
Bruno Lazzari de Lima - USP · A Síndrome de Marfan (SMF) é uma doença de tecido conjuntivo, com...
115
Instituto de Biociências Universidade de São Paulo Caracterização da variabilidade fenotípica em um modelo animal para Síndrome de Marfan Bruno Lazzari de Lima São Paulo 2011
Transcript of Bruno Lazzari de Lima - USP · A Síndrome de Marfan (SMF) é uma doença de tecido conjuntivo, com...

Instituto de Biociências Universidade de São Paulo
Caracterização da variabilidade fenotípica em um modelo animal para Síndrome de Marfan
Bruno Lazzari de Lima
São Paulo 2011

Bruno Lazzari de Lima
Caracterização da variabilidade fenotípica em um modelo animal para Síndrome de Marfan
Characterization of the phenotypic variability in a mouse model for Marfan Syndrome
Tese apresentada ao Instituto de Biociências da Universidade de São Paulo, para a obtenção de Título de Doutor em Ciências, na Área de Biologia / Genética. Orientadora: Dra. Lygia da Veiga Pereira Carramaschi.
São Paulo 2011

COMISSÃO JULGADORA
____________________________ ____________________________
Prof(a). Dr(a). Prof(a). Dr(a).
Instituição. Instituição.
____________________________ ____________________________
Prof(a). Dr(a). Prof(a). Dr(a).
Instituição. Instituição.
__________________________________________
Profa. Dra. Lygia da Veiga Pereira Carramaschi
Orientadora
Lima, Bruno Lazzari de Caracterização da variabilidade fenotípica em um modelo animal para Síndrome de Marfan.
114 páginas Tese de Doutorado - Instituto de Biociências da Universidade de São Paulo. Departamento de Genética e Biologia Evolutiva. Área de concentração: Biologia/Genética 1. Síndrome de Marfan 2. Fibrilina-1 3. Modelos animais Universidade de São Paulo. Instituto de Biociências. Departamento de Genética e Biologia Evolutiva

Dedico este trabalho,
- aos meus pais, Ana Maria e João Antônio, por
sempre estarem presentes para me ouvir,
aconselhar e incentivar em todos os momentos da
minha vida;
- à minha tia e madrinha Marcia Cristina, minha
segunda mãe.

AGRADECIMENTOS
____________________________________________________

Meus sinceros votos de agradecimento, Aos meus pais, meus orientadores na vida. Por toda sua dedicação, carinho,
apoio e tudo aquilo que somente pais podem nos dar. Pela sua paciência nos meus momentos de estresse, pelas palavras certas nos momentos difíceis e por sempre estarem lá. Pelo beijinho de boa noite, pelo dinheirinho da balada, pela ajuda na lição de casa, pelo passeio do final de semana...
À Dra. Lygia, pela oportunidade de realizar esse projeto. Pela sua paciência, dedicação e conhecimentos repassados durante todo o desenvolvimento do mesmo. Foi muito bom trabalhar com alguém que tem tanta empolgação com aquilo que faz.
À Dra. Mariz Vainzof, minha madrinha dentro da ciência, tendo me ajudado e contribuído para o meu desenvolvimento desde o princípio da minha carreira acadêmica.
Aos demais professores do departamento de genética e biologia evolutiva, pelos conhecimentos compartilhados.
À Dra Silvia pela sua colaboração com a manutenção dos animais, tão importantes para o desenvolvimento deste projeto, e por toda sua ajuda.
À minha tia Marcia Cristina, sem dúvida nenhuma, a principal influência para que eu quisesse um dia me tornar um doutor. E por todos os inesquecíveis momentos da minha vida.
Aos meus familiares, tio Jose, tia Vera e tio Silvio, meus primos Thiago, André, Carolina, Rodrigo, Virginia, Guilherme e Henrique, meu avô Antônio e minha avó Gentil (i.m.) Por serem alguns dos pilares da minha vida.
Aos meus antigos amigos de faculdade, em especial ao Alex, Marcelo (Sinhá), Vitor Hugo (Vitão), Gledsley (Gepeto), Bruno (Nelas), Natale (Nani), Ana, Sabrina (Timão). Por todos os momentos alegres e divertidos, que são tão importantes na vida de uma pessoa.
À Natássia, uma pessoa muito especial na minha vida. Pelo seu companheirismo e por toda a sua colaboração pessoal e profissional durante todos esses anos.
A todos os meus colegas de laboratório que, de alguma maneira, sempre colaboraram e me ajudaram. Em especial ao Gustavo, pela sua fundamental colaboração neste trabalho, e à Ana Maria, Joana, Érica e Raquel, pelas conversas e conselhos.
A todos os meus colegas de departamento, vizinhos de laboratório e funcionários do IB-USP.
Aos meus novos amigos, em especial à Camila, Fernanda Donata, Fernanda Raquel, Flavio, João, Larissa, Manuela, Marcelle, Micheli, Natália, Priscilla, Rafael, Taísa, Waleska e Kalana. Passar as manhãs com vocês me mantém feliz pelo resto do dia.
À Laura, que mesmo estando a tão pouco tempo na minha vida, tem me proporcionado tanto bons e inesquecíveis momentos. Obrigado por tudo que você tem feito, pelo seu carinho e sua paciência.
À Mauy (i.m.), Victória (i.m.), Batata (i.m.), Jeep (i.m.) e Thyla, meus irmãos de quatro patas. “Um cachorro não se importa se você é rico ou pobre, inteligente ou idiota, esperto ou burro... dê seu coração a ele, e ele lhe dará o dele... De quantas pessoas você pode falar isso?” (J. Grogan).
À FAPESP pelo apoio financeiro.

“A vida é aquilo que acontece enquanto fazemos
planos para o futuro.”
(John Lennon)

RESUMO
____________________________________________________

A Síndrome de Marfan (SMF) é uma doença de tecido conjuntivo, com caráter autossômico dominante, que acomete cerca de 1 em 5.000 indivíduos. As principais manifestações clínicas incluem aneurismas e rompimento da aorta, crescimento excessivo dos ossos, escoliose e deformidades torácicas. Mutações no gene FBN1, que codifica a proteína de matriz extracelular fibrilina-1, foram relacionadas à doença, fazendo com que essa fosse classificada no grupo das fibrilinopatias. Mais de 500 mutações já foram identificadas e, com exceção de um pequeno grupo de mutações recorrentes, as mutações são únicas, sendo encontradas em famílias isoladas. A doença caracteriza-se pela grande variabilidade tanto intra quanto interfamilial, não sendo possível fazer uma correlação precisa entre genótipo e fenótipo. Este trabalho visa discutir os mecanismos responsáveis pela variabilidade clínica inter e intra familiar da SMF através da caracterização qualitativa e quantitativa da variabilidade fenotípica observada no modelo murino para SMF mgΔloxPneo . Neste sentido, caracterizamos o modelo mgΔloxPneo em duas linhagens de camundongos diferentes, C57BL/6 e 129/Sv. Os animais mutantes de ambas as linhagens apresentaram deficiência na deposição de microfibrilas, cifose de coluna, enfisema pulmonar e degeneração da parede aórtica. Contudo, a idade de início dos sinais fenotípicos mostrou-se mais tardia em animais da linhagem C57BL/6 em comparação com os animais 129/Sv, indicando a presença de genes modificadores entre as duas linhagens. Além disso, caracterizamos uma grande variabilidade fenotípica entre os animais 129/Sv mutantes, o que é sugestivo do envolvimento de fatores epigenéticos na gravidade da doença. Finalmente, demonstramos uma forte correlação negativa entre os níveis globais de transcrição do gene Fbn1 e a gravidade do fenótipo. Esses resultados corroboram a hipótese de que o nível de expressão da proteína normal está relacionado com a gravidade do quadro clínico da SMF em humanos. Com base nisso, o trabalho também visa o estudo de novas estratégias terapêuticas para a SMF nesse mesmo modelo.

ABSTRACT
____________________________________________________

The Marfan syndrome (MFS) is an autosomal dominant disease of connective tissue, which affects 1 in 5,000 individuals. The main clinical manifestations include aneurysms and aortic disruption, excessive growth of bones, scoliosis and thoracic deformities. Mutations in the FBN1 gene, which encodes the fibrillin-1 protein, were genetically linked to the MFS, classifying this disease in the fibrilinopathies group. Over 500 mutations have been identified and, except for a small group of recurrent mutations, the mutations are unique, being found in unrelated families. The disease is characterized by a wide clinical variability both within and between families, and it is not possible to make a precise genotype-phenotype correlation. This work concerns the analysis of the mechanisms associated with the clinical variability present within and between MFS families, by qualitative and quantitative characterization of the phenotypic variability observed in the mgΔloxPneo model for MFS. We characterize the model mgΔloxPneo, in two different mouse strains, the C57BL/6 and the 129/Sv strain. Mutant animals from both strains present defective microfibrillar deposition, emphysema, deterioration of aortic wall and kyphosis. However, the onset of a clinical phenotypes is earlier in the 129/Sv than in C57BL/6 background, indicating the existence of genetic modifiers of MFS between these two mouse strains. In addition, we characterized a wide clinical variability within the 129/Sv heterozygotes, suggesting involvement of epigenetic factors in disease severity. Finally, we show a strong negative correlation between overall levels of Fbn1 expression and the severity of the phenotypes. These results corroborated with studies, using animal models, as well with MFS patients, where the levels of normal fibrillin-1 seem to have the potential to modulate the clinical severity of the disease. In addition, the study also aims to evaluate new treatment possibilities for MFS in this same model.

SUMÁRIO
I. INTRODUÇÃO ................................................................................................... 17
1.1. Um breve histórico .............................................................................................................18
1.2. A Síndrome de Marfan .......................................................................................................20
1.3. O gene FBN1 e a SMF ......................................................................................................24
1.4. A Fibrilina-1 e os possíveis mecanismos patogênicos ........................................................26
1.5. O efeito das mutações do gene FBN1 sobre a citosina TGF-β1 .........................................30
1.6. As proteínas associadas à via de sinalização de TGF-β1 ...................................................32
1.7. A utilização de fármacos no tratamento da síndrome de Marfan .........................................34
1.8. Os modelos murinos para a síndrome de Marfan ...............................................................35
1.9. O modelo mgΔloxPneo
..........................................................................................................40
II. OBJETIVOS ....................................................................................................... 44
2.1. Objetivo Geral ....................................................................................................................45
2.2. Objetivos Específicos .........................................................................................................45
III. MATERIAL E MÉTODOS ................................................................................... 46
3.1. Animais .............................................................................................................................47
3.2. Genotipagem dos animais .................................................................................................47
3.3. Análise fenotípica dos animais ...........................................................................................49
3.3.1. Análise radiográfica .......................................................................................................49
3.3.2. Análises histopatológicas ...............................................................................................50
3.4. Análise imunohistoquímica ................................................................................................51
3.5. Administração da droga Ramipril em animais heterozigotos da linhagem C57BL/6.............51
3.6. Real Time PCR (RT-PCR) .................................................................................................52
3.7. Western-Blot para o estudo da proteína phospho-smad2 ...................................................53
IV. RESULTADOS ................................................................................................... 56
4.1. Fenótipo ............................................................................................................................57
4.1.1. Caracterização fenotípica do modelo mgΔloxPneo
.............................................................57
4.1.2. Quantificação das alterações fenotípicas .......................................................................58
4.1.3. A variabilidade fenotípica da linhagem 129/Sv................................................................63
4.1.4. Imunohistoquímica .........................................................................................................66
4.2. Análise dos níveis de transcrição do gene Fbn1 por Real Time PCR (RT-PCR) .................68
4.2.1. Comparação da transcrição do alelo Fbn1 x Fbn1mgΔloxPneo
em fibroblastos fetais............68
4.2.2. Comparação da transcrição do alelo Fbn1 x Fbn1mgΔloxPneo
em animais adultos e a sua
correlação com a variação de fenótipo ..........................................................................................69

4.2.2.1. A diferença de transcrição entre as linhagens C57BL/6 e 129/Sv ...............................69
4.2.2.2. A relação entre a variação de transcrição do gene Fbn1 com a idade.........................71
4.2.2.3. Comparação da transcrição do gene Fbn1 entre animais da linhagem 129/Sv com
diferentes fenótipos .......................................................................................................................72
4.2.3. Efeito da droga Ramipril sobre a transcrição global do gene Fbn1 em animais C57BL/6
mutantes 74
4.3. Avaliação da atividade de TGF-β1 nos tecidos afetados ....................................................76
V. DISCUSSÃO ...................................................................................................... 78
5.1. Caracterização fenotípica do modelo mgΔloxPneo
.................................................................80
5.2. Avaliação da atividade de TGF-β1 nos tecido aórtico e pulmonar do modelo mgΔloxPneo
.....82
5.3. As alterações presentes na imunohistoquímica de fibroblastos fetais em cultura ................84
5.4. A diferença fenotípica entre os animais mutantes das linhagens 129/Sv e C57BL/6 ...........85
5.5. A variabilidade fenotípica entre os animais mutantes da linhagem 129/Sv ..........................87
5.6. Os mecanismos de patogênese relacionados com a Síndrome de Marfan .........................88
VI. CONCLUSÕES .................................................................................................. 94
VII. REFERÊNCIAS .................................................................................................. 97
VIII. ANEXO ............................................................................................................. 105

LISTA DE FIGURAS
FIGURA 1. O pediatra francês Atonine-Bernard Marfan e sua primeira paciente a ser descrita como portadora da dolicoestenomelia, Gabrielle P...................................... 18
FIGURA 2. Algumas personalidades das quais se suspeita que fossem portadoras da SMF ...................................................................................................................... 20
FIGURA 3. Características clínicas mais freqüentes da SMF ................................... 21
FIGURA 4. Cirurgia profilática visando o reparo da aorta por meio da substituição da região do aneurisma por uma prótese. ...................................................................... 22
FIGURA 5. Outras características clínicas da SMF).................................................. 23
FIGURA 6. Uma criança com a forma neonatal da SMF ........................................... 25
FIGURA 7. Dois indivíduos com uma mesma mutação no gene FBN1, porém com fenótipos diferentes ................................................................................................... 26
FIGURA 8. Estrutura primária da fibrilina-1 ............................................................... 28
FIGURA 9. A relação entre a fibrilina-1 e a LTBP-1. ................................................. 31
FIGURA 10. Representação esquemática para a via de sinalização da citosina TGF-β1 .............................................................................................................................. 33
FIGURA 11. Reversão do fenótipo aórtico do modelo murino C1039G para SMF tratado com Losartan................................................................................................. 34
FIGURA 12. O modelo mgΔ. ..................................................................................... 36
FIGURA 13. O modelo mgR. A. ................................................................................ 37
FIGURA 14. O modelo C1039G ................................................................................ 39
FIGURA 15. O modelo Tight Skin ............................................................................. 40
FIGURA 16. Representação esquemática do alelo Fbn1mgΔloxPneo ............................ 41
FIGURA 17. Heredograma dos camundongos heterozigotos fundadores da linhagem mgΔloxPneo .................................................................................................................. 42
FIGURA 18. Mensurações utilizadas no cálculo de KR. ........................................... 49
FIGURA 19. Análise histológica dos pulmões (marcação por HE) ............................ 59
FIGURA 20. Análise histológica da aorta torácica (coloração de Weigert)................ 60

FIGURA 21. Análise radiográfica. ............................................................................. 61
FIGURA 22. Gráficos comparativos dos valores de Lm, espessura da parede da aorta (EPA) e KR, entre animais mutantes das linhagens 129/Sv e B6 aos três meses de idade. ........................................................................................................ 62
FIGURA 23. Correlação entre a progressão dos fenótipos pulmonar (Lm), aórtico (Espessura da aorta - EPA) e ósseo (KR) com a idade (meses) em animais heterozigotos da linhagem 129/Sv ............................................................................ 63
FIGURA 24. Análise comparativa da variabilidade fenotípica na linhagem 129/Sv ... 65
FIGURA 25. Variabilidade fenotípica presente na linhagem 129/Sv ......................... 66
FIGURA 26. Imunofluorescência de fibroblastos fetais em cultura, com marcação para fibrilina-1 ............................................................................................................ 67
FIGURA 27. Quantificação relativa dos níveis totais de mRNA do gene Fbn1 em fibroblastos fetais selvagens, heterozigotos e homozigotos por Real Time PCR ...... 68
FIGURA 28. Quantificação relativa dos níveis totais de mRNA do gene Fbn1 em animais selvagens ..................................................................................................... 70
FIGURA 29. Quantificação dos níveis mRNA referentes ao alelo selvagem do gene Fbn1 em animais selvagens e mutantes heterozigotos, aos três e seis meses de idade.......................................................................................................................... 71
FIGURA 30. Correlação entre a transcrição total do gene Fbn1 e a idade de animais B6 e 129/Sv ............................................................................................................... 72
FIGURA 31. Correlação entre a transcrição do gene Fbn1 e a gravidade do fenótipo em animais heterozigotos da linhagem 129/Sv ......................................................... 73
FIGURA 32. Gráfico comparativo da transcrição do gene Fbn1 em animais B6 selvagens, heterozigotos sem tratamento e heterozigotos tratados com a droga Ramipril ..................................................................................................................... 74
FIGURA 33. Análise dos efeitos do tratamento com Ramipril em animais B6 .......... 75
FIGURA 34. Western-blot para a proteína phospho-Smad2 (P-smad2) a partir de amostras protéicas pulmonares e aórticas de animais B6 de três e seis meses de idade.......................................................................................................................... 77

LISTA DE ABREVIATURAS
ACE - Angiotensin-converting enzyme.
Ang II - Angiotensina II.
AT1 - Angiotensin receptor type I.
C57BL/6 - B6.
cbEGF - Calcium-binding EGF-precursor-like.
dNTP - desorribonuleotídeo trifosfatado.
EGF - Epidermal growth factor.
EPA - Espessura da parede da aorta.
ERK1 - Extracellular signal-regulated kinase 1.
ERK2 - Extracellular signal-regulated kinase 2.
Het - Heterozigoto.
JNK - C - Jun N - terminal kinase.
Kb - Kilobases.
kDa - Kilodaltons.
KO - Knockout.
KR - Kifosis ratio.
kV - Kilovoltagem.
Lm - Mean linear intercept.
LTBP - Latent TGF-β binding protein.
µm - Micrômetro.
mA - Microamperes.
MAPK - MAP Kinases.
MEC - Matriz extracelular.
n - Número.
neoR - Cassete de resistência à neomicina.
ng - Nanogramas.
nm - Nanomolar.
pb - Pares de base.
PBS - Phosphate buffered saline.
PCR - Polimerase chain reaction.
PI3K-Akt - Phosphoinositide 3-kinase-Akt.
P-smad2 - Phospho-Smad2.

R-Smads - Receptor-regulated Smads.
RT-PCR - Real-Time PCR.
SMF - Síndrome de Marfan.
TGF-β1 - Transforming growth factor β1.
Tm - Temperatura de melting.
Tsk - Tight Skin.
UV - Ultravioleta.
WT - Wild-type.

I. INTRODUÇÃO ____________________________________________________

INTRODUÇÃO 18
1.1. Um breve histórico
Em 1896, Antonine-Bernard Marfan, professor de pediatria em Paris,
descreveu uma menina com cinco anos de idade, Gabrielle P (Fig.1), a qual
apresentava membros e dedos desproporcionalmente longos e magros,
característica denominada por ele como pattes d´araignée ou patas de aranha. A
paciente apresentava ainda contraturas nas articulações dos dedos e joelhos, um
crânio estreito e alongado (dolicocefalia), além de uma estatura muito elevada para
sua idade. Embora seus olhos, coração e intelecto fossem normais, exames
radiográficos indicaram a presença de cifoescoliose tóraco-lombar, deformidade na
região esternal (pectus excavatum) e enfisema pulmonar (PYERITZ, 2000).
FIGURA 1. O pediatra francês Atonine-Bernard Marfan (esquerda) e sua primeira paciente a ser descrita como portadora da dolicoestenomelia, Gabrielle P (direita) (Fonte: http://www.pifo.uvsq.fr/hebergement/marfan/).

INTRODUÇÃO 19
Em quarenta anos após esse primeiro relato clínico, Marfan avaliou mais de
150 casos similares, os quais foram denominados como dolicoestenomelia, termo
preferido pelo médico devido aos membros excessivamente longos. Nessa ocasião,
outras alterações associadas já haviam sido reconhecidas, como subluxação do
cristalino (ectopia lentis), malformação da valva átrio-ventricular esquerda (valva
mitral) e complicações aórticas de dissecação e dilatação (SCHORR et al., 1951).
Marfan notou que ambos os sexos eram igualmente afetados, e que as alterações
podiam ser recorrentes em gerações seqüenciais de uma mesma família,
características compatíveis com um padrão de herança mendeliana dominante
(PYERITZ, 2000).
Atualmente, vários atletas jovens morrem por complicações aórticas da
Síndrome de Marfan (SMF), alguns dos quais foram diagnosticados somente através
de retrospectos clínicos. A morte de uma estrela do voleibol olímpico norte-
americano em 1986, Flo Hyman (Fig.2), aumentou a consciência e a informação
entre o público geral, atletas, treinadores e médicos ao longo do mundo (BAYLES,
2002). Até os dias de hoje, continua o debate a respeito do diagnóstico de SMF do
ex-presidente norte-americano Abraham Lincoln (Fig.2), pois esse era alto,
dolicostenomélico e apresentava hiperflexibilidade articular (SCHWARTZ, 1964 ;
LATTIMER, 1981). Contudo, casos de personalidades famosas não se restringem
aos séculos atuais, pois se acredita que o faraó Akhenaten (Fig.2), o qual governou
o Egito antigo de 1350 a 1334 a.C. e fora pai do faraó Tutankhamun, possuísse
várias das características relacionadas à síndrome (ALDRED & SANDISON, 1962).

