies surfactantes (mecanismo cooperativo). Entretanto, esses dois modelos são insuficientes para o entendimento mecanistico de formação da estrutura mesoporosa.
Posteriormente, Stucky e colaboradores38 publicaram um modelo baseado no mecanismo cooperativo, que explica detalhadamente a formação da mesofase. De acordo com esse modelo, a formação da estrutura mesoporosa ocorre através de três etapas: interação entre as espécies silicato e as espécies surfactantes na fase micelar; polimerização preferencial de espécies silicato na região interfacial silicato/surfactante em detrimento da polimerização em solução e, equilíbrio entre as densidades de carga do silicato e do surfactante.
A compensação da carga superficial das micelas por anions silicato é o parâmetro mais importante na formação das diferentes fases me-soporosas, uma vez que variando algumas condições de síntese, tais como pH, temperatura, etc, pode-se influenciar no grau de polimeri-zação e, consequentemente, na densidade de carga do silicato.39 Em presença de espécies silicato muito despolimerizadas, há a tendência de formação de estruturas lamelares ou cúbicas, que possuem menor raio de curvatura e fator de empacotamento mais elevado, resultando em maior densidade de carga positiva. Desta forma, estes arranjos são capazes de compensar a elevada densidade de carga negativa das espécies de silicato mais despolimerizadas. Por outro lado, as espécies mais polimerizadas tendem a formar estruturas hexagonais com menor densidade de carga.
A natureza da mesofase obtida também depende da concentração do surfactante,37,40 como mostrado na Figura 2S, material suple-mentar. Em concentrações mais baixas as moléculas de surfactante se encontram isoladas, enquanto que aumentando a concentração, as moléculas do surfactante se agregam formando micelas. Se a concentração continua aumentando, são formados os arranjos hexagonais empacotados formando a fase hexagonal. A próxima etapa, nesse processo de formação micelar, é a coalescência dos cilindros adjacentes para formar a fase lamelar. Em alguns casos, forma-se previamente a fase cúbica. A natureza da fase presente na solução aquosa do surfactante depende, não apenas da concentração, mas também da natureza do surfactante, bem como dos parâmetros de síntese.40
Existem três tipos principais de peneiras moleculares mesopo-rosas, que são obtidos através de diferentes rotas de formação da mesofase. O primeiro tipo é a família M41S de silicatos ou alumi-nossilicatos mesoporosos, que inclui as fases hexagonal (MCM-41), cúbica (MCM-48) e lamelar (MCM-50)37,40 mostradas na Figura 2a. A preparação destes materiais envolve o uso de surfactantes iônicos, como um molde (template) e um mecanismo de organização iônica representado por S+I-. De acordo com este mecanismo, ocorre a formação de um par iônico entre as espécies inorgânicas aniônicas e o surfactante catiônico. Este método foi estendido a uma série de mecanismos de organização eletrostática, incluindo a rota reversa S-I+, em que as espécies inorgânicas são catiônicas e as espécies sur-factantes, aniônicas. Outro mecanismo refere-se às rotas auxiliadas por contraíons, representadas por S+X-I+ e S-M+I-, em que as espécies inorgânicas interagem com as espécies do surfactante por intermédio de um contraíon.41 O segundo tipo de peneiras moleculares mesopo-rosas envolve uma interação entre espécies surfactantes e espécies inorgânicas neutras (S0I0).7 Entretanto, as sílicas mesoporosas hexa-gonais (HMS e MSU) obtidas através desta rota são menos ordenadas do que as peneiras moleculares mesoporosas preparadas via meca-nismo iônico.42 O último tipo de peneiras moleculares mesoporosas, que inclui a SBA-15 (Figura 2b), é obtido através de uma nova rota de síntese, empregando copolímeros di e triblocos anfifílicos, como moldes (templates).8
A principal diferença entre os materiais mesoporosos e as zeólitas consiste no ordenamento da estrutura resultante. Nas estruturas zeolí-ticas, os tetraedros TO4 (T = Si, Al) estão espacialmente ordenados e, portanto, é possível criar uma estrutura cristalina a partir da repetição no espaço de uma unidade elementar, denominada célula unitária. Nos materiais mesoporosos, não é possível definir uma célula unitária e nem as posições cristalográficas. A única organização é a geometria tetraédrica dos átomos T. A partir da unidade TO4 não existe um ar-ranjo definido de átomos. Esses materiais, portanto, possuem paredes amorfas.43 Desta forma, os materiais mesoporosos apresentam apenas um ordenamento dos mesoporos de longo alcance, enquanto que as zeólitas possuem uma organização de curto alcance, característica de materiais cristalinos.

Grecco et al.134 Quim. Nova
A família M41S: estruturas e métodos de preparação
A família M41S inclui as estruturas MCM-41, MCM-48 e MCM-50 (Figura 2a). A estrutura MCM-41 é formada por um arranjo hexagonal de mesoporos uniformes e unidimensionais, enquanto a MCM-48 possui um arranjo cúbico de mesoporos interconectados, resultando em um sistema de poros tridimensional. Por outro lado, a estrutura MCM-50 possui um arranjo lamelar constituído por uma camada dupla do surfactante alternada por camadas de sílica; após a remoção do surfactante, se formará um sistema de poros bidimensio-nal, cuidando-se para que a estrutura seja devidamente estabilizada, por exemplo, via o processo de pilarização.
Entre as estruturas da família M41S, a estrutura MCM-41 é a que tem sido mais investigada, uma vez que as demais estruturas são instáveis termicamente ou difíceis de sintetizar.44,45 Este material possui área superficial específica superior a 1000 m2 g-1 e diâmetro de mesoporos na faixa de 1,5 a 10 nm,40,45 que podem ser controla-dos através da escolha cuidadosa do molde (template), da adição de compostos orgânicos auxiliares ou modificando-se os parâmetros de síntese.37 Além disso, seus mesoporos são uniformes, o que resulta em uma distribuição estreita de diâmetros de poros.45 Entretanto, as paredes dos mesoporos são finas, com uma espessura que varia de 1 a 1,5 nm, o que impõe limitações na estabilidade térmica e hidro-térmica do material.
O padrão de difração da MCM-41 (Figura 3Sa, material suple-mentar) apresenta apenas reflexões na região de baixos ângulos mas, em muitos casos, observa-se apenas um pico de difração em 2q igual a 2°.40 Diferentemente das zeólitas, estas reflexões são provenientes do ordenamento dos mesoporos, que resulta em uma organização de longo alcance nestes materiais amorfos.
A MCM-41 apresenta um grande potencial de aplicação como
catalisador ou suporte catalítico, especialmente em reações de conversão de moléculas volumosas, devido às suas características, principalmente a área superficial específica elevada e a estrutura de poros com canais de tamanho e forma definidos. Entretanto, os ma-teriais mesoporosos contendo apenas silício apresentam acidez baixa, atribuída aos grupos silanóis, presentes na superfície.42
Como nas zeólitas, a substituição de átomos de silício por átomos de alumínio na estrutura da MCM-41 cria cargas negativas que, ao serem compensadas por prótons, geram os sítios ácidos de Brönsted. Comparada às zeólitas, a MCM-41 contendo alumínio exibe uma forte tendência à desaluminação durante a remoção do surfactante. Este processo ocorre, principalmente, devido à hidrólise do alumínio da rede pelo vapor d’água gerado durante a combustão do surfactante.40 Além disso, independentemente da quantidade de alumínio existente na estrutura, os materiais obtidos apresentaram acidez de Brönsted mais baixa quando comparados às zeólitas.46
Assim como as zeólitas, a MCM-41 é obtida a partir de uma mistura reacional contendo o surfactante e uma fonte de silício ([Si]-MCM-41), podendo também conter uma fonte de alumínio ([Al]-MCM-41). Esta mistura é submetida a um tratamento hidrotérmico, em temperaturas que variam entre 70 e 150 °C, por um período de tempo que pode variar entre 1 a 10 dias.40,44 O rendimento da síntese e suas propriedades físico-químicas são determinadas pela temperatura, pH, tempo de cristalização, composição molar da mistura reacional e natureza dos materiais precursores.44
A família SBA: estruturas e métodos de preparação
Em 1998, foi sintetizada uma nova família de silicatos ou alu-minossilicatos mesoporosos altamente ordenados em meio ácido, empregando copolímeros triblocos como molde (template). Os co-polímeros apresentam a fórmula geral OEnOPmEOn e são formados por blocos de óxido de etileno (OE)n e de óxido de propileno (OP)m.8 Diversas estruturas foram preparadas apresentando diferentes arranjos periódicos dos mesoporos, tais como SBA-1 (arranjo cúbico), SBA-11 (arranjo cúbico), SBA-12 (arranjo hexagonal com sistema de poros tridimensional), SBA-14 (arranjo lamelar), SBA-15 (arranjo hexago-nal com sistema de poros bidimensional) e SBA-16 (arranjo cúbico centrado em uma cavidade mesoporosa).45 Entre elas, a SBA-15 é a estrutura que tem atraído mais atenção, devido às suas propriedades, tais como estabilidade térmica e hidrotérmica mais elevadas quando comparada aos outros materiais mesoporosos.
A estrutura SBA-15 é formada por um arranjo hexagonal de mesoporosos similar à estrutura MCM-41, porém com espessuras da parede dos mesoporos superiores (entre 3 e 6 nm) e diâmetros de poros na faixa de 4 a 14 nm.45 Devido à maior espessura da parede, a SBA-15 possui uma estabilidade térmica e hidrotérmica maior do que a MCM-41, o que aumenta o seu potencial de aplicação em catálise e em processos industriais. Essas paredes espessas são formadas por uma matriz de sílica microporosa. A SBA-15 é um material composto por microporos e mesoporos, como mostrado na Figura 2b. Essa estrutura apresenta um arranjo mesoporoso altamente ordenado e o diâmetro dos microporos pode variar entre 0,5 e 3 nm, dependendo das condições empregadas na síntese.45
Os copolímeros di e triblocos proporcionam a formação da es-trutura da SBA-15, uma vez que as cadeias do polímero de óxido de etileno são hidrofílicas, enquanto as cadeias do polímero de óxido de propileno são hidrofóbicas; isto conduz à formação de micelas cilíndricas, com as cadeias do polímero de óxido de etileno localizadas no lado externo das micelas. Desta forma, as cadeias do polímero de óxido de etileno podem ser ocluídas nas paredes do silicato, como mostrado na Figura 2b. Após a calcinação, a estrutura resultante possui mesoporos e microporos formados durante a remoção do
Figura 2. Ilustração da estrutura, (a) MCM-41, MCM-48 e MCM-50 e (b) SBA-15. POP= Polímero do óxido de propileno; POE= polímero do óxido de etileno. Adaptada das ref. 37 (a) e 47 (b)

Zeólitas hierarquicamente estruturadas 135Vol. 36, No. 1
polímero de óxido de etileno e do polímero de óxido de propileno, respectivamente. Entretanto, Garlaneau e colaboradores47 observaram que a formação de uma estrutura de poros tridimensional, na qual os mesoporos estão interconectados através dos microporos, só é possível quando se utiliza temperaturas inferiores a 130 °C. Acima desta temperatura, o óxido de etileno não permanece ocluído nas paredes do silicato.
A SBA-15 apresenta apenas reflexões na região de baixos ângulos no seu padrão de difração de raios X (Figura 3Sb, material suple-mentar), o que evidencia apenas uma organização de longo alcance dos mesoporos. Nenhuma reflexão pode ser observada na região de altos ângulos, devido à natureza amorfa das paredes dos poros. Pelo fato da SBA-15 ser obtida em meio fortemente ácido, a incorporação de alumínio nas suas paredes é dificultada, o que prejudica a possi-bilidade de se introduzir prótons na sua estrutura, inviabilizando sua aplicação como catalisador ácido. As dificuldades de incorporação de alumínio nestes materiais se devem à fácil dissociação da ligação Al-O-Si sob condições hidrotérmicas ácidas e à diferença entre as taxas de hidrólise dos precursores de silício e de alumínio. Várias estratégias foram empregadas para solucionar estes problemas, tais como a pré-hidrólise dos alcoxissilanos antes da adição do alcóxido de alumínio48 ou diminuindo a taxa de hidrólise dos precursores de alumínio por complexação com agentes quelantes, tais como o acetoacetato de etila.49 O problema também pode ser resolvido adicionando-se alguns catalisadores, tais como fluoreto, para acelerar as taxas de hidrólise do precursor de silício.50 Apesar destes esforços, estes materiais também possuem acidez mais baixas que as zeólitas.
Na primeira vez em que a SBA-15 foi sintetizada, Zhao e cola-boradores8 empregaram um copolímero tribloco, conhecido como Pluronic P123, que apresenta a fórmula geral OE20OP70OE20. Foram empregadas concentrações diluídas do Pluronic P123 e meio ácido (pH < 1), variando-se a concentração do copolímero e a temperatura de mesoestruturação. Observou-se que, usando concentrações do copolímero superiores a 6% ou inferiores a 0,5%, se formava apenas um silicato não ordenado. Por outro lado, em temperaturas inferiores a 35 °C e superiores a 80 °C era produzido, também, um silicato pouco ordenado. A partir desses resultados, concluiu-se que as condições mais adequadas para a formação de uma estrutura mesoporosa or-denada eram concentrações do copolímero na faixa de 0,5 a 6% e temperatura de mesoestruturação entre 35 e 80 °C.
Outra importante variável estudada, na formação de sólidos do tipo SBA, foi a relação entre o número de unidades do óxido de etileno e do óxido de propileno, que controla o tipo da estrutura mesoporosa que será obtida (fase lamelar, cúbica, hexagonal e outras). Quando são empregadas relações óxido de etileno/óxido de propileno mais baixas, a formação da fase hexagonal é favorecida.51,52 Por outro lado, alterando o comprimento da cadeia do polímero de óxido de propileno, pode-se controlar o diâmetro dos mesoporos.52 O diâmetro de poros também pode ser controlado através da adição de aditivos, tais como cossurfactantes, agentes expansivos, eletrólitos e sais.45
MATERIAIS ZEOLÍTICOS COM ESTRUTURA HIERÁRQUICA DE POROS
Comparados às zeólitas, os aluminossilicatos mesoporosos apre-sentam acidez e estabilidades hidrotérmica e mecânica mais baixas, o que limita suas aplicações industriais, especialmente em catálise.42
Uma vez que as estabilidades térmica, hidrotérmica e mecânica, assim como a acidez elevada, são parâmetros fundamentais para as aplicações catalíticas industriais, muitos esforços têm sido realizados com o intuito de obter novos materiais que combinem as vantagens das zeólitas e dos materiais mesoporosos, principalmente em relação à elevada acidez e mesoporosidade. Dessa forma, diversas metodologias
de síntese têm sido desenvolvidas para aumentar a acessibilidade aos sítios ácidos das zeólitas. As estratégias envolvem a geração de mesoporos dentro do cristal da zeólita ou a síntese de nanocristais. Os materiais resultantes, que constituem as zeólitas com estrutura hierárquica de poros, foram classificados em três tipos diferentes,53 como mostrado na Figura 3, que podem ser denominados cristais hierárquicos, cristais nanométricos e cristais suportados de zeólitas.
O primeiro tipo de material, cristais hierárquicos de zeólita, possui um sistema de mesoporos intracristalinos e um sistema de macroporos intercristalinos (porosidade que se gera pela aglome-ração de cristais de zeólita), além da microporosidade intrínseca das zeólitas. O segundo tipo, cristais nanométricos de zeólita, por definição, inclui todos os cristais de zeólitas com tamanhos de até 1000 nm.54 Entretanto, na maioria dos trabalhos, têm sido obtidos cristais de zeólitas com tamanhos inferiores a 100 nm, que são muito menores do que aqueles apresentados pelas zeólitas convencionais. Esses materiais apresentam um sistema de microporos bem definido e, adicionalmente, um sistema de mesoporos intercristalinos como resultado do empacotamento dos cristais nanométricos. No último tipo de materiais, cristais suportados de zeólita, os cristais da zeólita estão dispersos ou suportados no sistema de poros de outro material. Neste caso, o sólido resultante não é uma zeólita pura, mas um sólido constituído pelo sistema de microporos da zeólita e um sistema de mesoporos e macroporos intercristalinos, cujo diâmetro de mesoporos é, principalmente, determinado pelo suporte (material não zeolítico). Esses sólidos têm apresentado uma acidez relativamente mais baixa quando comparados com a zeólita pura, o que limita sua aplicação como catalisador em processos da indústria de refino de petróleo, petroquímica e outras aplicações potenciais na indústria química.
Estes sólidos podem ser obtidos através de diferentes métodos de síntese, usando ou não a técnica de nanomoldagem; alguns podem ser usados para produzir diferentes tipos de materiais. Geralmente, os métodos que empregam a técnica de nanomoldagem tornam possível ajustar o tamanho dos poros através do uso de um molde de mesopo-ros com um tamanho característico que, ao ser removido, permite a formação de poros com o mesmo tamanho e forma do molde. Estes
Figura 3. Ilustração dos cristais e poros de materiais zeolíticos com estrutura hierárquica de poros. Adaptada da ref. 53

