
Relatório de final de iniciação científica Processo · de sódio ou borohidreto de sódio...
19
Relatório de final de iniciação científica Processo: Determinação voltamétrica do lapachol na presença de lapachonas e extrato etanólico Tabebuia impetiginosa utilizando um eletrodo de epóxi-grafite Aluna: Ana Beatriz Azevedo Orientador: Ricardo Queiroz Aucélio Participação: Carlos Alberto Toloza Toloza e Joseany de Moraes Santos Almeida Departamento de Química Pontifícia Universidade Católica do Rio de Janeiro (PUC-Rio) Resumo O projeto teve como objetivo a determinação do lapachol através da voltametria anódica por onda quadrada (VOQ), utilizando um eletrodo de epóxi-grafite feito no laboratório. O meio eletrolítico consiste em uma solução aquosa contendo o surfactante catiônico CTAB (1,2 × 10 -4 mol L -1 ), tampão fosfato (4,0 × 10 -2 mol L -1 ; pH 6,0) e KNO 3 (1,0 mol L -1 ). O surfactante catiônico conseguiu melhorar a difusão e a interação do eletrodo com o analito, produzindo um processo reversível que melhorou a detecção da corrente total através da VOQ. O sinal do lapachol foi detectado em -470 mV, após uma pré-concentração em 400 mV durante 140 s, usando uma frequência de 30 Hz e uma amplitude de pulso de 40 mV e com um passo potencial de 20 mV. O limite de detecção instrumental foi de 0,029 mg L -1 e a faixa dinâmica linear foi na ordem de duas grandezas. Na presença de β-lapachona (isômero estrutural) a seletividade foi obtida na derivada de primeira ordem do sinal da VOQ. A determinação do lapachol no extrato etanólico presente no cerne da planta Tabebuia Impetiginosa foi obtida através da separação do analito dos componentes complexos da amostra, por cromatografia de camada fina (TLC). Os resultados concordaram, em um nível de confiança de 95%, com os obtidos utilizando a cromatografia líquida de alta eficiência (HPLC) com detecção absorciométrica. Palavras-chave: eletrodo de epóxi-grafite; lapachol; lapachonas; voltametria de onda quadrada; extrato do cerne.
Transcript of Relatório de final de iniciação científica Processo · de sódio ou borohidreto de sódio...

Relatório de final de iniciação científica
Processo:
Determinação voltamétrica do lapachol na presença de lapachonas e extrato etanólico
Tabebuia impetiginosa utilizando um eletrodo de epóxi-grafite
Aluna: Ana Beatriz Azevedo
Orientador: Ricardo Queiroz Aucélio
Participação: Carlos Alberto Toloza Toloza e Joseany de Moraes Santos Almeida
Departamento de Química
Pontifícia Universidade Católica do Rio de Janeiro (PUC-Rio)
Resumo
O projeto teve como objetivo a determinação do lapachol através da voltametria anódica
por onda quadrada (VOQ), utilizando um eletrodo de epóxi-grafite feito no laboratório. O meio
eletrolítico consiste em uma solução aquosa contendo o surfactante catiônico CTAB (1,2 × 10-4
mol L-1
), tampão fosfato (4,0 × 10-2
mol L-1
; pH 6,0) e KNO3 (1,0 mol L-1
). O surfactante
catiônico conseguiu melhorar a difusão e a interação do eletrodo com o analito, produzindo um
processo reversível que melhorou a detecção da corrente total através da VOQ. O sinal do
lapachol foi detectado em -470 mV, após uma pré-concentração em 400 mV durante 140 s,
usando uma frequência de 30 Hz e uma amplitude de pulso de 40 mV e com um passo potencial
de 20 mV. O limite de detecção instrumental foi de 0,029 mg L-1
e a faixa dinâmica linear foi na
ordem de duas grandezas. Na presença de β-lapachona (isômero estrutural) a seletividade foi
obtida na derivada de primeira ordem do sinal da VOQ. A determinação do lapachol no extrato
etanólico presente no cerne da planta Tabebuia Impetiginosa foi obtida através da separação do
analito dos componentes complexos da amostra, por cromatografia de camada fina (TLC). Os
resultados concordaram, em um nível de confiança de 95%, com os obtidos utilizando a
cromatografia líquida de alta eficiência (HPLC) com detecção absorciométrica.
Palavras-chave: eletrodo de epóxi-grafite; lapachol; lapachonas; voltametria de onda quadrada;
extrato do cerne.

Resumo da produção científica
Artigo científico
Selective determination of naphtoquinones by square-wave voltammetry usig the graphite-epoxi
electrode, Azevedo A.B, Alnmeida, J.M.S., Toloza C.A.T., Aucelio R.Q., em preparação para
submissão em 2017.
1 Introdução
A flora tropical é uma abundante fonte de diferentes substâncias biológicas, como por
exemplo as naftoquinonas que são comumente encontradas na família de plantas Bignoniaceae 1.
O Lapachol (Fig. 1) é uma para-naftoquinona que possui compostos biológicos responsáveis por
combater diferentes bactérias2, o vírus HIV-1
3, parasitas molusco-hospedeiros
4 e protozoários
como o Trypassoma cruzi5. Oliveira e outros coautores demonstraram que o lapachol também
possui atividade contra o mosquito Aedes aegypti, principal transmissor do vírus da Dengue e
Zika6. Os isômeros α-lapachona e β-lapachona (Fig. 1) também possuem espécies da família
Bignoniaceae, entretanto tem quantidades significativamente menores quando comparadas ao
lapachol, o qual contém até 7% do cerne da planta1,7
.
(2) O
O
O
(1)
O
O
OH
CH3
CH3
O
O
O
(3)
Fig. 1: Estruturas química do (1) lapachol, (2) α-lapachona e (3) β-lapachona
A principal abordagem na determinação do lapachol e de outras naftoquinonas presentes
em extratos de plantas é baseada na cromatografia líquida de troca iônica (HPLC) e na
fotometria de absorção e detecção UV8,9
. Steinert et al. propôs a cromatografia líquida de alta
eficiência na região do ultravioleta (HPLC-UV) para separar e determinar as naftoquinonas em
extratos de Tabebuia avellanedae (Bignoniaceae). Porém, o autor falhou em detectar lapachol
em extratos aquosos8. O lapachol e outras naftoquinonas foram isoladas com extrato etanólico da
raiz da Zeyheria montana (genus Tabeluia) e foi quantificado, com sucesso, através da HPLC-
UV, indicando a presença de 0,001% m/m de lapachol na amostra9.
A determinação indireta baseada na fotoluminescência de supressão de pontos quânticos
(QDs) na presença de lapachol, foi recentemente relatada10,11
. Demonstrou-se que o ácido 3-
mercaptopropiônico-CdTe QDs foi usado como sonda para determinar lapachol em amostras de
urinas (previamente limpas utilizando um cartucho com polímero de acrílico) com um limite de
detecção (LOD) de 1,9 mg L-1
. A supressão da fotoluminescência do ácido 3-
mercaptopropiônico-CdTe QDs promovido pelo lapachol, β-lapachona, α-lapachona ou ácido 3-
sulfônico-β-lapachona tornou-se melhor em uma dispersão aquosa contendo brometo de

