
1. Perceber o interesse e as limitações inerentes … de pares de valores de concentração vs...
70
1 1. Perceber o interesse e as limitações inerentes aos modelos farmacocinéticos Para uma descrição adequada da evolução temporal dos níveis do fármaco recorre-se a modelos nos quais se expressam matematicamente as velocidades dos diferentes processos e que levam a equações que permitem descrever e predizer as quantidades/concentrações do fármaco no corpo em função do tempo. Nota: Os modelos compartimentais são os mais utilizados. O número de parâmetros necessários para descrever um modelo dependerá da complexidade dos processos implicados e da via de administração considerada; Por outro lado, devido ao facto da determinação dos parâmetros ser feita a partir da informação proveniente de pares de valores de concentração vs tempo, a limitação do número de dados disponíveis é determinante sempre e quando se pretenda uma determinação fiável de parâmetros farmacocinéticos. Um compartimento representa uma fracção de material biológico no qual se supõe um fármaco uniformemente distribuído e no qual apresenta as mesmas propriedades cinéticas. Assim, um compartimento tende a agrupar zonas orgânicas semelhantes mas, na realidade, trata-se apenas de um conceito cinético cuja entidade não é necessariamente fisiológica no sentido estrito apesar de abarcar zonas com constantes cinéticas semelhantes. Têm como principais funcões: 1) Prever concentrações plasmáticas, tecidulares e urinárias associadas a qualquer regime medicamentoso; 2) Calcular o regime de classificação óptimo para cada doente, permitindo a individualização posológica; 3) Avaliar uma possível acumulação no organismo do fármaco e/ou dos seus metabolitos; 4) Correlacionar concentrações de fármaco com o efeito farmacológico e/ou toxicológico; 5) Avaliar diferenças na biodisponibilidade e bioequivalência das formulações; 6) Explicar interacções farmacológicas É de realçar que os modelos farmacocinéticos, independentemente da sua complexidade, são apenas simplificações dos sistemas biológicos, desta forma deverá existir precaução na sua aplicação especialmente se não foram devidamente validados. 2. Diferenciar farmacocinética linear e não linear Modelo farmacocinético linear- Se todos os processos de transferência/reacção do modelo proposto forem processos de ordem 1, isto é, se as velocidades de transferência/reacção forem directamente proporcionais à concentração ou à diferença de concentrações entre os compartimentos.
Transcript of 1. Perceber o interesse e as limitações inerentes … de pares de valores de concentração vs...

1
1. Perceber o interesse e as limitações inerentes aos modelos farmacocinéticos
Para uma descrição adequada da evolução temporal dos níveis do fármaco recorre-se a modelos nos quais se
expressam matematicamente as velocidades dos diferentes processos e que levam a equações que permitem
descrever e predizer as quantidades/concentrações do fármaco no corpo em função do tempo. Nota: Os
modelos compartimentais são os mais utilizados.
O número de parâmetros necessários para descrever um modelo dependerá da complexidade dos
processos implicados e da via de administração considerada;
Por outro lado, devido ao facto da determinação dos parâmetros ser feita a partir da informação
proveniente de pares de valores de concentração vs tempo, a limitação do número de dados
disponíveis é determinante sempre e quando se pretenda uma determinação fiável de parâmetros
farmacocinéticos.
Um compartimento representa uma fracção de material biológico no qual se supõe um fármaco
uniformemente distribuído e no qual apresenta as mesmas propriedades cinéticas.
Assim, um compartimento tende a agrupar zonas orgânicas semelhantes mas, na realidade, trata-se
apenas de um conceito cinético cuja entidade não é necessariamente fisiológica no sentido estrito
apesar de abarcar zonas com constantes cinéticas semelhantes.
Têm como principais funcões:
1) Prever concentrações plasmáticas, tecidulares e urinárias associadas a qualquer regime
medicamentoso;
2) Calcular o regime de classificação óptimo para cada doente, permitindo a individualização posológica;
3) Avaliar uma possível acumulação no organismo do fármaco e/ou dos seus metabolitos;
4) Correlacionar concentrações de fármaco com o efeito farmacológico e/ou toxicológico;
5) Avaliar diferenças na biodisponibilidade e bioequivalência das formulações;
6) Explicar interacções farmacológicas
É de realçar que os modelos farmacocinéticos, independentemente da sua complexidade, são apenas
simplificações dos sistemas biológicos, desta forma deverá existir precaução na sua aplicação especialmente
se não foram devidamente validados.
2. Diferenciar farmacocinética linear e não linear
Modelo farmacocinético linear- Se todos os processos de transferência/reacção do modelo proposto forem
processos de ordem 1, isto é, se as velocidades de transferência/reacção forem directamente proporcionais à
concentração ou à diferença de concentrações entre os compartimentos.

2
A linearidade de um modelo farmacocinético é demonstrada pela relação linear existente entre a
dose administrada pela via intravenosa e a concentração plasmática observada num determinado
momento a área sob a curva da concentração plasmática em função do tempo.
Se a dose for duplicada ou triplicada, a concentração plasmática, num determinado momento (ou a
área sob a curva) aumentará proporcionalmente.
A equação representa a taxa de variação da concentração do fármaco no sangue ou plasma ao
longo do tempo e Kel é a constante de velocidade de eliminação de 1ªordem do corpo.
As equações das taxas são escritas em relação à quantidade de fármaco num compartimento especial.
Modelo farmacocinético não-linear
Quando a velocidade de transferência/reacção é de ordem 0 o modelo é não linear Deixa de existir relação
linear entre a dose administrada e a área sob a curva da concentração plasmática em função do tempo
(ex.:saturação das enzimas de biotransformação). A taxa não está, portanto relacionada linearmente com a
concentração.
1. No caso de saturação de uma via metabólica: a área sob a curva concentração plasmática - tempo
aumenta não-linearmente com a dose e, muitas vezes, apresenta um acentuado aumento nas concentrações
plasmáticas que pode conduzir a toxicidade.
2. No caso de elevada afinidade às proteínas do plasma: a concentração total do fármaco no plasma
também aumenta e a fracção de fármaco livre pode aumentar porque o local de ligação plasmático está
saturado. Acima do nível de saturação de ligação, ocorre a distribuição não-linear que pode levar à toxicidade.
Não relacionado à não-linearidade está o caso em que um segundo fármaco pode ser administrado com
afinidade para o mesmo sítio de ligação, levando à deslocação do fármaco ligado e aumentar marcadamente
a concentração de fármaco livre e também levar à toxicidade (ex: varfarina e outros fármacos
anticoagulantes)
3. Aprender a calcular a AUC
Método 1: estimativa numérica simples da área pela “regra do trapézio”.
A vantagem deste método é que apenas requer uma simples extensão de uma tabela com dados
experimentais.
Outros métodos envolvem uma enorme complexidade numérica ou equações convenientes para as
observações e cálculo da área por integração da equação adaptada.
Caso geral
Considere os dados concentração no sangue-tempo (as duas primeiras colunas da tabela A-1) obtidos após a
administração oral de 50mg de fármaco. Qual a AUC?
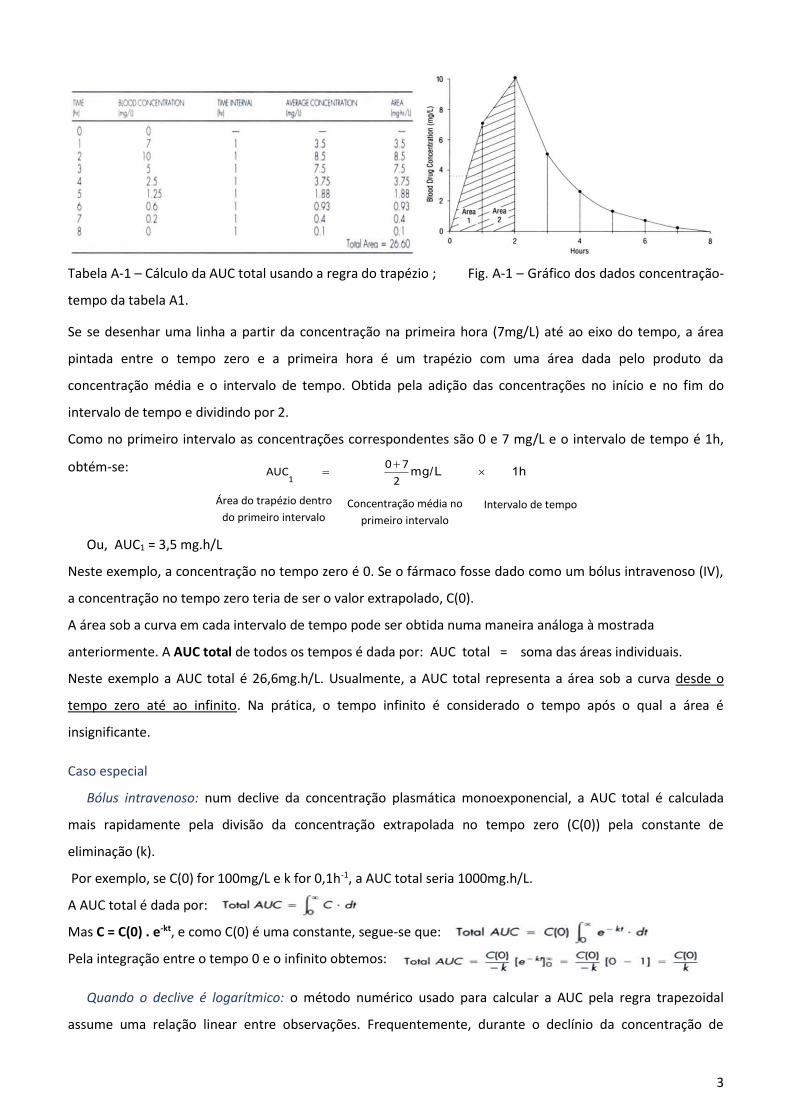
3
Tabela A-1 – Cálculo da AUC total usando a regra do trapézio ; Fig. A-1 – Gráfico dos dados concentração-
tempo da tabela A1.
Se se desenhar uma linha a partir da concentração na primeira hora (7mg/L) até ao eixo do tempo, a área
pintada entre o tempo zero e a primeira hora é um trapézio com uma área dada pelo produto da
concentração média e o intervalo de tempo. Obtida pela adição das concentrações no início e no fim do
intervalo de tempo e dividindo por 2.
Como no primeiro intervalo as concentrações correspondentes são 0 e 7 mg/L e o intervalo de tempo é 1h,
obtém-se:
1h L/mg
2
7 0 AUC
1
Ou, AUC1 = 3,5 mg.h/L
Neste exemplo, a concentração no tempo zero é 0. Se o fármaco fosse dado como um bólus intravenoso (IV),
a concentração no tempo zero teria de ser o valor extrapolado, C(0).
A área sob a curva em cada intervalo de tempo pode ser obtida numa maneira análoga à mostrada
anteriormente. A AUC total de todos os tempos é dada por: AUC total = soma das áreas individuais.
Neste exemplo a AUC total é 26,6mg.h/L. Usualmente, a AUC total representa a área sob a curva desde o
tempo zero até ao infinito. Na prática, o tempo infinito é considerado o tempo após o qual a área é
insignificante.
Caso especial
Bólus intravenoso: num declive da concentração plasmática monoexponencial, a AUC total é calculada
mais rapidamente pela divisão da concentração extrapolada no tempo zero (C(0)) pela constante de
eliminação (k).
Por exemplo, se C(0) for 100mg/L e k for 0,1h-1, a AUC total seria 1000mg.h/L.
A AUC total é dada por:
Mas C = C(0) . e-kt, e como C(0) é uma constante, segue-se que:
Pela integração entre o tempo 0 e o infinito obtemos:
Quando o declive é logarítmico: o método numérico usado para calcular a AUC pela regra trapezoidal
assume uma relação linear entre observações. Frequentemente, durante o declínio da concentração de
Área do trapézio dentro
do primeiro intervalo Concentração média no
primeiro intervalo
Intervalo de tempo

4
fármaco, a queda é exponencial. Assim, o método mais rigoroso para calcular a área durante o declínio é o log
da regra do trapézio.
Considerarando duas observações consecutivas C(ti) e C(ti+1) aos tempos ti e ti+1 respectivamente. Estas
observações estão relacionadas por:
Onde ki é a constante que permite que a concentração diminua de forma exponencial
de C(ti) para C(ti+1) no intervalo de tempo ti+1 - ti, ou seja, Δti. O valor de ki é dado
colocando o log em ambos os lados da equação anterior e rearranjando-a.
A AUC durante um intervalo de tempo Δti é a diferença entre as áreas totais desde o ti até o infinito e desde
ti+1 até ao infinito, respectivamente. Pode-se então concluir que:
E por uma substituição apropriada de ki na equação anterior obtém-se:
Este cálculo é depois repetido para todas as equações que fiquem
antes do pico da concentração.
Na prática uma maior discrepância aparece entre o método acima descrito e o que utiliza a regra
trapezoidal (linear) apenas quando observações consecutivas diferem para mais do dobro.
4. Entender os princípios subjacentes à análise farmacocinética compartimental
4.1 Entender a noção de compartimento e as propriedades respectivas. Diferenciar modelo
compartimental linear e não linear.
Modelos Compartimentais
Consideram o organismo como sendo constituído por um ou mais compartimentos reversivelmente ligados
entre si e baseiam-se em sistemas físico-químicos de transferência de matéria, regidos por constantes de
velocidade. É assumida para cada compartimento uma incorporação (entrada de fármaco no compartimento)
instantânea e homogénea cujo resultado se traduz na existência de uma concentração que, para qualquer
ponto do mesmo, é representativa do resto do compartimento.
O modelo compartimental mais simples é aquele que considera o organismo como um
compartimento único modelo monocompartimental.
O organismo pode ser dividido em vários compartimentos (modelos bicompartimentais, …,
multicompartimentais), nos quais o fármaco apresenta um comportamento cinético diferente.
Na prática clínica, utilizam-se essencialmente os modelos mono e bicompartimentais os modelos mais
complexos são de difícil aplicação.
Geralmente, os processos cinéticos considerados são do tipo linear, embora seja possível incluir termos ou
elementos matemáticos que descrevem processos cinéticos não-lineares implicados na disposição de fármaco
no organismo (ex. metabolismo de acordo com a equação de Michaelis-Menten).
Propriedades de um Compartimento Farmacocinético Clássico
Cinética homogénea um compartimento contém tecidos agrupados de acordo com propriedades
cinéticas semelhantes permitindo ao fármaco uma rápida distribuição reversível entre os tecidos.

5
As barreiras entre os compartimentos, (representadas pelas
setas na figura), são limitadas pela difusão e podem representar
o transporte através de barreiras anatómicas (ex.: epitélio do
TGI ou os glomérulos no rim), conversão metabólica, ou, se
desconhecido, o transporte para dentro ou fora de um grupo de
tecidos com diferentes propriedades cinéticas.
Os compartimentos encontram-se relacionados por constantes
de primeira ordem. As constantes de saída e entrada podem ser
de ordem zero.
Compartimento
É definido por sectores aquosos que ocupam um determinado volume
(V) e que contêm uma quantidade determinada de fármaco (Q). Assim, a
concentração de fármaco (C) no compartimento é dada por: C = Q/V.
Uma representação esquemática de um compartimento pode ser feita
mediante um quadrado/rectângulo com setas que indicam a
entrada/saída do fármaco. Exemplo: representação de um
compartimento com saída do fármaco de acordo com uma
cinética de 1ª ordem será a seguinte: , sendo k a constante de
velocidade de primeira ordem.
Atenção: nem sempre um compartimento engloba uma entidade
fisiológica, tratando-se de uma unidade conceptual, e zonas próximas e relacionadas do organismo podem
pertencer a compartimentos diferentes.
Modelos Farmacocinéticos Lineares e Não-Lineares
Modelo Farmacocinético Linear- Os processos cinéticos respondem a uma cinética de 1ª ordem.
Desta forma, os valores dos parâmetros farmacocinéticos não devem variar com a dose.
A linearidade de um modelo farmacocinético é demonstrada pela relação linear existente entre a dose
administrada pela via intravenosa e a concentração do fármaco num determinado momento a área sob a
curva da concentração plasmática em função do tempo.
Modelo Farmacocinético Não – Linear- A velocidade de transferência/reacção é de ordem 0 e o modelo é não
linear.Observa-se que ao variar a dose de um fármaco, o valor de um ou mais parâmetros farmacocinéticos
varia e a concentração num determinado momento não é directamente proporcional à variação da dose.
FIGURE 1.1 The three most commonly used pharmacokinetic
models in explaining the pharmacokinetic behavior of drugs.
The symbols C, P, S, and D represent central, peripheral,
shallow, and deep compartments, whereas the first-order
rate constants, symbolized by kij, represent drug transport
from compartment i to compartment j. DIV, ka, and k0
represent a bolus IV dose, the absorption rate constant, and
constant rate infusion, respectively.
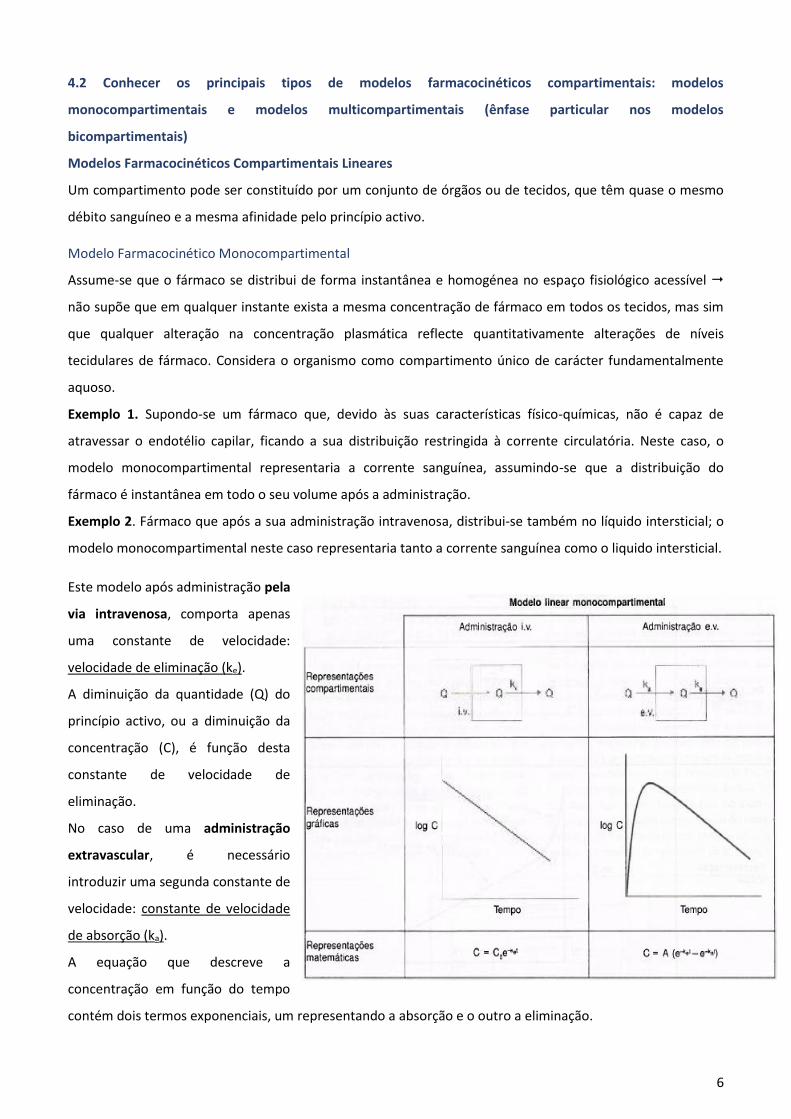
6
4.2 Conhecer os principais tipos de modelos farmacocinéticos compartimentais: modelos
monocompartimentais e modelos multicompartimentais (ênfase particular nos modelos
bicompartimentais)
Modelos Farmacocinéticos Compartimentais Lineares
Um compartimento pode ser constituído por um conjunto de órgãos ou de tecidos, que têm quase o mesmo
débito sanguíneo e a mesma afinidade pelo princípio activo.
Modelo Farmacocinético Monocompartimental
Assume-se que o fármaco se distribui de forma instantânea e homogénea no espaço fisiológico acessível
não supõe que em qualquer instante exista a mesma concentração de fármaco em todos os tecidos, mas sim
que qualquer alteração na concentração plasmática reflecte quantitativamente alterações de níveis
tecidulares de fármaco. Considera o organismo como compartimento único de carácter fundamentalmente
aquoso.
Exemplo 1. Supondo-se um fármaco que, devido às suas características físico-químicas, não é capaz de
atravessar o endotélio capilar, ficando a sua distribuição restringida à corrente circulatória. Neste caso, o
modelo monocompartimental representaria a corrente sanguínea, assumindo-se que a distribuição do
fármaco é instantânea em todo o seu volume após a administração.
Exemplo 2. Fármaco que após a sua administração intravenosa, distribui-se também no líquido intersticial; o
modelo monocompartimental neste caso representaria tanto a corrente sanguínea como o liquido intersticial.
Este modelo após administração pela
via intravenosa, comporta apenas
uma constante de velocidade:
velocidade de eliminação (ke).
A diminuição da quantidade (Q) do
princípio activo, ou a diminuição da
concentração (C), é função desta
constante de velocidade de
eliminação.
No caso de uma administração
extravascular, é necessário
introduzir uma segunda constante de
velocidade: constante de velocidade
de absorção (ka).
A equação que descreve a
concentração em função do tempo
contém dois termos exponenciais, um representando a absorção e o outro a eliminação.

