2 avaliação ecotoxicológica do fármaco cloridrato de fluoxetina e do ...

Farmacocinética – UP4
87
Dependendo do objectivo de estudo, da capacidade de
analisar o fármaco (e metabolitos) nos fluídos biológicos,
da farmacodinâmica da substância medicamentosa, da
via de administração e da natureza do medicamento.
3.2. Conhecer os diferentes métodos para a determinação da biosdisponibilidade.
São usados métodos directos e indirectos
Para determinar a Biodisponibilidade são
usados:
- Parâmetros farmacocinéticos e/ou
farmacodinâmicos
- Observações clínicas
- Estudos in vitro
Concentração de Fármaco no Plasma A medida das concentrações plasmáticas no sangue, plasma, ou soro depois do fármaco ser
administrado é o dado mais directo e objectivo para determinar a biodisponibilidade sistémica do fármaco.
Com amostras apropriadas de sangue e usando um ensaio validado do fármaco, pode ser obtida uma
descrição precisa das concentrações plasmáticas do fármaco versus o tempo da substância terapeuticamente
activa.
tmax Cmax
Corresponde ao tempo necessário para chegar à
concentração máxima após administração do
fármaco;
• Em tmax, verifica-se o pico de absorção do
fármaco e a taxa de absorção é igual à taxa de
eliminação;
• A absorção do fármaco continua depois de tmax
ser alcançado, mas a um ritmo mais lento. tmax
pode utilizar-se como indicador aproximado da
taxa de absorção.
• tmax será menor se a taxa de absorção for mais
rápida.
Representa o máximo de concentração plasmática
obtida após administração oral
• Para muitos fármacos, existe uma relação entre
o efeito farmacodinâmico e a concentração
plasmática
• Fornece indicações se o fármaco é
suficientemente absorvido sistemicamente
para produzir uma resposta terapêutica
• Prevê os possíveis níveis tóxicos do fármaco.
AUC
É uma medida do grau de biodisponibilidade do fármaco. Reflecte a quantidade total de fármaco activo que
atinge a circulação sistémica.
AUC é a área sob o gráfico do nível plasmático de fármaco vs tempo, de t = 0 a t = , e é igual à
quantidade de fármaco inalterado que alcança a circulação geral, dividido pela clearance.
F = Fracção de dose absorvida
D0 = dose
k = constante da taxa de eliminação
VD = Volume de Distribuição
Farmacocinética – UP4
88
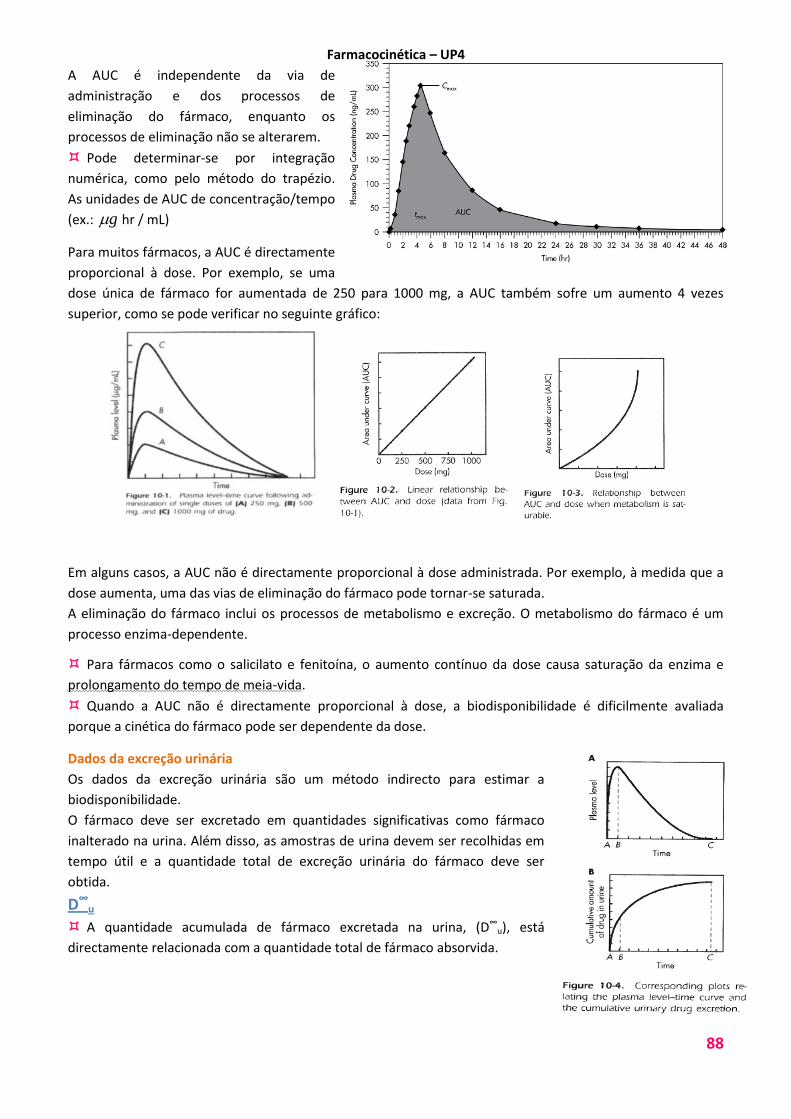
A AUC é independente da via de
administração e dos processos de
eliminação do fármaco, enquanto os
processos de eliminação não se alterarem.
Pode determinar-se por integração
numérica, como pelo método do trapézio.
As unidades de AUC de concentração/tempo
(ex.: g hr / mL)
Para muitos fármacos, a AUC é directamente
proporcional à dose. Por exemplo, se uma
dose única de fármaco for aumentada de 250 para 1000 mg, a AUC também sofre um aumento 4 vezes
superior, como se pode verificar no seguinte gráfico:
Em alguns casos, a AUC não é directamente proporcional à dose administrada. Por exemplo, à medida que a
dose aumenta, uma das vias de eliminação do fármaco pode tornar-se saturada.
A eliminação do fármaco inclui os processos de metabolismo e excreção. O metabolismo do fármaco é um
processo enzima-dependente.
Para fármacos como o salicilato e fenitoína, o aumento contínuo da dose causa saturação da enzima e
prolongamento do tempo de meia-vida.
Quando a AUC não é directamente proporcional à dose, a biodisponibilidade é dificilmente avaliada
porque a cinética do fármaco pode ser dependente da dose.
Dados da excreção urinária
Os dados da excreção urinária são um método indirecto para estimar a
biodisponibilidade.
O fármaco deve ser excretado em quantidades significativas como fármaco
inalterado na urina. Além disso, as amostras de urina devem ser recolhidas em
tempo útil e a quantidade total de excreção urinária do fármaco deve ser
obtida.
D∞u
A quantidade acumulada de fármaco excretada na urina, (D∞u), está
directamente relacionada com a quantidade total de fármaco absorvida.

Farmacocinética – UP4
89
Experimentalmente, as amostras de urina são recolhidas periodicamente após a administração de um
medicamento.
Cada amostra de urina é analisada para o fármaco livre com um ensaio específico.
Um gráfico é construído relacionando a quantidade acumulada de fármaco excretado com o intervalo
de colheita (fig 10-4 B).
A relação entre a quantidade acumulada de fármaco excretada na urina e a curva nível plasmático-
tempo é mostrada na fig. 10-4.
Quando o fármaco é quase totalmente eliminado (ponto C), a concentração plasmática aproxima-se
de zero e a quantidade máxima de fármaco excretado na urina, D∞u, é obtida.
dDu/dt
Como a maioria dos fármacos são eliminados por um processo de ordem 1, a taxa de fármaco excretado
depende da constante k de ordem 1 e da concentração de fármaco no plasma, CP.
Na figura 10.4, a taxa máxima de excreção do fármaco é verificada no ponto B, enquanto a taxa mínima de
excreção do fármaco pode ser verificada nos pontos A
e C.
Assim, um gráfico que compara a taxa de excreção
de fármacos em relação ao tempo deve ser similar na
forma como na curva do nível plasmático-tempo para
o fármaco. (fig,10-5)
t∞ O tempo total para o fármaco para ser excretado
é t∞.
A inclinação da curva do segmento A-B está relacionada com a taxa de absorção do fármaco, enquanto que o
ponto C está relacionado com o tempo total necessário após a administração do fármaco, para que este seja
completamente absorvido e excretado (t = ∞). O t ∞ é um parâmetro útil em estudos de bioequivalência.
Efeito agudo farmacodinâmico
Em alguns casos, a avaliação quantitativa de um fármaco no plasma ou urina não tem um ensaio com uma
precisão e /ou reprodutibilidade suficiente.
Um efeito agudo farmacodinâmico (ex.: efeito no diâmetro das pupilas, taxa cardíaca, ou pressão sanguínea)
pode ser usado como um índice de biodisponibilidade do fármaco, sendo medido durante um período de
tempo após a administração do medicamento.
As medições devem ser feitas com frequência suficiente para permitir uma estimativa razoável para
um período de tempo, pelo menos, 3 vezes o tempo de meia-vida.
O uso de um efeito agudo farmacodinâmico para determinar a biodisponibilidade geralmente requer a
demonstração de uma curva dose-resposta.
A biodisponibilidade é determinada pela caracterização da curva dose-resposta.
Parâmetros farmacodinâmicos que são obtidos incluindo a área total abaixo da curva efeito
farmacodinâmico-tempo, pico do efeito farmacodinâmico, e tempo para o pico do efeito
farmacodinâmico.
O tempo de início e duração do efeito farmacocinético também podem ser incluídos na análise dos
dados.
A utilização de parâmetros farmacodinâmicos para a determinação de biodisponibilidade e bioequivalência é
muito mais variável do que a medição de plasma ou concentrações do fármaco na urina.

