







Línguas
Páginas
Legal


Gabriela Ribeiro Silva
ESTUDO DE ALCALOIDES DOS FRUTOS DE Passiflora alata E DE Passiflora
edulis POR SBSE, CLAE-Flu E IDENTIFICAÇÃO POR CLUE-EM.
Dissertação apresentada ao Instituto de Química
de São Carlos da Universidade de São Paulo
como parte dos requisitos para obtenção do título
de Mestre em Ciências.
Área de concentração: Química Orgânica e Bioquímica
Orientadora: Profa. Dra. Janete Harumi Yariwake
São Carlos
2015
Exemplar revisado
O exemplar original encontra-se em
acervo reservado na Biblioteca do IQSC-USP

Dedico este trabalho aos meus pais,
Ronaldo e Inez, que com tanto esforço,
amor e vontade educaram a mim e
minha irmã, Bárbara. Nos ensinaram o
valor do conhecimento, mas também a
importância da generosidade e do amor.
Obrigada por tudo sempre.

Agradecimentos
À Deus, por tantas bençãos, pela vida, saúde e força, pelas maravilhosas
pessoas que colocou em meu caminho.
Aos meus pais, Ronaldo e Inez, pelo amor, compreensão, apoio e por tantos
ensinamentos, que estiveram sempre ao meu lado, sendo meu apoio e meu porto
seguro.
À minha irmã, Bárbara, pelo amor, amizade, alegrias e responsabilidades que
assumiu tão forte.
Aos meus amigos, Ana Paula, Nati, Ju, Lindo, Dani, pelas conversas, risadas,
carinho, ombro amigo e momentos especiais.
Ao meu amigo e namorado, Vinícius, sempre me incentivando a ver além do
que se vê. E à sua família, que me acolheu como parte dela.
À minha família de sangue e de coração, sempre tão presentes e amorosos.
À professora Janete, pelos ensinamentos, seu auxílio na construção do meu
conhecimento, que como orientadora me concedeu a oportunidade de aprender
tanto, e poder crescer academica e pessoalmente.
Ao Bene, pelos ensinamentos práticos, além de sua companhia, ajuda diária,
generosidade e amizade.
À Renatinha e a Tanare, por toda atenção, preocupação e disponibilidade
quando tive dúvidas e problemas. E também ao Leandro Arriveti, pelo auxílio no LC-
MS, sempre tão solicito.
À CNPq pela bolsa concedida e a FAPESP, Processo 2013/21886-7, pelo
auxilio financeiro.
Ao IQSC pelo suporte acadêmico e por ser minha outra casa desde 2008.
E à todos que direta ou indiretamente estiveram ao meu lado e me ajudaram
nesta caminhada, meu muito obrigada!

“É exatamente disso que a vida é feita, de momentos. Momentos que temos que passar, sendo bons
ou ruins, para o nosso próprio aprendizado. Nunca esquecendo do mais importante: nada nessa vida
é por acaso. Absolutamente nada. Por isso temos que nos preocupar em fazer a nossa parte, da
melhor forma possível. A vida nem sempre segue a nossa vontade, mas ela é perfeita naquilo que
tem que ser.”
Chico Xavier

RESUMO
GABRIELA, R. S. Estudo de alcaloides dos frutos de Passiflora alata e de
Passiflora edulis por SBSE, CLAE-Flu e identificação por CLUE-EM. 2015.
Dissertação (Mestrado) Instituto de Química de São Carlos – Universidade de São
Paulo, São Paulo, 2015.
O maracujá, nome popular atribuído ao fruto das diversas espécies do gênero
Passiflora, da família Passifloraceae, é amplamente comercializado e consumido no
mundo, sendo o Brasil um dos maiores produtores do fruto. Alguns estudos apontam
possível toxicidade relacionada às espécies de Passiflora, principalmente P.
incarnata. No entanto, há pouco conhecimento acerca das espécies P. edulis e P.
alata, sobretudo em relação à polpa e sementes. Os extratos da polpa e das
sementes dos frutos dessas duas espécies de “maracujá”, Passiflora alata e
Passiflora edulis, foram estudados com o objetivo de identificar alcaloides
harmânicos, pelo preparo das amostras por extração por sorção em barra magnética
recoberta com polidimetilsiloxano (SBSE-PDMS) e SBSE recoberta com
polietilenoglicol silicone (SBSE-EG Silicone) e análise por cromatografia líquida de
alta eficiência com detector por fluorescência (CLAE-Flu) e cromatografia líquida de
ultra eficiência acoplada à espectrometria de massas sequencial (CLUE-EM/EM). A
análise dos alcaloides harmana e harmina nos extratos da polpa de P. alata foi feita
por meio do método de adição de padrão e mostrou menor quantidade destes
alcaloides, em comparação com os resultados da análise dos extratos da polpa dos
frutos de P. edulis, no trabalho de Pereira e colaboradores. As análises CLUE-EM e
CLUE-EM/EM possibilitaram a identificação dos alcaloides nos extratos: nas
sementes de P. alata, os alcaloides harmana, harmina, harmol, harmalol e harmalina
foram identificados, enquanto que na polpa, harmana e harmina tiveram a
confirmação da sua presença. Nos extratos da polpa dos frutos de P. edulis
observou-se os alcaloides harmana, harmina e harmalina. E nas sementes de P.
edulis harmina foi encontrada, porém há indícios da presença de harmana. A
literatura sobre as barras de SBSE-EG Silicone Twister® não relata nenhum estudo
relacionado ao seu uso para extração e concentração de alcaloides harmânicos nos
extratos de P. alata e P. edulis. Por isso foi proposto inicialmente a aplicação do
planejamento fatorial fracionário para otimização do método de extração, utilizando
os padrões comerciais dos alcaloides harmana e harmina. O planejamento

experimental revelou as variáveis principais e seus níveis de importância, e a partir
destes resultados foi realizado o estudo cinético dos tempos de extração e de
dessorção das barras de SBSE-EG Silicone. Porém, os resultados mostraram que
as barras de SBSE-EG Silicone não são adequadas para a extração dos alcaloides
harmana e harmina, uma vez que a recuperação obtida foi baixa, na ordem de 30%.
Palavras-chave: Passiflora alata, Passiflora edulis, alcaloides harmânicos,
SBSE/CLAE-Flu, CLUE-EM/EM.

ABSTRACT
GABRIELA, R. S. Alkaloids studies from Passiflora alata and Passiflora edulis
fruits analyzed by SBSE, CLAE-Flu, and identified by CLUE-EM. 2015. (Master of
Science) Instituto de Química de São Carlos – Universidade de São Paulo, São
Paulo, 2015.
“Maracujá” is the popular name given to the fruit of several species of Passiflora
genus, from Passifloraceae family, it is widely commercialized and consumed around
the world, and Brazil is one of the largest producers of this fruit. Some studies
pointed out the possible toxicity related to Passiflora species, mainly P. incarnata.
Although, there is a lack of knowledge about the P. edulis and P. alata species,
especially with regards to the pulp and seeds. The extracts of pulp and seeds from
the “maracujá” species Passiflora alata and Passiflora edulis, were studied in order to
identify harman alkaloids. The samples were prepared by extraction with sorptive stir
bar coated with polydimethylsiloxane (SBSE-PDMS) and SBSE coated with
polyethylene glycol silicon (SBSE-EG Silicone). The samples were analyzed by high
performance liquid chromatography with fluorescence detector (HPLC-Flu), and ultra-
high pressure liquid chromatography coupled to tandem mass spectrometry (UHPLC-
MS/MS). Harmane and harmine alkaloids in P. alata pulp extracts were analyzed
using the standard addition method and the results showed a lower amount of these
alkaloids, compared with the test results for the extracts from the P. edulis pulp in the
work of Pereira et al. UHPLC-MS and UHPLC-MS/MS analysis enabled to identify
the alkaloids amount present in the extracts. In the P. alata seeds extract the
following alkaloids were identified harmane, harmine, harmol, harmalol and
harmaline, while in the pulp extract, harmane and harmine were confirmed. In the
extracts of P. edulis pulp the alkaloids identified were harmane, harmine and
harmaline. And in the P. edulis seeds extract the harmine alkaloid was found, some
indications of the presence of harmana were observed. The literature about SBSE-
EG Silicone Twister® bars reports no study related to their use for extraction and
concentration of harman alkaloids in P. alata and P. edulis. Thus, it was initially
proposed the application of fractional factorial design to optimize the extraction
method using commercial standards of harmane and harmine alkaloids. The
experimental design revealed the main variables and their importance levels, and

from these results kinetic studies were performed for the extraction and desorption
times of SBSE-EG Silicone bars. However, the results showed SBSE-EG Silicone
bars are not suitable for the extraction of harmane and harmine alkaloids, since the
recovery obtained was low, on the order of 30%.
Keywords: Passiflora alata, Passiflora edulis, harman alkaloids, SBSE/HPLC-Flu,
UHPLC-MS/MS.

LISTA DE FIGURAS
FIGURA 1 - IMAGENS DOS FRUTOS DE (A) P EDULIS (MARACUJÁ AZEDO) E (B) P.ALATA
(MARACUJÁ DOCE). ................................................................................................ 19
FIGURA 2 - ESTRUTURA DOS ALCALOIDES HARMÂNICOS: (A) HARMANA (B) HARMINA (C)
HARMALINA (D) HARMALOL E (E) HARMOL. .............................................................. 21
FIGURA 3 - REPRESENTAÇÃO DO MÉTODO DE EXTRAÇÃO POR SORÇÃO, UTILIZANDO BARRAS
MAGNÉTICAS RECOBERTAS COM POLIDIMETILSILOXANO (PDMS) (A) E OS PROCESSOS
DE DESSORÇÃO LÍQUIDA (B). ................................................................................... 22
FIGURA 4 - REPRESENTAÇÃO ESQUEMÁTICA DE UMA BARRA COMERCIAL PARA SBSE DE (A)
PDMS E (B) .......................................................................................................... 25
FIGURA 5 - BARRA DE EXTRAÇÃO DE EG-SILICONE TWISTER®”. ...................................... 26
FIGURA 6 - ILUSTRAÇÃO DE UM SISTEMA ON-LINE CLAE-EM, COM A SEQUÊNCIA DE
ELEMENTOS. ......................................................................................................... 29
FIGURA 7 - CURVA DA EQUAÇÃO DE VAN DEEMTER, PARA PARTÍCULAS DE 10, 5, 3 E ≤ 2µM.
............................................................................................................................ 31
FIGURA 8 - FLUXOGRAMA DO FUNCIONAMENTO DO SISTEMA DE DETECÇÃO DE
ESPECTROMETRIA DE MASSAS. ............................................................................... 33
FIGURA 9 - PROCESSO DE FORMAÇÃO DAS GOTÍCULAS DE ANALITO E SOLVENTE, E COMO
SÃO CONDUZIDAS PELO CAMPO ELÉTRICO APLICADO. ................................................ 35
FIGURA 10 - INTERFACE DO PROCESSO DE ELETRONEBULIZAÇÃO DOS ANALITOS, ENTRE A
SAÍDA DO SISTEMA CROMATOGRÁFICO E A INJEÇÃO NO ESPECTRÔMETRO DE MASSAS. . 36
FIGURA 11 - REPRESENTAÇÃO DE UM ESPECTÔMETRO DE MASSAS, PARA MOSTRAR O
MODELO DE ANALISADOR DO TIPO QUADRUPOLO. ..................................................... 38
FIGURA 12 - REPRESENTAÇÃO DO ANALISADOR DE MASSAS ORBITRAP, COM SEU ELETRODO
EXTERNO (A) E O ELETRODO CENTRAL (B), ONDE, R E Z CORRESPONDEM ÀS
COORDENADAS CILÍNDRICAS. .................................................................................. 39
FIGURA 13 - ESQUEMA DO ANALISADOR ORBITRAP. ........................................................ 40
FIGURA 14 - CROMATOGRAMAS DO EXTRATO DA POLPA DE P. ALATA OBTIDOS POR
SBSE/CLAE-FLU DUAL: (A) DETECÇÃO NO COMPRIMENTO DE ONDA ESPECÍFICO PARA
O ALCALOIDE HARMANA E (B) DETECÇÃO NO COMPRIMENTO DE ONDA ESPECÍFICO PARA
HARMINA. .............................................................................................................. 49
FIGURA 15 - COMPARAÇÃO DOS CROMATOGRAMAS DO EXTRATO DA POLPA DE P. ALATA COM
(A) PADRÃO DE HARMANA 20 µG L-1 E (B) PADRÃO DE HARMINA 20 µG L-1, OBTIDOS POR
CLAE-FLU (ΛEXCITAÇÃO = 254 NM E ΛEMISSÃO = 425 NM PARA HARMANA E ΛEXCITAÇÃO = 254
NM E ΛEMISSÃO = 410 NM PARA HARMINA. ................................................................... 51
FIGURA 16 - COMPARAÇÃO DOS ESPECTROS DE FLUORESCÊNCIA DOS PICOS
CROMATOGRÁFICOS DOS ALCALOIDES (A) HARMINA (ΛEMISSÃO = 410 NM) E (B) HARMANA
(ΛEMISSÃO = 425 NM) PRESENTES NO EXTRATO DA POLPA DE P. ALATA, IDENTIFICADOS
POR SBSE/CLAE-FLU DUAL, E OS ESPECTROS DE FLUORESCÊNCIA DOS PICOS
CROMATOGRÁFICOS OBTIDOS PELA ANÁLISE CLAE-FLU DAS SOLUÇÕES DE PADRÕES
COMERCIAIS DE (C) HARMINA E (D) HARMANA. .......................................................... 52
FIGURA 17 - CURVAS ANALÍTICAS OBTIDAS PELO MÉTODO SBSE/CLAE-FLU POR ADIÇÃO DE
PADRÃO PARA QUANTIFICAÇÃO DE (A) HARMANA E (B) HARMINA NOS EXTRATOS DA
POLPA DE P ALATA. ................................................................................................ 53

FIGURA 18 - (A) CROMATOGRAMA DO ÍON TOTAL E (B) ESPECTRO CLUE-EM, IONIZAÇÃO
ELETROSPRAY, MODO POSITIVO: ÍON [M+H]+ = 183 DO PADRÃO HARMANA, TR = 2,05 MIN
(SOLUÇÃO 100 MG L-1). ......................................................................................... 56
FIGURA 19 - ESPECTRO CLUE-EM/EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO: ÍON
[M+H]+ = 183, TR = 2,05 MIN, DO ALCALOIDE HARMANA. ........................................... 57
FIGURA 20 - (A) CROMATOGRAMA DO ÍON TOTAL E (B) ESPECTRO CLUE-EM, IONIZAÇÃO
ELETROSPRAY, MODO POSITIVO: ÍON [M+H]+ = 213 DO PADRÃO HARMINA, TR = 2,65 MIN
(SOLUÇÃO 100 MGL-1). .......................................................................................... 58
FIGURA 21 - ESPECTRO CLUE-EM/EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO: ÍON
[M+H]+ = 213, TR = 2,65 MIN, ALCALOIDE HARMINA. ................................................. 59
FIGURA 22 - CAMINHO DE FRAGMENTAÇÃO DO ÍON MOLECULAR DO ALCALOIDE HARMINA. .. 60
FIGURA 23 - (A) CROMATOGRAMA DO ÍON TOTAL E (B) ESPECTRO CLUE-EM, IONIZAÇÃO
ELETROSPRAY, MODO POSITIVO: ÍON [M+H]+ = 199 DO PADRÃO HARMOL, TR = 1,81 MIN
(SOLUÇÃO 100 MG L-1). ......................................................................................... 61
FIGURA 24 - ESPECTRO CLUE-EM/EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO:
FRAGMENTAÇÃO DO [M+H]+ DO ALCALOIDE HARMOL. ............................................... 62
FIGURA 25 - CAMINHO DE FRAGMENTAÇÃO DO ÍON MOLECULAR DO ALCALOIDE HARMOL. ... 62
FIGURA 26 - (A) CROMATOGRAMA DO ÍON TOTAL E (B) ESPECTRO CLUE-EM, IONIZAÇÃO
ELETROSPRAY, MODO POSITIVO: ÍON [M+H]+ = 201 DO PADRÃO HARMALOL, TR = 1,60
MIN (SOLUÇÃO 100 MG L-1)..................................................................................... 63
FIGURA 27 - ESPECTRO CLUE-EM/EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO:
FRAGMENTAÇÃO DO [M+H]+ DO ALCALOIDE HARMALOL. ............................................ 64
FIGURA 28 - CAMINHOS DE FRAGMENTAÇÃO DO ÍON MOLECULAR DO ALCALOIDE HARMALOL.
............................................................................................................................ 65
FIGURA 29 - (A) CROMATOGRAMA DO ÍON TOTAL E (B) ESPECTRO CLUE-EM, IONIZAÇÃO
ELETROSPRAY, MODO POSITIVO: ÍON [M+H]+ = 215 DO PADRÃO HARMALINA, TR = 2,44
MIN (SOLUÇÃO 100 MG L-1)..................................................................................... 66
FIGURA 30 - ESPECTRO CLUE-EM/EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO:
FRAGMENTAÇÃO DO [M+H]+ DO ALCALOIDE HARMALINA. ........................................... 67
FIGURA 31 - CAMINHO DE FRAGMENTAÇÃO DO ÍON MOLECULAR DO ALCALOIDE HARMALINA.
............................................................................................................................ 67
FIGURA 32 - CROMATOGRAMA DO ÍON TOTAL DO BRANCO (METANOL), OBTIDO POR CLUE-
EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO. ...................................................... 69
FIGURA 33 - ESPECTRO CLUE-EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO, DO BRANCO
(METANOL). ........................................................................................................... 69
FIGURA 34 - CROMATOGRAMA DO ÍON TOTAL (TIC) DO EXTRATO TOTAL DA POLPA DE P.
ALATA, OBTIDO POR CLUE-EM, IONIZAÇÃO POR ELETROSPRAY, NO MODO POSITIVO. (A)
CROMATOGRAMA DO ÍON MOLECULAR (TIC) E CROMATOGRAMA DO ÍON EXTRAÍDO (B)
M/Z 183, (C) M/Z 199, (D) M/Z 201, (E) M/Z 213 E (F) M/Z 215. ................................ 70
FIGURA 35 - ESPECTRO CLUE-EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO: DOS PICOS
DO ÍONS (A) M/Z 183, (B) M/Z 199 E (C) M/Z 201, PRESENTES NOS CROMATOGRAMAS
DOS ÍONS EXTRAÍDOS DO EXTRATO DA POLPA DE P. ALATA FIGURA 34. ....................... 71

