
Síndromes talassêmicas: epidemiologia e...
36
Sandra Regina Loggetto Mestre em Pediatria, área de Hematologia Pediátrica Síndromes talassêmicas: epidemiologia e diagnóstico
Transcript of Síndromes talassêmicas: epidemiologia e...

Sandra Regina Loggetto
Mestre em Pediatria, área de Hematologia Pediátrica
Síndromes talassêmicas:
epidemiologia e diagnóstico
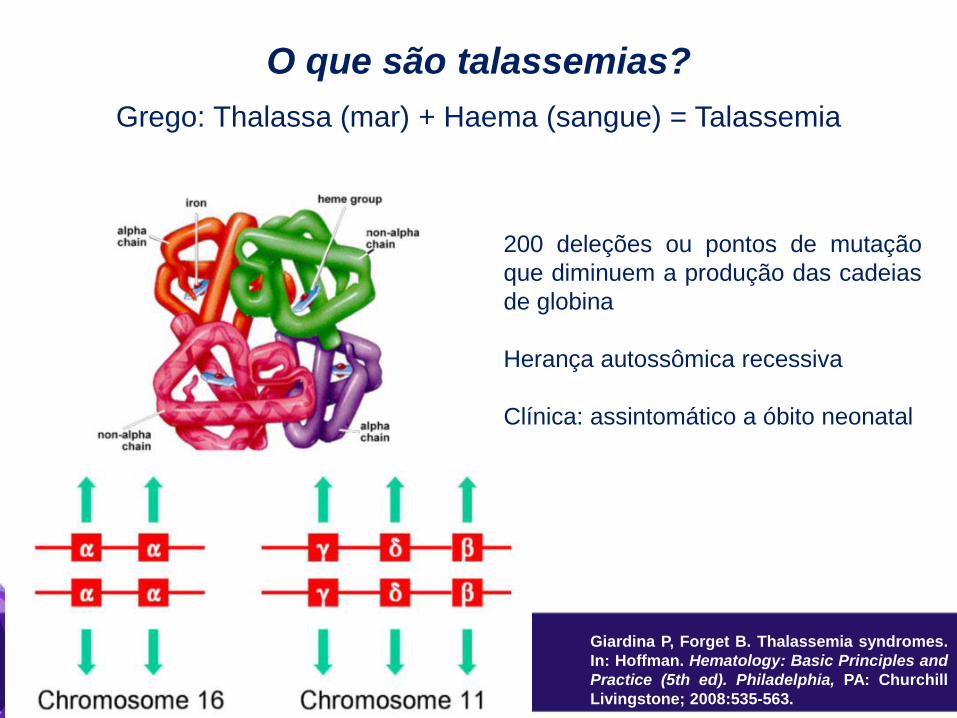
O que são talassemias?
Grego: Thalassa (mar) + Haema (sangue) = Talassemia
200 deleções ou pontos de mutação
que diminuem a produção das cadeias
de globina
Herança autossômica recessiva
Clínica: assintomático a óbito neonatal
Giardina P, Forget B. Thalassemia syndromes.
In: Hoffman. Hematology: Basic Principles and
Practice (5th ed). Philadelphia, PA: Churchill
Livingstone; 2008:535-563.