Farmacocinética – UP4
90
Observações clínicas
Em seres humanos, ensaios clínicos bem controlados estabelecem a segurança e a eficácia dos medicamentos
e podem ser usados para determinar a biodisponibilidade. No entanto, a abordagem de ensaios clínicos é o
menos preciso, menos sensível e menos reprodutível dos critérios gerais para a determinação da
biodisponibilidade in vivo.
Segundo a EMEA, em casos em que a bioequivalência não possa ser demonstrada utilizando concentrações
do fármaco, em circunstâncias excepcionais, poderá ser necessário recorrer a dados farmacodinâmicos ou
clínicos No entanto, esta situação está fora do alcance da diretriz e o leitor é remetido para orientações
terapêuticas específicas.
Um exemplo desta abordagem é a determinação da bioequivalência de 2 produtos antifúngicos tópicos
por diferentes fabricantes contendo o mesmo agente antifúngico activo (ex. cetoconazol).
Estudos in vitro
Estudos de dissolução do fármaco sob certas condições dão uma indicação da biodisponibilidade do fármaco.
Estudos de dissolução são geralmente realizados em várias formulações teste do mesmo fármaco.
A formulação teste que mostra a taxa mais rápida de dissolução do fármaco in vitro geralmente tem a
taxa mais rápida de biodisponibilidade do fármaco in vivo.
3.3 – Perceber a necessidade da demonstração de bioequivalência entre formulações, aspecto inerente ao
conceito de medicamento genérico
3.3.1. Conhecer os aspectos a considerar no planeamento e na execução de estudos de bioequivalência e na
demonstração de bioequivalência terapêutica
Estudos de Bioequivalência:
Diferenças na resposta clínica prevista ou efeitos adversos podem ser devido a diferenças na comportamento
farmacocinético e/ou farmacodinâmico do fármaco entre indivíduos ou diferenças na biodisponibilidade do
fármaco.
Medicamentos bioequivalentes que têm a mesma biodisponibilidade sistémica terão a mesma resposta.
As respostas clínicas variáveis entre indivíduos que não estão relacionados com a biodisponibilidade pode ser
devido a diferenças na farmacocinética. Diferenças na farmacodinâmica estão relacionadas a diferenças na
sensibilidade dos receptores para o fármaco.
Vários fatores que afetam farmacodinâmica comportamento pode incluir idade, tolerância,
interações medicamentosas, e fatores fisiopatológicos desconhecidos.
A biodisponibilidade de um fármaco pode ser facilmente reproduzível quando calculada para indivíduos
que se encontram em jejum em estudos altamente controlados, mas quando é utilizado num regime diário, a
dieta do indivíduo pode afectar a concentração plasmática.
Doentes com uma dieta rica em hidratos de carbono têm mais tempo de semi-vida de teofilina, devido à
menor depuração metabólica do fármaco (t1/2, 18,1 h), em comparação com doentes com dieta normal (t1 / 2 =
6,76 h).
A maior concentração plasmática de fármaco resultante de uma dieta de hidratos de carbono pode submeter
o doente a um maior risco de intoxicação por fármacos como teofilina.
Bases para determinar a bioequivalência
A bioequivalência é estabelecida se a biodisponibilidade in vivo de um fármaco em teste (normalmente o
produto genérico) não diferir significativamente na velocidade e extensão de absorção, como determinado
por comparação dos parâmetros medidos, em relação ao material de referência.

Farmacocinética – UP4
91
Um fármaco que difere do material de referência na sua velocidade de absorção mas não na extensão da
mesma, pode ser considerado bioequivalente se a diferença na velocidade de absorção for intencional e/ou
se não afecta negativamente a segurança e eficácia do fármaco.
Medicamentos com possíveis problemas de Biodisponibilidade e Bioequivalência
Existe suspeita de falta de biodisponibilidade ou bioequivalência quando:
1. Evidências de que vários fármacos não originam efeitos terapêuticos semelhantes.
2. Evidências que o fármaco exibe um rácio terapêutico estreito e concentrações mínimas efectivas no
sangue e que a utilização segura e eficaz do fármaco requer dosagem cuidadosa e monitorização dos
pacientes.
3. Origem a efeitos adversos sérios no tratamento e prevenção de uma doença ou condição grave.
4. Evidências físico-químicas de:
i. O princípio activo tem uma baixa hidrosolubilidade (menos do que 5 mg/mL);
ii. A taxa de dissolução dos produtos é lenta;
iii. O tamanho das partículas e/ou a área de superfície do princípio activo é critico na determinação da
biodisponibilidade;
iv. Certas formas estruturais do princípio activo (formas polimórficas, complexos, etc...) dissolvem-se
pobremente, afectando a absorção.
v. Os fármacos possuem um alto rácio de princípio activo para excipentes (superior a 5 para 1).
vi. Ingredientes inactivos específicos (excipientes hidrofílicos ou hidrofóbicos e lubrificantes) podem ser
necessários para a absorção do princípio activo ou pode interferir com a mesma absorção.
5. Evidência farmacocinética do seguinte:
i. O princípio activo ou o seu precursor, é absorvido em grande quantidade num segmento particular
do TGI ou é absorvido de um sítio localizado.
ii. O grau de absorção do princípio activo é pobre (menos do que 50% quando comparado com uma
dose intravenosa), mesmo quando é administrado na sua forma pura (ex: em solução).
iii. Existe um metabolismo rápido do fármaco na parede intestinal ou no fígado durante o processo de
absorção, de forma que a velocidade de absorção não é muito importante para o seu efeito
terapêutico ou tóxico.
iv. O fármaco é rapidamente metabolizado ou excretado, necessitando de uma dissolução e absorção
rápida para ser eficaz.
v. O fármaco é instável em porções específicas do TGI e precisa de formulações especiais
(revestimentos entéricos, tampões, etc…) para assegurar uma absorção adequada.
vi. O fármaco encontra-se sujeito a cinéticas dependentes da dose dentro ou próxima da margem
terapêutica e a velocidade e extensão de absorção são importantes para a bioequivalência.
Critérios para abdicar da evidência para biodisponibilidade in vivo:
Para determinado fármacos, a biodisponibilidade in vivo pode ser óbvia ou irrelevante para este atingir os
seus objectivos.
1. O fármaco é uma solução projectada para apenas ser administrada por via intravenosa e contém um
princípio activo combinado com o mesmo solvente e na mesma concentração que uma solução intravenosa
que foi alvo de uma aprovação completa.
2. O fármaco é uma preparação para aplicação tópica (creme, gel, etc..)
3. O fármaco encontra-se disponível para administração oral mas não se pretende que seja absorvido (ex:
antiácido).
4. O fármaco cumpre ambas as condições seguintes:
a. É administrado por inalação com um gás ou vapor;
b. Contém um princípio activo na mesma forma de dosagem que um produto que já foi aprovado.

Farmacocinética – UP4
92
5. O fármaco cumpre todas as condições seguintes:
a. É uma solução oral, elixir, xarope, tintura ou outra forma solubilizada semelhante.
b. Contém um princípio activo na mesma forma de dosagem que um produto que já foi aprovado.
c. Não contém nenhum excipiente que se saiba afectar significativamente a absorção do princípio activo.
Design e avaliação de estudos de bioequivalência:
As técnicas estatísticas deve ser sensíveis o suficiente para detectar diferenças na taxa e extensão de
absorção que não são atribuíveis à variabilidade do individuo.
Design
O design básico para um estudo de bioequivalência é determinado por:
a) a questão científica a que se pretende responder
b) a natureza do material de referência e a forma de dosagem a ser testada
c) a disponibilidade de métodos analíticos
d) considerações de benefício-risco relativamente aos testes em humanos.
EMEA oferece diretrizes gerais para a realização dos estudos mas o investigador principal deve preparar um
protocolo detalhado para o estudo.
Para os estudos de bioequivalência, tanto a formulação do fármaco em teste como o de referência contêm
um princípio activo equivalente na mesma dosagem e forma (ex: libertação imediata ou controlada) e ambos
são dados pela mesma via de administração.
Antes de iniciar, o estudo deve ser aprovado pelo o IRB (Institutional Review Board). O IRB é composto
tanto profissionais e leigos com diversas formações sendo da sua responsabilidade salvaguardar o bem-estar
e os direitos dos seres humanos que se vão submeter ao estudo.
Geralmente, o estudo é realizado em voluntários saudáveis normais, homens e mulheres que deram o
consentimento informado para a o estudo.
o Pacientes criticamente doentes não estão incluídos em estudos de biodisponibilidade in vivo, a
menos que o atendimento médico determina que existe um benefício potencial para o paciente.
O número de indivíduos no estudo depende da variabilidade interindividual do mesmo e são escolhidos de
acordo com alguns critérios pré-estabelecidos.
Os indivíduos são sujeitados a um período de jejum de cerca de 10-12 horas antes da administração do
fármaco e podem continuar em jejum 2-4 horas após a mesma.
Métodos Analíticos

Farmacocinética – UP4
93
O método analítico deve demonstrar exatidão e sensibilidade suficiente para medir com precisão adequada,
a concentração real do fármaco activo ou porção terapêutica do fármaco ou do seu metabolito activo, obtido
no corpo.
Para estudos de biodisponibilidade, tanto o fármaco e seus principais metabólitos ativos são geralmente
medido. Para estudos de bioequivalência, apenas o fármaco é medido.
Padrão de Referência
→ Uma formulação do fármaco é escolhida para padrão de referência com o qual todas as outras
formulações testadas devem ser comparadas.
O fármaco de referência deve ser dado pela mesma via de administração daqueles que estão a ser testados a
menos que seja necessária uma via de administração alternativa para se poderem tirar certas conclusões
acerca da sua farmacocinética.
Exemplo: se um fármaco possui baixa biodisponibilidade oral, pode ser comparado com um solução
oral ou uma injecção intravenosa.
→ O padrão de referência, geralmente é o fármaco de marca original para o qual já existem dados válidos
acerca da sua segurança e eficácia.
→ Antes de se iniciar um estudo de bioequivalência in vivo o conteúdo total de princípio activo no produto
genérico não deve diferir em mais do que 5% em relação ao produto de referência.
→ Para além disso, é normal realizarem-se estudos comparativos de dissolução ou libertação do fármaco in
vitro em várias condições especiais.
Formulações de libertação prolongada
O objectivo de um estudo de biodisponibilidade in vivo envolvendo um medicamento de libertação
prolongada é para determinar se:
(1) O produto satisfaz as reivindicações de libertação controlada para ele concebidos;
(2) O perfil de biodisponibilidade estabelecido para o fármaco exclui a ocorrência de qualquer dose
dumping;
(3) Desempenho do medicamento no steady-state é equivalente ao de uma formulação de libertação não
prolongada atualmente comercializada;
(4) A formulação de produto proporciona um desempenho consistente farmacocinético entre as
unidades de dosagem individuais;
O estudo de biodisponibilidade desta comparação é utilizado para o desenvolvimento de um novo produto de
fármaco de libertação prolongada em que o medicamento de referência pode ser uma solução ou suspensão
do composto activo ou um medicamento atualmente comercializado de libertação não controlada tais como
um comprimido ou cápsula.
Para uma bioequivalência, o estudo de um novo medicamento genérico de liberação prolongada, o
medicamento de referência é atualmente comercializado como produto de liberação prolongada.
A combinação de fármacos
O objectivo de um estudo de biodisponibilidade in vivo, envolvendo um produto de combinação de fármacos
contendo mais do que uma substância activa é para determinar se a taxa e a extensão de absorção de cada
substância activa, fracção terapêutica ou composto no produto de combinação de fármacos é equivalente à
taxa e extensão de absorção de cada substância ativa ou grupo terapêutico quando administrado
separadamente em formulações de uma única substância activa.
O material de referência deverá ser de dois ou mais fármacos atualmente comercializados, em formulações
com uma única substância activa.

