
APLICAÇÃO DA CROMATOGRAFIA LÍQUIDA DE ALTA EFICIÊNCIA …
36
1 UNIVERSIDADE DE SÃO PAULO FACULDADE DE CIÊNCIAS FARMACÊUTICAS Trabalho de Conclusão do Curso de Farmácia-Bioquímica APLICAÇÃO DA CROMATOGRAFIA LÍQUIDA DE ALTA EFICIÊNCIA EM FASE REVERSA PARA A DETERMINAÇÃO DA NATEGLINIDA EM MEDICAMENTOS Aluno: Renato Biscain Orientadora: Profa. Dra. María Segunda Aurora Prado São Paulo 2020
Transcript of APLICAÇÃO DA CROMATOGRAFIA LÍQUIDA DE ALTA EFICIÊNCIA …

1
UNIVERSIDADE DE SÃO PAULO
FACULDADE DE CIÊNCIAS FARMACÊUTICAS
Trabalho de Conclusão do Curso de Farmácia-Bioquímica
APLICAÇÃO DA CROMATOGRAFIA LÍQUIDA DE ALTA
EFICIÊNCIA EM FASE REVERSA PARA A DETERMINAÇÃO DA
NATEGLINIDA EM MEDICAMENTOS
Aluno: Renato Biscain
Orientadora: Profa. Dra. María Segunda Aurora Prado
São Paulo
2020

2
SUMÁRIO
Lista de Figuras …………………………………………………………………….....3
Lista de Tabelas………………………………………………………………….........4
Lista de abreviaturas…………………………………………………………………..5
RESUMO……………………………………………………………………………….6
1. INTRODUÇÃO..........................................................................................7
2. REVISÃO DA LITERATURA....................................................................8
3. OBJETIVOS............................................................................................14
3.1 Objetivo Geral........................................................................................14
3.2 Objetivos Específicos.............................................................................14
4. MATERIAL E MÉTODO.........................................................................14
4.1 MATERIAIS............................................................................................14
4.1.1 Reagentes, padrões, solventes e soluções.......................................15
4.1.2 Equipamentos....................................................................................16
4.2 MÉTODO................................................................................................16
5. RESULTADO E DISCUSSÃO................................................................22
6. CONCLUSÃO.........................................................................................29
7. REFERÊNCIAS......................................................................................31
ANEXOS

3
LISTA DE FIGURAS
Figura 1.
Figura 2.
Figura 3.
Figura 4.
Estrutura química da nateglinida.
Cromatograma da solução do padrão de nateglinida (50 µg
mL-1).
Cromatogramas da solução padrão de nateglinida, diluente,
placebo e amostra.
Curva analítica da solução padrão de nateglinida na faixa de
concentração de 40 a 60 µg mL-1

4
Tabela 1.
Tabela 2.
Tabela 3.
Tabela 4.
Tabela 5.
Tabela 6.
Tabela 7.
Tabela 8.
Tabela 9.
Tabela 10.
.
LISTA DE TABELAS
Placebo de nateglinida.
Concentração das soluções tampão de fosfato de sódio
monobásico com seus correspondentes pHs.
Composição das Fases móveis.
Teste de Youden.
Composição das fases móveis.
Dados estatísticos.obtidos na curva analítica. Método HPLC.
Precisão do método (repetitividade e precisão intermediária).
Recuperação da solução padrão de nateglinida. Método
HPLC.
Resultados obtidos em oito condições para avaliação da
robustez.
Efeitos dos parâmetros analíticos no teor e tempo de
retenção (TR).

5
ANVISA
ATP
DM
DPP4
DPR
g
GLP-1
HPLC
HPLTC
ICH
KATP
LD
LQ
mg
mm
mM
MM
min
mL
NA
nm
pH
RDC
SDS
TR
UV
Vis
λ
µg
LISTA DE ABREVIATURAS
Agência Nacional de Vigilância Sanitária
Adenosina trifosfato
Diabetes Mellitus
Inibidores da dipeptidil peptidase 4
Desvio padrão relativo
Grama
Peptídeo semelhante a glucagon 1
High performance liquid chromatography
High-performance thin-layer chromatography
International Council for Harmonisation
Canal de potássio sensível ao ATP
Limite de detecção
Limite de Quantificação
Miligrama
Milímetro
Milimolar
Massa molar
Minuto
Mililitro
Nateglinida
Nanômetro
Potencial hidrogeniônico
Resolução da Diretoria Colegiada
Dodecil sulfato de sódio
Tempo de retenção
Ultravioleta
Visível
Comprimento de onda
Micrograma

6
RESUMO
O diabetes mellitus é uma doença crônica onde a concentração de glicose
encontra-se anormalmente elevada (hiperglicemia) e está associada a diversas
disfunções metabólicas. Atualmente, o diabetes representa grande ameaça à
saúde global estando relacionado dentre os principais motivos ao estilo de vida
moderno das grandes metrópoles e cuja doença acomete milhões de pessoas
a cada ano. Um dos fármacos utilizados no tratamento do diabetes mellitus, a
Nateglinida da classe das metiglinidas age como anti-hiperglicêmico. Objetiva-
se com esse trabalho desenvolver e validar uma metodologia analítica que seja
rápida, exata e precisa para a determinação da nateglinida em comprimidos
através da cromatografia líquida de alta eficiência em microemulsão. Foi
utilizada uma coluna C18 Nova-Pak (150 × 3.9 mm, 4 μm) e fase móvel
constituída por 3,4 g dodecil sulfato de sódio (SDS), 6,6 g butanol, 0,8 g octano
e 89,2 g de tampão fosfato 20 mM pH 2,6, a vazão foi de 1,0 mL/min,
temperatura de forno de 25 ºC. A Detecção foi realizada em 215 nm. O método
foi validado de acordo com as diretrizes dos guias ICH, Farmacopeia
Americana e resolução RDC N° 166/2017 da ANVISA. O método demonstrou
ser linear na faixa de concentração de 40 à 60 µg mL-1 com R2 = 0,9975. A
nateglinida foi detectada em 2,7 min. Os limites de detecção e quantificação
foram 2,30 µg mL-1 e 6,98 µg mL-1, respectivamente, a precisão expressa como
desvio padrão relativo foi menor que 2%. A exatidão, expresso em termos de
percentagem de recuperação de nateglinida foi 100,6%. O método mostrou ser
linear, específicopreciso, exato, e robusto para a quantificação da nateglinida
em medicamentos. Não foi observada interferência por parte dos excipientes
com a nateglinida. O método proposto pode ser utilizado para análises de
rotina em laboratórios de controle de qualidade.
BISCAIN, R. Aplicação da cromatografia líquida de alta eficiência em fase
reversa para a determinação da nateglinida em medicamentos. no. 35 f. 38
Trabalho de Conclusão de Curso de Farmácia e Bioquímica – Faculdade de
Ciências Farmacêuticas – Universidade de São Paulo, São Paulo, 2020.
Palavras-chave: Diabetes tipo 2, HPLC, validação.