Grecco et al.136 Quim. Nova
métodos podem ser classificados como moldagem sólida, supramo-lecular e indireta.53
No método de moldagem sólida, a cristalização controlada da fase zeolítica ocorre em presença de um material sólido, que é posterior-mente removido para gerar a porosidade. Portanto, o sólido atua como um molde dos mesoporos e pode ser removido por combustão ou por dissolução. Por outro lado, na moldagem supramolecular, emprega--se um agregado supramolecular de surfactantes como molde dos mesoporos, que pode ser removido por combustão ou por extração com solventes. No caso da moldagem indireta, ocorre a transformação parcial de um material mesoporoso não zeolítico, previamente sinte-tizado em uma estrutura zeolítica hierárquica mesoporosa. Uma vez que o molde não está presente quando a zeólita cristaliza, o efeito da moldagem é considerado como indireto. A deposição controlada de uma zeólita, no sistema de poros de um material mesoporoso, tam-bém pode ser considerada como um método de moldagem indireta. A maioria das abordagens está relacionada com uma cristalização (secundária) parcial dos materiais mesoporosos ordenados em uma estrutura zeolítica.53,55 Geralmente, os materiais obtidos através da moldagem indireta podem ser classificados como cristais suportados de zeólitas e, portanto, não são objeto desta revisão.
As zeólitas mesoporosas hierárquicas também podem ser ob-tidas na ausência de um molde, através da desmetalização ou de uma cristalização controlada. Na desmetalização, geralmente, uma zeólita é submetida a um tratamento pós-síntese para a remoção de um elemento metálico específico da estrutura. Quando uma zeólita é submetida a um tratamento térmico em presença de vapor d’água ou a um lixiviamento ácido, ocorre a remoção preferencial de átomos de alumínio da rede (desaluminização). Embora os tratamentos térmicos sem vapor também possam criar defeitos na estrutura da zeólita, o uso do vapor aumenta muito a mobilidade das espécies de alumínio e silício. Entretanto, esse procedimento apresenta alguns problemas, tal como o fato de que a remoção seletiva do alumínio da rede altera a razão Si/Al e, consequentemente, modifica as propriedades ácidas e de troca iônica da zeólita. Além disso, nas estruturas zeolíticas ricas em silício, o baixo conteúdo de alumínio limita a geração de mesoporos, uma vez que serão criados poucos sítios de defeitos. Desta forma, a extração seletiva do silício da rede através de um tratamento alcali-no (dessilicação) constitui um método mais conveniente para gerar mesoporos nas zeólitas ricas em silício, tais como ZSM-5 e zeólita beta. Porém, a extração do metal, independente do método, conduz à formação de mesoporos intracristalinos não uniformes e com uma larga distribuição de diâmetros.
Os cristais nanométricos zeolíticos também podem ser obtidos na ausência de moldes, através do controle das condições de cristali-zação da zeólita. Entretanto, os materiais obtidos apresentam apenas mesoporos interparticulares.54
Zeólitas com estrutura hierárquica de poros preparadas via moldagem sólida
Diversos materiais sólidos podem ser empregados como molde de mesoporos, o que torna este método muito versátil na síntese dos diferentes tipos de materiais zeolíticos mesoporosos hierárquicos. Entre eles, podem ser destacados os seguintes materiais: nanopartí-culas de carbono,11 nanotubos de carbono,12 nanofibras de carbono,13 aerogéis de carbono,14 peneiras moleculares de carbono,15,16 polímeros catiônicos17 e polímeros silanizados.18
A primeira metodologia para a obtenção desses materiais foi desenvolvida em 1999, por pesquisadores da Haldor Topsøe11 e consistiu na preparação de cristais nanométricos de ZSM-5, através da cristalização da zeólita dentro do sistema de poros de uma matriz de carbono, como mostrado na Figura 4a. O procedimento consistiu
na impregnação úmida incipiente de um carvão ativo, com uma solução contendo hidróxido de tetrapropilamônio (TPAOH), etanol, isopropóxido de alumínio e água destilada. Após a evaporação do etanol à temperatura ambiente, o carvão foi impregnado com tetrae-tilortossilicato (TEOS) e, posteriormente, submetido a um tratamento hidrotérmico com vapor saturado, a 180 °C. A matriz de carbono foi removida por combustão a 550 °C, por 6 h, obtendo-se cristais de ZSM-5 com diâmetros médios na faixa de 20 a 75 nm. Uma vez que esses cristais crescem dentro da estrutura porosa das partículas de carbono, as distribuições de tamanho do cristal são governadas pelo tamanho dos poros do carvão. Desta forma, é possível controlar o tamanho dos cristais da zeólita pela escolha adequada de um molde de carbono. Diferentes zeólitas têm sido preparadas em escala nano-métrica, a partir da síntese em espaço confinado, tais como zeólita beta (7-30 nm), zeólita X (22–60 nm), zeólita A (25–37 nm).56 As etapas fundamentais na síntese em espaço confinado consistem em54
restringir a cristalização da zeólita dentro do sistema de poros da matriz de carbono, o que pode ser alcançado através da impregnação úmida incipiente do carbono com o gel de síntese da zeólita e, evitar a difusão das espécies de silicatos/aluminossilicatos presentes no gel de síntese para o exterior dos poros do molde, o que pode ser obtido impedindo o contato direto entre a matriz de carbono impregnada e a água, durante o tratamento hidrotérmico.
Os moldes de carbono também foram empregados na síntese de cristais hierárquicos contendo mesoporosidade intrazeolítica.57 Utilizando um excesso do gel de síntese da zeólita, observou-se que
Figura 4. Ilustração (a) das etapas de síntese de cristais zeolíticos nanomé-tricos em espaço confinado em um suporte poroso e (b) do crescimento dos cristais zeolíticos em torno das nanopartículas de carbono. Adaptada das ref. 54 (a) e 57 (b)

Zeólitas hierarquicamente estruturadas 137Vol. 36, No. 1
os cristais da zeólita cresciam em torno das partículas do carvão ativo, resultando em cristais de zeólita embebidos com um material de carbono, como mostrado na Figura 4b. Isso significa que os cris-tais da zeólita são nucleados dentro do sistema de poros da matriz de carbono e, posteriormente, devido ao excesso do gel de síntese, esses cristais crescem o suficiente para encapsular as partículas de carbono. A remoção das partículas de carbono, por combustão, conduz à formação de cristais hierárquicos zeolíticos contendo um sistema de mesoporosos intracristalinos na faixa de 10 a 100 nm. Portanto, através da escolha adequada do molde de carbono, é possível controlar o sistema mesoporoso.
A formação de cristais nanométricos ou de cristais zeolíticos mesoporosos é determinada pelos mesmos fatores experimentais que influenciam as taxas de nucleação e de crescimento do cristal. Os cristais nanométricos são obtidos quando as velocidades de nuclea-ção são mais elevadas que as velocidades de crescimento. Os fatores chave para a formação dos nanocristais não agregados são a elevada supersaturação e a estabilidade estérica dos núcleos. Estas condições, geralmente, são obtidas através da utilização de quantidades abundan-tes do direcionador de estrutura, além de uma quantidade reduzida dos cátions alcalinos para limitar a agregação das partículas. Além disso, devem ser empregadas temperaturas relativamente moderadas. Por outro lado, empregando velocidades de crescimento mais elevadas, a formação de cristais zeolíticos mesoporosos é favorecida.58
Em outros trabalhos, os pesquisadores da Haldor Topsøe empregaram nanotubos de carbono e nanofibras de carbono como moldes na síntese de zeólitas mesoporosas.12,13,59 Entretanto, os nanotubos e as nanofibras de carbono foram encapsulados apenas parcialmente durante o crescimento dos cristais zeolíticos. Desta forma, a remoção seletiva destes materiais conduziu à formação de mesoporos intracristalinos. Quando se empregou nanotubos de carbono, foram obtidos cristais com tamanhos de 100 a 500 nm, contendo mesoporos uniformes e retos, com diâmetros de 12 a 30 nm. Observou-se que a quantidade de mesoporos é determinada pela quantidade de nanotubos de carbono em relação à quantidade do gel de síntese da zeólita. Por outro lado, podem ser obtidos cristais zeolíticos mesoporosos mais econômicos, usando nanofibras de carbono, que são de custo muito mais baixo que os nanotubos de carbono; entretanto, os mesoporos do sólido obtido podem variar em uma faixa mais ampla de tamanhos.
Posteriormente, Tao e colaboradores14 empregaram, pela primeira vez, aerogéis de carbono ou aerogéis orgânicos como moldes de car-bono, na síntese de zeólitas mesoporosas. Os aerogéis de carbono são obtidos, principalmente, por pirólise de aerogéis orgânicos em tempe-raturas acima de 500 °C,60 sendo aqueles de resorcinol-formaldeído e de melamina-formaldeído os mais amplamente estudados. Durante a pirólise, os aerogéis de carbono, obtidos a partir dos aerogéis de resorcinol-formaldeído, mantêm a área superficial específica elevada e o elevado volume de mesoporos dos materiais precursores. Uma vez que os aerogéis de carbono possuem mesoporos uniformes que são facilmente ajustáveis, por exemplo, variando a razão molar dos materiais de partida (resorcinol e formaldeído), constituem impor-tantes moldes na preparação de zeólitas com porosidade hierárquica.
Tao e colaboradores14,61 também empregaram um aerogel de resorcinol-formaldeído carbonizado na preparação de zeólitas me-soporosas, obtendo cristais de ZSM-5 e de zeólita Y nanométricos, com uma estrutura de poros bimodal compreendendo mesoporos de tamanhos uniformes. Durante a etapa de cristalização, os cristais da zeólita crescem no espaço confinado do aerogel de carbono, resultando na formação de cristais nanométricos. As distribuições de diâmetro de mesoporos são bastante estreitas, com um máximo próximo a 11 nm, que correspondem à espessura da parede dos poros do aerogel de carbono usado (cerca de 10 nm). Portanto, variando a
estrutura zeolítica e a porosidade do aerogel de carbono, é possível obter diferentes materiais zeolíticos com mesoporosidade controlada.
Em 2004, dois grupos de pesquisa, independentemente, empre-garam peneiras moleculares mesoporosas de carbono como moldes na formação de zeólitas mesoporosas hierárquicas.15,16 Estes sólidos carbonáceos mesoporosos, denominados materiais CMK, podem ser sintetizados através da carbonização de diferentes moléculas orgânicas nos poros de materiais mesoporosos ordenados.62 Em se-guida, os poros do carbono são preenchidos com um gel de síntese da zeólita e o sistema é submetido à cristalização hidrotérmica e à remoção da matriz de carbono por combustão. Se o gel precursor for convertido em uma fase zeolítica contínua dentro dos poros do carbono mesoporoso, podem ser obtidos cristais nanométricos com mesoporosidade interzeolítica. Entretanto, os canais mesoporosos são muito estreitos, o que dificulta uma cristalização controlada no espaço confinado do carbono mesoporoso e, portanto, o gel tende a migrar, parcial ou completamente, para a superfície externa encapsu-lando as partículas do carbono mesoporoso, durante a cristalização.63 Após a remoção do molde, são obtidos materiais zeolíticos com mesoporos intracristalinos desordenados, em forma de caverna, ao invés de ocorrer uma replicação da forma dos poros do carbono. Estes resultados sugerem que os carbonos mesoporosos não atuam como um verdadeiro molde.64
De modo a controlar a cristalização dentro dos poros do carbono, investigou-se o método de conversão do gel seco na moldagem com carbonos mesoporosos.63,65 Observou-se que, quando a umidade relativa foi muito elevada (> 95%), o gel precursor migrou para a superfície externa formando uma fase zeolítica. Por outro lado, em-pregando valores de umidade entre 80 e 85%, formou-se uma zeólita com mesoporos altamente ordenados. Além disso, observou-se que a formação de uma estrutura zeolítica com mesoporos ordenados não dependia apenas da umidade, mas também do diâmetro de poros e da rigidez das paredes do carbono mesoporoso.63
Posteriormente, Xiao e colaboradores17 empregaram polímeros catiônicos em mesoescala, como molde de mesoporos, na síntese de zeólitas mesoporosas hierárquicas. O procedimento consistiu na adição de um polímero catiônico ao gel de síntese da zeólita beta. Após a cristalização hidrotérmica e calcinação, foram obtidas zeólitas hierárquicas com uma distribuição de diâmetro de mesoporos na faixa de 5 a 40 nm, que corresponde ao tamanho do polímero. A mesopo-rosidade intrazeolítica obtida pode ser ajustada através da quantidade adicionada do polímero catiônico. Outros materiais zeolíticos, ou com características zeolíticas, foram preparados através desta meto-dologia, tais como a ZSM-5, zeólita Y e TS-1 (titanossilicato).66 No mesmo período, Pinnavaia e colaboradores18 empregaram polímeros funcionalizados com grupos silanos, na preparação das zeólitas hie-rárquicas. A principal vantagem desse método consiste na obtenção de cristais zeolíticos com mesoporos intracristalinos com diâmetros pequenos e uniformes, na faixa de 2,0 a 3,0 nm.
Outras matrizes de carbono também têm sido estudadas, porém em menor extensão. Por exemplo, em 2005, Katsuki e colaboradores67
empregaram casca de arroz carbonizada como molde de carbono. Estes materiais apresentam um elevado teor de silício e, por isso, foram também utilizados como fonte de sílica na preparação dos cristais de zeólitas com mesoporos intracristalinos. Por outro lado, em 2007, Kustova e colaboradores68 desenvolveram um método de nanomoldagem com materiais de carbono preparados in situ, através da decomposição de um carboidrato, diretamente, na sílica usada como material de partida na obtenção da zeólita. Na preparação de zeólitas hierárquicas com morfologias diferentes, alguns materiais biológicos também têm sido usados como moldes de carbono.69 Eles representam uma alternativa atrativa em relação aos demais moldes, uma vez que são abundantes e frequentemente de baixo custo.