7
A principal característica deste modelo é o facto de se considerar instantânea a distribuição do fármaco.
Desta forma, a representação esquemática deste modelo é a seguinte:
A equação de velocidade de eliminação do fármaco é a seguinte:
Dividindo ambos os membros por Vd, obtém-se a expressão da velocidade em função da concentração do
fármaco no compartimento:
Quando a administração é extravascular, existe uma fase durante o qual se absorve fármaco. O modelo,
neste caso, é o seguinte:
Qa – quantidade de fármaco que permanece no local de absorção num determinado momento, após a
administração; Ka – constante de absorção (que se supõe ser de primeira ordem)
Neste caso a equação da variação da quantidade de fármaco no organismo:
ka . Qa -representa a velocidade de absorção do fármaco; e kel . Q - velocidade de eliminação.
Modelo Farmacocinético Bicompartimental
Modelo constituído por dois compartimentos apresenta boa capacidade predictiva para uma grande
quantidade de fármacos mas peca pelo número de concentrações necessárias para uma correcta
determinação dos parâmetros que o caracterizam.
Ao contrário do que acontecia com o modelo monocompartimental, não se assume a existência de um
equilíbrio instantâneo na distribuição do fármaco a todos os tecidos ao qual ele tem capacidade de acesso.
Assim, considera-se a existência de:
Compartimento central (Q1): inclui o sangue e todos os tecidos fortemente irrigados e que se caracteriza por
um volume específico (Vc – volume do compartimento central).
Compartimento periférico (Q2): agrupa os tecidos menos acessíveis ao fármaco;caracterizado por um volume
aparente de distribuição diferente (Vp – volume do compartimento periférico).
O equilíbrio entre os compartimentos central e periférico é regulado por microconstantes de transferência k12
e k21, que por sua vez, dependem das propriedades físico-quimicas do fármaco:
k12 : caracteriza a velocidade de distribuição do compartimento central para o periférico
k21: caracteriza a velocidade de distribuição do compartimento periférico para o central.
A constante de velocidade de eliminação do compartimento central é representada por k10.
Div – dose de fármaco administrada por via intravenosa
Vd – volume de distribuição do fármaco
C – concentração do fármaco no compartimento num determinado
momento
Q – quantidade de fármaco no compartimento nesse mesmo momento
Kel – constante de eliminação (primeira ordem)
Qel – quantidade de fármaco eliminada

8
A equação que descreve a concentração
plasmática em função do tempo contém os
expoentes α e β, que são macroconstantes,
ou constantes híbridas, que compreendem
as microconstantes k12, k21 e k10.
Uma vez alcançado o equilíbrio de
distribuição entre ambos os
compartimentos, completa-se dessa forma
a denominada fase α ou fase de rápida
disposição; no caso da eliminação ser linear
(mais frequente), esta irá decorrer de
acordo com um processo
monoexponencial, o que indica uma
homogeneidade cinética em todos os
fluidos e tecidos do organismo (fase β ou
fase de lenta disposição).
O esquema que representa este modelo é o seguinte:
A equação da variação da quantidade de fármaco no compartimento central:
Caso a administração seja feita por via extravascular e a absorção seja de 1ª
ordem, o modelo é o seguinte:
A equação de velocidade é a seguinte:
Apesar de se poderem aplicar modelos bem mais complexos (com três ou mais compartimentos), na prática, o
modelo bicompartimental permite uma adequada descrição farmacocinética da maioria dos fármacos.
Qc- quantidade de fármaco existente no comp. Central
Vc- volume do comp. Central
Qp- quantidade de fármaco existente no comp. Periférico
K12- constante de distribuição do fármaco desde o comp. central até ao
comp. periférico
K21- constante de distribuição do fármaco desde o comp. periférico até
ao comp. central
Kel- constante de eliminação

9
Modelos Farmacocinéticos Compartimentais Não Lineares
Os processos de absorção, distribuição e eliminação dos fármacos não são sempre de 1ª ordem, e em alguns
casos podem tratar-se de processos saturáveis.
A absorção pode ter lugar mediante um processo activo de transporte.
A distribuição pode ser afectada pela natureza saturável da união do fármaco às proteínas
plasmáticas, a união saturável aos tecidos e o transporte saturável através das membranas celulares.
A eliminação pode ser afectada pela união saturável às proteínas plasmáticas ou pela natureza dos
mecanismos próprios de eliminação que respondem a cinéticas de Michaelis-Menten, como a
secreção tubular passiva e o metabolismo.
Na maioria dos casos, a não linearidade deve-se a um processo de eliminação saturável com cinética de
Michaelis-Menten. Assim:
Modelo monocompartimental Modelo bicompartimental
Dado que as equações de velocidade estão expressas em função da quantidade de fármaco no organismo
(modelo monocompartimental) ou no compartimento central (modelo bicompartimental), a constante de
Michaelis (Km) tem, nestas equações, unidades de massa e Vm, unidades de massa x tempo-1.
4.3 Entender o porquê da larga aplicação do modelo monocompartimental na clínica.
A principal vantagem do modelo monocompartimental reside na sua boa capacidade preditiva aliada a uma
simplicidade matemática, embora não lhe possa ser atribuída qualquer tipo de realidade fisiológica. Mais
usada também devido à escassez de informação disponível-> neste modelo qualquer alteração na
concentração plasmática reflecte quantitativamente alterações de níveis tecidulares de fármaco.
Apesar da biofase residir nos tecidos (podendo ou não residir no compartimento central), as concentrações
plasmáticas foram escolhidas para a medição devido à facilidade de amostragem e porque são referência para
uma dose apropriada, através do estabelecimento da janela terapêutica.

10
5. Estudar a farmacocinética de dose única mediante a administração intravenosa rápida (tipo
bólus), administração intravenosa contínua (perfusão contínua) e administração extravascular.
5.1 Entender a aplicação do modelo farmacocinético monocompartimental para descrever a
cinética da administração intravenosa rápida (tipo bólus) de fármacos: curva da concentração
plasmática em função do tempo, estimação dos parâmetros farmacocinéticos e o seu significado.
Ct – concentração plasmática ao tempo t; D – dose administrada
Vd – volume de distribuição; Ke – constante de velocidade de eliminação
Constante de eliminação
Geralmente, o fármaco parental ou activo é medido no compartimento vascular.
A remoção total ou eliminação do fármaco parental do compartimento vascular é afectado pelo
metabolismo (biotransformação) e excreção.
A constante de eliminação representa a soma de cada um destes processos:
Cada um destes processos tem a sua própria constante de eliminação.
A expressão para a figura 3.1 é:
Justificação para o uso clínico do modelo monocompartimental, em vez do modelo multicompartimental,
teoricamente mais correcto:
1. O fármaco distribui-se rapidamente apenas para os tecidos perfundidos e fluidos circulantes localizados no
compartimento central (ex. fármacos hidrofílicos, como as penicilinas e as cefalosporinas)
2. O fármaco distribui-se tanto para os tecidos centrais como periféricos; contudo, a concentração do fármaco
no compartimento periférico encontra-se abaixo da janela terapêutica (ex. aminoglicosídeos)
3. O fármaco distribui-se ao compartimento periférico em concentrações significativas. Ignorando a
distribuição nos compartimentos periféricos e utilizando as equações do modelo monocompartimental, o
erro encontra-se dentro dos limites aceitáveis )ex. teofilina).
DB – fármaco no corpo no tempo t
D0B – fármaco no corpo no tempo t=0
Km – taxa de 1ª ordem do processo de metabolismo
Ke – taxa de 1ª ordem do processo de excreção
Parâmetros derivados:
Semi-vida de eliminação:
Clearance:

11
Esta expressão mostra que a taxa de eliminação do fármacono corpo é
um processo de 1ª ordem. A sua integração é:
A equação 3.3 também pode ser expressa por:
Volume de distribuição aparente
Representa o volume que deve ser considerado para estimar a
quantidade de fármaco no corpo a partir da concentração do fármaco encontrada no compartimento
de amostra. É também o volume aparente (VD) no qual o fármaco está dissolvido (equação 3.5)
A quantidade de fármaco no corpo não é determinada directamente. Em vez disso, uma amostra de
sangue é colhida em intervalos periódicos e analisada a concentração de fármaco.
O VD relaciona a concentração de fármaco no plasma (Cp) e a quantidade de fármaco no corpo (DB):
Substituindo na equação 3.3:
A equação 3.6 também pode ser expressa por:
EXEMPLO: Exactamente 1g de fármaco é dissolvido num volume de água desconhecido. Após o ensaio, a
concentração nesta solução é 1 mg/mL. Qual é o volume original desta solução?
O volume original da solução pode ser obtido pela seguinte proporção e lembrando que 1g = 1000mg
Portanto, o volume original é 1000 mL ou 1 L.
Se no exemplo acima o volume de solução fosse 1L e a concentração da solução 1 mg/mL, para calcular a
quantidade total de fármaco presente:
Portanto, a quantidade total de fármaco presente na solução é 1000 mg ou 1 g.
Do exemplo anterior, se o volume de solução no qual o fármaco está dissolvido e a concentração do fármaco
na solução fossem conhecidos, a concentração total de fármaco presente na solução podia ser calculada.
Esta relação entre a concentração de fármaco, volume e a concentração total de fármaco presente é dada
pela seguinte expressão:
O corpo pode ser considerado um sistema de volume constante. Assim, o volume de distribuição aparente
para um dado fármaco é geralmente constante.
Cp – concentração do fármaco no plasma no tempo t
C0p – concentração do fármaco no plasma no tempo t=0

12
Cálculo do volume de distribuição (eq.3.8)
Quando determinado por extrapolação, C0p, representa a concentração
instantânea de fármaco (concentração do fármaco a t=0) após o fármaco
ter tempo para se equilibrar no corpo.
A dose de fármaco dada por bólus IV (injecção IV rápida) representa a
quantidade de fármaco no corpo, D0B, a t=0.
Porque C0p e D0
B são conhecidos a t=0, o volume aparente de distribuição,
VD, pode ser calculado a partir da expressão 3.8.
O volume aparente de distribuição, VD, pode também ser calculado a
partir do conhecimento da dose, constante de eliminação e AUC deste T=0 a t=∞:
A partir da expressão 3.2, a constante de eliminação do fármaco é:
Através da substituição da equação 3.5 na equação 3.2:
Após rearranjo da eq.3.10:
Como k e VD são constantes, a equação pode ser integrada:
A equação 3.12 mostra que uma pequena alteração no tempo (dt) resulta numa pequena alteração na
quantidade de fármaco no corpo, DB
, que é a área debaixo da curva de t=0 a t=∞, O é O integral representa o
usualmente estimado pela regra do trapézio.
Após integração, a equação 3.12 fica:
Rearranjada:
O cálculo do VD por esta expressão é um método independente do modelo uma vez que nenhum modelo
farmacocinético é considerado e a AUC é determinada directamente pela regra do trapézio.
Significado do volume aparente de distribuição
A equação 3.8 mostra que o VD aparente é dependente da C0p
• A partir da equação 3.8 podemos deduzir:
1. Se C0p for pequena -> o VD é alto -> O fármaco está mais concentrado no tecido extravascular
e menos concentrados no intravascular.
2. Se C0p for alta (no caso de ligação às proteínas plasmáticas ou se o fármaco permanecer na
região vascular) -> o VD é pequeno.
Expressão do VD em termos de percentagem do peso corporal:

13
1 L de volume é assumido ser equivalente ao peso de 1 Kg. Por exemplo, se o VD for 3500 mL para uma
pessoa com um peso de 70 Kg, o VD será expresso como percentagem de peso corporal da seguinte forma:
Expressando o VD em termos de percentagem do peso corporal, os valores de VD podem corresponder a
verdadeiros valores anatómicos (tabela 3.1). Contudo pode ser apenas ocasional que o valor do VD aparente
para um fármaco seja o mesmo que o volume anatómico real-> é necessária depois uma investigação para
testar esta hipótese.
Em certos casos patológicos, o
VD aparente do fármaco pode
ser alterado se a distribuição
deste for alterada:
1. Em condições edematosas (água corporal e extracelular total aumentam aumento do VD)
2. Alterações do peso corporal total e perda de massa corporal (normalmente com a idade)
Clearance do fármaco no modelo monocompartimental
• Volume de plasma que é “limpo” de fármaco por unidade de tempo ou a fracção de fármaco removido
por unidade de tempo.
Eliminação do fármaco expressa como QUANTIDADE por unidade de tempo (mg/min, mg/hr)
a) Para um processo de eliminação de ordem 0:
A expressão da taxa de eliminação do fármaco como massa
por unidade de tempo é conveniente porque a taxa é
constante (Fig. 3-4A).
b) Para um processo de eliminação de ordem 1:
O processo de eliminação não é constante e altera em
relação à concentração de fármaco no corpo.
A clearance é expressa como volume por unidade de tempo
(L/h ou mL/min) porque é constante.
Eliminação do fármaco expressa como VOLUME por
unidade de tempo
Comum na farmácia:
Fig 3-4 – Diagrama que ilustra 3 diferentes formas de eliminação do fármaco após a injecção IV de uma dose de 100 mg num volume de 10 mL
Se o valor de VD for um número grande (isto é, > 100% do
peso do corpo) então pode-se assumir que o fármaco está
concentrado em certos compartimentos do tecido. Deste
modo, o VD aparente é um parâmetro útil em considerar as
quantidades relativas do fármaco nos tecidos vascular e
extravascular.

14
Exemplo: a um paciente pode ser dada uma dose na proporção de 2 colheres de chá (teaspoonsful)
(10 mL) de um medicamento líquido diariamente (10 mg/mL), ou, em alternativa, uma dose (peso) de
100 mg do mesmo fármaco diariamente.
Uma vez que um volume constante de plasma (cerca de 120 mL/min em humanos) é filtrado através dos
glomérulos, a taxa de fármaco removido é dependente da concentração do fármaco no plasma em todos os
tempos. A clearance para um processo de 1ª ordem é constante independentemente da concentração do
fármaco porque a clearance é expressa em volume por unidade de tempo.
Matematicamente a taxa de eliminação do fármaco é semelhante à equação 3.10:
Rácio da clearance e volume de distribuição, Cl/VD
Exemplo: 100 mg de fármaco são dissolvidos em 10 mL de fluido e 10 mg do fármaco são removidos no
primeiro minuto. O processo de eliminação do fármaco pode ser descrito como:
a) mg de fármaco eliminado por minuto.
b) mL de fluído de fármaco ‘cleared’ por minuto.
c) Fracção de fármaco eliminado por minuto
Se a concentração de fármaco é Cp, a taxa de eliminação do fármaco é:
Para um processo de primeira ordem:
Equacionando as duas expressões:
Equação em termos de Cl e VD - Modelo de um compartimento.
dDB/dt– taxa de eliminação do fármaco no corpo em relação ao
tempo (mg/hr)
Cp – concentração do fármaco no plasma (mg/L)
k – constante de 1ª ordem (hr-1 ou 1/hr)
VD – volume de distribuição aparente
Cl – Clearance (L/hr , neste exemplo)
O sinal negativo refere-se à saída do fármaco do corpo
Mostra que a clearance é uma
constante porque tanto o VD como o k
são ambos constantes.
Dividindo a expressão em ambos os lados por Cp
rearranjo da equação 3.15

15
A equação 3.20 pode ser reescrita em termos de clearance e volume de distribuição pela substituição de k
por Cl/V.
Quando apenas uma amostra está disponível, isto é, Cp é conhecida a determinado tempo t após uma dose
dada, a equação não pode ser determinada sem ambiguidadade, uma vez que dois parâmetros
desconhecidos devem ser resolvidos, isto é, Cl e VD. Na prática, os valores médios para Cl e VD de um
fármaco são obtidos através de valores teóricos da população (posteriormente ajustados usando um
programa de computador). A razão Cl/VD pode ser calculada descuidando o tipo de modelo compartimental,
usando as mínimas amostras de plasma.
Practical Focus
O método cinético mais exacto para determinar o VD e a cinética de distribuição de um fármaco num paciente
é dar o fármaco por um bólus único IV. A equação de dose única IV (3.22) pode ser modificada para calcular a
constante de eliminação ou o tempo de meia vida de um fármaco num paciente quando duas amostras
plasmáticas e os seus tempos de colheita são conhecidos:
Se a primeira amostra plasmática for recolhida a t1, em vez de a t0 e corresponder à concentração plasmática
do fármaco, então C2 é a concentração ao tempo t2 e t = (t2-t1)
Rearranjando:
Na prática clínica, várias doses do fármaco podem ter sido administradas ao paciente e os tempos das doses
prévias podem não ser exactamente conhecidos.
Se o farmacêutico julgar que o fármaco no organismo está numa fase de declínio (i.e., a absorção está
completa), esta equação pode ser usada para determinar o tempo de meia vida de um fármaco num paciente
recolhendo duas amostras de plasma temporalmente afastadas e registando os tempos de recolha.
Clearance a partir de tecidos eliminadores de fármaco
Desde que esteja envolvido um processo de eliminação de primeira ordem a clearance total representa a
soma das clearances para cada órgão eliminador de fármaco:
A ClNR é principalmente devida à clearance hepática (ClH) na ausência de outras clearances não significativas
tais como a eliminação através do pulmão ou da bílis tal como se mostra na equação 3.27:
t1 = tempo da primeira colheita
c1 = concentração plasmática em t1
t2 = tempo da segunda colheita
c2= concentração plasmática em c2
Onde:
ClR - a clearance renal.
ClNR - a clearance não renal, através de
outros órgãos.

16
Alternativamente, ClT pode ser definida como a taxa de eliminação dividida pela concentração plasmática.
Rearranjando a equação 3.29, obtém-se a equação
3.30:
Onde: ClT é a constante para um fármaco específico e representa o declive da recta obtida traçando dDE/dT vs
CP.
Para fármacos que seguem uma eliminação de 1ª ordem, a taxa de eliminação é dependente da quantidade
de fármaco remanescente no corpo:
Substituindo a taxa de eliminação na equação 3.30 por kCPVD na equação 3.31 e resolvendo em ordem a ClT:
Esta equação mostra que a clearance é o produto de VD e k (similar à equação 3.19)
À medida que a concentração plasmática de fármaco diminui, dDE/dT também diminui, mas a clearence
permanece constante (desde que a taxa de eliminação seja um processo de 1ª ordem).
Para alguns fármacos, o processo de taxa de eliminação é mais complexo e para calcular certos parâmetros
farmacocinéticos como a clearance pode ser usado um método não-compartimental.
A clearance pode ser determinada directamente a partir das concentrações plasmáticas de fármaco versus
curva de tempo:
Onde, D0 – é a dose e
Nenhum modelo compartimental é assumido porque é calculada a partir da concentração
plasmática de fármaco versus a curva de tempo a partir de 0 ao usando a regra do trapézio.
Contudo para extrapolar os dados para o infinito para obter a ou é usualmente
assumida uma eliminação de primeira ordem.
Neste caso, se o fármaco segue uma cinética de modelo monocompartimental, a ClT é numericamente similar
ao produto de VD e o k obtido ajustando os dados para um modelo monocompartimental.
Cálculo de k a partir de dados da excreção urinária
O termo ke é a constante de excreção renal e Du é a quantidade de fármaco excretada na urina
Onde: DE é a quantidade de fármaco
eliminado e dDE/dT é a taxa de
eliminação.