Farmacocinética – UP4
94
Estudos de design
Estudos necessários para as formas farmacêuticas sólidas orais:
1. Estudos em jejum
Este estudo é requerido para todas as formas orais de libertação imediata e modificada.
São colhidas amostras sanguíneas prévias (no tempo zero) à administração e a intervalos apropriados após
a dose para obter uma descrição adequada da concentração plasmática versus tempo.
Os intervenientes devem estar em jejum antes da administração do fármaco e devem assim continuar até
4H após a administração.
Por norma não é dada qualquer medicação aos intervenientes até uma semana antes do estudo
2. Estudos com intervenção de alimentos
Comparação entre administrações a indivíduos em jejum e imediatamente após uma refeição padrão rica
em gorduras
Este estudo é requerido para todas as formulações de libertação modificada e pode ser requerido para
formulações de libertação imediata se a biodisponibilidade for afectada pelos alimentos
3. Estudos de dose múltipla (steady-state)
Requeridos para formulações de libertação controlada em acréscimo aos estudos anteriores.
Devem ser medidas três concentrações (Cmin) em 3 dias consecutivos para se ter a certeza que os
intervenientes se encontram no steady state.
A última dose matinal é dada aos intervenientes após jejum nocturno, continuando por pelo menos mais 2
horas após a administração.
Designs Crossover
Os indivíduos que cumprem os critérios de estudo de inclusão e exclusão são seleccionados
aleatoriamente. Cada indivíduo recebe o fármaco em teste e o produto de referência.
Tabela 10.3 e 10.4 → exemplos de “ Latin Square crossover designs” para estudos de bioequivalência em
humanos voluntários, comparando 3 formulações diferentes (A, B, C) ou 4 (A, B, C, D).
O design “latin square” planeia os testes clínicos de forma a que cada indivíduo receba cada fármaco
apenas uma vez, com tempo adequado entre as administrações de forma que exista tempo suficiente para
ocorrer a eliminação do fármaco anterior. Neste design, cada indivíduo é o seu próprio controlo o que reduz a
variabilidade inter-individual.
Além disso, também se reduz as variações devido a sequência, período e tratamento (formulação) uma vez
que os vários pacientes não recebem o mesmo fármaco ou pela mesma ordem. Assim, o fármaco B pode ser
seguido do fármaco A, D ou C.
Depois de cada indivíduo ter recebido um fármaco, são recolhidas amostras de sangue a intervalos de
tempo apropriados de forma a se obter uma curva de concentração-tempo válida.

Farmacocinética – UP4
95
Os intervalos de tempo devem ser espaçados de forma a que o pico de concentração, a AUC total e as
fases de absorção e eliminação sejam bem descritas.
Nota: em alguns casos pode ser necessária a medição do fármaco em amostras de urina.
→ O período refere-se ao período de tempo no qual o estudo é realizado. (ex: o estudo de 2 períodos significa
que o estudo foi realizado em 2 dias diferentes, separados por um tempo durante o qual a maior parte do
fármaco é eliminado do organismo – geralmente cerca de 10 tempos de meia-vida).
→ Uma sequência refere-se ao número de ordens diferentes nos grupos de tratamento de um estudo.
Avaliação dos dados
Método Analítico
Tem de ser validado relativamente à exactidão, precisão, sensibilidade e especificidade.
O uso de mais do que um método analítico pode não ser válido, dado que métodos diferentes podem
produzir resultados diferentes.
Deve ser disponibilizada a curva concentração de fármaco vs tempo.
Avaliação farmacocinética dos dados
Para estudos de doses individuais, incluindo um estudo em jejum, as análises farmacocinéticas incluem o
cálculo de cada parâmetro da AUC para a última concentração quantificável (AUC0-t) e para o infinito (AUC0-∞),
Tmax e Cmax.
Além disso, podem estimar-se a constante de velocidade de eliminação (k), a meia vida de eliminação e
outros parâmetros.
Para estudos de doses múltiplas, a análise farmacocinética inclui o cálculo da AUC no steady-state (AUC0-t),
Tmax, Cmin, Cmax e da percentagem de flutuação:
Avaliação estatística dos dados
Para se provar a bioequivalência, não pode haver diferença estatística entre a biodisponibilidade do produto-
teste e do produto de referência.
São usadas abordagens estatísticas para comparar a biodisponibilidade do fármaco.
Muitas abordagens estatísticas assumem que os dados estão distribuídos de acordo com uma distribuição
normal (curva em forma de sino).
A distribuição de alguns parâmetros biológicos (ex: Cmax, AUC) tem uma cauda maior que a observada
numa distribuição normal. Além disso, pode ser difícil acertar a verdadeira distribuição desses parâmetros,
devido ao pequeno número de parâmetros estudados.
Se os dados forem convertidos em valores logarítmicos, a distribuição aproximar-se-á mais de uma
distribuição normal. Por isso, procede-se à logaritmização dos dados de biodisponibilidade antes da avaliação
estatística dos dados para determinação da bioequivalência.
Análise da variância (ANOVA)
É um procedimento estatístico usado para testar os dados e averiguar a diferença entre os grupos de
tratamento e de controlo.
Os parâmetros estudados: AUC0-24, AUC0-∞, tmax e Cmax, sendo obtidos para cada tratamento ou forma de
dosagem.
Utiliza-se para avaliar a variabilidade em sujeitos, grupos de tratamento, período de estudo, formulação e
outras variáveis dependentes do design do estudo.

Farmacocinética – UP4
96
Se a variabilidade de dados for grande, a diferença na média para cada parâmetro farmacocinético
pode ser mascarada e o investigador pode concluir que os 2 produtos do fármaco são bioequivalentes.
A diferença estatística entre parâmetros farmacocinéticos é considerada significativa se a
probabilidade de esses resultados terem acontecido for menor ou igual a 0,05 ( ).
Se P > 0,05 As diferenças entre 2 produtos do fármaco não são consideradas estatisticamente
significativas.
Para reduzir a possibilidade de erro, realiza-se um power test para calcular a probabilidade de a conclusão do
ANOVA ser válida. A capacidade do teste depende do tamanho da amostra, da variabilidade dos dados e do
nível de significância desejado. Geralmente o Power é estabelecido a 0.80 com α=0.2 e um nível de
significância de 0.05.
Procedimento dos testes “two one-sided” Abordagem do intervalo de confiança.
Usado para demonstrar se a biodisponibilidade do fármaco a partir de uma formulação teste é demasiado
baixa ou alta em comparação com o produto de referência do fármaco.
Tem por objectivo de determinar se há grande diferenças (>20%) entre as amostras médias.
O intervalo estimado baseia-se na distribuição t de Student dos dados. Neste teste, um intervalo de
confiança de 90% em relação ao ratio dos 2 produtos do fármaco deve ser para a medida da
velocidade e extensão da biodisponibilidade do fármaco.
O intervalo de confiança de 90% mínimo para o ratio não pode ser menor que 0,80. O intervalo de
confiança de 90% máximo para o ratio não pode ser maior que 1,20.
Quando se utilizam os dados logaritmizados, o intervalo de confiança estabelecido é 80-125%, uma vez que
log0.8 = 125% .
O intervalo de confiança de 90% é função do tamanho da amostra e da variabilidade do estudo.
Para uma dose única e estudo em jejum, o ANOVA é geralmente realizado para valores logaritmizados de
AUC e Cmax. Não deve haver diferenças entre a AUC e a Cmax do teste (genérico) e do fármaco de referência.
Além disso, os valores médios de AUC e Cmax do fármaco-teste não devem ser menores que 0,80 (80%) nem
maiores que 1,25 (125%) que os do produto de referência.
Exemplo de bioequivalência
Um estudo hipotético de biodisponibilidade no qual 3 comprimidos diferentes foram comparados com uma
solução de fármaco administrada em igual dose. A biodisponibilidade das 3 formulações de comprimido foi
maior que 80% da da solução.
De acordo com o ANOVA, os valores médios de AUC não foram estatisticamente diferentes uns dos outros ou
diferentes dos da solução. Contudo, o intervalo de confiança de 90% para a AUC mostrou que, para o
comprimido A, a biodisponibilidade foi menor que 80% (ie, 74%), comparado com a solução.

Farmacocinética – UP4
97
Consideremos um fármaco que foi preparado na mesma dose em 3 formulações (A, B e C). Estas formulações
foram administradas a um grupo de voluntários. Para cada indivíduo, foram obtidos o nível de fármaco no
plasma e dados relativos à excreção urinária do fármaco. Com estes dados, é possível observar a relação entre
parâmetros do plasma e da excreção urinária e da biodisponibilidade do fármaco.
A velocidade de absorção do fármaco a partir da formulação A é mais rápida que a partir da
formulação B, uma vez que o tmax é menor para a formulação A.
Dado que a AUC da formulação A é idêntica à AUC da formulação B, a extensão da biodisponibilidade
a partir de ambas as formulações é a mesma.
Contudo, a Cmax de A é maior que a de B, uma vez que a velocidade de absorção do fármaco é maior.
A Cmax é geralmente maior quando a biodisponibilidade do fármaco é maior.
A velocidade de absorção de fármaco da formulação C é igual à da formulação A, mas a
biodisponibilidade é menor. A Cmax é menor para a formulação C do que para a formulação A. A
diminuição na Cmax da formulação C é proporcional à diminuição na AUC, em comparação com os
níveis plasmáticos de fármaco da formulação A.
Tabela 10.7modo como os parâmetros de biodisponibiidade no plasma e na urina se alteram quando a
extensão e a velocidade da biodisponibilidade mudam, respectivamente.
Submissão do estudo
A Tabela 10.8 mostra os parâmetros que deve contemplar um estudo completo de biodisponibilidade. (era
uma tabela enorme que a sotra disse que no era preciso)
Antes de o estudo ser realizado, o investigador deve assegurar-se de que o estudo foi adequadamente
planeado, de que os objectivos estão claramente definidos e de que o método de análise foi validado. Além
disso, o protocolo do estudo pode ser submetido à EMEA para discussão e aprovação.
Após a realização do estudo, os resultados são analisados estatística e farmacocineticamente.