INTRODUÇÃO 20
FIGURA 2. Algumas personalidades das quais se suspeita que fossem portadoras da SMF. A jogadora de vôlei norte-americana Flo Hyman (esquerda), o ex-presidente norte-americano Abraham Lincoln (centro) e o faraó Akhenaten (Fonte: http://lewiscrusade.wordpress.com/category/marfan-syndrome/).
1.2. A Síndrome de Marfan
A SMF (OMIM 154700) (SCHORDERET, 1991) é uma doença pleitrópica,
com manifestações clínicas diversas, e aparentemente independentes, em muitos
tecidos e órgãos (PYERITZ, 1989 ; 1993). Trata-se de uma doença com caráter
autossômico dominante, que acomete cerca de 1 em 5.000 indivíduos (ADES,
2007). As manifestações clínicas incluem aneurismas e rompimento da aorta,
descolamento da retina, subluxação do cristalino, crescimento excessivo dos ossos,
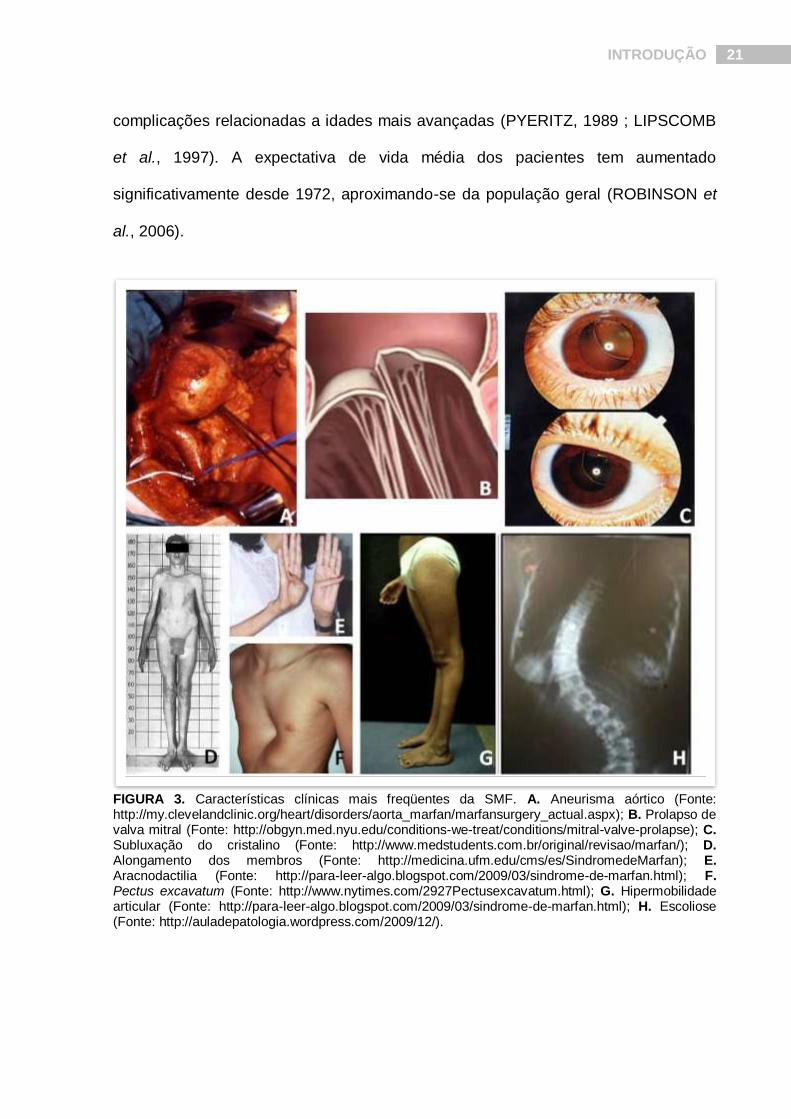
hipermobilidade articular, escoliose e deformidades torácicas (Fig.3) (SILVERMAN et
al., 1995a ; SILVERMAN et al., 1995b) . Essa gama de pleiotropismo vem
aumentando devido a intervenções terapêuticas modernas, que possibilitam uma
maior longevidade aos indivíduos afetados, permitindo a visualização de
INTRODUÇÃO 21
complicações relacionadas a idades mais avançadas (PYERITZ, 1989 ; LIPSCOMB
et al., 1997). A expectativa de vida média dos pacientes tem aumentado
significativamente desde 1972, aproximando-se da população geral (ROBINSON et
al., 2006).
FIGURA 3. Características clínicas mais freqüentes da SMF. A. Aneurisma aórtico (Fonte: http://my.clevelandclinic.org/heart/disorders/aorta_marfan/marfansurgery_actual.aspx); B. Prolapso de valva mitral (Fonte: http://obgyn.med.nyu.edu/conditions-we-treat/conditions/mitral-valve-prolapse); C. Subluxação do cristalino (Fonte: http://www.medstudents.com.br/original/revisao/marfan/); D. Alongamento dos membros (Fonte: http://medicina.ufm.edu/cms/es/SindromedeMarfan); E. Aracnodactilia (Fonte: http://para-leer-algo.blogspot.com/2009/03/sindrome-de-marfan.html); F. Pectus excavatum (Fonte: http://www.nytimes.com/2927Pectusexcavatum.html); G. Hipermobilidade articular (Fonte: http://para-leer-algo.blogspot.com/2009/03/sindrome-de-marfan.html); H. Escoliose (Fonte: http://auladepatologia.wordpress.com/2009/12/).

INTRODUÇÃO 22
Embora algumas intervenções terapêuticas, como a utilização de β-
bloqueadores visando uma diminuição stress hemodinâmico, e cirurgias profiláticas
para o reparo da aorta (Fig.4), possibilitem um aumento significativo na expectativa
de vida dos pacientes, essas manifestações ainda são responsáveis por uma
elevada taxa de mortalidade dos mesmos (SILVERMAN et al., 1995a).
FIGURA 4. Cirurgia profilática visando o reparo da aorta por meio da substituição da região do aneurisma por uma prótese (Fonte: http://my.clevelandclinic.org/).
Até pouco tempo, a descrição da SMF, bem como seus critérios
diagnósticos, estavam focados nas avaliações referentes aos três principais órgãos
e sistemas envolvidos: o sistema esquelético, os olhos e o coração juntamente com
a aorta. Contudo, a pele, a face, os pulmões, os músculos esqueléticos e tecido
adiposo, estão claramente envolvidos (Fig.5). Ocasionalmente o envolvimento de
qualquer uma desses sistemas pode ser classificado como a principal manifestação
clínica de um determinado paciente, assim, para que seja realizado um diagnóstico
adequado, faz-se necessário uma avaliação muito criteriosa (PYERITZ, 2000).

INTRODUÇÃO 23
FIGURA 5. Outras características clínicas da SMF. A. Deformidades faciais; B. Enfisema pulmonar; C. Elevação do palato; D. Lesões de pele (Fonte: http://para-leer-algo.blogspot.com/2009/03/sindrome-de-marfan.html).
Dilemas diagnósticos podem ocorrer devido à grande variabilidade clínica
inter e intrafamilial da SMF. Além disso, características como prolapso da valva
mitral, escoliose, deslocamento do cristalino etc., podem estar presentes em outras
doenças do tecido conjuntivo (ROBINSON & GODFREY, 2000). Assim o diagnóstico
da SMF deve seguir as recomendações estabelecidas pelo Comitê Internacional de
Nosologia (Ghent nosology) (PYERITZ, 2000). De acordo com essas
recomendações, em um caso isolado é necessário que pelo menos dois dos
sistemas cardinais na síndrome (ocular, ósseo e cardiovascular) apresentem
alterações, como luxação do cristalino, e dilatação ou dissecção da aorta, bem como
o envolvimento de um terceiro sistema. Nos casos familiares, o indivíduo deve
apresentar a principal manifestação descrita em sua família, associada a uma
alteração em dos sistemas cardinais, e o envolvimento de um terceiro sistema.

INTRODUÇÃO 24
A descoberta da relação entre mutações entre o gene FBN1 com o
desenvolvimento das manifestações fenotípicas da SMF, permitiu uma maior
exatidão no diagnóstico de determinados casos (LOEYS et al., 2010). Novas
técnicas moleculares permitem a detecção de mutações no gene FBN1 em até 97%
dos pacientes com SMF que preencheram todos os critérios estabelecidos pelo
comitê (LOEYS et al., 2001 ; LOEYS et al., 2004).
1.3. O gene FBN1 e a SMF
Em 1991, mutações no gene FBN1 (OMIM 134797), que codifica a proteína
de matriz extracelular (MEC) fibrilina-1, foram relacionadas à SMF, fazendo com que
essa doença fosse classificada no grupo das fibrilinopatias (DIETZ et al., 1991 ;
KAINULAINEN & PELTONEN, 1991). Trata-se de um gene relativamente grande,
localizado na região 15q21.1, com aproximadamente 230 kb e composto por 65
exons (PEREIRA et al., 1993). Mais de 500 mutações, distribuídas em quase todos
os exons do gene, já foram identificadas e, com exceção de um pequeno grupo de
mutações recorrentes, as mutações são únicas, sendo encontradas em famílias
isoladas (BOILEAU et al., 2005).
Aproximadamente 25% de todas as mutações já identificadas no gene FBN1
em pacientes com SMF resultam em inserções ou deleções de bases na seqüência
do gene que levam a uma troca no quadro de leitura do mesmo e a um códon de
parada de tradução prematuro (BOILEAU et al., 2005).

INTRODUÇÃO 25
Embora ainda não seja possível fazer uma correlação precisa entre genótipo
e fenótipo, mutações em determinadas regiões do gene parecem estar relacionadas
com a gravidade da doença. Mutações descritas na região entre os exons 24 e 32
foram associadas à forma mais grave da doença, a forma neonatal de SMF (Fig.6)
(ROBINSON & GODFREY, 2000). Mutações descritas na região entre os exons 59 e
65 foram associadas à forma intermediária da doença (PALZ et al., 2000). Porém
essas associações não possuem valor preditivo da gravidade do fenótipo.
FIGURA 6. Uma criança com a forma neonatal da SMF (modificado de TEKIN et al., 2007).
A grande maioria das mutações foi descrita como apresentando penetrância
completa (PYERITZ, 1986), embora alguns indivíduos portadores de mutações no
gene FBN1 não apresentem qualquer um dos sinais clínicos mais evidentes da SMF.
Dentre esses casos raros, nos chama mais atenção um caso familiar, onde dois
indivíduos, um filho de 18 anos de idade e sua mãe de 41 anos de idade, eram
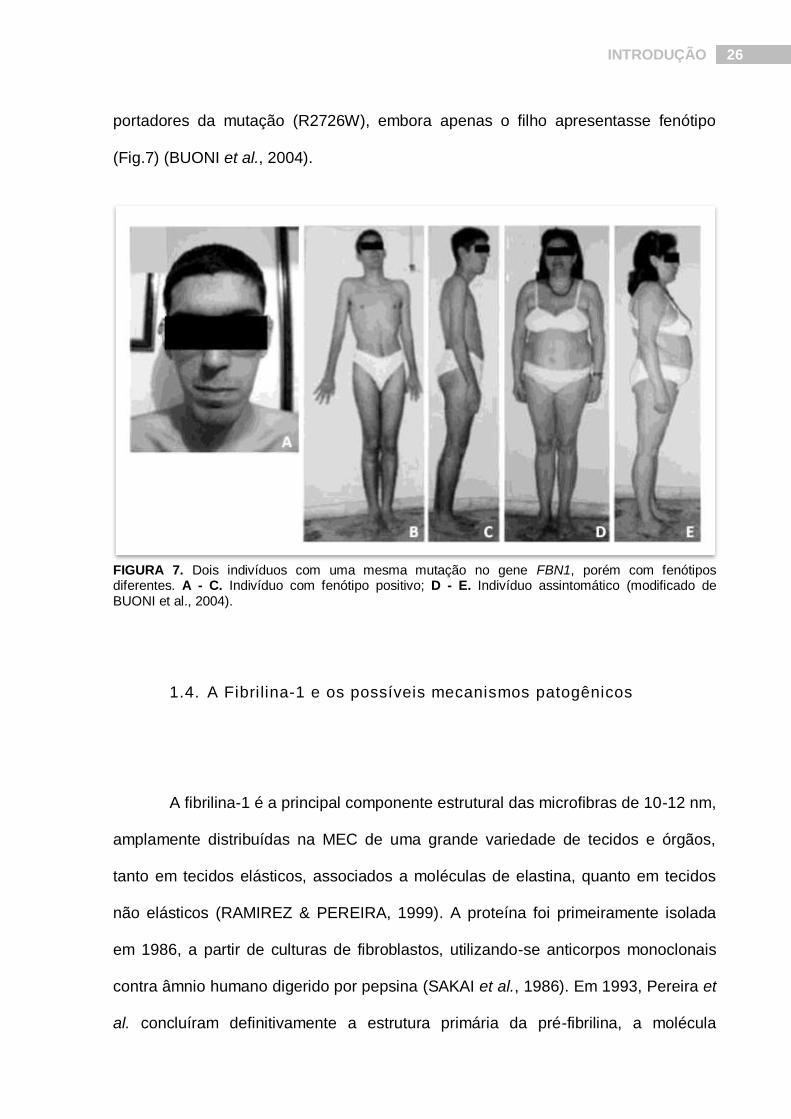
INTRODUÇÃO 26
portadores da mutação (R2726W), embora apenas o filho apresentasse fenótipo
(Fig.7) (BUONI et al., 2004).
FIGURA 7. Dois indivíduos com uma mesma mutação no gene FBN1, porém com fenótipos diferentes. A - C. Indivíduo com fenótipo positivo; D - E. Indivíduo assintomático (modificado de BUONI et al., 2004).
1.4. A Fibrilina-1 e os possíveis mecanismos patogênicos
A fibrilina-1 é a principal componente estrutural das microfibras de 10-12 nm,
amplamente distribuídas na MEC de uma grande variedade de tecidos e órgãos,
tanto em tecidos elásticos, associados a moléculas de elastina, quanto em tecidos
não elásticos (RAMIREZ & PEREIRA, 1999). A proteína foi primeiramente isolada
em 1986, a partir de culturas de fibroblastos, utilizando-se anticorpos monoclonais
contra âmnio humano digerido por pepsina (SAKAI et al., 1986). Em 1993, Pereira et
al. concluíram definitivamente a estrutura primária da pré-fibrilina, a molécula

INTRODUÇÃO 27
precursora da fibrilina-1, caracterizando-a como um peptídeo composto por 2871
aminoácidos e com um peso molecular estimado de 347 kDa.
A fibrilina-1 é uma glicoproteína cujas moléculas apresentam em sua
estrutura diversas repetições homólogas, ricas em cisteínas, de forma semelhante à
encontrada em outras proteínas, como as fibulinas-1 e 2 e a proteína de ligação ao
TGF-β1 (LTBP). Essas repetições são classificadas como unidades calcium-binding
EGF-precursor-like (cbEGF), por serem semelhantes aos já descritos módulos EGF
(epidermal growth factor), e apresentam algumas funções críticas, como proteção
contra as ações proteolíticas extracelulares, mediação da interação entre
monômeros, favorecimento do empacotamento lateral de microfibras, organização
de macroagregados, entre outras (KIELTY & SHUTTLEWORTH, 1995). A fibrilina-1
é composta basicamente de quarenta e sete módulos EGF, sendo que quarenta e
três destes módulos associados ao cálcio (cbEGF), sete módulos TB (módulos com
oito cisteínas, primeiramente descritos na LTBP) e três módulos Fib, módulos
híbridos que possuem características tanto dos domínios EGF quanto dos domínios
TB (Fig.8) (PEREIRA et al., 1993).

INTRODUÇÃO 28
FIGURA 8. Estrutura primária da fibrilina-1. EGF (epidermal growth factor); TB (LTBP - latent transforming growth factor-β binding protein); cbEGF (calcium binding epidermal growth factor) ; Fib (hibridos de EGF e TB); N-glycos (sítio de glicosilação); furina/PACE (sítio de endoprotease); RGD (seqüência Arg-Gly-Asp) (SANTOS, 2005).
A maior parte da síntese de fibrilina-1 parece estar associada a processos
tardios de morfogênese. Estudos sugerem que as fibrilinas estão relacionadas ao
processo de eslatogênese, pois as microfibras estão presentes na MEC antes da
deposição de tropoelastinas. Por esse motivo, uma deficiência na síntese ou na
estrutura da fibrilina-1 poderia prejudicar a formação de fibras elásticas, levando a
processos patológicos como a dissecção da aorta (ZHANG et al., 1995).
As microfibras podem desempenhar diversas funções, em diferentes tecidos.
Em tecidos não elásticos, elas apresentam a função de ancoragem, como por
exemplo, a ligação entre as lentes e o corpo ciliar dos olhos. Em tecidos elásticos,
elas promovem a adesão entre as fibras elásticas, e dessas as outras estruturas e a

INTRODUÇÃO 29
componentes celulares. A relação funcional entre a fibrilina-1 e o esqueleto pode
estar ligada às forças de tensão que regulam o crescimento ósseo. Microfibras ricas
em fibrilina-1, presentes no periósteo, podem controlar negativamente o crescimento
dos mesmos, pela manutenção de tensões periósteas, e pela força de
tensionamento dos ligamentos e tendões (ZHANG et al., 1995).
Durante a sua secreção celular as moléculas de profibrilina-1 são
processadas e agregadas à MEC junto com outras proteínas, formando assim as
microfibras (KIELTY et al., 2002). Esse processo ainda não está completamente
compreendido, e por isso o mecanismo patogênico da SMF permanece
desconhecido. Contudo, por tratar-se de uma proteína multimérica, uma hipótese
sugere que os monômeros de fibrilina-1 mutados poderiam exercer um efeito
dominante negativo sobre os monômeros normais em pacientes heterozigotos
(DIETZ et al., 1993a). Pacientes com um menor nível de expressão de proteína
mutada parecem apresentar um quadro clínico da SMF mais leve, corroborando com
este modelo dominante negativo (DIETZ et al., 1993b).
Uma segunda hipótese sugere que um mecanismo de haploinsuficiência
possa estar associado com a doença. Em estudos realizados com modelos animais,
assim como em pacientes com SMF, o nível de expressão da proteína normal
mostrou estar relacionado com a gravidade do quadro clínico da doença
(HUTCHINSON et al., 2003 ; JUDGE et al., 2004). Em um estudo foi descrito um
indivíduo com alterações esqueléticas que possuía a mutação R2726W no gene
FBN1 (MILEWICZ et al., 1995). Essa mutação gera uma alteração na região C-
terminal da proteína fibrilina-1, impedindo que os monômeros mutantes possam se
agregar às microfibras – na prática o equivalente a um alelo nulo do FBN1. Isso

INTRODUÇÃO 30
sugere que esse indivíduo deve apresentar um fenótipo associado a um modelo de
haploinsuficiência.
Uma vez que os níveis de expressão da fibrilina-1 normal parecem
apresentar o potencial de modular os efeitos patogênicos das proteínas mutadas, é
possível que uma variação no nível de expressão dos alelos normais do gene FBN1
possa explicar parte da heterogeneidade clínica encontrada em pacientes com SMF
(HUTCHINSON et al., 2003).
1.5. O efeito das mutações do gene FBN1 sobre a citosina TGF-β1
Algumas das características da SMF, como alterações no crescimento dos
músculos esqueléticos e no metabolismo de gorduras, e o desenvolvimento de
enfisema pulmonar, não parecem estar relacionadas com falhas na função estrutural
desempenhada pela fibrilina-1 (BYERS, 2004). Essas alterações parecem estar mais
relacionadas com perturbações de desenvolvimento, do que com falhas
simplesmente mecânicas (NEPTUNE et al., 2003).
Após a sua síntese e processamento, as moléculas de fibrilina-1 passam a
compor a matriz extracelular, associando-se a outras proteínas, como a LTBP-1
(BYERS, 2004). As LTBPs são muito importantes por propiciarem um sítio de ligação
para as moléculas latentes de TGF-β1 (transforming growth factor β1), até que essas
sejam liberadas e ativadas, devido a perturbações nos elementos constituintes da
MEC (ANNES et al., 2003). Elas são membros da família de proteínas LTBP/fibrilina,

INTRODUÇÃO 31
a qual compreende a fibrilina-1, fibrilina-2 e fibrilina-3, e as LTBP-1, LTBP-2, LTPB-3
e LTBP-4 (NAKAJIMA et al., 1999 ; BYERS, 2004). A similaridade da seqüência e da
estrutura entre as LTBPs e as fibrilinas (Fig.9A) sugeriu que a fibrilina-1 também
poderia prover um sítio de ligação para moléculas latentes de TGF-β1 (NAKAJIMA et
al., 1999). Contudo, a fibrilina-1 não parece ligar-se diretamente a essa proteína
(SAHARINEN & KESKI-OJA, 2000). Por outro lado, a porção N-terminal da fibrilina-1
está ligada à porção C-terminal da LTPB-1 (Fig.9B), permitindo que alterações na
MEC levem à liberação de moléculas latentes de TGF-β1, passando para sua forma
ativa (KAARTINEN & WARBURTON, 2003).
FIGURA 9. A relação entre a fibrilina-1 e a LTBP-1. A. A Similaridade entre as duas proteínas (NEPTUNE et al., 2003); B. A ligação da porção N-terminal da fibrilina à porção C-terminal da LTPB-1 (BYERS, 2004).
O TGF-β1 é uma importante citosina moduladora de crescimento celular,
processos inflamatórios, síntese da MEC, ativação de proteinases e apoptose
(TAIPALE et al., 1998). Mudanças no padrão de atividade de TGF-β1 estão
associadas com várias alterações, como crescimento de células tumorais, fibroses e
doenças autoimunes (BLOBE et al., 2000). Este padrão de atividade é

INTRODUÇÃO 32
primariamente definido pela conversão das moléculas latentes de TGF-β1 para sua
forma ativa (ANNES et al., 2003).
A hipótese de que a fibrilina-1 é mais do que apenas uma proteína estrutural,
e que poderia estar participando no controle da sinalização de TGF-β1 foi testada
em camundongos com mutações do tipo knockdown no gene Fbn1 (NEPTUNE et
al., 2003). Estes animais apresentam níveis aumentados de TGF-β1 ativo nos seus
pulmões, assim como um aumento do espaço alveolar, e desenvolvimento de
enfisema durante o seu envelhecimento. Essa alteração foi revertida a partir do
tratamento desses animais com anticorpos para TGF-β1, ainda em fase uterina.
1.6. As proteínas associadas à via de sinalização de TGF-β1
As proteínas Smad são componentes essenciais das vias de sinalização
intracelular dependentes de TGF-β1, e participam da formação de fibroses induzidas
por TGF-β1 (Fig.10) (MASSAGUE & CHEN, 2000). Após sua ativação, a citosina
TGF-β1 transmite seu sinal através da membrana plasmática, ancorando-se a 2
receptores serina/treonina kinases específicos, dos tipos I e II. Ao ancorar-se ao
receptor tipo II, o qual ativa o receptor tipo I, faz com que este último fosforile duas
receptor-regulated Smads (R-Smads), Smad2 e Smad3. Ambas acabam por se
dissociar do complexo receptor, para formar um novo complexo heterotrimérico com
a Smad4. Por fim esses complexos são translocados para o núcleo, atuando como
reguladores transcricionais de genes alvos (MASSAGUE & CHEN, 2000 ;
JAVELAUD & MAUVIEL, 2004). Assim, a quantificação da proteína Smad2

INTRODUÇÃO 33
fosforilada é um bom método para a determinação do nível de atividade da citosina
TGF-β1 em tecidos.
FIGURA 10. Representação esquemática para a via de sinalização da citosina TGF-β1 (COHN et al., 2007).
A proteína Angiotensina II (Ang II) é uma reguladora de crescimento celular,
processos inflamatórios e fibrose, contribuindo para o surgimento de danos
vasculares. Em diferentes doenças e processos celulares, Ang II tem o papel
regulatório sobre a expressão de TGF-β1, o qual pode mediar algumas respostas à
Ang II. Drogas inibidoras da ACE (Angiotensin-converting enzyme) e antagonistas de
AT1 (receptor tipo I para angiotensina-II) diminuem a expressão de TGF-β1 e
formação de fibroses, assim como o bloqueio de TGF-β1 diminui a produção de Ang
II na matriz extracelular (BORDER & NOBLE, 1998 ; RUIZ-ORTEGA et al., 2003 ;
RODRIGUEZ-VITA et al., 2005).

INTRODUÇÃO 34
Baseados nesta nova função da fibrilina-1, de moduladora dos níveis de
TGF-β1 ativo na matriz, novas estratégias terapêuticas para a SMF foram testadas.
A administração de uma droga antagonista de AT1, Losartan, em um modelo murino
para SMF foi capaz de diminuir a atividade de TGF-β1, resultando em reversão dos
fenótipos pulmonares e aórticos desses animais (Fig.11) (HABASHI et al., 2006).
Atualmente um tratamento similar em seres humanos está em teste clínico em vários
países (DETAINT et al., 2010 ; RADONIC et al., 2010).
FIGURA 11. Reversão do fenótipo aórtico do modelo murino C1039G para SMF tratado com Losartan (HABASHI et al., 2006).
1.7. A utilização de fármacos no tratamento da síndrome de Marfan
Devido aos problemas vasculares agudos presentes em pacientes
portadores da SMF, como principalmente o rompimento da artéria aorta, um dos
tratamentos mais utilizados para a síndrome baseia-se na administração de drogas
anti-hipertensivas. Em geral tais drogas reduzem a rigidez das paredes arteriais por
ação passiva, diminuindo a pressão arterial. Pesquisas recentes vêm demonstrando
que algumas das drogas mais utilizadas, em particular as inibidoras da ACE e as
antagonistas de AT1, podem ter um efeito adicional sobre a constituição da parede

INTRODUÇÃO 35
arterial, o que pode contribuir na redução da sua rigidez (AHIMASTOS et al., 2005).
Dentre essas drogas, encontramos o Losartan, descrito anteriormente, e o Ramipril.
Ahimastos et al. (2005) descreveram que a droga Ramipril parece ter grande
influência na estrutura das paredes arteriais. Nesse estudo os pesquisadores
demonstraram que células humanas de músculo liso tratadas em cultura com
Ramipril apresentam um decréscimo na deposição de colágeno da ordem de 50%,
assim como um aumento na deposição de elastina e fibrilina-1 da ordem de três
vezes e quatro vezes, respectivamente. Além disso, a droga leva a uma redução da
transcrição dos genes MMP-2 e MMP-3, que codificam as metaloproteinases 2 e 3,
envolvidas na degradação de proteínas da MEC (BOOMS et al., 2000).
1.8. Os modelos murinos para a síndrome de Marfan
Um dos temas de estudos emergentes é o desenvolvimento de
camundongos mutantes para o estudo de doenças genéticas. Os camundongos
(Mus musculus) mostram-se uma espécie com grandes vantagens para a realização
de pesquisas científicas. São animais de fácil armazenamento e manipulação, com
uma elevada taxa de reprodução, atingem idade reprodutiva rapidamente e possuem
um período curto de gestação. Além disso, atualmente já se encontra disponível, em
bancos de dados públicos, a seqüência genômica da espécie, e existem diversas
técnicas bem estabelecidas de manipulação do genoma desses animais.