17
A partir da equação 3.34, DB pode ser substituído por
Colocando o logaritmo natural em ambos os lados e transformando em logaritmos comuns:
Uma linha recta é obtida a partir desta equação num traçado semilogaritmico dDU/dt em relação ao tempo
(Figura 3-5 e 3-6)
O declive desta curva é igual a e y intercepta-a em
Para uma administração IV rápida DB0 é igual a dose D0. Assim, se DB
0 for conhecido, a constante de excreção
renal (ke) pode ser obtida.
ke e k podem ser determinados por este método e a constante não-renal (Knr) para qualquer via de eliminação
além da excreção renal pode ser encontrada por:
Contudo, como a eliminação do fármaco é normalmente afectada pela excreção renal e pelo metabolismo
(biotransformação) do fármaco:
A substituição de km por knr na equação 3.37 origina a equação 3.1
A taxa de excreção urinária de um fármaco (dDu/dt) não pode ser determinada experimentalmente a
qualquer instante. Assim, a taxa média de excreção urinária de um fármaco, Du / t é traçada versus o tempo
médio, t* para a amostra colhida de urina.
PROBLEMAS PRÁTICOS
1- Uma única dose IV de um AB é dada a uma mulher de 50kg com uma dose de 20 mg/kg. Amostras de
sangue e de urina foram removidas periodicamente e ensaiadas para o fármaco parental. Foram obtidos
os seguintes dados:

18
SOLUÇÃO
Configurando a seguinte tabela:
t* - ponto médio do período de colheita
t – intervalo de tempo para a colheita da amostra de
urina
Construindo um gráfico numa escala semilogaritmica de Du/t versus t*. O declive desta linha mostra-se igual a
–k /2.3. O t1/2 de eliminação é mais fácil determinar-se directamente a partir da curva e depois calculado o k a
partir:
Aqui, t1/2=1,0 hr e k = 0,693 hr-1. Um gráfico semelhante dos valores de Cp versus t pode originar uma curva
com um declive com o mesmo valor do derivado da curva anterior. Note-se que o declive da constante de
excreção log é uma função da constante de eliminação k e não da constante de excreção urinária ke (Figura 3-
6).
Um método alternativo para calcular a constante de eliminação k a partir dos valores de excreção urinária é o
método sigma-minus ou o método quantidade de fármaco que falta excretar. O método sigma-minus é por
vezes preferido em relação ao método anterior porque as flutuações na taxa de eliminação são minimizadas.
A quantidade de fármaco não alterada na urina pode ser expressa como uma função do tempo através da
seguinte equação:
Onde Du é a quantidade cumulativa de fármaco não alterado excretado na urina.
A quantidade de fármaco não alterada que é, em última instância, excretada na urina pode ser
determinada tornando o tempo igual ao infinito. Assim, o termo fica desprezível e é obtida a
seguinte expressão:
Substituindo por na equação 3.39 e rearranjando:
A equação 3.41 pode ser escrita na forma logarítimica para obter uma equação linear:
A equação 3.42 descreve a relação entre a quantidade de fármaco que falta excretar
versus o tempo.
É obtida uma curva linear através do gráfico em escala logaritmica
da quantidade de fármaco não alterada ainda a ser eliminada log
versus o tempo. O declive desta curva é –k /2.3 e a
intercepção de y é
2- Usando os dados no problema anterior, determine a constante de
eliminação
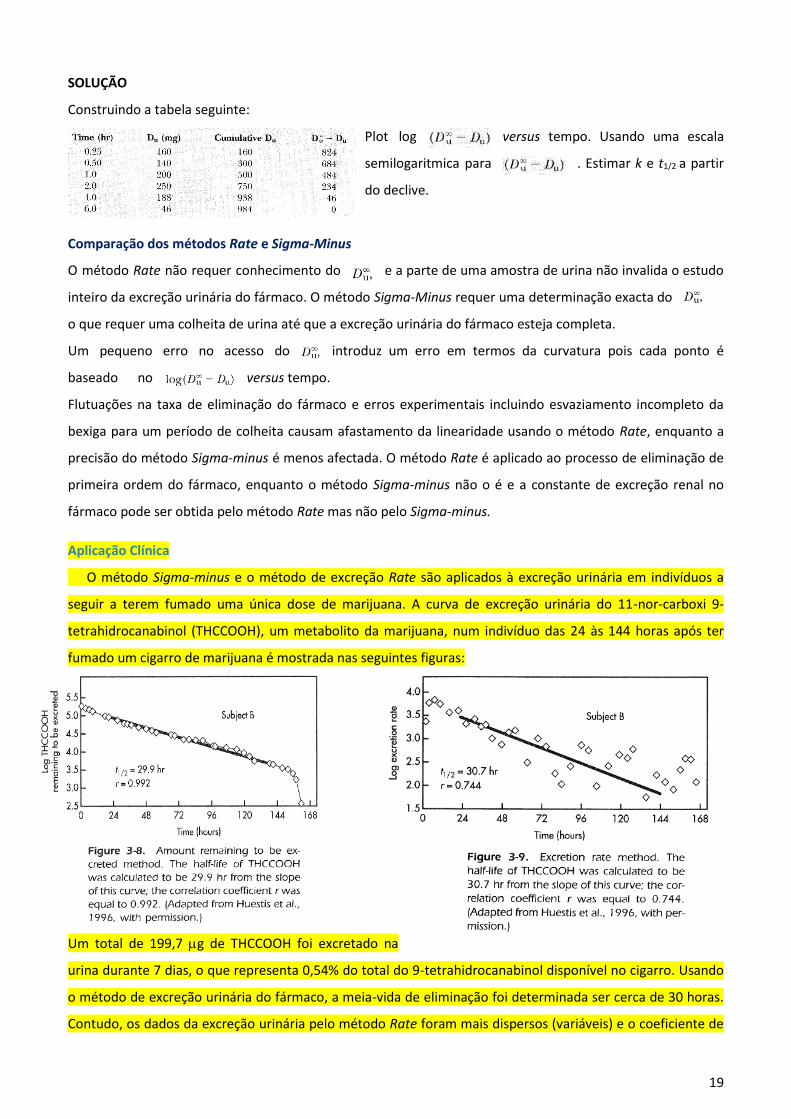
19
SOLUÇÃO
Construindo a tabela seguinte:
Plot log versus tempo. Usando uma escala
semilogaritmica para . Estimar k e t1/2 a partir
do declive.
Comparação dos métodos Rate e Sigma-Minus
O método Rate não requer conhecimento do e a parte de uma amostra de urina não invalida o estudo
inteiro da excreção urinária do fármaco. O método Sigma-Minus requer uma determinação exacta do
o que requer uma colheita de urina até que a excreção urinária do fármaco esteja completa.
Um pequeno erro no acesso do introduz um erro em termos da curvatura pois cada ponto é
baseado no versus tempo.
Flutuações na taxa de eliminação do fármaco e erros experimentais incluindo esvaziamento incompleto da
bexiga para um período de colheita causam afastamento da linearidade usando o método Rate, enquanto a
precisão do método Sigma-minus é menos afectada. O método Rate é aplicado ao processo de eliminação de
primeira ordem do fármaco, enquanto o método Sigma-minus não o é e a constante de excreção renal no
fármaco pode ser obtida pelo método Rate mas não pelo Sigma-minus.
Aplicação Clínica
O método Sigma-minus e o método de excreção Rate são aplicados à excreção urinária em indivíduos a
seguir a terem fumado uma única dose de marijuana. A curva de excreção urinária do 11-nor-carboxi 9-
tetrahidrocanabinol (THCCOOH), um metabolito da marijuana, num indivíduo das 24 às 144 horas após ter
fumado um cigarro de marijuana é mostrada nas seguintes figuras:
Um total de 199,7 g de THCCOOH foi excretado na
urina durante 7 dias, o que representa 0,54% do total do 9-tetrahidrocanabinol disponível no cigarro. Usando
o método de excreção urinária do fármaco, a meia-vida de eliminação foi determinada ser cerca de 30 horas.
Contudo, os dados da excreção urinária pelo método Rate foram mais dispersos (variáveis) e o coeficiente de

20
correlação r foi igual a 0,744 (Figura 3-9) comparando o coeficiente de correlação r de 0,992 usando o método
Sigma-minus.
Problemas na obtenção de dados válidos da excreção urinária
Certos factores podem tornar difícil a obtenção de dados válidos da excreção urinária. Alguns destes
factores são:
1. Uma fracção significante de fármaco não alterado pode ser excretada na urina.
2. A técnica de ensaio pode ser específica para o fármaco não alterado e não deve ter interferências devido
aos metabolitos dos fármacos que têm semelhantes estruturas químicas.
3. São necessárias amostras frequentes para a descrição de uma boa curva.
4. Amostras de urina devem ser colhidas periodicamente até quase todo o fármaco ser excretado. Um
gráfico do fármaco cumulativo excretado versus tempo originará uma curva que se aproxima a uma
assimptota ao tempo infinito. Na prática, aproximadamente 7 meias vidas de eliminação são necessárias para
que 99% do fármaco seja eliminado.
5. Variações no pH urinário e volume podem causar variações significativas nas taxas de excreção urinária.
Os sujeitos devem ser cuidadosamente instruídos quanto à necessidade de uma amostra completa de urina
(i.e. esvaziamento completo da bexiga).
5.2 Entender a aplicação do modelo farmacocinético monocompartimental para descrever a cinética da
administração intravenosa contínua (perfusão contínua) de fármacos: curva da concentração plasmática em
função do tempo (concentração no estado de equilíbrio estacionário e tempo para a alcançar), estimação
dos parâmetros farmacocinéticos e o seu significado. Perceber o conceito e o interesse de uma dose de
carga (bólus) seguida de perfusão contínua.
Para fármacos com uma margem terapêutica estreita, a infusão IV mantém
uma concentração plasmática do fármaco eficaz, eliminando as grandes
flutuações entre o pico (máximo) e o mínimo da concentração plasmática do
fármaco.
Porque não existe fármaco no corpo para t=0, os níveis de fármaco
aumentam a partir da concentração zero e gradualmente se torna constante
quando um determinado nível (“plauteau” ou “steady-state” ) é atingido.
No steady-state, a taxa de fármaco que é eliminada iguala a taxa de fármaco que entra no corpo (taxa de
infusão). A taxa de mudança da concentração plasmática do fármaco é dada por: dCp/dt=0
Perfusão contínua (via intravenosa)
No modelo monocompartimental, os fármacos seguem uma cinética de
infusão de ordem zero e uma cinética de eliminação de ordem 1. Assim
sendo, uma alteração da quantidade de fármaco no corpo num dado
momento é igual à velocidade de infusão menos a velocidade de eliminação.

21
Integrando esta equação, e substituindo DB por CPVD vem que:
A próxima equação dá-nos a concentração plasmática do fármaco a qualquer tempo durante a infusão IV.
A eliminação segue uma cinética de ordem 1 → as concentrações do fármaco decrescem de acordo com uma
constante de eliminação de primeira ordem, sendo o declive da recta –k/2.3 (fig. 14-2)
Parâmetros derivados: t1/2= o,693 /ke ; semi-vida de eliminação
CL= Vd . ke ; clearance
Se a administração for interrompida antes do estado estacionário (steady
state/ SS) ser atingido, o declive da curva permanece igual (fig.14-2 B)
Matematicamente: todas as infusões são interrompidas antes do estado
estacionário porque teoricamente, este só é atingido após um tempo de
infusão infinito (porque a eliminação do fármaco é de ordem 1).
Na prática clínica: a concentração plasmática de um fármaco a aproximar-se do estado estacionário é
considerada a concentração plasmática do fármaco no SS.
À medida que o fármaco é infundido, t aumenta; enquanto aproxima-se de zero.
Concentração do fármaco no estado estacionário (Css) e tempo necessário para o atingir
O tempo requerido para se atingir o estado estacionário da concentração plasmática do fármaco depende da
constante de eliminação do fármaco para um volume constante de distribuição.
DB – quantidade de fármaco no corpo R – taxa de infusão (ordem zero) k – taxa de eliminação constante (ordem 1)
Ct - concentração plasmática a tempo “t” k0 – velocidade de perfusão Vd – volume de distribuição ke - constante de velocidade de eliminação t – qualquer tempo após o início da perfusão T – tempo real da perfusão (t-T) - tempo ocorrido após o término da perfusão

22
Processo de ordem zero: se a velocidade de infusão é maior que a velocidade de eliminação, a
concentração no plasma aumenta e não é atingido nenhum estado estacionário.
Nota: Isto é potencialmente perigoso devido à saturação do processo metabólico.
Processo de ordem 1: se a infusão é constante (e, portanto, a velocidade de infusão é constante),
enquanto que a velocidade de eliminação aumenta gradualmente até se atingir o estado
estacionário (CP aumenta até se atingir SS).
A concentração plasmática do fármaco no estado estacionário está relacionada com a taxa de infusão e
inversamente relacionada com a clerance (eq.5.5)
Na prática clínica, a actividade do fármaco será observada quando a concentração de fármaco está perto da
concentração plasmática desejada, que é geralmente a concentração no estado estacionário.
O tempo para atingir 90, 95 e 99% do estado estacionário pode ser calculado (tabela 14.1)
Depois de uma infusão IV de fármaco para 5 meias-vidas a concentração plasmática estará entre
95% (4,32 t1/2) e 99% (6,65 t1/2) do estado estacionário.
O tempo para o fármaco cujo t1/2 é 6 h para alcançar >95% do estado estacionário será 5 t1/2 ou
5x6h = 30h.
Um aumento na velocidade de infusão não encurta o tempo para se atingir a CSS , se
o fármaco é administrado mais rapidamente, é obtido um nível mais elevado no
estado estacionário, mas o tempo para o atingir é igual.
No estado estacionário, a velocidade de infusão equivale à velocidade de eliminação,
por isso a velocidade de alteração da concentração plasmática é igual a zero.
Exemplo 1:
Um antibiótico tem um volume de distribuição de 10 L e k=0,2h-1. É desejado um estado estacionário de 10
µg/mL. A taxa de infusão necessária para manter esta concentração pode ser determinada:
CSS depende do volume de distribuição,
constante da velocidade de eliminação e
velocidade de infusão. A alteração de
qualquer um destes factores afecta a CSS.

23
Assumindo que o doente tinha uma condição urémica e a constante de eliminação diminuiu para 0,1h-1, para
manter o estado estacionário é necessário determinar uma nova taxa de infusão:
Quando a constante de eliminação diminui, a taxa de infusão deve diminuir proporcionalmente para manter a
mesma CSS. Contudo, porque a constante de eliminação é pequena (ie, o t1/2 é longo), o tempo para alcançar
CSS será longo.
Exemplo 2:
É necessário um tempo infinito para alcançar o estado estacionário. Contudo, na prática isto é bastante
aceitável para alcançar 99% CSS (ie, 99% do estado estacionário).
Então, 99% do estado estacionário é dado por:
Substituindo na eq.5.2 por CP, podemos encontrar o tempo necessário para atingir o estado estacionário,
resolvendo em ordem a t.
Fazendo o logaritmo natural em ambos os lados:
Substituindo (o,693/t1/2) por k,
Usando cálculos similares, o tempo necessário para alcançar qualquer percentagem do estado estacionário
pode ser obtido (tabela 14.1). A infusão IV pode ser usada para determinar a clearance se a taxa de infusão e
o nível do estado estacionário forem conhecidos:
Porque a clearance total, CLT, é igual para VDk:
Exemplo 3:
Um doente está a tomar um antibiótico (t1/2 = 6h) por infusão IV constante a uma taxa de 2mg/h. Ao fim de 2
dias, a concentração de fármaco no soro era 10mg/L. Calcular a clearance total, CLT, para este antibiótico.
A clearance total pode ser estimada a partir da equação:
A amostra de soro foi obtida 2 dias (ou 48h) após infusão, o qual representa 8 t1/2. A concentração de fármaco
no soro aproxima-se de CSS.
O tempo necessário para alcançar o estado
estacionário não é dependente da taxa de infusão
mas é-o da meia vida de eliminação.

24
Método de infusão para calcular o tempo de meia vida de eliminação
A equação 5.2 pode ser usada para calcular k, ou indirectamente o tempo de meia
vida de eliminação do fármaco num doente. O conhecimento da meia vida na
população ajuda a determinar se a amostra é colhida no estado estacionário do paciente.
Como:
Substituindo a equação acima na equação 5.2:
Rearranjando e colocando o log em ambos os lados:
Exemplo 1
Um antibiótico tem um tempo de meia vida de 3 a 6 h na população em geral. Um doente recebe uma infusão
IV de um antibiótico a uma taxa de infusão de 15 mg/h. Amostras de sangue são recolhidas às 8 e 24h e as
concentrações plasmáticas eram 5,5 e 6,5 mg/h, respectivamente. Estimar o tempo de meia vida de
eliminação do fármaco neste doente.
Porque a segunda amostra é recolhida às 24h, ou 24/6=4 meias vidas depois da infusão, a concentração
plasmática do fármaco na amostra é aproximadamente 95% da verdadeira concentração plasmática no
estado estacionário, assumindo um caso extremo de t1/2=6 horas.
Substituindo na equação 5.8:
O tempo de meia vida de eliminação calculado desta forma não é tão preciso quanto ao cálculo do t1/2 usando
vários pontos do tempo de concentração do fármaco após uma dose única em bólus IV ou após a interrupção
da perfusão IV. Como a segunda amostra é recolhida mais perto do estado estacionário, a precisão deste
método melhora. Às 30h, por exemplo, a concentração plasmática deverá ser 99% do verdadeiro valor do
estado estacionário (correspondendo a 30/6 ou 5 meias vidas de eliminação), e um menor erro poderia
resultar na aplicação da equação 5.8. Quando esta equação é usada,como no exemplo acima para calcular o
t1/2 do fármaco no doente, a segunda concentração plasmática do fármaco é assumida teoricamente como
CSS.
Note que CSS é o mesmo que a concentração obtida às 24h no
exemplo acima.
Na prática, antes do início da infusão IV, uma taxa de infusão (R) apropriada é geralmente calculada a partir
da equação 5.8. São recolhidas 2 amostras de plasma e os tempos são registados. Se o tempo de meia vida de
eliminação calculado confirmar que a segunda amostra foi recolhida no estado estacionário, a concentração
plasmática é simplesmente assumida como o estado estacionário e uma nova taxa de infusão pode ser
calculada.
CP – concentração no plasma no
tempo t
CSS – concentração no estado
estacionário

25
Exemplo 2
Se a concentração plasmática terapêutica desejada é 8 mg/L para o doente do exemplo 1, qual é a taxa de
infusão adequada para o doente?
Para o exemplo 1, a taxa de infusão era 15 mg/h. Assumindo que a segunda amostra de sangue está no
estado estacionário, 6,5µg/L, a clearance do doente é:
A nova taxa de infusão pode ser:
Neste exemplo, o t1/2 deste doente é um pouco mais curto, cerca de 3h comparando com as 3 a 6h reportadas
para a população em geral. A taxa de infusão pode aumentar um pouco a fim de manter o nível de estado
estacionário desejado de 15µg/L.
Dose de carga e perfusão contínua
A dose de carga DL, ou bólus inicial de fármaco, é usado para obter rapidamente um estado estacionário. A
concentração de fármaco no corpo para o modelo monocompartimental depois de um bólus IV é descrita por:
A concentração por infusão a uma taxa R é dada por:
Assumindo que um bólus IV, DL, de fármaco é administrado e que uma infusão IV é iniciada ao mesmo tempo,
a concentração total, CP, às t horas depois do início da infusão seria igual a C1 + C2 devido à soma das
contribuições do bólus e da infusão, ou:
A dose de carga (DL) é igual à quantidade de fármaco no corpo no estado
estacionário:
Como e pela equação temos que:
Substituindo na equação 5.12 na equação 5.11 a expressão entre parênteses é
igual a zero. Logo:
Se uma dose de carga IV de R/k é dada por infusão IV, as CSS são obtidas e mantidas imediatamente.
Por diferenciação no estado estacionário:

26
Para manter o nível do SS constante instantâneo , a dose de carga deve ser
igual a R/k.
Curva b: níveis sanguíneos após dose de carga de R/k mais infusão da qual se obtém o SS.
Curva a: dose de carga é aumentada->concentração plasmática demora mais tempo a
atingir o SS.
Se a dose de carga é diminuída, a concentração plasmática vai aumentar lentamente
até ao SS, mas mais rapidamente que sem qualquer dose de carga (curva c).
Outro modo para calcular a dose de carga é baseado no conhecimento da CSS
desejada e o volume aparente de distribuição:
Para muitos fármacos, CSS é descrito na literatura como sendo a concentração
terapêutica efectiva.
EXERCÍCIOS
1- Um médico quer administrar um anestésico a uma taxa de 2 mg/h por infusão IV. A constante de
eliminação é 0,1 h-1 e o volume de distribuição (monocompartimento) é 10 L. Qual a dose a
administrar de o médico pretender alcançar uma concentração imediata de 2µg/mL?
Solução:
Para alcançar CSS imediatamente,
2- Qual é a concentração de fármaco 6 h depois da administração de uma dose de carga de 10 mg e
simultaneamente uma infusão de 2mg/h (o fármaco tem um t1/2 de 3h e um volume de distribuição
de 10L)? Solução:
3- Calcular a concentração de fármaco no sangue após a infusão ser tido parada.
Solução: Esta concentração pode ser calculada em 2 partes (ver fig. 14.2-A). Primeiro, calcular a
concentração do fármaco durante a infusão, e segundo, calcular a concentração presente, C0. Em seguida,
utiliza-se a equação do bólus IV (C=C0e-kt) para cálculos a qualquer outro ponto do tempo. Por conveniência,
as 2 equações podem ser combinadas. ( equação igual à da página 21 Ct para cada tempo)
b – extensão do tempo do período de infusão t – tempo total (infusão e pós-infusão) (t-b) – extensão do tempo depois da infusão ser parada