Farmacocinética – UP4
98
O ramo biofarmacêutico da EMEA, após receber o estudo
completo, procederá à revisão detalhada do estudo com
base nos parâmetros indicados na Tabela, tomando
posteriormente a decisão de aprovar ou não o estudo da
biodisponibilidade do fármaco.
Testes de disssolução in situ de estudos de Biodisponibilidade
Se houver uma forte correlação entre a dissolução e a biodisponibilidade do fármaco, os testes de dissolução
comparativa entre o produto-teste e o produto de referência deverão ser suficientes para demonstrar a
bioequivalência.
Para a maioria dos fármacos (comprimidos e cápsulas de libertação rápida) não se verifica forte
correlação e são exigidos estudos de bioequivalência in vivo.
Para formas sólidas orais, pode ser necessário um estudo de bioequivalência para apoiar pelo menos
uma dose do produto. Normalmente, é necessário um estudo de bioequivalência in vivo para a força da
dose mais elevada.
Se a dose mais baixa do produto-teste for proporcionalmente semelhante nos ingredientes activos e
inactivos, pode ser usada apenas uma dissolução in vitro de comparação entre as formulações teste e de
referência.
Por exemplo, para um fármaco disponível em doses de 50 mg, 75 mg e 100 mg, realiza-se um estudo in vivo
para a dose de 100 mg e estudos de dissolução comparativos para as doses 50 mg e 75 mg.
O Sistema de classificação biofarmacêutica (BCS)
Desenvolveu-se a base teórica para correlacionar a dissolução do fármaco in-vitro com biodisponibilidade in
vivo baseia-se na solubilidade do fármaco e a permeação do fármaco atravésdo TGI. O sistema de
classificação é baseada na primeira lei de Fick, aplicado a uma membrana:
Jw onde é o fluxo de fármaco (em massa/área /hora) através da parede intestinal, em qualquer posição e tempo, Pw
representa a permeabilidade da membrana e Cw é a concentração de fármaco na superfície da membrana intestinal.
Assume que nenhum dos outros componentes na formulação afectam a permeabilidade da membrana e/ ou
transporte intestinal.
Estudou-se a solubilidade e as características de permeabilidade de vários fármacos representativos e obteve-
se uma classificação biofarmacêutica de fármacos para prever a dissolução in-vitro de medicamentos sólidos
orais de libertação imediata com absorção in-vivo.
O FDA pode dispensar a realização de uma biodisponibilidade in vivo ou estudo de bioequivalência para certos
produtos sólidos orais de libertação imediata que atendam a critérios muito específicos, ou seja:
permeabilidade, solubilidade e dissolução do fármaco. Estas características incluem a dissolução in vitro do

Farmacocinética – UP4
99
fármaco em diversos meios, permeabilidade, e assumindo o comportamento ideal do medicamento,
dissolução do fármaco e a absorção no TGI.
Solubilidade
Um objectivo da abordagem BCS é determinar o equilíbrio de solubilidade de um fármaco sob condições
aproximadamente fisiológicas. É sugerido uma gama de pH de 1-8.
A classe de solubilidade é determinado calculando que volume de um meio aquoso é suficiente para dissolver
a maior dose prevista.
Um fármaco é considerado altamente solúvel quando a maior a dose solúvel em 250 ml ou menos
do meio aquoso ao longo da gama de pH 1-8.
Permeabilidade
A extensão da absorção em seres humanos ou métodos de permeabilidade intestinal podem ser usados para
determinar a classe de permeabilidade de um fármaco.
Para ser classificado como altamente permeável, um fármaco em ensaio deve ter uma extensão da absorção>
90% em seres humanos. Informação de suporte sobre as características de permeabilidade do fármaco
também deve ser proveniente de suas propriedades físico-químicas (ex.: octanol: partição da água
coeficiente).
Métodos para determinar a permeabilidade de um fármaco a partir do tracto gastrointestinal:
a) Estudos in vivo de perfusão intestinal em seres humanos;
b) Estudos in vivo ou in situ, de perfusão intestinal em animais;
c) Experiências in vitro de permeação, utilizando tecidos intestinais humanas ou animais excisadas;
d) Experiências in vitro de permeação através de uma monocamada de células intestinais humanas
cultivadas.
Ao usar esses métodos, o Dados experimentais de permeabilidade deve ser correlacionado com os dados de
ponto-de-absorção conhecidos em seres humanos.
Dissolução
A classe de dissolução baseia-se na taxa de dissolução in vitro de fármaco de libertação imediata em
condições de teste específicas e destina-se a indicar a rápida dissolução in vivo em relação à taxa média de o
esvaziamento gástrico nos seres humanos, em condições de jejum.
Um medicamento de liberação imediata é considerado rapidamente dissolvido quando não menos
que 85% da quantidade de fármaco usada se dissolve dentro de 30 minutos, usando Aparelho USP I (ver a), a
100 rpm ou Aparelho II a 50 rpm em um volume de 900 mL ou menos, em cada um dos seguintes meios: (1)
meios ácidos, como HCl 0,1 N ou USP fluido gástrico simulado sem enzimas, (2) um pH 4,5 tampão, e (3) um
tampão de pH 6,8 ou fluido intestinal simulado USP sem enzimas.
Fármacos para os quais a Biodisponibilidade e Bioequivalência são evidentes
A melhor medida do desempenho de um fármaco é determinar a biodisponibilidade in vivo do mesmo. Para
alguns medicamentos bem caracterizados e para certos medicamentos em que a biodisponibilidade é
evidente (ex.: soluções para injecção), estudos de biodisponibilidade in-vivo podem ser desnecessários ou
sem importância atingir os fins pretendidos do produto.
No entanto, podem existir requisitos específicos para certos medicamentos, e a divisão apropriado da FDA
deve ser consultado.
1. O fármaco:
a. É uma solução destina-se exclusivamente para a administração intravenosa

Farmacocinética – UP4
100
b. contém um composto activo ou porção terapêutica combinada com o mesmo solvente e na
mesma concentração como numa solução intravenosa cujo requerimento de novo fármaco
foi aprovado.
2. O fármaco é uma preparação de aplicação tópica (ex.: creme, pomada, gel ou destinados a locais
efeito terapêutico). A FDA publicou orientações para a realização de estudos de bioequivalência em
corticosteróides tópicos e antifúngicos. A FDA também está a considerar realizar estudos derma-
farmacocinética (DPK) noutros medicamentos de aplicação tópica. Além disso,são necessários estudos
de libertação do fármaco in vitro e de difusão.
3. O fármaco está numa forma de dosagem oral que não se destina a ser absorvida (ex.: um anti-ácido).
Estudos específicos in-vitro de bioequivalência podem ser exigidos pela FDA.
Exemplo:a bioequivalência de resina de colestiramina é demonstrada in vitro por meio da
ligação de ácidos biliares para a resina.
4. O fármaco satisfaz ambas as seguintes condições:
a. É administrado por inalação, como um gás ou vapor;
b. Contém um composto activo ou porção terapêutica na mesma forma de dosagem que um
fármaco cujo requerimento de novo fármaco foi aprovado.
5. O fármaco satisfas todas as seguintes condiçõe:
a. Trata-se uma solução oral, elixir, xarope, tintura, ou outra forma semelhante solubilizado.
b. Contém um composto activo ou porção terapêutica na mesma concentração que um fármaco
cujo requerimento de novo fármaco foi aprovado.
c. Não contém compostos inactivos conhecidos para afectarem significativamente a absorção
do fármaco activa ou fracção terapêutica.
Significado clínico dos estudos de Bioequivalência
A bioequivalência equivalência no que se refere à velocidade e extensão da absorção sistêmica do fármaco.
Geralmente, 2 formulações cujas velocidade e extensão de absorção diferem em 20% ou menos são
consideradas bioequivalentes.
Estudos clínicos de efectividade têm dificuldade em detectar diferenças na dose de 50-100%.
Consequentemente, observa-se uma variação normal na prática médica e os níveis de fármaco no plasma
podem variar mais de 20% entre indivídiduos.
Quando se consideram os objectivos terapêuticos do fármaco, deve ser obtida uma resposta clínica
equivalente a partir das formas dosagem de comparação se as concentrações de fármaco no plasma
permanecerem acima da MEC (concentração mínima efectiva) para um intervalo apropriado e não atinjam a
MTC (concentração tóxica mínima).
Populações especiais (ex: idosos, pessoas submetidas a quimioterapia) não são usadas em estudos de
bioequivalência, preferindo-se voluntários saudáveis. O uso de indivíduos saudáveis minimiza a variabilidade
inter e intraindividual nos estudos de biodisponibilidade.
É teoricamente possível que os excipientes de uma forma teste possam causar problemas em pessoas que
usam a forma genérica.
Na manufactura de uma forma de dosagem, pretende-se que haja uniformidade, devendo os
procedimentos de controlo de qualidade minimizar a variabilidade produto-produto por diferentes
fabricantes e a variabilidade lote-a-lote do mesmo fabricante.

Farmacocinética – UP4
101
Preocupações especiais nos estudos de Biodisponibilidade e Bioequivalência
O design e avaliação dos estudos de bioequivalência gerais podem ser usados para fármacos absorvidos
sistemicamente e formas orais convencionais. Contudo, para alguns fármacos, a biodisponibilidade e
bioequivalência sistêmicas são difíceis de acertar.
Os fármacos são considerados altamente variáveis se a variabilidade intraindividual em parâmetros de
biodisponibilidade >30% por análise da variação do coeficiente de variância.
De acordo com a EMEA, o número de indivíduos para demonstrar a bioequivalência dos fármacos não
deverá ser inferior a 12.
A variabilidade intraindividual pode dever-se ao fármaco em si, à formulação ou a ambos.
Para fármacos com meias vida de eliminação muito longa ou uma fase de eliminação complexa, pode ser
difícil obter a curva concentração plasmática de fármaco VS tempo completa para estudos de
bioequivalência. Para estes fármacos pode ser prática uma curva mais curta/truncada (AUC0-t). O uso da
AUC truncada permite a medição do pico de absorção e diminuirá o tempo e o custo da realização do
estudo de biequivalência.
Alguns fármacos são estereoisómeros e cada isômero produz uma resposta farmacodinâmica diferente e
pode ter uma velocidade de biotransformação diferente. Pode ser difícil medir a biodisponibilidade dos
isômeros individuais.
Alguns fármacos (ex: tioridazina, selegileno) têm metabolitos activos, que deve ser quantificados, bem
como o fármaco que lhes deu origem.
Por outro lado, alguns fármacos, como a procainamida, têm um metabolito activo, N-acetilprocainamida. A
acetilação da procainamida demonstra polimorfismo genético com 2 grupos de indivíduos, havendo
acetiladores rápidos e acetiladores lentos. Para diminuir a variabilidade interindividual, um estudo de
bioequivalência deve ser realizado apenas em um dos fenótipos.
Alguns fármacos (ex: benzocaína, hidrocortisona) destinam-se a um efeito local, não possuindo
biodisponibilidade sistêmica significativa a partir do local de administração. Pode ser difícil determinar a
biodisponibilidade de fármacos que não são absorvidos sistemicamente a partir do local de aplicação.
Para esses fármacos é necessário um marcador para determinar a bioequivalência. Por exemplo, a
capacidade neutralizante dos ácidos de um anti-ácido oral tem sido usada como marcador “surrogate”
in situ em estudos de bioequivalência in vivo.
Vários sistemas de entrega e novas formas de dosagem são planeados para conduzir o fármaco por uma
via não oral, podendo produzir apenas biodisponibilidade sistêmica parcial. A inalação de fármacos para
o tratamento da asma tem sido usada para maximizar o fármaco nas vias respiratórias e para diminuir
os efeitos sistêmicos laterais.
Dermatocinética é o estudo da captação de fármaco através das camadas da pele após administração
tópica. As medições da concentração de fármaco são efectuadas na pele em diferentes períodos de
tempo após a administração.
Fármacos de substituição hormonal (ex: levotiroxina) ou suplementos de potássio são administrados
oralmente e podem não produzir os parâmetros biodisponibilidade (AUC, Cmax, tmax) habituais. Para
estes fármacos TAM que ser usados métodos mais indirectos para acertar a bioequivalência. Por
exemplo, a excreção urinária de potássio permite medir a biodisponibilidade dos suplementos de
potássio. Contudo, para a levotiroxina em indivíduos com hipotiroidismo, é mais adequado medir a
concentração de hormonas no steady-state, os níveis de TSH e os pontos farmacodinâmicos terminais.