INTRODUÇÃO 36
No caso da SMF, três modelos animais foram criados visando o estudo (1)
da doença nos diferentes sistemas afetados; (2) e da função da proteína fibrilina-1
durante o desenvolvimento embrionário e a vida pós-natal (PEREIRA et al., 1999).
O primeiro modelo, a linhagem mgΔ, possui uma deleção em fase dos exons
19 a 25 do gene Fbn1 (Fig.12), substituídos por um cassete de expressão do gene
de resistência à neomicina (neoR) (PEREIRA et al., 1997). Surpreendentemente, os
animais heterozigotos não apresentam nenhum fenótipo, enquanto que os
homozigotos morrem com até duas semanas de vida pós-natal por falhas no sistema
cardiovascular. A análise detalhada do alelo Fbn1mgΔ revelou que este produz
somente 10% dos níveis de mRNA produzidos pelo alelo normal, sugerindo que de
fato o fenótipo da SMF seja decorrente da presença de quantidades significativas de
fibrilinas mutantes, de acordo com o modelo dominante-negativo. A baixa
quantidade de moléculas de fibrilinas mutantes naqueles animais não seria
suficiente para causar a desorganização das microfibras.
FIGURA 12. O modelo mgΔ. A. Representação esquemática do alelo Fbn1
mgΔ. Os retângulos vazios
representam os exons do gene Fbn1 e o retângulo cinza o cassete NeoR; B. Análise por Northern-blot desmontando que o alelo Fbn1
mgΔ (direita) produz somente 10% dos níveis de mRNA produzidos
pelo alelo normal (PEREIRA et al., 1997).

INTRODUÇÃO 37
A linhagem mgR é resultado de uma recombinação aberrante entre o vetor
de recombinação homóloga e a seqüência alvo do gene Fbn1 (Fig.13) (PEREIRA et
al., 1999). De maneira similar ao alelo Fbn1mgΔ, o alelo Fbn1mgR apresenta o cassete
neoR inserido no intron 18, no entanto a seqüência endógena, correspondente aos
exons 19 a 25, não foi deletada, e assim o alelo Fbn1mgR produz fibrilina-1 normal.
Como os animais mg, os mgR heterozigotos não possuem nenhum fenótipo. Já os
as animais mgR homozigotos apresentam uma longevidade superior aos da
linhagem mgΔ, embora também apresentem alterações vasculares. A análise do
alelo Fbn1mgR revelou que este produz 20% dos níveis de mRNA do alelo normal, e
assim o fenótipo dos mgR homozigotos é devido à diminuição dos níveis de fibrilina-
1 normal (PEREIRA et al., 1999).
FIGURA 13. O modelo mgR. A. Representação esquemática do alelo Fbn1
mgR. Os retângulos
brancos representam os exons do gene Fbn1, as formas cinza representam os exons deletados na linhagem mgΔ , e o retângulo cinza grande representa o cassete neoR; B. Análise por Northern-blot demonstrando que o alelo Fbn1
mgR (direita) produz somente 20% dos níveis de mRNA produzidos
pelo alelo normal (PEREIRA et al., 1999).
Nesses modelos foram identificados entre outros: o papel da fibrilina-1 no
suporte estrutural da fibra elástica na vida pós-natal e não na formação desta
durante o desenvolvimento embrionário (PEREIRA et al., 1997); a contribuição da
resposta inflamatória na progressão das lesões vasculares (PEREIRA et al., 1999); o

INTRODUÇÃO 38
papel da fibrilina-1 no controle da ativação de TGF-β1 no pulmão, e a contribuição
desta citosina nas manifestações da SMF (NEPTUNE et al., 2003).
Apesar de serem instrumentos de pesquisa importantes, esses dois modelos
animais apresentaram o fenótipo somente em homozigose. Aparentemente, a
presença do cassete neoR no intron 18 leva à baixa expressão dos alelos mutantes
Fbn1mgΔ e Fbn1mgR, impedindo a manifestação do efeito dominante-negativo
característico da SMF (PEREIRA et al., 1997 ; PEREIRA et al., 1999). Assim, para a
geração de um modelo animal para a SMF, seria necessário utilizar o sistema de
recombinação CRE-loxP (SUNAGA et al., 1997) para eliminar o cassete de
expressão neoR e assim obter um alelo mutante Fbn1 com nível de expressão
equivalente ao do alelo selvagem.
O terceiro modelo animal para a SMF, a linhagem de camundongos
C1039G, é caracterizado pela substituição de uma cisteína por uma glicina na
posição 1039, em um domínio EGF da fibrilina-1, sendo este o tipo mais comum de
mutação em FBN1 em indivíduos com SMF. Animais heterozigotos apresentam
deficiência na deposição de microfibrilas na MEC, alterações esqueléticas e
progressiva deterioração da arquitetura da parede aórtica (Fig.14) (JUDGE et al.,
2004).

INTRODUÇÃO 39
FIGURA 14. O modelo C1039G. A. Imagem radiográfica de um animal heterozigoto C1039G; B. Comparação histológica da parede aórtica de um animal selvagem (WT) e um animal heterozigoto C1039G. À esquerda coloração HE e à direita coloração de Weigert, específica para a visualização de fibras elásticas. A flecha indica um ponto de calcificação (JUDGE et al., 2004).
Além dos modelos desenvolvidos em laboratório, temos ainda o modelo
Tight Skin (Tsk), um mutante natural, que por possuir uma mutação no gene Fbn1,
apresenta algumas características da SMF, incluindo o crescimento exagerado dos
ossos longos e enfisema pulmonar. A mutação desses animais caracteriza-se pela
duplicação da região dos exons 19 a 24. Tal alteração resulta na produção de uma
da fibrilina-1 mutada contendo 984 aminoácidos a mais, gerando uma glicoproteína
de 418 kDa. O animal Tsk heterozigoto apresenta um aumento na produção de
tecido ósseo, enfisema pulmonar e um aumento do tecido conjuntivo (Fig.15).
Diferente dos modelos anteriores, este animal não apresenta complicações
cardiovasculares características da SMF. Quando em homozigose, o modelo Tsk
morre ainda no estágio embrionário de pré-implantação (GAYRAUD et al., 2000).

INTRODUÇÃO 40
FIGURA 15. O modelo Tight Skin. Comparação de cortes histológicos de pulmão de um animal selvagem e um animal mutante, indicando a presença de enfisema em animais Tsk (GREEN et al., 1976).
1.9. O modelo mgΔ loxPneo
O modelo mgΔloxPneo é uma variante do modelo mgΔ, no qual o mesmo alelo
mutante está presente, porém com o cassete neoR flanqueado por duas seqüência
lox-P (Fig.16), permitindo a deleção do cassete de resistência através da proteína
CRE-recombinase (SANTOS, 2005). O objetivo da criação deste alelo do gene Fbn1
foi o desenvolvimento de um modelo onde fosse possível controlar a expressão do
alelo mutante, quando esses animais são cruzados com animais transgênicos para a
proteína CRE-recombinase.

INTRODUÇÃO 41
FIGURA 16. Representação esquemática do alelo Fbn1
mgΔloxPneo. Os triângulos vermelhos
representam a seqüência loxP flanqueando o gene neoR. Os retângulos vazios representam os exons do gene Fbn1 e os retângulos cinza representam os exons 19-24 deletados no alelo mgΔ
loxPneo.
As barras azuis representam as sondas de DNA utilizadas para identificação da mutação por Southern-blot.
Como observado no modelo mgΔ original, os animais mgΔ loxPneo
heterozigotos da primeira geração (F1) não apresentavam nenhum fenótipo.
Contudo, em cruzamentos subseqüentes, foram obtidos alguns animais, também
heterozigotos, com um fenótipo muito grave da SMF, caracterizado por
deformidades na coluna vertebral e alterações pulmonares (enfisema) (Fig.17). De
um total de 47 animais heterozigotos, quatro apresentaram as características
mencionadas, e morreram aos três meses de idade por causas desconhecidas,
porém a presença de hemotórax sugeriu ruptura da aorta (SANTOS, 2005).

INTRODUÇÃO 42
FIGURA 17. Heredograma dos camundongos heterozigotos fundadores da linhagem mgΔ
loxPneo. Os
animais híbridos (129/Sv / CD1) eram assintomáticos até que em F3 um animal gravemente afetado foi encontrado (flecha). A radiografia desse animal encontra-se na figura (SANTOS, 2005).
Todos estes animais eram derivados de cruzamentos entre duas linhagens
murinas diferentes, a linhagem outbred CD-1 e a linhagem isogênica 129/Sv. Esse
fato foi sugestivo de que a variabilidade fenotípica observada poderia estar
relacionada com diferenças no background genético dos animais. Para testar essa
hipótese, o alelo Fbn1mgΔloxPneo foi colocado em duas linhagens isogênicas, a
linhagem C57BL/6 e a 129/Sv (SANTOS, 2005).
Os animais heterozigotos da linhagem C57BL/6 mostraram-se
assintomáticos, enquanto que os animais heterozigotos da linhagem 129/Sv podiam
apresentar desde um fenótipo leve até um fenótipo muito grave, reproduzindo a
variabilidade fenotípica interfamilial observada na SMF. Assim o modelo mgΔ loxPneo
mostrou-se adequado não só para o estudo dos mecanismos de patogênese

INTRODUÇÃO 43
relacionados à SMF, como potencialmente para o estudo da variabilidade clínica da
síndrome (SANTOS, 2005).

II. OBJETIVOS ____________________________________________________

OBJETIVOS 45
2.1. Objetivo Geral
Este trabalho visa a análise dos mecanismos responsáveis pela variabilidade
clínica inter e intra familiar da SMF através da caracterização qualitativa e
quantitativa da variabilidade fenotípica observada no modelo mgΔloxPneo em duas
linhagens murinas diferentes. Além disso, o trabalho também visa o estudo de novas
possibilidades de tratamento para a SMF nesse mesmo modelo.
2.2. Objetivos Específicos
1. Caracterizar a variabilidade fenotípica nos sistemas ósseo, pulmonar e
cardiovascular.
2. Comparar o padrão de expressão do gene Fbn1 entre animais da linhagem
C57BL/6 e 129/Sv e correlacionar esta expressão com a variabilidade
fenotípica observada em animais da linhagem 129/Sv.
3. Definir o mecanismo patogênico relacionado com o fenótipo dos animais
mutantes (Dominante negativo X Haploinsuficiência).
4. Testar os efeitos de Ramipril sobre a transcrição do gene Fbn1 e o fenótipo
dos animais mutantes.

III. MATERIAL E MÉTODOS ____________________________________________________

MATERIAL E MÉTODOS 47
3.1. Animais
Todos os animais utilizados foram mantidos no biotério de linhagens
isogênicas do Departamento de Imunologia do ICB-USP, sobre condições
controladas de luz e temperatura, e em ambiente livre de agentes potencialmente
patogênicos. Obtivemos a aprovação do comitê de Ética em Pesquisa Animal,
protocolo CEA/IBUSP 020/2004.
Foram analisados 45 animais mutantes e 20 animais selvagens, em três
idades diferentes (três, seis e nove meses), pertencentes a duas linhagens murinas
congênicas. A linhagem C57BL/6 (B6) foi escolhida por ser a linhagem murina mais
utilizada em estudos genéticos, além de ser a linhagem de todos os modelos
murinos para SMF (PEREIRA et al., 1997 ; SAITO et al., 1999 ; JUDGE et al., 2004).
A linhagem 129/Sv foi selecionada porque as células embrionárias utilizadas durante
o desenvolvimento da linhagem mgΔloxPneo eram derivadas desta linhagem
(SANTOS, 2005).
3.2. Genotipagem dos animais
Amostras de DNA foram extraídas de fragmentos de cauda medindo 0,5 cm
de comprimento, como previamente descrito (ZANGALA, 2007). Cada amostra foi
submetida a duas PCRs, para a identificação da presença do alelo Fbn1mgΔloxPneo e
do alelo selvagem, o qual foi utilizado como controle interno para a reação.

MATERIAL E MÉTODOS 48
Primers para amplificação do alelo Fbn1mgΔloxPneo (433bp; Tm 57°C):
Neo F: 5’ -GAG GCT ATT CGG CTA TGA CT – 3’
Neo R: 5’ – CTC TTC GTC CAG ATC ATC CT – 3’
Condições de amplificação: Etapa 1 (1 ciclo) - 94oC x 2,5 minutos. Etapa 2
(30 ciclos) - 94oC x 1 minuto; 55oC x 1 minuto; 72oC x 1 minuto. Etapa 3 (1 ciclo) -
72oC x 10 minutos.
Primers para amplificação do alelo selvagem (600bp; Tm 55°C):
20F: 5’ – AAA CCA TCA AGG GCA CTT GC – 3’
CC-R: 5’ – CAC ATT GCG TGC CTT TAA TTC – 3’
Condições de amplificação: Etapa 1 (1 ciclo) - 94oC x 2,5 minutos. Etapa 2
(30 ciclos) - 94oC x 1 minuto; 55oC x 1 minuto; 72oC x 1 minuto. Etapa 3 (1 ciclo) -
72oC x 10 minutos.
As PCRs foram montadas adicionando-se 10 ng de cada primer, 1 l de
solução tampão 10x (Invitrogen), 1,6 mM de dNTP, 1 unidade da enzima taq DNA
polimerase (Invitrogen) e água deionizada para um volume final de 10 l. Após o
termino da etapa de amplificação, as amostras foram separadas por corrida
eletroforética em gel de agarose 1,5% e visualizadas em luz ultra violeta (UV).
MATERIAL E MÉTODOS 49
3.3. Análise fenotípica dos animais
3.3.1. Análise radiográfica
Os animais foram sacrificados por inalação de CO2 e posteriormente fixados
em cassetes contendo filme para radiografia. Seus membros posteriores e anteriores
foram distendidos, para que pudéssemos avaliar o grau de escoliose da coluna.
Posteriormente foram expostos a 4,0 Kv de raio-x durante o período de 3,0 ms
(milissegundos) (Tecno-Design – 500Ma/ 125 Kv).
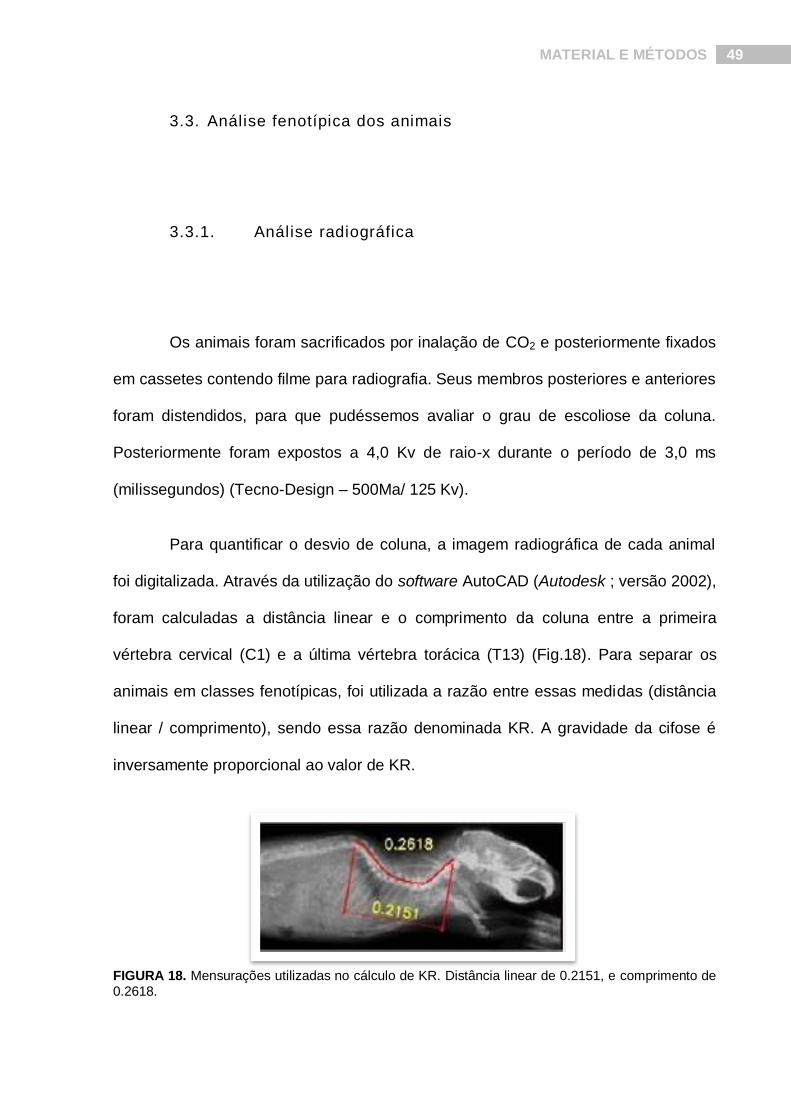
Para quantificar o desvio de coluna, a imagem radiográfica de cada animal
foi digitalizada. Através da utilização do software AutoCAD (Autodesk ; versão 2002),
foram calculadas a distância linear e o comprimento da coluna entre a primeira
vértebra cervical (C1) e a última vértebra torácica (T13) (Fig.18). Para separar os
animais em classes fenotípicas, foi utilizada a razão entre essas medidas (distância
linear / comprimento), sendo essa razão denominada KR. A gravidade da cifose é
inversamente proporcional ao valor de KR.
FIGURA 18. Mensurações utilizadas no cálculo de KR. Distância linear de 0.2151, e comprimento de 0.2618.

MATERIAL E MÉTODOS 50
3.3.2. Análises histopatológicas
Os pulmões e aorta foram submetidos às técnicas rotineiras de fixação,
desidratação, diafanização, inclusão em parafina e microtomia (ANDRIKOPOULOS
et al., 1995). Cortes medindo 5 µm de espessura foram corados com hematoxilina-
eosina (HE), e cortes adjacentes foram corados por coloração de Weigert, específica
para a visualização de fibras elásticas. Os cortes corados foram examinados e
fotografados utilizando-se o equipamento Axiovert 200 (Carl Zeiss).
Parede aórtica: As amostras foram fotografadas em ampliações de 50x e
100x. Foram realizadas mensurações do perímetro interno e externo da aorta com o
auxílio do software ImageJ (MURAISHI, 2010). Com o valor do perímetro calculamos
o raio (R) do vaso (Perímetro=2πR) e então o diâmetro do mesmo. A espessura do
vaso é calculada pela metade do valor obtido pela subtração do perímetro externo
pelo interno.
Tecido pulmonar: As amostras foram fotografadas na ampliação de 50x. O
tamanho dos espaços aéreos alveolares foi avaliado pela mensuração do diâmetro
alveolar médio pulmonar (Lm) como previamente descrito (DUNNILL, 1962).

MATERIAL E MÉTODOS 51
3.4. Análise imunohistoquímica
Culturas de fibroblastos fetais foram estabelecidas a partir de fetos com 13-
14 dias de gestação, seguindo-se protocolos previamente estabelecidos (HOGAN et
al., 1994).
As células foram fixadas em paraformaldeído 4% em PBS (Phosphate
buffered saline) durante 20 minutos a 4°C. Em seguida passaram pelo processo de
permeabilização em Triton X-100 0,05% em PBS durante 5 minutos e pelo processo
de bloqueio de marcações inespecíficas com BSA 10% em PBS durante 1 hora à
temperatura ambiente. Por fim as células foram incubadas com o anticorpo primário
pAb9543 (diluição 1:1000 em PBS) para a marcação de fibrilina-1 (REINHARDT et
al., 1996) overnight a 4°C, e a seguir com o anticorpo secundário associado a Cy3
por uma hora à temperatura ambiente. Os sinais fluorescentes foram avaliados com
o equipamento Axiovert 200 (Carl Zeiss) e com o software ApoTome imaging system
(Carl Zeiss).
3.5. Administração da droga Ramipril em animais heterozigotos da
linhagem C57BL/6
Seis animais mutantes, pertencentes à linhagem B6, receberam a
administração oral da droga Ramipril, durante o período de 24 semanas. A droga foi
diluída em uma razão de 15 mg/litro em água, e diariamente oferecida aos animais

MATERIAL E MÉTODOS 52
de forma livre. Seis animais controles de mesma linhagem, idade e sexo foram
utilizados para a comparação dos efeitos da droga sobre a transcrição do gene
Fbn1, in vivo, por Real time PCR. Após o período de tratamento os animais foram
sacrificados, e amostras teciduais de pulmão e aorta foram coletadas para análise.
3.6. Real Time PCR (RT-PCR)
Utilizando-se o regente TRIzol (Invitrogen) extraímos o RNA total de
amostras pulmonares, bem como de fibroblastos fetais mantidos em cultura, de
acordo com os protocolos do fabricante. Em seguida, 1 µg de RNA total,
previamente tratado com DNase (Invitrogen), foi reversamente transcrito em cDNA ,
através da utilização da enzima SuperScript III First-Strand Synthesis System
(Invitrogen).
Os níveis de mRNA selvagem, mutante e total do gene Fbn1 foram
determinados pelo equipamento de Real Time PCR (7500 Real Time System;
Applied Biosystems). O aparelho foi ajustado e programado com as condições de
PCR pré-estabelecidas: Etapa 1 (1 ciclo): 50°C x 2 minutos. Etapa 2 (1 ciclo): 95°C x
10 minutos. Etapa 3 (40 ciclos): 95°C x 15 segundos ; 60°C x 1 minuto. As amostras
foram preparadas para um volume total de 25 μl, contendo a amostra de cDNA a ser
estudada (50 ng), os primers (3 mM), e o tampão de amostra Universal PCR master
mix (Applied Biosystems). Os níveis de mRNA foram normalizados pelos níveis de
mRNA do gene Actb, de acordo com os protocolos previamente descritos (36).

MATERIAL E MÉTODOS 53
A seguir estão as seqüências de todos os primers e sondas, desenvolvidos
com o auxílio do software PrimerExpress (Applied Biosystems):
Alelo Selvagem: WT F 5´ – ACA TAA CTG GGA AAA ACT GTG TCG
ATA – 3´, WT R 5´ – TTC CAG GTG TGT TTC GAC ATT GT – 3´,
Sonda WT 5´ – TGT GCT GAA CAG TCT ACT – 3´.
Alelo Mutante: KO F 5´ – GGG ATA TGA AGT AGA CAT AAC TGG
GAA A– 3´, KO R 5´ – GAG GCT GGG TAT CAT CTT GCA – 3´,
Sonda KO 5´ – ACT GTG TCG ATA TCA ATG – 3´.
Global: Fbn1Total F 5´ – CCT GTG CTA TGA TGG GTT CA – 3´,
Fbn1Total R 5´ – AGG TCC CAC TAA GGC AGA TGT – 3´.
Controle endógeno: ACTB F 5´ – ACGGCCAGGTCATCACTATTG –
3´, ACTB R 5´ – CAAGAAGGAAGGCTGGAAAAGA – 3´.
3.7. Western-Blot para o estudo da proteína phospho-smad2
Quatrocentos μl de solução de extração de proteínas (1% de SDS, 0,3% de
EDTA, 1,2% Tris, DTT) foram adicionados ao total de células de uma garrafa de
cultura celular T75 ou a 5 mg de tecido. As amostras foram aquecidas e agitadas até
serem completamente solubilizadas, sendo em seguida quantificadas por
espectrofotometria, e igualmente diluídas em tampão de corrida eletroforética
(VAINZOF et al., 2003).
Cada uma das amostras foi fervida por dois minutos, e aplicada em gel de
poliacrilamida, passando em seguida por um processo eletroforético, para a

MATERIAL E MÉTODOS 54
separação das proteínas totais. Em seguida as amostras foram transferidas para
uma membrana de nitrocelulose, utilizando-se para isso tampão de transferência
(142,6 g glicina, 24,2 g Tris, 10 g SDS, água destilada para 1L), após serem
submetidas a uma corrente de 300 mA durante uma hora. Posteriormente a
membrana foi seca em estufa 37ºC overnight, lavada com água destilada e corada
com solução de Ponceu, para que pudéssemos verificar a presença de proteínas
(VAINZOF et al., 2003).
Para a localização da proteína em estudo, foi utilizado o anticorpo #3104
(Cell Signaling) para a proteína phospho-Smad2. A proteína β-actina foi utilizada
como normalizadora das análises comparativas. Por fim a imagem da membrana foi
digitalizada, e a quantificação da intensidade das bandas de proteínas foi feita com o
auxilio do software ImageJ (MURAISHI, 2010).
3.8. Análises estatísticas
O coeficiente de correlação de Pearson (R) foi utilizado para a determinação
do nível de correlação entre a expressão do gene Fbn1 com a gravidade dos
fenótipos; correlação fraca 0≤|R|≤0,29, correlação moderada 0,3≤|R|≤0,69 e
correlação forte |R|≥0,70. O nível de significância (P) foi fixado em 0,05.
O teste não paramétrico de Mann Whitney foi utilizado para avaliarmos a
significância estatística das comparações aqui apresentadas. Todas as análises

MATERIAL E MÉTODOS 55
estatísticas foram realizadas com o auxílio do software MINITAB (R14). O nível de
significância (P) foi fixado em 0,05.

