
RASTREAMENTO DE MUTAÇÕES NO GENE RAS EM … · sou. OBRIGADA meus amores, minha vida, meu tudo! 8...
138
1 INSTITUTO NACIONAL DE CÂNCER – INCA CENTRO DE PESQUISA – CPq PÓS-GRADUAÇÃO STRICTO SENSU EM ONCOLOGIA PROGRAMA DE HEMATOLOGIA E ONCOLOGIA PEDIÁTRICOS RASTREAMENTO DE MUTAÇÕES NO GENE RAS EM LEUCEMIAS AGUDAS EM LACTENTES VICTORIA EUGENIA GIMÉNEZ PÉREZ Rio de Janeiro 2010
Transcript of RASTREAMENTO DE MUTAÇÕES NO GENE RAS EM … · sou. OBRIGADA meus amores, minha vida, meu tudo! 8...

1
INSTITUTO NACIONAL DE CÂNCER – INCA
CENTRO DE PESQUISA – CPq
PÓS-GRADUAÇÃO STRICTO SENSU EM ONCOLOGIA
PROGRAMA DE HEMATOLOGIA E ONCOLOGIA PEDIÁTRICOS
RASTREAMENTO DE MUTAÇÕES NO GENE RAS EM LEUCEMIAS
AGUDAS EM LACTENTES
VICTORIA EUGENIA GIMÉNEZ PÉREZ
Rio de Janeiro
2010

2
RASTREAMENTO DE MUTAÇÕES NO GENE RAS EM LEUCEMIAS
AGUDAS EM LACTENTES
VICTORIA EUGENIA GIMÉNEZ PÉREZ
Sob a orientação da Dra. Maria do Socorro Pombo de Oliveira
e co-orientação da Dra. Mariana Emerenciano
Rio de Janeiro
2010
Dissertação de Mestrado apresentada ao
Curso de Pós-Graduação do Instituto
Nacional de Câncer, como requisito
parcial para obtenção do título de
Mestre em Oncologia

3
P438r Pérez, Victoria Eugenia Giménez.
Rastreamento de mutações no gene RAS em leucemias agudas em lactentes / Victoria Eugenia Giménez Pérez. - Rio de Janeiro: INCA, 2010.
138 f.
Dissertação (Mestrado) - Instituto Nacional de Câncer. Programa de Pós-graduação Stricto Sensu em Oncologia do Instituto Nacional de Câncer (INCA-RJ), 2010. Orientador: Maria do Socorro Pombo de Oliveira. Co-Orientador: Mariana Emerenciano.
1. Leucemia Mielóide Aguda. 2. Leucemia Linfóide Aguda. 3. Genes RAS/rastreamento. 4. Lactente. 5. Polimorfismo de Fragmento de Restrição. 6. Tabagismo. I. Oliveira, Maria do Socorro Pombo de. II. Emerenciano, Mariana. III. Título.
CDD 616.99419027

4
RASTREAMENTO DE MUTAÇÕES NO GENE RAS EM LEUCEMIAS
AGUDAS EM LACTENTES
Victoria Eugenia Giménez Pérez
ORIENTADORA: Dra. Maria do Socorro Pombo de Oliveira
CO-ORIENTADORA: Dra. Mariana Emerenciano
Aprovada em 26 de Fevereiro de 2010
BANCA EXAMINADORA:
Dr. Claudio Gustavo Stefanoff
(INCA)
Dr. Luiz Claudio Santos Thuler
(INCA)
Dra. Mihoko Yamamoto
(UNIFESP)
Dra. Claudete Esteves Nogueira Pinto Klumb
(INCA)

5
A todas as crianças que são o nosso estímulo e nossa força para nos empenharmos
e não desistirmos de trabalhar para lhes oferecer uma vida melhor

6
AGRADECIMENTOS Em primeiro lugar agradeço ao Instituto Nacional de Câncer, especialmente ao programa de
pós-graduação Stricto Sensu pela oportunidade de realizar o meu projeto e descobrir a minha
verdadeira vocação.
Agradeço à Dra. Maria do Socorro Pombo de Oliveira por me orientar e ter aberto as portas
do seu laboratório para poder realizar este trabalho.
Ao Ministério da Saúde e todas as agencias de fomento que financiaram o meu mestrado já
que sem sua ajuda teria sido impossível chegar até aqui.
Aos colegas do Programa de Hematologia e Oncologia Pediátricos pela ajuda que sempre
me ofereceram.
A Dra. Mariana Emerenciano que me acolheu, me ensinou, me ajudou e praticamente foi
um anjo da guarda nessa jornada, sem ela nada disso seria possível.
A Dra. Rosane e ao Diogo da divisão de Farmacologia pela grande ajuda e paciência que
tiveram para que o meu trabalho tivesse os resultados que hoje posso mostrar.
A Isabelle Small que foi indispensável para obter os dados estatísticos do meu trabalho e que
acabou se tornando uma pessoa muito querida para mim.

7
A minha amiga Camilla Andrade que foi a minha companheira, irmã, pano de lagrimas e que
sempre soube me dar uma palavra de apoio e me deu muita força para conseguir chegar até
aqui.
Ao Dr. Juliano Javert que foi o meu suporte, meu amigo e quem soube me ouvir e me
aconselhar no começo desta jornada.
A aqueles companheiros de laboratório que de um jeito ou de outro acabaram se tornando
meus amigos e que sempre estiveram nos momentos bons e não tão bons.... a vocês Lilian,
Eliane e Juliane meu muito obrigada!
A Kelly Cristina que começou como uma simples colega e que em pouco tempo foi
conquistando a minha amizade e o meu carinho, devo muito a você amiga. Muito obrigada
pela sua amizade e por todos os momentos de risadas que tivemos.
A todas aquelas pessoas que de um jeito ou de outro foram muito importantes para alcançar
esta vitoria, são tantas que não teria espaço para colocar o nome de cada uma delas, mas cada
uma sabe o quanto significam para mim.
E por último, mas aos mais importantes quero agradecer aos meus pais. Se não fosse pela
força, pela dedicação, pelo imenso esforço que fizeram para que eu continuasse lutando e pro
acreditarem tanto em mim eu não teria conseguido metade do que consegui nem seria o que
sou. OBRIGADA meus amores, minha vida, meu tudo!

8
“Si la naturaleza se opone lucharemos contra ella y haremos que nos obedezca”
Simón Antonio de la Santísima Trinidad Bolívar y Palacios

9
ÍNDICE DE FIGURAS
Figura 1. Ontogenia das células sanguíneas............................................................................23
Figura 2. Desenho esquemático do FLT3................................................................................32
Figura 3. Agentes mutagênicos presentes na fumaça do cigarro.............................................38
Figura 4. Localização citogenética dos genes NRAS, KRAS e HRAS......................................41
Figura 5. Via de sinalização da RAS.......................................................................................43
Figura 6. Gel de agarose 1,5% da amplificação do KRAS.......................................................62
Figura 7. Géis de agarose de 3% para digestão do KRAS........................................................63
Figura 8. Gel de agarose 1,5% da amplificação do NRAS.......................................................64
Figura 9. Cromatogramas do controle e caso analisados por dHPLC.....................................66
Figura 10. Gráfico dos resultados do KRAS demonstrando os resultados percentuais da
análise........................................................................................................................................71
Figura 11. Gráfico dos resultados do NRAS demonstrando os resultados percentuais da
análise........................................................................................................................................74

10
ÍNDICE DE TABELAS
Tabela 1. Classificação FAB das leucemias mielóides agudas........…....................................27
Tabela 2. Principais características demográficas e clínicas das leucemias infantis, Brasil,
1998-2008.................................................................................................................................70
Tabela 3. Análises das principais variáveis demográficas e status do KRAS em leucemias
agudas no Brasil........................................................................................................................72
Tabela 4. Análises das principais alterações laboratoriais e status do KRAS em leucemias
agudas no Brasil entre 1998-2008.............................................................................................73
Tabela 5. Análises das principais variáveis demográficas e status do NRAS em leucemias
agudas no Brasil, entre 1998-2008............................................................................................75
Tabela 6. Análises das principais alterações laboratoriais e status do NRAS em leucemias
agudas no Brasil, entre 1998-2008............................................................................................76
Tabela 7. Características clínicas e coexistência de alterações genéticas com os genes RAS (K
e N) mutados e selvagens..........................................................................................................77
Tabela 8. Principais características demográficas das mães de crianças com leucemias de
lactentes no Brasil, entre 1998-2008.........................................................................................78
Tabela 9. Relação entre o status do KRAS nas crianças com leucemias e o hábito de fumar na
família.......................................................................................................................................80
Tabela 10. Relação entre o status do NRAS nas leucemias e o hábito de fumar na
família.......................................................................................................................................81
Tabela 11. Descrição sumaria dos casos com KRAS e NRAS mutados e as principais variáveis
analisadas..................................................................................................................................82

11
LISTA DE ABREVIATURAS
AF4 – gene parceiro do MLL na t(4;11)(q21;q23)
AF5α – gene parceiro do MLL na t(5;11)(q12;q23)
AF6 – gene parceiro do MLL na t(6;11)(q27;q23)
AF9 – gene parceiro do MLL na t(9;11)(p22;q23)
AKT – família de proteínas cujos membros também são chamados de proteína quinase B
Asp-12 – ácido aspártico 12
ATP – adenosine triphosphate (adenosina trifosfato)
BCR-ABL – gene híbrido resultante da translocação recíproca dos cromossomos 9 e 22
BCSGIAL – Brazilian Colaboratory Study Group of Infant Acute Leukemia
BstNI – enzima de restrição proveniente do Bacillus stearothermophilus N I
CEP – comitê de ética em pesquisa
dHPLC – denaturing high-performance liquid chromatography (cromatografia liquida
desnaturante de alta performance)
DNA – ácido desoxirribonucléico
dNTP – desoxinucleotídeos
EDTA – ácido etilenodiaminotetraacético
EGFR – epidermal growth factor receptor (receptor do fator de crescimento epidermal)
EGIL – European Group for Immunophenotyping Leukaemia
Elk-1 – proteína da família oncogênica ETS
ENL – proteína nuclear capaz de ativar a transcrição de genes reporters sintéticos em ambas
as células mielóides e linfóides
ERK – extracellular signal-regulated kinases (quinase extracelular receptor-estimulada)

12
FAB – French-American-British group (grupo Franco-Americano-Britânico)
FLT3 – tirosina quinase Fms-like 3
GAPs – GTPase activating proteins (proteínas ativadores de GTPase)
GDP – guanosina difosfato
GRB2 – growth factor receptor-bound protein 2 (receptor do fator de crescimento ligado à
proteína 2)
GTP – guanosina trifosfato
HRAS – v-Harvey RAS
HSCs – hematopoetic stem cells (células-tronco hematopoéticas)
IGF-1 – insulin-like growth factor 1 (fator de crescimento tipo insulina 1)
ITD – internal tandem duplication (duplicação em repetição interna)
kb – kilobase (quilobase)
KCl – cloreto de potássio
KIT – gene que codifica o homologo do proto-oncogene c-KIT
KRAS – Kirsten RAS
LAL – leucemia aguda em lactente
LLA – leucemia linfoblástica aguda
LLA-c – leucemia linfoblástica aguda comum
LMA – leucemia mielóide aguda
LMC – leucemia mielóide crônica
LMMC – leucemia mielomonocítica crônica
MAPK – mitogen-activated protein kinase (proteína quinase receptor-estimulada)
MEK – mitogen regulated kinase (quinase regulada por metiletilcetona-mitogénio)
MgCl2 – cloreto de magnésio

13
MLL – mixed lineage leukemia (leucemia de linhagem mista)
MO – medula óssea
MUT – mutante
NaOH – hidróxido de sódio
NF1 – gene localizado no cromossomo 17 que codifica a proteína neurofibrina 1 (NF1)
NRAS – neuroblastoma RAS
PAH – polycyclic aromatic hydrocarbons (hidrocarbonetos aromáticos policíclicos)
PBX1 – pre-B-cell leukemia homeobox 1 gene
PCR – polymerase chain reaction (reação em cadeia da polimerase)
PflMI – enzima de restrição proveniente do Pseudomonas fluorescens M I
PKC412 – substância estudada no tratamento de leucemia que pertence à família de drogas
inibidoras de proteína quinases
PTPN11 – gene que codifica a proteína tirosino fosfatase
RAF – protein tyrosine receptor protein kinase (receptor com atividade tirosina quinase)
RAS – gene inicialmente identificado em sarcoma de “ratos” envolvido em diversas
neoplasias
RFLP – restriction fragment lenght polymorphism (fragmentação por enzima de restrição
polimorfismo-específica)
rpm – rotações por minuto
RTK – receptor tirosina quinase
RT-PCR – reverse transcriptase polymerase chain reaction (reação em cadeia da polimerase
por transcriptase reversa)
SH2 – SRC homology 2 (homólogo SRC 2)
SH3 – SRC homology 3 (homólogo SRC 3)

14
SNC – sistema nervoso central
SOS – son of sevenless (proteína filha daquela com perda do fotoreceptor R7)
SP – sangue periférico
SSCP – Single-Strand Conformational Polymorphism
Taq – enzima Thermus aquaticus
TCLE – termo de consentimento livre e esclarecido
TE Buffer – Tris-EDTA buffer (tampão Tris-EDTA)
TKD – tyrosine kinase domain (dominio tirosino quinase)
TLH – tampão de lise de hemácias
TLN – tampão de lise de núcleos
Tris-HCl – Tris-hydrochloride
WT – wild type (selvagem)

15
RESUMO
As proteínas RAS ativam vias de sinalização para promover proliferação,
diferenciação, sobrevida e apoptose dependendo das condições celulares. Alelos mutantes das
isoformas do RAS (N-, K-, e H-) foram relacionados com subtipos de leucemias pediátricas e
alguns estudos sugerem a ocorrência da mutação RAS associada a exposições químicas,
especificamente carcinógenos do tabaco. Para rastrear as mutações no RAS e avaliar as
mutações em leucemias de lactentes (LLs), foi realizada uma investigação em 138 DNAs de
amostras de LL do Brazilian Collaborative Study Group of Infant Acute Leukemia (Pombo-
de-Oliveira e cols., 2006). Ao diagnóstico das leucemias, as mães foram entrevistadas,
respondendo questionário com questões sobre o hábito de fumar durante a gravidez.
As mutações no KRAS foram detectadas através da técnica Restriction Fragment
Lenght Polimorphism (RFLP) e no NRAS por denaturing High-Performance Liquid
Chromatography (dHPLC). Um total de 102 casos de leucemia linfoblástica aguda (LLA), 36
de leucemia mielóide aguda (LMA) foram analisados. A idade média na época do diagnóstico
foi de 11,9 meses e entre os casos de LLA foi observada a predominância do imunofenótipo
pró-B e nas LMAs o subtipo M4. As mutações KRAS foram detectadas em 26,1% (36/138),
enquanto NRAS em 15,2% (10/66). Em contraste com resultados anteriores, encontramos um
número maior de mutações do KRAS em pacientes com LLA (26,47%) quando comparado
com LMA (19,44%). Não foi encontrada nenhuma associação estatística significativa com a
idade da criança, sexo, cor da pele, peso ao nascer ou rearranjos no gene MLL. No entanto, foi
encontrada uma relação estatisticamente significativa com leucometria elevada [NRAS
(P=0,05)] e idade materna <30 [NRAS (P=0,03)]. O hábito materno de fumar antes ou durante
a gravidez não foi associado à ocorrência de LLs com mutações do RAS. No entanto, uma

16
associação de chance de risco entre as mutações no KRAS e as mães que relataram haver
fumantes em casa (pai e outros) ou no ambiente de trabalho durante a gravidez foi de OR 2,27
CI (1,13-4,57) (P=0,02). Portanto, ainda há questões a serem exploradas em relação à
exposição indireta ao tabaco, como a ocorrência do hábito familiar de fumar, bem como as
informações importantes sobre exposições maternas e LLs.

17
ABSTRACT
RAS proteins activate several pathways to promote proliferation, differentiation,
survival, and apoptosis depending on cellular conditions. Mutant alleles RAS isoforms (N-, K-
and H-) have correlated to pediatric leukemias subtypes and several studies suggest RAS-
mutation associated with chemical exposures specifically tobacco. In order to screen RAS
mutations and evaluate these mutations in infant leukemias (IL), 138 DNAs of ILs from the
Brazilian Collaborative Study Group of IAL (Pombo-de-Oliveira et al., 2006) were analyzed.
Mothers were interviewed answering a questionnaire with related questions about the
smoking habits during pregnancy. Restriction Fragment Length Polymorphism (RFLP)
detected KRAS mutations and denaturing High Performance Liquid Chromatography
(dHPLC) analysis detected NRAS mutations. A total of 102 ALL and 36 AML were analyzed.
The mean age at diagnosis was 11.9 months and among ALL cases was observed
predominance of pro-B immunophenotype and M4 in AML. KRAS mutations were detected in
26,1% (36/138) while 15.2% (10/66) in NRAS. Differing from previous results, a higher
number of KRAS mutations was found in patients with ALL (26,47%) when compared to
AML (19,44%). No significant statistical association was found with the age at diagnosis,
gender, skin color, birth weight or MLL rearrangements. Thus, a significant statistical
association was found in WBC count of NRAS (P=0,05) and maternal age of NRAS (P=0,03).
Maternal smoking habit before or during pregnancy was not associated to ILs with RAS
mutations occurrence. Thus, the risk association found between KRAS mutations and the
mothers that related having smokers at home (father or others) or at work was of OR 2,27 CI
(1,13-4,57) (P=0,02). That is why there still questions to be explored related to the tobacco

18
exposures as the occurrence of the smoking familiar habits and the important information
about maternal exposures and ILs.

19
SUMÁRIO
Índice de figuras..........................................................................................................................9
Índice de tabelas........................................................................................................................10
Lista de abreviaturas.................................................................................................................11
Resumo......................................................................................................................................15
Abstract.....................................................................................................................................17
1. Introdução..........................................................................................................................22
1.1. Leucemia de lactentes........................................................................................................24
1.1.1. Aspectos clínicos das leucemias linfoblástica agudas....................................................25
1.1.2. Aspectos clínicos das leucemias mielóides agudas........................................................26
1.1.3. Patogênese das leucemias com alterações do MLL na banda cromossômica 11q23......28
1.1.4. Interferência com a DNA topoisomerase II na gênese das translocações......................30
1.1.5. Patogênese das leucemias com mutações no gene FLT3...............................................31
1.1.6. Exposição a fatores ambientais e risco de leucemia aguda em lactentes.......................34
1.1.6.1. Leucemias agudas e sua relação à exposição à fumaça do cigarro.............................38
1.2. Gene RAS...........................................................................................................................39
1.2.1. Localização do RAS........................................................................................................40
1.2.2. Função do RAS................................................................................................................41
1.2.3. Alterações do RAS e sua associação com leucemias......................................................44
1.2.3.1. Coexistência de alterações nos genes MLL e RAS......................................................45
1.2.3.2. Coexistência de alterações nos genes FLT3 e RAS.....................................................46
1.2.4. Mutações no gene RAS e a sua associação com fatores ambientais...............................47
2. Justificativa do estudo.....................................................................................................50

20
3. Objetivos...........................................................................................................................54
4. Material e métodos..........................................................................................................56
4.1. Desenho do estudo.............................................................................................................57
4.2. Casuística...........................................................................................................................57
4.3. Extração de DNA para realização dos PCRs do gene RAS................................................58
4.4. Quantificação do DNA.......................................................................................................60
4.5. PCR para a amplificação do KRAS....................................................................................61
4.6. Digestão do gene KRAS com endonucleases de restrição..................................................62
4.7. PCR para a amplificação do NRAS....................................................................................63
4.8. Denaturing high pressure liquid cromatography na detecção das mutações do NRAS.....65
4.9. Análise estatística...............................................................................................................67
5. Resultados..........................................................................................................................68
5.1. Análises de frequências......................................................................................................69
5.1.1. Características clínicas das amostras..............................................................................69
5.2. Rastreamento de mutações no gene KRAS.........................................................................70
5.2.1. Características clínicas das amostras com as alterações genéticas no gene KRAS.........71
5.3. Rastreamento de mutações no gene NRAS.........................................................................74
5.3.1. Características clínicas das amostras com as alterações genéticas no gene NRAS.........74
5.4. Análises epidemiológicas com as características demográficas e hábito de fumar na
família de crianças com leucemias em lactentes.......................................................................78
6. Discussão............................................................................................................................85
7. Conclusão...........................................................................................................................96
8. Referências bibliográficas................................................................................................98
9. Anexos..............................................................................................................................128

21
9.1. Ficha de encaminhamento................................................................................................129
9.2. TCLE................................................................................................................................130
9.3. Géis de RFLP...................................................................................................................133
9.4. Cromatogramas do dHPLC..............................................................................................136
1. INTRODUÇÃO

22
Fisiologicamente, a medula óssea (MO) é responsável pela formação de novas células
sanguíneas (hematopoese). Norteados pelas citosinas e pelos fatores de crescimento
23
hematopoéticos, todos os tipos celulares sanguíneos derivam de um reservatório comum de
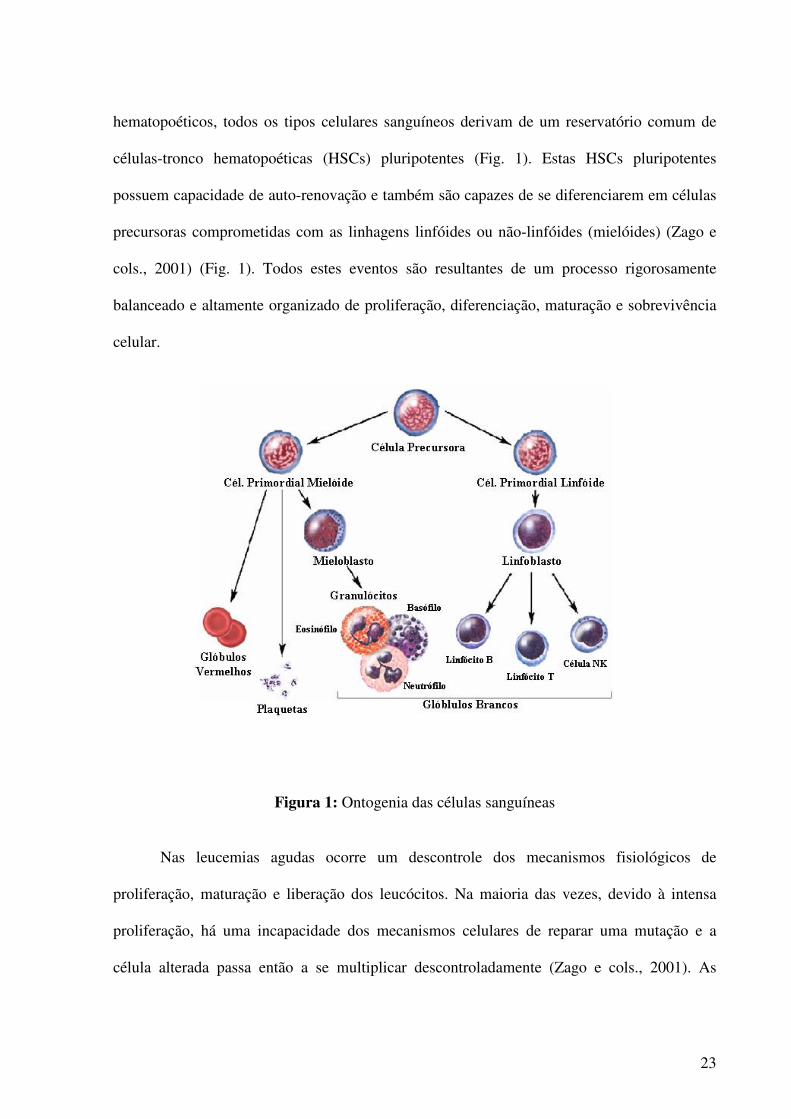
células-tronco hematopoéticas (HSCs) pluripotentes (Fig. 1). Estas HSCs pluripotentes
possuem capacidade de auto-renovação e também são capazes de se diferenciarem em células
precursoras comprometidas com as linhagens linfóides ou não-linfóides (mielóides) (Zago e
cols., 2001) (Fig. 1). Todos estes eventos são resultantes de um processo rigorosamente
balanceado e altamente organizado de proliferação, diferenciação, maturação e sobrevivência
celular.
Figura 1: Ontogenia das células sanguíneas
Nas leucemias agudas ocorre um descontrole dos mecanismos fisiológicos de
proliferação, maturação e liberação dos leucócitos. Na maioria das vezes, devido à intensa
proliferação, há uma incapacidade dos mecanismos celulares de reparar uma mutação e a
célula alterada passa então a se multiplicar descontroladamente (Zago e cols., 2001). As