Grecco et al.138 Quim. Nova
Zeólitas com estrutura hierárquica de poros preparadas via moldagem supramolecular
A primeira metodologia relacionada à obtenção de zeólitas com estrutura hierárquica de poros, por moldagem supramolecular, foi desenvolvida por Kloetstra e colaboradores,70 que prepararam peneiras moleculares com distribuição de poros bimodal, combinando micro-poros e mesoporos. O método consistiu na preparação de materiais compósitos micro e mesoporosos através da cristalização simultânea da zeólita FAU e da MCM-41 ou através da deposição de MCM-41 sobre cristais da zeólita FAU. Os resultados de microscopia eletrônica de transmissão mostraram que os materiais consistiam de uma fina camada de MCM-41 cobrindo a superfície dos cristais da zeólita FAU.
Posteriormente, Karlsson e colaboradores71 desenvolveram uma nova metodologia de preparação de aluminossilicatos mesoporosos, que consistiu na adição simultânea dos agentes direcionadores das estruturas microporosa (molde molecular) e mesoporosa (molde supramolecular). Esse método é baseado na ideia de que um molde molecular direciona a cristalização da estrutura da zeólita, enquanto que, simultaneamente, um agregado supramolecular de surfactantes (template) conduz à formação da estrutura mesoporosa. Entretanto, observou-se que os moldes atuam de modo competitivo, resultando na formação de uma fase zeolítica pura, uma fase mesoporosa pura ou uma mistura física do material mesoporoso com a zeólita.
Uma nova metodologia de preparação de zeólitas com estrutura hierárquica de poros foi desenvolvida em 2000, por Pinnavaia e co-laboradores,10 que consistiu na organização dos precursores zeolíticos que normalmente nucleiam a cristalização de zeólitas microporosas. Estes precursores, também conhecidos como sementes de zeólitas, se assemelham às unidades secundárias de construção da estrutura zeolítica. O acoplamento destes precursores em uma estrutura mesoporosa hexagonal do tipo MCM-41, utilizando o brometo de cetiltrimetilamônio (CTAB), confere estabilidade hidrotérmica e acidez à estrutura mesoporosa. O material denominado [Al]-MSU-S (MSU - Michigan State University) foi submetido a um tratamento com vapor d’água a 800 °C e manteve a sua estrutura mesoporosa hexagonal ordenada. O método não se limita apenas a sementes de zeólita Y; diversos trabalhos foram desenvolvidos empregando a mesoestruturação de sementes de zeólitas para obter novos materiais hierárquicos zeolíticos, tais como os compósitos beta/MCM-41,72 beta/MCM-48,73 ZSM-5/MCM-41,74 ZSM-5/MCM-48,75 mordenita/MCM-4176 e mordenita/MCM-48.77 Outros materiais também foram obtidos, através do acoplamento de precursores zeolíticos em uma estrutura mesoporosa hexagonal do tipo SBA-15, tais como os com-pósitos Y/SBA-15, ZSM-5/ SBA-15 e beta/SBA-15.78
Diversas estratégias de síntese foram empregadas na obtenção de zeólitas mesoporosas hierárquicas através da moldagem supramole-cular.79 As observações experimentais mostraram que o emprego de precursores zeolíticos, como blocos de construção da rede mesoporo-sa, produzia os materiais mais estáveis. Entretanto, foi observado que a curvatura da micela do surfactante não favorece o empacotamento dessas nanopartículas de construção. Por exemplo, em estruturas do tipo MCM-41 que possuem espessura da parede mesoporosa na ordem de 1 nm, seria muito difícil acomodar essas nanopartículas para formar paredes cristalinas altamente condensadas (Figura 5a). Por outro lado, pode-se propor um agrupamento de agregados de sílica/direcionador, que conduz a unidades primárias amorfas na ordem de 1 a 3 nm que melhor se ajustam à parede mesoporosa, como mostrado na Figura 5b.
Entretanto, os materiais obtidos através da mesoestruturação de precursores zeolíticos são, provavelmente, compostos por uma matriz de sílica/alumina amorfa contendo domínios zeolíticos na superfície ou dentro dos poros, ou seja, são materiais híbridos das estruturas
mesoporosa ordenada e zeolítica.80
Com a expectativa de melhorar a acessibilidade em zeólitas lamelares, Corma e colaboradores9 desenvolveram um método de preparação que consistiu na deslaminação de seus precursores, de maneira similar ao que é realizado com uma estrutura de camadas de argila. Inicialmente, as lamelas de um precursor zeolítico, MCM-22 (P), foram expandidas pela adição de CTAB. Esse processo de expansão pode ser monitorado através do aumento da distância entre as lâminas, observado por difração de raios X. Nesse processo, as la-melas do precursor zeolítico são separadas, através de sonicação, mas, a estrutura zeolítica é preservada nas camadas individuais. Após a re-moção do agente de expansão, as lâminas são reempilhadas, de modo desorganizado, resultando em um aluminossilicato com elevada área superficial específica (ITQ-2). Os mesoporos e macroporos formados entre as lamelas desorganizadas proporcionam maior acessibilidade aos sítios ativos, localizados dentro das lâminas finas de zeólita. Após a deslaminação, a estrutura resultante apresenta uma importante per-da de microporosidade. Por outro lado, ocorre um aumento da área externa e, consequentemente, a formação de superfícies fortemente hidroxiladas.81 Durante o processo de deslaminação, também ocorre desaluminização, conduzindo a uma diminuição da acidez do sóli-do.81 Observou-se que as zeólitas deslaminadas adsorveram menor quantidade de piridina quando comparadas a seu precursor. Por outro lado, as medidas de acidez por adsorção de 2,6-di-terc-butilpiridina mostraram um comportamento inverso, revelando que o processo de deslaminação permite o acesso de moléculas volumosas aos sítios ativos. A restrição desta metodologia, entretanto, é que sua aplicabi-lidade se limita a estruturas zeolíticas lamelares.
De modo a obter materiais com mesoporos bem definidos, em 2006, Ryoo e colaboradores19 desenvolveram uma estratégia para a síntese direta de zeólitas, usando organossilanos anfifílicos, [(CH3O)3SiC3H6N(CH3)2CnH2n+1]Cl, como molde supramolecular. Estes compostos são formados por um grupo amônio quaternário, responsável pelo ordenamento da estrutura zeolítica, uma fração de organossilano ligada ao grupo amônio e uma cadeia alquila. Espera-se que o organossilano anfifílico possa interagir fortemente com a estru-tura zeolítica durante a cristalização, através da formação de ligações covalentes entre as espécies silicato ou aluminato com a fração de organossilano. Dessa maneira, as moléculas de surfactante não são re-movidas da esfera de aluminossilicato ou do aluminofosfato, durante o processo de cristalização, o que torna possível produzir cristais com uma estrutura mesoporosa intracristalina desordenada (Figura 6a). O
Figura 5. Ilustração da possível organização de precursores zeolíticos com uma fase micelar do surfactante. (a) organização das nanopartículas do precursor zeolítico coloidal; (b) organização de agregados amorfos mediada pelo direcionador. Adaptada da ref. 79

Zeólitas hierarquicamente estruturadas 139Vol. 36, No. 1
comprimento da cadeia alquila promove a formação de mesoporos de tamanho relativamente uniforme. Desta forma, diferentes estruturas zeolíticas com mesoporosos ajustáveis podem ser obtidas variando--se o comprimento da cadeia alquila. No trabalho pioneiro de Ryoo e colaboradores,19 os organossilanos anfifílicos foram adicionados diretamente ao gel de síntese da zeólita MFI, contendo o direcionador de estrutura. Os cristais obtidos apresentaram diferentes diâmetros médios de mesoporos, que variaram em função do comprimento da cadeia alquila do organossilano ou da temperatura da síntese hidrotér-mica. Por exemplo, as zeólitas mesoporosas apresentaram diâmetros médio de poros de 2,1; 3,1 e 3,9 nm, quando o número de átomos de carbono da cadeia alquila foi 12, 16 e 18, respectivamente. Entretanto, podem ser obtidos sólidos com diâmetros de mesoporos acima de 20 nm sob condições de síntese especificas, tais como a adição de copolímeros tribloco ao gel de síntese ou de agentes de expansão dos poros.82 Usando organossilanos anfifílicos, diversas outras estruturas contendo mesoporosidade intracristalina podem ser preparadas, dentre as quais podem-se citar as zeólitas LTA,19,83 a sodalita84 e a mordenita85 e os aluminofosfatos,86 titanossilicatos87 e zirconossili-catos.88 A presença de mesoporos usando organossilanos anfifílicos foi investigada através de microscopia eletrônica de varredura de alta resolução e por simulação de Monte Carlo de crescimento do cristal.83 Os cristais da zeólita LTA apresentaram morfologia cúbica, típica da zeólita, que se tornou progressivamente arredondada à medida que a concentração do organossilano foi aumentada. Além disso, a estrutura mesoporosa foi preservada durante um período de cristalização de 6 dias. Estes resultados sugerem um mecanismo de geração de meso-poros intracristalinos, em que as micelas do organossilano anfifílico permanecem ocluídas no interior do cristal da zeólita.
Recentemente, Ryoo e colaboradores89-91 desenvolveram novas metodologias empregando surfactantes bifuncionais, que podem di-recionar a formação de ambas as estruturas, micro e mesoporosa, da zeólita hierárquica. Na primeira delas,89 empregaram um surfactante, C22H45-N
+(CH3)2-C6H12-N+(CH3)2-C6H13, contendo um grupo alquil de
cadeia longa (C22H45) e dois grupos amônio quaternário separados por dois grupos alquílicos.89 O grupo diamônio foi responsável pelo ordenamento da estrutura zeolítica do tipo MFI, enquanto a intera-ção hidrofóbica das cadeias longas (C22H45) induziu a formação de uma mesoestrutura micelar. As cadeias hidrofóbicas restringem o crescimento da estrutura zeolítica na parte hidrofílica das micelas, resultando na formação de lamelas de zeólita MFI em escala nano-métrica (espessura de 2 nm). Dependendo das condições de síntese, as nanolamelas podem se auto-organizar em um arranjo unilamelar desordenado ou um arranjo multilamelar ordenado bidimensional.89,90 As camadas lamelares também podem ser submetidas a um processo de pilarização,90 no qual as lamelas são separadas por pilares de sílica que, posteriormente, são convertidos em uma parte da estrutura zeo-lítica; portanto, a mesoporosidade interlamelar é preservada, após a remoção do surfactante. Em outro trabalho, Ryoo e colaboradores91 empregaram uma série de surfactantes de poliamônio quaternário, que podem atuar como direcionador de diferentes estruturas hierár-quicas ordenadas. Um membro especialmente importante desta série é o surfactante C18H37-N
+(CH3)2-C6H12-N+(CH3)2-C6H12-N
+(CH3)2-C18H37(Br-)3, responsável pela formação de uma peneira molecular mesoporosa ordenada hexagonal, que possui paredes microporosas cristalinas com características zeolíticas,92 como mostrado na Figura 6b. Os mesoporos em arranjo hexagonal são gerados pelos agregados do surfactante, enquanto os diferentes grupos poliamônio quaternário atuam como direcionadores das diferentes estruturas zeolíticas. A espessura da parede, o tipo de estrutura zeolítica e o diâmetro de mesoporos podem ser controlados usando surfactantes com diferentes grupos, envolvendo um número maior de grupos amônio quaternário e a presença de anéis fenil. Embora normalmente o ordenamento dos
mesoporos diminua com o aumento da espessura da parede, observou--se que a espessura da parede foi uniforme, variando na faixa de 2,3 a 5,1 nm. O diâmetro de mesoporos (3,8 a 21 nm) pode ser controlado através da adição de agentes de expansão dos poros.91
Também em 2006, Serrano e colaboradores93 apresentaram uma nova metodologia de preparação de cristais nanométricos. Ela con-sistiu na perturbação do crescimento dos cristais da zeólita através da silanização de sementes, de modo a impedir uma posterior agre-gação destes precursores. A síntese de zeólitas a partir da silanização de sementes consiste de três etapas, mostradas na Figura 7:93,94 (i) formação dos núcleos da zeólita, que ocorre durante uma etapa de pré-cristalização. Esses núcleos devem ser considerados como entidades pseudocristalinas, uma vez que apresentam propriedades intermediárias entre os aluminossilicatos amorfos e as zeólitas. Esses núcleos são obtidos nos primeiros estágios de cristalização (na etapa de pré-cristalização) da zeólita e são amorfos aos raios X. Por outro lado, foram observadas vibrações da estrutura zeolítica através de bandas no espectro de infravermelho; (ii) acoplamento do agente de silanização, em presença ou ausência de alcoóis, na
Figura 6. (a) Ilustração do mecanismo proposto para a síntese de cristais de aluminofosfatos hierárquicos, empregando organossilanos anfifílicos como molde supramolecular. (b) Ilustração esquemática da estrutura hierárquica obtida usando surfactantes bifuncionais. Adaptada das ref. 86 (a) e 91 e 92 (b)