FIGURA 36 - CROMATOGRAMAS OBTIDOS ATRAVÉS DAS ANÁLISES REALIZADAS POR CLUE-
EM/EM DO PADRÃO DE HARMANA E DO EXTRATO DA POLPA DE P. ALATA. (A) SOLUÇÃO
PADRÃO DE HARMANA M/Z 183, TR = 2,05 MIN E (B) EXTRATO DA POLPA DE P. ALATA. . 72
FIGURA 37 - ESPECTROS DE MASSAS DA ANÁLISE CLUE-EM/EM DO ÍON [M+H]+ DE M/Z
183, TR = 2,05 MIN: (A) DO PADRÃO COMERCIAL DE HARMANA E (B) EXTRATO DA POLPA
DE P. ALATA. ......................................................................................................... 73
FIGURA 38 - CROMATOGRAMAS OBTIDOS ATRAVÉS DAS ANÁLISES REALIZADAS POR CLUE-
EM/EM DO PADRÃO DE HARMINA E DO EXTRATO DA POLPA DE P. ALATA. (A) SOLUÇÃO
PADRÃO DE HARMINA M/Z 213, TR = 2,83 MIN E (B) EXTRATO DA POLPA DE P. ALATA. .. 74
FIGURA 39 - ESPECTROS DE MASSAS DA ANÁLISE CLUE-EM/EM DO ÍON [M+H]+ DE M/Z
213, TR = 2,85 MIN: (A) DO PADRÃO COMERCIAL DE HARMINA E (B) EXTRATO DA POLPA
DE P. ALATA. ......................................................................................................... 75
FIGURA 40 - CROMATOGRAMA DO ÍON TOTAL (A) DO EXTRATO DAS SEMENTES DE P. ALATA,
OBTIDO POR CLUE-EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO. CROMATOGRAMA
DO ÍON EXTRAÍDO (B) M/Z 183, (C) M/Z 199, (D) M/Z 201, (E) M/Z 213 E (F) M/Z 215. . 76
FIGURA 41 - ESPECTROS CLUE-EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO: DOS PICOS
DOS ÍONS [M+H]+ (A) M/Z 183 (TR = 2,03 MIN), (B) M/Z 199 (TR = 1,68 MIN), (C) M/Z
201 (TR = 1,38 MIN), (D) M/Z 213 (TR = 2,80 MIN) E (E) M/Z 215 (TR = 2,54 MIN),
PRESENTES NOS CROMATOGRAMAS DOS ÍONS EXTRAÍDOS DO EXTRATO DAS SEMENTES
DE P. ALATA. ......................................................................................................... 77
FIGURA 42 - CROMATOGRAMA DO ÍON TOTAL (A) DO EXTRATO DAS SEMENTES DE P. EDULIS,
OBTIDO POR CLUE-EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO. CROMATOGRAMA
DO ÍON EXTRAÍDO (B) M/Z 183, (C) M/Z 199, (D) M/Z 201, (E) M/Z 213 E (F) M/Z 215. . 79
FIGURA 43 - CROMATOGRAMA DO ÍON TOTAL (A) DO EXTRATO DA POLPA DE P. EDULIS,
OBTIDO POR CLUE-EM, IONIZAÇÃO ELETROSPRAY, MODO POSITIVO. CROMATOGRAMA
DO ÍON EXTRAÍDO (B) M/Z 183, (C) M/Z 199, (D) M/Z 201, (E) M/Z 213 E (F) M/Z 215. . 80
FIGURA 44 - CROMATOGRAMAS OBTIDOS ATRAVÉS DAS ANÁLISES REALIZADAS POR CLUE-
EM/EM DO PADRÃO DE HARMALINA M/Z 215 (TR = 2,60 MIN) E DO EXTRATO DA POLPA
DE P. EDULIS (A) PADRÃO DE HARMALINA, (B) EXTRATO DA POLPA DE P. EDULIS E (C)
EXTRATO DAS SEMENTES DE P. EDULIS.................................................................... 81
FIGURA 45 - ESPECTROS DE MASSAS DA ANÁLISE CLUE-EM/EM DO ÍON [M+H]+ DE M/Z
215, TR = 2,60 MIN: (A) DO PADRÃO COMERCIAL DE HARMALINA E (B) EXTRATO DA
POLPA DE P. EDULIS. ............................................................................................. 82
FIGURA 46 - CROMATOGRAMAS OBTIDOS ATRAVÉS DAS ANÁLISES REALIZADAS POR CLUE-
EM/EM DO PADRÃO DE HARMINA M/Z 213 (TR = 2,85 MIN) E DO EXTRATO DA POLPA DE
P. EDULIS: (A) PADRÃO DE HARMINA (B) EXTRATO DAS SEMENTES DE P. EDULIS E (C)
EXTRATO DA POLPA DE P. EDULIS. .......................................................................... 83
FIGURA 47 - ESPECTROS DE MASSAS DA ANÁLISE CLUE-EM/EM DO ÍON [M+H]+ DE M/Z
213, TR = 2,85 MIN: (A) PADRÃO COMERCIAL DE HARMINA, (B) EXTRATO DA POLPA DE P.
EDULIS E (C) EXTRATO DAS SEMENTES DE P. EDULIS. ............................................... 84
FIGURA 48 - CROMATOGRAMAS OBTIDOS ATRAVÉS DAS ANÁLISES REALIZADAS POR CLUE-
EM/EM DO PADRÃO DE HARMANA M/Z 183 (TR = 2,09 MIN) E DO EXTRATO DA POLPA DE
P. EDULIS: (A) PADRÃO DE HARMANA (B) EXTRATO DA POLPA DE P. EDULIS, (C)
EXTRATO DAS SEMENTES DE P. EDULIS E (D) EXTRATO DA POLPA DE P. ALATA. .......... 85

FIGURA 49 - ESPECTROS DE MASSAS DA ANÁLISE CLUE-EM/EM DO ÍON [M+H]+ DE M/Z
183, TR = 2,05 MIN: (A) PADRÃO COMERCIAL DE HARMANA, (B) EXTRATO DA POLPA DE
P. EDULIS E (C) EXTRATO DAS SEMENTES DE P. EDULIS E (D) EXTRATO DA POLPA DE P.
ALATA. .................................................................................................................. 86
FIGURA 50 - GRÁFICOS DE PARETO PARA VISUALIZAÇÃO DOS EFEITOS DAS VARIÁVEIS
QUÍMICAS (% MEOH, PH, TEMPO DE EXTRAÇÃO (TEXTR), NACL E TEMPO DE
DESSORÇÃO (TDESS)) SOBRE A EXTRAÇÃO POR SBSE-EG SILICONE, PARA OS
ALCALOIDES (A) HARMANA E (B) HARMINA. ............................................................... 89
FIGURA 51 - GRÁFICOS DOS EFEITOS PRINCIPAIS PARA VISUALIZAÇÃO DA INFLUÊNCIA DOS
NÍVEIS DAS VARIÁVEIS QUÍMICAS (% MEOH, PH, TEMPO DE EXTRAÇÃO (TEXTRAÇ),
NACL E TEMPO DE DESSORÇÃO (TDESSOR)) SOBRE A EXTRAÇÃO POR SBSE-EG
SILICONE, PARA OS ALCALOIDES (A) HARMANA E (B) HARMINA. ................................... 90
FIGURA 52 – FORMAS IÔNICAS E MOLECULAR DO ALCALOIDE HARMANA EM SOLUÇÃO
AQUOSA. ............................................................................................................... 91
FIGURA 53 – FORMAS IÔNICAS E MOLECULAR DO ALCALOIDE HARMINA EM SOLUÇÃO AQUOSA.
............................................................................................................................ 91
FIGURA 54 - GRÁFICOS DA VARIAÇÃO DA ÁREA DO PICO CROMATOGRÁFICO DOS ALCALOIDES
(A) HARMINA E (B) HARMANA EXTRAÍDOS POR SBSE-EG SILICONE, EM FUNÇÃO DOS
DIFERENTES TEMPOS DE EXTRAÇÃO SOB AGITAÇÃO. ................................................. 93
FIGURA 55 - GRÁFICOS DA VARIAÇÃO DA ÁREA DO PICO CROMATOGRÁFICO DOS ALCALOIDES
(A) HARMINA E (B) HARMANA NA EXTRAÇÃO SBSE-EG SILICONE, EM FUNÇÃO DOS
DIFERENTES TEMPOS DE DESSORÇÃO NO ULTRASSOM. ............................................. 94
FIGURA 56 - CURVA DA PORCENTAGEM DE RECUPERAÇÃO DO ALCALOIDE HARMANA EM
FUNÇÃO DAS CONCENTRAÇÕES DA SOLUÇÃO DE EXTRAÇÃO, POR SBSE COM BARRAS
MAGNÉTICAS EG-SILICONE. ................................................................................... 96
FIGURA 57 - CURVA DA PORCENTAGEM DE RECUPERAÇÃO DO ALCALOIDE HARMINA EM
FUNÇÃO DAS CONCENTRAÇÕES DA SOLUÇÃO DE EXTRAÇÃO, POR SBSE-EG SILICONE.
............................................................................................................................ 97

LISTA DE TABELAS
TABELA 1 - PROCEDIMENTO DE EXTRAÇÃO DOS ALCALOIDES HARMÂNICOS HARMANA E
HARMINA A PARTIR DA POLPA DE P. EDULIS E P. ALATA. ............................................ 43 TABELA 2 - PROCEDIMENTO DE EXTRAÇÃO DOS ALCALOIDES HARMÂNICOS HARMANA E
HARMINA A PARTIR DAS SEMENTES SECAS DE P. EDULIS E P. ALATA........................... 43 TABELA 3 - NÍVEIS E FATORES AVALIADOS NO PLANEJAMENTO FATORIAL 25-1. ................... 44 TABELA 4 - MATRIZ DE EXPERIMENTOS (FATORES E SEUS RESPECTIVOS NÍVEIS) DO
PLANEJAMENTO FATORIAL FRACIONÁRIO 25-1. ........................................................... 45 TABELA 5 - CONDIÇÕES CROMATOGRÁFICAS DE ANÁLISE DOS ALCALOIDES HARMÂNICOS EM
CLAE-FLU. .......................................................................................................... 46 TABELA 6 - DETERMINAÇÃO QUANTITATIVA POR CLAE-FLU DOS ALCALOIDES EM POLPA DE
P. ALATA. .............................................................................................................. 54 TABELA 7 - ALCALOIDES HARMÂNICOS ESTUDADOS, SEUS RESPECTIVOS TEMPO DE
RETENÇÃO, ÍON MOLECULAR E ÍONS FILHOS. ............................................................ 55 TABELA 8 - AVALIAÇÃO DA IDENTIFICAÇÃO DOS ALCALOIDES HARMÂNICOS NOS EXTRATOS DA
POLPA E DAS SEMENTES DE P. ALATA E P. EDULIS. ................................................... 87

LISTA DE ABREVIATURAS
[M+H]+ - íon molecular protonado
APCI – atmospheric pressure chemical ionization (Ionização Química à Pressão
Atmosférica)
CG – Cromatografia Gasosa
CLAE – Cromatografia Líquida de Alta Eficiência; em inglês HPLC – High Pressure
Liquid Chromatography
CLAE-EM – Cromatografia Líquida de Alta Eficiência acoplada à Espectrometria de
Massas
CLAE-Flu – Cromatografia Líquida de Alta Eficiência acoplada a Detector de
Fluorescência
CLUE – Cromatografia Líquida de Ultra Eficiência; em inglês UHPLC – Ultra High
Pressure Liquid Chromatography
CLUE-EM – Cromatografia Líquida de Ultra Eficiência acoplada à Espectrometria de
Massas
Da – Daltons
EFS – extração em fase sólida
EG-Silicone – silicone modificado com etileno glicol
ELL – extração líquido-líquido
ESI – electrospray ionization (Ionização por Eletronebulização)
FE – fase estacionária
FM – fase móvel
H – altura de prato
KOW – coeficiente de partição em octanol/água
KPDMS/água – coeficiente de partição em PDMS e água
LD – liquid desorption (Dessorção Líquida)
m/z – relação massa carga
ODS – Octadecil silano
PAHs – Polycyclic aromatic hydrocarbon (Hidrocarbonetos Policíclicos aromáticos)
PDA – Photodiode Array (Arranjo de diodos)
PDMS – polidimetilsiloxano
SBSE – stir bar sorptive extraction (Extração Sortiva em Barra de Agitação)
SPME – solid phase micro extraction (Microextração por Fase Sólida)

TD – thermal desorption (Dessorção Térmica)
UV-Vis – Ultravioleta visível

SUMÁRIO
1 INTRODUÇÃO ...................................................................................17
1.1 Análise Fitoquímica: Importância e Aplicações ............................................................. 17
1. 2 Passiflora ................................................................................................................................. 18
1. 2. 1. Alcaloides harmânicos .................................................................................................. 20
1. 3 Stir Bar Sorption Extraction – SBSE .................................................................................... 21
1. 3. 1 Otimização do Método de Extração por SBSE .......................................................... 27
1. 4 Técnicas Cromatográficas de Análise ................................................................................. 28
1. 4. 1 Cromatografia Líquida de Alta Eficiência ................................................................... 28
1. 4. 2 Cromatografia Líquida de Ultra Eficiência acoplada à Espectrometria de Massas
....................................................................................................................................................... 32
2 OBJETIVOS .......................................................................................41
3 PARTE EXPERIMENTAL ..................................................................41
3. 1. Material Vegetal ..................................................................................................................... 41
3. 2 Extração dos analitos do material vegetal .......................................................................... 42
3. 2. 1 Materiais utilizados: ........................................................................................................ 42
3. 2. 2 Procedimento de extração: ........................................................................................... 42
3. 2. 2. 1 SBSE – PDMS........................................................................................................ 42
3. 2. 2. 2 Desenvolvimento SBSE – EG Silicone ............................................................... 44
3. 3 Análise por Cromatografia Líquida de Alta Eficiência com Detector de Fluorescência46
3. 3. 1 Análise quantitativa dos alcaloides em P. alata ........................................................ 46
3. 4 Análise por Cromatografia Líquida de Ultra Eficiência acoplada a Espectrometria de
Massas ............................................................................................................................................. 47
4 RESULTADOS E DISCUSSÃO .........................................................48
4. 1 Análise quantitativa dos alcaloides em polpa de P. alata ................................................ 48
4. 2 Análise por Cromatografia Líquida de Ultra Eficiência acoplada à Espectrometria de
Massas ............................................................................................................................................. 55
4. 2. 1 Análise dos extratos de Passiflora alata e Passiflora edulis ................................... 68
4. 2. 1. 1 Análise dos extratos de Passiflora alata ............................................................ 70
4. 2. 1. 2 Análise dos extratos de Passiflora edulis ........................................................... 79
4. 3 Extração de alcaloides harmânicos por SBSE-EG Silicone. ........................................... 88
5 CONCLUSÕES ..................................................................................98
REFERÊNCIAS .....................................................................................99

17
1 INTRODUÇÃO
1.1 Análise Fitoquímica: Importância e Aplicações
A biodiversidade tem oferecido aos seres humanos saúde e bem estar, por
oferecer elementos essenciais para a vida. A profusão de componentes genéticos e
bioquímicos presentes nos vegetais têm sido aproveitados na garantia de fonte de
alimentos e medicamentos (SHARMA et al., 2013).
A fitoquímica, tem grande importância na identificação dos compostos
químicos presentes nas estruturas dos vegetais. Compostos fitoquímicos são
moléculas produzidas pelos tecidos das plantas, durante o metabolismo primário ou
secundário, e estão associados ao mecanismo de proteção contra insetos, doenças,
radiação ultravioleta, contaminantes ambientais e ameaças que estes organismos
possam sofrer. Os metabólitos das plantas também apresentam características de
proteção para os consumidores humanos, por exemplo os flavonoides que possuem
atividade antioxidante (HAJNOS; SHERMA, 2011).
A área de aplicação mais importante dos métodos de investigação
fitoquímicos é a farmacognosia, campo de estudos de medicamentos derivados de
fontes naturais. Embora a maioria dos estudos estejam direcionados às plantas,
outros tipos de organismos também são de interesse, principalmente
microrganismos. A farmacognosia tem seus estudos divididos em: etnobotânica, que
estuda as plantas tradicionalmente utilizadas para tratamentos medicamentosos;
etnofarmacologia, que estuda as propriedades farmacológicas de substâncias
medicinais; fitoterapia, que estuda o uso medicinal de extratos vegetais; e
fitoquímica, que estuda os compostos químicos obtidos das plantas (HAJNOS;
SHERMA, 2011).
As plantas têm sido extensivamente estudadas, como fonte de substâncias
com propriedades medicinais, pela produção de moléculas bioativas, na grande
maioria envolvidas no sistema de defesa química contra predadores e infecções
(RIPA et al., 2009). Segundo a Organização Mundial de Saúde (OMS, ou WHO,
sigla em inglês), as plantas medicinais são a matriz para obtenção de uma variedade
de medicamentos (YADAV et al., 2011).
Os compostos fitoquímicos responsáveis por ação fisiológica específica no
organismo humano, são produzidos no organismo vegetal pelo metabolismo primário
e, mais comumente, secundário. Estes compostos estão associados a grande

18
variedade de terapias, humanas e veterinárias, à agricultura e pesquisas científicas.
Alguns destes compostos biologicamente ativos são taninos, alcaloides,
carboidratos, terpenoides, esteroides e flavonoides. Os compostos encontrados nas
raízes, sementes, frutos, folhas, flores, galhos, hastes, dentre outras partes das
plantas, são utilizados como princípio ativo nos medicamentos fitoterápicos há
muitos anos. Assim, o conhecimento da estrutura e função destas substâncias é de
grande valor para a síntese de produtos químicos mais complexos e mais efetivos
(YADAV et al., 2011).
Os extratos de plantas medicinais constituem-se de uma mistura complexa de
substâncias fitoquímicas, em que suas características diferem consideravelmente
entre as diferentes espécies. Até mesmo os extratos de uma mesma planta podem
diferir em sua composição de acordo com as condições climáticas durante o
crescimento, a origem, o processo de desidratação, entre outros fatores (YANG et
al., 2009).
Muitos medicamentos, provenientes de plantas, são utilizados e preferidos por
sua alta efetividade e baixa toxicidade, e estão presentes em produtos comerciais.
Materiais vegetais são obtidos de fontes naturais e cultivados para utilização como
fontes de substâncias medicinais, utilizados na indústria farmacêutica. Devido a
esses fatores há uma necessidade crescente do controle de pureza do material
vegetal, e pesquisas complementares para identificação e determinação da
composição química destes vegetais, para a obtenção do efeito terapêutico
desejado (HAJNOS; SHERMA, 2011).
1. 2 Passiflora
O gênero Passiflora compreende aproximadamente 500 espécies de plantas
da família Passifloraceae e é o de maior importância, com um maior número de
espécies cultivadas e consumidas. Encontradas na área que corresponde à América
Latina, principalmente América do Sul, as plantas são conhecidas como maracujá, e
em inglês denominadas “passion fruit” ou “passion flower” (ZUCOLOTTO et al.,
2012). Existem inúmeras formas de consumo do “maracujá”, onde a polpa do seu
fruto pode ser utilizada na produção de sucos e chás, e suas folhas têm aplicação na
produção de medicamentos.
Dentre todas as espécies conhecidas desse gênero, apenas 30 são descritas
como próprias para o consumo, e as mais comumente cultivadas e comercializadas

19
são o maracujá-azedo ou amarelo (Figura 1 a) (Passiflora edulis Sims, f. flavicarpa
Degener) e o maracujá-doce (Figura 1 b) (Passiflora alata Dryander). O Brasil é o
maior produtor de maracujá do mundo, com uma distribuição geográfica
principalmente localizada no Centro-Norte, e as principais formas de ingestão deste
fruto são por meio de chás, sucos e preparações alimentícias. Na Farmacopéia
Brasileira há relatos acerca das propriedades ansiolíticas atribuídas às folhas de P.
alata. Entretanto, estudos apontam possível toxicidade relacionada às espécies de
Passiflora, principalmente P. incarnata. Contudo, pouco conhecimento se tem das
espécies P. edulis e P. alata, sobretudo em relação à polpa e sementes (PEREIRA
et al., 2012).
A medicina popular usa, desde muito tempo, as folhas de várias espécies de
Passiflora como medicamento ansiolítico, sedativo e tranquilizante. Mesmo
possuindo registros na Farmacopéia Brasileira, como é o caso das folhas de
Passiflora alata Curtis, existem poucos estudos no que se refere à química dos
compostos ativos, tornando difícil a padronização do uso deste material vegetal para
produção de medicamentos. Experimentos in vivo foram realizados com o objetivo
de definir precisamente a atividade farmacológica relacionada às espécies de
Passiflora; contudo, seus efeitos não puderam ser atribuídos a somente um
composto, tendo sido apresentados dados que os conferem aos alcaloides e
flavonoides presentes em sua matriz (MACHADO et al., 2010).
Apesar do extensivo consumo, faltam informações acerca das reais
aplicações medicinais que apresentam. Os diversos estudos apontam que os
compostos majoritários destas espécies são alcaloides, flavonoides, glicosídeos,
(a) (b)
Figura 1 - Imagens dos frutos de (a) P edulis (maracujá azedo) e (b) P.alata (maracujá doce).