24
leucemias agudas podem ser classificadas de acordo com a origem da linhagem celular, em
leucemia linfoblástica aguda (LLA) originada de precursores da linhagem linfóide e a
leucemia mielóide aguda (LMA) originada de precursores da linhagem mielóide.
Variações genéticas contribuem para a transformação leucêmica das células-tronco
hematopoéticas e de seus progenitores comprometidos, alterando suas funções celulares. Elas
comprometem processos regulatórios essenciais, mantendo ou intensificando uma capacidade
de auto-renovação ilimitada, subvertendo os controles de proliferação normal, bloqueando a
diferenciação e promovendo resistência ao sinal de morte (apoptose) (Hanahan e Weinberg,
2000).
1.1. Leucemia de lactentes
A leucemia é a malignidade pediátrica mais comum no Brasil, contando por
aproximadamente 18% a 41% de todos os novos diagnósticos de câncer abaixo dos 19 anos de
idade (de Camargo e cols., 2010). Para lactentes abaixo dos 12 meses de idade, as leucemias
compreendem aproximadamente 16% de todas as malignidades neoplásicas (depois do
neuroblastoma) com uma taxa geral de incidência de aproximadamente 36 casos por milhão
de lactentes. Possui características epidemiológicas, biológicas e clínicas únicas (Ross, 2008).
A maioria dos casos de LLA e LMA em lactentes é caracterizada por uma contagem
leucocitária elevada, doença extramedular volumosa, uma propensão à expressão de
marcadores fenotípicos linfóides e mielóides, e alterações cromossômicas do gene MLL na
banda cromossômica 11q23 (Pui e cols., 1995; Reaman e cols., 1999; Biondi e cols., 2000).
As LLAs são mais freqüentes que as LMAs representando aproximadamente 80-85%
das leucemias (Chowdhury e Brady, 2008). A LLA em lactentes é tipicamente apresentada

25
com um fenótipo imaturo de células pró-B, algumas vezes com antígenos de marcadores
mielóides. Já as LMAs em lactentes, se apresentam principalmente com uma classificação
morfológica M4 ou M5 (Chessells, 1992). O pico de incidência anual de 10,6 casos por
milhão de LMAs pediátricas ocorre durante o primeiro ano de vida. Em contraste, o pico de
incidência anual de 76,6 casos por milhão ocorre entre os 2 e 3 anos de vida (Gurney e cols.,
1995). Ambas a LLA e a LMA ocorrem com maior freqüência em meninas do que em
meninos (Gurney e cols., 1995) e as taxas em lactentes brancos são mais elevadas que as dos
lactentes não brancos (Gurney e cols., 1995).
Como os rearranjos recíprocos envolvendo o gene MLL são as características genéticas
mais comuns nas leucemias agudas em lactentes (LALs) é importante entender como os genes
de fusão podem ter sido originados possivelmente como um efeito de exposições,
principalmente transplacentária.
1.2.5. Aspectos clínicos das leucemias linfoblásticas agudas em lactentes
No diagnóstico inicial, a LLA em lactentes está caracterizada por uma contagem
leucocitária médiana de >50X109/l, hepatoesplenomegalia frequente, e envolvimento do
sistema nervoso central (SNC) (Reaman e cols., 1999). De 14% a 41% dos lactentes tem
doença do SNC no diagnóstico, comparado com aproximadamente 5% das crianças com LLA
(Reaman e cols., 1999). Embora o fenótipo imaturo de linhagem B ocorre com maior
freqüência do que o de linhagem T, a linhagem é ambígua ou mista, com ambas as populações
linfóide e mielóide presentes na maioria das leucemias de lactentes (Matamoros e cols.,
1994). A translocação t(4;11)(q21;q23) é a mais freqüente (Behm e cols., 1996), seguida pela
t(11;19)(q23;p13) (Rubnitz e cols., 1996). Enquanto a t(5;15)(p15;q11-q13) vem sendo

26
descrita como importante, pois t(4;11)(q21;q23) confere um prognóstico ruim e resistência
terapêutica (Behm e cols., 1996; Pui e cols., 1996; Reaman e cols., 1999; Pui e cols., 2003)
enquanto t(5;15)(p15;q11-q13) confere uma boa sobrevida em 5 anos..
1.2.6. Aspectos clínicos das leucemias mielóides agudas
A LMA é uma doença bastante heterogênea com consideração nas características
clínicas e alterações genéticas adquiridas, ambas detectáveis microscopicamente como
aberrações cromossômicas estruturais e numéricas, e aquelas detectadas como mutações
genéticas sub-microscópicas e mudanças na expressão genética (Tenen, 2003; Mrózek e
Bloomfield, 2006). Caracteriza-se por um acúmulo de precursores granulocíticos e
monocíticos na MO e no sangue (Tenen, 2003). Nos lactentes se caracteriza por
hepatoesplenomegalia, leucemia cútis, doença no SNC, e uma elevada contagem leucocitária
ao diagnóstico (Lampkin, 1997). Apesar da elevada carga tumoral, os lactentes apresentam
menos acidentes cardiovasculares e leucostase pulmonar do que as crianças. Os diferentes
subtipos são categorizados no estagio no qual a diferenciação normal foi bloqueada nos
blastos leucêmicos (Bennett e cols., 1976) (tabela 1).
Tabela 1: Sumário da classificação das leucemias mielóides agudas, de acordo com os
critérios FAB
Subtipos Descrição

27
M0 Leucemia mieloblástica aguda indiferenciada
M1 Leucemia mieloblástica aguda sem maturação
M2 Leucemia mieloblástica aguda com maturação
M3 Leucemia promielocítica aguda ou promielocítica
M4 Leucemia mielomonocítica aguda
M5 Leucemia monoblástica aguda
M6 Leucemia eritróide aguda ou Eritroleucemia
M7 Leucemia megacarioblástica aguda
A translocação t(9;11)(p22;q23) é a mais comum nas LMAs de lactentes, seguida da
t(10;11)(p12;q23) (Meyer e cols., 2009). Os casos de LMAs M4 ou M5 sem translocações no
MLL incluem leucemias com inv(16), frequentemente as M4 com eosinofilia (M4eo), e
leucemias com monossomia do 7, anormalidades citogenéticas aleatórias, ou cariótipos
normais (Kalwinsky e cols., 1990).
No estudo de Jones e cols. (Jones e cols., 2010) assim como em estudos prévios
(Goemans e cols., 2005; Boissel e cols., 2006; Cairoli e cols., 2006) as mutações no KIT,
FLT3, e no RAS estão associadas com contagem leucocitária elevada, consistente com o papel
destas mutações na via do crescimento celular na leucemia e um desfecho adverso (Nguyen e
cols., 2002; Care e cols., 2003; Meshinchi e cols., 2003; Schlenk e cols., 2004; Wang e cols.,
2005; Paschka e cols., 2006; Schnittger e cols., 2006; Kim e cols., 2008). Recentemente
alguns estudos demonstraram resultados controversos entre a associação do estado mutacional
de NRAS/KRAS e o prognóstico dos pacientes adultos. (Boissel e cols., 2006; Jones e cols.,
2010). O desfecho dos pacientes com LMA com a t(9;11)(p22;q23) pode depender tanto da
idade quanto da contagem leucocitária, uma vez que um grande estudo sobre o prognóstico de
LMA, encontrou o melhor desfecho para pacientes entre 1 a 9 anos de idade com baixa
contagem leucocitária (Swansbury e cols., 1998). Há um consenso de que as translocações no

28
MLL com determinados genes parceiros confere um prognóstico desfavorável também em
LMA (Lillington e cols., 1998; Martineau e cols., 1998; Moorman e cols., 1998). Em
contraste à LLA, na LMA o fato de ser lactente por si só não é um prognóstico desfavorável
(Woods e cols., 1996; Smith, 1997). Esta diferença pode ser devido à elevada incidência de
translocações no MLL e morfologias M4/M5 em crianças até 4 anos de idade, à fusão do MLL
com parceiros que sejam menos favoráveis e o desfecho relativamente ruim para a maioria
das LMAs fora da população de lactentes (Hann e cols., 1997).
Durante a última década, vários estudos mostraram que a presença ou ausência de
mutações genéticas específicas e/ou mudanças na expressão gênica tem afetado no
prognóstico dos pacientes (Mrózek e cols., 2006). Até o momento, provavelmente o fator
prognóstico mais importante na LMA citogeneticamente normal é a duplicação em repetição
interna (ITD) do FLT3.
Em ambos os casos de leucemias de lactentes (LLA e LMA), cujos prognósticos ainda
são sombrios, as características da patogênese com os rearranjos do MLL como fator
preponderante, podem ser mais bem exploradas com investigações relevantes quanto a
compreensão da interação de fatores que induzem a leucemogênese.
1.2.7. Patogênese das leucemias com alterações do MLL na banda cromossômica
11q23
O MLL desempenha um papel importante na hematopoese maligna, regulando a
diferenciação hematopoética (Ernst e cols., 2002; Emerenciano e cols., 2005; Popovic e
Zeleznik-Le, 2005; Mullighan e cols., 2007). O MLL tem 90kb de comprimento, contém 38
éxons, e codifica 3969 aminoácidos protéicos (Rasio e cols., 1996). A proteína MLL contém

29
motivos estruturais que sugerem uma função na regulação da transcrição de outros genes
(Domer e cols., 1993). Os rearranjos do MLL incluem deleções, duplicações, inversões e
translocações recíprocas no 11q23, e estes estão implicados em aproximadamente 5-10% de
todas as leucemias em crianças e adultos (Chowdhury e Brady, 2008). Os pontos de quebra
das translocações na leucemia de lactentes e nos casos relacionados aos inibidores da DNA
topoisomerase II geralmente se encontram mesma região de freqüentes quebras (bcr) 8,3kb
entre os éxons 5 e 11 do MLL (Felix e cols., 1998).
Recentemente as alterações envolvendo o MLL foram amplamente estudadas e 104
diferentes rearranjos envolvendo a região 11q23 e 64 diferentes genes já foram caracterizados
(Borkhardt e cols., 1997; Hillion e cols., 1997; Megonigal e cols., 1998; Meyer e cols., 2006;
Meyer e cols., 2009). No entanto, as translocações mais comuns são aquelas envolvendo
genes localizados nos cromossomos 4, 6, 9, ou 19 (Ross, 2008). Os genes parceiros codificam
produtos protéicos de vários tipos diferentes e podem ser distintos quanto ao tipo de
leucemias. Por exemplo, o gene AF4 na banda cromossômica 4q21, o qual é membro de uma
família de genes transcricionais, é o parceiro mais comum do MLL nas LLAs (Nilson e cols.,
1997; Pui e cols., 2003). O AF9 na banda cromossômica 9p22 e o ENL na banda
cromossômica 19p13 também são genes parceiros comuns e codificam fatores de transcrição
(Rubnitz e cols., 1994; Pui e cols., 2003) e encontrados na LLA e LMA. No que se refere as
LMAs, os rearranjos do MLL podem ocorrer por translocações complexas que fusionam MLL
com 2 genes parceiros diferentes como o AF6 e AF5α, típicos da LMA-M5 em lactentes (Taki
e cols., 1996). Além das numerosas translocações, duplicações parciais de diversos éxons do
MLL também ocorrem dentro do bcr (Schichman e cols., 1995).
Rearranjos do MLL únicos e idênticos foram primeiramente detectados em casos de
LLA (Ford e cols., 1993; Gill-Super e cols., 1994; Mahmoud e cols., 1995), e depois em casos

30
de LMA (Megonigal e cols., 1998). Eles caracterizaram a t(4;11)(q21;q23) e a
t(11;19)(q23;p13) na maioria dos casos de LLA, enquanto que na LMA num par de gêmeos
lactentes mostraram t(11;22)(q23;q11) (Megonigal e cols., 1998). Evidências adicionais para
eventos pré-natais na gênese das leucemias em lactentes provem dos estudos de Gale e cols.
(Gale e cols., 1997). Pela amplificação por reação em cadeia de polimerase (PCR) do DNA
genômico das amostras neonatais de sangue (cartão de filtro), estes pesquisadores mostraram
que as translocações leucemo-específicas t(4;11)(q21;q23) já estavam presentes no momento
do nascimento nos pacientes diagnosticados com LLA de 5 meses até os 2 anos de idade
(Gale e cols., 1997).
1.2.8. Interferência com a DNA topoisomerase II na gênese das translocações
A DNA topoisomerase II catalisa o relaxamento do anelamento do DNA através de um
mecanismo de clivagem e re-ligamento de ambas as fitas da dupla-hélice (Corbett e Osheroff,
1993; Bromberg e cols., 2003). As epipodofilotoxinas são chamadas de inibidores da
topoisomerase II porque diminuem a taxa de re-ligamento, causando uma quebra
cromossômica (Chen e Liu, 1994). Entre os inibidores da topoisomerase II estão o benzeno,
metabólitos, como benzoquinona, isoflavonas, antraquinona, e antibióticos quinolonas (Ross e
cols., 1994). O reparo da quebra cromossômica mediado pela topoisomerase II também pode
resultar em translocações (Felix e cols., 1995). A análise da seqüência genômica mostrou
sítios de reconhecimento pela topoisomerase II co-localizados aos pontos de quebra
genômicos do MLL (Gu e cols., 1994). Os sítios abásicos (sítios com ausência de uma purina
ou pirimidina) também aumentam a clivagem da topoisomerase II o que levou a Kingma e

31
cols. a propor que os sítios abásicos também podem romper a topoisomerase II e promover
translocações (Kingma e cols., 1997).
A interferência na topoisomerase II, seja pelas drogas anti-câncer topoisomerase II
alvo-específicas, substâncias da dieta, ou sítios abásicos espontâneos, pode ser a característica
compartilhada entre a leucemia ao tratamento e à leucemia em lactentes (Felix e Lange,
1999).
As duplicações e inserções no DNA foram observadas nas junções dos pontos de
quebra das translocações do MLL (Reichel e cols., 1998). Estes achados sugerem um
mecanismo complicado de translocação que envolve a possibilidade de uma quebra
cromossômica pela topoisomerase II, um processamento, e ligação das regiões terminais
quebradas pelo reparo do DNA (Reichel e cols., 1998).
1.2.9. Patogênese das leucemias com mutações no gene FLT3
Experimentos na ablação de genes estabeleceram que o FLT3 tem um papel
importante no desenvolvimento hematopoético (Mackarehtschian e cols., 1995). Estudos mais
recentes mostraram que o FLT3 está mutado e ativo constitutivamente em aproximadamente
30% dos casos de LMA (Gilliland e Griffin, 2002). Devido ao elevado nível de expressão do
FLT3 nos pacientes com MLL rearranjado e seu perfil de expressão gênica na hematopoese,
Armstrong e cols. (Armstrong e cols., 2003) realizaram um estudo com a técnica de
microarranjo, destacando FLT3 como um exemplo de um potencial alvo terapêutico, com uma
superexpressão do gene selvagem, característica distinguível de LLA MLL+ sobre LLA MLL
-
e LMA.

32
O FLT3 é um receptor tirosino quinase expresso por células hematopoéticas imaturas e
que junto com o seu ligante, é importante para o desenvolvimento normal das células tronco e
o sistema imune (Fig. 2). Enquanto a superexpressão do FLT3 selvagem com o seu ligante é
um mecanismo potencial do envolvimento do FLT3 nas leucemias, a presença de mutações (a
maioria pontuais) ativas do FLT3, incluindo a duplicação em repetição interna (ITD) ou o
loop de ativação do domínio quinase, destacam a importância deste gene (Chowdhury e
Brady, 2008). Recentemente, as mutações FLT3 têm sido encontradas em pacientes com LLA
(1-3% dos pacientes), mielodisplasia (5-10%) e LMA (15-35%), fazendo do FLT3 um dos
genes mais freqüentemente mutados nas malignidades hematológicas (Stirewalt e Radich,
2003).
Figura 2: Desenho esquemático do FLT3. Modificado de
http://www.fleury.com.br/Medicos/SaudeEmDia/RevistaMedicinaESaude.aspx
Mutações no FLT3 em leucemias MLL foram achadas em 8 (18%) de 44 lactentes com
LLA com rearranjos no MLL (Taketani e cols., 2004). A expressão do FLT3 em 41 lactentes
com rearranjos no MLL encontrou-se significativamente elevada quando comparada às
linhagens germinativas do MLL em LLAs de lactentes e não lactentes, e as células leucêmicas

33
de MLL em lactentes foram significativamente mais sensíveis ao PKC412, inibidor da FLT3,
do que as células de LLAs dos não lactentes (Stam e cols., 2005). Enquanto os testes clínicos
de inibidores da FLT3 têm incluído principalmente pacientes adultos e a eficácia clínica ainda
tem que ser determinada, eles podem ter uma aplicação potencial também nos casos
pediátricos.
A forma mais comum da mutação no FLT3 é a ITD nos éxons 14 e 15 (anteriormente
conhecidos como éxons 11 e 12), que ocorre em 15-35% dos pacientes com LMA (Abu-
Duhier e cols., 2000; Kottardis e cols., 2001; Meshinchi e cols., 2001; Stirewalt e cols., 2001;
Schnittger e cols., 2002; Thiede e cols., 2002) e 5-10% dos pacientes com mielodisplasia
(Yokota e cols., 1997). As FLT3-ITDs são formadas quando um fragmento da seqüência
codificante do domínio justamembrana é duplicado e inserido. O comprimento do ITD varia
de 3 a ≥400pb, e a estrutura de leitura do transcrito sempre é preservada, seja pela duplicação
“in-frame” ou pela inserção de nucleotídeos na junção ITD para manter a estrutura original de
leitura (Schnittger e cols., 2002).
A segunda mutação mais comum do FLT3 é a mutação pontual missense no éxon 20
(previamente conhecido como o éxon 17) do domínio tirosino quinase (TKD). As mutações
no TKD ocorrem em pacientes com LMA (5-10%), SMD (2-5%) e LLA (1-3%) (Abu-Duhier
e cols., 2001; Yamamoto e cols., 2001; Thiede e cols., 2002), e a substituição do nucleotídeo
(GAT→TAT) que muda um acido aspártico por uma tirosina. Outras mutações no códon 835,
como deleções, também tem sido descritas, mas são menos comuns. Um único paciente pode
ocasionalmente ter ambas mutações pontuais ITD e TKD no FLT3, mas a maioria dos
pacientes só tem um tipo de mutação (Thiede e cols., 2002; Meshinchi e cols., 2003). No
total, aproximadamente 25-45% dos pacientes com LMA terão alguma forma de mutação no
FLT3 (Stirewalt e Radich, 2003).

34
Nas LMAs, as FLT3-ITDs geralmente são encontradas em pacientes com t(15;17). Foi
hipotetizado que o desenvolvimento de LMA requer pelo menos dois tipos de anormalidades
genéticas que disrompem independentemente pelo menos dois processos regulatórios nas
células hematopoéticas (Stirewalt e Radich, 2003). Esta teoria deriva da hipótese dos “dois
eventos” de Knudson que propõe que a maioria dos cânceres requer pelo menos duas
mutações, a primeira podendo ser tanto germinativa quanto somática, enquanto que a segunda
sempre é somática (Knudson e cols., 1975). Porém, apesar deste conceito ser intelectualmente
satisfatório, ainda não está claro como as mutações no FLT3 realmente cooperam no
desenvolvimento de câncer. Por exemplo, as mutações no FLT3 alteram a diferenciação,
proliferação e apoptose (Hayakawa e cols., 2000; Mizuki e cols., 2000; Meshinchi e cols.,
2001; Zheng e cols., 2002), e os componentes destas vias regulatórias apresentam elevada
redundância e o “cross-talk”. Então, uma única mutação não poderá acometer totalmente uma
via completa ou sua função regulatória, ou pode não afetar uma única via funcional. Em
alguns casos, deve haver um efeito de “dosagem da via”, através do qual as diferentes
mutações devem agir na mesma via regulatória para promover uma disrupção mais completa
da sua atividade normal (Stirewalt e Radich, 2003).
1.2.10. Exposição a fatores ambientais e risco de leucemia aguda em lactentes
Não há dúvidas de que o risco de desenvolver LAL é modulado por interações
complexas entre predisposições herdadas, exposições ambientais a agentes danificadores e
eventos ao acaso (Spector e cols., 2005). A LAL constitui um grupo único que serve como
modelo para investigações epidemiológicas, pois o período de exposição a agentes relevantes
é curto e conhecido (Ross e cols., 1994). Uma vez que exposições ocupacionais ou ambientais

35
a radiação ionizante, solventes, derivados do petróleo e pesticidas foram correlacionadas a um
risco aumentado de LMA em adultos, os primeiros estudos epidemiológicos em leucemias
pediátricas focaram nestes agentes (Buckley, 1992).
A maioria dos estudos que avaliam a relação entre a exposição a pesticidas de uso
doméstico e leucemias em lactentes sugerem que o risco aumentado de leucemia está
associado com a exposição in utero e pós-natal a pesticidas (Zaham e Ward, 1998; Meinert e
cols., 2000). Estudos mostram que o risco na exposição durante a gravidez é maior que os
riscos para exposições após o nascimento. O uso doméstico freqüente de pesticidas durante o
período pré-natal também foi associado ao aumento no risco de desenvolver LLA (Infante-
Rivard e cols., 1999; Ma e cols., 2002). Pode-se inferir que o embrião ou feto é especialmente
sensível ou susceptível a carcinógenos no ambiente. Há evidências que a LLA se inicia in
utero (Ford e cols., 1993), e tem se mostrado que algumas translocações cromossômicas
relacionadas à incidência desta leucemia em lactentes também é de origem pré-natal
(Wiemels e cols., 1999a; Wiemels e cols., 1999b).
A exposição parental a químicos relacionada à ocupação, como pesticidas e solventes,
também foi sugerida como um fator de risco para a leucemia de lactentes (Meinert e cols.,
2000). As crianças que habitam em comunidades agrícolas também podem ser expostas a
pesticidas de uso agrícola (Lu e cols., 2000). Estudos caso-controle conduzidos entre 1980 e
1984 pelo Children’s Cancer Group encontraram que as exposições parentais peri-natais a
pesticidas e marihuana, e o uso materno de etanol durante a gravidez, estavam
significativamente associadas à LMA M4 e M5 em crianças e lactentes (Buckley, 1992;
Severson e cols., 1993).
Semelhanças clínicas e moleculares observadas entre leucemias em lactentes e casos
induzidos por epipodofilotoxina serviram de base para o estudo epidemiológico no qual Ross

36
e cols. determinaram que o consumo materno de inibidores da topoisomerase II pode ser um
fator nas LMAs, mas não nas LLAs em lactentes (Ross e cols., 1996). A dieta materna com
inibidores de topoisomerase II incluía cafeína, quercitina em frutas e vegetais frescos, e
catequinas no cacau (Ross e cols., 1996). Como já foi mostrado, as associações entre o
consumo de álcool durante a gravidez e LMAs em lactentes foram sugeridas. Além disso, os
metabólitos podem interferir na topoisomerase II (Reynolds, 1998). Fórmulas a base de soja
podem expor os lactentes a altos níveis de isoflavonas, incluindo daidzeína e genisteína,
outros inibidores da topoisomerase II (Corbett e Osheroff, 1993; Setchell e cols., 1997),
levantando questões sobre a função potencial da topoisomerase II nas leucemias de lactentes.
O alto peso ao nascer também foi correlacionado com um risco aumentado de LLA e
LMA. Resultados concordantes foram observados no Grupo Brasileiro de Estudo
Colaborativo da Leucemia Aguda em Lactente (BCSGIAL), usando a informação sobre o
peso obtida a partir de questionários de 202 casos de LAL e 440 controles (Koifman e cols.,
2008). Como o fator de crescimento insulina tipo 1 (IGF-1) é importante na formação e
regulação sanguínea e foi demonstrado que é capaz de estimular o crescimento tanto de
células mielóides quanto linfóides em cultura, foi postulado que altos níveis de IGF-1 podem
gerar bebês grandes e contribuir com o desenvolvimento da leucemia (Ross e cols., 1996;
Koifman e cols., 2008).
Outro fator de risco determinado em diversos estudos epidemiológicos foi o baixo
status socioeconômico, medido pelos ingressos e a educação materna. Este fator foi associado
a um risco aumentado de LALs (Ross e cols., 1997; Pombo-de-Oliveira e cols., 2006). Estes
resultados contrastam com o de crianças com <24 meses de idade, de classe média e alta, que
tendem a ter um risco aumentado de desenvolver LLA-c associada à superproteção e aos altos
padrões de vida (Gale e cols., 1997). Esta diferença é plausível e pode ser explicada pela

37
exposição negligente a componentes ambientais nocivos entre as mães com menos educação,
baixa renda e geralmente menos conhecimento sobre os fatores de risco à saúde em geral
(Pombo-de-Oliveira e cols., 2006).
O estudo realizado por Pombo-de-Oliveira e cols. (Pombo-de-Oliveira e cols., 2006)
sugeriu que algumas exposições ambientais durante a gravidez pode levar a um aumento no
risco de LAL na descendência. Uma significância estatística notavelmente elevada foi
observada para uma exposição hormonal durante a gravidez. A associação positiva entre a
exposição materna a dipirona e a LAL oferece um suporte para estudos prévios (Alexander e
cols., 2001; Ma e cols., 2002). O consumo de dipirona durante a gravidez foi previamente
associada a tumor de Wilms’ (Sharpe e cols., 1996) e no estudo (Pombo-de-Oliveira e cols.,
2006) também se encontrou uma associação às LLAs com rearranjos no MLL, sugerindo que a
dipirona pode ser considerada nociva durante a gravidez, especialmente no Brasil onde é um
composto baixo custo e pode oferecer um risco elevado para malignidades infantis (Sharpe e
cols., 1996; Alexander e cols., 2001).
Em relação ao hábito de fumar dos pais antes ou durante a gestação ser considerado
como um fator de risco para o desenvolvimento de leucemia em lactentes é controverso
(Belson e cols., 2007). Dois estudos encontraram que a freqüência, quantidade, e duração do
hábito de fumar do pai antes da concepção foram relacionadas a um risco significativamente
elevado (Sorahan e cols., 1995; Ji e cols., 1997). Porém, outros estudos não encontraram
nenhuma associação entre a doença e o hábito paterno de fumar em algum momento
(Brondum e cols., 1999).
Estudos relacionados ao fumo passivo durante a infância, apesar de menos numerosos,
também não conseguiram mostrar uma relação com a leucemia (Sasco e Vainio, 1999).
Apesar dos resultados negativos parecerem consistentes até o momento, o hábito de fumar