Grecco et al.140 Quim. Nova
superfície externa dessas entidades, que ocorre durante a etapa de silanização, com um agente direcionador de estrutura ocupando os microporos e (iii) cristalização hidrotérmica das sementes fun-cionalizadas, obtendo-se materiais com características cristalinas e com porosidade hierárquica, uma vez que a presença do agente de silanização impede a agregação das entidades nanométricas. Esta estratégia foi bem sucedida na preparação das zeólitas ZSM-5 e beta nanométricas, usando fenilaminopropil-trimetoxissilano como agente de funcionalização.93-95 Por outro lado, essa metodologia pode ser também aplicada na preparação de outras nanozeólitas, tais como mordenita, faujasita e materiais com características zeolíticas, como os titanossilicatos.96 No caso da ZSM-5 nanométrica, foram obtidas partículas de 300 a 400 nm, que são formadas por aglomerados de cristais pequenos da ZSM-5 (10-20 nm).93,95 Por outro lado, a zeólita beta consistiu de macropartículas (500 a 600 nm), formadas por aglomerados de nanocristais de cerca de 15 nm.97 O efeito da natureza do agente de silanização sobre as características dos sólidos também foi estudado por esses pesquisadores,95 empregando quatro organossilanos diferentes: 3-aminopropil-trimetoxissilano (APTMS), isobutil-trietoxissilano (IBTES), fenilaminopropil-trimetoxissilano (PHAPTMS) e octadecil-trimetoxissilano (ODTMS). O PHAPTMS foi o agente de silanização mais eficiente, tanto no seu acoplamento à superfície externa dos núcleos quanto na perturbação do cresci-mento do cristal da zeólita. Foi observado que as propriedades das nanozeólitas obtidas através da silanização dependem não apenas da natureza do agente de silanização, mas também da concentração desse agente, das condições de pré-cristalização e da adição de alcoóis na etapa de silanização.94,98
CONCLUSÕES
Durante a última década, as zeólitas mesoporosas hierárquicas têm se apresentado como uma importante classe de materiais em ci-ência e tecnologia. A presença de mesoporos assegura a acessibilidade e um transporte eficiente dos reagentes e produtos nos poros, durante uma reação catalítica, enquanto os microporos conduzem à seletivi-dade desejada. Portanto, a combinação de microporos e mesoporos, em um cristal da zeólita, possibilita a otimização do seu desempenho catalítico, especialmente em reações envolvendo moléculas orgânicas com diâmetro cinético superior ao diâmetro médio dos microporos.
As duas principais estratégias de síntese das zeólitas mesopo-rosas hierárquicas focam a síntese de zeólitas com estruturas que
minimizem as restrições impostas pelos microporos à difusão de moléculas de reagentes e produtos. O princípio dessas estratégias consiste em limitar a cristalização através de agentes e condições específicas de síntese adequada à cada finalidade desejada. Uma das estratégias consiste na geração de mesoporos dentro do cristal da zeólita, enquanto que outra envolve a síntese de nanocristais. Ambas podem ser conduzidas através da técnica de nanomoldagem, além de outras; os sólidos resultantes possuem mesoporosidade intrazeolítica ou interzeolítica, além da microporosidade intrínseca das zeólitas. Dentre as metodologias bem sucedidas na produção de zeólitas ultra-finas (com espessura de aproximadamente 5 nm), pode-se mencionar a deslaminação de precursores zeolíticos lamelares e a síntese direta de zeólitas empregando surfactantes bifuncionais.
Através do método de deslaminação, pode-se obter zeólitas com estabilidade térmica e/ou hidrotérmica e acidez elevadas e com es-trutura porosa capaz de permitir maior acessibilidade de moléculas volumosas aos sítios ácidos, tornando esses materiais mais atrativos para muitas aplicações catalíticas. Entretanto, a aplicação desse mé-todo na preparação de zeólitas hierárquicas é muito limitada, uma vez que existe um número reduzido de zeólitas lamelares.
Por outro lado, a metodologia empregando surfactantes bifun-cionais tem se mostrado como uma rota promissora na preparação de diferentes estruturas zeolíticas hierárquicas ordenadas. Esses materiais apresentaram atividade catalítica mais elevada, em várias reações de conversão de moléculas volumosas, quando comparados às zeólitas convencionais ou materiais mesoporosos ordenados com paredes amorfas.
Apesar do aumento de trabalhos dedicados a esse tema, ainda não existem estudos conclusivos sobre o efeito das diferentes variáveis de síntese hidrotérmica, tais como a composição do gel e outros parâ-metros de síntese, sobre as propriedades de materiais zeolíticos com estrutura hierárquica de poros. Existe a expectativa de que diferentes estruturas zeolíticas possam ser obtidas, empregando diferentes gru-pos poliamônio quaternário, com diâmetros médios de mesoporos que podem ser ajustados, variando-se a composição da mistura reacional, o comprimento da cadeia alquila mais longa do surfactante, as condi-ções hidrotérmicas ou empregando-se agentes de expansão de poros, combinados ou não. Espera-se que a otimização das metodologias de preparação desses materiais possa proporcionar novas oportunidades de obtenção de catalisadores projetados especialmente para atuar seletivamente em reações envolvendo moléculas volumosas.
MATERIAL SUPLEMENTAR
Está disponível em http://quimicanova.sbq.org.br, em arquivo pdf, com acesso livre.
AGRADECIMENTOS
S. T. F. Grecco agradece ao CNPq pela bolsa concedida. Os au-tores agradecem à FINEP e ao CNPq pelo apoio financeiro.
REFERÊNCIAS
1. Escobar, M. F. A.; Batista, M. S; Urquieta-González, E. A.; Quim. Nova 2000, 23, 303; Dimitrova, R.; Gunduz, G.; Dimitrov, L.; Tsoncheva, T.; Yialmaz, S.; Urquieta-Gonzalez, E. A.; J. Mol. Catal. A: Chem. 2004, 214, 265; Grecco, S. T. F.; Gomes, L. P.; Reyes, P.; Oportus, M.; Rangel, M. C.; Stud. Surf. Sci. Catal. 2005, 158, 1937; Ramos, M. S.; Grecco, S. T.; Gomes, L. P.; Oliveira, A. C.; Reyes, P.; Oportus, M.; Rangel, M. C.; Stud. Surf. Sci. Catal. 2005, 156, 809; Pereira, A. L. C.; González-Carballo, J. M.; Pérez-Alonso, F. J.; Rojas, S.; Fierro, J. L. G.; Rangel, M. D. C.; Top. Catal. 2011, 54, 179; Fonseca, J. D. S. L.; Júnior, A. D.
Figura 7. Ilustração das etapas de síntese de nanocristais de zeólita a partir da silanização de sementes. Adaptada da ref. 94

Zeólitas hierarquicamente estruturadas 141Vol. 36, No. 1
C. F.; Grau, J. M.; Rangel, M. D. C.; Appl. Catal. A 2010, 386, 201. 2. Corma, A.; Chem. Rev. 1995, 95, 559; Corma, A.; Chem. Rev. 1997, 97,
2373. 3. Majano, G.; Mintova, S.; Ovsitser, O.; Mihailova, B.; Bein, T.;
Microporous Mesoporous Mater. 2003, 80, 227. 4. Rangel, M. C.; Valentini, A.; Oliveira, A. S.; David, J. M.; Marques
Britto, J.; Domingues, S. M.; Reyes, P.; Quim. Nova 2003, 26, 305; Guisnet, M.; Magnoux, P.; Appl. Catal. 1989, 54, 1.
5. Beck, J. S.; Vartuli, J. C.; Roth, W. J.; Leonowicz, M. E.; Kresge, C. T.; Schmitt, K. D.; Chu, C. T.-W.; Olson, D. H.; Sheppard, E. W.; McCul-len, S. B.; Higgins, J. B.; Schlenker, J. L.; J. Am. Chem. Soc. 1992, 114, 10834.
6. Kresge, C. T.; Leonowicz, M. E.; Roth, W. J.; Vartuli, J. C.; Beck, J. S.; Nature 1992, 359, 710.
7. Tanev, P. T.; Chibwe, M.; Pinnavaia T. J.; Nature 1994, 368, 321. 8. Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G. H.; Chmelka,
B. F.; Stucky G. D.; Science 1998, 279, 548; Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G. H.; Chmelka, B. F.; Stucky, G. D.; J. Am. Chem. Soc. 1998, 120, 6024.
9. Corma, A.; Fornes, V.; Pergher, S. B.; Maesen, Th. L. M.; Buglass, J. G.; Nature 1998, 396, 353.
10. Liu, Y.; Zhang, W.; Pinnavaia, T. J.; J. Am. Chem. Soc. 2000, 122, 8791. 11. Madsen, C.; Jacobsen, C. J. H.; Chem. Commun. 1999, 673. 12. Schmidt, I.; Boisen, A.; Gustavsson, E.; Stahl, K.; Pehrson, S.; Dahl, S.;
Carlsson, A.; Jacobsen, C. J. H.; Chem. Mater. 2001, 13, 4416; Borges, S. M. S.; Rangel, M. C.; Curr. Top. Catal. 2012, 57.
13. Janssen, A. H.; Schmidt, I.; Jacobsen, C. J. H.; Koster, A. J.; De Jong, K. P.; Microporous Mesoporous Mater. 2003, 65, 59.
14. Tao, Y.; Kanoh, H.; Kaneko, K.; J. Am. Chem. Soc. 2003, 125, 6044. 15. Sakthivel, A.; Huang, S. J.; Chen, W. H.; Lan, Z. H.; Chen, K. H.; Kim,
T. W.; Ryoo, R.; Chiang, A. S. T.; Liu, S. B.; Chem. Mater. 2004, 16, 3168.
16. Yang, Z.; Xia, Y.; Mokaya, R.; Adv. Mater. 2004, 16, 727. 17. Xiao, F.-S.; Wang, L.; Yin, C.; Lin, K.; Di, Y.; Li, J.; Xu, R.; Su, D. S.;
Schlögl, R.; Yokoi, T.; Tatsumi, T.; Angew. Chem., Int. Ed. 2006, 45, 3090.
18. Wang, H.; Pinnavaia, T. J.; Angew. Chem., Int. Ed. 2006, 45, 7603. 19. Choi, M.; Cho, H. S.; Srivastava, R.; Venkatesan, C.; Choi, D.; Ryoo,
R.; Nat. Mater. 2006, 5, 718. 20. Klein, C.; Hurlbut, C. S.; Manual of Mineralogy, 21st ed., Jonh Wiley &
Sons: New York, 1977. 21. Breck, D. W.; Zeolite Molecular Sieves: Structure, Chemistry and Use,
Robert E. Krieger Publishing Company: Malabar, 1984. 22. Pérez-Pariente, J. Em Materiales Zeolíticos: Síntesis, Propriedades
y Aplicaciones; Martínez, J. G.; Pérez-Pariente, J., eds.; Universidad Alicante: San Vicente, 2002, chap. 1.
23. Braga, A. A. C.; Morgon, N. H.; Quim. Nova 2007, 30, 178. 24. http://www.iza-structure.org/databases, acessada em Novembro 2012. 25. Yang, R. T.; Adsorbents: Fundamentals and Applications, John Wiley
& Sons: New Jersey, 2003. 26. Payra, P.; Dutta, P. K. Em Handboook of Zeolites Science and Technol-
ogy; Auerboch, S. M.; Carrado, K. A.; Dutta, P. K., eds.; Marcel Dekker Inc: New York, 2003, chap. 1.
27. Luna, F. J.; Schuchardt, U.; Quim. Nova 2001, 24, 885. 28. Corma, A.; J. Catal. 2003, 216, 298. 29. Moreno, E. L.; Rajagopal, K.; Quim. Nova 2009, 32, 538. 30. Guisnet, M.; Ribeiro, F. R.; Zeólitos: Um Nanomundo ao Serviço da
Catálise, Fundação Calouste Gulbenkian: Lisboa, 2004. 31. Barrer, R. M.; J. Chem. Soc. 1948, 127; Milton, R. M.; U.S. pat.
2,882,243 1959. 32. Barrer, R. M.; Hydrothermal Chemistry of Zeolites, Academic Press:
London, 1982. 33. Cundy, C. S.; Paul, C.; Chem. Rev. 2003, 103, 663.
34. Cundy, C. S.; Paul, C.; Microporous Mesoporous Mater. 2005, 82, 1. 35. Wadlinger, R. L.; Kerr, G. T.; Rosinski, E. J.; U.S. pat. 3,308,069 1967. 36. Davis, M. E.; Katz, A.; Ahmad, W. R.; Chem. Mater. 1996, 8, 1820. 37. Vartuli, J. C.; Roth, W. J.; Beck, J. S.; Mccullen, S. B.; Kresge, C. T. Em
Molecular Sieves; Karge, H. G.; Weitkamp, J., eds.; Springer-Verlag: Berlin, 1998, chap. 4.
38. Monnier, A.; Schuth, F.; Huo, Q.; Kumar, D.; Margolese, D.; Stucky, G. D.; Krishnamurty, M.; Petroff, P.; Firouzi, A.; Janicke, M.; Ghmelka, B. F.; Science 1993, 261, 1299.
39. Rey, F.; Jordá, L. J. Em ref. 22, chap. 3. 40. Zhao, X. S.; Lu, G. K.; Millar, G. J.; Ind. Eng. Chem. Res. 1996, 35,
2075. 41. Huo, Q.; Margolese, D. I.; Stucky, G. D.; Chem. Mater. 1996, 8, 1147. 42. Trong On, D.; Desplantier-Giscard, D.; Danumah, C.; Kaliaguine, S.;
Appl. Catal., A 2003, 253, 545. 43. Mascarenhas, A. J. S.; Oliveira, E. C.; Pastore, H. O.; Cadernos
Temáticos de Química Nova na Escola 2001, No. 2, 25. 44. Sayari, A.; Chem. Mater. 1996, 8, 1840. 45. Meynen, V.; Cool, P.; Vansant, E. F.; Microporous Mesoporous Mater.
2009, 125, 170. 46. Corma, A.; Fornés, V.; Navarro, M. T.; Pérez-Pariente, J.; J. Catal. 1994,
148, 569. 47. Galarneau, A.; Cambon, H.; Renzo, F. D.; Fajula, F.; Langmuir 2001,
17, 8328. 48. Yoldas, B. E.; Partlow, D. P. J.; Mater. Sci. 1998, 23, 1895. 49. Pierre, A. C.; Elaloui, E.; Pajonk, G. M.; Langmuir 1998, 14, 66. 50. Zhang, W. H.; Lu, J.; Han, B.; Li, M.; Xiu, J.; Ying, P.; Li, C.; Chem.
Mater. 2002, 14, 3413; Li, Y.; Zhang, W.; Zhang, L.; Yang, Q.; Wei, Z.; Feng, Z.; Li, C.; J. Phys. Chem. B 2004, 108, 9739.
51. Impéror-Clerc, M.; Davidson, P.; Davidson, A.; J. Am. Chem. Soc. 2000, 122, 11925.
52. Kipkemboi, P.; Fodgen, A.; Alfredsson, V.; Flodström, K.; Langmuir 2001, 17, 5398.
53. Egeblad, K.; Christensen, C. H.; Kustova, M.; Christensen, C. H.; Chem. Mater. 2008, 20, 946.
54. Tosheva, L.; Valtchev V. P.; Chem. Mater. 2005, 17, 2494. 55. Campos, A. A.; Martins, L.; Oliveira, L. L.; Silva, C. R.; Wallau,
M.; Urquieta-Gonzalez, E. A.; Stud. Surf. Sci. Catal. 2004, 154, 541; Campos, A. A.; Martins, L.; Oliveira, L. L.; Silva, C. R.; Wallau, M.; Urquieta-González, E. A.; Catal. Today 2005, 107-108, 759.
56. Schmidt, I.; Madsen, C.; Jacobsen, C. J. H.; Inorg. Chem. 2000, 39, 2279.
57. Jacobsen, C. J. H.; Madsen, C.; Houzvicka, J.; Schmidt, I.; Carlsson, A.; J. Am. Chem. Soc. 2000, 122, 7116.
58. Jacobsen, C. J. H.; Houzvicka, J.; Carlsson, A.; Schmidt, A.; Stud. Surf. Sci. Catal. 2001, 135, 167.
59. Boisen, A.; Schmidt, I.; Carlsson, A.; Dahl, S.; Brorson M.; Jacobsen, C. J. H.; Chem. Commun. 2003, 958.
60. Pierre, A. C.; Pajonk, G. M.; Chem. Rev. 2002, 102, 4243. 61. Tao, Y.; Kanoh, H.; Kaneko, K.; J. Phys. Chem. B 2003, 107, 10974;
Tao, Y.; Kanoh, H.; Hanzawa, H.; Kaneko, K.; Colloids Surf., A 2004, 241, 75; Tao, Y.; Hattori, Y.; Matumoto, A.; Kanoh, H.; Kaneko, K.; J. Phys. Chem. B 2005, 109, 194; Tao, Y.; Kanoh, H.; Kaneko, K.; Lang-muir 2005, 21, 504.
62. Wallau, M.; Silva, C. R.; Urquieta-González, E. A.; Stud. Surf. Sci. Catal. 2006, 162, 409; Veloso, C. M.; Rangel, M. C.; Quim. Nova 2009, 32, 2133.
63. Cho, H. S.; Ryoo, R.; Microporous Mesoporous Mater. 2012, 151, 107. 64. Li, H.; Sakamoto, Y.; Liu, Z.; Ohsuna, T.; Terasaki, O.; Thommes, M.;
Che, S.; Microporous Mesoporous Mater. 2007, 106, 174. 65. Zhang, Y.; Okubo T.; Ogura, M.; Chem. Commun. 2005, 2719. 66. Xiao, F.-S.; Wang, L.; Yin, C.; Lin, K.; Di, Y.; Li, J.; Xu, R.; Su, D.;
Schlögl, R.; Yokoi, T.; Tatsumi, T.; Angew. Chem., Int. Ed. 2006, 45,