7
1. INTRODUÇÃO
O Diabetes mellitus, também chamado de Diabetes tipo 2 é uma doença
crônica onde a concentração sérica de glicose encontra-se anormalmente
elevada (hiperglicemia). [1] Vários processos patogênicos estão envolvidos no
desenvolvimento do diabetes, os quais dois são mais comuns: a diminuição da
capacidade da insulina em atuar nos tecidos periféricos (resistência à insulina)
e a disfunção das células β pancreáticas [2] representada pela incapacidade de
produzir quantidade suficiente de insulina para superar a resistência à insulina
nos tecidos periféricos. [3]
Estima-se que cerca de 425 milhões de pessoas possuem diabetes e a
projeção para 2045 é que esse número cresça em torno de 48% entre pessoas
de 20 a 79 anos de idade em todo o mundo. [4] Estudos apontam que os
maiores aumentos ocorrerão em países de renda média e baixa. No Brasil, a
doença acomete cerca de 12,5 milhões de pessoas, sendo o quarto país com o
maior número de afetados. [4, 5] As razões para a crescente epidemia de
diabetes mellitus são múltiplas, incluindo o envelhecimento da população, a
crescente prevalência da obesidade e do sedentarismo, e os processos de
urbanização são considerados os principais fatores relacionados ao aumento
da incidência e prevalência da doença. [6, 7]
Atualmente, existem diversos medicamentos que podem ser utilizados
no tratamento da diabetes tipo 2 individualmente ou em associações. São
utilizados sulfonilureias de segunda geração (glibenclamida), biguanidas
(metformina), inibidores de α-glicosidase (acarbose, miglitol), inibidores de
DPP-4 (alogliptina), metiglinidas (nateglinida, repaglinida) entre outros. [8, 9] A
Nateglinida é um agente insulinotrópico de ação rápida e curta duração que
reduz os níveis de glicose no sangue, pela sua ligação ao canal KATP das
células β pancreáticas para estimular a liberação de insulina. [10, 11] Esses
medicamentos têm grande importância no controle da glicose sanguínea e no
tratamento da DM.
A literatura revela alguns trabalhos para a determinação de nateglinida
por cromatografia líquida de alta eficiência em fluidos biológicos como plasma
humano [12], em plasma de coelho [13, 14], em intestino de rato [15] e em

8
medicamentos [16, 17]. Outros trabalhos também foram realizados utilizando-
se as técnicas analíticas como eletroforese capilar [18, 19], cromatografia de
camada delgada de alto desempenho (HPTLC) [20]. Foi encontrado apenas um
trabalho que utilizou a fase móvel composta por microemulsões [21].
As microemulsões são definidas como fluidos macroscopicamente
homogêneos, opticamente transparentes, compostas por gotículas dispersas,
na maioria das vezes carregadas negativamente, do tamanho de nanômetros
de um solvente orgânico lipofílico imiscível. [22] Para formar essa mistura, a
tensão interfacial entre o óleo (como octano) e a água deve estabilizada pela
adição de um surfactante e um co-surfactante [23, 24] Podem ser classificadas
como óleo em água (O / A), ou água em óleo (A / O) [22]. O presente trabalho
visa o uso da cromatografia líquida em microemulsão para determinar a
nateglinida em medicametos.
2. REVISÃO DA LITERATURA
Diabetes
O Diabetes é uma doença metabólica caracterizada por hiperglicemia
resultante de defeitos na secreção de insulina, ação da insulina ou ambos.
Vários processos patogênicos estão envolvidos no desenvolvimento do
diabetes. Estes variam desde a destruição autoimune das células β
pancreáticas com consequente deficiência de insulina até anormalidades que
resultam em resistência à ação da insulina. [2] O diabetes está associado ao
aumento da mortalidade, bem como à morbidade por cegueira, insuficiência
renal que requer diálise, amputações não traumáticas dos membros inferiores e
doença vascular aterosclerótica. O custo do gerenciamento do diabetes é muito
alto e mais da metade é gasta no gerenciamento de complicações associadas
ao DM consumindo uma grande quantidade de recursos de saúde pública.
Existem três tipos principais de diabetes: tipo 1, tipo 2 (mais comum, constitui
cerca de 90% dos casos) e diabetes gestacional. [25]

9
Apesar dessa prevalência alarmante de DM, ainda existe uma falta de
diagnóstico que afeta cerca de 193 milhões de pessoas em todo o mundo e
desconhecem a doença devido ao desenvolvimento silencioso de sintomas e
sinais mínimos e a falta de acesso aos cuidados de saúde. [26]
Os fatores de risco para DM tipo 2 incluem uma combinação de fatores
genéticos e metabólicos que contribuem para sua prevalência. Fatores não
modificáveis que incluem etnia, histórico familiar e idade avançada; além dos
fatores modificáveis, como obesidade, alimentação não saudável,
sedentarismo e tabagismo. [26, 27]
Tratamento da diabetes
Em geral, o controle do DM e a prevenção de suas complicações são
obtidas através do controle da dieta e do exercício físico ou do uso de
antidiabéticos ou da combinação de ambos. [28]
Existem no momento diversas opções terapêuticas, incluindo
secretagogos de sulfonilureia, secretagogos de não sulfonilureia, biguanidas,
inibidores da α-glucosidase e Inibidores da dipeptidil peptidase-4. As classes de
medicamentos listadas acima constituem a espinha dorsal da terapia
medicamentosa padrão ou tradicional para o diabetes tipo 2. [28]
Sulfonilureias: Derivam da condensação do grupo arilsulfona com a porção
uréia. [28] São secretagogos que atuam desencadeando a secreção endógena
de insulina das nos canais de potássio sensíveis a ATP das células β
pancreáticas. [29] A segunda geração inclui gliburida, glipizida e glimeprida [28]
Biguanidas: Apresentam dois núcleos de guanida condensados. Três
medicamentos estão incluídos nessa classe: metformina, buformina e
fenformina. [28] A metformina é o medicamento antidiabético mais prescrito,
especialmente usado em indivíduos obesos e com sobrepeso. Este
medicamento ainda é a melhor escolha para monoterapia. Atuam aumentando
a sensibilidade à insulina, aumentando a captação de glicose. [29]