38
ainda permanece como uma exposição de interesse devido ao seu potencial carcinogênico em
vários órgãos, a habilidade de vários constituintes do cigarro de atravessar a barreira
placentária e sua alta prevalência durante a gravidez (Menegaux e cols., 2005).
1.2.10.1. Leucemias agudas e sua relação à exposição à fumaça do cigarro
A fumaça do cigarro possui milhares de componentes incluindo vários agentes
mutagênicos tais como, hidrocarbonetos aromáticos policíclicos (PAH) e nitrosaminas (Fig.
3), esta mistura é considerada como um carcinogênico completo (Belpomme, 2005).
Figura 3: Agentes mutagênicos presentes na fumaça do cigarro.
O papel do hábito de fumar dos pais é pouco conhecido, mas sabe-se que é
biologicamente plausível. Recém-nascidos de mães fumantes mostraram um aumento na
freqüência de anomalias cromossômicas (Pluth e cols., 2000), assim como também se
observou associação em danos oxidativos e aneuploidias dos espermatozóides dos pais
fumantes (Shi e cols., 2001).
Um estudo de caso-controle na China no qual nenhuma das mães fumou mostrou que
o risco de leucemia aguda infantil aumentou se o pai fumou 5 ou mais maços de cigarros por
ano antes da concepção (Ji e cols., 1997). Um grande estudo de caso-controle no Reino Unido
baseado em 1630 casos de leucemia e 6987 controles mostrou uma tendência ao aumento não-
significativo na associação de leucemia infantil com o hábito de fumar do pai antes da

39
concepção e uma tendência decrescente quando a mãe fumou durante a gravidez (Pang e
cols., 2003).
Em contraste, um grande estudo caso-controle realizado nos Estados Unidos realizado
em aproximadamente 2.500 casos de leucemia e 2.500 controles não encontrou evidências de
uma associação entre o hábito de fumar do pai e da mãe antes ou durante a gravidez e a
leucemia infantil (Brondum e cols., 1999). Recentemente, um estudo populacional com uma
coorte de 1.440.542 crianças Suecas indicou que o hábito de fumar da mãe entre a oitava e a
décima segunda semana de gestação estava associado a um risco significativamente reduzido
para LLA e elevado para LMA (Mucci e cols., 2004).
Estes modelos de estudos podem explicar parcialmente os resultados inconsistentes da
relação do hábito de fumar dos pais com as leucemias em lactentes (Chang e cols., 2006).
1.3. Gene RAS
As alterações no genoma celular que afetam a expressão ou função de genes que
controlam o crescimento e diferenciação da célula são consideradas a principal causa de
câncer. A pesquisa molecular do câncer tem como propósito identificar genes que são
alterados nos vários tipos de tumores e desvendar o papel destes genes na carcinogênese. A
família de genes que freqüentemente resguarda uma mutação nos tumores humanos é a dos
genes RAS (Barbacid, 1987).
O primeiro oncogene foi isolado do tumor de bexiga humano e foi quase idêntico ao
gene humano normal e ao gene presente no vírus tumoral que infectou os camundongos
utilizados num estudo anterior. Este gene foi denominado RAS, porque foi originalmente
isolado de “ratos” (camundongos) com sarcoma e também causou sarcomas nos humanos. A

40
única diferença significativa encontrada entre o gene humano normal, o gene do tumor de
bexiga e o gene viral do roedor foi a mudança no códon 12 e, conseqüentemente uma
mudança nos aminoácidos (Singer, 1992). Logo se estabeleceu que os agentes ativos na
transformação do DNA da linhagem celular de câncer de bexiga humano eram os homólogos
do HRAS e do KRAS, oncogenes encontrados no retrovírus Harvey e Kirsten, respectivamente
(Der e cols., 1982) de sarcoma de camundongo. Eles demonstraram pela primeira vez que os
tumores humanos possuíam oncogenes ativados, relacionados a aqueles retirados de retrovírus
dos seus genomas hospedeiros.
Depois de alguns meses, os mesmos grupos encontraram que a diferença entre o gene
humano HRAS normal e a forma oncogênica encontrada nos tumores era uma única mutação
pontual. Rapidamente tornou-se claro que uma grande proporção de tumores humanos possuía
tais mutações ativadas nos oncogenes RAS (Malumbres e Barbacid, 2003; Denayer e cols.,
2008). Assim começou a pesquisa nas três proteínas intimamente relacionadas, H-, K-, e
NRAS, que coletivamente são referidas como RAS (Downward, 2006).
1.3.1. Localização do RAS
O NRAS foi localizado no braço curto do cromossomo 1 (Davis e cols., 1983). Uma
banda escura foi observada logo acima do centrômero na banda 1p13, mas foi em 1995 que
Mitchell e cols. precisaram a localização do NRAS na banda 1p13.2 (Fig. 4a). O mapeamento
do KRAS foi realizado pelo método da fragmentação por enzima de restrição polimorfismo-
específica (RFLP) que confirmou a localização aproximada do KRAS no 12p12.1 (O’Connell
e cols., 1985) (Fig. 4b). Já o HRAS foi mapeado, por hibridização de células somáticas, no
cromossomo 11p15.5 (Junien e cols., 1984) (Fig. 4c).

41
Figura 4: Localização citogenética dos genes NRAS, KRAS e HRAS
1.3.2. Função do RAS
As vias de controle de sinalização das proteínas RAS são reguladores chaves do
crescimento celular normal. Aproximadamente 20-30% de todos os tumores possuem uma
mutação de ativação em um dos RAS. Nestes tumores, a proteína RAS ativa contribui
significativamente em vários aspectos do fenótipo maligno, incluindo a desregulação do
crescimento celular, morte celular programada e invasão, e a habilidade para neo-
angiogênese. Outra importante função da RAS e dos seus efetores a jusante é seu
envolvimento nos processos neuronais como aprendizado, memória e plasticidade sináptica
(Denayer e cols., 2008).
As RAS são proteínas ligadas ao nucleotídeo guanosina que ciclam entre uma
conformação GTP-ligante ativa e GDP-ligante inativa (Fig. 5). Elas podem ser ativadas
quando um fator de crescimento se liga a um receptor tirosino quinase, como o receptor do
fator de crescimento epidermal (EGFR). Isso resulta em uma dimerização e auto-fosforilação

42
do receptor, que se liga ao domínio SH2 da proteína adaptadora GRB2. Através do seu
domínio SH3, a GRB2 se liga a SOS, que é assim recrutada para a membrana plasmática (Fig.
5). As proteínas SOS (SOS1 e SOS2) são RAS-GEFs (fatores de intercâmbio do nucleotídeo
guanosina) importantes que catalisam o intercâmbio de GDP ligada a RAS pela GTP
(Denayer e cols., 2008).
A proximidade aumentada de SOS à membrana ligada a RAS resulta em um aumento do
intercâmbio de nucleotídeos na RAS. Muitos outros tipos de receptores, incluindo os
receptores emparelhados de proteína G, podem ativar RAS através da estimulação dos fatores
de intercâmbio (Denayer e cols., 2008) (Fig. 5).
RAS-GTP tem moléculas efetoras diferentes das quais a RAF quinase serina-treonina é a
mais importante. As RAF quinases ativadas fosforilam MEK, que por sua vez fosforila e ativa
ERK. ERK/MAPK possui vários substratos nucleares e citosólicos, tais como os fatores de
transcrição e proteínas de sinalização. O resultado da sinalização por esta via é uma mudança
no padrão da expressão gênica que pode estimular a proliferação celular, promover
sobrevivência celular ou controlar a diferenciação celular (Denayer e cols., 2008) (Fig. 5).
A sinalização através desta cascata termina quando a GTP é hidrolisada à GDP e fosfato
inorgânico seja pela atividade GTPase intrínseca do RAS (lenta), ou pelas proteínas que
ativam as GTPase (GAPs; rápida) (Denayer e cols., 2008) (Fig. 5).

43
Figura 5: Via de sinalização da RAS. A ligação de fatores de crescimento aos receptores
tirosino quinase estimula a auto-fosforilação de tirosinas específicas dos receptores. O
receptor fosforilado logo se liga a uma proteína adaptadora chamada GRB2 que, por sua vez,
recruta SOS à membrana plasmática. SOS é um fator de intercâmbio de nucleotídeo guanina
que desloca GDP do RAS, subseqüentemente permitindo a ligação do GTP (RAS já está
ancorada à membrana plasmática pelos lipídeos pós-transcricionais agregados, identificados
por uma linha vermelha). RAS GTP-ligante recruta e ativa Raf. Raf inicia a cascata de
fosforilação protéica fosforilando primeiro MEK. A MEK fosforilada por sua vez fosforila
ERK. A ERK fosforilada se desloca do citoplasma até o núcleo onde subseqüentemente
fosforila uma quantidade de fatores de transcrição, incluindo o fator de transcrição específico
chamado Elk-1. Os fatores de transcrição fosforilados acionam a transcrição (expressão
gênica) de uma série específica de genes alvo. A atividade de RAS é limitada pela hidrolise da
GTP de volta á GDP pelas proteínas que ativam a GTPase (GAP). Outras abreviações são:
MEK (MAPK/ERK quinase), ERK (quinase extracelular receptor-estimulada), MAPK
(proteína quinase receptor-estimulada). As quinases são enzimas que agregam fosfatos a
moléculas usando ATP. Mitógenos são fatores (ex. fatores de crescimento) que estimulam a
divisão celular.

44
1.3.3. Alterações do RAS e sua associação com leucemias
As mutações somáticas pontuais que introduzem substituições de aminoácidos nos
códons 12, 13 (éxon 1) ou 61 (éxon 2) são encontradas em aproximadamente 30% dos
cânceres humanos (Braun e Shannon, 2008). Estes códons localizam-se entre duas regiões de
homologia, que aparentemente formam componentes importantes dos sítios da GTP-ligante
(McCormick e cols., 1985).
As mutações gênicas, cuja forma mais comum são as mutações pontuais, são
altamente prevalentes em cânceres pancreáticos (>80%), colo-retais (40%-50%), endometriais
(40%), pulmonares (30%), e cervicais (20%-30%) (Schubbert e cols., 2007). Já nas
malignidades hematopoéticas, estas mutações foram observadas numa prevalência variável,
inclusive na LMA e na LLA (Liang e cols., 2006). As mutações no RAS ocorrem em
aproximadamente 20% dos pacientes com LLA (Browett e Norton, 1989) e aproximadamente
em 30% dos pacientes com LMA (Barletta e cols., 2004). Diferentemente de outros cânceres
humanos, onde as mutações no KRAS geralmente são predominantes, as mutações no NRAS
são as mais freqüentes nas neoplasias hematológicas (Usher e cols., 2009).
Os sítios das mutações são principalmente nos códons 12, 13 e 61 do NRAS e no
códon 12 do KRAS; e, com muita pouca freqüência, no HRAS (Guo e cols., 1998). O NRAS é
duas vezes mais mutado que o KRAS nas leucemias, e 60% das mutações são transições G→A
no códon 12, resultando na substituição de glicina por ácido aspártico (Jekic e cols., 2004). A
incidência das substituições do Asp-12 pode refletir o envolvimento de um agente particular
na leucemogênese. Por exemplo, a mutação G→A é a mais freqüentemente encontrada com
agentes alquilantes (Farr e cols., 1998).

45
1.3.3.1. Coexistência de alterações nos genes MLL e RAS
O rearranjo do MLL é uma das mutações genéticas que bloqueia a diferenciação das
células leucêmicas; por outro lado, as mutações do RAS levam à proliferação dessas células.
Na literatura, apenas um estudo mostra a coexistência do rearranjo do MLL e as
mutações no RAS (Mahgoub e cols, 1998). Estes autores analisaram 32 amostras com
leucemias pediátricas que, por citogenética e análise molecular, mostraram possuir um
rearranjo no cromossomo 11q23. Embora as mutações no RAS sejam as mais comuns nas
LMAs, ambas as leucemias mielóides e linfóides foram incluídas no estudo (Pui e cols, 1995).
Este estudo mostrou padrões normais de KRAS e NRAS em 23 dos 25 casos com leucemias de
novo. Como todos os casos estudados, sem mutações detectadas, possuíam uma elevada
contagem de blastos e também foram rastreados por diferentes técnicas (single-strand
conformational polymorphism [SSCP] e allele-specific restriction enzime assay), estes dados
sugeriram que a aquisição das mutações no RAS não tem um papel principal nas leucemias
com rearranjos no MLL (Mahgoub e cols, 1998).
Uma questão importante na patogênese das leucemias com translocações no MLL é a
necessidade de mutações em outros oncogenes para o surgimento da doença. A ausência de
mutações no RAS nas 13 LLAs estudadas com translocações no MLL é similar a achados
prévios da falta de freqüência de mutações no RAS em LLAs pré-B com t(1;19)(q23;p13)
(Kawamura e cols., 1995). Estas observações sugeriram que as mutações no RAS não são
essenciais na patogênese das LLAs com translocações cromossômicas que envolve a quebra
do PBX1 ou MLL. Porém, juntas, as mutações no RAS e as translocações no MLL podem
contribuir ocasionalmente ao desenvolvimento de leucemias mielóides (Mahgoub e cols.,
1998). Os autores sugeriram também que estudos adicionais deviam ser realizados para

46
determinar a existência de formas alternativas de ativação da via da RAS no caso de
rearranjos no MLL sem mutações no RAS. Por exemplo, na leucemia mielóide crônica (LMC),
onde as mutações no RAS geralmente estão ausentes, a via de sinalização da RAS é
desregulada pela proteína de fusão BCR-ABL, que induz níveis elevados da RAS GTP-ligante
ativa (Pendergast e cols., 1993). Também, em aproximadamente metade dos pacientes com
NF1 que desenvolvem leucemia, há uma deleção do alelo normal do gene supressor de tumor
NF1 (Shannon e cols., 1994). A neurofibrina, o produto do gene NF1, ativa a GTPase e dessa
forma acelera a hidrolise do GTP nas proteínas RAS (Shannon e cols., 1994). Então, neste
contexto, a transformação leucêmica não é independente da RAS (Mahgoub e cols., 1998).
Estudos adicionais de casos podem gerar uma estimativa mais confiável da freqüência
de mutações no RAS nas leucemias com rearranjos no MLL. Apesar disso, o estudo do
Mahgoub e cols. serviu como suporte no papel das mutações do RAS nas leucemias de novo
que possuem translocações no MLL numa freqüência similar a aquela encontrada em outras
formas de LMA.
1.3.3.2. Coexistência de alterações nos genes FLT3 e RAS
Estudos tentaram relacionar as mutações no FLT3 e no RAS, já que são os genes mais
comuns nas leucemias agudas, porém sugeriram que as mutações do FLT3 e do NRAS
ocorrem de maneira independente, apesar de que não se pode descartar a possibilidade que
existam interações fracas adversas entre o FLT3 mutante e o NRAS mutante. Como ambas as
alterações genéticas estão associadas à transdução de sinais aberrantes, estas mutações podem
ser um complemento ou estarem sinergicamente associadas à progressão leucêmica. (Kiyoi e
cols., 1999).

47
Sabe-se que FLT3 ativa vários efetores a jusante, incluindo RAS, ERK, proteína
quinase B (PKB/AKT) e STAT5 (Hayakawa e cols., 2000; Mizuki e cols., 2000) e resultados
de estudos mostram uma prevalência surpreendente de mutações em ambas LMAs pediátricas
e de adultos, com mutações de alguns componentes da via ocorrendo em >50% dos casos.
Como esperado, as mutações de dois membros da mesma via (FLT3 e RAS) na mesma
leucemia são raras, assim como uma mutação adicional nesta via numa célula já mutada não
se esperaria que conferisse nenhuma vantagem de seleção (Stirewalt e Radich, 2003).
Evidências recentes de que as mutações no FLT3 podem ser também um evento
complementar na leucemia hiperdiplóide (Armstrong e cols., 2004; Taketani e cols., 2004)
são compatíveis com os resultados do Wiemels e cols. (Wiemels e cols., 2005) onde se mostra
que o FLT3 sinaliza em parte através da via RAS. Além disto, mutações em outras vias do
gene RAS, PTPN11, estão restritas geneticamente às leucemias que não possuem mutações no
RAS, e também são encontradas nas mutações TEL, no RAS, e FLT3 que são exclusivas a cada
uma delas nas LLAs infantis.
1.3.4. Mutações no gene RAS e a sua associação com fatores ambientais
Recentemente, mutações no RAS foram associadas a diferentes subgrupos de
leucemias cujas mães foram expostas a determinadas substâncias durante a gestação da
criança. Mutações no NRAS têm sido associadas com a exposição, em qualquer momento, a
produtos derivados do óleo ou carvão antes do diagnóstico do caso ou durante a gravidez, a
exposição paterna a materiais plásticos antes do início da gravidez ou outros hidrocarbonetos
durante o período pós-natal, e ao uso de psicotrópicos pelos pais. Mutações no KRAS têm sido

48
associadas à exposição materna a solventes e materiais plásticos durante a gravidez e depois
dela, e ao uso paterno de anfetaminas ou pílulas dietéticas (Shu e cols., 2004).
Estudos prévios mostraram que as mutações no RAS podem estar associadas com
exposições químicas. Dois estudos epidemiológicos tem ligado as mutações no RAS nas
LMAs em adultos a ocupações de “alto risco” para a leucemogênese (Taylor e cols., 1992;
Barletta e cols., 2004). Outro estudo caso-caso de leucemias pediátricas sugeriu um papel para
as exposições a hidrocarbonetos parentais incluindo alguns específicos para o pai para as
leucemias com mutações no RAS comparadas com aquelas que não possuíam mutações (Shu e
cols., 2004).
O estudo de Wiemels e cols. (Wiemels e cols., 2005) onde foram estudados 157 casos
diagnosticados com LLA, 31 com LMA e 2 com LMC, com mutações no RAS presentes em
20% (38/191) de todas as leucemias incluindo 17 KRAS, 18 NRAS, e 3 mutações em ambos K
e NRAS, não detectou uma prevalência elevada do hábito de fumar dos pais entre os casos
positivos para mutações no RAS comparados com os casos negativos para mutações no RAS
ou subtipos. Além disso, químicos mutagênicos do hábito de fumar da mãe atravessam a
placenta aumentando a plausibilidade de um efeito do hábito de fumar dos pais no risco de
desenvolvimento de leucemias pediátricas (Milunsky e cols., 2000). O estudo do Wiemels e
cols. (Wiemels e cols., 2005) mostrou uma associação significativa entre o hábito de fumar do
pai e a leucemia hiperdiplóide quando comparada aos outros subtipos leucêmicos. Como o
hábito de fumar dos pais não foi significativamente associado com a ocorrência das leucemias
com mutação no RAS, isto sugere que outro subtipo molecular de leucemia possa estar
associado positivamente a este hábito. Futuros estudos devem se esforçar em avaliar o papel
do hábito de fumar dos pais e outros hidrocarbonos nas leucemias pediátricas entre os vários
subtipos genéticos tumorais chave (Wiemels e cols., 2005).

49
Apesar dos mecanismos específicos das mutações do RAS ainda serem desconhecidos,
estudos epidemiológicos e em animais sugerem que mutações pontuais específicas nos genes
RAS podem ser induzidas também por certos carcinógenos químicos (Barletta e cols., 2004).
As associações encontradas servem como modelo de estudos etiológicos futuros, que
terão como objetivo encontrar as vias que causam a leucemia infantil e enfatizar a função
crítica de subgrupos de tumores genéticos em estudos epidemiológicos de leucemia pediátrica
(Wiemels e cols., 2005). Por isso, levantou-se a hipótese: as alterações no gene RAS e no
FLT3 podem ser complementares e/ou associadas a leucemogênese (com rearranjos no
MLL) e estas estarem relacionadas à exposição com os diferentes fatores de risco
avaliados?

50
2. JUSTIFICATIVA DO ESTUDO

51
É relevante lembrar que as leucemias agudas na primeira infância compreendem um
grupo de leucemias caracterizadas pelo diagnóstico de LLA ou LMA durante os primeiros
anos de vida. O termo LAL usualmente é aplicado quando o diagnóstico é feito entre os
primeiros 12 meses após o nascimento. Todavia, estudos encontraram uma percentagem
relevante de características típicas das LALs (MLL+) em crianças entre 12-23 meses
(Emerenciano e cols., 2005), assim sustentando a nossa inclusão destas crianças no presente
estudo. Apesar de não ser tão freqüente quanto o neuroblastoma, a leucemia é a causa
principal de morte de doenças neoplásicas durante o período perinatal. Embora seja a segunda
malignidade mais comum no primeiro ano de vida, ela é uma doença bastante rara em
crianças pequenas, ocorrendo com maior freqüência nas crianças mais velhas. As leucemias
são doenças progressivas que, na ausência de intervenção médica, provocam a morte em
poucos meses. Para uma intervenção cada vez mais precisa e eficaz, é necessário maior
entendimento dos mecanismos da leucemogênese. Um estudo epidemiológico-molecular já
vem sendo desenvolvido no Programa de Hematologia e Oncologia Pediátricos com o
objetivo de explorar a hipótese de que certas exposições ambientais podem aumentar o risco
das LALs, principalmente com rearranjos no MLL. Como o melhor modelo para estudo desta
doença são as leucemias que ocorrem no primeiro ano de vida (Gurney e cols., 1995),
utilizou-se material da coorte já existente, de pacientes com LALs, como objeto do nosso
estudo. Consideramos este grupo para o estudo já que o intervalo entre a exposição a fatores
de risco putativos e o começo da leucemia é mais curto, a identificação de exposição a fatores
de risco nas crianças é mais fácil, e a quantidade de fatores ambientais, aos quais os lactentes
são expostos, são menores que os associados com as leucemias em adultos (Belson e cols.,
2007).

52
Entre os eventos genéticos mais comuns que ocorrem nas crianças com menos de 12
meses de vida, tanto nas LLAs como nas LLAs, estão os rearranjos no MLL no cromossomo
11q23, que pode ser de até 85% dependendo da técnica aplicada. Os lactentes diagnosticados
com leucemia aguda que possuem um rearranjo 11q23 possuem um prognóstico
particularmente ruim quando comparadas às outras crianças com leucemias agudas. Além
disso, estudos como o de Armstrong e cols. (Armstrong e cols., 2003) demonstraram que o
receptor FLT3-TK era o gene mais comumente superexpresso nas leucemias com rearranjos
no MLL, desta forma levantando a hipótese de que um sinal constitutivo da FLT3 pode estar
relacionado com o desenvolvimento e manutenção do MLL.
Levando em consideração esses dados e seguindo a linha de estudo das pesquisas
realizadas para explorar a influência das exposições ambientais no aumento do risco de
desenvolvimento de LALs, analisamos os aspirados de MO e SP de lactentes que formam
parte do banco de amostras do laboratório para determinar possíveis mutações no RAS, que
sabemos que é um gene mutado numa grande percentagem de tumores humanos e que as suas
mutações estão correlacionadas ás leucemias infantis. Sendo assim, o nosso estudo foi
conduzido ao relacionamento das LALs, que apresentaram mutações no RAS, com possíveis
mutações no FLT3 e rearranjos no MLL e a relação destas mutações com o hábito de fumar
dos pais antes e durante à gestação e a exposição da mãe à fumaça do cigarro no ambiente de
trabalho e dentro de casa durante a gravidez. Este tipo de exposição foi do nosso interesse já
que se tem muitos estudos relacionando o cigarro a diversos tipos de câncer, mas pouco se
sabe sobre a relação entre a exposição do embrião ou do feto a este agente carcinogênico, que
é muito comum na nossa sociedade.
Fizemos a opção de utilizar a técnica de denaturing high pressure liquid
chromatography (dHPLC) por ser uma técnica bastante segura e que tem se mostrado eficaz,

53
para análises de varias amostras, num curto período de tempo com uma relação custo-
efetividade bastante econômica. A dHPLC tem uma alta sensibilidade e especificidade
estimada entre 96% e 100% nas análises. Ela tem sido empregada com sucesso na detecção de
mutações em diferentes genes de interesse médico, mostrando-se mais eficiente que outras
técnicas rotineiramente utilizadas para este fim. Além da alta sensibilidade na detecção de
mutações, esta técnica permite um alto grau de automação e não requer nenhum tratamento
especial para o produto de PCR. Desta forma, facilita a análise de um grande número de
amostras. A técnica de Restriction Fragment Length Polymorphism (RFLP) foi realizada para
consolidação dos resultados.

54
3. OBJETIVOS

55
3.1. Objetivo principal:
Rastrear as mutações no gene RAS em lactentes portadores de leucemias agudas
3.2. Objetivos Secundários:
• Estimar o papel das mutações no RAS como fatores de risco associados às leucemias
de lactentes;
• Correlacionar as possíveis mutações no gene RAS com os achados epidemiológicos
referentes às exposições maternas ao fumo durante a gestação, visando à identificação
de possíveis interações entre gene-ambiente.