Grecco et al.142 Quim. Nova
3090; Liu, S.; Cao, X.; Li, L.; Li, C.; Ji, Y.; Xiao, F.-S.; Coll. Surf., A 2008, 318, 269.
67. Katsuki, H.; Furuta, S.; Watari, T.; Komarneni, S.; Microporous Meso-porous Mater. 2005, 86, 145.
68. Kustova, M.; Egeblad, K.; Zhu, K.; Christensen C. H.; Chem. Mater. 2007, 19, 2915.
69. Davis, S. A.; Burkett, S. L.; Mendelson, N. L.; Mann, S.; Nature. 1997, 385, 420; Davis, S. A.; Patel, H. M.; Mayes, E. L.; Mendelson, N. L.; Franco, G.; Mann, S.; Chem. Mater. 1998, 10, 2516; Zhang, B.; Davis, S. A.; Mendelson, N. H.; Mann, S.; Chem. Commun. 2000, 781.
70. Kloetstra, K. R.; Zandbergen, H. W.; Jansen J. C.; van Bekkum H.; Microporous Mesoporous Mater. 1996, 6, 287.
71. Karlsson, A.; Stocker, M.; Schmidt, R.; Microporous Mesoporous Mater. 1999, 27, 181.
72. Grecco, S. T. F.; Nobre, P. S. S.; Urquieta-González, E. A; Rangel, M. C. Em Nanoporous Materials: Proceedings of the 5th International Symposium; Sayari, A.; Jaroniec, M., eds.; World Scientific Publishing Co. Pte. Ltd.: New Jersey, 2008. p 599; Guo, W.; Huang, L.; Deng, P.; Xue, Z.; Li, Q.; Microporous Mesoporous Mater. 2001, 44-45, 427; Guo, W.; Xiong, C.; Huang, L.; Li, Q.; J. Mater. Chem. 2001, 11, 1886; Liu, Y.; Zhang, W.; Pinnavaia, T. J.; Angew. Chem., Int. Ed. 2001, 40, 1255; Zhang, Z.; Han, Y.; Xiao, F. S.; Qiu, S.; Zhu, L.; Wang, R.; Yu, Y.; Zhang, Z.; Zou, B.; Wang, Y.; Sun, H.; Zhao, D.; Wei, Y. J. Am. Chem. Soc. 2001, 123, 5014.
73. Shih, P. C.; Wang, J. H.; Mou, C. Y.; Catal. Today 2004, 93, 365. 74. Gonçalves, M. L.; Dimitrov, L. D.; Wallau, M.; Urquieta-Gonzalez, E.
A.; Stud. Surf. Sci. Catal. 2006, 162, 323; Gonçalves, M. L.; Dimitrov, L. D.; Jordão, M. H.; Wallau, M.; Urquieta-González, E. A.; Catal. Today 2008, 133-135, 69.
75. Xia, Y.; Mokaya, R.; J. Mater. Chem. 2004, 14, 863. 76. Wang, S.; Dou, T.; Li, Y.; Zhang, Y.; Li, X.; Yan Z.; J. Solid State Chem.
2004, 177, 4800. 77. Dimitrov, L.; Wallau, M.; Urquieta-González, E. A.; Abstracts, 13th An-
nual Meeting of the Brazilian Catalysis Society, Foz do Iguaçu, Brazil, 2005.
78. Liu, Y.; Pinnavaia, T. J.; Chem. Mater. 2002, 14, 3. 79. Pérez-Pariente, J.; Díaz, I.; Agúndez, J.; C. R. Chim. 2005, 8, 569.
80. Grecco, S. T. F.; Urquieta-González, E. A.; Reyes, P.; Oportus, M.; Ran-gel M. C.; Abstracts, 20th Annual Meeting of Iberoamerican Federation of Catalysis Societies, Viña del Mar, Chile, 2010.
81. Corma, A.; Fornés, V. Em ref. 22, chap. 11. 82. Cho, K.; Cho, H. S.; Ménorval, L. C.; Ryoo, R.; Chem. Mater. 2009, 21,
5664. 83. Cho, K.; Ryoo, R.; Asahinab, S.; Xiao, C.; Klingstedt, M.; Umemura,
A.; Anderson, M. W.; Terasaki, O.; Solid State Sci. 2011, 13, 750. 84. Shanbhag, G. V.; Choi, M.; Kim, J.; Ryoo, R.; J. Catal. 2009, 264, 88. 85. Li, X.; Prinsa, R.; van Bokhoven, J. A.; J. Catal. 2009, 262, 257. 86. Choi, M.; Srivastava, R.; Ryoo, R.; Chem. Commun. 2006, 4380. 87. Cheneviere, Y.; Chieux, F.; Caps, V.; Tuel, A.; J. Catal. 2010, 269, 161. 88. Zhao, Z.; Liu, Y.; Wu, H.; Li, X.; He, M.; Wu, P.; Microporous
Mesoporous Mater. 2009, 123, 324. 89. Choi, M.; Na, K.; Kim, J.; Sakamoto, Y.; Terasaki, O.; Ryoo, R.; Nature
2009, 461, 246. 90. Na, K.; Choi, M.; Park, W.; Sakamoto, Y.; Terasaki, O.; Ryoo, R.; J. Am.
Chem. Soc. 2010, 132, 4169. 91. Na, K.; Jo, C.; Kim, J.; Cho, K.; Jung, J.; Seo, Y.; Messinger, R. J.;
Chmelka, B. F.; Ryoo, R.; Science 2011, 333, 328. 92. Möller K.; Bein, T.; Science 2011, 333, 328. 93. Serrano, D. P.; Aguado, J.; Escola, J. M.; Rodríguez, J. M.; Peral, A.;
Chem. Mater. 2006, 18, 22462. 94. Serrano, D. P.; Aguado, J.; Escola, J. M.; Peral, A.; Morales, G.; Abella
E.; Catal. Today 2011, 168, 86. 95. Serrano, D. P.; Aguado, J.; Escola, J. M.; Rodríguez, J. M.; Peral, A.; J.
Mater. Chem. 2008, 18, 4210. 96. Aguado, J.; Serrano, D. P.; Escola, J. M.; Peral, A.; J. Anal. Appl. Pyrol.
2009, 85, 352; Vuonga, G.-T.; Hoanga, V.-T.; Nguyenb, D.-T.; Do, T.-O.; Appl. Catal. A 2010, 382, 231; Sanza, R.; Serrano, D. P.; Pizarro, P.; Moreno, I.; Chem. Eng. J. 2011, 171, 1428.
97. Aguado, J.; Serrano, D. P.; Escola, J. M.; Rodríguez, J. M.; Peral, A.; Microporous Mesoporous Mater. 2009, 115, 504.
98. Serrano, D. P.; Aguado, Morales, G.; Rodríguez, J. M.; Peral, A.; Thommes, M.; Epping, J. D.; Chmelka, B. F.; Chem. Mater. 2009, 21, 641.
Quim. Nova, Vol. 36, No. 1, S1, 2013
Mat
eria
l Sup
lem
enta
r
*e-mail: [email protected]
ZEÓLITAS HIERARQUICAMENTE ESTRUTURADAS
Saulo de Tarso Figueiredo Grecco e Maria do Carmo Rangel* Instituto de Química. Universidade Federal da Bahia. Campus Universitário de Ondina, 40170-290 Salvador – BA, BrasilErnesto Antonio Urquieta-GonzálezDepartamento de Engenharia Química, Universidade Federal de São Carlos, 13565-905 São Carlos – SP, Brasil
Figura 3S. Padrão de difração de raios X: (a) MCM-41 e (b) SBA-15. Adaptada da ref. 47
Figura 2S. Ilustração esquemática das diferentes estruturas do surfactante em uma solução aquosa em função da concentração. Adaptada da ref. 62
Figura 1S. Esquema mostrando os caminhos mecanísticos possíveis de formação da MCM-41: (1) fase cristal líquido forma-se inicialmente; (2) os ânions silicato interagem inicialmente com as micelas do surfactante (cooperativo). Adaptada da ref. 5