10
Inibidores da α-glicosidase: A α-glicosidase é uma enzima responsável pelo
metabolismo dos carboidratos no intestino. Os inibidores dessa enzima
dificultam a absorção dos carboidratos no trato gastrointestinal e com a
secreção de insulina ativada, a glicemia é reduzida no organismo. [30] As
estruturas químicas dos membros da classe indicam moléculas semelhantes a
açúcar, projetadas para ação local no trato gastrointestinal sem absorção
sistêmica. Estão disponíveis três inibidores da enzima: acarbose, miglitol e
voglibose. [28]
Inibidores da dipeptidil peptidase-4 (DPP4): Os inibidores da DPP4
(dipeptidil peptidase 4) comumente denominados gliptinas, impedem a
inativação de GLP-1 que promove a secreção da insulina), resultando na
potencialização da sinalização pancreática dessa enzima o que aumenta o
consumo de glicose e reduz a produção de glicose hepática. Fármacos
exemplares dessa classe são a vildagliptina, sitagliptina e saxagliptina. [31]
Metiglinidas: são um grupo de medicamentos hipoglicemiantes indicados no
tratamento inicial da diabetes mellitus tipo 2. Dentre os representantes dessa
classe são a repaglinida e nateglinida. [28] Sua ação se dá pelo bloqueio dos
canais de KATP na membrana celular das células β pancreática induzindo a
secreção de insulina. O mecanismo do canal iônico é altamente seletivo para
os tecidos, com baixa afinidade para o músculo cardíaco e esquelético. [10]
Nateglinida
A Nateglinida é um pó branco ou quase branco cristalino, inodoro de
sabor amargo. [32] É um ácido orgânico fraco, cujo pka é de 3,1. [21] A NA é
altamente solúvel em metanol, etanol, clorofórmio, éter, acetona e pouco
solúvel em acetonitrila, octanol, enquanto praticamente insolúvel em água. [32,
33]. Quimicamente é Ácido (2R) -3-fenil-2 - [(4-propan 2-il ciclo-hexano
carbonil) amino]. Sua fórmula molecular é C19H27NO3, com massa molecular
de 317,429 mol/g [11]. A Figura 1 mostra a estrutura química da nateglinida:

11
Figura 1. Estrutura química da nateglinida.
Fonte: Site da tcichemicals. [34]
Usos
A nateglinida (starlix) é um agente anti-hiperglicêmico oral usado no
tratamento do diabetes mellitus não dependente de insulina que age ligando-se
às células β do pâncreas para estimular a liberação de insulina. Seu efeito na
redução da glicose pós-prandial é bastante específico e apenas modestamente
reduz a glicemia no jejum. Portanto, a nateglinida é mais apropriadamente
usada quando os níveis de glicose em jejum são moderadamente elevados no
cenário de diabetes precoce ou em combinação com sensibilizadores de
insulina ou com insulina noturna prolongada. É um fármaco de curta duração e
seu efeito ocorre poucos minutos após a ingestão de alimentos, na primeira
fase da liberação de insulina. A dose habitual é de 120 mg devendo ser tomada
antes das refeições. Os efeitos adversos do Starlix incluem ganho de peso,
hiperinsulinemia e hipoglicemia. [35, 11]
Validação de métodos analíticos
De modo a garantir eficácia e segurança dos medicamentos, é de
enorme importância o controle de qualidade de medicamentos nas indústrias
farmacêuticas, as quais precisam cumprir as determinações impostas pela

12
Agência Nacional de Vigilância Sanitária (ANVISA) [36, 40]. Para tal, é
necessário a escolha de um método analítico adequado e validado. O
desenvolvimento do método analítico, a adaptação ou implementação de
método conhecido, envolve processo de avaliação que estime sua eficiência na
rotina do laboratório visando confirmar a confiabilidade e capacidade de
desempenho do mesmo. Esse processo que costuma ser denominado de
validação é atividade prioritária para o cumprimento das práticas de controle,
permitindo níveis apropriados de exatidão, precisão e confiabilidade. [37, 38]
Em âmbito internacional a regulamentação é realizada de acordo com as
exigências do ICH, representado por Estados Unidos, Japão e Europa, que em
comum acordo definem os parâmetros, metodologias e os requerimentos para
a validação de métodos analíticos. No Brasil a ANVISA por através da RDC Nº
166, DE 24 DE JULHO DE 2017, estabelece os critérios para validação de
métodos analíticos utilizados em produtos biológicos, insumos farmacêuticos e
medicamentos. [39] Os parâmetros de validação para uma metodologia
analítica são seletividade/especificidade, linearidade/faixa de trabalho,
precisão, exatidão, robustez, limite de detecção e limite de quantificação. [40]
Especificidade/seletividade: Especificidade é a capacidade de avaliar
inequivocamente o analito na presença de componentes que podem estar
presentes. Normalmente, estes podem incluir impurezas, produtos de
degradação, matriz, etc. A seletividade é definida como sendo a capacidade de
detecção de substâncias. [40 - 42]
Linearidade: corresponde à capacidade de obter respostas analíticas
diretamente proporcionais à concentração de um analito em uma amostra,
dentro de uma determinada faixa de aplicação. Para o estabelecimento da
linearidade, deve-se utilizar, no mínimo, 5 (cinco) concentrações diferentes da
substância química de referência para as soluções preparadas em, no mínimo,
triplicata. O coeficiente de determinação (R2) deve estar acima de 0,990. [40 -
42]
Precisão: avalia a proximidade de concordância entre os resultados de
análises individuais quando o procedimento é aplicado diversas vezes em uma

13
mesma amostra homogênea, em idênticas condições de ensaio. Deve ser
expressa por meio da repetibilidade, da precisão intermediária ou da
reprodutibilidade. É recomendado que a precisão seja estimada empregando-
se no mínimo, nove determinações de três concentrações, uma alta, uma
média e uma em baixas concentrações em triplicata, ou seis determinações da
concentração nominal.
Repetibilidade expressa a precisão nas mesmas condições de operação
em um curto intervalo de tempo. A repetibilidade também é denominada
precisão intra-ensaio.
A precisão intermediária expressa variações dentro dos laboratórios: dias
diferentes, analistas diferentes, equipamentos diferentes. [40 - 42]
Exatidão: O grau de concordância entre os resultados individuais do método
em estudo em relação a um valor aceito como verdadeiro é definido pela
exatidão e é calculada como a porcentagem de recuperação através do ensaio
de uma quantidade conhecida da substância em análise adicionada a um meio
de composição definida. Deve ser verificada a partir de, no mínimo, nove
determinações, contemplando o intervalo linear do método analítico, ou seja,
três concentrações: baixa, média e alta, com três réplicas em cada nível. [40 –
42]
Limite de detecção: é a menor concentração da substância em exame que
pode ser detectada, mas não necessariamente quantificada, com certo limite
de confiabilidade. [40 - 42]
Limite de quantificação: é a menor concentração de uma substância que
pode ser determinada quantitativamente com precisão e exatidão adequadas.
O limite de quantificação é um parâmetro de ensaios quantitativos para baixos
níveis de compostos em matrizes de amostras e é usado particularmente para
a determinação de impurezas e / ou produtos de degradação. [40 - 42]
Robustez: A capacidade de um método de permanecer inalterado pequenas e
deliberadas variações como composição da fase móvel, pH, força iônica é
medida pela robustez. Fornece uma indicação de sua confiabilidade durante o