IV. RESULTADOS ____________________________________________________

RESULTADOS 57
4.1. Fenótipo
4.1.1. Caracterização fenotípica do modelo mgΔ loxPneo
Os camundongos heterozigotos de ambas as linhagens possuem uma
longevidade e capacidade reprodutiva normais. Contudo, o cruzamento entre
animais heterozigotos resultou em 42% (n=30) de animais selvagens e 58% (n=41)
de animais heterozigotos, indicando que quando em homozigose, o alelo
Fbn1mgΔloxPneo torna-se letal. Embriões com esse genótipo foram identificados até o
13º dia de desenvolvimento embrionário.
Os animais heterozigotos apresentaram algumas das principais
características fenotípicas da SMF. Dentre essas, destacamos as alterações
pulmonares, que incluem o alargamento dos espaços aéreos (bronquíolos
respiratórios e alvéolos), assim como a degeneração da estrutura das paredes
alveolares, caracterizando um quadro de enfisema. Também pudemos detectar uma
grande quantidade de células mononucleares, indicativas de um processo
inflamatório crônico nos pulmões. O fenótipo vascular incluiu o espessamento da
túnica média da aorta, e a degradação e ruptura de suas fibras elásticas. Entretanto
não foram detectadas células inflamatórias nesse tecido. Finalmente, os animais
mutantes também apresentaram manifestações esqueléticas, representadas pela
cifose e alterações morfológicas do gradil costal.

RESULTADOS 58
4.1.2. Quantificação das alterações fenotípicas
Com o objetivo de caracterizarmos a variabilidade fenotípica nas duas
linhagens, o que nos permitiria separar os animais em grupos (assintomáticos, leve
e grave), o fenótipo encontrado nos sistemas afetados foi quantificado.
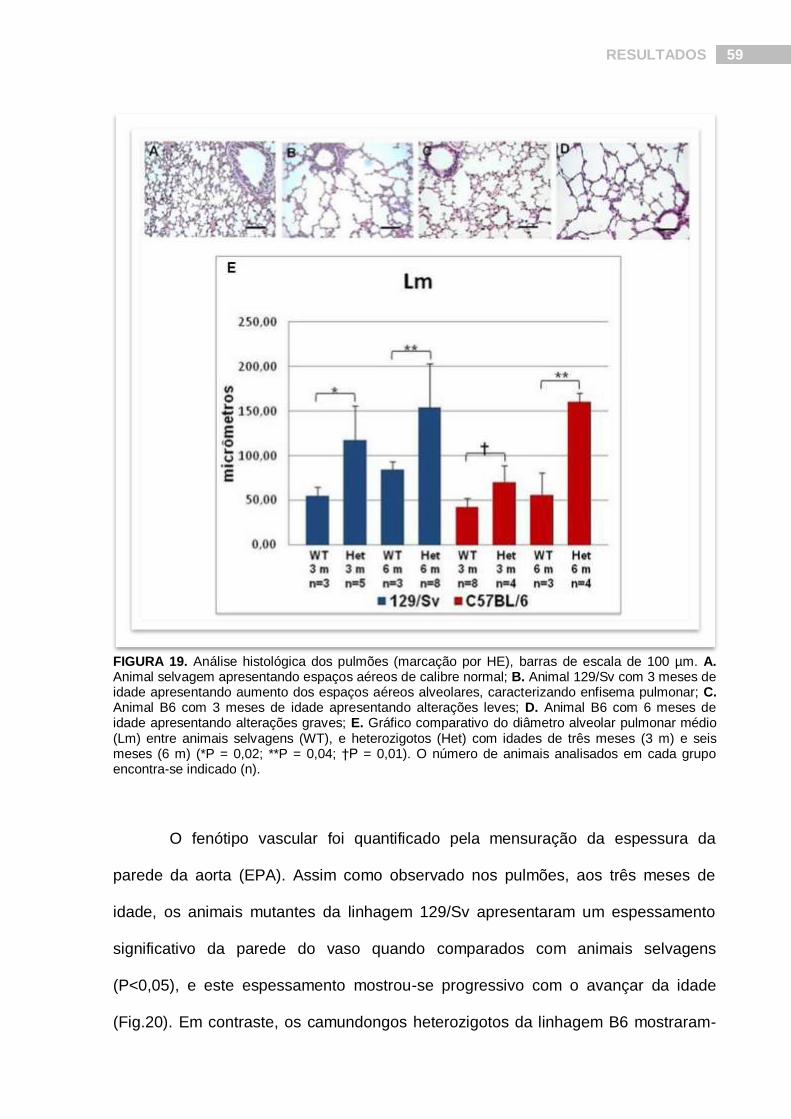
A análise do diâmetro alveolar pulmonar médio (Lm) revelou alterações no
tamanho dos alvéolos de animais heterozigotos da linhagem 129/Sv (Fig.19). Aos
três meses de idade, esses animais apresentaram um Lm significativamente elevado
quando comparado a animais selvagens (p<0,02), e esta diferença tornou-se mais
pronunciada com o avançar da idade. Por outro lado, animais B6 com três meses de
idade apresentaram um fenótipo pulmonar leve (embora significativo, p<0,01), o qual
se tornou grave apenas após os seis meses de idade (Fig.19).
RESULTADOS 59
FIGURA 19. Análise histológica dos pulmões (marcação por HE), barras de escala de 100 µm. A. Animal selvagem apresentando espaços aéreos de calibre normal; B. Animal 129/Sv com 3 meses de idade apresentando aumento dos espaços aéreos alveolares, caracterizando enfisema pulmonar; C. Animal B6 com 3 meses de idade apresentando alterações leves; D. Animal B6 com 6 meses de idade apresentando alterações graves; E. Gráfico comparativo do diâmetro alveolar pulmonar médio (Lm) entre animais selvagens (WT), e heterozigotos (Het) com idades de três meses (3 m) e seis meses (6 m) (*P = 0,02; **P = 0,04; †P = 0,01). O número de animais analisados em cada grupo encontra-se indicado (n).
O fenótipo vascular foi quantificado pela mensuração da espessura da
parede da aorta (EPA). Assim como observado nos pulmões, aos três meses de
idade, os animais mutantes da linhagem 129/Sv apresentaram um espessamento
significativo da parede do vaso quando comparados com animais selvagens
(P<0,05), e este espessamento mostrou-se progressivo com o avançar da idade
(Fig.20). Em contraste, os camundongos heterozigotos da linhagem B6 mostraram-

RESULTADOS 60
se assintomáticos aos três meses de idade, e somente apresentaram alterações
vasculares aos nove meses, sendo estas idênticas às alterações presentes em
animais 129/Sv de mesma idade (Fig.20).
FIGURA 20. Análise histológica da aorta torácica (coloração de Weigert), barras de escala de 100 µm. A. Animal selvagem; B. Animal 129/Sv com 3 meses de idade; C. Animal B6 assintomáticos com 3 meses de idade; D. Animal B6 com 9 meses de idade apresentando alterações graves. Podemos notar fibras elásticas rompidas na região da túnica média do vaso (flechas); E. Gráfico comparativo da espessura da parede da aorta entre animais selvagens (WT), e heterozigotos (Het) com idades de três meses (3 m), seis meses (6 m) e nove meses (9 m) (*P < 0,05; **P = 0,007; †P = 0,18; ††P 0,03). O número de animais analisados em cada grupo encontra-se indicado (n).
A quantificação das manifestações esqueléticas baseou-se no cálculo da
razão entre a distância linear e o comprimento do espaço entre a primeira vértebra
cervical (C1) e a última vértebra torácica (T13), sendo essa razão denominada KR.

RESULTADOS 61
Como observado nos demais sistemas, os animais heterozigotos da linhagem
129/Sv apresentaram um fenótipo esquelético grave mais precocemente do que os
animais da linhagem B6 (Fig.21).
FIGURA 21. Análise radiográfica. A. Animal selvagem; B. Animal 129/Sv com 3 meses de idade; C. Animal B6 com 3 meses de idade; D. Animal B6 com 6 meses de idade; E. Gráfico comparativo da KR média entre animais selvagens (WT), e heterozigotos (Het) com idades de três meses (3 m) e seis meses (6 m) (*P = 0,01 ; †P = 0,04). O número de animais analisados em cada grupo encontra-se indicado (n).
Nos gráficos a seguir (Fig.22) vemos que as alterações pulmonares e
aórticas são significativamente mais graves em animais 129/Sv aos três meses do
que em animais B6. Contudo os gráficos também nos demonstram diferenças não
significativas quando analisamos as alterações em coluna e aorta, o que é explicado
pela variabilidade fenotípica presente em animais da linhagem 129/Sv (resultados
mais adiante).

RESULTADOS 62
FIGURA 22. Gráficos comparativos dos valores de Lm (A), espessura da parede da aorta (EPA) (B) e KR (C), entre animais mutantes das linhagens 129/Sv (n=5) e B6 (n=4) aos três meses de idade (*P = 0,03; **P = 0,18; ***P = 0,25).
Após os seis meses de idade todos os animais mutantes, de ambas as
linhagens, apresentaram um fenótipo pulmonar, vascular e esquelético grave,
tornando-se ainda mais acentuado após os nove meses. Isso indica que a gravidade
do fenótipo observada nesses animais é progressiva. Essa avaliação encontra-se
representada nos gráficos da figura 23, onde correlacionamos a idade do animal
com a gravidade do fenótipo. As correlações variam de moderadas a fortes,
dependendo do sistema analisado.

RESULTADOS 63
FIGURA 23. Correlação entre a progressão dos fenótipos pulmonar (Lm), aórtico (Espessura da aorta - EPA) e ósseo (KR) com a idade (meses) em animais heterozigotos da linhagem 129/Sv. Podemos notar uma correlação variando de moderada (0,3≤|R|≤0,69) a forte (|R|≥0,70). Todos os valores são estatisticamente significativos com P ≤ 0,05.
Os resultados indicam que a diferença principal observada nas
manifestações fenotípicas entre as duas linhagens é a idade de início dos sintomas,
a qual é mais avançada nos animais B6.
4.1.3. A variabilidade fenotípica da linhagem 129/Sv
Enquanto os animais da linhagem B6 apresentaram um padrão homogêneo
para os diferentes fenótipos avaliados, os animais da linhagem 129/Sv, com idade

RESULTADOS 64
inferior a seis meses, mostraram uma grande variabilidade de fenótipo, variando de
assintomático a grave. Isso é demonstrado pelas barras de desvio padrão presentes
nos gráficos das figuras 21, 22 e 23.
Tendo como base a média de Lm, nós classificamos 13 animais 129/Sv
heterozigotos, com idades variando entre três e seis meses, em três categorias
quando comparados com animais selvagens (Lm = 66,75 ± 18,25 µm): 15% foram
classificados como assintomáticos (Lm = 68,45 ± 5,2 µm; P = 0,42); 38% como
portadores de alterações leves (Lm = 116,44 ± 5,46 µm; P < 0,0006) e 46% como
apresentando alterações graves (Lm = 183,86 ± 19,84 µm; P < 0,006) (Fig.24A).
O fenótipo vascular também apresentou variabilidade em animais 129/Sv
heterozigotos, aos três meses de idade, nos permitindo classificar-los em duas
categorias: 60% foram definidos como assintomáticos (EPA = 52,21 ± 6,46 µm
contra 61,61 ± 11,08 µm em animais selvagens; P = 0,33) e 40% foram classificados
como apresentando alterações leves (EPA = 77,75 ± 2,59 µm; P = 0,04). Aos seis
meses de idade, 33% dos animais heterozigotos foram classificados como
assintomáticos (EPA = 79,10 ± 3,43 µm contra 70,34 ± 2,72 µm em animais
selvagens; P = 0,80) e 66% foram classificados como gravemente afetados (EPA =
101,85 ± 6,92 µm; P = 0,01) (Fig.24B).
Uma variabilidade fenotípica similar pode ser observada no fenótipo
esquelético de animais 129/Sv, onde os heterozigotos com três e seis meses de
idade mostraram diferentes graus de cifose, permitindo a classificação dos mesmos
em animais com alterações leves e graves (Fig.24C).
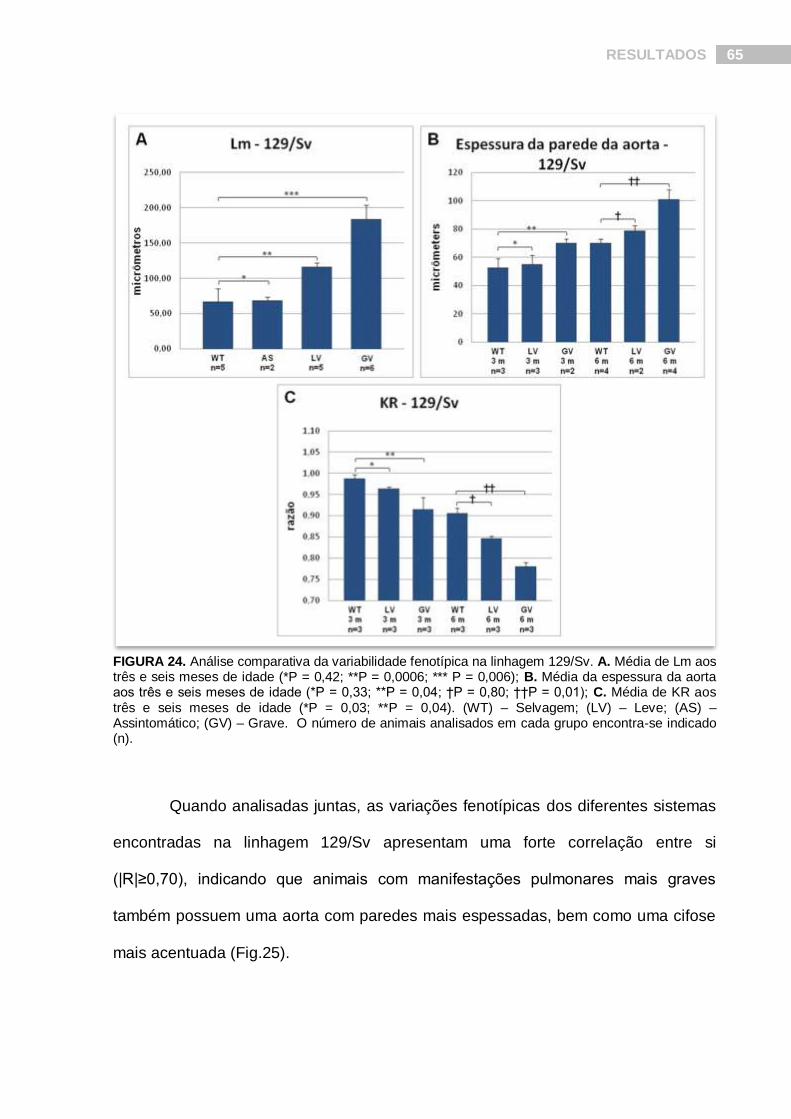
RESULTADOS 65
FIGURA 24. Análise comparativa da variabilidade fenotípica na linhagem 129/Sv. A. Média de Lm aos três e seis meses de idade (*P = 0,42; **P = 0,0006; *** P = 0,006); B. Média da espessura da aorta aos três e seis meses de idade (*P = 0,33; **P = 0,04; †P = 0,80; ††P = 0,01); C. Média de KR aos três e seis meses de idade (*P = 0,03; **P = 0,04). (WT) – Selvagem; (LV) – Leve; (AS) – Assintomático; (GV) – Grave. O número de animais analisados em cada grupo encontra-se indicado (n).
Quando analisadas juntas, as variações fenotípicas dos diferentes sistemas
encontradas na linhagem 129/Sv apresentam uma forte correlação entre si
(|R|≥0,70), indicando que animais com manifestações pulmonares mais graves
também possuem uma aorta com paredes mais espessadas, bem como uma cifose
mais acentuada (Fig.25).

RESULTADOS 66
FIGURA 25. Variabilidade fenotípica presente na linhagem 129/Sv. Comparação de cortes histológicos (pulmão e aorta) e imagens radiográficas de um animal selvagem (+/+) com dois animais heterozigotos mutantes (+/-). (AS) – assintomático; (GV) Grave. As barras de escala equivalem a 100 µm.
4.1.4. Imunohistoquímica
A avaliação imunohistoquímica de fibroblastos fetais, derivados de animais
B6, revelou diferenças qualitativas do padrão imunoreativo de fibrilina-1 entre células
controle (selvagens) e células portadoras do alelo Fbn1mgΔloxPneo (Fig.26). Em
contraste com a elaborada rede filamentosa de fibrilina-1 observada em linhagens
celulares controle, as células heterozigotas apresentaram algumas estruturas
filamentosas dispersas sobre um fundo imunoreativo difuso, enquanto que as
culturas de células homozigotas mostraram-se com um padrão puramente difuso.

RESULTADOS 67
Esse padrão é semelhante ao observado em cultura de fibroblastos fetais
homozigotos para alelo Fbn1mgΔ e heterozigotos para o alelo Fbn1TSK (PEREIRA et
al., 1997 ; GAYRAUD et al., 2000).
FIGURA 26. Imunofluorescência de fibroblastos fetais em cultura, com marcação para fibrilina-1 (barras de escala com 50 µm). A. Selvagens; B. Heterozigotos; C. Homozigotos. Em contraste com a elaborada rede filamentosa imunorreativa de fibrilina-1 observada em células normais, as células mutantes apresentam um padrão difuso; D. Campo com maior aumento mostrando a deposição intracelular de proteína mutante (barra de escala com 25 µm).
Também foi possível observar uma aparente deposição intracelular de
proteínas mutantes nas células homozigotas para o alelo Fbn1mgΔloxPneo, o que é
evidenciado pela delimitação da região nuclear por um material difuso (Fig.26C, D).

RESULTADOS 68
4.2. Análise dos níveis de transcrição do gene Fbn1 por Real Time
PCR (RT-PCR)
4.2.1. Comparação da transcrição do alelo Fbn1 x
Fbn1mgΔloxPneo em fibroblastos fetais
A análise quantitativa (RT-PCR) do cDNA obtido a partir de fibroblastos
fetais (aos 13 dias), de ambas as linhagens, mostrou que as células homozigotas
para o alelo Fbn1mgΔloxPneo apresentam 47% ± 5 (P ≤ 0,05) da transcrição do gene
Fbn1 quando comparadas com células selvagens, enquanto que as células
heterozigotas apresentam 78% ± 10 (P ≤ 0,05) (Fig.27). Por tanto, a expressão do
alelo Fbn1mgΔloxPneo é de aproximadamente 50% do alelo selvagem,
significativamente mais elevada do que o nível de 10% observado no alelo Fbn1mgΔ
original (PEREIRA et al., 1997). A maior expressão do alelo Fbn1mgΔloxPneo pode
justificar as diferenças fenotípicas observadas entre os dois modelos.
FIGURA 27. Quantificação relativa dos níveis totais de mRNA do gene Fbn1 em fibroblastos fetais selvagens (SV), heterozigotos (HE) e homozigotos (HO) por Real Time PCR (P ≤ 0,05).

RESULTADOS 69
4.2.2. Comparação da transcrição do alelo Fbn1 x
Fbn1mgΔloxPneo em animais adultos e a sua correlação com a variação de
fenótipo
Foram realizadas análises comparativas entre indivíduos adultos, nas
diferentes idades, com o objetivo de verificarmos o efeito de variações
transcricionais do gene Fbn1 sobre a heterogeneidade fenotípica observada entre
animais de uma mesma linhagem e entre linhagens. Para tal foram utilizados primers
e sondas específicas para os alelos selvagens e mutantes, bem como primers
capazes de ligar-se a ambos, de forma a analisar a transcrição total do gene Fbn1.
Todos os experimentos de RT-PCR foram realizados a partir de cDNA
sintetizado de RNA de amostras pulmonares.
4.2.2.1. A diferença de transcrição entre as linhagens C57BL/6 e
129/Sv
Os resultados, obtidos a partir da avaliação da transcrição total do gene
Fbn1, demonstraram que os animais selvagens da linhagem B6 apresentam uma
transcrição 33% superior à linhagem 129/Sv (P = 0,0004) (Fig.28). Essa variação
pode estar associada com os padrões fenotípicos mais brandos e tardios
observados na linhagem B6.

RESULTADOS 70
FIGURA 28. Quantificação relativa dos níveis totais de mRNA do gene Fbn1 em animais selvagens (*P = 0,0004).
Quando utilizamos primers e sondas específicas para o alelo selvagem,
verificamos que animais heterozigotos da linhagem B6 quando comparado a animais
selvagens têm uma redução significativa na transcrição do gene Fbn1 de
aproximadamente 50% aos três meses (n = 4; P = 0,03) e seis meses de idade (n =
5; P ≤ 0,05), o que já era esperado devido a hemizigose do alelo selvagem (Fig.29).
Contudo, ainda avaliando apenas o alelo selvagem, verificamos que animais
heterozigotos da linhagem 129/Sv, aos três e seis meses de idade, possuem uma
variabilidade de transcrição muito grande, apresentando inclusive valores médios
superiores (ainda que não significativos; P > 0,05) aos dos animais selvagens
(Fig.29).
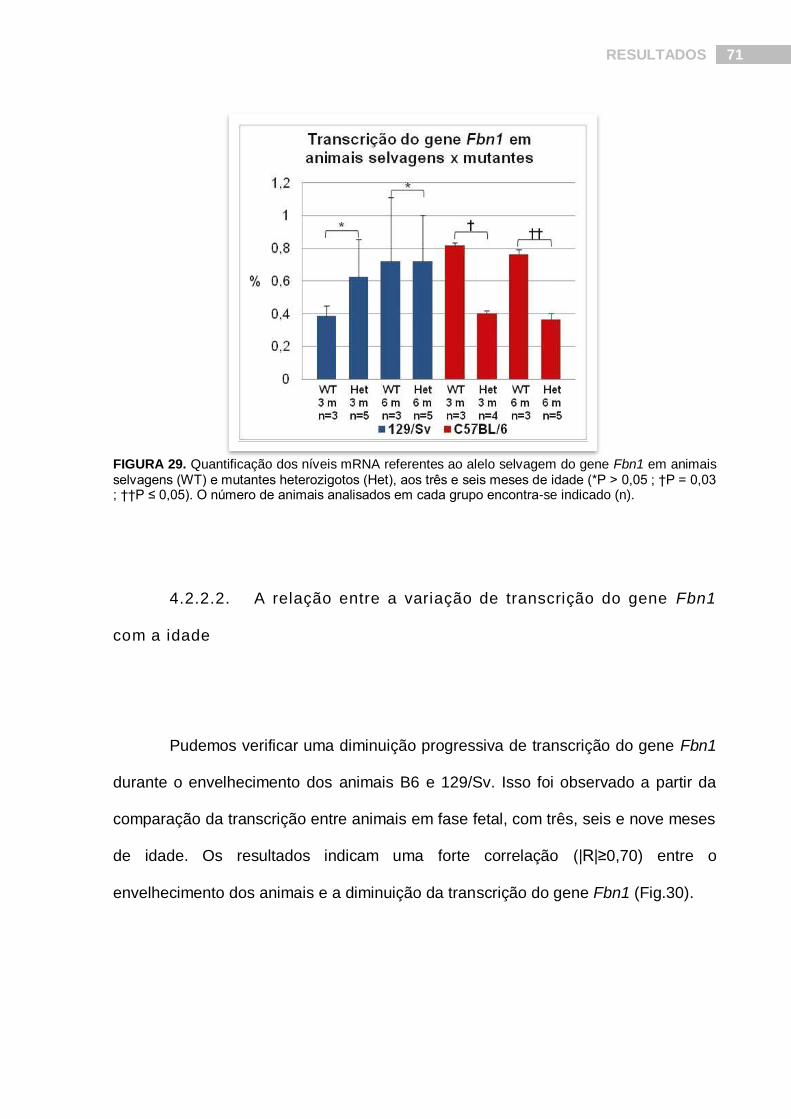
RESULTADOS 71
FIGURA 29. Quantificação dos níveis mRNA referentes ao alelo selvagem do gene Fbn1 em animais selvagens (WT) e mutantes heterozigotos (Het), aos três e seis meses de idade (*P > 0,05 ; †P = 0,03 ; ††P ≤ 0,05). O número de animais analisados em cada grupo encontra-se indicado (n).
4.2.2.2. A relação entre a variação de transcrição do gene Fbn1
com a idade
Pudemos verificar uma diminuição progressiva de transcrição do gene Fbn1
durante o envelhecimento dos animais B6 e 129/Sv. Isso foi observado a partir da
comparação da transcrição entre animais em fase fetal, com três, seis e nove meses
de idade. Os resultados indicam uma forte correlação (|R|≥0,70) entre o
envelhecimento dos animais e a diminuição da transcrição do gene Fbn1 (Fig.30).