Fenótipos

Alfa-Talassemia Harteveld and Higgs Orphanet Journal of Rare Diseases 2010, 5:13
http://www.ojrd.com/content/5/1/13
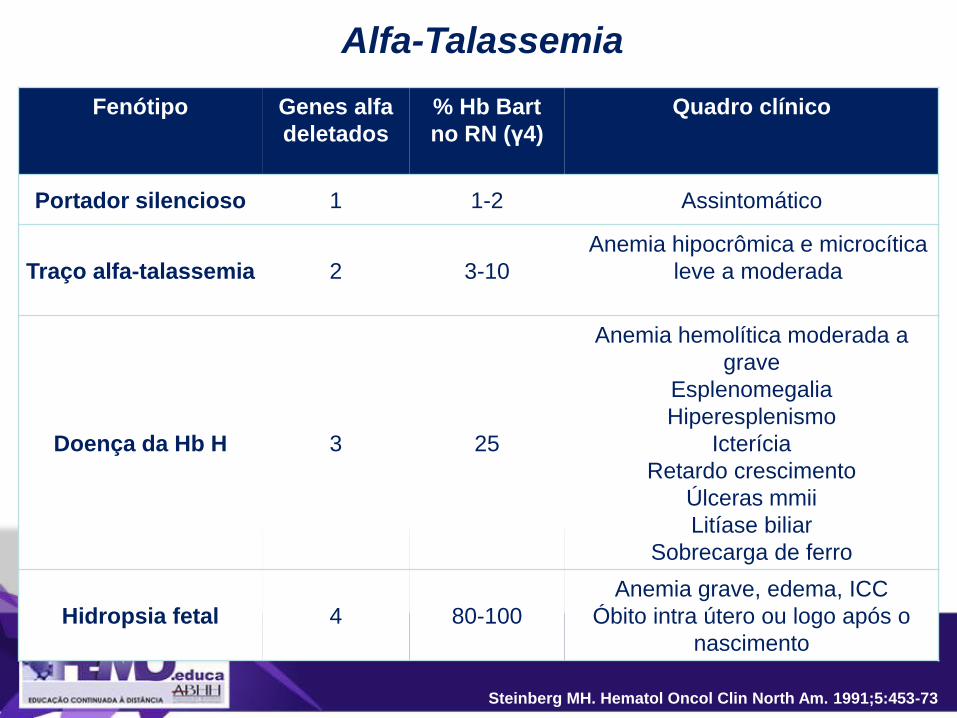
Alfa-Talassemia
Fenótipo Genes alfa
deletados
% Hb Bart
no RN (γ4)
Quadro clínico
Portador silencioso 1 1-2 Assintomático
Traço alfa-talassemia 2 3-10
Anemia hipocrômica e microcítica
leve a moderada
Doença da Hb H 3 25
Anemia hemolítica moderada a
grave
Esplenomegalia
Hiperesplenismo
Icterícia
Retardo crescimento
Úlceras mmii
Litíase biliar
Sobrecarga de ferro
Hidropsia fetal 4 80-100
Anemia grave, edema, ICC
Óbito intra útero ou logo após o
nascimento
Steinberg MH. Hematol Oncol Clin North Am. 1991;5:453-73

