
UNIVERSIDADE DO BRASIL - Livros Grátislivros01.livrosgratis.com.br/cp098456.pdf · CENTRO DE...
183
i UNIVERSIDADE DO BRASIL CENTRO DE CIÊNCIAS MATEMÁTICAS E DA NATUREZA Instituto de Química Departamento de Química Orgânica CROMATOGRAFIA GASOSA DE ALTA RESOLUÇÃO ACOPLADA A ESPECTROMETRIA DE MASSAS APLICADA AO ESTUDO DE AGENTES ANABÓLICOS E CANDIDATOS A FÁRMACOS MODULADORES DE RECEPTORES DOPAMINÉRGICOS EM FLUIDOS BIOLÓGICOS MONICA COSTA PADILHA Tese de Doutorado
Transcript of UNIVERSIDADE DO BRASIL - Livros Grátislivros01.livrosgratis.com.br/cp098456.pdf · CENTRO DE...

i
UNIVERSIDADE DO BRASIL
CENTRO DE CIÊNCIAS MATEMÁTICAS E DA NATUREZA
Instituto de Química
Departamento de Química Orgânica
CROMATOGRAFIA GASOSA DE ALTA RESOLUÇÃO ACOPLADA A ESPECTROMETRIA DE MASSAS APLICADA AO ESTUDO DE AGENTES
ANABÓLICOS E CANDIDATOS A FÁRMACOS MODULADORES DE RECEPTORES DOPAMINÉRGICOS EM FLUIDOS BIOLÓGICOS
MONICA COSTA PADILHA Tese de Doutorado

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

ii
Rio de Janeiro 2007
CROMATOGRAFIA GASOSA DE ALTA RESOLUÇÃO ACOPLADA A ESPECTROMETRIA DE MASSAS APLICADA AO ESTUDO DE AGENTES
ANABÓLICOS E CANDIDATOS A FÁRMACOS MODULADORES DE RECEPTORES DOPAMINÉRGICOS EM FLUIDOS BIOLÓGICOS
Monica Costa Padilha
Tese submetida ao corpo docente do Instituto de Química da Universidade do Brasil-UFRJ, como parte dos requisitos necessários à obtenção do grau de Doutor em Ciências Aprovado por: Prof. Francisco Radler de Aquino Neto (IQ/UFRJ)
Prof. Carlos Alberto Manssour Fraga (IQ/UFRJ)
Prof. Ângelo da Cunha Pinto (IQ/UFRJ)
Prof. Eliezer de Jesus Barreiro (FF/UFRJ)
Profa. Paula Fernandes de Aguiar (IQ/UFRJ)
Prof. Tanus Jorge Nagem (DEQUI/UFOP)
Rio de Janeiro, dezembro de 2007

iii
FICHA CATALOGRÁFICA
Padilha, Monica Costa CROMATOGRAFIA GASOSA DE ALTA RESOLUÇÃO ACOPLADA AESPECTROMETRIA DE MASSAS APLICADA AO ESTUDO DE AGENTESANABÓLICOS E CANDIDATOS A FÁRMACOS MODULADORES DERECEPTORES DOPAMINÉRGICOS EM FLUIDOS BIOLÓGICOS xx, f. Tese (Doutorado em Ciências) – Universidade do Brasil – UFRJ. Centro de CiênciasMatemáticas e da Natureza, Instituto de Química, Departamento de QuímicaOrgânica, 2007 Orientadores: Francisco Radler de Aquino Neto e Carlos Alberto Manssour Fraga 1. Anabolizantes 2. Protótipos para receptores D2 3. Cromatografia gasosa de altaresolução 4. Espectrometria de massas 5. Validação de metodologia – Teses. I.Francisco Radler de Aquino Neto e Carlos Alberto Manssour Fraga (orientadores). II.Universidade do Brasil. Centro de Ciências Matemáticas e da Natureza. Instituto deQuímica. Departamento de Química Orgânica. III. Título.

iv
Tese realizada no Departamento de Química Orgânica do
INSTITUTO DE QUÍMICA DA UNIVERSIDADE DO BRASIL-
UFRJ, como parte dos requisitos necessários para a obtenção
do Grau de Doutor em Ciências, sob a orientação dos
Professores Francisco Radler de Aquino Neto e Carlos Alberto
Manssour Fraga.

v
AGRADECIMENTOS
Ao professor Francisco Radler de Aquino Neto, pela enorme contribuição na minha
formação profissional, pela parceria, confiança, pelas melhores oportunidades de
aprendizado e pelo incentivo demonstrado na convivência ao longo de todos esses anos.
Ao professor Carlos Alberto Manssour Fraga, pela importante contribuição científica
e enorme incentivo ao longo da realização desse trabalho.
Aos meus queridos amigos Renata S. L. Raices, Flavio L. Vicente, a Júlia R. L.
Vicente, Rebecca S. Nocolich e Valter Luiz Gonçalves pelo total apoio e amizade
demonstrados ao longo do desenvolvimento desta tese.
Aos companheiros de trabalho do LAB DOP, Henrique, Rafael, Professor Luiz
Nelson e Tarcísio, pela união nos momentos difíceis e pelo apoio e incetivo frente aos
desafios que envolvem trabalhar com controle de dopagem no Brasil.
A VARIAN pelo apoio, amizade e enorme contribuição em minha carreira.
Aos professores da PGQO, em especial aos professores Pierre Mothé Esteves,
Ângelo da Cunha Pinto e Cláudia Moraes de Rezende pela enorme contribuição em
minha carreira.
A professora Paula Fernandes de Aguiar pela amizade, motivação e por
compartilhar comigo seus conhecimentos em estatística.
Aos amigos da PGQO, em especial a turma de 2004/1, por todos os momentos que
passamos juntos ao longo desses anos.
Aos técnicos da triagem IV, Isadora, Márcia, Sabrina, Roberto, Glauco e Mariana
pelo apoio, compreensão e incentivo ao longo desses anos.

vi
A todos os companheiros do LAB-DOP - LADETEC (Instituto de Química, UFRJ) e
a todos que contribuíram de forma direta ou indireta na execução desta tese.
Aos professores da CEFETEQ-RJ, pelo incentivo e amizade ao longo de todos
esses anos de convivência.
Ao Dr. Ivan Costas Barros pela pergunta certa em um momento difícil, pelo
incentivo e amizade ao longo de todos esses anos de convivência.
À minha mãe e ao meu pai pela força, carinho e incentivo, a minha irmã Luciana e
ao meu cunhado Marcelo pelo apoio e compreensão.

vii
“A imaginação é mais importante que o conhecimento”
Albert Einstein

viii
RESUMO
A pesquisa de protótipos de candidatos a fármacos e o desenvolvimento de
métodos de controle de dopagem no esporte exigem, invariavelmente, o estudo do perfil
de biotransformação da substância de interesse. Para identificar esses possíveis
metabólitos, a cromatografia gasosa de alta resolução acoplada a espectrometria de
massas (CGAR-EM) tem-se mostrado uma técnica analítica confiável e versátil,
permitindo sua aplicação a diferentes tipos de moléculas orgânicas.
Um grupo de protótipos candidatos a fármacos com propriedades antipsicóticas foi
sintetizado pelo LASSBio/FF-UFRJ em 2004. Entretanto, para esses protótipos N-fenil-
piperazínicos, ainda não havia sido feita a caracterização detalhada por espectrometria de
massas e a investigação de possíveis metabólitos usando modelos animais. Por outro
lado, em função dos jogos Pan-Americanos 2007, o LABDOP/IQ-UFRJ teve de incluir
diversos agentes anabólicos no escopo de seu trabalho, o que requer o controle não só
dos fármacos mas também de seus metabólitos.
Tendo em vista essas duas necessidades prementes, o objetivo deste trabalho foi
desenvolver, e validar metodologias analíticas, baseadas em CGAR-EM, para o estudo
dos metabóliots de N-fenilpiperazinas e de esteróides anabolizantes em fluidos biológicos.
Para os protótipos candidatos a fármacos, a caracterização por ESI-EM/EM indicou
um único fragmento dominante para os pirazóis e quatro fragmentos majoritários para os
triazóis. Em relação a investigação de metabólitos para o composto LASSBio 581, não foi
possível a detecção de algum sinal que pudesse ser atribuído a um possível metabólito.
No caso dos agentes anabólicos, foi possível a inclusão na rotina do laboratório
dos metabólitos de Tamoxifeno, Clomifeno, Ciclofenil, Letrozol, Metilnortestosterona,
Metasterona, Tibolona, Zeranol e dos fármacos Formestano e Hiperdrol, assim como a
validação da metodologia empregada. Adicionalmente foi possível desenvolver um
método que tanto atendeu a demanda dos jogos – permitindo, inclusive, liberação de
resultados em 24 horas – quanto cumpriu as exigências da Agência Mundial Anti-
dopagem. Assim, com um tempo total de corrida de 6,25 min para análise de Clembuterol,
Epimetendiol, 3α,5β-Metiltestosterona, 3’OH-estanozolol e Norandorsterona, a técnica
proporcionou a detecção no limite de desempenho requerido pela agência reguladora.

ix
ABSTRACT The research for prototypes to drug candidates and the methods development for
doping control in sports invariably demand the study of the biotransformation profile of the
interest substance. In order to identify these possible metabolites, high resolution gas
chromatography coupled with mass spectrometry (HRGC-MS) has been shown to be
reliable and versatile, allowing its application to different types of organic molecules.
A group of prototypes drug candidates for schizophrenia control was synthesized by
LASSBio/FF-UFRJ in 2004. However, for these N-phenylpiperazinic prototypes, there had
not been conducted the characterization by mass spectrometry and the investigation about
possible metabolites, in animal models. On the other hand, due Pan-American Games
2007, LABDOP/IQ-UFRJ had to include several anabolic steroids in the work scope, which
would require the control of not only the parent drugs but also their metabolites.
In the face of these two pressing necessities, the objective of this work is to develop
and validate analytical methodologies based on HRGC-MS for the study of N-
phenylpiperazines and anabolic steroids metabolites in biological fluids.
For the prototypes drug candidates, the characterization was performed by
electrospray ionization mass and tandem mass spectrometry indicating a dominant
fragment for the pyrazole series and four major fragments for the triazole series.
Regarding to metabolites of LASSBio 581, was not possible detect any signal which could
be related with possible metabolites.
In the case of the steroids, was possible include in screening of laboratory
metabolites of Tamoxifen, Clomiphene, Cyclofenil, Letrozole, Methylnortestosterone,
Methasterone, Tibolone, Zeranol the drugs Formestane and Hyperdrol, as well the
validation of this method used. In addition was possible to develop a method that both
fulfilled the demand of the Games – allowing the release of results within 24 hours indeed
– and complied with the requirements from the World Anti-Doping Agency. Then, with a
total running time of 6.25 min to analysis of Clembuterol, Epimetendiol, 3α,5β-
Methiltestosterone, 3’OH-stanozolol and Norandrosterone, the technique also provide the
minimum required performance limits according regulatory agency.

x
ÍNDICE GERAL
Capítulo 1. INTRODUÇÃO Página 1.1. CROMATOGRAFIA GASOSA DE ALTA RESOLUÇÃO 1
1.2. ESPECTROMETRIA DE MASSAS 3
1.2.1. Analisadores do tipo “armadilha de íons” – Ion trap 6
1.2.1.1. Modos de operação no Ion trap 6
1.3. TÉCNICAS DE PREPARAÇÃO DE AMOSTRA 6
1.3.1. Precpitação 7
1.3.2. Extração por fase sólida Hidrólise 7
1.3.3. Extração líquido-líquido 8
1.3.4. Derivatização 9
1.4. CONTROLE DE DOPAGEM NO ESPORTE - UM BREVE HISTÓRICO 10
1.5. AGENTES ANABÓLICOS NO CONTEXTO DO CONTROLE DE DOPAGEM NO
ESPORTE 11
1.5.1. Mecanismo de ação e uso clínico 13
1.5.2. Abuso de esteróides 13
1.5.3. Metabolismo 14
1.5.4. Estrutura química 19
1.5.4.1. Anabolizantes 21
1.5.4.1.1. Estanozolol 21
1.5.4.1.2. Metandienona 23
1.5.4.1.3. Nandrolona 24
1.5.4.1.4. Metiltestosterona 25
1.5.4.1.5. Metilnortestosterona 26
1.5.4.1.6. Metasterona 27
1.5.4.2. Agentes anabólicos 27
1.5.4.2.1. Clembuterol 27
1.5.4.2.2. Tibolona 28
1.5.4.2.3. Zeranol 29
1.5.4.3. Agentes com atividade anti-estrogênica 30
1.5.4.3.1. Tamoxifeno 32
1.5.4.3.2. Clomifeno 34
1.5.4.3.3. Ciclofenil 35

xi
1.5.4.3.4. Letrozol 35
1.5.4.3.5. Formestano 36
1.5.4.3.6. Hiperdrol 36
1.5.5. Estratégia do exame antidopagem para agentes anabólicos 37
1.5.6. Métodos analíticos aplicados a agentes anabólicos 38
1.5.6.1. Cromatografia gasosa de alta resolução acoplada
a espectrometria de massas (CG-EM) 38
1.5.6.2. Critérios de identificação para ensaios qualitativos
envolvendo a Cromatografia e a Espectrometria de massas 38
1.6. PROTÓTIPOS PARA RECEPTORES DOPAMINÉRGICOS 40
1.6.1. Planejamento estrutural de novos heterocíclicos N-fenilpiperazínicos
a partir da clozapina 40
1.6.2. Processo de desenvolvimento de fármacos 42
1.6.3. Metabolismo de N-fenilpiperazínicos 43
Capítulo 2. OBJETIVOS 44
Capítulo 3. EXPERIMENTAL 3.1. SUBSTÂNCIAS DE REFERÊNCIA 46
3.2. SOLVENTES E REAGENTES 46
3.3. MEDICAMENTOS 47
3.4. AMOSTRAS BIOLÓGICAS 47
3.5. MATERIAIS 47
3.6. EQUIPAMENTOS 47
3.7. LIMPEZA DA VIDRARIA 48
3.8. PREPARO DAS SOLUÇÕES 48
3.8.1. Soluções padrão estoque 48
3.8.2. Soluções padrão de trabalho 49
3.8.3. Padrão interno (ISTD) 50
3.8.4. Solução de D5-androsterona-glicuronídeo 50
3.8.5. Solução de KOH 50
3.8.6. Solução derivatizante 51
3.8.7. Limpeza e silanização dos liners (encamisamento de vidro) de quartzo e / ou vidro
borossilicato (método LADETEC n°5106) 51

xii
3.9. MÉTODOS DE EXTRAÇÃO EM MATRIZES BIOLÓGICAS 52
3.9.1. Extração de agentes anabólicos em urina humana 52
3.9.1.1. Método de triagem (método LADETEC n°5070) 52
3.9.2. Extração de LASSBio 581 em plasma de rato wistar 54
3.10. CONDIÇÕES DE ANÁLISE CROMATOGRÁFICA (CGAR-EM) DAS AMOSTRAS
BIOLÓGICAS 56
3.10.1. Análise de agentes anabólicos em urina humana 56
3.10.2. Análise de LASSBio 581 em plasma de rato Wistar 56
3.11. METABOLISMO DO COMPOSTO LASSBio 581 57
3.11.1. Animais 57
3.11.2. Preparo e administração das doses 57
3.11.3. Coleta das amostras de plasma e urina 57
3.12. VALIDAÇÃO DE MÉTODOS 58
3.12.1. Recuperação do processo de extração 60
3.12.2. Protocolo de validação para análise qualitativa 61
3.12.2.1. Especificidade 61
3.12.2.2. Limite de detecção 62
3.12.2.3. Precisão intra-ensaio (repetitividade) 62
3.12.2.4. Valores aberrantes 62
3.12.3. Protocolo de validação para análise semi-quantitativa 63
3.12.3.1. Especificidade 63
3.12.3.2. Contaminação entre amostras (arraste) 63
3.12.3.3. Limite de detecção (LD) 64
3.12.3.4. Limite de quantificação (LQ) 64
3.12.3.5. Precisão intra-ensaio (repetitividade) 64
3.12.3.6. Valores aberrantes 65
3.12.4. Protocolo de validação para análise quantitativa 65
3.12.4.1. Especificidade 66
3.12.4.2. Contaminação entre amostras (arraste) 66
3.12.4.3. Valores aberrantes 66
3.12.4.4. Homocedasticidade / Heterocedasticidade 66
3.12.4.5. Curva de calibração / Linearidade 67
3.12.4.6. Limite de detecção (LD) e limite de quantificação (LQ) 68
3.12.4.7. Precisão e exatidão inter-ensaio 68

xiii
3.12.4.8. Cálculo da incerteza expandida (U) 69
3.13. MONITORAMENTO DO TEMPO NECESSÁRIO PARA HIDRÓLISE DE AGENTES
ANABÓLICOS 71
Capítulo 4. RESULTADOS E DISCUSSÃO 4.1. DETERMINAÇÃO DAS CONDIÇÕES DE ANÁLISE CROMATOGRÁFICA E DA
DETECÇÃO DE AGENTES ANABÓLICOS 72
4.1.1. Padronização interna e controle da qualidade do método 75
4.2. IMPLEMENTAÇÃO DE AGENTES ANABÓLICOS A PARTIR DE URINAS DE
EXCREÇÃO 76
4.2.1. Metilnortestosterona 76
4.2.2. Metasterona 81
4.2.3. Hiperdrol 86
4.3. IMPLEMENTAÇÃO DE AGENTES ANABÓLICOS E ESTUDO DA CINÉTICA DA
HIDRÓLISE 89
4.3.1. Clomifeno 90
4.3.2. Tamoxifeno 93
4.3.3. Ciclofenil 97
4.3.4. Cinética da hidrólise 99
4.4. IMPLEMENTAÇÃO DE AGENTES ANABÓLICOS A PARTIR DE PADRÕES DE
REFERÂNCIA 100
4.4.1. Validação qualitativa do método analítico 100
4.4.1.1. alfa-Zeranol 102
4.4.1.2. beta-Zeranol 105
4.4.1.3. Tibolona 109
4.4.1.4. Letrozol 111
4.4.1.5. Formestano 115
4.4.2. Validação semi-quantitativa do método analítico 118
4.4.2.1. Determinação do método de análise por CGAR-ITD – VARIAN 4000 121
4.4.3. Validação quantitativa do método analítico 126
4.5. DERIVADOS N-FENILPIPERAZINAS 134
4.5.1. Caracterização química dos derivados N-fenilpiperazinas 134
4.5.2. Desenvolvimento de metodologia para análise do composto LASSBio 581 134

xiv
Capítulo 5. CONCLUSÃO 136 Capítulo 6. BIBLIOGRAFIA 137
ANEXOS 145

xv
ÍNDICE DE FIGURAS Página Figura 1: Metabolismo no anel A dos EAA: redução 5α e 5β de esteróides 3-ceto-4-eno. 16 Figura 2: Metabolismo no anel A dos EAA: redução do grupo 3-ceto-4-eno. 16 Figura 3: Metabolismo no anel B dos EAA: 6β-hidroxilação. 17 Figura 4: Metabolismo no anel D dos EAA: oxidação do grupo hidroxila em 17β. 17 Figura 5: Metabolismo no anel D dos EAA: hidroxilação em C16 e formação de metabólitos 16-ceto. 18 Figura 6: Estrutura química da testosterona (Androst-4-en-17β-ol-3-ona) e estrutura química geral dos esteróides. 19 Figura 7: Estrutura de diferentes esteróides anabólicos. 21 Figura 8: Principais metabólitos do estanozolol em humanos. 22 Figura 9: Principais metabólitos da metandienona em humanos. 24 Figura 10: Principais metabólitos da nandrolona em humanos. 25 Figura 11: Principais metabólitos da metiltestosterona em humanos. 26 Figura 12: Estrutura química da metilnortestosterona. 26 Figura 13: Estrutura química da metasterona. 27 Figura 14: Estrutura química do clembuterol. 28 Figura 15: Principais metabólitos da tibolona em humanos. 29 Figura 16: Principais metabólitos do zeranol em humanos. 30 Figura 17: Estrutura química de agentes com atividade anti-estrogênica. 32 Figura 18: Principais metabólitos do tamoxifeno em humanos. 33 Figura 19: Principal metabólito do clomifeno em humanos. 34 Figura 20: Principal metabólito do ciclofenil em humanos. 35 Figura 21: Principais metabólitos do letrozol em humanos. 35 Figura 22: Estrutura química do formestano. 36 Figura 23: Estrutura química do hiperdrol. 36 Figura 24: Novas famílias de compostos heterocíclicos. 41 Figura 25: Estrutura química da clozapina. 42 Figura 26: Proposta do metabolismo de MeOPP em rato wistar. 43 Figura 27: Fluxograma do método de extração de agentes anabólicos em urina humana. 53 Figura 28: Fluxograma do método de extração de LASSBio 581 em plasma de rato wistar. 55

xvi Figura 29: Gaiola metabólica. 58 Figura 30: Diagrama de causa-efeito (espinha de peixe). 70 Figura 31: Fluxograma das condições de derivatização. 73 Figura 32: Análise da metiltestosterona (PI) por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) espectro de massas da metiltestosterona-bis-TMS. 74 Figura 33: Reação de derivatização da metiltestosterona. 75 Figura 34: Controle de derivatização. (A) Amostra de urina não derivatizadas, (B) Amostra de urina derivatizada. 76 Figura 35: Possível metabólito da metilnortestosterona. 77 Figura 36: Proposta de possíveis metabólitos da metilnortestosterona. 77 Figura 37: (A) cromatograma de íons totais, (B) espectro de massas do metabólito da metilnortestosterona-bis-TMS. 79 Figura 38: Análise do fragmentograma da metilnortestosterona e dos dez brancos de urina avaliados. 81 Figura 39: Possíveis metabólitos da metasterona. 81 Figura 40: Análise da metasterona e seu metabólito por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) e (B1) espectro de massas da metasterona-bis-TMS e Metasterona-met.-bis-TMS. 83 Figura 41: Análise do fragmentograma da metasterona e dos dez brancos de urina avaliados. 86 Figura 42: Análise do hiperdrol por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) espectro de massas do hiperdrol-bis-TMS. 87 Figura 43: Análise do fragmentograma do hiperdrol e dos dez brancos de urina avaliados para cada substância. 88 Figura 44: Proposta de possíveis metabólitos do clomifeno. 90 Figura 45: Análise do clomifeno por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) espectro de massas do hidroxi-clomifeno-mono-OTMS. 91 Figura 46: Análise do fragmentograma do hidroxiclomifeno e dos dez brancos de urina avaliados para cada substância. 93 Figura 47: Análise do tamoxifeno por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B1) espectro de massas do Metoxi-hidroxi- tamoxifeno-mono-OTMS (possível metabólito), (B2) espectro de massas do carboxi-tamoxifeno-mono-OTMS (possível metabólito). 94 Figura 48: Análise do fragmentograma dos metabólitos do tamoxifeno e dos dez brancos de urina avaliados. 97 Figura 49: Análise de ciclofenila por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) espectro de massas de ciclofenila-mono-OTMS. 98 Figura 50: Análise do fragmentograma do metabólito de ciclofenila e dos dez brancos de urina avaliados. 99 Figura 51: Análise do alfa-zeranol por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) espectro de massas do alfa-zeranol-tris-TMS. 105

xvii Figura 52: Análise do beta-zeranol por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) espectro de massas do beta-zeranol-tris-TMS. 108 Figura 53: Análise de hidroxi-tibolona por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) espectro de massas de hidroxi-tibolona. 111 Figura 54: Análise do letrozol por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) espectro de massas de letrozol-bis-TMS. 114 Figura 55: Análise do formestano por CGAR-EM (varredura linear). (A) cromatograma de íons totais, (B) espectro de massas de formestano-mono-TMS. 117 Figura 56: Cromatograma de íons (A) Clembuterol, (B) Norandrosterona, (C) Epimetendiol, (D) 3α,5β-Metiltestosterona, (E) Metiltestosterona (PI) e (F) 3’OH-estanozolol. 125 Figura 57: Curva de calibração de 3’OH-estanozolol. 130 Figura 58: Curva de calibração de 3’OH-estanozolol. 130 Figura 59: Cromatograma de íons totais de 3’OH-estanozolol na concentração de 0,2 ng/mL. 132

xviii
ÍNDICE DE TABELAS Página Tabela 1: Histórico da cromatografia gasosa de alta resolução. 1
Tabela 2: Histórico da espectrometria de massas. 4
Tabela 3: Histórico do uso de agentes anabólicos no esporte. 12
Tabela 4: Medicamentos utilizados para estudos de excreção. 47
Tabela 5: Composição da solução de padrão interno. 50
Tabela 6: Protocolo para a validação do método. 66
Tabela 7: Cinética da hidrólise após adição de enzima. 100
Tabela 8: Valores de G crítico para identificação de 1 valor aberrante pelo teste de Grubbs
em diferentes níveis de confiança. 102
Tabela 9: Resultados da recuperação para o alfa-zeranol. 103
Tabela 10: Valores para determinação do limite de detecção e repetitividade. 104
Tabela 11: Resultados da recuperação para o beta-zeranol. 106
Tabela 12: Valores para determinação do limite de detecção e repetitividade. 107
Tabela 13: Resultados da recuperação para o 3α-hidroxi-tibolona. 109
Tabela 14: Valores para determinação do limite de detecção e repetitividade. 110
Tabela 15: Resultados da recuperação para o letrozol. 112
Tabela 16: Valores para determinação do limite de detecção e repetitividade. 113
Tabela 17: Resultados da recuperação para formestano. 115
Tabela 18: Valores para determinação do limite de detecção e repetitividade. 116
Tabela 19: Valores estimados de LD e LQ. 118
Tabela 20: Valores para determinação do limite de detecção e repetitividade. 119
Tabela 21: Valores para determinação do limite de detecção e repetitividade. 119
Tabela 22: Valores para determinação do limite de detecção e repetitividade. 120
Tabela 23: Valores para determinação do limite de detecção e repetitividade. 120
Tabela 24: Valores para determinação do limite de detecção e repetitividade. 120
Tabela 25: Parâmetros de CG-EM-EM utilizados para fragmentação do íon pai. 122
Tabela 26: Tempo de retenção e fragmentos gerados a partir do EM-EM. 122
Tabela 27: Valores para o teste de Grubbs. 127
Tabela 28: Valores de C crítico para nível de confiança de 0,05. 128
Tabela 29: Valores para o teste de Cochran. 129
Tabela 30: Precisão e exatidão inter-ensaio do 3’OH-estanozolol para 1ª curva. 131
Tabela 31: Precisão e exatidão inter-ensaio do 3’OH-estanozolol para 2ª curva. 131
Tabela 32: Valores de recuperação do composto LASSBio 581. 133

xix
ABREVIAÇÕES E SÍMBOLOS 11-OHA 11β-hidroxiandrosterona
11-OHE 11β-hidroxietiocolanolona
a Inclinação da reta
Adiol 5α-androstano-3α,17β-diol
b Intercepto com o eixo y
Bdiol 5β-androstano-3α,17β-diol
BSTFA N,O-bis-Trimetilsililtrifluoracetamida
CG Cromatografia Gasosa
CGAR Cromatografia Gasosa de Alta Resolução
CGARAT Cromatografia Gasosa de Alta Resolução e Alta Temperatura
CGAR-EM Cromatografia Gasosa de Alta Resolução acoplada a
Espectrometria de Massas
CGAR-EM-IE Cromatografia Gasosa de Alta Resolução acoplada a
Espectrometria de Massas por impacto de elétrons
CGAR-EM-IQ Cromatografia Gasosa de Alta Resolução acoplada a
Espectrometria de Massas por ionização química
CLAE Cromatografia Líquida de Alta Eficiência
CLAE-EM Cromatografia Líquida de Alta Eficiência acoplada a
Espectrometria de Massas
CLAE-EM-EM Cromatografia Líquida de Alta Eficiência acoplada a
Espectrometria de Massas acoplada a Espectrometria de
Massas
COI Comitê Olímpico Internacional
CV% Coeficiente de variação

xx
Da Daltons, unidade de massa atômica
DIC Dissociação induzida por colisão
DQO / IQ / UFRJ Departamento de Química Orgânica do Instituto de Química da
Universidade Federal do Rio de Janeiro
EC Energia de colisão
EFS Extração por fase sólida
ELL Extração líquido-líquido
EM Espectrometria de Massas
EMAR Espectrometria de massas de alta resolução
EMn Espectrometria de massas em tandem; Espectrometria de
massas acoplada a espectrometria de massas
Epit Epitestosterona
EUA Estados Unidos da América
FA Fortificado antes
FD Fortificado depois
FDA Food and Drug Administration-USA, Administração para Drogas
e Alimentos dos EEUU
IE Ionização por impacto de elétrons
IQ Ionização química
INMETRO Instituto Nacional de Metrologia, Normalização e Qualidade
Industrial
IP Identification Point, Ponto de identificação
IQ / UFRJ Instituto de Química da Universidade Federal do Rio de Janeiro
LIQ Limite inferior de quantificação
LD Limite de detecção
LMR Limite máximo de resíduo permitido

xxi
LMDR Limite mínimo de desempenho requerido
M Molar
m/z Razão massa / carga
MAPA Ministério da Agricultura, Pecuária e Abastecimento
MeOH Metanol
MRM Multiple reaction monitoring, monitoramento de reações
múltiplas
MSTFA N-metil-N-trimetilsililtrifluoracetamida
OMS Organização Mundial de Saúde
PI Padrão interno
ppb Parte por bilhão
ppt Parte por trilhão
RBC Rede Brasileira de Calibração
r Coeficiente de correlação
r2 Coeficiente de determinação
RSD Desvio padrão relativo
rpm Rotação por minuto
S / R Razão sinal / ruído
SIM Single reaction monitoring, monitoramento de reação única
t1/2 Tempo de meia-vida
tmax Tempo em que Cmax é alcançada
WADA World Anti-doping Agency, Agência Mundial Antidopagem,
AMA
WHO World Health Organization, Organização Mundial da Saúde,
OMS

xxii
ESTRUTURAS DOS COMPOSTOS CITADOS
H
H Estrano (3)
H
H
H Gonano (2)
OH
O
12
34
56
7
89
10
11 12 13
14 1516
17
18
19
A B
C D
Testosterona (1)
H
H
H
Androstano (4) Pregnano (5) Colestano (6)
Clostebol (8)
O
OH
Cl H
OH
O
H3C
O
O
OH
H
Oxandrolona (12)
OH
O
Nandrolona (11)
Drostanolona (9)
OHCH3
CH3O
Bolasterona (7)
OH
OH
CH3
Metenolona (10)

xxiii
O
OH
OHN
N
CH3
CH3OH
CH3
H
H
HO
3’OH-estanozolol (15)
H
OHCH3
NNH
Estanozolol (14) Oximesterona (13)
4β-OH-estanozolol (16) 3’OH-17-epiestanozolol (17) 16α-OH-estanozolol (18)
NN
CH3
CH3OH
CH3
H
H
OH
NN
CH3
CH3OH
CH3
H
H
OH
NN
CH3
CH3CH3
OH
H
H
HO
16β-OH-estanozolol (19)
NN
CH3
CH3OH
CH3
H
H
OH
OCH3
O
SO3H
Metandienona 17β-sulfato (21)Metandienona (20)
OHCH3
O
CH3OH
O
O
CH3
CH3OH
CH3
OH
18-nor-17,17-dimetilandrosta-1,4,13-
trien-3-ona (22) 17-Epimetandienona (23) 17β-hidroxi-17α-metil-5β-androst-1-eno-3-ona (24)

xxiv
17α-metil-5β-androst-1-eno-3α,17β-diol (25) Epimetendiol (26) 18-nor-17,17-dimetil-5β-
androstano-1,13-dien-3α-ol (27)
17α-metil-5β-androstano-3α,17β-diol (28)
OHCH3
HOH
CH3OH
HOH
CH3CH3
HOH
OHCH3
HOH
HHO
O
H
O
HO
3β-hidroxi-5α-estran-17-ona (30) Norandrosterona (29)
Noretiocolanolona (31)
HHO
O
Metiltestosterona (32)
OHCH3
HHO
OHCH3
O
17α-metil-5α-androstano-3α,17β-diol (33)
OHCH3
HOH
H3C
O
CH3
CH3OH
CH3
H
Metasterona (36)
OH
CH3
O
7α-metil-5β-androstano-3α,17β-diol (34) Metilnortestosterona (35)

xxv
N
OH
Cl
H2N
Cl
H
Clembuterol (37) Tibolona (38) 3α-OH-tibolona (39)
7α-metil-noretisterona (40) 3β-OH-tibolona (41)
O
OH OH
HO
OH
HO
O
O
OOH
HO
Zearalanona (42)
O
OH
O
N
H3C
α-Zeranol (43)
O
OOH
HO OH
O
OOH
HO OH
β-Zeranol (44) Tamoxifeno (45)
Cl
O
NCH3
CH3
Clomifeno (46)
NN
N
CN
CN
Letrozol (47)
O
O
OH
CH3 H
HH
CH3
Formestano (48)

xxvi
S
O
N
O
OH
HO
N
NH2
O
O
CH3
HN
N
N
CH3
CN
CH3
H3C CH3
NC
Aminoglutetimida (49) Anastrozol (50) Raloxifeno (51)
O
N
H3C
O
Toremifeno (53)
O
N
CH3
CH3
ClO
O
CH2
Examestano (52)
Tamoxifeno-N-óxido (54)
N-demetiltamoxifeno (55) 4-OH-Tamoxifeno (56) N-dedimetiltamoxifeno (57)
O
NH
H
H3C
O
N
H3C
OH
O
N
H
H3C

xxvii
4-OH-clomifeno (59)
Cl
O
NCH3
CH3
HO Ciclofenil (60)
O
N
H
H3C
OH
4-hidroxi-N-demetiltamoxifeno (58)
CN
CN
HO
OH
OH
Hidroxi-bis-desacetil-ciclofenil (61)
NNH
N
Triazol (62) Bis-4-cianofenil-metanol (63)
N
Hiperdrol (65)
O
Br
O
N
N
N
NN
H
N
N
CH3
N
N
H
Cl
Clozapina (68)
LASSBio 580 (66)Formestano (64)
O
O
OH
CH3 H
HH
CH3
N
N
N
N
ClLASSBio 581 (67)

xxviii
1-(4-metoxifenil)piperazina (69) 1-(4-hidroxifenil)piperazina (70) N-(4-metoxifenil)etilenodiamina (71)
4-metoxianilina (72) 4-hidroxianilina (73)
N
NHO
N
NHHO
N
O
NH2
NH2
O
NH2
HO
N
HO
O
N-acetil-4-hidroxianilina (74)

CAPÍTULO 1
INTRODUÇÃO

Introdução -1-
1. INTRODUÇÃO 1.1. CROMATOGRAFIA GASOSA DE ALTA RESOLUÇÃO
A cromatografia em coluna foi inicialmente desenvolvida pelo químico de petróleo D.
T. Day em 1900. M. S. Tswett, o botânico polonês, usou em 1906, as colunas de adsorção
nas suas investigações de pigmentos de plantas. Ele criou o termo cromatografia, não se
sabe se por ter separado pigmentos de colorações diferentes ou porque Tswett significa
“cor” em russo. Até 1930, esse método não tinha sido extensivamente estudado pelos
químicos. No início da década de 50, James e Martin introduziram a cromatografia gasosa
(Aquino Neto & Souza Nunes, 2003) (Tabela 1).
Tabela 1: Histórico da cromatografia gasosa de alta resolução*. Ano Fato relevante
1951 Desenvolvimento da cromatografia gás-sólido.
1952 Desenvolvimento da cromatografia gás-líquido.
1956 1° Simpósio de cromatografia gasosa da American Chemical Society.
1957 Desenvolvimento das colunas capilares de metal.
1959 Coluna capilar de vidro, o desenvolvimento tinha o objetivo de reduzir o custo de obtenção
do capilar; mas Desty abandonou os esforços após dois anos de tentativas de recobrir o
vidro com a fase estacionária.
1961 K. Grob reinicia na Suíça os esforços de Destsy.
1978 É dominada a confecção de colunas capilares (inertes), além das técnicas de introdução de
amostras, principalmente devido aos esforços da família Grob.
1979 Surgimento das colunas capilares de sílica fundida.
1983/84 Imobilização de fases estacionárias com OH terminal.
1985 Sistematização da confecção de colunas capilares de alta resolução e alta temperatura.
* Tabela retirada na íntegra de Pereira & Aquino Neto, 2000.
Dentre os modernos métodos de análise, a cromatografia ocupa, sem dúvida, um
lugar de merecido destaque no que concerne à separação, identificação e quantificação de
espécies químicas.
Cromatografia é um método físico de separação, no qual os componentes a serem
separados são distribuídos entre duas fases: uma fase fixa de grande área superficial
denominada fase estacionária, e a outra um fluido que percola através dela sendo, por isso,
denominada fase móvel. Portanto, a cromatografia gasosa é um processo utilizado para
separar uma amostra em seus componentes individuais.