hexadeciltrimetilamônio (CTAB). A presença do surfactante CTAB promoveu uma rápida
estabilização dos pontos quânticos depois das interações com as naftoquinonas e melhorou a
magnitude da supressão de fluorescência. O limite de detecção foi de 0,04 mg L-1
para o
lapachol e ele foi determinado, com sucesso, em um extrato etanólico do cerne de T.
impetiginosa11
. Outro método baseado na luminescência dependeu do sinal gerado depois da
redução das naftoquinonas, usando hidrosulfito de sódio, para gerar a derivatização da
fluorescência do lapachol, a qual é instável na presença de oxigênio12
. A redução com o ditionito
de sódio ou borohidreto de sódio permitiu a determinação fluorimétrica do lapachol (alcance do
LOD é de μg L-1
) e de outras quinonas em formulações farmacêuticas. Entretanto, o uso de
diferentes derivatizações dos produtos influenciaram à precisão do método13
.
A voltametria foi empregada em estudos de redução e determinação de naftoquinonas14-
17. Estudos utilizando a voltametria cíclica (VC) feitos por Goulart et al., usando um eletrodo de
carbono vítreo (GCE), mostraram que o radical aniônico do lapachol interage com o oxigênio e
provoca a desprotonação do lapachol e de radicais peroxil16
. A redução eletroquímica das
lapachonas foi estudada por Oliveira-Brett et al., utilizando a VC, voltametria de onda quadrada
(VOQ) e voltametria de pulso diferencial em um meio hidroalcóolico. O processo de redução,
promovido pelo GCE, para a β-lapachona, ácido 3-sulfônico-β-lapachona e β-lapachona 3-
bromo-β-lapachona foi reversível e dependente do pH. Entretanto, para a α-lapachona o processo
foi irreversível no pH 4,5 e quasi-reversível para pH 7,0. Outro estudo para a redução
eletroquímica da β-lapachona e o seu ácido, derivado, 3-sulfônico, em meio aquoso, foi feito
utilizando GCE. Os resultados indicaram um processo reversível e dependente do pH e
evidências de uma interação entre β-lapachona e a topoisomerase17
. A voltametria de pulso
diferencial foi aplicada na determinação do ácido 3-sulfônico-β-lapachona com LOD de 0,41 mg
L-1 15
.
Os eletrodos feitos de carbono são amplamente utilizados como sensores eletroquímicos
devido à sua janela de potencial operacional favorável em uma ampla faixa de pH. Eles são
dispositivos de baixo custo e fáceis de modificar e manusear18
. Adams, em 1958, introduziu os
compósitos, os quais são constituídos de uma fase condutiva mista de carbono e um material
isolante, como resinas, polímeros e óleos18-20
. Os compósitos são microscopicamente
heterogêneos e apresentam propriedades de materiais precursores21
. Os eletrodos compósitos
podem ser aplicados em uma ampla faixa de pH e potencial e também apresentam uma boa
condutividade, baixo custo, estabilidade mecânica e versatilidade na maneira em que podem ser
preparados. Mudanças químicas, principalmente com surfactantes, podem melhorar a
sensibilidade e a seletividade em analitos específicos. Além disso, mudanças reprodutíveis
podem ser facilmente feitas na a superfície do eletrodo22,23
.
Semaan et al., desenvolveu um compósito de poliuretano-grafite para determinação de
furosemida em fármacos através de SWV. A resposta do analito foi linear até 6,95 mg L-1
. Não
houve necessidade de renovação constante da superfície do eletrodo, uma vez que o analito não
adsorveu no material do eletrodo23
. Mais recentemente, Balbin-Tamayo et al., validou o
compósito de epóxi-grafite para ser usado como sensor de DNA24
. Eletrodos feitos com
diferentes proporções de epóxi/grafite foram caracterizados por VC, espectroscopia de
impedância eletroquímica e microscopia eletrônica de varredura por emissão de campo. A
melhor resposta eletroquímica, usando SWV, para a guanosina monofosfato e a adenina foi
obtida usando uma resina endurecedora de epóxi-grafite nas proporções 3,3/2,5/1m/m/m.
Neste trabalho, um método SWV sensível para determinar o lapachol, utilizando o
eletrodo compósito de epóxi-grafite, foi desenvolvido. A resposta analítica foi melhor em um

meio contendo o surfactante catiônico CTAB. Uma simples separação por cromatografia de
camada fina permitiu determinar a seletividade do lapachol no extrato etanólico do cerne de T.
impetiginosa.
2 Experimental
2.1. Instrumentação
O método voltamétrico foi desenvolvido utilizando-se um potenciostato / galvanostato
(m-AUTOLAB Type III, Metrohm, Holanda) interligado a um computador e operando no modo
de onda quadrática de análise voltamétrica e voltametria cíclica. O eletrodo de trabalho foi de
epóxi-grafite e foi feito de acordo com o trabalho de Balbin-Tamayo et al. 24
. Utilizou-se o Ag /
AgCl (KClsat) como eletrodo de referência do sistema eletroquímico e um fio de platina como
eletrodo auxiliar. O GCE também foi usado para comparar experimentos. Uma célula
eletroquímica de 15 mL, feita de borosilicato, foi utilizada com uma tampa de Teflon, a qual
proporcionou o acesso dos três eletrodos até a solução. As medidas de pH foram feitas em um
pHmetro (modelo mPA-210, MS Tecnopon, Brasil) usando um eletrodo de vidro combinado
com um eletrodo de referência Ag / AgCl (KClsat). As análises de cromatografia foram feitas em
um cromatógrafo líquido de alta eficiência (modelo 1200, Agilent Technologies, Japão),
equipado com detecção de absorção fotométrica, em um forno de coluna (mantido a 30oC) e em
um Agilent Eclipse XDB–C18 com coluna (250 × 4.6 mm e tamanho médio de partícula de 5
μm, EUA).
2.2. Reagentes e materiais
Todas as soluções foram preparadas utilizando água deionizada (resistividade inferior a
18 Mcm) obtida a partir de um purificador de água Milli-Q Gradient Sistem A10, Millipore
(EUA). O lapachol (142-143oC), α-Lapachona (140
oC), β-lapachona (155
oC) e ácido sulfônico-
β-lapachona (158-160ºC) foram obtidos segundo os procedimentos obtidos na literatura25-27
.
Todas as naftoquinonas foram purificadas e caracterizadas por métodos espectroscópicos e os
resultados concordaram com os dados da literatura28-30
. O brometo de hexadeciltrimetilamônio
(CTAB) e o ácido acético (grau analítico) foram comprados da Sigma-Aldrich (EUA). O nitrato
de potássio, fosfato de sódio monobásico e o fosfato de sódio dibásico foram obtidos na Merck
(Alemanha). O hexano, álcool metílico e etanol (todos com grau HPLC) foram obtidos na Tedia
(Brasil). O clorofórmio foi comprado na Isofar (Brasil), o ferrocianeto de potássio foi obtido na
Autolabor (Brasil) e o tampão Tris.HCl foi comprado na Synth, Brasil.
O pó de grafite de grau espectroscópico foi adquirido na Ringsdorff-Werke GMBH
(Alemanha). A resina epóxi foi obtida a partir de um kit comercial de pasta Araldite®. A pasta
de alumínio (1 m) foi adquirida da Fortel (Brasil). O gás nitrogênio, marca comercial, foi
comprado na Linde-gases (Brasil). As placas de gel de sílica (60 F254), para cromatografida de
camada fina, foram adquiridas da Merck. Os filtros de seringa de PTFE (0,45 μm) foram
comprados na Whatman (Reino Unido).
O extrato comercial (Pau d'arco, supostamente da T. impetiginosa) foi comprado na
farmácia e o cerne natural da T. impetiginosa (Ipê Rosa) foi comprado em um mercado local.