20
fenóis, além de compostos voláteis (DHAWAN et al., 2004). Os extratos das
espécies P. incarnata, P. edulis e P. alata têm sido os de maior interesse e alvo de
pesquisas, que revelaram a presença de flavonoides C-glicosídicos, saponinas e
alcaloides. Sendo as duas primeiras espécies as que possuem maior número de
informações sobre sua constituição fitoquímica (ZUCOLOTTO et al., 2012).
1. 2. 1. Alcaloides harmânicos
A atividade antioxidante, correlacionada aos flavonoides, dos frutos de
maracujá já foi objeto de pesquisa de vários trabalhos. Contudo, poucos
apresentaram a relação dos seus constituintes com o efeito ansiolítico do suco de
maracujá, sendo atribuído à presença de pequenas quantidades dos alcaloides
harmânicos. Estes alcaloides são do tipo indólico, caracterizados como o segundo
maior grupo de alcaloides conhecidos (ZERAIK et al., 2010).
Encontrados em extratos de Passiflora, os alcaloides harmânicos são um
grupo de alcaloides β-carbolínicos presentes em diversas famílias de plantas e, em
menor concentração, em alimentos cozidos. Inúmeras atividades farmacológicas têm
sido atribuídas a estes alcaloides, o que os torna alvo de intensa pesquisa
(ABOURASHED et al., 2003).
Em Passiflora edulis, foram identificados os alcaloides: harmana, harmina,
harmalina e harmalol (Figura 2), e a maior concentração apresenta-se nas folhas da
planta (DHAWAN et al., 2004). Estudos apontam que a harmina (Figura 2b) é o
principal constituinte da classe dos alcaloides harmânicos que apresenta efeitos
farmacológicos (ONISHI et al., 2012).
Os alcaloides harmânicos estão descritos como constituintes em diversos
tipos de alimentos de fonte vegetal, sendo encontrados também em carnes muito
cozidas e substâncias provenientes de fumaças de queima de vegetais, além de
possuir atividade psicotrópica - podem interagir de forma irreversível com as
enzimas monoamina oxidase A e inibição da enzima acetilcolinesterase -,
apresentam ação antitumoral, efeitos analgésicos, atividade vasorrelaxante e
antimicrobiana. Alguns estudos, ainda, relatam que os alcaloides β-carbolínicos
atuam como substâncias co-mutagênicas na presença de aminas aromáticas, por
exemplo, anilina e toluidina (ZHAO et al., 2012; ONISHI et al., 2012).

21
As espécies de maracujá P. edulis e P. alata apresentam os alcaloides
harmânicos e devido à sua atividade inibidora sobre as enzimas monoamina
oxidases, têm sido considerados como as principais substâncias bioativas presentes
nas espécies de Passiflora (DHAWAN et al., 2004).
1. 3 Stir Bar Sorption Extraction – SBSE
Anteriormente à análise e detecção dos compostos orgânicos presentes em
amostras alimentares e vegetais, é necessária uma etapa de extração. A técnica de
preparação da amostra, composta pela extração, “clean up” e concentração de
analitos, reduz os efeitos de matriz, que podem influenciar na exatidão, precisão e
robustez do método bioanalítico (WU et al., 2013). A partir de matrizes aquosas de
alimentos, o processo de extração pode ser realizado por meio de técnicas
tradicionais como extração líquido-líquido (ELL) e extração em fase sólida (EFS), as
quais demandam, principalmente a primeira, o uso de grandes quantidades de
solvente, uma quantidade relativamente grande de amostra, e ainda pode ser
necessária a realização de uma etapa de “clean up” (ZUIN et al., 2005).
O tipo de extração que se deseja realizar é o critério mais importante na
comparação e seleção da técnica de extração. O processo de extração pode ser
baseado no equilíbrio entre líquido-gás, extração dinâmica líquido-gás, partição ou
adsorção líquido-líquido, e extração por sorção. Esta última tem sido vista como uma
alternativa ecologicamente favorável, principalmente à extração líquido-líquido. Na
extração por sorção os analitos da fase líquida ou gasosa entram na matriz de uma
fase líquida imiscível (fase extratora), ao invés de se aderirem a um sítio específico
ou à superfície do adsorvente (DAVIS et al., 2007).
(a) (b) (c)
(d) (e)
Figura 2 - Estrutura dos alcaloides harmânicos: (a) Harmana (b) Harmina (c) Harmalina (d) Harmalol e (e) Harmol.

22
O cenário atual, dentro das análises químicas, busca à simplificação,
miniaturização, fácil manipulação dos aparelhos analíticos, redução ou extinção do
uso de solventes orgânicos, além de baixos volumes de amostra, de acordo com os
princípios da “química verde” (NOGUEIRA, 2012). Assim, novas técnicas para
preparação de amostras têm se tornado um dos enfoques da química analítica,
oferecendo a possibilidade de automação, seletividade, boa eficiência e rapidez.
Além de serem consideradas “limpas” ou “ambientalmente corretas”, deseja-se que
tenham custo reduzido, fácil manipulação e utilizem pouco ou nenhum solvente
(VIÑAS et al., 2008).
Um exemplo desse tipo de técnica é a microextração em fase sólida, SPME
(solid phase micro extraction), que promove a concentração de compostos voláteis
ou não voláteis em amostras líquidas simplesmente por imersão direta ou
“headspace”. Outra técnica, que utiliza pouquíssima quantidade de solvente para a
extração de compostos orgânicos, é conhecida como extração por sorção em barra
magnética, SBSE (stir bar sorptive extraction). Constitui-se de uma barra magnética
recoberta por um polímero, em resumo (VIÑAS et al., 2008). A SBSE é considerada
uma técnica “verde” de preparação de amostra, pois sua aplicação é capaz de
eliminar ou reduzir a utilização de solventes, por dessorção térmica e dessorção
líquida, respectivamente (PLOTKA et al., 2013). A figura 3 mostra como ocorre o
processo de sorção dos analitos de interesse durante a etapa de extração (a),
seguida pela etapa de dessorção líquida (b).
Figura 3 - Representação do método de extração por sorção, utilizando barras magnéticas recobertas com polidimetilsiloxano (PDMS) (a) e o processo de dessorção líquida (b).
(Fonte: Adaptado de RODRIGUEZ et al., 2013).
Na SBSE, o líquido imiscível no qual os analitos se difundem é conhecido
como fase extratora. O material mais amplamente utilizado nesta função é o
polidimetilsiloxano (PDMS), que também tem grande aplicação como fase
estacionária na técnica de cromatografia gasosa; é termoestável, podendo ser
utilizado dentro de uma larga faixa de temperaturas. Ainda, o PDMS como fase

23
extratora garante o enriquecimento da amostra, ausência dos efeitos de
deslocamento, inércia e a rápida dessorção térmica em temperaturas amenas. Após
a extração, o analito pode ser dessorvido por dessorção térmica (TD, thermal
desorption) ou por dessorção líquida (LD, liquid desorption), usando um solvente
orgânico (DAVIS et al., 2007).
Arthur e Pawliszyn propuseram o método de microextração por sorção em
PDMS, denominado SPME, no qual a fase estacionária sortiva de
polidimetilsiloxano, aderido à uma fibra, é colocada em contato com a amostra. No
entanto, algumas complicações com relação ao baixo volume de fase extratora
(aproximadamente 0,5 µL) recobrindo a fibra e a polaridade dos analitos levaram os
pesquisadores a procurar por outros métodos que fossem mais eficientes. Desta
forma, desenvolveram-se as barras de agitação recobertas de uma fina camada de
polidimetilsiloxano, que em agitação na amostra aquosa possibilitava a extração e o
enriquecimento dos solutos na cobertura de PDMS. Esta técnica recebeu o nome de
extração por sorção em barra magnética, SBSE em inglês. A técnica de SBSE
emprega o mesmo princípio de extração por sorção que a técnica de SPME, no
entanto possui um volume de polidimetilsiloxano entre 25 e 125 µL, que é 50 a 250
vezes maior na SPME (DAVIS et al., 2007; CALDAS et al., 2011).
O processo de extração por sorção ocorre por equilíbrio: em amostras
aquosas, o controle dos analitos que são extraídos do meio ocorre por meio do
coeficiente de partição dos solutos entre a fase extratora e a fase aquosa. Este
coeficiente de partição está relacionado ao coeficiente de distribuição octanol-água,
por meio do qual se pode predizer se, e quão bem, um determinado soluto poderá
ser extraído pela técnica de SPME ou SBSE (DAVIS et al., 2007).
O coeficiente de partição do analito entre a fase extratora e a fase aquosa
corresponde à razão entre a concentração deste soluto na fase de
polidimetilsiloxano e a concentração em água, quando em equilíbrio. Este valor
corresponde à massa de analito presente na fase extratora dividida pela massa na
fase aquosa, multiplicada pela razão de fase β, conforme mostra a equação [1] a
seguir.

24
Koctanol/água ≈ KPDMS/água = 𝐶𝑃𝐷𝑀𝑆
𝐶á𝑔𝑢𝑎 =
𝑚𝑃𝐷𝑀𝑆
𝑚á𝑔𝑢𝑎
𝑉á𝑔𝑢𝑎
𝑉𝑃𝐷𝑀𝑆 =
𝑚𝑃𝐷𝑀𝑆
𝑚á𝑔𝑢𝑎𝛽 [1]
CPDMS = concentração do analito no PDMS
Cágua = concentração do analito em água
mPDMS = massa do analito no PDMS
mágua = massa do analito na água
Vágua = volume de água
VPDMS = volume de PDMS
β = Vágua/VPDMS
A quantidade de analito que pode ser recuperada é calculada pela razão entre
a massa de soluto na fase de polidimetilsiloxano e a massa inicial
(m0 = mágua + mPDMS), que correspondente a 𝐾𝑃𝐷𝑀𝑆/á𝑔𝑢𝑎 𝛽⁄
1+(𝐾𝑃𝐷𝑀𝑆/á𝑔𝑢𝑎 𝛽)⁄.
Quanto maior o valor do coeficiente de partição do analito (KPDMS/água), mais
eficiente será a extração, ou seja, maior será a recuperação do analito. Então,
analitos mais polares serão menos retidos pelo polidimetilsiloxano. Além disso, a
razão de fase β é outro parâmetro importante: maiores quantidades de PDMS levam
a menores valores de β, e maior será a eficiência da extração. Estudos realizados
com PAHs (Hidrocarbonetos Aromáticos Policíclicos) e pesticidas, extraídos de
soluções aquosas usando SBSE e SPME, mostraram a maior recuperação obtida
pela aplicação da técnica de extração por SBSE (DAVIS et al., 2007).
Na análise de quantidades muito pequenas, a recuperação teórica - que pode
ser calculada a partir do volume de amostra, das dimensões da barra de extração e
das características do soluto por meio de um programa de software - é tão
importante quanto o fator de enriquecimento e o total de analito que pode ser
introduzido e detectado (DAVIS et al., 2007).
Além dos parâmetros termodinâmicos, os critérios que afetam a cinética do
processo de extração são de extrema importância e devem ser analisados e
utilizados de forma a obter-se uma extração eficiente. A quantidade de soluto que
migra para a cobertura de polidimetilsiloxano é determinada pela constante de
difusão, pela condição de agitação, volume de amostra, entre outros. No entanto,
quanto maior o volume de amostra e de PDMS, mais tempo é necessário para que a
extração seja eficiente como se espera. Então, uma boa correlação entre o volume
de amostra e o volume de fase extratora pode ser alcançada, resultando em um

25
tempo aplicável de análise. Após a etapa de dessorção dos analitos presentes na
fase extratora, as barras de agitação poderão ser novamente utilizadas (DAVIS et
al., 2007).
Os compostos retidos na fase extratora podem ser dessorvidos termicamente,
antes da análise em cromatógrafo gasoso, por meio de uma unidade de dessorção
térmica; ou por dessorção líquida, quando o método de análise aplicado for
cromatografia líquida (CACHO et al., 2013). A técnica LD é considerada menos
onerosa e fácil de trabalhar, já que não é necessário um dipositivo no equipamento
de cromatografia para dessorção dos analitos (MAGI et al., 2012).
As barras de extração de PDMS comercializadas possuem comprimento entre
1 e 2 cm, recobertas com uma camada entre 0,5 ou 1 mm de polímero. O bastão
magnético deve estar protegido com uma camada de vidro, onde a cobertura de
polidimetilsiloxano é aderida, uma vez que o contato direto com a barra pode levar à
rápida degradação do polímero, como pode ser visto na figura 4 (a) (DAVIS et al.,
2007).
Figura 4 - Representação esquemática de uma barra comercial para SBSE de (a) PDMS e (b) de uma barra do tipo “dual phase”.
(a) barra convencional (“Twister”) (b) “dual-phase twister”
Fonte: Adaptado de BICCHI et al., 2005.
As barras de extração recobertas com PDMS apresentam algumas limitações,
assim novos materiais poliméricos têm sido estudados e utilizados com o objetivo de
melhorar a flexibilidade e seletividade das barras, de forma que sua capacidade de
concentração seja mantida e suas aplicações possam ser expandidas (NOGUEIRA,
2012). Barras de extração de duas fases foram propostas experimentalmente, e
consistem em uma cobertura de PDMS do lado de fora com um material de carbono
adsorvente recobrindo a parte interna (figura 4 b). Esta estrutura possibilita o
aumento de recuperação de analitos polares em amostras aquosas (DAVIS et al.,
2007).

26
Recentemente foram lançadas barras de extração recobertas por fase
extratora mais polar, de silicone modificado com etileno glicol, comercializadas como
“Ethylene Glycol (EG)-Silicone Twister®” (Figura 5). Por serem mais polares que o
PDMS, extraem compostos polares, com menores valores de log Ko/w, mais
eficientemente, como, por exemplo, os alcaloides harmânicos harmalina (Log Ko/w =
2.57), harmol (Log Ko/w = 1.82) e harmalol (Log Ko/w = 2.19). A base de silicone
garante também a extração eficiente de compostos não polares, com valores de log
Ko/w altos.
Figura 5 - Barra de extração de EG-Silicone Twister®”.
(Fonte: Gerstel, 2011).
O estudo comparativo entre as barras magnéticas recobertas por EG-Silicone
e PDMS concluiu que a primeira é capaz de realizar uma extração mais eficiente de
compostos polares. Na extração de fenóis, furanos, alcoóis e ácidos as barras EG-
Silicone Twister apresentam extração mais eficiente. Já para espécies não polares
como terpenos e ésteres etílicos, as barras EG-Silicone Twister fornecem uma
extração similar às das barras recobertas com PDMS (NIE et al., 2011).
A técnica de SBSE é influenciada fortemente pela complexidade da matriz,
que afeta a recuperação dos analitos alvo. Entretanto, seguindo os métodos
analíticos desenvolvidos, a técnica de SBSE tem sido amplamente utilizada na
análise de substâncias presentes em concentrações muito baixas, em diferentes
áreas relacionadas à sociedade, tais como: ambiental, alimentar e sensorial,
biomédica, forense e farmacêutica (NOGUEIRA, 2012).
Uma vez que a concentração dos alcaloides harmânicos harmana e harmina,
presentes nas sementes secas de maracujá azedo (P. edulis), é muito pequena, na
ordem de nanogramas (RODRIGUES, 2013), a técnica de pré-concentração é
adequada para a preparação das amostras de sementes e polpas de Passiflora alata
e Passiflora edulis. A técnica de SBSE, desde o seu desenvolvimento em 1999, é

27
uma das mais populares técnicas de pré-concentração, por sua simplicidade e
robustez (CACHO et al., 2013).
As aplicações da técnica de SBSE em análises alimentares são classificadas
dentro de três grupos: análise de compostos minoritários (compostos voláteis e
aditivos), determinação de compostos de aroma não desejados (off-flavours) e
determinação de compostos de contaminantes em baixas concentrações (DAVIS et
al., 2007). Matrizes alimentares foram analisadas utilizando a técnica de SBSE,
como por exemplo, análise de compostos voláteis ou semi-voláteis de pesticidas em
frutas e vegetais, contaminantes e compostos de aroma em bebidas (DAVIS et al.,
2007; NOGUEIRA, 2012). Barras de SBSE são submetidas a diferentes tratamentos
de forma a comparar sua eficiência de extração para compostos de diferentes
polaridades. Em um estudo realizado por Xu e colaboradores, diferentes métodos de
SBSE foram desenvolvidos para a extração de conservantes com diferentes
polaridades de amostras alimentares (XU et al., 2013).
A técnica de extração por SBSE é recente, mas o número de artigos e
trabalhos publicados mostrando sua aplicação principalmente na análise de
compostos orgânicos contidos em pequenas concentrações indicam as vantagens
em comparação às outras técnicas de extração por sorção. Além disso, artigos de
revisão apontam as possibilidades de métodos e aplicações da técnica de SBSE
(NOGUEIRA, 2012).
1. 3. 1 Otimização do Método de Extração por SBSE
Para que o método de extração por SBSE seja eficiente e reprodutível é
necessária uma etapa de otimização do método. A SBSE envolve diversas variáveis
que interferem simultaneamente no método. A avaliação da influência dessas
diversas variáveis (fatores) que determinam a resposta de um processo químico é
uma das tarefas de indústrias e laboratórios de pesquisa, com o objetivo de
reconhecer valores (níveis) para esses fatores de forma que a resposta seja
maximizada ou minimizada, dependendo da proposta do procedimento (ZERAIK,
2010).
Os sistemas de planejamento fatorial, com análise multivariada, permitem
avaliar simultaneamente o efeito de um grande número de variáveis, com menor
número de ensaios experimentais, comparativamente aos métodos de otimização

28
univariada, pois investiga a influência de todas as variáveis de interesse e os efeitos
que as interações têm sobre a resposta ou respostas (TEOFILO; FERREIRA, 2006).
Quando a combinação de k fatores é analisada em dois níveis, por exemplo, o
planejamento fatorial consiste de 2k ensaios. Os níveis são designados por (-), para
o nível mais baixo, e (+), para o nível mais alto. Um nível zero pode ser inserido no
centro dos ensaios, e neste ponto todas as variáveis estão em seu valor médio. Os
pontos centrais (nível zero, sendo a média dos níveis superior e inferior) dos
planejamentos permitem identificar relações não lineares no intervalo, e a estimativa
do erro experimental, sem a necessidade de replicatas em todos os pontos do
planejamento.
Utiliza-se os planejamentos fatoriais fracionários, quando o k > 4, pois os
efeitos podem ser não significativos, desse modo a realização de todos os ensaios
para estimar tais efeitos de interação pode ser irrelevante. Assim, é necessário um
número menor de experimentos, mas sem perder informações importantes sobre os
efeitos no sistema (TEOFILO; FERREIRA, 2006).
1. 4 Técnicas Cromatográficas de Análise
1. 4. 1 Cromatografia Líquida de Alta Eficiência
A análise de alcaloides β-carbolínicos nas diversas matrizes complexas
(alimentos de origem animal e vegetal) exige, como primeira etapa, sua extração,
seguido pela identificação, anteriormente às análises quantitativas. No entanto,
como estas substâncias estão presentes em pequenas concentrações, as técnicas
analíticas que têm apresentado melhores resultados são a cromatografia gasosa
(CG) e a cromatografia líquida de alta eficiência (CLAE). A utilização de
cromatografia gasosa apresenta a desvantagem de requerer a derivatização química
da amostra, na geração de β-carbolinas voláteis. O detector ultravioleta e sistemas
eletroquímicos de detecção podem ser utilizados para a técnica de CLAE, mas os
detectores por fluorescência (Flu) e espectrômetro de massas (EM) são preferidos,
devido à seletividade e sensibilidade de detecção (HERRAIZ, 2000).
A utilização de detectores de fluorescência, para quantificação de alcaloides
harmânicos, está fundamentada na sua emissão fluorescente. Uma vez que estes
alcaloides contêm um grupo β-carbolínico, alguns estudos relatam a intensa
fluorescência na região do azul (entre 400 e 500 nm) (AMJADI et al., 2014).

