Classificação e Processo Regulamentar de ... - PTCRIN · No prazo máximo de 30 dias úteis o...
34
Classificação e Processo Regulamentar de Dispositivos Médicos nos ensaios Intervencionais Sónia Cardoso 23 anos de serviço público com valores e ética
Transcript of Classificação e Processo Regulamentar de ... - PTCRIN · No prazo máximo de 30 dias úteis o...

Classificação e Processo Regulamentar de
Dispositivos Médicosnos ensaios Intervencionais
Sónia Cardoso
23 anos de serviço público com valores e ética

Objetivos
Processo Regulamentar
Critérios de Classificação
Consequências na aplicação da Lei nº 21/2014
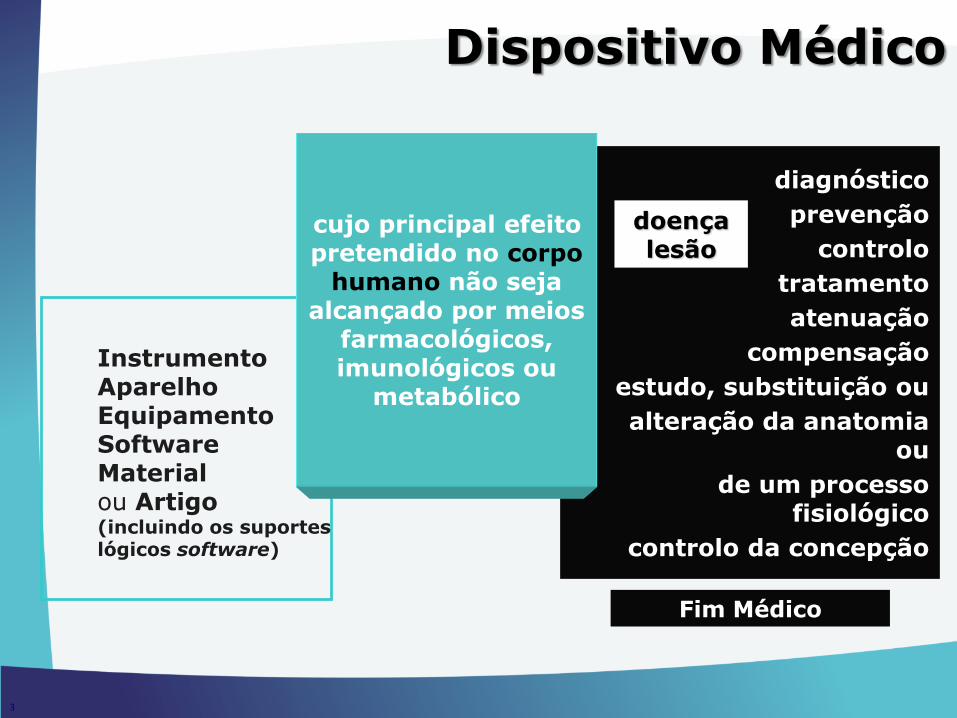
InstrumentoAparelhoEquipamentoSoftwareMaterialou Artigo (incluindo os suportes lógicos software)
diagnóstico
prevenção
controlo
tratamento
atenuação
compensação
estudo, substituição ou
alteração da anatomia ou
de um processo fisiológico
controlo da concepção
doençalesão
Fim Médico
cujo principal efeito pretendido no corpo
humano não seja alcançado por meios
farmacológicos, imunológicos ou
metabólico
3
Dispositivo Médico