RESULTADOS 72
FIGURA 30. Correlação entre a transcrição total do gene Fbn1 e a idade de animais B6 (A) e 129/sv (B). Notamos que existe uma diminuição progressiva da transcrição do gene com o envelhecimento dos animais.
4.2.2.3. Comparação da transcrição do gene Fbn1 entre animais
da linhagem 129/Sv com diferentes fenótipos
Os níveis de transcrição do gene Fbn1, bem como a razão desses níveis
entre alelos selvagens e mutantes, têm sido associados com a gravidade do fenótipo
da SMF em humanos e modelos murinos (HUTCHINSON et al., 2003 ; JUDGE et al.,
2004). Assim, nós analisamos a transcrição do gene Fbn1 em animais da linhagem
129/Sv para avaliar se existiria alguma correlação com a variabilidade fenotípica
observada nesta linhagem.
A análise desses animais revelou que a razão entre a transcrição do alelo
selvagem e mutante é constante, ou seja, quando comparamos dois animais, aquele
cuja transcrição do alelo selvagem é maior, possui uma transcrição do alelo mutante
proporcionalmente maior (Fig.31A). Pudemos verificar ainda uma forte correlação
negativa (|R|≥0,75; P = 0,04) entre a expressão total do gene Fbn1 (soma do

RESULTADOS 73
transcrito selvagem e mutante) e a gravidade do fenótipo, indicando que
heterozigotos com uma menor transcrição do gene Fbn1 apresentam fenótipos mais
graves nos três sistemas avaliados (Fig.31B-D).
FIGURA 31. Correlação entre a transcrição do gene Fbn1 e a gravidade do fenótipo em animais heterozigotos da linhagem 129/Sv. A. Análise por Real-time PCR dos alelos selvagem (SV) (gráfico à esquerda) e mutante (gráfico à direita) em animais heterozigotos 129/Sv. Os indivíduos estão representados por barras de cores diferentes. Notamos que apesar da variação de transcrição, a razão entre os transcritos selvagens e mutantes é constante; B-D. Correlação entre os níveis transcricionais do gene Fbn1 e a gravidade dos fenótipos pulmonares (Lm) (B), aórtico (EPA) (C) e ósseo (KR) (D). Os pontos correspondem aos animais presentes em A. Notamos uma forte correlação entre a transcrição do gene e a gravidade do fenótipo.

RESULTADOS 74
4.2.3. Efeito da droga Ramipril sobre a transcrição global do
gene Fbn1 e o fenótipo de animais C57BL/6 mutantes
Os animais mutantes pertencentes à linhagem B6, que foram tratados com a
droga Ramipril durante o período de 24 semanas, apresentaram um aumento
significativo de aproximadamente 35% (P = 0,016) da expressão global do gene
Fbn1, quando comparados ao grupo controle não tratado (Fig.32).
FIGURA 32. Gráfico comparativo da transcrição do gene Fbn1 em animais B6 selvagens (WT), heterozigotos sem tratamento (Controle) e heterozigotos tratados com a droga Ramipril (Ramipril) (* P > 0,05 ; **P = 0,016).
Além das mudanças transcricionais, os animais tratados com Ramipril
apresentaram melhora fenotípica. No sistema ósseo, os animais tratados
apresentaram reversão completa das alterações, tendo atingido valores similares ao
observado em animais selvagens. No sistema pulmonar os animais tratados

RESULTADOS 75
apresentaram uma melhora significativa quando comparados com animais mutantes
controle, porém não tendo atingido valores normais (Fig.33). A aorta não pode ser
analisada por problemas durante o processamento do material, sendo necessária
uma nova análise.
FIGURA 33. Análise dos efeitos do tratamento com Ramipril em animais B6. (A-D) Análise radiográfica. A. Animal selvagem; B. Animal heterozigoto não tratado; C. Animal heterozigoto tratado com Ramipril; D. Gráfico comparativo da KR média entre animais selvagens (WT), heterozigotos não tratados (controle) e heterozigotos tratados com Ramipril (Ramipril) (*P > 0,05; **P = 0,003). (E-H) Análise histológica dos pulmões (marcação por HE), barras de escala de 100 µm. E. Animal selvagem apresentando espaços aéreos de calibre normal; F. Animal heterozigoto não tratado com aumento no espaçamento alveolar; G. Animal heterozigoto tratado com Ramipril, apresentando melhora das alterações; H. Gráfico comparativo do diâmetro alveolar pulmonar médio (Lm) entre animais selvagens (WT), heterozigotos não tratados (controle) e heterozigotos tratados com Ramipril (Ramipril) (*P = 0,018; **P = 0,019). Todos os animais desta figura possuem 6 meses de idade. O número de animais analisados em cada grupo encontra-se indicado (n).

RESULTADOS 76
4.3. Avaliação da atividade de TGF-β1 nos tecidos afetados
A quantificação da proteína Smad2 em sua forma fosforilada (phospho-
Smad2), a partir de amostras protéicas pulmonares e aórticas, foi realizada a partir
de amostras de animais B6 de três e seis meses de idade, pois a ausência de
variação de fenótipo nessa linhagem nos garante a presença de fenótipo nos tecidos
avaliados (Fig.34A). Além disso, poderíamos verificar se os tecidos de animais mais
velhos, com um fenótipo mais grave, apresentariam uma maior atividade da citosina.
Porém, essa análise não indicou aumento significativo dessa proteína nos tecidos de
animais heterozigotos quando comparadas com amostras de animais selvagens
(Fig.34B).
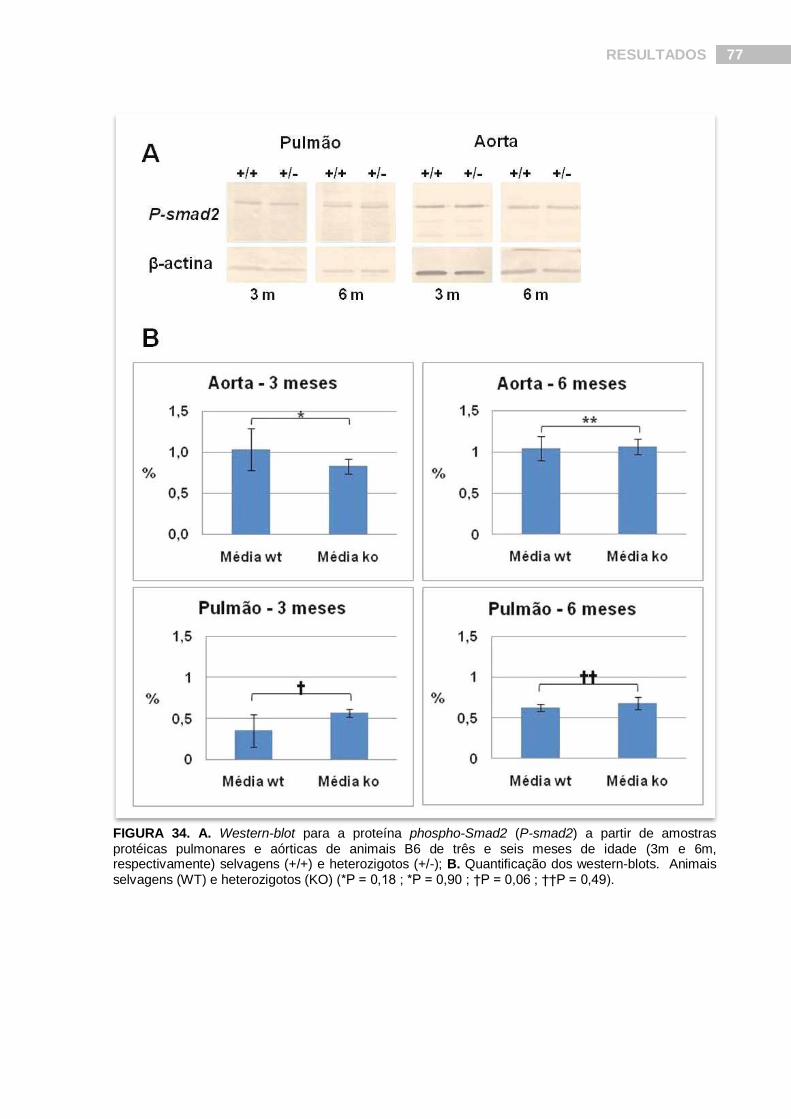
RESULTADOS 77
FIGURA 34. A. Western-blot para a proteína phospho-Smad2 (P-smad2) a partir de amostras protéicas pulmonares e aórticas de animais B6 de três e seis meses de idade (3m e 6m, respectivamente) selvagens (+/+) e heterozigotos (+/-); B. Quantificação dos western-blots. Animais selvagens (WT) e heterozigotos (KO) (*P = 0,18 ; *P = 0,90 ; †P = 0,06 ; ††P = 0,49).

V. DISCUSSÃO ____________________________________________________

DISCUSSÃO 79
A Síndrome de Marfan caracteriza-se por ser uma doença multisistêmica do
tecido conjuntivo que apresenta um alto grau de variabilidade clínica tanto intra
como interfamilial. Sendo uma doença genética relativamente freqüente em
humanos, onde apresenta uma elevada morbidade e mortalidade devido a
complicações cardiovasculares, a SMF tem sido alvo de diversos estudos tanto
clínicos como moleculares, especialmente através da utilização de modelos murinos.
Os primeiros modelos murinos para a SMF eram animais homozigotos para
alelos hipomórficos do gene Fbn1: os modelos mgΔ e mgR (PEREIRA et al., 1997 ;
PEREIRA et al., 1999). Enquanto o modelo mgΔ expressava baixos níveis de uma
fibrilina-1 truncada, o que resultava em letalidade pós-natal, os animais mgR
homozigotos produziam baixo níveis de fibrilina-1 normal, e recapitulavam a forma
letal adulta da SMF (PEREIRA et al., 1997 ; PEREIRA et al., 1999). Um terceiro
modelo, o C1039G, é caracterizado pela substituição de uma cisteína por uma
glicina na posição 1039 (PEREIRA et al., 1999 ; JUDGE et al., 2004). Os animais
heterozigotos apresentam deficiência na deposição de microfibrilas na MEC,
alterações esqueléticas e deteriorização progressiva da arquitetura da parede
aórtica. Os três modelos murinos foram caracterizados exclusivamente na linhagem
isogênica C57BL/6 (PEREIRA et al., 1997 ; PEREIRA et al., 1999 ; JUDGE et al.,
2004).
O mgΔloxPneo é um novo modelo para SMF que, além de recapitular alguns
dos sinais mais importantes da doença em humanos, apresenta a variabilidade
fenotípica característica da síndrome. A única diferença entre esse novo alelo e o
alelo Fbn1mgΔ original é a presença da seqüência loxP flanqueando o cassete neoR,
já que os dois alelos mutantes resultam na síntese de um transcrito com a deleção

DISCUSSÃO 80
dos exons 19 ao 24 (PEREIRA et al., 1997). Contudo, o alelo Fbn1mgΔloxPneo
apresentou um aumento de 47% nos níveis de transcrição, quando comparado com
o alelo Fbn1mgΔ, o que de acordo com o modelo dominante negativo para a SMF,
pode explicar as manifestações fenotípicas nos animais heterozigotos mgΔ loxPneo. Os
motivos pelos quais o alelo Fbn1mgΔloxPneo apresentou esse aumento de transcrição
permanecem indefinidos.
5.1. Caracterização fenotípica do modelo mgΔ loxPneo
Os camundongos mgΔloxPneo heterozitogos apresentam alterações
esqueléticas, caracterizada pela deformidade da coluna cervical e torácica, com um
padrão compatível com cifose. Além disso, podemos notar uma evidente
deformidade da caixa torácica, com alterações morfológicas dos arcos costais.
Essas alterações são compatíveis com as alterações presentes nos demais modelos
murinos para a SMF (GREEN et al., 1976 ; PEREIRA et al., 1999 ; JUDGE et al.,
2004), assim como com as manifestações clínicas de seres humanos afetados
(LOEYS et al., 2010). No entanto, não foi observada mudança no comprimento e
espessura de ossos longos (fêmur, úmero, rádio etc.) (dados não apresentados),
sendo essa uma característica clássica da SMF e que pode ser vista no modelo TSK
(GREEN et al., 1976 ; GAYRAUD et al., 2000).
Os mecanismos patogênicos relacionado com as manifestações
esqueléticas da SMF ainda são desconhecidos (BYERS, 2004). Contudo temos a
hipótese de que a frouxidão de ligamentos e tendões, bem como uma diminuição da

DISCUSSÃO 81
tensão exercida pelo periósteo, poderia estar relacionada com o crescimento
exagerado dos ossos e deformidades da coluna (ZHANG et al., 1995). Além disso,
sabemos que a ocorrência de escoliose e cifose, pode resultar em deformidades
secundárias da caixa torácica (LOEYS et al., 2010). Essas alterações são todas
decorrentes de uma falha na correta organização da MEC, nesses casos, devido a
mutações na fibrilina-1. Assim parece bastante plausível que as manifestações
presentes no modelo mgΔloxPneo estejam relacionadas com uma maior quantidade de
proteína mutante em sua MEC, quando comparada com o modelo mgΔ. A ausência
de ossos mais alongados em nosso modelo ainda não pode ser explicada, pedindo
um estudo mais detalhado.
Os camundongos heterozitogos mgΔloxPneo apresentam um fenótipo muito
similar ao descrito no modelo C1039G (JUDGE et al., 2004), especialmente no que
se refere a manifestações aórticas, incluindo a conservação no número de lamelas
elásticas na túnica média da aorta, apesar do espessamento da mesma. Contudo o
modelo mgΔloxPneo apresenta um processo inflamatório crônico em seus pulmões,
caracterizado pela presença de um grande número de células mononucleadas no
parênquima desse tecido (monócitos), similar ao observado nos animais
homozigotos do modelo mgR (PEREIRA et al., 1999).

DISCUSSÃO 82
5.2. Avaliação da atividade de TGF-β1 nos tecido aórtico e
pulmonar do modelo mgΔ loxPneo
Sabemos que as alterações pulmonares e aórticas presentes na SMF são
decorrentes do aumento de atividade da citosina TGF-β1, cujas principais funções
incluem o desenvolvimento de processos inflamatórios (ativação de monócitos etc.)
e a replicação de células de músculo liso, presentes nas paredes das artérias
(ANNES et al., 2003 ; KAARTINEN & WARBURTON, 2003 ; NEPTUNE et al., 2003 ;
HABASHI et al., 2006). Essa teoria é sustentada por diversos trabalhos envolvendo
modelos animais, onde esses fenótipos puderam ser revertidos, tanto pela
neutralização direta de TGF-β1, quanto pela interferência em sua via de sinalização
por ação da droga Losartan (JUDGE et al., 2004 ; NG et al., 2004). No entanto, para
nossa surpresa, ao analisarmos a atividade de TGF-β1 através da quantificação de
phospho-Smad2 em nosso modelo mgΔloxPneo, não verificamos diferenças
significativas entre a atividade da citosina nos tecidos de animais heterozigotos e
selvagens.
A via canônica de sinalização da citosina TGF-β1, ou via das smads, sempre
foi reconhecida como via de predileção para o desenvolvimento das alterações
associadas ao aumento de atividade dessa citosina. Assim, o aumento de smad2
fosforilada foi tradicionalmente utilizado como forma de se avaliar o aumento da
atividade de TGF-β1, inclusive nos trabalhos que associam perturbações na fibrilina-
1 com este aumento (NEPTUNE et al., 2003 ; HABASHI et al., 2006).

DISCUSSÃO 83
Atualmente outras vias, classificadas como não canônicas, foram
descobertas como alternativas independentes das smads2 e 3 para a sinalização de
TGF-β1 (ZHANG, 2009). Dentre essas vias temos as vias das MAP Kinases
(MAPK), que incluem a extracellular signal-regulated kinase 1 e 2 (ERK1 e ERK2)
(FUNABA et al., 2002), a p38 mitogen-activated protein kinase (p38-MAPK)
(HANAFUSA et al., 1999), a c-Jun N-terminal kinase (JNK) (HOCEVAR et al., 1999)
e a phosphoinositide 3-kinase-Akt (PI3K-Akt) (CHEN et al., 1998).
Em um trabalho recente, Doyle et al. (2009) mostraram que as vias não
canônicas das MAPK (ERK1/2 e JNK) são as mais importantes para o
desenvolvimento das manifestações aórticas presentes em um modelo para SMF, e
ainda, que a via das smads não é fundamental para o desenvolvimento desse
fenótipo. Nesse trabalho os pesquisadores cruzaram o modelo C1039G com um
camundongo haploinsuficiente para Smad4 (S4+/-), e verificaram que os animais
duplos mutantes (S4+/- : C1039G/+) apresentaram degeneração da parede aórtica
com morte por sua ruptura. A análise por Western-blot revelou que esses animais
apresentavam um aumento da marcação para smad2 fosforilada, porém este era
muito discreto quando comparado com o aumento na concentração de proteínas
relacionadas à via de ativação ERK1/2. Além disso, por apresentar
haploinsuficiência para smad4, a ativação da via canônica não poderia explicar o
fenótipo vascular. Finalmente, a utilização de antagonistas para JNK foi capaz de
reverter o desenvolvimento do fenótipo.
Assim, é possível que a ausência de um aumento na marcação para smad2
fosforilada verificada no modelo mgΔloxPneo seja decorrente da não ativação dessa
via nesse animais, e que a via utilizada na sinalização da citosina TGF-β1 seja a via

DISCUSSÃO 84
das MAPK. Por tanto, é importante que novas análises sejam realizadas, com
anticorpos específicos para as proteínas dessas vias alternativas.
5.3. As alterações presentes na imunohistoquímica de fibroblastos
fetais em cultura
Os mecanismos moleculares que resultam em alterações nas microfibrilas
mostram-se muito difíceis de serem identificados, não sendo possível correlacioná-
los com as diferentes mutações no gene FBN1. As análises por imunohistoquímica
realizadas em fibroblastos dermais de pacientes com SMF, mostram uma grande
variedade de alterações na síntese, secreção e deposição de fibrilina-1 na MEC
(AOYAMA et al., 1994 ; SCHRIJVER et al., 1999). Contudo, embora essas análises
mostrem alterações qualitativas e quantitativas na marcação de fibrilina-1, o que
sugere mecanismos do tipo dominante-negativo e de haploinsuficiência
respectivamente, elas não identificam se esse efeito ocorre na fase intra ou
extracelular da proteína (WHITEMAN & HANDFORD, 2003).
Algumas alterações estruturais da fibrilina-1, derivadas da substituição de
cisteínas por outros aminoácidos (C1117Y, C1129Y e G1127S), podem dificultar o
transporte dos monômeros de fibrilina-1 dentro das células, o que acarreta em um
atraso em sua secreção (WHITEMAN & HANDFORD, 2003). Essas substituições
causam a retenção dos monômeros mutantes no interior do retículo endoplasmático,
impedindo que esses cheguem ao aparelho Golgiense para serem adequadamente
glicosilados. A retenção dessas proteínas pode resultar em sua agregação, tanto

DISCUSSÃO 85
com outros monômeros mutantes quando com normais, e posterior degradação
(WHITEMAN & HANDFORD, 2003).
Pudemos identificar sinais de que a proteína mutante do alelo Fbn1mgΔloxPneo
permanece retida no interior da célula. Isso é evidenciado pela delimitação das
regiões referentes aos núcleos celulares pela marcação com anticorpos para
fibrilina-1, o que sugere ainda que essa proteína esteja retida dentro do retículo
endoplasmático. Uma vez que os monômeros mutantes permanecem retidos dentro
da célula, esses podem resultar tanto em um efeito de haploinsuficiência, quanto em
um efeito dominante-negativo intracelular, onde os monômeros normais acabam
sendo degradados por associarem-se com monômeros mutantes. Nesse caso
podemos dizer que temos um efeito de haploinsuficiência funcional, pois embora a
proteína esteja sendo produzida, ela permanece retida no interior da célula, e o
resultado prático disso é similar ao de uma ausência de proteína (WHITEMAN &
HANDFORD, 2003).
5.4. A diferença fenotípica entre os animais mutantes das
linhagens 129/Sv e C57BL/6
Por apresentarem variações de fenótipo para uma mesma mutação, de
acordo com o background genético, o estudo de modelos animais pode ajudar no
esclarecimento do mecanismo envolvido em doenças humanas, as quais
apresentam dificuldade em se determinar uma relação entre genótipo e fenótipo.
Dessa forma, o entendimento das bases moleculares e celulares envolvidas nessas

DISCUSSÃO 86
variações, pode possibilitar o desenvolvimento de novas intervenções terapêuticas
para essas doenças.
Nós demonstramos o efeito do background genético sobre a gravidade do
fenótipo de SMF através da avaliação dos efeitos do alelo Fbn1mgΔloxPneo em duas
linhagens murinas congênicas diferentes, onde animais heterozigotos da linhagem
129/Sv apresentaram fenótipo mais precocemente do que os animais da linhagem
B6. Essa variabilidade inter-linhagens poderia ser atribuída a diferenças no efeito
exercido pelo cassete neoR na expressão do alelo mutante entre as duas linhagens
congênicas. Contudo, nós demonstramos que o nível de expressão do alelo mutante
corresponde a 47% do alelo selvagem em ambas as linhagens. Portanto, fatores
genéticos diferentes do gene Fbn1 são relevantes para a patogênese da SMF no
modelo, e esses modulam as diferenças fenotípicas entre as duas linhagens.
A influência do background genético sobre o fenótipo tem sido relatada em
camundongos modelo do tipo knockout para diversas doenças humanas, incluindo a
fibrose cística e a doença de Huntington (SWEET et al., 1992 ; YANG et al., 2005 ;
OTSURU et al., 2010). Além disso, em alguns casos, esses animais têm sido
utilizados com sucesso para a identificação dos respectivos genes modificadores
(DIETRICH et al., 1993 ; WHEELER et al., 2005 ; PU, 2009). A variabilidade
fenotípica do modelo mgΔloxPneo pode permitir a identificação de modificadores
genéticos também relacionados com à heterogeneidade clínica da SMF em
humanos.