29
Estudos na área de análise de produtos naturais, que aplicam a cromatografia
líquida acoplada à espectrometria de massas, são cada mais comuns, uma vez que
a capacidade de detecção e separação dos analitos têm sido aumentadas e
melhoradas nos equipamentos de CLAE-EM. Para a análise de compostos
fitoquímicos a técnica de CLAE de fase reversa, utilizando colunas C18 ou ODS
(Octadecil silano), é a mais aplicada. De acordo com o propósito da separação, as
características químicas do material de empacotamento e os aspectos físicos da
coluna devem ser levados em consideração para a definição da melhor alternativa
(WU et al., 2013).
O sistema analítico de CLAE-EM é composto pelo sistema de injeção de
amostras (autosampler), o sistema de CLAE de bombas e coluna cromatográfica,
detector UV e o espectrômetro de massas (Figura 6), estando todos os elementos
conectados a um sistema computacional, que permite a coleta e análise dos
resultados obtidos.
Figura 6 - Ilustração de um sistema on-line CLAE-EM, com a sequência de elementos.
(Fonte: Adaptado de CASS; BARREIRO, 2011).
A cromatografia líquida instrumental tem seu início nos anos 50, e os
desenvolvimentos significativos que ocorreram desde então são resultado do intenso
uso da CLAE, que tornou-se a técnica analítica mais desenvolvida e amplamente
utilizada em laboratórios químicos, farmacêuticos e médicos. Uma história que
começou com colunas recheadas com partículas irregulares de 100-200 µm e hoje
em dia utiliza partículas de fase estacionária com diâmetros inferiores a 2 µm. A
busca por análises mais rápidas e com melhor rendimento cromatográfico
impulsionaram as pesquisas. Inicialmente, a alternativa mais simples, visando a
diminuição do tempo de análise, seria a aplicação de colunas de menor

30
comprimento e vazões elevadas de fase móvel (FM). Entretanto, apesar desta
estratégia ter sido utilizada com colunas contendo partículas de 3-3,5 µm, as
separações não apresentavam melhor desempenho cromatográfico, além de
perderem resolução (MALDANER et al., 2009).
A eficiência do sistema de separação da CLAE pode ser observada pela
equação [2] a seguir, conhecida como equação de van Deemter:
𝐻 = 𝐴𝑑𝑝 + 𝐵𝐷𝑀
µ+
𝐶𝑑𝑝2µ
𝐷𝑀 [2]
H = altura de prato
µ = velocidade linear da fase móvel
dp = tamanho da partícula de empacotamento
DM = coeficiente de difusão do analito
A = constante dos diferentes caminhos seguidos pelas moléculas do analito.
B = constante da difusão longitudinal ou difusão do soluto na FM.
C = constante da transferência de massa do analito entre FM e fase
estacionária (FE).
A base da evolução da CLAE está no constante desenvolvimento de colunas
com partículas cada vez menores, como explicado pela análise da equação de van
Deemter. O termo A está relacionado ao alargamento dos picos cromatográficos
devido aos diferentes percursos que as moléculas do analito podem seguir dentro da
coluna. Assim, a influência deste termo pode ser reduzida utilizando colunas com
diâmetros menores, partículas menores e mais uniformes. O termo B corresponde à
difusão longitudinal ou difusão do analito na FM, a partir da aplicação de velocidades
lineares mais altas da FM, a contribuição deste termo pode ser minimizada. E o
termo C é atribuído à transferência de massa do analito entre a FM e a FE
(MALDANER et al., 2009).
Uma coluna mais eficiente terá uma altura de prato menor e,
consequentemente, maior número de pratos por comprimento. O valor mais baixo de
H na curva de van Deemter, representada na figura 7, corresponde à vazão ótima da
FM que resultará na máxima eficiência da coluna. Partículas menores,
principalmente <2 µm, apresentam valor de H menor a uma vazão maior, e esse
resultado está relacionado aos termos A, B e C. As partículas com tamanhos
inferiores possibilitam troca mais rápida do soluto entre a FM e os poros das
partículas, já que os poros possuem menor profundidade, e este comportamento é
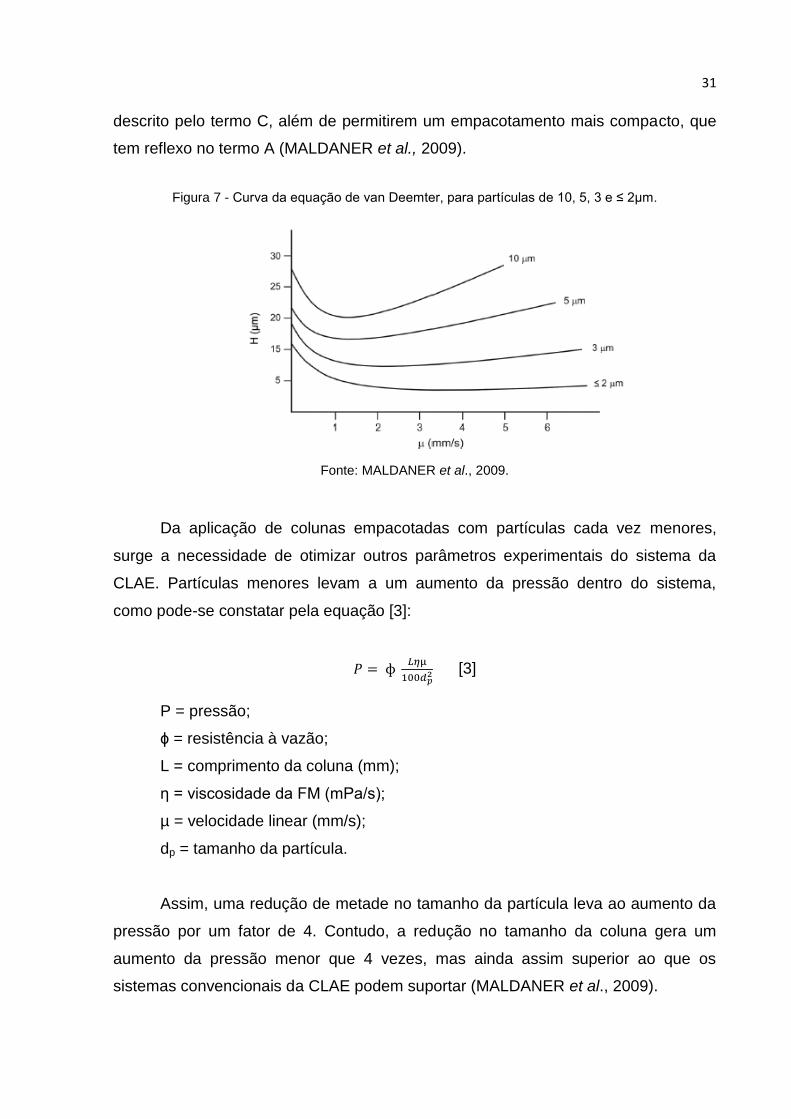
31
descrito pelo termo C, além de permitirem um empacotamento mais compacto, que
tem reflexo no termo A (MALDANER et al., 2009).
Figura 7 - Curva da equação de van Deemter, para partículas de 10, 5, 3 e ≤ 2µm.
Fonte: MALDANER et al., 2009.
Da aplicação de colunas empacotadas com partículas cada vez menores,
surge a necessidade de otimizar outros parâmetros experimentais do sistema da
CLAE. Partículas menores levam a um aumento da pressão dentro do sistema,
como pode-se constatar pela equação [3]:
𝑃 = ɸ 𝐿𝜂µ
100𝑑𝑝2 [3]
P = pressão;
ɸ = resistência à vazão;
L = comprimento da coluna (mm);
η = viscosidade da FM (mPa/s);
µ = velocidade linear (mm/s);
dp = tamanho da partícula.
Assim, uma redução de metade no tamanho da partícula leva ao aumento da
pressão por um fator de 4. Contudo, a redução no tamanho da coluna gera um
aumento da pressão menor que 4 vezes, mas ainda assim superior ao que os
sistemas convencionais da CLAE podem suportar (MALDANER et al., 2009).

32
As colunas cromatográficas “eXtented Performance XP”, da Waters, são
empacotadas com partículas de fase estacionária com 2,5 µm de diâmetro. Foram
desenvolvidas para atuar na transição do sistema CLAE para o sistema de
cromatografia líquida de ultra eficiência (CLUE - UHPLC, Ultrahigh performance
liquid chromatography, em inglês), e são compatíveis com ambos os sistemas de
cromatografia líquida, com o objetivo de aumentar a produtividade do sistema
submetido a contra-pressões intermediárias. A possibilidade de aplicação de colunas
com partículas menores aumenta a qualidade das separações cromatográficas,
aumenta a velocidade de análise, o que diminui o tempo e os custos relativos, pois
utiliza quantidades menores de FM. A coluna BEH Shield RP18 XP 2.5 µm, é da
linha XBridge, desenvolvida para suportar grandes diferenças de pH, aumentando o
tempo de uso (WATERS, 2012, 2014).
1. 4. 2 Cromatografia Líquida de Ultra Eficiência acoplada à
Espectrometria de Massas
Para análises quantitativas, a CLAE utiliza detectores como indíce de
refração, ultravioleta, espalhamento de luz e fluorescência, entre outros, por meio de
softwares dedicados, que permitem a quantificação de substâncias em
concentrações muito baixas. Apesar do uso do tempo de retenção na tentativa de
identificar compostos, uma vez que estes possuem tempos de retenção
característicos, esta é uma prática que pode levar a resultados errôneos. Diferentes
compostos podem apresentar o mesmo tempo de retenção numa mesma condição
cromatográfica. Mesmo depois da aplicação de sistemas de detecção fotométrica,
com detectores UV-VIS de comprimento de onda variável e com arranjos de diodos
(PDA, Photodiode Array), a identificação dos compostos presentes nos picos
cromatográficos não é tarefa fácil. O desenvolvimento de sistemas que permitiram o
acoplamento da CLAE e o espectrômetro de massas trouxe uma nova forma de
identificação de compostos (LANÇAS, 2009). A figura 8 apresenta um fluxograma
das etapas do sistema de detecção usando espectrômetro de massas. Quando o
objetivo é a identificação dos compostos, um espectrômetro de massas acoplado ao
cromatógrafo líquido de alta eficiência é considerado o melhor sistema hifenado,
uma vez que geram análises altamente seletivas e específicas (HERRAIZ, 2000).

33
Figura 8 - Fluxograma do funcionamento do sistema de detecção de espectrometria de massas.
Fonte: Adaptado de LANÇAS, 2009.
No fim dos anos 90, dentro de laboratórios acadêmicos, utilizando sistemas
de fabricação própria para cromatografia líquida de ultra alta eficiência (CLUAE),
como era denominado anteriormente, surgiram os primeiros estudos com resultados
relevantes. Então, o potencial de separação em um sistema de CLUAE despertou o
interesse em pesquisas para o desenvolvimento de instrumentação adequada, com
pressões elevadas, partículas com diâmetro inferior a 2 µm e colunas semelhantes
às de CLAE. O sistema CLUE, desenvolvido comercialmente, possui capacidade de
trabalhar sob altas pressões (100 Mpa), os volumes internos têm de ser muito
menores (conexões, alça de amostragem, cela de detector, bombas) com celas de
detector que não tenham dispersão e alta taxa de aquisição, e melhoramento no
sistema de controle e de dados, enquanto que as colunas têm de ser resistentes às
altas pressões e com menor volume morto, bem como injetores capazes de trabalhar
com faixa de volumes muito pequenos (de 0,1 a 50 µL) (MALDANER et al., 2009).
A cromatografia líquida de ultra eficiência é uma técnica versátil e pode ser
usada para aumentar o rendimento de análises, principalmente correspondentes a
amostras complexas, como extratos vegetais e seus metabólitos, utilizando colunas
empacotadas com partículas de tamanho inferior a 2 µm, operando sob altas
pressões, separações eficientes e superiores podem ser obtidas em menores
tempos (WU et al., 2013).

34
Estudos revelam que a aplicação da CLUE gera análises mais rápidas, com
menor consumo de solventes e maior detectabilidade. Além disso, há a possibilidade
de transferência de um método CLAE para CLUE, e seu menor volume de eluição é
adequado à utilização do espectrômetro de massas como detector. Para essa
transferência, alguns parâmetros devem ser observados, as fases estacionárias das
colunas devem ser similares, enquanto o volume de injeção e a vazão da fase móvel
deverão ser otimizados (MALDANER et al., 2009).
O desenvolvimento de métodos de ionização sob pressão atmosférica
permitiram a conexão da cromatografia líquida de alta eficiência ao espectrômetro
de massas, e são capazes de transferir a amostra que sai da coluna cromatográfica
para a fase gasosa e a ionização desta. Os modos de ionização química sob
pressão atmosférica (APCI, atmospheric pressure chemical ionization, em inglês) e
ionização por eletronebulização (ESI, electrospray ionization, em inglês) são as
técnicas mais aplicadas no acoplamento entre CLAE e EM. Na técnica de APCI, o
eluente da coluna cromatográfica, solventes e analitos, são submetidos à
vaporização e em seguida sofrem o processo de ionização. Este ocorre pela
aplicação de uma descarga elétrica sob o fluido que está em contato com um
condutor energizado eletricamente. É considerada uma técnica adequada para
análise de compostos neutros, além disso, requer menos “clean-up” da amostra e
comporta facilmente o fluxo típico (1 mL min-1) das colunas cromatográficas. A
sensibilidade do método é dependente da volatilidade dos compostos eluídos
(YOUDIM et al., 2010).
A técnica de ionização mais comumente utilizada, no acoplamento entre
CLAE e EM, é a ESI. O processo ocorre, inicialmente, pela dissolução da amostra
em um solvente, geralmente não polar, que é pressurizado em um tubo capilar de
aço inox. Uma voltagem entre 3000 e 5000 V é aplicada sob esse tubo capilar,
fazendo o líquido emergir do capilar à pressão atmosférica, como um aerossol.
Estas gotículas, formadas pela aspersão do líquido, são dessolvatadas
continuamente dentro do ambiente sob vácuo. À medida que as gotículas perdem
solvente, aumentam as forças repulsivas resultantes da carga imposta, ocorrendo
“explosões” coulômbicas, gerando íons dos analitos. Estes íons migram para o
analisador de massas, induzidos pela atração eletrostática. Estes fenômenos estão
representados na figura 9, ilustrando o caminho do analito. A eletronebulização pode
ser operada no modo positivo ou negativo, controlando-se apenas o sinal da tensão
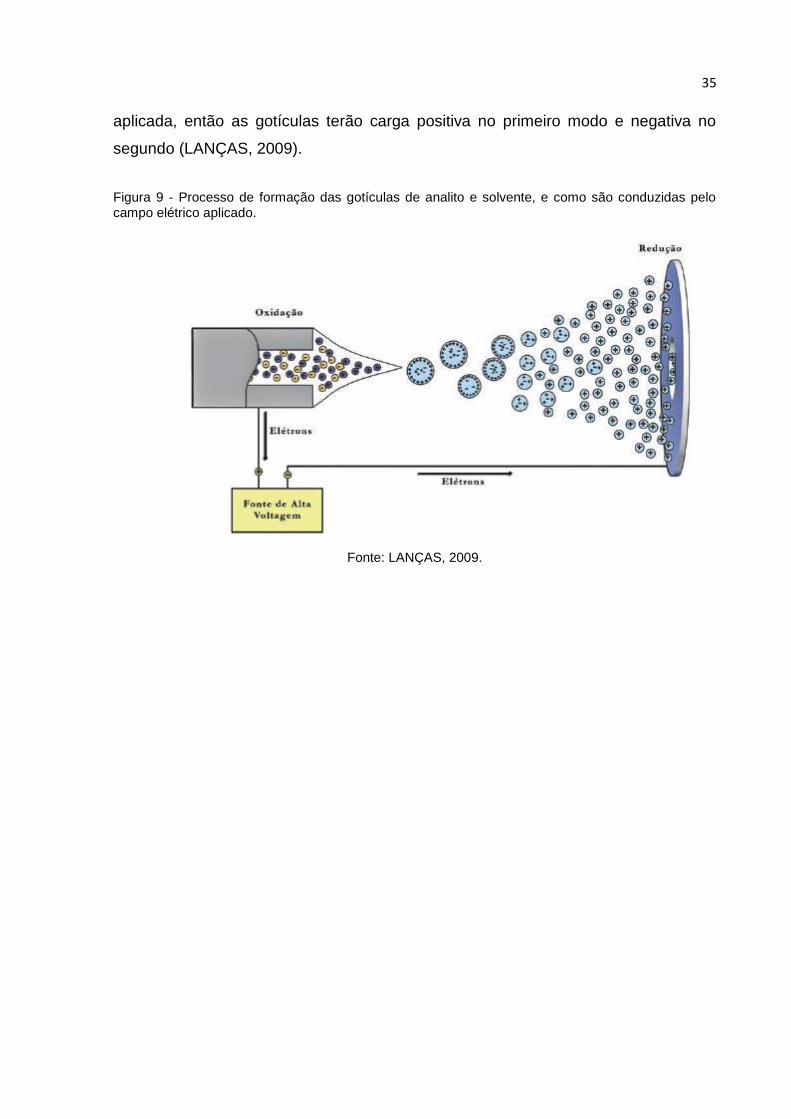
35
aplicada, então as gotículas terão carga positiva no primeiro modo e negativa no
segundo (LANÇAS, 2009).
Figura 9 - Processo de formação das gotículas de analito e solvente, e como são conduzidas pelo campo elétrico aplicado.
Fonte: LANÇAS, 2009.