DIR 2003/12/CE – Reclassificação de Implantes Mamários - DL nº 259/2003
DIR 2005/50/CE – Reclassificação de Próteses Internas - DL nº 258/2007
DIR 90/385/CEE – DM Implantável Activo (DMIA)
DIR 93/42/CEE – Dispositivos Médicos (DM) DL nº 145/2009
DIR 2007/47/CE
DIR 98/79/CE – DMs para diagnóstico in vitro (DIV) DL nº 189/2000
Quadro Legislativo
DIR 2000/70/CE – DMs com derivados estáveis do Sangue – DL Nº 145/2009
REG UE 722/2012 – DMs com derivados de origem Animal
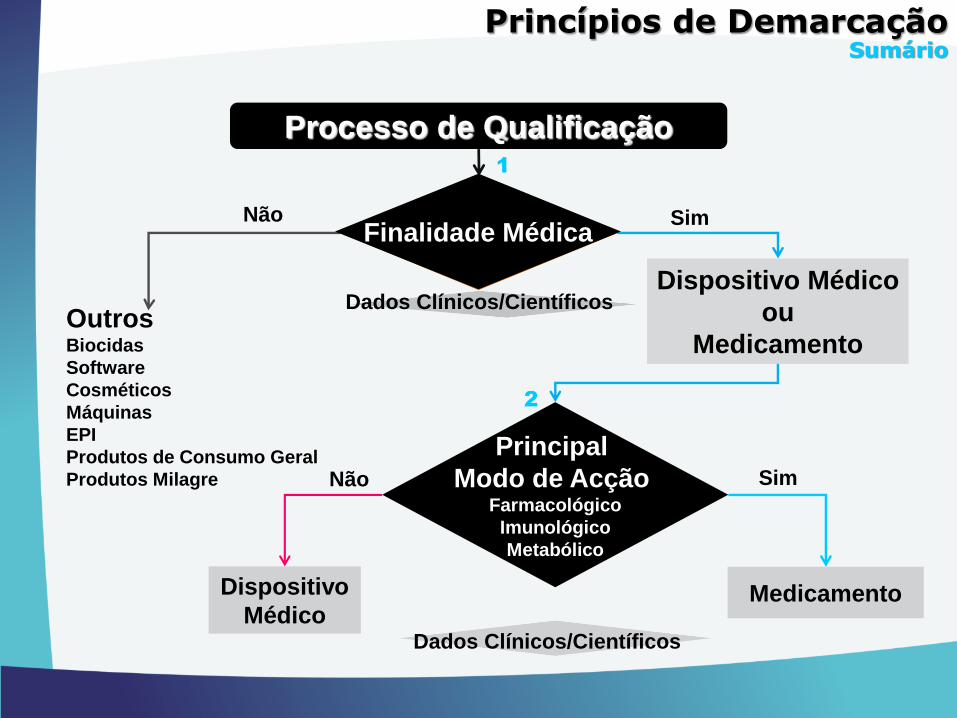
Princípios de DemarcaçãoSumário
Finalidade
Medica
Não Sim
Dados Clínicos/CientíficosOutrosBiocidas
Software
Cosméticos
Máquinas
EPI
Produtos de Consumo Geral
Produtos Milagre
Dispositivo Médico
ou
Medicamento
Principal
Modo de Acção Farmacológico
Imunológico
Metabólico
MedicamentoDispositivo
Médico
Não
Processo de Qualificação
Sim
Dados Clínicos/Científicos
Finalidade Médica
1
2

Medicamentos
Autorização de Introdução
no Mercado
Autoridade Reguladora
Dispositivos Médicos
Procedimento de Avaliação
de Conformidade
Fabricante
Organismo Notificado
Livre Circulação: UE, Países da EFTA e Turquia
Procedimento Centralizado
Procedimento de Reconhecimento
Mútuo
Procedimento Descentralizado
Procedimento Nacional
Consequências da QualificaçãoProcesso Regulamentar

Elementos da Conformidade no Pré Mercado
Dispositivo
Fabricante
Organismo Notificado
Autoridade de Designação
Avaliação Conjunta (ADs + COM)
DM - RE’s / Normas Europeias Harmonizadas
FAB - Sistemas da Qualidade / Ensaios ao Produto
ON - Capacidade e Competência Técnica na Avaliação da Conformidade
AD - Gerir os Sistemas de Designação / Notificação de Forma Credível
AC - Ganhar Transparência, Competência e Imparcialidade