27
4- Um doente está com uma infusão à 6h (k=0,01h-1; Vd=10L) a uma taxa de 2mg/h. Qual é a
concentração de fármaco no corpo 2 h depois do término da infusão?
Solução:
Alternativamente, quando a
infusão pára, CP´ é calculado:
5- Um homem adulto asmático (78 kg, 48 anos) com uma história de tabagismo intenso recebe uma
infusão IV de aminofilina a uma taxa de 0,5mg/kg por h. Uma dose de carga de 6 mg/kg é
administrada num bólus IV apenas antes do início da infusão. 2 h depois do início da infusão IV, a
concentração plasmática de teofilina foi medida, sendo de 5,8 L/kg. O VD aparente para a teofilina é
0,45 L/kg. Aminofilina é o sal etilenodiamina da teofilina e contém 80% da base de teofilina.
O doente não tem respondido bem à terapia com aminofilina e, portanto, o médico aumentou a
concentração plasmática de teofilina no doente para 10µg/mL. Qual a recomendação de dosagem
que daria ao médico? Recomendaria outra carga de dose?
Solução: Se não fosse dada outra carga de dose e a taxa de infusão IV fosse aumentada, o tempo para
alcançar o estado estacionário seria cerca de 4 a 5 t1/2 para alcançar 95% de CSS. Uma segunda carga de dose
deve ser recomendada para aumentar rapidamente a concentração plasmática de teofilina para 10µg/mL. A
taxa de infusão pode também ser aumentada para manter a CSS desejada.
O cálculo da carga de dose DL pode ser considerado como a concentração plasmática de teofilina presente.
Para a aminofilina S é igual a 0,80 e para um bólus IV F é igual a 1.
A manutenção da taxa de infusão IV pode ser calculada depois da estimativa da clearance do doente, CLT.
porque uma dose de carga e uma infusão IV de 0,5 mg/h por kg pode ser dada ao doente, a concentração
plasmática de teofilina de 5,8 mg/L está no estado estacionário CSS. A clearance total pode ser estimada por:
A normal CLT para um adulto, não fumador com asma não complicada é aproximadamente 0,65 mL/min
por kg. Tabagismo intenso aumenta a CLT da teofilina.
Uma nova taxa de infusão, R´, é calculada por:
S – sal do fármaco
F – fracção de fármaco biodisponível

28
6- A um homem adulto (43 anos, 80kg) é administrada um antibiótico por infusão IV. De acordo com a
literatura, o antibiótico tem um t1/2 de 2h, VD de 1,25 L/kg, e é efectivo a uma concentração
plasmática de 14mg/L. O fármaco é fornecido em ampolas de 5 mL contendo 150 mg/mL.
a. Recomenda-se uma taxa de infusão inicial em miligramas por litros por hora.
Solução: Assume-se que a concentração plasmática efectiva é a concentração do fármaco alvo ou CSS.
Porque o fármaco é fornecido a uma concentração de 150 mg/mL,
Então,
b. Amostras de sangue são recolhidas do doente às 12, 16 e 24 h depois do início da infusão.
Para este dado adicional, calcular a clearance total CLT do fármaco no doente.
Solução: Porque as concentrações plasmáticas às 12, 16 e 24h são semelhantes,
o estado estacionário foi atingido. Nota: um aumento contínuo nas concentrações plasmáticas
pode ser causado por acumulação do fármaco para um segundo compartimento, ou por variação da quantidade de fármaco.
Assumindo um CSS de 16,3 mg/mL, CLT é calculado.
c. A partir dos dados acima, estimar a meia vida de eliminação para o antibiótico no doente.
Solução: Geralmente, o volume aparente de distribuição (VD) varia menos que o t1/2. Assumindo que o
valor da literatura para VD é 1,25 L/kg, então o t1/2 pode ser estimado a partir da CLT.
Deste modo, o t1/2 para o antibiótico no doente é 2,32 h, que está mais ou menos de acordo com o valor da
literatura de 2h.
d. Depois de rever a farmacocinética do antibiótico no doente, a taxa de infusão para o antibiótico deve
ser mudada?
Solução: Para decidir correctamente se a taxa de infusão deve ser mudada, o farmacêutico deve ter em
conta a farmacodinâmica e a toxicidade do fármaco. Assumindo que o fármaco tem uma grande janela
terapêutica e não tem grandes efeitos adversos, a taxa de infusão de 485,1 mg/h, calculada de acordo com os
valores farmacocinéticos da literatura para o fármaco, parece ser correcta.

29
Estimativa da clearance do fármaco e VD a partir de dados de infusão
A concentração plasmática de um fármaco durante uma infusão constante é descrita em termos de volume
de distribuição a constante de eliminação k na equação 5.2.
Alternativamente, a equação pode ser escrita em termos de clearance substituindo k=CL/VD
na equação:
Neste modelo, o tempo para o estado estacionário e a concentração neste é dependente da clearance e do
volume de distribuição.
Quando o volume de distribuição é constante, o tempo para o estado estacionário está inversamente
relacionado com a clearance.
5.3. Entender a aplicação do modelo farmacocinético monocompartimental para descrever a cinética da
administração extravascular (oral, rectal,…) de fármacos: curva da concentração plasmática em função do
tempo (absorção de ordem um), estimação dos parâmetros farmacocinéticos, incluindo a constante de
velocidade de absorção, e seu significado.
Farmacocinética da absorção oral do fármaco
A taxa de absorção do fármaco pode ser descrita matematicamente como
um processo de primeira ordem ou de ordem zero (maioritariamente
como 1ª ordem). A taxa de mudança da quantidade de fármaco no corpo,
DdB/dt, depende das taxas de absorção e eliminação. A taxa de acumulação do fármaco no corpo, a qualquer
tempo, é igual à taxa de absorção menos a taxa de
eliminação:
Durante a fase de absorção a sua taxa é maior que a taxa de
eliminação.
No pico de concentração no plasma, a taxa de absorção é igual à taxa de
eliminação, e não há mudança na quantidade de fármaco no corpo.
Imediatamente depois do pico de absorção, algum fármaco pode ainda estar no sítio de absorção (ie, no
tracto GI). Contudo, a taxa de eliminação é, neste tempo, maior que a taxa de
absorção.
Quando o fármaco no sítio de absorção se esgota, a taxa de absorção aproxima-se de zero, ou dDGI/dt =
0.
Durante a fase de eliminação a taxa de mudança da quantidade de fármaco no corpo é descrito como
um processo de ordem 1.
Onde k é a constante de eliminação de ordem 1.
Modelo de absorção de ordem zero

30
Neste modelo, o fármaco no TGI, DGI, é absorvido sistemicamente a uma
taxa constante, k0. O fármaco é eliminado do corpo por um processo de
ordem 1, com uma constante de ordem 1, k->A taxa de eliminação, a
qualquer tempo, é igual a DBk.
A mudança de quantidade de fármaco por unidade de tempo pode ser expressa por:
Integrando esta equação com substituição de VDCp por
DB:
O tempo em que a absorção do fármaco é contínua é igual a DGI/k0 -> depois deste tempo, o fármaco já
não está disponível para absorção no intestino, e a equação 7.7 já não faz sentido.
Modelo de absorção de ordem 1
Este modelo assume como primeira ordem o processo de absorção
através da parede intestinal e o processo de eliminação. Nota:Fármacos
administrados por injecções intramusculares aquosas também pode ser descrito como
um processo de ordem 1.
A taxa de desaparecimento do fármaco do tracto GI é descrita como:
Integrando a equação diferencial 7.8 obtém-se:
A taxa de eliminação é igual a –kDB.
A taxa de fármaco no corpo, dDB/dt, é, portanto, a taxa de fármaco dentro, menos a taxa de fármaco
fora, como demonstrado pela equação diferencial 7.10:
Desde que o fármaco no TGI também siga um processo de declínio de ordem 1, a quantidade de
fármaco no TGI é igual a D0e-kat. Nota: F pode variar de 1, para fármacos absorvido completamente, a zero, para fármacos que
não são absorvidos.
Esta equação pode ser integrada para originar uma equação geral para a absorção oral para calcular a
concentração de fármaco (Cp) no plasma a qualquer tempo t:
O tempo necessário para alcançar a Cmáx é
independente da dose e é dependente das
constantes de absorção (ka) e eliminação (k) (equação 7.13a).
Onde ka é a constante de absorção de ordem 1 a partir do
tracto GI, F é a fracção absorvida, e DGI é a quantidade de
fármaco em solução no tracto GI a qualquer tempo t.
D0 é a dose do fármaco.
F é a fracção de fármaco absorvido
sistemicamente
Ct – concentração plasmática a tempo “t” F – fracção absorvida D – dose administrada ka – constante de velocidade de absorção Vd – volume de distribuição ke – constante de velocidade de eliminação

31
A taxa de mudança de concentração na Cmáx é igual a zero → pode ser obtida
pela diferenciação da equação 7.11:
Simplificando,
O tempo para a concentração máxima, tmáx, é dependente apenas das constantes de absorção (ka) e
eliminação (k) (eq.7.13a).
Para calcular o pico da concentração plasmática, o valor para tmáx é determinado pela equação 7.13a e
depois substituindo-se na equação 7.11, resolve-se em ordem a Cmáx. A equação 7.11 mostra que a Cmáx é
directamente proporcional à dose administrada (D0) e à fracção de fármaco absorvido (F).
Intervalos de tempo mais tarde, quando a absorção do fármaco está completa , a
equação 7.11 reduz-se à expressão seguinte:
Colocando o logaritmo natural na expressão:
Substituição dos logaritmos comuns:
Com esta equação, o gráfico construído pelo traçado do log Cp versus o tempo originará uma recta com uma
inclinação de –k/2,3 (fig. 9-6A).

32
A taxa de excreção de fármaco depois de uma dose única oral de fármaco origina:
O gráfico construído pelo traçado de dDu/dt versus o tempo originará uma curva idêntica à curva nível
plasmático – tempo (fig. 9-7B)
Depois da absorção do fármaco estar virtualmente completa, aproxima-se de zero, e a
equação 7.17 reduz-se para a expressão seguinte:
Fazendo o logaritmo natural em ambos os lados da expressão e substituindo
os termos dos
logaritmos
comuns:
Quando o log (dDu/dt) é traçado em função do tempo, o gráfico resulta numa
recta com declive –k/2,3 (fig. 9-6B).
Porque a taxa de excreção urinária do fármaco, dDu/dt, não pode ser
determinada directamente, obtém-se uma média, e este valor é traçado
contra o ponto médio do período de colheita para cada amostra de urina.
Para obter a excreção cumulativa do fármaco na urina, a equação 7.17 deve
ser integrada:
O traçado de Du vs tempo origina a curva de excreção urinária do fármaco (fig. 9-8).
Quando todo o fármaco já foi excretado, a t = ∞, a equação 9.20 reduz-se a:
Determinação das constantes de absorção para dados de absorção oral
Método dos resíduos
dDu/dt = taxa de excreção urinária do fármaco, ke =
constante de ordem 1 da excreção renal, e F = fracção
de dose absorvida
Onde é a quantidade máxima de
fármaco excretado.

33
Assumindo na equação 7.11, o valor para a segunda exponencial pode tornar-se
insignificantemente pequeno com o tempo e pode ser omitido → neste caso a absorção
é virtualmente completa.
A equação 7.11 reduz-se à equação 7.22:
Para isto, pode-se também obter a intercepção com o eixo dos y (fig. 9-9).
, onde A é uma constante.
A equação 7.22 torna-se:
O declive para o traçado gráfico da equação é igual a –k/2,3.
O valor para ka pode ser obtido usando o método do resíduos ou uma técnica de revestimento. É obtido pelo
seguinte procedimento:
• Traçar o gráfico da concentração (logaritmo) do fármaco vs tempo;
• Obter o declive da fase terminal (linha BC, fig. 9-9) por extrapolação;
• Tomar quaisquer pontos da linha BC (p.e., x´1, x´2, x´3,…) e baixar
verticalmente para obter os pontos correspondentes na curva (p.e., x1, x2, x3,…);
• Ler os valores de x1 e x´1 e x2 e x´2, …. Traçar os valores das diferenças nos
pontos correspondentes ∆1, ∆2, ∆3, … obtém-se uma linha com declive de –k/2,3
(fig. 9-9).
No método dos resíduos devem-se usar, no mínimo, 3 pontos para traçar a recta.
Se a absorção do fármaco começa imediatamente após a administração oral, as
linhas residuais obtidas por “feathering” da curva nível plasmático – tempo (fig.
9-9) intersecta o eixo dos y no ponto A → este valor representa uma constante
híbrida composta por ka, k, VD e FD0 (equação 7.23).
Tempo de atraso (atraso na absorção)

34
O tempo decorrido antes do início da absorção de ordem 1 é conhecido como tempo de atraso-> observado
se duas linhas residuais obtidas por “feathering” do nível plasmático de absorção
oral-tempo se intersectarem num ponto depois de t=0 no eixo dos x -> o tempo
no ponto de intersecção no eixo dos x é o tempo de atraso (fig. 9-10).
O tempo de atraso, t0, representa o início da absorção, e não deve ser
confundido com o tempo de latência, que representa o tempo necessário para o
fármaco alcançar o efeito terapêutico mínimo.
A curva pode ser descrita por duas equações. Numa o t0 é subtraído a cada ponto
do tempo:
Onde é o valor de y no ponto de intersecção das linhas
residuais na fig. 9-10.
A segunda equação descreve a curva omitindo o tempo de
retardação:
Onde A e B representam os pontos de intercepção no eixo dos y depois da extrapolação das linhas residuais
para a absorção e eliminação, respectivamente.
Flip-flop de ka e k
Usando o método dos resíduos para obter uma estimativa de ka e k, a fase terminal da curva de absorção oral
é geralmente representada pela constante de eliminação (k) já o declive é representado pela constante de
absorção (ka) (fig. 9-11). Em poucos casos, k obtido pelos dados da absorção oral não estão de acordo com os
obtidos após a injecção de um bólus IV.
Por exemplo: o k obtido depois de uma injecção de um bólus IV de um broncodilatador era 1,72 h-1;
considerando que o k calculado depois da administração oral era 0,7 h-1 (fig. 9-11). Quando ka é obtido pelo
método dos resíduos, resulta, surpreendentemente, num ka igual a 1,72 h-1.
Aparentemente, o ka e o k obtidos pelo método dos resíduos têm sido trocados. Este fenómeno é chamado de
flip-flop ou reversão das constantes ka e k e pode ocorrer sempre que ka e k são estimados a partir de dados
de absorção oral. Depois de uma injecção IV o declive nos níveis plasmáticos de fármaco ao longo do tempo

35
representa a verdadeira constante de eliminação. Nota: A maioria dos fármacos que têm características flip-flop são
fármacos com rápida eliminação (ie, k>ka). Os fármacos usados oralmente com longas meias vidas de eliminação, podem usar a fase
terminal da curva na fig. 9-11 como constante de eliminação.
Para fármacos com K elevado (k>0,69h-1), a mudança para flip-flop de ka e k é muito maior-> são considerados
inadequados como fármaco oral, devido à sua grande constante de eliminação, correspondendo a um
pequeno t1/2 de eliminação.
Fármacos de libertação prolongada podem retardar a absorção, tal que o ka seja mais pequeno que o k,
produzindo uma situação flip-flop.
Determinação de ka pela percentagem de fármaco não absorvido versus o (método de Wagner-Nelson)
Após uma única dose oral de um fármaco, o total deve ser contabilizado a partir da quantidade presente no
corpo, a quantidade presente na urina e a quantidade presente no TGI. A dose (D0) é expressa da seguinte
forma:
Seja Ab = DB + Du = à quantidade de fármaco absorvida e Ab ∞ = à quantidade de fármaco absorvida quando t
=∞. Para qualquer tempo a fracção de fármaco absorvida seria Ab / Ab∞, e da fracção não absorvida seria 1 -
(Ab / Ab∞ ).
A quantidade de fármaco excretada em qualquer tempo t pode ser calculado através:
A quantidade de fármaco no corpo (DB), em qualquer momento = CpVD. A qualquer tempo t, a quantidade de
fármaco absorvida (Ab) é:
Em t =∞ , Cp ∞= 0 (i.e, a concentração plasmática é insignificante) e a quantidade total de fármaco absorvida
é:
A fracção de fármaco absorvida em qualquer momento é:
A fracção de fármaco não absorvida em qualquer
momento t é:
Porque o fármaco restante no trato gastrointestinal em qualquer momento t é:
Portanto, a fracção do fármaco restante é:
Porque DGI/D0 é a fracção de fármaco não absorvida, ou seja, 1 - (Ab / Ab ∞) - gráfico de 1 - (Ab / Ab ∞)
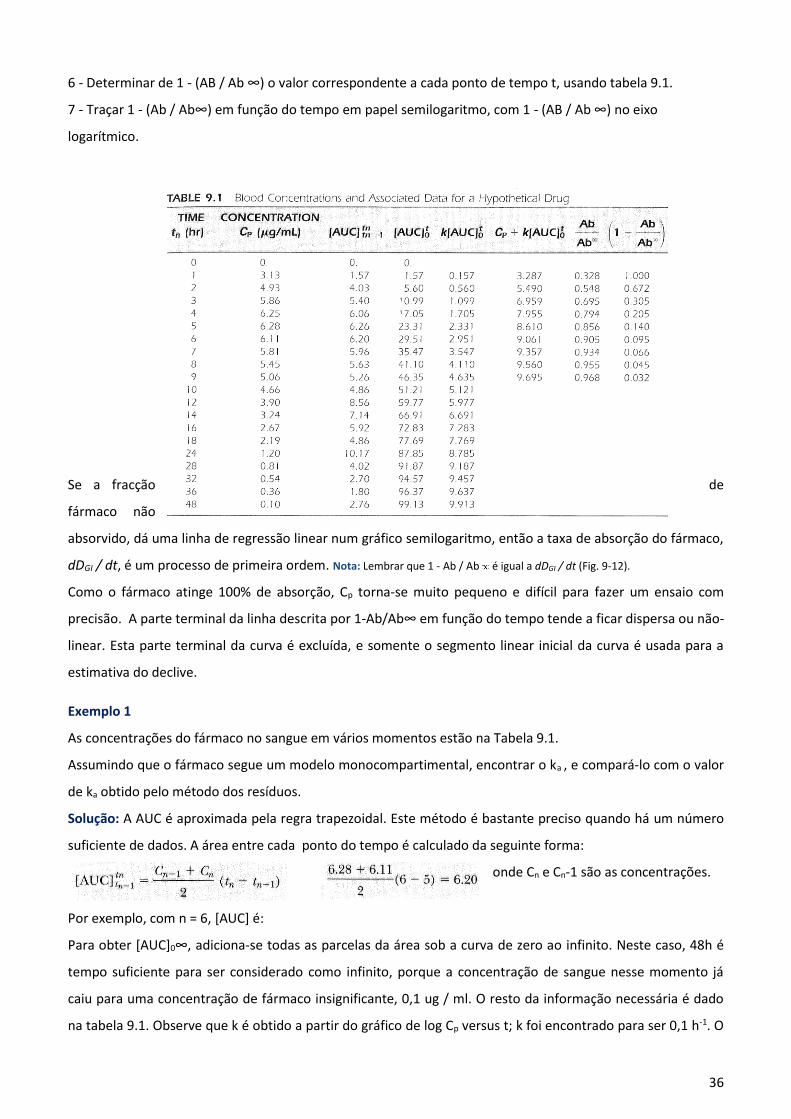
versus tempo dá - ka / 2,3 como o declínio (fig. 9-12).
Os passos seguintes devem ser úteis na determinação de Ka:
1-Traçar log da concentração do fármaco versus o tempo.
2-Encontrar k da parte terminal de declive, quando a inclinação = - k/2.3
3 - Encontrar [AUC]t0 para traçar Cp versus t.
4 - Encontrar k [AUC]t0 pela multiplicação de cada [AUC] por k.
5 -Encontrar [AUC]0∞ pela soma de todas as partes de [AUC] a partir de
t = 0 para t = ∞.
36
6 - Determinar de 1 - (AB / Ab ∞) o valor correspondente a cada ponto de tempo t, usando tabela 9.1.
7 - Traçar 1 - (Ab / Ab∞) em função do tempo em papel semilogaritmo, com 1 - (AB / Ab ∞) no eixo
logarítmico.
Se a fracção de
fármaco não
absorvido, dá uma linha de regressão linear num gráfico semilogaritmo, então a taxa de absorção do fármaco,
dDGI / dt, é um processo de primeira ordem. Nota: Lembrar que 1 - Ab / Ab é igual a dDGI / dt (Fig. 9-12).
Como o fármaco atinge 100% de absorção, Cp torna-se muito pequeno e difícil para fazer um ensaio com
precisão. A parte terminal da linha descrita por 1-Ab/Ab∞ em função do tempo tende a ficar dispersa ou não-
linear. Esta parte terminal da curva é excluída, e somente o segmento linear inicial da curva é usada para a
estimativa do declive.
Exemplo 1
As concentrações do fármaco no sangue em vários momentos estão na Tabela 9.1.
Assumindo que o fármaco segue um modelo monocompartimental, encontrar o ka , e compará-lo com o valor
de ka obtido pelo método dos resíduos.
Solução: A AUC é aproximada pela regra trapezoidal. Este método é bastante preciso quando há um número
suficiente de dados. A área entre cada ponto do tempo é calculado da seguinte forma:
onde Cn e Cn-1 são as concentrações.
Por exemplo, com n = 6, [AUC] é:
Para obter [AUC]0∞, adiciona-se todas as parcelas da área sob a curva de zero ao infinito. Neste caso, 48h é
tempo suficiente para ser considerado como infinito, porque a concentração de sangue nesse momento já
caiu para uma concentração de fármaco insignificante, 0,1 ug / ml. O resto da informação necessária é dado
na tabela 9.1. Observe que k é obtido a partir do gráfico de log Cp versus t; k foi encontrado para ser 0,1 h-1. O