36
A figura 10 ilustra a interface no acoplamento do sistema de CLAE e o
espectrômetro de massas, quando a ionização da amostra é feita por ESI.
Figura 10 - Interface do processo de eletronebulização dos analitos, entre a saída do sistema cromatográfico e a injeção no espectrômetro de massas.
Fonte: Adaptado de ZERAIK, 2010.
A ionização por eletronebulização é utilizada para compostos polares e
termicamente sensíveis, os quais são ionizados em solução antes de serem
introduzidos na fonte de íons. É amplamente utilizada nas pesquisas envolvendo
fármacos, devido às propriedades físico-químicas destas substâncias, às quais a
técnica é adequada. A solução contendo os íons é emitida para a fase gasosa, sem
aplicação de calor. A possibilidade de utilizar essa técnica em análises de
macromoléculas biológicas é uma vantagem para a ionização por eletronebulização,
uma vez que os compostos não são fragmentados (YOUDIM et al., 2010).
A ionização por eletronebulização gera três tipos de íons: 1) os íons
moleculares, a partir de reações redox, (M+•) ou (M-•); 2) moléculas
protonadas/desprotonadas (íons quase-moleculares), provenientes de reações
ácido/base, [M+H]+ - moléculas protonadas ou [M-H]- - moléculas desprotonadas; e
3) moléculas cationizadas ou anionizadas, resultado da coordenação dos
fragmentos com cátions (geralmente, família 1A) ou ânions (principalmente cloretos),
[M+Na]+, [M+K]+, [M+Cl]- (CROTTI et al., 2006).

37
Ainda, no processo de detecção dos analitos pelo espectrômetro de massas,
uma de suas partes fundamentais é o analisador, responsável pela seleção e/ou
separação dos íons, levando em consideração a relação massa/carga (m/z) de cada
íon. Diante de tantos equipamentos disponíveis, deve-se observar os objetivos da
análise, qual a faixa de massas desejada, a resolução que se espera dos espectros
e o preço que se pode pagar. O analisador de massas do tipo quadrupolo é o mais
popular, pela sua simplicidade, preço relativamente baixo, boa linearidade em
análises quantitativas e de fácil utilização. Sua resolução pode variar de 1000 a
4000, dependendo das condições aplicadas, com exatidão na faixa de 0,1 e 0,2 Da e
faixa de massas entre 10 e 4000 Da (LANÇAS, 2013).
O analisador de quadrupolo constitui-se de quatro barras, geralmente
confeccionadas em metal e cilíndricas, dispostas em dois pares. Um dos pares é
mantido em um potencial elétrico positivo e outro a um potencial negativo. Às barras
é aplicada uma combinação de corrente contínua (DC) e radiofrequência (Rf). O par
de barras com potencial positivo irá filtrar as maiores massas, enquanto que as de
potencial negativo filtram as massas menores. A razão entre Rf e DC é mantida
constante na operação do quadrupolo, resultando em uma resolução constante.
Determinando-se uma amplitude para as voltagens de Rf e DC, apenas os íons com
uma razão m/z definida (íons ressonantes), em ressonância com o campo aplicado,
passarão pelas barras e atingirão o detector. Os que divergirem da relação m/z
escolhida (íons não ressonantes) não passarão pelas barras, atingindo-as, sendo
descartados pela bomba de vácuo. A figura 11 apresenta a disposição do
quadrupolo e sua inserção no sistema do espectrômetro de massas, bem como
ilustra o comportamento dos íons (LANÇAS, 2013).

38
Figura 11 - Representação de um espectômetro de massas, para mostrar o modelo de analisador do tipo quadrupolo.
Fonte: LANÇAS, 2013.
Outro tipo de analisador utilizado no espectrômetro de massas, acoplado à
cromatografia líquida, é o orbitrap. Já por volta de 1923, Kingdon prôpos o princípio
do aprisionamento orbital. Depois de algumas décadas de estudos, demonstrou-se
que partículas carregadas poderiam ficar aprisionadas sob campos eletrostáticos
(LANÇAS, 2013).
A representação do equipamento é apresentada na figura 12, e o dispositivo
efetua aprisionamento orbital em um campo eletrostático com distribuição potencial.
O analisador de massas orbitrap é construido a partir de um eletrodo externo, similar
a uma barra (a) e um eletrodo central que é posicionado ao longo do eixo (b)
(LANÇAS, 2013).

39
Figura 12 - Representação do analisador de massas orbitrap, com seu eletrodo externo (a) e o eletrodo central (b), onde, r e z correspondem às coordenadas cilíndricas.
Fonte: LANÇAS, 2013.
No orbitrap, os íons são submetidos a um laser pulsado de nitrogênio, na
direção da ponta da sonda de aço inox, são introduzidos em um acelerador de lentes
eletrostático, constiuido de duas aberturas definidas e uma abertura entre elas.
Estas lentes focalizam o feixe de íons em um restritor de condutividade de 1 mm de
diâmetro interno e 10 mm de comprimento, separando o volume de aprisionamento
interno do exterior. Uma miniatura de defletor está posicionado na saída do restritor,
para otimizar o ângulo de entrada do feixe de íons. Os íons, então, são inseridos na
armadilha por um estreito canal de injeção, tangencialmente ao eletrodo externo. Um
eletrodo adicional, com voltagem ajustável, é utilizado como um transformador de
campo, a fim de minimizar a distorção do campo tridimensional formado pela injeção
de íons (MAKAROV, 2000). Este processo é esquematizado na figura 13, onde
estão representados os componentes do analisador orbitrap.

40
Figura 13 - Esquema do analisador orbitrap.
Fonte: Adaptado de MAKAROV, 2000.
No analisador orbitrap, a relação m/z é obtida da frequência das oscilações
harmônicas dos íons, ao longo do eixo do campo. As vantagens do orbitrap são sua
alta resolução de massas (acima de 100000-200000) e eficiência na determinação
da massa, faixa dinâmica e superior limite de massas (MAKAROV, 2000).
Estudos relacionados à identificação e quantificação de alcaloides β-
carbolínicos em amostras complexas (com muitos compostos numa mesma matriz)
mostram resultados satisfatórios na aplicação da técnica de CLUE-EM
(MCLLHENNY et al., 2010; BUSQUETS et al., 2006). Ainda, utilizando a mesma
técnica, as aminas heterocíclicas aromáticas, cuja estrutura molecular é similar à dos
alcaloides β-carbolínicos, foram analisadas, identificadas e quantificadas com
sucesso (ZHANG et al., 2012). Segundo Lumbreras e colaboradores, a CLUE-EM é
um método adequado para a análise de aminas heterocíclicas aromáticas, incluindo
o alcaloide harmana, melhorando o tempo de análise, a resolução dos picos e o
limite de detecção. Além disso, a baixa taxa de fluxo leva ao uso de menor
quantidade de solventes orgânicos, produzindo volumes baixos de resíduos,
fazendo-a uma técnica ecologicamente correta, de custo reduzido e mais adequada
ao acoplamento com detecção por espectrômetro de massas (LUMBRERAS et al.,
2010).

41
2 OBJETIVOS
Identificação dos alcaloides presentes no extrato da polpa e das sementes
dos frutos de duas espécies de “maracujá”, Passiflora alata e Passiflora edulis,
comumente consumidos pela população brasileira, aplicando a técnica de extração
por sorção utilizando barras de extração recobertas por polidimetilsiloxano (PDMS) e
o desenvolvimento da metodologia para extração com as barras de polietilenoglicol
(EG Silicone Twister®), SBSE.
Analisar os extratos de alcaloides presentes na polpa e sementes dos frutos
de Passiflora alata e Passiflora edulis, por meio da técnica cromatográfica de CLUE-
EM e por CLAE-Flu (Cromatografia Líquida de Alta Eficiência acoplada a detector de
Fluorescência), utilizando metodologia desenvolvida por Pereira; Santos; Yariwake,
(2013), SBSE/CLAE-Flu dual, quantificar os alcaloides harmana e harmina nas
polpas de P. alata.
3 PARTE EXPERIMENTAL
3. 1. Material Vegetal
Os frutos dos maracujás azedo (Passiflora edulis Sims f. Flavicarpa Degener)
e doce (Passiflora alata Curtis), dos quais foram utilizadas as sementes e a polpa
neste trabalho, foram comprados no comércio local de São Carlos e São Paulo, no
estado de São Paulo, Brasil.
Após aquisição do material vegetal, os frutos foram separados, lavados e
cortados. Para separação da polpa das sementes utilizou-se peneira comum, com
abertura de 1,4 mm. Posterior à separação, as sementes foram espalhadas sobre
fôrma de alumínio, devidamente coberta com folha de papel alumínio, e levada à
estufa, sob temperatura de 40 – 45 ºC por 72 horas, aproximadamente. E a polpa,
sem sementes, fora acondicionada em recipiente com tampa e levada ao
refrigerador.
As sementes secas foram processadas em liquidificador doméstico e
posteriormente fracionadas utilizando jogo de peneiras para determinação de
granulometria. As peneiras possuiam as seguintes características: 16 – 32 mesh, ou
ABNT 18 = 1,0 mm de abertura; 32 – 65 mesh, ou ABNT 35 = 0,25 a 0,59 mm de
abertura. As frações foram acondicionadas em diferentes recipientes plásticos com
tampa e estocadas ao abrigo de luz, umidade e calor.

42
A fração de sementes processadas, utilizadas para preparação das amostras
analisadas neste trabalho, possui granulometria de 1,0 – 0,5 mm, 16 mesh.
3. 2 Extração dos analitos do material vegetal
3. 2. 1 Materiais utilizados:
SOLVENTES E REAGENTES: Foram utilizados acetonitrila e metanol grau
CLAE (Tedia, Fairfield, OH, EUA), ácido fórmico grau p.a. (Merck, Darmstadt,
Alemanha), cloreto de sódio grau analítico (Spectrum, Gardena, CA, EUA), água
purificada pelo sistema Milli-Q (Millipore, Bedford, MA, EUA) e padrões analíticos
dos alcaloides harmânicos: harmana, harmina, harmalina, harmol e harmalol, com
98% de pureza Sigma-Aldrich (Steinheim, Germany).
BARRAS DE AGITAÇÃO PARA SBSE: as barras de agitação utilizadas neste
trabalho, barras comerciais (TwisterTM), para extração sortiva da Gerstel (Mulheim an
der Ruhr, Germany). As de PDMS têm 20 mm de comprimento com 0.5 mm de filme
de PDMS, volume de 55 µL – LOTE 0101071110 – e as de EG-Silicone têm 10 mm
de comprimento com 32 µL de volume de fase de PDMS e Etileno Glicol – LOTE
01690400100.
3. 2. 2 Procedimento de extração:
3. 2. 2. 1 SBSE – PDMS
Utilizando o método desenvolvido e otimizado por Pereira e colaboradores
para as barras magnéticas de PDMS, o procedimento de extração dos alcaloides
harmânicos, a partir da polpa de P. alata e P. edulis seguiu os seguintes passos:
para extração de harmana - 1 mL de polpa de maracujá, 2 mL de NaOH 1 mol L-1
(pH 13), 5 g de NaCl e 7 mL de água Milli-Q foram colocados em um frasco de 10
mL juntamente com a barra de extração. O alcaloide harmina foi extraído pela
adição de 1 mL de polpa de maracujá, 0,250 mL de NH4OH 6 mol L-1 (pH 10), 5 g de
NaCl e 8,750 mL de água Milli-Q e adicionados em um recipiente de 10 mL contendo
a barra de extração (Tabela 1) (PEREIRA et al., 2012).
Para extração dos alcaloides a partir das sementes foi usado o seguinte
procedimento: extração de harmana - 1 g de sementes trituradas de maracujá, 1 mL
de NaOH 1 mol L-1 (pH 13), 5 g de NaCl e 9 mL de água Milli-Q foram colocados em
um frasco de 10 mL juntamente com a barra de extração. O alcaloide harmina foi

43
extraído pela adição de 1 g de sementes de maracujá, 0,100 mL de NH4OH 6 mol L-1
(pH 10), 5 g de NaCl e 9,9 mL de água Milli-Q e adicionados em um recipiente de 10
mL contendo a barra de extração (Tabela 2) (RODRIGUES, 2013).
As amostras foram colocadas simultaneamente (em paralelo) sob agitação
durante 120 minutos, a 1000 rpm à temperatura ambiente. Após a agitação as
barras de SBSE, foram retiradas da solução e colocadas em um mesmo “vial” com
150 μL e sob sonicação por 60 minutos (PEREIRA et al., 2012).
Tabela 1 - Procedimento de extração dos alcaloides harmânicos Harmana e Harmina a partir da polpa de P. edulis e P. alata.
Extração dos alcaloides a partir da Polpa Harmana Harmina
Material vegetal 1mL de polpa 1 mL de polpa
pH 13 (2 mL NaOH 1M) 10 (0,250 mL NH4OH 6M)
NaCl 5 g 5 g
Volume de H2O Milli-Q 7 mL 8,750 mL
Tempo de extração 120 min
Dessorção 150 µL de MeOH – 60 min
Análise Injeção de 10 µL – CLAE-Flu/CLUE-EM
Tabela 2 - Procedimento de extração dos alcaloides harmânicos Harmana e Harmina a partir das sementes secas de P. edulis e P. alata.
Extração dos alcaloides a partir das
Sementes Secas
Harmana Harmina
Material vegetal 1 g de sementes 1 g de sementes
pH 13 (1 mL NaOH 1M) 10 (0,1 mL NH4OH 6M)
NaCl 5 g 5 g
Volume de H2O Milli-Q 9 mL 9,9 mL
Tempo de extração 120 min
Dessorção 150 µL de MeOH – 60 min
Análise Injeção de 10 µL – CLAE-Flu/CLUE-EM
Então, o “vial” com o metanol contendo os compostos dessorvidos das barras
de extração é utilizado diretamente para análise no equipamento de CLAE-Flu ou
CLUE-EM (PEREIRA et al., 2012).
Após cada etapa de extração e dessorção líquida, as barras magnéticas de
PDMS foram submetidas a um procedimento de limpeza, para garantir a remoção de
analitos restantes e evitar interferência destes nas extrações posteriores. A limpeza
das barras foi feita utlizando uma mistura de metanol e acetonitrila (50:50 v/v), as

44
barras eram colocadas individualmente sob agitação por 3 h, em 30,0 mL desta
solução em um béquer.
3. 2. 2. 2 Desenvolvimento SBSE – EG Silicone
A metodologia de extração aplicada para as barras magnéticas de EG-
Silicone foi definida utilizando-se um planejamento fatorial fracionário 25-1. Com o
objetivo de selecionar os fatores e suas interações que interferem significativamente
na resposta final (PEREIRA et al., 2014).
O planejamento experimental possui cinco variáveis independentes (fatores),
que são: tempo de extração, quantidade de NaCl, pH da solução, tempo de
dessorção e porcentagem de metanol (MeOH) para dessorção. A variável
dependente (resposta) foi a área relativa do pico dos padrões dos alcalóides
harmana e harmina. As respostas foram obtidas analisando cromatogramas por
meio do programa Empower (Waters Associates, Milford, MA, EUA) (PEREIRA et al.,
2014).
No planejamento fatorial fracionário 25-1, de dois níveis, cinco fatores e um
ponto central. Cada fator possui um nível inferior e superior, representados pelos
sinais (-) e (+), respectivamente (Tabela 3).
Tabela 3 - Níveis e fatores avaliados no planejamento fatorial 25-1
.
O planejamento fatorial fracionário foi elaborado utilizando o software
MiniTab® (Minitab Inc, USA), versão 13 , resultando em 19 experimentos (16 ensaios
e 1 ponto central em triplicata) (Tabela 4). O mesmo software foi usado para o
cálculo dos efeitos e suas interações, bem com a construção do gráfico de Pareto.
Para execução dos experimentos do planejamento fatorial fracionário, uma alíquota
de 10 µL de cada um dos padrões de harmana e harmina (solução de 20 µg L-1 em
metanol 100%) foi diluída com volume adequado de água Milli-Q, para concentração
final de 20 ng L-1 em 10 mL de solução. Em seguida, para o teste do nível máximo
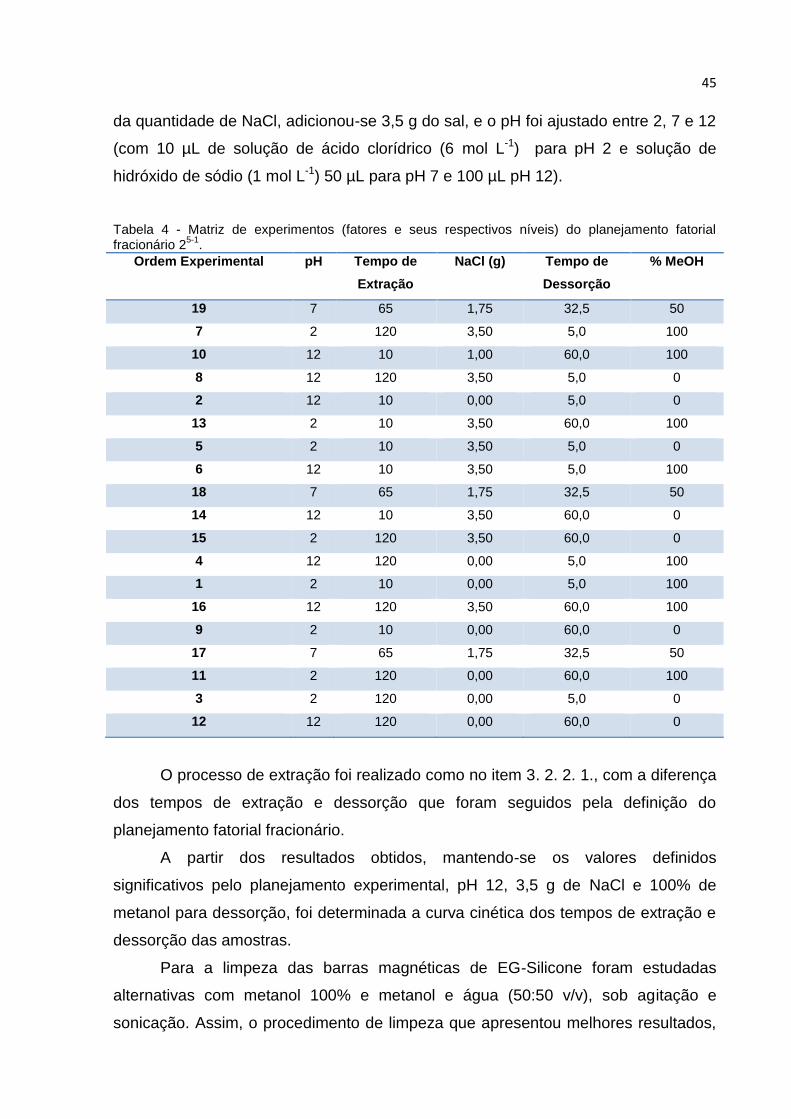
45
da quantidade de NaCl, adicionou-se 3,5 g do sal, e o pH foi ajustado entre 2, 7 e 12
(com 10 µL de solução de ácido clorídrico (6 mol L-1) para pH 2 e solução de
hidróxido de sódio (1 mol L-1) 50 µL para pH 7 e 100 µL pH 12).
Tabela 4 - Matriz de experimentos (fatores e seus respectivos níveis) do planejamento fatorial fracionário 2
5-1.
Ordem Experimental pH Tempo de
Extração
NaCl (g) Tempo de
Dessorção
% MeOH
19 7 65 1,75 32,5 50
7 2 120 3,50 5,0 100
10 12 10 1,00 60,0 100
8 12 120 3,50 5,0 0
2 12 10 0,00 5,0 0
13 2 10 3,50 60,0 100
5 2 10 3,50 5,0 0
6 12 10 3,50 5,0 100
18 7 65 1,75 32,5 50
14 12 10 3,50 60,0 0
15 2 120 3,50 60,0 0
4 12 120 0,00 5,0 100
1 2 10 0,00 5,0 100
16 12 120 3,50 60,0 100
9 2 10 0,00 60,0 0
17 7 65 1,75 32,5 50
11 2 120 0,00 60,0 100
3 2 120 0,00 5,0 0
12 12 120 0,00 60,0 0
O processo de extração foi realizado como no item 3. 2. 2. 1., com a diferença
dos tempos de extração e dessorção que foram seguidos pela definição do
planejamento fatorial fracionário.
A partir dos resultados obtidos, mantendo-se os valores definidos
significativos pelo planejamento experimental, pH 12, 3,5 g de NaCl e 100% de
metanol para dessorção, foi determinada a curva cinética dos tempos de extração e
dessorção das amostras.
Para a limpeza das barras magnéticas de EG-Silicone foram estudadas
alternativas com metanol 100% e metanol e água (50:50 v/v), sob agitação e
sonicação. Assim, o procedimento de limpeza que apresentou melhores resultados,