Introdução - 2 -
A separação entre dois, ou mais, componentes, resultará da diferença de suas
constantes de equilíbrio de distribuição (KD) entre as duas fases. Simplificando, quando mais
tenazmente um componente é preso pela fase estacionária, mais alta é a porcentagem das
moléculas daquele componente que nela ficam retidas provocando um retardamento na
migração da mesma. Um segundo componente menos retido na fase estacionária terá uma
porcentagem mais alta de moléculas na fase móvel em relação ao primeiro componente.
Assim na média, o conjunto das moléculas do componente que está menos fortemente
preso se moverá através do sistema (na direção do fluxo) a uma velocidade mais alta que o
outro, resultando na migração dos componentes em regiões separadas (bandas) da fase
estacionária. Essa migração dos analitos através do leito de fase estacionária, pela
passagem da fase móvel, recebe o nome de eluição (LANÇAS & McNAIR, 1983).
Devido a grande diversidade de fases líquidas disponíveis, a cromatografia gás-
líquido (fase estácionária é um filme delgado líquido, o qual recobre um suporte sólido inerte)
torna-se a mais versátil e seletiva forma de cromatografia em fase gasosa. Isto permite que
sejam analisadas amostras sólidas, líquidas ou gasosas desde que sejam voláteis ou
possam ser volatilizadas sem sofrerem decomposição no cromatógrafo (AQUINO NETO &
CARDOSO, 1985).
Na CGAR a introdução de amostra é crucial para garantir a alta resolução. Como o
processo cromatográfico só tende a alargar picos, quanto menor for a largura inicial, melhor
será a resolução no final do cromatograma. A escolha apropriada das condições de injeção
(tais como o volume de amostra, temperatura do injetor e tipo de injeção) dependem em
larga escala, do estado físico da amostra. A grande maioria das amostras líquidas requer,
para sua rápida volatilização, que a temperatura do injetor esteja 20 a 30ºC acima da
temperatura de ebulição do componente menos volátil. O elevado coeficiente de expansão
dos líquidos, quando vaporizados, permite que sejam injetados pequenos volumes, o que
maximiza a resolução do sistema e confere uma forma ideal aos picos eluídos. A câmara de
vaporização deverá estar suficientemente quente para vaporizar rapidamente a amostra,
evitando perda da eficiência devido à injeção, mas de tal forma a evitar que haja
decomposição térmica ou rearranjos na amostra. A seleção da técnica de injeção mais
adequada para trabalho com colunas capilares é função exclusivamente da amostra, sua
natureza e concentração. São três as principais técnicas de introdução de amostras em
colunas capilares: injeção a quente em vaporizadores (com ou sem divisão de fluxo) e
injeção da amostra “a frio” diretamente no interior da coluna (CARDOSO & AQUINO NETO,
1989; AQUINO NETO & CARDOSO, 1992).

Introdução - 3 -
A separação efetiva dos componentes da amostra é efetuada na coluna
cromatográfica, onde a natureza do tubo, do suporte sólido, o tipo e a quantidade da fase
líquida, o método de recheio, o comprimento e a temperatura são fatores importantes para
se ter à resolução desejada (AQUINO NETO & CARDOSO, 1985).
Foram desenvolvidos muitos tipos de colunas tubulares abertas para a cromatografia
gasosa. O material dos tubos é geralmente sílica fundida, cobre, aço inoxidável, alumínio e
vidro borossilicato. O material usado como suporte inerte deve ter granulometria uniforme,
ter boas características operacionais (resistência suficiente para não quebrar durante a
operação) e ser capaz de constituir um leito uniforme na coluna. A área específica do
material deve ser elevada a fim de promover a distribuição pelicular da fase líquida e
assegurar o rápido equilíbrio entre as fases estacionária e móvel (AQUINO NETO &
CARDOSO, 1985).
Nas colunas capilares, a fase estacionária é depositada na forma de um filme fino e
uniforme na parede interna do tubo, deixando a parte central oca. São denominadas colunas
tubulares abertas. As características principais das colunas capilares atuais são: capilares
de vidro ou sílica fundida com diâmetro interno menor do que 0,3 mm, expessura de filme de
fase estacionária (df) menor do que 0,5 mm e desativados com agentes silanizantes ou
similares, que permitem a compatibilização do filme da fase estacionária com o suporte.
A CGAR, já é uma das técnicas mais populares e poderosas para separação de
misturas, podendo ser utilizada em diversos campos da ciência como por exemplo: na
química de polímeros, na geoquímica orgânica, na química de alimentos, de produtos
naturais, na arqueologia, no controle de qualidade de produtos industriais, na química
ambiental e medicinal, na toxicologia dentre outras. A grande versatilidade da técnica é
baseada intrinsecamente no grande poder de resolução e extrema inércia química das
colunas capilares, além da facilidade de utilização de vários sistemas de detecção e de
contar com sistemas de transferência de amostra não discriminatórios (PEREIRA & AQUINO
NETO, 2000; PEREIRA et. al., 2004).
1.2. ESPECTROMETRIA DE MASSAS
A espectrometria de massas teve início no final do século XIX. A espectrometria de
massas é uma técnica analítica poderosa utilizada para identificar compostos
desconhecidos, quantificar materiais conhecidos e elucidar as propriedades químicas e
estruturais de moléculas; pode ser realizada com quantidades bem pequenas (ao nível do
picograma) e a concentrações bem baixas em misturas quimicamente complexas (ng/L)

Introdução - 4 -
(James Barker, 1999). A tabela 2, apresenta um breve histórico do desenvolvimento da
espectrometria de massas.
Tabela 2: Histórico da Espectrometria de Massas. Ano Fato relevante 1886 Descobrimento de íons positivos por Eugene Goldstein.
1898 Wilhelm Wien analizou íons positivos por deflecção magnética (W. Wien em 1911 ganhou o
Prêmio Nobel de Física pela descoberta das leis de irradiação do calor – Lei de Wien.
1901 Walther Kaufmann analizou raios catódicos usando campos eletromagnéticos paralelos.
1911 Joseph John Thomson descobre duas massas diferentes do Neônio (20 e 22), um ano mais
tarde obteve o espectro de massas de O2, N2, CO, CO2 e COCl2. Para muitos cientistas que a
espectrometria de massas teve início de fato com os trabalhos de J. J. Thomson, que em 1906
ganhou o Prêmio Nobel de física.
1913 J. J. Thomson observa pela primeira vez a dissociação de íons moleculares.
1918 A. J. Dempster desenvolve o primeiro espectrômetro de massas com setor magnético com
formato de 180° em direção ao foco, servindo este setor magnético como analisador de
massas.
1919 Francis William Aston (estudante de Thomson) desenvolve o primeiro espectrômetro com
seleção de velocidade. Nesse mesmo ano, F. W. Aston confirma que os íons de Neônio
descobertos por Thomson eram realmente isótopos usando o primeiro espectrômetro de
massas (na época chamado espectrógrafo de massas). A confirmação dos dois isótopos é tida
como o primeiro experimento de relação isotópica de um gás por espectrometria de massas.
Em 1922, F. W. Aston ganha o Prêmio Nobel de física.
1930 R. Conrad aplica pela primeira vez a espectrometria de massas a química orgânica.
1940 A. O. Nier mostra que o U253 é fissionável (separa U235 e U238). Fato decisivo para o projeto
Manhattan, que envolveu o desenvolvimento de separação isotópica e a construção de um
reator nuclear.
1948 Cameron descobre a medida de tempo de vôo de íons como princípio de análise.
1952 As teorias de “quasi-equilibrium” (QET – sobre velocidade de reação em condições de
pressões infinitamente baixas) e RRKM (descreve quantitativamente as velocidades das
reações como função das energias internas das espécies envolvidas) formularam modelos
que explicam a fragmentação monomolecular de íons. O acrônimo RRKM vem das iniciais dos
nomes Rice, Ramsperger, Kassel e Marcus, sendo que este último em 1992 recebeu o Prêmio
Nobel.
1953 Wolfgang Paul e H. S. Steinwedel desenvolvem o analisador de massas do tipo quadrupolar,
que usa campo elétrico para focalizar e separar íons. Pouco tempo depois descreveram o “Íon
trap”, na época conhecido como quadrupolo tridimensional. Em 1989, W. Paul ganha o Prêmio
Nobel de física.
1956 McLafferty fez o primeiro acoplamento entre um cromatógrafo a gás e um espectrômetro de
massas.
1966 Munson e Field descobrem a ionização química.
1968 Primeiro CGEM comercial com colunas capilares. Equipamento rapidamente difundido na

Introdução - 5 -
maioria dos laboratórios de análise de compostos orgânicos no mundo inteiro.
1969 Malcom Dole ionização por ESI
1973 G. Cooks desenvolveu o Tandem MS/MS ou espectrometria de massas seqüencial de duplo
estágio, onde um íon precursor tem sua massa selecionada e é tipicamente fragmentado por
CID seguido por análise de massas dos produtos resultantes.
1978 Yost e Enke desenvolvem o primeiro triplo quadrupolo.
1979 Comisarow e Marshall adaptaram os métodos de transformada de Fourier à ICRMS e
construíram o FTMS.
1981 Barber desenvolve o FAB.
1983 Blakely desenvolve o Termospray.
1985 Hillenkamp e Karas desenvolvem a técnica de MALDI-MS.
1988 Fenn desenvolve o eletrospray ligado ao espectrômetro de massas.
O espectrômetro de massas é um instrumento analítico, através do qual os íons
produzidos a partir de uma amostra, são separados por campos elétricos e / ou magnéticos
de acordo com a razão massa-carga (m/z) dos constituintes da amostra. Todas as moléculas
ionizadas a princípio são passíveis de serem analisadas por espectrometria de massas,
tornando esta uma técnica universal para análise química.
Vários tipos de espectrômetros de massas tem sido descritos incluindo analisadores
por setor de massas (único ou duplo foco), analisadores tipo quadrupolo, analisadores por
aprisionamento de íons (Ion trap), analisadores por tempo de vôo, dentre outros. Quadrupolo
e aprisionamento ou armadilha de íons (Ion trap) são os tipos mais utilizados. A grande
maioria dos experimentos descritos neste trabalho foi realizada nesses dois tipos de
espectrômetros.
É possível atingir um elevado grau de certeza na identificação molecular pelo
acoplamento de sistemas cromatográficos ou afins, a diversos espectrômetros. As técnicas
combinadas principais são a cromatografia gasosa com a espectrometria por infravermelho
com transformada de Fourier e as cromatografias gasosa ou líquida e eletroforese capilar
acopladas com a espectrometria de massas. O efluente do sistema de separação é
transferido através de uma interface para a fonte de íons. A extração das espécies iônicas é
efetuada de acordo com o analisador de íons e em seguida ocorre a detecção pelo
“multiplicador de elétrons” (Aquino Neto & Souza Nunes, 2003).

Introdução - 6 -
1.2.1. Analisadores do tipo “armadilha de íons”- Ion trap
A armadilha de íons consiste de dois eletrodos idênticos (endcap), o eletrodo de
entrada e o eletrodo de saída, e o eletrodo intermediário (ring electrode). Os eletrodos são
isolados por espaçadores de quartzo, criando uma cavidade na qual os íons são
aprisionados. Os íons passam para dentro (ion injection) e para fora da armadilha (trap)
através de um orifício presente nos eletrodos endcaps. Na teoria os íons podem permanecer
indefinidamente dentro do trap através da oscilação do campo elétrico. Entretanto, para ser
detectado, o íon precisa ser varrido pelo sistema de detecção, ou seja, o íon precisa ser
ejetado. Para tanto aumenta-se a freqüência do eletrodo intermediário fazendo com que os
íons sejam ejetados do trap para o detector (VARIAN, 2004).
1.2.1.1. Modos de operação no Ion trap
O espectrômetro de massas do tipo aprisionamento (armadilha) de íons pode ser
operado nos diferentes modos:
Varredura linear (Full scan): Todos os íons são coletados e depois ejetados, resultando em
um espectro que apresenta cada massa que inicialmente entrou no trap.
Monitoramento de íons selecionados (MIS): Neste caso, os íons também são coletados, mas
durante o tempo em que os íons são mantidos no trap, voltagens são alteradas para isolar
um único íon ou janela de íons, assim somente o íon ou os íons de interesse são retidos. No
momento da ejeção, somente o(s) íon(s) isolado é então escaneado(s).
MS2: Neste modo, os íons são coletados e um único íon é isolado, assim como em MIS. Em
seguida, aplica-se uma voltagem para excitar e fragmentar o íon precursor em íons produto
que então serão escaneados. Uma característica importante do Ion trap é que esta técnica
permite múltiplos estágios de espectrometria de massas (MSn). Um íon precursor pode ser
fragmentado sequencialmente.
1.3. TÉCNICAS DE PREPARAÇÃO DE AMOSTRA
As técnicas mais comumente utilizadas na extração e purificação de fármacos em
matrizes biológicas, para a análise por cromatografia gasosa ou líquida acoplada a
detectores universais ou específicos, são a extração líquido-líquido (ELL) e a extração por
fase sólida (EFS). A escolha do solvente ideal para essas técnicas pode ser feita

Introdução - 7 -
empiricamente, com base em observações práticas, ou teoricamente, com base nas
propriedades termodinâmicas do solvente e do soluto. Entretanto, a avaliação teórica da
solubilidade de uma substância – especialmente quando está presente em níveis residuais
(5 ng/mL a 1 pg/mL) – pode ser pouco prática, ou até mesmo impossível, dada a
complexidade de certas matrizes. Desta forma, a escolha empírica do solvente de extração
tem sido bastante utilizada. O objetivo principal do pré-tratamento da amostra é viabilizar a
extração dos compostos de interesse presentes na matriz ou fluido biológico. Para isto,
estes fluidos biológicos são submetidos a vários procedimentos individuais ou combinações
de diferentes procedimentos de pré-tratamento.
1.3.1. Precipitação
A técnica de precipitação é usada para a remoção de proteínas por desnaturação das
mesmas, em amostras de plasma ou sangue total. A precipitação de proteínas ocorre pela
adição de ácidos (ácido tricloroacético, ácido perclórico, etc.), solventes orgânicos (acetona,
metanol, etanol, acetonitrila), dentre outros, seguido da filtração ou centrifugação.
Entretanto, essa técnica apresenta certas desvantagens: analitos fortemente ligados às
proteínas podem precipitar resultando em uma recuperação baixa e pouco reprodutível;
alguns interferentes não-protêicos podem permanecer na solução (McDOWALL, 1989).
1.3.2. Extração por fase sólida
A extração por fase sólida é uma técnica de isolamento amplamente empregada em
diversos campos, como no controle de qualidade de produtos farmacêuticos, no
monitoramento terapêutico de drogas, em estudos farmacocinéticos, na quantificação de
compostos endógenos para fins de diagnóstico de abuso de drogas, análise forense, entre
outros (KRISHNAN & IBRAHAM, 1994; MOORS et. al., 1994; HENNION, 2000).
Os princípios da EFS são semelhantes aos da ELL, envolvendo a partição dos
compostos entre duas fases. Na EFS a amostra para ser extraída é dividida entre a fase
sólida e a líquida (como se fosse entre dois líquidos imiscíveis na ELL). O material de
interesse da amostra terá maior ou menor afinidade pela fase sólida, sendo adsorvido.
Posteriormente é extraído por um outro solvente que tenha maior afinidade com o material
de interesse do que com a fase sólida; ou ainda, o material pode não ser retido, eluindo
imediatamente pela fase sólida, enquanto que a maioria dos interferentes ficam presos na
resina. Os mecanismos de retenção e eluição ocorrem através de forças intermoleculares

Introdução - 8 -
entre a amostra e a superfície das fases, envolvendo interações tipo Van der Waals,
interações tipo eletrostáticas, dipolo induzido - dipolo permanente, dipolo permanente -
dipolo permanente, íons – dipolo induzido, dipolo permanente – íons, íons – íons e
interações como ligação de hidrogênio.
Na extração por fase sólida, o adsorvente ou absorvente (fase sólida) é fixado num
cartucho de polietileno, entre dois discos compactando essa fase, na extremidade superior e
inferior. A fase líquida é eluída sob pressão ou vácuo. O processo consiste de 5 etapas: 1)
Ativação do adsorvente através da passagem de um solvente que condicione a superfície da
fase sólida. 2) Remoção do solvente responsável pela ativação. 3) Aplicação da amostra.
3.a) A substância de interesse e a matriz ficam retidas na fase sólida. 3.b) A substância de
interesse fica retida e parte da matriz passa pela fase sólida. 3.c) A substância de interesse
passa pela fase sólida e a matriz fica retida. Nesse caso, a fração de interesse é
imediatamente coletada. 4) Etapa de lavagem: 4.a) a fase sólida é lavada com um solvente
adequado retirando a matriz ou parte dela, sem eliminar os compostos de interesse. 4.b) A
substância de interesse continua na fase sólida enquanto ocorre a lavagem com um solvente
apropriado, eliminando o que restou da matriz ou parte dela. 5) Eluição da substância de
interesse do adsorvente, com um solvente apropriado, que será utilizado no próximo passo
da análise geral (HENNION, 2000).
As fases sólidas usadas em EFS são semelhantes às utilizadas nas colunas para
cromatografia líquida. De acordo com o grupo funcional preso à sílica ou ao polímero, a fase
resultante é classificada como polar, não polar ou de troca iônica.
O processo de EFS pode ser utilizado tanto “on-line” quanto “off-line”. Na
configuração em linha, a substância de interesse eluída do cartucho é introduzida no
cromatógrafo líquido através de uma válvula de injeção. O módulo do cartucho de extração é
inserido como parte do equipamento cromatográfico, conectado diretamente na linha que a
fase móvel percorre. A extração por fase sólida em linha é uma técnica de pré-tratamento
usada principalmente associada à cromatografia líquida de alta eficiência.
1.3.3. Extração líquido-líquido
A extração líquido-líquido (ELL) é uma técnica simples, rápida, de baixo custo e muito
útil para separar analitos de interferentes presentes na matriz através da partição da amostra
entre dois líquidos imiscíveis ou duas fases. A ELL tem sido o método tradicional para o
isolamento de fármacos de matrizes biológicas, principalmente para métodos que utilizam a
cromatografia gasosa como técnica analítica.

Introdução - 9 -
Na ELL, uma das fases é geralmente aquosa e a segunda fase constituída por um
solvente orgânico. Analitos extraídos na fase orgânica são facilmente recuperados pela
evaporação do solvente.
Uma vez que a extração é um processo de equilíbrio com eficiência limitada,
quantidades significativas do analito podem permanecer em ambas as fases. O equilíbrio
químico envolve mudanças no pH, no pareamento iônico, na concentração, e pode ser
usado para aumentar a recuperação do analito e / ou a eliminação de interferentes. Um bom
solvente para ELL, deve ter as seguintes características: baixa solubilidade em água (inferior
a 10%), volatilidade que permita fácil remoção e concentração após a extração, polaridade e
propriedades que permitam a formação de ligações de hidrogênio a fim de aumentar a
recuperação do analito na fase orgânica e alta pureza para minimizar a contaminação da
amostra (CARDOSO, 2002).
Qualquer espécie será distribuída entre dois solventes imiscíveis, sendo a razão entre
a concentração do analito entre as duas fases uma constante. O processo pode ser
traduzido pela equação: KD = Co / Caq, onde KD é a constante de distribuição, Co a
concentração do analito na fase orgânica e Caq a concentração do analito na fase aquosa.
Os solventes orgânicos ou mistura de, mais utilizados são terc-butilmetiléter, acetato
de etila, acetona:clorofórmio (1:1), tolueno, diclorometano:acetona (2:1), tolueno:acetato de
etila (4:1), diclorometano:isopropanol:acetato de etila (1:1:3),
clorofórmio:isopropanol:heptano (60:14:26), clorofómio, acetato de butila e éter etílico
(DRUMMER, 1999). Existem algumas diferenças em relação ao poder extrativo de cada um
desses solventes ou mistura de solventes, entretanto é preciso enfatizar que a escolha por
sistemas mais polares freqüentemente gera grande ruído de fundo (“background”) na
cromatografia.
1.3.4. Derivatização
O desenvolvimento, nos últimos anos, de espectrômetros de massas de baixo custo,
promoveu um aumento significativo na aplicação de sistemas de CG-EM na análise de
substâncias ilícitas para uso em humanos. Drogas e / ou xenobióticos são metabolizados in
vivo através de um caminho que os torne mais polares. Dessa forma a derivatização desses
metabólitos é praticamente inevitável para viabilizar sua análise por CG-EM (SEGURA et.
al., 1998).
Avanços nas técnicas de derivatização empregadas em testes para o controle de
dopagem no esporte, tem sido relevantes particularmente para os agentes anabólicos,

Introdução - 10 -
diuréticos e glicocorticóides. Nesse sentido, têm sido aplicados métodos como a metilação,
alquilação e acilação, de forma que a volatilidade e estabilidade térmica sejam garantidas
para análise por CG-EM (PEREIRA, 2004).
1.4. CONTROLE DE DOPAGEM NO ESPORTE – UM BREVE HISTÓRICO
Durante toda a história, atletas têm procurado alimentos e poções para aumentar o
desempenho físico. Os gladiadores romanos faziam uso de uma mistura de estimulantes
com álcool para superar a fadiga e injúrias, os guerreiros escandinavos comiam cogumelos
halucinógenos para se prepararem para as batalhas (DELBEKE, 2000). Os primeiros atletas
acusados de doping, foram nadadores em uma competição em Amsterdam no ano de 1860.
No final do século XIX, ciclistas europeus estavam usando substâncias como a cafeína
revestida por açúcar, na forma de cubos, com o objetivo de reduzir a dor e retardar a fadiga,
infelizmente isto ocorreu quarenta anos antes do primeiro teste para controle de dopagem
ser introduzido (VERROKEN, 2000). Este teste pode ser considerado simples, tratava-se de
CGAR acoplada a detector de nitrogênio e fósforo e imunoensaios, se comparado com as
técnicas disponíveis atualmente. Dessa forma, atletas não tinham medo de serem pegos no
exame antidopagem o que fez com que o uso de métodos e substâncias proibidas com o
intuito de aumentar a performance durante uma competição fosse difundido mundialmente.
Pouco tempo depois da segunda guerra mundial, estimulantes e drogas contra a
fadiga, desenvolvidos para o exército soviético, se popularizaram, resultando em vários
casos fatais. Uma das vítimas mais conhecidas desse período foi o ciclista inglês Tom
Simpson que morreu em 1967 em função do uso da combinação de anfetamina, álcool e
diurético. Tom Simpsom foi a primeira vítima fatal do uso excessivo de doping na história do
Tour de France. O abuso exagerado de estimulantes na década de 60, com a
disponibilidade das anfetaminas sintéticas, preparadas durante a segunda grande guerra,
levou ao estabelecimento, pelo Comitê Olímpico Internacional (COI), do controle de
dopagem em 1967. Em 1968 atletas foram testados pela primeira vez, por ocasião dos jogos
olímpicos na cidade do México (AQUINO NETO, 2001). Embora nesse primeiro teste o foco
principal fossem os estimulantes, a detecção de outros compostos também se fez
necessária. Nos anos de 1950 e 1960, em função do avanço da química orgânica, uma
grande variedade de compostos farmacologicamente ativos, incluindo diuréticos, beta-
bloqueadores, corticosteróides e esteróides anabólicos foram sintetizados. Estes compostos
foram extensivamente utilizados por atletas durante os anos 70. No final dos anos 70 e
principalmente durante os anos 80 a biotecnologia avançou significativamente. A reação de

Introdução - 11 -
polimerização em cadeia (PCR) e modificações genéticas in vitro, permitiram a produção em
quantidade razoável de proteínas para uso na medicina, dentre elas o hormônio do
crescimento (GH), insulina e seus derivados, hormônio adrenocorticotrópico (ACTH) e
eritopoetina, também conhecida como EPO (LASNE et. al., 2002). Essas proteínas,
utilizadas na prática médica para auxiliar a terapia de doenças diversas, passaram a ser
também empregadas na dopagem. Em relação ao GH, sua detecção ainda está sob
investigação. A EPO já está sendo monitorada desde os jogos olímpicos de Sydney em
2000. Em função do sucesso na batalha contra o doping, aumentando o risco para os atletas
serem flagrados, estes passam a tentar “velhas” formas para se doparem, e.g. a transfusão
sangüínea usada por ciclistas na volta ciclística da França de 2004.
Outro caminho para tentar burlar o exame foi a utilização de esteróides projetados,
que nada mais são que esteróides anabólicos quimicamente modificados, que foram
desenvolvidos entre os anos de 1960 e 1970 (RIVIER, 2002). Estas substâncias não eram
detectadas pelos laboratórios responsáveis pelo controle de dopagem até 2003, quando o
esteróide projetado tetrahidrogestrinona (THG) foi encontrado através de informação
anônima (MARQUES et. al., 2007, CATLIN et. al., 2002, CATLIN et. al. 2004, SEKERA et.
al., 2005).
No passado recente cientistas obtiveram sucesso com a terapia genética para
tratamento de doenças ligadas ao enfraquecimento muscular. Dessa forma, a terapia
genética pode também ser usada por atletas na tentativa de aumentar a força muscular em
músculos específicos. Embora nenhuma aplicação no controle de dopagem tenha sido
avaliada até hoje, a agência mundial anti-dopagem (AMA), incluiu o doping genético na lista
de métodos proibidos desde janeiro de 2003.
É difícil prever quais novas substâncias serão utilizadas no futuro com o intuito de
dopagem. O abuso das mesmas ou de métodos com esse intuito é alertado pelas
autoridades do controle de dopagem, através de rumores ou informações anônimas. No
entanto as autoridades do controle de dopagem estão cuidadosamente observando e
avaliando os avanços científicos a fim de identificar o que pode vir a ser usado com a
finalidade de dopagem no futuro.
1.5. AGENTES ANABÓLICOS NO CONTEXTO DO CONTROLE DE DOPAGEM NO
ESPORTE
Com o intuito de aumentar seu desempenho físico, atletas são encorajados a fazer uso
de substâncias químicas sintéticas ou meios artificiais proibidos no esporte. O controle de

Introdução - 12 -
dopagem mundial está sob a supervisão da Agência Mundial Anti-dopagem (AMA), a qual
mantém o código mundial anti-dopagem que inclui uma lista de substâncias e métodos não
lícitos à prática do esporte. O abuso de drogas é controlado pela análise em laboratórios
acreditados pela AMA, amostras de urina ou sangue dos atletas são coletadas antes ou
durante uma competição.
Esteróides anabólicos androgênicos (EAA) são um grupo de substâncias naturais e
sintéticas que são quimicamente similar e mimetizam a ação da testosterona endógena. EAA
têm sido usados por uma grande variedade de atletas por mais de cinqüenta anos, com o
objetivo de aumentar sua capacidade de treinamento, resistência e desempenho. O uso de
EAA por atletas foi proibido desde meados dos anos 70, mas ainda são a principal classe de
substâncias utilizada no esporte (MARCOS, et. al., 2002). Já nos anos 20, amostras de tecido
de testículos de macaco eram enxertados em atletas e, em conseqüência da “organoterapia”
no final dos anos 80 difundiu-se o consumo de urina de mulheres grávidas como fonte de
anabolizantes (CATLIN & HATTON, 1991). A tabela 3 apresenta um breve histórico do uso de
agentes anabólicos no esporte.
Tabela 3: Histórico do uso de agentes anabólicos no esporte. Ano Fato relevante 1953 Anabolizantes sintéticos entram no mercado.
1964 Olimpíadas de Tóquio apresentaram atletas com musculatura surpreendente, lançando a suspeita
de abuso de anabolizantes.
1976 Nadadoras alemães nitidamente “fabricadas” por doping, nas olimpíadas de Montreal.
1980 Novamente as nadadoras alemães se destacaram.
1984 Martti Vainio (atletismo) é flagrado pelo uso de metenolona.
1986 Salviano Domingues (atletismo) é flagrado pelo uso de nandrolona.
1988 Ben Johnson é flagrado pelo uso de estanozolol, um anabolizante sintético de última geração.
Florence Griffith-Joyner, nitidamente moldada por anabolizantes, não é flagrada.
1988 Andor Szanyl (Levantamento de peso) é flagrado pelo uso de estanozolol.
Anos 90 Internet banaliza o acesso e uso de anabolizantes e “complementos nutricionais”.
1992 Berenice Pereira (atletismo) é flagrada pelo uso de nandrolona.
1994 Sueli Pereira dos Santos (lançamento de dardo) é flagrada pelo uso de nandrolona.
1994 Maureen Maggi (salto triplo) é flagrada pelo uso de Clostebol.
1996 Iva Prandzheva (salto triplo) é flagrada pelo uso de metandienona.
A detecção de EAA no contexto do controle de dopagem no esporte é historicamente
realizada por Cromatografia Gasosa de Alta Resolução acoplada a Espectrometria de Massas
(quadrupolo único). Esta análise tem a urina como fluido biológico de escolha e exige a

Introdução - 13 -
detecção de grande número de esteróides diferentes e seus metabólitos em baixa
concentração (10 ng/mL - 2 ng/mL).
1.5.1. Mecanismo de ação e uso clínico
EAA são derivados sintéticos da testosterona, hormônio natural masculino responsável
pelos efeitos anabólicos e androgênicos observados nos homens durante a adolescência e a
vida adulta. Nos homens a testosterona é sintetizada nas células de Leyding e testículos,
enquanto nas mulheres está presente em pequenas quantidades sendo sintetizada nos
ovários e glândula adrenal (SCHANZER, 1996).
A principal importância fisiológica da testosterona se dá a partir da sua ligação com
receptores citoplasmáticos, promovendo assim a transcrição do gene e a tradução em
proteína. O principal alvo para ação anabólica da testosterona são o músculo esquelético, o
tecido muscular do osso e a hematopoiese. Além disso, alguns efeitos anabólicos são
mediados também indiretamente: EAA são antagonistas de glicocorticóides endógenos e
inibem processos protéicos catabólicos. Também podem estimular a formação do hormônio
de crescimento e do fator de crescimento semelhante a insulina.
EAA tem muitas indicações clínicas. Por muito tempo foram usados por homens para o
tratamento de hipergonadotrofia. Outras terapias onde são utilizados incluem a contracepção
e o tratamento de doenças crônicas como a doença pulmonar obstrutiva e a infecção por HIV.
EAA tem sido usados para promover a deposição muscular depois de queimaduras, cirurgia e
terapia com radiação. Eles também podem ser usados no tratamento da osteoporose,
doenças hepáticas, na cura de ferimentos, anemia e algumas desordens psiquiátricas. As vias
de administração de EAA são: oral, intramuscular, nasal, transdérmica ou ainda absorvido
pela boca.
1.5.2. Abuso de esteróides
EAA têm sido usados por décadas pelos atletas. Entretanto o abuso não é limitado a
atletas profissionais, uma grande parte da população que freqüenta academias de ginástica
também utiliza EAA. A incidência do abuso por atletas tem sido estudada intensivamente
durante competições ou fora de competição. Os resultados encontrados dependem do
esporte, do gênero e idade dos atletas. De acordo com estatísticas oficiais da AMA, em 2004
cerca de 0,7% das 170.000 amostras apresentaram resultado analítico adverso para agentes

Introdução - 14 -
anabólicos, onde em 86% dos casos os esteróides utilizados foram: testosterona, estanazolol
e metandienona (AYOTTE, 1996, WADA, 2007).
Estudos recentes mostram que doses supraterapêuticas de EAA combinadas com
treinamento intenso, têm um efeito saudável no homem. O mecanismo de ação ainda não foi
totalmente esclarecido, mas postula-se ser mediado indiretamente. Altas doses de EAA
podem também aumentar o número de receptores androgênios nas células. Uma parte dessa
ação pode ser psicológica uma vez que com o abuso de esteróides atletas sentem-se com
mais energia e assim treinam mais intensamente.
O abuso de EAA difere significativamente de seu uso clínico. Essas drogas vêm
freqüentemente do mercado negro e algumas são somente para uso na medicina veterinária.
A dose total utilizada é geralmente supraterapêutica e são tomadas em ciclos de um até três
meses com completa abstinência da medicação entre os ciclos, para tentar minimizar os
efeitos colaterais. Mais que um esteróide é usado simultaneamente como um “stack” para
evitar tolerância. A dosagem é inicialmente pequena, depois é gradativamente aumentada e
no final do ciclo é descendente para evitar os sintomas da retirada da droga (abstinência). A
maioria dos usuários de EAA usam concomitantemente outras drogas para prevenir os efeitos
indesejáveis dos EAA.
Os efeitos adversos dos EAA estão geralmente associados com o abuso dos mesmos,
principalmente por longos períodos de tempo em doses supraterapêuticas. EAA em homens
podem reduzir a fertilidade (azoospermia), provocar a diminuição dos testículos, a impotência,
ginecomastia e o estreitamento da uretra; em mulheres podem provocar a masculinização
(para alguns efeitos irreversível), como por exemplo a excessiva pilosidade corporal, calvície
de padrão masculino, hipertrofia de clitóris, irregularidade ou ausência do ciclo menstrual, voz
rouca e acne. Outros efeitos adversos incluem: problemas cardiovasculares (infarto agudo do
miocárdio e cardiopatias), disfunção hepática (icterícia), tumores no fígado (adenona,
carcinoma), desordens psiquiátricas (aumento da agressividade, psicose, disforia, depressão),
acidente vascular cerebral e embolia pulmonar (KUHN, 2002). Casos de dependência química
também são conhecidos.
1.5.3. Metabolismo
O metabolismo dos EAA é extenso e segue o caminho usualmente evidenciado para a
testosterona. Dessa forma, o metabolismo da testosterona pode ser discutido como base para
a rota metabólica de todos os EAA sintéticos. As enzimas que convertem testosterona em
seus diferentes metabólitos também são ativas para conversão de EAA com grupos e

Introdução - 15 -
configurações similares àquelas do principal androgênio endógeno. O metabolismo da
testosterona foi investigado em diversos tecidos in vivo e in vitro em vários modelos animais e
através de estudos clínicos com humanos. Vários desses estudos foram feitos utilizando
testosterona deuterada para permitir a identificação de forma inequívoca de seus possíveis
metabólitos. Os principais metabólitos da testosterona excretados na urina são: androsterona
(3α-hidroxi-5α-androstan-17-ona), etiocolanolona (3α-hidroxi-5β-androstan-17-ona),
epiandrosterona (3β-hidroxi-5α-androstan-17-ona), 5α-androstano-3α,17β-diol, 5β-
androstano-3α,17β-diol e 5α-androstano-3β,17β-diol (KUHN, 2002).
A rota de metabolização dos EAA inclui oxidação, redução, hidroxilação e epimerização
(reações de fase I), e reações de conjugação formando glicuronídeos e sulfatos (reações de
fase II). Os metabólitos formados são excretados na urina e nas fezes (SCHANZER &
DONIKE, 1993).
Os principais alvos para o metabolismo de fase I são os anéis A, B e D. Similarmente
ao perfil evidenciado para a testosterona, a ligação dupla entre C4 e C5 dos EAA é reduzida
produzindo os isômeros 5α e 5β. Essa redução é o passo limitante no metabolismo de
esteróides do tipo 3-ceto-4-eno. A redução produz um centro assimétrico em C5, podendo
apresentar configuração 5α e 5β (Fig. 1). As enzimas que catalisam este processo, 5α-
redutase e 5β-redutase, estão localizadas principalmente no fígado. A 5α-redutase se localiza
no retículo endoplasmático e a 5β-redutase no citoplasma. Ambas as enzimas necessitam de
NADPH como cofator. A proporção dos diferentes isômeros produzidos depende do padrão
estrutural do esteróide. EAA que possuem em sua estrutura 1,4-dieno, como a metandienona,
não produzem o isômero 5α. Em geral, a estrutura 1,4-dieno confere ao anel A alguma
resistência às reduções metabólicas, uma vez que o grupo 1-eno é estável. Quando a dupla
ligação é reduzida, o grupo 3-ceto é imediatamente transformado (BASARIA et. al., 2001,
KICMAN, et al., 2003, SCHANZER & DONIKE, 1993, SCHANZER, 1996).