Farmacocinética – UP4
102
Substituição genérica
Para conter os custos dos medicamentos, a maioria dos estados têm aprovado leis de substituição
genérica para permitir que os farmacêuticos distribuam um medicamento genérico por um medicamento de
marca que tenha sido prescrito.
Alguns adoptam um formulário positivo, que lista os medicamentos terapeuticamente equivalentes ou
intercambiáveis que os farmacêuticos podem dispensar. Outros usam um formulário negativo, que lista os
medicamentos que não são terapeuticamente equivalentes e /ou a transferência de que é proibido.
Dois medicamentos que contenham a mesma substância activa são considerados bioequivalentes se eles
são equivalentes ou alternativas farmacêuticas e a sua biodisponibilidade (velocidade e extensão) após a
administração na mesma dose molar é aceitável dentro de certos limites predefinidos. Esses limites são
definidos para assegurar o desempenho in vivo, ou seja, semelhança em termos de segurança e eficácia.
Selecionados os parâmetros farmacocinéticos e os pré-limites de aceitação permite a decisão final sobre a
bioequivalência dos produtos testados.
Em estudos de bioequivalência, a curva concentração plasmática-tempo é geralmente usada para avaliar a
taxa e extensão da absorção.
o AUC, área sob a curva, reflete o grau de exposição. C max , a concentração plasmática máxima ou a
exposição de pico, e o tempo de concentração plasmática máxima, t max , são parâmetros que são
influenciados pela taxa de absorção.
Design, realização e avaliação dos estudos de bioequivalência
O nº de estudos e o design do estudo depende das características físico-químicas da substância, das suas
propriedades farmacocinéticas e proporcionalidade na composição, que devem ser justificadas
convenientemente.

Farmacocinética – UP4
103
Em particular, isto pode ser necessário para direccionar a linearidade da farmacocinética, a necessidade para estudos tanto na alimentação e jejum, a necessidade para nalises enantioselectiva e a possibilidade de renúncia de formas adicionais.
Design do estudo
O estudo deve ser desenhado de forma a que o efeito da formulação seja distinto de outros efeitos.
Design padrão
Se duas formulações são comparadas, é recomendado uma randomização, 2 períodos, 2 sequências de dose
única.
Os períodos de tratamento devem ser separados por um período de lavagem suficiente para garantir que
as concentrações do fármaco estão abaixo do limite inferior de quantificação bioanalítico em todas as
disciplinas no início do segundo período.
Normalmente, pelo menos, 5 semi-vidas são necessárias para alcançar este objectivo.
Projectos alternativos
Condução de um estudo de dose múltipla em doentes é aceitável se um estudo de dose única não pode ser
realizado em voluntários saudáveis, por razões de tolerabilidade, e um estudo de dose única não é factível em
doentes.
Na situação rara em que os problemas de sensibilidade do método analítico impede as medições precisas
da concentração plasmática após a administração de dose única e onde as concentrações no estado
estacionário são suficientemente elevadas para ser mensurado com fiabilidade, um estudo de dose múltipla
pode ser aceite como uma alternativa para o estudo de dose única.
No entanto, dado que um estudo de dose múltipla é menos sensível para detectar diferenças na Cmax, isso só
será aceitável se o requerente justificar que a sensibilidade do método analítico não pode ser melhorada e
que não é possível mensurar fiavelmente o composto após a administração de dose única, tendo em conta
também a opção de usar uma dose supra-terapêutica no estudo de bioequivalência.
Devido ao recente desenvolvimento da metodologia bioanalítica, é incomum que o fármaco não possa ser
medido com rigor e precisão. Assim, a utilização de um estudo de dose múltipla, em vez de uma única dose,
devido à limitada sensibilidade do método analítico, só serão aceites em casos excepcionais.
Em estudos de estado estacionário, o período de lavagem do tratamento anterior pode sobrepor-se à
acumulação do segundo tratamento, desde que o período de acumulação seja suficientemente longo (pelo
menos 5 vezes o meia-vida)
Referência e produto teste (a prof não deu importância)
Referência do Produto
A escolha do produto de referência utilizado no estudo de bioequivalência deve ser baseada em conteúdo e
dados de ensaio de dissolução e é de responsabilidade do candidato. Salvo motivo justificado, o conteúdo
ensaiado do lote utilizado como produto de teste não deve diferir mais de 5% do valor do lote utilizado como
produto de referência determinado com o método analítico proposto para o teste de qualidade de rotina do
teste de produto.
O candidato deve documentar como um grupo representativo do produto de referência no que diz
respeito à dissolução e teor de ensaio foi selecionado.
É aconselhável a investigar mais de um único lote do medicamento de referência na escolha do lote
do produto de referência para o estudo de bioequivalência.

Farmacocinética – UP4
104
Produto teste (não)
O produto teste utilizado no estudo deve ser representativo do produto a ser comercializado e isso deve ser
discutido e justificado pelo requerente.
Por exemplo, para formas sólidas orais de acção sistémica:
a) O produto de teste deve normalmente ter origem de um lote de pelo menos 1 / 10 da escala de
produção ou 100.000 unidades, o que for maior, salvo motivo justificado.
b) A produção de lotes utilizados devem proporcionar um elevado nível de garantia de que o produto e
processo será viável em escala industrial.
No caso de um lote de produção menor do que 100 mil unidades, um lote de produção total será
necessário.
c) A caracterização e especificação de atributos críticos de qualidade do medicamento, tais como
dissolução, devem ser estabelecidas a partir do lote de ensaio, ou seja, o grupo clínico para o qual
bioequivalência tem sido demonstrada.
d) Amostras do produto-piloto adicionais e/ou escala de produção em lotes, submetidos a apoiar o
pedido, devem ser comparados com os do estudo de bioequivalência lote de ensaio, e deve mostrar
similar perfis de dissolução in vitro, quando do emprego de testes de dissolução adequado condições.
Para formas farmacêuticas de libertação imediata de acção sistémica, a justificação da natureza
representativa do lote de ensaio deve ser igualmente estabelecida.
Embalagem dos produtos do estudo(não)
A referência e teste de produtos devem ser embalados para cada pessoa e fase, quer antes da sua
transferência para o centro de ensaio, ou no próprio sítio de julgamento.
Deveria ser possível identificar de forma inequívoca a identidade do produto administrado para cada
sujeito em cada período experimental.
Embalagem, rotulagem e administração dos produtos aos temas que deve ser documentado em detalhes.
Esta documentação deve incluir todas as precauções tomadas para evitar e identificar o potencial de erros de
dosagem. O uso de etiquetas com uma porção de corte é recomendado.
Indivíduos (sim)
Número de indivíduos
O número de temas a serem incluídos no estudo deve ser baseado num cálculo adequado do tamanho.
O número de indivíduos avaliados no estudo de bioequivalência não deve ser inferior a 12.
Seleção de temas
A população objeto de estudo de bioequivalência devem ser selecionada com o intuito de permitir a detecção
de diferenças entre os produtos farmacêuticos.
A fim de reduzir a variabilidade não relacionada com diferenças entre os produtos, os estudos devem ser
normalmente realizados em voluntários saudáveis, a menos que o fármaco traga problemas de segurança que
fazem deste antiético.
Este modelo, in vivo de voluntários saudáveis, é considerado adequado na maioria dos casos para detectar
diferenças de formulação e permitir a extrapolação dos resultados para as populações para as quais o
medicamento de referência seja aprovado (a idosos, crianças,
doentes com insuficiência renal ou hepática, etc.)
Os critérios de inclusão/exclusão deverão ser claramente indicados no protocolo. Os doentes devem ter 18
anos ou mais de idade, e de preferência ter um Índice de Massa Corporal entre 18,5 e 30 kg/m2.

Farmacocinética – UP4
105
Os indivíduos devem ser rastreados para a adequação por meio de testes de laboratório clínico, uma
anamnese e exame físico.
Dependendo da classe terapêutica do fármaco e do perfil de segurança especial,
investigações médicas e precauções podem ter de ser efectuados antes, durante e após a conclusão do
estudo.
Os indivíduos podem pertencer a ambos os sexos, porém, o risco para mulheres em idade fértil devem
ser considerados.
Devem ser preferencialmente não-fumadores e sem história de abuso de álcool ou drogas.
Fenotipagem e/ou genotipagem dos indivíduos podem ser considerados para a segurança ou por razões
farmacocinéticas.
Em estudos de desenho paralelo, os grupos de tratamento devem ser comparáveis em todas as variáveis
conhecidas que podem afectar a farmacocinética da substância activa ( idade, peso corporal, sexo, origem
étnica, tabagismo, extensivo/mau estado metabólico). Este é um pré-requisito essencial para dar validade aos
resultados de tais estudos.
Se a substância activa investigada é conhecido por ter efeitos adversos, e os efeitos farmacológicos ou os
riscos são considerados inaceitáveis para os voluntários saudáveis, pode ser necessário para incluir doentes
em vez disso, sob cuidados e supervisão adequadas.
Condução do estudo (sim)
Normalização
As condições de ensaio devem ser padronizadas, a fim de minimizar a variabilidade dos factores envolvidos,
excepto as dos produtos testados.
Recomenda-se a padronização da dieta, ingestão de líquidos e exercícios físicos.
A hora do dia para a ingestão deve ser especificada.
Os doentes devem estar em jejum pelo menos oito horas antes da administração de produtos, salvo
motivo justificado.
Como a ingestão de líquidos pode influenciar a passagem gástrica para as formas de administração
oral, os produtos teste e referência devem ser administrados com um volume de líquido
padronizado (pelo menos 150 ml).
Recomenda-se que água seja permitido como desejado, excepto por uma hora antes e uma hora
após a administração de fármacos e alimentos não são permitidos, pelo menos, quatro horas após
a dose.
Refeições feitas após a dose deve ser padronizadas em relação à composição e tempo de
administração, durante um período de tempo adequado (por exemplo, 12 horas).
No caso do estudo ser realizado na presença de alimento:
O horário da administração do fármaco em relação à ingestão de alimentos é recomendado ser de
acordo com o SmPC do autor do produto. Se não houver recomendação específica, é recomendado
que o indivíduo inicie a refeição 30 min antes da administração do fármaco e comer esta refeição
em 30 min.
Como a biodisponibilidade de uma parte activa de uma forma de dosagem pode ser dependente
tempos de trânsito gastrointestinal e regional dos fluxos sanguíneos, a postura e actividade física
podem precisar de padronização.
Os indivíduos devem abster-se de alimentos e bebidas, que podem interagir com o aparelho
circulatório, função gastrointestinal, hepática ou renal (ex: bebidas alcoólicas ou sumos de frutas,
tais como certos sumos de toranja), durante um período adequado antes e durante o estudo.