In: Xylenes ISBN: 978-1-62808-342-2 Editor: Michael Olawale Daramola © 2013 Nova Science Publishers, Inc.
Chapter 4
INFLUENCE OF PLATINUM ON THE PERFORMANCE
OF BETA ZEOLITE-BASED CATALYSTS FOR
XYLENES PRODUCTION
Maria do Carmo Rangel1,*
, Saulo de Tarso Figueiredo Grecco1,
Diego Rodrigues de Carvalho1,
Ernesto Antonio Urquieta-González2, Javier Mario Grau
3,
Patricio Reyes4 and Marcelo Oportus
4
1GECCAT Grupo de Estudos em Cinética e Catálise, Instituto de Química, Universidade Federal da Bahia, Campus Universitário de Ondina, Salvador, Ba, Brazil
2Departamento de Engenharia Química, Universidade Federal de São Carlos, São Carlos, São Paulo, Brazil
3Instituto de Investigaciones en Catálisis y Petroquímica (INCAPE), (FIQ, UNL-CONICET), Santa Fe, Argentina
4Facultad de Ciências Químicas, Universidad de Concepción, Casilla 3-C Concepción, Chile
ABSTRACT
Xylenes are important raw materials with considerable application for chemicals and petrochemicals industry such as the production of synthetic fibers, plastics, resins and other products. The major sources of these compounds, naphtha reforming and gasoline pyrolysis, generate an appreciable amount of toluene and trimethylbenzene. The production of xylenes can be optimized by transalkylation of these compounds or by toluene disproportionation. The commercial process uses large pore zeolites which, however, deactivate with time mainly due to coke formation. In order to obtain new catalysts for these reactions, catalysts based on beta zeolite containing 0.5 and 1.0% platinum were synthesized and evaluated in toluene disproportionation. Zeolite-based materials were prepared by a two-step procedure and the metal was incorporated in the catalysts by wetness impregnation or by introducing platinum in the zeolite synthesis gel.
* E-mail: [email protected].
Complimentary Contributor Copy

Maria do Carmo Rangel, Saulo de Tarso Figueiredo Grecco et al. 58
The catalysts were characterized by differential thermal analysis, thermogravimetry, Fourier transformed infrared spectroscopy, X-ray diffraction, and adsorption/desorption of nitrogen and transmission electron microscopy. The activities of metallic and acidic sites were evaluated by cyclohexane dehydrogenation, a model reaction for both kinds of sites. All catalysts were more active than the commercial mordenite in toluene disproportionation, carried out at 470 ºC and 1 atm. Platinum decreased the activity but increased the xylene selectivity, especially p-xylenes. The most promising catalyst was prepared by incorporating platinum (0.5%) by impregnation, which produces the highest amount of xylenes, mainly p-xylenes and m-xylenes, high value chemicals.
Keywords: Catalysis, para-xylenes, meta-xylenes,zeolite catalyts
INTRODUCTION Aromatics play an important role in chemical and petrochemical industries, being used as
raw materials for the manufacture of several high value chemicals. Benzene, toluene and xylenes (BTX) are among the most important ones, being extensively used to produce the majority of the intermediates used in the production of synthetic fibers, plastics and resins [1]. Among them, xylenes are by far the most valuable ones due to their wide range of applications in various fields, such as solvents and components in several final products. The individual isomers are even more valuable because of their applications. p-Xylene, for instance, is the precursor for the manufacture of terephthalic acid and dimethyl terephthalate both starting materials for the production of polyethylene terephthalate (PET), used in the manufacture of plastic bottles and polyester clothing. On the other hand, o-xylene is used for manufacturing phthalic anhydride and plasticizers, which are responsible for the flexibility of polyvinyl chloride (PVC). In addition, m-xylene can be oxidized to isophthalic acid to produce polyurethane and Nomex aramid polymer, [2-4]. Currently, p-xylene is responsible for about 83% of world demand for xylenes, which is higher than 42 million t/y [5].
Xylenes are commercially produced by naphtha reforming and as a by-product of naphtha cracking for ethylene production (gasoline pyrolysis), in which large amounts of C7 and C9 aromatics are also produced [1, 6, 7]. By upgrading these low value streams through aromatic transalkylation or disproportionation reactions, xylene production can be optimized, with the advantage of developing environmental friendly processes at low operating costs [7].
The toluene disproportionation occurs in equilibrium with isomerization and transalkylation of aromatics such as xylenes, trimethylbenzene and others [8]. However, the control of kinetic parameters, the reactivity of aromatics, the kind of catalytic sites and of the morphology and texture of the catalyst can affect the equilibrium shift towards the desired product. Therefore, these factors determine the occurrence of disproportionation or transalkylation reactions [9-11]. The disproportionation of aromatics leads to products of higher commercial value, mainly p-xylene. Toluene has the lowest market demand while benzene and xylenes present an annual growth rate of about 10% [12]. The process thus represents a promising alternative for the production of xylenes and benzene and may be performed in the presence of liquid or solid acid catalysts [1].
Complimentary Contributor Copy

Influence of Platinum on the Performance of Beta Zeolite-Based Catalysts … 59
The first commercial process of alkylbenzenes disproportionation had begun in 1953, using a catalyst based on hydrofluoric acid and boron fluoride (HF-BF3) [13]. In 1964, the process was improved by replacing the catalyst by a Friedel-Crafts type one, based on aluminum chloride [1]. However, due to environmental and technological problems related to the use of liquid acid catalysts, solid acids have become promising candidates to replace liquid systems in various industrial processes [14, 15]. Currently, most commercial processes for toluene disproportionation use catalysts based on ZSM-5, which deactivate with time mainly due to coke deposition. This process occurs due to pore blockage, since the first coke molecules formed in the large channels are retained because of their low volatility. Therefore, one coke molecule is enough to inhibit the diffusion of the reactant to the active sites of the channels [16, 17].
Due to the susceptibility of ZSM-5 catalyst to deactivate by coke deposition, new zeolites with larger pores (mordenite, Y, beta, SSZ-33 and TNU-9) have been extensively studied [18-20] as well as silica [21] and others. For most cases, the concentrations of xylene isomers produced were close to the thermodynamic equilibrium ratios. Since the amount of p-xylene obtained directly from disproportionation can not satisfy the market requirements, there is an urgent need to maximize the production of this isomer.
Several studies have shown [22-24] that the selectivity of p-xylene isomer was increased by optimizing the crystal diameter, Si/Al ratio, activation and pelletization methods for the zeolites. The most common method for improving p-xylene selectivity is to modify the catalysts with magnesium, phosphorous, boron, calcium, lanthanum and/or cerium oxides. These dopants act by adjusting the pore radii and/or the protecting the external surface sites against coke deposition. In general, these changes increase the p-xylene selectivity but decrease the activity resulting in low p-xylene yield.
Therefore, other catalysts have been studied for this reaction, such as micro-mesoporous composites based on zeolites [24-26] and metal-containing zeolites [27-31]. The micro-mesoporous composites have attracted much attention for toluene disproportionation to produce benzene and xylenes, since the reaction involves a bulky intermediate which requires a large pore size to be accommodated [26]. On other hand, zeolite-supported metals have been successfully used to improve toluene disproportionation. The highly dispersed metal on zeolite is known to have electron donor/acceptor ability, which stabilizes the conversion values and suppresses coke deposition. Bhavani et al. [29], for instance, have studied the toluene disproportionation and transalkylation of aromatic C9+ in the range between 370 and 550°C, in the presence of hydrogen using copper and/or palladium catalysts. They have found that the best composition for the catalyst is 8.7 wt% Cu and 0.69 wt% Pd supported on mordenite, which led to a conversion of 53% [29]. In another works [30, 31], we have studied the influence of nickel, palladium and platinum in transalkylation of trimethylbenzene with benzene and have found that the nickel-based catalysts produced mainly ethylbenzene while the platinum-based ones produced large amount of xylenes, being the best metal to improve the catalyst.
In the present work, we studied platinum-supported beta zeolite containing 0.5 and 1.0% platinum for toluene disproportionation, in order to obtain catalysts more selective to p-xylenes than the commercial one.
Complimentary Contributor Copy

Maria do Carmo Rangel, Saulo de Tarso Figueiredo Grecco et al. 60
EXPERIMENTAL
Synthesis of Beta/MCM-41 Composite The beta/MCM-41 composite was prepared by mesostructuration/crystallization of beta
zeolite seeds, using a method based on the methodology previously reported but with some changes [25]. The beta zeolite seeds gel (1.47 Na2O: Al2O3: 30 SiO2: 8.23 TEAOH: 368 H2O) was obtained by mixing silica, sodium aluminate, sodium hydroxide, tetraethylammonium hydroxide (TEAOH) and water. The gel produced was submitted to a hydrothermal treatment at 140°C, for 192 h. The seeds gel was mesostructured with cetyltrimethylammonium bromide (CTAB) by adjusting the pH between 9.0 and 9.5 and then was undergone to a hydrothermal treatment at 140°C, for 48 h. After this step, the sample was centrifuged, washed with water and dried at 60°C, for 24 h. The solid underwent to ion exchange with 300 mL of an aqueous solution of ammonium chloride (0.1 mol.L-1) at 65°C, for 3 h. After this, the suspension was centrifuged, the solid was washed with water and dried at 60°C, for 24 h. The sample was then calcined at 550°C for 6 h for obtaining the zeolite in the protonic form (B sample).
Synthesis of Catalyst The catalysts were obtained by dispersing the supports previously prepared in a
hexachloroplatinic acid solution and keeping the suspension at rest, for 24 h. In order to obtain catalysts with 1.0% platinum, approximately 2 mL of the hexachloroplatinic acid solution (0.025 mol.L-1) per gram of the support were used. Then, the solid was dried at 120°C, for 12 h and calcined at 350°C, for 3 h producing a platinum supported on beta/MCM-41 composite (1%PtB sample). For the material containing 0.5% platinum a different concentration (0.0125 mol.L-1) of the hexachloroplatinic acid solution was used keeping the same conditions described, in order to obtain the 0.5%PtB sample.
Incoporation of Platinum in the Zeolite Synthesis Gel The catalyst with 1% platinum was also prepared by adding the platinum precursor in the
zeolite synthesis gel. In this case, the solid was prepared by adding 0.16 g of nitrate tetraminplatinum (II) dissolved in water during the preparation of zeolite seeds, just before of the addition of cetyltrimethylammonium bromide (CTAB). The same methodology previously described was then followed for getting the 1%PtBG sample.
Samples Characterization The support was characterized by thermogravimetry, differential thermal analysis,
Fourier transform infrared spectroscopy, X-ray diffraction and specific surface area and porosity measurements. The catalysts were characterized by specific surface area and porosity
Complimentary Contributor Copy

Influence of Platinum on the Performance of Beta Zeolite-Based Catalysts … 61
measurements, X-ray diffraction and transmission electron microscopy. The activities of metallic and acidic sites were evaluated by cyclohexane dehydrogenation, a model reaction for both kinds of sites.
The decomposition and the removal of the templates from the solids were monitored by thermogravimetry (TG) and differential thermal analysis (DTA). These experiments were carried out in a Mettler Toledo model TGA/SDTA 851e equipment, using 5 mg of the sample which was placed in a specimen holder of alumina and heated (10°C.min-1) from room temperature to 1000°C under air flow (50 mL.min-1).
The removal of the template and the production of the zeolite structure were also followed by Fourier transform infrared spectroscopy (FTIR). The experiments were performed in a Perkin Elmer model Spectrum One spectrophotometer. For each spectrum 32 scans were obtained in the range between 400 and 4000 cm-1 with a resolution of 4.0 cm-1. The sample (about 50 mg) was mixed with 200 mg of potassium bromide and placed in a metal sample holder, with approximately 10 mm in diameter and 2.3 mm thick. All experiments were carried out at room temperature.
The formation of hexagonal mesoporous structure of MCM-41 type and of the microporous structure of beta zeolite in the samples was investigated through X-ray diffraction (XRD), performed in a Shimadzu model XRD 6000 equipment, using CuKα radiation generated at 40 kV and 30 mA and a monochromator. The analysis was performed in the 2θ scanning range from 1 to 5° with a goniometer speed of 0.5°.min-1, for the mesoporous phase identification and from 5 to 45° with a goniometer speed of 2.0°.min-1, for the microporous phase identification.
The textural properties of the catalysts were determined by nitrogen adsorption experiments performed at 77 K in a Micromeritics model ASAP 2020 apparatus. Before analysis, approximately 0.20 g of the sample was heated (10°C.min-1) up to 250°C for 4 h under vacuum and then submitted to a pressure increase up to 10 μmHg, for the removal of water adsorbed on the sample and in the zeolite pore system. During analysis, the samples were submitted to pulses of nitrogen until a maximum increase of pressure of 925 mmHg.
The transmission electron microscopy and electron diffraction analysis were performed in a Jeol model JEM 1200 EXII equipment. The samples were dispersed in ethanol and one drop of the suspension was placed on carbon-coated copper grids (150 mesh). In the electron diffraction experiments, an accelerating voltage and the focal length 120 kV and 60 cm respectively were used.
The activities of the metallic and acidic sites were measured by the model reaction of cyclohexane dehydrogenation. The experiments were performed in a glass reactor at 300°C and 1 bar. Before reaction, the catalyst (0.1 g) was reduced under hydrogen flow (80 cm3. min-1) at 500°C, for 1 h. The products were analyzed in a Varian model 3400 CX chromatograph equipped with a FID and a packed column of FFAP on Chromosorb.
Catalysts Evaluation The catalysts were evaluated in toluene disproportionation. The experiments were carried
out in a tubular microreactor operating at 470 ºC and 1 atm, using H2/hydrocarbon (molar)= 4 and WHSV= 1.0 h-1. Prior to the tests, the catalyst (0.5 g) was heated at 500 ºC, under
Complimentary Contributor Copy