Introdução - 16 -
Isômero 5β Isômero 5α
O
HO O
H
5β-redutase5α-redutase
Figura 1: Metabolismo no anel A dos EAA: redução 5α e 5β de esteróides 3-ceto-4-eno.
O grupo ceto em C3 é também facilmente reduzido, pelas enzimas 3α-hidroxiesteróide
desidrogenase e 3β-hidroxiesteróide desidrogenase, produzindo predominantemente o
isômero 3α-hidroxila (Fig. 2). Dependendo dos substituintes no anel A, outros metabólitos
podem também ser bio formados.
HO
HO O
H HO O
H
H HHO
HHO H
HO
3β-hidroxi esteróide desidrogenase
3α-hidroxi esteróide desidrogenase
Metabólitos 3α-hidroxila Metabólitos 3β-hidroxila
Figura 2: Metabolismo no anel A dos EAA: redução do grupo 3-ceto.
A modificação mais comum no anel B é a 6β-hidroxilação que ocorre para muitos
esteróides que têm em sua estrutura 1,4-dieno (Fig. 3). Para alguns esteróides como a

Introdução - 17 -
fluoximesterona, que possui um átomo de flúor em C9 a 6β-hidroxilação também ocorre
preferencialmente.
O
O
OH
O
F
O
F
OH
6β-hidroxilação
Figura 3: Metabolismo no anel B dos EAA: 6β-hidroxilação.
No anel D, o oxigênio na posição C-17 é muito sensível a reações de oxiredução.
Muitos 17β-hidroxiesteróides são oxidados para 17-cetoesteróides, pela enzima 17β-hidroxi
esteróide desidrogenase. Entretanto o grupo 17α-alquila impede esta reação. A oxidação do
grupo 17β-hidroxila é reversível e a extensão do equilíbrio depende de outras reações
metabólicas, como a conjugação na hidroxila 17β e a redução do anel A. A conversão
enzimática para 17α-hidroxi não é comum e ocorre somente menos frequentemente em
alguns esteróides.
OHH
O HOH
17α-hidroxi esteróide
desidrogenase
17β-hidroxi esteróide
desidrogenase
Figura 4: Metabolismo no anel D dos EAA: Oxidação do grupo hidroxila em 17β.
EAA também podem ser metabolizados através da hidroxilação de C16, gerando os
isômeros 16α e 16β, cuja proporção da formação dos dois isômeros difere de acordo com os
diferentes EAA. Não existe uma regra geral, e em alguns casos somente um dos isômeros é
excretado. Para o estanozolol, o isômero 16β-hidroxi é um dos principais metabólitos.

Introdução - 18 -
OH
R
OH
ROH
OH
RO
OH
CH3
OOH
O
16α/16β hidroxila 16-ceto
16α e 16β hidroxila
R = H
16α/16β-hidroxilação
R = H, CH3
17-oxidação para R=H Hidroxilação 16α/16β
Figura 5: Metabolismo no anel D dos EAA: Hidroxilação em C16 e formação de metabólitos
16-ceto.
Muitos dos metabólitos de fase I, são posteriormente metabolizados por reações de
fase II. A conjugação com ácido glicurônico uridinadifosfato e fosfoadenosina fosfosulfato
produzem glicuronídeos e sulfatos, respectivamente. No homem, a glicuronidação parece ser
mais importante que a sulfatação. A conjugação ocorre principalmente para os anéis A e D.
3α-hidroxiesteróides são principalmente conjugados com ácido glicurônico e 3β-
hidroxiesteróides com sulfato. Por sua vez, 17β-hidroxiesteróides podem ser conjugados com
ácido glicurônico e sulfato. No caso do estanozolol, n-glicuronidação também pode ocorrer. A
glicuronidação do grupo 17β-hidróxi dos 17α-metilesteróides não é comum em função do
impedimento estérico. 17-metilesteróides são capazes de formar conjugados com sulfato.
Entretanto, 17-sulfatos são entretanto, instáveis e decompõem-se resultando na formação dos
epímeros 17α-hidroxi-17β-metil e 18-nor-17,17-dimetil-13(14)-esteróides. Muitos esteróides
17-alquilados e seus metabólitos são parcialmente resistentes ao metabolismo de fase II e
são excretados na urina na forma livre.

Introdução - 19 -
1.5.4. Estrutura química
EAA sintetizados quimicamente são estruturalmente relacionados com a molécula da
testosterona. Eles são desenhados para aumentar o efeito anabólico protêico da testosterona
com redução dos efeitos androgênicos indesejados, e para melhorar as propriedades
farmacológicas da molécula. São compostos lipídicos com um sistema de anel peridro-1,2-
ciclopentanofenantreno, sendo classificados em seis grupos de acordo com o número de
átomos de carbono: gonano (C19), estrano (C18), androstano (C19), pregnano (C21), colano
(C24) e colestano (C27) (Fig. 6) (CARDOSO, 2002). EAA podem ser divididos em três grupos:
I) análogos produzidos pela via da esterificação do grupo 17β-hidróxido, II) análogos
alquilados na posição 17α e III) análogos onde os anéis A, B ou C são modificados. Vários
compostos pertencem a cada um desses grupos, como ilustra a figura 6.
Figura 6: Estrutura química da testosterona (androst-4-en-17β-ol-3-ona) e estrutura
química geral dos esteróides.
A esterificação do grupo hidroxila em 17β de alguns esteróides com ácido carboxílico
viabiliza seu uso como fármaco pela via intramuscular. Os derivados 17β-ésteres mais
OH
O
12
34
56
7
89
10
11 12 13
14 1516
17
18
19
A B
C D
Testosterona
H
H
H
Gonano (C17)
H
H
Estrano (C18)
H
H
Colestano (C27)
HAndrostano (C19) Pregnano (C21)

Introdução - 20 -
comuns são derivados da testosterona, como por exemplo cipionato, proprionato, enantionato
e undecanoato.
A alquilação na posição 17α protege esteróides contra o efeito de primeira passagem
do metabolismo e permite seu uso por via oral. Muitos dos derivados 17-alquilados são
metilados, mas também existem derivados etilados e etinilados. A grande maioria dos EAA
tem modificações estruturais nos anéis A, B ou C. Estes esteróides estão freqüentemente
disponíveis para uso por via oral. A maioria das modificações são a introdução de uma dupla
ligação entre C1 e C2 e redução da dupla ligação entre C4 e C5. Outras modificações incluem
a introdução de um grupo metila em C1, ou de um grupo metila ou metoxila em C2 ou sua
substituição por oxigênio, a introdução de pirazola ou outra estrutura cíclica no anel A através
de C2 e C3, a introdução de um cloro ou hidroxila em C4 e a introdução de um grupo metila
em C7. Nandrolona é o análogo mais comum da testosterona, tem a estrutura da testosterona
com exceção do grupo metila em C19 entre os anéis A e B.
Juntamente aos EAA sintéticos, existem muitos esteróides que são vendidos em alguns
países livremente como suplementos nutricionais ou pró-hormônios. Deidroepiandrosterona
(DHEA ou prasterona) e 4-androstenodiona são esteróides endógenos. Outras substâncias
quimicamente relacionadas a eles incluem 5-androstenodiona, 4-androstenodiol, 5-
androstenodiol, 19-norandrost-4-enodiona, 19-norandrost-5-enodiol e 19-norandrost-4-enodiol.
Muitos desses esteróides não têm nenhuma indicação clínica conhecida.
Uma nova tendência no abuso de EAA são os esteróides projetados, os quais não são
comercialmente disponíveis e a intenção é que não sejam detectados no teste anti-dopagem.
O primeiro a ser descoberto foi a norboletona. Dois outros esteróides, tetrahidrogestrinona
(THG) e “madol” (DMT ou desoximetiltestosterona; 17α-metil-5α-androst-2-eno-17β-ol)
também foram encontrados. Assim como muitos pró-hormônios sua ação e toxicidade ainda
não estão claras.

Introdução - 21 -
Bolasterona
OHCH3
CH3O
H
OH
O
H3C
Drostanolona O
OH
Cl Clostebol
OH
OH
CH3
OH
O
O
O
OH
H
CH
O
OHC
OH
Oximesterona
Metenolona
Figura 7: Estrutura de diferentes esteróides anabólicos.
Oxandrolona
H
OHCH3
NNH
Estanozolol
Nandrolona
1.5.4.1. Anabolizantes 1.5.4.1.1. Estanozolol
Estanozolol, 17β-hidroxi,17α-metil-5α-androst-2-eno(3,2-c)-pirazola, foi sintetizado em
1959, suas propriedades terapêuticas como a estimulação da síntese protéica e o
tratamento da osteoporose têm sido descritas por vários autores. Uma vez que seus efeitos
anabólicos foram reconhecidos, estanozolol tem sido um dos agentes anabólicos mais
freqüentemente utilizados no esporte, gerando numerosos casos de resultados analíticos
adversos, no controle de dopagem no esporte. Desde as olimpíadas de Seoul em 1988
foram reportados casos de dopagem pelo uso de estanozolol. Também tem sido
frequentemente reportado seu uso em eqüinos com a finalidade de dopagem e na engorda
de gado para consumo interno e exportação. Efeitos colaterais sobre o fígado, como hepatis
peliosis, colestasis ou tumores hepáticos, assim como doenças cardiovasculares e

Introdução - 22 -
desordens neurológicas têm sido relatadas após o abuso deste esteróide, principalmente em
usuários que iniciam o abuso de esteróides por estanozolol.
Em humanos os principais produtos do metabolismo de fase I do estanozolol, são os
derivados monohidroxilados 4β-OH-estanozolol, 16β-OH-estanozolol e 3’-OH-estanozolol,
assim como seus análogos epimerizados em C17. O metabólito 3’-OH-estanozolol é o
analito alvo no controle de dopagem no esporte, por ser o metabólito de excreção mais
prolongada.
Estanozolol e seus metabólitos são estruturalmente diferentes da maioria dos
esteróides anabólicos e são particularmente difíceis de se detectar em urina: esses
compostos têm um comportamento ruim em cromatografia gasosa e as concentrações
geralmente encontradas na urina são muito baixas devido a sua lenta taxa de excreção. De
fato, somente 16% dos metabólitos do estanozolol são excretados na urina durante o
primeiro dia, enquanto 40-60% são excretados nas fezes. Após a administração, estanozolol
é rapidamente metabolizado e seus metabólitos podem ser detectados na urina por até seis
dias, de acordo com a dose administrada e metabolismo individual (THEVIS, et. al., 2006).
Os metabólitos do estanozolol (Fig. 8), são excretados na urina principalmente sob a forma
conjugada.
Figura X: Figura X: Metabolismo da Metandienona. Metabolismo do estanozolol.
NN
CH3
CH3OH
CH3
H
H
NN
CH3
CH3OH
CH3
H
H
HO
NN
CH3
CH3OH
CH3
H
H
OH
NN
CH3
CH3OH
CH3
H
H
OH
NN
CH3
CH3CH3
OH
H
H
HO
NN
CH3
CH3OH
CH3
H
H
OH
3’OH-17-epiestanozolol
16α-OH-estanozolol
16β-OH-estanozolol
4β-OH-estanozolol3’OH-estanozolol
Estanozolol
Figura 8: Principais metabólitos do estanozolol em humanos.

Introdução - 23 -
O limite mínimo de desempenho requerido, determinado pela AMA através de
documento técnico (TD2004MRPL) para o metabólito 3’-hidroxiestanozolol é 2 ng/mL. Nesta
concentração sua detecção é particularmente difícil por CG-EM, método analítico de escolha
para triagem de agentes anabólicos, mesmo no modo SIM (HUENERBEIN, 2003). Dessa
forma, faz-se necessário a utilização de CG-EMAR ou CG-EMn para análise desses
compostos de acordo com os critérios estabelecidos pela AMA.
1.5.4.1.2. Metandienona
Metandienona, 17α-metil-androsta-1,4-dien-17β-ol-3-ona, foi sintetizada em 1955, por
Vischer e colaboradores através da desidrogenação microbiológica da metiltestosterona. Em
1956, Meystere e colaboradores relataram a desidrogenação da metiltestosterona com dióxido
de selênio. O metabolismo, parâmetros farmacocinéticos, assim como os efeitos anabólicos e
androgênicos da metandienona em humanos foram investigados detalhadamente
(SCHANZER et. al., 2006). 6β-Hidroximetandienona foi identificado como sendo o metabólito
principal, sendo excretado livre na urina. Em 1963, verificou-se que 6β-hidroximetandienona
pode ser, também, excretada como um conjugado lábil de estrutura não conhecida. Em 1971
foi identificado outro metabólito, também excretado na forma livre, a 17-epimetandienona.
Mais tarde constatou-se que este metabólito é resultado da degradação e rearranjo de um
metabólito excretado na forma de 17β-sulfato. Em 1991, Schanzer e colaboradores
publicaram a identificação de metabólitos excretados na urina sob a forma glicoconjugada.
Tais metabólitos são fruto da redução do anel A da metandienona, sendo estes 17α-metil-5β-
androst-1-eno-3α,17β-diol, seu epímero na posição 17, 17β-metil-5β-androst-1-eno-3α,17α-
diol (epimetendiol, EMD) e 17α-metil-5β-androstano-3α,17β-diol (SCHANZER, et. al., 1991).
Posteriormente, outro metabólito descrito foi o 18-nor-17,17-dimetil-5β-androst-1,13-dien-3α-ol
(18-nor epimetendiol), o qual junto com o epimetendiol foram identificados como metabólitos
de mais longa excreção, sendo portanto alvo analíticos para o controle de dopagem do
esporte (SCHANZER, et al., 1992).

Introdução - 24 -
OHCH3
O
OHCH3
O
OH
OCH3
O
SO3H
CH3OH
O
CH3CH3
O
OHCH3
OH
OHCH
HOH
OHCH
HOH
CH3OH
HOH
CH3CH3
HOH
17α-metil-5β-androstano-3α,17β-diol
18-nor-17,17-dimetilandrosta-1,4,13-
trien-3-ona
17β-metil-5β-androst-1-eno-3α,17α-diol (EMD – Epimetendiol) 18-nor-17,17-dimetil-5β-
androstano-1,13-dien-3α-ol
17α-metil-5β-androst-1-eno-3α,17β-diol17β-hidroxi-17α-metil-5β-androst-1-eno-3-ona
17-Epimetandienona
6β-hidroximetandienonaMetandienona
Metandienona 17β-sulfato
Figura 9: Principais metabólitos da metandienona em humanos.
1.5.4.1.3. Nandrolona
Nandrolona (17β-hidroxiestr-4-en-3-ona) foi sintetizada em 1950, por Birch e seu
metabolismo foi investigado por Engel e colaboradores em 1958, quando foram identificados
os metabólitos 3α-hidroxi-5α-estran-17-ona (norandrosterona) e 3α-hidroxi-5β-estran-17-ona
(noretiocolanolona). Em 1988, Schanzer e colaboradores identificaram o isômero 3β-hidroxi,
3β-hidroxi-5α-estran-17-ona, que também é excretado pela urina conjugado com sulfato na
hidroxila em 3β (VAN EENOO, et. al., 1999).

Introdução - 25 -
Figura 10: Principais metabólitos da nandrolona em humanos.
OHH
O
HHO
O
H
O
HO
HHO
O
3β-hidroxi-5α-estran-17-ona
Noretiocolanolona
Norandrosterona
Nandrolona
1.5.4.1.4. Metiltestosterona
Metiltestosterona (17β-hidroxi-17α-metilandrost-4-en-3-ona) foi sintetizada em 1935,
por Ruzicka. O metabolismo da metiltestosterona em humanos foi investigado em 1962 por
Rongone e Segaloff. Nesse estudo foram identificados 17α-metil-5α-androstano-3α,17β-diol e
17α-metil-5β-androstano-3α,17β-diol como sendo os principais metabólitos. O metabolismo da
metiltestosterona segue o metabolismo do anel A da testosterona. Foi identificado também o
epímero na posição 17, i.e. o metabólito 17β-hidroxi-17α-metil, sendo este excretado em
pequena quantidade (cerca de 2 a 3,5%) (SCHANZER et. al., 1995).

Introdução - 26 -
OHCH3
O
OHCH3
HOH
OHCH3
HHO
Metiltestosterona
17α-metil-5β-androstano-3α,17β-diol17α-metil-5α-androstano-3α,17β-diol
Figura 11: Principais metabólitos da metiltestosterona em humanos.
1.5.4.1.5. Metilnortestosterona
Metilnortestosterona (17β-hidroxi-17α-metil-estr-4-en-3-ona) é um esteróide projetado
encontrado recentemente em complemento nutricional comercializado pela internet. Também
é conhecido pelos nomes: metilestrenolona, normetandrolona ou ainda normetandrona.
OHCH3
OMetilnortestosterona
Figura 12: Estrutura química da metilnortestosterona.

Introdução - 27 -
1.5.4.1.6. Metasterona
Metasterona (2α,17α-dimetil-etiocolan-3-ona-17-ol), também conhecido como superdrol
é um esteróide projetado encontrado recentemente em um suplemento nutricional
denominado metilmasterdrol, comercializado pela internet.
H3C
O
CH3
CH3OH
CH3
HMetasterona
Figura 13: Estrutura química da metasterona.
1.5.4.2. Agentes anabólicos 1.5.4.2.1. Clembuterol
O clembuterol (4-amino-3,5-dicloro-α-[[(1,1-dimetiletil)amino]metil]benzenometanol) é
um β2-agonista extremamente potente. A farmacocinética e o metabolismo do clembuterol têm
sido extensivamente estudados em animais com o intuito de, entre outras razões, discriminar
entre o seu uso terapêutico ou ilícito em animais cuja carne é utilizada para o consumo
humano. Estudos realizados em gado, aves, pescado, coelhos, cavalos e ratos utilizando
diferentes tipos de matrizes, descrevem a farmacocinética e o metabolismo deste fármaco.
Segundo YAMAMOTO et al. (1985), após administração de 20, 40 e 80 µg de cloridrato de
clembuterol em voluntários sadios, as concentrações plasmáticas alcançam valores máximos
de 0,1, 0,2 e 0,35 ng / mL, respectivamente, em cerca de 2,5 horas após a ingestão do
fármaco. No mesmo estudo, após dosagens de 20 µg deste β2-agonista em homens sadios
durante vários dias (duas vezes ao dia), foi observado que a concentração plasmática do
composto não modificado atinge um platô de 0,2 a 0,3 ng / mL após o quarto dia da
administração do fármaco. Sua união às proteínas plasmáticas é estimada entre 89-98% após

Introdução - 28 -
administração de dose única de 80 µg de cloridrato de clembuterol, e a meia-vida no plasma é
estimada em torno de 35 horas.
Com relação ao metabolismo, 34 a 43% da dose administrada de clembuterol é
eliminado principalmente por via renal, na forma não modificada. Por não possuir hidroxila
fenólica, o clembuterol não é conjugado com sulfato. Estudo realizado em urina mostrou que
após administração oral de 80 µg de cloridrato de clembuterol, foi possível detectar a
presença do fármaco não modificado no período entre 2 e 48 horas, sendo a concentração
máxima encontradas em torno de 7 horas após a administração. Outro estudo indica que
cerca de 20% da dose administrada é detectada em urina até 72 h depois como fármaco
inalterado.
N
OH
Cl
H2N
Cl
H
Clembuterol
Figura 14: Estrutura química do clembuterol.
1.5.4.2.2. Tibolona
Tibolona é um esteróide sintético estruturalmente relacionado com os 19-nor-
esteróides, como por exemplo a norestisterona. Em função de sua potencia estrogênica e
atividade androgênica, pode ser usada por atletas com a finalidade de dopagem. Tibolona é
rapidamente metabolizada em 3α e 3β-hidróxi-tibolona pela enzima desidrogenase, no
intestino e no fígado. Estes metabólitos são estrógenos fracos e possuem tempo de meia vida
de aproximadamente 7 horas. No endométrio, 3-OH-tibolona é metabolizada pela isomerase
3-OH-desidrogenase ao ∆4-isômeros (7α-metil-noretisterona), que apresenta atividade
progestagen e androgem de fraca a moderada (MARQUES, et. al., 2004).

Introdução - 29 -
7α-metil-noretisterona
O CH3
OH
CH3
OH
HOCH3
OH
HO
O CH3
OH3α-OH-tibolona
3β-OH-tibolona
Tibolona
Figura 15: Principais metabólitos da tibolona em humanos.
1.5.4.2.3. Zeranol
Zeranol é um hormônio não esteroidal largamente utilizado nos EUA, representando a
classe das lactonas resorcíclicas. Quimicamente derivado da micotoxina zearalenona,
produzida pelo fungo Gibberella zeae. Apresenta efeitos muito similares aos do 17β-
estradiol.
A principal via metabólica das lactonas resorcíclicas envolve redução a cetona ou
oxidação na hidroxila do carbono 7(C7), podendo originar metabólitos que são isômeros. Por
exemplo o taleranol (17β-zeranol) é formado a partir do zeranol via intermediário
zearalanona. Os isômeros α e β-zeranol são resultantes da redução metabólica da
zearalenona (MARQUES, 1992).

Introdução - 30 -
O
O
OOH
HO
O
OOH
HO OH
O
OOH
HO OH
Metabólitos conjugados
β-zeranol α-zeranol
Zearalanona
Figura 16: Principais metabólitos do zeranol em humanos. 1.5.4.3. Agentes com atividade anti-estrogênica
Citrato de clomifeno, foi o primeiro anti-estrogênico aprovado pelo FDA nos Estados
Unidos em 1967. Este fármaco tinha como alvo terapêutico a indução da ovulação e o
tratamento para câncer de mama dependente de hormônio. Em meados de 1970, tamoxifeno
foi aprovado para uso no tratamento do câncer de mama e mais tarde foi a vez do toremifeno.
Outro anti-estrogênico que hoje começa a ser experimentado para prevenção do câncer de
mama é o raloxifeno, até então utilizado para o tratamento de osteopenia e osteoporose.
Todos esses medicamentos exercem forte efeito antagonista de estrogênio em certos tecidos
e atividade como agonista parcial do estrogênio em outros tecidos, sendo portanto
considerados moduladores seletivos para o receptor de estrogênio (LEWIS & JORDAN,
2005). Mais recentemente antiestrogênios “puros”, ou seja, que não apresentam propriedades
agonistas foram desenvolvidos e estão sendo testados em ensaios clínicos.
Pesquisadores, em 1960 desenvolveram o conceito do uso de inibidores da aromatase
para tratamento de uma variedade de processos estrogênio dependentes. Aromatase é uma
enzima citocromo p450 que catalisa a conversão de androgênios para estrogênios.
Aromatização é o passo limitante na formação de estrogênios e portanto um alvo lógico para o

Introdução - 31 -
desenvolvimento de agentes para bloquear a síntese de estradiol. Um pouco mais tarde,
experiências clínicas com a primeira geração de inibidores da aromatase, como por exemplo,
com aminoglutetimida, comprovou que inibidores da aromatase poderiam ser usados para o
tratamento de câncer de mama hormônio dependente (SANTEN, 2OO3).
Comparações clínicas diretas com o antiestrogênio, tamoxifeno, demonstraram igual
eficácia para esta indicação. A vantagem em relação ao uso do tamoxifeno, esta relacionada
aos efeitos adversos e toxicidade apresentados pela aminoglutetimida, que diminuem seu
uso. Nos últimos trinta anos, foram desenvolvidos inibidores da aromatase, menos tóxicos,
mais potentes e seletivos (SANTEN, 2OO3).
A terceira geração de agentes, agora aprovado para uso, no mundo todo, são quase
completamente seletivos para a enzima aromatase e entre 1000 e 10000 vezes mais potente
que aminoglutetimida, além de serem muito melhor tolerados. Estes novos inibidores
compreendem duas categorias com diferentes mecanismos de ação: os inibidores
competitivos os quais se ligam ao sítio ativo da enzima aromatase e bloqueiam a formação de
estradiol e os inativadores que são modificados pelos efeitos catalíticos da aromatase para
formar compostos reativos os quais se ligam covalentemente ao sítio ativo da enzima e
destroem irreversivelmente sua ação enzimática. Mais tarde, denominados inativadores
suicidas. Três novos compostos fazem parte da terceira geração de inibidores da aromatase,
sendo estes: anastrozol e letrozol (inibidores competitivos) e examestano (inativador)
(SERALINI & MOSLEMI, 2001).
Durante o processo de desenvolvimento e testes em pacientes com câncer de mama, a
segunda geração (fadrozol, 4-OH-androstenodiona ou formestano) de inibidores
demonstraram igual, mas não superior eficácia clínica quando comparado com os
moduladores seletivos para o receptor de estrogênio. Dessa forma, foi surpreendente para os
pesquisadores quando a terceira geração de inibidores de aromatase pareceram ser superior
ao tamoxifeno para o tratamento de câncer de mama hormônio dependente. Evidências
preliminares também sugerem superioridade no cenário adjuvante para prevenção de câncer
de mama e talvez também para disfunções ovulatórias em mulheres inférteis (INGLE &
SUMAN, 2005, LEWIS & JORDAN, 2005).
Anti-estrogênicos podem causar um aumento da produção endógena de androgênios.
Atletas tem sido encorajados a tratar os efeitos adversos do abuso extensivo de esteróides
anabólicos androgênicos, como por exemplo, ginecomastia e supressão de androgênios com
o uso de anti-estrogênicos. Sendo assim, o uso de agentes com atividade anti-estrogênica
foram proibidos pelo COI e AMA para atletas do sexo masculino e feminino desde setembro
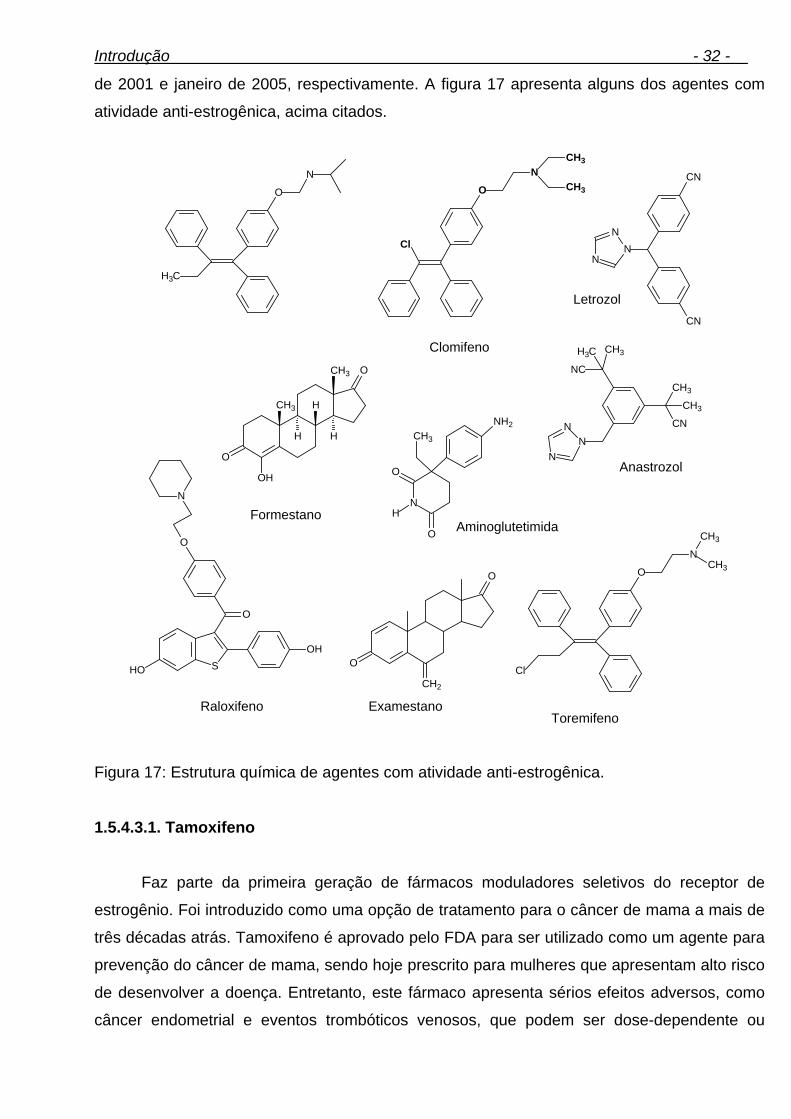
Introdução - 32 -
de 2001 e janeiro de 2005, respectivamente. A figura 17 apresenta alguns dos agentes com
atividade anti-estrogênica, acima citados.
Tamoxifeno
Cl
ON
CH3
CH3O
N
H3CN
NN
CN
CN
S
O
N
O
OH
HO
O
N
CH3
CH3
Cl
NN
N
CH3
CN
CH3
H3C CH3
NC
N
NH2
O
O
CH3
H
O
O
CH2
O
O
OH
CH3 H
HH
CH3
Examestano
Formestano
Raloxifeno Toremifeno
Aminoglutetimida
Anastrozol
Clomifeno
Letrozol
Figura 17: Estrutura química de agentes com atividade anti-estrogênica.
1.5.4.3.1. Tamoxifeno Faz parte da primeira geração de fármacos moduladores seletivos do receptor de
estrogênio. Foi introduzido como uma opção de tratamento para o câncer de mama a mais de
três décadas atrás. Tamoxifeno é aprovado pelo FDA para ser utilizado como um agente para
prevenção do câncer de mama, sendo hoje prescrito para mulheres que apresentam alto risco
de desenvolver a doença. Entretanto, este fármaco apresenta sérios efeitos adversos, como
câncer endometrial e eventos trombóticos venosos, que podem ser dose-dependente ou
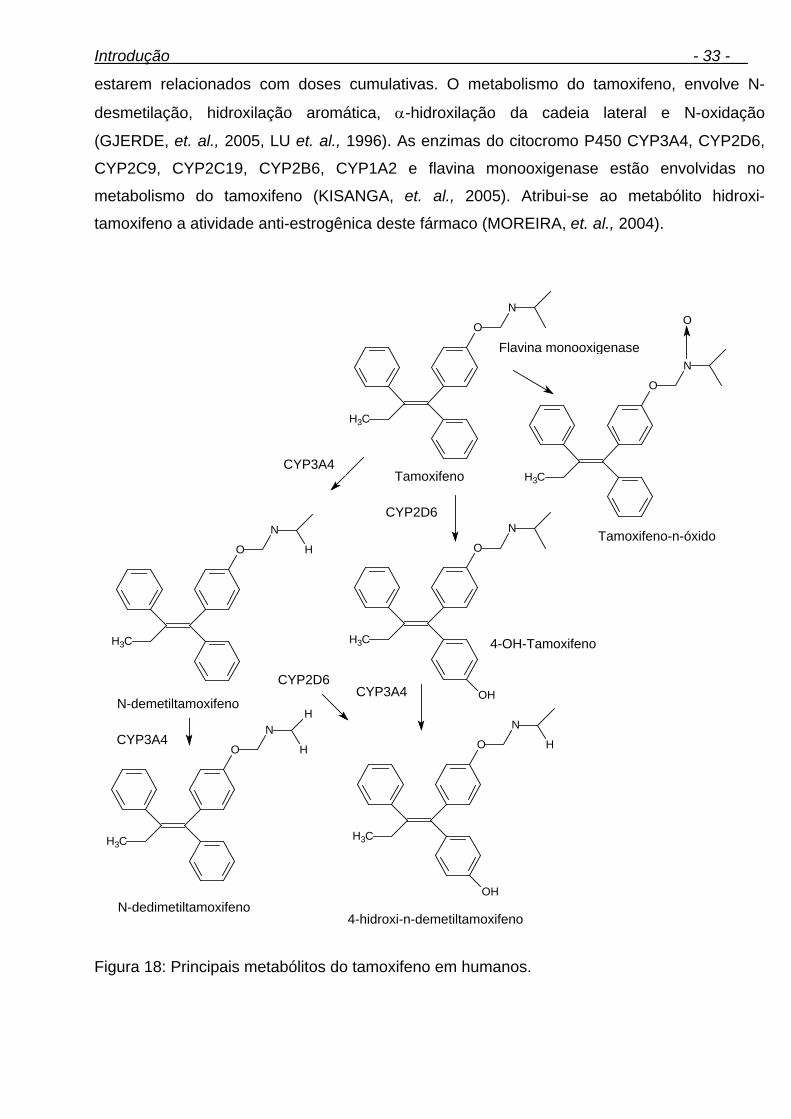
Introdução - 33 -
estarem relacionados com doses cumulativas. O metabolismo do tamoxifeno, envolve N-
desmetilação, hidroxilação aromática, α-hidroxilação da cadeia lateral e N-oxidação
(GJERDE, et. al., 2005, LU et. al., 1996). As enzimas do citocromo P450 CYP3A4, CYP2D6,
CYP2C9, CYP2C19, CYP2B6, CYP1A2 e flavina monooxigenase estão envolvidas no
metabolismo do tamoxifeno (KISANGA, et. al., 2005). Atribui-se ao metabólito hidroxi-
tamoxifeno a atividade anti-estrogênica deste fármaco (MOREIRA, et. al., 2004).
4-hidroxi-n-demetiltamoxifeno
O
N
H3C
O
N
H3C
O
O
N
H
H3C
O
N
H3C
OH
O
N
H
H3C
OH
O
NH
H
H3C
CYP3A4CYP2D6
CYP3A4
CYP3A4
N-dedimetiltamoxifeno
N-demetiltamoxifeno
4-OH-Tamoxifeno
Tamoxifeno-n-óxido
Flavina monooxigenase
CYP2D6
Tamoxifeno
Figura 18: Principais metabólitos do tamoxifeno em humanos.