Alfa-Talassemia
Fenótipo Genes alfa deletados
(genótipo)
Quadro clínico
Síndrome ATR16
(síndrome α-talassemia/
retardo mental)1,2
Deleções muito grandes do
cromossomo 16, envolvendo o gene
da alfa globina e genes a sua volta
Atraso mental
Síndrome ATR-X
(síndrome do retardo
mental ligada ao X
associada a
α-talassemia)1,3-6
Mutações no gene ATR do
cromossomo X
Atraso mental mais
grave
Síndrome ATMDS1,7 Mutação adquirida no gene ATRX Homens idosos com
SMD
RARAS
1. Higgs. Disorders of Hemoglobin. 2009.; 2. Wilkie. Am J Hum Genet 1990, 46:1112-1126.
3. Gibbons R. Orphanet J Rare Dis 2006, 1:15.; 4. Gibbons. Cell 1995, 80:837-845.
5. Wilkie. Am J Hum Genet 1990, 46:1127-1140.. 6. Wilkie. J Med Genet 1991, 28:738-741.
7. Gibbons. Nat Genet 2003, 34:446-449
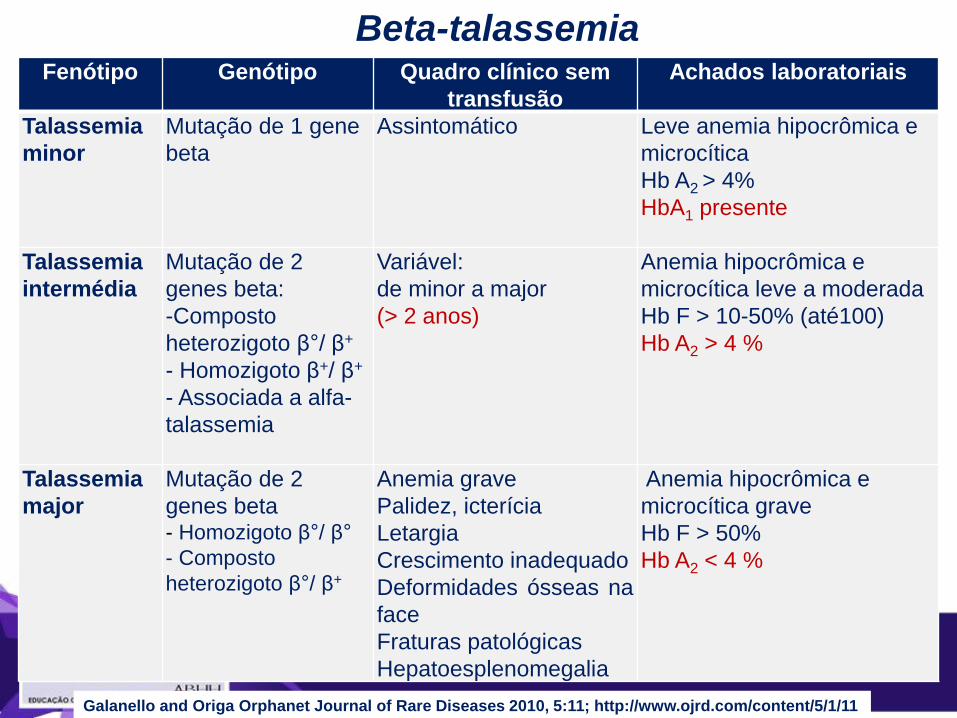
Fenótipo Genótipo Quadro clínico sem
transfusão
Achados laboratoriais
Talassemia
minor
Mutação de 1 gene
beta
Assintomático Leve anemia hipocrômica e
microcítica
Hb A2 > 4%
HbA1 presente
Talassemia
intermédia
Mutação de 2
genes beta:
-Composto
heterozigoto β°/ β+
- Homozigoto β+/ β+
- Associada a alfa-
talassemia
Variável:
de minor a major
(> 2 anos)
Anemia hipocrômica e
microcítica leve a moderada
Hb F > 10-50% (até100)
Hb A2 > 4 %
Talassemia
major
Mutação de 2
genes beta - Homozigoto β°/ β°
- Composto
heterozigoto β°/ β+
Anemia grave
Palidez, icterícia
Letargia
Crescimento inadequado
Deformidades ósseas na
face
Fraturas patológicas
Hepatoesplenomegalia
Anemia hipocrômica e
microcítica grave
Hb F > 50%
Hb A2 < 4 %
Beta-talassemia
Galanello and Origa Orphanet Journal of Rare Diseases 2010, 5:11; http://www.ojrd.com/content/5/1/11

Fenótipo Genótipo Quadro clínico
Beta-talassemia
com outras Hb
anômalas
HbC/Beta-talassemia
Assintomático ou
Anemia e esplenomegalia
HbE/Beta-talassemia Leve: 15% casos, assintomática
Moderadamente grave: >ria dos
casos, talassemia intermédia
Grave: talassemia major
HbS/Beta-talassemia Clínica de anemia falciforme
PHHF e beta-
talassemia
Normal a talassemia intermédia
Formas
autossômicas
dominantes
Heterozigoto para beta dominante Fenótipo de talassemia em
pacientes heterozigotos
Hb instaveis que se precipitam e
causam hematopoiese ineficaz
Beta-talassemia
com outras
manifestações
Beta-talassemia-tricotiodistrofia
Talassemia com trombocitopenia
ligada ao X
Beta-talassemia
Galanello and Origa Orphanet Journal of Rare Diseases 2010, 5:11
http://www.ojrd.com/content/5/1/11

Epidemiologia

A distribuição mundial das hemoglobinopatias se sobrepõe a distribuição geográfica da
malária. A prevalência aumentou em outras regiões devido ao fluxo migratório, escravos,
comércio e colonização
Harteveld and Higgs Orphanet Journal of Rare Diseases 2010, 5:13
http://www.ojrd.com/content/5/1/13
Epidemiologia da talassemia Hemoglobinopatias e Malária no Velho Mundo

Epidemiologia da talassemia Frequência de portadores do gene (%)
2 – 60 0 0 – 13 Pacífico Ocidental
10 – 50 0 0 – 12 África Sub-
saariana
3 – 40 1 – 30 0 – 11 Sudeste da Ásia
0 – 12 1 – 2 0 – 19 Europa
1 – 60 0 – 2 2 – 18 Mediterrâneo
Oriental
0 – 40 0 – 5 0 – 3 Américas
0-Talassemia -Talassemia Região
Weatherall D et al. Inherited Disorders of Hemoglobin’ 2006.
Disease Control Priorities in Developing Countries (2nd Edition).
http://www.dcp2.org/pubs/DCP
+-Talassemia