2.3. Construção e caracterização do eletrodo de epóxi-grafite
Para a construção do eletrodo de epóxi-grafite foi utilizado o procedimento descrito na
literatura24
, com pequenos ajustes. A resina epóxi e o endurecedor (do kit de Araldite) foram
misturados em quantidades iguais, antes da adição do pó de grafite. Esses componentes foram
misturados a fim de se obter um compósito homogêneo constituído de 3%m/m de uma mistura
resina-endurecedor e 97% m/m de grafite. Antes do compósito de epóxi-grafite endurecer, ele foi
introduzido na ponta de um capilar de vidro (aproximadamente 10mm), onde já tinha um fio de
cobre, estabelecendo o contato elétrico entre o fio de cobre e o compósito. Após 24h, a superfície
do compósito foi polida usando uma lixa (1200 e 600 grão) e depois realizou-se um polimento
final utilizando alumina em suspensão (1 μm). A imagem microscópica do compósito polido é
mostrada na (Fig. 2). A área ativa de superfície do eletrodo foi determinada usando medidas de
VC (de -250 até +650 mV na faixa de varredura de 20-100 mV s-1
) de uma solução de K4[Fe
(CN)6] (1,0 × 10-3
mol L-1
) usando uma solução de KNO3 (0,5 mol L-1
) como eletrólito suporte.
Fig. 2: Imagem de microscopia eletrônica de varredura com emissão de campo das superfícies polidas de eletrodos
compósitos de epóxi-grafite (tensão de aceleração de 1,0k; resolução de 10 μm).
2.4. Soluções Estoque e soluções padrão
Quantidades apropriadas de cada uma das naftoquinonas (lapachol, α-lapachona e β-
lapachona) foram usadas para preparar as soluções estoque de 1,0 × 10-2
mol L-1
e 1,0 × 10-4
mol
L-1
em metanol. Soluções mais diluídas de naftoquinonas foram preparadas para diluir nas
soluções estoque com metanol. As concentrações finais dos componentes presentes na solução
eletrolítica de trabalho foram: tampão fosfato (4,0 × 10-2
mol L-1
; pH 6,0), nitrato de potássio
(1,0 mol L-1
) e o surfactante catiônico CTAB (1,2 × 10-4
mol L-1
) em água deionizada. A
presença de lapachol diminuiu a concentração micelar crítica do CTAB (normalmente em 9,2 ×
10-4
mol L-1
31
), o que torna o sistema eletrolítico rico em micelas e estruturas pré-micelas.
2.5. Preparação da Amostra
Como descrito por Lima et al. 11
, o Pau d'arco comercial (250 mg do interior da casca em
pó por cápsula) foi extraído, utilizando etanol, em um banho de ultrassom (5 min) e depois o
extrato foi passado por um filtro seringa (0,45 μm) e coletado em um balão volumétrico de 10,00
mL. Ao final, ele foi guardado no escuro e sob refrigeração. O cerne da T. impetiginosa (Ipê
Rosa) foi triturado finamente (aproximadamente 1 g). Depois, porções de 125 mg do pó

resultante foi extraído com etanol (como descrito acima) e guardado em um balão volumétrico de
10,00 mL. Todas os extratos da amostra foram realizados em triplicata.
2.6. Cromatografia de Camada Fina
O procedimento de cromatografia de camada fina (TLC) utilizado foi o descrito por Lima
e co-autores11
. Alíquotas de 20 μL de soluções estoque de naftoquinonas (1,0 × 10-2
mol L-1
ou
1,0 × 10-3
mol L-1
) ou alíquotas de 40 μL de amostras de extratos alcóolicos foram pingadas na
placa de TLC e deixadas para secar a temperatura ambiente. Depois, as placas foram colocadas
em uma câmara de revelação contendo uma fase móvel (clorofórmio:hexano 8:2 v/v). Misturas
contendo lapachol, α-lapachona e β-lapachona (proporção equimolar) foram pingadas na placa
TLC para mostrar a separação do lapachol das outras naftoquinonas. O lapachol apresentou o
maior fator de retenção (FR = 0,78), sendo completamente separado das outras naftoquinonas. A
área da placa de TLC contendo o pingo de lapachol foi removida e extraída com 3 mL de
metanol (em 5 min de ultrassom). A solução eluída foi filtrada em um filtro seringa. Depois,
colocou-se 5 mL de metanol no filtro para lavá-lo. A solução coletada foi evaporada (em um
banho quente) até secar. Depois, o resíduo foi redissolvido em metanol (1,00 mL) e coletou-se
uma alíquota de 250 μL para ser adicionada na célula eletrolítica a fim de se realizar medidas
voltamétricas. Os outros pontos da placa de TLC não usada (aproximadamente 1g) também
foram tratados do mesmo modo para avalia qualquer interferência potencial causada pelo
fluoróforo da placa removida.
2.7. Medições Voltamétricas do Lapachol
Todos os experimentos voltamétricos foram realizados depois da purga da solução na
célula eletrolítica (aproximadamente 2 min) e antes da medição. Também foram realizados
experimentos de VC para avaliar o processo redox na faixa de -900 mV a +600 mV, usando 24
mg L-1
de lapachol na solução eletrolítica de trabalho que continha ou não CTAB. Medições de
SWV foram realizadas, usando a solução eletrolítica de trabalho, para avaliar o processo redox e
também para determinar analiticamente o lapachol. Realizaram-se experimentos de diagnóstico
redox na faixa de +600 mV a -1000 mV. Determinações analíticas foram obtidas com uma pré-
concentração analítica em +400 mV por 140 s (sob regime de transporte convectivo e purga com
gás nitrogênio). Após o tempo de equilíbrio (1 min) sem agitação e purga, as medidas de +600
mV até -900 mV foram realizadas usando 30 Hz, 20 mV de passo potencial e 40 mV de
amplitude de pulso. O sinal analítico foi um pico em -470 mV.
2.8. Análise da Cromatografia
As amostras extraídas foram analisadas por HPLC-UV sob uma eluição isocrática com
metanol/solução 5% de ácido acético (80/20% v/v)11
. A vazão da fase móvel foi 1 mL min-1
e o
volume de amostra injetado foi 10 μL. Sob essas condições, o tempo de retenção do lapachol foi
de 5,7 min. As curvas analíticas foram construídas através da injeção de 20 μL de soluções
estoque de lapachol com 24 mg L-1
a 96,9 mg L-1
. As análises foram realizadas em triplicata com
detecção absorciométrica em 278 nm.
3 Resultados e Discussão
3.1 Estudos preliminares