Introdução - 34 -
1.5.4.3.2. Clomifeno
Há mais de 40 anos citrato de clomifeno foi introduzido na prática clínica, sendo um dos
fármacos mais prescrito para a indução da ovulação. A resposta farmacológica do clomifeno é
mediada pela sua concentração no tecido ovariano e seus efeitos na glândula pituitária. A
concentração ovariana esta relacionada com a concentração da droga no sangue, sendo
determinada pelos processos de absorção, distribuição, metabolismo e excreção. Diferenças
individuais em qualquer um desses processos pode resultar em diferentes concentrações
plasmáticas de clomifeno após ministrar a mesma dose a diferentes indivíduos. Cada tablete
do comprimido contém uma mistura dos dois isômeros na forma do sal de citrato. As formas E
e Z são chamadas enclomifeno e zuclomifeno, respectivamente, constituindo
aproximadamente 62% e 38% do conteúdo do tablete. Zuclomifeno é muito mais potente na
indução da ovulação quando comparado ao enclomifeno (HODJEGAN, et. al., 2004).
Clomifeno é bem absorvido, e os isômeros e seus metabólitos são excretados
principalmente nas fezes. Em ratos e coelhos, os principais produtos metabólicos resultam da
4-hidroxilação, N-deetilação e N-oxidação pelo citocromo P450. Em humanos a rota
metabólica dos isômeros do clomifeno parecem ser muito similares as do tamoxifeno. Em
urina é possível detectar após uma única dose de 50mg de citrato de clomifeno, o metabólito
4-hidróxi clomifeno.
Cl
O
NCH3
CH3
Cl
O
NCH3
CH3
HO
Hidroxilação
CYP2D6
4-OH-clomifeno Clomifeno
Figura 19: Principal metabólito do clomifeno em humanos.

Introdução - 35 -
1.5.4.3.3. Ciclofenila
Assim como o clomifeno, ciclofenila inibe a ligação do estrogênio na adenohipófise,
causando aumento na secreção de GnRH e das gonadotropinas. Isto resulta em acentuada
estimulação e aumento dos ovários, com secreção aumentada de estrogênio. O principal
efeito de sua ação antiestrogênica na hipófise consiste em induzir a ovulação, sendo utilizado
no tratamento da infertilidade devido à falta de ovulação. Em urina é possível detectar após
uma dose única de 400 mg, o metabólito hidroxi-bis-desacetil-ciclofenil (Mareck et.al., 2001).
OH
OH
Hidroxi-bis-desacetil-ciclofenilCiclofenil
Figura 20: Principal metabólito do ciclofenil em humanos.
1.5.4.3.4. Letrozol
Letrozol (1-(bis-(4-cianofenil)metil)-1,2,4-triazol), inibidor da aromatase não esteroidal, é
terapeuticamente utilizado no tratamento do câncer de mama, em mulheres na pós
menopausa. A maior rota de eliminação do letrozol, via metabolismo, é a formação do
metabólito inativo bis-4-cianofenil-metanol e de triazol (MARECK, et. al., 2005).
NN
N
CN
CNN
NHN
CN
CN
HO
Bis-4-cianofenil-metanolTriazolLetrozol
Figura 21: Principais metabólitos do letrozol em humanos.

Introdução - 36 -
1.5.4.3.5. Formestano Formestano (4-Hidroxiandrost-4-eno-3,17-diona), inibidor da aromatase, foi introduzido
no mercado em 1993, com indicação para o tratamento de câncer de mama em estágio
avançado em mulheres na pós-menopausa. Formestano pode ser administrado pela via
intramuscular ou oral. Entretanto apresenta baixa biodisponibilidade, quando administrado
pela via oral. É rapidamente metabolizado, sendo eliminado na forma glicoconjugada
(LONNING, 1998). O
O
OH
CH3 H
HH
CH3
Formestano
Figura 22: Estrutura química do formestano.
1.5.4.3.6. Hiperdrol
Hiperdrol (6ξ-bromo-androst-4-eno-3,17-diona), ainda não explicitamente incluído na
lista de substâncias proibidas da AMA. Os dois isômeros, 6α e 6β-bromo-androstenodiona
tem sido reportados como inibidores da aromatase. O isômero 6α- bromo-androstenodiona é
considerado inibidor irreversível. O metabolismo dessa substância até o momento só foi
estudado ”n vitro”, portanto nada se sabe sobre seu metabolismo em humanos. Hiperdrol ou
Restore é comercializado, via internet, sob a forma do complemento nutricional Anabolic
Xtreme, onde no rótulo do produto recomenda-se 1 a 3 cápsulas por dia durante quatro
semanas. O
Br
OHiperdrol
Figura 23: Estrutura química do Hiperdrol.

Introdução - 37 -
1.5.5. Estratégia do exame antidopagem para Agentes Anabólicos EAA no teste antidopagem são analisados somente a partir da urina. Em geral são
avaliados somente de forma qualitativa, mas a detecção do abuso de esteróides endógenos e
compostos para os quais foram definidas concentrações de corte (threshold) exigem métodos
semi-quantitativos de análise. Todos os métodos para determinação de EAA são baseados na
cromatografia acoplada a espectrometria de massas. Normalmente os alvos analíticos são um
ou dois metabólitos principais de cada EAA monitorado. Dependendo do esteróide, o limite
mínimo de desempenho necessário (MRPL, “Minimum Required Performance Limit” AMA
2004) é 2 ng/mL. Até o presente momento, as amostras analisadas rotineramente pelos
laboratórios acreditados pela AMA, avaliam a presença de esteróides livres ou
glicoconjugados, não abordando os sulfoconjugados.
A análise de EAA, assim como de outras substâncias banidas, é dividida em duas
categorias, a análise em triagem e métodos de confirmação. O objetivo da triagem é apontar
amostras suspeitas para posterior análise. Um método de triagem ideal deve ser simples,
rápido, seletivo, sensível e gastar o menor volume possível de urina. Em função do grande
número de substâncias banidas, métodos de triagem multi-resíduos são desenvolvidos com o
objetivo de englobar todas as substâncias em um menor número possível de triagens.
Todas as amostras suspeitas encontradas são confirmadas a partir da re-análise de
uma nova alíquota da urina em um método altamente específico e sensível. Durante o
processo de preparação da amostra, controles positivos são preparados a partir da
contaminação de brancos de urina (previamente avaliados) com padrão do analito alvo livre
ou conjugado se disponível ou através da análise de urinas de excreção. Além obviamente, do
controle negativo, no caso o branco de urina. Todas as amostras (amostra suspeita e
controles) devem ser preparadas no mesmo momento. O resultado analítico adverso é
determinado pela comparação entre o tempo de retenção e o espectro de massas do analito
na amostra suspeita com o material de referência. Por se tratar de compostos presentes na
urina, na grande maioria dos casos, em baixas concentrações (cerca de 10 ng/mL) são
permitidos métodos de confirmação utilizando o monitoramento seletivo de íons (SIM). Os
critérios de identificação são regulamentados pela AMA através de documentos técnicos a fim
de harmonizar os procedimentos entre os trinta e quatro laboratórios de controle de dopagem
acreditados no mundo, gerando resultados defensáveis.

Introdução - 38 -
1.5.6. Métodos analíticos aplicados a agentes anabólicos 1.5.6.1. Cromatografia Gasosa de Alta Resolução acoplada a Espectrometria de Massas (CG-EM) O primeiro método para detectar EAA em urina foi desenvolvido por Donike e
colaboradores, e explora um espectrômetro de massas, com analisador tipo quadrupolo e
impacto de elétrons como modo de ionização (monitoramento de íons positivos) acoplado a
cromatógrafo a gás equipado com coluna capilar. A extração de EAA das amostras de urina
foi otimizada passando a mesma por colunas de extração por fase sólida do tipo XAD-2,
seguida de hidrólise dos metabólitos conjugados usando enzima β-glicuronidase e extração
líquido-líquido com éter dietílico em pH alcalino. Antes da injeção da amostra no CG-EM
usando monitoramento seletivo de íons, a amostra era derivatizada com uma mistura de N-
metil-N-(trimetilsilil)trifluoracetamida (MSTFA), trimetiliodosilano e um antioxidante para
converter grupos hidroxila e ceto em trimetilsililéter e enol éter. Neste primeiro momento os
esteróides excretados livres na urina eram analisados separadamente pela extração direta da
amostra ou mesmo depois de purificação usando XAD-2, com dietiléter em pH básico,
seguido de derivatização seletiva resultando na formação de derivados O-TMS. Modificações
no método original incluem a extração por fase sólida utilizando cartuchos C18, a extração
líquido-líquido utilizando terc-bútilmetiléter e a combinação durante o processo de pré-
tratamento da amostra da fração livre (esteróides não conjugados) com a fração conjugada
(esteróides glicoconjugados). A última modificação no método inclui hidrólise direta da urina
seguida de extração líquido-líquido. O método para análise de esteróides por CG-EM-MIS tem
tempo total que pode variar entre 20 e 30 minutos. O limite de detecção depende do esteróide
e pode variar entre 2 e 30 ng/mL.
1.5.6.2. Critérios de identificação para ensaios qualitativos envolvendo a Cromatografia e a Espectrometria de Massas.
De acordo com o documento técnico da AMA (TD2003IDCR), a identificação de
compostos utilizando a espectrometria de massas de baixa resolução deve estar baseada no
tempo de retenção (TR) ou tempo de retenção relativo (TRR), e na comparação de íons
diagnósticos. A primeira condição é que a diferença em termos de TR (ou TRR) do analito,
não deva ser superior a 1%, quando comparada com a mesma substância presente em uma
urina controle positiva. Entende-se como íon diagnóstico aquele íon cujo valor de m/z

Introdução - 39 -
corresponde ao íon molecular ou ao produto de uma fragmentação cuja presença e
abundância são características da substância, podendo, desse modo, servir para a sua
identificação.
Com relação à espectrometria de massas, a avaliação deve incluir um mínimo de três
íons diagnósticos. Se os três íons diagnósticos não estiverem disponíveis, um segundo
derivado deverá ser preparado ou uma segunda técnica de ionização deverá ser utilizada. Em
todo caso, um mínimo de dois íons diagnósticos devem ser comparados em espectro de
massas. As abundâncias relativas dos íons na amostra problema não devem diferir por mais
do que 5% (absoluto) ou 20% (relativo), qualquer que seja o maior valor encontrado, dos
mesmos íons da amostra de urina controle positiva. Entende-se por abundância relativa, a
abundância de um íon particular relativa a outro íon monitorado mais abundante, expressa
como valor percentual (DAMASCENO, 2001). Um melhor entendimento sobre este critério
pode ser dado pelas seguintes definições:
- diferenças máximas permitidas em termos de abundância relativa – referem-se às
diferenças máximas permitidas entre a abundância relativa de um íon particular obtido na
amostra e a obtida na amostra de urina controle positiva. Estas diferenças entre abundância
relativas podem ser expressas em termos absolutos ou relativos.
- diferença em termos absolutos – calculada como o valor absoluto da diferença entre
as abundâncias relativas obtidas para o íon estudado da amostra problema e da amostra de
controle positiva. Como exemplo, se a abundância relativa de um íon na amostra de controle
positiva é de 45%, e na amostra problema é de 50%, a diferença entre as abundâncias
relativas, em termos absolutos, é de 5%.
- diferença em termos relativos – calculada como o quociente entre a diferença em
termos absolutos e a maior das abundâncias relativas obtidas (na amostra problema ou na
amostra de controle positiva), e expressa como valor percentual. Como exemplo, se a
abundância relativa de um íon na amostra de controle positiva é 45%, e na amostra problema
é 50%, então a diferença entre as abundâncias relativas, em termos relativos seria de 10%,
isto é, (5/50)*100 = 10%.
Como último critério, a concentração do analito na urina controle positiva não deve
diferir por um fator maior do que 5 vezes da concentração do composto na urina problema.

Introdução - 40 -
1.6. PROTÓTIPOS PARA RECEPTORES DOPAMINÉRGICOS 1.6.1. Planejamento estrutural de novos derivados heterocíclicos n-fenilpiperazínicos a partir da clozapina. A busca de novos candidatos a protótipos de fármacos de origem sintética é um
importante capítulo da Química Medicinal, uma vez que seu desdobramento leva a
substâncias terapêuticas, apresentando diversos níveis de complexidade estrutural, como o
analgésico e anti-inflamatório Aspirina® e o fármaco semi-sintético taxol (Menegatti, 2002). No
âmbito de uma linha de pesquisa que visa o planejamento, síntese e avaliação farmacológica,
de novos candidatos a protótipos de fármacos neuroativos, tendo como alvo patológico a
esquizofrenia, duas novas famílias de compostos heterocíclicos foram construídas e
previamente planejadas para apresentarem funcionalidade seletiva sobre receptores
dopaminérgicos (Menegatti, 2001).
A dopamina é o neurotransmissor catecolaminérgico mais importante do sistema
nervoso central dos mamíferos e está envolvida na etiologia de diversas desordens
neuropsiquiátricas, incluindo a doença de Parkinson, a depressão e a esquizofrenia. Foram
identificados 5 tipos de receptores dopaminérgicos no cérebro, todos eles acoplados a
proteínas G e divididos em duas famílias farmacológicas denominadas D1-like (subtipos D1 e
D5) e D2-like (subtipos D2, D3 e D4). Diferentes fármacos são usados em doenças relacionadas
a dopamina, porém os problemas oriundos da terapia, especialmente a ação frente a
diferentes receptores, podem limitar o tratamento (Menegatti, 2001, Menegatti,2003).
A esquizofrenia é uma desordem que afeta o sistema nervoso central e incide sobre
cerca de 1-2% da população mundial. De modo geral, se conhece que em um quadro
esquizofrênico típico as funções cognitivas e emocionais, são divididas em dois subgrupos de
sintomas, os positivos e os negativos. Postula-se que os sintomas positivos, como a
desilusão, alucinações, psicoses, paranóia, pensamento desordenado e fala desorganizada,
ocorram devido a uma hiperatividade dopaminérgica na área mesolímbica do cérebro de
pacientes esquizofrênicos. Já os sintomas negativos, como a desmotivação, comportamento
emocional violento, isolamento social, deficiência cognitiva e fala lenta, seriam oriundos de
uma hipoatividade dopaminérgica no córtex pré-frontal (Menegatti et al., 2004).
Devido à necessidade de novos agentes que fossem ligantes dopaminérgicos seletivos
e eficazes frente a patologias mediadas pelo sistema dopaminérgico, foram planejados por
bioisosterismo e sintetizados pelo Laboratório de Avaliação e Síntese de Substâncias

Introdução - 41 -
bioativas (LASSBio – FF – UFRJ), diferentes compostos heterocíclicos N-fenilpiperazínicos,
tendo como alvo patológico a ezquizofrenia.
O planejamento destes novos candidatos a protótipos incluiu, entre outras, a
abordagem terapêutica, onde se utilizou como composto de partida a clozapina , por
apresentar elevada afinidade por receptor dopaminérgico do subtipo D4. A clozapina é um
antipsicótico atípico e foi utilizada como protótipo para subseqüentes modificações estruturais,
objetivando-se a separação dos efeitos sanguíneos, indesejáveis, daqueles centrais. A
clozapina, após ter sofrido modificações moleculares, deu origem a duas famílias de
compostos pirazóis e triazóis (Fig. 24). Tais modificações compreenderam a contração do
anel B e a transposição do núcleo aromático D para o átomo de nitrogênio distal do anel
piperazínico C (Fig. 25) (Menegatti, 2001).
N
N Y
N
NN
W
N
N Y
NN
W
LASSBio 622 W=H e Y=fenil LASSBio 623 W=H e Y=4-clorofenil LASSBio 624 W=H e Y=4-fluorfenil LASSBio 579 W=Cl e Y=fenil LASSBio 617 W=Cl e Y=4-clorofenil LASSBio 628 W=Cl e Y=4-metoxifenil LASSBio 664 W=F e Y=fenil LASSBio 667 W=F e Y=4-fluorfenil
pirazóis
1,2,3-triazóis LASSBio 580 W=H e Y=fenil LASSBio 618 W=H e Y=4-clorofenil LASSBio 620 W=H e Y=4-fluorfenil LASSBio 581 W=Cl e Y=fenil LASSBio 619 W=Cl e Y=4-clorofenil LASSBio 698 W=Cl e Y=4-metoxofenil LASSBio 701 W=Cl e Y=2-pirimidinila LASSBio 586 W=F e Y=fenil LASSBio 621 W=F e Y=4-fluorfenil LASSBio 666 W=NO2 e Y=4-fenil LASSBio 696 W=NH2 e Y=4-fenil LASSBio 697 W=NHAc e Y=4-fenil
Figura 24: Novas famílias de compostos heterocíclicos.

Introdução - 42 -
N
N
CH3
N
N
H
Cl
A
BDC
Figura 25: Estrutura química da clozapina.
1.6.3. Processo de desenvolvimento de fármacos
O processo de desenvolvimento de fármacos impõem a necessidade de interação entre
numerosos grupos de indivíduos que trabalham em áreas abrangendo desde as ciências
básicas até as profissões médicas e legais. O custo e o tempo total médio envolvido foi
estimado em 114 milhões de dólares e 10 anos de trabalho ou mais. O processo de
desenvolvimento é demorado em virtude da necessidade de diversos testes que asseguram
segurança e eficácia do fármaco, a fim de ser aprovado pelos órgãos regulamentadores.
Quando o fármaco atinge o estágio de teste em seres humanos, sua farmacocinética,
farmacodinâmica e propriedades tóxicas já foram avaliadas in vitro e em várias espécies
animais. Procede-se, então, aos estudos clínicos, constituídos por quatro fases: fase I, fase II,
fase III e fase IV. Se os resultados das investigações clínicas dão suporte aos efeitos
terapêuticos como sendo úteis e como tendo mínimos efeitos adversos, os dados pré-clínicos
e clínicos são encaminhados para aprovação e posterior registro (TASSO, 2003).
Os ensaios farmacológicos pré-clínicos, também chamado de fase não clínica ou de
fase zero, envolvem a avaliação da eficácia e segurança do fármaco sob investigação em
estudos in vitro e em animais experimentais. Os testes em animais, que determinam a dose
eficaz, dose tóxica, metabolismo, farmacocinética, efeitos teratogênicos, entre outras
informações, servem de base para a pesquisa nos seres humanos que vem a seguir (TASSO,
2003). As transformações enzimaticamente promovidas na estrutura química dos fármacos
podem acarretar profundas alterações na resposta biológica, uma vez que modificações
moleculares, ainda que singelas, podem alterar significativamente o farmacóforo, dificultando
sua interação com o biorreceptor original, ou ainda, favorecendo novas interações com outras
biomacromoléculas, correspondendo a novos e distintos efeitos biológicos, algumas vezes
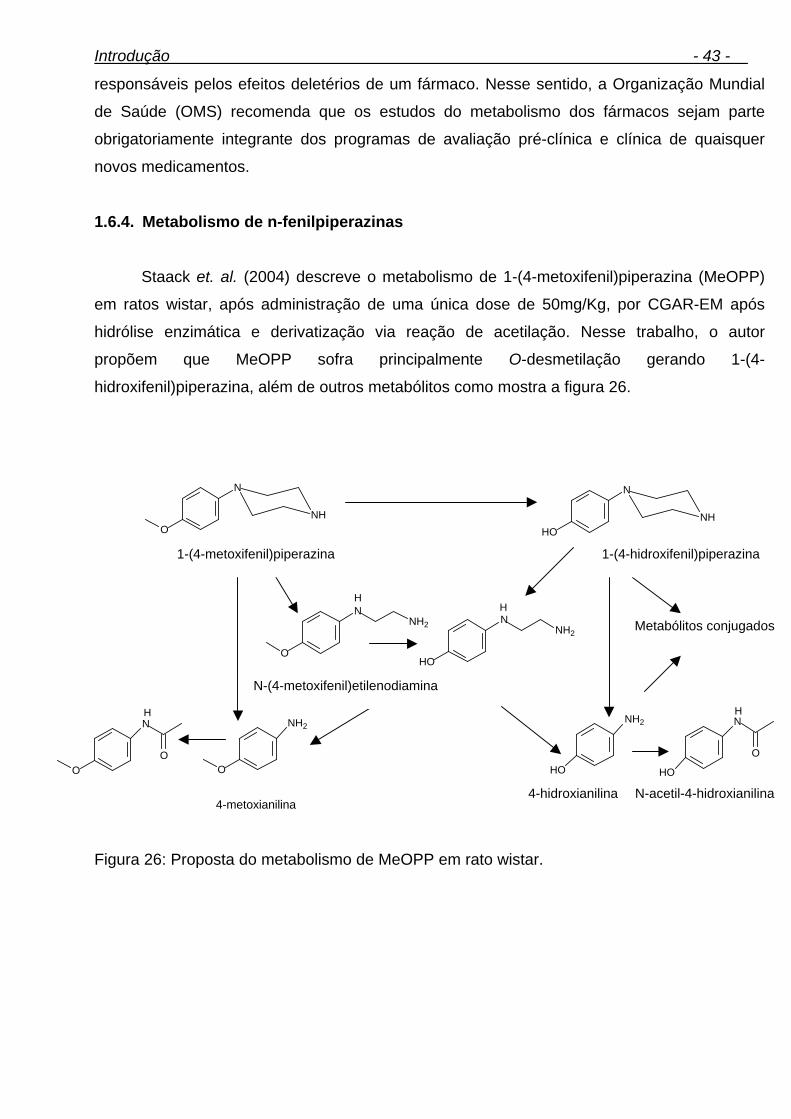
Introdução - 43 -
responsáveis pelos efeitos deletérios de um fármaco. Nesse sentido, a Organização Mundial
de Saúde (OMS) recomenda que os estudos do metabolismo dos fármacos sejam parte
obrigatoriamente integrante dos programas de avaliação pré-clínica e clínica de quaisquer
novos medicamentos.
1.6.4. Metabolismo de n-fenilpiperazinas
Staack et. al. (2004) descreve o metabolismo de 1-(4-metoxifenil)piperazina (MeOPP)
em ratos wistar, após administração de uma única dose de 50mg/Kg, por CGAR-EM após
hidrólise enzimática e derivatização via reação de acetilação. Nesse trabalho, o autor
propõem que MeOPP sofra principalmente O-desmetilação gerando 1-(4-
hidroxifenil)piperazina, além de outros metabólitos como mostra a figura 26.
4-hidroxianilina N-acetil-4-hidroxianilina4-metoxianilina
N
NHO
N
NHHO
N
O
NH2
H
N
HO
NH2
H
NH2
HO
N
HO
O
HNH2
O
N
OO
H
N-(4-metoxifenil)etilenodiamina
1-(4-metoxifenil)piperazina 1-(4-hidroxifenil)piperazina
Metabólitos conjugados
Figura 26: Proposta do metabolismo de MeOPP em rato wistar.

CAPÍTULO 2
OBJETIVOS

Objetivos - 44- 2. OBJETIVOS A presente tese tem como principais objetivos:
- Caracterização por espectrometria de massas in tandem das novas famílias de compostos
heterocíclicos (1,2,3-triazóis e pirazóis) planejados e sintetizados pelo Laboratório de
Avaliação e Síntese de Substâncias Bioativas.
- Desenvolver metodologia analítica para extração e análise por CGAR-EM, do composto
LASSBio 581 e possíveis metabólitos , em plasma de rato Wistar.
- Incluir na análise de rotina do LAB DOP, agentes anabólicos e / ou metabólitos que no
presente ano e no próximo passam a integrar a lista de substâncias proibidas da Agência
Mundial Anti-dopagem (AMA).
- Validar o procedimento analítico referente a análise qualitativa dos agentes anabólicos
incluídos nas análises de rotina do laboratório.
- Desenvolver procedimento analítico de triagem para monitoramento de agentes anabólicos
cujo LMDR é equivalente ou menor que 2 ng/mL, no equipamento VARIAN 4000, para suprir
os jogos PAN-AMERICANOS 2007.
- Validar o procedimento analítico desenvolvido para detecção de agentes anabólicos em
baixa concentração, utilizando a técnica de cromatografia gasosa de alta resolução acoplada
a espectrometria de massas (CGAR-EM).
- Validar de forma quantitativa, 3’-OH-estanozolol a fim de avaliar a resposta (linearidade,
homocedasticidade / heterocedasticidade, precisão, exatidão, robustez) do equipamento
VARIAN 4000 adquirido em função dos jogos PAN-AMERICANOS 2007, frente a esse
composto.

CAPÍTULO 3
EXPERIMENTAL

Experimental 45
3. EXPERIMENTAL
A maior parte dos procedimentos analíticos descritos na presente tese foram
realizados no Laboratório de Controle de Dopagem do IQ / UFRJ, credenciado pelo
INMETRO, tanto para ensaios quanto para calibração segundo a norma ISO – 17025. O
laboratório também é credenciado pela Agência Mundial Antidopagem (AMA) para o controle
de dopagem em atletas, pelo Ministério da Agricultura, Pecuária e Abastecimento (MAPA),
para análise de resíduos em alimentos e pela Agência Nacional de Vigilância Sanitária
(ANVISA – MS) para análises toxicológicas. Ainda sendo integrado a rede de laboratórios
acreditados RBLE (nº ANALI 029), RBC (nº 328) e REBLAS. O laboratório possui projeto
aprovado pelo Comitê de Ética em Pesquisa (CEP) do Hospital Universitário Clementino
Fraga Filho (HUCFF-UFRJ) (número do protocolo: 020/00) para realização de estudos de
excreção. O laboratório ainda é registrado no Conselho Regional de Química – terceira
região para atividades de análise química, físico-química, química-biológica, toxicológica,
bromatológica e legal, padronização e controle da qualidade de produtos químicos. Vistoria,
perícia, avaliação, arbitragem, elaboração de pareceres, laudos e atestados da
especialidade.
Para realização do presente trabalho também foi possível contar com a infra-estrutura
do Laboratório de Avaliação e Síntese de Substâncias Bioativas (LASSBio FF / UFRJ),
laboratório de pesquisa em Química Medicinal, área do conhecimento farmacêutico
essencial a inovação terapêutica. O LASSBio tem como meta a capacitação de alunos,
desde jovens graduandos até pós-doutores, no desenho racional, síntese, avaliação
farmacológica e otimização de novos protótipos de fármacos de diversas classes
terapêuticas.
Além do LASSBio, contamos com o laboratório Thomson Espectrometria de Massas e
com o laboratório de controle de dopagem do Instituto de Bioquímica (Instiut für Biochemie)
da Universidade do Esporte em Colonia (Deutsche Sporthochschule Köln), na Alemanha.
O laboratório Thomson Espectrometria de Massa, localiza-se no Instituto de Química
da Universidade Estadual de Campinas (UNICAMP) e se destina a utilização da
espectrometria de massas na solução de problemas de diversas áreas científicas, incluindo
as ciências dos materiais, ciências biológicas, ambientais, de alimentos e farmacêuticas,
além da química orgânica, inorgânica, analítica e físico-química.
O laboratório de controle de dopagem da Universidade do Esporte em Colonia,
destina-se assim como o LAB DOP, ao controle de dopagem no esporte, fazendo um total
de onze mil amostras por ano. Esse laboratório tem reconhecida experiência na análise de
anabolizantes, assim como na síntese química dos mesmos.

Experimental 46
3.1. SUBSTÂNCIAS DE REFERÊNCIA
Os padrões de referência 17β-hidroxi,17α-metil-5α-androst-2-eno(3,2-c)-pirazol, 17β-
metil-5β-androst-1-eno-3α,17α-diol, norandrosterona, 17α-metil-5β-androstano-3α,17β-diol,
4-amino-3,5-dicloro-α-[[(1,1-dimetiletil)amino]metil]benzenometanol, foram adquiridos da
Sigma (St. Louis, MO, USA), da Serva (Heidelberg, NY, USA) e da Steraloids (NewPort,
EUA).
3.2. SOLVENTES E REAGENTES Reagentes: O reagente de derivatização N-metil-N-trimetilsililtrifluoracetamida (MSTFA) foi
adquirido da Chem Fabrik (Waldstetten, Germany). A enzima utilizada para a hidrólise das
amostras de urina, β-glicuronidase de E. Coli, foi adquirida da Sigma Chemical CO (St.
Louis, MO, USA). Iodeto de amônio e 2-mercaptoetanol foram adquiridos da Sigma
Chemical CO (St. Louis, MO, USA), hidróxido de potássio p.a., pentóxido de fósforo p.a.,
fosfato de sódio monobásico monohidratado p.a. e bicarbonato de potássio p.a. foram
adquiridos da Merck (Darmstald, Germany).
Solventes: Acetona, metanol e terc-butilmetiléter (TBME) foram adquiridos da Tedia
(Fairfield, USA) com grau pesticida para análise de resíduos. Água bidestilada foi utilizada
para o preparo das soluções tampão e solução de KOH.
3.3. MEDICAMENTOS
Os medicamentos utilizados, para estudo de excreção, encontram-se listados na
tabela abaixo (tabela 4).
Tabela 4: Medicamentos utilizados para estudo de excreção. Medicamento Fabricante Composição
Clomid 50 mg, 10 comprimidos por caixa Medley Citrato de clomifeno
Menopax 200 mg, 20 comprimidos por
caixa
Ache Ciclofenila
Nolvadex 10 mg, 30 comprimidos por caixa Astrazen-h Citrato de tamoxifeno
Femara 2,5 mg, 28 comprimidos por caixa Novartis-h Letrozol

Experimental 47
3.4. AMOSTRAS BIOLÓGICAS
As amostras de urina foram coletadas de doadores usuários crônicos de
medicamento ou foram doação do laboratório de controle de dopagem de Colônia
(Alemanha). Utilizou-se urinas contendo os seguintes compostos: 17β-hidroxi-17α-metil-estr-
4-en-3-ona, 2α,17α-dimetil-etiocolan-3-ona-17-ol, 4-hidroxiandrost-4-eno-3,17-diona, 6ξ-
bromo-androst-4-eno-3,17-diona, tamoxifeno, clomifeno, ciclofenil, letrozol.
O laboratório detém aprovação do comitê de Ética em Pesquisa do Hospital
Universitário Clementino Fraga Filho da UFRJ (CEP / HU - CFF – UFRJ, protocolado sob o
n° 020/00).
3.5. MATERIAIS
Os termômetros e todo o material volumétrico utilizado ao longo do desenvolvimento
desse trabalho foram calibrados no LABCAL - LADETEC - IQ / UFRJ acreditado em 2006,
fazendo parte da RBC (Rede Brasileira de Calibração lab. n° 328), tendo seus
procedimentos aprovados pelo Inmetro, segundo a norma ISO / IEC 17025.
3.6. EQUIPAMENTOS
O processo de preparação das amostras de plasma e urina requer os seguintes
equipamentos: agitador de tubos (vórtex)-Barnsted / Thermolyne (Dubuque, Iowa, USA),
agitador orbital Fanem shaker (São Paulo, SP, Brasil), centrifuga Excelsa Baby II, modelo
206-R, série HI6730 (Fanem, Brasil), evaporador Reach-Therm III (Pierce, USA). Demais
equipamentos utilizados: Ultra-som THORNTON INPEC ELETRONICA S.A. – modelo T7
(EUA), Estufa – QUIMIS (Brasil), pHmetro – Corning-modelo 320-série C3642 (Brasil),
densímetro – ATAGO CO. LTD – série 9711050 (Japão), balança – SARTORIUS AG
GOTTINGEN – modelo BP210D – 5 casas decimais (Germany) e balança – DENVER
INSTRUMENT COMPANY – modelo AA-200DS – 4 casas decimais (EUA), geladeira Frost
Free metalfrio – ANTHONY CAT 770 REFLECTIVE – modelo 7000 (Brasil) e câmara fria a –
20°C para a estocagem das amostras biológicas. Para a análise das amostras utilizou-se um
Cromatógrafo Gasoso de Alta Resolução (CGAR) acoplado a Espectrômetro de Massas
(EM), Agilent (CGAR 6890 / EM 5973) e um Cromatógrafo Gasoso de Alta Resolução
(CGAR) acoplado a Espectrômetro de Massas (EM), VARIAN (CGAR 3800 / EM 4000).

Experimental 48
3.7. LIMPEZA DA VIDRARIA
Todo o material de vidro utilizado no presente trabalho foi limpo para limpeza de
material de laboratório, que descreve a seguinte seqüência de lavagem:
a) lavagem com água em abundância (todo o material que esteve em contato com
derivatizante deve ser rinsado com metanol antes desta etapa de lavagem);
b) aquecimento em água com detergente comercial neutro, por no mínimo 2 horas;
c) lavagem com água em abundância;
d) imersão em solução de Extran neutro 5% (Merck, Alemanha), por no mínimo 24 horas;
e) lavagem com água em abundância;
f) rinsagem com água destilada;
g) secagem em estufa a aproximadamente 60°C (exceção para material volumétrico que
deve ser seco a temperatura ambiente).
3.8. PREPARO DAS SOLUÇÕES 3.8.1. Soluções padrão estoque
Foram preparadas soluções padrão de estoque na concentração de 1mg / mL em
metanol grau pesticida, a partir de cada padrão de referência (método LADETEC n°5042).
Para tanto, pesou-se 10 mg do padrão de referência em balança analítica com precisão de 5
casas decimais, devidamente calibrada, em balão volumétrico de 10 mL e o volume foi
completado com metanol pesticida até a marca de referência. A solução resultante foi
homogeneizada em vórtex por 20 segundos obtendo-se, assim, uma solução de estoque de
1mg / mL, chamada de solução mãe. Todas as soluções foram armazenadas em frascos de
vidro âmbar com tampa e mantidos sob refrigeração a -20°C, apresentando validade inicial
de 3 anos.
3.8.2. Soluções padrão de trabalho
Solução padrão de 10 ng / µL: em um balão volumétrico de 10 mL adicionou-se
quantitativamente 100 µL da solução mãe (1mg / mL) e o volume foi completado com
metanol grau pesticida até a marca de referência. A solução resultante foi homogeneizada
em vórtex por 20 segundos obtendo-se, assim, uma solução de 10 ng / mL (método
LADETEC n°5154).