Maria do Carmo Rangel, Saulo de Tarso Figueiredo Grecco et al. 62
hydrogen flow, for 2 h. The gaseous effluent was analyzed by on line gas chromatography, using a Thermofinnigan model Trace GC instrument with FFAP capillary column and ionization detector. A commercial mordenite was also evaluated to be used as a reference. After each experiment, the reactor with the catalyst was cooled under nitrogen up to room temperature. The spent catalysts were analyzed by chemical analysis (carbon amount) in a CS-200 LECO model equipment using a ceramic crucible containing 0.20 g of sample, 1.25 g of tungsten compound (Lecocel) and 1.25 g of accelerator to help the combustion.
RESULTS AND DISCUSSION The thermogravimetry curve of the beta/MCM-41 composite is shown in Figure 1a. In
agreement with Kleitz et al. [32], this curve displayed four main stages of weight loss. The first one, at temperatures below 100°C, is related to the desorption of water while the second and third steps, between 150 and 500°C, can be associated to the decomposition and combustion of the templates in zeolite (TEAOH) and in MCM-41 (CTAB) structures. The weight loss at higher temperatures (near 600°C) is attributed to dehydroxylation of silanol groups and to combustion of residual coke.
The curve of differential thermal analysis (Figure 1b) shows that the decomposition of the templates involves three steps, in accordance with previous work [32]. It can be observed an endothermic peak near 250°C, related to the elimination of trimethylamine by Hofmann degradation, which leads to hydrocarbons formation. In the range from 250 to 350°C, they go into decomposition to produce lower molecular weight compounds. At this stage, it can be observed two exothermic events, each one associated to the decomposition of the hydrocarbon chain from each template. Finally, most of the oxidation processes occurs between 350 to 450°C, transforming the low molecular weight organic compounds into carbon dioxide, water and carbonaceous residues (exothermic process). The decomposition and the removal of the templates from the samples during calcination were also followed through FTIR. The spectra of the samples before calcination (Figure 2a) show bands related to symmetrical and asymmetrical stretching of C-H bonds near 2923 and 2853 cm-1, respectively. These bonds are characteristic of CH2 and CH3 groups from the templates. After calcination, those bands disappeared, indicating that the calcination process was efficient for the template removal [33]. It can also be observed the band of angular deformation of CH bonds at 1454 cm-1 (CH3) and 1394 cm-1 (CH2) [33]. After calcination, these bands disappeared, indicating that the calcination process was effective for removing these compounds.
Figure 2b shows the spectra enlargement in the wavenumber range from 1400 to 400 cm-1 in order to make the visualization of the characteristic bands of amorphous and crystalline materials easier. It can be observed a band nearby 460 cm-1, assigned to the internal vibrations of the aluminosilicate tetrahedra, which is insensitive to structural variations and thus is characteristic of both zeolitic and amorphous materials [34]. The spectra of the samples also exhibit two absorption band between 500 and 600 cm-1, indicating the presence of a ring-like structure with five members of siloxan bond, T-O-T (T= Si, Al), typical of beta zeolite seeds [34]. The high crystallinity of the solids after calcination was detected by X-ray diffraction, as
Complimentary Contributor Copy
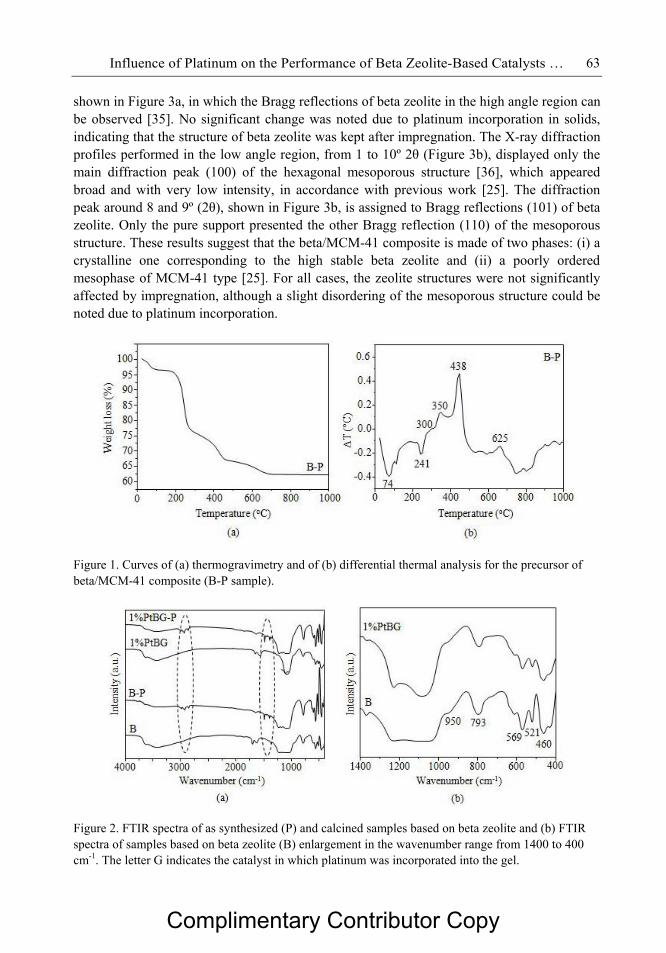
Influence of Platinum on the Performance of Beta Zeolite-Based Catalysts … 63
shown in Figure 3a, in which the Bragg reflections of beta zeolite in the high angle region can be observed [35]. No significant change was noted due to platinum incorporation in solids, indicating that the structure of beta zeolite was kept after impregnation. The X-ray diffraction profiles performed in the low angle region, from 1 to 10º 2θ (Figure 3b), displayed only the main diffraction peak (100) of the hexagonal mesoporous structure [36], which appeared broad and with very low intensity, in accordance with previous work [25]. The diffraction peak around 8 and 9º (2θ), shown in Figure 3b, is assigned to Bragg reflections (101) of beta zeolite. Only the pure support presented the other Bragg reflection (110) of the mesoporous structure. These results suggest that the beta/MCM-41 composite is made of two phases: (i) a crystalline one corresponding to the high stable beta zeolite and (ii) a poorly ordered mesophase of MCM-41 type [25]. For all cases, the zeolite structures were not significantly affected by impregnation, although a slight disordering of the mesoporous structure could be noted due to platinum incorporation.
Figure 1. Curves of (a) thermogravimetry and of (b) differential thermal analysis for the precursor of beta/MCM-41 composite (B-P sample).
Figure 2. FTIR spectra of as synthesized (P) and calcined samples based on beta zeolite and (b) FTIR spectra of samples based on beta zeolite (B) enlargement in the wavenumber range from 1400 to 400 cm-1. The letter G indicates the catalyst in which platinum was incorporated into the gel.
Complimentary Contributor Copy
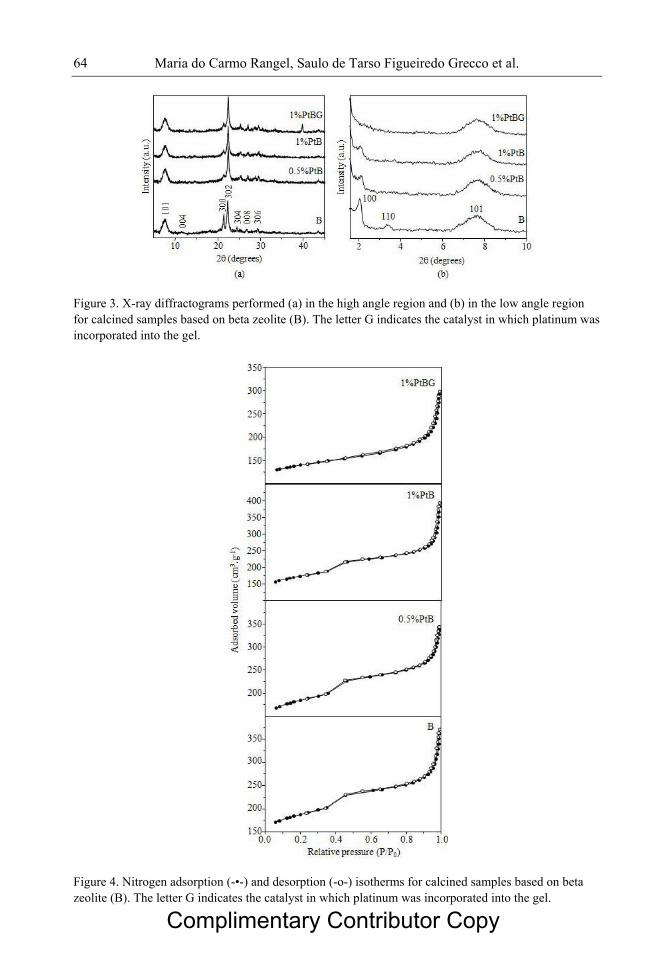
Maria do Carmo Rangel, Saulo de Tarso Figueiredo Grecco et al. 64
Figure 3. X-ray diffractograms performed (a) in the high angle region and (b) in the low angle region for calcined samples based on beta zeolite (B). The letter G indicates the catalyst in which platinum was incorporated into the gel.
Figure 4. Nitrogen adsorption (-�-) and desorption (-o-) isotherms for calcined samples based on beta zeolite (B). The letter G indicates the catalyst in which platinum was incorporated into the gel.
Complimentary Contributor Copy