56
4. MATERIAL E MÉTODOS

57
4.1. Desenho do estudo
O desenho do estudo utilizado foi o caso-caso. O estudo de caso trata-se de uma
abordagem metodológica de investigação especialmente adequada quando procuramos
compreender, explorar ou descrever acontecimentos e contextos complexos, nos quais estão
simultaneamente envolvidos diversos fatores num mesmo grupo de casos previamente
selecionados.
Nós exploramos as vantagens de uma análise pré-estabelecida do tipo caso-controle e
comparamos as magnitudes do risco das mutações no gene RAS usando a abordagem do tipo
caso-orientado (casos de leucemias em lactentes).
4.2. Casuística
Amostras de material diagnostico (aspirado de medula óssea ou sangue periférico)
provenientes de 138 lactentes foram encaminhadas por diversos hospitais de diferentes
estados do país (Rio de Janeiro, Bahia, São Paulo, Rio Grande do Sul, Distrito Federal, Minas
Gerais, Pernambuco e Santa Catarina) ao laboratório no Centro de Pesquisas do Instituto
Nacional de Câncer (INCA), Rio de Janeiro, no período de 1998 a 2008 foram selecionados
para este estudo. Estas amostras foram devidamente congeladas a -80°C após exames
diagnósticos. Juntamente ao material biológico foram enviados os dados demográficos e
clínicos. Todas as informações avaliadas neste estudo encontram-se nos Anexos e fazem parte
de um estudo epidemiológico do tipo caso-controle de base-hospitalar onde as informações
foram obtidas através de questionários bem estruturados e já analisados parcialmente (Pombo-
de-Oliveira e cols., 2006; Koifman e cols., 2008).

58
A primeira parte do questionário foi dedicado ao nascimento da criança e aos seus
cuidados e a segunda parte foi dedicada às exposições durante a gravidez. Perguntas sobre a
demografia incluíram o ingresso familiar, idade materna, e nível de educação. O histórico de
doenças maternas e o motivo do uso de medicamentos foram obtidos através de perguntas
relacionadas ao uso de diversos tipos de drogas devido a doenças infecciosas, tratamento por
perda fetal previa, anemia, etc., assim como a exposição a pesticidas, consumo de álcool e o
hábito de fumar dela, do pai da criança e exposição a fumaça de cigarro. Estas análises
incluíram informação sobre as exposições maternas durante o período de 3 meses antes da
gestação, durante a gravidez e depois da gravidez (Pombo-de-Oliveira e cols., 2006).
Portanto, utilizamos como critérios de seleção para este estudo: diagnóstico de LLA e
LMA em crianças, cujas mães concordaram com o estudo e responderam ao questionário
epidemiológico entre 1998-2008, cujas questões sobre o hábito de fumar estavam completas
nos questionários.
A autorização para a utilização destas amostras em pesquisas foi solicitada ao
responsável legal da criança, através da assinatura de um “Termo de Consentimento Livre e
Esclarecido” (TCLE). O projeto foi aprovado em 2008 pelo Comitê de Ética em Pesquisa
(CEP) do INCA sob o número de protocolo #005/2008. A cópia da carta de aprovação do
CEP e o TCLE se encontram nos Anexos.
4.3. Extração de DNA para realização dos PCRs do gene RAS
A maioria das amostras selecionadas estava devidamente congelada como células a -
80oC ou -180oC e os DNAs já extraídos, estavam conservados em câmera fria a 4ºC.

59
Adicionalmente, material obtido de esfregaços de sangue periférico (SP), ou aspirados de
medula óssea (MO) foram adicionados para complementação do estudo.
O método de sal que não utiliza fenol-cloroformio foi então empregado para extração
de DNA (Muller, 1988). No início do procedimento foi adicionado tampão de lise de
hemácias (TLH) previamente gelado, de maneira que fique 3,5 vezes do volume total de
sangue, foi bastante homogeneizado com a pipeta e colocado em banho com gelo por 30 min.
Logo, centrifugou-se a 3000 rpm por 20 min em centrífuga refrigerada a 4°C (se possível). O
sobrenadante foi descartado e o grumo lavado com 300µL de TLH a fim de retirar o excesso
de hemácias junto aos leucócitos. Homogeneizou-se bem até soltar o grumo e ficar limpo e foi
centrifugado a 3000rpm por 10 min. Após a centrifugação, ressuspendeu-se o grumo em
300µL de tampão de lise de núcleo (TLN) e dissolveu-se completamente o grumo. Logo em
seguida, foi adicionado 1,0µL de Proteinase K (concentração final de 50µg/mL) e
homogeneizou-se para posterior incubação da amostra durante a noite a 56°C. No dia
seguinte, foram adicionados 60,0µL de NaCl 5,0M e centrifugou-se a 6000 rpm por 30 min a
4°C. Ao término da centrifugação foi retirado o sobrenadante e passado para um novo tubo
onde se adicionaram aproximadamente 900,0µL de etanol absoluto o que tornou o DNA
insolúvel e assim provocou a precipitação do mesmo. Verteu-se o tubo várias vezes e foi
incubado a –70°C por 30 min. Após a incubação, o tubo foi centrifugado a 10000 rpm por 10
min de preferência em centrífuga refrigerada a 4°C. O tubo foi invertido para a retirada do
etanol e foram adicionados 1000,0µL de etanol 70% para a retirada do excesso de sal. Foi
centrifugado novamente a 10000 rpm por 5 min a 4°C (se possível) e finalizou-se o processo
com a adição de 30-50µL de TE Buffer (Tris-EDTA) ou água milli-Q autoclavada para
posterior incubação a 68ºC por 30 min. O DNA foi armazenado na geladeira ou câmara fria a
4°C.

60
Outra metodologia utilizada para o protocolo de extração de DNA foi a do reagente
Trizol® onde após ter removido a fase aquosa foram adicionados 300,0µL de etanol 100%
(para cada 1,0ml de Trizol® utilizado) para a precipitação do DNA. Foi misturado por
inversão e incubado por 3 min a temperatura ambiente, logo se centrifugou a 2000rpm por 5
min em centrífuga refrigerada a 4°C e, após a retirada do sobrenadante, lavou-se o grumo de
DNA com citrato de sódio 0,1 M em 10% de etanol (usou-se 1,0mL de citrato de sódio para
cada 1,0 de Trizol®).
Deixou-se incubado a temperatura ambiente (15-30°C) por 30 min, homogeneizando-
se periodicamente, e centrifugou-se a 2000 rpm por 5 min em centrífuga refrigerada a 4°C.
Repetiu-se o processo 2 vezes (para lavagem) e o DNA foi ressuspendido em 1500µL de
etanol 75% e incubou-se por 15 min homogeneizando periodicamente. Após uma nova
centrifugação a 2000 rpm por 5 min em centrífuga refrigerada a 4°C, desprezou-se o
sobrenadante e secou-se o DNA com o tubo aberto por 5-15 min e se dissolveu em 50µL de
NaOH 8mM. Foi centrifugado a 12000 rpm por 10 min para remoção do DNA e o
sobrenadante contendo o DNA foi transferido para um novo tubo. O DNA foi estocado a 4°C.
4.4. Quantificação do DNA
Após a extração, o DNA foi quantificado. Para isto foi utilizado um espectrofotômetro
(NanoDrop 1000), que possui um programa específico para quantificação de DNA onde o
fator de diluição foi ajustável, a unidade de concentração foi ng/µL, a absorbância utilizada
foi de 260nm. Para a realização da quantificação foi utilizado 1,0µL de DNA e a concentração
foi dada de forma automática pela máquina, de acordo com o programa já existente nela.

61
4.5. PCR para a amplificação do KRAS
O éxon 1 foi amplificado, de acordo com os critérios do protocolo utilizado por
Bornholdt e cols. (Bornholdt e cols., 2008) sendo que este éxon contém 2 códons: 12 e 13,
onde se localizam as possíveis mutações do gene em estudo. Para os dois códons foi
necessária a realização de uma reação única.
Foram utilizadas 2 amostras diagnosticadas com leucemia mielomonocítica crônica
(LMMC) por serem portadoras de mutações no RAS, uma delas com mutação no códon 12 e a
outra no 13. Foram utilizadas como controles positivos para as reações de PCR para o RFLP.
As condições da reação de PCR, para ambos os códons 12 e 13, foram desenhadas
para um volume final de 25,0µL, utilizando tampão de PCR (50mM KCl, 20nM Tris-HCl pH
8,4); MgCl2 a uma concentração final de 2,5mM; dATP, dTTP, dCTP e dGTP a uma
concentração final de 0,2mM. O substrato foi sempre de DNA (100,0-150,0 ng/µL) e a
quantidade de enzima foi de 0,75U de Taq DNA polimerase.
O procedimento da reação de PCR foi realizado em um termociclador GeneAmp®
PCR System 9700 (Applied Biosystems), onde os perfis térmicos da reação seguiram as
seguintes condições: Uma etapa inicial de desnaturação a 94°C por 1 min, 35 ciclos de
desnaturação a 94°C por 30 seg, temperatura e tempo de anelamento de 55°C por 45 seg, e
extensão a 72°C por 45 seg; um ciclo final de extensão a 72°C por 10 min e uma etapa final a
4°C. A reação foi realizada com os oligonucleotídeos KRAS12-F e KRAS1213-R, e KRAS13-F
e KRAS1213-R, sendo que as seqüências destes oligos são:
KRAS12-F: 5’-ACT GAA TAT AAA CTT GTG GTA GTT GGA CCT-3’ 30pb
KRAS13-F: 5’-TCA AAG AAT GGT CCT GGA CC-3’ 20pb
KRAS1213-R: 5’-ATA TAA ACT TGT GGT AGT TCC AGC TGG-3’ 27pb

62
A revelação da reação deste PCR foi feita através de gel de agarose 1,5% corado com
brometo de etídeo (Fig. 6).
1 2 3 4 5 6 7 8 9 10 11
Figura 6: Gel de agarose 1,5% corado com BE para amplificação do KRAS. (1) padrão de
peso molecular de 100pb; (2) controle positivo: amostra de LMMC; (3) controle negativo
(água e mix de PCR); (4-11) amostras de pacientes com leucemias com banda amplificada.
4.6. Digestão do gene KRAS com endonucleases de restrição
Após a amplificação, o produto do PCR foi digerido por duas enzimas de restrição
BstNI e PflMI (Prodimol) para os códons 12 e 13, respectivamente.
A técnica de digestão é chamada de Restriction Fragment Length Polymorphism (RFLP) na
qual 5,0µL da amostra amplificada para o códon 12 foram digeridos por 10U da enzima BstNI
a 60ºC overnight e 5,0µL da amostra amplificada para o códon 13 foram digeridos por 2U da
enzima PflMI a 37°C por 6 horas (Bornholdt e cols., 2008). Os fragmentos gerados na
digestão foram separados por eletroforese em gel de agarose 3% e analisados em comparação
com um padrão molecular de 25pb (Invitrogen) (Fig. 7).

63
Códon 12 Códon 13
1 2 3 4 5 1 2 3 4 5 6 7 8 9 10 11
(a) (b)
Figura 7: Géis de agarose de 3% corados com BE. Gel (a): (1) padrão de peso molecular de
25pb; (2 e 4) PCRs do gene KRAS de amostras de pacientes com leucemia; (3) amostra do
paciente 2 após digestão com enzima BstNI com padrão de duas bandas (padrão mutado); (5)
amostra do paciente 4 após digestão com enzima BstNI com padrão de uma banda (padrão
selvagem). Gel (b): (1,3,5,7,9) PCRs do gene KRAS de amostras de pacientes com leucemia;
(2,6,10) amostra dos pacientes 1,5,9 respectivamente, após digestão com enzima PflMI com
padrão de duas bandas (padrão selvagem); (4 e 8) amostra dos pacientes 3 e 7
respectivamente, após digestão com enzima PflMI com padrão de uma banda (padrão
mutado); (11) padrão de peso molecular de 25pb.
4.7. PCR para a amplificação do NRAS
O éxon 1 foi amplificado, de acordo com os critérios do protocolo utilizado por Liang
e cols. (Liang e cols., 2006) sendo que este éxon contém os códons 12/13, onde se localizam
as possíveis mutações do gene em estudo.
Foram utilizadas 2 amostras diagnosticadas com leucemia mielomonocítica crônica
(LMMC) por serem comprovadamente portadoras de mutações no códon 12/13 do RAS.
Foram utilizadas como controles positivos para as reações de PCR no dHPLC.
As condições da reação de PCR foram desenhadas para um volume final de 50,0µL,
utilizando tampão de PCR (50mM KCl, 20nM Tris-HCl pH 8,4); MgCl2 a uma concentração

64
final de 1,5mM; dATP, dTTP, dCTP e dGTP a uma concentração final de 0,2mM. O substrato
foi sempre de DNA (100,0-150,0 ng/µL) e a quantidade de enzima foi de 0,625U de Taq DNA
polimerase.
O procedimento da reação de PCR foi realizado em um termociclador Gene Amp®
PCR System 9700 (Applied Biosystems), onde os perfis térmicos da reação seguiram as
seguintes condições: Uma etapa inicial de desnaturação a 94°C por 5 min, 40 ciclos de
desnaturação a 94°C por 1min, temperatura e tempo de anelamento de 55°C por 1min, e
extensão a 72°C por 1min; um ciclo final de extensão a 72°C por 10 min e uma etapa final a
4°C. A reação foi realizada com os oligonucleotídeos NRAS12/13-F e NRAS12/13-R, sendo
que as seqüências destes oligos são:
NRAS12/13-F: 5’-GAC TGA GTA CAA ACT GGT GG-3’ 20pb
NRAS12/13-R: 5’-TGC ATA ACT GAA TGT ATA CCC-3’ 21pb
A revelação da reação deste PCR foi feita através de gel de agarose 1,5% corado com
brometo de etídeo (Fig. 8).
1 2 3 4 5 6 7 8 9
Figura 8: Gel de agarose 1,5% corado com BE para amplificação do NRAS. (1) padrão de
peso molecular de 100pb; (2) controle positivo: amostra de LMMC; (3) controle negativo
(água e mix de PCR); (4-5,7-9) amostras de pacientes com leucemias com banda amplificada;
(6) amostra de paciente com leucemia cuja banda não região esperada não amplificou.

65
4.8. Denaturing high pressure liquid cromatography na detecção das mutações
do NRAS
A detecção de mutações no éxon 1 do NRAS foi executada através de PCRs
submetidos à desnaturação por denaturing high pressure liquid chromatography (dHPLC)
que usou um sistema de análise de fragmentos por ondas de DNA, sendo o aparelho equipado
com uma coluna DNASep HT.
Os amplicóns para a análise das mutações foram gerados pela combinação dos
nucleotídeos citados acima. Antes da análise por dHPLC, os produtos gerados pelo PCR
foram desnaturados a 95°C por 5 minutos e passaram por uma graduação de temperatura
reduzida até 50°C usando um termociclador. O objetivo foi a formação de heteroduplexes nas
amostras em heterozigose para um alelo mutante.
Os fragmentos gerados pela PCR (40,0µL por amostra) foram injetados na coluna de
DNASep HT para a análise. Os produtos foram eluídos em um taxa de fluxo constante de
1,5mL/min com uma gradiente linear de acetonitrila determinado pelo programa
computacional Navigator (Transgenomic) baseado no tamanho e no conteúdo GC dos
amplicons. O gradiente foi produzido combinando o tampão acetato de 0,10 M (TEAA, pH 7)
(Transgenomic) e o tampão B (0,10 M TEAA com 25% acetonitrila) (Transgenomic).
Os perfis de eluição dos fragmentos de DNA, monitorados pelo sistema UV de
detecção, foram usados para gerar cromatogramas. Os picos dos homo e dos heteroduplexes
foram detectados entre o pico de injeção inicial, produzido por nucleotídeos residuais e por
oligonucleotídeos em reação, e pelo estágio de lavagem (pico de saída). Os perfis de
anelamento para os produtos do PCR (de 241 pares de bases) foram construídos usando o
programa computacional Navigator.

66
Padrão de análise: As amostras com a sequência selvagem eluiram como um único
pico (Figura 9a), enquanto picos múltiplos indicativos de heteroduplexes apareceram nas
amostras heterozigotas mutadas (Figura 9b).
(a)
(b)
Figura 9: (a) Cromatograma do controle negativo utilizando a linhagem celular HL60, com a
temperatura de formação de heteroduplex padronizada em 60.2°C como ótima para a
cromatografia por dHPLC.
(b) Cromatograma do controle positivo, paciente com LMMC e mutação no gene
NRAS com temperatura de 60.2°C.

67
4.9. Análise estatística
As análises estatísticas foram realizadas inicialmente com testes de freqüência,
buscando a significância estatística entre os diferentes grupos estudados, calculados utilizando
o teste do qui-quadrado (pearson) e o teste exato de Fisher. A associação entre a exposição às
variáveis ambientais e as LALs durante os períodos antes, durante e após a gravidez foi
realizada por o Statistical Package for the Social Sciences version 13 (SPSS, Chicago, IL).
Algumas análises foram ajustadas por sexo, idade do lactente, leucometria, diagnóstico da
LAL, se possuía rearranjo no MLL e mutação no FLT3. As análises do RAS foram ajustadas
por idade materna e por peso da criança ao nascer. Os resultados foram expressos como razão
de chances (odds ratios,OR) com intervalos de confiança de 95% (IC 95%), e todos os P
valores são bilaterais. A associação entre as mutações no RAS e as variáveis selecionadas
foram certificadas usando ORs e IC 95% ajustado para as variáveis previamente mencionadas.

68
5. RESULTADOS

69
5.1. ANÁLISES DE FREQUÊNCIAS
5.1.1. Características clínico-laboratoriais das amostras
No presente estudo, foram analisados 138 casos de leucemias agudas infantis dos quais
102 eram LLAs (73,9%), 36 eram LMAs (26,1%). As diferenças de idade variaram de 1 mês
a 24 meses com uma média de idade de 11,5 meses; com predominância do sexo masculino
56,5% e 54,3% com a cor da pele definida como branca (Tabela 2). A respeito da contagem
leucocitária verificamos que 48,5% dos casos eram hiperleucocitários, sendo igual ou superior
a 50x109/l. Também foi possível observar que a média do peso ao nascer dos pacientes foi de
3,400Kg.
Neste estudo, 125 casos foram previamente analisados por FISH e RT-PCR quanto à
presença do rearranjo no MLL, destes 55 foram positivos (39,9%) e 70 foram negativos
(50,7%). As leucemias agudas sem rearranjos no MLL apresentavam outras alterações
adquiridas como a fusão gênica TEL/AML1 ou hiperdiploidia nos casos de LLA e de
AML1/ETO nos casos de LMA.
O rastreamento em busca de mutações no gene FLT3 foi realizado previamente em 80
amostras do nosso estudo. Nestas amostras foram identificadas mutações no FLT3 em 4
(2,9%) dos 80 casos.

70
Tabela 2: Principais características demográficas e clínicas das leucemias infantis, Brasil,
1998-2008
Características das LALs Grupo Total
n (%)
Idade 0-12 76 (55,1) 13-24 62 (44,9) Sexo Feminino 60 (43,5) Masculino 78 (56,5) Cor da pele Branca 75 (54,3) Não-branca 49 (35,5) Sem informação 14 (10,2) Peso ao nascer (gr) ≤2500 7 (5,0) 2501-3499 57 (41,3) ≥3500 43 (31,2) Sem informação 31 (22,5) Leucometria (x109/l) <50 64 (46,4) ≥50 67 (48,5) Sem informação 7 (5,1) Diagnóstico de leucemia aguda Leucemia linfoblástica aguda 102 (73,9) Leucemia mielóide aguda 36 (26,1) MLL Positivo 55 (39,9) Negativo 70 (50,7) Sem informação 13 (9,4) FLT3 Mutado 4 (2,9) Selvagem 76 (55,1) Sem informação 58 (42,0) Total 138 (100)
5.2. Rastreamento de mutações no gene KRAS
No grupo total, dentre 138 casos de leucemias, 36 (26,1%) apresentaram mutações no
KRAS conforme ilustrado na Figura 10. Ressaltamos que entre os casos que foram definidos

71
como do tipo selvagens, 21 (15,2%), só puderam ser analisados para o códon 12; 81 (58,7%)
restantes foram considerados selvagens após rastreamento de mutações nos 2 códons (12 e
13).
Figura 10: Gráfico dos resultados do KRAS demonstrando os resultados percentuais da
análise. MUT= casos mutados; WT= casos selvagens.
5.2.1. Características clínicas das amostras com as alterações genéticas no gene KRAS
Analisamos as freqüências de mutações KRAS nas leucemias agudas e as variáveis
demográficas. Conforme a Tabela 3, observamos a presença de mutação no KRAS em 79,4%
(n=27) de LLAs e 20,6% (n=7) em LMAs, com percentuais diferentes, porém sem valor
estatisticamente significativos. Da mesma forma, não houve diferenças estatisticamente
significativas nos casos com mutação entre lactentes e crianças com idade superior a 12 meses
de idade. O sexo feminino parece ter uma maior tendência de mutações do KRAS [OR 1,64
CI (0,91-2,96)]. As demais variáveis tais como cor da pele, peso ao nascer e leucometria não
apresentaram valores estatisticamente significativos (Tabela 3).

72
Tabela 3: Análises das principais variáveis demográficas e status do KRAS em leucemias
agudas no Brasil
KRAS+ KRAS- OR (IC 95%)
Diagnóstico de LA
LLA (n=102) 27 (79,4%) 75 (72,1%)
LMA (n=36) 7 (20,6%) 29 (27,9%)
1,36 (0,65-2,85)
Idade
≤12 meses (n=76) 18 (52,9%) 58 (55,8%)
>12 meses (n=62) 16 (47,1%) 46 (44,2%)
0,92 (0,51-1,65)
Sexo
Feminino (n=60) 19 (55,9%) 41 (39,4%)
Masculino (n=78) 15 (44,1%) 63 (60,6%)
1,65 (0,92-2,96)
Raça
Brancos (n=75) 20 (58,8%) 55 (52,9%)
Não-Brancos (n=49) 14 (41,2%) 35 (33,6%)
Ignorados (n=14) 0 (0,0%) 14 (13,5%)
0,93 (0,52-1,66)
Peso ao nascer
≤2500gr (n=7) 0 (0,0%) 7 (6,7%)
2501-3499gr (n=57) 15 (44,1%) 42 (40,4%) 0,66 (0,25-1,72)
≥3500gr (n=43) 13 (38,2%) 30 (28,9%)
Ignorados (n=31) 6 (17,7%) 25 (24,0%)
Leucometria
<50x109/l (n=64) 16 (47,1%) 48 (46,2%)
≥50x109/l (n=67) 18 (52,9%) 49 (47,1%) 0,93 (0,52-1,66)
Ignorados (n=7) 0 (0,0%) 7 (6,7%)
Total (n=138) 34 (24,6%) 104 (75,4%)
OR= odds ratio; IC= intervalo de confiança

73
As análises moleculares foram realizadas com os marcadores mais importantes da
leucemogênese, como rearranjos no MLL e mutações no FLT3 (Tabela 4). Neste estudo, 125
casos foram analisados quanto à presença do rearranjo do MLL e 55 foram positivos (44,0%);
dentre as LALs com rearranjos no MLL, 16 casos (47,1%) apresentaram mutação no KRAS.
Em 4 casos com rearranjos do MLL foi identificada também, a presença da mutação do FLT3.
No entanto, em nenhum destes foi detectada mutação no KRAS, com possível tendência de
efeito protetor contra as duas mutações RAS e FLT3 concomitantemente (OR 0,76; IC 95%,
0,67-0,86) (Tabela 4).
Tabela 4: Análises das principais alterações moleculares e status do KRAS em leucemias
agudas no Brasil entre 1998-2008
KRAS+ KRAS- OR (IC 95%)
MLL
Positivo (n=55) 16 (47,1%) 39 (37,5%)
Negativo (n=70) 16 (47,1%) 54 (51,9%)
Ignorados (n=13) 2 (5,8%) 11 (10,6%)
1,27 (0,70-2,31)
FLT3
Mutado (n=4) 0 (0,0%) 4 (3,8%) 0,76 (0,67-0,86)
Selvagem (n=76) 18 (52,9%) 58 (55,8%)
Ignorados (n=58) 16 (47,1%) 42 (40,4%)
Total (n=138) 34 (24,6%) 104 (75,4%)
OR= odds ratio; IC= intervalo de confiança