Experimental 49
Solução padrão de 1 ng / µL: em um balão volumétrico de 10 mL adicionou-se
quantitativamente 1mL da solução padrão de trabalho de 10 ng / mL e o volume foi
completado com metanol grau pesticida até a marca de referência. A solução resultante foi
homogeneizada em vórtex por 20 segundos obtendo-se, assim, uma solução de 1 ng / µL
(método LADETEC n°5153).
Solução padrão de 0,1 ng / µL: em um balão volumétrico de 10 mL adicionou-se
quantitativamente 100 µL da solução padrão de trabalho de 10 ng / µL e o volume foi
completado com metanol grau pesticida até a marca de referência. A solução resultante foi
homogeneizada em vórtex por 20 segundos obtendo-se, assim, uma solução de 0,1 ng / µL
(método LADETEC n°5149).
Todas as soluções de trabalho foram armazenadas em frascos de vidro âmbar com
tampa, devidamente rotulados, e mantidos refrigerados a -20°C. Antes do uso, aguarda-se
ao menos 20 minutos antes que os frascos sejam abertos, de modo a atingirem a
temperatura ambiente.
3.8.3. Padrão interno (ISTD)
Solução de padrão interno (ISTD): A solução de padrão interno foi adquirida do
laboratório de Colônia (Alemanha), 20 µL dessa solução são adicionados a 2 mL de urina.
Tabela 5: Composição da solução de padrão interno.
Compostos Concentração da solução (ng/mL)
Concentração em urina (ng/mL)
Etiocolanolona-d4 25 µg/mL 500 ng/mL
17α-metiltestosterona 25 µg/mL 500 ng/mL
Testosterona-d3 4,5 µg/mL 90 ng/mL
Epitestosterona-d3 0,75 µg/mL 15 ng/mL
3.8.4. Solução de D5-androsterona-glicuronídeo
Para preparação do padrão interno conjugado, 20 µL da solução de D5-androsterona-
glicuronídeo são adicionados a cada 2 mL de urina (concentração em urina = 500 ng/mL).
Solução tampão carbonato: bicarbonato de potássio 20% (pH 10): solubilizou-se 40,0 g
de K2CO3 e 40,0 g de KHCO3 em 320 mL de água destilada à temperatura ambiente. A

Experimental 50
solução tampão foi armazenada a temperatura ambiente, em lugar fresco e ventilado,
apresentando validade de 3 meses (método LADETEC n°5052).
Solução tampão fosfato de sódio 0,8 M (pH 7): solubilizou-se 17,6 g de NaH2PO4H2O e
34,6 g de Na2HPO4 em 460 mL de água destilada à temperatura ambiente. A solução
tampão foi armazenada a temperatura ambiente, em lugar fresco e ventilado, apresentando
validade de 3 meses (método LADETEC n°5108).
Solução tampão fosfato de sódio 0,2 M: foi preparada a partir da diluição de 1 parte da
solução tampão fosfato de sódio 0,8 M para 3 partes de água destilada, homogeneizando a
solução, em chapa de aquecimento, com auxílio de magneto até ebulição. Esta solução
deve ser preparada na hora de ser utilizada, sendo desprezada após o uso (método
LADETEC n°5053).
3.8.5. Solução de KOH A solução de KOH 0,5 M foi preparada pela dissolução de 1,4 g de KOH em 50 mL de água
destilada, ajustando-se o pH para 12 com solução de HCl 1M. A solução foi armazenada à
temperatura ambiente, apresentando validade de uma semana (método LADETEC n°5059).
3.8.6. Solução derivatizante Solução mãe: Em um frasco de vidro âmbar com tampa pesar 20 mg de NH4I e adicionar 1
mL de MSTFA e 60 µL de 2-marcaptoetanol. Tampar devidamente o frasco, agitar em vórtex
por 20 segundos e aquecer até solubilizar todo o NH4I. Armazenar em dessecador. O prazo
de validade da solução é de 15 dias (método LADETEC n°5045).
Solução de uso: Transferir 1 mL da solução mãe para frasco de vidro âmbar, com tampa de
vidro esmerilhada. Adicionar 9 mL de MSTFA. Tampar o frasco e agitar em vórtex por 20
segundos. Rotular o frasco com o nome da solução [MSTFA:NH4I:2-mercaptoetanol
(1000:2:6, v:w:v)]. Armazenar em dessecador, contendo sílica gel azul. O prazo de validade
da solução é de 15 dias (método LADETEC n°5044).

Experimental 51
3.8.7. Limpeza e silanização dos liners (encamisamento de vidro) de quartzo e / ou vidro borossilicato (método LADETEC n°5106)
A limpeza e silanização dos liners utilizados no presente trabalho foram realizadas do
seguinte modo:
- Imersão em mistura sulfocrômica por 30 minutos;
- Aplicação de ultra-som no material ainda submerso na mistura sulfocrômica por
aproximadamente 30 minutos;
- Lavagem com água destilada;
- Lavagem com metanol;
- Secagem em estufa a 100°C por 1 hora;
- Recheio com lã de vidro (0,02 g);
- Silanização com hexametildisilazano (HMDS) por 1 hora;
- Aquecimento dos liners em estufa a 100°C por duas horas.

Experimental 52
3.9. MÉTODOS DE EXTRAÇÃO EM MATRIZES BIOLÓGICAS 3.9.1. Extração de agentes anabólicos em urina humana 3.9.1.1. Método de triagem (método LADETEC n°5070) A maioria dos agentes anabólicos apresenta metabólitos livres e conjugados na urina.
A metodologia descrita a seguir propicia a extração de todos estes metabólitos e da droga
mãe (quando presente), a partir da mesma alíquota, envolvendo as frações aquosas e
orgânicas.
A 2 mL de urina adicionar 20µL de ISTD e 20µL de D5-androsterona-glicuronídeo,
agitar em vórtex por 20 segundos. Adicionar 750 µL de tampão fosfato 0,8 M pH 7,0, 50 µL
de β-glicuronidase de E. Coli , agitar em vórtex por 20 segundos e submeter a hidrólise a
50°C por uma hora. Adicionar 500 µL de solução tampão carbonato de potássio a 20% pH 9
– 10. Adicionar 5 mL de TBME e agitar por 5 min e em seguida centrifugar a 3000 rpm por 5
minutos. Transferir a fase orgânica, com auxílio de pipeta Pasteur, para outro tubo, evaporar
o solvente sob fluxo de N2 a 40°C e manter os tubos em dessecador a vácuo contendo
pentóxido de fósforo lentilhas de hidróxido de potássio, por 30 minutos para posterior
derivatização. Adicionar 100 µL da solução derivatizante de uso (item 3.8.6) e aquecer por
20 minutos a 60°C e injetar em CGAR-EM. Nessa fração encontram-se os metabólitos livres,
conjugados e a droga mãe (quando for o caso) dos agentes anabólicos alvo do trabalho. A
figura 26 apresenta o fluxograma do método descrito acima.

Experimental 53
Separar as fases
Injetat 3µL no CGAR-EM
Derivatizar com 100µL de MSTFA:NH4I:2-mercaptoetanol (1000:2:6, v:w:v)
Manter o resíduo em dessecador contendo P2O5 / KOH por 30 min
Evaporar a fase orgânica sob N2 / 40°C
Descartar a fase aquosa
Agitar em shaker por 5 min e centrifugar a 3000 rpm por 5min
Adicionar 5 mL de TBME
Adicionar 500 µL de tampão carbonato de potássio a 20% pH9-10
Hidrólise à 50°C por 1 hora
Homogenizar em vórtex por 20s
Adicionar 50 µL de β-glicuronidase de E.coli
Adicionar 750 µL de tampão fosfato de sódio 0,8M pH 7,0
Fortificar com o ISTD e com solução padrão de D5-Androsterona Glicuronídeo
2 mL de urina humana
Figura 27: Fluxograma do método de extração de agentes anabólicos em urina humana.

Experimental 54
3.9.2. Extração de LASSBio 581 em plasma de rato wistar A metodologia de extração de LASSBio 581 em plasma de rato wistar consiste de
uma etapa de extração líquido-líquido, empregando uma mistura de terc-butilmetiléter
(TBME) e terc-butanol (1:1) como solvente de extração. Esta metodologia foi aplicada em
amostras de branco de plasma fortificadas com solução padrão de LASSBio 581,
empregadas para a etapa de desenvolvimento do método. Para tanto, em tubos de ensaio,
fortificar alíquotas de 200µL de branco de plasma com 2000 ng de padrão interno (LASSBio
580) e com o padrão de LASSBio 581 na concentração de 10 ng/µL. Homogeneizar em
vórtex por 20 segundos. Em seguida adicionar 4 mL de TBME/t-ButOH, agitar em “shaker”
por 5 minutos e centrifugar à 3000rpm por 5 min. Separar a fase orgânica e evaporar o
solvente sob fluxo de N2 à 40°C. Manter as amostras em dessecador a vácuo contendo
hidróxido de sódio e pentóxido de fósforo por 1 hora. Derivatizar com 100 µL de BSTFA
durante 30 minutos à 60°C e injetar em CGAR-EM. A figura 27 apresenta o fluxograma do
método de extração do LASSBio 581 em plasma de rato wistar.

Experimental 55
Separar as fases
Injetat 3µL no CGAR-EM
Derivatizar com 100µL de MSTFA:NH4I:2-mercaptoetanol (1000:2:6, v:w:v)
Manter o resíduo em dessecador contendo P2O5 / KOH por 30 min
Evaporar a fase orgânica sob N2 / 40°C
Descartar a fase aquosa
Centrifugar à 3000 rpm por 5min
Agitar em shaker por 20 min
Adicionar 4mL de TBME/t-BuOH (1:1)
Fortificar com o padrão interno (LASSBio 580 – 10 ng/mL) e com padrão de LASSBio
581 ng/mL)
200 µL de plasma de rato wistar
Figura 28: Fluxograma do método de extração de LASSBio 581 em plasma de rato wistar.

Experimental 56
3.10. CONDIÇÕES DE ANÁLISE CROMATOGRÁFICA (CGAR-EM) DAS AMOSTRAS BIOLÓGICAS 3.10.1. Análise de agentes anabólicos em urina humana As análises de agentes anabólicos em urina humana foram realizadas em um sistema
de cromatografia acoplada a espectrometria de massas (CG-EM) Agilent (Palo Alto, EUA)
equipado com um injetor automático modelo 7683 com controle eletrônico de pressão.
Temperaturas de operação do detector de massas quadrupolar modelo 5973; linha de
transferência, 280°C; fonte iônica, 220°C; interface, 280°C; quadrupolo, 150°C. Detecção por
monitoramento de íons seletivos (MIS) com um tempo de varredura de 20 milisegundos.
Ionização por impacto de elétrons a 70eV. Condições de operação do cromatógrafo modelo
6890: Injetor, 280°C; coluna, 140°C (temperatura inicial, 0 min) gradiente de 40°C/min até
180°C; 3,0°C/min até 230°C; 40°C/min até 300°C; gás carreador He, fluxo total, 18,4 ml/min
(fluxo constante); pressão, 16,0 psi; velocidade linear média, 38 cm/s; 3µL de amostra
injetada com divisão de fluxo na razão 1:10. Pulso de pressão, 25 psi (0,85 min).
Encamisamento de vidro (“liner”) da HP de 6 mm x 1 mm e volume interno de 0,9 mL foi
usado. Dentro do encamisamento, 0,017 mg de lã de vidro desativada foram compactadas
entre 23 e 33 mm a partir do topo. Coluna capilar de sílica fundida HP-1 (17,0 m x 0,2 mm x
0,11 µm de expessura de filme de fase estacionária).
3.10.2. Análise de LASSBio 581 em plasma de rato Wistar
As análises de LASSBio 581 em plasma de rato wistar foram realizadas em um
sistema de cromatografia acoplada a espectrometria de massas (CG-EM) Agilent (Palo Alto,
EUA) equipado com um injetor automático modelo 7683 com controle eletrônico de pressão.
Temperaturas de operação do detector de massas quadrupolar modelo 5973; linha de
transferência, 280°C; fonte iônica, 230°C; interface, 280°C; quadrupolo, 150°C. Detecção por
monitoramento de íons seletivos (MIS) com um tempo de varredura de 20 milisegundos.
Ionização por impacto de elétrons a 70eV. Condições de operação do cromatógrafo modelo
6890: Injetor, 280°C; coluna, 200°C (temperatura inicial, 0 min) gradiente de 15°C/min até
300°C; por 5 minutos; gás carreador He, fluxo total, 18,4 ml/min (fluxo constante); pressão,
16,0 psi; velocidade linear média, 38 cm/s; 3µL de amostra injetada com divisão de fluxo na
razão 1:10. Pulso de pressão, 25 psi (0,85 min Encamisamento de vidro (“liner”) da HP de 6
mm x 1 mm e volume interno de 0,9 mL foi usado. Dentro do encamisamento, 0,017 mg de
lã de vidro desativada foram compactadas entre 23 e 33 mm a partir do topo. Coluna capilar

Experimental 57
de sílica fundida HP-1 (17,0 m x 0,2 mm x 0,11 µm de expessura de filme de fase
estacionária).
3.11. METABOLISMO DO COMPOSTO LASSBio 581 3.11.1. Animais Os animais, ratos machos Wistar, com massa corporal variando entre 400 e 500 g
foram usados nos experimentos para investigação de metabólitos em plasma e urina. Os
animais foram mantidos em ciclo artificial de luminosidade (12 h claro – 12 h escuro) à
temperatura média de 22 ± 2 °C e umidade relativa em torno de 64%. Alimentação (ração) e
água estiveram disponíveis até vinte e quatro horas antes da administração do composto
LASSBio 581 por via oral. Para investigação de metabólitos do composto LASSBio 581 tanto
em plasma quanto em urina, três ratos, para cada matriz, foram selecionados.
3.11.2. Preparo e administração das doses O composto LASSBio 581 foi preparado na dose de 30 mg/kg para ser administrado
por via oral. A dose foi administrada na forma de suspensão e foram preparadas como se
segue: 15,0 mg do composto LASSBio 581 em bécher e adicionou-se 50 µL de Tween 80®.
Após a mistura, adicionou-se solução salina isotônica e sonicou-se por cinco minutos. Em
seguida, adicionou-se 2450 µL de solução salina 0,9%. A suspensão foi agitada
imediatamente antes do uso e administradas logo após o preparo.
A dose foi administrada por gavagem, com uma sonda siliconizada, conectada em
uma seringa de 3 mL. Os animais utilizados nos experimentos não foram anestesiados.
3.11.3. Coleta das amostras de plasma e urina Após a administração, os ratos permaneceram sem alimentação, sendo somente
oferecida água após quatro horas da administração da dose. Doze horas após a
administração, foram coletadas amostras de sangue através da decaptação dos ratos, o
sangue foi coletado em tubos falcon previamente heparinizados. A amostra de sangue
obtida foi submetida à centrifugação a 5000 rpm, por 10 min. O plasma separado, foi
acondiconado em tubo falcon e congelado a -20°C até o momento da extração (item 3.9.2) e

Experimental 58
posterior análise por CG-EM (item 3.10.2). O mesmo procedimento para obtençãoa partir da
decaptação foi utilizado para obtenção de branco de plasma.
Para coleta de urina os ratos foram mantidos em gaiola metabólica (Fig. 29), que
separa a urina das vezes dos animais, o que tem grande importância para a pesquisa de
metabólitos, uma vez que bactéria presentes nas fezes podem transformar possíveis
metabólitos presentes na urina. Também para a coleta da urina os ratos a alimentação foi
retirada vinte e quatro horas antes da administração e água foi oferecida quatro horas após
a administração. Antes da administração do composto LASSBio 581 toda a urina dos três
ratos wistar foi coletada, para ser utilizada como branco de urina e para o desenvolvimento
do método de extração.
Reservatório para coleta de urina
Reservatório para ração
Reservatório para água
Local para acomodação das fezes
Figura 29: Gaiola metabólica.

Experimental 59
3.12. VALIDAÇÃO DE MÉTODOS Ainda na etapa de desenvolvimento de um método analítico, parâmetros como a
robustez e a recuperação do analito submetido ao processo de extração devem ser
mensurados.
Entende-se por robustez de um método, uma característica de desempenho do
mesmo, capaz de medir a sensibilidade que um método apresenta frente a pequenas
variações que possam ocorrer no momento da execução do mesmo. Tal parâmetro deve ser
avaliado durante o desenvolvimento do método, desde que o profissional em função de
conhecimento prévio, possa determinar quais etapas são as mais críticas. Uma vez tendo
definido estas etapas, as mesmas devem ser modificadas (para mais e para menos) e o
resultado final deve ser avaliado. Com o avanço da metrologia, modelos empíricos como o
descrito acima passaram a não ser bem aceitos, fazendo-se necessário a utilização de
testes estatísticos, que transformem em números a simples declaração: o método é robusto.
O INMETRO, por exemplo, sugere a utilização do teste de Youden que permite não só
avaliar a robustez do método, como também ordenar a influência de cada uma das
variações nos resultados finais, indicando qual o tipo de influência de cada uma dessas
variações.
A recuperação de um método é outro parâmetro que deve ser avaliado durante o
desenvolvimento do método. Trabalhar com padrão na ausência de matriz é aceitável
apenas em uma primeira etapa de desenvolvimento de um método cromatográfico. É
indispensável a observação da existência ou não de interferentes provenientes da matriz,
próximos ao analito alvo, durante a otimização.
No caso da detecção utilizando armadilha de íons, isso se torna crucial, uma vez que
a energia de excitação do íon, assim como seu tempo de ionização podem variar na
presença de matriz. Quando trabalhamos no modo massas-massas podemos escolher íons
“pais” (ou íons precursores) que não estejam presentes na matriz e que portanto só
verificaríamos a presença dos mesmos, se durante a otimização trabalhássemos com matriz
fortificada e não apenas padrão derivatizado, no caso da análise por CGAR-EM.
A respeito da recuperação do método analítico, ainda podemos comentar que
podemos trabalhar com valores baixos (entre 5% e 20%) de recuperação, desde que o limite
de detecção do método em questão seja muito menor que o limite mínimo de desempenho
requerido, ou seja, que a quantidade do analito alvo que deve ser detectado segundo
critérios de qualidade pré-estabelecidos.
Após a avaliação desses dois critérios, o próximo passo seria a validação do método
analítico.

Experimental 60
A validação é o conjunto de operações necessárias para demonstrar que um
procedimento analítico é adequado para aplicação pretendida. Essas operações variam de
acordo com a natureza do método, podendo este ser qualitativo, semi-quantitativo ou
quantitativo. Métodos qualitativos destinam-se a responder a seguinte pergunta: O composto
X está presente ou ausente na amostra? Dessa forma, apenas os parâmetros limite de
detecção, seletividade / especificidade e repetitividade devem ser avaliados durante a
validação qualitativa. No caso do controle de dopagem no esporte a grande maioria das
substâncias monitoradas apresenta apenas LMDR (limite mínimo de desempenho
requerido), devendo o método analítico para análise das mesmas, ser validado de forma
qualtitativa. Poucas substâncias apresentam valor de corte, ou seja, para concentrações
maiores ou iguais a determinado valor, um resultado analítico adverso pode ser declarado.
Nesse caso, quando trabalhamos em torno de determinado valor, o método passa a ser
semi-quantitativo e a pergunta nesse caso seria: O composto X esta presente na amostra,
acima ou abaixo da concentração de corte? Para um método semi-quantitativo as
características de desempenho que devem ser avaliadas são: limite de detecção,
seletividade / especificidade, repetitividade, e limite de quantificação.
Para confirmação de substâncias que apresentam valor de corte, a AMA recomenda a
construção de uma curva de calibração num intervalo de 50% a 200% do valor de corte,
construída a partir da análise de três amostras (matriz fortificada) para cada ponto, devendo
a amostra suspeita ser também preparada em triplicata. Nesse caso a pergunta seria: Qual a
concentração do composto X na amostra e qual a incerteza do método? Os seguintes
parâmetros de desempenho devem ser avaliados na validação quantitativa: linearidade,
especificidade / seletividade, exatidão, precisão, limite de detecção, limite de quantificação e
incerteza expandida.
3.12.1. Recuperação do processo de extração
A recuperação do processo de extração deve ser avaliada antes da validação do
método seja este qualitativo, semi-quantitativo ou quantitativo. O protocolo descrito a seguir
abrange todos esses três protocolos.
A recuperação do processo de extração consiste na relação existente entre a
resposta obtida quando se analisa uma quantidade definida de analito presente em matriz
biológica seguindo o procedimento analítico completo, e a resposta obtida quando se analisa
a mesma quantidade de analito, sendo este adicionado à matriz após todas as estapas do
procedimento analítico, exceto aquelas onde o analito necessariamente já deve estar
presente (p. ex., evaporação, derivatização).

Experimental 61
Amostras de urina previamente avaliadas frente à presença dos analitos alvo, foram
fortificadas antes (6 amostras) e depois (6 amostras) do processo de extração no LMDR.
Após essas fortificações, as amostras foram preparadas conforme o procedimento
estabelecido, e analisadas por CGAR-EM.
A recuperação mede a eficiência do procedimento de extração do método analítico
dentro de um limite de variação. Porcentagens de recuperação próximas a 100% são
desejáveis porém admite-se valores menores (50 a 60%), desde que a recuperação seja
precisa e exata.
Recuperação = [(área analito (FA) / área padrão interno) / (área analito (FD) / área
padrão interno)] x 100
Onde:
FA = fortificado antes do processo de extração
FD = fortificado depois do processo de extração
3.12.2. Protocolo de validação para análise qualitativa O protocolo de validação de métodos para análise qualitativa consiste em uma série
de experimentos, que devem ser realizados em um único dia, nos quais são avaliados os
seguintes parâmetros: seletividade / especificidade, limite de detecção, precisão intra-ensaio
(repetitividade), além da avaliação de valores aberrantes. No presente trabalho, este
protocolo foi aplicado aos agentes anabólicos cujo limite mínimo de desempenho requerido é
10 ng/mL e foi possível obter o padrão analítico. Sendo estes: tibolona, zeranol, letrozol e
formestano.
3.12.2.1. Especificidade / seletividade O teste de especificidade serve para assegurar que não hajam interferentes na região
do analito, ou seja, o valor medido provém somente do analito. Determina a capacidade de
um método em determinar um analito na presença de outros compostos presentes na matriz
da amostra. Foram analisados dez brancos de urina, escolhidos aleatoriamente, e os
resultados dessas análises foram comparados com os obtidos a partir da urina fortificada
com o analitos na concentração equivalente ao limite mínimo de desempenho requerido. No
caso de urinas de excreção, os resultados dessas análises foram comparados com os
obtidos a partir da urina de excreção extraída e analisada no modo monitoramento seletivo
de íons e varredura linear.

Experimental 62
3.12.2.2. Limite de detecção É a concentração mínima de analito que pode ser detectada com precisão aceitável.
O LD de um composto é definido como o menor nível fortificado para o qual, em todas as
amostras de urina, o composto alvo pode ser detectado com uma relação sinal / ruído maior
ou igual a três. A determinação do limite de detecção é descrita pela Eurachem e, de acordo
com esse guia, para determinação do LD, para métodos de análise qualitativa, dez replicatas
independentes devem ser fortificadas com padrões em diferentes níveis de concentração.
Isso significa fortificar dez brancos de urina de origens diferentes (escolhidos
aleatoriamente), em três níveis de concentração diferentes, sendo estes: ½ x LMDR, LMDR,
2 x LMDR. Um total de trinta amostras são preparadas e analisadas.
3.12.2.3. Precisão intra-ensaio (repetitividade) É a medida do erro aleatório expressa pela dispersão obtida em uma série de
medidas repetidas realizadas durante um mesmo ensaio (lote experimental).
A precisão foi determinada através da repetitividade dos resultados em todos os
níveis de concentração testados, correspondente a ½ x LMDR, LMDR, 2 x LMDR dos
analitos alvo, sendo expressa como o coeficiente de variação (CV%), não se admitindo
valores superiores a 20%.
3.12.2.4. Valores aberrantes
Valores aberrantes são aqueles que possuem um desvio maior do que o aceitável em
torno de uma média. Os testes de rejeição são utilizados para centrar os valores ao redor da
média. O teste de Grubbs apresenta os valores que devem ser rejeitados em relação à
estimativa do desvio padrão e foi aplicado com nível de confiança 95%.
Este teste avalia os valores dispersos maiores ou menores que aparecem no grupo
de medidas. A ausência de valores aberrantes é observada quando o valor de G calculado é
inferior ao valor de G tabelado.
Os valores de G calculados para as respostas superiores e inferiores obtidas para as
replicatas em cada nível de concentração nas amostras de calibração, foram obtidos do
seguinte modo:
S
xxGcal
−=
*

Experimental 63
onde:
Gcal - corresponde ao valor de G calculado;
x* - corresponde ao valor suspeito aberrante (maior ou menor);
x - corresponde a média dos valores analisados;
S - corresponde ao desvio padrão dos valores analisados.
O teste de Grubbs é o mais indicado para avaliação de valores aberrantes segundo
recomendação das normas ISO e AOAC (KELLY, 1990; HARTMANN et al., 1998).
3.12.3. Protocolo de validação para análise semi-quantitativa
O protocolo de validação de métodos para análise semi-quantitativa consiste em uma
série de experimentos, que podem ser realizados em um mesmo dia, no qual são avaliados
os seguintes parâmetros: seletividade / especificidade, contaminação entre amostras
(arraste), limite de detecção (LD), limite de quantificação (LQ), e precisão intra-ensaio
(repetitividade). Este protocolo foi aplicado aos agentes anabólicos cujo limite mínimo de
desempenho requerido é 2 ng/mL e 1 ng/mL. Sendo estes: clembuterol, metandienona,
metiltestosterona, norandrosterona e estanozolol. Dentre estes somente norandrosterona
apresenta valor de corte, sendo este igual a 2 ng/mL. Clembuterol, metandienona,
metiltestosterona e estanozol apresentam LMDR igual a 2 ng/mL enquanto norandrosterona
apresenta LMDR igual a 1 ng/mL.
3.12.3.1. Especificidade Idem item 3.12.2.1.
3.12.3.2. Contaminação entre amostras (arraste)
Consiste na capacidade do método em produzir resultados referentes a uma amostra,
independentemente da amostra analisada anteriormente.

Experimental 64
3.12.3.3. Limite de detecção (LD)
O limite de detecção corresponde à menor quantidade de um analito que pode ser
detectada, porém, não necessariamente quantificada.
Na prática o limite de detecção pode ser determinado como a menor concentração do
analito que pode ser diferenciada do ruído do sistema com segurança. O ruído (N, noise) é a
amplitude esperada da linha base, a qual inclui todas as variações aleatórias do sinal do
detector cuja freqüência esteja na ordem de um ou mais ciclos por minuto.
Um procedimento comum é aceitar como limite de detecção a concentração ou massa
do analito que gera um sinal três vezes maior do que o ruído do sistema, ou seja, LD = 3N.
Outra forma é definir o LD como a concentração ou massa do analito que produz um sinal
igual a 3s, em que s é o desvio padrão do ruído médio empregando-se um branco em vez do
sinal do equipamento.
Para determinação do limite de detecção, utilizando o protocolo de validação semi-
quantitativa, dez brancos de urina foram injetados e o desvio padrão do ruído médio foi
determinado.
3.12.3.4. Limite de quantificação (LQ)
O limite de quantificação corresponde à menor quantidade de um analito que pode ser
quantificada com precisão e exatidão desejável. Pode ser definido também em relação ao
ruído empregando-se o branco como referência. Nesse caso, seria o desvio padrão do sinal
gerado empregando-se um branco e o limite de quantificação seria o valor correspondente a
10s.
Para determinação do limite de quantificação, utilizando o protocolo de validação
semi-quantitativa, adotou-se o mesmo procedimento para determinação do LD para o
cálculo do desvio padrão. Em seguida esse valor foi multiplicado por 10 e o LQ foi
determinado.
3.12.3.5. Precisão intra-ensaio (repetitividade)
A precisão, neste caso, foi determinada através da repetitividade dos resultados de
urinas fortificadas na concentração igual ao valor mínimo de desempenho requerido. O que
corresponde a 2 ng/mL para clembuterol, metiltestosterona, metandienona, estanozolol e

Experimental 65
1ng/mL para norandrosterona. A repetitividade foi expressa como o coeficiente de variação
(CV%), não se admitindo valores superiores a 20%.
3.12.3.6. Valores aberrantes
Idem item 3.12.2.4.
3.12.4. Protocolo de validação para análise quantitativa
O protocolo de validação de métodos para análise quantitativa consiste em uma série
de experimentos, que devem ser realizados em ao menos dois dias consecutivos, nos quais
são avaliados os seguintes parâmetros: seletividade / especificidade, contaminação entre
amostras (arraste), curva de calibração / linearidade, limite de detecção (LD), limite de
quantificação (LQ), precisão e inter-ensaio, exatidão inter-ensaio e incerteza expandida de
medição. Segundo o ISL não é necessário a aplicação de protocolo de validação para
análise quantitativa a nenhum dos resíduos monitorados no controle de dopagem. Para a
maior parte dos analitos deve-se apenas provar que podem ser detectados segundo
rigorosos critérios de qualidade no valor do LMDR e para um pequeno grupo deve-se provar
que podem ser detectados acima de determinado valor de corte. Entretanto com o objetivo
de verificar a possibilidade de uma análise quantitativa no equipamento VARIAN 4000, foi
proposta a validação quantitativa de 3’OH-estanozolol (metabólito do estanozolol), de acordo
com o esquema apresentado abaixo (tabela 6):
Tabela 6: Protocolo para a validação do método. Experimento 1 Experimento 2 Experimento 3
Especificidade
Contaminação entre
amostras
Limite de detecção
Precisão inter-ensaio
Exatidão inter-ensaio
Limite de quantificação
Incerteza de medição

Experimental 66
3.12.4.1. Especificidade
Idem item 3.12.2.1.
3.12.4.2. Contaminação entre amostras (arraste) Idem item 3.12.3.2.
3.12.4.3. Valores aberrantes
Idem item 3.12.2.4.
3.12.4.4. Homocedasticidade / Heterocedasticidade
Consiste na comprovação da independência/dependência da variância das respostas
do método em relação à concentração do analito nas amostras analisadas.
Para avaliar o grau de dependência ou independência da variância das respostas,
com a concentração do analito ao longo das curvas de calibração nos diferentes ensaios foi
realizada uma prova de homogeneidade de variâncias, ou seja, o teste de Cochran (α=0,05).
Este teste consiste na comparação da razão entre a maior variância obtida na curva de
calibração e o somatório das variâncias calculadas para todos os pontos da curva.
∑=
= n
i
calculado
js
sC
1
2
2 max
onde:
s2max = maior variância entre todos os pontos da curva;
s2j = somatório de todas as variâncias relativas a todos os níveis da curva.
Quando Ccalculado menor que Ccrítico (tabelado), o método é considerado
homocedastico. Quando Ccalculado maior que Ccrítico, o método é considerado heterocedastico.

Experimental 67
3.12.4.5. Curva de calibração / Linearidade
A linearidade do sistema de CGAR-EM foi testada através da injeção de amostras de
urina fortificadas com soluções-padrão dos analitos alvo, em concentrações crescentes, ou
seja, através da construção de curvas de calibração.
As curvas de calibração foram usadas para o estabelecimento da linearidade, para a
quantificação de amostras e para a avaliação da incerteza expandida. Recomenda-se que a
curva de calibração seja realizada por meio da análise de amostras extraídas (branco de
urina fortificado com analitos alvo no início do procedimento), no mínimo com 5
concentrações diferentes, dentro da faixa de trabalho requerida. Para o presente trabalho as
curvas de calibração foram compostas por 5 níveis de concentração, 0,2, 0,4, 0,5 1,0 e 2,0
(primeira curva) e 10, 25, 50, 75 e 100 (segunda curva) ng por 1 mL de branco de urina
humana. Foi realizada a análise de cada nível de concentração em uma única replicata
(n=1). As curvas de calibração foram construídas nos três dias de validação por analistas
diferentes.
A linearidade consiste na demonstração de que a relação entre a concentração do
analito (x) e a medida da resposta (y) se ajusta a uma equação do tipo y = ax + b, onde a e b
são os parâmetros próprios da equação obtida.
Para a avaliação da linearidade deve-se calcular o desvio entre a concentração obtida
e a esperada e devem ser observados os critérios descritos abaixo.
Critérios de aceitação dos resultados:
• Desvio de, no máximo, 20% para o ponto de concentração próxima ao limite de
quantificação do método (LQ);
• Desvio de, no máximo, 15% para as outras concentrações;
• O valor do coeficiente de correlação deve ser r > ou = a 0,99.

Experimental 68
3.12.4.6. Limite de detecção (LD) e limite de quantificação (LQ)
O LD é estabelecido por meio da análise de soluções de concentrações conhecidas e
decrescentes do alvo analítico, até o menor nível detectável. Recomenda-se que o LD seja 3
vezes superior ao ruído da linha de base.
O LQ é a concentração mínima do analito que pode ser quantificada com a exatidão e
a precisão adequadas.
O pico de resposta do analito no LQ deve ser identificável e reprodutível de acordo
com os seguintes critérios:
Precisão de, no mínimo, 20%;
Exatidão entre 80 e 120 %.
3.12.4.7. Precisão e exatidão inter-ensaio
Precisão inter-ensaio (Precisão intermediária)
É a medida do erro aleatório expressa pela dispersão obtida em uma série de
medidas repetidas, realizadas durante diversos lotes experimentais, processados em dias
distintos.
Exatidão inter-ensaio
É o grau de concordância entre o valor de uma medição (expresso como a média de
uma série de medidas durante diversos lotes experimentais) e o valor nominal.
Para avaliação da exatidão e da precisão utiliza-se o valor dos controles, preparados
para construção da curva de calibração.
A precisão foi determinada através da repetitividade dos resultados da concentração
correspondente ao LMDR dos analitos alvo, sendo expressa como o coeficiente de variação
(CV%), não se admitindo valores superiores a 15%.
A exatidão do método foi estabelecida através da relação entre a concentração média
obtida experimentalmente e a concentração teórica correspondente, expressa conforme a
equação abaixo:
Exatidão = [(concentração média experimental – concentração teórica) / concentração
teórica] x 100
A exatidão deve estar compreendida no intervalo de 80 a 120%.