37
gráfico de 1-(AB/Ab∞) vs t no papel semilogaritmo é mostrado na Figura 9-12.
Um método mais completo de obter [AUC]0∞ é estimar a área residual da última concentração de plasma,
Cpn, no tn de tempo igual ao infinito.
Esta equação é:
O total [AUC]0∞ é a soma das áreas obtidas pela regra do trapézio, [AUC]t0, [AUC]t
0 e a área residual [AUC]t∞,
conforme descrito na seguinte expressão:
Estimativa do ka a partir dos dados urinários
A constante de absorção também pode ser estimada a partir dos dados de excreção
urinária, através de uma percentagem de fármaco não absorvido em função do tempo. Para um modelo
monocompartimental:
De = quantidade total de fármaco excretado (fármaco e metabolitos).
O diferencial da equação de 9.38 em relação ao tempo dá:
Assumindo uma cinética de primeira ordem de eliminação com a eliminação renal constante, ke,
Assumindo um modelo monocompartimental:
Substituindo VDCp na equação 9.39,
O rearranjo da equação 7.40:
Substituindo por dCp/ dt na equação 7.41 e kDu / ke por De
Quando a expressão acima é integrada a partir de zero ao
tempo t,
A t = , todo o fármaco que é finalmente absorvido é expresso como Ab∞, e dDu/ dt = 0. A quantidade
total de fármaco absorvido é a seguinte:
Onde Du∞ é a quantidade total de fármaco excretado na urina na forma inalterada.
A fracção de fármaco absorvido em qualquer tempo t é igual à quantidade de fármaco absorvido no tempo,
(Ab)t, dividido pela quantidade total de fármaco absorvido, Ab .
O gráfico da fracção do fármaco não absorvido [1 - (Ab / Ab ∞)] em função do tempo dá -ka / 2,3 como declive
(eq.7.34).
Ab= DB + DE (7.38)
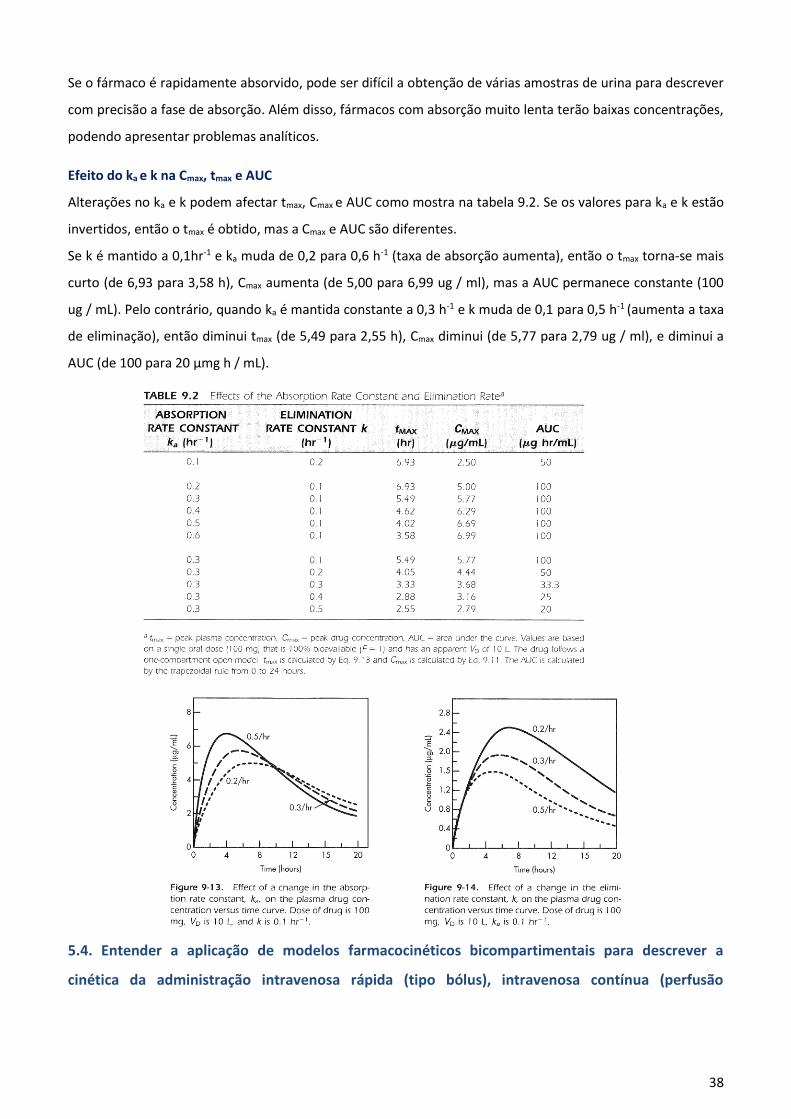
38
Se o fármaco é rapidamente absorvido, pode ser difícil a obtenção de várias amostras de urina para descrever
com precisão a fase de absorção. Além disso, fármacos com absorção muito lenta terão baixas concentrações,
podendo apresentar problemas analíticos.
Efeito do ka e k na Cmax, tmax e AUC
Alterações no ka e k podem afectar tmax, Cmax e AUC como mostra na tabela 9.2. Se os valores para ka e k estão
invertidos, então o tmax é obtido, mas a Cmax e AUC são diferentes.
Se k é mantido a 0,1hr-1 e ka muda de 0,2 para 0,6 h-1 (taxa de absorção aumenta), então o tmax torna-se mais
curto (de 6,93 para 3,58 h), Cmax aumenta (de 5,00 para 6,99 ug / ml), mas a AUC permanece constante (100
ug / mL). Pelo contrário, quando ka é mantida constante a 0,3 h-1 e k muda de 0,1 para 0,5 h-1 (aumenta a taxa
de eliminação), então diminui tmax (de 5,49 para 2,55 h), Cmax diminui (de 5,77 para 2,79 ug / ml), e diminui a
AUC (de 100 para 20 µmg h / mL).
5.4. Entender a aplicação de modelos farmacocinéticos bicompartimentais para descrever a
cinética da administração intravenosa rápida (tipo bólus), intravenosa contínua (perfusão
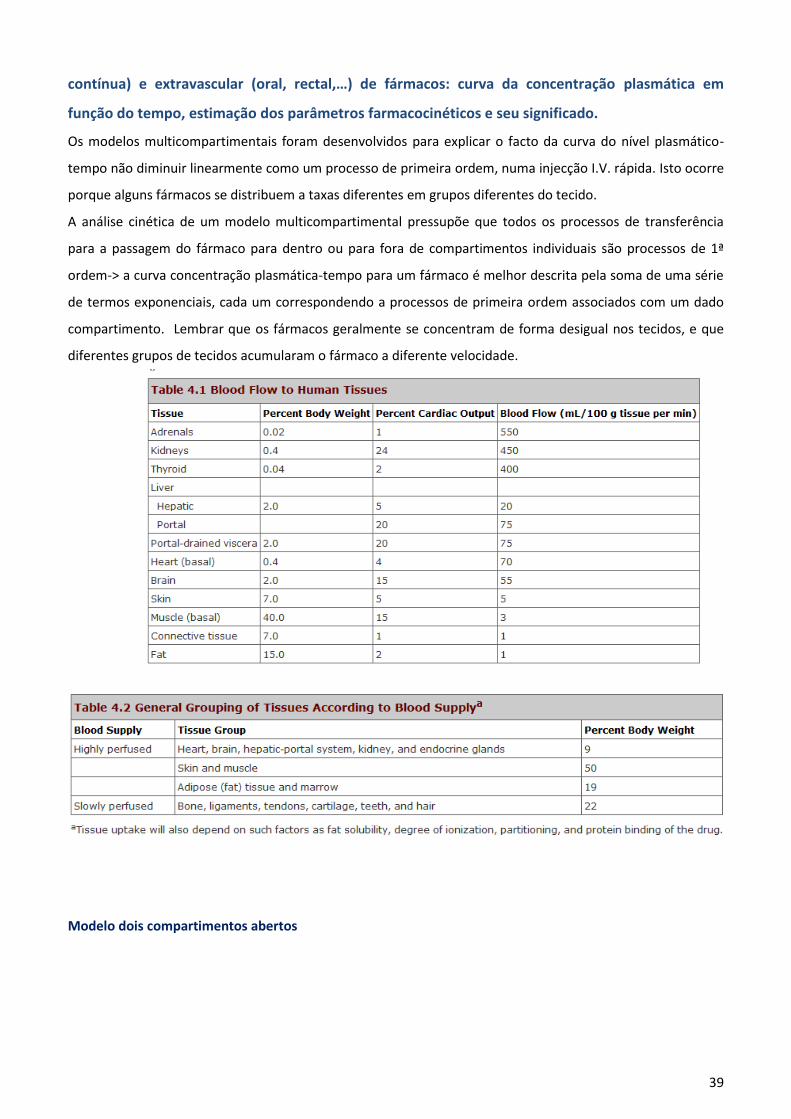
39
contínua) e extravascular (oral, rectal,…) de fármacos: curva da concentração plasmática em
função do tempo, estimação dos parâmetros farmacocinéticos e seu significado.
Os modelos multicompartimentais foram desenvolvidos para explicar o facto da curva do nível plasmático-
tempo não diminuir linearmente como um processo de primeira ordem, numa injecção I.V. rápida. Isto ocorre
porque alguns fármacos se distribuem a taxas diferentes em grupos diferentes do tecido.
A análise cinética de um modelo multicompartimental pressupõe que todos os processos de transferência
para a passagem do fármaco para dentro ou para fora de compartimentos individuais são processos de 1ª
ordem-> a curva concentração plasmática-tempo para um fármaco é melhor descrita pela soma de uma série
de termos exponenciais, cada um correspondendo a processos de primeira ordem associados com um dado
compartimento. Lembrar que os fármacos geralmente se concentram de forma desigual nos tecidos, e que
diferentes grupos de tecidos acumularam o fármaco a diferente velocidade.
Modelo dois compartimentos abertos
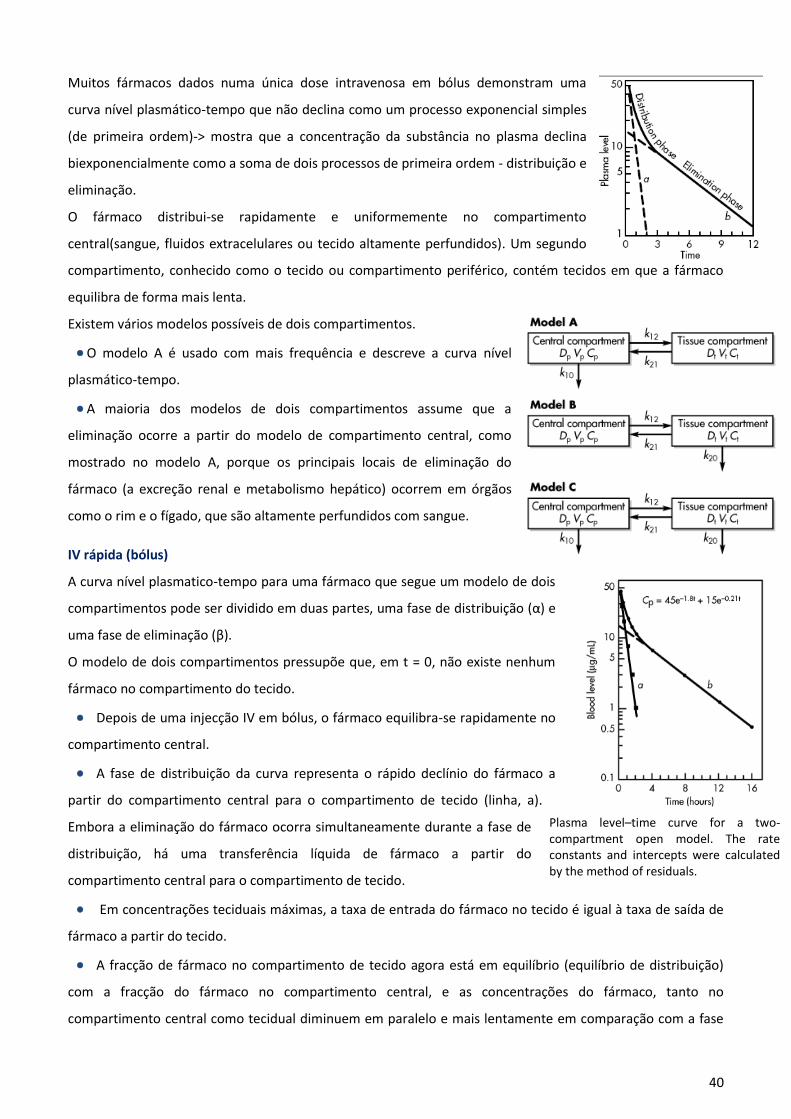
40
Muitos fármacos dados numa única dose intravenosa em bólus demonstram uma
curva nível plasmático-tempo que não declina como um processo exponencial simples
(de primeira ordem)-> mostra que a concentração da substância no plasma declina
biexponencialmente como a soma de dois processos de primeira ordem - distribuição e
eliminação.
O fármaco distribui-se rapidamente e uniformemente no compartimento
central(sangue, fluidos extracelulares ou tecido altamente perfundidos). Um segundo
compartimento, conhecido como o tecido ou compartimento periférico, contém tecidos em que a fármaco
equilibra de forma mais lenta.
Existem vários modelos possíveis de dois compartimentos.
O modelo A é usado com mais frequência e descreve a curva nível
plasmático-tempo.
A maioria dos modelos de dois compartimentos assume que a
eliminação ocorre a partir do modelo de compartimento central, como
mostrado no modelo A, porque os principais locais de eliminação do
fármaco (a excreção renal e metabolismo hepático) ocorrem em órgãos
como o rim e o fígado, que são altamente perfundidos com sangue.
IV rápida (bólus)
A curva nível plasmatico-tempo para uma fármaco que segue um modelo de dois
compartimentos pode ser dividido em duas partes, uma fase de distribuição (α) e
uma fase de eliminação (β).
O modelo de dois compartimentos pressupõe que, em t = 0, não existe nenhum
fármaco no compartimento do tecido.
Depois de uma injecção IV em bólus, o fármaco equilibra-se rapidamente no
compartimento central.
A fase de distribuição da curva representa o rápido declínio do fármaco a
partir do compartimento central para o compartimento de tecido (linha, a).
Embora a eliminação do fármaco ocorra simultaneamente durante a fase de
distribuição, há uma transferência líquida de fármaco a partir do
compartimento central para o compartimento de tecido.
Em concentrações teciduais máximas, a taxa de entrada do fármaco no tecido é igual à taxa de saída de
fármaco a partir do tecido.
A fracção de fármaco no compartimento de tecido agora está em equilíbrio (equilíbrio de distribuição)
com a fracção do fármaco no compartimento central, e as concentrações do fármaco, tanto no
compartimento central como tecidual diminuem em paralelo e mais lentamente em comparação com a fase
Plasma level–time curve for a two-compartment open model. The rate constants and intercepts were calculated by the method of residuals.

41
de distribuição. Este declínio é um processo de primeira ordem e é chamado a fase de eliminação ou β (linha
b). As concentrações da substância no plasma fornecem alguma indicação sobre a concentração do fármaco
no tecido.
Uma típica curva de nível de fármaco nos tecidos após uma única dose
intravenosa é apresentada no gráfico ao lado. As concentrações do fármaco no
tecido são apenas teóricas.
As diferenças na concentração do fármaco no tecido é reflectido na fracção K12 /
K21. A eliminação do fármaco do compartimento do tecido pode não ser o
mesmo que a eliminação do compartimento central. Por exemplo, se K12.Cp é
maior do que K21. Ct (taxa in> taxa out), as concentrações teciduais do fármaco
irão aumentar e as concentrações plasmáticas de fármacos vão diminuir.
A curva do nível plasmático-tempo dos fármacos representa uma fase de equilíbrio inicial rápido com o
compartimento central (a fase de distribuição), seguido por uma fase de eliminação após o compartimento de
tecido também estar em equilíbrio com o fármaco.
No modelo descrito acima, K12 e K21 são constantes de velocidade de primeira ordem que regem a taxa de
variação de fármaco dentro e fora dos tecidos:
A relação entre a quantidade de fármaco em cada compartimento e a concentração do fármaco no
compartimento é mostrado pelas equações 4.2 e 4.3:
Resolvendo as equações 4.4 e 4.5 dará as equações 4.6 e 4.7, que descrevem a variação da concentração
do fármaco no sangue e nos tecidos em relação ao tempo.
Dp = quantidade de fármaco no
compartimento central, Dt = quantidade
de fármaco no compartimento de tecido,
Vp = volume de fármaco no
compartimento central, e Vt = volume
de fármaco no compartimento de tecido.
D 0 p= dose administrada por via
intravenosa, t = tempo após a
administração da dose, e a e b são
constantes que dependem
exclusivamente de K ..12, K 21, e K.