3.1.1. Área eletroativa
As quantidades de resina epóxi e grafite usadas para preparar o compósito de epóxi-
grafite foram ajustadas de acordo com Tamayo-Balbin et al. 24
. A proporção escolhida (97% de
grafite e 3% de resina m/m) apresentou excelentes propriedades mecânicas e produziu correntes
de pico de difusão acentuadas a partir da guanina com sinal de base de corrente baixa. A corrente
de difusão do lapachol encontrada foi intensa. A caracterização da área eletroativa foi realizada
usando VC (de -250mV a +650mV) com uma célula eletroquímica contendo [Fe(CN)6]3-
(1,0 ×
10-3
mol L-1
) em KNO3 (0,5 mol L-1
). Varreduras sequenciais geraram uma corrente de pico (Ip)
que aumenta linearmente em função da faixa de varredura (v), de 20 a 100 mV s-1
. Como a
concentração de espécies eletroativas (C) foi 1,00 × 10-6
mol cm-3
, foi possível estimar a área
eletroativa como 0,0078 cm2 pela simplificação da equação de Randles-Sevcik: A = Ip/(2,686 ×
105 v
1/2 n
3/2 C D
1/2 ), onde D é 6,32 × 10
-6 cm
2 s
-1 e n=1. Para o GCE, a área eletroativa foi 0,019
cm2.
3.1.2. Influência do CTAB no processo eletroanalítico para diferentes valores de pH
Os surfactantes representam um importante papel no processo eletroquímico, pois eles
conseguem mediar a interação entre as espécies eletroativas e o eletrodo32,33
. Os surfactantes
também podem influenciar processos redox e minimizar a passivação da superfície do eletrodo
causada pela adsorção de produtos eletrogerados ou por componentes da matriz da amostra 34,35
.
Além disso, eles melhoram a solubilidade de analitos hidrofóbicos e facilitam o transporte de
massa 36,37
.
Guin et al., revelou a área eletroquímica de redução de quinonas, em diferentes meios de
comunicação 38
. Em meio aquoso tamponado, o par redox quinona-hidroquinona envolve a
transferência de dois elétrons, em potenciais que variam com pH, com etapas que envolvem
diferentes espécies reduzidas e oxidadas com diferentes valores de pKa. Em pH ácido, devido à
rápida cinética e disponibilidade de prótons, o processo de protonação em duas fases afeta ambos
grupos carboxílicos e pode ser visto como um processo de apenas uma fase, envolvendo dois
elétrons e dois prótons. Em pH básico, o processo de redução ocorre, unicamente, devido aos
elétrons, enquanto em pH neutro, a redução envolve ou um próton e dois elétrons ou apenas dois
elétrons38
. Para o lapachol, Ngameni et al,.14
mostrou que o aumento do pH causa um
alargamento dos picos anódicos (em maior extensão devido à dificuldade da re-oxidação do
hidrolapachol) e catódicos na VC, o que é um reflexo da cinética dos diferentes processos que
estão ocorrendo. Em condições básicas, as espécies não-protonadas hidrofílicas (devido ao
próton perdido no grupo hidroxílico do sistema enol) são dominantes em relação às protonadas.
O comportamento, no meio básico aquoso (usando Tris-HCl com pH 9), confirmou, neste
trabalho, que o processo de redução não é favorável, devido à falta de prótons, entretanto o
processo de oxidação ocorre, provavelmente envolvendo apenas elétrons (voltamograma b da
Fig. 3A). Na presença de CTAB (perto da CMC), o pico de redução foi reestabelecido,
provavelmente, por causa da estabilização, reduzindo a energia necessária para produzir radicais.
Uma melhora na oxidação foi claramente notada, provavelmente, ocorrendo em diferentes
estágios, como é mostrado no voltamograma cíclico, e além do pico principal da oxidação,
percebem-se picos menores em potenciais positivos (voltamograma a da Fig. 3A).
No voltamograma obtido em pH 6 (tampão fosfato) em meio aquoso, o pico de
oxidação foi menor do que o de redução, indicando um processo quasi-reversível (voltamograma
a da Fig. 3B). Nestas condições, o lapachol teve um valor de pKa de 6 (o que se refere a

ionização do enol39
), portanto, a proporção entre as espécies protonadas e as não-protonadas é de
aproximadamente 1. Em contraste, quando o surfactante catiônico foi incluído na solução de pH
6, um comportamento reversível apareceu, com correntes de pico mais intensas (voltamograma b
da Fig. 3B). Em um meio contendo CTAB, o pKa de soluções ácidas, como o lapachol, diminui
em aproximadamente duas unidades 40,41
, fazendo com que as espécies não-protonadas e
hidrofílicas sejam predominantes em pH 6. Em meios aquosos com pH 4 (tampão acetato), um
comportamento quasi-reversível foi observado e a intensidade de ambas correntes de pico de
redução foi menor que as de pH 6 (voltamograma a da Fig. 3C). No pH 4, as espécies não-
protonadas foram predominantes, mas quando se colocou CTAB, a proporção não-
protonado/protonado dessas espécies ficou próxima a 1 e o pico de oxidação aumentou até o
ponto que o processo eletroquímico se tornou reversível (voltamograma b da Fig. 3C). O CTAB
também deslocou o pico de oxidação para um potencial mais anódico e o pico de redução para
um potencial mais catódico. Apesar disso, a intensidade do pico de corrente foi
significantemente menor que os observados em pH 6.
A presença de CTAB pareceu melhorar a difusão do lapachol, já que o eletrodo realizou
um melhor transporte e uma reação redox mais eficiente. As micelas e os agregados pré-micelas
confinaram os reagentes e produtos envolvidos na reação (especialmente na de oxidação)
fazendo a concentração local (dentro da micela normal ou entre os monômeros que formam a
estrutura micelar) ficar muito alta, mas melhorando a cinética da reação. A formação de radicais
também é favorecida, pois eles são estabilizados pelo sistema de micelas.
É importante ressaltar que os processos redox intensos e reversíveis costumam levar a
picos SWV muito intensos (que contribuem para um sinal para ambas correntes de difusão de
redução e oxidação), levando a um maior poder de detecção em análises quantitativas. Assim, o
meio tamponado a pH 6 e contendo CTAB foi escolhido para estabelecer o método quantitativo
voltamétrico.
Como mencionado na seção anterior, o estudo qualitativo do processo eletroquímico do
lapachol (24 mg L-1
) na presença de CTAB por VC produziu um único pico de redução (máximo
em -450 mV) na direção catódica e também um único pico de oxidação (máximo em -154 mV)
na direção anódica. Uma outra adição de lapachol (para produzir uma concentração final de 72,7
mg L-1
) produziu um resultado similar, mas com picos mais intensos. Esse comportamento é
típico de processos reversíveis (taxa de transferência de carga elevada e transferência de massa
controlada por difusão) o que possibilitou um equilíbrio dinâmico na interface do eletrodo42
.
Varreduras VC sequenciais foram feitas para checar a estabilidade do processo (taxa de
varredura foi e 100 mV s-1
, de -900 mV a 700 mV e sem transporte convectivo forçado de
massa). Mesmo depois do décimo ciclo (Fig. 4A) não existiam evidências de decréscimo da
magnitude do pico, indicando que as espécies na interface solução-eletrodo são prontamente e
repetidamente convertidas nas formas oxidada e reduzida. Além disso, a razão calculada entre a
corrente do pico anódico (Ipa) e a corrente do pico catódico (Ipc), na velocidade de varredura de -
100 mV s-1
, foi de 0,83. A literatura mostra que em sistemas redox mais simples, essa razão
tende a se aproximar de 1 à medida que o pH fica mais ácido e a disponibilidade de H+ garante
uma rápida reoxidação14
.Os valores de razão perto ou igual a 1, independentemente da taxa de
varredura, são típicos de sistemas reversíveis, como é observado no lapachol na presença de
CTAB (Fig. 4B)20,43
. A diferença entre os potenciais de pico anódico e catódico (ΔEp = Epa -
Epc) manteve-se constante com a mudança da taxa de varredura (de 10 a -100 mV s-1
), o que é
uma indicação de reversibilidade. A reversibilidade do processo foi confirmada, pois a raiz
quadrada da taxa de varredura (v), de 10 a 1000 mV s-1
, produziu uma resposta linear (R2 =