14
uso normal. Considera-se que um método é robusto quando ele não é afetado
por uma modificação pequena e deliberada em seus parâmetros. [40 - 42]
A robustez de um procedimento analítico é uma medida de sua
capacidade de permanecer inalterada por variações pequenas, mas
deliberadas, nos parâmetros do método e fornece uma indicação de sua
confiabilidade durante o uso normal. [40 - 42]
3. OBJETIVOS
3.1 Objetivo geral
O presente trabalho tem como objetivo desenvolver e validar uma
metodologia analítica rápida, precisa e exata para a determinação da
nateglinida em comprimidos através da cromatografia líquida de alta eficiência
em microemulsão.
3.2 Objetivos específicos
- Desenvolver um método analítico que seja rápido, preciso e exato por
cromatografia líquida de alta eficiência em microemulsão.
- Validar a metodologia analítica desenvolvida visando à aplicação em
laboratórios de controle de qualidade.
- Aplicar da metodologia desenvolvida para a quantificação da nateglinida
em medicamentos.
4. MATERIAL E MÉTODO
4.1 MATERIAIS
4.1.1 Reagentes, padrões, solventes e soluções

15
Padrão
Padrão de nateglinida, (pureza de 99,9%) (Novartis, São Paulo).
Amostra
Comprimidos revestidos contendo 120 mg de nateglinida, Starlix® (Novartis,
São Paulo).
Placebo
O placebo da amostra comercial constituído de lactose monohidratada,
celulose microcristalina, povidona, croscarmelose de sódio, estearato de
magnésio, óxido de ferro amarelo, hipromelose, dióxido de titânio, talco,
macrogol, sílica coloidal anidra.
Reagentes e Solventes
- Metanol, J.T Baker® ;
- Água purificada Milli-Q;
- Octano, Merck®;
- n-Butanol, Merck®;
- Dodecil Sulfato de Sódio (SDS), Sigma Aldrich®;
- Fosfato de Sódio Monobásico, J.T Baker®;
- Ácido Fosfórico, Sigma Aldrich®;
Vidrarias e acessórios
- Pipeta automática (Eppendorf®);
- Balões volumétricos âmbar;
- Vidraria âmbar;
- Sistema de filtração a vácuo;
- Unidade filtrante PTFE Millex 0,45 µm 13mm (Millipore®);
- Membrana filtrante HV Millex 0,45 µm 47mm (Millipore®);
- Béquer de vidro;
- Bastão de vidro;
- Gral e pistilo;
- Vials

16
4.1.2 Equipamentos
- Balança analítica (METTLER-TOLEDO®)
- Aparelho de ultrassom (UNIQUE®) - modelo USC 1450;
- Aparelho medidor de pH (Digimed®)
- Cromatógrafo à Líquido (Agilent®), 1100® com bomba quaternária,
degaseificador online, amostrador automático, aquecedor/resfriador de
coluna e detector UV.
- Software OpenLab Plus
- Purificador de água Milli-Q (Millipore®)
- Coluna Nova-Pak C18 (4 μm, 150 mm × 3.9 mm)
4.2 MÉTODO
Preparação da solução padrão estoque
A solução padrão foi obtida pesando-se cerca de 5 mg do padrão
nateglinida em uma balança analítica devidamente calibrada e transferindo seu
conteúdo para um balão âmbar de 5 mL. Foi adicionado cerca de 2,5 mL de
metanol e foi sonicado por 2 minutos para dissolução. Esfriou-se e o volume foi
completado com metanol, obtendo-se uma concentração de 1000 µg mL-1. A
solução foi filtrada usando membrana de filtro PTFE de 45 µm.
Preparação da solução de trabalho
Foi transferido 250 µL da solução padrão estoque para um balão âmbar
de 5 ml e completou-se o volume com metanol, obtendo-se concentração final
de 50 µg mL-1. A solução foi filtrada usando membrana de filtro PTFE de 45
µm.
Preparação da solução da amostra estoque
Foram pesados 20 comprimidos e triturados em gral de porcelana
utilizando-se um pistilo até obter um pó fino e homogêneo. Logo depois, pesou-
se uma quantidade do pó (equivalente a 5 mg de nateglinida) e transferiu-se
para um balão volumétrico âmbar de 5 ml e foi adicionado cerca de 2,5 ml de
metanol. A amostra foi sonicada por 5 minutos para completa dissolução, foi

17
resfriada e completado o volume com metanol, obtendo-se uma concentração
de 1000 µg mL-1 de nateglinida. A solução foi filtrada usando membrana de
filtro PTFE de 45 µm.
Preparo das soluções estoque de placebo
A solução placebo foi obtida pesando-se o equivalente de placebo a 5
mg de nateglinida em uma balança analítica devidamente calibrada e
transferindo seu conteúdo para um balão âmbar de 5 mL. Foi adicionado cerca
de 2,5 mL de metanol e foi sonicado por 5 minutos para completa dissolução.
Esfriou-se e o volume foi completado com metanol. A solução foi filtrada
usando membrana de filtro PTFE de 45 µm. A constituição do placebo segue
as proporções da Tabela 1. [43]
Tabela 1: Placebo de nateglinida.
Fonte: Autoria própria.
Preparo das soluções estoque de tampão
Quatro soluções estoque de fosfato de sódio monobásico (MM 119,997
g mol-1) [44] foram preparadas. Duas soluções estoque de tampão 20 mM e
duas de 16 mM. Para a obtenção de uma concentração de 20 mM, foi pesado
aproximadamente cerca de 1,20 g de fosfato de sódio monobásico e transferido
para dois balões volumétricos de 500 ml. Em outros dois balões de 500 ml
foram pesados aproximadamente 0,9599 g deste sal. Os balões foram

18
completados com água MilliQ® e para cada concentração de tampão fosfato o
pH foi ajustado para 2,6 e 2,8 com ácido fosfórico. Obteve-se dessa forma
quatro soluções tampão segundo a Tabela 2:
Tabela 2. Concentração das soluções tampão de fosfato de sódio monobásico
com seus correspondentes pHs.
Fonte: Autoria própria.
Preparo das Fases Móveis
As fases móveis constituídas de microemulsões foram preparadas a
partir da mistura de dodecil sulfato de sódio (SDS), butanol, octano com as
Soluções 1, 2, 3 e 4 contidas na Tabela 3. As soluções foram sonicadas por 30
minutos em ultrassom. Filtrou-se cada fase móvel através de uma membrana
de 0,45 µm.
Foram obtidas quatro fases móveis: A, B, C e D, conforme a Tabela 3.