DISCUSSÃO 87
5.5. A variabilidade fenotípica entre os animais mutantes da
linhagem 129/Sv
Observamos uma grande e inesperada variabilidade fenotípica entre
heterozigotos da linhagem 129/Sv. A variabilidade fenotípica em animais congênicos
já foi relatada em diferentes trabalhos, e em alguns casos foi associada a
modificadores epigenéticos de um loci específico (RAKYAN et al., 2002). Portanto,
nós propomos que fatores epigenéticos devem estar contribuindo na variabilidade
fenotípica do modelo mgΔloxPneo, quando esse encontra-se na linhagem 129/Sv,
assim como pode estar acontecendo na variabilidade clínica de pacientes humanos
de uma mesma família.
Neste sentido, mostramos uma forte correlação negativa entre os níveis de
transcrição global do gene Fbn1 e a gravidade do fenótipo, enquanto que a razão
entre a transcrição dos alelos de mutante e normal não variou entre os animais
diferentemente afetados. O efeito protetor exercido pelos níveis de fibrilina-1 normal
sobre a gravidade do fenótipo já foram sugeridos por Hutchinson et al. (2003), que
mostraram uma correlação entre altos níveis na transcrição do gene FBN1 normal
com fenótipos mais leves em três indivíduos de uma mesma família. Contudo, a
mutação no gene FBN1 presente nessa família era do tipo nonsence, levando à
degradação da proteína mutante, e impossibilitando que esta pudesse exercer um
efeito dominante-negativo sobre as proteínas normais. Em contraste, os animais
heterozigotos da linhagem 129/Sv apresentam este efeito protetor exercido pelos
altos níveis de fibrilina-1 normal mesmo na presença de monômeros mutantes.

DISCUSSÃO 88
Assim, propomos que alterações epigenéticas no gene Fbn1 estão
relacionadas com as diferenças fenotípicas observadas na linhagem 129/Sv. No
momento, essa hipótese está sendo avaliada a partir de análises de metilação da
região promotora do gene Fbn1 dos animais 129/Sv diferentemente afetados.
5.6. Os mecanismos de patogênese relacionados com a Síndrome
de Marfan
Diversas observações sugerem um tradicional mecanismo dominante-
negativo para a patogênese da SMF. Nesse caso, proteínas anormais derivadas de
um alelo mutante interagem e interferem com as proteínas derivadas do alelo
normal, resultando em uma perda de função deste. Algumas evidências que
sustentam essa hipótese incluem: o padrão de herança da doença (autossômico
dominante), o padrão multimérico da proteína (SAKAI et al., 1986) e a dramática
escassez de microfibrilas extracelulares nos tecidos derivados de pacientes, cujos
níveis são muito inferiores aos 50% previstos a partir de uma simples perda de
participação do alelo mutante (GODFREY et al., 1990a ; GODFREY et al., 1990b ;
HOLLISTER et al., 1990).
Outra observação que sustenta o tradicional mecanismo dominante-negativo
para a SMF provém da caracterização clínica de pacientes portadores de alelos
nonsense que leva à rápida degradação dos transcritos mutantes pela via de
degradação mediada por códon de terminação (nonsense-mediated mRNA decay),
limitando a produção de quantidades substanciais de fibrilina-1 mutante (HEWETT et

DISCUSSÃO 89
al., 1994 ; FRISCHMEYER & DIETZ, 1999). Curiosamente, de acordo com a
hipótese dominante-negativa, a maioria dos alelos nonsense resulta em
manifestações clínicas extremamente leves, quase sempre incompatíveis com os
critérios diagnósticos para SMF (DIETZ et al., 1993a ; SCHRIJVER et al., 2002).
Contudo, alguns pacientes portadores dessas mutações instáveis, apresentam os
clássicos sinais da SMF (NIJBROEK et al., 1995 ; SCHRIJVER et al., 2002 ;
HUTCHINSON et al., 2003). Assim, surgiu a possibilidade de que um mecanismo de
haploinsuficiência poderia estar relacionado com a síndrome.
Como já apresentado anteriormente, um estudo envolvendo um modelo
animal, sugere que um mecanismo de haploinsuficiência possa estar associado com
a doença (JUDGE et al., 2004). Judge et al. (2004) utilizaram um cromossomo
artificial para super-expressar uma forma mutante de fibrilina-1 humana (C1663R)
em uma linhagem isogênica selvagem de camundongo. Esses animais não
apresentaram nenhum sinal fenotípico, embora as análises por Northern-blot e
imunohistoquímica indicassem altos níveis de proteína mutante interagindo com a
proteína murina selvagem. A hipótese levantada pelo grupo era a de que a presença
de dois alelos normais (endôgenos) era capaz de impedir o desenvolvimento de
fenótipo, o que vai a favor de um mecanismo de haploinsuficiência. Contudo, a
utilização de um gene exógeno é uma variável muito importante, e que pode estar
mascarando os efeitos deletérios dos monômeros mutantes. Finalmente, quando
uma mutação similar foi inserida em um alelo murino, os animais apresentam
diversos fenótipos da SMF (JUDGE et al., 2004).
A maioria dos estudos envolvendo modelos animais para SMF tem nos
guiado em direção ao clássico modelo dominante-negativo. Os resultados obtidos

DISCUSSÃO 90
com os modelos mgΔ e mgR são bastante sugestivos para essa hipótese (PEREIRA
et al., 1997 ; PEREIRA et al., 1999). Embora esses modelos apresentem algum nível
de expressão dos alelos mutantes (10% e 20% respectivamente), a ausência de
fenótipo em animais portadores de alelos hipomórficos é um indicativo da
importância de uma proteína mutante, em quantidades significativas para o
desenvolvimento da doença. Os resultados obtidos nesse trabalho corroboram com
isso, pois o aumento da atividade do alelo mutante resultou na presença dos
diversos sinais aqui apresentados.
Finalmente, o desenvolvimento do modelo Fbn1null, portador de um alelo
nulo do gene Fbn1, vem a confirmar a necessidade de uma proteína mutante para o
desenvolvimento da SMF (MERKEL, 2006). Nesse modelo, o vetor de recombinação
homóloga foi construído de maneira a promover a substituição de 700 pb da região
5´ do gene Fbn1, incluindo parte do exon 1 que codifica o peptídeo sinal e o ATG
para início de tradução. Os animais homozigotos, não sobrevivem mais do que duas
semanas e apresentam alterações estruturais do parênquima pulmonar, com ductos
alveolares aumentados. Os animais heterozigotos não apresentam nenhuma
alteração fenotípica aparente, embora estudos histológicos tenham demonstrado
pequenas alterações no parênquima pulmonar e na matriz elástica de vasos em
animais em idade avançada.
Devemos salientar que a observação uma melhora fenotípica relacionada
com valores mais elevados de expressão de fibrilina-1 não são contraditórios com a
hipótese de um mecanismo dominante-negativo. Embora a presença de monômeros
mutantes possa aumentar o dano tecidual encontrado em indivíduos portadores da
SMS (BOOMS et al., 2000), os níveis de fibrilina-1 normal podem ter um potencial de

DISCUSSÃO 91
modular os efeitos negativos das proteínas mutantes, pois a maior disponibilidade de
monômeros normais pode suplantar sua degeneração. Isso reforça a hipótese de
que tratamentos que busquem aumentar a expressão do gene FBN1 podem ser
estratégias terapêuticas eficientes para a SMF.
Finalmente, devemos avaliar a importância dos resultados relacionados com
a diminuição da transcrição do gene Fbn1 ao longo do envelhecimento. Estudos
recentes, com amostras humanas, mostram que diversos genes apresentam um
aumento de sua metilação acompanhando a idade do indivíduo, o que resulta na
diminuição da transcrição (CONNEELY et al., 2010). Dentre esses genes, destacam-
se aqueles cuja região promotora é rica em ilhas CpG, como o gene FBN1. É
possível que essa redução na transcrição do gene ao longo da vida, associada a
uma maior suscetibilidade à proteólise dos monômeros mutantes de fibrilina-1,
possa ser uma dos motivos da SMF ser uma doença progressiva. Talvez indivíduos
normais, ainda que já estejam produzindo menos proteína em idades mais
avançadas, sofram uma menor degradação de suas fibras do que indivíduos
portadores de mutações missense.
5.7. O efeito de Ramipril no fenótipo da SMF
A melhora fenotípica associada com o aumento transcricional de Fbn1 em
animais tratados com Ramipril vem a confirmar a relação fundamental da quantidade
de proteína com a modulação do fenótipo em SMF, embora não deva ser a única
causa.

DISCUSSÃO 92
Trabalhos anteriores já haviam descrito a capacidade de Ramipril em
aumentar a transcrição do gene Fbn1 em até quatro vezes em células em cultura.
Contudo esses trabalhos também descrevem que outros genes, como o da elastina,
do colágeno e das metaloproteinases 2 e 3 também têm suas transcrições alteradas
após o tratamento com a droga (AHIMASTOS et al., 2005). Sendo estas proteínas
muito importantes nos sistemas aqui avaliados, é fundamental que mais análises,
envolvendo esses genes, sejam realizadas para que possamos compreender toda a
complexidade associada a essas variações de fenótipo.
O fato das alterações dos sistemas ósseo e pulmonar não terem
apresentado a mesma melhora, tendo o primeiro obtido uma total reversão, mostra
que diferentes mecanismos devem atuar em cada um dos sistemas. Nossos
resultados são compatíveis com os de trabalhos anteriores, onde animais tratados
com com losartan apresentaram melhoras pulmonares e aórticas, porém sem
nenhum efeito sobre o sistema ósseo (ADACHI et al., 2006 ; HABASHI et al., 2006).
Possivelmente a quantidade de proteína normal seja muito mais importante
em um sistema onde a fibrilina-1 exerça um papel fundamentalmente mecânico
como o ósseo, do que em outros sistemas onde além da função mecânica, a
proteína seja importante na modulação de TGF-β1, associado a processos
inflamatórios e degenerativos. Por esse motivo observamos resultados diferentes
entre animais tratados com Losartan e com Ramipril. A primeira droga atua
especificamente na neutralizando da ação de TGF-β1, por ser antagonista de AT1,
enquanto que o Ramipril atua aumentando a transcrição do gene Fbn1, não tendo
por tanto um efeito direto sobre a atividade de TGF-β1.

DISCUSSÃO 93
Finalmente devemos salientar que em todos os trabalhos onde Losartan foi
administrado em animais modelos para SMF, as concentrações utilizadas da droga
eram muito maiores do que a dose indicada para humanos. Por esse motivo em
testes clínicos envolvendo pacientes com SMF não foram obtidos resultados
satisfatórios, como observado nos testes com animais. Em nosso experimento com
Ramipril, a dose administrada nos animais é a mesma já utilizada em humanos.

VI. CONCLUSÕES ____________________________________________________

CONCLUSÕES 95
1. Caracterizamos a presença de variabilidade fenotípica no modelo
animal mgΔloxPneo para SMF, em animais mutantes das linhagens murinas 129/Sv e
B6, estabelecendo assim um sistema experimental para a identificação de genes
modificadores do fenótipo da doença;
2. Caracterizamos a variabilidade fenotípica do modelo mgΔloxPneo dentro
da linhagem congênica 129/Sv, cujo fenótipo variou de assintomático a grave.
Propomos então que fatores epigenéticos modulam a gravidade do fenótipo nesses
animais;
3. Observamos em animais heterozigotos da linhagem 129/Sv a
existência de uma forte correlação negativa entre a variabilidade fenotípica e
variabilidade transcricional global do gene Fbn1. Assim, podemos inferir que a
quantidade de proteína fibrilina-1 pode modular a gravidade da doença. Outro dado
que sustenta essa hipótese é o fato dos animais B6, os quais não mostram
variabilidade fenotípica, também serem bastante homogêneos no que se refere à
transcrição do gene Fbn1;
4. Verificamos uma diminuição gradativa na quantidade de fibrilina-1
produzida pelo organismo durante o seu envelhecimento. Essa diminuição pode
estar relacionada com a característica progressiva da doença. Em indivíduos
heterozigotos a taxa de degradação da proteína pode estar superando a capacidade
de reposição da mesma;
5. Sendo a quantidade de proteína um fator importante para a
determinação da gravidade dos fenótipos, bem como para sua progressão,
propomos que novas estratégias terapêuticas para a SMF sejam baseadas no
aumento da síntese de fibrilina-1;

CONCLUSÕES 96
6. O tratamento de animais mutantes com a droga Ramipril, com uma
dosagem equivalente a já utilizada em humanos, resultou em um aumento de
aproximadamente 35% da transcrição global dom gene Fbn1, quando comparado
com animais mutantes não tratados. Além disso, os animais tratados apresentaram
melhora fenotípica nos sistemas ósseo e pulmonar, tendo atingido níveis normais no
primeiro. Esses resultados reforçam a hipótese da importante relação entre a
quantidade de fibrilina-1 com a variação de fenótipo, e indicam outros fatores
modulam o fenótipo nos diferentes sistemas.

VII. REFERÊNCIAS ____________________________________________________

98
ADACHI, Y., H. OYAIZU, et al. Treatment and transfer of emphysema by a new bone marrow transplantation method from normal mice to Tsk mice and vice versa. Stem Cells, v.24, n.9, Sep, p.2071-7. 2006. ADES, L. Guidelines for the diagnosis and management of Marfan syndrome. Heart Lung Circ, v.16, n.1, Feb, p.28-30. 2007. AHIMASTOS, A. A., A. K. NATOLI, et al. Ramipril reduces large-artery stiffness in peripheral arterial disease and promotes elastogenic remodeling in cell culture. Hypertension, v.45, n.6, Jun, p.1194-9. 2005. ALDRED, C. e A. T. SANDISON. The Pharaoh Akhenaten. A problem in Egyptology and pathology. Bull Hist Med, v.36, Jul-Aug, p.293-316. 1962. ANDRIKOPOULOS, K., X. LIU, et al. Targeted mutation in the col5a2 gene reveals a regulatory role for type V collagen during matrix assembly. Nat Genet, v.9, n.1, Jan, p.31-6. 1995. ANNES, J. P., J. S. MUNGER, et al. Making sense of latent TGFbeta activation. J Cell Sci, v.116, n.Pt 2, Jan 15, p.217-24. 2003. AOYAMA, T., U. FRANCKE, et al. Quantitative differences in biosynthesis and extracellular deposition of fibrillin in cultured fibroblasts distinguish five groups of Marfan syndrome patients and suggest distinct pathogenetic mechanisms. J Clin Invest, v.94, n.1, Jul, p.130-7. 1994. BAYLES, J. Sounding the alarm. If undetected, Marfan syndrome can be a silent killer. J Ark Med Soc, v.99, n.4, Oct, p.106-11. 2002. BLOBE, G. C., W. P. SCHIEMANN, et al. Role of transforming growth factor beta in human disease. N Engl J Med, v.342, n.18, May 4, p.1350-8. 2000. BOILEAU, C., G. JONDEAU, et al. Molecular genetics of Marfan syndrome. Curr Opin Cardiol, v.20, n.3, May, p.194-200. 2005. BOOMS, P., F. TIECKE, et al. Differential effect of FBN1 mutations on in vitro proteolysis of recombinant fibrillin-1 fragments. Hum Genet, v.107, n.3, Sep, p.216-24. 2000. BORDER, W. A. e N. A. NOBLE. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension, v.31, n.1 Pt 2, Jan, p.181-8. 1998. BUONI, S., R. ZANNOLLI, et al. The FBN1 (R2726W) mutation is not fully penetrant. Ann Hum Genet, v.68, n.Pt 6, Nov, p.633-8. 2004. BYERS, P. H. Determination of the molecular basis of Marfan syndrome: a growth industry. J Clin Invest, v.114, n.2, Jul, p.161-3. 2004. CHEN, R. H., Y. H. SU, et al. Suppression of transforming growth factor-beta-induced apoptosis through a phosphatidylinositol 3-kinase/Akt-dependent pathway. Oncogene, v.17, n.15, Oct 15, p.1959-68. 1998. COHN, R. D., C. VAN ERP, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med, v.13, n.2, Feb, p.204-10. 2007.

99
CONNEELY, K. N., J. W. SCHROEDER, et al. Global and Gene-Specific Changes in DNA Methylation Associated with Aging. 2010 ASHG Annual Meeting Washington, DC, 2010. p. DETAINT, D., P. AEGERTER, et al. Rationale and design of a randomized clinical trial (Marfan Sartan) of angiotensin II receptor blocker therapy versus placebo in individuals with Marfan syndrome. Arch Cardiovasc Dis, v.103, n.5, May, p.317-25. 2010. DIETRICH, W. F., E. S. LANDER, et al. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell, v.75, n.4, Nov 19, p.631-9. 1993. DIETZ, H. C., G. R. CUTTING, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature, v.352, n.6333, Jul 25, p.337-9. 1991. DIETZ, H. C., I. MCINTOSH, et al. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics, v.17, n.2, Aug, p.468-75. 1993a. DIETZ, H. C., D. VALLE, et al. The skipping of constitutive exons in vivo induced by nonsense mutations. Science, v.259, n.5095, Jan 29, p.680-3. 1993b. DUNNILL, M. S. Quantitative Methods in the Study of Pulmonary Pathology. Thorax, v.17, p.320-28. 1962. FRISCHMEYER, P. A. e H. C. DIETZ. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet, v.8, n.10, p.1893-900. 1999. FUNABA, M., C. M. ZIMMERMAN, et al. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J Biol Chem, v.277, n.44, Nov 1, p.41361-8. 2002. GAYRAUD, B., D. R. KEENE, et al. New insights into the assembly of extracellular microfibrils from the analysis of the fibrillin 1 mutation in the tight skin mouse. J Cell Biol, v.150, n.3, Aug 7, p.667-80. 2000. GODFREY, M., V. MENASHE, et al. Cosegregation of elastin-associated microfibrillar abnormalities with the Marfan phenotype in families. Am J Hum Genet, v.46, n.4, Apr, p.652-60. 1990a. GODFREY, M., S. OLSON, et al. Unilateral microfibrillar abnormalities in a case of asymmetric Marfan syndrome. Am J Hum Genet, v.46, n.4, Apr, p.661-71. 1990b. GREEN, M. C., H. O. SWEET, et al. Tight-skin, a new mutation of the mouse causing excessive growth of connective tissue and skeleton. Am J Pathol, v.82, n.3, Mar, p.493-512. 1976. HABASHI, J. P., D. P. JUDGE, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science, v.312, n.5770, Apr 7, p.117-21. 2006. HANAFUSA, H., J. NINOMIYA-TSUJI, et al. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J Biol Chem, v.274, n.38, Sep 17, p.27161-7. 1999.

100
HEWETT, D., J. LYNCH, et al. Differential allelic expression of a fibrillin gene (FBN1) in patients with Marfan syndrome. Am J Hum Genet, v.55, n.3, Sep, p.447-52. 1994. HOCEVAR, B. A., T. L. BROWN, et al. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J, v.18, n.5, Mar 1, p.1345-56. 1999. HOGAN, A., R. BEDDINGTON, et al. Manipulating the mouse Embryo: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. 1994 HOLLISTER, D. W., M. GODFREY, et al. Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N Engl J Med, v.323, n.3, Jul 19, p.152-9. 1990. HUTCHINSON, S., A. FURGER, et al. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet, v.12, n.18, Sep 15, p.2269-76. 2003. JAVELAUD, D. e A. MAUVIEL. [Transforming growth factor-betas: smad signaling and roles in physiopathology]. Pathol Biol (Paris), v.52, n.1, Feb, p.50-4. 2004. JUDGE, D. P., N. J. BIERY, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest, v.114, n.2, Jul, p.172-81. 2004. KAARTINEN, V. e D. WARBURTON. Fibrillin controls TGF-beta activation. Nat Genet, v.33, n.3, Mar, p.331-2. 2003. KAINULAINEN, K. e L. PELTONEN. Marfan gene discovered. Ann Med, v.23, n.4, Oct, p.395-6. 1991. KIELTY, C. M., C. BALDOCK, et al. Fibrillin: from microfibril assembly to biomechanical function. Philos Trans R Soc Lond B Biol Sci, v.357, n.1418, Feb 28, p.207-17. 2002. KIELTY, C. M. e C. A. SHUTTLEWORTH. Fibrillin-containing microfibrils: structure and function in health and disease. Int J Biochem Cell Biol, v.27, n.8, Aug, p.747-60. 1995. LATTIMER, J. K. Lincoln did not have the Marfan Syndrome; documented evidence. N Y State J Med, v.81, n.12, Nov, p.1805-13. 1981. LIPSCOMB, K. J., J. CLAYTON-SMITH, et al. Evolving phenotype of Marfan's syndrome. Arch Dis Child, v.76, n.1, Jan, p.41-6. 1997. LOEYS, B., J. DE BACKER, et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat, v.24, n.2, Aug, p.140-6. 2004. LOEYS, B., L. NUYTINCK, et al. Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med, v.161, n.20, Nov 12, p.2447-54. 2001. LOEYS, B. L., H. C. DIETZ, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet, v.47, n.7, Jul, p.476-85. 2010.

101
MASSAGUE, J. e Y. G. CHEN. Controlling TGF-beta signaling. Genes Dev, v.14, n.6, Mar 15, p.627-44. 2000. MERKEL, C. A. Geração e análise de um animal "knockout" para a fibrilina-1, o produto gênico envolvido na Síndrome de Marfan. Instituto de Ciência Biomédicas, Universidade de São Paulo, São Paulo, 2006. MILEWICZ, D. M., J. GROSSFIELD, et al. A mutation in FBN1 disrupts profibrillin processing and results in isolated skeletal features of the Marfan syndrome. J Clin Invest, v.95, n.5, May, p.2373-8. 1995. MURAISHI, H. [Fundamental of medical image processing with personal computer system--development of Plugins by ImageJ]. Nippon Hoshasen Gijutsu Gakkai Zasshi, v.66, n.3, Mar 20, p.260-4. 2010. NAKAJIMA, Y., K. MIYAZONO, et al. Immunolocalization of latent transforming growth factor-beta binding protein-1 (LTBP1) during mouse development: possible roles in epithelial and mesenchymal cytodifferentiation. Cell Tissue Res, v.295, n.2, Feb, p.257-67. 1999. NEPTUNE, E. R., P. A. FRISCHMEYER, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet, v.33, n.3, Mar, p.407-11. 2003. NG, C. M., A. CHENG, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest, v.114, n.11, Dec, p.1586-92. 2004. NIJBROEK, G., S. SOOD, et al. Fifteen novel FBN1 mutations causing Marfan syndrome detected by heteroduplex analysis of genomic amplicons. Am J Hum Genet, v.57, n.1, Jul, p.8-21. 1995. OTSURU, S., T. J. HOFMANN, et al. Osteopoietic engraftment after bone marrow transplantation: Effect of inbred strain of mice. Exp Hematol, May 3. 2010. PALZ, M., F. TIECKE, et al. Clustering of mutations associated with mild Marfan-like phenotypes in the 3' region of FBN1 suggests a potential genotype-phenotype correlation. Am J Med Genet, v.91, n.3, Mar 20, p.212-21. 2000. PEREIRA, L., K. ANDRIKOPOULOS, et al. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet, v.17, n.2, Oct, p.218-22. 1997. PEREIRA, L., M. D'ALESSIO, et al. Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. Hum Mol Genet, v.2, n.10, Oct, p.1762. 1993. PEREIRA, L., S. Y. LEE, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A, v.96, n.7, Mar 30, p.3819-23. 1999. PU, W. T. Identification of a cardiac disease modifier gene using forward genetics in the mouse. PLoS Genet, v.5, n.9, Sep, p.e1000643. 2009. PYERITZ, R. E. The Marfan syndrome. Am Fam Physician, v.34, n.6, Dec, p.83-94. 1986.