0,982) em função de Ip (catódico). A linearidade (R2 = 0,990) foi encontrada em relação ao log v
em função do log Ip, produzindo um valor de inclinação de 0,6, o que sugere que o processo
envolvido é pincipalmente difusional em vez de adsortivo (Fig. 4C)44
.
-1200 -800 -400 0 400 800-2
-1
0
1A b
a
I (
A)
Potential x Ag/AgCl (mV)
-1200 -800 -400 0 400 800-2
-1
0
1B
b
a
I (
A)
Potential x Ag/AgCl (mV)
-1200 -800 -400 0 400 800-2
-1
0
1 C
ba
I (
A)
Potential x Ag/AgCl (mV)
Fig. 3: Voltametria Cíclica do processo redox do lapachol em meio aquoso em diferentes pHs na (a) presença e (b)
ausência de CTAB. A- tampão Tris-HCl (4,0 × 10-2
mol L-1
, pH 9); B- tampão fosfato (4,0 × 10-2
mol L-1
, pH 6) e C-
tampão acetato (4,0 × 10-2
mol L-1
, pH 4). v = 100 mV s-1

3.1.3. Estudo dos mecanismos em meio aquoso contendo CTAB
-1200 -800 -400 0 400 800
-2
-1
0
1
2A
Potential x Ag/AgCl (mV)
I (
A)
a
k
a
k
-1200 -800 -400 0 400 800
-1,8
-0,9
0,0
0,9
1,8 B 100 mV s-1
10 mV s-1
I (
A)
Potential x Ag/AgCl (mV)
1,0 1,5 2,0 2,5 3,0-6,4
-6,0
-5,6
-5,2C
Lo
g I
p
Log v
Fig. 4: A – Análises cíclicas sequenciais (linha a-k) para o lapachol a 100 mV s
-1 em 72,7 mg L
-1 de lapachol. B –
Taxa de varredura cíclica de 10-100 mV s-1
para o lapachol na presença de CTAB 1,2 × 10-4
mol L-1
para checar a
reversibilidade do processo redox. C – Estudo do log v × log Ip (29,1 mg L-1
) do lapachol; v = 10 a 1000 mV s-1
.
Condições Experimentais: tampão fosfato (4 × 10-2
mol L-1
; pH 6), 1,2 × 10-4
mol L-1
CTAB, KNO3 (1 mol L-1
) em
meio aquoso com v = 100 mV s-1
.
A SWV também forneceu informações importantes sobre o processo. A dependência da
frequência (na verdade log f com f de 10 a 90 Hz) em relação ao potencial de pico (Ep) foi linear
(R2 = 0,994) como mostrado na Fig. 5A. Além disso, o aumento de Ip em função de f (Fig. 5B)
foi diretamente proporcional e linear (R2 = 0,964), o que é uma indicação que o processo foi
controlado pela difusão dos reagentes43
.
Na presença de CTAB, o Ep mudou para valores negativos à medida que o pH do
eletrólito suporte foi aumentando (Fig. 5C). Esses são processos típicos influenciados pela
concentração hidrogeniônica do meio, a qual é confirmada pela relação dada por Ep (mV) = (48,0
± 1,3) + (-68,5 ± 0,18) pH, que relaciona a equação de Nernst e a transferência de dois
elétrons43,45
. Isso é compatível com a redução de dois grupos carboxílicos, um deles perto do
grupo hidroxil, que pode promover a ligação de hidrogênio auxiliada pela estabilização dos

radicais gerados durante a redução15
. A concentração de CTAB (8,1 × 10-4
mol L-1
), no eletrólito
suporte, estava ligeiramente abaixo da concentração micelar crítica teórica (CMC de 1,0 × 10-3
mol L-1
) e foi escolhida com base em testes preliminares.
Fig. 5: A – Estudo do log f × Ep na faixa de 10 - 90 Hz; B – Estudo de f × Ip na faixa de 10 a 90 Hz; C – Estudo do
pH × Ep na faixa de pH de 5 a 9, usando SWV e 29,1 mg L-1
de lapachol.
3.2 Otimização de condições experimentais e instrumentais para análise usando SWV
Foi descoberto que o lapachol acumula na superfície do eletrodo (sob transporte
convectivo forçado e purga com N2) na mesma faixa não importando o potencial aplicado (na
faixa de +600 a -100 mV), o que permite uma pré-concentração do analito antes da
quantificação. A corrente de redissolução medida após diferentes tempos de pré-concentração
pode ser vista na Fig. 6A, onde a saturação é alcançada após 140 s. Para fins quantitativos, 160 s
de acumulação do analito em +400 mV foram usados para medir a SWV da corrente de
redissolução do analito.
Os parâmetros instrumentais para a voltametria de onda quadrada (amplitude de pulso de
10 a 100 mV e frequência de 10 a 60 Hz) foram ajustados usando um passo potencial de 20 mV.
Apesar do fato de que a magnitude da corrente de redissolução medida foi diretamente
proporcional à frequência ou à amplitude de pulso, as distorções na forma dos picos
voltamétricos também foram consideradas na escolha dos valores finais: amplitude de pulso de
40 mV e frequência de 30 Hz (Fig. 6B e 6C).
1.0 1.2 1.4 1.6 1.8 2.0
-350
-400
-450
-500
-550A
Ep (
mV
)Log f
3 4 5 6 7 8 9 10-3,5
-4,2
-4,9
-5,6
-6,3
-7,0B
f-1/2
I (
A)
5 6 7 8 9
-300
-360
-420
-480
-540
-600C
Ep (
mV
)pH

Fig. 6: Sinal SWV (medido usando eletrodo de epóxi-grafite) dependendo do: A - tempo de deposição; B –
amplitude de pulso (a) e C – frequência de pulso (f). Eletrólito suporte: CTAB (1,2 × 10-4
mol L-1
), tampão fosfato
(4,0 × 10-2
mol L-1
; pH 6) e KNO3 (1,0 mol L-1
) em meio aquoso. Concentração de lapachol de 0,388 mg L-1
.
3.3 Determinação analítica do lapachol
3.3.1. Características analíticas
Figuras analíticas de mérito foram obtidas usando condições experimentais selecionadas
para determinar o lapachol (Tabela 1). O sinal analítico (corrente de pico) foi diretamente e
linearmente proporcional à concentração de lapachol na célula eletroquímica (R2 = 0,996). A
equação da curva de adição padrão foi Ip (μA) = (2,5 × 10-7
± 7,0 × 10-9
) Clapachol (mg L-1
) + (3,8
× 10-6
± 3,4 × 10-8
) (R2 = 0,999), cobrindo o intervalo até 0,49 mg L
-1) de analito (Fig. 7A). As
medidas foram realizadas em triplicata e os erros associados a sensibilidade e coeficiente linear
foram calculados como desvio-padrão. O LD instrumental de 0,029 mg L-1
e o LQ instrumental
de 0,097 mg L-1
foram calculados usando, respectivamente, 3sb/m e 10sb/m, onde sb é o desvio-
padrão de dez medições consecutivas de sinal da menor concentração de analito na curva de
adição analítica e m é a sensibilidade dessa curva. A precisão instrumental de 2% foi obtida a
partir de dez medições consecutivas de sinal (medidas após a pré-concentração) produzido pelos
padrões analíticos em duas diferentes concentrações (0,097 mg L-1
e 0,580 mg L-1
).
Para estabelecer uma comparação, a curva de adição analítica foi realizada usando
também o eletrodo de GCE (área eletroativa de aproximadamente 0,019 cm2), SWV e as mesmas
condições estabelecidas neste trabalho. A curva de adição padrão (R2 = 0,996) foi Ip (μA) = (9,9
× 10-7
± 9,2 × 10-8
) Clapachol (mg L-1
) + (2,7 × 10-5
± 7,7× 10-7
) e ela cobriu, aproximadamente, a
mesma gfaixa de concentrações com quase a mesma sensibilidade, apesar da área eletroativa ser
cerca de três vezes maior que a área eletroativa do eletrodo compósito de epóxi-grafite (Fig. 7B).