46
20,0 mL de metanol 100% sob sonicação por 30 min, foi utilizado para assegurar
que não restassem analitos na matriz da barra magnética entre uma extração e
outra.
3. 3 Análise por Cromatografia Líquida de Alta Eficiência com
Detector de Fluorescência
Foi feita a análise qualitativa (“perfil cromatográfico”) para avaliar o perfil
cromatográfico da amostra, em cromatógrafo líquido (Waters Alliance 2695)
conectado a um detector UV/DAD (Waters, PDA 2996) e a detector de fluorescência
(Waters 2475). A coluna cromatográfica utilzada, X-Terra® C18 (250 mm x 4,6 mm i.
d., 5µm) e coluna de guarda X -Terra® C18 (20 mm x 4,6 mm i. d., 5µm), ambas da
Waters (Waters Associates, Milford, MA, EUA).
A tabela 5 apresenta as condições cromatográficas utilizadas para as análises
das amostras vegetais e os padrões comerciais. A detecção, utilizando detector de
fluorescência, operou na faixa de varredura λemissão = 390 nm – 490 nm.
Tabela 5 - Condições cromatográficas de análise dos alcaloides harmânicos em CLAE-Flu.
3. 3. 1 Análise quantitativa dos alcaloides em P. alata
A quantificação dos alcaloides harmana e harmina na polpa do fruto de P.
alata foi realizada por meio do método de adição de padrão. À cada amostra de
extração (realizada em triplicata) foi adicionada uma alíquota da solução-estoque de
cada um dos padrões dos alcaloides harmana e harmina (20 µg L-1 em metanol
100%) a fim de obter as concentrações finais: 0,1; 1,25; 2,5; 3,75 e 5 µg L-1, as

47
curvas analíticas de adição de padrão foram construídas em triplicata (RODRIGUES,
2013).
3. 4 Análise por Cromatografia Líquida de Ultra Eficiência acoplada
a Espectrometria de Massas
A identificação foi realizada em espectrômetro de massas de alta resolução
LTQ Orbitrap Velos (Thermo Scientific, Waltham, MA, EUA) equipado com interface
de ionização por eletrospray, nanoeletrospray (nanoESI) e ionização química à
pressão atmosférica. O espectrômetro de massas está hifenado a um cromatógrafo
líquido de ultra eficiência Accela High Speed LC (Thermo Scientifc, Waltham, MA,
EUA). Foi utilizada uma coluna cromatográfica X-BridgeTM BEH Shield RP18 (150 mm
x 2,1 mm x 2,5 µm) com pré coluna (12 mm x 2,1 mm x 2,5 µm), ambas da Waters.
Das condições cromatográficas utilizadas para as análises em CLAE-Flu (item 4.3),
os solventes e o gradiente da fase móvel foram mantidos, contudo o fluxo de
solvente utilizado foi 0,3 mL min-1 e o volume de injeção de 10 μL.
As condições do espectrômetro de massas foram definidas a partir de estudos
de ZHAO, T. e colaboradores, para análises de alcaloides harmânicos: voltagem do
eletrospray + 3,7 kV, o fluxo de gás (N2) de nebulização 35 (unidades arbitrárias),
fluxo de gás auxiliar (N2) 10 (unidades arbitrárias) e temperatura do capilar aquecido
a 300 oC (ZHAO, et al. 2012). A aquisição de dados no modo positivo, foi feita em
full-scan, dentro do intervalo de m/z 50-250, bem como a seleção de íons (SIM,
seletive ions monitoring) e o modo de EM/EM. Todas as operações foram
controladas pelo software XcaliburTM (Thermo Scientific, Waltham, MA, EUA).

48
4 RESULTADOS E DISCUSSÃO
4. 1 Análise quantitativa dos alcaloides em polpa de P. alata
Trabalhos anteriores mostram a presença dos alcaloides harmana e harmina
nos extratos da polpa e das sementes dos frutos de P. edulis. Assim, foi proposto o
estudo analítico do extrato da polpa dos frutos de P. alata, utilizando a metodologia
de extração por SBSE com barra magnética recoberta com PDMS, desenvolvida por
Pereira e colaboradores. Os extratos foram analisados por CLAE-Flu, com método
adequado para a detecção dos alcaloides, com λexcitação = 254 nm e λemissão = 425 nm
para harmana e λexcitação = 254 nm e λemissão = 410 nm para harmina; a partir destas
informações, para a detecção destes alcaloides na polpa de P. alata foram mantidas
estas condições (PEREIRA et al., 2013).
A figura 14 apresenta os cromatogramas obtidos pela análise SBSE/CLAE-Flu
dual nas condições cromatográficas definidas pela metodologia desenvolvida para o
estudo de alcaloides harmânicos.
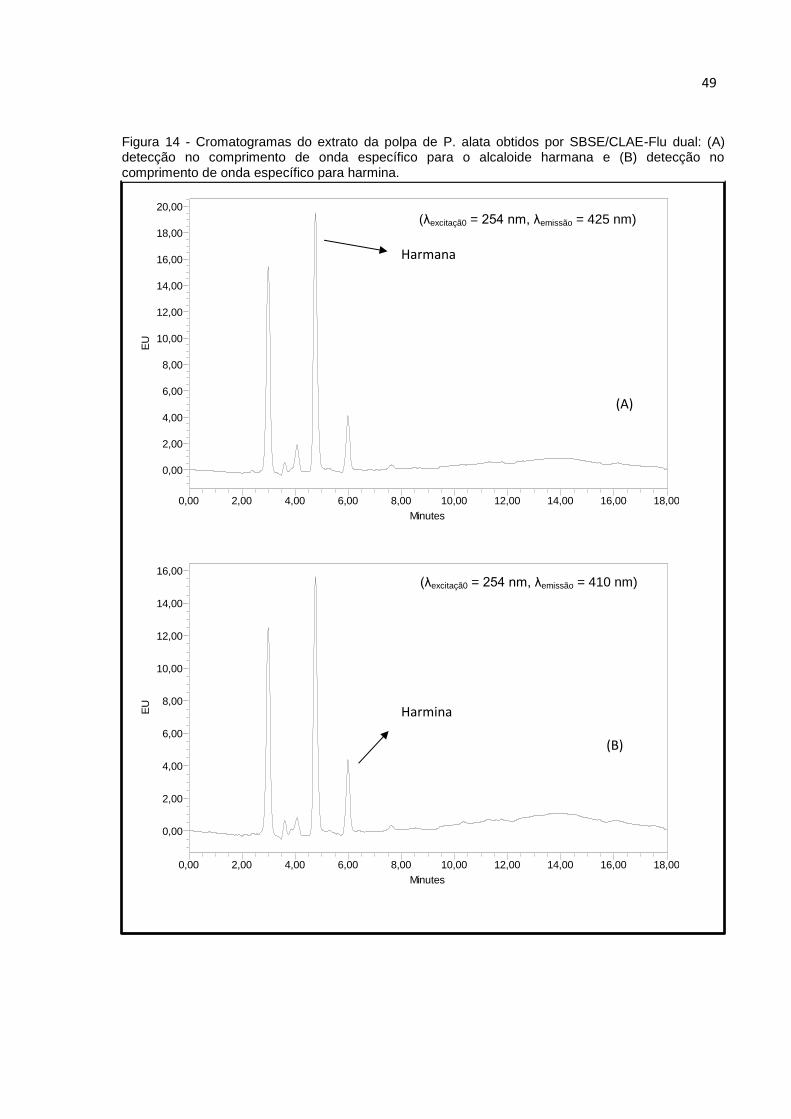
49
EU
0,00
2,00
4,00
6,00
8,00
10,00
12,00
14,00
16,00
18,00
20,00
Minutes
0,00 2,00 4,00 6,00 8,00 10,00 12,00 14,00 16,00 18,00
EU
0,00
2,00
4,00
6,00
8,00
10,00
12,00
14,00
16,00
Minutes
0,00 2,00 4,00 6,00 8,00 10,00 12,00 14,00 16,00 18,00
(λexcitaçã0 = 254 nm, λemissão = 425 nm)
Harmina
Harmana
(λexcitaçã0 = 254 nm, λemissão = 410 nm)
(A)
(B)
Figura 14 - Cromatogramas do extrato da polpa de P. alata obtidos por SBSE/CLAE-Flu dual: (A) detecção no comprimento de onda específico para o alcaloide harmana e (B) detecção no comprimento de onda específico para harmina.

50
Foram analisadas soluções dos padrões comerciais dos alcaloides harmana e
harmina, dentro das mesmas condições cromatográficas. A figura 15 mostra o
cromatograma do extrato da polpa de P. alata sobreposto ao cromatograma da
solução padrão de 20 µg L-1 da harmana (Figura 15 A) e da harmina (Figura 15 B) e
observa-se a similaridade dos tempos de retenção dos picos cromatográficos,
atribuídos à harmana (tR = 4,757 min) e à harmina (tR = 5,985 min), no
cromatograma do extrato da polpa, com os picos dos cromatogramas das soluções
padrões de harmana (tR = 4,772 min) e harmina (tR = 5,845 min).
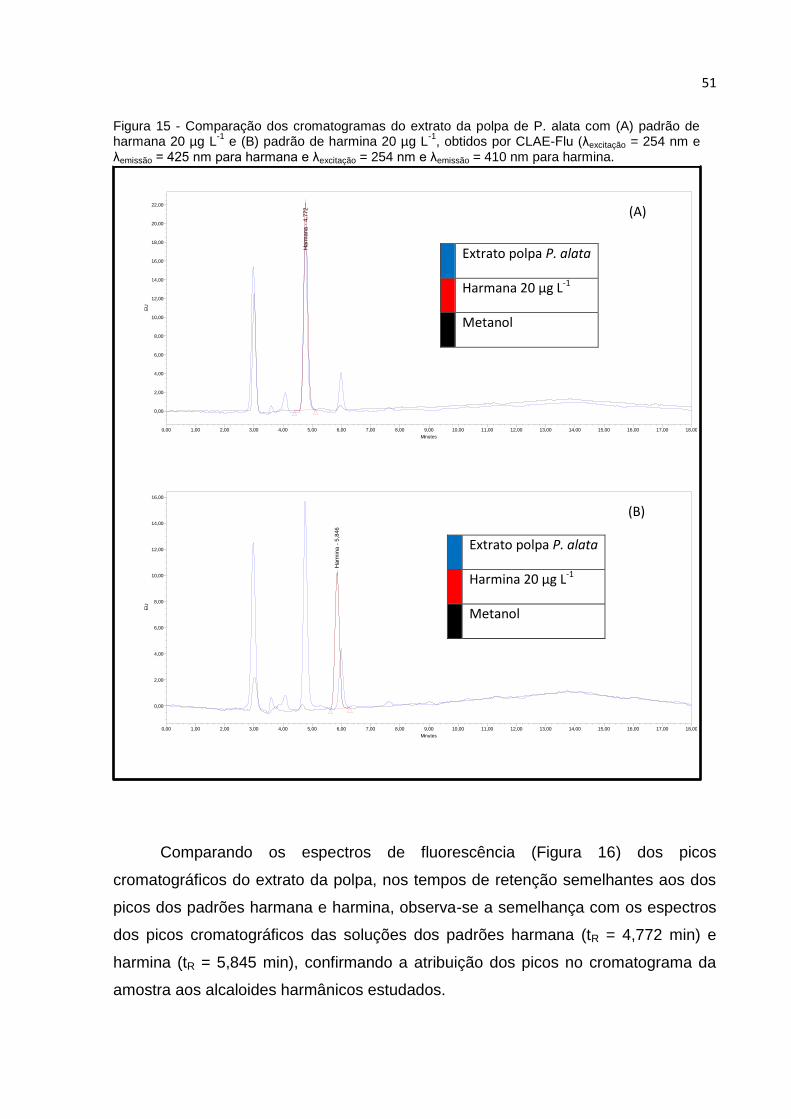
51
Comparando os espectros de fluorescência (Figura 16) dos picos
cromatográficos do extrato da polpa, nos tempos de retenção semelhantes aos dos
picos dos padrões harmana e harmina, observa-se a semelhança com os espectros
dos picos cromatográficos das soluções dos padrões harmana (tR = 4,772 min) e
harmina (tR = 5,845 min), confirmando a atribuição dos picos no cromatograma da
amostra aos alcaloides harmânicos estudados.
Harm
ana -
4,7
72
EU
0,00
2,00
4,00
6,00
8,00
10,00
12,00
14,00
16,00
18,00
20,00
22,00
Minutes
0,00 1,00 2,00 3,00 4,00 5,00 6,00 7,00 8,00 9,00 10,00 11,00 12,00 13,00 14,00 15,00 16,00 17,00 18,00
Harm
ina -
5,8
46
EU
0,00
2,00
4,00
6,00
8,00
10,00
12,00
14,00
16,00
Minutes
0,00 1,00 2,00 3,00 4,00 5,00 6,00 7,00 8,00 9,00 10,00 11,00 12,00 13,00 14,00 15,00 16,00 17,00 18,00
Extrato polpa P. alata
Harmana 20 µg L-1
Metanol
Extrato polpa P. alata
Harmina 20 µg L-1
Metanol
(B)
(A)
Figura 15 - Comparação dos cromatogramas do extrato da polpa de P. alata com (A) padrão de harmana 20 µg L
-1 e (B) padrão de harmina 20 µg L
-1, obtidos por CLAE-Flu (λexcitação = 254 nm e
λemissão = 425 nm para harmana e λexcitação = 254 nm e λemissão = 410 nm para harmina.
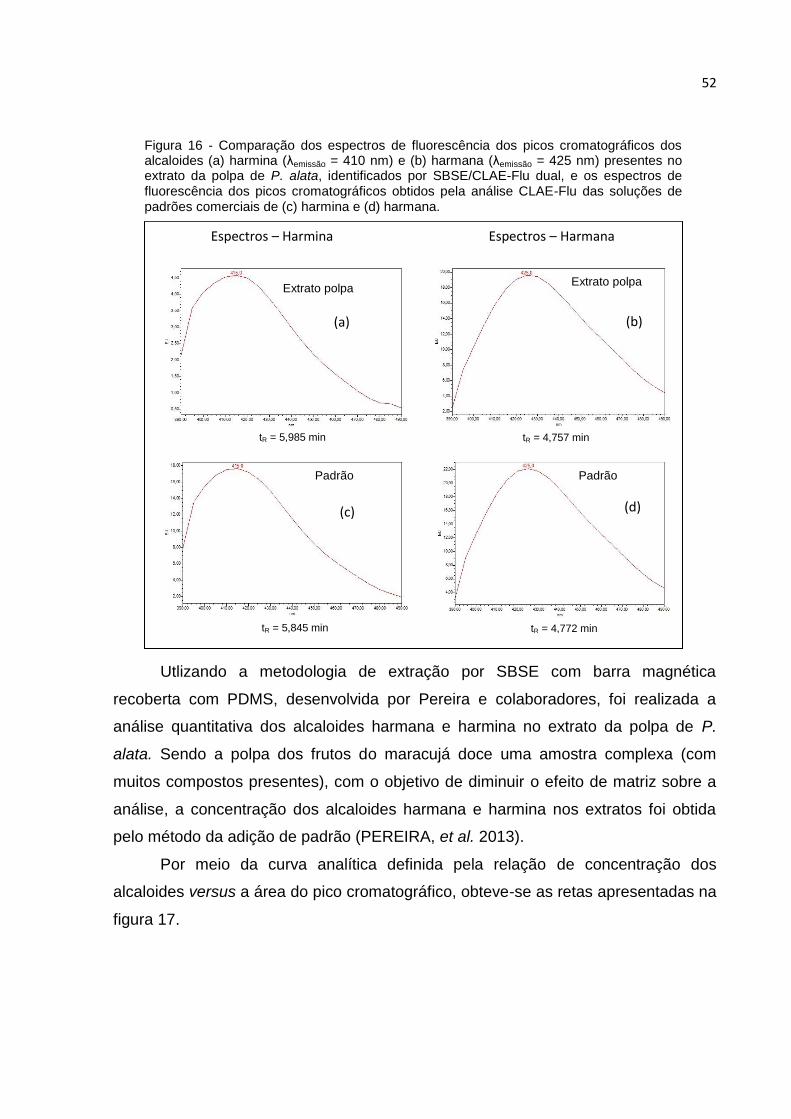
52
v
Utlizando a metodologia de extração por SBSE com barra magnética
recoberta com PDMS, desenvolvida por Pereira e colaboradores, foi realizada a
análise quantitativa dos alcaloides harmana e harmina no extrato da polpa de P.
alata. Sendo a polpa dos frutos do maracujá doce uma amostra complexa (com
muitos compostos presentes), com o objetivo de diminuir o efeito de matriz sobre a
análise, a concentração dos alcaloides harmana e harmina nos extratos foi obtida
pelo método da adição de padrão (PEREIRA, et al. 2013).
Por meio da curva analítica definida pela relação de concentração dos
alcaloides versus a área do pico cromatográfico, obteve-se as retas apresentadas na
figura 17.
Espectros – Harmina Espectros – Harmana
Extrato polpa Extrato polpa
Padrão Padrão
tR = 4,757 min
tR = 4,772 min
tR = 5,985 min
tR = 5,845 min
(a) (b)
(c) (d)
Figura 16 - Comparação dos espectros de fluorescência dos picos cromatográficos dos alcaloides (a) harmina (λemissão = 410 nm) e (b) harmana (λemissão = 425 nm) presentes no extrato da polpa de P. alata, identificados por SBSE/CLAE-Flu dual, e os espectros de fluorescência dos picos cromatográficos obtidos pela análise CLAE-Flu das soluções de padrões comerciais de (c) harmina e (d) harmana.
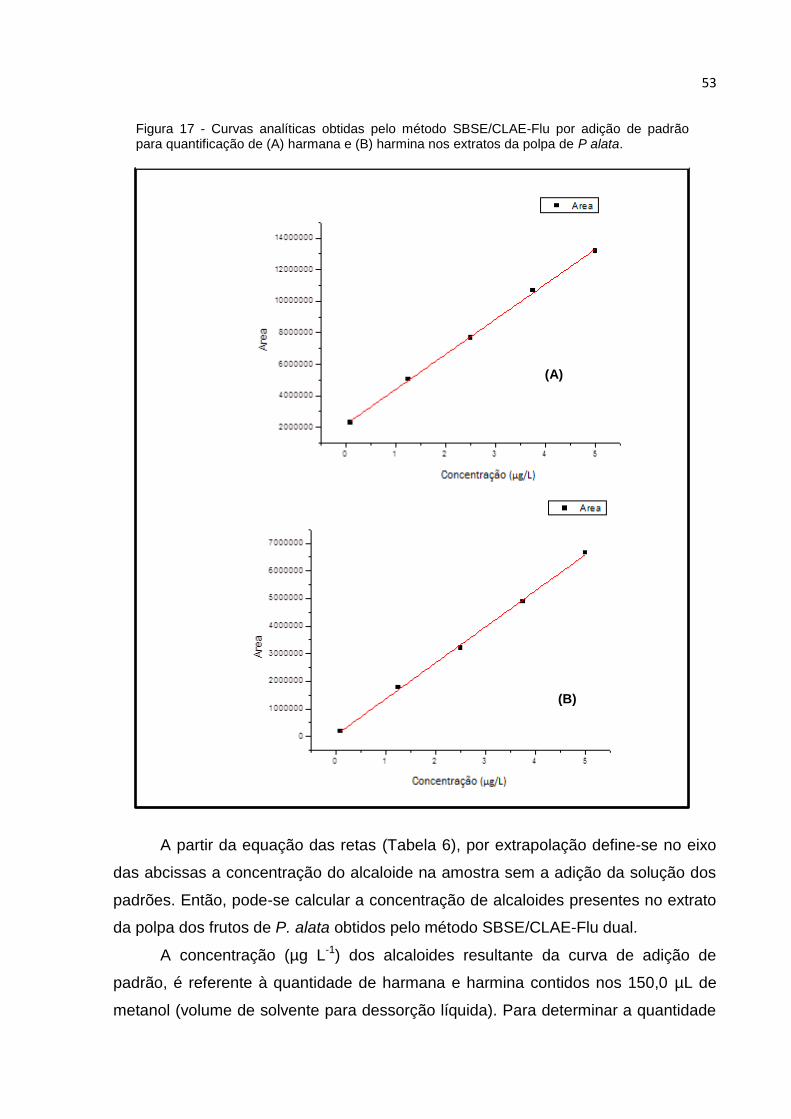
53
A partir da equação das retas (Tabela 6), por extrapolação define-se no eixo
das abcissas a concentração do alcaloide na amostra sem a adição da solução dos
padrões. Então, pode-se calcular a concentração de alcaloides presentes no extrato
da polpa dos frutos de P. alata obtidos pelo método SBSE/CLAE-Flu dual.
A concentração (µg L-1) dos alcaloides resultante da curva de adição de
padrão, é referente à quantidade de harmana e harmina contidos nos 150,0 µL de
metanol (volume de solvente para dessorção líquida). Para determinar a quantidade
(A)
(B)
Figura 17 - Curvas analíticas obtidas pelo método SBSE/CLAE-Flu por adição de padrão para quantificação de (A) harmana e (B) harmina nos extratos da polpa de P alata.