102
PYERITZ, R. E. Pleiotropy revisited: molecular explanations of a classic concept. Am J Med Genet, v.34, n.1, Sep, p.124-34. 1989. PYERITZ, R. E. Marfan syndrome: current and future clinical and genetic management of cardiovascular manifestations. Semin Thorac Cardiovasc Surg, v.5, n.1, Jan, p.11-6. 1993. PYERITZ, R. E. The Marfan syndrome. Annu Rev Med, v.51, p.481-510. 2000. RADONIC, T., P. DE WITTE, et al. Losartan therapy in adults with Marfan syndrome: study protocol of the multi-center randomized controlled COMPARE trial. Trials, v.11, p.3. 2010. RAKYAN, V. K., M. E. BLEWITT, et al. Metastable epialleles in mammals. Trends Genet, v.18, n.7, Jul, p.348-51. 2002. RAMIREZ, F. e L. PEREIRA. The fibrillins. Int J Biochem Cell Biol, v.31, n.2, Feb, p.255-9. 1999. REINHARDT, D. P., D. R. KEENE, et al. Fibrillin-1: organization in microfibrils and structural properties. J Mol Biol, v.258, n.1, Apr 26, p.104-16. 1996. ROBINSON, P. N., E. ARTEAGA-SOLIS, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet, v.43, n.10, Oct, p.769-87. 2006. ROBINSON, P. N. e M. GODFREY. The molecular genetics of Marfan syndrome and related microfibrillopathies. J Med Genet, v.37, n.1, Jan, p.9-25. 2000. RODRIGUEZ-VITA, J., E. SANCHEZ-LOPEZ, et al. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation, v.111, n.19, May 17, p.2509-17. 2005. RUIZ-ORTEGA, M., M. RUPEREZ, et al. Molecular mechanisms of angiotensin II-induced vascular injury. Curr Hypertens Rep, v.5, n.1, Feb, p.73-9. 2003. SAHARINEN, J. e J. KESKI-OJA. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol Biol Cell, v.11, n.8, Aug, p.2691-704. 2000. SAITO, S., H. NISHIMURA, et al. Characterization of mutated protein encoded by partially duplicated fibrillin-1 gene in tight skin (TSK) mice. Mol Immunol, v.36, n.3, Feb, p.169-76. 1999. SAKAI, L. Y., D. R. KEENE, et al. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol, v.103, n.6 Pt 1, Dec, p.2499-509. 1986. SANTOS, E. Desenvolvimento de um modelo animal para a Síndrome de Marfan através da manipulação do genoma do camundongo. Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, 2005. SCHORDERET, D. F. Using OMIM (On-line Mendelian Inheritance in Man) as an expert system in medical genetics. Am J Med Genet, v.39, n.3, Jun 1, p.278-84. 1991.

103
SCHORR, S., K. BRAUN, et al. Congenital aneurysmal dilatation of the ascending aorta associated with arachnodactyly; an angiocardiographic study. Am Heart J, v.42, n.4, Oct, p.610-6. 1951. SCHRIJVER, I., W. LIU, et al. Cysteine substitutions in epidermal growth factor-like domains of fibrillin-1: distinct effects on biochemical and clinical phenotypes. Am J Hum Genet, v.65, n.4, Oct, p.1007-20. 1999. SCHRIJVER, I., W. LIU, et al. Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet, v.71, n.2, Aug, p.223-37. 2002. SCHWARTZ, H. Abraham Lincoln and the Marfan Syndrome. JAMA, v.187, Feb 15, p.473-9. 1964. SILVERMAN, D. I., K. J. BURTON, et al. Life expectancy in the Marfan syndrome. Am J Cardiol, v.75, n.2, Jan 15, p.157-60. 1995a. SILVERMAN, D. I., J. GRAY, et al. Family history of severe cardiovascular disease in Marfan syndrome is associated with increased aortic diameter and decreased survival. J Am Coll Cardiol, v.26, n.4, Oct, p.1062-7. 1995b. SUNAGA, S., K. MAKI, et al. Efficient removal of loxP-flanked DNA sequences in a gene-targeted locus by transient expression of Cre recombinase in fertilized eggs. Mol Reprod Dev, v.46, n.2, Feb, p.109-13. 1997. SWEET, A., R. P. ERICKSON, et al. A potential animal model for studying CF heterozygote advantage: genetic variation in theophylline-inducible colonic chloride currents among inbred strains of mice. Biochem Med Metab Biol, v.47, n.1, Feb, p.97-102. 1992. TAIPALE, J., J. SAHARINEN, et al. Extracellular matrix-associated transforming growth factor-beta: role in cancer cell growth and invasion. Adv Cancer Res, v.75, p.87-134. 1998. VAINZOF, M., M. R. PASSOS-BUENO, et al. Immunological methods for the analysis of protein expression in neuromuscular diseases. Methods Mol Biol, v.217, p.355-78. 2003. WHEELER, F. C., L. FERNANDEZ, et al. QTL mapping in a mouse model of cardiomyopathy reveals an ancestral modifier allele affecting heart function and survival. Mamm Genome, v.16, n.6, Jun, p.414-23. 2005. WHITEMAN, P. e P. A. HANDFORD. Defective secretion of recombinant fragments of fibrillin-1: implications of protein misfolding for the pathogenesis of Marfan syndrome and related disorders. Hum Mol Genet, v.12, n.7, Apr 1, p.727-37. 2003. YANG, T., Y. G. HUANG, et al. Influence of genetic background and gender on hypertension and renal failure in COX-2-deficient mice. Am J Physiol Renal Physiol, v.288, n.6, Jun, p.F1125-32. 2005. ZANGALA, T. Isolation of genomic DNA from mouse tails. J Vis Exp, n.6, p.246. 2007. ZHANG, H., W. HU, et al. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J Cell Biol, v.129, n.4, May, p.1165-76. 1995.

104
ZHANG, Y. E. Non-Smad pathways in TGF-beta signaling. Cell Res, v.19, n.1, Jan, p.128-39. 2009.

VIII. ANEXO ____________________________________________________

A New Mouse Model for Marfan Syndrome PresentsPhenotypic Variability Associated with the GeneticBackground and Overall Levels of Fbn1 ExpressionBruno L. Lima1., Enrico J. C. Santos1.¤a, Gustavo R. Fernandes1, Christian Merkel1¤b, Marco R. B.
Mello3¤c, Juliana P. A. Gomes1, Marina Soukoyan1, Alexandre Kerkis1¤a, Silvia M. G. Massironi2, Jose A.
Visintin3, Lygia V. Pereira1*
1 Laboratorio de Genetica Molecular do Departamento de Genetica e Biologia Evolutiva, Universidade de Sao Paulo, Sao Paulo, Brazil, 2 Departamento de Imunologia do
Instituto de Ciencias Biomedicas, Universidade de Sao Paulo, Sao Paulo, Brazil, 3 Departamento de Reproducao Animal da Faculdade de Veterinaria e Zootecnia,
Universidade de Sao Paulo, Sao Paulo, Brazil
Abstract
Marfan syndrome is an autosomal dominant disease of connective tissue caused by mutations in the fibrillin-1 encodinggene FBN1. Patients present cardiovascular, ocular and skeletal manifestations, and although being fully penetrant, MFS ischaracterized by a wide clinical variability both within and between families. Here we describe a new mouse model of MFSthat recapitulates the clinical heterogeneity of the syndrome in humans. Heterozygotes for the mutant Fbn1 allelemgDloxPneo, carrying the same internal deletion of exons 19–24 as the mgD mouse model, present defective microfibrillardeposition, emphysema, deterioration of aortic wall and kyphosis. However, the onset of a clinical phenotypes is earlier inthe 129/Sv than in C57BL/6 background, indicating the existence of genetic modifiers of MFS between these two mousestrains. In addition, we characterized a wide clinical variability within the 129/Sv congenic heterozygotes, suggestinginvolvement of epigenetic factors in disease severity. Finally, we show a strong negative correlation between overall levelsof Fbn1 expression and the severity of the phenotypes, corroborating the suggested protective role of normal fibrillin-1 inMFS pathogenesis, and supporting the development of therapies based on increasing Fbn1 expression.
Citation: Lima BL, Santos EJC, Fernandes GR, Merkel C, Mello MRB, et al. (2010) A New Mouse Model for Marfan Syndrome Presents Phenotypic VariabilityAssociated with the Genetic Background and Overall Levels of Fbn1 Expression. PLoS ONE 5(11): e14136. doi:10.1371/journal.pone.0014136
Editor: Gisela Nogales-Gadea, University Hospital Vall d’Hebron, Spain
Received July 26, 2010; Accepted November 4, 2010; Published November 30, 2010
Copyright: � 2010 Lima et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permitsunrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: This work was funded by Fundacao de Amparo Pesquisa do Estado de Sao Paulo (FAPESP: www.fapesp.br) and Conselho Nacional de DesenvolvimentoCientifico e Tecnologico (CNPq: www.cnpq.br). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of themanuscript.
Competing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected]
¤a Current address: Celltrovet – Applied Genetics, Veterinary Activities Ltda., Sao Paulo, Brazil¤b Current address: Instituto do Coracao e Fundacao Faculdade de Medicina da Universidade de Sao Paulo, Sao Paulo, Brazil¤c Current address: Departamento de Reproducao e Avaliacao Animal, Instituto de Zootecnia da Universidade Federal Rural do Rio de Janeiro, Seropedica, Brazil
. These authors contributed equally to this work.
Introduction
Marfan Syndrome (MFS; OMIM#154700) is an autosomal
dominant disorder with pleiotropic phenotype variations involving
the skeletal, ocular and cardiovascular systems [1,2,3]. The disease
incidence is 1 in 10,000 with 25% of cases being sporadic [4]. In
1991, mutations in the FBN1 gene (OMIM 134797), which
encodes the fibrillin-1 protein, were genetically linked to the MFS
phenotype [5,6,7]. Although MFS is completely penetrant [8], it
presents a wide clinical variability [9]. Genotype-phenotype
correlations in MFS have been complicated by the large number
of unique mutations reported, as well as by clinical heterogeneity
among individuals with the same mutation; this extensive clinical
variability, even within families, suggests the presence of modifier
genes [10].
In 1997, a murine model for MFS - mgD - was created to mimic
the dominant-negative effect of fibrillin-1 mutations seen in MFS
patients [11]. Approximately 6 kb of the Fbn1 gene encompassing
exons 19–24 were replaced by a neomycin-resistance expression
cassette (neoR), resulting in a protein monomer missing 272
residues. Surprisingly, heterozygote animals were histologically
indistinguishable from wild-type mice, suggesting absence of the
dominant-negative effects seen in MFS patients. In contrast,
homozygote animals died before the second week of life, due to
cardiovascular failure. Expression analysis showed that the mgDallele had a 90% lower transcript level compared to the normal
Fbn1 allele. Accordingly, it was postulated that the neoR cassette
sequence had interfered with the mutant allele expression,
consequently restricting the dominant-negative effect of the
mutation.
Here we report the generation of a novel variant of the mgDmouse model in which the same mutant Fbn1 allele is present, but
with neoR flanked by lox-P sequences (mgDloxPneo), allowing Cre-
recombinase-mediated deletion of the resistance cassette [12].
Unexpectedly, this construct now resulted in heterozygous
mgDloxPneo mice presenting some aspects of the MFS phenotype,
PLoS ONE | www.plosone.org 1 November 2010 | Volume 5 | Issue 11 | e14136

including aortic, skeletal and respiratory system manifestations,
before the removal of neoR sequence. Moreover, these phenotypes
differ significantly between two different isogenic mouse strains,
C57BL/6 (B6) and 129/Sv, and also vary within the 129/Sv
background. Therefore, in addition to modeling the clinical
manifestations of MFS disease, the mgDloxPneo mouse model is an
experimental system in which both the genetic background and
epigenetic contributions to MFS clinical variability can be
evaluated.
Results
Animal developmentCells from correctly targeted ES cell clones were aggregated to
morulas, and used to produce chimeric male mice that, after mating
with CD1 females, resulted in transmission of the mgDloxPneo Fbn1
allele to a proportion of the F1 generation (Figure 1A). RT-PCR
and sequencing analysis of skin RNA from heterozygous animals
confirmed the in-frame deletion of exons 19–24 in transcripts
derived from the targeted allele (Figure 1B–C). As observed in the
original mgD strain, the heterozygous mgDloxPneo from the F1 did
not present any apparent phenotype. However, in subsequent
crosses between F1 heterozygotes, we obtained some heterozygous
animals with a very severe phenotype, characterized by deformities
of the spine at 2 months of age (Figure 1 D). From a total of 47
heterozygous mice, four presented the mentioned feature, and died
by 3 months of age of unknown causes presenting hemothorax,
suggestive of aortic rupture. We also obtained four homozygous
animals that died between 4 and 8 days of age. All these animals
came from mixed 129/Sv and CD-1 backgrounds.
These results suggested that the phenotype variability could be
associated with the different genetic backgrounds of the animals. To
test this hypothesis, the mgDloxPneo allele was put into two different
isogenic backgrounds, namely the 129/Sv and B6 strains. Haplotype
analysis of a large panel of microsatellite markers confirmed the
congenic status of mice after 14 generations (data not shown).
Real-time RT-PCR analysis of embryonic day 13 fibroblasts
from both strains showed 47%65 (P,0.05) of Fbn1 mRNA levels
in homozygous, and 78%610 (P,0.05) in heterozygous fibro-
blasts when compared to wild-type cells (Figure 1E). Therefore,
the level of expression of the mgDloxPneo allele is significantly
higher than the 10% level observed in the original mgD allele [11],
which may explain the differences in phenotypes seen in
heterozygotes between the two models.
Immunohistochemical comparison of cultured fetal fibroblasts
from B6 animals revealed qualitative differences in fibrils between
wild-type and mutant cells (Figure 2). In contrast to the elaborate
network of immunoreactive fibrillin-1 seen in control lines
(Figure 2A), heterozygous cells present fibers spread over a diffuse
background (Figure 2B), while homozygous cell cultures show a
diffuse pattern of immunoreactive material (Figure 2C) similar to
that reported in cultured fibroblasts from homozygous mgD, and
heterozygous Tight Skin models [11,13]. In the homozygous
mutant fibroblasts we also observed an apparent intracellular
deposition of mutant protein, evidenced by the visualization of
cells with nuclear regions delimitated by marked protein
(Figure 2D). The changes observed in the immunofluorescence
assays indicate that a portion of the mutant molecules may be
retained inside the cell, which in turn, could be a significant factor
contributing to the pathogenesis of MFS [14].
PhenotypesHeterozygous mice from both strains have normal lifespan and
reproductive capacity, but display some of the classic MFS
phenotypes (Figure 3). Pulmonary alterations include enlargement
of peripheral air space (respiratory bronchioles and alveoli), and
destruction of alveolar wall structures, characterizing pulmonary
emphysema (Figure 3A–D). We also detected a large amount of
infiltrating mononuclear cells, indicating a chronic inflammation
process in the lung. The cardiovascular phenotypes include
thickening of the aortic media, disruption/degradation of the
elastic fibers (Figure 3F–I), but no inflammatory cells were
detected. Finally, mutant animals also presented skeletal manifes-
tations, mostly kyphosis (Figure 3K–N).
In order to characterize the phenotypic variability in the two
strains, we quantified the phenotypes in the three affected organ
systems. Analysis of the mean linear intercept (Lm) revealed
changes on the average size of alveoli in 129/Sv heterozygotes
(Figure 3E). By 3 months of age, these animals present an Lm
significantly higher (p,0.02) than wild-type mice, and this
difference became more pronounced with age. In contrast, B6
mice at 3 months of age presented milder (although significant,
p,0.01) pulmonary alterations, and only at 6 months of age did
the alterations become severe.
The vascular phenotype was quantified by measuring the
thickness of the aortic media in heterozygous animals. As observed
in the lung, at 3 months of age 129/Sv heterozygotes exhibited a
significantly enlarged media when compared to wild-type, and this
difference increased with age (Figure 3J). In contrast, in the B6
background heterozygous mgDloxPneo mice were asymptomatic at
3 months of age, whereas by 9 months they presented severe
alterations indistinguishable from those of 129/Sv heterozygotes at
the same age.
Quantification of the skeletal manifestations was performed by
calculating the ratio between the linear distance and the length
from the first cervical vertebrae to the last thoracic vertebrae (KR)
(Figure 3N). As with the other phenotypes, heterozygous animals
from the 129/Sv strain manifested a more severe skeletal
phenotype earlier than those from the B6 background
(Figure 3O). Together, these data show that the main differences
observed in disease manifestation between the two strains is the
age of onset of symptoms, which is delayed in B6 animals.
Finally, when present in homozygocity the mutation is lethal
during gestation in both strains. Crossings between heterozygotes
produced 30 (42%) wild-type animals and 41 (58%) heterozygous
offsprings. Homozygous mutant embryos were identified only
prior to embryonic day 13.
129/Sv phenotypic variabilityInterestingly, while the different phenotypes are homogeneous
among B6 heterozygotes (Figure 3E, J and O), animals from the
129/Sv strain present a wide clinical variability before 6 months of
age, with phenotypes varying from mild to very severe (Figure 4A).
Based on the average Lm in alveoli, we scored thirteen 129/Sv
heterozygotes between 3 to 6 months of age in 3 clinical categories
(Figure 4B): compared with wild-type animals (Lm =
66.75618.26 mm), 15% were classified as asymptomatic
(Lm = 68.4565.2 mm; P,0,42); 38% as carrying moderate
alterations (Lm = 116.4465.46 mm; P,0.0006) and 46% as
presenting severe alterations (Lm = 183.86619.84 mm; P,0.006).
The aortic phenotype also exhibited variability in the 129/Sv
heterozygotes at 3 months of age (Figure 4C), allowing the
classification of phenotypes in 2 categories: 60% were defined as
asymptomatic (55.2166.46 mm versus 61.61611.08 mm in wild
types; P = 0.33), and 40% were classified as presenting moderate
changes (77.7562.59 mm; P,0.04). At 6 months of age, 33% of
the heterozygotes were classified as asymptomatic (79.106
mgDloxPneo Mouse Model for MFS
PLoS ONE | www.plosone.org 2 November 2010 | Volume 5 | Issue 11 | e14136

3.43 mm versus 70.3462.72 mm in wild types; P = 0.80); and 66%
were scored as severely affected (101.8566.92 mm; P,0.01).
A similar variability was observed in the skeletal phenotype in
the 129/Sv strain, where heterozygotes with 3 months of age
displayed different degrees of kyphosis, allowing the classification
of heterozygotes into distinct classes of mild and severe, according
to the animal age (Figure 4D).
When analyzed together, the phenotypic variations in the 129/
Sv background show a strong correlation (|R|$0.70), i.e., animals
with more severe manifestations in the pulmonary tissue also have
a more thickened aortic wall, as well as a more severe kyphosis
(Figure 4A).
Molecular analysisThe levels of normal and mutant fibrillin-1 transcripts have
been associated with the severity of the disease in humans and
mice [15,16]. Therefore, we analyzed the expression of the normal
and mgDloxPneo-mutant Fbn1 alleles in differently affected animals
of the 129/Sv strain to evaluate if there was any correlation with
the corresponding phenotypes. Real-time RT-PCR from lung
Figure 1. Targetting of Fbn1. (A) Scheme of Fbn1 targeting. From top to bottom, fibrillin1 protein with the internally deleted region (residues 770–1042); schematic representation of the corresponding Fbn1 gene region with the probes (I and III) used in the Southern-blot analysis, and the sizes ofBamHI (b) and HindIII (h) fragments; and targeted Fbn1mgDloxPneo allele with the neoR flanked by lox-P sequences (triangles). (B) Southern blot ofgenomic DNA from correctly targeted ES clone digested with BamHI, and hybridized to probe I. (C) sequence of the cDNA from the Fbn1mgDloxPneo
allele showing the junction of exons 18 and 25; (D) Heredogram of founder heterozygotes in a mixed 129/Sv/CD-1 background. Hetreozygotes werephenotypically normal until the F3, where one severely affected heterozygote was found (arrow), whose X-ray is shown. (E) Relative quantification oftotal Fbn1 mRNA levels in wild-type (WT), heterozygous (HE) and homozygous (HO) fetal fibroblasts by Real Time RT-PCR analysis (P,0.05).doi:10.1371/journal.pone.0014136.g001
mgDloxPneo Mouse Model for MFS
PLoS ONE | www.plosone.org 3 November 2010 | Volume 5 | Issue 11 | e14136

RNA showed a wide variability in the levels of expression of
normal and mutant Fbn1 alleles in 129/Sv heterozygotes
(Figure 5A). Furthermore, the analysis of heterozygotes in the
129/Sv background revealed that the ratio between mutant and
normal Fbn1 transcripts is similar among animals with different
phenotypes (Figure 5A). However, there is a strong negative
correlation (|R|$0.75) between the overall Fbn1 expression and
the severity of the phenotypes, i.e., mice with lower expression of
Fbn1 gene tend to have more severe phenotypes in the three
affected systems (Figure 5B–D).
Discussion
The first knockout mice to model MSF were homozygous for
hypomorphic alleles of Fbn1 gene: the mgD and mgR models [11].
While the mgD expresses a truncated form of fibrillin-1 at very low
levels resulting in early post-natal lethality, mgR homozygotes
produce low levels of normal fibrillin-1, and recapitulate the adult
lethal form of MFS [17]. A third model, the C1039G, is
characterized by substitution of a cysteine by a glycine at residue
1039, in an EGF domain of fibrillin-1, one of the most common
type of mutations in humans [16,18]. Heterozygotes show
deficiency in the deposition of microfibrils in the extracellular
matrix, skeletal disorders, and progressive deterioration of the
architecture of the aortic wall. The three mouse models have been
characterized exclusively in the B6 inbred strain [11,14,24].
The mgDloxPneo is a new mouse model for MFS that, in addition
to recapitulating some important phenotypes of the human
disease, presents the clinical variability characteristic of the
syndrome. The main difference between this new mutation and
the original mgD is the presence in the former of two loxP sites
flanking neoR – both mutant alleles result in Fbn1 transcripts with
an in frame deletion of exons 19 through 24. Nevertheless, the
mgDloxPneo allele has a 47% increase in the level of expression
when compared to the mgD mutant allele which, according to the
dominant-negative model of pathogenesis for MFS, can explain
the manifestation of the disease in mgDloxPneo heterozygotes. We
are currently breeding these animals with CRE-transgenic mice in
order to eliminate neoR and possibly increase the level of mutant
transcripts to that of the normal mRNA in the heterozygotes.
The mgDloxPneo heterozygotes have phenotypes similar to the
C1039G model [16], including conserved number of elastic
lamellae in the aortic media despite overall enlargement of the
aorta. However, this new model presents a chronic inflammation
process in the lung, characterized by the presence of large number
of mononuclear cell in the lung parenchyma, as observed in the
hypomorphic homozygous mgR model [19].
We demonstrated the effect of the genetic background on the
severity of the MFS phenotype by backcrossing the mgDloxPneo allele
into two different inbred mouse strains, and showing the earlier
onset of the disease in heterozygotes in the 129/Sv background
when compared to the B6 strain. This inter-strain phenotypic
variability could be attributed to differences in the extent of
interference of the neoR cassete in the expression of the mutant allele
between the two congenic strains. However, we show that the level
of expression of the mutant allele is 47% of the wild type Fbn1 in
both strains. Therefore, we believe that genetic factors modulating
the difference in phenotype expression between the two strains can
be relevant for the pathogenesis of MFS. Interestingly, the most
severe phenotypes, including death at 3 months of age, were
Figure 2. Immunofluorescence of cultured fetal fibroblasts. (A) Wild-type, (B) heterozygous and (C) homozygous fibroblasts immune-stainedfor fibrillin-1 (scale bars 50 mm). In contrast to the elaborate network of immunoreactive fibrillin-1 seen in control lines, heterozygous andhomozygous cell cultures show a diffuse pattern of immunoreactive material. (D) Representative field at higher magnification to show intracellulardeposition of mutant protein (scale bar 25 mm).doi:10.1371/journal.pone.0014136.g002
mgDloxPneo Mouse Model for MFS
PLoS ONE | www.plosone.org 4 November 2010 | Volume 5 | Issue 11 | e14136