Só podem ser colocados no mercado e entrar emserviço os dispositivos que satisfaçam os requisitosessenciais estabelecidos no anexo I do Decreto-Lei145/2009, de 17 de junho
Requisitos para a colocação no mercado– Art 5º, DL 145/2009
Requisitos Essenciais
“6.1 - A demonstração da conformidade com osrequisitos essenciais deve incluir uma avaliaçãoclínica nos termos do anexo XVI”
Ponto 6.1. do Anexo I, DL 145/2009
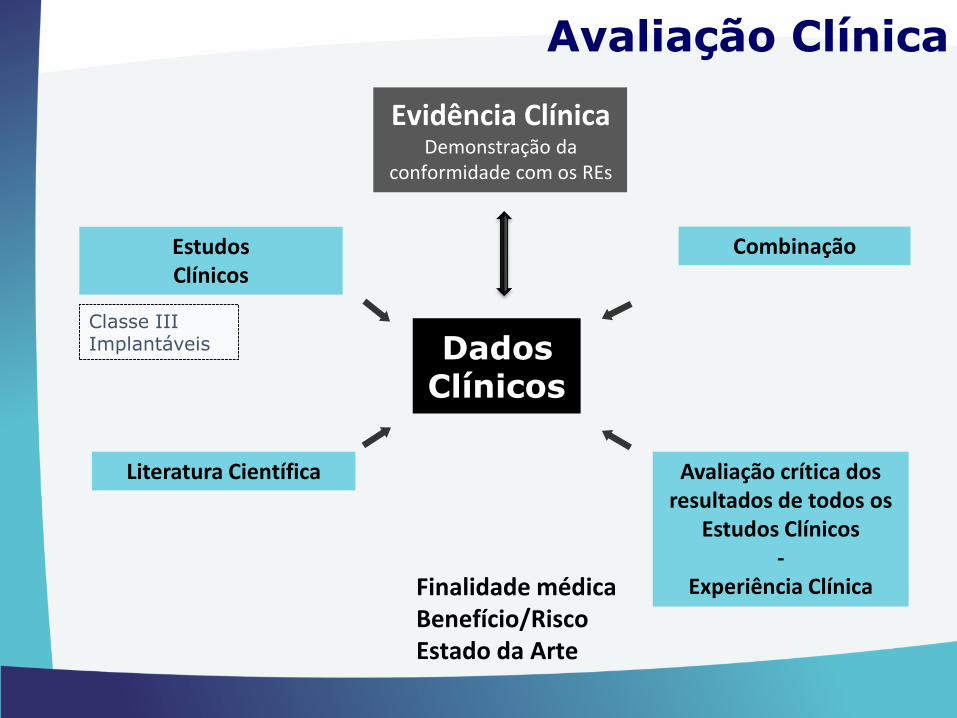
Evidência ClínicaDemonstração da
conformidade com os REs
Finalidade médicaBenefício/Risco Estado da Arte
Avaliação Clínica
Literatura Científica Avaliação crítica dos resultados de todos os
Estudos Clínicos-
Experiência Clínica
Estudos Clínicos
Combinação
Classe IIIImplantáveis Dados
Clínicos

Demonstração de Equivalência
Clínica
• Finalidade
• Condições clínicas de uso
• Local de aplicação
• População semelhante
• Desempenho clínico
Técnica
• Especificações e propriedades semelhantes
• Concepção semelhante
• Princípios de operação semelhantes
Biológica
• Os mesmos materiais em contacto com os tecidos e fluidos corporais (biocompatibilidade)
Avaliação Clínica

Objectivo:
Estabelecer um sistema de classificação tendo em conta avulnerabilidade do corpo humano perante uma potencialfalha ou mau funcionamento do produto;
Adequar a avaliação da conformidade ao risco envolvidona utilização do dispositivo [Regras de proporcionalidade];
Adequar a informação a ser cedida ao risco associado aodispositivo médico.
Classificação de DM
Intervenção de Organismo Notificado
Procedimento de avaliação adequado
Exigência do Procedimento
Classe III
Classe IIb
Classe IIa
Classe I
RISCO

1. Fim de Destino do Dispositivo Médico
- Documentação Técnica
- Rotulagem e Instruções de Utilização (FI)
- Materiais Promocionais
2. Duração – Tempo de contacto(utilização real ininterrupta do dispositivo para a finalidade prevista – uso continuado)
3. Invasivo vs. Não Invasivo
4. Anatomia afectada
5. Utilização potencialmente
perigosa
- Debilidade da população alvo
- Localizações Anatómicas Críticas
- Riscos envolvidos na utilização
TemporárioCurta
Duração
Longa
Duração
Implantáveis absorvíveis
Implantáveis
Cirurgicamente invasivos
Invasivo
(Orifícios naturais do corpo e
estomas)
Não invasivo
Duração
Invasibilidade
Classificação de DMCritérios