74
5.3. Rastreamento de mutações no gene NRAS
Identificamos mutações no NRAS em 10 (15,2%) dos 65 casos (Fig. 11).
Figura 11: Gráfico representativo dos resultados percentuais demonstrando 15,2% de
mutações no NRAS
5.3.1. Características clínicas das amostras com as alterações genéticas no gene NRAS
Analisamos a relação entre as características clínicas das leucemias agudas e as mutações
no gene NRAS (Tabela 5). Encontramos 7 LLAs com mutação no gene NRAS (70,0%) e
dentre as LMAs 30,0% (n=3) apresentavam NRAS mutado. Crianças com idade inferior a 12
meses apresentaram uma freqüência maior de NRAS mutado (70,0%) quando comparados
com aqueles pacientes tinham com idade superior a 12 meses de idade (30,0%). Entretanto,
nenhuma das análises foi estatisticamente significativa.
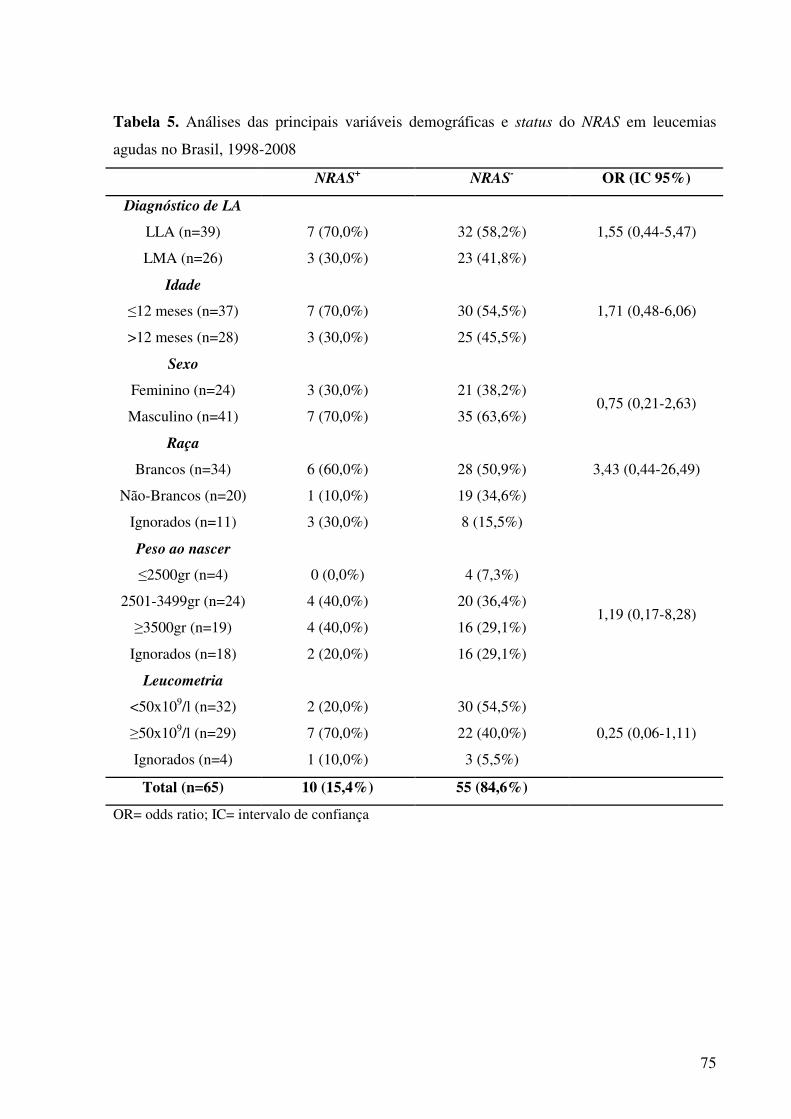
75
Tabela 5. Análises das principais variáveis demográficas e status do NRAS em leucemias
agudas no Brasil, 1998-2008
NRAS+ NRAS- OR (IC 95%)
Diagnóstico de LA
LLA (n=39) 7 (70,0%) 32 (58,2%) 1,55 (0,44-5,47)
LMA (n=26) 3 (30,0%) 23 (41,8%)
Idade
≤12 meses (n=37) 7 (70,0%) 30 (54,5%)
>12 meses (n=28) 3 (30,0%) 25 (45,5%)
1,71 (0,48-6,06)
Sexo
Feminino (n=24) 3 (30,0%) 21 (38,2%)
Masculino (n=41) 7 (70,0%) 35 (63,6%) 0,75 (0,21-2,63)
Raça
Brancos (n=34) 6 (60,0%) 28 (50,9%)
Não-Brancos (n=20) 1 (10,0%) 19 (34,6%)
Ignorados (n=11) 3 (30,0%) 8 (15,5%)
3,43 (0,44-26,49)
Peso ao nascer
≤2500gr (n=4) 0 (0,0%) 4 (7,3%)
2501-3499gr (n=24) 4 (40,0%) 20 (36,4%)
≥3500gr (n=19) 4 (40,0%) 16 (29,1%) 1,19 (0,17-8,28)
Ignorados (n=18) 2 (20,0%) 16 (29,1%)
Leucometria
<50x109/l (n=32) 2 (20,0%) 30 (54,5%)
≥50x109/l (n=29) 7 (70,0%) 22 (40,0%) 0,25 (0,06-1,11)
Ignorados (n=4) 1 (10,0%) 3 (5,5%)
Total (n=65) 10 (15,4%) 55 (84,6%)
OR= odds ratio; IC= intervalo de confiança
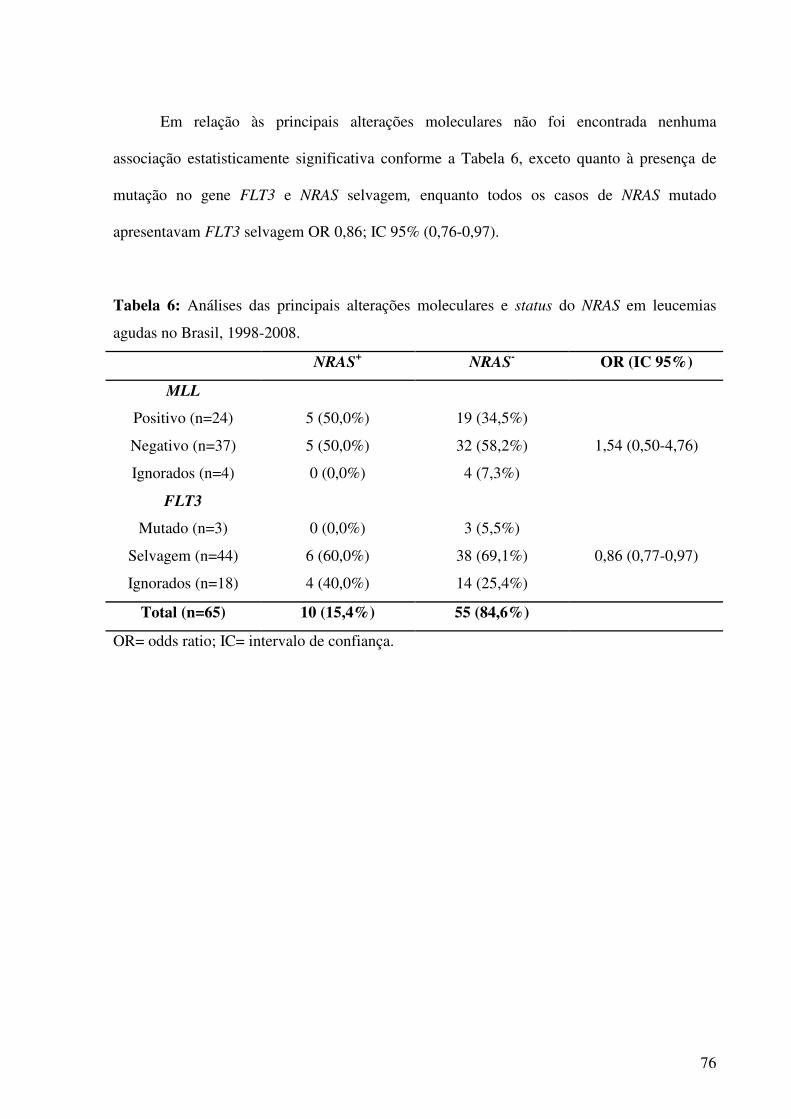
76
Em relação às principais alterações moleculares não foi encontrada nenhuma
associação estatisticamente significativa conforme a Tabela 6, exceto quanto à presença de
mutação no gene FLT3 e NRAS selvagem, enquanto todos os casos de NRAS mutado
apresentavam FLT3 selvagem OR 0,86; IC 95% (0,76-0,97).
Tabela 6: Análises das principais alterações moleculares e status do NRAS em leucemias
agudas no Brasil, 1998-2008.
NRAS+ NRAS- OR (IC 95%)
MLL
Positivo (n=24) 5 (50,0%) 19 (34,5%)
Negativo (n=37) 5 (50,0%) 32 (58,2%)
Ignorados (n=4) 0 (0,0%) 4 (7,3%)
1,54 (0,50-4,76)
FLT3
Mutado (n=3) 0 (0,0%) 3 (5,5%)
Selvagem (n=44) 6 (60,0%) 38 (69,1%) 0,86 (0,77-0,97)
Ignorados (n=18) 4 (40,0%) 14 (25,4%)
Total (n=65) 10 (15,4%) 55 (84,6%)
OR= odds ratio; IC= intervalo de confiança.

77
Tabela 7: Características clínicas e coexistência de alterações genéticas com os genes RAS (K e N) mutados e selvagens
KRAS + n(%)
KRAS - n(%)
p-valor*
NRAS+ n(%)
NRAS- n(%)
p-valor*
Idade (meses) 0-12 18 (52,9) 58 (55,8) 7 (70,0) 30 (54,5) 13-24 16 (47,1) 46 (44,2)
0,46 3 (30,0) 25 (45,5)
0,29
Sexo Feminino 19 (55,9) 41 (39,4) 3 (30,0) 21 (38,2) Masculino 15 (44,1) 63 (60,6)
0,07 7 (70,0) 35 (63,6)
0,46
Peso ao nascer (gr) ≤2500 0 (0,0) 7 (6,7) 0 (0,0) 4 (7,3) 2501-3499 15 (44,1) 42 (40,4) 4 (40,0) 20 (36,4) ≥3500 13 (38,2) 30 (28,9) 4 (40,0) 16 (29,1) Ignorados 6 (17,7) 25 (24,0)
0,31
2 (20,0) 16 (29,1)
0,69
Leucometria (x109/mm) <50 16 (47,1) 48 (46,2) 2 (20,0) 30 (54,5) ≥50 18 (52,9) 49 (47,1) 7 (70,0) 22 (40,0) Ignorados 0 (0,0) 7 (6,7)
0,29 1 (10,0) 3 (5,5)
0,05
Cor da pele Branca 20 (58,8) 55 (52,9) 6 (60,0) 28 (50,9) Não-branca 14 (41,2) 35 (33,6) 1 (10,0) 19 (34,6) Ignorados 0 (0,0) 14 (13,5)
0,07 3 (30,0) 8 (15,5)
0,23
Diagnóstico LA LLA 27 (79,4) 75 (72,1) 7 (70,0) 32 (58,2) LMA 7 (20,6) 29 (27,9)
0,27 3 (30,0) 23 (41,8)
0,37
MLL Positivo 16 (47,1) 39 (37,5) 5 (50,0) 19 (34,5) Negativo 16 (47,1) 54 (51,9) 5 (50,0) 32 (58,2) Ignorados 2 (5,8) 11 (10,6)
0,52 0 (0,0) 4 (7,3)
0,50
FLT3 status Mutado 0 (0,0) 4 (3,8) 0 (0,0) 3 (5,5) Selvagem 18 (52,9) 58 (55,8) 6 (60,0) 38 (69,1) Ignorados 16 (47,1) 42 (40,4)
0,45 4 (40,0) 1 (25,4)
0,52
Mãe fumante Sim 9 (26,5) 23 (22,1) 2 (20,0) 12 (21,8) Não 20 (58,8) 59 (56,7) 6 (60,0) 29 (52,7) Ignorados 5 (14,7) 22 (21,2)
0,68 2 (20,0) 14 (25,5)
0,91
Fumou 3 meses antes da gestação
Sim 5 (14,7) 17 (16,3) 2 (20,0) 7 (12,7) Não 24 (70,6) 65 (62,5) 6 (60,0) 34 (61,8) Ignorados 5 (14,7) 22 (21,2)
0,66 2 (20,0) 14 (25,5)
0,81
Fumou durante a gestação Sim 3 (8,8) 10 (9,6) 1 (10,0) 4 (7,3) Não 26 (76,5) 72 (69,2) 7 (70,0) 37 (67,3) Ignorados 5 (14,7) 22 (21,2)
0,69 2 (20,0) 14 (25,4)
0,91
Pai fumante Sim 3 (2,9) 1 (2,0) 0 (0,0) 4 (7,3) Não 26 (82,4) 77 (74,0) 7 (70,0) 36 (65,4) Ignorados 5 (14,7) 25 (24,0)
0,11 3 (30,0) 15 (27,3)
0,68
Fumou 3 meses antes da gestação
Sim 1 (2,9) 1 (2,0) 0 (0,0) 2 (3,6) Não 28 (82,4) 77 (74,0) 7 (70,0) 37 (67,3) Ignorados 5 (14,7) 25 (24,0)
0,34 3 (30) 16 (29,1)
0,83
Fumantes em casa durante a gestação
Sim 19 (55,9) 33 (31,7) 4 (40,0) 17 (30,9) Não 9 (26,5) 47 (45,2) 4 (40,0) 22 (40,0) Ignorados 6 (17,6) 24 (23,1)
0,02 2 (20,0) 16 (29,1)
0,79
Total 34 (24,6) 104 (75,4) 10 (15,4) 55 (84,6)

78
5.4. Análises epidemiológicas com as características demográficas e hábito de fumar na
família de crianças com leucemias em lactentes
De acordo com os dados extraídos dos questionários maternos, a maioria das crianças
desta série de casos são provenientes de regiões urbanas, principalmente de cidades com
população superior a 1 milhão de pessoas (tabela 8). A maioria das mães tem distribuição de
idade entre 18 e 30 anos de idade. O nível socioeconômico predominante foi de casos
oriundos de classe média-baixa, cujas mães apresentavam um baixo índice de escolaridade.
Tabela 8: Principais características demográficas das mães de crianças com leucemias de
lactentes no Brasil, entre 1998-2008*
n(%)
Idade materna
<18 3 (2,14)
18-30 92 (65,72)
>31 45 (32,14)
Densidade populacional
<40.000-99.999 23 (16,43)
100.000-499.000 43 (30,71)
500.000-1.000.000 16 (11,43)
>1.000.000 58 (41,43)
Educação Materna (anos)
<4 72 (51,43)
5-9 51 (36,43)
>10 17 (12,14)
Renda Familiar (R$ por mês)
<400,00 69 (49,29)
500,00-999,00 35 (25,00)
1.000,00-1.999,00 24 (17,14)
>2.000,00 12 (8,57)
*As variáveis utilizadas são do estudo original tipo caso-controle (Pombo-de-Oliveira e cols., 2006)

79
De acordo com os questionários maternos, 32 mães de crianças com leucemias
(28,8%) eram fumantes, das quais 9 (26,5%) tiveram filhos com mutação no gene KRAS
(tabela 9); 22 mães relataram que só fumaram 3 meses antes da gestação. Não foi encontrada
nenhuma associação de risco entre o hábito materno de fumar e LAL com mutação no KRAS.
Em relação à exposição materna ao tabaco, pais fumantes ou outros familiares
fumantes na residência apresentaram associações de riscos (Tabela 9) estatisticamente
significativas OR 2,27 IC 95% (1,13-4,56). Os mesmos resultados foram observados em
relação às mutações encontradas no NRAS, com menor magnitude das análises de risco OR
1,20, IC 95% (1,04-1,35) (Tabela 10).

80
Tabela 9: Relação entre o status do KRAS nas crianças com leucemias e o hábito de fumar na
família
KRAS+ KRAS- OR (IC 95%)
Mãe fumante
Sim (n=32) 9 (26,5%) 23 (22,1%)
Não (n=79) 20 (58,8%) 59 (56,7%)
Ignorados (n=27) 5 (14,7%) 22 (21,2%)
1,11 (0,57-2,17)
Mãe fumou 3 meses antes da gestação
Sim (n=22) 5 (14,7%) 17 (16,3%)
Não (n=89) 24 (70,6%) 65 (62,5%)
Ignorados (n=27) 5 (14,7%) 22 (21,2%)
0,84 (0,36-1,96)
Mãe fumou durante a gravidez
Sim (n=13) 3 (8,8%) 10 (9,6%)
Não (n=98) 26 (76,5%) 72 (69,2%)
Ignorados (n=27) 5 (14,7%) 22 (21,2%)
0,87 (0,31-2,47)
Pai fumante
Sim (n=5) 3 (8,8%) 2 (2,0%)
Não (n=103) 26 (76,5%) 77 (74,0%)
Ignorados (n=30) 5 (14,7%) 25 (24,0%)
2,38 (1,08-5,23)
Pai fumou 3 meses antes da gestação
Sim (n=2) 1 (2,9%) 1 (1,0%)
Não (n=105) 28 (82,4%) 77 (74,0%)
Ignorados (n=31) 5 (14,7%) 26 (25,0%)
1,87 (0,45-7,77)
Fumantes em casa
Sim (n=52) 19 (55,9%) 33 (31,7%) 2,27 (1,13-4,57)
Não (n=56) 9 (26,5%) 47 (45,2%)
Ignorados (n=30) 6 (17,6%) 24 (23,1%)
Total (n=138) 34 (24,6%) 104 (75,4%)
OR= odds ratio; IC= intervalo de confiança.

81
Tabela 10: Relação entre o status do NRAS nas leucemias e o hábito de fumar na família.
NRAS+ NRAS- OR (IC 95%)
Mãe fumante
Sim (n=14) 2 (20,0%) 12 (21,8%)
Não (n=35) 6 (60,0%) 29 (52,7%)
Ignorados (n=18) 2 (20,0%) 14 (25,5%)
0,83 (0,19-3,64)
Mãe fumou 3 meses antes da gestação
Sim (n=9) 2 (20,0%) 7 (12,7%)
Não (n=40) 6 (60,0%) 34 (61,8%)
Ignorados (n=16) 2 (20,0%) 14 (25,5%)
1,48 (0,35-6,18)
Mãe fumou durante a gravidez
Sim (n=5) 1 (10,0%) 4 (7,3%)
Não (n=44) 7 (70,0%) 37 (67,3%)
Ignorados (n=16) 2 (20,0%) 14 (25,5%)
1,26 (0,19-8,24)
Pai fumante
Sim (n=4) 0 (0,0%) 4 (7,3%)
Não (n=43) 7 (70,0%) 36 (65,4%)
Ignorados (n=18) 3 (30,0%) 15 (27,3%)
1,19 (1,05-1,36)
Pai fumou 3 meses antes da gestação
Sim (n=2) 0 (0,0%) 2 (3,6%)
Não (n=44) 7 (70,0%) 37 (67,3%)
Ignorados (n=) 3 (30,0%) 16 (29,1%)
1,19 (1,05-1,35)
Fumantes em casa
Sim (n=21) 4 (40,0%) 17 (30,9%) 1,24 (0,35-4,37)
Não (n=26) 4 (40,0%) 22 (40,0%)
Ignorados (n=) 2 (20,0%) 16 (29,1%)
Total (n=65) 10 (15,4%) 55 (84,6%)
OR= odds ratio; IC= intervalo de confiança.
Na tabela 11 mostramos os casos mutados no KRAS e NRAS, cada um deles descritos
de acordo com a idade, o diagnóstico da leucemia e seu subtipo, as alterações adicionais (MLL
ou FLT3) e qual o tipo de exposição ao fumo, seja pela mãe, pelo pai ou pelos fumantes em
casa.
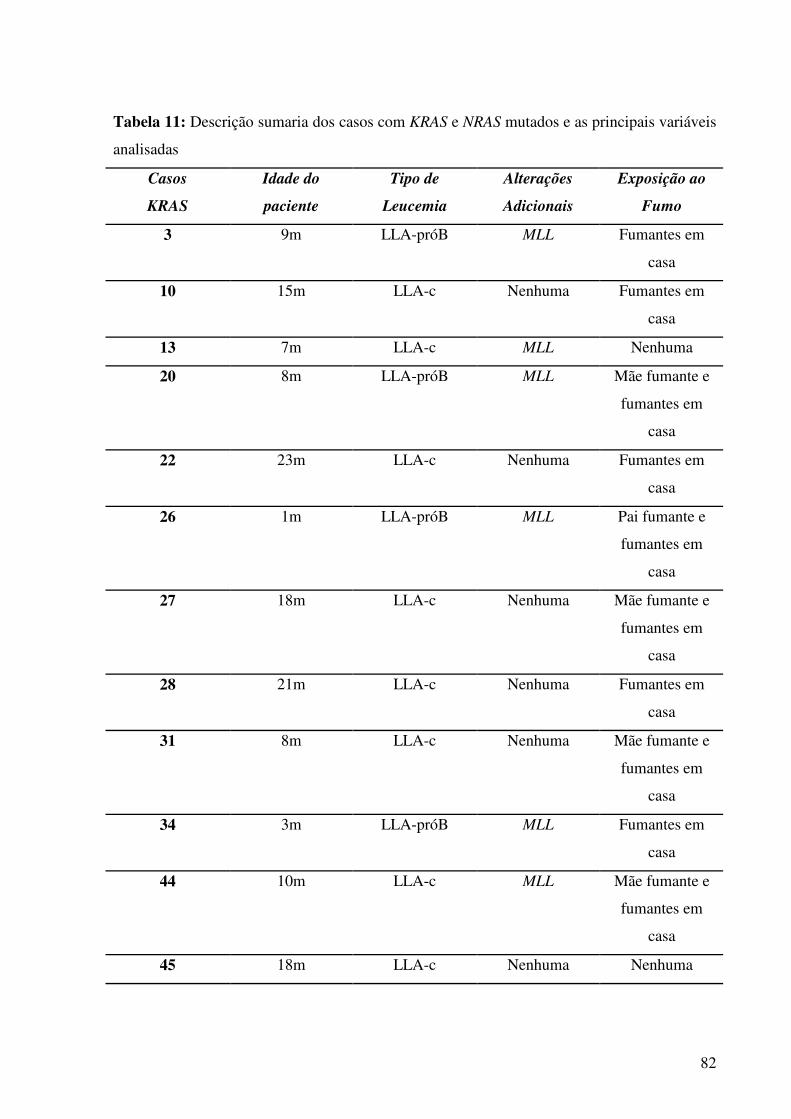
82
Tabela 11: Descrição sumaria dos casos com KRAS e NRAS mutados e as principais variáveis
analisadas
Casos
KRAS
Idade do
paciente
Tipo de
Leucemia
Alterações
Adicionais
Exposição ao
Fumo
3 9m LLA-próB MLL Fumantes em
casa
10 15m LLA-c Nenhuma Fumantes em
casa
13 7m LLA-c MLL Nenhuma
20 8m LLA-próB MLL Mãe fumante e
fumantes em
casa
22 23m LLA-c Nenhuma Fumantes em
casa
26 1m LLA-próB MLL Pai fumante e
fumantes em
casa
27 18m LLA-c Nenhuma Mãe fumante e
fumantes em
casa
28 21m LLA-c Nenhuma Fumantes em
casa
31 8m LLA-c Nenhuma Mãe fumante e
fumantes em
casa
34 3m LLA-próB MLL Fumantes em
casa
44 10m LLA-c MLL Mãe fumante e
fumantes em
casa
45 18m LLA-c Nenhuma Nenhuma

83
50 7m LLA-próB MLL Mãe fumante e
fumantes em
casa
61 16m LLA-préB Nenhuma Nenhuma
63 5m LLA-próB MLL Fumantes em
casa
64 7m LMA-M4 MLL Pais fumantes e
fumantes em
casa
65 8m LLA-próB MLL Fumantes em
casa
70 19m LLA-c Sem informação Fumantes em
casa
78 19m LMA-M5 Nenhuma Fumantes em
casa
54 17m LMA-M4 MLL Nenhuma
81 16m LMA-M4 Nenhuma Mãe fumante
86 11m LMA-M7 MLL Sem informação
101 21m LLA-c Nenhuma Sem informação
109 14m LLA-c Nenhuma Mãe fumante e
fumantes em
casa
117 2m LLA-próB MLL Nenhuma
121 9m LLA-próB MLL Sem informação
122 5m LLA-próB MLL Sem informação
128 2m LLA-próB Nenhuma Sem informação
129 23m LLA-próB Nenhuma Mãe fumante
131 10m LMA-M4 Nenhuma Nenhuma
132 0.1m (3 dias) LLA-c Nenhuma Fumantes em
casa
133 23m LLA-próB Nenhuma Nenhuma

84
139 22m LLA Nenhuma Fumantes em
casa
136 17m LMA Sem informação Nenhuma
Casos NRAS
37 9m LLA-c MLL Fumantes em casa
51 7m LLA-c Nenhuma Nenhuma 52 5m LLA-próB MLL Fumantes em
casa 55 2m LLA-próB MLL Nenhuma 94 12m LMA-M7 Nenhuma Nenhuma
105 6m LLA-próB MLL Fumantes em casa
109 14m LLA-c Nenhuma Mãe fumante e fumantes em
casa 119 8m LMA MLL Mãe fumante 123 18m LLA-c Nenhuma Sem informação 130 24m LMA-M5 Nenhuma Sem informação

85
6. DISCUSSÃO

86
Os fatores exógenos ou ambientais responsáveis pela leucemogênese ainda não foram
totalmente comprovados, porém há evidências epidemiológicas que demonstram que o
desenvolvimento de leucemias infantis está associado à predisposição hereditária, exposições
ambientais, bem como com eventos ao acaso. A ocorrência de alterações genéticas, que
envolvam genes chave para o sistema hematopoético está diretamente associada a estes
fatores (Greaves, 2002). Atualmente, estas alterações genéticas são identificadas como
marcadores importantes no diagnóstico, para o conhecimento da etiopatogênese da leucemia,
e para análises de sobrevida pós-tratamento.
Mutações gênicas, cuja forma mais comum são as mutações pontuais, são altamente
prevalentes em diversos tipos de neoplasias. As mutações no gene RAS foram identificadas
em aproximadamente 20% dos pacientes com LLA e em aproximadamente 30% dos pacientes
com LMA (Browett e Norton, 1989; Barletta e cols., 2004). Portanto, resultados encontrados
no nosso estudo coincidem na freqüência de mutações no RAS nas LMAs que se apresentaram
em aproximadamente 25-30% dos casos.
Estudos realizados mostram que diferentemente de outros cânceres humanos, onde
predominam as mutações no RAS, as que ocorrem no NRAS são as mais freqüentes nas
neoplasias hematológicas, principalmente nas LMAs (Ritter e cols., 2004; Bowen e cols.,
2005; Usher e cols., 2009). Um estudo realizado por Clementino e cols. em 2001, analisou
101 DNAs de pacientes com menos de 15 anos de idade, diagnosticados com LLA. Utilizando
as técnicas de SSCP e dot blot, eles detectaram 7% (7/101) mutações no gene NRAS neste
grupo de crianças brasileiras com LLA. Porém, a presença da mutação não foi associada
estatisticamente à idade do paciente, sexo, leucometria ou status nutricional. Nossos
resultados apresentam uma predominância de mutações no NRAS (30,0% nas LMAs) em
comparação às mutações no KRAS (20,6% nas LMAs). Uma análise crítica destes resultados