Influence of Platinum on the Performance of Beta Zeolite-Based Catalysts … 65
As shown in Figure 4, all solids show isotherms, which are intermediates between I type, typical of microporous materials and IV type, typical of mesoporous materials [37]. As mentioned before, the presence of microporous is related to the structure of highly crystalline beta zeolites. Otherwise, the presence of mesoporous can be related to the formation of a poorly ordered mesophase during the mesostructuration step in the presence of CTAB and from mesopores among the particles, which are able to adsorb nitrogen at high relative pressures. This behavior shows that the synthesis of beta zeolites in two steps, used in the present work, results in a solid formed by small zeolite crystals between amorphous silica particles. In the first step, the synthesis conditions allow the formation of beta zeolite seeds whose sizes make their accommodation unfeasible around the CTAB micelles during mesostructuration; therefore, a less ordered mesoporous structure is generated, which become even lesser ordered as the crystals grow during the crystallization step. These results were confirmed by transmission electron microscopy that showed: (i) the presence of particles with irregular shape and heterogeneous characteristics, indicating that the solids are made of more than one phase (Figure 5a) and (ii) the dominion of mesophase (Figure 5b). The electron diffraction image corresponds to the crystalline dominion of beta zeolite (Figure 6).
The textural properties of the catalysts are shown in Table 1. As expected, they showed high specific surface areas and presented a narrow pore size distribution (Figure 7) with an average diameter of around 3.1 nm as shown by its pore size distribution determined by the BJH method from the adsorption branch of the nitrogen isotherms [38].
Figure 5. TEM images of particles of beta/MCM-41 composite showing (a) the different phases of the solids and (b) the domain of mesophase.
Figure 6. Electron diffractogram of the crystalline phase of beta zeolite for the beta/MCM-41 composite.
Complimentary Contributor Copy
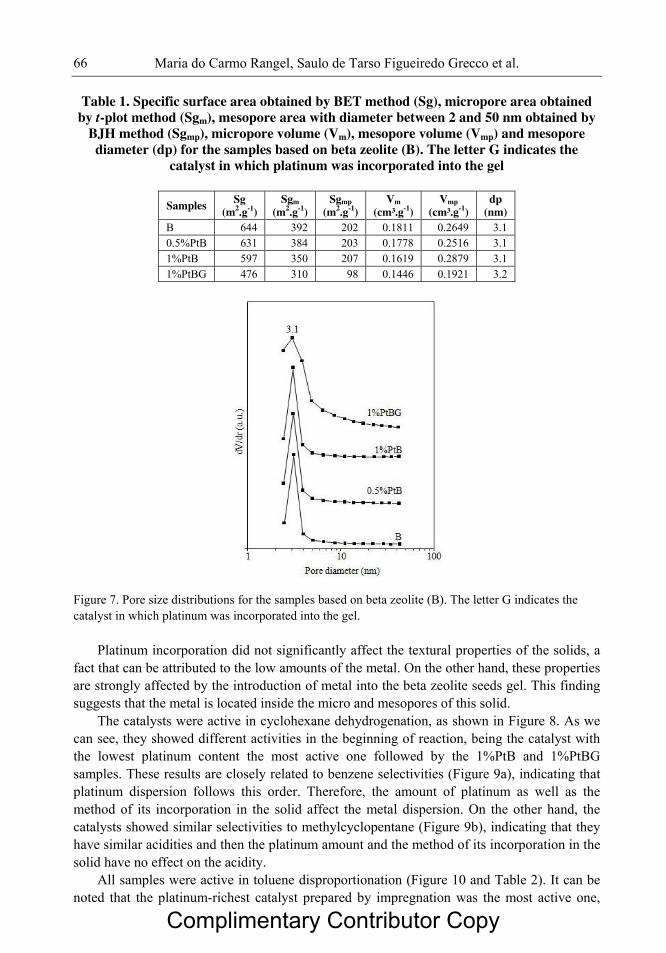
Maria do Carmo Rangel, Saulo de Tarso Figueiredo Grecco et al. 66
Table 1. Specific surface area obtained by BET method (Sg), micropore area obtained
by t-plot method (Sgm), mesopore area with diameter between 2 and 50 nm obtained by
BJH method (Sgmp), micropore volume (Vm), mesopore volume (Vmp) and mesopore
diameter (dp) for the samples based on beta zeolite (B). The letter G indicates the
catalyst in which platinum was incorporated into the gel
Samples Sg
(m2.g
-1)
Sgm
(m2.g
-1)
Sgmp
(m2.g
-1)
Vm
(cm³.g-1
)
Vmp
(cm³.g-1
)
dp
(nm)
B 644 392 202 0.1811 0.2649 3.1
0.5%PtB 631 384 203 0.1778 0.2516 3.1
1%PtB 597 350 207 0.1619 0.2879 3.1
1%PtBG 476 310 98 0.1446 0.1921 3.2
Figure 7. Pore size distributions for the samples based on beta zeolite (B). The letter G indicates the catalyst in which platinum was incorporated into the gel.
Platinum incorporation did not significantly affect the textural properties of the solids, a
fact that can be attributed to the low amounts of the metal. On the other hand, these properties are strongly affected by the introduction of metal into the beta zeolite seeds gel. This finding suggests that the metal is located inside the micro and mesopores of this solid.
The catalysts were active in cyclohexane dehydrogenation, as shown in Figure 8. As we can see, they showed different activities in the beginning of reaction, being the catalyst with the lowest platinum content the most active one followed by the 1%PtB and 1%PtBG samples. These results are closely related to benzene selectivities (Figure 9a), indicating that platinum dispersion follows this order. Therefore, the amount of platinum as well as the method of its incorporation in the solid affect the metal dispersion. On the other hand, the catalysts showed similar selectivities to methylcyclopentane (Figure 9b), indicating that they have similar acidities and then the platinum amount and the method of its incorporation in the solid have no effect on the acidity.
All samples were active in toluene disproportionation (Figure 10 and Table 2). It can be noted that the platinum-richest catalyst prepared by impregnation was the most active one,
Complimentary Contributor Copy
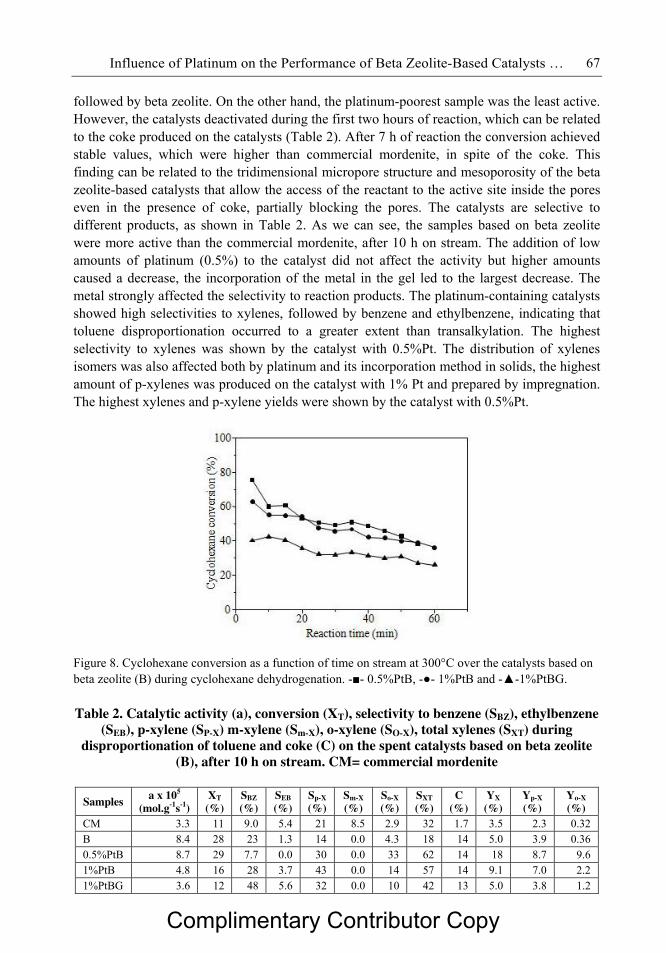
Influence of Platinum on the Performance of Beta Zeolite-Based Catalysts … 67
followed by beta zeolite. On the other hand, the platinum-poorest sample was the least active. However, the catalysts deactivated during the first two hours of reaction, which can be related to the coke produced on the catalysts (Table 2). After 7 h of reaction the conversion achieved stable values, which were higher than commercial mordenite, in spite of the coke. This finding can be related to the tridimensional micropore structure and mesoporosity of the beta zeolite-based catalysts that allow the access of the reactant to the active site inside the pores even in the presence of coke, partially blocking the pores. The catalysts are selective to different products, as shown in Table 2. As we can see, the samples based on beta zeolite were more active than the commercial mordenite, after 10 h on stream. The addition of low amounts of platinum (0.5%) to the catalyst did not affect the activity but higher amounts caused a decrease, the incorporation of the metal in the gel led to the largest decrease. The metal strongly affected the selectivity to reaction products. The platinum-containing catalysts showed high selectivities to xylenes, followed by benzene and ethylbenzene, indicating that toluene disproportionation occurred to a greater extent than transalkylation. The highest selectivity to xylenes was shown by the catalyst with 0.5%Pt. The distribution of xylenes isomers was also affected both by platinum and its incorporation method in solids, the highest amount of p-xylenes was produced on the catalyst with 1% Pt and prepared by impregnation. The highest xylenes and p-xylene yields were shown by the catalyst with 0.5%Pt.
Figure 8. Cyclohexane conversion as a function of time on stream at 300°C over the catalysts based on beta zeolite (B) during cyclohexane dehydrogenation. -■- 0.5%PtB, -●- 1%PtB and -▲-1%PtBG.
Table 2. Catalytic activity (a), conversion (XT), selectivity to benzene (SBZ), ethylbenzene
(SEB), p-xylene (SP-X) m-xylene (Sm-X), o-xylene (SO-X), total xylenes (SXT) during
disproportionation of toluene and coke (C) on the spent catalysts based on beta zeolite
(B), after 10 h on stream. CM= commercial mordenite
Samples a x 10
5
(mol.g-1
s-1
)
XT
(%)
SBZ
(%)
SEB
(%)
Sp-X
(%)
Sm-X
(%)
So-X
(%)
SXT
(%)
C
(%)
YX
(%)
Yp-X
(%)
Yo-X
(%)
CM 3.3 11 9.0 5.4 21 8.5 2.9 32 1.7 3.5 2.3 0.32
B 8.4 28 23 1.3 14 0.0 4.3 18 14 5.0 3.9 0.36
0.5%PtB 8.7 29 7.7 0.0 30 0.0 33 62 14 18 8.7 9.6
1%PtB 4.8 16 28 3.7 43 0.0 14 57 14 9.1 7.0 2.2
1%PtBG 3.6 12 48 5.6 32 0.0 10 42 13 5.0 3.8 1.2
Complimentary Contributor Copy
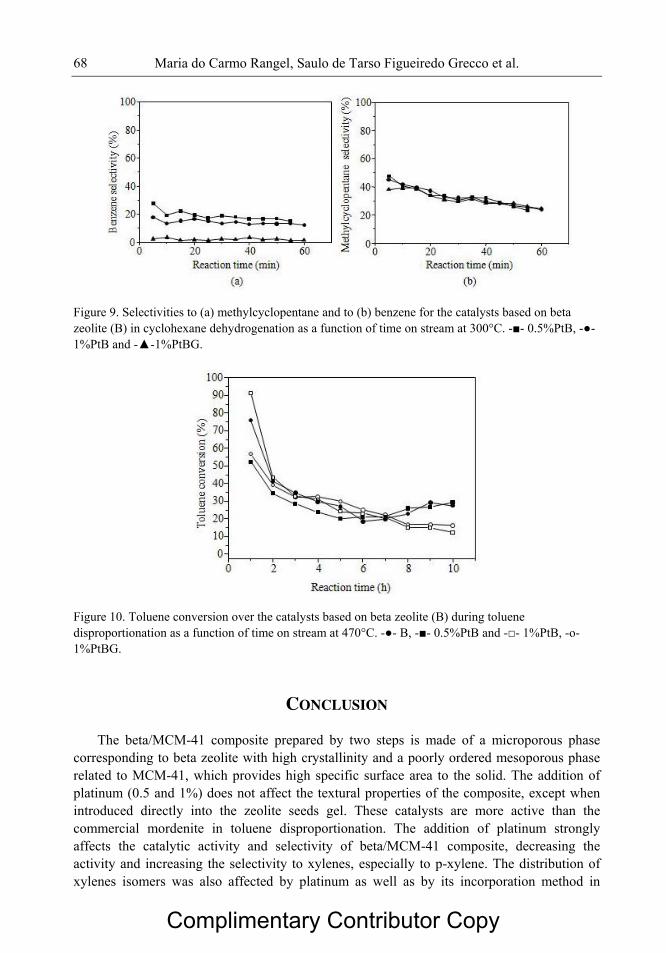
Maria do Carmo Rangel, Saulo de Tarso Figueiredo Grecco et al. 68
Figure 9. Selectivities to (a) methylcyclopentane and to (b) benzene for the catalysts based on beta zeolite (B) in cyclohexane dehydrogenation as a function of time on stream at 300°C. -■- 0.5%PtB, -●- 1%PtB and -▲-1%PtBG.
Figure 10. Toluene conversion over the catalysts based on beta zeolite (B) during toluene disproportionation as a function of time on stream at 470°C. -●- B, -■- 0.5%PtB and -□- 1%PtB, -o-1%PtBG.
CONCLUSION The beta/MCM-41 composite prepared by two steps is made of a microporous phase
corresponding to beta zeolite with high crystallinity and a poorly ordered mesoporous phase related to MCM-41, which provides high specific surface area to the solid. The addition of platinum (0.5 and 1%) does not affect the textural properties of the composite, except when introduced directly into the zeolite seeds gel. These catalysts are more active than the commercial mordenite in toluene disproportionation. The addition of platinum strongly affects the catalytic activity and selectivity of beta/MCM-41 composite, decreasing the activity and increasing the selectivity to xylenes, especially to p-xylene. The distribution of xylenes isomers was also affected by platinum as well as by its incorporation method in
Complimentary Contributor Copy

Influence of Platinum on the Performance of Beta Zeolite-Based Catalysts … 69
solids; in general, the catalysts were more selective to p-xylene, followed by o-xylene, the most valuable isomers and did not produce m-xylene. The most suitable catalyst to produce xylenes, especially p-xylenes, was prepared by incorporating platinum (0,5%) by impregnation.
REFERENCES
[1] Tsai, TC; Liu, SB; Wang, I. Appl. Catal. A: General, 181, (1999), 355-398. [2] Olah, GA; Molnár, A. Hydrocarbon Chemistry, John Wiley & Sons, New Jersey, 2003. [3] Sherman, JD. Proc. Natl. Acad. Sci. USA, 96, (1999), 3471-3478. [4] Hocking, MB. Handbook of Chemical Technology and Pollution Control, third ed.,
Academic Press, New York, 2005. [5] Balasamy, RJ; Odedairo, T; Al-Khattaf, S. Appl. Catal. A: General, 409-410, (2011),
223-233. [6] Tsai, TC; Chen, WH; Liu, SB; Tsai, CH; Wang, I; Ku, CS. Catal. Today, 73, (2002),
39-47. [7] Forni, L; Cremona, G; Missineo, F; Bellusi, G; Perego, C; Pazzuconi, G. Appl. Catal.
A: General, 121, (1995), 261-272. [8] Serra, JM; Guillon, E; Corma, A. J. of Catal. 227, (2004), 459-469. [9] Tsai, TC. Appl. Catal. A: General, 301, (2006), 292-298. [10] Tsai, TC; Chen, WH; Lai, CS; Liu, SB; Wang, I; Ku, CS. Catal. Today, 97, (2004),
297-302. [11] Mavrodinova, V; Popova, M; Mihályi, RM; Pál-Borbély, G; Minchev, C. J. of Molec.
Catal. A: Chemical, 220, (2004), 239-246. [12] Harvan, RG. Oil Gas J. 30, (1998), 56-61. [13] Lien, AP; Mccaulay, DA. J. Am. Chem. Soc. 75, (1953), 2407-2410. [14] Degnan Jr., TF; Smith, CM; Venkat, CR. Appl. Catal. A: General, 221, (2001),
283-294. [15] Sulikowski, B; Rachwalik, R. Appl. Catal. A: General, 256, (2003), 173-182. [16] Rangel, MC; Valentini, A; Oliveira, AS; David, JM; Britto, JM; Domingues, SM;
Reyes, P. Quim. Nova, 26, (2003), 305-308. [17] Guisnet, M; Magnoux, P. Appl. Catal. A: General, 212, (2001), 83-96. [18] Cavani, F; Corazzari, M; Bencini, E; Goffredi, G. Appl. Catal. A: General, 226, (2002),
31-40. [19] Oliveira, AC; Essayem, N; Tuel, A; Clacens, J-M; Taarit, YB; Rangel, MC. Stud. Surf.
Sci. Catal. 158, (2005), 1677-1684. [20] Baduraig, A; Odedairo, T; Al-Khattaf, S. Top. Catal, 53, (2010), 1446-1456. [21] Dias, JA; Rangel, MC; Dias, SCL; Caliman, E; Garcia, FAC. Appl. Catal. A: General,
328, (2007), 189-194. [22] Uguina, MA; Sotelo, JL; Serrano, DP. Appl. Catal. 76, (1991), 183-198. [23] Uguina, MA; Sotelo, JL; Serrano, DP; van Grieken, R. Ind Eng Chem Res, 31, (1992),
1875-1880. [24] Mitra, B; Kunzru, D. Catal. Lett. 141, (2011), 1569-1579.
Complimentary Contributor Copy

Maria do Carmo Rangel, Saulo de Tarso Figueiredo Grecco et al. 70
[25] Grecco, STF; Nobre, PSS; Urquieta-González, EA; Rangel, MC. in: A. Sayari, M. Jaroniec, (Eds.), Nanoporous Materials: Proceedings of the 5th International Symposium, World Scientific Publishing Co. Pte. Ltd. New Jersey, 2008, pp. 599-606.
[26] Ungureanu, A; Hoang, TV; Trong On, D; Dumitriu, E; Kaliaguine, S. Appl. Catal. A:
General, 294, (2005), 92-105. [27] Oliveira, AC; Rangel, MC; Fierro, JLG; Reyes, P; Oportus, M. Quim. Nova, 28, (2005),
37-41. [28] Oliveira, AC; Reyes, P; Oportus, M; Rangel, MC. Stud. Surf. Sci. Catal. 154, (2004),
886-893. [29] Bhavani, AG; Karthekayen, D; Rao, AS; Lingappanet, N. Catal. Lett. 103, (2005),
89-100. [30] Ramos, MS; Grecco, ST; Gomes, LP; Oliveira, AC; Reyes, P; Oportus, M; Rangel,
MC. Stud. Surf. Sci. Catal. 156, (2005), 809-814. [31] Grecco, STF; Gomes, LP; Reyes, P; Oportus, M; Rangel, MC. Stud. Surf. Sci. Catal.
158, (2005), 1937-1944. [32] Kleitz, F; Schimidt, W; Schüth, F. Microporous Mesoporous Mater. 65, (2003), 1-29. [33] Selvaraj, M; Pandurangan, A; Seshadri, KS; Sinha, PK; Lal, KB. Appl. Catal. A:
General, 242, (2003), 347-364. [34] Flanigen, EM. in: J.A. Rabo, (Eds.), Zeolite Chemistry and Catalysis, American
Chemical Society Washington, DC, 1976, 80-117. [35] Treacy, MMJ; Higgins, JB. Collection of Simulated XRD Powder Patterns for Zeolites,
fifth ed., Elsevier, Amsterdam, 2007. [36] Beck, JS; Vartuli, JC; Roth, WJ; Leonowicz, ME; Kresge, CT; Schmitt, KD; Chu, CT-
W; Olson, DH; Sheppard, EW; McCullen, SB; Higgins, JB; Schlenker, JL. J. Am.
Chem. Soc. 114, (1992), 10834-10843. [37] Schneider, P. Appl. Catal. A, 129, (1995), 157-165. [38] Barrett, EP; Joyner, LG; Halenda, PP. J. Am. Chem. Soc. 73, (1951), 373-380.
Complimentary Contributor Copy


