Experimental 69
3.12.4.8. Cálculo da incerteza expandida (U)
A incerteza de medição é uma medida útil para demonstrar qualidade de resultados
analíticos, isto é, demonstrar a adequação ao uso, dando um intervalo de confiança que
pode ser atribuído a um resultado, incluindo a faixa que um resultado pode concordar com
outros resultados.
Segundo o Vocabulário Internacional de Metrologia (VIM), a incerteza de medição
pode ser definida como um parâmetro, associado ao resultado de uma medição, que
caracteriza a dispersão dos valores que podem ser razoavelmente atribuídos a um
mensurando.
Para que a incerteza de medição possa ser calculada é preciso, inicialmente que
sejam determinados os fatores que influenciam a medida em questão (ou seja, as fontes de
incerteza que afetam o resultado). Várias são as fontes de incerteza: amostragem, efeitos de
matriz e interferentes, condições ambientais, incertezas de pesos, equipamentos
volumétricos e padrões de referência além de variações aleatórias.
Na estimativa da incerteza total, é necessário tratar separadamente cada fonte de
incerteza para determinar sua contribuição. Cada uma das contribuições separadas de
incerteza é tratada como um componente de incerteza. Quando expressa como um desvio
padrão, um componente da incerteza é conhecido como incerteza padrão, µ(y).
Para um resultado de medição y, a incerteza total ou incerteza padrão combinada,
µc(y), é uma estimativa do desvio padrão combinado igual à raiz quadrada positiva da
variância total obtida pela combinação de todos os componentes de incerteza, avaliados,
usando a lei da propagação de incerteza.
A incerteza expandida (U) representa o intervalo dentro do qual o valor do
mensurando é acreditado com um nível particular de confiança. A incerteza expandida (U) é
obtida pela multiplicação de µc(y), incerteza padrão combinada, por um fator de abrangência
K. A escolha do fator K é baseada no nível de confiança desejado. Para um nível de
aproximadamente 95%, K é 2.
No presente trabalho, foram consideradas como fontes de incerteza: a incerteza
associada a variância média relacionada ao desvio padrão experimental das n replicatas de
três níveis da curva de calibração (QCA, QCM e QCB) , a incerteza associada a curva de
calibração e a incerteza associada ao padrão de referência (Fig. 30).

Experimental 70
Repetitividade
Medida
Padrão
Curva de calibração
Figura 30: Diagrama de causa-efeito (espinha de peixe).
A incerteza padrão combinada foi determinada pela raiz quadrada da soma dos
quadrados de todas as fontes de incerteza.
222 scsspc ++=µ
Onde:
sp2 corresponde à incerteza associada ao padrão de referência utilizado na análise. Este
valor é expresso em termos do desvio padrão da pureza do material de referência. Este
valor é retirado diretamente do certificado de calibração. Se o certificado de análise do
padrão de referência apresentar a incerteza do padrão ao invés do desvio padrão, este valor
é transformado em desvio padrão, através da divisão do mesmo pelo fator de abrangência
K, geralmente presente no certificado de calibração. Uma vez ausente assumi-se K = 3,
considerando tratar-se de uma distribuição retangular.
sc2 corresponde à incerteza da curva de calibração determinada pela equação:
( )1
1
2
2
−
−=
∑=
n
xxiSc
n
i
2s corresponde a variância média obtida através do desvio padrão experimental (S2),
segundo a equação:
nSS
22=
Onde:
n representa o número de replicatas, por nível, utilizadas para construção da curva de
calibração.
Incerteza expandida (U)
A incerteza expandida é determinada através da incerteza padrão combinada (µc),
segundo a equação abaixo:

Experimental 71
%95,VeffctU µ=
Veff = graus de liberdade efetivos
∑=
=n
i i
i
cVefft
1
4
4
νµ
µ
µi representa o desvio padrão das replicatas que geram o maior valor de µc.
νi representa os graus de liberdade de cada fonte de incerteza (i).
( ) ⎥⎦
⎤⎢⎣
⎡+⎥
⎦
⎤⎢⎣
⎡−
=⎟⎟⎠
⎞⎜⎜⎝
⎛∑ 501
44Pi
i
i Snµ
νµ
Utliza-se 50 como número de graus de liberdade para Sp porque é um número que
permite considerar K=2.
3.13. MONITORAMENTO DO TEMPO NECESSÁRIO PARA HIDRÓLISE DE AGENTES ANABÓLICOS A investigação da cinética da hidrólise de metabólitos conjugados de agentes
anabólicos foi investigada a partir da extração de urinas de excreção onde a etapa de
hidrólise foi controlada.
Alíquotas de 2 mL de amostras de urina foram fortificadas com solução de padrão
interno (ISTD - item 3.8.3) e 50 µL da enzima β-glicuronidase de E. coli. As amostras foram
incubadas a 50°C. A hidrólise foi interrompida após diferentes períodos de tempo: 0, 5, 15,
30, 120 e 240 minutos. Após interrupção da hidrólise as amostras foram preparadas de
acordo com o procedimento descrito no item 3.9.1.1 e analisada de acordo com o item
3.10.1.
As amostras de urina também foram extraídas sem a adição da enzima, para checar a
existência de metabólitos excretados livres (não conjugados). Essas amostras foram
extraídas de acordo com o item 3. 9.1.1 com exceção da etapa da hidrólise e analisados de
acordo com o item 3.10.1, mas nesse caso no modo de aquisição varredura linear (SCAN).

CAPÍTULO 4
RESULTADOS E DISCUSSÃO

Resultados e Discussão 72
4. RESULTADOS E DISCUSSÃO 4.1. Determinação das condições de análise cromatográfica e da detecção de agentes anabólicos. A escolha da técnica CGAR-EM para a análise de agentes anabólicos baseou-se na
necessidade da incorporação de novos resíduos ao método já existente no laboratório. Cabe
ressaltar que inúmeras publicações enfatizam a aplicação dessa técnica para análise de
resíduos de fármacos, destacando-se agentes anabólicos em urina e plasma de atletas. O
espectrômetro de massas é um dos detectores mais utilizados para análise de traços em
amostras biológicas, por sua alta sensibilidade e especificidade, favorecendo assim sua
aplicação para análise de agentes anabólicos, que em sua grande maioria são encontrados
a nível de ng / mL em matrizes biológicas.
A cromatografia gasosa é uma técnica de separação de compostos voláteis e
termicamente estáveis. Entretanto muitos compostos de interesse biomédico e ambiental
contém grupos funcionais polares, dificultando assim sua análise por CG. Esses compostos
normalmente apresentam baixa volatilidade, são instáveis à temperatura requerida para
análises cromatográficas, ou ainda podem interagir fortemente com a fase estacionária de
uma coluna cromatográfica. Assim, a obtenção de derivados menos polares e mais voláteis,
estáveis termicamente, torna-se imprescindível para análises cromatográficas. Esses
derivados são obtidos pelas reações de derivatização. A análise de agentes anabólicos por
CG requer uma etapa de derivatização antes da injeção no sistema cromatográfico, devido a
presença de grupos funcionais polares (hidroxilas, cetonas e ácidos carboxílicos). No caso
do método em questão foram preparados derivados trimetilsilila (TMS), uma vez que estes
apresentam estabilidade térmica e química além de alta volatilidade. N-metil-n-
trimetilsililtrifluoracetamida (MSTFA) é o reagente mais empregado na derivatização dos
esteróides anabolizantes, sendo aplicado em combinação com catalisadores como
carbonato de potássio, trimetilsilil-imidazol (TMSIm), iodeto de trimetilsilila (TMSI) ou iodeto
de amônio (NH4I) + agente redutor (etanotiol ou 2-mercaptoetanol).
As condições de derivatização empregadas para obtenção dos derivados TMS para
os agentes anabólicos em estudo, seguiu a metodologia descrita na figura 30.

Resultados e Discussão 73
Amostra (resíduo seco)
Adicionar 100 µL da solução derivatizante MSTFA : NH4I : 2-mercaptoetanol
(1000:2:6)
Colocar em banho termostatizado a 60°C durantes 20 min
Figura 31: Fluxograma das condições de derivatização. A AMA através do código mundial anti-dopagem (ISL versão 4.0, agosto de 2004),
recomenda que na ausência de substância de referência, seja realizado um estudo de
excreção através da administração do fármaco ou suplemento nutricional que
conhecidamente apresente em sua composição o alvo analítico ou que este possa ser
gerado através do metabolismo humano. Quando for inviável a realização de estudo de
excreção, pode-se trabalhar com urina de usuários crônicos de algum medicamento e / ou
complemento nutricional. Para comprovar que na urina humana o analito alvo esta ausente
recomenda-se realizar a análise de dez brancos de urina coletados aleatoriamente
juntamente com a análise da urina que contém a(s) substância(s) de interesse.
Para implementação da análise dos agentes anabólicos metilnortestosterona,
metasterona, hiperdrol, tamoxifeno, clomifeno, ciclofenila, letrozol, alfa e beta zeranol,
tibolona e formestano utilizou-se o padrão interno metiltestosterona. A utilização de um
padrão interno evita erros de quantificação provocados por problemas na preparação das
amostras (métodos de extração), variações instrumentais e erros na introdução da amostra
no sistema cromatográfico. A padronização interna consiste em adicionar uma quantidade
conhecida de uma substância padrão nas amostras a serem analisadas, e relacionar as
duas áreas obtidas (padrão interno e analito alvo).
A figura 32 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), do padrão interno metiltestosterona, após
etapa de derivatização.
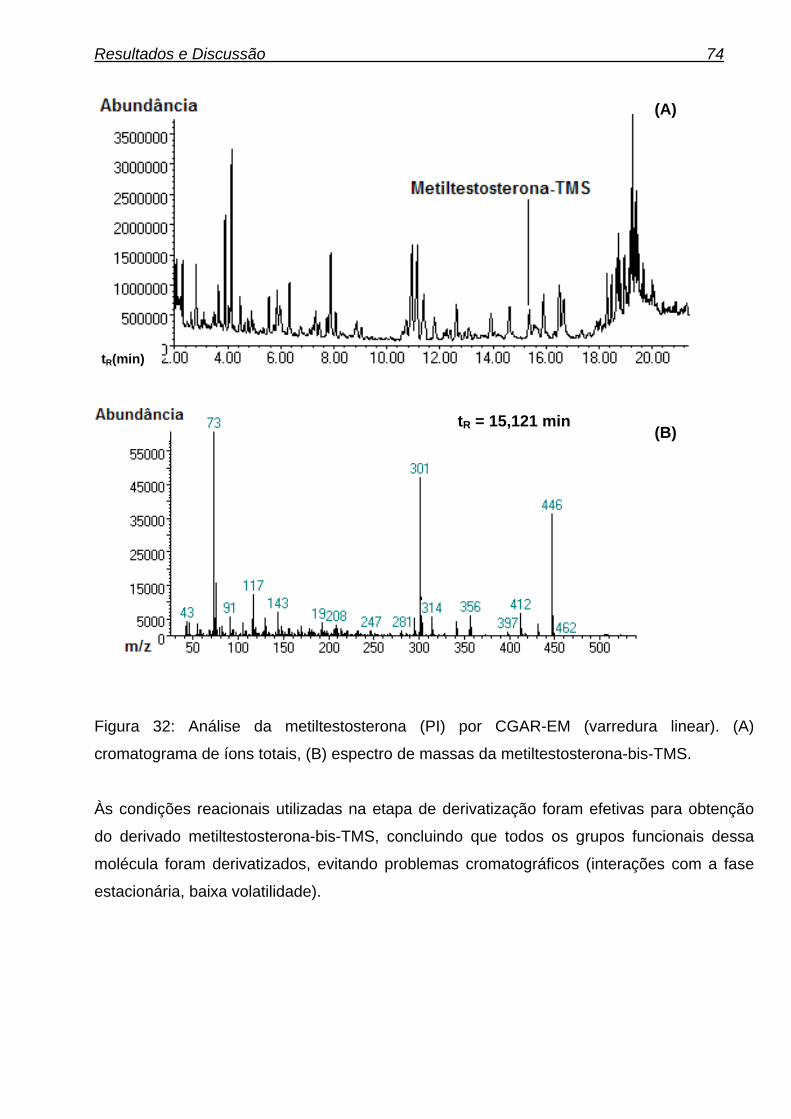
Resultados e Discussão 74
tR(min)
(A)
(B)tR = 15,121 min
Figura 32: Análise da metiltestosterona (PI) por CGAR-EM (varredura linear). (A)
cromatograma de íons totais, (B) espectro de massas da metiltestosterona-bis-TMS.
Às condições reacionais utilizadas na etapa de derivatização foram efetivas para obtenção
do derivado metiltestosterona-bis-TMS, concluindo que todos os grupos funcionais dessa
molécula foram derivatizados, evitando problemas cromatográficos (interações com a fase
estacionária, baixa volatilidade).

Resultados e Discussão 75
O
CH3
CH3OH
CH3
+ I Si(CH3)3
CH3
CH3O
CH3
OSiH3C
CH3
CH3
Si
CH3H3C
CH3
Figura 33: Reação de derivatização da metiltestosterona. 4.1.1. Padronização interna e controle da qualidade do método
A todas as amostras foram adicionados metiltestosterona-P, testosterona-D3,
epitestosterona-D3 e D4-etiocolanolona (solução de padrão interno - ISTD) e padrão D5-
androsterona glicuronídeo. D5-androsterona glicuronídeo funciona para verificar a eficiência
da hidrólise, quando menos que 25% de D5-androsterona é detectado, a amostra é re-
preparada, pois considera-se que também para os analitos alvos a hidrólise pode não ter
sido efetiva. D4-etiocolanolona é utilizada como padrão interno para alguns esteróides
endógenos (androsterona e etiocolanolona).
Através da observação dos derivados Androsterona mono e bis TMS, realiza-se o
controle da eficiência da reação de derivatização. Quando é possível a observação de mais
que 5% do derivado androsterona mono-TMS em relação a androsterona bis-TMS, a
amostra é re-derivatizada. Se o problema persistir a amostra é re-preparada.
tR(min)
Androsterona bis-TMS
(A)
Androsterona mono-TMS
tR(min)

Resultados e Discussão 76
tR(min)
Androsterona mono-TMS
(B)
Androsterona bis-TMS
tR(min)
Figura 34: Controle de derivatização. (A) Amostra de urina não derivatizada, (B) Amostra de
urina derivatizada.
4.2. Implementação de agentes anabólicos a partir de urinas de excreção Seis compostos foram incluídos na rotina da triagem de agentes anabólicos, a partir
de urinas de excreção: Metilnortestosterona, Metasterona, Hiperdrol, Tamoxifeno, Clomifeno
e Ciclofenila.
4.2.1. Metilnortestosterona
Metilnortestosterona, foi apontada pela AMA, pela primeira vez como substância
proibida em 2006. Desde então, esforços foram feitos na tentativa de obtenção de urina de
excreção ou material de referência para implementação desse composto na rotina da
triagem responsável pelo monitoramento de agentes anabólicos do LABDOP.
Este esteróide nos dias de hoje é comercializado através da internet (mercado negro)
com forte apelo por apresentar atividade anabólica preponderante sobre a androgênica. Em
fevereiro de 2006 o departamento federal de saúde do Canadá alertou os consumidores
para o potencial risco que esse composto apresenta, no sentido de causar desordens no
fígado e endurecimento de artérias.

Resultados e Discussão 77
O
OHCH3
OHCH3
HHO
Metilnortestosterona-met. Metilnortestosterona
Figura 35: Possível metabólito da Metilnortestosterona.
Podemos supor que metilnortestosterona sofra as reações metabólicas clássicas do
anel A de um EAA. Nesse caso a ligação dupla entre C4 e C5 seria reduzida produzindo os
isômeros 5α e 5β. Em seguida o grupo ceto em C3 seria reduzido, formando
predominantemente o isômero 3α-hidróxido. A partir desse raciocínio os seguintes
metabólitos poderiam ser formados.
O
OHCH3
OHCH3
HHO
OHCH3
HHO
OHCH3
HHO
OHCH3
HHO
Figura 36: Proposta de possíveis metabólitos da Metilnortestosterona.
A urina de excreção foi submetida ao procedimento para extração de agentes
anabólicos em urina (item 3.9.1.1), o extrato final foi derivatizado e injetado em CGAR-EM
(item 3.10.1) (aquisição no modo varredura linear – SCAN), na tentativa de detectar a droga
mãe e seu metabólito hidroxilado proposto acima. Os íons m/z 432 (íon molecular) e m/z 143
(fragmento característico da quebra do anel D com adicional abstração de hidrogênio)
referentes a 17α-metilnortestosterona foram procurados, não tendo sido possível encontrar
nenhum sinal referente a esse composto. Partiu-se para tentativa de identificação do

Resultados e Discussão 78
metabólito hidroxilado, utilizando-se os íons m/z 143 e m/z 421. Foi possível detectar um
sinal a aproximadamente 12,10 min no modo varredura linear, que acreditamos tratar-se do
metabólito 17α-metil-5ξ-estran-3ξ,17β-diol.
A figura 37 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), de metilnortestosterona, após etapa de
derivatização.
Os íons obtidos na ionização por IE do derivado metilnortestosterona-bis-TMS
(espectro de massas, figura 35B), sugerem fragmentação indicada na figura. O pico de m/z
421 foi atribuído ao íon molecular menos uma metila. O pico de m/z 143 foi atribuído ao
fragmento iônico obtido a partir de um rearranjo do derivado metlnortestosterona bis-TMS,
seguido pela cisão entre as ligações C-14 e C-15 do anel D. Os picos de m/z 421 e m/z 143
apresentam intensidades no espectro de massas de 66% e 100%, respectivamente.

Resultados e Discussão 79
Figura X: Análise da metilnortestosterona por CGAR-EM (varredura linear).
tR(min)
(A)
(B)
M+• - 15
OCH3
OH
Si
Si
CH3H3C
CH3
H3C
H3C
CH3
tR = 12,115 min
m/z 143
Figura 37: (A) cromatograma de íons totais, (B) espectro de massas do metabólito da
metilnortestosterona -bis-TMS.
Para inclusão de metilnortestosterona na rotina da triagem IV, assim como para todos
os outros compostos estudados, foram analisados dez brancos de urina (Fig. 37), doados
por voluntários de ambos os sexos escolhidos aleatoriamente, com idade entre 20 e 30
anos. Os íons m/z 143 e 421 foram inseridos no método de análise de agentes anabólicos
por CG-EM. O íon de maior intensidade (pico base), m/z 73, não foi selecionado por ser um
fragmento comum presente em derivados TMS (fragmento referente ao grupo trimetilsilila).
Em função de ser este um método multi-resíduo que já apresenta muitos íons por grupo,

Resultados e Discussão 80
onde cada grupo é dividido em função do tempo de retenção comportando não mais que
trinta íons (por grupo), somente dois íons diagnósticos foram inseridos.
tR(min)

Resultados e Discussão 81
Figura 38: Análise do fragmentograma da metilnortestosterona e dos dez brancos de urina
avaliados.
4.2.2. Metasterona
Assim como Metilnortestosterona, Metasterona passou a fazer parte da lista de
substâncias proibidas da AMA em 2006, sendo comercializada da mesma forma e
apresentando os mesmos riscos potencias a saúde.
Podemos supor que metasterona sofra as reações metabólicas clássicas que os
esteróides que apresentam o grupo ceto em C3. Este grupo é facilmente reduzido
produzindo predominantemente o isômero 3α-hidróxido (Fig. 39).
OHCH3
HO
H3C
OHCH3
H
H3C
HO
OHCH3
H
H3C
HO
Figura 39: Possíveis metabólitos da Metasterona.

Resultados e Discussão 82
Após submeter a urina de excreção ao procedimento para extração de agentes
anabólicos em urina (item 3.9.1.1.), o extrato final foi derivatizado e injetado em CGAR-EM,
na tentativa de detectar a droga mãe e seu metabólito hidroxilado proposto acima. Os íons
m/z 462 (íon molecular) e m/z 143 (fragmento característico da quebra do anel D com
adicional abstração de hidrogênio) referentes à metasterona (droga mãe) foram procurados.
Foi identificado um sinal a 15,65 min que pode ser atribuído a metasterona. Partiu-se para
identificação do metabólito hidroxilado, utilizando-se os íons m/z 464 e m/z 143. Não foi
possível a detecção do íon m/z 464, entretanto em 13,25 min foi possível a visualização de
um sinal que possuía o íon m/z 143 e 449. Acredita-se que este seja correspondente ao
metabólito hidroxilado (2α,17α-dimetil-5α-androstano-3α,17β-diol) da metasterona e que o
íon m/z 449, corresponda a perda de uma metila a partir do íon molecular (m/z 464).
No caso da metasterona os picos m/z 462 e m/z 143 apresentam intensidades no
espectro de massas de 51% e 86%, respectivamente. Para o metabólito hidroxilado os picos
m/z 449 e m/z 143 apresentam intensidades no espectro de massas de 17% e 100%,
respectivamente. O íon m/z 73 correspondente ao fragmento do grupo trimetilsilila refere-se
ao pico base.
A figura 40 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), de metasterona e seu metabólito, após
etapa de derivatização.

Resultados e Discussão 83
tR(min)
(A)
Figura 40: Análise da metasterona e seu metabólito por CGAR-EM (varredura linear). (A)
cromatograma de íons totais, (B) e (B1) espectro de massas da metasterona -bis-TMS e
metasterona-met.-bis-TMS.
M+•
M+•
m/z 464
M+• - 15
(B1)
OSi
H3C
H3C
CH3
O
CH3
H
H3C
Si
CH3
CH3
H3C
tR = 13,289 min
m/z 143
(B)
O
CH3
H
H3C
Si
CH3
CH3
H3C
OSi
H3C
H3C
CH3
tR = 15,645 min
m/z 143

Resultados e Discussão 84
A figura 41 apresenta o fragmentograma da metasterona, de seu metabólito e dos dez
brancos avaliados no estudo.
Metasterona

Resultados e Discussão 85
Metabólito da Metasterona

Resultados e Discussão 86
Figura 41: Análise do fragmentograma da metasterona, seu metabólito e dos dez brancos de urina
avaliados para cada substância.
Para inclusão de metasterona na rotina da triagem IV, procedeu-se como no caso da
metilnortestosterona. Os íons incluídos nesse caso foram o m/z 143 e o m/z 449. Por se
tratar o m/z 73 um fragmento comum presente em derivados TMS, o mesmo não foi incluído
no método. Cabe ressaltar, que nesse caso, deve-se realizar um novo estudo de excreção a
fim de identificar a espécie de mais longa excreção e se está sendo excretada na forma livre
ou na forma conjugada.
4.2.3. Hiperdrol
Assim como metilnortstosterona e metasterona, hiperdrol é comercializado através da
internet sob a forma de complemento alimentar denominado Superdrol. Passará a fazer
parte da lista de substâncias proibidas pela AMA a partir de janeiro de 2008. Foi possível a
obtenção de urina de excreção, através de um estudo realizado a partir do consumo de uma
única dose (1 cápsula) do suplemento (até o momento não comercializado no Brasil).
Após submeter a urina de excreção ao procedimento para extração de agentes
anabólicos em urina (item 3.9.1.1.), o extrato final foi derivatizado e injetado em CGAR-EM,
na tentativa de detectar a droga mãe. Os íons m/z 514 e m/z 512 (isótopos do Bromo) foram
procurados, tendo sido encontrado um sinal a 16,50 min que acreditamos tratar-se do
hiperdrol (droga mãe).

Resultados e Discussão 87
No caso do hiperdrol os picos m/z 512 e m/z 514 apresentam intensidades no
espectro de massas de 84% e 93%, respectivamente.
A figura 42 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), de hiperdrol, após etapa de derivatização.
tR(min)
(A)
(B)
M+•
O
Si
CH3H3C
CH3
OSi
H3C
CH3
CH3Br
tR = 16,579 min
Figura 42: Análise do hiperdrol por CGAR-EM (varredura linear). (A) cromatograma de íons
totais, (B) espectro de massas do hiperdrol-bis-TMS.

Resultados e Discussão 88
Figura 43: Análise do fragmentograma do hiperdrol e dos dez brancos de urina avaliados para cada
substância.

Resultados e Discussão 89
Para inclusão deste composto na triagem IV, procedeu-se como descrito para
metilnortestosterona e metastesrona. Também para Hiperdrol, deve-se realizar um estudo
de excreção a fim de verificar a existência ou não de metabólitos e se a droga mãe é
excretada livre ou conjugada.
4.3. Implementação de agentes anabólicos e estudo da cinética da hidrólise
Para clomifeno, tamoxifeno, ciclofenil e letrozol (validação qualitativa em função da
disponibilidade de padrão – item 3.12.2) foi possível um estudo para avaliação da cinética da
hidrólise dos metabólitos conjugados dos quatro fármacos. Além da investigação sobre a
possibilidade de excreção de metabólitos livres.
A inclusão destes compostos na rotina da triagem responsável pelo monitoramento de
agentes anabólicos, foi possível em função da doação de urina de tamoxifeno e letrozol de
pacientes mulheres que iniciam tratamento para evitar a recidiva de câncer de mama. A
amostra foi coletada após administração da primeria dose (40 mg de tamoxifeno e 2,5 mg de
letrozol). Nesse momento ambas as pacientes não haviam iniciado o uso de outros
medicamentos. Realizou-se um estudo de excreção através da administração de uma única
dose terapêutica de clomifeno (100 mg) e ciclofenila (400mg) a indivíduos do sexo masculino
com idade entre 20 e 30 anos. Nos dois primeiros dias todas as amostras de urina foram
coletadas.
As amostras de urina foram preparadas de acordo com o procedimento para extração
de agentes anabólicos em urina (item 3.9.1.1.), o extrato final foi derivatizado e injetado em
CGAR-EM.

Resultados e Discussão 90
4.3.1. Clomifeno
Monohidroxiclomifeno foi identificado como sendo o principal metabólito do clomifeno.
Foi possível a detecção do íon molecular m/z 493 correspondente ao derivado mono-TMS e
m/z 495 referente aos isótopos do cloro. O pico base m/z 86, corresponde a clivagem α-
nitrogênio e o fragmento m/z 100 corresponde a clivagem β-nitrogênio. O íon molecular
diagnóstico apresenta-se muito pouco abundante, os íons m/z 86 e 100 embora abundantes,
apresentam massa baixa, não sendo considerados “bons” íons diagnósticos. Em função da
pobre fragmentação do clomifeno foram propostos novos metabólitos (Figua 42), e o íon
molecular de cada estrutura proposta foi procurado após injeção no modo varredura linear
(SCAN). Nenhum outro metabólito foi encontrado, para consolidar esse resultado faz-se
necessário uma concentração maior da amostra, extração com alíquotas de 8 mL, para
posterior análise.
Cl
ON
CH3
CH3
Cl
O
NH
CH3
HO
Cl
O
NCH3
CH3
HO
Cl
HO
O
NH
CH3
OH
Cl
O
NCH3
CH3
O OH
H3C
Cl
HO
ON
CH3
CH3
OH Figura 44: Proposta de possíveis metabólitos do clomifeno.
Di-hidroxi-clomifeno
Mono-hidroxi-N-desacetil-clomifeno
Mono-hidroxi-metoxi-clomofeno
Di-hidroxi-N-desacetil-clomifeno
Mono-hidroxi-clomifeno
Clomifeno

Resultados e Discussão 91
Os picos m/z 86, m/z 100 e m/z 493, referentes a possível metabólito hidroxi-
clomifeno, apresentam intensidades no espectro de massas de 100%, 20% e 5%,
respectivamente.
A figura 45 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), de hidroxi-clomifeno, após etapa de
derivatização.
(B)
tR(min)
(A)
M+•
Cl
O
NCH3
CH3
O
SiH3C
H3C
CH3
tR = 19,484 min m/z 86
m/z 100
Figura 45: Análise do clomifeno por CGAR-EM (varredura linear). (A) cromatograma de íons
totais, (B) espectro de massas do hidroxi-clomifeno-mono-OTMS.

Resultados e Discussão 92

Resultados e Discussão 93
Figura 46: Análise do fragmentograma de hidroxiclomifeno e dos dez brancos de urina avaliados
para cada substância.
4.3.2. Tamoxifeno
Hidroxi-metoxi-tamoxifeno foi identificado como sendo o principal metabólito do
tamoxifeno. Foi possível a identificação do íon molecular m/z 489, do pico base m/z 58 e de
um segundo fragmento m/z 72. O íon m/z 58 corresponde a clivagem α-nitrogênio e o m/z 72
a clivagem β-nitrogênio.
Os picos m/z 58, m/z 72 e m/z 489, referentes a possível metabólito metoxi-hidroxi-
tamoxifeno, apresentam intensidades no espectro de massas de 100%, 55% e 20%,
respectivamente. Para o metabólito carboxi-tamoxifeno os picos m/z 58, m/z 72 e m/z 473,
apresentam intensidades no espectro de massas de 100%, 30% e 10%, respectivamente.
A figura 47 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), de metoxi-hidroxi-tamoxifeno e carboxi-
tamoxifeno, após etapa de derivatização.
tR(min)
(A)

Resultados e Discussão 94
M+•
O
H3C
O
O CH3
SiCH3
CH3
H3C
N
CH3
CH3
tR = 18,961 min
m/z 72
m/z 58
(B1)
M+•
tR = 18,961 min
ON
TMSO
O m/z 72
m/z 58
(B2)
Figura 47: Análise do tamoxifeno por CGAR-EM (varredura linear). (A) cromatograma de
íons totais, (B1) espectro de massas metoxi-hidroxi-tamoxifeno-mono-OTMS (possível
metabólito) e (B2) espectro de massas do carboxi-tamoxifeno-mono-OTMS (possível
metabólito).
Carboxi-tamoxifeno

Resultados e Discussão 95

Resultados e Discussão 96
Metoxi-hidroxi-tamoxifeno

Resultados e Discussão 97
Figura 48: Análise do fragmentograma dos metabólitos do tamoxifeno e dos dez brancos de
urina avaliados para cada substância
4.3.3. Ciclofenila
Hidroxi-bis-desacetil-ciclofenil foi identificado como sendo o principal metabólito. Foi
possível a detecção do íon molecular m/z 512 correspondente ao derivado ciclofenil-tris-
TMS. O pico base m/z 422 corresponde ao íon molecular menos 90 (M+ - 90). O terceiro
fragmento mais abundante corresponde ao íon m/z 343.
Os picos m/z 422, m/z 343 e m/z 512 apresentam intensidades no espectro de
massas de 100%, 30% e 20%, respectivamente.
A figura 49 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), do ciclofenil, após etapa de derivatização
tR(min)
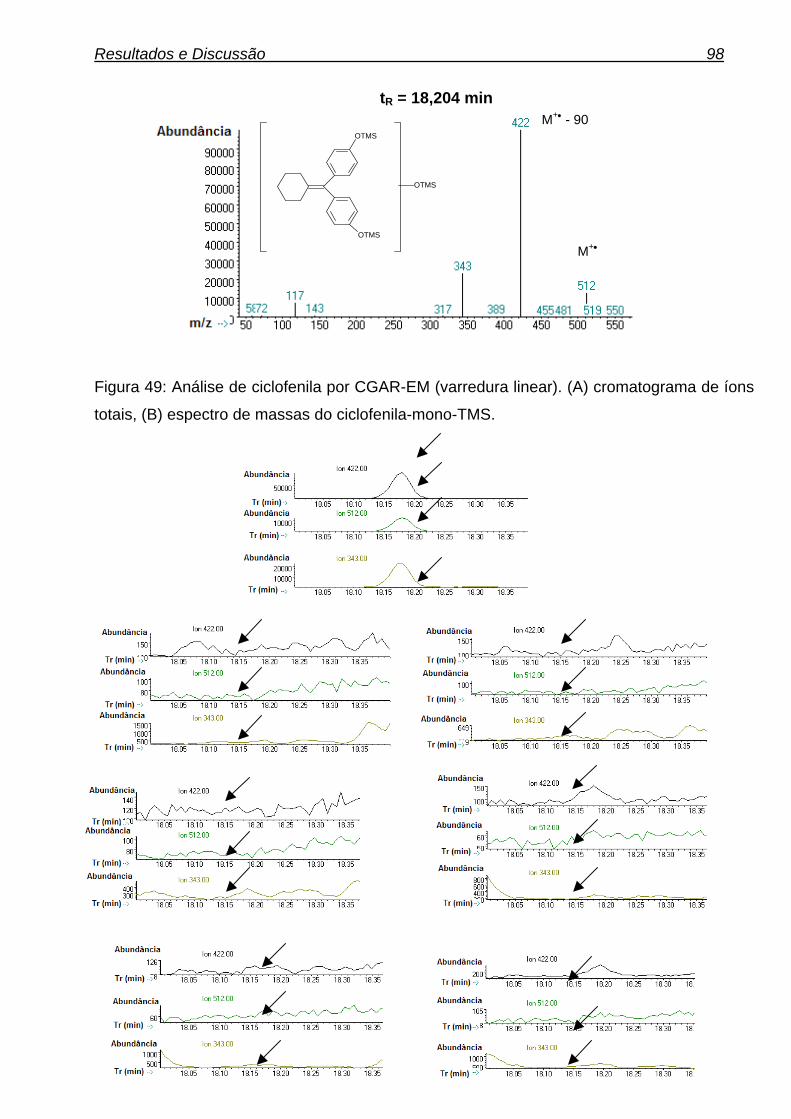
Resultados e Discussão 98
tR = 18,204 min
OTMS
OTMS
OTMS
M+• - 90
M+•
Figura 49: Análise de ciclofenila por CGAR-EM (varredura linear). (A) cromatograma de íons
totais, (B) espectro de massas do ciclofenila-mono-TMS.

Resultados e Discussão 99
Figura 50: Análise do fragmentograma do metabólito do ciclofenil e dos dez brancos de urina
avaliados para cada substância.
4.3.4. Cinética da hidrólise
As amostras de urina, provenientes do estudo de excreção, foram preparadas de
acordo com o procedimento para extração de agentes anabólicos em urina (item 3.9.1.1), o
extrato final foi derivatizado e injetado em CGAR-EM. A hidrólise foi interrompida após zero
(sem incubação), 5, 15, 30, 60, 120 e 240 minutos. A fim de verificar a presença de
metabólitos livres, uma amostra de cada urina foi preparada sem adição de enzima.
Tempo de incubação
(min)
Clomifeno (hidroxi
clomifeno) (%)
Tamoxifeno (metoxi-hidroxi-
tamoxifeno) (%)
Ciclofenil (%)
Letrozol (%)
Zero 10 0 5 0
5 30,8 0 15,2 98,7
15 32,4 10,4 98,5 97,6
30 35 10,2 98,9 98,4
60 100,4 40,4 100,3 99,8
120 104 60,4 100,1 99,7
240 105 61 100,4 100,1
Tabela 7: Cinética da hidrólise após adição de enzima.