42
A quantidade de fármaco remanescente no plasma e compartimento de tecido, a qualquer momento pode
ser descrita de forma realista pelas equações 4.8 e 4.9.
As microconstantes não podem ser determinados por medição directa, mas podem ser estimados por um
método gráfico. As derivadas após a integração das eq. 4.4 e 4.5:
A equação 4.6 pode ser transformada na seguinte expressão:
As constantes A e B são interceptadas no eixo y para cada segmento da curva exponencial na Equação 4.12.
Estes valores podem ser obtidos graficamente pelo método de resíduos ou por computador. Intercepções A e
B são actualmente constantes híbridos, como mostrado nas equações 4.13 e 4.14, e não tem significado
fisiológico real.
Método dos resíduos
O método dos resíduos (também conhecidas como franjas ou peeling) é um procedimento útil para ajustar
uma curva de dados experimentais de um farmaco, o que demonstra a necessidade de um modelo
multicompartimental.
Por exemplo, 100 mg de um fármaco foi administrado por injecção IV
rápida a um adulto homem saudável de 70kg. Amostras de sangue foram
recolhidas periodicamente após a administração do fármaco, e a fração de
plasma de cada amostra foi analisada para a fármaco. Os dados obtidos
estão na tabela ao lado.
Quando estes dados são plotados em papel de gráfico semi-logarítmico,
observa-se uma linha curva.A relação da linha curva entre o logaritmo da
concentração plasmática e do tempo indica que o fármaco é distribuído
em mais de que um compartimento.
A fase de distribuição rápida é confirmada com a constante a ser maior do que a taxa constante b. Portanto,
algum tempo depois, o termo Ae– at se aproximará de zero,
enquanto Be–bt continua a ter um valor. Neste tempo, a equação
4.12 reduz-se a:
que, em logaritmos comum, é:
Da equação 4.16, a constante de velocidade pode ser obtida a partir do declive (-b/2.3) de uma linha reta que
representa a fase terminal exponencial.
O t 1/2 para a fase de eliminação pode ser derivada da seguinte relação:
No caso da amostra aqui considerada, b mostrou ser 0,21 h-1.
43
A partir desta informação a linha de regressão exponencial para a fase terminal ou b é extrapolada para o eixo
y; o intercepto y é igual a B, ou 15 g / mL. Valores da linha extrapolada são então subtraídos aos pontos
originais dos dados experimentais e uma linha recta é obtida. A nova linha obtida graficamente do logaritmo
da concentração plasmática residual (Cp – C'p) em função do tempo representa a fase a.
O valor de a é de 1,8 h-1, e o valor da intercepção de y é 45 µg/mL. A eliminação t1/2b é calculado a partir de B
pelo uso da equação 4.17 e tem o valor de 3,3 hr.
Um certo número de parâmetros farmacocinéticos podem ser derivados por substituição adequada das
constantes de velocidade a e b e y intercepta A e B nas equações seguintes:
Simulação do nível plasmático e tecidual de um modelo de dois compartimentos do fármaco digoxina em
pacientes normais e com falha renal
Uma vez que os parâmetros farmacocinéticos são
determinados para um indivíduo, a quantidade de
fármaco remanescente no plasma e
compartimento de tecido pode ser calculado
utilizando as equações 4.8 e 4.9.
Os dados farmacocinéticos da digoxina foram
calculados em sujeitos normais e com deficiência
renal, com 70-kg utilizando os parâmetros da
tabela 4.4 conforme relatado na literatura.
O montante remanescente de digoxina no plasma e compartimento de tecido estão tabelados e plotados
abaixo.
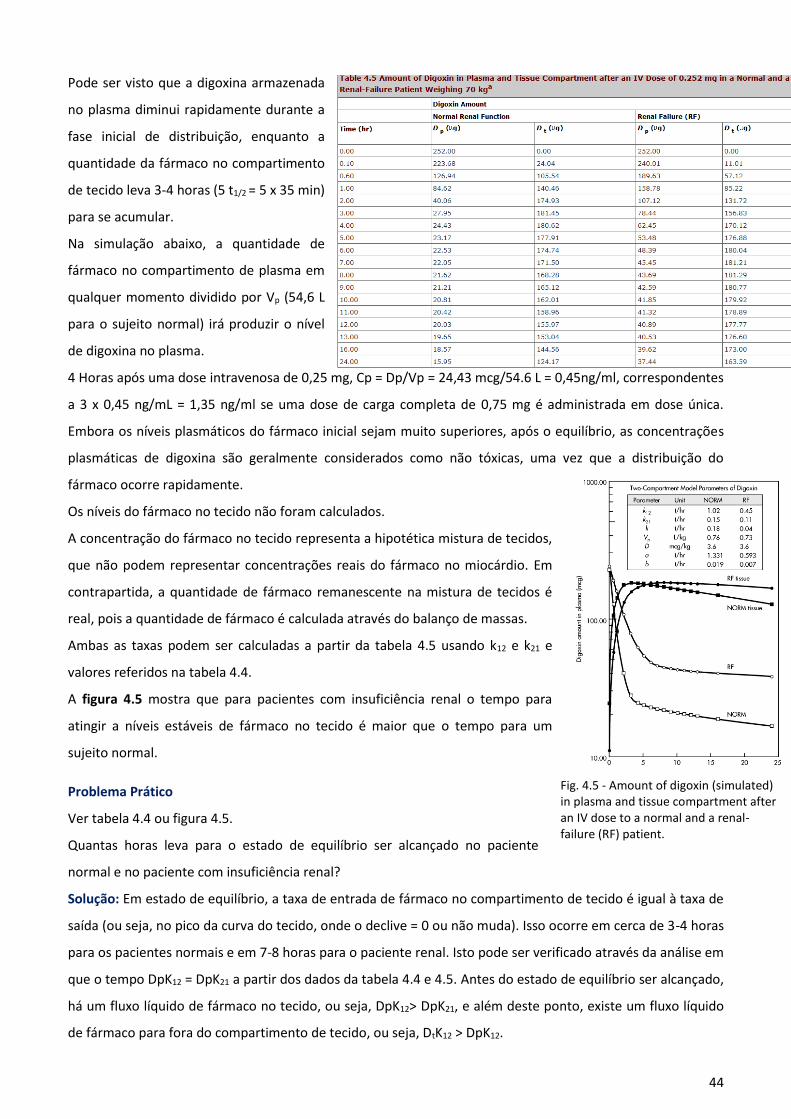
44
Pode ser visto que a digoxina armazenada
no plasma diminui rapidamente durante a
fase inicial de distribuição, enquanto a
quantidade da fármaco no compartimento
de tecido leva 3-4 horas (5 t1/2 = 5 x 35 min)
para se acumular.
Na simulação abaixo, a quantidade de
fármaco no compartimento de plasma em
qualquer momento dividido por Vp (54,6 L
para o sujeito normal) irá produzir o nível
de digoxina no plasma.
4 Horas após uma dose intravenosa de 0,25 mg, Cp = Dp/Vp = 24,43 mcg/54.6 L = 0,45ng/ml, correspondentes
a 3 x 0,45 ng/mL = 1,35 ng/ml se uma dose de carga completa de 0,75 mg é administrada em dose única.
Embora os níveis plasmáticos do fármaco inicial sejam muito superiores, após o equilíbrio, as concentrações
plasmáticas de digoxina são geralmente considerados como não tóxicas, uma vez que a distribuição do
fármaco ocorre rapidamente.
Os níveis do fármaco no tecido não foram calculados.
A concentração do fármaco no tecido representa a hipotética mistura de tecidos,
que não podem representar concentrações reais do fármaco no miocárdio. Em
contrapartida, a quantidade de fármaco remanescente na mistura de tecidos é
real, pois a quantidade de fármaco é calculada através do balanço de massas.
Ambas as taxas podem ser calculadas a partir da tabela 4.5 usando k12 e k21 e
valores referidos na tabela 4.4.
A figura 4.5 mostra que para pacientes com insuficiência renal o tempo para
atingir a níveis estáveis de fármaco no tecido é maior que o tempo para um
sujeito normal.
Problema Prático
Ver tabela 4.4 ou figura 4.5.
Quantas horas leva para o estado de equilíbrio ser alcançado no paciente
normal e no paciente com insuficiência renal?
Solução: Em estado de equilíbrio, a taxa de entrada de fármaco no compartimento de tecido é igual à taxa de
saída (ou seja, no pico da curva do tecido, onde o declive = 0 ou não muda). Isso ocorre em cerca de 3-4 horas
para os pacientes normais e em 7-8 horas para o paciente renal. Isto pode ser verificado através da análise em
que o tempo DpK12 = DpK21 a partir dos dados da tabela 4.4 e 4.5. Antes do estado de equilíbrio ser alcançado,
há um fluxo líquido de fármaco no tecido, ou seja, DpK12> DpK21, e além deste ponto, existe um fluxo líquido
de fármaco para fora do compartimento de tecido, ou seja, DtK12 > DpK12.
Fig. 4.5 - Amount of digoxin (simulated) in plasma and tissue compartment after an IV dose to a normal and a renal-failure (RF) patient.

45
Volume do compartimento central
O volume do compartimento central (geralmente > que 3L) é útil para determinar a concentração de fármaco
logo após uma injecção intravenosa no corpo assim como a CL. Também conhecido como Vi (volume inicial de
distribuição).
Para muitos fármacos polares, um volume inicial de 7-10 L pode ser interpretada como uma rápida
distribuição de fármacos no plasma e outros fluidos extracelulares. Por exemplo, o Vp de intervalos de
moxalactam entre 0,14-0,15 L/kg, corresponde a cerca de 9,8-10,5L para um típico paciente com 70 kg. Em
contraste, o Vp hidromorfona é de cerca de 24 L, possivelmente por causa da sua rápida saída do plasma para
os tecidos, mesmo durante a fase inicial.
No tempo zero, nenhum fármaco é eliminado, D0 =Vp.Cp. O pressuposto básico do modelo é: a concentração
do fármaco no plasma é representativa da concentração do fármaco dentro do fluido de distribuição. Se esta
afirmação é verdadeira, então o volume de distribuição será de 3L e, se ele não for, então a distribuição de
fármaco pode ocorrer também fora da rede vascular.
No tempo zero (t = 0), todo o fármaco está no compartimento central. C0p pode ser igual a A+B pela seguinte
equação.
Em t = 0, e0 = 1. Portanto,
Vp é determinado pela equação 4.24 medindo A e B, após a difusão da curva, como discutido anteriormente:
Alternativamente, o volume do compartimento central pode ser calculada a partir da [AUC]∞0 de um modo
semelhante ao cálculo do VD aparente no modelo de um compartimento.
Para o modelo de um compartimento,
Em contraste, [AUC]∞0, para o modelo de dois compartimentos é:
Rearranjo da equação:
Volume aparente de distribuição no estado de equilíbrio
Estas taxas de transferência de fármacos são descritas pelas seguintes expressões:
Como a quantidade de fármaco no compartimento central, Dp, é igual a VpCp, por substituição na equação
acima,
A quantidade total de fármaco no corpo em estado de equilíbrio é igual à soma da quantidade de fármaco no
compartimento de tecido, Dt, e à quantidade de fármaco no compartimento central, Dp.

46
Portanto, o volume aparente de fármacos no estado de equilíbrio (VD )SS pode ser calculado dividindo a
quantidade total de fármaco no corpo pela concentração de fármaco no compartimento central no estado de
equilíbrio:
Pela substituição da equação 4.30 na equação 4.31, e expressando Dp como VpCp, uma equação mais útil
para o cálculo (VD)ss é obtida:
o que reduz a:
Volume extrapolado de distribuição
O volume extrapolado de distribuição (VD)exp é calculado pela seguinte equação:
Porque o intercepto y é um híbrido constante, como mostrado pela equação 4.14, (VD)exp também pode ser
calculado pela seguinte expressão:
Esta equação mostra que uma mudança na distribuição de um fármaco, que é observada por uma mudança
no valor de Vp, será reflectida numa mudança do (VD)exp.
Volume de Distribuição por Área
O (VD) área, também conhecido como (VD)β, é obtido através de cálculos similares aos utilizados para
encontrar Vp, excepto no facto da taxa constante b ser usada em vez da Ktotal.
(VD)β é frequentemente calculado a partir da clearance corporal total dividida por b e é influenciada pela
eliminação da fármaco na fase β. A redução da clearance de fármacos do organismo pode aumentar a AUC,
tal que (VD)β é reduzido ou sem alterações, dependendo do valor de b, como mostrado pela equação 4.36.
Geralmente, a redução da clearance do fármaco é acompanhada por uma diminuição da constante b (ou seja,
um aumento da meia-vida de eliminação de b). Porque a clearance corporal total é igual a D0/[AUC]∞0, (VD)β
pode ser expresso em termos de clearance e da taxa constante b:
Pela substituição de kVp para a clearance na equação 4.37, obtém-se:
Teoricamente, o valor de b pode permanecer inalterado em pacientes que mostram diversos graus de
insuficiência renal moderada. Neste caso, uma redução de (VD)β pode ser responsável por todas as
diminuições da Cl, enquanto b é inalterada na equação 4.37. Dentro do corpo, a redistribuição de fármacos
entre o plasma e o tecido vai mascarar o declínio esperado em b.
B é o intercepto y obtido por
extrapolação da fase b da curva de
nível plasmático para o eixo y.

47
O exemplo a seguir de dois pacientes mostra que a taxa de eliminação constante b permanece a mesma,
enquanto a constante de velocidade de distribuição muda. Curiosamente, Vp é inalterado, enquanto o (VD)β
que seria elevado mudou no exemplo simulado.
Problema Prático
Simulando a concentração do fármaco no plasma após uma dose IV em bolus
(100mg) de um antibiótico em dois pacientes – paciente 1 com um k normal, e
o paciente 2, com um K reduzido, como é mostrado na figura. Os dados nos
dois pacientes foram simulados com parâmetros usando a equação do modelo
de dois compartimentos. Os parâmetros utilizados são os seguintes:
- Paciente 1: k = 0.3 hr– 1, V p = 10 L, Cl = 3 L/hr; k 12 = 5 hr– 1, k 21 = 0.2 hr– 1
- Paciente 2: k = 0.1 hr– 1, V p = 10 L, Cl = 1 L/hr ; k 12 = 2 hr– 1, k 21 = 0.25 hr– 1
1. Será uma redução na Cl do fármaco, geralmente acompanhada por aumento na sua [] plasmática,
independentemente do modelo compartimental seguido pelo fármaco?
2. Será uma redução na Cl do fármaco geralmente acompanhada por um aumento do seu t1/2 de eliminação
b? [Localizar (VD)β usando a eq. 4.37, e depois b usando eq. 4.38.]
3. Muitos antibióticos seguem perfis multiexponenciais de concentração plasmática, mostrando a distribuição
do fármaco em compartimentos teciduais. Na farmacocinética clínica, a meia-vida terminal é frequentemente
determinada com dados recentes limitados. Com base nos dados da simulação, qual o doente que tem maior
meia-vida terminal?
Solução: 1. A concentração plasmática do fármaco deve ser maior em indivíduos com reduzida clearance em
comparação com indivíduos com uma clearance normal, independentemente de qual modelo de
compartimento é usado.
2. A Cl no modelo de dois compartimentos é afectada pela taxa constante de eliminação, b, e pelo volume de
distribuição na fase β, o que se reflecte nos dados. Uma diminuição no (VD)β com b inalterado é possível,
embora este não seja o caso comum. Quando isto acontece, os dados do pico concluem que a semi-vida de
eliminação dos pacientes 1 e 2 é a mesma, devido ao b similar. Na verdade, a semi-vida real de eliminação do
fármaco deriva de k que é um parâmetro muito melhor, desde que k reflicta a evolução da função renal, mas
não b, que permanece inalterado uma vez que é mascarado pelas mudanças do (VD)β.
3. Ambos os pacientes têm o mesmo valor de b (b = 0,011 h-1); os picos do declive são idênticos. Ignorando os
pontos de início e tendo em conta apenas dados terminais, levaria a uma conclusão errónea de que o
processo de eliminação renal é inalterada, enquanto o volume de distribuição do paciente com insuficiência
renal é menor. Neste caso, o paciente com insuficiência renal tem uma clearance de 1 L/h em comparação
com 3L/h para o sujeito normal. A rápida distribuição do fármaco no tecido do sujeito normal provoca uma
fase de distribuição mais íngreme e longa. Mais tarde, a redistribuição de fármaco para fora dos tecidos

48
mascara o efeito da rápida eliminação do fármaco através dos rins. No paciente com insuficiência renal, a
distribuição do tecido é reduzida, como resultado, pouco fármaco é redistribuído para fora do tecido na fase
β. Assim, as fases β aparentam ser idênticas nos dois pacientes.
Significado dos volumes de distribuição
A magnitude dos vários volumes de distribuição aparente tem as seguintes relações entre
si:
(VD) ss é muito maior do que Vp; aproxima-se de (VD)β, mas difere um pouco em termos de valor, em função
das constantes de transferência. Um aumento da AUC de um cardiotónico num paciente CHF deve-se a uma
redução na clearance de fármacos, uma vez que o volume de distribuição não foi alterado. A meia-vida de
eliminação foi reduzida devido à redução da clearance do fármaco.
O fluxo sanguíneo é um parâmetro independente que irá afectar tanto a clearance como a distribuição.
Para os fármacos que seguem uma cinética de modelo de dois compartimentos, as mudanças em estados da
doença não podem resultar em diferentes parâmetros farmacocinéticos.
Fármaco no compartimento de tecido
O volume aparente do compartimento de tecido (Vt) é um volume apenas conceitual e não representa os
verdadeiros volumes anatómicos. O Vt pode ser calculado a partir do conhecimento das constantes de taxa de
transferência e Vp:
O cálculo da quantidade de fármaco no compartimento de tecido não implica o uso de Vt e fornece uma
estimativa para a acumulação do fármaco nos tecidos do corpo-> informação vital para estimar a toxicidade
crónica e relatar a duração da actividade farmacológica da dose.
Juntamente com VP e Cp, que calcula a quantidade de fármaco no plasma, o modelo de compartimento
fornece informações de balanço de massa. Além disso, a actividade farmacodinâmica pode-se relacionar
melhor com a curva concentração do fármaco no tecido-tempo.
Para calcular a quantidade de fármaco no compartimento tecidual,
Dt:
Problema práctico: A concentração plasmática terapêutica da digoxina é entre 1 e 2 ng / mL e como tem uma
semi-vida longa leva muito tempo para chegar a um nível estável. A dose de carga geralmente é dada no início
da terapêutica com digoxina. Considere as implicações da dose de 1 mg sugerido para um sujeito com 70-kg.
A fonte clínica cita que um volume de distribuição aparente para a digoxina é de 7,3 l / kg. Use os parâmetros
farmacocinéticos da digoxina da tabela 4.4.
Solução: A dose de carga foi calculada considerando-se o corpo como um
compartimento durante o estado de equilíbrio, momento em que o fármaco
também penetra no compartimento de tecido. O volume de distribuição

49
(VD)β da digoxina é muito maior do que Vp, ou o volume do compartimento de plasma.
Clearance do fármaco
Clearance é frequentemente calculada por uma abordagem monocompartimental,
como na equação 4.41, em que a dose IV em bólus é dividida pela área abaixo da curva
de concentração plasmática-tempo de zero ao infinito, [AUC] ∞ 0.
Equação 4.41 pode ser rearranjada à equação 4.42 para mostrar que a CL no modelo de dois compartimentos,
é o produto de (VD)β e b.
Se ambos os parâmetros são conhecidos, então a determinação da clearance é simples e mais precisa do que
utilizando a regra do trapézio para obter a área.
Eliminação Constante
Por causa da redistribuição de fármaco para fora do compartimento de tecido, a curva do nível plasmático
diminui mais lentamente na fase b. Assim b é menor do que k que é uma verdadeira constante de eliminação,
enquanto b é uma constante da taxa de eliminação híbrida sendo influenciado pela taxa de transferência de
fármacos dentro e fora do compartimento de tecido.
Quando for impraticável determinar k, b é calculado a partir do declive b. O t1/2β é frequentemente usado
para calcular a dose do fármaco.
Modelo de dois compartimentos: relação entre a distribuição e a semi-vida aparente (ß)
Um tecido com pouco suprimento de sangue não pode atingir uma concentração de fármaco suficiente (
quando existe uma eliminação rápida) para exercer o seu impacto e influência global no perfil de
concentração plasmática. Em contraste, fármacos como a digoxina tem uma semi-vida longa, e concentração
do fármaco diminui lentamente para permitir mais tempo para a distribuição aos tecidos.
Determinação do ka de dados de absorção oral de dois compartimentos (método de Loo-Riegelman)
Plotting a percentagem de fármaco absorvida vs tempo para determinar o ka pode ser calculado para um
fármaco que exibe um modelo de dois compartimentos cinéticos.
Após a administração oral de uma dose de um fármaco que apresenta uma cinética de modelo de dois
compartimentos, a quantidade de fármaco absorvida é calculada como a soma das quantidades de fármaco
no compartimento central (Dp) e no compartimento de tecido (Dt) e a quantidade de fármaco eliminada por
todas as vias (Du).
Cada um destes termos pode ser expressa em termos de constantes cinéticas e das concentrações da
substância no plasma, como se segue:

50
Substituindo a expressão acima para Dp e Du na equação 7.46:
Ao dividir essa equação por Vp expressar a equação em
concentrações de fármaco, obtemos:
Em t = ∞ esta equação torna-se:
A equação 7.53 dividida pela equação 7.54 dá a fracção de
fármaco absorvido a qualquer momento.
A parcela da fracção do fármaco não absorvido, [1 - Ab / Ab ∞], em função do tempo dá -ka/2.3 como o
declive do qual o valor para a taxa de absorção constante é obtido.
Cp e [AUC] t 0 são calculados a partir de uma parcela de Cp em função do tempo.
Valores de (Dt /Vp) podem ser aproximados pelo método de Loo Riegelman, como se segue:
O cálculo dos valores de Ct é mostrado na tabela 7.3, utilizando um conjunto típico de dados de absorção oral.
Após o cálculo dos valores de Ct, a percentagem do fármaco não absorvido é calculada com a equação 7.54,
como mostrado na tabela 7.4. Um gráfico de percentagem de fármaco não absorvido em função do tempo dá
ka de aproximadamente 0,5 h-1.
Para o cálculo do ka por este método, o fármaco deve ser administrado por via intravenosa para permitir a
avaliação da distribuição e das constantes de velocidade de eliminação.
Ct é t Dt/Vp-> concentração de tecido aparente; t = tempo
de amostragem para amostra n, tn-1 = tempo de
amostragem para o ponto de amostragem anterior à
amostra n, e (Cp)tn-1 = concentração de fármaco no
compartimento central para a amostra n – 1.