Também pode-se notar que o sinal de fundo gerado usando o eletrodo epóxi-grafite foi
significantemente menos intenso que o associado com o eletrodo GCE, o que reflete na relação
sinal-ruído, que por sua vez afeta a capacidade de detectar baixas concentrações de lapachol (Fig.
7C).
Tabela 1: Condições para determinação de lapachol por SWV.
Parâmetro Valor
Mistura Eletrolítica CTAB (1,2 × 10-4
mol L-1
) / tampão fosfato (4 × 10-2
mol L-1
; pH 6) / KNO3 (1 mol L
-1)
Potencial de Deposiçao 400 mV
Tempo de Deposição 140 s
Amplitude 40 mV
Passo Potencial 20 mV
Frequência 30 Hz
Sinal Monitorado em 447 mV
Alcance do Potencial de
Varredura
600 a -900 mV
Fig. 7: Curva de adição analítica SWV do lapachol cobrindo a faixa de concentrações de 0,097 mg L-1
a 0,485 mg L-
1 usando: A – eletrodo de epóxi-grafite e B – eletrodo de carbono vítreo (GCE). Condições experimentais da tabela
1. C – Sinal de fundo medido usando eletrodo de epóxi grafite (GEE) e eletrodo de carbono vítreo (GCE).
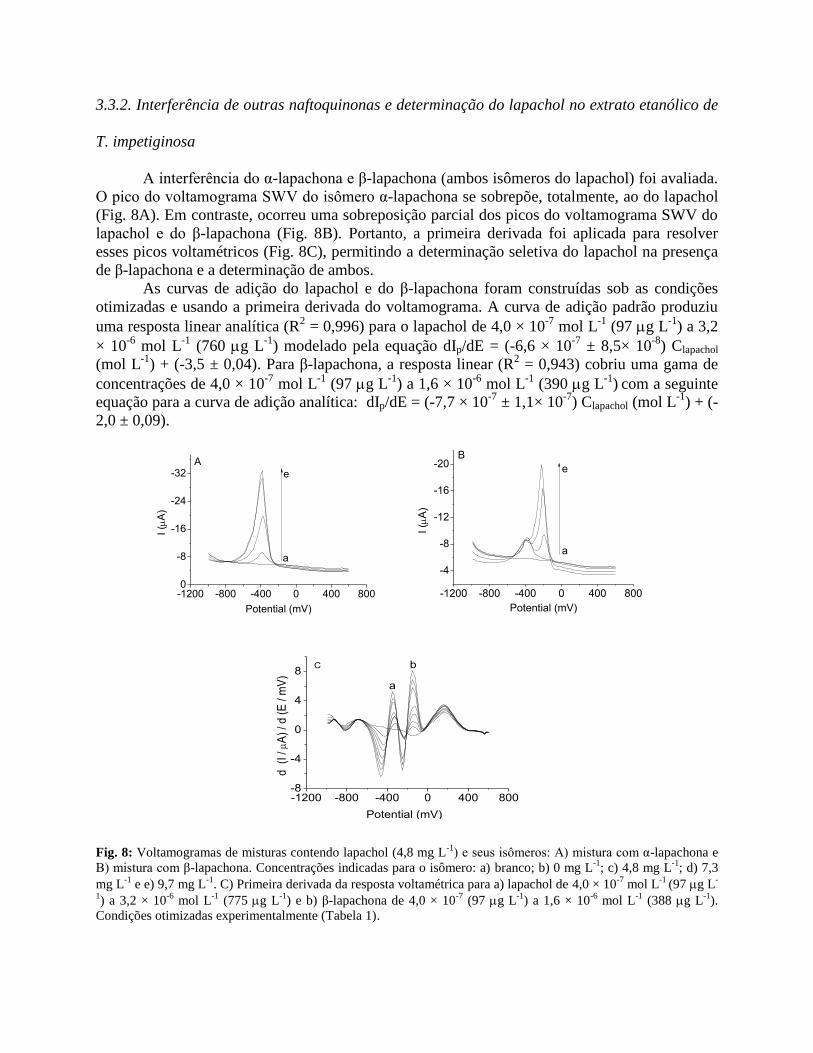
3.3.2. Interferência de outras naftoquinonas e determinação do lapachol no extrato etanólico de
T. impetiginosa
A interferência do α-lapachona e β-lapachona (ambos isômeros do lapachol) foi avaliada.
O pico do voltamograma SWV do isômero α-lapachona se sobrepõe, totalmente, ao do lapachol
(Fig. 8A). Em contraste, ocorreu uma sobreposição parcial dos picos do voltamograma SWV do
lapachol e do β-lapachona (Fig. 8B). Portanto, a primeira derivada foi aplicada para resolver
esses picos voltamétricos (Fig. 8C), permitindo a determinação seletiva do lapachol na presença
de β-lapachona e a determinação de ambos.
As curvas de adição do lapachol e do β-lapachona foram construídas sob as condições
otimizadas e usando a primeira derivada do voltamograma. A curva de adição padrão produziu
uma resposta linear analítica (R2 = 0,996) para o lapachol de 4,0 × 10
-7 mol L
-1 (97 g L
-1) a 3,2
× 10-6
mol L-1
(760 g L-1
) modelado pela equação dIp/dE = (-6,6 × 10-7
± 8,5× 10-8
) Clapachol
(mol L-1
) + (-3,5 ± 0,04). Para β-lapachona, a resposta linear (R2 = 0,943) cobriu uma gama de
concentrações de 4,0 × 10-7
mol L-1
(97 g L-1
) a 1,6 × 10-6
mol L-1
(390 g L-1
) com a seguinte
equação para a curva de adição analítica: dIp/dE = (-7,7 × 10-7
± 1,1× 10-7
) Clapachol (mol L-1
) + (-
2,0 ± 0,09).
-1200 -800 -400 0 400 8000
-8
-16
-24
-32A
e
a
I (
A)
Potential (mV) -1200 -800 -400 0 400 800
-4
-8
-12
-16
-20B
e
a
I (
A)
Potential (mV)
Fig. 8: Voltamogramas de misturas contendo lapachol (4,8 mg L-1
) e seus isômeros: A) mistura com α-lapachona e
B) mistura com β-lapachona. Concentrações indicadas para o isômero: a) branco; b) 0 mg L-1
; c) 4,8 mg L-1
; d) 7,3
mg L-1
e e) 9,7 mg L-1
. C) Primeira derivada da resposta voltamétrica para a) lapachol de 4,0 × 10-7
mol L-1
(97 g L-
1) a 3,2 × 10
-6 mol L
-1 (775 g L
-1) e b) β-lapachona de 4,0 × 10
-7 (97 g L
-1) a 1,6 × 10
-6 mol L
-1 (388 g L
-1).
Condições otimizadas experimentalmente (Tabela 1).
-1200 -800 -400 0 400 800-8
-4
0
4
8C b
a
d (I
/
A)
/ d (
E / m
V)
Potential (mV)