54
dos alcaloides em 1 mL de polpa do fruto de P. alata foram realizados os seguintes
cálculos:
Intercepto da curva de adição de padrão (µg) – 1000 mL
X (µg) – 150 µL
Considerando a concentração de alcaloides definida pelo intercepto das
curvas de adição de padrão para a harmana = 0,97032 µg L-1 e para a harmina =
0,03609 µg L-1, a tabela 6 apresenta os valores de massa dos alcaloides por 1 mL
de polpa.
Tabela 6 - Determinação quantitativa por CLAE-Flu dos alcaloides em polpa de P. alata.
Alcaloides Equação da Reta Coeficiente de
Correlação (r2)
Massa de alcaloide/
volume de polpa
(pg mL-1
) ± d.p.*
Harmana y = 2,24687.10
6 x + 2,18019.10
6 0,99884 145,54500 ± 0,00085
Harmina y = 1,35385.10
6 x + 4,88652.10
4 0,99855 5,41350 ± 0,00006
*d.p. = desvio padrão.
De acordo com Pereira e colaboradores, na polpa de P. edulis foram
encontrados (3,00 ± 0,04) µg L-1 do alcaloide harmana e (2,72 ± 0,02) µg L-1 do
alcaloide harmina (Pereira et al., 2014). A partir desses resultados, pode-se
constatar que os frutos de P. alata possuem menor quantidade dos alcaloides
harmana e harmina em sua polpa, comparada às concentrações determinadas em
extratos da polpa de P. edulis.
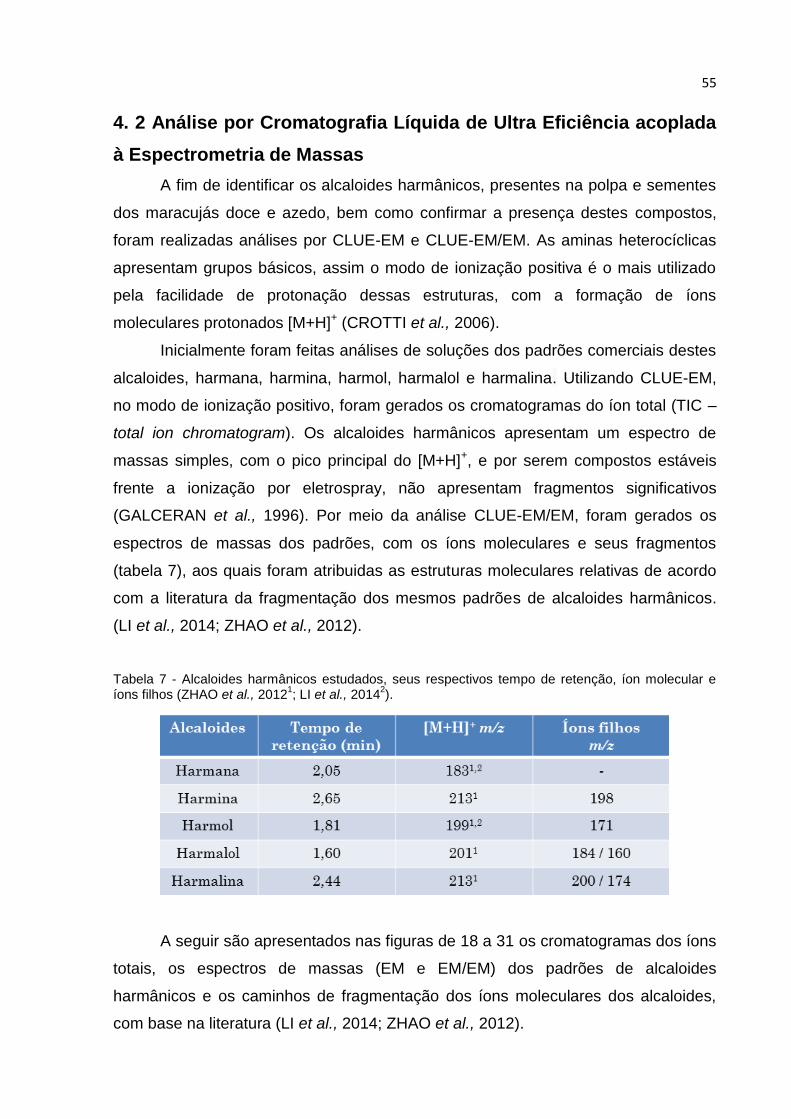
55
4. 2 Análise por Cromatografia Líquida de Ultra Eficiência acoplada
à Espectrometria de Massas
A fim de identificar os alcaloides harmânicos, presentes na polpa e sementes
dos maracujás doce e azedo, bem como confirmar a presença destes compostos,
foram realizadas análises por CLUE-EM e CLUE-EM/EM. As aminas heterocíclicas
apresentam grupos básicos, assim o modo de ionização positiva é o mais utilizado
pela facilidade de protonação dessas estruturas, com a formação de íons
moleculares protonados [M+H]+ (CROTTI et al., 2006).
Inicialmente foram feitas análises de soluções dos padrões comerciais destes
alcaloides, harmana, harmina, harmol, harmalol e harmalina. Utilizando CLUE-EM,
no modo de ionização positivo, foram gerados os cromatogramas do íon total (TIC –
total ion chromatogram). Os alcaloides harmânicos apresentam um espectro de
massas simples, com o pico principal do [M+H]+, e por serem compostos estáveis
frente a ionização por eletrospray, não apresentam fragmentos significativos
(GALCERAN et al., 1996). Por meio da análise CLUE-EM/EM, foram gerados os
espectros de massas dos padrões, com os íons moleculares e seus fragmentos
(tabela 7), aos quais foram atribuidas as estruturas moleculares relativas de acordo
com a literatura da fragmentação dos mesmos padrões de alcaloides harmânicos.
(LI et al., 2014; ZHAO et al., 2012).
Tabela 7 - Alcaloides harmânicos estudados, seus respectivos tempo de retenção, íon molecular e íons filhos (ZHAO et al., 2012
1; LI et al., 2014
2).
A seguir são apresentados nas figuras de 18 a 31 os cromatogramas dos íons
totais, os espectros de massas (EM e EM/EM) dos padrões de alcaloides
harmânicos e os caminhos de fragmentação dos íons moleculares dos alcaloides,
com base na literatura (LI et al., 2014; ZHAO et al., 2012).

56
RT: 0.00 - 19.97
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
1.22
2.05
8.86 10.42 12.5411.7210.118.73 12.76 13.896.57 15.026.662.56 19.4815.11 17.265.99 18.372.83 3.59 5.914.35
0.23
NL:5.42E7
TIC MS harmana(+)_141218194313
harmana(+)_141218194313 #43-52 RT: 1.88-2.26 AV: 10 SB: 443 0.01-1.84 , 2.30-19.88 NL: 1.71E7T: FTMS + p ESI Full ms [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
183.09107
C 12 H11 N2 = 183.09167
-3.33103 ppm
(A)
(B) NH
NH
harmana
Figura 18 - (A) Cromatograma do íon total e (B) Espectro CLUE-EM, ionização eletrospray, modo positivo: íon [M+H]
+ = 183 do padrão harmana, tR = 2,05 min (solução 100 mg L
-1).
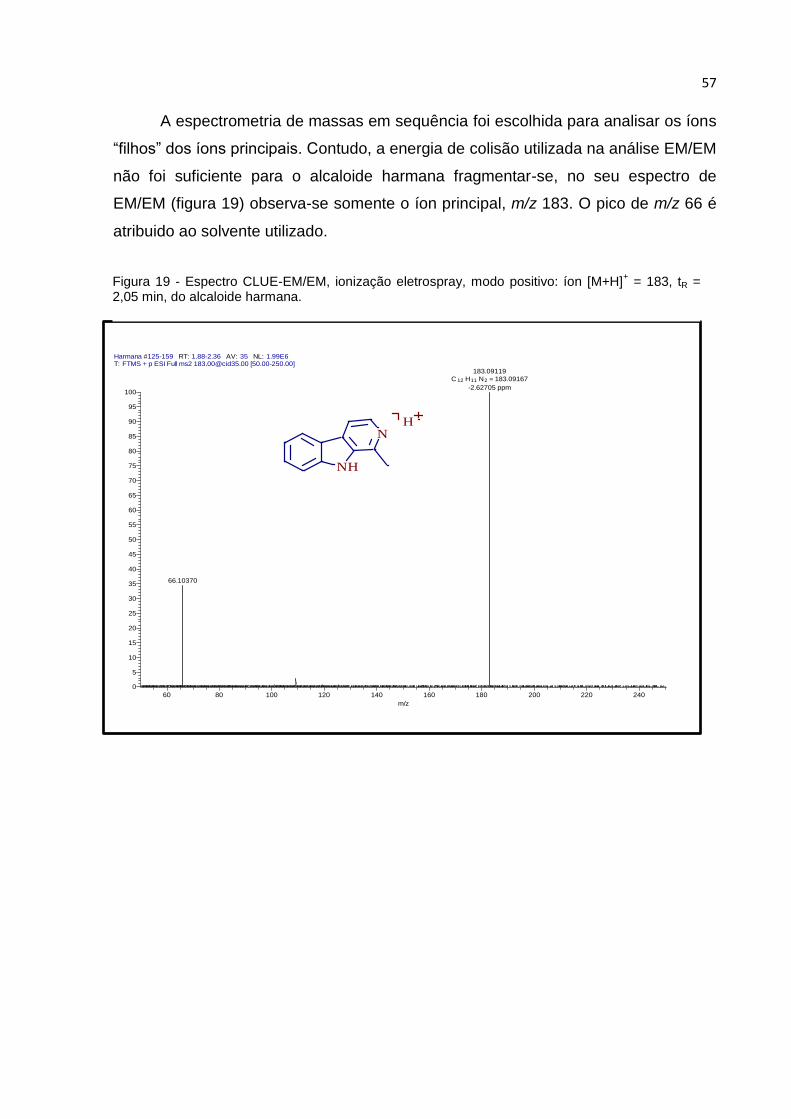
57
A espectrometria de massas em sequência foi escolhida para analisar os íons
“filhos” dos íons principais. Contudo, a energia de colisão utilizada na análise EM/EM
não foi suficiente para o alcaloide harmana fragmentar-se, no seu espectro de
EM/EM (figura 19) observa-se somente o íon principal, m/z 183. O pico de m/z 66 é
atribuido ao solvente utilizado.
Harmana #125-159 RT: 1.88-2.36 AV: 35 NL: 1.99E6T: FTMS + p ESI Full ms2 [email protected] [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
183.09119
C 12 H11 N2 = 183.09167
-2.62705 ppm
66.10370
NH
NH
Figura 19 - Espectro CLUE-EM/EM, ionização eletrospray, modo positivo: íon [M+H]+ = 183, tR =
2,05 min, do alcaloide harmana.
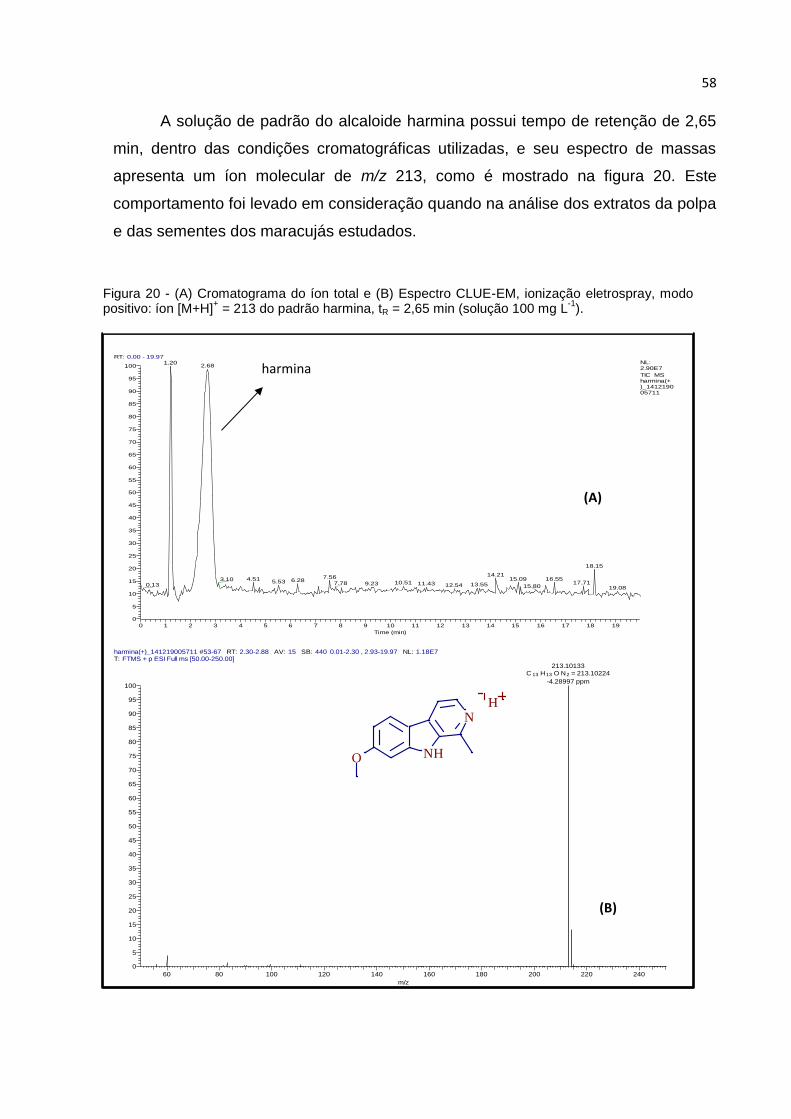
58
A solução de padrão do alcaloide harmina possui tempo de retenção de 2,65
min, dentro das condições cromatográficas utilizadas, e seu espectro de massas
apresenta um íon molecular de m/z 213, como é mostrado na figura 20. Este
comportamento foi levado em consideração quando na análise dos extratos da polpa
e das sementes dos maracujás estudados.
RT: 0.00 - 19.97
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
1.202.68
18.15
14.217.564.51 16.5515.093.10 6.285.53 17.7110.517.78 9.23 11.43 13.550.13 12.54 15.80 19.08
NL:2.90E7
TIC MS harmina(+)_141219005711
harmina(+)_141219005711 #53-67 RT: 2.30-2.88 AV: 15 SB: 440 0.01-2.30 , 2.93-19.97 NL: 1.18E7T: FTMS + p ESI Full ms [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lativ
e A
bu
nd
an
ce
213.10133
C 13 H13 O N2 = 213.10224
-4.28997 ppm
(A)
(B)
NH
N
O
H
harmina
Figura 20 - (A) Cromatograma do íon total e (B) Espectro CLUE-EM, ionização eletrospray, modo positivo: íon [M+H]
+ = 213 do padrão harmina, tR = 2,65 min (solução 100 mg L
-1).