Farmacocinética – UP4
106
Não devem tomar qualquer medicação concomitante (incluindo remédios de ervas) num intervalo
adequado antes, bem como durante o estudo. Contudo, anticoncepcionais são permitidos.
o Em casos raros, o uso de uma medicação concomitante é necessária para todas as fases, por
razões de segurança ou tolerabilidade (ex: antagonistas opióides, anti-eméticos). Nesse
cenário, o risco de uma potencial interacção ou interferência bioanalítico afectando os
resultados devem ser abordadas.
Os medicamentos que, segundo o autor SmPC, devem ser utilizados explicitamente em combinação com
outro produto (por exemplo, os inibidores da protease em combinação com certos ritonavir) podem ser
estudados tanto como a combinação aprovada ou sem o produto recomendado para ser administrado
concomitantemente.
Em estudos de bioequivalência de substâncias endógenas, factores que podem influenciar os níveis de
base endógena deve ser controlados (por exemplo, controle rigoroso da ingestão).
Tempos de amostragem (sim)
Um número suficiente de amostras para descrever adequadamente o plasma perfil temporal de concentração
deverão ser recolhidos.
O calendário deve incluir a amostragem frequente em torno de tmax previsto para fornecer uma estimativa
confiável de exposição de pico. Em particular, o cronograma de amostragem deve ser planeado para evitar
Cmax sendo o primeiro ponto de uma curva de tempo de concentração.
O cronograma de amostragem deve abranger também a curva tempo-concentração plasmática, o
suficiente para fornecer uma estimativa confiável do grau de exposição que pode ser conseguido se AUC (0-t)
abrange pelo menos 80% das AUC (0 - ∞).
Pelo menos três a quatro amostras são necessários durante a fase terminal log-linear, a fim de estimar
confiantemente a taxa de terminal constante (o que é necessário para uma estimativa fiável da AUC (0 - ∞)).
AUC incompleta às 72 horas (AUC(0-72h)) pode ser usada como uma
alternativa para a AUC (0-t) para comparação da extensão da exposição, a fase de absorção tem sido
coberto por 72 h para formulações de libertação imediata.
Um período de amostragem superior a 72 h não é considerada necessária para qualquer formulação de
libertação imediata, independentemente da meia vida do fármac.
Em estudos de dose múltipla, a pré-dose deve ser tomada imediatamente antes (dentro de 5 minutos)
da administração e a última amostra é recomendada para ser tomada no prazo de 10 minutos do
tempo nominal para o intervalo de dosagem para garantir uma determinação precisa da AUC(0-τ).
Se a urina é usada como fluido de amostragem biológica, esta deve ser normalmente recolhida ao longo de
pelo menos três vezes a semi-vida de eliminação terminal. No entanto, em consonância com as
recomendações de amostras de plasma, urina, não precisam ser recolhidos por mais de 72 h. Se a taxa de
excreção é determinada, os intervalos de colecta deve ser tão curtos quanto possível durante a fase de
absorção.
No caso de substâncias endógenas, o calendário de amostragem deve permitir a caracterização do perfil
de linha de base endógena de cada indivíduo em cada período. Muitas vezes, é determinada uma linha de
base de 2-3 amostras colhidas antes do fármaco ser administrado. Noutros casos, a amostragem em
intervalos regulares ao longo de 1-2 dias (s) antes da administração podem ser necessárias para dar conta de
flutuações na linha de base endógena, devido aos ritmos circadianos.

Farmacocinética – UP4
107
Condições de jejum ou com alimentos (sim)
Em geral, um estudo de bioequivalência deve ser realizado sob condições de jejum, pois este é considerado o
estado mais sensível para detectar uma diferença de potencial entre as formulações.
Para os produtos onde o SmPC recomenda a ingestão do medicamento de referência, com o estômago vazio
ou, independentemente da ingestão de alimentos, o estudo de bioequivalência deverá ser conduzido sob
condições de jejum. Para os produtos onde o SmPC recomenda a ingestão do medicamento de referência só
no estado alimentado, o estudo de bioequivalência deve ser realizado nestas condições.
Porém, para formulação de produtos com características específicas (ex: microemulsões, dispersões sólidas),
estudos de bioequivalência realizados em ambos, condições de jejum e com alimentos podem ser necessárias.
Estudos realizados em condições de alimentação, a composição da refeição é recomendado para ser de
acordo com a SmCP do produto de origem.
Se não houver recomendação específica, a refeição deve ter um alto teor de gordura (cerca de 50 por
cento do valor calórico total da refeição) e alto valor calórico (cerca de 800-1000 kcal). Esta refeição deve
derivar de ensaio cerca de 150, 250 e 500-600 kcal de proteína, hidratos de carbono e gordura,
respectivamente.
A composição da refeição deve ser descrita com relação à proteína, hidrato de carbono e gordura
(especificado em gramas, calorias e teor calórico em relação (%)).
Características a investigar (sim)
Parâmetros farmacocinéticos
O tempo real de amostragem deve ser utilizado na estimativa dos parâmetros farmacocinéticos.
Em estudos para determinar a bioequivalência, após uma única dose, a AUC (0-t), a AUC (0 - ∞), a área
residual, Cmax e tmax deve ser determinada.
Em estudos com um período de amostragem de 72 h, e onde a concentração às 72 horas é quantificável, a
AUC (0 - ∞) e a área residual não precisa ser relatado, é suficiente para relatar AUC incompleta às 72h, a
AUC (0 - 72h).
Parâmetros adicionais incluem a taxa de terminal constante, λz e t1/2.
Em estudos para determinar a bioequivalência de formulações de libertação imediata no estado de
equilíbrio, a AUC (0-τ), Cmax,ss, e tmax,ss devem ser determinados.
Ao usar dados urinários, Ae (0-t) e, se aplicável, Rmax devem ser determinados.
Os métodos não-compartimentais devem ser utilizado para determinação de parâmetros farmacocinéticos
em estudos de bioequivalência. A utilização de métodos compartimentais para estimação de parâmetros não
é aceitável.
Composto original ou metabolitos (não)
Recomendações gerais
Em princípio, a avaliação da bioequivalência deve ser baseada em medidas das concentrações de compostos
pai.
A razão para isto é que Cmax de um composto-pai é geralmente mais sensível para detectar diferenças
entre as formulações em velocidade de absorção de Cmax do metabolito.
Pró-fármaco inativo
→ Também para a pró-fármacos inativos, a demonstração de bioequivalência para a substância activa é
recomendado.

Farmacocinética – UP4
108
O metabolito ativo não precisa ser medido. No entanto, alguns pró-fármacos podem ter baixas concentrações
plasmáticas e ser eliminados rapidamente, resultando em dificuldades na demonstração de bioequivalência
para a substância activa. Nesta situação, é aceitável para demonstrar a bioequivalência o principal metabolito
ativo sem medição do composto original.
O uso de um metabolito como um substituto para um composto activo-pai não é incentivada. Isso só
pode ser considerada se o candidato poder justificar que a sensibilidade do método analítico para a
medição do composto original não pode ser melhorado e que não é possível mensurar fiavelmente o
composto-pai após a administração de dose única, tendo em conta também a opção de com uma
maior dose no estudo de bioequivalência.
Devido aos desenvolvimentos recentes na metodologia bioanalítica, é incomum que o fármaco-pai não
possa ser medido com rifor e precisão. Assim, a utilização de um metabolito como um substituto para
a substância activa deverá ser aceite apenas em casos excepcionais.
Enantiómeros
A utilização de métodos bioanalíticos aquiral é geralmente aceitável. No entanto, os enantiomeros individuais
devem ser medidos quando as seguintes condições são satisfeitas:
(1) os enatiómeros têm diferente farmacocinética.
(2) os enantiómeros têm uma diferença pronunciada na farmacodinâmica.
(3) a relação exposição (AUC) dos enantiômeros é modificada por uma diferença na taxa de absorção.
Os enantiómeros individuais também devem ser medidos se as condições acima são satisfeitas, ou se são
desconhecidos.
Se é um enantiómero farmacologicamente activo e o outro é inactivo ou tem uma baixa contribuição para a
actividade, é suficiente para demonstrar a bioequivalência do enantiómero activo.
Utilização de dados urinários
A utilização de dados de excreção urinária como um substituto para a concentração plasmática pode ser
aceitável na determinação do grau de exposição onde não é possível medir fiavelmente o perfil plasmático
temporal de concentração do composto-pai.
→ No entanto, a utilização de dados urinários deve ser cuidadosamente justificada, quando utilizado para
estimar a exposição de pico. Se um Cmax plasmático pode ser determinado, este deve ser combinado com
dados urinários sobre a extensão da exposição para a avaliação de bioequivalência.
→ Ao usar dados urinários, o requerente deve apresentar todos os dados disponíveis da excreção urinária
de apoio que irá refletir a exposição plasma.
Substâncias endógenas
→ Se a substância a ser estudada é endógena, o cálculo dos parâmetros farmacocinéticos devem ser
realizados utilizando a correcção da linha de base para que os parâmetros farmacocinéticos calculados
referentes às concentrações adicionais fornecidas pelo tratamento.
A administração de doses supra-terapêuticas pode ser considerada em estudos de bioequivalência de
medicamentos endógenos, desde que a dose seja bem tolerada, de modo que a concentração adicional do
valor inicial previsto pelo tratamento podem ser determinados com fiabilidade.
→ Em casos raros, onde um aumento substancial em relação ao início dos níveis endógenos são vistos, a
correcção da linha de base pode não ser necessária.

Farmacocinética – UP4
109
Dosagem para ser investigado (sim)
Se várias dosagens de um produto de teste são pedidas, pode ser suficiente para estabelecer a
bioequivalência em apenas um ou dois pontos fortes, dependendo da proporcionalidade na composição dos
diferentes pontos fortes e as questões relacionadas com outros produtos.
A dosagem (s) para avaliar depende da linearidade na farmacocinética da substância activa.
Em caso de farmacocinética não-linear pode haver uma diferença entre doses diferentes em relação à
sensibilidade para detectar diferenças de potencial entre as formulações.
No contexto, a farmacocinética é considerado linear, se a diferença na dose ajustada AUC média há
mais de 25% quando se compara a dose de estudo (ou a dose do estudo de bioequivalência
planejado) e doses para os quais renúncia é considerada.
o Para avaliar a linearidade, o candidato deverá considerar todos os dados disponíveis em
domínio público no que diz respeito à proporcionalidade da dose e analisar os dados críticos.
Avaliação da linearidade analisará se as diferenças na AUC dose ajustada atendem a um
critério de ± 25%.
Se foi demonstrada a bioequivalência com as doses que são mais sensíveis para detectar uma diferença de
potencial entre os produtos, estudos de bioequivalência in vivo para a outras dosagens podem ser
dispensados.
Critérios biowaiver gerais(não)
Os seguintes critérios gerais devem ser cumpridos quando um levantamento de força adicional (s) é
reivindicada:
a) os produtos farmacêuticos são fabricados pelo mesmo processo de fabrico;
b) a composição qualitativa dos diferentes pontos fortes é a mesma;
c) a composição das forças são quantitativamente proporcionais, ou seja, a relação entre a quantidade
de cada excipiente à quantidade da substância activa (s) é a mesma para todas as forças (por
medicamentos de liberação imediata, os componentes do revestimento, revestimento da cápsula, os
agentes de cores e sabores não são obrigados a seguir esta regra).
Se houver algum desvio da composição quantitativa proporcional, condição c ainda é considerada
cumprida se a condição de i) e ii) ou i) e iii) a seguir aplicam-se a força usada no
estudo de bioequivalência ea força (s) para que a renúncia é considerada
i. a quantidade de substância activa (s) é inferior a 5% do peso do núcleo de comprimido, o
peso, do conteúdo da cápsula
ii. as quantidades dos diferentes excipientes de núcleo ou o conteúdo da cápsula é a mesma
para o forças envolvidas e apenas a quantidade de substância activa é alterada
iii. iii. a quantidade de material de enchimento é alterado para dar conta da mudança na
quantidade de ativos substância. As quantidades de outros excipientes de núcleo ou o
conteúdo da cápsula deverá ser o mesmo para as forças envolvidas
d) dados apropriados de dissolução in vitro deve confirmar a adequação da renúncia adicional in vivo
testes de bioequivalência.
Farmacocinética linear (sim)
Para os produtos em que todas as condições acima estão preenchidas, é suficiente para estabelecer
bioequivalência com apenas uma força.
→ O estudo de bioequivalência deve ser realizado em geral na maior força. Para os produtos com a
farmacocinética linear e em que o fármaco é altamente solúvel, a selecção de uma intensidade menor do que
o mais alto também é aceitável.