87
deve levar em consideração o pequeno número de amostras, as características biológicas dos
casos e o desenho do estudo onde foram selecionadas as amostras.
Outro aspecto para a análise crítica seria a escolha das técnicas para as análises
mutacionais disponíveis com capacidade de identificação precisa de mutações pontuais em
clones leucêmicos. Entre estas técnicas se destacam, para o rastreamento genético, a utilização
do single strand PCR polymorfism, RFLP, dHPLC e sequenciamento direto. Todas estas
técnicas possuem vantagens e desvantagens. Para estudos com um grande tamanho amostral
se preconiza a combinação das diferentes técnicas, iniciando-se com aquela com maior
sensibilidade, seguida de técnica mais especificas e confirmatórias dos resultados.
O RFLP é uma técnica com comprovadas aplicações bem sucedidas, uma técnica
rápida, de baixa complexidade e baixo custo, porém, apresenta algumas desvantagens tais
como a necessidade de DNA relativamente puro.
A técnica por dHPLC é uma ferramenta que vem sendo utilizada como um método de
rastreamento altamente automatizado para a detecção de novos polimorfismos. O principio
científico da técnica se baseia na retenção diferencial de homo- e heteroduplexes de DNA por
cromatografia por pares de íons sob condições de desnaturação parcial. As moléculas
heteroduplex são geradas em amostras heterozigóticas quando as fitas simples, do DNA
homólogo, carregam uma mutação pontual ou pequenos re-anelamentos de inserções ou
deleções. Sob condições de desnaturação parcial, as moléculas heteroduplex geralmente são
eluidas à frente das homoduplex, produzindo pelo menos um pico adicional. Os perfis de
eluição de tais amostras são diferentes daqueles que possuem seqüências homozigóticas,
fazendo com que a identificação das amostras, abrangendo polimorfismos ou mutações, um
procedimento fácil e direto. Os mutantes homozigotos podem ser detectados pela adição de
uma quantidade aproximadamente igual de DNA selvagem às espécies mutantes. Estudos

88
prévios mostraram que o dHPLC não é só uma ferramenta que proporciona uma metodologia
altamente sensitiva e automatizada que pode ser usada como uma técnica de rastreamento
poderosa para a detecção de polimorfismos (Oefner e Underhill, 1995; Liu e cols., 1998;
Schaeffeler e cols., 2001), mas também pode ser utilizada para genotipagem (Underhill e
cols., 1997; Gross e cols., 2000; Kaler e cols., 2000). A sensibilidade do dHPLC depende das
condições HPLC utilizadas para a análise. Outro passo crítico é a análise dos perfis de
eluição. Para uma máxima sensibilidade, qualquer desvio do perfil de eluição das amostras
controles deve ser considerado. A sensibilidade da dHPLC tem sido investigada em vários
estudos, com alguns deles apresentando precisão para detecção de mutações de até 100%.
Porém, em alguns casos diferentes, as mutações renderam perfis de eluição similares, o que
levou a uma má interpretação (Gross e cols., 2000; Spiegelman e cols., 2000).
Case e cols. realizaram um estudo em 2008 onde 86 casos de LLA foram analisados
com o dHPLC. A incidencia de mutações de NRAS e KRAS identificada neste estudo, ao
diagnóstico, foi de 31% e está considerada como a mais elevada quando comparada com
outros estudos e certamente é devido à sensibilidade aumentada da metodologia do dHPLC
(Case e cols., 2008).
Com estas considerações teóricas e análises críticas quanto às escolhas de ferramentas
metodológicas, no nosso estudo realizamos inicialmente uma comparação com os métodos já
usados para genotipagem e bem estabelecidos como RFLP. Nós concluímos que a dHPLC,
mostrou ser uma técnica menos laboriosa, mais rápida e com alta sensibilidade nos resultados
de rastreamento. Porém, a alta sensibilidade desta ferramenta pode criar desvantagens como
fragmento pequeno que não necessariamente é uma anomalia do DNA, por este motivo é
aconselhável complementar esta metodologia com o RFLP ou sequenciamento direto.
Portanto, acreditamos que os resultados globais de frequência de mutações nos K- e NRAS em

89
diferentes subtipos de leucemias agudas de lactentes, no nosso estudo, não foram afetados por
possíveis problemas técnicos.
Estudos prévios sustentam a hipótese de que as leucemias agudas em lactentes, com
rearranjos MLL podem ser causadas pelas exposições transplacentários de substâncias lesivas
ao DNA (Alexander e cols., 2001; Spector e cols., 2005; Pombo-de-Oliveira e cols., 2006).
Porém, o nosso estudo é um dos primeiros a relacionar as mutações do RAS com as leucemias
agudas infantis com rearranjos MLL e com as variáveis epidemiológicas numa série de
leucemias.
As hipóteses sobre os mecanismos da leucemogênese nas LALs especulam que o feto
em desenvolvimento é mais sensível aos efeitos de danos ao DNA durante o início da
gravidez. A fusão gênica resultante de translocações cromossômicas aleatórias parece ser o
primeiro evento na leucemogênese (Greaves, 1997; Greaves e Wiemels, 2003). Há evidências
de que os rearranjos recíprocos do MLL são comuns nas LALs e de que possivelmente podem
ter sido originadas como efeitos das exposições a substâncias inibidoras de topoisomerase II
que atravessaram a barreira transplacentária (Alexander e cols, 2001; Pombo-de-Oliveira e
cols., 2006). Sendo assim, é importante buscar o entendimento de como estas fusões anômalas
podem ter sido causadas.
De acordo com a teoria de Knudson (Knudson, 1975) um segundo evento deve ocorrer
para o desenvolvimento de tumores de origem embrionária, por isso, nós especulamos que as
mutações no RAS podem favorecer ao possível “acúmulo de eventos” na cadeia biológica de
eventos ao acaso relacionados às LALs. Mahgoub e cols., em um estudo realizado em 1998,
analisaram 32 leucemias pediátricas que previamente apresentaram rearranjos no MLL e não
encontraram evidências de interação funcional entre estes genes. Neste estudo 23 de 25 casos
mostraram padrões normais de KRAS e NRAS. No entanto, as nossas análises não mostraram

90
uma relação estatisticamente significativa, com o risco relativo entre as mutações no RAS e os
rearranjos MLL, onde 47,1% dos casos com mutação no KRAS possuíam rearranjos no MLL
(OR 1,27 IC 95% 0,70-2,31), e nos casos com mutação no NRAS 50,0% possuíam rearranjos
do MLL (OR 1,54 IC 95%, 0,50-4,76). Em ambas circunstâncias, as análises não foram
estatisticamente significativas. Por isso, estudos adicionais devem ser realizados para
determinar se podem existir modelos adicionais da ativação da via do RAS nos casos com
rearranjos MLL sem mutações no RAS. Por exemplo, nas leucemias mielóides crônicas
(LMC), onde as mutações no RAS geralmente estão ausentes, a via de sinalização RAS é
desregulada pela proteína de fusão BCR-ABL, que induz a um aumento nos níveis de GTP-
ligante ativa (Pendergast e cols., 1993; Mahgoub e cols., 1998). A neurofibrina, produto do
NF1, é uma proteína ativadora de GTPase que acelera a hidrolise GTP nas proteínas RAS
(Shannon e cols., 1994). Assim, a transformação leucêmica não é independente da via RAS.
Assim como na presença de rearranjos no MLL nas LALs, um estudo experimental
com cobaia realizado recentemente (Usher e cols., 2009) sugere que o RAS pode sim agir
como um evento inicial ou secundário numa neoplasia. Este estudo tentou relacionar o
acúmulo de efeitos das mutações do FLT3 e das mutações no RAS, como fatores
correlacionados com as leucemias agudas. Estas mutações podem ocorrer de maneira
independente, embora não se deve descartar a possibilidade de interações fracas adversas
entre o FLT3 mutante e o RAS mutante pela associação das mesmas na transdução de sinais
aberrantes. Não encontramos nenhum caso com concomitância das duas mutações em FLT3 e
RAS.
Um dado clínico que apresentou uma relação estatisticamente significativa foi a
leucometria elevada. Esta relação significativa com a mutação no NRAS contrasta com dados
encontrados na literatura, onde pacientes com mutações no NRAS apresentaram uma média de

91
leucometria de 28,0x109/l nos pacientes sem mutações NRAS (P<0,001) (Bacher e cols.,
2006). Porém, sabe-se que os lactentes com leucemias agudas se caracterizam por uma
contagem leucocitaria mediana de ≥50x109/l, sendo assim nossos dados de NRAS não fogem
do denominados comum das LALs e sim os do KRAS. Resta então saber se as mutações no
RAS em lactentes podem ser associadas ao prognóstico e a resposta terapêutica.
Nosso estudo foi totalmente motivado por questões referentes aos fatores de riscos
associados à exposição materna a substâncias carcinógenos que estão associadas ao RAS.
Apesar dos mecanismos específicos das mutações do RAS ainda serem desconhecidos,
estudos epidemiológicos em humanos e em animais sugerem que mutações pontuais
específicas nos genes RAS podem ser induzidas também por certos carcinógenos químicos
(Barletta e cols., 2004). Entre estes carcinógenos se encontra o cigarro que possui vários
agentes mutagênicos tais como hidrocarbonetos aromáticos policíclicos e nitrosaminas. No
entanto, os diversos estudos epidemiológicos realizados tentando demonstrar à associação
entre o hábito de fumar dos pais e as leucemias infantis encontraram resultados inconsistentes.
Por exemplo, um grande estudo caso-controle realizado nos Estados Unidos analisou 1842
casos de LLA e 517 casos de LMA através de entrevistas por telefone, e seus controles foram
questionados sobre seus hábitos de fumar no período antes, durante e depois da gravidez. O
risco de desenvolver leucemia foi estudado de acordo com o tipo histológico, a idade da
criança ao diagnostico e imunofenótipo. Este estudo mostrou que o risco de LLA não estava
associado com o fato do pai sequer ter fumado alguma vez (OR 1,04 IC 95% 0,90-1,20) ou a
mãe sequer ter fumado alguma vez (OR 1,04 IC 95% 0,91-1,19). Da mesma forma, nenhum
risco significativo de LMA foi observado para o hábito de fumar do pai (OR 0,88 IC 95%
0,67-1,16) ou da mãe (OR 0,95 IC 95% 0,74-1,22). O risco relativo de leucemia não foi
diferentemente significativo do nulo para o hábito de fumar dos pais em qualquer período

92
durante ou ao redor da gravidez, nem foi relacionado ao numero de cigarros, anos de fumo, ou
numero de maços por ano. Eles concluíram que hábito de fumar dos pais durante a gravidez
ou a exposição ao fumo por um curto período após o nascimento aparentemente não contribui
de maneira substancial no risco de desenvolvimento de leucemias pediátricas (Brondum e
cols., 1999).
Em contraste, um estudo caso-controle realizado na China em 1997 por Ji e cols.,
investigou a relação do hábito de fumar do pai, particularmente no período pré-concepção,
com cânceres infantis entre a descendência das mães que não tinham o hábito de fumar. Esta
pesquisa surgiu do fato que o hábito de fumar pode aumentar o dano oxidativo do DNA nas
células espermáticas humanas e de que a maioria dos estudos de cânceres infantis se foca
primeiramente no hábito de fumar da mãe. O estudo incluiu 642 casos de crianças com
neoplasias e seu controle, sendo que a informação relativa ao hábito de fumar foi obtida
diretamente com os pais. Foi concluído que o hábito de fumar dos pais antes da concepção
estava relacionado ao risco elevado de desenvolvimento de cânceres em crianças,
particularmente nas leucemias agudas e linfomas. Comparadas com as crianças cujos pais
jamais fumaram, as crianças com pais que tinham o hábito de fumar mais de 5 maços de
cigarro por ano antes da sua concepção mostraram um risco relativo (OR 3,8 IC 95% 1,3-
12,3) para LLAs. Os riscos estatisticamente significativos foram restritos às crianças com <5
anos de idade ao diagnostico, cujos pais fumaram durante todo o período de 5 anos até a
concepção (Ji e cols., 1997).
Recentemente num estudo do tipo caso-controle realizado nos Estados Unidos por
Chang e cols., foram avaliados 327 casos de leucemias agudas infantis (281 LLAs e 46
LMAs) e eles demonstraram que o hábito de fumar das mães não estava associado ao risco
elevado de desenvolver leucemias, seja LLAs ou LMAS. Já o hábito de fumar do pai antes da

93
concepção foi associado significativamente a um aumento no risco de LMA (OR 3,84 IC 95%
1,04-14,17). Estes resultados sugeriram que a exposição ao fumo paterno antes da concepção
e/ou o fumo passivo pós-natal pode ser importante no risco de leucemias infantis (Chang e
cols., 2006). Um estudo prévio realizado por Shu e cols. (Shu e cols.,1996) mostrou que o
hábito de fumar do pai um mês antes da gravidez também estava associado a um aumento no
risco (OR 1,56 IC 95% 1,03-2,36) nas LLAs em lactentes.
Wiemels e cols. realizaram um estudo em 2005, onde foram analisadas 161 amostras
de leucemias agudas pediátricas que possuíam aspirados de MO criopreservados obtidos do
serviço clínico que tinha previamente diagnosticado o caso. As características demográficas e
a informação sobre o hábito de fumar dos pais foram providenciadas pelas mães dos casos
(97,5%) ou pelos pais (2,5%) através de entrevistas pessoais na casa da família. As
informações do hábito de fumar dos pais foram estratificadas de acordo com o período deste
hábito tanto no pai quanto na mãe. As mutações no RAS foram relacionadas com as
informações dos pais que fumaram 3 meses antes da gravidez [12% (4/191) (P=0,13)], as
mães que fumaram 3 meses antes da gravidez (19% (4/191) (P=0,89)] e as mães que fumaram
durante a gravidez [15% (2/191) (P=0,65)]. Com estes resultados foi sugerido que não há uma
prevalência elevada do hábito de fumar dos pais entre os casos positivos para as mutações
RAS quando comparados aos casos não mutados no RAS. Também, químicos mutagênicos
provenientes do cigarro do hábito de fumar das mães atravessam a placenta levando a um
possível efeito do hábito de fumar parental no risco de leucemia pediátrica (Wiemels e cols.,
2005).
Corroborando com estas investigações anteriores, nós observamos resultados
consistentes onde as leucemias de lactentes com mutações no KRAS estão associadas com o

94
hábito de fumar do pai, antes da gestação ou durante a gravidez. Enquanto o hábito de fumar
das mães não apresenta uma relação estatisticamente significativa.
Nossa interpretação é de que estes dados precisam ser re-avaliados em estudos mais
robustos, utilizando marcadores de aferição de produtos do metabolismo do cigarro nas mães,
já que as respostas a questionários podem ser respondidas sob um efeito de censura. Por
exemplo, uma informação bastante interessante é a relação estatisticamente significativa entre
as leucemias com mutações no RAS, especificamente no KRAS, e as mães que estiveram
expostas à fumaça de cigarro durante a gravidez devido à presença de fumantes em casa ou no
ambiente de trabalho [(19/138) (P=0,02)]. Além desta relação, observamos um risco relativo
bastante significativo para esta exposição e a aparição da mutação nas LALs (OR 2,27, 95%
1,13-4,567). Não encontramos estudos na literatura que mostrassem dados desta relação,
porém podemos justificar esta relação com uma possível sensibilidade das mães à fumaça do
cigarro e, como foi mostrado nos estudos anteriores, os agentes carcinógenos da fumaça
atravessam a placenta podendo ativar mutações in utero em certas vias, como a via da RAS,
levando a um possível risco de leucemia pediátrica.
Finalmente, as conclusões deste estudo levantam diversas questões sobre fatores
ambientais associados às leucemias de lactentes, e destaca o potencial papel interativo da
mutação do RAS nas leucemias, idade materna, leucometria e o hábito de fumar dos pais antes
e durante a gravidez, assim como a exposição materna a fumaça de cigarro em casa ou no
ambiente de trabalho. As associações significativas encontradas neste trabalho devem guiar o
desenho de estudos futuros, que tenham como objetivo principal desvendar as possíveis
causas das leucemias infantis.

95
7. CONCLUSÃO

96
Com este trabalho concluímos que as mutações no gene RAS apresentam potencial papel
interativo em:
• Diagnóstico de leucemias
• Leucometria
• Idade materna
• Hábito de fumar do pai (antes ou durante a gravidez)
• Exposição materna à fumaça do cigarro

97
8. REFERENCIAS BIBLIOGRÁFICAS

98
Abu-Duhier,F.M., Goodeve,A.C., Wilson,G.A., Care,R.S., Peake,I.R., Reilly,J.T. (2001)
Genomic structure of human FLT3: implications for mutational analysis. British Journal of
Haematology, 113(4), 1076-1067.
Abu-Duhier,F.M., Goodeve,A.C., Wilson,G.A., Carr,R.S., Peake,I.R., Reilly,J.T. (2002)
FLT3 internal tandem duplication mutations are rare in agnogenic myeloid metaplasia. Blood,
100(1), 364.
Alexander,F.E., Patheal,S.L., Biondi,A., Brandalise,S., Cabrera,M.E., Chan,L.C., Chen,Z.,
Cimino,G., Cordoba,J.C., Gu,L.J., Hussein,H., Ishii,E., Kamel,A.M., Labra,S.,
Magalhães,I.Q., Mizutani,S., Petridou,E., de Oliveira,M.P., Yuen,P., Wiemels,J.L.,
Greaves,M.F. (2001) Transplacental chemical exposure and risk of infant leukemia with MLL
gene fusion. Cancer Research, 61(6), 2542-2546.
Armstrong,S.A., Kung,A.L., Mabon,M.E. (2003) Inhibition of FLT3 in MLL. Validation of a
therapeutic target identified by gene expression based classification. Cancer Cells, 3, 173-
183.
Armstrong,S.A., Mabon,M.E., Silverman,L.B., Li,A., Gribben,J.G., Fox,E.A., Sallan,S.E.,
Korsmeyer,S.J. (2004) FLT3 mutations in childhood acute lymphoblastic leukemia. Blood,
103(9), 3544-3546.
Bacher,U., Haferlach,T., Schooch,C., Kern,W., Schnittger,S. (2006) Implications of NRAS
mutations in AML: a study of 2502 patients. Blood, 107, 3847-3853.

99
Barbacid,M. (1987) RAS genes. Annual Review of Biochemistry, 56, 779-827.
Barletta,E., Gorini,G., Vineis,P., Miligi,L., Davico,L., Mugnai,G., Ciolli,S., Leoni,F.,
Bertini,M., Matullo,G., Costantini,A.S. (2004) RAS gene mutations in patients with acute
myeloid leukaemia and exposure to chemical agents. Carcinogenesis, 25, 749-755.
Behm,F.G., Raimondi,S.C., Frestedt,J.L., Liu,Q., Crist,W.M., Downing,J.R., Rivera,G.K.,
Kersey,J.H., Pui,C.H. (1996) Rearrangement of the MLL gene confers a poor prognosis in
childhood acute lymphoblastic leukemia, regardless of presenting age. Blood, 87(7), 2870-
2877.
Belpomme,D. (2005) The contribution of the physic-chemical environment to the genesis of
cancer: what extent and how to measure it? Bulletin et mémoires de l’Académie royale de
médecine de Belgique, 160(3-4),163-180.
Belson,M., Kingsley,B., Holmes,A. (2007) Risk factors for acute leukemia in children: a
review. Environmental Health Perspectives, 115, 138-145.
Béne,M.C., Castoldi,G., Knapp,W., Ludwig,W.D., Matutes,E., Orfao,A., van’t Veer,M.B.
(1995) Proposals for the immunological classification of acute leukemias. European Group
for the Immunological Characterization of Leukemias (EGIL). Leukemia, 9, 1783-1786.

100
Bennett,J.H. (1845) Two cases of disease and enlargement of the spleen in which death takes
place from the presence of purulent matter in the blood. Edinburgh Medical and Surgical
Journal.
Bennett,J.M., Catovsky,D., Daniel,M.T., Flandrin,G., Galton,D.A., Gralnick,H.R., Sultan,C.
(1976) Proposals for the classification of the acute leukemias. British Journal of
Haematology, 33(4), 451-458.
Biondi,A., Cimino,G., Pieters,R., Pui,C.H. (2000) Biological and therapeutic aspects of infant
leukemia. Blood, 96, 24-33.
Boissel,N., Leroy,H., Brethon,B., Philippe,N., de Botton,S., Auvrignon,A., Raffoux,E.,
Leblanc,T., Thomas,X., Hermine,O., Quesnel,B., Baruchel,A., Leverger,G., Dombret,H.,
Preudhomme,C. (2006) Incidence and prognostic impact of c-Kit, FLT3, and Ras gene
mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia, 20, 965–
970.
Borkhardt,A., Repp,R., Haas,O., Leis,T., Harbott,J., Kreuder,J., Hammermann,J., Henn,T.,
Lampert,F. (1997) Cloning and characterization of AFX, the gene that fuses to MLL in acute
leukemias with a t(X;11)(q13;q23). Oncogene, 14, 195-202.

101
Bornholdt,J., Hansen,J., Steiniche,T., Dictor,M., Antonsen,A., Wolff,H., Schlünssen,V.,
Holmila,R., Luce,D., Vogel,U., Husgafvel-Pursiainen,K., Wallin,H. (2008) K-RAS mutations
in sinonasal cancers in relation to wood dust exposure. BMC Cancer, 8, 53.
Bowen,D., Frew,M., Hills,R., Gale,R., Wheatley,K., Groves,M.J., Langabeer,S.E.,
Kottaridis,P.D., Moorman,A.V., Burnett,A.K., Linch,D.C. (2005) RAS mutation in acute
myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence
outcome in patients younger than 60 years. Blood, 106, 2113-2119.
Braun,B.S. and Shannon,K. (2008) Targeting RAS in Myeloid Leukemias. Clínical Cancer
Research, 14(8), 2249-2252.
Bromberg,K.D., Burgin,A.B., Osheroff,N. (2003) A two-drug model for etoposide action against
human topoisomerase IIalpha. The Journal of Biological Chemistry, 278(9), 7406-7412.
Brondum,J., Shu,X.O., Steinbuch,M., Severson,R.K., Potter,J.D., Robison,L.L. (1999)
Parental cigarette smoking and the risk of acute leukemia in children. Cancer. 85(6), 1380-
1388.
Browett,P.J. and Norton,J.D. (1989) Analysis for RAS gene mutations and methylation state in
human leukemias. Oncogene, 4, 1029-1036.
Buckley,J.D. (1992) The aetiology of cancer in the very young. The British Journal of
Cancer. Supplement., 18, S8-12.

102
Cairoli,R., Beghini,A., Grillo,G., Nadali,G., Elice,F., Ripamonti,C.B., Colapietro,P.,
Nichelatti,M., Pezzetti,L., Lunghi,M., Cuneo,A., Viola,A., Ferrara,F., Lazzarino,M.,
Rodeghiero,F., Pizzolo,G., Larizza,L., Morra,E. (2006) Prognostic impact of c-KIT mutations
in core binding factor leukemias: An Italian retrospective study. Blood, 107, 3463–3468.
Care,R.S., Valk,P.J., Goodeve,A.C., Abu-Duhier,F.M., Geertsma-Kleinekoort,W.M.,
Wilson,G.A., Gari,M.A., Peake,I.R., Löwenberg,B., Reilly,J.T. (2003) Incidence and
prognosis of c-KIT and FLT3 mutations in core binding factor (CBF) acute myeloid
leukaemias. British Journal of Haematology, 121(5), 775-777.
Case,M., Matheson,E., Minto,L., Hassan,R., Harrison,C.J., Bown,N., Bailey,S., Vormoor,J.,
Hall,A.G., Irving,J.A.E. (2008) Mutation of genes affecting the RAS pathway is common in
childhood acute lymphoblastic leukemia. Cancer Research, 68, 6803-6809.
Chang,S.H., Lee,N.Y., Kim,D.H., Sohn,S.K., Suh,J.S. (2006) FLT3 Gene Mutations as a
Prognostic Factor for Acute Myeloid Leukemia. The Korean journal of laboratory medicine,
26(4), 233-240.
Chen,A.Y. and Liu,L.F. (1994) DNA topoisomerases: essential enzymes and lethal targets.
Annual Review of Pharmacology and Toxicology, 84, 191-218.
Chessells,J.M. (1992) Leukaemia in the young child. The British Journal of Cancer.
Supplement., 18, S54 – S57.