Local n
População estudada Talassemia alfa
heterozigótica
Campinas1 339 Adultos sem anemia,
com hipocromia e
microcitose
49,9%
Goiás2 404 Adultos alunos de
universidade
5,2%
Salvador3 451 Crianças com doença
falciforme
23%
Minas Gerais4 221 Crianças com doença
falciforme
29,3%
Epidemiologia da alfa-talassemia no Brasil Frequência de portadores do gene (%)
1.Borges. J Med Biol Res. 2001 Jun;34(6):759-62.
2. Melo-Reis. J Bras Patol Med Lab. 2006;42(6):425-430
3. Fonseca. 2009
4. Belisário. Hemoglobin. 2010;34(6):516-29.

Epidemiologia da beta-talassemia Prevalência do gene
Países do Mediterrâneo, Oriente Médio, Ásia
Central, Índia, Sul da China, Extremo Oriente,
Norte da África, América do Sul
Frequência do gene 1,5% da população do mundo
= 80-90 milhões de pessoas
Doentes 60.000 nascimentos/ano
Maioria nos países em desenvolvimento
Incidência anual de
doentes
1 / 100.000 pessoas no mundo
1 / 10.000 pessoas na Europa
TIF (Thalassemia
International Federation)
Apenas 200.000 pacientes com talassemia major
recebem tratamento regular
Flint J. Bailliere's Clinical Hematology 1998, 11:1-50
Vichinsky EP. Ann N Y Acad Sci 2005, 1054:18-24.
Thalassemia International Federation. 2008 [http://www.thalassemia.org.cy]

Local Frequência (%)
Rio Grande do Sul1 1,1
Minas Gerais2 0,13
São José do Rio Preto3 1,29
Ribeirão Preto4 0,8
Campinas e Região5 1,3
Salvador6 0,3
Recife7 0,68
Goiás8 0,7
Epidemiologia da beta-talassemia no Brasil Frequência de portadores do gene (%)
1– Freitas & Rocha, 1983; 2- Melo et al., 2000;
3-Viana-Baracioli et al., 2001; 4- Zago et al., 1983;
5- Ramalho et al., 1999; 6- Fonseca etal., 2005;
7- Almeida et al., 2005; 8- Melo-Reis. 2006

UF TI TM TOTAL
AC 0 0 0
AL 0 03 03
AP 0 0 0
AM 01 01 02
BA 02 01 03
CE 14 01 15
DF 02 02 04
ES 01 06 07
GO 03 12 15
MA 02 05 07
MG 12 21 33
MS 03 02 05
MT 01 03 04
PA 13 02 15
UF TI TM TOTAL
PB 04 01 05
PE 56 19 75
PI 0 0 0
PR 11 26 37
RJ 10 18 28
RN 05 03 08
RO 05 10 14
RR 01 0 01
RS 05 05 10
SE 00 00 00
SC 03 09 12
SP 89 159 248
TO 0 01 01
Total 243 310 553
Fonte: ABRASTA 2010
Distribuição da beta-talassemia major e intermédia
no Brasil