Farmacocinética – UP4
110
→ Selecção de uma intensidade menor também pode ser justificada se a maior resistência não pode ser
administrado a voluntários saudáveis, por razões de segurança / tolerância.
Além disso, os problemas de sensibilidade do método analítico podem impedir medidas precisas de
concentração plasmática após administração de dose única de alta resistência, assim, uma dose mais elevada
pode ser seleccionado (de preferência com vários comprimidos de alta resistência). A dose seleccionada pode
ser maior do que a maior dose terapêutica, desde que esta dose seja bem tolerada em voluntários saudáveis e
que não haja absorção ou limitações de solubilidade com esta dose.
Farmacocinética não-linear(sim)
Para medicamentos com farmacocinética não-linear caracterizada por um aumento mais que proporcional da
AUC com o aumento da dose no intervalo de dose terapêutica, o estudo de bioequivalência deve ser realizado
em geral na maior força.
Quanto aos medicamentos com uma farmacocinética linear menor que a força pode ser justificada se
a maior resistência não pode ser administrado a voluntários saudáveis, por razões de segurança /
tolerância.
Da mesma forma uma dose mais elevada pode ser utilizado em caso de problemas de sensibilidade do
método analítico, em conformidade com as recomendações dadas para os produtos com a
farmacocinética linear acima.
Para os medicamentos com menos de um aumento proporcional da AUC com o aumento da dose no intervalo
de doses terapêuticas, a bioequivalência deve, na maioria dos casos, ser estabelecida tanto na maior força e
ao menor força (ou uma resistência na faixa linear), ou seja, nesta situação dois estudos de bioequivalência
são necessários.
→ Se a não-linearidade não é causada por solubilidade limitada, mas é devido, por exemplo, saturação dos
transportadores de captação e desde que as condições acima são preenchidos e os produtos teste e
referência não contêm qualquer um dos excipientes que podem afectar a motilidade gastrintestinal ou o
transporte de proteínas, que é suficiente para demonstrar a bioequivalência com a dosagem mais baixa (ou
uma resistência na faixa linear).
→ Selecção de outras forças pode ser justificada se houver problemas sensibilidade analítica impedindo um
estudo da menor resistência, ou se a maior resistência não poder ser administrada a voluntários saudáveis,
por razões de segurança / tolerância.
Abordagem “bracketing”(não)
Caso a avaliação de bioequivalência em mais de duas forças seja necessária ex: por causa de desvio de
composição proporcional, uma abordagem de escalonamento pode ser usado.
Nesta situação, pode ser aceitável realizar dois estudos de bioequivalência, se as forças seleccionadas
representam os extremos, por exemplo, a maior e a menor força ou as duas forças mais diferentes na
composição, de modo que as diferenças na composição de forças restante é coberto pelos dois estudos
realizados.
→ Caso a avaliação de bioequivalência seja necessário, tanto em jejum e em estado alimentado e em dois
pontos fortes devido à absorção não linear ou desvio de composição proporcional, pode ser suficiente para
avaliar a bioequivalência em jejum e alimentados em apenas um dos pontos fortes.

Farmacocinética – UP4
111
Combinações
As condições relativas à composição proporcional deverão ser preenchidas por todas as substâncias activas de
combinações fixas. Ao considerar a quantidade de cada substância activa, numa combinação fixa de outra
substância activa (s) pode ser considerado como excipientes.
No caso de comprimidos bicamada, cada camada pode ser considerada de forma independente.
Metodologia bioanalítica(não)
A parte de ensaios de bioequivalência bioanalítico deve ser realizada em conformidade com os princípios das
Boas Práticas de Laboratório (BPL). No entanto, como estudos humanos bioanalítico fora do âmbito de GLP,
os locais que realizam os estudos não são obrigados a ser monitorados, como parte de um programa nacional
de cumprimento das BPL.
Os métodos utilizados bioanalítico devem ser bem caracterizados, plenamente validados e documentados
para obter resultados confiáveis, que podem ser interpretados de forma satisfatória.
Dentro do estudo de validação deve ser realizada através de amostras de controle de qualidade em cada
corrida analítica.
As principais características de um método bioanalítico que é essencial para garantir a aceitabilidade do
desempenho e da confiabilidade dos resultados analíticos são: seletividade, limite de quantificação, a função
de resposta (desempenho da curva de calibração), exatidão, precisão e estabilidade.
O limite inferior de quantificação deve ser de 1/20 de Cmax ou inferior, em concentrações pré-dose deve
ser detectável em 5% do Cmax ou inferior.
Reanálise das amostras do estudo deve ser pré-definida em protocolo de estudo (e / ou SOP) antes do
início efectivo da análise das amostras. Normalmente a reanálise de amostras de por causa de uma razões
farmacocinéticas não é aceitável. Isto é especialmente importante para estudos de bioequivalência, pois este
viés pode o resultado desse estudo. Análise das amostras deve ser conduzida sem informação sobre o
tratamento.
Avaliação(não)
Em estudos de bioequivalência, os parâmetros farmacocinéticos não devem, em geral, ser ajustado para
as diferenças no conteúdo analisado do teste e de referência do lote.
No entanto, em casos excepcionais, quando um grupo de referência, com um teor de ensaio que difira
menos de 5% do produto de teste não pode ser encontrada a correcção de conteúdo poderá ser aceite. Se a
correcção de conteúdo deve ser usado, este deve ser pré-especificados no protocolo e justificado pela
inclusão dos resultados do teste do teste e referência no protocolo.
Razões para exclusão (não)
A avaliação imparcial dos resultados de estudos randomizados exige que os sujeitos sejam observados e
tratados de acordo com as mesmas regras. Estas regras devem ser independentes do tratamento ou
resultado.
Em consequência, a decisão de excluir um tema a partir da análise estatística deve ser feita antes
bioanalise. Em princípio, qualquer razão para a exclusão é válida, desde que seja especificado no protocolo e a
decisão de exclusão seja feita antes bioanalise.
No entanto, a exclusão de dados devem ser evitados, como o poder do estudo será reduzida e um mínimo de
12 indivíduos avaliados é necessário.

Farmacocinética – UP4
112
→ Exemplos de motivos para excluir os resultados de um sujeito em um determinado período são eventos
como vómitos e diarreia, que poderia tornar o tempo de concentração plasmática de perfil não-confiáveis. Em
casos excepcionais, o uso de medicação concomitante poderia ser uma razão para excluir o assunto.
→ As razões para a exclusão permitida devem ser pré-especificados no protocolo.
→ Exclusão dos indivíduos com base nesses critérios pré-especificados devem ser claramente descritos e
constantes do relatório do estudo.
→ Exclusão de dados não pode ser aceite com base na análise estatística ou por razões farmacocinéticas
sozinho, porque é impossível distinguir os efeitos de formulação de outros efeitos que influenciam a
farmacocinética.
As excepções são:
1) A falta de individuos com qualquer concentração mensurável ou apenas as concentrações plasmáticas
muito baixas para o medicamento de referência. Um tema é considerado de ter concentrações
plasmáticas muito baixas, se a sua AUC é inferior a 5% do medicamento de referência AUC média
geométrica (que deve ser calculado sem a inclusão dos dados do assunto periféricas). A exclusão de
dados devido a este motivo, só serão aceitos em casos excepcionais, pode questionar a validade do
julgamento.
2) Indivíduos com concentrações diferentes de zero inicial> 5% do Cmax. Esses dados devem ser
excluídos do cálculo de bioequivalência (ver carry-over effects abaixo).
A descrição acima pode, por formulações de liberação imediata, ser o resultado de sujeito não-conformidade
e um período de wash-out insuficiente, respectivamente, e devem, na medida do possível, ser evitada
“check” da boca das pessoas após a ingestão da medicação em estudo para garantir que os individuos
ingerirão a medicação em estudo e pela elaboração do estudo com um período de wash-out suficiente.
→ A AUC (0-t) deve cobrir pelo menos 80% das AUC (0 - ∞). Assuntos não devem ser excluídos da análise
estatística se AUC (0-t) cobre menos de 80% da AUC (0 - ∞), mas se a percentagem é inferior a 80% em mais
de 20% das observações, então a validade do o estudo pode ter de ser discutido. Isto não se aplica se o
período de amostragem é de 72 h ou mais e AUC (0-72h) é usado em vez de AUC (0-t).
Parâmetros a serem analisados e limites de aceitação (sim)
Em estudos para determinar a bioequivalência, após uma única dose, os parâmetros a serem analisados são
AUC (0-t), ou, quando for caso disso, a AUC (0-72h) e Cmax.
→ Para estes parâmetros o intervalo de confiança de 90% para o rácio entre o produto teste e referência
devem estar contidos dentro do intervalo de aceitação de 80,00-125,00%.
→ Para estar dentro do intervalo de aceitação do limite inferior deve ser ≥ 80,00% quando arredondados
para duas casas decimais, o limite superior deve ser ≤ 125,00% quando arredondado para duas casas
decimais.
→ Para os estudos para determinar a bioequivalência de formulações de liberação imediata no estado de
equilíbrio, a AUC (0-τ) e Cmax, ss devem ser analisados usando o mesmo intervalo de aceitação como indicado
acima.
→ Nos raros casos em que os dados urinária tem sido utilizado, Ae (0-t) devem ser analisados usando o
mesmo intervalo de aceitação como dito acima para a AUC (0-t). Rmax devem ser analisados utilizando o
intervalo de aceitação mesmo que para Cmax.
→ A avaliação estatística dos tmax não é necessária. No entanto, se a libertação rápida é reivindicada a ser
clinicamente relevantes e de importância para o início da acção ou está relacionada a eventos adversos, não