Invasibilidade do corpo humano:
- através de um orifício natural do corpo
- através de uma abertura criada cirurgicamente
Dispositivo Invasivo
O dispositivo que penetra parcial ou totalmente no corpo por um
dos seus orifícios, ou atravessando a sua superfície
Orifício corporal
qualquer abertura natural do corpo , bem como a
superfície externa do globo ocular, ou qualquer abertura
artificial permanente, como, por exemplo, um estoma.
Dispositivos invasivo do tipo cirúrgicoo dispositivo invasivo que penetra no corpo por meio de
uma intervenção cirúrgica ou no contexto de uma
intervenção cirúrgica.Anexo IX DIR 93/42/CEE
Classificação de DMConceitos e Definições (1/2)

Dispositivo Implantável:
dispositivo destinado a ser introduzido totalmente no corpo
humano, ou a substituir uma superfície epitelial ou a superfície
do olho através de uma intervenção cirúrgica e que se destina a
ser conservado no local após a intervenção.
Dispositivo Activo:
dispositivo cujo funcionamento depende de uma fonte de energia
eléctrica ou outra, não gerada pelo corpo humano ou pela
gravidade e que actua por conversão dessa energia.
Duração do Contacto (Uso contínuo ininterrupto)
Temporária ≤ 60 minutos
Curta Duração > a 60 minutos ≤ a 30 dias
Longa Duração > 30 dias
Classificação de DMConceitos e Definições (2/2)
Anexo IX DIR 93/42/CEE
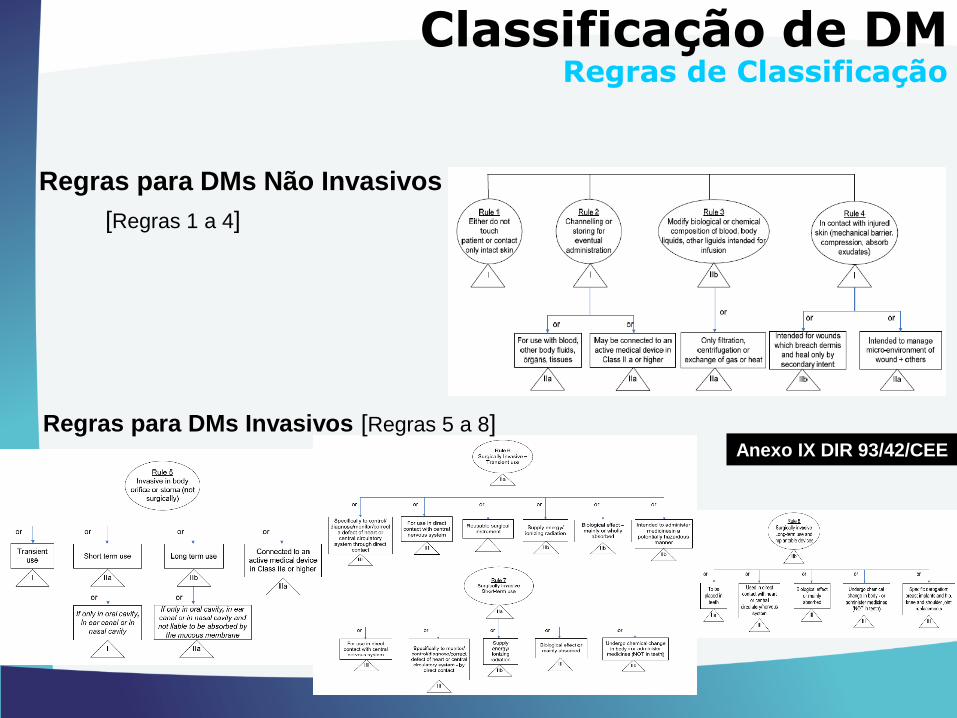
Regras para DMs Não Invasivos
[Regras 1 a 4]
Regras para DMs Invasivos [Regras 5 a 8]
Classificação de DMRegras de Classificação
Anexo IX DIR 93/42/CEE
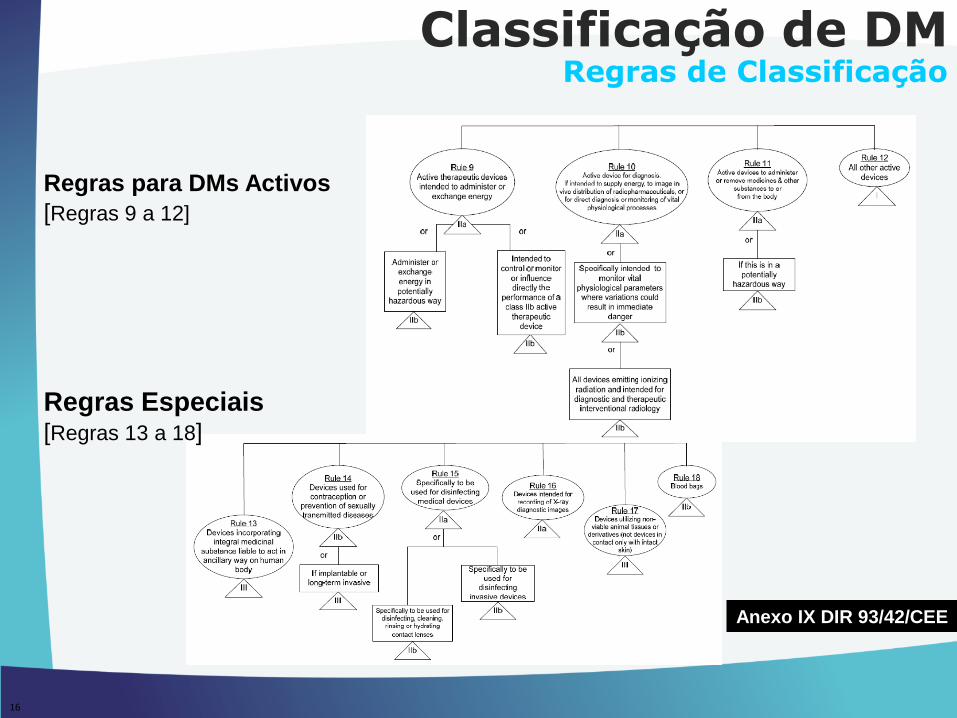
16
Regras para DMs Activos
[Regras 9 a 12]
Regras Especiais[Regras 13 a 18]
Classificação de DMRegras de Classificação
Anexo IX DIR 93/42/CEE