Resultados e Discussão 100
Não foi possível detectar nenhum metabólito livre, proveniente da extração sem
adição de enzima. Foi observado que a hidrólise começa sem incubação, no tempo zero
10% de hidroxi-clomifeno foi detectado. Após uma hora a hidrólise foi considerada completa,
100% do metabólito estava presente, o mesmo foi observado após duas e quatro horas.
O metabólito metoxi-hidroxi-tamoxifeno não é hidrolisado sem incubação. Após meia-
hora apenas cerca de 10% do metabólito foi hidrolisado e após uma hora cerca de 40%.
Após quatro horas apenas 60% do metabólito hidrolisado foi detectado, o que indica a
necessidade de realizar um novo experimento incluindo 24 e 48 horas de incubação.
A hidrólise para o metabólito de ciclofenil apresentou-se extremamente rápida. Após
cinco minutos cerca de 5% do metabólito foi hidrolisado e após 15 minutos a hidrólise é
completa. Cerca de 2,2 % de ciclofenil foi detectado livre (sem adição de enzima).
Não foi possível detectar o metabólito glicoconjugado do letrozol sem adição de
enzima. Após cinco minutos de incubação a hidrólise já estava completa.
4.4. Implementação de agentes anabólicos a partir de padrões de referência
4.4.1. Validação qualitativa do método analítico A validação deve demonstrar que um método analítico cumpre os critérios aplicáveis
a características de desempenho relevantes. Na validação qualitativa os parâmetros
avaliados foram: especificidade, recuperação do processo de extração, valores aberrantes,
limite de detecção (LD), precisão intra-ensaio (repetitividade), contaminação entre amostras
(arraste). Em função da existência de padrões de referência foi possível aplicar esse ensaio
ao analitos: alfa e beta zeranol, tibolona, letrozol e formestano.
Especificidade
Os resultados para todos os alvo analíticos mostraram que não há interferência na
região cromatográfica de interesse, seja de substâncias endógenas presentes na urina.
Recuperação do analito A recuperação do analito foi calculada durante o primeiro dia de experimento, através
da análise de seis verdadeiras replicatas fortificadas no LMDR, no caso 10 ng/mL. As áreas
dos picos dos analitos, obtidas das amostras fortificadas antes do processo de extração,

Resultados e Discussão 101
foram comparadas com as áreas dos picos dos analitos obtidas das amostras fortificadas
depois do processo de extração, especificamente antes da evaporação do solvente. A
recuperação do analito corresponde à média dos valores de recuperação (n=6) para esta
concentração.
Identificação de valores aberrantes
Antes que qualquer informação fosse retirada dos dados obtidos na validação, estes
foram submetidos ao teste de Grubbs para identificação de valores aberrantes.
Teste de Grubbs para identificação de 1 valor aberrante
Este teste foi aplicado para verificar se o menor ou o maior valor de um conjunto de
dados de determinada concentração em estudo era aberrante.
Primeiramente, foi calculado o valor de G:
s
yyiG
−=
Onde:
“yi” é o valor suspeito de ser aberrante (pode ser o menor ou o maior valor dentre os
obtidos).
“ y ” é a média dos valores obtidos para uma determinada concentração.
“s” é o desvio padrão dos valores obtidos.
Se o valor de Gcalculado fosse superior ao valor de Gcrítico (Tabela 8) específico para o
número de replicatas (n) de cada concentração de estudo, o valor investigado era
considerado aberrante e, portanto, não incluído no cálculo para determinação do limite de
detecção. O nível de confiança utilizado para esse tipo de teste costuma ser α = 0,05.

Resultados e Discussão 102
Tabela 8: Valores de G crítico para identificação de 1 valor aberrante pelo teste de Grubbs
em diferentes níveis de confiança. n Valor de Gcrítico
α = 0,15 α = 0,10 α = 0,05
3 1,15 1,15 1,15
4 1,44 1,46 1,48
5 1,64 1,67 1,71
6 1,77 1,82 1,89
7 1,83 1,94 2,02
8 1,95 2,03 2,13
9 2,04 2,11 2,21
10 2,10 2,18 2,29
Limite de detecção (LD)
O limite de detecção foi determinado como descrito no item 3.12.2.2.
Precisão intra-ensaio (repetitividade)
Os resultados de precisão encontram-se nas tabelas 10 para alfa-zeranol; 12 para
beta-zeranol; 14 para tibolona; 16 para letrozol e 18 para formestano.
Os valores obtidos contemplam os critérios de aceitação descritos no item 3.12.2.3, ou seja,
a precisão mostrou desvio < 20% em todos os casos.
Contaminação entre amostras (arraste) Foi verificada a ausência de resposta para o analito em uma amostra de branco de
urina analisada imediatamente após uma amostra de controle na concentração de 100
ng/mL.
4.4.1.1. alfa-Zeranol Recuperação
A tabela 9 mostra os dados referentes à recuperação do alfa-zeranol. Segundo o
critério para avaliação dos resultados, o valor recomendável para a recuperação do analito
deve ser superior a 70%.

Resultados e Discussão 103
Tabela 9: Resultado da recuperação para o alfa-zeranol. Fortificado antes Fortificado depois
Resposta padrão analito (PI)
Resposta analito
Resposta analito / PI
Resposta padrão analito (PI)
Resposta analito
Resposta analito / PI
796736 23016 0,02889 785225 24543 0,03126 862314 20789 0,02411 771599 26317 0,03411 854311 21681 0,02538 786859 27770 0,03529 811731 22417 0,02762 738704 26936 0,03646 780633 23342 0,02990 737981 27575 0,03737 770826 22285 0,02891 727063 26131 0,03594
Recuperação (%) Recuperação média (%)
Desvio padrão (%) CV (%)
92,4234 70,6844 71,9092 75,7361 80,0241 80,4400
78,54 7,9 10,06
A recuperação do método para a determinação de alfa-zeranol foi de 78,54 ± 10,06%.
Limite de detecção, teste de Grubbs e repetitividade
A tabela 10 mostra os valores referentes ao experimento para determinação do limite
de detecção, a este conjunto de dados foi aplicado o teste de Grubbs e foi calculada a
repetitividade para todos os níveis de concentração.

Resultados e Discussão 104
Tabela 10: Valores para determinação do limite de detecção e repetitividade. Nivel de estudo Área analito Área PI Razão analito / PI G calculado CV%
10637 907657 0,01172 1,4822 8197 898979 0,00912 5844 845244 0,00691 7143 920826 0,00776 7873 850874 0,00925 9652 853465 0,01131 6046 925777 0,00653 1,5097 8740 947213 0,00923 10098 956398 0,01056
5,0 ng/mL
8052 884750 0,00910
18,9554
média 0,00915 desvio padrão 0,00173
20363 910582 0,02236 24391 913378 0,02670 23319 911747 0,02558 16809 831810 0,02021 2,1632 21168 846862 0,02500 23254 889887 0,02613 23577 869479 0,02712 1,1621 23058 932229 0,02473 23039 919549 0,02505
10,0 ng/mL
22020 912301 0,02414
8,4106
média 0,02470 desvio padrão 0,00208
41415 844125 0,04906 41885 917291 0,04566 45805 906310 0,05054 41448 904206 0,04584 46343 870606 0,05323 46257 888297 0,05207 44937 908231 0,04948 49677 930499 0,05339 1,1636 45416 887708 0,05116
20,0 ng/mL
36654 849328 0,04316 1,7917
7,0138
média 0,04936 desvio padrão 0,00346 Não foram encontrados valores aberrantes para os três níveis de concentração, dessa
forma nenhum dos resultados foi descartado e pode-se afirmar que o limite de detecção do
método é menor que 5,0 ng/mL. Os valores para repetitividade (precisão intra-ensaio),
contemplam os critérios de aceitação descritos no item 3.12.2.3, ou seja, a precisão mostrou
desvio menor que 20%.
No caso do alfa-zeranol os picos m/z 433, m/z 523 e m/z 538 apresentam
intensidades no espectro de massas de 100%, 21% e 17%, respectivamente.
A figura 51 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), do alfa-zeranol, após etapa de
derivatização.

Resultados e Discussão 105
tR(min)
M+• - 15
M+•
O
OO
O OSi
CH3
CH3
H3C
Si
CH3
CH3H3C
SiH3C CH3
H3C
tR = 17,246 min
Figura 51: Análise do alfa zeranol por CGAR-EM (varredura linear). (A) cromatograma de
íons totais, (B) espectro de massas do alfa zeranol.
4.4.1.2. beta-Zeranol Recuperação
A tabela 11 mostra os dados referentes à recuperação do beta-zeranol. Segundo o
critério para avaliação dos resultados, o valor recomendável para a recuperação do analito
deve ser superior a 70%.

Resultados e Discussão 106
Tabela 11: Resultado da recuperação para o beta-zeranol.
Fortificado antes Fortificado depois Resposta padrão
analito (PI) Resposta
analito Resposta analito / PI
Resposta padrão analito (PI)
Resposta analito
Resposta analito / PI
796736 16138 0,02026 785225 20307 0,02586 862314 17527 0,02033 771599 22503 0,02916 854311 18337 0,02146 786859 21015 0,02671 811731 18152 0,02236 738704 20972 0,02839 780633 18142 0,02324 737981 21454 0,02907 770826 17262 0,02239 727063 21116 0,02904
Recuperação (%) Recuperação média (%)
Desvio padrão (%) CV (%)
78,3220 69,6937 80,3674 78,7668 79,9420 77,1072
77,37 3,9 5,09
A recuperação do método para a determinação de beta-zeranol foi de 77,37 ± 5,09%.
Limite de detecção, teste de Grubbs e repetitividade
A tabela 12 mostra os valores referentes ao experimento para determinação do limite
de detecção, a este conjunto de dados foi aplicado o teste de Grubbs e foi calculada a
repetitividade para todos os níveis de concentração.

Resultados e Discussão 107
Tabela 12: Valores para determinação do limite de detecção e repetitividade. Nivel de estudo Área analito Área PI Razão analito / PI G calculado CV%
8389 907657 0,00924 8860 898979 0,00986 7241 845244 0,00857 1,2033 9713 920826 0,01055 8012 850874 0,00942 8123 853465 0,00952 9995 925777 0,01080 9996 947213 0,01055 13297 956398 0,01390 2,2147
5,0 ng/mL
10666 884750 0,01206
14,9468
média 0,01045 desvio padrão 0,00156
29244 910582 0,03212 26460 913378 0,02897 30551 911747 0,03351 30447 831810 0,03660 1,2023 23442 846862 0,02768 26388 889887 0,02965 25393 869479 0,02920 0,6631 25609 932229 0,02747 31120 919549 0,03384
10,0 ng/mL
35854 912301 0,03930
12,4585
média 0,03183 desvio padrão 0,00397
33504 844125 0,03969 0,5639 37528 917291 0,04091 37207 906310 0,04105 36659 904206 0,04054 34827 870606 0,04000 36017 888297 0,04055 37631 908231 0,04143 39339 930499 0,04228 32693 887708 0,03683
20,0 ng/mL
36654 849328 0,04316 1,4855
4,1608
média 0,04064 desvio padrão 0,00169
Também para o beta-zeranol não foram encontrados valores aberrantes para os três
níveis de concentração, dessa forma nenhum dos resultados foi descartado e pode-se
afirmar que o limite de detecção do método é menor que 5,0 ng/mL. Os valores para
repetitividade (precisão intra-ensaio), contemplam os critérios de aceitação descritos no item
3.12.3.5, ou seja, a precisão mostrou desvio menor que 20%.
No caso do beta-zeranol os picos m/z 433, m/z 523 e m/z 538 apresentam
intensidades no espectro de massas de 100%, 22% e 20%, respectivamente.
A figura 52 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), do beta-zeranol, após etapa de
derivatização.

Resultados e Discussão 108
tR(min)
M+•
M+• - 15
O
OO
O
Si
CH3
CH3H3C
SiH3C CH3
H3C OSi
CH3
CH3
H3C
tR = 17,561 min Figura 52: Análise do beta-zeranol por CGAR-EM (varredura linear). (A) cromatograma de
íons totais, (B) espectro de massas do beta-zeranol.

Resultados e Discussão 109
4.4.1.3. Tibolona
É preciso enfatizar que os dados abaixo apresentados são referentes a 3α-hidroxi-
tibolona, metabólito da tibolona encontrado majoritariamente na urina.
Recuperação
A tabela 13 mostra os dados referentes à recuperação da tibolona. Segundo o critério
para avaliação dos resultados, o valor recomendável para a recuperação do analito deve ser
superior a 70%.
Tabela 13: Resultado da recuperação para 3α-hidroxi-tibolona.
Fortificado antes Fortificado depois Resposta padrão
analito (PI) Resposta
analito Resposta analito / PI
Resposta padrão analito (PI)
Resposta analito
Resposta analito / PI
796736 11183 0,01404 785225 11383 0,01450 862314 10851 0,01258 771599 13791 0,01787 854311 11082 0,01297 786859 12714 0,01616 811731 11256 0,01387 738704 13408 0,01815 780633 11657 0,01493 737981 13681 0,01854 770826 11597 0,01504 727063 12689 0,01745
Recuperação (%) Recuperação média
(%) Desvio padrão (%) CV (%)
96,8236 70,4045 80,2818 76,3974 80,5503 86,2053
81,78 9,0 11,05
A recuperação do método para a determinação de tibolona foi de 81,78 ± 11,05%.
Limite de detecção, teste de Grubbs e repetitividade
A tabela 14 mostra os valores referentes ao experimento para determinação do limite
de detecção, a este conjunto de dados foi aplicado o teste de Grubbs e foi calculada a
repetitividade para todos os níveis de concentração.

Resultados e Discussão 110
Tabela 14: Valores para determinação do limite de detecção e repetitividade. Nivel de estudo Área analito Área PI Razão analito / PI G calculado CV%
5201 907657 0,00573 1,1441 5464 898979 0,00608 5988 845244 0,00708 1,6172 5356 920826 0,00582 5179 850874 0,00609 5605 853465 0,00657 6420 925777 0,00693 5493 947213 0,00580 6389 956398 0,00668
5,0 ng/mL
5428 884750 0,00614
7,7956
média 0,00629 desvio padrão 0,00049
11136 910582 0,01223 11748 913378 0,01286 12544 911747 0,01376 1,7874 10600 831810 0,01274 10317 846862 0,01218 1,5948 11582 889887 0,01302 11424 869479 0,01314 12088 932229 0,01297 12148 919549 0,01321
10,0 ng/mL
11995 912301 0,01315
3,6040
média 0,01293 desvio padrão 0,00047
19777 844125 0,02343 2,0425 22761 917291 0,02481 25160 906310 0,02776 22777 904206 0,02519 23554 870606 0,02705 23672 888297 0,02665 24654 908231 0,02715 25209 930499 0,02709 23886 887708 0,02691
20,0 ng/mL
23822 849328 0,02805 1,1235
5,5246
média 0,02641 desvio padrão 0,00146
Assim como para o alfa e para o beta-zeranol, também não foram encontrados
valores aberrantes para os três níveis de concentração, dessa forma nenhum dos resultados
foi descartado e pode-se afirmar que o limite de detecção do método é menor que 5,0
ng/mL. Os valores para repetitividade (precisão intra-ensaio), contemplam os critérios de
aceitação descritos no item 3.12.3.5, ou seja, a precisão mostrou desvio menor que 20%.
No caso da tibolona os picos m/z 443 e m/z 353 apresentam intensidades no espectro
de massas de 100% e 33%, respectivamente.
A figura 53 apresenta o espectro de massas (impacto de elétrons) obtido por CGAR-
EM, varredura linear (SCAN), de hidroxi-tibolona, após etapa de derivatização.

Resultados e Discussão 111
tR(min)
(A)
TMSO
TMSO
(B)
tR = 12,509 min
m/z 196
m/z 263
Figura 53: Análise de hidroxi-tibolona por CGAR-EM (varredura linear). (A) cromatograma de
íons totais, (B) espectro de massas de hidroxi-tibolona.
4.4.1.4. Letrozol
Para este composto foi possível realizar além da validação qualitativa, o estudo para
avaliação da cinética da hidrólise, uma vez que obtivemos urina de um usuário crônico do
medicamento Femara® (5 mg). Trata-se de um indivíduo do sexo feminino, de
aproximadamente 50 anos, que iniciava o tratamento com Femara® para evitar a recidiva de

Resultados e Discussão 112
um câncer de mama. Com a urina além da cinética enzimática foi possível a observação em
relação a presença ou não de metabólitos livres.
O metabólito do letrozol bis-4-cianofenil-metanol, já foi descrito na literatura. Outro
metabólito também descrito seria o triazol, entretanto em função de seu baixo ponto de
ebulição, não é possível a análise por CGAR-EM.
A validação qualitativa foi realizada com padrão de referência do metabólito bis-4-
cianofenil-metanol.
Recuperação
A tabela 15 mostra os dados referentes à recuperação do letrozol. Segundo o critério
para avaliação dos resultados, o valor recomendável para a recuperação do analito deve ser
superior a 70%.
Tabela 15: Resultado da recuperação para letrozol.
Fortificado antes Fortificado depois Resposta padrão
analito (PI) Resposta
analito Resposta analito / PI
Resposta padrão analito (PI)
Resposta analito
Resposta analito / PI
796736 21681 0,02721 785225 22056 0,02809 862314 24571 0,02849 771599 26873 0,03483 854311 23338 0,02732 786859 24758 0,03146 811731 22763 0,02804 738704 25285 0,03423 780633 21808 0,02794 737981 26441 0,03583 770826 23303 0,03023 727063 23077 0,03174
Recuperação (%) Recuperação média
(%) Desvio padrão (%) CV (%)
96,8796 81,8150 86,8218 81,9266 77,9716 95,2463
86,78 7,7 8,92
A recuperação do método para a determinação de letrozol foi de 86,78 ± 8,92%.
Limite de detecção, teste de Grubbs e repetitividade.
A tabela 16 mostra os valores referentes ao experimento para determinação do limite
de detecção, a este conjunto de dados foi aplicado o teste de Grubbs e foi calculada a
repetitividade para todos os níveis de concentração.

Resultados e Discussão 113
Tabela 16: Valores para determinação do limite de detecção e repetitividade.
Nivel de estudo Área analito Área PI Razão analito / PI G calculado CV%
4625 907657 0,00510 6000 898979 0,00667 5312 845244 0,00628 5648 920826 0,00613 5986 850874 0,00704 4739 853465 0,00555 5105 925777 0,00551 7782 947213 0,00822 7146 956398 0,00747 1,1078
5,0 ng/mL
5398 884750 0,00610 0,3194
14,9876
média 0,00641 desvio padrão 0,00096
19728 910582 0,02167 1,3846 32272 913378 0,03533 2,1060 24188 911747 0,02653 21739 831810 0,02613 21279 846862 0,02513 22515 889887 0,02530 27682 869479 0,03184 24304 932229 0,02607 22481 919549 0,02445
10,0 ng/mL
25929 912301 0,02842
14,4554
média 0,02709 desvio padrão 0,00392
35877 844125 0,04250 1,7668 45487 917291 0,04959 45259 906310 0,04994 40297 904206 0,04457 42905 870606 0,04928 42353 888297 0,04768 40975 908231 0,04512 47240 930499 0,05077 45563 887708 0,05133 0,8967
20,0 ng/mL
44837 849328 0,05279
6,8514
média 0,04836 desvio padrão 0,00331
Não foram encontrados valores aberrantes para os três níveis de concentração, dessa
forma nenhuns dos resultados foram descartados e podemos afirmar que o limite de
detecção do método é menor que 5,0 ng/mL. Os valores para repetitividade (precisão intra-
ensaio), contemplam os critérios de aceitação descritos no item 3.12.3.5, ou seja, a precisão
mostrou desvio menor que 20%.
No caso do metabólito do letrozol os picos m/z 217, m/z 291 e m/z 190 apresentam
intensidades no espectro de massas de 100%, 62% e 43%, respectivamente.
A figura 54 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtido por CGAR-EM, varredura linear (SCAN), do letrozol, após etapa de derivatização.

Resultados e Discussão 114
(A)
tR(min)
M+•
M+• - 15
CN
CN
TMSO
m/z 217
m/z 203
tR = 6,657 min (B)
Figura 54: Análise do letrozol por CGAR-EM (varredura linear). (A) cromatograma de íons
totais, (B) espectro do metabólito letrozol-bis-TMS.

Resultados e Discussão 115
4.4.1.5. Formestano
A tabela 17 mostra os dados referentes à recuperação do formestano. Segundo o
critério para avaliação dos resultados, o valor recomendável para a recuperação do analito
deve ser superior a 70%.
Tabela 17: Resultado da recuperação para formestano.
Fortificado antes Fortificado depois Resposta padrão
analito (PI) Resposta
analito Resposta analito / PI
Resposta padrão analito (PI)
Resposta analito
Resposta analito / PI
796736 45151 0,05667 785225 55616 0,07083 862314 45387 0,05263 771599 52868 0,06852 854311 46856 0,05485 786859 57028 0,07248 811731 42322 0,05214 738704 57719 0,07814 780633 45962 0,05888 737981 57623 0,07808 770826 47182 0,06121 727063 56126 0,07720
Recuperação (%) Recuperação média
(%) Desvio padrão (%) CV (%)
80,0106 76,8183 75,6760 66,7276 75,4052 79,2917
75,65 4,8 6,29
A recuperação do método para a determinação de formestano foi de 75,65 ± 6,29%.
Limite de detecção, teste de Grubbs e repetitividade.
A tabela 18 mostra os valores referentes ao experimento para determinação do limite
de detecção, a este conjunto de dados foi aplicado o teste de Grubbs e foi calculada a
repetitividade para todos os níveis de concentração.

Resultados e Discussão 116
Tabela 18: Valores para determinação do limite de detecção e repetitividade. Nível de estudo Área analito Área PI Razão analito / PI G calculado CV%
142570 5082868 0,02805 1,3094 126069 5242415 0,02405 140415 5134324 0,02735 137273 5143033 0,02669 144423 5510301 0,02621 142191 5569032 0,02553 149316 5500438 0,02715 137483 5710012 0,02408 115389 5225147 0,02208 1,9504
1/2 X LMDR
134009 5287795 0,02534
7,1341
média 0,02565 desvio padrão 0,00183
187256 5189381 0,03608 195236 5161770 0,03782 202209 5086269 0,03976 1,2574 196704 5654784 0,03479 214467 5860624 0,03659 183811 5898042 0,03116 1,9987 209003 5574299 0,03749 206545 5247550 0,03936 177400 5085739 0,03488
LMDR
166102 4824827 0,03443
7,2409
média 0,03644 desvio padrão 0,00264
280788 5451162 0,05151 294430 5253589 0,05604 343959 5207840 0,06605 1,0441 342895 5502319 0,06232 345038 5380152 0,06413 335658 5465834 0,06141 276613 5088073 0,05436 328549 5035141 0,06525 283028 5657919 0,05002 1,7367
2 X LMDR
300297 5072893 0,05920
9,5987
média 0,06003 desvio padrão 0,00576
Não foram encontrados valores aberrantes para os três níveis de concentração, dessa
forma nenhum dos resultados foram descartados e podemos afirmar que o limite de
detecção do método é menor que 5,0 ng/mL. Os valores para repetitividade (precisão intra-
ensaio), contemplam os critérios de aceitação descritos no item 3.12.3.5, ou seja, a precisão
mostrou desvio menor que 20%.
No caso do formestano os picos m/z 518, m/z 143 e m/z 503 apresentam intensidades
no espectro de massas de 100%, 24% e 4%, respectivamente.
A figura 55 apresenta o cromatograma e espectro de massas (impacto de elétrons)
obtidos por CGAR-EM, varredura linear (SCAN), do formestano, após etapa de
derivatização.

Resultados e Discussão 117
tR(min)
CH3
CH3
H
HH
O
Si
CH3
CH3H3C
O
O
Si
Si
CH3
CH3
H3C
H3C CH3
H3C
M+•tR = 17,374 min
Figura 55: Análise do formestano por CGAR-EM (varredura linear). (A) cromatograma de
íons totais, (B) espectro de massas do formestano-mono-TMS.

Resultados e Discussão 118
4.4.2. Validação semi-quantitativa do método analítico O protocolo de validação semi-quantitativa impõem que sejam avaliados critérios de
desempenho como limite de detecção, limite de quantificação, especificidade, contaminação
entre amostras, repetitividade e identificação de valores aberrantes.
Especificidade
Dez brancos foram injetados e cada segmento referente aos alvos analíticos,
clembuterol, epimetendiol, norandrosterona, metil-M2, 3’OH-estanozolol foram monitorados.
Não houve interferência na região cromatográfica de interesse.
Contaminação entre amostras Uma amostra de branco de urina fortificada com os analitos na concentração de 100
ng/mL foi injetada e em seguida foi injetada uma amostra de branco de urina livre dos
analitos. Foi verificada ausência de resposta para todos os alvo analíticos.
Limite de detecção (LD) e limite de quantificação (LQ) O limite de detecção foi estimado a partir de três vezes o desvio padrão do ruído,
calculado a partir da injeção de dez brancos de urina. Adotou-se o mesmo procedimento
para estimar o valor do limite de quantificação, onde o desvio padrão do ruído foi
multiplicado por dez. Os resultados para LD e LQ são apresentados na tabela 19.
Tabela 19: Valores estimados de LD e LQ. Resíduos monitorados
Clembuterol Norandrosterona Epimetendiol Metiltestosterona-M2 3’OH-estanozolol
Desvio padrão do
ruído (Counts)
140 512 99 360 363
LD (ng/mL) 0,1 0,3 0,2 0,6 0,04
LQ (ng/mL) 0,3 0,8 0,7 2,0 0,1
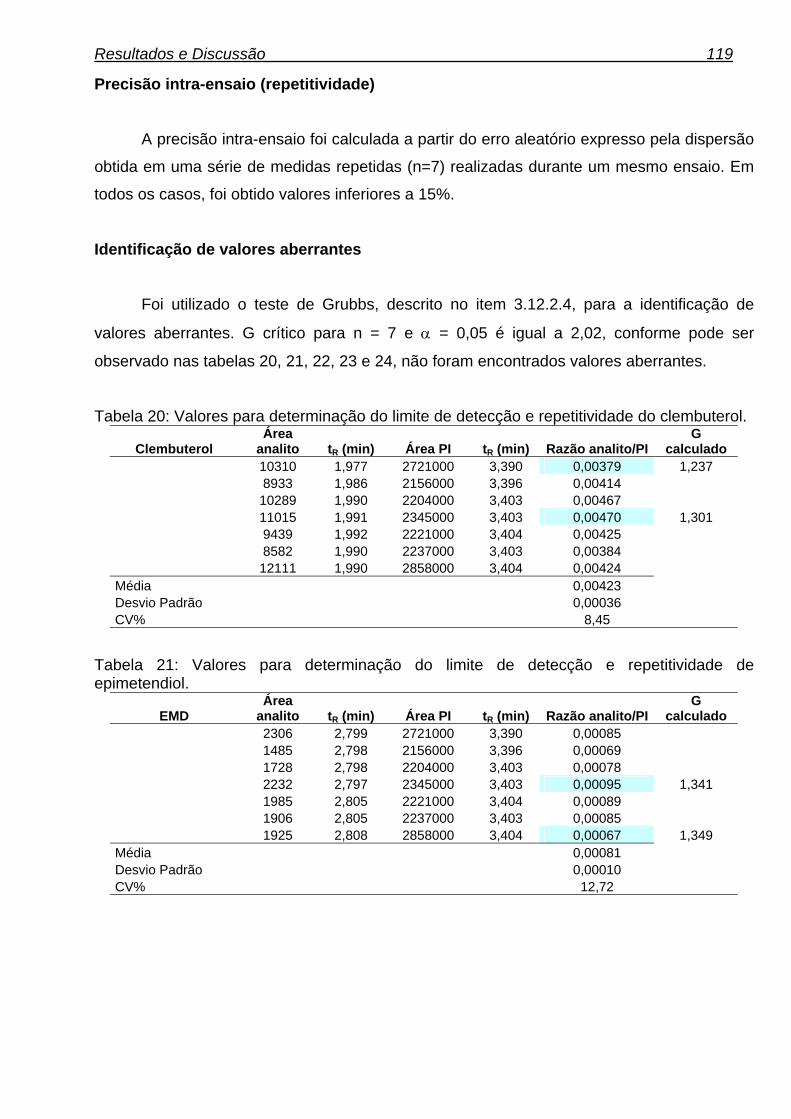
Resultados e Discussão 119
Precisão intra-ensaio (repetitividade) A precisão intra-ensaio foi calculada a partir do erro aleatório expresso pela dispersão
obtida em uma série de medidas repetidas (n=7) realizadas durante um mesmo ensaio. Em
todos os casos, foi obtido valores inferiores a 15%.
Identificação de valores aberrantes
Foi utilizado o teste de Grubbs, descrito no item 3.12.2.4, para a identificação de
valores aberrantes. G crítico para n = 7 e α = 0,05 é igual a 2,02, conforme pode ser
observado nas tabelas 20, 21, 22, 23 e 24, não foram encontrados valores aberrantes.
Tabela 20: Valores para determinação do limite de detecção e repetitividade do clembuterol.
Clembuterol Área
analito tR (min) Área PI tR (min) Razão analito/PI G
calculado 10310 1,977 2721000 3,390 0,00379 1,237 8933 1,986 2156000 3,396 0,00414 10289 1,990 2204000 3,403 0,00467 11015 1,991 2345000 3,403 0,00470 1,301 9439 1,992 2221000 3,404 0,00425 8582 1,990 2237000 3,403 0,00384 12111 1,990 2858000 3,404 0,00424 Média 0,00423 Desvio Padrão 0,00036 CV% 8,45
Tabela 21: Valores para determinação do limite de detecção e repetitividade de epimetendiol.
EMD Área
analito tR (min) Área PI tR (min) Razão analito/PI G
calculado 2306 2,799 2721000 3,390 0,00085 1485 2,798 2156000 3,396 0,00069 1728 2,798 2204000 3,403 0,00078 2232 2,797 2345000 3,403 0,00095 1,341 1985 2,805 2221000 3,404 0,00089 1906 2,805 2237000 3,403 0,00085 1925 2,808 2858000 3,404 0,00067 1,349 Média 0,00081 Desvio Padrão 0,00010 CV% 12,72

Resultados e Discussão 120
Tabela 22: Valores para determinação do limite de detecção e repetitividade de 3’-hidroxi-estanozolol.
3'OH-STAN Área
analito tR (min) Área PI tR (min) Razão analito/PI G
calculado 15837 4,147 2721000 3,390 0,00582 2,009 17581 4,156 2156000 3,396 0,00815 17127 4,161 2204000 3,403 0,00777 20373 4,161 2345000 3,403 0,00869 1,191 17028 4,166 2221000 3,404 0,00767 16482 4,161 2237000 3,403 0,00737 22509 4,166 2858000 3,404 0,00788 Média 0,00762 Desvio Padrão 0,001 CV% 11,76
Tabela 23: Valores para determinação do limite de detecção e repetitividade de norandrosterona.
Norandrosterona Área
analito tR (min) Área PI tR (min) Razão analito/PI G calculado 3747 2,740 2721000 3,390 0,00138 3693 2,746 2156000 3,396 0,00171 1,440 3044 2,761 2204000 3,403 0,00138 3633 2,744 2345000 3,403 0,00155 3636 2,744 2221000 3,404 0,00164 2866 2,753 2237000 3,403 0,00128 1,356 4273 2,744 2858000 3,404 0,00150 Média 0,00149 Desvio Padrão 0,00015 CV% 10,36
Tabela 24: Valores para determinação do limite de detecção e repetitividade de 17α-metil-5β-androstano-3α,17β-diol.
Metil-M2 Área
analito tR (min) Área PI tR (min) Razão analito/PI G calculado 1117 3,106 2721000 3,390 0,00041 1,186 1320 3,111 2156000 3,396 0,00061 1,808 931 3,116 2204000 3,403 0,00042 1188 3,117 2345000 3,403 0,00051 1038 3,120 2221000 3,404 0,00047 1147 3,116 2237000 3,403 0,00051 1432 3,117 2858000 3,404 0,00050 Média 0,00049 Desvio Padrão 0,00007 CV% 13,74

Resultados e Discussão 121
4.4.2.1. Desenvolvimento do método de análise por CGAR-ITD – VARIAN
O primeiro passo para o desenvolvimento do método foi a otimização de uma
programação de temperatura que permitisse uma análise rápida sem perda de resolução. O
equipamento VARIAN 4000 foi adquirido em função da necessidade de um método analítico
capaz de detectar clembuterol, 3’OH-estanozolol, epimetendiol, metiltestosterona-M2 e
norandorsterona, conhecidos como resíduos de baixa concentração, em 2ng/mL (LMDR)
com exceção de norandrosterona cujo LMDR é 1ng/mL. Em um primeiro momento
acreditava-se que só seria possível contar com este equipamento para análise de todas as
amostras dos jogos PAN AMERICANOS de 2007, uma vez que o quadrupolo não consegue
detectar estas substâncias neste nível de concentração. Dessa forma seria preciso uma
corrida analítica rápida, mas que permitisse a divisão efetiva de cada substância, ou seja a
separação pela cromatografia e não apenas pela diferença da massa de íons pai que
eventualmente apresentassem o mesmo tempo de retenção. O objetivo era dividir,
segmentar o método, para que em cada segmento fosse incluído o menor número possível
de substância para aumentar com isso a sensibilidade da detecção. Dessa forma, as
condições do trap seriam otimizadas de maneira quase que específica para cada substância.
Aquino Neto et. al. em 2003 descreveram a necessidade de manter a temperatura
inicial da coluna, na análise de agentes anabólicos por CG-EM, em 140°C, por causa de
analitos eluindo próximo ao solvente, em especial o clembuterol. Partindo de 140°C, foi
proposta uma corrida cromatográfica de 6,25 min. Temperatura inicial de 140°C em seguida
passando para 310°C, mantidos por 2 minutos, com uma taxa de aquecimento de 40°C /
min. Foi empregado injetor aquecido do tipo com divisão de fluxo, razão de divisão 1:5 e
após 1 minuto 1:100, para evitar a cristalização de derivatizante na saída do split e
conseqüentemente problemas na pressão da cabeça da coluna.
Para o desenvolvimento do método de aquisição, cada substância foi injetada
separadamente no modo “FULL SCAN”, para que os tempos de retenção fossem verificados
e o método segmentado. Entre cada injeção de padrão injetou-se um branco de urina, que
servia para capear possívies sítios ativos presentes na coluna. Após segmentar o método,
brancos de urina fortificados com 100ng/mL, 10ng/mL , 2ng/mL e 1ng/mL foram preparados.
O método de detecção foi desenvolvido na presença de matriz para evitar a escolha de íons
pai presentes na urina e para modular as condições do trap, em função do efluente real
proveniente da coluna que chegaria no trap no tempo de retenção do analito alvo, evitando
com isso o re-trabalho.