19
Tabela 3. Composição das Fases móveis.
Fonte: Autoria própria.
Validação do método analítico
Para validação do método foram avaliados os parâmetros de
seletividade, linearidade, limites de detecção e quantificação, precisão,
exatidão e robustez, seguindo as especificações da ANVISA e recomendações
do International Conference on Harmonization (ICH) e Farmacopeia Americana.
[40 - 42]
Seletividade/Especificidade
Com intuito de avaliar a Seletividade/Especificidade realizaram-se
injeções do diluente, placebo, padrão e amostra. Todas as soluções foram
filtradas através de membrana filtrante de de 45 µm.
Linearidade
Foram pegas 5 alíquotas da solução padrão estoque de nateglinida e
transferidas para 5 balões volumétricos âmbar de 5 mL. Foi adicionado metanol
até a metade dos balões e sonicados por 2 minutos até completa dissolução. O
volume foi completado com metanol. Filtrou-se as soluções através de um filtro
de 45 µm. Obteve-se concentrações de 40 µg mL-1, 45 µg mL-1, 50 µg mL-1, 55
µg mL-1 e 60 µg mL-1 de nateglinida (80% a 120% da concentração de

20
trabalho). As soluções foram corridas no equipamento de HPLC em triplicata.
As áreas obtidas foram plotadas versus as concentrações.
Precisão (repetitibilidade e precisão intermediária)
A repetitibilidade do método foi determinada através da preparação da
amostra a uma concentração 100% da concentração de trabalho a partir da
solução estoque da amostra. Foram pegas alíquotas da solução estoque da
amostra, previamente preparada, e transferidas para 6 balões volumétricos
âmbar de 5 mL. Os volumes foram completados com metanol e sonicados por
5 min. Filtrou-se as soluções através de um filtro de 45 µm. Obteve-se a
concentração de 60 µg mL-1 de nateglinida.
De maneira análoga foi utilizado o padrão de nateglinida. As amostras
foram quantificadas por HPLC, no mesmo dia, pelo mesmo analista e a mesma
instrumentação. A precisão do método foi expresso em termos de porcentagem
do desvio padrão relativo (% DPR).
Precisão Intermediária
A precisão intermediária foi realizada seguindo o mesmo procedimento
descrito na repetitividade. As amostras foram quantificadas pelo método de
HPLC, em dias diferentes, com mesmo analista e mesma instrumentação. A
precisão do método foi expresso em termos de porcentagem do desvio padrão
relativo (% DPR).
Exatidão
A exatidão foi determinada através da preparação de nove soluções de
placebo fortificadas com o padrão e testadas a três níveis de concentração
(80%, 100% e 120% da concentração de trabalho). Os resultados são
expressos na forma de porcentagem de recuperação das amostras.

21
Eq.(1)
Limites de detecção e quantificação
Os limites de detecção e quantificação foram calculados a partir da
curva analítica, segundo as equações a seguir:
Onde:
σ = estimativa do desvio padrão da resposta;
S = coeficiente angular da curva analítica.
Robustez
A robustez foi realizada usando o teste de Youden, que envolve a
variação de parâmetros analíticos combinados em oito experimentos. Para
determinar a variação de um fator, foram encontrados os quatro valores
correspondentes às letras maiúsculas e minúsculas, comparando-se as médias
dos dois grupos [45]. Para calcular as variações que cada parâmetro exerce no
método foram utilizadas as equações a seguir:
Eq. (2)
Eq. (3)

22
Onde : A/a, B/b, C/c e D/d são os efeitos do pH da fase móvel, temperatura da
coluna, fluxo da fase móvel e concentração de tampão respectivamente. As
letras de s a z identificam os resultados obtidos em cada ensaio.
A Tabela 4 mostra os fatores que foram modificados para a determinação da
robustez. Foram denominados os fatores nominais por letras maiúsculas de A a
D e as variações, por letras minúsculas correspondentes. Estes fatores e suas
variações foram escolhidos pelo analista de acordo com testes preliminares.
Tabela 4. Teste de Youden.
Fonte: Dias, Silva e Bonifácio (2017, p. 4). [46 ]
O impacto em percentual (%) de cada fator é expresso dividindo a
diferença obtida pela média global de 8 resultados. Essa porcentagem
demonstra a possível mudança de sistema capacidade de resposta quando a
variável em questão é alterada no intervalo indicado.
Eq. (4)
Eq. (5)
Eq. (6)
Eq. (7)

23
5. RESULTADOS E DISCUSSÃO
Desenvolvimento do método analítico
Seleção do Comprimento de onda
Uma solução do padrão de nateglinida foi preparada na concentração de
50 µg mL-1 e lida num espectrofotômetro UV/Vis na faixa comprimento de onda
de 200 a 400 nm. Obteve-se uma máxima em 215 nm. Este comprimento de
onda foi usado neste trabalho.
DESENVOLVIMENTO DO MÉTODO POR HPLC
Foram testados diferentes fases móveis com o intuito de otimizar o
método (Tabela 5).
Tabela 5. Composição das fases móveis
Fonte: Autoria própria.
As análises foram realizadas em temperatura ambiente controlada (25,0
± 1,0 ºC) utilizando vazões de 1,0 mL min-1 e 2,0 mL min-1 e volume de injeção
de 10 µL. Após estes testes obteve se um sistema otimizado constituído por:
Fase móvel: 3,4 g SDS, 6,6 g butanol, 0,8 g octano e 89,2 g de tampão fosfato
20 mM; coluna: Nova-Pak C18 (150 × 3.9 mm, 4 μm); vazão: 1,0 mL min-1;
volume de injeção: 10 µL; temperatura: 25 ºC e comprimento de onda de 215
nm. A nateglinida foi determinada em 2,7 min (Figura 2).

24
Figura 2. Cromatograma da solução do padrão de nateglinida (50 µg mL-1).
Condições: Fase móvel: 3,4 g SDS, 6,6 g butanol, 0,8 g octano e 89,2 g de
tampão fosfato 20 mM; coluna: Nova-Pak C18 (150 × 3.9 mm, 4 μm); vazão:
1,0 mL min-1; volume de injeção: 10 µL; temperatura: 25 ºC e detecção em 215
nm. Fonte: Autoria própria.
VALIDAÇÃO DO MÉTODO ANALÍTICO
Seletividade/Especificidade
A Figura 3 apresenta os cromatogramas de diluente, placebo, padrão e
amostra. Pode-se observar que não houve interferência por parte dos
excipientes da amostra e do diluente com o pico principal da nateglinida,
portanto, o método foi específico.