Ligaduras
Pensos
Pensos/Fraldas
para
Incontinência
Algodão
Hidrófilo
Meias de
Compressão
Cadeira de
Rodas
Macas
Camas
Hospitalares
Estetoscópio
Instrumentos
Cirúrgicos
Reutilizáveis
Dispositivos MédicosClasse I

Luvas de Exame
Sistemas
de Perfusão
Termómetro
Seringas
(sem agulha)
Dispositivos MédicosClasse I estéreis ou com função de medição

CLASSE I C/ FUNÇÃO
DE MEDIÇÃO
Seringas
(com agulha)
Compressas
Lentes de
Contacto
Equipamento de
Ressonância
Magnética
Aparelho de
Ultra-sons
Fios Ortodônticos
Lancetas e Agulhas
Dispositivos MédicosClasse IIa

Preservativos
Lentes de
Contacto
Lentes
intraoculares
Laser Cirúrgico
Incubadoras
Desfibrilhadores Externos
Ventiladores
Sacos de Sangue
Dispositivos MédicosClasse IIb
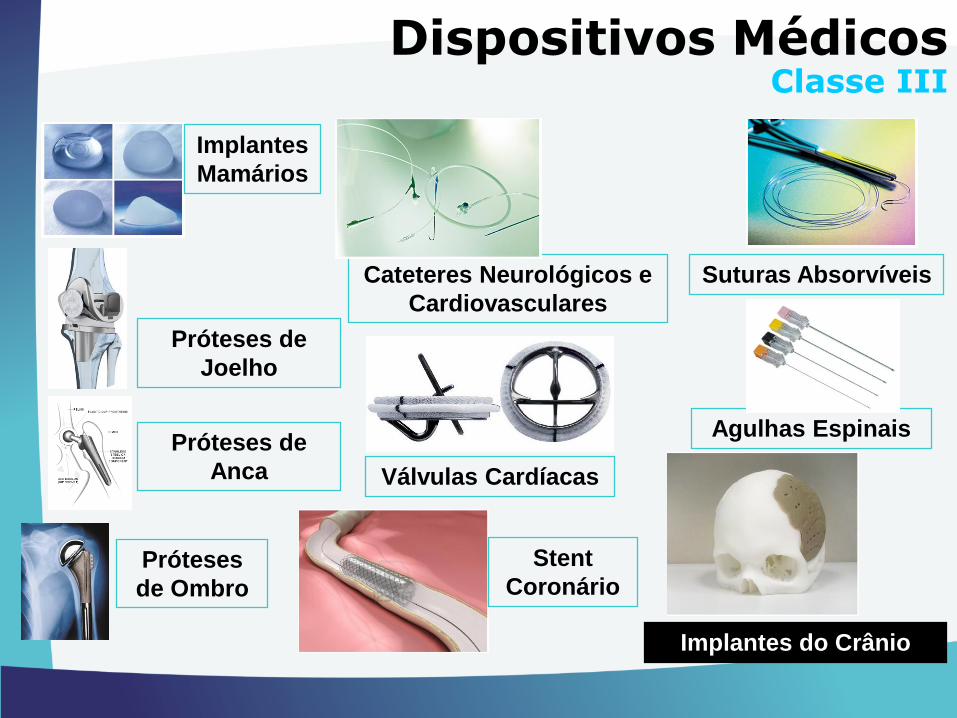
Cateteres Neurológicos e
Cardiovasculares
Implantes
Mamários
Próteses de
Joelho
Suturas Absorvíveis
Válvulas Cardíacas
Agulhas Espinais
Implantes do Crânio
Stent
Coronário
Próteses de
Anca
Próteses
de Ombro
Dispositivos MédicosClasse III

Pacemakers
Dispositivos Médicos Implantáveis ativos
Neuroestimuladores
Implantes Cocleares
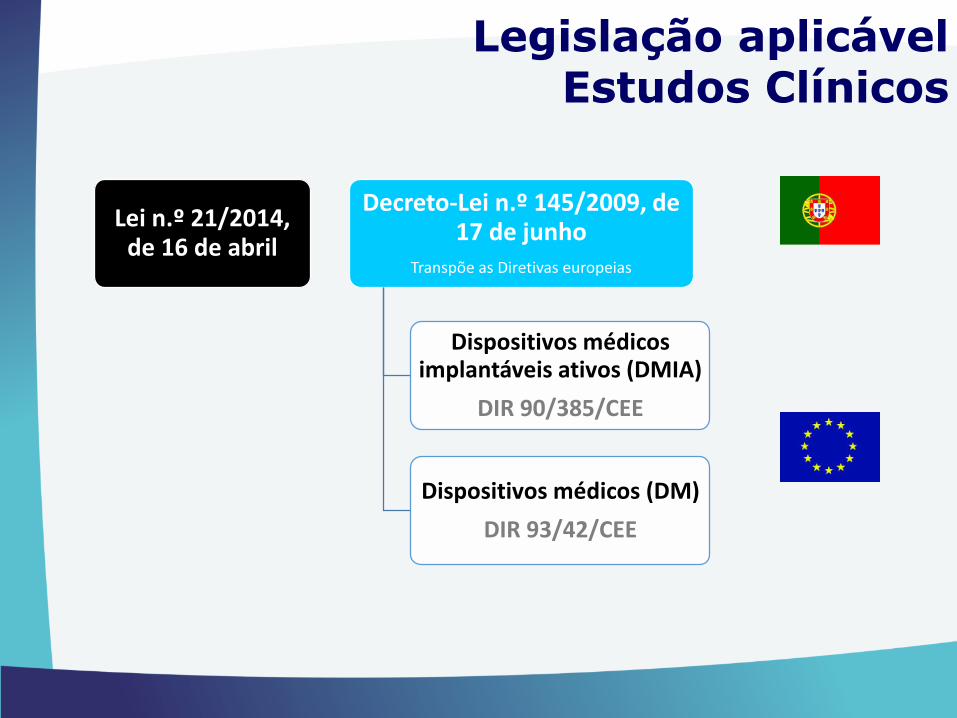
Lei n.º 21/2014, de 16 de abril
Decreto-Lei n.º 145/2009, de 17 de junho
Transpõe as Diretivas europeias
Dispositivos médicos implantáveis ativos (DMIA)
DIR 90/385/CEE
Dispositivos médicos (DM)
DIR 93/42/CEE
Legislação aplicável Estudos Clínicos