Resultados e Discussão 122
Após as condições de análise estabelecidas foi injetado branco fortificado em 10
ng/mL e em seguida em 2 ng/mL e 1ng/mL para norandrosterona, onde somente poucos
parâmetros foram alterados de modo a maximizar a detecção.
A maioria das substâncias apresentou diferenças entre seu espectro de massas
obtido com o quadrupolo e o espectro obtido em ion trap, ambos no modo varredura linear
(SCAN). Fragmentos com maior razão m/z geralmente são mais abundantes no ion trap que
no quadrupolo, em alguns casos a proporção entre os fragmentos encontram-se alteradas
devido ao tempo de permanência do íon no trap.
Os íons pai foram escolhidos com o objetivo de obter a maior seletividade possível,
em especial que não estivessem presentes no background, nem em interferentes
provenientes da matriz. Para confirmar informações estruturais obtidas no espectro,
procurou-se selecionar para íons pai os fragmentos mais abundantes no espectro (maior
m/z).
A fragmentação do íon precursor foi realizada a partir de dissociação induzida por
colisão (CID) com moléculas de hélio (o gás carreador). CID foi realizada verificando a
adequação para cada substância em relação ao modo de excitação ressonante e não-
ressonante. Uma vez o modo de excitação selecionado, os principais parâmetros que
determinam o comportamento do íon são o tempo de excitação, a energia de dissociação e
a radiofreqüência ideal para estocar o íon no trap. A energia de dissociação e a
radiofreqüência são parâmetros que devem ser cuidadosamente otimizados para garantir o
melhor desempenho da técnica. A tabela 23 apresenta os parâmetros utilizados para
fragmentação dos íons precursores. A tabela 24 apresenta os íons filhos gerados e suas
respectivas proporções.
Tabela 25: Parâmetros de CG-EM-EM utilizados para fragmentação do íon pai. Parâmetros para CID
Substância com
efeito anabólico
Substância monitorada (principal
substância excretada)
Íon pai
(m/z) Amplitude
(Volts)
Energia de
excitação
(m/z)
Tipo de onda
Clembuterol Clembuterol (droga mãe) 300 90,0 90,0 Não-ressonante
Metandienona Epimetendiol 358 1,40 157,7 Ressonante
Nandrolona Norandrosterona 405 0,49 178,4 Ressonante
Metiltestosterona 17α-metil-5β-androstano-3α,17β-diol 345 0,80 152,0 Ressonante
Estanozolol 3’OH-estanozolol 545 1,0 240,1 Ressonante
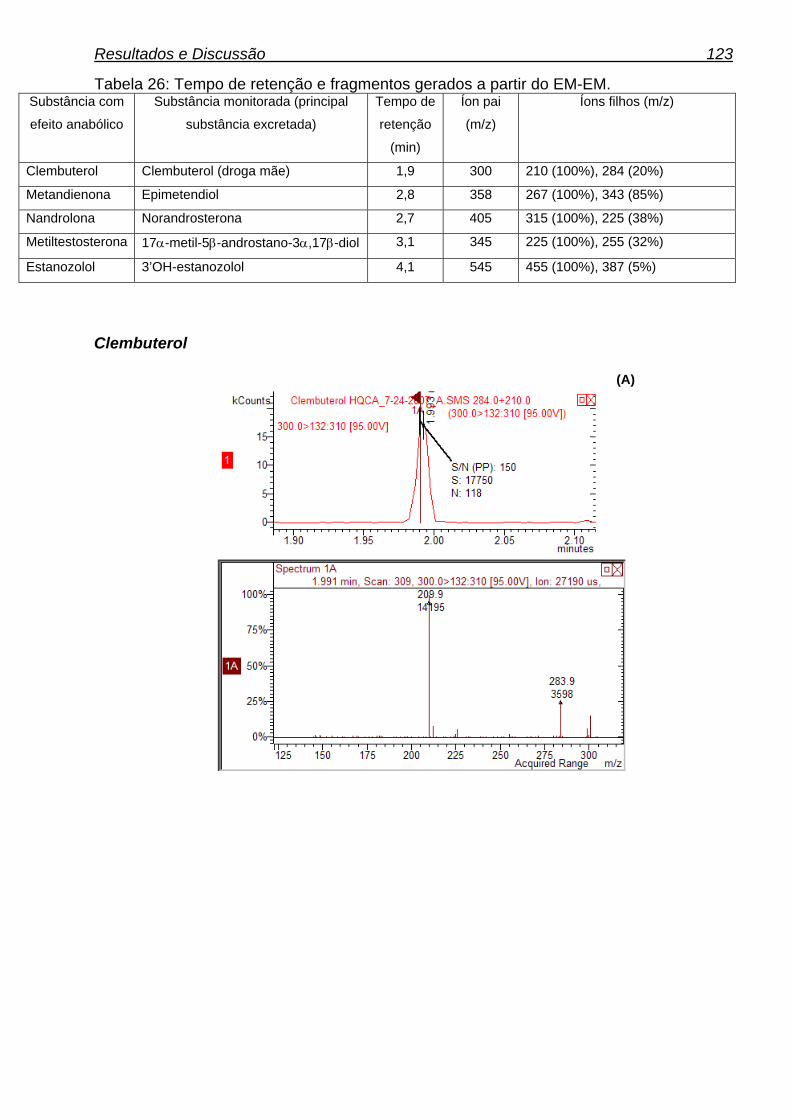
Resultados e Discussão 123
Tabela 26: Tempo de retenção e fragmentos gerados a partir do EM-EM. Substância com
efeito anabólico
Substância monitorada (principal
substância excretada)
Tempo de
retenção
(min)
Íon pai
(m/z)
Íons filhos (m/z)
Clembuterol Clembuterol (droga mãe) 1,9 300 210 (100%), 284 (20%)
Metandienona Epimetendiol 2,8 358 267 (100%), 343 (85%)
Nandrolona Norandrosterona 2,7 405 315 (100%), 225 (38%)
Metiltestosterona 17α-metil-5β-androstano-3α,17β-diol 3,1 345 225 (100%), 255 (32%)
Estanozolol 3’OH-estanozolol 4,1 545 455 (100%), 387 (5%)
Clembuterol
(A)

Resultados e Discussão 124
Norandrosterona
(B)
EMD (C)

Resultados e Discussão 125
3α,5β-Metiltestosterona
Metiltestosterona (Padrão interno)
(E)
(D)

Resultados e Discussão 126
3’OH-estanozolol (F)
Figura 56: Cromatograma de íons (A) Clembuterol, (B) Norandrosterona, (C) EMD, (D)
3α,5β-Metiltestosterona, (E) Metiltestosterona, (F) 3’OH-estanozolol.
4.4.3. Validação quantitativa do método analítico O protocolo de validação consistiu de três ensaios realizados em dias distintos,
conforme descrito no item 3.12.4. Cada ensaio representou uma etapa do processo de
validação.
A determinação do limite de quantificação para a construção da curva de calibração
foi efetuada após estimativa do limite de detecção (relação sinal-ruído maior do que 3),
através da análise de urinas fortificadas (2 mL) com o analito nas concentrações de 0,05;
0,1; 0,2; 0,4; 0,5; 1,0 e 2,0 ng/mL. O limite de quantificação (LQ) foi definido, então, como
0,2 ng/mL (Fig.59), sendo esta concentração quatro vezes acima do limite de detecção (0,05
ng/mL) para análise de urina utilizando o volume de 2 mL.
Foram construídas duas curvas de calibração com faixas de concentração diferentes
para verificar a linearidade do método e se o mesmo era homocedástico ou heterocedástico,
na primeira os pontos escolhidos foram 0,2; 0,4; 0,5; 1,0 e 2,0 ng/mL e na segunda os
pontos escolhidos foram 10,0; 25,0; 50,0; 75,0 e 100,0 ng/mL.

Resultados e Discussão 127
As amostras foram preparadas de acordo com o protocolo para análise de esteróides
em urina, descrito no item 3.9.1.1. Em todos os três dias as amostras preparadas foram
injetadas uma única vez, para cada nível de concentração.
Especificidade A especificidade é a capacidade de um método distinguir o analito das demais
substâncias. Para determinar a especificidade do método, dez matrizes isentas de analito
foram analisadas.
Os resultados das análises dos brancos de urina, para o segmento referente ao 3’OH-
estanozolol, mostraram que não há interferência na região cromatográfica de interesse, seja
de substâncias endógenas presentes na urina.
Identificação de valores aberrantes A presença de valores aberrantes nas respostas para cada nível de concentração
para as duas curvas de calibração foram testadas pelo teste de Grubbs no nível de
confiança α=0,05. As respostas foram obtidas como o quociente entre os valores de área do
analito e do padrão interno.
A maneira de calcular os valores aberrantes foi descrito no item 3.12.2.4. Não foram
encontrados valores aberrantes para nenhuma das concentrações estudadas (Tabela 27).

Resultados e Discussão 128
Tabela 27: Valores para o teste de Grubbs. Gcrítico=1,15
Conc. ng/ml
Razão analito/PI STD Média Gcalculado 0,00113 0,00121 0,2 0,00110
0,0000543 0,00115 1,10
0,00187 0,00211 0,4 0,00193
0,0001244 0,00197 1,12
0,00251 0,00273 0,5 0,00207
0,0003390 0,00244 0,87
0,00500 0,00451 1,0 0,00455
0,0002740 0,00469 1,15
0,01005 0,00918 2,0 0,01000
0,0004858 0,00974 0,63
0,04738 0,04700 10,0 0,05183
0,0026869 0,04874 1,15
0,12406 0,11384 25,0 0,10799
0,0081305 0,11530 1,08
0,23128 0,22515 50,0 0,21494
0,0082522 0,22379 0,16
0,34628 0,37127 75,0 0,34707
0,0142065 0,35487 1,15
0,50106 0,52873 100,0 0,51401
0,0138422 0,51460 1,02
Contaminação entre amostras (arraste) Foi verificada a ausência de resposta para 3’OH-estanozolol em uma amostra isenta
de analito, analisada imediatamente após uma amostra de controle na concentração de 100
ng/mL, denominada como amostra de concentração superior.
Homocedasticidade / heterocedasticidade Para avaliar o grau de dependência ou independência da variância das respostas com
a concentração do analito ao longo das curvas de calibração nos diferentes ensaios, foi
realizada uma prova de homogeneidade de variâncias, ou seja, o teste de Cochran (α =
0,05).
O teste de Cochran consiste na razão entre a maior variância de todos os níveis da
curva de calibração e o somatório de todas as variâncias.

Resultados e Discussão 129
∑=
= n
i
calculado
jS
SC
1
2
2 max
Onde:
S2max = maior variância entre todos os pontos da curva;
S2j = somatório de todas as variâncias relativas a todos os níveis da curva.
Quando Ccalculado menor que Ccrítico (tabelado), o método é considerado
homocedastico. Quando Ccalculado maior que Ccrítico, o método é considerado heterocedastico.
Tabela 28: Valores de C crítico para nível de confiança de 0,05.
λ Valor de Ccrítico
n = 3
2 0,975
3 0,871
4 0,768
5 0,684
6 0,616
7 0,561
8 0,516
9 0,478
10 0,445
11 0,417
12 0,392
13 0,371
14 0,352
A condição de homocedasticidade foi confirmada para as duas curvas de calibração
(tabela 29). O valor de Ccrítico encontrado para a primeira curva (0,2 – 2,0 ng/mL) foi de
0,531, menor que Ccrítico (0,684), o mesmo sendo observado para a segunda curva (10,0 –
100,0 ng/mL) onde o Ccrítico foi de 0,377 (tabela 29).

Resultados e Discussão 130
Tabela 29: Valores para o teste de Cochran. Ccrítico=0,684
Conc. ng/ml Razão analito/PI STD Variância Cochran
0,00113 0,00121 0,2 0,00110
0,0000543 2,94594E-09 0,531
0,00187 0,00211 0,4 0,00193
0,0001244 1,54671E-08
0,00251 0,00273 0,5 0,00207
0,0003390 1,14894E-07
0,00500 0,00451 1,0 0,00455
0,0002740 7,50515E-08
0,01005 0,00918 2,0 0,01000
0,0004858 2,35962E-07
0,04738 0,04700 10,0 0,05183
0,0026869 7,21952E-06 0,377
0,12406 0,11384 25,0 0,10799
0,0081305 6,61051E-05
0,23128 0,22515 50,0 0,21494
0,0082522 6,80985E-05
0,34628 0,37127 75,0 0,34707
0,0142065 0,000201825
0,50106 0,52873 100,0 0,51401
0,0138422 0,000191606
Linearidade A relação entre a resposta do equipamento, expressa como a razão entre as áreas do
analito e do padrão interno, e a concentração do analito na matriz foi estudada por análise
de regressão.
O ajuste dos valores obtidos ao modelo linear do tipo:
y = a + bx
onde:
“y” é a variável dependente que corresponde à resposta do detector (neste caso, a razão
entre as áreas do analito e do padrão interno);
“x” é a variável independente que corresponde à concentração de analito contida nas
amostras de calibração;
“a” é o coeficiente linear da reta;

Resultados e Discussão 131
“b” é o coeficiente angular;
A regressão linear foi feita pelo método dos mínimos quadrados. A partir dos valores
das concentrações e do quociente das áreas obtidas para o 3’OH-estanozolol e o padrão
interno, foram construídas as duas curvas de calibração. Os resultados obtidos para os
coeficientes angular e linear, foram calculados. O modelo linear foi investigado através do
cálculo do coeficiente de correlação de Pearson (r).
Curva de calibração 3'OH-estanozolol
y = 0,0048x + 6E-05R2 = 0,9928
0,00000
0,00200
0,00400
0,00600
0,00800
0,01000
0,01200
0 0,5 1 1,5 2 2,
Conc. ng/mL
Raz
ão á
rea
anal
ito/P
I
5
Figura 57: Curva de calibração 3’OH-estanozolol.
Curva de calibração 3'OH-estanozolol
y = 0,0051x - 0,0144R2 = 0,9906
0,00000
0,10000
0,20000
0,30000
0,40000
0,50000
0,60000
0,0 20,0 40,0 60,0 80,0 100,0 120,0
Conc. (ng/mL)
Raz
ão á
rea
anal
ito/P
I
Figura 58: Curva de calibração 3’OH-estanozolol.
Nos dois casos, o coeficiente de correlação linear foi maior do que 0,99. Isso
demonstra que o modelo linear é adequado para expressar a relação entre a razão área do

Resultados e Discussão 132
analito / área do PI. Todas as duas curvas de calibração cumprem o critério r maior que
0,99.
Limite de detecção (LD) e limite de quantificação (LQ) O limite de detecção foi calculado a partir de injeções sucessivas de branco de urina
(2 mL) fortificado com o analito nas concentrações de 0,05; 0,1; 0,2; 0,4; 0,5; 1,0 e 2,0
ng/mL. O LD foi de 0,05 ng/mL, guardando relação sinal / ruído maior que 3.
O limite de quantificação foi determinado como sendo 0,2 ng/mL (0,22 ± 0,011 ng/mL
com exatidão de 109.08%).
Precisão e exatidão inter-ensaio
Os resultados de precisão e exatidão encontram-se na tabela 30 e 31. Os valores
obtidos contemplam os critérios de aceitação descritos no item 3.12.4.7, ou seja, a precisão
mostrou desvio menor que 15% em todos os casos e exatidão compreendida entre 80 –
120%.
Tabela 30: Precisão e exatidão interensaio do 3’OH-estanozolol para 1ª curva. Concentração obtida para uma
Conc. replicata (ng/ml) CV Exatidãong/ml dia 1 dia 2 dia 3 média (%) (%)
0,2 0,22 0,23 0,21 0,22 ± 0,011 4,7 109,1 0,4 0,37 0,42 0,38 0,39 ± 0,026 6,3 97,3 0,5 0,50 0,55 0,41 0,49 ± 0,071 13,9 97,3 1,0 1,02 0,92 0,93 0,96 ± 0,057 5,8 95,6 2,0 2,07 1,89 2,06 2,01 ± 0,101 5,0 100,5
Tabela 31: Precisão e exatidão interensaio do 3’OH-estanozolol para 2ª curva. Concentração obtida para uma
Conc. replicata (ng/ml) CV Exatidãong/ml dia 1 dia 2 dia 3 média (%) (%) 10,0 9,60 9,53 10,48 9,87 ± 0,527 5,5 98,7 25,0 24,64 22,64 21,49 22,92 ± 1,594 7,1 91,7 50,0 45,66 44,46 42,46 44,19 ± 1,618 3,7 88,4 75,0 68,21 73,11 68,37 69,90 ± 2,786 4,0 93,2
100,0 98,56 103,99 101,10 101,22 ± 2,714 2,7 101,2

Resultados e Discussão 133
Incerteza expandida (U)
A incerteza expandida do método é 2,64 ng/mL (dados obtidos a partir da segunda
curva de calibração). Utilizando os dados da primeira curva o valor obtido para U foi de 0,92
ng/mL.
Figura 59: Cromatograma de íons de 3’OH-estanozolol na concentração de 0,2 ng/mL.

Resultados e Discussão 134
4.5. Derivados N-fenilpiperazinas 4.5.1. Caracterização química dos derivados N-fenilpiperazinas
As famílias 1,2,3-triazóis e pirazóis foram caracterizadas por espectrometria de
massas de alta resolução (artigo em anexo).
4.5.2. Desenvolvimento de metodologia para análise do composto LASSBio 581
Foi desenvolvida metodologia analítica para extração do composto LASSBio 581,
utilizando LASSBio 580 como padrão interno, em plasma de rato wistar. Os solventes
testados para extração do alvo analítico do plasma foram terc-butilmetileter (TBME), hexano,
diclorometano (DCM), TBME / DCM (1:1), acetato de etila, metanol, TBME / terc-butanol
(1:1). Os resultados obtidos encontram-se na tabela 28.
Tabela 32: Valores de recuperação do composto LASSBio 581. Solvente % Recuperação
TBME 83
Hexano 25
DCM 49
TBME / DCM (1:1) 68
Acetato de etila 57
Metanol 88
TBME / t-BUOH 78
Embora a recuperação do composto LASSBio 581, tenha sido de 88% para metanol e
83% para TBME, verificou-se nos respectivos cromatogramas a presença de interferentes,
provenientes da matriz, próximos ao alvo analítico. Em função destes resultados optou-se
por utilizar a mistura de solventes TBME / t-BUOH na proporção 1:1.

Resultados e Discussão 135
(A)LASSBio 580 (Padrão interno)
tR(min)
N
N
N
NN
H
(B1)tR = 5,254 min
LASSBio 581
N
N
N
NN
Cl
(B2)tR = 6,100 min

Resultados e Discussão 136
Figura 60: Análise de LASSBio 581 por CGAR-EM (varredura linear). (A) cromatograma de
íons totais, (B1) espectro de massas LASSBio 580 e (B2) espectro de massas LASSBio 581.
Os metabólitos propostos para o composto LASSBio 581 (Fig. 57) foram procurados
após extração do plasma rato Wistar que recebeu a substância. Não foi detectado nenhum
sinal no cromatograma que pudesse ser atribuído a um possível metabólito. O procedimento
será repetido, para confirmarmos esta informação.
N
N
N
NN
H
Cl
N
N
N
NN
OH
N
N
N
NN
OH
Cl
HN
N
N
NN
CH3
Cl
Figura 61: Proposta para possíveis metabólitos para LASSBio 581.

CAPÍTULO 5
CONCLUSÕES

Conclusão 136
5. CONCLUSÕES Foi desenvolvida metodologia analítica para a detecção de clembuterol,
norandrosterona, epimetendiol, 3α,5β-metiltestosterona e 3’OH-estanozolol no limite mínimo
de desempenho requerido pela Agência Mundial Anti-dopagem. O método desenvolvido
apresenta robustez suficiente para se tornar um método de rotina, mais de 1500 amostras
foram processadas sem que fosse necessária nenhuma intervenção no equipamento.
Foi possível também a constatação de que é de fato possível a análise quantitativa
em diferentes faixas de concentração em nível traço,utilizando ion trap.
Foram incluídos na rotina do laboratório a análise de metilnortestosterona,
metasterona, hiperdrol, letrozol, formestano, α-zeranol, β-zeranol, tamoxifeno, clomifeno e
ciclofenil. A metodologia foi validada garantindo que os métodos atendem as exigências das
aplicações analíticas, assegurando a confiabilidade dos resultados.
Foi avaliada a cinética da hidrólise para os anti-estrogênicos letrozol, clomifeno,
ciclofenil e tamoxifeno, onde foi constatada a necessidade da adição de enzima em todos os
casos. Foram encontrados para o tamoxifeno dois possíveis metabólitos diferentes daqueles
que tiveram sua estrutura caracterizada.
Foi desenvolvida metodologia para análise do composto LASSBio 581 em plasma,
não sendo encontrado, a princípio algum metabólito.
As novas famílias de N-fenilpiperazinas foram caracterizadas detalhadamente por
ESI-MS-MS.

CAPÍTULO 6
BIBLIOGRAFIA

Referências 137
6. BIBLIOGRAFIA AQUINO NETO, F. R. O papel do atleta na sociedade e o controle de dopagem no esporte.
Revista Brasileira de Medicina do Esporte, v.7, p.138-148, 2001.
AQUINO NETO F. R.; CARDOSO J. N. Cromatografia com fase gasosa de alta resolução
(CGAR) em colunas capilares de vidro e sílica fundida. Química Nova, p.272-274, 1985.
AQUINO NETO F. R.; CARDOSO J. N. Lacunas de retenção em cromatografia gasosa de alta
resolução. Química Nova, v.15, p.224-227, 1992.
AQUINO NETO F. R.; SOUZA MARQUES D. S. Cromatografia Princípios Básicos e Técnicas Afins, Rio de Janeiro: Interciência, 2003.
AYOTTE, C.; GOUDREAULT, D.; CHARLEBOIS. A. Testing for natural and synthetic anabolic
agents in human urine. Journal of Chromatography B, v.687, p.3-25, 1996.
BASARIA, S.; WAHLSTROM, J. T.; DOBS, A. S. Anabolic-androgenic steroid in the treatment of
chronic diseases. Journal Clinical Endocrinology Metabolism, v.86, p.5108-5117, 2001.
CARDOSO, C. R. ANÁLISE DE OXIMETOLONA POR CGAR-EM EM PLASMA, SALIVA E URINA DE HUMANOS APLICAÇÃO PARA ESTUDOS FARMACOCINÉTICOS, 2002, 242 f.
Tese (Doutorado em Química Orgânica) – Instituto de Química, Universidade Federal do Rio de
Janeiro, Rio de Janeiro.
CARDOSO J. N; AQUINO NETO F. R. Seleção da técnica de injeção de amostra em CGAR.
Química Nova, v.12, p.13-18, 1989.
CATLIN, D. H.; AHRENS, B. D.; KUCHEROVA, Y. Detection of norbolethone, an anabolic
steroid never marketed, in athletes’ urine. Rapid Communications in mass spectrometry,
v.16, p.1273-1275, 2002.

Referências 138
CATLIN, D. H.; HATTON, C. K. Use and abuse of anabolic and other drugs for athletic
enhancement. Advances in Internal Medicine, v.36, p. 399-424, 1991.
CATLIN, D. H.; SEKERA, M. H.; AHRENS, B. D.; STARCEVIC, B.; CHANG, Y. C.; HATTON, C.
K. Tetrahydrogestrinone: discovery, synthesis, and detection in urine. Rapid Communications in mass spectrometry, v.18, p.1245-1249, 2004.
DAMASCENO, L. M. P. DESENVOLVIMENTO DE PROCEDIMENTOS ANALÍTICOS PARA
DETECÇÃO DE OMPOSTOS β2-AGONISTAS EM AMOSTRAS DE URINA, 2001, Tese
(Doutorado em Química Orgânica) – Instituto de Química, Universidade Federal do Rio de
Janeiro, Rio de Janeiro.
DELBEKE, F. T. From Aminata Muscaria to somatropine: The doping story. Biology of Sport, v.17, p. 81-85, 2000.
DRUMMER, O. H. Chromatographic screening techniques in systematic toxicological analysis.
Journal of Chromatography B, v.733, p.27-45, 1999.
GJERDE, J.; KISANGA, E. R.; HAUGLID, M.; HOLM, P. I.; MELLGREN, G.; LIEN, E. A.
Identification and quantification of tamoxifen and four metabolites in serum by liquid
chromatography-tandem mass spectrometry. Journal of Chromatography A, v.1082, p.6-14,
2005.
GEISLER, J.; LONNING, P. E. Aromatase inhibitors as adjuvant treatment of breast cancer.
Oncology Hematology, v.57, p.53-61, 2006.
HENNION, M. C.; Contribution of theory to method development in solid-phase extraction.
Journal of Chromatography A, v.885, p.17-39, 2000.
HODJEGAN, A. R.; LENNARD, M. S.; TUCKER, G. T.; LEDGER, W. L. Monitoring plasma
concentrations to individualize treatment with clomiphene citrate. Fertility and Sterility, v.81,
p.1187-1193, 2004.

Referências 139
HUENERBEIN, A.; MARQUES, M. A. S.; PEREIRA, A. P.; AQUINO NETO, F. R. Improvement
in steroid screening for doping control with special emphasis on stanozolol. Journal of Chromatography A, v.985, p.375-386, 2003.
INGLE, J. N.; SUMAN, V. J. Aromatase inhibitors for therapy of advanced breast cancer.
Steroid Biochemistry & Molecular Biology, v.95, p. 113-119, 2005.
JAMES BARKER. Mass spectrometry, Canada: John Wiley & Sons, 1999.
KISANGA, E. R.; MOI, L. L. H.; GJERDE, J.; MELLGREN, G.; LIEN, E. A. Induction of hepatic
drug-metabolising enzymes and tamoxifen metabolite profile in relation to administration route
during low-dose treatment in nude rats. Steroid Biochemistry & Molecular Biology, v.94, p.
489-498, 2005.
KRISHNAN, T. R.; IBRAHAM, I. Solid-phase extraction technique for the analysis of biological
samples. Journal of Pharmaceutical & Biomedical Analysis, v.12, p.287-294, 1994.
KUHN, C. M. Anabolic steroids. Recent Progress in Hormone Research, v.57, p.411-434,
2002.
LANÇAS F. M.; McNAIR H. M. Cromatografia em fase gasosa 1. Teoria elementar. Química Nova, p.6-13, 1983.
LASNE, F.; MARTIN, L.; CREPIN, N.; CEAURRIZ. J. Detection of isoelectric profiles of
erythropoietin in urine: differentiation of natural and administered recombinant hormones.
Analytical Biochemistry., v.311, p.119-123, 2002.
LEWIS, J. S.; JORDAN, V. C. Selective estrogen receptor modulators (SERMs): Mechanisms of
anticarcinogenesis and drug resistance. Fundamental and Molecular Mechanisms of Mutagenesis, v.591, p.247-263, 2005.

Referências 140
LONNING, E. Pharmacological profiles of exemestane and formestane, steroidal aromatase
inhitors used for treatment of postmenopausal breast cancer. Breast cancer research and treatment, v.49, p.45-52, 1998.
LU, W.; POON, G. K.; CARMICHAEL, P. L.; COLE, R. B. Analysis of tamoxifen and its
metabolites by on-line capillary electrophoresis-electrospray ionization mass spectrometry
employing nonaqueous media containing surfactants. Analytical Chemistry, v.68, p.668-674,
1996.
MARCOS, J.; PASCUAL, J. A.; DE LA TORRE, X.; SEGURA, J. Fast screening of anabolic and
other banned doping substances in human urine by gás chromatography/tandem mass
spectrometry. Journal of Mass Spectrometry, v.37, p. 1059-1073, 2002.
MARECK, U.; SIGMUND, G.; OPFERMANN, G.; GEYER, H.; THEVIS, M.; SCHANZER, W.
Identification of the aromatase inhibitor letrozol in urine by gas chromatography/mass
spectrometry. Rapid Communications in mass spectrometry, v. 19, p.3689-3693, 2005.
MARECK, U.; SIGMUND, G.; OPFERMANN, G.; GEYER, H.; SCHANZER, W. Screening for
Tamoxifen, Clomiphene and Cyclofenil in Doping Analysis. In: COLOGNE WORKSHOP ON
DOPE ANALYSIS, 9., 2001, Cologne. Proceedings of the Manfred Donike Workshop
Cologne: Sport and Buch Strauβ, 2001, p.53-61.
MARQUES, M. A. S. Método de triagem de resíduos de anabolizantes em rebanho bovino brasileiro por CG-EM/C, 1992, Tese (Mestrado em Química Orgânica) – Instituto de Química,
Universidade Federal do Rio de Janeiro, Rio de Janeiro.
MARQUES, M. A. S.; PEREIRA, A. S.; DE OLIVEIRA, M. C.; TALHAS, I. B.; AQUINO NETO, F.
R. Validation of the determination of tibolone in human urine and saliva using GC-MS for doping
control and clinical studies. In: COLOGNE WORKSHOP ON DOPE ANALYSIS, 12., 2004,
Cologne. Proceedings of the Manfred Donike Workshop Cologne: Sport and Buch Strauβ,
2004, p.403-407.

Referências 141
MARQUES, M. A. S.; PEREIRA, H. M. G.; PADILHA, M. C.; AQUINO NETO, F. R. Analysis of
synthetic 19-norsteroids trenbolone, tetrahydrogestrinone and gestrinone by gas
chromatography-mass spectrometry. Journal of Chromatography A, v.1150, p.215-225, 2007
McDOWALL, R. D. Sample preparation for biomedical analysis. Journal of Chromatography,
v.492, p.3-58, 1989.
MENEGATTI, R. PLANEJAMENTO, SÍNTESE E AVALIAÇÃO FARMACOLÓGICA DE NOVOS CANDIDATOS A PROTÓTIPOS DE AGENTES ANTIPSICÓTICOS, 2002, Dissertação
(Mestrado em Química Orgânica) - Instituto de Química, Universidade Federal do Rio de
Janeiro, Rio de Janeiro.
MENEGATTI, R.; CUNHA, A. C.; FERREIRA, V. F.; PEREIRA, E. F. R.; EL-NABAWI, A.;
ELDEFRAWI, A. T.; ALBUQUERQUE, E. X.; NEVES, G.; RATES, S. M. K.; FRAGA, C. A. M.;
BARREIRO, E. J. Design, Synthesis and Pharmacological Profile of Novel Dopamine D2
Receptor Ligands. Bioorg. Méd. Chem., v.11, p.4807-4813, 2003.
MENEGATTI, R.; FRAGA, C. A. M.; BARREIRA, E. J.; LIMA, V. L. E.; RAYES, S. M. K.; DALLA
COSTA, T. Schizophrenia: forty years of the dopaminergic hypothesis under medicinal
chemistry point of view. Química Nova, v.27, p.447-455, 2004.
MILLER J. C.; MILLER, J. N. Statistics for Analytical Chemistry. New York: Ellis Howood,
1984.
MOORS , M. Analyte isolation by solid-phase extraction (SPE) on silica bonded phase. Pure and Applied Chemistry, v.66, p.277-304, 1994.
MOREIRA, P. I.; CUSTÓDIO, J. B.; OLIVEIRA, C. R.; SANTOS, M. S. Hydroxytamoxifen
protects against oxidative stress in brain mitochondria. Biochemical Pharmacology, v. 68, p.
195-204, 2004.
PEREIRA, H. M. G. DESENVOLVIMENTO DE MÉTODO ANALÍTICO PARA A DETECÇÃO DE GLICOCORTICOSTERÓIDES EM URINA HUMANA. AVALIAÇÃO INICIAL DO PERFIL

Referências 142
ENDÓGENO DE ATLETAS BRASILEIROS, 2004, Tese (Doutorado em Química Orgânica) –
Instituto de Química, Universidade Federal do Rio de Janeiro, Rio de Janeiro.
PEREIRA A. S.; AQUINO NETO F. R. Estado da arte da cromatografia gasosa de alta
resolução e alta temperatura. Química Nova, v.23, p.370-379, 2000.
PEREIRA A. S.; PADILHA M. C.; AQUINO NETO F. R. Two decades of high temperature gas
chromatography (1983 – 2003): what’s next? Microchemical Journal, v.77, p.141-149, 2004.
RIVIER, L. New trends in doping analysis. Analytical Chemistry at Forensic Institutes, v.56,
p.84-90, 2002.
STAACK, R. F.; MAURER, H. H. New designer drug 1-(3,4-methylenedioxybenzil)piperazine
(MDBP): studies on its metabolism and toxicological detection in rat urine using gas
chromatography / mass spectrometry. Journal of Mass Spectrometry, v.39, p.255-261.
SANTEN, R. J. Inhibition of aromatase: insights from recent studies. Steroids, v. 68, p.559-567,
2003.
SCHANZER, W. Metabolism of anabolic androgenic steroids. Clinical Chemistry, v.42, p.1001-
1020, 1996.
SCHANZER, W.; DONIKE, M. Metabolism of anabolic steroids in man:synthesis and use of
reference substances for identification of anabolic steroid metabolites. Analytica Chimica Acta,
v.275, p.23-48, 1993.
SCHANZER, W.; GEYER, H.; DONIKE, M. Metabolism of metandienone in man: Identification
and synthesis of conjugated excreted urinary metabolites, determination of excretion rates and
gas chromatographic-mass spectrometric identification of bis-hydroxylated metabolites. The
Journal of Steroid Biochemistry and Molecular Biology, v.38, p.441-464, 1991.

Referências 143
SCHANZER, W.; GEYER, H.; FUßHÖLLER, G.; HALATCHEVA, N.; KOHLER, M.; PARR, M.;
GUDDAT, S.; THOMAS, A.; THEVIS, M. Rapid Communications in mass spectrometry, v.
20, p.2252-2258, 2006.
SCANZER, W.; HORNING, S.; DONIKE, M. Metabolism of anabolic steroids in humans:
Synthesis of 6β-hydroxy metabolites of 4-chloro-1,2-dehydro-17α-methyltestosterone,
fluoxymesterone, and metandienone. Steroids, v.60, p.353-366, 1995.
SCHANZER, W.; OPFERMANN, G.; DONIKE, M. 17-Epimerization of 17α-methyl anabolic
steroids in humans: metabolism and synthesis of 17α-hydroxy-17β-methyl steroids. Steroids, v.
57, p. 537-550, 1992.
SEGURA, J.; VENTURA, R.; JURABO, C. Derivatization procedures for gás chromatographic-
mass spectrometric determination of xenobiotics in biological samples, with special attention to
drugs of abuse and doping agents. Journal of Chromatography B, v.713, p.61-90, 1998.
SEKERA, M. H.; AHRENS, B. D.; CHANG, Y. C.; STARCEVIC, B.; GEORGAKOPOULOS, C.;
CATLIN, D. H. Another designer steroid: discovery, synthesis, and detection of ‘madol’ in urine.
Rapid Communications in mass spectrometry, v.19, p.781-784, 2005.
SERALINI, G. E.; MOSLEMI, S. Aromatase inhibitors: past, present and future. Molecular and Cellular Endocrinology, v.178, p.117-131, 2001.
THEVIS, M.; FUßHÖLLER, G.; GEYER, H.; RODCHENKOV, G.; MARECK, U.; SIGMUND, G.;
KOCH, A.; THOMAS, A.; SCHANZER, W. Detection of stanozolol and its major metabolites in
human urine by liquid chromatography-tandem mass spectrometry. Chromatographia, v.64, p.
441-446, 2006.
VAN EENOO, P.; DELBEKE, F. T.; DE JONG, F. H.; DE BACKER, P. Urinary metabolites of
endogenous nandrolone in women: A case study. In: COLOGNE WORKSHOP ON DOPE
ANALYSIS, 6., 1998, Cologne. Proceedings of the Manfred Donike Workshop Cologne:
Sport and Buch Strauβ, 1999, p.105-117.

Referências 144
VARIAN, 4000 MS Users Guide Internal Ionization, p.1-41, 2004.
VERROKEN, M. Drug use and abuse in sport. Clinical Endocrinology Metabolism, v.14, p1-
23, 2000.
World Anti-Doping Agency. 2004 Adverse analytical findings reported by accredited laboratories.
Disponível em: http://www.wada-ama.org. Acessado em Outubro de 2007.
World Anti-Doping Agency. Minimum required performance limits for detection of prohibited
substances. WADA Technical Document – TD2004MRPL versão 1.0. Disponível em:
http://www.wada-ama.org
World Anti-Doping Agency. Identification criteria for qualitative assays incorporating
chromatography and mass spectrometry. WADA Technical Document – TD2003IDCR versão
1.2. Disponível em: http://www.wada-ama.org
YAMAMOTO, I.; IWATA, K.; NAKASHIMA, M. Pharmacokinetics of plasma and urine clenbuterol
in man, rat, and rabbit. Journal of Pharmacobiodyn, v.8, p.385-391, 1985.

Referências 145

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo


