25
Figura 3. Cromatogramas da solução padrão de nateglinida, diluente, placebo
e amostra. Condições: Fase móvel: 3,4 g SDS, 6,6 g butanol, 0,8 g octano e
89,2 g de tampão fosfato 20 mM; coluna: Nova-Pak C18 (150 × 3.9 mm, 4 μm);
vazão: 1,0 mL min-1; volume de injeção: 10 µL; temperatura: 25 ºC e detecção
em 215 nm. Fonte: Autoria própria, 2020.
26
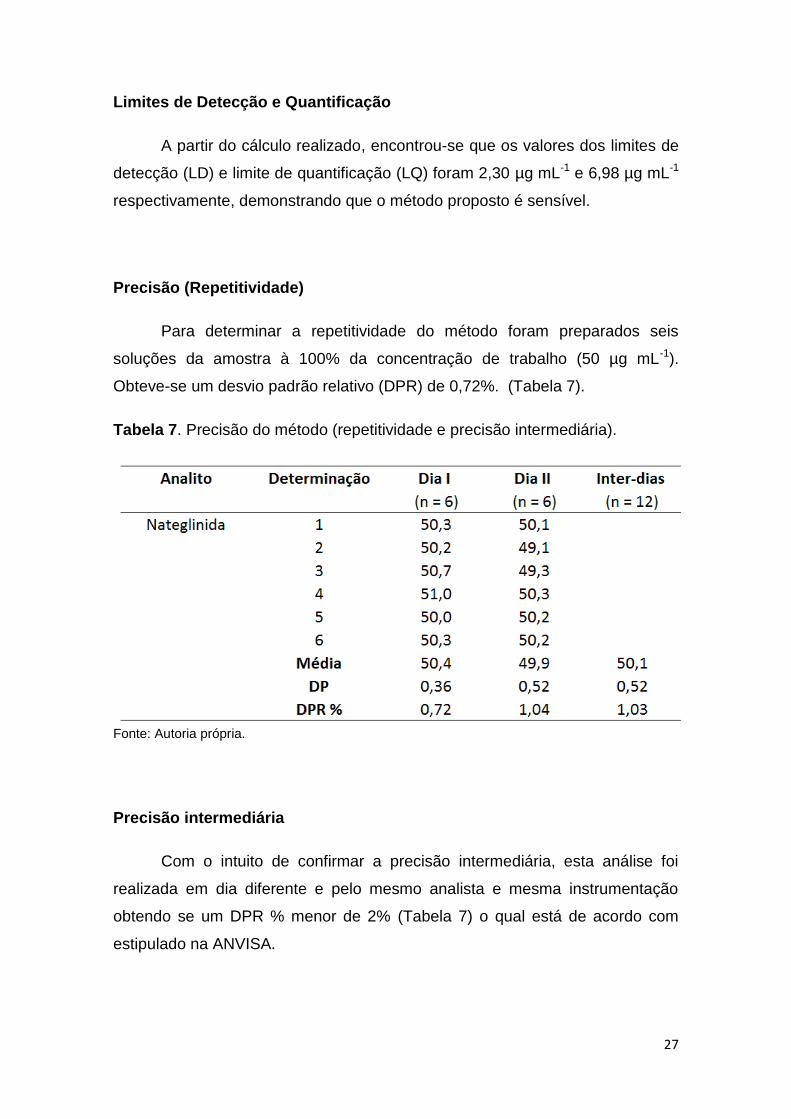
Linearidade
A linearidade foi realizada a fim de garantir que os resultados obtidos
são proporcionais à concentração do padrão de nateglinida, de acordo com o
intervalo definido no item 4.2. De acordo com a resolução da RDC Nº 166/17 o
coeficiente de determinação deve ser maior que 0,99.
O método proposto foi linear na faixa de concentração de 40 a 60 µg
mL-1. Obteve se um coeficiente de determinação maior que 0,99 (Tabela 6 e
Figura 4).
Tabela 6. Dados estatísticos obtidos da curva analítica. Método HPLC.
Fonte: Autoria própria
Figura 4. Curva analítica da solução padrão de nateglinida na faixa de
concentração de 40 a 60 µg mL-1. Método HPLC.
Fonte: Autoria própria.
27
Limites de Detecção e Quantificação
A partir do cálculo realizado, encontrou-se que os valores dos limites de
detecção (LD) e limite de quantificação (LQ) foram 2,30 µg mL-1 e 6,98 µg mL-1
respectivamente, demonstrando que o método proposto é sensível.
Precisão (Repetitividade)
Para determinar a repetitividade do método foram preparados seis
soluções da amostra à 100% da concentração de trabalho (50 µg mL-1).
Obteve-se um desvio padrão relativo (DPR) de 0,72%. (Tabela 7).
Tabela 7. Precisão do método (repetitividade e precisão intermediária).
Fonte: Autoria própria.
Precisão intermediária
Com o intuito de confirmar a precisão intermediária, esta análise foi
realizada em dia diferente e pelo mesmo analista e mesma instrumentação
obtendo se um DPR % menor de 2% (Tabela 7) o qual está de acordo com
estipulado na ANVISA.

28
Exatidão
A exatidão do método foi obtida através da percentagem de
recuperação. Obteve-se 100,6% de recuperação do padrão de nateglinida,
pode-se constatar que a percentagem de recuperação de nateglinida se
encontra dentro do intervalo estipulado, de acordo com o critério de aceitação
(98 a 102%) [40]. Deste modo, conclui-se que o método é exato (Tabela 8);
Tabela 8: Recuperação da solução padrão de nateglinida. Método HPLC.
* média de 3 determinações. Fonte: Autoria própria.
Robustez
Os resultados obtidos nas oito execuções para a amostra de nateglinida
são demonstrados na Tabela 9. Os valores apresentados na tabela
representam a média de três injeções da solução.
Para avaliar o efeito de cada parâmetro, a média dos quatro valores
correspondentes às condições alteradas foi subtraída da média dos quatro
valores obtidos nas condições nominais, conforme demonstrado nas Eqs (4),
(5), (6) e (7). Os efeitos das variações dos parâmetros nos resultados da
análise são apresentados na Tabela 10.

29
Tabela 9. Resultados obtidos em oito condições para avaliação da robustez.
* média de 3 determinações. ** média de 6 determinações (teste de precisão intermediária).
Fonte: César e Pianetti (2009, p. 238) [45]
Tabela 10. Efeitos dos parâmetros analíticos no teor e tempo de retenção (TR).
* média dos valores nominais - média das variações. Fonte: César e Pianetti (2009, p. 238) [45]
Usando os critérios do teste de Youden, o método cromatográfico
mostrou-se robusto em relação ao conteúdo de nateglinida, quando foram
introduzidas variações nos quatro parâmetros analíticos. A maior variação
obtida foi de 1,51% quando a temperatura da coluna foi alterada de 25 ºC para
30 ºC, um valor consideravelmente baixo. O resultado obtido na tabela 10
demonstrou que não existe uma tendência clara dos fatores que podem afetar
os resultados do teor e do tempo de retenção, pois, além da variação ser
consideravelmente baixa, alguns ensaios obtiveram resultados maiores tanto
na variável nominal quanto na variável alterada, a exceção foi o fluxo da fase
móvel, onde todos os ensaios com um fluxo mais alto resultaram em um tempo
de retenção menor devido a maior eluição do composto.

30
6. CONCLUSÃO
Neste trabalho foi desenvolvido e validado, com sucesso, um método
analítico, por HPLC em microemulsão. A partir dos resultados obtidos na
validação pode-se concluir que o método é específico, seletivo, exato, linear e
preciso para a quantificação da nateglinida em medicamentos, fornecendo
resultados de acordo com os parâmetros preconizados pelo ICH, Farmacopeia
Americana e ANVISA.
A fase móvel que se mostrou mais adequada para análises por HPLC foi
composta por 3,4 g SDS, 6,6 g butanol, 0,8 g octano e 89,2 g de tampão
fosfato ajustado a um pH de 2,6 a um fluxo de 1,0 mL min-1, onde o tempo de
análise foi de 2,7 minutos.
Em comparação com o único método encontrado na literatura para
quantificação da nateglinida em microemulsão houve redução de 7,1 para 2,7
minutos no tempo de retenção da nateglinida, sendo portanto, o método
proposto muito mais rápido. Em Controle de qualidade é muito importante o
uso de metodologias rápidas, econômicas e eficientes.
O método proposto pode ser utilizado para análises de rotina em
laboratórios de controle de qualidade.