Lei nº 21/2014 (LIC) Definições – Artigo 2º
– Estudo Clínico com Dispositivos Médicos - Art.º 2º, r)qualquer estudo com dispositivos médicos ou respetivosacessórios, que integram o âmbito de aplicação do Decreto-
Lei n.º 145/2009, de 17 de junho, e cujo objetivo inclua:
i) Verificar o nível de desempenho do dispositivo ou
ii) Determinar eventuais efeitos secundários indesejáveis em condições normais de utilização e avaliar se constituem riscos em função da utilização prevista para o dispositivo segundo a legis artis ou
iii) Realizar o acompanhamento clínico pós-comercialização - PMCF

Acompanhamento Clínico no Pós-mercado
Estudo levado a cabo após marcação CEde um dispositivo de forma a darrespostas a questões específicasrelacionadas com a segurança oudesempenho clínico (ex: riscos residuais)do dispositivo quando usado de acordocom o previsto.
PMCF
Avaliação Clínica

Estudos indicados nas seguintes situações:
- DMs inovadores;
- Após aprovação de um nova indicação/finalidade;
- DMs de classe de risco elevada;
- Questões sem resposta quanto à segurança e desempenho a longo termo;
- Riscos identificados na literatura ou outra fonte de dados relativos a DMs semelhantes colocados no mercado;
- Sensibilidade da população alvo;
- Quando a marcação CE foi baseada em equivalência, etc.
Avaliação Clínica
Acompanhamento Clínico no Pós-mercado
PMCF

Avaliação dos Estudos Clínicos - LIC
Autorização - Notificação
INFARMED I.P.
(Artº 25º)
Parecer (favorável prévio) de CEC (Comissão de Ética
Competente) no prazo de 30 dias (úteis) – (Art.16º)
a CES que funciona no centro de estudo clínico envolvido
a CEIC ou uma CES por ela designada (se centro não dispõe de CES)
(Art.16º)
Para todosos estudos clínicos
Restantes estudos
a CEIC ou – (Art.16º)
uma CES (desig. pela CEIC)
(Art.16º)
+EC comintervenção DM (e ensaios Clínicos)
Submissão simultânea (Artº 25º)

Autorização da Autoridade Competente (INFARMED,I.P.):
DM classe III
DM implantáveis ativos
DM implantáveis
DM invasivos IIa e IIb para utilização por longos prazos
No prazo máximo de 30 dias úteis o conselho diretivo do INFARMED, I.P.,
delibera sobre o pedido de autorização.
Para as restantes classes o início do estudo depende apenas da notificação
à Autoridade Competente e do parecer favorável da Comissão de Ética
Competente (CEC).
Procedimento administrativo
Art. 33.º, Lei n.º 21/2014
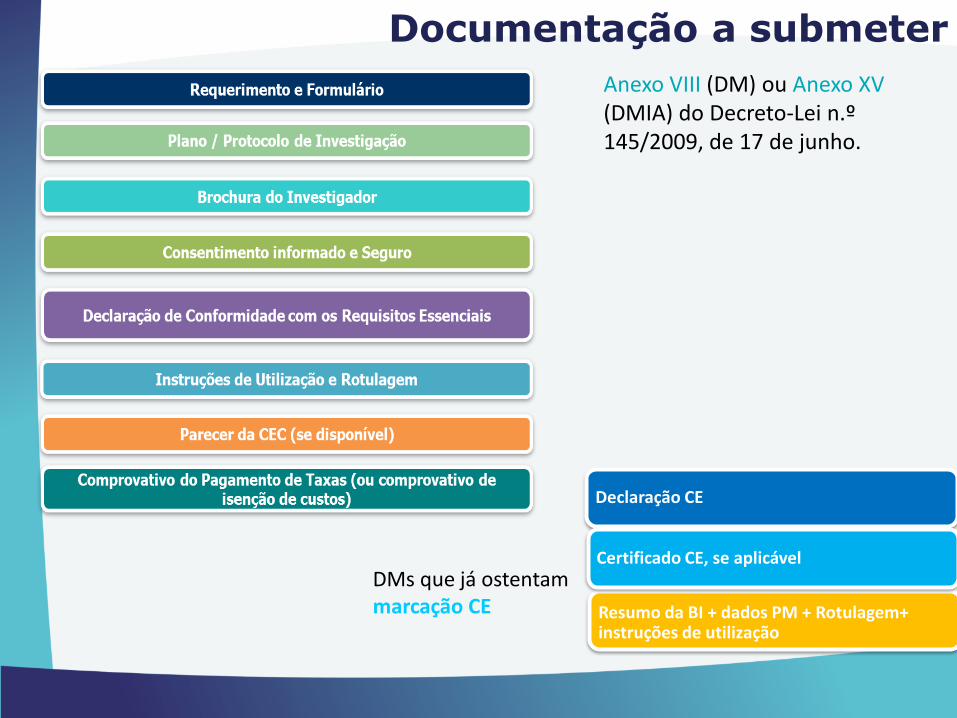
Documentação a submeter
Anexo VIII (DM) ou Anexo XV (DMIA) do Decreto-Lei n.º 145/2009, de 17 de junho.
DMs que já ostentam marcação CE
Declaração CE
Resumo da BI + dados PM + Rotulagem+ instruções de utilização
Certificado CE, se aplicável