51
Para os fármacos que não podem ser administrado por via IV, o ka não pode ser calculado pelo
método de Loo Riegelman-> fármacos administradas somente por via oral, usa-se o método de
Wagner-Nelson para fornecer uma estimativa inicial de ka.
Se o medicamento for administrado por via intravenosa, não há maneira de saber se há alguma
variação nos valores da taxa de eliminação, k, e a taxa de distribuição constante k12 e k21.
Tais variações alteram as constantes de velocidade. Portanto, um modelo de um compartimento é usado para
ajustar a curva plasmática após uma dose oral ou intramuscular.
O nível de plasma previsto a partir do ka obtido por este método desvia-se do nível de plasma real. No
entanto, em muitos casos, esse desvio não é significativo.
Fracção relativa cumulativa absorvida
A fracção de fármaco absorvida em qualquer tempo t que pode ser resumida ou acumulada para cada período
de tempo. Da equação 7.31, o termo Ab /Ab ∞ torna-se a fracção cumulativa relativa absorvida (CRFA).
Na equação de Wagner-Nelson, Ab / Ab ∞ ou CRFA irá eventualmente igualar à unidade, ou 100%, embora a
fármaco não possa estar 100% sistemicamente biodisponível.
A percentagem de fármaco absorvido é baseada na quantidade total de fármaco absorvida (Ab∞) em
vez da dose D.
Como a quantidade de fármaco absorvido, em última instância, Ab ∞, é igual a k[AUC]∞0, o numerador
será sempre igual ao denominador, se a fármaco é de 10, 20 ou 100% biodisponível.
A percentagem de fármaco absorvido com base em Ab/Ab ∞ é, portanto, diferente da real
percentagem de fármaco absorvido a menos que F = 1. No entanto, para o cálculo do ka, o método é
aceitável.
Cpt é a concentração plasmática no tempo t.

52
Um medicamento de referência foi administrado e as concentrações da substância no plasma foram
determinadas ao longo do tempo. Tendo em conta que: Ab é o montante acumulado de fármaco absorvido
do medicamento e Abref∞ é o montante acumulado final do fármaco absorvido de uma forma dose de
referência; calculou-se CRFA dividindo Ab/Abref∞.
Onde Kref e [AUC] ∞ ref são a constante de eliminação e a área sob a
curva determinada a partir do produto de referência. Os termos no numerador da equação 7.57 referem-se
ao produto, como na equação 7.56.
Cada fracção do fármaco absorvida é acumulada e plotted em função do intervalo de tempo em que a
amostra da substância no plasma foi obtida.
Fraction of drug absorbed.
(Wagner–Nelson method).
Mean cumulative relative
fractions of tolazamide
absorbed as a function of time.
Mean serum tolazamide levels as a
function of time.
Significado da Taxa de Absorção Constante
O processo real de absorção do fármaco pode ser de ordem 0, 1ª ordem, ou uma combinação de processos
taxa que são dificilmente quantificados.
Para muitas formas farmacêuticas de libertação imediata, o processo de absorção é de 1ª ordem,
devido à natureza física da difusão de fármacos.
Para certos medicamentos de libertação controlada, a taxa ordem 0 constante será mais adequada.
O conhecimento do ka e K permite a previsão de picos e concentrações mínimas de fármacos no plasma após
doses múltiplas. Em estudos de bioequivalência, os medicamentos são dados em doses químicas equivalentes
e as respectivas taxas de absorção sistémica não podem diferir muito. Então, tmax, ou o tempo do pico da
concentração do fármaco, pode ser muito útil na comparação das respectivas taxas de absorção de um
fármaco de um produto quimicamente equivalente.
IV contínua
Muitos fármacos, administradas por infusão IV seguem cinética de dois compartimentos.

53
Equação que descreve a concentração do fármaco no plasma em função do tempo:
No estado de equilíbrio, (ou seja, t = ∞), a equação 5.22 reduz-se a:
Rearranjando esta equação, a taxa de infusão para a concentração desejada de fármaco no plasma no estado
de equilíbrio é calculada por:
IV-Modelo bicompartimental
A concentração plasmática de um fármaco que segue este tipo de modelo,
após várias doses de carga é mostrada na figura 5.6.
Não é possível manter instantaneamente, um nível sanguíneo estável no
estado de equilíbrio para um fármaco que segue uma cinética de dois
compartimentos com uma taxa de infusão de ordem 0. Portanto, uma dose
de carga produz inicialmente um nível de sangue ligeiramente superior
ou inferior ao nível constante de sangue no estado de equilíbrio.
Para superar este problema: várias injecções IV bólus administrado
como infusão IV curta intermitente pode ser usado como um método
para administrar uma dose de carga para o paciente.
6 .Entender os princípios subjacentes à análise farmacocinética não-compartimental
6.1. Comparar a análise farmacocinética não-compartimental com a análise farmacocinética
compartimental
Abordagens para a modelagem de dados farmacocinéticos
Existem 3 abordagens básicas para modelar dados farmacocinéticos: modelos compartimentais tradicionais,
modelos não compartimentais e modelos baseados na fisiologia.
Modelos compartimentais tradicionais
São escolhidos compartimentos para representar o corpo, como já foi abordado (rever).
Modelo não-compartimental
Neste modelo a estrutura que suporta compartimentos e os parâmetros correspondentes não é modelada, mas a
resposta é-> Baseado em sistemas lineares para modelar os dados.
a e b são constantes de velocidade híbridas e
R é a taxa de infusão.
Fig.5.6-Plasma drug level after various loading doses and rates of infusion for a drug that follows a two-compartment model: a, no loading dose; b, loading dose = R/k (rapid infusion); c, loading dose = R/b (slow infusion); and d, loading dose = R/b (rapid infusion).

54
O modelo simplificado na Figura 13.2 assume que o fármaco entra num espaço cinético central; algum
fármaco deixa irreversivelmente o espaço e abandona o sistema (corpo), enquanto algum fármaco adicional
deixa o espaço central e vai para o compartimento
periférico onde abandona irreversivelmente o corpo;
Para a maioria dos fármacos isto despreza-se,
excepto para anestésicos inalatórios e fármacos
com alta excreção biliar; Algum fármaco deixa o
compartimento central e mais tarde volta depois de
passar através do sistema periférico;
O fármaco é distribuído através de processos estocásticos
(aleatórios). Existem 2 suposições inerentes a esta abordagem:
Sobreposição Tempo invariável
Quando um input origina uma certa resposta e é
dado outro input com outra resposta-> nestas
circunstâncias quando ambos os inputs são dados
juntos, a resposta vai ser a soma das 2 respostas
separadas;
Ex.: se uma dose IV e uma dose oral foram dadas e a
resposta a isto é conhecida, depois quando são
dadas simultaneamente, a resposta será a soma das
duas respostas em separado.
É o princípio usado na determinação da resposta a
múltiplas doses;
Uma certa dose é dada e uma certa resposta é
obtida, então se essa dose é dada noutro tempo, a
mesma resposta devia ser observada.
Para a maioria dos fármacos é verdade, mas alguns
fármacos que têm farmacocinética dependente do
tempo não obedecem a esta suposição-> uma dose
dada de manhã pode não produzir o mesmo
resultado quando dada à noite.
Também é possível que a taxa de eliminação se
altere se a eliminação hepática se tornar saturada.
Modelos baseados na fisiologia
Nestes modelos os compartimentos são escolhidos para representar compartimentos fisiológicos do corpo, a
figura 13.3 é uma simplificação desta abordagem. Alguns
pressupostos aplicados aos compartimentos dos modelos
tradicionais podem também ser aplicados neste modelo.
O fluxo sanguíneo pode ser conhecido ou estimado
através de cada compartimento e a quantidade de
fármaco que entra e sai do compartimento pode ser determinada.
Cada sistema de órgãos forma um compartimento separado-> O fármaco é homogeneamente e
instantaneamente distribuído (ln C vs. t).

55
Contudo, o compartimento é ‘bem movimentado’ e a concentração de fármaco que deixa o órgão está em
equilíbrio com a concentração de fármaco no órgão. Assim, uma constante de coeficiente de partição no
equilíbrio pode ser determinada a partir da concentração do fármaco no tecido em comparação com a
concentração no sangue.
Barreiras entre os compartimentos (sistemas fisiológicos):
A transferência é dependente da taxa de fluxo sanguíneo através do compartimento fisiológico, sendo que
cada compartimento tem uma taxa de clearance característica. Contudo, é necessário saber ou determinar a
taxa de fluxo sanguíneo em cada órgão ou sistema de tecidos.
Fármaco: A eliminação é apenas a partir de certos compartimentos que são específicos no modelo (ex.: fígado
e rins). Não existe ligação irreversível do fármaco aos tecidos.
Escolha do modelo
A escolha depende primariamente dos objectivos do estudo. Porém, conhecendo os pressupostos de cada
modelo, a resposta às seguintes questões pode influenciar a selecção da abordagem:
1. O que foi feito experimentalmente?
2. Que dados foram obtidos?
3. Podem os pressupostos do modelo ser validados para estes dados?
Objectivos de estudo
Existem 4 objectivos principais de estudo para a análise dos dados:
1. Sumarizar a cinética do fármaco (farmacocinética descritiva);
2. Quantificar 1 ou mais processos cinéticos de fármacos;
3. Explicar a farmacocinética;
4. Fazer predições farmacocinéticas.
Modelo compartimental tradicional
Uma das dificuldades é achar a relevância estrutural do modelo e dos parâmetros resultantes dele->
Existe uma pequena relevância fisiológica ou anatómica dos compartimentos.
Os parâmetros determinados matematicamente, têm em conta o efeito da distribuição do fármaco,
mas contribuem pouco para identificar a estrutura específica do processo cinético e muitas das
suposições são difíceis de verificar.
Esta aproximação pode ser usada para desenvolver a farmacocinética descritiva para o fármaco,
contudo não é significativo sumarizar em termos de parâmetros específicos da estrutura que não têm
importância fisiológica ou anatómica.
Os parâmetros que não são específicos da estrutura (Tmax, Cmax,AUC…) podem ser calculados ajustando
equações específicas do modelo. Porém esses parâmetros são mais prontamente obtidos usando
equações empíricas adequadas numa aproximação não compartimental.

56
O objectivo de estudo para quantificar um processo farmacocinético ou explicar a farmacocinética,
pode ser feita com esta abordagem. Contudo, porque os pressupostos relevantes ou a falta de uma
estrutura relevante são difíceis de verificar, estes objectivos tornam-se impraticáveis. Ex.: Quando
tentamos elucidar a clearance hepática, o modelo clássico não será a melhor escolha.
As predições farmacocinéticas podem ser feitas mais facilmente usando uma abordagem
independente do modelo, quando a estrutura do modelo não é importante. No entanto, uma
aproximação clássica pode ser por vezes usada quando se conhecem limitações do modelo.
Modelo não compartimental
Processo cinético específico (absorção, distribuição ou eliminação)-> Podem-se fazer predições mais
fortes.
Esta aproximação tenta modular a resposta em vez da estrutura do processo, sendo difícil explicar a
razão pela qual o fármaco exibe um certo perfil cinético (ex: sabe-se que a concentração evolui mas
não se sabe porquê).
Os efeitos totais dos processos cinéticos são colocados juntamente em funções como um impulso
unitário ou resposta característica-> função distribuição e função eliminação. Como resultado os
processos cinéticos individuais não são descritos.
Se o objectivo de estudo é sumarizar a cinética, quantificar um processo farmacocinético ou fazer
predições farmacocinéticas, esta aproximação pode ser útil porque tem uma natureza mais geral.
Se o objectivo de estudo é explicar a farmacocinética, então esta aproximação não é muito útil.
Comparado com o modelo clássico mais estruturado, o grau de complexidade e diferenciação estrutural nesta
aproximação parece ter um balanço mais realista com a quantidade e qualidade dos dados tipicamente
obtidos em estudos farmacocinéticos.
6.2 - Conhecer que parâmetros cinéticos podem ser determinados a partir das curvas
concentração-tempo mediante análise farmacocinética não-compartimental
Análise do modelo não-compartimental
O modelo não-compartimental é frequentemente utilizado para farmacocinética descritiva, para quantificar
um certo processo farmacocinético ou fazer predições. A distribuição do fármaco geralmente ocorre por 2
processos ocasionais estocásticos (aleatórios):
1. Convecção – fármaco é transportado através do sangue ou outro fluído com a corrente;
2. Difusão – fármaco move-se através de vários tecidos e membranas.

57
A distribuição do fármaco pode ser estocástica se ocorrerem interacções competitivas com substâncias
endógenas, se há um feedback fisiológico, ou se há transformações bioquímicas. Sendo assim 2 suposições
precisam de ser verificadas nesta aproximação: sobreposição e tempo.
A. Para verificar a sobreposição pode-se experimentar dar a um sujeito pelo menos 2 doses diferentes de
um fármaco.
B. A invariável tempo é validada dando a mesma dose a dois tempos diferentes, assumindo que no
intervalo das administrações o fármaco sofreu washout (“eliminação”) suficiente. Isto ajudará a
verificar mudanças diurnas na taxa de eliminação.
Esta aproximação não-compartimental procura modelar a resposta, não uma estrutura modelo, como tal,
uma curva adaptada é usada para ajustar os dados a um modelo de resposta adequado.
A estrutura simplificada na Figura 13.2, representa a entrada do fármaco no espaço central.
Não há tentativa de descrever o que estará a acontecer ao fármaco fora do espaço central. O modelo para
isto é mostrado na seguinte equação:
c(t) - resposta à concentração; f(t) - função input; cδ(t) - função característica do espaço central, conhecida
como a resposta característica ou resposta de impulso unitária.
O f(t) não é a taxa de libertação do fármaco mas é composto por pelo menos 2 processos:
1. Taxa de libertação do fármaco
2. Extensão da degradação e biotransformação antes do fármaco alcançar o espaço central.
Dados farmacocinéticos que têm sido recolhidos podem ser analisados adaptando os mesmos a uma soma de
exponenciais. Um programa curve-fitting (ajuste da curva) é usado para encontrar o nº óptimo de
exponenciais, e a resposta será:
Os termos pré-exponenciais, Ci, e os termos exponenciais, λi, são usados na análise não-compartimental para
calcular um nº de termos farmacocinéticos descritivos que descrevem a disposição do fármaco. Cada termo
exponencial tem uma meia vida associada:
Quando a resposta ao fármaco é poliexponencial, com eliminação somente a partir do compartimento
central, é possível calcular a área sobra a curva (AUC) e a área sobre a curva no momento (AUMC):

58
E a concentração inicial (C0) é:
Estes termos são usados para calcular/estimar as descrições farmacocinéticas independentes de modelos. A
clearance total é definida como:
Em adição, vários termos de biodisponibilidade periférica podem ser calculados para descrever a extensão em
que o fármaco é distribuído fora do espaço central.
Biodisponibilidade periférica
No modelo não-compartimental, há 2 espaços cinéticos de interesse, o espaço cinético central e o espaço
cinético periférico. A extensão média de tempo que todas as moléculas de fármaco passam num espaço
cinético específico é o tempo de residência médio desse espaço cinético.
Parâmetros de tempo médio
tb (MRTb) – tempo de residência médio para o fármaco no corpo – intervalo de tempo médio de todas as
moléculas que passam no corpo.
Ts (MRTs) – tempo de residência médio para o fármaco no espaço central – intervalo de tempo médio que
todas as moléculas passam no espaço central antes de serem eliminadas do corpo.
Tp (MRTp) – tempo de residência médio para o fármaco no sistema periférico – intervalo de tempo médio que
todas as moléculas passam no espaço cinético periférico.
Os tempos médios desses espaços cinéticos podem ser calculados quando o fármaco entra no espaço cinético
central e o fármaco que está a sair do sistema (corpo) pelo espaço cinético periférico é insignificante.
Vc - volume de distribuição do espaço central
Vss – volume de distribuição no estado de equilíbrio
FAUC ou RTPCt/c - a biodisponibilidade periférica
AUC é o rácio de quantidade de amostra no estado
de equilíbrio (steady state) no sistema periférico e
quantidade de amostra no espaço central; isto é
também referido como coeficiente de tempo de
partição residente.

59
Se o fármaco é dado por via extravascular e não é dada nenhuma dose IV, a fracção biodisponível da dose, F,
não será conhecida. Consequentemente, os cálculos resultantes das equações 13.7 até 13.9 determinam Cl/F,
Vc/F e Vss/F, para as quais F não é conhecido.
Tempos de meia vida, parâmetros de tempo médio e o coeficiente de partição do tempo de permanência são
independentes da biodisponibilidade.
Alguns programas de software farmacocinéticos, realizam a análise não-compartimental sem ajustar/adaptar
a curva de resposta completa. Estes programas, calculam a constante da taxa de eliminação (λ) para a fase de
eliminação terminal dos dados, e depois usam uma regra do trapézio com esta constante para calcular a AUC
e AUMC. Com estes termos, a clearance total, o volume de distribuição steady-state e o tempo de
permanência médio no corpo podem ser calculados.
Sem o C0, não é possível calcular o volume de distribuição do compartimento central ou o tempo de
residência médio no compartimento da amostra. Não se determina com precisão estes parâmetros de valores
e depende de uma adaptação imparcial e restrita dos dados às equações 13.2 e 13.6. É possível usar
resultados IV e extravasculares para “estudar” a função input pela função da resposta característica para
estimar parâmetros de tempo médios que examinam a chegada do fármaco ao compartimento da amostra.
7.Entender os princípios subjacentes aos modelos farmacocinéticos fisiológicos.
Os modelos farmacocinéticos baseados na fisiologia (PB/PK) podem ser considerados uma aproximação única
graças aos seus focos singulares de investigação na disposição dos fármacos nos tecidos.
As investigações combinadas de vários indivíduos estabeleceram a ideia de que a distribuição de
fármacos para órgãos específicos depende de variáveis anatómicas e fisiológicas relevantes.
Em contraste aos modelos compartimentais clássicos, os compartimentos de tecido representam
volumes anatómicos específicos conectados pela circulação sanguínea.
A captação de fármaco para os órgãos depende de parâmetros termodinâmicos (coeficiente de
partição, ligação as proteínas plasmáticas) e transporte membranar (coeficiente de transferência de
massa).
Os órgãos individuais normalmente possuem uma estrutura de modelo limitado pela perfusão ou pela
permeabilidade da membrana -> A forma como cada órgão é representado depende do fármaco e do animal
sobre o qual o modelo se debruça.
Existem 3 regiões anatómicas distintas de cada órgão: vascular; intersticial e espaços intracelulares.

60
A estrutura de um órgão num modelo PB/PK pode variar de um simples compartimento para uma estrutura
com 3 subcompartimentos dependendo da extensão da aglomeração.
Qualquer compartimento, quer seja um subcompartimento ou um compartimento, é assumido como um
espaço homogéneo que possui uma única concentração de
fármaco. A estrutura de um tecido monocompartimental é
referida como limitada pela perfusão uma vez que este
modelo assume que a corrente sanguínea é o passo limitante
na captação de fármaco para o tecido-> barreira vascular é
uma pequena resistência à captação de fármaco.
Fármacos altamente lipofílicos e órgãos com uma
baixa corrente sanguínea contribuem mais facilmente
para os modelos limitados pela perfusão.
A estrutura de um órgão limitado pela
permeabilidade é comprimida em dois
subcompartimentos homogéneos ambos com
configuração extracelular-intracelular ou vascular-
extravascular.
Em qualquer dos casos o transporte do fármaco é limitado pelo
transporte através da membrana.
O arranjo vascular - extravascular é necessário para representar
o transporte na BHE e junta os espaços intersticial e intracelular
num compartimento homogéneo baseado no facto que não há gradiente de concentração entre estes meios.
O arranjo extracelular - intracelular é utilizado para outros tecidos que não o cérebro.
Os espaços vascular e intersticial formam o compartimento extracelular e por isso é assumido que o fármaco
é equilibrado instantaneamente entre os espaços vascular e intersticial.
O nível de complexidade do órgão vai depender do número de espaços no tecido que podem ser identificados
com os parâmetros de transporte de fármaco.
Fig. 14.1-Representações do modelo PB/PK do orgão individual em três subcompartimentos (A), dois subcompartimentos (B), ou apenas ligado por membrana e limitado pela perfusão (C).