31
7. REFERÊNCIAS
1. BRUTTI, B.; FLORES, J.; HERMES, J.; MARTELLI, G.; PORTO, D. S.;
ANVERSA, E. T. R. Diabete Mellitus: definição, diagnóstico, tratamento
e mortalidade no Brasil, Rio Grande do Sul e Santa Maria, no período de
2010 a 2014. Brazilian Journal of health Review, Curitiba, v. 2, n. 4, p.
3174-3182, jun. 2019.
2. Diagnosis and Classification of Diabetes Mellitus. AMERICAN
DIABETES ASSOCIATION, V. 37, p. 81 - 90.
3. PALICKA, V. Pathophysiology of Diabetes Mellitus. The Jornal of
International Federation of Clinical Chemistry and Laboratory
Medicine, v. 5, n. 1, p. 140-144, mar. 2017.
4. LIMA, J. E. B. F. Estudo do estresse oxidativo e influência da
hiperglicemia crônica nos perfis de expressão gênica em pacientes
com diabetes mellitus tipo 2. 2019. 84f. Dissertação de Mestrado–
Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo,
Ribeirão Preto, 2019.
5. ZHENG, Y.; LEY, S. H.; HU, F. B. Global aetiology and epidemiology of
type 2 diabetes mellitus and its complications. Nature Reviews
Endocrinology, v.14, p.88 – 98, 2017.
6. COSTA, A. F.; FLOR, L. S.; CAMPOS, M. R.; OLIVEIRA, A. F. D.;
COSTA, M. D. F. D. S.; SILVA, R. S. D.; LOBATO, L. C. D. P.;
SCHRAMM, J. M. D. A. Carga do diabetes mellitus tipo 2 no Brasil.
Cadernos de Saúde Pública, Rio de Janeiro, v. 33, n. 2, p. 1-14, mar.
2017.
7. GRILLO, M. F. F.; GORINI, M. I. P. C. Caracterização de pessoas com
Diabetes Mellitus tipo 2. Revista Brasileira de Enfermagem, Brasília, v.
60, n. 1, p.49-59, Fev. 2007.
8. KAHN, S.R.; COOPER, M.E; DEL PRATO, S. Pathophysiology and
treatment of type 2 diabetes: perspectives on the past, present, and
future. The Lancet, v. 383, p.1068–1083, mar. 2014.
9. ARAÚJO, L. M. B.; BRITTO, M. M. dos S.; PORTO DA CRUZ, T. R.
Tratamento do diabetes mellitus do tipo 2: novas opções. Arquivos

32
Brasileiros de Endocrinologia & Metabologia, Salvador, v. 44 p.509–
518, dez. 2000
10. METIGLINIDES. GLOBALRPH. Disponível em:
<https://globalrph.com/drugs/diabetes-meglitinides/>. Acesso em: 02 fev.
2020.
11. NATEGLINIDE. DRUGBANK. Disponível em:
<www.drugbank.ca/drugs/DB00731>. Acesso em 20 Ago. 2019.
12. BAUER, S.; STÖRMER, E.; KIRCHHEINER, J.; MICHAEL, C.;
BROCKMÖLLER, J.; ROOTS, I. Rapid and simple method for the
analysis of nateglinide in human plasma using HPLC analysis with UV
detection. Journal of Pharmaceutical and Biomedical Analysis, v. 31,
n. 3, p. 551–555, mar. 2003.
13. PANI, N. R.; NATH, L.; SINGH, A. V.; MAHAPATRA, S. K. Development
and validation of analytical method for the estimation of nateglinide in
rabbit plasma. Journal of Pharmaceutical Analysis, v. 2, n. 6, p.492–
498, dez. 2012.
14. SAHOO, R. K.; BISWAS, N.; GUHA, A.; KUOTSU, K. Maltodextrin
Based Proniosomes of Nateglinide: Bioavailability Assessment.
International Journal of Biological Macromolecules, Bengala
Ocidental, India, v. 69, p. 430-434, mar. 2014.
15. MADDI, S.; KESHETTY, S.; MOHAN EGA, C.; RAO YAMASANI, M.;
SCRIBA, G.K.E. Development and validation of a stereoselective HPLC
method for the determination of the in vitro transport of nateglinide
enantiomers in rat intestine. Journal of Separation Science, Warangal,
India, 30, n. 12, p.1875-1880. Ago. 2007.
16. HACIOGLU, A. Development and validation of an HPLC method for
determination of nateglinide in drug substances. MARMARA
PHARMACEUTICAL JOURNAL, Istambul, v. 2, n. 19, p. 103-108, mar.
2015.
17. PATHARE, D. B.; JADHAV, A. S.; SHINGARE, M. S. A Validated
Stability Indicating LC Method for Nateglinide. Drug Development
and Industrial Pharmacy, v. 33 n. 5, p. 551-557, maio. 2007.
18. YAN, H.; YANG, G.; QIAO, F.; CHEN, Y. Determination of nateglinide in
animal plasma by micellar electrokinetic chromatography and on-line

33
sweeping technique. Journal of Pharmaceutical and Biomedical
Analysis, v. 36, n.1, p. 169-174, maio. 2004.
19. PHAN, T. D.; NGUYEN, N. H.; Kim, D. J.; Lee, Y. J.; CHOI, S.H.; KIM,
K. H. Determination of the L-enantiomer of nateglinide in pharmaceutical
formulations by micellar electrokinetic chromatography. Archives of
Pharmacal Research, Chuncheon, Coréia do Sul, v. 33, n. 12, p. 2017-
2024, set. 2010.
20. THOMAS, A. B.; PATIL, S. D.; NANDA, R. K.; KOTHAPALLI, L. P.;
BHOSLE, S. S.; Deshpande, A. D. Stability-indicating HPTLC method for
simultaneous determination of nateglinide and metformin hydrochloride
in pharmaceutical dosage form. Saudi Pharmaceutical Journal, Riade,
Arábia Saudita, v. 19, n. 4, p. 221-231, jun. 2011.
21. EL-WASSEEF, D. R. Simultaneous Determination of Metformin,
Nateglinide and Gliclazide in Pharmaceutical Preparations Using Micellar
Liquid Chromatography. International Journal of Biomedical Science,
Almançora, Egito, v. 8, n. 2 , jun. 2012.
22. EL-SHERBINY, D. T. M.; El-ASHRY, S. M.; MUSTAFA, M. A.; ABD-EL-
RAHMAN EL-EMAM, A.; HANSEN, S. H. Evaluation of the use of
microemulsions as eluents in high-performance liquid chromatography.
Journal of Separation Science, v. 26 n. 6-7, p.503-509, maio de 2003.
23. MCEVOY, E.; DONEGAN, S.; POWER, J.; ALTRIA, K. Optimisation and
validation of a rapid and efficient microemulsion liquid chromatographic
(MELC) method for the determination of paracetamol (acetaminophen)
content in a suppository formulation. Journal of Pharmaceutical and
Biomedical Analysis, v. 44, n. 1, p. 137-143, maio. 2007.
24. RYAN, R.; DONEGAN, S.; POWER, J.; MCEVOY, E.; ALTRIA, K.
MICROEMULSION HPLC. 2008.
25. KHOO, C. M. Diabetes Mellitus Treatment. International Encyclopedia
of Public Health, 2. ed, v. 1, p. 288–293.
26. GLOVACI, D.; FAN, W.; WONG, N. D. Epidemiology of Diabetes Mellitus
and Cardiovascular Disease. Current Cardiology Reports, v. 21, n. 4,
mar. 2019.
27. BELLO, L. M. G; LIMA, A. M. S; ALVES, K. C. G; SOUSA, S. T. K;
SILVA, A. D. MORTALIDADE PRECOCE POR DIABETES MELLITUS E