Farmacocinética – UP4
113
deve haver nenhuma diferença aparente no tmax mediana e sua variabilidade entre o teste e o medicamento
de referência.
Fármacos de janela terapêutica estreita
O intervalo de aceitação para AUC deve ser apertado para 90,00-111,11%. Onde Cmax é de particular
importância para a segurança, a eficácia do fármaco ou nível de acompanhamento do intervalo de aceitação
90,00-111,11% também deve ser aplicada para este parâmetro.
Não é possível definir um conjunto de critérios para classificar os medicamentos como drogas estreito índice
terapêutico (NTIDs) e deve ser decidido caso a caso, se uma substância activa é um NTID com base em
considerações clínicas.
Fármacos altamente variáveis (sim)
Medicamentos altamente variáveis (HVDP) são aqueles cuja variabilidade intra-sujeito para um parâmetro é
maior do que 30%.
Se o candidato suspeitar que um medicamento pode ser considerado como altamente variável na sua
frequência e/ou extensão da absorção, um estudo sobre design pode ser realizado.
Aqueles HVDP para que uma maior diferença na Cmax é considerado clinicamente relevante com base numa
sólida justificação clínica pode ser avaliada com um intervalo de aceitação alargada.
Se este for o caso, os critérios de aceitação para Cmax pode ser alargado até um máximo de 69,84-
143,19%. Para que o intervalo de aceitação possa ser ampliado o estudo de bioequivalência deve ter idêntico
design onde se demonstrou que a variabilidade intra-sujeito para Cmax do composto de referência no estudo
é> 30%.
A extensão da ampliação é definida com base na variabilidade intra-sujeito visto no
estudo de bioequivalência utilizando escala média-bioequivalência de acordo com [U, L] = exp [± K · sWR], onde
U é o limite superior do intervalo de aceitação, L é o limite inferior do intervalo de aceitação, k é a constante
reguladora conjunto de cabos de aço e 0,760 é o desvio-padrão sujeito dos valores log-transformados de
Cmax do produto de referência.
A tabela abaixo apresenta exemplos de como diferentes níveis de variabilidade levar à aceitação dos limites
diferentes utilizando esta metodologia.
A razão da média geométrica (GMR) deve situar-se dentro do intervalo de aceitação convencional 80,00-
125,00%.
A possibilidade de alargar os critérios de aceitação como base a variabilidade intra-sujeito alto, não se aplica a
AUC, onde o intervalo de aceitação deve permanecer em 80,00-125,00%, independentemente da
variabilidade.

Farmacocinética – UP4
114
Testes de dissolução in vitro(não)
Os resultados dos testes de dissolução in vitro de três buffers diferentes (normalmente, pH 1.2, 4.5 e 6.8) e os
meios destinados a libertação do fármaco (media QC), obtidos com os lotes de produtos teste e referência
que foram utilizados no estudo de bioequivalência deve ser relatados.
Formas de dosagem especial como ODT (comprimidos orais dispersíveis) podem exigir investigações
usando diferentes condições experimentais.
No caso dos resultados comparativos de dissolução in vitro do “biobatches” não refletirem a
bioequivalência, como demonstrado in vivo, o último prevalece. No entanto, as possíveis razões para a
discrepância devem ser abordadas e justificadas.
Definições(sim)
Equivalência Farmacêutica – produtos farmacêuticos são farmacologicamente equivalentes se contiverem a
mesma quantidade da(s) mesma(s) substância(s) activa(s), nas mesmas formas de dosagem que atendam aos
mesmos ou a padrões comparáveis.
A equivalência farmacêutica não implica necessariamente que bioequivalência como diferenças nos
excipientes e/ou no processo de manufactura, possa levar a dissolução e/ou absorção mais rápida ou mais
lenta.
Alternativas Farmacêuticas – são produtos farmacêuticos com diferentes sais, éteres, isómeros, misturas de
isómeros, complexos ou derivados de uma porção activa, ou aqueles que diferem na forma de dosagem ou
força.
Parâmetros Farmacocinéticos:
Ae(0-t) Excreção urinária cumulativa de fármaco inalterado desde a administração até tempo t. AUC(0-t) - Área sob a curva da concentração plasmática desde a administração até à última concentração observada ao tempo t AUC(0-∞) : Área sob a curva da concentração plasmática extrapolada até tempo infinito;
AUC(0-): AUC durante o intervalo de dosagem no steady state; AUC(0-72h) AUC desde a administração até às 72 h;
Cmax: Concentração plasmática máxima; Cmax,ss: Concentração plasmática máxima no steady state; Residual area - Área extrapolada (AUC(0-∞) - AUC(0-t))/ AUC(0-∞); Rmax Taxa máxima de excreção urinária; tmax: Tempo até Cmax ser atingida; tmax,ss: Tempo até Cmax,ss ser atingida; t1/2: Tempo de meia-vida plasmática;
z: Terminal rate constant; SmPC – Sumário das características do produto.
Testes de Dissolução e Semelhança dos Perfis de Dissolução (não)
1. Aspectos Gerais de Dissolução relacionados com biodisponibilidade
Durante o desenvolvimento de um produto farmacêutico um teste de dissolução é usado como
ferramenta para identificar factores de formulação que influenciam e que podem ter um efeito crucial na
biodisponibilidade do fármaco.
Tão cedo quanto a composição e o processo de fabrico são definidos, um teste de dissolução é usado no
controlo de qualidade de scale-up e dos lotes de produção para garantir a coerência de lote para lote e que os
perfis de dissolução são semelhantes aos do ensaio clínico. Além disso, em certos casos, um teste de
dissolução pode ser usado para dispensar um estudo de bioequivalência.

Farmacocinética – UP4
115
Estudos de dissolução podem servir vários objectivos:
i - Ensaios sobre a qualidade dos produtos
Para obter informações sobre os lotes de ensaio usados em biodisponibilidade/bioequivalência e
estudos clínicos pivot para apoiar as especificações para controlo de qualidade.
Para ser usado como uma ferramenta no controlo de qualidade para demonstrar a coerência na
produção
Para obter informações sobre o produto de referência usado em estudos de biodisponibilidade /
bioequivalência e estudos clínicos pivot.
ii - Inferência da bioequivalência substituta (ATENÇÃO! Aqui não faço a menor ideia de tradução)
• Para demonstrar a semelhança entre diferentes formulações de uma substância activa e do
medicamento de referência (biowaivers como por exemplo, variações, alterações durante o
desenvolvimento e medicamentos genéricos)
• Para verificar a homogeneidade, lote por lote, dos produtos (teste e referência) para ser usado como
base para a selecção de lotes adequados para o estudo in vivo.
Os métodos de análise devem ser desenvolvidos de produtos relacionados com base no geral e / ou em
requisitos específicos da farmacopeia. No caso de esses requisitos se revelarem insatisfatórios e / ou que não
reflictam a dissolução em vivo (ie. biorelevância), métodos alternativos podem ser considerados quando se
justifique que estas sejam discriminatórios e capazes de diferenciar entre os lotes com desempenho aceitável
e não aceitável do produto in vivo. Devido às informações actuais, deve ser sempre considerada a interacção
de características derivadas da classificação BCS e forma farmacêutica.
Os tempos da amostra devem ser suficientes para obter perfis de dissolução significativa, e pelo menos a
cada 15 minutos. Amostras mais frequentes durante o período de maior alteração no perfil de dissolução são
recomendadas. Para produtos de dissolução rápida, onde a dissolução completa é de 30 minutos, a criação de
um perfil adequado de amostragem intervalos de 5 ou 10 minutos pode ser necessária.
Se uma substância activa é considerada altamente solúvel, é razoável esperar que ela não causará
quaisquer problemas de biodisponibilidade além disso, o sistema de dosagem é rapidamente dissolvido na
janela de pH fisiológico e os excipientes não são conhecidos por afectarem a biodisponibilidade.
Em contraste, se uma substância activa é considerada como tendo uma solubilidade limitada ou
reduzida, o passo limitante para a absorção pode ser a dissolução da forma de dosagem. Este é também o
caso quando excipientes estão a controlar a liberação e posterior dissolução da substância activa. Nestes
casos, uma variedade de condições de teste é recomendada e amostragem adequada deve ser executada.
2. Semelhança dos perfis de dissolução
• Ensaio de dissolução da similaridade de perfis e todas as conclusões obtidas a partir dos resultados (por
exemplo, a justificação para um biowaiver) podem ser considerados válidos somente se o perfil de dissolução
foi satisfatoriamente caracterizado por um número suficiente de tempos. Para formulações de libertação
imediata, após a orientação dada na secção 1 acima, a comparação aos 15 min é essencial para saber se a
dissolução completa é atingida antes do esvaziamento gástrico. Quando mais de 85% do fármaco é dissolvido
em 15 minutos, perfis de dissolução podem ser aceites como semelhantes, sem avaliação matemática
posteriores.
• No caso de mais de 85% não ser dissolvida em 15 minutos, mas dentro de 30 minutos, pelo menos três
tempos são necessários: o primeiro tempo, antes dos 15 minutos, o segundo aos 15 minutos e o terceiro
quando a libertação está próxima 85%.
• Para os produtos de libertação modificada, o conselho dado na orientação relevante deve ser seguido.
Semelhança de dissolução pode ser determinada através da estatística ƒ2 como se segue:

Farmacocinética – UP4
116
Nesta equação ƒ2 é o factor de semelhança, n é o número de pontos no tempo, R (t) é a média percentual de
fármaco de referência, dissolvida no tempo t após o início do estudo, T (t) é a média percentual do fármaco
em ensaio, dissolvida no tempo t após o início do estudo. Para ambas as formulações, referência e teste, a
dissolução percentual deve ser determinada.
A avaliação do factor de semelhança baseia-se nas seguintes condições:
• Um mínimo de três tempos (zero excluído).
• Os pontos de tempo devem ser os mesmos para as duas formulações.
• Doze valores individuais para cada tempo, para cada formulação.
• Não mais do que um valor médio > 85% dissolvido para qualquer das formulações.
• O desvio padrão relativo ou coeficiente de variação de qualquer produto deve ser inferior a 20% para o
primeiro ponto e menos de 10% do segundo para o último ponto.
Um valor de f2 entre 50 e 100 sugere que os dois perfis de dissolução são semelhantes.
Quando a estatística ƒ2 não é adequada, a semelhança poderá ser comparada usando métodos modelo-
dependentes ou modelo-independentes, como por exemplo comparação estatística multivariada dos
parâmetros da função Weibull ou a percentagem dissolvida em diferentes tempos.
Os métodos alternativos para a estatística ƒ2 para demonstrar a semelhança de dissolução, são
considerados aceitáveis, se estatisticamente válida e satisfatoriamente justificada.
Os limites de aceitação de semelhança deve ser pré-definidos e justificados, não podem ser superiores a
uma diferença de 10%. Além disso, a variabilidade de dissolução do teste e os dados do produto de referência
também devem ser semelhantes, no entanto, uma menor variabilidade do produto de teste pode ser
aceitável.
A evidência de que o software estatístico foi validado também deve ser fornecida.
Uma descrição clara e explicação das medidas tomadas na aplicação do procedimento devem ser fornecidas,
com tabelas-resumo adequadas.