A adição de uma alíquota do extrato etanólico do cerne da T. impetiginosa diretamente
dentro da célula eletroquímica produziu um sinal amplo e largo, o que indica que a matriz
interfere na análise. A separação do lapachol dos componentes da matriz da amostra foi feita
utilizando cromatografia em camada fina (TLC) a fim de eliminar qualquer interferência
potencial durante a análise. A separação por TLC foi muito simples e relativamente rápida,
levando a uma percentagem de recuperação do lapachol maior que 95% e um FR de 0,78. Isso
foi verificado por duas analistas diferentes, cada uma utilizando três réplicas da solução padrão
de lapachol. Ao detectar uma massa de lapachol de 97 mg, a perda média de analito durante o
processo de separação foi de 3,3 ± 0,3% (para o analista 1) e 3,5 ± 0,4% (para o analista 2), o que
permitiu uma recuperação de 93,7 mg, levando em conta o percentual de perda média de 3,4%
do analito. Esse resultado pode ser considerado satisfatório, mas, quando necessário, a
percentagem de perda média pode ser utilizada para corrigir o resultado final da análise do
extrato de planta.
A precisão intermediária do método SWV foi também calculada levando em
consideração os resultados das análises das amostras obtidas por ambos analistas. A precisão
intermediária considerou a variabilidade do procedimento da TLC. Dois níveis do analito foram
usados para avaliação utilizando 40 μL de um extrato da amostra. Os resultados (em massa
absoluta) obtidos foram 3,10 ± 0,05 μg para o analista 1 e 3,05 ± 0,04 μg para o analista 2. O
desvio padrão combinado (analista 1 e analista 2) produziu a precisão intermediária de 0,06μg ou
2% da média geral.
Amostras do extrato do cerne da T. impetiginosa foram analisados, com sucesso, a fim de
determinar o lapachol. A análise de diferentes alíquotas do extrato (diluídas 500 vezes, por causa
da sensibilidade do método) foram feitas em três dias diferentes e os resultados (média das três
replicatas) estão mostrados na Tabela 2. A concentração média do lapachol (com limite de
confiança de 95% e n=9) no extrato do cerne de T. impetiginosa foi de 340 ± 10 mg L-1
.
Tabela 2: Concentração de lapachol (analisada em três dias, com n=3 replicatas) na madeira da T. impetiginosa (Ipê
Rosa) e no extrato comercial após separação por TLC
Amostra Dia Concentração
(mg L-1
)
Extrato de T. impetiginosa
(Ipê Rosa)
1 a(0,34 ± 0,016)
2 a(0,32 ± 0,009)
3 a(0,36 ± 0,02)
Extrato Comercial 1 < LD
2 < LD
3 < LD a Resultados dos extratos das amostras diluídas 500 vezes
As mesmas amostras do extrato também foram analisadas por HPLC e os resultados (três
amostras feitas em três replicatas) são mostrados na Tabela 3 junto com os obtidos pelo método
proposto por SWV. Os resultados são estatisticamente concordantes entre si, pois o teste t
Student (usando o valor da média geral) deu um valor para tcalculado =1,2 e tcrítico = 2,0, em um
nível de confiança de 95% (n1 = n2 = 24). A análise da variância (fator único) usando cada valor

médio como um resultado independente produziu um Fcalculado de 1,3 e Fcrítico de 4,3, em um nível
de confiança de 95% e n1 = n2 = 24.
O método voltamétrico proposto foi usado também para determinar lapachol em uma
formulação comercial de pó de vegetal (Pau d'arco), supostamente contendo T.impetiginosa. O
lapachol não foi detectado por esse método e nenhuma indicação do analito foi encontrada no FR
da placa TLC.
Tabela 3: Determinação de lapachol em amostras do cerne do Ipê Rosa
Amostra Método Concentração no extrato
(mg L-1
)
Conteúdo de lapachol
na madeira
(%)
1 SWV a,b
(0,1696 ± 0,0121) 1,36 ± 0,10
2 HPLC-UV c(0,17686 ± 0,00848) 1,41 ± 0,07
a Concentração corrigida pelo fator de diluição de 500
b Procedimento de TLC para separar analito
c Solução de amostra não diluída
4 Conclusão
Um método voltamétrico simples foi desenvolvido para quantificar o lapachol e foi
aplicado em análises de extratos do cerne da T.impetiginosa. Um eletrodo de epóxi-grafite de
laboratório, fácil de fazer e barato, possibilitou a melhor resposta linear e baixo ruído quando
comparado com o eletrodo de carbono vítreo. A utilização de CTAB no eletrólito suporte (a pH
6) possibilitou picos redox intensos e reversíveis para o lapachol, o qual produziu um intenso
sinal SWV. O LD instrumental de 0,029 mg L-1
foi alcançado com um intervalo linear de duas
ordens de grandeza. A determinação do lapachol, na presença de β-lapachona, foi realizada com
êxito usando a primeira derivada do sinal e o procedimento da TLC possibilitou a separação do
lapachol de componentes presentes em extratos de plantas, permitindo assim determinações
exatas.
5 Referências
1 H. Hussain, K. Krohn, V. G. Ahmad, G. A. Miana, I. R. Greend, Lapachol: an overview, ARKIVOC ii (2007) 145-
171.
2 P. Guiraud, R. Steiman, G. M. Campos-Takaki, F. Seigle-Murandi, M. S. de Buochberg, Comparison of
antibacterial and antifungal activities of lapachol and betalapachone, Planta Med. 60 (1994) 373-374.
3 C. J. Li, L. J. Zhang, B. J. Dezube, C. S. Crumpacker, A. B. Pardee, Three inhibitors of type 1 human
immunodeficiency virus long terminal repeat-directed gene expression and virus replication, Proc. Natl. Acad. Sci.
U. S. A. 9 (1993) 1839-1842.

4 N. M. F. Lima, C. S. Correia, P. A. L. Ferraz, A.V. Pinto, M. C. R. F. Pinto, A. E. G. Santana, M. O. F. Goulart,
Molluscicidal Hydroxynaphthoquinones and Derivatives: Correlation Between their Redox Potential and Activity
Against Biomphalaria glabrata, J. Braz. Chem. Soc. 13 (2002) 822-829.
5 C.O. Salas, M. Faúndez, A. Morello, J.D. Maya, R.A. Tapia, Natural and synthetic naphthoquinones active against
Trypanosoma cruzi: an initial step towards new drugs for Chagas disease, Curr. Med. Chem. 18 (2011) 144–161.
6 M. F. Oliveira, T. L. G. Lemos, M. C. de Mattos, T. A. Segundo, G. M. P. Santiago, R. Braz-Filho, New enamine
derivatives of lapachol and biological activity, An. Acad. Bras. Cienc. 74 (2002) 211-221.
7 E. J. Hoffman. Cancer and the Search for Selective Biochemical Inhibitors, second ed., CRC Press, New York,
EUA, 2007, pp 308.
8 J. Steinert, H. Khanlaf, M. Rimpler, HPLC separation and determination of naphtho[2,3 b]furan-4,9-diones and
related compounds in extracts of Tabebuia avellanedae (Bignoniaceae), J. Chromatogr. A 693 (1995) 281–287.
9 R. L. R. P. Jácome, A. B. de Oliveira, D. S. Raslan, A. Müller, H. Wagner, Análise de naftoquinonas em extratos
brutos de raízes de Zeyheria montana M. (bolsa-depastor), Quim. Nova 22 (1999) 175-177.
10 R. Q. Aucélio, A. I. Peréz-Cordovés, J. L. X. Lima, A. B. B. Ferreira, A. M. E. Guas, A. R. da Silva,
Determination of lapachol in the presence of other naphthoquinones using 3MPA-CdTe quantum dots fluorescente
probe, Spectrochim. Acta Part A 100 (2013) 155–160.
11 J. L. X. Lima, A. Peréz-Gramatges, R. Q. Aucélio, A. R. da Silva, Improved quantum dots fluorescence
quenching using organized medium: A study of the effect of naphthoquinones aiming the analysis of plant extracts,
Microchem. J. 110 (2013) 775–782.
12 J.M. Finkel, S.D. Harrison Jr., Fluorometric method for the determination of lapachol in serum, Anal. Chem. 41
(1969) 1854–1855.
13 S.K.B. Alcanfôr, S.V. Cardoso, C.G. de Lima, Fluorimetric studies of some quinones and quinonoid compounds
after reduction reaction, Anal. Chim. Acta 289 (1994) 273–290.
14 E. Ngameni, I. K. Tonle, C. P. Nanseu, R. Wandji, Voltammetry Study of 2-Hydroxy-3 isopropenyl-1,4-
naphthoquinone Using a Carbon Paste Electrode, Electroanalysis 12 (2000) 847-852.
15 F. C. Abreu, M. O. F. Goulart, A. M. O. Brett, Reduction of Lapachones in Aqueous Media at a Glassy Carbon
Electrode, Electroanalysis 14 (2002) 29-34.
16 M. O. F. Goulart, P. Falkowski, T. Ossowski, A. Liwo, Electrochemical study of oxygen interaction with lapachol
and its radical anions, Bioelectrochem. 59 (2003) 85–87.
17 A. M. Oliveira-Brett, M. O. F. Goulart, F. C. Abreu, Reduction of lapachones and their reaction with L-cysteine
and mercaptoethanol on glassy carbon electrodes, Bioelectrochem. 56 (2002) 53– 55.
18 R. N. Adams, Carbon paste electrodes, Anal. Chem. 30 (1958) 1576 – 1576.
19 J. Wang, Analytical electrochemistry, third ed., John Wiley e Sons, Hoboken, 2006, pp 154.
20 C. M. A. Brett, A. M. O. Brett, Electrochemistry: principles, methods and applications, first ed., Oxford
University Press, Oxford, 1993, pp133.
21 G. W. Milton, The Theory of Composites, first ed., Cambridge University Press, Cambridge, 2002, chapter 1.