103
Chowdhury,T. and Brady,H. (2008) Insights from clínical studies into the role of the MLL
gene in infant and childhood leukemia. Blood Cells, Molecules, and Diseases, 40, 192–199.
Clementino,N.C.D., Yamamoto,M., Viana,M.B., Figueiredo,M.S., Kerbauy,S., Saad,S.T.O.,
Costa,F.F. (2001) Lack of association between N-ras gene mutations and clinical prognosis in
brazilian children with acute lymphoblastic leukemia. Leukemia and Lymphoma, 42, 473-479.
Corbett,A. and Osheroff,N. (1993) When good enzymes go bad: conversion of topoisomerase
II to a cellular toxin by antineoplastic drugs. Chemical Research in Toxicology, 6, 585-597.
Davis,M., Malcolm,S., Hall,A., Marshall,C.J. (1983) Localisation of the human N-RAS
oncogene to chromosome 1cen-1p21. Embo Journal. 12, 2281–2283.
de Camargo,B., de Oliveira Santos,M., Rebelo,M.S., de Souza Reis,R., Ferman,S., Noronha,C.P.,
Pombo-de-Oliveira,M.S. (2010) Cancer incidence among children and adolescents in Brazil: first
report of 14 population-based cancer registries. International Journal of Cancer, 126(3):715-720.
Denayer,E., de Ravel,Th., Legius,E. (2008) Clínical and molecular aspects of RAS related
disorders. Journal of Medical Genetics, 45, 695-703.
Der,C.J., Krontiris,T.G., Cooper,G.M. (1982) Transforming genes of human bladder and lung
carcinoma cell lines are homólogous to the RAS genes of Harvey and Kirsten sarcoma viruses.
Proceeding of the National Academy of Sciences of the United States of America, 79, 3637-
3640.

104
Domer,P.H., Fakharzadeh,S.S., Chen,C-S., Jockel,J., Johansen,L., Silverman,G.A.,
Kersey,J.H., Korsmeyer,S.J. (1993) Acute mixedlineage leukemia t(4;11)(q21;q23) generates
an MLL-AF4 fusion product. Proceeding of the National Academy of Sciences of the United
States of America, 90, 7884-7888.
Downward,J. (2006) Signal transduction. Prelude to an anniversary for the RAS oncogene.
Science, 314(5798), 433-434.
Emerenciano,M., Agudelo Arias,D.P., Coser,V.M., de Brito,G.D., Macedo Silva,M.L.,
Pombo-de-Oliveira,M.S., Brazilian Collaborative Study Group of Infant Acute Leukemia.
(2005) Molecular cytogenetic findings of acute leukemia included in the Brazilian
Collaborative Study Group of Infant Acute Leukemia. Pediatric Blood e Cancer, 47(5), 549-
554.
Ernst,P., Wang,J. , Korsmeyer,S.J. (2002) The role of MLL in hematopoiesis and leukemia.
Current Opinion in Hematology, 9(4), 282–287.
Farr,C.J., Saiki,R.K., Erlich,H.A., McCormick,F., Marshall,C.J. (1998) Analysis of RAS gene
nucleotide probes. Proceeding of the National Academy of Sciences of the United States of
America, 85, 1629-1633.
Felix,C.A., Lange,B.J., Hosler,M.R., Fertala,J., Bjornsti,M.A. (1995) Chromosome band
11q23 translocation breakpoints are DNA topoisomerase II cleavage sites. Cancer Research,
55, 4287-4292.

105
Felix,C.A., Hosler,M.R., Slater,D.J., Parker,R.I., Masterson,M., Whitlock,J.A., Rebbeck,T.R.,
Nowell,P.C., Lange,B.J. (1998) MLL genomic breakpoint distribution within the breakpoint
cluster region in de novo leukemia in children. Journal of Pediatric Hematology/Oncology,
20, 299-308.
Felix,C.A. and Lange,B.J. (1999) Leukemia in Infants. The Oncologist, 4, 225-240.
Ford,A.M., Ridge,S.A., Cabrera,M.E., Mahmoud,H., Steel,C.M., Chan,L.C., Greaves,M.
(1993) In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature,
363, 358-360.
Gale,K.B., Ford,A.M., Repp,R., Borkhardt,A., Keller,C., Eden,O.B., Greaves,M.F. (1997)
Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal
blood spots. Proceedings of the National Academy of Sciences of the United States of
America, 94(25), 13950-13954.
Gill-Super,H.J., Rothberg,P.G., Kobayashi,H., Freeman,A.I., Diaz,M.O., Rowley,J.D. (1994)
Clonal, nonconstitutional rearrangements of the MLL gene in infant twins with acute
lymphoblastic leukemia: in utero chromosome rearrangement of 11q23. Blood, 83, 641-644.
Gilliland,D.G. and Griffin,J.D. (2002) The roles of FLT3 in hematopoiesis and leukemia.
Blood, 100, 1532-1542.

106
Goemans,B.F., Zwaan,C.M., Miller,M., Zimmermann,M., Harlow,A., Meshinchi,S.,
Loonen,A.H., Hählen,K., Reinhardt,D., Creutzig,U., Kaspers,G.J., Heinrich,M.C. (2005)
Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid
leukemia. Leukemia, 19(9), 1536-42.
Greaves,M.F. (1997) Etiology of acute leukemia. Lancet, 349, 344-349.
Greaves,M.F. (2002) Childhood leukemia. BMJ, 324, 283-287.
Greaves,M.F. and Wiemels,J. (2003) Origins of chromosome translocations in childhood
leukaemia. Nature Reviews. Cancer, 3(9):639-49.
Gross,E., Arnold,N., Pfeifer,K., Bandick,K., Kiechle,M. (2000) Identification of specific
BRCA1 and BRCA2 variants by DHPLC. Human Mutation, 16, 345–53.
Gu,Y., Alder,H., Nakamura,T., Schichman,S.A., Prasad,R., Canaani,O., Saito,H., Croce,C.M.,
Canaani,E. (1994) Sequence analysis of the breakpoint cluster region in the ALL-1 gene
involved in acute leukemia. Cancer Research, 54, 2327-2330.
Guo,W., Tang,B., Xu,S., Yao,Y., Ye,D. (1998) N-RAS mutations in 43 Chinese cases of acute
myeloid leukemia. Chinese Medical Journal, 111, 343–345.
Gurney,J.G., Severson,R.K., Davis,S., Robison,L.L. (1995) Incidence of cancer in children in
the United States. Cancer, 75, 2186-2195.

107
Hanahan,D. and Weinberg,R.A. (2000) The hallmarks of cancer. Cell, 100, 57-70.
Hann,I.M., Stevens,R.F., Golstone,A.H., Rees,J.K., Wheatley,K., Gray,R.G., Burnett,A.K.
(1997) Randomized comparison of DAT versus ADE as induction chemotherapy in children
and younger adults with acute myeloid leukemia: results of the Medical Research Council’s
10th AML trial (MRC AML 10). Blood, 89, 2311-2318.
Hayakawa,F., Towatari,M., Kiyoi,H., Tanimoto,M., Kitamura,T., Saito,H., Naoe,T. (2000)
Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces
autonomous cell growth in IL-3-dependent cell lines. Oncogene, 19(5), 624-631.
Heerema,N.A., Arthur,D.C., Sather,H., Albo,V., Feusner,J., Lange,B.J., Steinherz,P.G.,
Zeltzer,P., Hammond,D., Reaman,G.H. (1994) Cytogenetic features of infants less than 12
months of age at diagnosis of acute lymphoblastic leukemia: impact of the 11q23 breakpoint
on outcome: a report of the Childrens Cancer Group. Blood, 83, 2274-2284.
Hillion,J., Le Coniat,M., Jonveaux,P., Berger,R., Bernard,O.A. (1997) AF6q21, a novel
partner of the MLL gene in t(6;11)(q21;q23), defines a Forkhead transcriptional factor
subfamily. Blood, 9, 3714-3719.
Infante-Rivard,C., Labuda,D., Krajinovic,M., Sinnett,D. (1999) Risk of childhood leukemia
associated with exposure to pesticides and with gene polymorphisms. Epidemiology, 10(5),
481-487.

108
Jekic,B., Novakovic,I., Lukovic,L., Kuzmanovic,M., Popovic,B., Pastar,I., Milasin,J.,
Bunjevacki,G., Bunjevacki,V. (2004) Low frequency of NRAS and KRAS2 gene mutations in
childhood myelodysplastic syndromes. Cancer Genetics and Cytogenetics, 154, 180-182.
Ji,B.T., Shu,X.O., Linet,M.S., Zheng,W., Wacholder,S., Gao,Y.T., Ying,D.M. Jin,F. (1997)
Paternal cigarette smoking and the risk of childhood cancer among offspring of nonsmoking
mothers. Journal of the National Cancer Institute, 89(3), 238–244.
Jones,D., Yao,H., Romans,A., Dando,C., Pierce,S., Borthakur,G., Hamilton,A.,Bueso-
Ramos,C., Ravandi,F., Garcia-Manero,G., Kantarjian,H. (2010) Modeling Interactions
Between Leukemia-Specific Chromosomal Changes, Somatic Mutations, and Gene
Expression Patterns During Progression of Core-Binding Factor Leukemias. Genes,
Chromosomes e Cancer, 49, 182–191.
Junien,C., Huerre,C., Despoisse,S., Gilgenkrantz,S., Lenoir,G.M. (1984) c-Ha-RAS1 is not
deleted in del(11p13) Wilms' tumor (WAGR) and maps to 11p15.1-11p15.5. Cytogenetics
and Cell Genetics, 37, 503.
Kaler,S.G., Devaney,J.M., Pettit,E.L., Kirshman,R., Marino,M.A. (2000) Novel method for
molecular detection of the two common hereditary hemochromatosis mutations. Genetic
Testing, 4, 125–129.

109
Kalwinsky,D.K., Raimondi,S.C., Shell,M.J., Mirro,J Jr., Santana,V.M., Behm,F., Dahl,G.V.,
Williams,D. (1990) Prognostic importance of cytogenetic subgroups in de novo pediatric
acute nonlymphocytic leukemia. Journal of Clínical Oncology, 8, 75-83.
Kawamura,M., Kikuchi,A., Kobayashi,S., Hanada,R., Yamamoto,K., Horibe,K., Shikano,T.,
Ueda,K., Hayashi,K., Sekiya,T., Hayashi,Y. (1995) Mutations of the p53 and RAS genes in
childhood t(1;19) acute lymphoblastic leukemia. Blood, 85, 2546-2552.
Kim,H.G., Kojima,K., Swindle,C.S., Cotta,C.V., Huo,Y., Reddy,V., Klug,C.A. (2008) FLT3-
ITD cooperates with inv(16) to promote progression to acute myeloid leukemia. Blood, 111,
1567–1574.
Kingma,P.A., Greider,C.A., Osheroff,N. (1997) Spontaneous DNA lesions poison human
topoisomerase IIa and stimulate cleavage proximal to leukemic 11q23 chromosomal
breakpoints. Biochemistry, 36, 5934-5939.
Kinlen,L.J. (1995) Epidemiological evidence for an infective basis in childhood leukemia.
British Journal of Cancer, 71, 1-5.
Kiyoi,H., Naoe,T., Nakano,Y., Yokota,S., Minami,S., Miyawaki,S., Asou,N., Kuriyama,K.,
Jinnai,I., Shimazaki,C., Akiyama,H., Saito,K., Oh,H., Motoji,T., Omoto,E., Saito,H.,
Ohno,R., Ueda,R. (1999) Prognostic implication of FLT3 and N-RAS gene mutations in acute
myeloid leukemia. Blood, 93(9), 3074-3080.

110
Knudson,A.G. Jr., Hethcote,H.W., Brown,B.W. (1975) Mutation and childhood cancer: a
probabilistic model for the incidence of retinoblastoma. Proceedings of the National Academy
of Sciences of the United States of America, 72(12), 5116-5120.
Koifman,S., Pombo-de-Oliveira,M.S., Brazilian Collaborative Study Group of Infant Acute
Leukemia. (2008) High birth weight as an important risk factor for infant leukemia. British
Journal of Cancer, 98, 664-667.
Kottaridis,P.D., Gale,R.E., Frew,M.E., Harrison,G., Langabeer,S.E., Belton,A.A., Walker,H.,
Wheatley,K., Bowen,D.T., Burnett,A.K., Goldstone,A.H., Linch,D.C. (2001) The presence of a
FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important
prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy:
analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12
trials. Blood, 98(6), 1752-1759.
Lampkin,B.C. (1997) The newborn infant with leukemia. The Journal of Pediatrics, 131, 176-
177.
Liang,D.C., Shih,L.Y., Fu,J.F., Li,H.Y., Wang,H.I., Hung,I.J., Yang,C.P., Jaing,T.H.,
Chen,S.H., Liu,H.C. (2006) K-RAS Mutations and N-RAS Mutations in Childhood Acute
Leukemias With or Without Mixed-Linage Leukemia Gene Rearrangements. Cancer, 106,
950-956.

111
Lillington,D.M., Young,B.D., Berger,R., Martineau,M., Moorman,A.V., Secker-Walker,L.M.
(1998) The t(10;11)(p12;q23) translocation in acute leukemia: a cytogenetic and clínical study
of 20 patients. Leukemia, 12, 801-804.
Liu,W., Smith,D.I., Rechtzigel,K.J., Thibodeau,S.N., James,C.D. (1998) Denaturing high
performance liquid chromatography (DHPLC) used in the detection of germline and somatic
mutations. Nucleic Acids Research, 26, 1396–1400.
Lu,C., Fenske,R.A., Simcox,N.J., Kalman,D. (2000) Pesticide exposure of children in an
agricultural community: evidence of household proximity to farmland and take home
exposure pathways. Environmental Research, 84(3), 290-302.
Ma,X., Buffler,P.A., Gunier,R.B., Dahl,G., Smith,M.T., Reinier,K., Reynolds,P. (2002) Critical
windows of exposure to household pesticides and risk of childhood leukemia. Environmental
Health Perspectives, 110(9), 955-960.
Mackarehtschian,K., Hardin,J.D., Moore,K.A., Boast,S., Goff,S.P., Lemischka,I.R. (1995)
Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic
progenitors. Immunity, 3(1), 147-161.
Mahgoub,N., Parker,RI., Hosler,MR., Close,P., Winick,NJ., Masterson,M., Shannon,KM.,
Felix,CA. (1998) RAS mutations in pediatric leukemias with MLL gene rearragments. Genes,
Chromosomes and Cancer, 21(3), 270-275.

112
Mahmoud,H.H., Ridge,S.A., Behm,F.G., Pui,C.H., Ford,A.M., Raimondi,S.C., Greaves,M.F.
(1995) Intrauterine monoclonal origin of neonatal concordant acute lymphoblastic leukemia
in monozygotic twins. Medical and Pediatric Oncology, 24, 77-81.
Malumbres,M. and Barbacid,M. (2003) RAS oncogenes: the first 30 years. Nature Reviews.
Cancer., 3, 459-465.
Martineau,M., Berger,R., Lillington,D.M., Moorman,A.V., Secker-Walker,L.M. (1998) The
t(6;11)(q27;q23) translocation in acute leukemia: a laboratory and clínical study of 30 cases.
Leukemia, 12, 788-791.
Matamoros,N., Matutes,E., Hernandex,N., Galmes,A., Perez-Payarols,J., Buccheri,V.,
Morilla,R., Catovsky,D., Healy,L.E., Ridge,S.A. (1994) Neonatal mixed lineage leukemia.
Leukemia, 8, 1236-1242.
McCormick,F., Clark,B.F., la Cour,T.F., Kjeldgaard,M., Norskov-Lauritsen,L. Nyborg,J.
(1985) A model for the tertiary structure of p21, the product of the RAS oncogene. Science,
230(4721), 78-82.
Megonigal,M.D., Rappaport,E.F., Jones,D.H., Williams,T.M., Lovett,B.D., Kelly,K.M.,
Lerou,P.H., Moulton,T., Budarf,M.L., Felix,C.A. (1998) t(11;22)(q23;q11.2) in acute myeloid
leukemia of infant twins fuses MLL with hCDCrel, a cell division cycle gene in the genomic
region of deletion in DiGeorge and velocardiofacial syndromes. Proceedings of the National
Academy of Sciences of the United States of America, 95, 6413-6418.

113
Meinert,R., Schüz,J., Kaletsch,U., Kaatsch,P., Michaelis,J. (2000) Leukemia and non-
Hodgkin's lymphoma in childhood and exposure to pesticides: results of a register-based case-
control study in Germany. American Journal of Epidemiology, 151(7), 639-650.
Menegaux,F., Steffen,C., Bellec,S., Baruchel,A., Lescoeur,B., Leverger,G., Nelken,B.,
Philippe,N., Sommelet,D., Hémon,D., Clavel,J. (2005) Maternal coffee and alcohol
consumption during pregnancy, parental smoking and risk of childhood acute leukaemia.
Cancer Detection and Prevention, 29(6), 487-493.
Meshinchi,S., Woods,W.G., Stirewalt,D.L., Sweetser,D.A., Buckley,J.D., Tjoa,T.K.,
Bernstein,I.D., Radich,J.P. (2001) Prevalence and prognostic significance of Flt3 internal
tandem duplication in pediatric acute myeloid leukemia. Blood, 97(1), 89-94.
Meshinchi,S., Stirewalt,D.L., Alonzo,T.A., Zhang,Q., Sweetser,D.A., Woods,W.G.,
Bernstein,I.D., Arceci,R.J., Radich,J.P. (2003) Activating mutations of RTK/ras signal
transduction pathway in pediatric acute myeloid leukemia. Blood, 102(4), 1474-1479.
Meyer,C., Schneider,B., Jakob,S., Strehl,S., Schnittger,S., Schoch,C., Jansen,M.W., van
Dongen,J.J., den Boer,M.L., Pieters,R., Ennas,M.G., Angelucci,E., Koehl,U., Greil,J.,
Griesinger,F., Zur Stadt,U., Eckert,C., Szczepański,T., Niggli,F.K., Schäfer,B.W.,
Kempski,H., Brady,H.J., Zuna,J., Trka,J., Nigro,L.L., Biondi,A., Delabesse,E., Macintyre,E.,
Stanulla,M., Schrappe,M., Haas,O.A., Burmeister,T., Dingermann,T., Klingebiel,T.,
Marschalek,R. (2006) The MLL recombinome of acute leukemias. Leukemia, 20, 777-784.

114
Meyer,C., Kowarz,E., Hofmann,J., Renneville,A., Zuna,J., Trka,J., Ben Abdelali,R.,
Macintyre,E., De Braekeleer,E., De Braekeleer,M., Delabesse,E., de Oliveira,M.P., Cavé,H.,
Clappier,E., van Dongen,J.J., Balgobind,B.V., van den Heuvel-Eibrink,M.M., Beverloo,H.B.,
Panzer-Grümayer,R., Teigler-Schlegel,A., Harbott,J., Kjeldsen,E., Schnittger,S., Koehl,U.,
Gruhn,B., Heidenreich,O., Chan,L.C., Yip,S.F., Krzywinski,M., Eckert,C., Möricke,A.,
Schrappe,M., Alonso,C.N., Schäfer,B.W., Krauter,J., Lee,D.A., Zur Stadt,U., Te Kronnie,G.,
Sutton,R., Izraeli,S., Trakhtenbrot,L., Lo Nigro,L., Tsaur,G., Fechina,L., Szczepanski,T.,
Strehl,S., Ilencikova,D., Molkentin,M., Burmeister,T., Dingermann,T., Klingebiel,T.,
Marschalek,R. (2009) New insights to the MLL recombinome of acute leukemias. Leukemia,
23, 1490-1499.
Milunsky,A., Carmella,S.G., Ye,M., Hecht,S.S. (2000) A tobacco-specific carcinogen in the
fetus. Prenatal Diagnosis, 20(4), 307-310.
Mitchell,E.L.D., Jones,D., White,G.R.M., Varley,J.M., Santibanez-Koref,M.F. (1995)
Determination of the gene order of the three loci CD2, NGFB, and NRAS at human
chromosome band 1p13 and refinement of their localisation at the subband level by
fluorescence in situ hybridisation. Cytogenetics and Cell Genetics, 70(3-4), 183-185.
Mizuki,M., Fenski,R., Halfter,H., Matsumura,I., Schmidt,R., Müller,C., Grüning,W., Kratz-
Albers,K., Serve,S., Steur,C., Büchner,T., Kienast,J., Kanakura,Y., Berdel,W.E., Serve,H.
(2000) Flt3 mutations from patients with acute myeloid leukemia induce transformation of
32D cells mediated by the RAS and STAT5 pathways. Blood, 96, 3907-3914.

115
Moorman,A.V., Hagemeijer,A., Charrin,C., Rieder,H., Secker-Walker,L.M. (1998) The
translocations, t(11;19)(q23;p13.1) and t(11;19)(q23;p13.3): a cytogenetic and clínical profile
of 53 patients. Leukemia, 12, 805-810.
Mrózek,K. and Bloomfield,C.D. (2006) Chromosome Aberrations, Gene Mutations and
Expression Changes, and Prognosis in Adult Acute Myeloid Leukemia. Hematology, 169-
177.
Mrózek,K., Marcucci,G., Paschka,P., Whitman,S.P., Bloomfield,C.D. (2006) Clínical
relevance of mutations and gene-expression changes in adult acute myeloid leukemia with
normal cytogenetics: are we ready for a prognostically prioritized molecular classification?
Blood, 109(2), 431-448.
Mucci,L.A., Granath,F., Cnattingius,S. (2004) Maternal smoking and childhood leukemia and
lymphoma risk among 1,440,542 Swedish children. Cancer Epidemiology, Biomarkers e
Prevention, 13(9), 1528-1533.
Mullighan,C.G., Goorha,S., Radtke,I., Miller,C.B., Coustan-Smith,E., Dalton,J.D.,
Girtman,K., Mathew,S., Ma,J., Pounds,S.B., Su,X., Pui,C.H., Relling,M.V., Evans,W.E.,
Shurtleff,S.A., Downing,J.R. (2007) Genome-wide analysis of genetic alterations in acute
lymphoblastic leukaemia. Nature, 446, 758-764.

116
Nguyen,S., Leblanc,T., Fenaux,P., Witz,F., Blaise,D., Pigneux,A., Thomas,X., Rigal-
Huguet,F., Lioure,B., Auvrignon,A., Fiere,D., Reiffers,J., Castaigne,S., Leverger,G.,
Harousseau,J.L., Socie,G., Dombret,H. (2002) A white blood cell index as the main
prognostic factor in t(8;21) acute myeloid leukemia (AML): A survey of 161 cases from the
French AML Intergroup. Blood, 99, 3517–3523.
Nilson,I., Reichel,M., Ennas,M.G., Greim,R., Knörr,C., Siegler,G., Greil,J., Fey,G.H.,
Marschalek,R. (1997) Exon/intron structure of the AF-4 gene, a member of the AF-4/LAF-
4/FMR-2 gene family coding for a nuclear protein with structural alterations in acute
leukaemia. British Journal of Haematology, 98, 157-169.
O'Connell,P., Leppert,M., Hoff,M., Kumlin,E., Thomas,W., Cai,G., Law,M. White,R. (1985)
A linkage map for human chromosome 12. The American Journal of Human Genetics, 37,
A169.
Oefner,P.J. and Underhill,P.A. (1995) Comparative DNA sequencing by denaturing high-
performance liquid chromatography (DHPLC). The American Journal of Human Genetics,
57(Suppl):A266.
Pang,D., McNally,R., Birch,J.M. (2003) Parental smoking and childhood cancer: results from
the United Kingdom Childhood Cancer Study. British Journal of Cancer, 88(3), 373-381.
Parikh,C., Subrahmanyam,R., Ren,R. (2006) Oncogenic NRAS rapidly and efficiently induces
CMML –and AML– like diseases in mice. Blood, 108(7), 2349-2357.

117
Paschka,P., Marcucci,G., Ruppert,A.S., Mrozek,K., Chen,H., Kittles,R.A., Vukosavljevic,T.,
Perrotti,D., Vardiman,J.W., Carroll,A.J., Kolitz,J.E., Larson,R.A., Bloomfield,C.D. (2006)
Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with
inv(16) and t(8;21): A Cancer and Leukemia Group B Study. Journal of Clínical Oncology,
24, 3904–3911.
Pendergast,A.M., Quilliam,LA., Cripe, L.D., Bassing,C.H., Zonghan,D., Nanxing,L.,
Batzer,A., Rabun,K.M., Der,C.J., Schlessinger,D., Gishizky,M.L. (1993) BCR-ABL-Induced
oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor
protein. Cell, 75, 175-185.
Pluth,J.M., Ramsey,M.J., Tucker,J.D. (2000) Role of maternal exposures and newborn
genotypes on newborn chromosome aberration frequencies. Mutation Research, 465(1-2),
101-111.
Pombo-de-Oliveira,M.S., Koifman,S., Brazilian Collaborative Study Group of Infant Acute
Leukemia (2006) Infant acute leukemia and maternal exposures during pregnancy. Cancer
Epidemiology, Biomarkers e Prevention, 15(12), 2336-2341.
Popovic,R., Zeleznik-Le,N.J. (2005) MLL: how complex does it get? Journal of Cellular
Biochemistry, 95(4), 234–242.
Pui,CH., Kane,J., Crist,W. (1995) Biology and treatment of infant leukemias. Leukemia, 9,
762-769.