Fisiopatologia da
talassemia

Alfa-talassemias
Diminuição da produção de cadeias de globina alfa
Excesso de cadeias de globina gama nos RN
Excesso de cadeias de globina beta nos adultos
Weatherall DJ & Clegg JB. Bull World Health Organ 2001;79:704–712
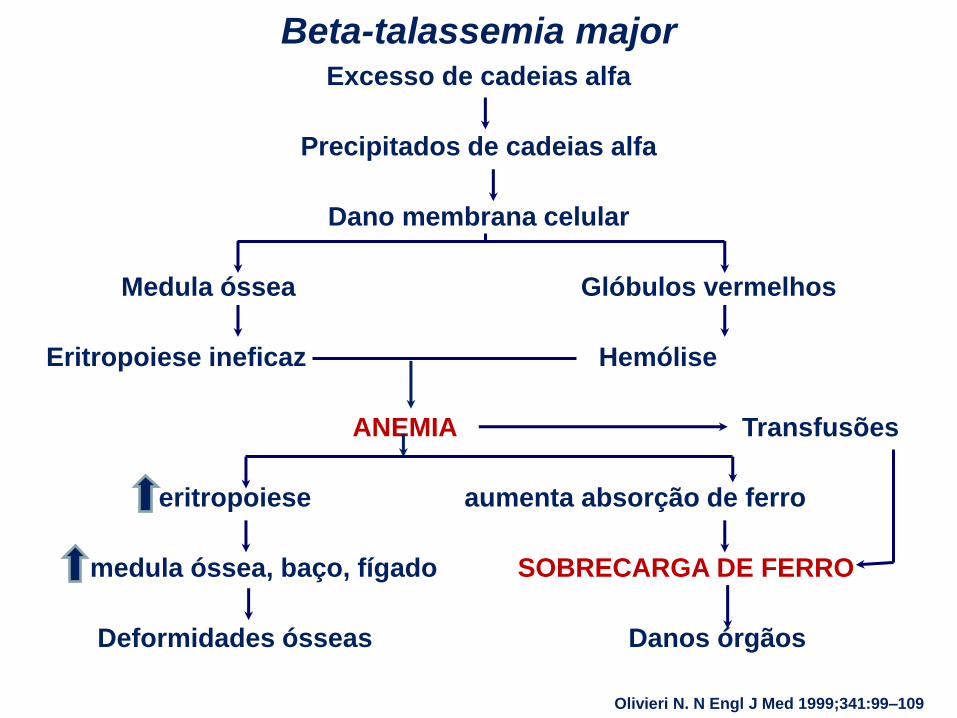
Excesso de cadeias alfa
Precipitados de cadeias alfa
Dano membrana celular
Medula óssea Glóbulos vermelhos
Eritropoiese ineficaz Hemólise
ANEMIA Transfusões
eritropoiese aumenta absorção de ferro
medula óssea, baço, fígado SOBRECARGA DE FERRO
Deformidades ósseas Danos órgãos
Beta-talassemia major
Olivieri N. N Engl J Med 1999;341:99–109

Beta-talassemia intermédia
Menos transfusões de hemácias,
mas também sobrecarga de ferro
Cossu P, Eur J Pediatr 1981;137:267-271.
Fiorelli G, Haematologica 1990;75(Suppl 5):89-95.
Borgna-Pignatti C. British J Hematol 2007:138,291-304.
Eritropoiese ineficaz
Hipertrofia importante da medula óssea e deformidades ósseas
Anemia crônica
absorção intestinal de ferro
Sobrecarga de ferro

Diagnóstico alfa-talassemia

Alfa-talassemia
Geralmente suspeita ocorre em HEMOGRAMA de rotina
Harteveld and Higgs Orphanet Journal of Rare Diseases 2010, 5:13
http://www.ojrd.com/content/5/1/13
Anemia, microcitose ( VCM) e hipocromia ( HCM) – relacionam-se
com o número de genes alfa deletados
Bonini-Domingos C.R. RBHH, 2000, 22(3): 388-394

HbH: Fração de movimento rápido Hb Bart desaparece rápido após o
nascimento
Alfa-talassemia EFHb por HPLC (High-performance liquid chromatography)
Harteveld and Higgs Orphanet Journal of Rare Diseases 2010, 5:13
http://www.ojrd.com/content/5/1/13
HbA2 normal ou pouco
Adulto --/-α RN --/αα

Coloração de sangue periférico com azul cresil brilhante 1%
Corpúsculos de inclusão nos glóbulos vermelhos
= precipitação dos tetrâmeros β4 na membrana celular, os quais lesam a
membrana e induzem hemólise
Como HbH é instável, deve ser investigada em amostra de sangue recente
Traço α0-thalassaemia tem bem menos inclusões do que doença HbH
Harteveld and Higgs Orphanet Journal of Rare Diseases 2010, 5:13
http://www.ojrd.com/content/5/1/13

Análise molecular (genótipo)
Chong SS. Blood 2000, 95:360-362.
Liu YT. Br J Haematol 2000, 108:295-299.
Tan ASC. Blood 2001, 98:250-251.
Alfa-talassemia
Análise molecular α+-talassemia α0-talassemia
Gap-PCR -α3.7
α-4.2
-(α)20.5
--SEA
-- Med I
-- Thai
- - Fil
Southern blotting para rearranjos desconhecidos
MLPA
Brasil: mais comum deleção α3.7 MLPA… Multiplex Ligation-dependent Probe Amplification