34
FATORES DE RISCO EM PALMAS, TOCANTINS. SOCIEDADE DE
PATOLOGIA DO TOCANTINS, Tocantins, v. 4, n. 01, p. 01-22, 2017.
28. MEHANNA, A. Antidiabetic agents: past, present and future. Future
Medical Chemistry, v. 5, n. 4, p. 411-430, abr. 2012.
29. TAN, S. Y.; MEI WONG, J. L.; SIM, Y. J.; WON, S. S.; MOHAMED
ELLHASSAN, S. A.; TAN, S. H.; LIM, G. P. L.; TAY, N. W. R.; ANNAN,
N. C; BRATTAMISRA, S.; CANDASAMY, M. Type 1 and 2 diabetes
mellitus: A review on current treatment approach and gene therapy as
potential intervention. Diabetes & Metabolic Syndrome: Clinical
Research & Reviews., v. 13, n. 1, fev. 2019.
30. NERES, L. V. Efeitos adversos no tratamento do diabetes tipo 2.
2018. 41 f. Trabalho de Conclusão de Curso (Bacharel) – Instituto de
Ciências Ambientais, Químicas e Farmacêuticas, Universidade Federal
de São Paulo, Diadema, 2018.
31. COPPOLINO, G.; LEPORINI, C.; RIVOLI, L.; URSINI, F.; DI PAOLA, E.
D.; CERNARO, V.; ANDREUCCI, M. Exploring the effects of DPP-4
inhibitors on the kidney from the bench to clinical trials.
Pharmacological Research, v. 129, p. 274-294. mar. 2018.
32. NATEGLINIDE. Chemical Book, 2020. Disponível em:
<https://www.chemicalbook.com/ChemicalProductProperty_EN_CB9472
923.htm>. Acesso em 21 jan. 2020.
33. NAIK, J.; LOKHANDE, A.; MISHRA, S.; KULKARNI, R. Preparation and
Characterization of Nateglinide Loaded Hydrophobic Biocompatible
Polymer Nanoparticles. Journal of The Institution of Engineers,
Maharashtra, India, serie D, v. 98 n. 2, p. 269-277, ago. 2016.
34. Nateglinide. TCI - Tokyo Chemical Industry. Disponível em:
<https://www.tcichemicals.com/AU/en/p/N0912>. Acesso em 20. jun.
2020.
35. DUNGAN K. M. Management of Type 2 Diabetes Mellitus.
Endocrinology: Adult and Pediatric, 2. ed, p. 839-853, 2016.

35
36. ROCHA, T. G.; GALENDE, S. B. A IMPORTÂNCIA DO CONTROLE DE
QUALIDADE NA INDÚSTRIA FARMACÊUTICA. Revista UNINGÁ, v.
20, n. 2, p. 97-103, out. 2014.
37. BRITO, N. M.; DE AMARANTE JUNIOR, O. P.; POLESE, L.; RIBEIRO,
M. L. VALIDAÇÃO DE MÉTODOS ANALÍTICOS: ESTRATÉGIA E
DISCUSSÃO. Pesticidas: Revista de Ecotoxicologia e Meio
Ambiente, v. 13, p. 129-146, dez. 2003.
38. LOMBARDO, M.; ESERIAN, J. K.; A análise da qualidade de
medicamentos e o papel do laboratório oficial no contexto da saúde
pública. Revista de Administração em Saúde, São Paulo, v. 17, n. 67,
Abr. – Jun. 2017.
39. DIAS, F. R. S. DESENVOLVIMENTO E VALIDAÇÃO DE MÉTODOS
ANALÍTICOS. 2019. 16 f. Programa de Pós-graduação em Fármacos e
Medicamentos Curso de Farmácia e Bioquímica, Universidade de São
Paulo (USP), 2019.
40. ANVISA – AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA.
Resolução RDC nº 166, de 24 de julho de 2017. Guia para validação de
métodos analíticos e bioanalíticos. Diário Oficial da República
Federativa do Brasil, Brasília, DF, de 25 de julho de 2017.
41. INTERNATIONAL CONFERENCE ON HARMONISATION OF
TECHNICAL REQUIREMENTS FOR REGISTRATION OF
PHARMACEUTICALS FOR HUMAN USE. Validation of Analytical
Procedures: Text and Methodology Q2(R1), 2005.
42. THE UNITED STATES PHARMACOPEIA. General Chapter, <1225>
Validation of compendial procedures, Rockville, Maryland, USA:
United States Pharmacopeial Convention.
43. MINISTERIO DE SALUD SECRETARÍA DE POLÍTICAS, REGULACIÓN
e INSTITUTOS. A.N. M.A.T. Disposición nº 1541. Novartis Argentina,
Buenos Aires, 12. fev. 2015. Disponível em:
<http://www.anmat.gov.ar/boletin_anmat/febrero_2015/Dispo_1541-
15.pdf>. Acesso em 12. out. 2019.
44. PUBCHEM. Monosodium phosphate. Disponível em:
<https://pubchem.ncbi.nlm.nih.gov/compound/Monosodium-phosphate>.
Acessado em: 01. dez. 2019.

36
45. CÉSAR, I. da C.; PIANETTI, G. A. Robustness evaluation of the
chromatographic method for the quantitation of lumefantrine using
Youden’s test. Brazilian Journal of Pharmaceutical Sciences, v. 45, n. 2,
p. 235-240, jun. 2009.
46. DIAS, D.C.S.; SILVA, N. C.; BONIFÁCIO, R. L. Robustness study in
SSNTD method validation: indoor radon quality. Brazilian National
Commission for Nuclear Energy, Poços de Caldas, 25 set. 2017.
_________________________ ________________________
Data e assinatura do aluno(a) Data e assinatura do orientador(a)
São Paulo, 26/06/2020 São Paulo, 26/06/2020


