22 D. E. Tallman, S. L. Petersen, Composite electrodes for electroanalysis: Principles and applications,
Electroanalysis 2 (1990) 499- 510.
23 F. S. Semaan, E. M. Pinto, E. T. G. Cavalheiro, C. M. A. Brett, A graphitepolyurethane composite electrode for
the analysis of furosemide, Electroanalysis 20 (2008) 2287- 2293.
24 A. I. Balbín-Tamayo, L. S. Riso, A. Pérez-Granates, P. A. M. Farias, A. M. Esteva- Guas, I. Tamanaca.
Electrochemical Characterization a New Epoxy Graphite Composite Electrode as Transducer for Biosensor, Sensors
& Transducers 202 (2016) 59-65.
25 E. Paternò, Ricerche sull'acido lapacico, Gazz. Chim. Ital. 12 (1882) 337–392.
26 S. C. Hooker, The constitution of lapachol and its derivatives. Part V. The structure of Paternò's
“isolapachone”1,2, J. Am. Chem. Soc. 58 (1936) 1190–1197.
27 L. F. Fieser, Naphthoquinone antimalarials. XVI. Water-soluble derivatives of alcoholic and unsaturated
compounds, J. Am. Chem. Soc. 70 (1948) 3232–3237.
28 C. A. C. Ferreira, V. F. Ferreira, A. V. Pinto, R. S. C. Lopes, M. C. R. Pinto, A. J. R. da Silva, 13CNMR Spectra
of natural products Part 5 - Naphthopyrandiones and naphthofurandiones, An. Acad. Bras. Cienc. 59 (1987) 5–8.
29 A. V. Pinto, M. C. F. R. Pinto, C. G. T. de Oliveira, Síntese das α- e β-norlapachonas, propriedades em meio ácido
e reações com N-bromosuccinimida, An. Acad. Bras. Cienc. 54 (1982) 107–114.
30 V. F. de Andrade-Neto, M. O. F. Goulart, J. F. da Silva Filho, M. J. da Silva, M. C. F. R. Pinto, A. V. Pinto, M. G.
Zalis, L. H. Carvalho, A. U. Krettli, Antimalarial activity of phenazines from lapachol, beta-lapachone and its
derivatives against Plasmodium falciparum in vitro and Plasmodium berghei in vivo, Bioorg. Med. Chem. Lett. 14
(2004) 1145–1149.
31 J. M. Neugebauer, Detergents: An Overview, Methods in Enzymology 182 (1990) 239-253.
32 M. Nitschke, G. M. Pastore, Biossurfactantes: propriedades e aplicações, Quim. Nova 25 (2002) 772-776.
33 T. F. Vandamme, Microemulsions as ocular drug delivery systems: recent developments and future challenges,
Prog. Retin. Eye Res. 21(2002) 15-34.
34 M. F. Bergamini, A. L. Santos, N. R. Stradiotto, M. V. B. Zanoni, A disposable electrochemical sensor for the
rapid determination of levodopa, J. Pharm. Bio. Anal. 39 (2005) 54-59.
35 M. F. Bergamini, M. V. B. Zanoni, Anodic stripping voltammetric determination of autothiomalate in urine using
a screen-printed carbon electrode, Electroanalysis 18 (2006) 1457-1462.
36 B. Hoyer, N. Jensen, Stabilization of the voltammetric serotonin signal by surfactants, Electrochem. Com. 8
(2006) 323-328.
37 P. Jara-Ulloa, L. J. Núñez-Vergara, J. A. Squella, Micellar effects on the reduction of 4 nitroimidazole derivative:
detection and quantification of the nitroradical anion, Electroanalysis 19 (2007) 1490-1495.
38 P. S. Guin, S. Das, P. C. Mandal, Electrochemical Reduction of Quinones in Different Media: A Review, Int. J.
Electrochem. 2011 (2011) 1-22.
39 T. Ossowski, M. O. F. Goulart, F. C. de Abreu, A. E. G. Sant’Ana, P. R. B. Miranda, C. O. Costa, A. Liwo, P.
Falkowski, D. Zarzeczanska, Determination of the pKa values of some biologically active and inactive
hydroxyquinones, J. Braz. Chem. Soc. 19 (2008) 175-183.

40 T. L. Ferreira, T. R. L. C. Paixão, E. M. Richter, O. A. El Seoud, Mauro Bertotti, Use of Microdevices To
Determine the Diffusion Coefficient of Electrochemically Generated Species: Application to Binary Solvent
Mixtures and Micellar Solutions, J. Phys. Chem. B 111 (2007) 12478-12484.
41 A.P. dos Reis, C.R.T. Tarley, N. Maniasso, L.T. Kubota, Exploiting micellar environment for simultaneous
electrochemical determination of ascorbic acid and dopamine,Talanta 67 (2005) 829–835.
42 D. de Souza, L. Codognoto, A. R. Malagutti, R. A. Toledo, V. A. Pedrosa, R. T. S. Oliveira, L. H. Mazo, L. A.
Avaca, S. A. S. Machado. Voltametria de onda quadrada. Segunda Parte: Aplicações, Quim. Nova 27 (2004) 790-
797.
43 M. F. Cabral, D. de Souza, C. R. Alves, S. A. S. Machado. Estudo do comportamento eletroquímico do herbicida
ametrina utilizando a técnica de voltametria de onda quadrada, Ecl. Quim. 28 (2003) 41-47.
44 D. K. Gosser. Cyclic Voltammetry: Simulation and Analysis of Reaction Mechanisms, first ed., WILEY-VCH,
New York, EUA, 1993, pp 43.
45 C. M. P. Vaz, S. Crestana, S. A. S. Machado, L. H. Mazo, L. A. Avaca. Electroanalytical Determination of the
Herbicide Atrazine in Natural Waters, Int. J. Environ. Anal. Chem. 62 (1996) 65-76.


