Diagnóstico beta-talassemia
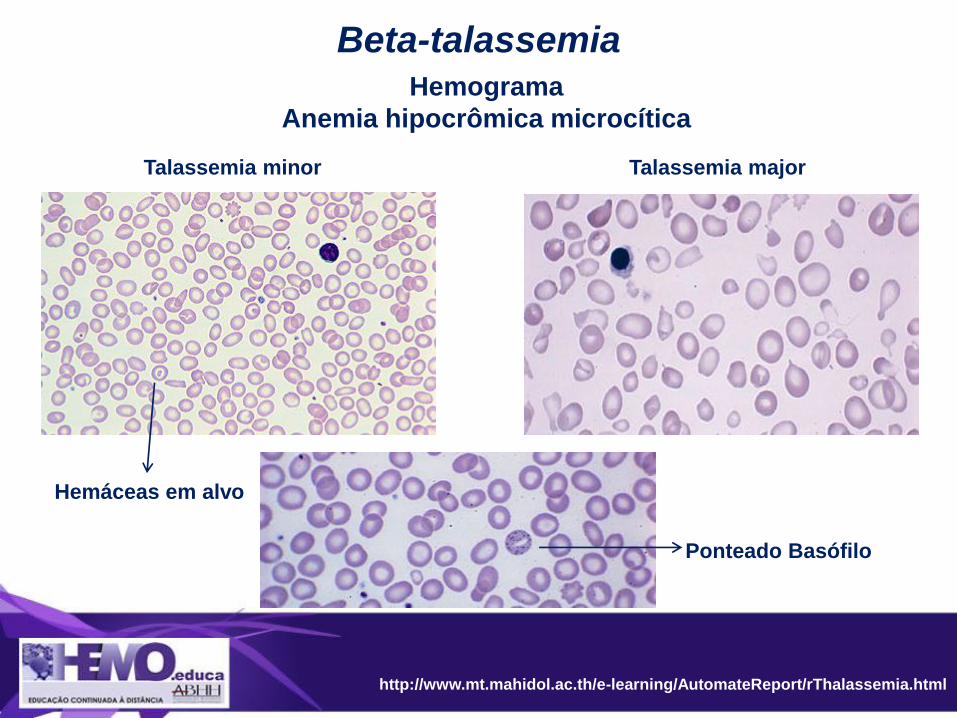
http://www.mt.mahidol.ac.th/e-learning/AutomateReport/rThalassemia.html
Beta-talassemia
Talassemia minor Talassemia major
Hemáceas em alvo
Hemograma
Anemia hipocrômica microcítica
Ponteado Basófilo