Figure 3. Phenotypic variability between strains. (A to D) Histologic analysis of lung (HE staining), scale bars 100 mm. (A) wild-type mice shownormal airspace caliber; (B) 129/Sv heterozygous mouse by 3 months of age shows diffuse distal airspace widening; B6 heterozygous mice present (C)mild alterations by 3 months of age, and (D) severe phenotype by 6 months of age; (E) Average mean linear intercept is greater for mutant mice thanfor wild-type (*P,0.02, **P,0.04, {P,0.01); (F–I) Histologic analysis of thoracic aorta (Weigert coloration), scale bars 100 mm. (F) wild-type, (G) 129/Svheterozygote at 3 months of age, B6 heterozygote at (H) 3 months and (I) 9 months of age. Note diffuse fragmentation of elastic fibers (arrows). (J)Average thickness of the thoracic aortic media (*P,0.05, **P,0.007, {P,0.18, {{P,0.03); (K–N) X-ray analysis. (K) Wild-type, (L) 3 months old 129/Svheterozygote, B6 heterozygote at (M) 3 months and (N) 9 months of age. (O) Average KR, used to quantify skeletal phenotype (*P,0.01, {P,0.04).The number of animals analyzed in each group (n) and the respective age (3 m, 6 m, and 9 m indicate 3, 6 and 9 months, respectively) are shown.doi:10.1371/journal.pone.0014136.g003
mgDloxPneo Mouse Model for MFS
PLoS ONE | www.plosone.org 5 November 2010 | Volume 5 | Issue 11 | e14136
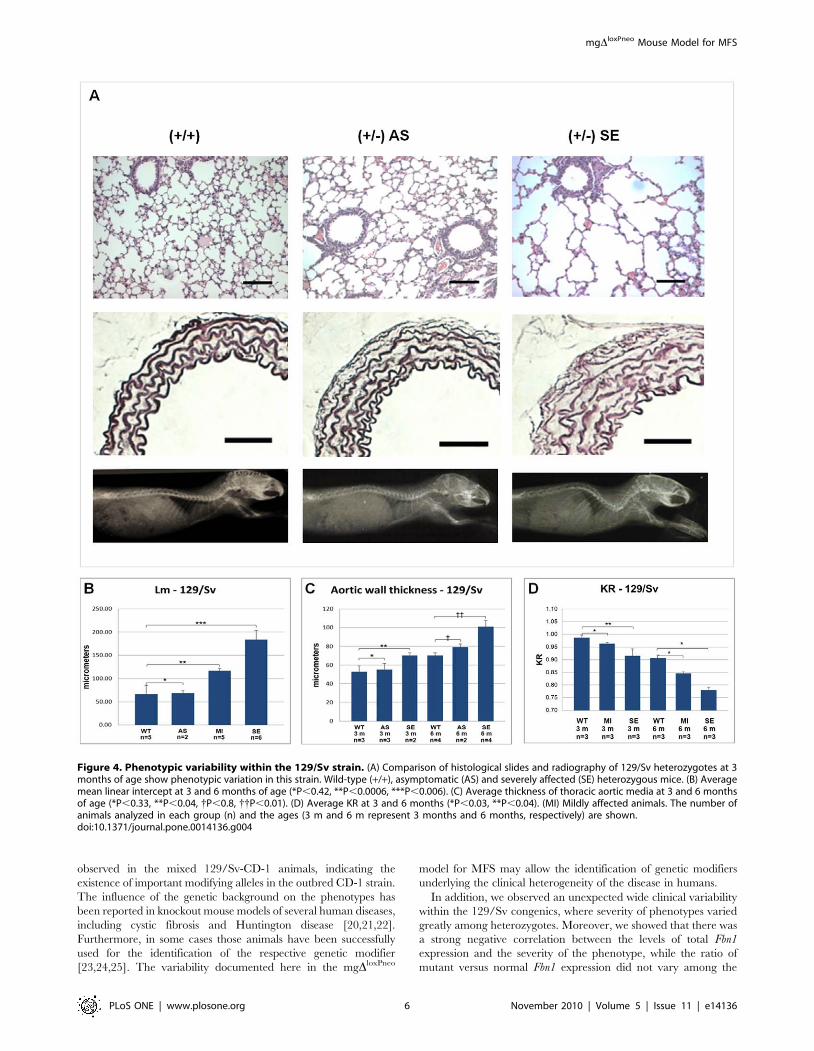
observed in the mixed 129/Sv-CD-1 animals, indicating the
existence of important modifying alleles in the outbred CD-1 strain.
The influence of the genetic background on the phenotypes has
been reported in knockout mouse models of several human diseases,
including cystic fibrosis and Huntington disease [20,21,22].
Furthermore, in some cases those animals have been successfully
used for the identification of the respective genetic modifier
[23,24,25]. The variability documented here in the mgDloxPneo
model for MFS may allow the identification of genetic modifiers
underlying the clinical heterogeneity of the disease in humans.
In addition, we observed an unexpected wide clinical variability
within the 129/Sv congenics, where severity of phenotypes varied
greatly among heterozygotes. Moreover, we showed that there was
a strong negative correlation between the levels of total Fbn1
expression and the severity of the phenotype, while the ratio of
mutant versus normal Fbn1 expression did not vary among the
Figure 4. Phenotypic variability within the 129/Sv strain. (A) Comparison of histological slides and radiography of 129/Sv heterozygotes at 3months of age show phenotypic variation in this strain. Wild-type (+/+), asymptomatic (AS) and severely affected (SE) heterozygous mice. (B) Averagemean linear intercept at 3 and 6 months of age (*P,0.42, **P,0.0006, ***P,0.006). (C) Average thickness of thoracic aortic media at 3 and 6 monthsof age (*P,0.33, **P,0.04, {P,0.8, {{P,0.01). (D) Average KR at 3 and 6 months (*P,0.03, **P,0.04). (MI) Mildly affected animals. The number ofanimals analyzed in each group (n) and the ages (3 m and 6 m represent 3 months and 6 months, respectively) are shown.doi:10.1371/journal.pone.0014136.g004
mgDloxPneo Mouse Model for MFS
PLoS ONE | www.plosone.org 6 November 2010 | Volume 5 | Issue 11 | e14136
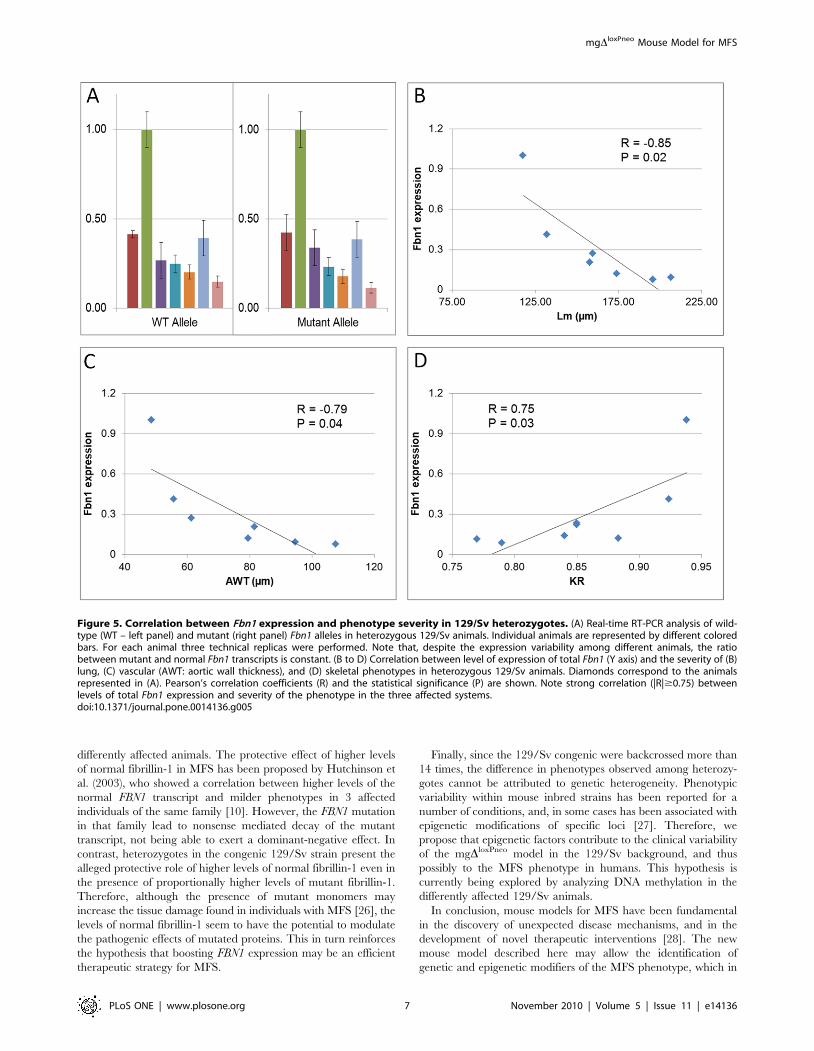
differently affected animals. The protective effect of higher levels
of normal fibrillin-1 in MFS has been proposed by Hutchinson et
al. (2003), who showed a correlation between higher levels of the
normal FBN1 transcript and milder phenotypes in 3 affected
individuals of the same family [10]. However, the FBN1 mutation
in that family lead to nonsense mediated decay of the mutant
transcript, not being able to exert a dominant-negative effect. In
contrast, heterozygotes in the congenic 129/Sv strain present the
alleged protective role of higher levels of normal fibrillin-1 even in
the presence of proportionally higher levels of mutant fibrillin-1.
Therefore, although the presence of mutant monomers may
increase the tissue damage found in individuals with MFS [26], the
levels of normal fibrillin-1 seem to have the potential to modulate
the pathogenic effects of mutated proteins. This in turn reinforces
the hypothesis that boosting FBN1 expression may be an efficient
therapeutic strategy for MFS.
Finally, since the 129/Sv congenic were backcrossed more than
14 times, the difference in phenotypes observed among heterozy-
gotes cannot be attributed to genetic heterogeneity. Phenotypic
variability within mouse inbred strains has been reported for a
number of conditions, and, in some cases has been associated with
epigenetic modifications of specific loci [27]. Therefore, we
propose that epigenetic factors contribute to the clinical variability
of the mgDloxPneo model in the 129/Sv background, and thus
possibly to the MFS phenotype in humans. This hypothesis is
currently being explored by analyzing DNA methylation in the
differently affected 129/Sv animals.
In conclusion, mouse models for MFS have been fundamental
in the discovery of unexpected disease mechanisms, and in the
development of novel therapeutic interventions [28]. The new
mouse model described here may allow the identification of
genetic and epigenetic modifiers of the MFS phenotype, which in
Figure 5. Correlation between Fbn1 expression and phenotype severity in 129/Sv heterozygotes. (A) Real-time RT-PCR analysis of wild-type (WT – left panel) and mutant (right panel) Fbn1 alleles in heterozygous 129/Sv animals. Individual animals are represented by different coloredbars. For each animal three technical replicas were performed. Note that, despite the expression variability among different animals, the ratiobetween mutant and normal Fbn1 transcripts is constant. (B to D) Correlation between level of expression of total Fbn1 (Y axis) and the severity of (B)lung, (C) vascular (AWT: aortic wall thickness), and (D) skeletal phenotypes in heterozygous 129/Sv animals. Diamonds correspond to the animalsrepresented in (A). Pearson’s correlation coefficients (R) and the statistical significance (P) are shown. Note strong correlation (|R|$0.75) betweenlevels of total Fbn1 expression and severity of the phenotype in the three affected systems.doi:10.1371/journal.pone.0014136.g005
mgDloxPneo Mouse Model for MFS
PLoS ONE | www.plosone.org 7 November 2010 | Volume 5 | Issue 11 | e14136

turn will lead to a better understanding of the clinical variability of
the disease, and of the physiology of pulmonary, cardiovascular
and skeletal systems.
Materials and Methods
AnimalsAll animals used were housed under controlled temperature and
light conditions in pathogen-free environment at the Immunology
Department of ICB USP experimentation housing facility. We
analyzed 45 mutant animals and 20 wild-type animals, at three
different ages and from two different mice strains. The C57BL/6
(B6) was chosen because it is the most widely used inbred strain,
and all other extant mouse models for MFS are in that
background. The 129/Sv was chosen because the ES cells used
for gene targeting were derived from this strain. All animal
experiments followed the protocols approved by the Institutional
Animal Care and Use Committee of the Instituto de Biociencias at
USP. Protocol ID: CEA/IBUSP 020/2004.
Development of the mgDloxPneo mouse modelThe murine ES cell line USP-1 was used for gene targeting
experiments [29]. Generation of positively targeted ES cell clones
and production of chimaeric mice were performed as previously
described [11]. The Fbn1mgDloxPneo targeting vector was a
modification of the previously described targeting vector used to
create the mgD and mgR models [11,17]. Two complementary
38-bp oligonucleotides with the loxP consensus sequence [12] were
synthesized, annealed, and cloned flanking the neoR expression
cassette, which was then used to replace the original neoR cassette
of the mgD vector. Correctly targeted ES clones were identified by
southern blot as described [11].
GenotypingDNA was extracted from a 0.5 cm piece of tail using Proteinase
K (Promega) as described [30]. Each sample was submitted to two
PCRs to identify the presence of the Fbn1mgDloxPneo allele and the
normal allele, which served as an internal reaction control.
Fbn1mgDloxPneo allele primers: forward 59 -GAG GCT ATT CGG
CTA TGA CT – 39, reverse 59 – CTC TTC GTC CAG ATC
ATC CT – 39. Cycling conditions were 94uC for 2.5 min, then
94uC, 57uC, 72uC for 1 min for 30 cycles in a 10 ml volume.
Fbn1wt allele primers: forward 59 – AAA CCA TCA AGG GCA
CTT GC – 39, reverse 59 – CAC ATT GCG TGC CTT TAA
TTC – 39. Cycling conditions were 94uC for 2.5 min primary
denaturation, then 94uC, 55uC, 72uC for 1 min for 30 cycles in a
10 ml volume.
ImmunofluorescenceMouse embryonic fibroblasts (MEFs) were prepared from
embryos at 13–14 days of gestation as described [31]. Cells were
fixed in 4% paraformaldehyde in PBS for 20 min at 4uC and
permeabilized in 0.05% Triton X-100 in PBS for 5 min. Non-
specific binding was blocked with 10% FBS in PBS for 1 h at room
temperature. Cells were incubated with pAb9543 (1:1000 dilution)
primary antibody [32] overnight at 4uC and with secondary
antibody coupled to Cy3 for 1 h at room temperature. The
fluorescence signals were examined using an Axiovert 200 (Carl
Zeiss) and an ApoTome imaging system (Carl Zeiss).
Histological analysisAnimals were sacrificed by cervical dislocation. Mouse tissues
were processed as previously described [33]. Five-micron sections
were stained with hematoxylin and eosin, and adjacent sections
were assayed for Weigert coloration, specific for elastic fiber
visualization. Slides were examined and photographed using an
Axiovert 200 (Carl Zeiss).
Phenotype quantificationSkeletal: A full body x-ray of each mouse was digitalized and the
follow measurements were taken using AutoCAD software 2002:
the cervical-thoracic segment length and the straight line distance
of the same segment. With those measures we were able to
establish a kyphosis ratio (straight distance/segment length), and
use the ratio to score the animals according to the severity of the
skeletal manifestation (the smaller ratio, the more severe the
manifestation).
Aortic wall: The histological samples were photographed at 506and 1006 magnification, and the length of the inner and outer
perimeters of the aorta were measured using the imageJ software
[34]. From this we could estimate the inner and outer radius, and
wall thickness of the aorta.
Lung: The size of alveolar airways was determined by
measuring the linear intercept on H&E-stained lungs as previously
described [35].
Real-time RT-PCRTotal RNA was extracted from mouse lung and from mouse
embryonic day 13 fibroblasts using TRIzol (Invitrogen Corp.).
The RNA was treated with DNase according to the manufacture’s
instructions (Invitrogen Corp.). A total of 1 mg of total RNA was
reverse-transcribed with SuperScriptTM III First-Strand Synthesis
System (Invitrogen Corp.). Wild type, mutant and total Fbn1
mRNA levels were determined using real-time RT-PCR sequence
detection (7500 Real Time System; Applied Biosystems). mRNA
levels were normalized to Actb mRNA, and fold expression
determined as previously described [36]. The following primers
and probes sequences were designed using the PrimerExpress
software (Applied Biosystems): WT forward 59 – ACA TAA CTG
GGA AAA ACT GTG TCG ATA – 39, WT reverse 59 – TTC
CAG GTG TGT TTC GAC ATT GT – 39, WT probe 59 –TGT
GCT GAA CAG TCT ACT– 39; KO forward 59 –GGG ATA
TGA AGT AGA CAT AAC TGG GAA A– 39, KO reverse 59 –
GAG GCT GGG TAT CAT CTT GCA – 39, KO probe 59 –
ACT GTG TCG ATA TCA ATG– 39; Fbn1Total forward 59 –
CCT GTG CTA TGA TGG GTT CA – 39, Fbn1Total reverse 59
– AGG TCC CAC TAA GGC AGA TGT – 39; ACTB forward 59
–ACGGCCAGGTCATCACTATTG – 39, ACTB reverse 59 –
CAAGAAGGAAGGCTGGAAAAGA– 39. Three technical rep-
licates of each reaction were performed.
Statistical AnalysesPearson’s Correlation Coefficient (R) was used to determine the
correlation between the Fbn1 gene expression and the severity of
the phenotype, reflecting the degree to which the variables are
related; weak correlation 0#|R|#0.29; moderate correlation
0.30#|R|#0.69; strong correlation |R|$0.70. P,0.05 was
deemed to be significant.
A nonparametric test, Mann Whitney test, was used to
determine statistical significance for all tests other than the
Pearson Correlation Coefficient. All statistical analyses were
performed using MINITAB (R14). P,0.05 was deemed to be
significant.
Acknowledgments
We wish to thank Dr Peter Pearson for his significant help on the
preparation of the manuscript. We also gratefully acknowledge our
mgDloxPneo Mouse Model for MFS
PLoS ONE | www.plosone.org 8 November 2010 | Volume 5 | Issue 11 | e14136

colleagues Joana C.M de Mello, Ana Maria Fraga, Raquel Stabellini, Erica
Sara Araujo, Fabiano T. de Araujo, Natassia M.S Vieira, Cynthia Q.
Cardoso and Dra. Mariz Vainzof for helpful suggestions.
Author Contributions
Conceived and designed the experiments: BLL EJCS SMGM JAV LVP.
Performed the experiments: BLL EJCS GRF CAM MRBM MS AK
SMGM JPAG LVP. Analyzed the data: BLL EJCS GRF CAM MRBM
SMGM JPAG JAV LVP. Wrote the paper: BLL LVP.
References
1. Pyeritz RE (1990) Marfan syndrome. N Engl J Med 323: 987–989.
2. Silverman DI, Gray J, Roman MJ, Bridges A, Burton K, et al. (1995) Familyhistory of severe cardiovascular disease in Marfan syndrome is associated with
increased aortic diameter and decreased survival. J Am Coll Cardiol 26:
1062–1067.3. Pyeritz RE, McKusick VA (1981) Basic defects in Marfan syndrome.
N Engl J Med 305: 1011–1012.4. Gray JR, Bridges AB, Faed MJ, Pringle T, Baines P, et al. (1994) Ascertainment
and severity of Marfan syndrome in a Scottish population. J Med Genet 31:51–54.
5. Kainulainen K, Peltonen L (1991) Marfan gene discovered. Ann Med 23:
395–396.6. Kainulainen K, Steinmann B, Collins F, Dietz HC, Francomano CA, et al.
(1991) Marfan syndrome: no evidence for heterogeneity in different populations,and more precise mapping of the gene. Am J Hum Genet 49: 662–667.
7. Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, et al. (1991) Marfan
syndrome caused by a recurrent de novo missense mutation in the fibrillin gene.Nature 352: 337–339.
8. Pyeritz RE (1986) The Marfan syndrome. Am Fam Physician 34: 83–94.9. Pyeritz RE, Murphy EA, McKusick VA (1979) Clinical variability in the Marfan
syndrome(s). Birth Defects Orig Artic Ser 15: 155–178.10. Hutchinson S, Furger A, Halliday D, Judge DP, Jefferson A, et al. (2003) Allelic
variation in normal human FBN1 expression in a family with Marfan syndrome:
a potential modifier of phenotype? Hum Mol Genet 12: 2269–2276.11. Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, et al. (1997) Targetting
of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfansyndrome. Nat Genet 17: 218–222.
12. Sunaga S, Maki K, Komagata Y, Ikuta K, Miyazaki JI (1997) Efficient removal
of loxP-flanked DNA sequences in a gene-targeted locus by transient expressionof Cre recombinase in fertilized eggs. Mol Reprod Dev 46: 109–113.
13. Gayraud B, Keene DR, Sakai LY, Ramirez F (2000) New insights into theassembly of extracellular microfibrils from the analysis of the fibrillin 1 mutation
in the tight skin mouse. J Cell Biol 150: 667–680.14. Whiteman P, Handford PA (2003) Defective secretion of recombinant fragments
of fibrillin-1: implications of protein misfolding for the pathogenesis of Marfan
syndrome and related disorders. Hum Mol Genet 12: 727–737.15. Buoni S, Zannolli R, Macucci F, Ansaldi S, Grasso M, et al. (2004) The FBN1
(R2726W) mutation is not fully penetrant. Ann Hum Genet 68: 633–638.16. Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, et al. (2004) Evidence for
a critical contribution of haploinsufficiency in the complex pathogenesis of
Marfan syndrome. J Clin Invest 114: 172–181.17. Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, et al. (1999)
Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A 96: 3819–3823.
18. Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, et al. (2004) TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan
syndrome. J Clin Invest 114: 1586–1592.
19. Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, et al. (2003)Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan
syndrome. Nat Genet 33: 407–411.
20. Sweet A, Erickson RP, Huntington C, Dawson D (1992) A potential animal model
for studying CF heterozygote advantage: genetic variation in theophylline-
inducible colonic chloride currents among inbred strains of mice. Biochem Med
Metab Biol 47: 97–102.
21. Otsuru S, Hofmann TJ, Rasini V, Veronesi E, Dominici M, et al. (2010)
Osteopoietic engraftment after bone marrow transplantation: Effect of inbred
strain of mice. Exp Hematol.
22. Yang T, Huang YG, Ye W, Hansen P, Schnermann JB, et al. (2005) Influence of
genetic background and gender on hypertension and renal failure in COX-2-
deficient mice. Am J Physiol Renal Physiol 288: F1125–1132.
23. Wheeler FC, Fernandez L, Carlson KM, Wolf MJ, Rockman HA, et al. (2005)
QTL mapping in a mouse model of cardiomyopathy reveals an ancestral
modifier allele affecting heart function and survival. Mamm Genome 16:
414–423.
24. Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, et al. (1993) Genetic
identification of Mom-1, a major modifier locus affecting Min-induced intestinal
neoplasia in the mouse. Cell 75: 631–639.
25. Pu WT (2009) Identification of a cardiac disease modifier gene using forward
genetics in the mouse. PLoS Genet 5: e1000643.
26. Booms P, Tiecke F, Rosenberg T, Hagemeier C, Robinson PN (2000)
Differential effect of FBN1 mutations on in vitro proteolysis of recombinant
fibrillin-1 fragments. Hum Genet 107: 216–224.
27. Rakyan VK, Blewitt ME, Druker R, Preis JI, Whitelaw E (2002) Metastable
epialleles in mammals. Trends Genet 18: 348–351.
28. Judge DP, Dietz HC (2008) Therapy of Marfan syndrome. Annu Rev Med 59:
43–59.
29. Sukoyan MA, Kerkis AY, Mello MR, Kerkis IE, Visintin JA, et al. (2002)
Establishment of new murine embryonic stem cell lines for the generation of
mouse models of human genetic diseases. Braz J Med Biol Res 35: 535–542.
30. Zangala T (2007) Isolation of genomic DNA from mouse tails. J Vis Exp. 246 p.
31. Hogan A, Beddington R, Constantine F, Lacy E (1994) Manipulating the mouse
Embryo: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor
Laboratory Press.
32. Reinhardt DP, Keene DR, Corson GM, Poschl E, Bachinger HP, et al. (1996)
Fibrillin-1: organization in microfibrils and structural properties. J Mol Biol 258:
104–116.
33. Andrikopoulos K, Liu X, Keene DR, Jaenisch R, Ramirez F (1995) Targeted
mutation in the col5a2 gene reveals a regulatory role for type V collagen during
matrix assembly. Nat Genet 9: 31–36.
34. Muraishi H [Fundamental of medical image processing with personal computer
system—development of Plugins by ImageJ]. Nippon Hoshasen Gijutsu Gakkai
Zasshi 66: 260–264.
35. Dunnill MS (1962) Quantitative Methods in the Study of Pulmonary Pathology.
Thorax 17: 320–328.
36. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using
real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:
402–408.
mgDloxPneo Mouse Model for MFS
PLoS ONE | www.plosone.org 9 November 2010 | Volume 5 | Issue 11 | e14136


