Outras notificações previstas na LIC
• Alterações ao Protocolo [Artº 18º]
O promotor deve solicitar à CEC a modificação substancial do protocolo, e notifica ainda o INFARMED, I. P.
• Conclusão do Estudo Clínico [Artº 19º]
O promotor notifica a CEC e o INFARMED,I.P. no prazo de 90 dias a contar da data de conclusão da participação do último participante no estudo clínico.
O relatório final deve ser apresentado no prazo de 12 meses.
No caso de antecipação da conclusão do estudo por motivos de segurança, a notificação é transmitida no prazo de 15 dias.
• Registo e notificação de acontecimentos adversos [Artº 22]

Acontecimentos adversos graves
Defeitos dos dispositivos
Notificação do investigador ao promotor
24 horas 24 horas
Notificação do promotor à AC e CEC
2 dias 7 dias
Artigo 22º, Lei n.º 21/2014
Durante a realização do estudo clínico com intervenção e até à suaconclusão, o promotor apresenta anualmente à CEC e à AutoridadeCompetente, uma lista de todas as suspeitas de acontecimentos adversosgraves ocorridos durante esse período, bem como um relatório relativo àsegurança dos participantes.
Notificação de Acontecimentos Adversos

Legislação Nacional
DL nº 145/2009, de 17 de junho
http://www.infarmed.pt/portal/page/portal/INFARMED/LEGISLACAO/LEGISLACAO_FARMACEUTICA_COMPILADA/TITULO_V/TITULO_
V_CAPITULO_II/122-A_DL_145_2009.pdf
DL nº 189/2000, de 12 de agosto
http://www.infarmed.pt/portal/page/portal/INFARMED/LEGISLACAO/LEGISLACAO_FARMACEUTICA_COMPILADA/TITULO_V/TITULO_
V_CAPITULO_II/125_DL_189_2000_VP.pdf
Lei n.º 21/2014, de 16 de abril
http://www.infarmed.pt/portal/page/portal/INFARMED/LEGISLACAO/LEGISLACAO_FARMACEUTICA_COMPILADA/TITULO_III/TITULO_
III_CAPITULO_I/036-B1_Lei_21_2014_1alt.pdf
Legislação e guidance Europeia
Diretivas Europeias:
http://ec.europa.eu/growth/sectors/medical-devices/regulatory-framework/index_en.htm
European Guidance - MEDDEVs
http://ec.europa.eu/growth/sectors/medical-devices/guidance/index_en.htm
Propostas da Presidência [Junho 2015] para os Regulamentos sobre DMs e DIVs
http://www.consilium.europa.eu/en/press/press-releases/2015/06/19-medical-devices-council-ready-talk-with-ep/
Leitura Recomendada

Obrigada pela vossa atenção

Sónia Cardoso
Direção de Produtos de Saúde
INFARMED, I.P.
www.infarmed.pt
http://m.infarmed.pt
Consulte também:
https://twitter.com/INFARMED_IP
http://www.linkedin.com/company/infarmed
23 anos de serviço público com valores e ética