EFHb: Utiliza corrente elétrica para separar os diferentes tipos de Hb
Também diagnostica as associações: HbC, HbE, HbS, etc
Beta-talassemia
HbA HbF HbA2
beta0-talassemia
--- 92-95% ---
beta+-talassemia homozigotos
beta+/beta0-talassemia
10-30% 70-90% variável
beta-talassemia minor < 96% > 4%
Eletroforese de Hb
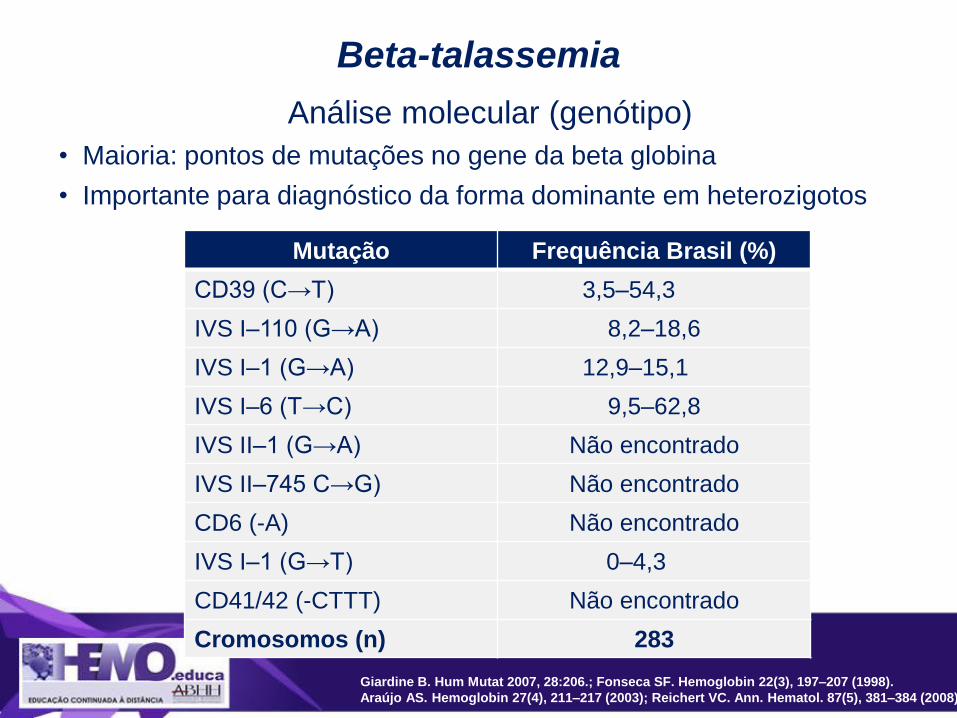
Mutação Frequência Brasil (%)
CD39 (C→T) 3,5–54,3
IVS I–110 (G→A) 8,2–18,6
IVS I–1 (G→A) 12,9–15,1
IVS I–6 (T→C) 9,5–62,8
IVS II–1 (G→A) Não encontrado
IVS II–745 C→G) Não encontrado
CD6 (-A) Não encontrado
IVS I–1 (G→T) 0–4,3
CD41/42 (-CTTT) Não encontrado
Cromosomos (n) 283
Beta-talassemia
Análise molecular (genótipo)
• Maioria: pontos de mutações no gene da beta globina
• Importante para diagnóstico da forma dominante em heterozigotos
Giardine B. Hum Mutat 2007, 28:206.; Fonseca SF. Hemoglobin 22(3), 197–207 (1998).
Araújo AS. Hemoglobin 27(4), 211–217 (2003); Reichert VC. Ann. Hematol. 87(5), 381–384 (2008).

HbA2 HbF EFHb
Alfa-
talassemia
Normal
ou
pouco (HbH)
normal Padrão AA
Beta-
talassemia
> 4% normal ou Padrão AA
Diagnóstico diferencial entre os traços
talassêmicos
HPLC ou Acetato de Celulose
Alfa: ideal análise molecular das deleções do gene da globina alfa
OU: hemograma dos pais
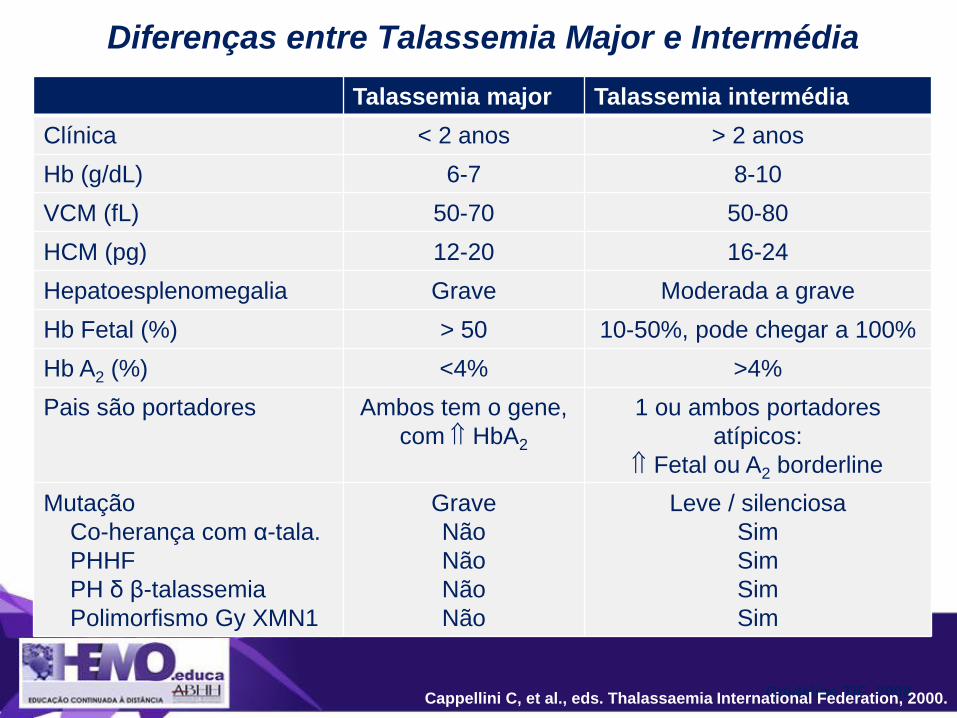
Talassemia major Talassemia intermédia
Clínica < 2 anos > 2 anos
Hb (g/dL) 6-7 8-10
VCM (fL) 50-70 50-80
HCM (pg) 12-20 16-24
Hepatoesplenomegalia Grave Moderada a grave
Hb Fetal (%) > 50 10-50%, pode chegar a 100%
Hb A2 (%) <4% >4%
Pais são portadores Ambos tem o gene,
com HbA2
1 ou ambos portadores
atípicos:
Fetal ou A2 borderline
Mutação
Co-herança com α-tala.
PHHF
PH δ β-talassemia
Polimorfismo Gy XMN1
Grave
Não
Não
Não
Não
Leve / silenciosa
Sim
Sim
Sim
Sim
Diferenças entre Talassemia Major e Intermédia
Guideline TIF 2008 Cappellini C, et al., eds. Thalassaemia International Federation, 2000.