61
Divisão do compartimento intracelular em subcompartimentos representando diferentes tipos de células->
um dos quais serve como receptor do fármaco.
A representação do corpo inteiro por órgãos individuais conectados anatomicamente pela corrente sanguínea
é referida como o modelo PB/PK “global”.
O modelo PB/PK “híbrido” foca-se num único órgão ou num pequeno grupo de órgãos. Os modelos de órgãos
são análogos àqueles que são encontrados no modelo global; no entanto, a concentração do fármaco no
sangue que entra no compartimento é descrita por uma equação exponencial ou por uma “forcing function”.
Esta é normalmente alcançada por uma equação poli-exponencial dada por dados “concentração do fármaco
no plasma versus tempo”, o que justifica o termo híbrido.
As vantagens do modelo híbrido são:
Poucos parâmetros têm que ser estimados;
Os dados da concentração de fármaco nos tecidos versus tempo são desnecessários, no entanto o
órgão de interesse mantém a sua representação fisiológica.
Os objectivos para o desenvolvimento de um modelo PB/PK podem ser variados.
A aceitação do modelo caracteriza a disposição do fármaco como linear ou não linear respeitando a
clearence, transporte membranar e a ligação às proteínas plasmáticas.
Os modelos PB/PK também podem ser uma ferramenta preditiva de alto potencial-> a capacidade
preditiva dos sistemas de transporte de fármacos, graças à sua capacidade para caracterizar a
disposição do fármaco no tecido ou na célula.
Uma vez que a captação no tecido e as interacções celulares vão ser cruciais no sistema de transporte, o
modelo PB/PK capaz de caracterizar estes parâmetros pode ser uma ferramenta quantitativa para avaliar o
sistema de distribuição.
Fig.14.2-Possíveis conexões na circulação sanguínea no modelo PB/PK: (A) retorno venoso incorporado numa
equação balanço de massa no pulmão; (B) compartimento venoso separado

62
Equações alométricas empíricas baseadas no peso corporal dos animais têm sido a principal base para
aproximar à escala humana, relativamente à corrente sanguínea e ao peso dos órgãos, assim como variáveis
macro-farmacocinéticas como a clearence e o volume de distribuição.
No entanto, poucas relações têm sido estabelecidas para parâmetros termodinâmicos e de transporte de
fármacos-> singularidade destes parâmetros para cada fármaco e tecido, e também para a percepção que
grandes quantidades de dados são necessárias para desenvolver relações entre escalas.
A utilização de sistemas in vitro de diferentes espécies para medir taxas metabólicas, coeficientes de
partição e parâmetros de transporte membranar pode levar a relações entre escalas e a uma
diminuição no número de experiências com animais.
Uma substituição de um modelo global para um modelo híbrido vai reduzir a magnitude do esforço
para se adaptar a escala, e pode levar a novos paradigmas da extrapolação de animais para humanos.
O estabelecimento de estudos in vitro optimizados em diferentes espécies de animais também pode
limitar o número de estudos necessários. A extrapolação de modelos PB/PK para humanos é um
benefício com riscos que se têm que tomar para providenciar bases para uma futura pesquisa.
Características dos Modelos PB/PK
Aproximações de balanço de massa para caracterizar a disposição de fármaco;
Utiliza equações diferenciais para descrever sistemas de modelos;
É racional ao considerar aproximação PB/PK apenas se o focus se encontra em tentar compreender a
disposição de fármacos nos tecidos;
Oferece um elevado potencial para predizer as concentrações de fármaco sob diferentes condições
fisiológicas e farmacológicas;
Pode ser adaptado de animais para humanos.
Formulações Matemáticas
O compartimento A da fig.14.1 ilustra um compartimento de tecido que não elimina fármaco, dividido em
três subcompartimentos anatomicamente relevantes, cada um deles homogéneo no que diz respeito à
concentração de fármaco. As equações de balanço de massa correspondentes são:
; ;
Ci1 = concentração no compartimento 1 do tecido i
Ci2 = concentração no compartimento 2 do tecido i
Ci3 = concentração no compartimento 3 do tecido i
Cb = concentração de sangue que entra no tecido i
ni12 = fluxo de massa do compartimento 1 para o 2,
quantidade/tempo
ni23 = fluxo de massa do compartimento 2 para o 3,
quantidade/tempo
Vi1 = volume do compartimento 1 no tecido i
Vi2 = volume do compartimento 2 no tecido i
Vi3 = volume do compartimento 3 no tecido
Nota: O compartimento 1 representa o espaço vascular do tecido e pode ser identificado como sangue, plasma ou espaço do soro.

63
Assumindo um transporte membranar do fármaco de 1ª ordem ou linear, o fluxo pode ser expresso por :
Os coeficientes de transferência de massa também podem ser expressos em unidades de tempo-1 através da
multiplicação pelo volume compartimental apropriado.
A eliminação irreversível de fármaco do tecido requer uma expressão adicional à equação que representa o
subcompartimento no qual a eliminação ocorre. Ex.: a eliminação hepática do fármaco pode ser descrita por
uma expressão linear ou não-linear adicionada à equação de balanço de massa do compartimento (fígado)
uma vez que este representa os hepatócitos.
O modelo de tecido dos três compartimentos é simplificado por aglomerar todos os três compartimentos,
apenas os subcompartimentos 1 e 2 ou 2 e 3. Estas simplificações resultam nos modelos limitados pela
perfusão (aglomeram-se os três subcompartimentos) e limitados pela permeabilidade (aglomerando qualquer
um dos dois subcompartimentos). (pág.300)
Assim, as equações de balanço de massa para um compartimento que não elimina e é limitado pela
permeabilidade são dadas por: ;
Combinando os subcompartimentos vascular e intersticial define-se o subcompartimento 1 como extracelular
e o 2 como intracelular.
Aglomerando os subcompartimentos intersticial e intracelular, o que deve ser feito para um compartimento
cerebral, define-se compartimento 1 como espaço vascular e 2 como extravascular.
Sob condições de transporte membranar linear:
(14.8)
O transporte de membrana saturável ou não linear é representado como:
(14.9)
ai = taxa de transporte máxima, quantidade/tempo; bi = concentração de fármaco a metade de ai
quantidade/volume
O transporte membranar de fármacos por mecanismos linear e não-linear é representado pela adição das
eq.14.8 e 14.9.
A eliminação de fármaco de um compartimento limitado pela permeabilidade necessita de subtrair a taxa de
eliminação, qi, da equação de balanço de massa apropriada, tipicamente do subcompartimento 2.
Para uma eliminação de fármaco linear ou de primeira ordem:
hi12 = coeficiente de transferência de massa (volume/tempo)
caracterizando o transporte do fármaco do compartimento 1 para o
2
hi23 = coeficiente de transferência de massa (volume/tempo)
caracterizando o transporte de fármaco do compartimento 2 para o
3
Ri12=fu
1/ fu2
Ri23=fu
3/ fu2
fu= fracção não ligada de fármaco no compartimento 1, 2, 3

64
Para uma eliminação não linear:
Aglomera-se os compartimentos 1, 2 e 3 num único compartimento homogéneo-> implica um modelo
limitado pela perfusão. A equação de balanço de massa para um órgão que não elimina é:
Os termos de eliminação de fármaco são agora:
para uma eliminação linear e não-linear respectivamente.
A eq.14.13 é frequentemente representada como:
Onde, Cli
1 = clearence total intrínseca do órgão
A conexão das circulações venosa e arterial pode ser feita de várias formas dependendo se se usa ou não
compartimentos diferentes para o sangue venoso e arterial ou se se separa os compartimentos do lado
direito e esquerdo do coração. Os dois métodos mais utilizados estão ilustrados na fig.14.2 para
compartimentos limitados pela perfusão. As equações de balanço de massa associadas ao modelo A dessa
figura são:
Para o modelo B as equações são:
; ;
Onde, v indica o compartimento do sangue venoso.
Dependendo da representação usada para o sistema arterial-venoso, o balanço da corrente sanguínea tem de
ser mantido, assim:
Para os modelos A e B, respectivamente.
O transporte membranar de fármacos por difusão passiva, apesar da função das concentrações de fármaco
livre, é normalmente expresso em termos de concentração total de fármaco, como apresentado nas eq.14.8 e
ki = constante de eliminação de primeira ordem,
tempo-1; Vm = taxa de eliminação máxima,
quantidade/tempo; km = constante de Michaelis,
igual à concentração de metade de Vm.
Ri = Ci/C0 (onde C0 é a concentração do efluente sanguíneo
venoso)
(3) (14.13)
Onde 1 e b se referem ao pulmão e ao
compartimento do sangue arterial, e n é igual ao
número de compartimentos que derivam do
retorno venoso.

65
14.9. Alteram-se estas equações em termos de concentrações totais o que requer uma reparametrização das
expressões do fluxo usando as concentrações do fármaco livre.
Por exemplo, a eq.14.8 chega a:
Onde, f se refere ao fármaco livre
Ri1 = 1 / f1
u
Ri2 = 1 / f2
u
Substituindo as equações (14.24) e (14.25) na (14.23) dá-nos:
Sendo que hi = hi12f / Ri
1 e Ri12 = Ri
2 / Ri1 permite que a equação seja escrita como:
Apesar dos fluxos de fármaco e das concentrações no tecido serem para propósitos práticos apresentados em
termos de concentrações totais de fármaco, a divisão entre concentração de fármaco livre e ligado às
proteínas é possível. Esta aproximação pode ser benéfica para um fármaco que tenha uma ligação às
proteínas não linear ou quando os problemas farmacodinâmicos são relevantes. Os problemas encontrados
na representação de equações de balanço de massa de fármaco livre e ligado têm sido discutidos e centrados
no uso de fracções proteicas efectivas capazes de ligar fármaco nos tecidos.
Funções que descrevam a taxa de dose administrada que entra no compartimento (função input) podem ter
várias formas dependendo da monitorização após a administração. A função input I(t), é a adicionada à
equação de balanço de massa apropriada e pode descrever qualquer administração de fármaco.
A absorção de 1ª ordem pode ser expressa como:
Onde, F = fracção de dose administrada que entra no compartimento
ka = taxa de absorção de 1ª ordem
Taxas constantes ou infusões de ordem zero são:
Onde, k0 = taxa de infusão constante de ordem zero, quantidade/tempo, T =
duração da infusão
A administração de um bólus é normalmente caracterizada por uma função de injecção normalizada que
efectivamente dá a dose para que esta possa ser representada numa curva em forma de sino. Uma vez que a
administração em bólus não é instantânea, a sua aproximação é considerada mais realista.
I(t) para uma administração em bólus é:
Onde, M = dose; g(t) = função de injecção normalizada e é 30λ(λt)2(1-λt)2; λ = inverso da
duração da injecção
As técnicas de análises têm sido usadas para gerar funções input para modelos PB/PK.
(14.24)
(14.23)
(14.25)

66
Comparações entre as concentrações previstas e observadas de fármaco são baseadas nas
concentrações totais de fármaco no tecido uma vez que a maioria dos métodos analíticos utilizam
tecidos totais homogéneos.
A diferenciação de concentrações celular e extracelular de fármaco não é possível com
homogeneização destrutiva de tecidos implementada rotineiramente no laboratório.
A quantificação nas concentrações de fármaco no subcompartimento torna-se mais realista à medida
que os métodos de quantificação não invasivos se tornam mais acessíveis e precisos.
A média do volume é calculada através da previsão das concentrações de fármaco no subcompartimento que
permitem comparações às concentrações de fármaco total no tecido observadas.
Para uma estrutura de 3 subcompartimentos como verificado na primeira figura, a concentração prevista
total, Ct é:
Onde, Vt = ao volume total no tecido.
As concentrações de fármaco numa estrutura de dois subcompartimentos são:
Os volumes no tecido podem ser expressos em unidades de massa ou volume: quando são utilizadas unidades
de volume, é normalmente assumida uma densidade de 1.
Predizer as concentrações de um compartimento limitado pela perfusão pode ser comparado directamente a
valores observados.
A diferença chave na aproximação híbrida é que a concentração de fármaco na corrente que chega ao órgão é
caracterizada por uma função empírica. Esta função é imediatamente caracterizada por uma equação poli-
exponencial como:
Onde, C = concentração de fármaco no plasma ou no sangue; Ai = intersecção
com o eixo do y; λi = constante da velocidade de disposição para o
compartimento i
A função que descreve a concentração input de fármaco (14.34), C, como uma função do tempo é substituída
nas equações de balanço de massa do órgão alvo por Cb, permitindo que as equações do órgão alvo possam
ser resolvidas analiticamente ou numericamente.
Desenvolvimento do Modelo
Perfusões e Volumes dos Órgãos
A perfusão num órgão pode ser medida por substâncias radioactivas, microesferas e clearence de compostos
altamente extraídos pelo órgão. A autoradiografia quantitativa tem sido utilizada para medir a corrente
sanguínea no órgão assim como a permeabilidade capilar usando marcadores standard marcados
radioactivamente.
(14.34)

67
Os volumes de tecido total são determinados pelo peso, assumindo a densidade como 1, e fraccionada em
volumes de subcompartimentos dependendo da estrutura do modelo.
Existem tabelas que são utilizadas para estimar os volumes de água e sangue dos órgãos para permitir
correcções de contaminação do sangue e para assistir na determinação efectiva das fracções ligadas a
proteínas-> Existem investigações independentes suficientes para desenvolver relações alométricas baseadas
no peso do animal.
A estimação desses parâmetros, particularmente a corrente sanguínea no órgão, varia imensamente, e não
podem ser ignoradas como fontes de erro no modelo.
Coeficientes de Partição
Têm sido muito utilizados métodos de estimação para coeficientes de partição tecido-sangue (Ri) graças à
necessidade deste parâmetro para a maioria dos modelos de órgãos (tanto a estimação de parâmetros in
vitro como in vivo são utilizadas).
Assumindo uma equação do tipo Langmuir, em que há uma igualdade de concentrações de fármaco ligado à
proteína livre no plasma, o coeficiente de partição, Ri, é:
Onde, αi1 = razão entre a concentração livre e ligada no plasma ou no tecido
i; λ = razão da concentração total no sangue e no plasma
As equações de Chen e Gross são utilizadas para calcular coeficientes de partição para compartimentos
limitados pela perfusão a partir não só da taxa de infusão (isto é, condições de steady-state) mas também do
regime intravenoso. Para um órgão que não elimina sob condições de steady-state,
E para um bólus intravenoso,
Gallo et all desenvolveram um método de calcular os coeficientes de partição tanto para compartimentos
limitados pela perfusão como para limitados pela permeabilidade através da AUC. A partir de uma
administração em bólus intravenoso, o coeficiente de partição para um compartimento que não elimina
limitado pela perfusão é:
AUCi = área total sob a curva concentração de fármaco no tecido – tempo
AUCp = área total sob a curva concentração-tempo
Sob as mesmas condições para um compartimento limitado pela permeabilidade, Ri é:
Vi = volume total no tecido; Vi1, Vi
2 = volumes para o tecido i nos
subcompartimentos 1 ou 2
Tanto os métodos de Chen e Gross como Gallo et all têm sido aplicados a compartimentos que eliminam.
Ambos os métodos são derivados de equações de balanço de massa específicas para o modelo dado.
Ciss = concentração de fármaco no steady-state no tecido i
Cpss = concentração de fármaco no steady-state no plasma
Ci0 = concentração de fármaco que intercepta o eixo dos y
Cp0 = concentração de fármaco inicial no plasma a t=0
α = declive da fase terminal do gráfico concentração-tempo

68
Coeficientes de Transferência de Massa
Os coeficientes de transferência de massa (hi) podem ser obtidos a partir de experiências in vitro utilizando
varias metodologias de permeabilidade de membranas. Isto inclui rápida mistura e separação, volume celular
e técnicas espectroscópicas. Uma abordagem geral baseia-se em adicionar o fármaco a uma suspensão de
células, mexer rapidamente e separar-las, e por fim medir a concentração de fármaco no meio de suspensão
após diferentes tempos de mistura. Esta experiência é utilizada para medir a taxa de influxo para a célula e se
o transporte for linear e bidireccional, a taxa de efluxo de fármaco é igual. Taxas específicas de efluxo podem
ser obtidas ao realizar o estudo com células que já possuem o fármaco.
Diferentes concentrações iniciais de fármaco devem ser utilizadas para examinar a possibilidade de um
transporte membranar não linear.
Outro método in vitro usa um compartimento receptor-dador separado por uma monocamada celular para
estimar os parâmetros do transporte membranar. Os coeficientes de permeabilidade, P, foram calculados por:
A taxa de fluxo é obtida pelo declive do gráfico concentração do soluto na câmara do receptor versus tempo.
O coeficiente de transferência de massa, hi, é igual a PA, onde A é a superfície da membrana que separa dois
subcompartimentos. Os coeficientes de transferência de massa também podem ser estimados a partir de
experiências in vivo. Sob as condições de steady-state do fármaco no sangue, um método é utilizado para
estimar um coeficiente de transferência de massa linear.
Outro método, referido como o método do momento, é derivado de Gallo et all para estimar coeficientes de
transferência de massa lineares para órgãos que não eliminam.
Clearances
As clearences dos órgãos estão baseadas mais em estudos in vivo do que in vitro.
O conhecimento da clearence total do fármaco e a contribuição de cada via de eliminação permite calcular as
clearences intrínsecas do fármaco e do órgão.
Em geral:
A clearence intrínseca do órgão, Clil, é obtida de:
Os parâmetros metabólicos do fármaco, Vm e Km, podem ser estimados a partir de
enzimas hepáticas in vitro e preparações de hepatócitos.
Ligação às Proteínas Plasmáticas
Os parâmetros de ligação às proteínas plasmáticas são normalmente obtidos a partir de experiências in vitro
utilizando filtração, centrifugação e diálise dinâmica ou de equilíbrio.
Mistura
K = taxa de fluxo do soluto, quantidade/volume; V = volume da
câmara onde existe o dador; A = área da superfície celular; C0 =
concentração inicial de soluto na câmara do dador
Cli = clearence do órgão
fi = fracção da dose eliminada pelo órgão i
Cl = clearence total sistémica

69
A “forcing function” pode ser utilizada para estimar quaisquer parâmetros associados com as equações de
balanço de massa. Os procedimentos podem ser aplicados num contexto tanto de modelos híbridos ou
globais PB/PK. O método requer uma regressão não linear baseada nos dados da concentração de fármaco no
sangue e no tecido – tempo. Estes dados vão servir para obter uma “forcing function”, normalmente uma
equação poli-exponencial, a qual é depois usada como uma função input na equação de balanço de massa
para o tecido. O modelo que descreve a disposição de fármaco no tecido individual é depois alterado para os
dados observados com um ou mais parâmetros a serem estimados.
A “forcing function” pode ser particularmente necessária para estimar parâmetros que a literatura
não pode avaliar.
A aplicação do método a um modelo PB/PK total vai requerer uma adaptação sequencial aos modelos
dos órgãos individuais para obter parâmetros estimados e depois reconstruir o modelo global. Para
um modelo PB/PK híbrido, a adaptação do modelo resulta num modelo final.
Funções paramétricas ou não paramétricas são adaptadas para observar dados concentração do
fármaco no sangue – tempo, depois são combinados com medidas concentração do fármaco – tempo
utilizados para obter uma função para a cinética do fármaco no tecido. A partir desta função, alguns
parâmetros de órgãos limitados pela perfusão podem ser obtidos.
Distinção entre modelos de órgãos limitados pela perfusão e pela permeabilidade
Um modelo limitado pela permeabilidade com dois ou três subcompartimentos é indicado se as
concentrações de fármaco no tecido não declinam em paralelo com as concentrações de fármaco no plasma
ou sangue. Um critério formal foi dado por Dedrick e Bischoff, como:
Depois o órgão pode ser representado como limitado pela perfusão.
Se esta condição não for verdadeira, o órgão é melhor representado como sendo limitado pela
permeabilidade. Qi/hi << 1, desde que Vi1/ RiVi
2 seja muito menor que 1.
Assim, determinar tanto um compartimento limitado pela perfusão ou pela permeabilidade é compatível com
os dados necessários para estimar Ri e hi.
Normalmente, quando não existem analises formais a representação que é assumida é a limitada pela
perfusão.
Validação do Modelo
Existem dois problemas aquando da validação do modelo PB/PK:
O primeiro problema é a exactidão com a qual o modelo prediz as concentrações verdadeiras do
fármaco.
Os dados verdadeiros concentração- tempo são mais utilizados para estimar certos parâmetros de
certos modelos.

70
Normalmente, a aceitação do modelo é baseada em avaliação subjectiva de comparações gráficas dos
valores de concentração observados e previstos.
O teste “Goodness-Of-Fit” pode ser um simples cálculo da soma de resíduos para cada órgão num modelo, ou
o cálculo do log de uma função semelhante.
Neste caso,
E no caso anterior,
Valores mais baixos de SSR e LL indicam um
acordo maior entre as concentrações previstas e as observadas para aquele órgão. O problema de validação
do segundo modelo relaciona a confirmação e a aceitação das previsões quando os dados que indicam se
existe limitação não estão disponíveis. A capacidade dos modelos PB/PK para predizer as concentrações de
fármaco sob diferentes condições das que são utilizadas para gerar dados experimentais, libertam o poder
preditivo dos modelos PB/PK. No entanto, pode ser necessário confirmar estas predições que limitam os
dados particularmente se o modelo for originalmente derivado de um set de dados que não são óptimos. A
avaliação do modelo PB/PK deve incluir uma examinação das suposições e da capacidade que os dados têm
para diferenciar entre processos cinéticos lineares ou não lineares, e entre estruturas limitadas pela perfusão
ou pela permeabilidade. Respostas favoráveis a este tipo de questões dão-nos, no mínimo, a capacidade para
aceitar as previsões do modelo.
Desenvolvimento de Modelos PB/PK
Técnicas in vitro e in vivo são ambas utilizadas para estimar os parâmetros dos modelos PB/PK;
Estudos de design e de consistência das aproximações dão-nos provavelmente um modelo mais
robusto;
Técnicas de validação do modelo não são aplicadas rigorosamente.
SSR = soma dos resíduos quadrados
N = número de observações
Ci0 = média da concentração observada
Cip = concentração prevista de fármaco
ni = número de repetições experimentais
Si2 = variância das concentrações observadas a cada ponto
de dados
LL = log da função semelhante


