118
Pui,C.H., Ribeiro,R.C, Campana,D., Raimondi,S.C., Hancock,M.L., Behm,F.G.,
Sandlund,J.T., Rivera,G.K., Evans,W.E., Crist,W.M., Krance,R. (1996) Prognostic factors in
the acute lymphoid and myeloid leukemias of infants. Leukemia,10, 952-956.
Pui,C.H., Chessells,J.M., Camitta,B., Baruchel,A., Biondi,A., Boyett,J.M., Carroll,A.,
Eden,O.B., Evans,W.E., Gadner,H., Harbott,J., Harms,D.O., Harrison,C.J., Harrison,P.L.,
Heerema,N., Janka-Schaub,G., Kamps,W., Masera,G., Pullen,J., Raimondi,S.C., Richards,S.,
Riehm,H., Sallan,S., Sather,H., Shuster,J., Silverman,L.B., Valsecchi,M.G., Vilmer,E.,
Zhou,Y., Gaynon,P.S., Schrappe,M. (2003) Clínical heterogeneity in childhood acute
lymphoblastic leukemia with 11q23 rearrangments. Leukemia, 17, 700-706.
Rasio,D., Schichman,S.A., Negrini,M., Canaani,E., Croce,C.M. (1996) Complete exon
structure of the ALL1 gene. Cancer Research, 56, 1766-1769.
Reaman,G., Sposto,R., Sensel,M., Lange,B., Feusner,J., Heerema,N., Leonard,M., Holmes,E.,
Sather,H., Pendergrass,T., Johnstone,H., O'Brien,R., Steinherz,P., Zeltzer,P., Gaynon,P.,
Trigg,M., Uckun,F. (1999) Treatment outcome and prognostic factors for infants with acute
lymphoblastic leukemia treated on two consecutive trials of the Children's Cancer Group.
Journal of Clínical Oncology, Vol 17, 445.
Reichel,M., Gillert,E., Nilson,I., Siegler,G., Greil,J., Fey,G.H., Marschalek,R. (1998) Fine
structure of translocation breakpoints in leukemic blasts with chromosomal translocation
t(4;11): the DNA damage-repair model of translocation. Oncogene, 17, 3035-3044.

119
Reynolds,T. (1998) Causes of childhood leukemia beginning to emerge. Journal of the
Natural Cancer Institute, 90, 8-10.
Ritter,M., Kim,T.D., Lisske,P., Thiede,C., Schaich,M., Neubauer,A. (2004) Prognostic
significance on N-RAS and K-RAS mutations in 232 patients with acute myeloid leukemia.
Haematologica, 89, 1397-1399.
Ross,J.A., Davies,S.M., Potter,J.D., Robison,L.L. (1994) Epidemiology of childhood
leukemia, with a focus on infants. Epidemiologic Reviews, 16, 243-272.
Ross,J.A., Parentesis,J.P., Robison,L.L., Davies,S.M. (1996) Big babies and infant leukemia:
a role for insulin-like growth factor-1? Cancer Causes Control, 7, 553-559.
Ross,J.A., Potter,J.D., Shu,X.O., Reaman,G.H., Lampkin,B., Robison,L.L. (1997) Evaluating
the relationships among maternal reproductive history, birth characteristics, and infant
leukemia: a report from the Children's Cancer Group. Annals of Epidemiology, 7(3), 172-179.
Ross,J.A. (2008) Environmental and Genetic Susceptibility to MLL-Defined Infant Leukemia.
Journal of the National Cancer Institute Monographs, 39, 83-86.
Rubnitz,J.E., Morrissey,J., Savage,P.A., Cleary,M.L. (1994) ENL, the gene fused with HRX in
t(11;19) leukemias, encodes a nuclear protein with transcriptional activation potential in
lymphoid and myeloid cells. Blood, 84, 1747-1752.

120
Rubnitz,J.E., Behm,F.G., Curcio-Brint,A.M., Pinheiro,R.P., Carroll,A.J., Raimondi,S.C.,
Shurtleff,S.A., Downing,J.R. (1996) Molecular analysis of t(11;19) breakpoints in childhood
acute leukemias. Blood, 87, 4804-4808.
Sasco,A.J. and Vainio,H. (1999) From in utero and childhood exposure to parental smoking
to childhood cancer: a possible link and the need for action. Human e Experimental
Toxicology, 18(4), 192-201.
Schaeffeler,E., Lang,T., Zanger,U.M., Eichelbaum,M., Schwab,M. (2001) High-throughput
genotyping of thiopurine s-methyltransferase by denaturing HPLC. Clínical Chemistry, 47(3),
548–555.
Schichman,S.A., Canaani,E., Croce,C.M. (1995) Self-fusion of the ALL1 gene. JAMA: The
Journal of the American Medical Association, 273, 571-576.
Schlenk,R.F., Benner,A., Krauter,J., Buchner,T., Sauerland,C., Ehninger,G., Schaich,M.,
Mohr,B., Niederwieser,D., Krahl,R., Pasold,R., Dohner,K., Ganser,A., Dohner,H., Heil,G.
(2004) Individual patient data-based meta-analysis of patients aged 16 to 60 years with core
binding factor acute myeloid leukemia: A survey of the German Acute Myeloid Leukemia
Intergroup. Journal of Clínical Oncology, 22, 3741–3750.

121
Schnittger,S., Schoch,C., Dugas,M., Kern,W., Staib,P., Wuchter,C., Löffler,H.,
Sauerland,C.M., Serve,H., Büchner,T., Haferlach,T., Hiddemann,W. (2002) Analysis of FLT3
length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics,
FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the
detection of minimal residual disease. Blood, 100, 59-66.
Schnittger,S., Kohl,T.M., Haferlach,T., Kern,W., Hiddemann,W., Spiekermann,K., Schoch,C.
(2006) KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired
event-free and overall survival. Blood, 107, 1791–1799.
Schubbert,S., Shannon,K., Bollag,G. (2007) Hyperactive Ras in developmental disorders and
cancer. Nature Reviews. Cancer., 7, 295-308.
Setchell,K.D., Zimmer-Nechemias,L., Cai,J., Heubi,J.E. (1997) Exposure of infants to phyto-
oestrogens from soy-based infant fórmula. Lancet, 350, 23-27.
Severson,R.K., Buckley,J.D., Woods,W.G., Benjamin,D., Robison,L.L. (1993) Cigarette
smoking and alcohol consumption by parents of children with acute myeloid leukemia: an
analysis within morphologic subgroups - a report from the Children’s Cancer Group. Cancer
Epidemiology, Biomarkers e Prevention, 2, 433-439.

122
Shannon,K., O’Conell,P., Martin,G., Paderanga,D., Olson,K., Dinndorf,P., McCormick,F.
(1994) Loss of the normal NF1 allele from the bone marrow of children with type 1
neurofibromatosis and malignant myeloid disorders. The New England Journal of Medicine,
330, 597-601.
Sharpe,C.R. and Franco,E.L. (1996) Use of dipyrone during pregnancy and risk of Wilms'
tumor. Brazilian Wilms' Tumor Study Group. Epidemiology, 7(5), 533-535.
Shi,Q., Ko,E., Barclay,L., Hoang,T., Rademaker,A., Martin,R. (2001) Cigarette smoking and
aneuploidy in human sperm. Molecular Reproduction and Development, 59(4), 417-421.
Shu,X.O., Ross,J.A., Pendergrass,T.W., Reaman,G.H., Lampkin,B., Robison,L.L. (1996) Parental
alcohol consumption, cigarette smoking, and risk of infant leukemia: a Children’s Cancer Group
study. Journal of the National Cancer Institute, 88(1), 24-31.
Shu,X.O., Perentesis,J.P., Wen,W., Buckley,J.D., Boyle,E., Ross,J.A., Robison,L.L. (2004)
Parental Exposure to Medications and Hydrocarbons and RAS Mutations in Children with
Acute Lymphoblastic Leukemia: A Report from the Children’s Oncology Group. Cancer
Epidemiology, Biomarkers e Prevention, 13, 1230-1235.
Singer,M. In: CONFERENCE AT “WINDING YOUR WAY THROUGH DNA”, 1992.
Smith,M. (1997) Considerations on a possible viral etiology for B-precursor acute
lymphoblastic leukemia of childhood. Journal of Immunotherapy, 20(2), 89-100.

123
Sorahan,T., Lancashire,R., Prior,P., Peck,I., Stewart,A. (1995) Childhood cancer and parental
use of alcohol and tobacco. Annals of Epidemiology, 5(5), 354-359.
Spector,L.G., Xie,Y., Robison,L.L., Heerema,N.A., Hilden,J.M., Lange,B., Felix,C.A.,
Davies,S.M., Slavin,J., Potter,J.D., Blair,C.K., Reaman,G.H., Ross,J.A. (2005) Maternal diet
and infant leukemia: the DNA topoisomerase II inhibitor hypothesis: a report from the
Children’s Oncology Group. Cancer Epidemiology, Biomarkers e Prevention, 14, 651-655.
Spiegelman,J.I., Mindrinos,M.N., Oefner,P.J. (2000) High-accuracy DNA sequence variation
screening by DHPLC. Biotechniques, 29, 1084–1092.
Stam,R.W., den Boer,M.L., Schneider,P., Nollau,P., Horstmann,M., Beverloo,H.B., van der
Voort,E., Valsecchi,M.G., de Lorenzo,P., Sallan,S.E., Armstrong,S.A., Pieters,R. (2005)
Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood,
106(7), 2484-2490.
Stirewalt,D.L., Kopecky,K.J., Meshinchi,S., Appelbaum,F.R., Slovak,M.L., Willman,C.L.,
Radich,J.P. (2001) FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid
leukemia. Blood, 97(11), 3589-3595.
Stirewalt,D.L. and Radich,J.P. (2003) The role of FLT3 in haematopoietic malignancies.
Nature Reviews. Cancer., 3(9), 650-665.

124
Stirewalt,D.L., Meshinchi,S., Kussick,S.J., Sheets,K.M., Pogosova-Agadjanyan,E.,
Willman,C.L., Radich,J.P. (2004) Novel FLT3 point mutations within exon 14 found in
patients with acute myeloid leukaemia. British Journal of Haematology, 124, 481-484.
Swansbury,G.J., Slater,R., Bain,B.J., Moorman,A.V., Secker-Walker,L.M. (1998)
Hematological malignancies with t(9;11)(p21-22;q23)-a laboratory and clínical study of 125
cases. European 11q23 Workshop participants. Leukemia, 12(5), 792-800.
Taketani,T., Taki.T., Sugita,K., Furuichi,Y., Ishii,E., Hanada,R., Tsuchida,M., Sugita,K.,
Ida,K., Hayashi,Y. (2004) FLT3 mutations in the activation loop of tyrosine kinase domain
are frequently found in infant ALL with MLL rearrangements and pediatric ALL with
hyperdiploidy. Blood, 105, 2631-2639.
Taki,T., Ida,K., Bessho,F., Hanada,R., Kikuchi,A., Yamamoto,K., Sako,M., Tsuchida,M.,
Seto,M., Ueda,R., Hayashi,Y. (1996) Frequency and clínical significance of MLL gene
rearrangements in infant acute leukemia. Leukemia, 10, 1303-1307.
Taylor,J.A., Sandler,D.P., Bloomfield,C.D., Shore,D.L., Ball,E.D., Neubauer,A.,
McIntyre,O.R., Liu,E.. (1992) RAS oncogene activation and occupational exposures in acute
myeloid leukemia. Journal of the Natural Cancer Institute, 84(21), 1626-1632.
Tenen,D.G. (2003) Disruption of differentiation in human cancer: AML shows the way.
Nature Reviews, 3, 89-101.

125
Thiede,C., Steudel,C., Mohr,B., Schaich,M., Schäkel,U., Platzbecker,U., Wermke,M.,
Bornhäuser,M., Ritter,M., Neubauer,A., Ehninger,G., Illmer,T. (2002) Analysis of FLT3-
activating mutations in 979 patients with acute myelogenous leukemia: association with FAB
subtypes and identification of subgroups with poor prognosis. Blood, 99(12), 4326-4335.
Underhill,P.A., Jin,L., Lin,A.A., Mehdi,S.Q., Jenkins,T., Vollrath,D., Davis,R.W, Cavalli-
Sforzal,L.L., Oefner,P.J (1997) Nucleotide detection of numerous Y chromosome biallelic
polymorphisms by denaturing high-performance liquid chromatography. Genome Research,
7, 996–1005.
Usher,S.G., Radford,A.D., Villiers,E.J., Blackwood,L. (2009) RAS, FLT3, and C-KIT
mutations in immunophenotyped canine leukemias. Experimental Hematology, 37, 65-77.
Wang,Y.Y., Zhou,G.B., Yin,T., Chen,B., Shi,J.Y., Liang,W.X., Jin,X.L., You,J.H., Yang,G.,
Shen,Z.X., Chen,J., Xiong,S.M., Chen,G.Q., Xu,F., Liu,Y.W., Chen,Z., Chen,S.J. (2005)
AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: implication in stepwise
leukemogenesis and response to Gleevec. Proceedings of the National Academy of Sciences of
the United States of America, 102(4), 1104-1109.
Wiemels,J.L., Cazzaniga,G., Daniotti,M., Eden,O.B., Addison,G.M., Masera,G., Saha,V.,
Biondi,A., Greaves,M.F. (1999a) Prenatal origin of acute lymphoblastic leukaemia in
children. Lancet, 354(9189), 1499-1503.

126
Wiemels,J.L., Ford,A.M., Van Wering,E.R., Postma,A., Greaves,M. (1999b) Protracted and
variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero.
Blood, 94(3), 1057-1062.
Wiemels,J.L., Zhang,Y., Chang,J., Zheng,S., Metayer,C., Zhang,L., Smith,M.T., Ma,X.,
Selvin,S., Buffer,P.A., Wiencke,J.K. (2005) RAS mutation is associated with hyperdiploidy
and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia, 19, 415-
419.
Woods,W.G., Kobrinsky,N., Buckley,J.D., Lee,J.W., Sanders,J., Neudorf,S., Gold,S.,
Barnard,D.R., DeSwarte,J., Dusenbery,K., Kalousek,D., Arthur,D.C., Lange,B.J. (1996)
Timed-sequential induction therapy improves postremission outcome in acute myeloid
leukemia: a report from the Children’s Cancer Group. Blood, 87, 4979-4989.
Yamamoto,Y., Kiyoi,H., Nakano,Y., Suzuki,R., Kodera,Y., Miyawaki,S., Asou,N.,
Kuriyama,K., Yagasaki,F., Shimazaki,C., Akiyama,H., Saito,H., Ueda,R., Ohno,R., Naoe,T.
(2001) Activating mutation of D835 within the activation loop of FLT3 in human hematologic
malignancies. Blood, 97, 2434-2439.
Yokota,S., Kiyoi,H., Nakao,M., Iwai,T., Misawa,S., Okuda,T., Sonoda,Y., Abe,T.,
Kahsima,K., Matsuo,Y., Naoe,T. (1997) Internal tandem duplication of the FLT3 gene is
preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various
hematological malignancies. A study on a large séries of patients and cell lines. Leukemia,
11(10), 1605-1609.

127
Zago,M.A., Falcao,R.P., Pasquini,R. (2001) Hematologia Fundamentos e Prática, pp. 15-73.
Atheneu, São Paulo.
Zahm,S.H. and Ward,M.H. (1998) Pesticides and childhood cancer. Environmental Health
Perspectives, 106 Suppl. 3, 893-908.
Zheng,R., Friedman,A.D., Small,D. (2002) Targeted inhibition of FLT3 overcomes the block to
myeloid differentiation in 32Dcl3 cells caused by expression of FLT3/ITD mutations. Blood,
100(12), 4154-4161.

128
9. ANEXOS

129
9.1. Ficha de encaminhamento

130
9.2. TCLE

131

132

133
9.3. Géis de RFLP
(a) (b)
Géis de agarose de 3% corados com BE. Gel (a): as setas indicam amostras de pacientes, após
digestão com enzima PflMI para o KRAS com padrão de uma banda (padrão mutado); Gel (b):
as setas indicam amostras de pacientes após digestão com enzima BstNI com padrão de duas
bandas (padrão mutado).
(c) (d)
Géis de agarose de 3% corados com BE. Gel (c): as setas indicam amostra de paciente após
digestão com enzima BstNI com padrão de duas bandas (padrão mutado) Gel (d): as setas
indicam amostras de pacientes após digestão com enzima PflMI com padrão de uma banda
(padrão mutado)
(e) (f)
Géis de agarose de 3% corados com BE. Géis (e) e (f): as setas indicam amostra de pacientes
após digestão com enzima PflMI com padrão de uma banda (padrão mutado).

134
(g) (h) Géis de agarose de 3% corados com BE. Gel (a): as setas indicam amostras de pacientes, após
digestão com enzima PflMI para o KRAS com padrão de uma banda (padrão mutado); Gel (b):
as setas indicam amostras de pacientes após digestão com enzima BstNI com padrão de duas
bandas (padrão mutado).
(i) (j)
Géis de agarose de 3% corados com BE. Géis (i) e (j): as setas indicam amostras de pacientes
após digestão com enzima BstNI com padrão de duas bandas (padrão mutado).
(k) (l)
Géis de agarose de 3% corados com BE. Gel (k): as setas indicam amostras de pacientes, após
digestão com enzima PflMI para o KRAS com padrão de uma banda (padrão mutado); Gel (l):
as setas indicam amostras de pacientes após digestão com enzima BstNI com padrão de duas
bandas (padrão mutado).

135
(m) (n) Géis de agarose de 3% corados com BE. Gel (m): (1) as setas indicam amostras de pacientes
após digestão com enzima BstNI com padrão de duas bandas (padrão mutado); Gel (n): as
setas indicam amostras de pacientes após digestão com enzima PflMI com padrão de uma
banda (padrão mutado).
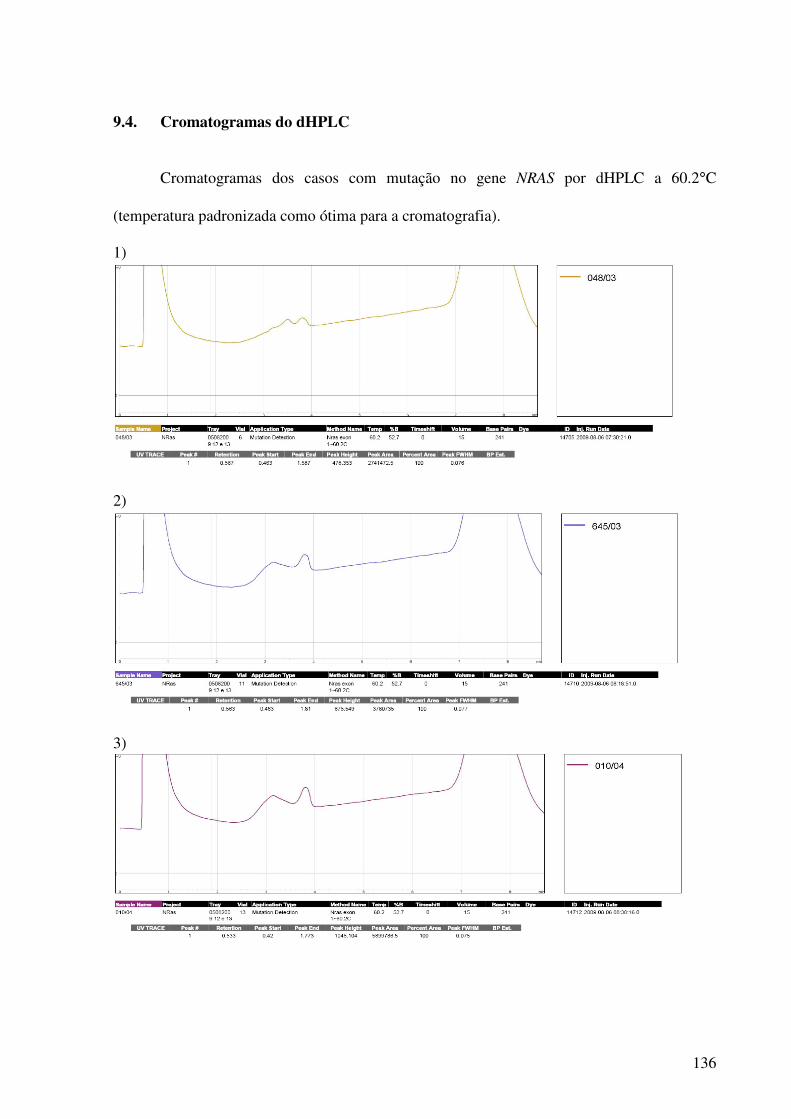
136
9.4. Cromatogramas do dHPLC
Cromatogramas dos casos com mutação no gene NRAS por dHPLC a 60.2°C
(temperatura padronizada como ótima para a cromatografia).
1)
2)
3)
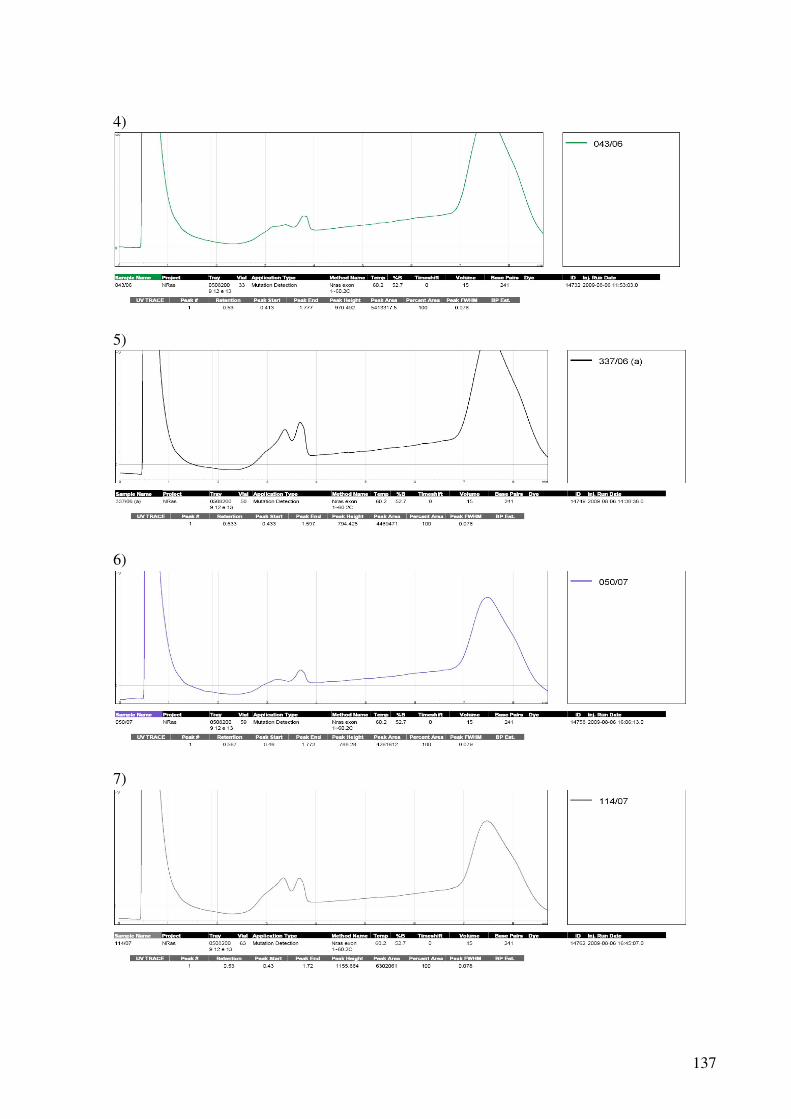
137
4)
5)
6)
7)
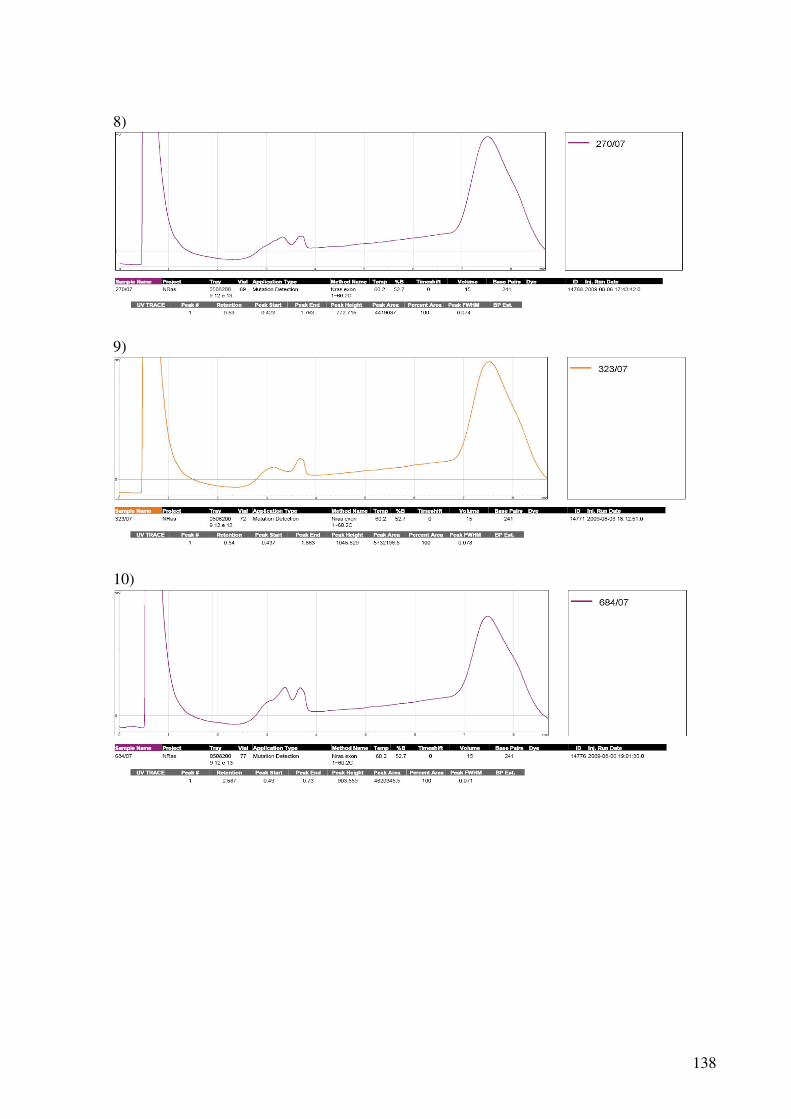
138
8)
9)
10)


