Ferro Ferritina TIBC Sat. RDW Diagnóstico
Talassemia
minor
Normal Normal Normal Normal Normal EFHb com
HbA2 > 4%
Traço
alfa-
talassemia
Normal Normal Normal Normal Normal EFHb normal
Ideal: análise
molecular das
deleções do gene
da globina alfa
Anemia
ferropriva
Identificar causa da
perda de ferro
Anemia da
inflamação
Normal
ou
Normal Normal Identificar a causa
Anemia
sideroblástica
Normal Normal Medula óssea com
sideroblastos em
anel
Investigação laboratorial
anemia hipocrômica microcítica

Aconselhamento Genético e Diagnóstico
Prénatal da beta-talassemia
• Identificação do estado de portador dos pais
• Casal portador: 25% chance de ter filho com talassemia
major por cada gestação
• Diagnóstico Prénatal:
análise de DNA das células fetais por amniocentese (15-
18 semanas gestação)
ou
biópsia de vilo coriônico (11 semanas de gestação)
Galanello and Origa Orphanet Journal of Rare Diseases 2010, 5:11
http://www.ojrd.com/content/5/1/11

Resultado Interpretação Quadro clínico
FA Normal Assintomático
FAS Traço falciforme Assintomático
FS Anemia falciforme (HbSS) ou
SBeta0-talassemia ou
S PHHF
Anemia hemolítica
FSA ou FS2 SBeta+-talassemia Anemia hemolítica
FSC Hemoglobinopatia SC Anemia hemolitica
FSD Hemoglobinopatia SD Anemia hemolitica
FSA + Bart S alfa-talassemia Anemia hemolitica
FAC Traço C Assintomático
FC Hemoglobinopatia C ou
Hemoglobinopatia C-beta0talassemia
Anemia hemolitica
FCA Hemoglobinopatia C-beta+talassemia Anemia hemolitica
FAD Traço D Assintomático
FD Hemoglobinopatia D Anemia hemolitica
FDA Hemoglobinopatia D-beta+talassemia Anemia hemolitica
FA+ Bart (1-5%) Portador silencioso da alfa-talassemia Assintomático
FA + Bart (5-10%) Traço alfa-talassemia Anemia leve
FA +Bart (25-50%) Doença HbH Anemia hemolitica
F 0-talassemia (talassemia major) - HPLC Anemia hemolitica
Resultados triagem neonatal (HPLC ou FIE)

Conclusões

Conclusões
• As síndromes talassêmicas estão presentes no mundo
todo.
• Tem grande heterogenicidade genética e fenotípica.
• É importante o diagnóstico diferencial entre os diferentes
tipos de talassemias e entre as anemias hipocrômicas
microcíticas para que o adequado tratamento possa ser
oferecido.

Bibliografia recomendada
• Harteveld and Higgs, Alfa-thalassaemia. Orphanet
Journal of Rare Diseases 2010, 5:13
• Galanello and Origa, Beta-thalassemia. Orphanet
Journal of Rare Diseases 2010, 5:11
• Weatherall D et al. Inherited Disorders of Hemoglobin’
2006. Disease Control Priorities in Developing Countries
(2nd Edition). http://www.dcp2.org/pubs/DCP