59
A literatura sobre espectrometria de massas traz a “regra do nitrogênio”, onde
afirma que uma molécula de peso molecular par ou não contém nitrogênio ou
contém um número par de átomos de nitrogênio, e moléculas com peso molecular
ímpar contém número ímpar de átomos de nitrogênio; uma consequência dessa
regra explica a fragmentação dos íons moleculares, onde a quebra de uma ligação
simples gera um fragmento iônico par a partir de um íon molecular ímpar, desde que
o fragmento mantenha todos os átomos de nitrogênio do íon molecular
(SILVERSTEIN et al., 2010).
O espectro de massas do alcaloide harmina apresenta os picos [M+H]+ m/z
213 e o íon “filho” m/z 198 (Figura 21). Zhao e colaboradores atribuiram o pico m/z
198 à desmetilação do íon molecular da harmina – no carbono 7 -, com eliminação
de 15 Da, correspondendo ao [M+H-15]+ = 198 observado na figura 22, o que pela
“regra do nitrogênio” se justifica, pois o [M+H]+ m/z 213 (ímpar) gera um fragmento
iônico de m/z 198 (par). Ainda, a fórmula molecular do íon “filho”, atribuído pelo
software Xcallibur, é correspondente à estrutura elucidada, figura 22 (ZHAO et al.,
2012).
MS2harmina #153-194 RT: 2.30-2.88 AV: 42 NL: 1.47E7T: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e Ab
unda
nce
198.07820
C 12 H10 O N2 = 198.07876
-2.87214 ppm
213.10160
C 13 H13 O N2 = 213.10224
-2.99169 ppm
66.10639
C 5 H6 = 66.04640
907.46327 ppm
NH
N
O
H
Figura 21 - Espectro CLUE-EM/EM, ionização eletrospray, modo positivo: íon [M+H]+ = 213, tR = 2,65 min,
alcaloide harmina.
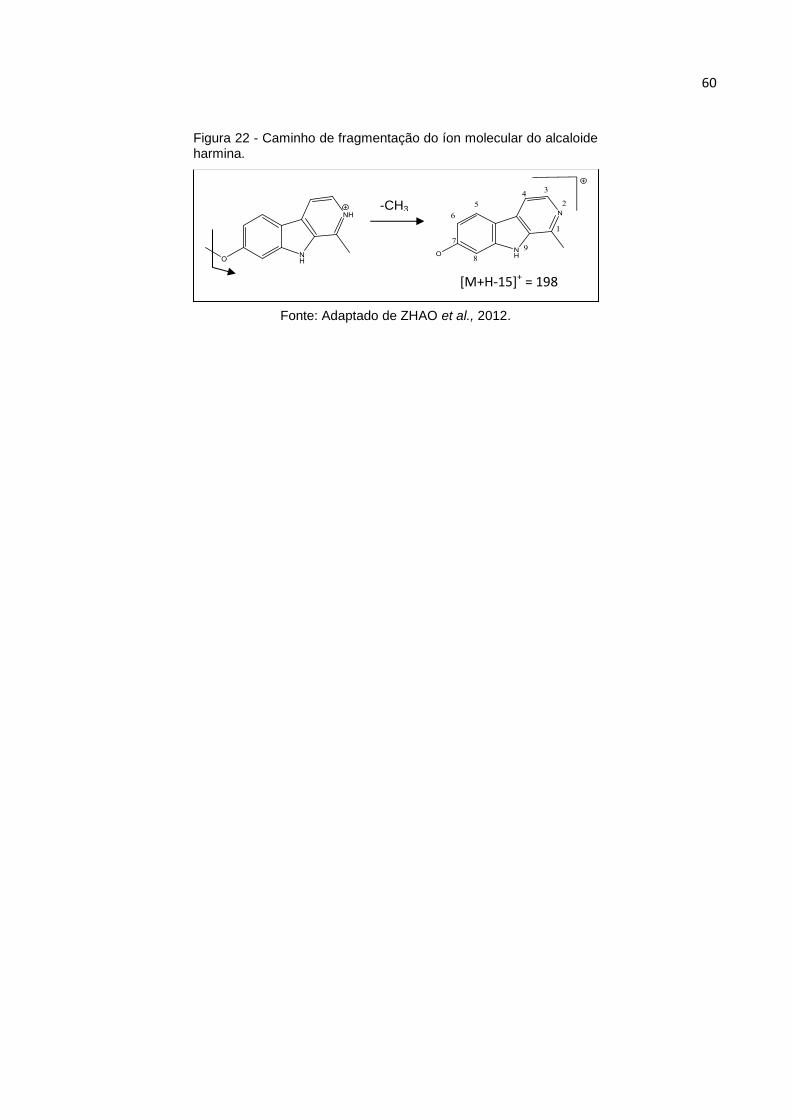
60
-CH3
[M+H-15]+ = 198
Fonte: Adaptado de ZHAO et al., 2012.
Figura 22 - Caminho de fragmentação do íon molecular do alcaloide harmina.
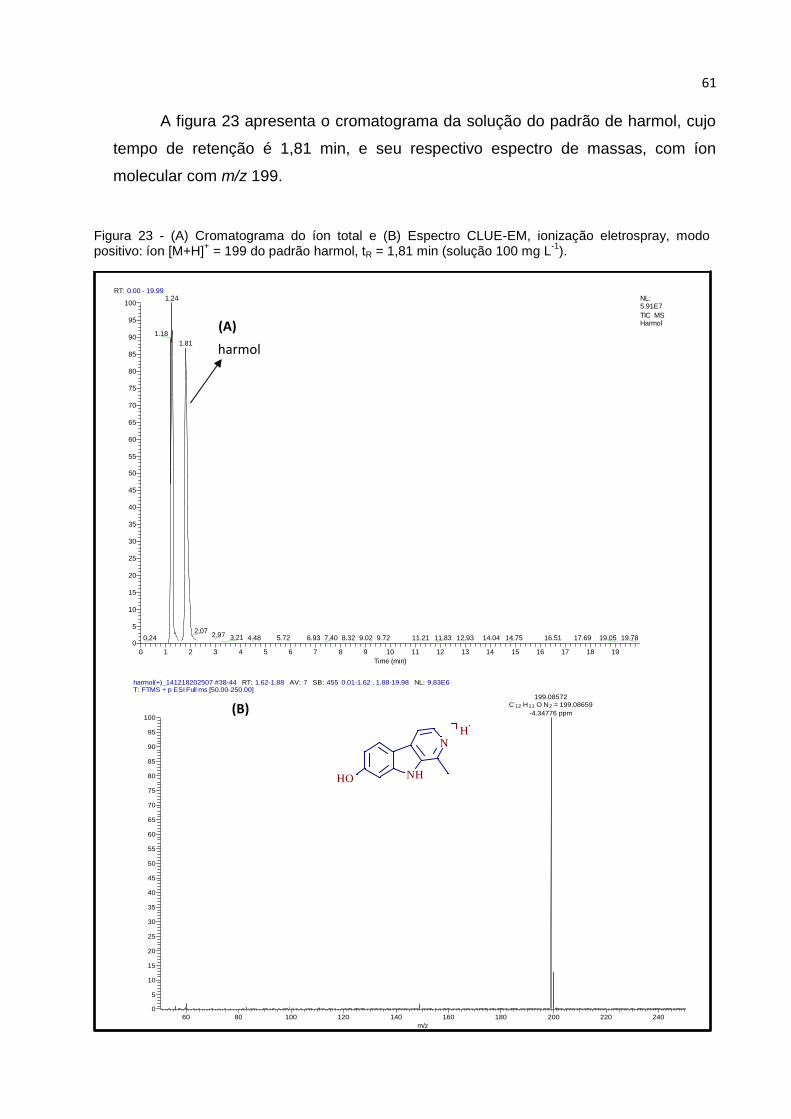
61
A figura 23 apresenta o cromatograma da solução do padrão de harmol, cujo
tempo de retenção é 1,81 min, e seu respectivo espectro de massas, com íon
molecular com m/z 199.
RT: 0.00 - 19.99
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
1.24
1.18
1.81
2.072.97 3.21 5.724.48 6.93 9.728.32 9.02 11.21 11.83 14.047.40 14.75 19.7812.93 16.51 19.0517.690.24
NL:5.91E7
TIC MS Harmol
harmol(+)_141218202507 #38-44 RT: 1.62-1.88 AV: 7 SB: 455 0.01-1.62 , 1.88-19.98 NL: 9.83E6T: FTMS + p ESI Full ms [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lativ
e A
bu
nd
an
ce
199.08572
C 12 H11 O N2 = 199.08659
-4.34776 ppm
(A)
(B)
NH
N
OH
H
harmol
Figura 23 - (A) Cromatograma do íon total e (B) Espectro CLUE-EM, ionização eletrospray, modo positivo: íon [M+H]
+ = 199 do padrão harmol, tR = 1,81 min (solução 100 mg L
-1).
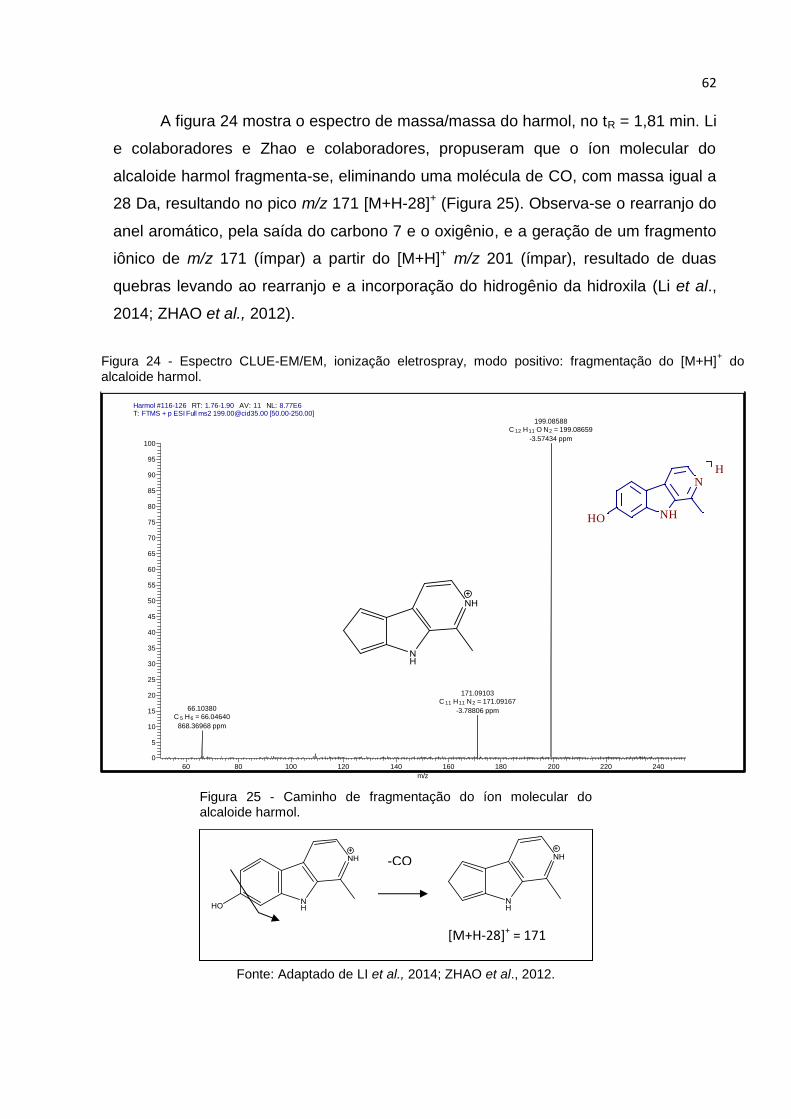
62
A figura 24 mostra o espectro de massa/massa do harmol, no tR = 1,81 min. Li
e colaboradores e Zhao e colaboradores, propuseram que o íon molecular do
alcaloide harmol fragmenta-se, eliminando uma molécula de CO, com massa igual a
28 Da, resultando no pico m/z 171 [M+H-28]+ (Figura 25). Observa-se o rearranjo do
anel aromático, pela saída do carbono 7 e o oxigênio, e a geração de um fragmento
iônico de m/z 171 (ímpar) a partir do [M+H]+ m/z 201 (ímpar), resultado de duas
quebras levando ao rearranjo e a incorporação do hidrogênio da hidroxila (Li et al.,
2014; ZHAO et al., 2012).
Harmol #116-126 RT: 1.76-1.90 AV: 11 NL: 8.77E6T: FTMS + p ESI Full ms2 [email protected] [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
199.08588
C 12 H11 O N2 = 199.08659
-3.57434 ppm
171.09103
C 11 H11 N2 = 171.09167
-3.78806 ppm66.10380
C 5 H6 = 66.04640
868.36968 ppm
NH
N
OH
H
-CO
[M+H-28]+ = 171
Fonte: Adaptado de LI et al., 2014; ZHAO et al., 2012.
Figura 24 - Espectro CLUE-EM/EM, ionização eletrospray, modo positivo: fragmentação do [M+H]+ do
alcaloide harmol.
Figura 25 - Caminho de fragmentação do íon molecular do alcaloide harmol.
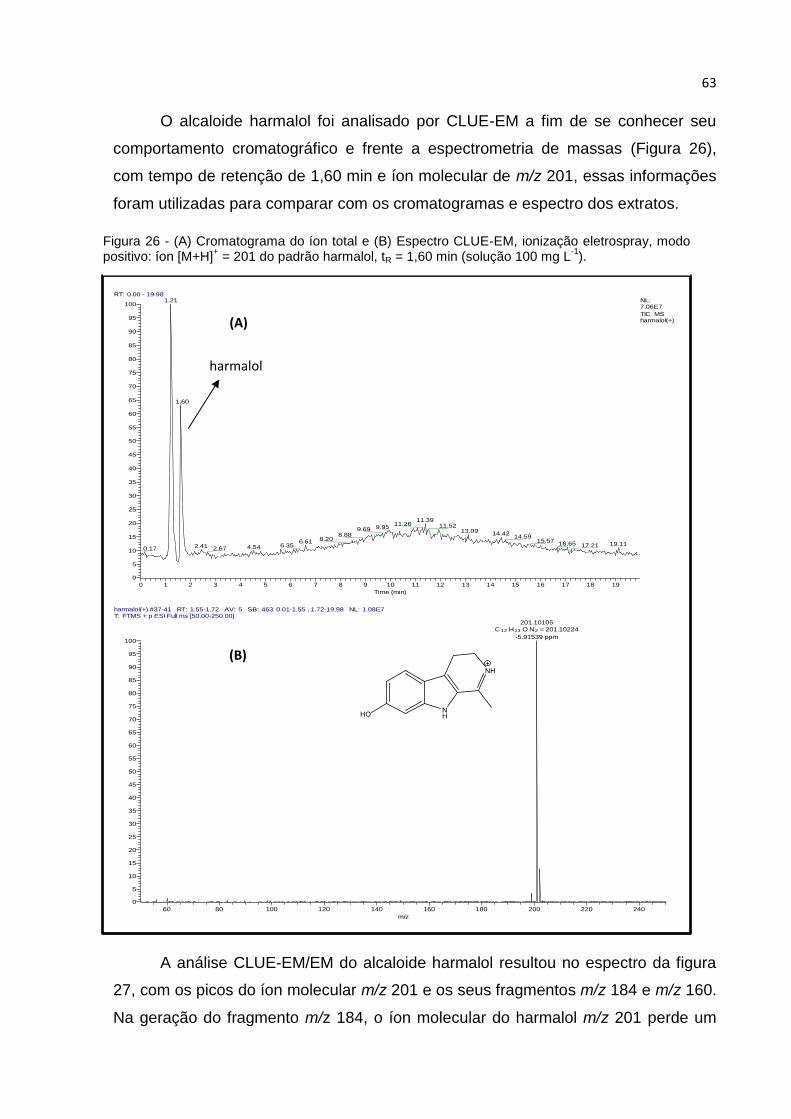
63
O alcaloide harmalol foi analisado por CLUE-EM a fim de se conhecer seu
comportamento cromatográfico e frente a espectrometria de massas (Figura 26),
com tempo de retenção de 1,60 min e íon molecular de m/z 201, essas informações
foram utilizadas para comparar com os cromatogramas e espectro dos extratos.
A análise CLUE-EM/EM do alcaloide harmalol resultou no espectro da figura
27, com os picos do íon molecular m/z 201 e os seus fragmentos m/z 184 e m/z 160.
Na geração do fragmento m/z 184, o íon molecular do harmalol m/z 201 perde um
RT: 0.00 - 19.98
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
1.21
1.60
11.3911.26 11.529.959.69 13.09 14.428.88 14.598.20 15.576.61 16.65 19.116.35 17.212.41 4.540.17 2.67
NL:7.06E7
TIC MS harmalol(+)
harmalol(+) #37-41 RT: 1.55-1.72 AV: 5 SB: 463 0.01-1.55 , 1.72-19.98 NL: 1.08E7T: FTMS + p ESI Full ms [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
201.10105
C 12 H13 O N2 = 201.10224
-5.91539 ppm
(A)
(B)
harmalol
Figura 26 - (A) Cromatograma do íon total e (B) Espectro CLUE-EM, ionização eletrospray, modo positivo: íon [M+H]
+ = 201 do padrão harmalol, tR = 1,60 min (solução 100 mg L
-1).

64
fragmento de massa 17 Da. Zhao e colaboradores atribuiram a perda de OH para
este pico (C12H12N2) (ZHAO et al., 2012) (Figura 28 a), que obedece a “regra do
nitrogênio”, pois um íon molecular ímpar sofrendo uma quebra simples resulta em
um íon “filho” par, mas apresenta um erro de -25 ppm entre a massa teórica e a
massa experimental. A fórmula molecular atribuída ao pico m/z 184 (C12H10ON), pelo
software utilizado, possui um átomo de O e um só átomo de N, com um erro de -8,9
ppm, com base nessa informação a estrutura da figura 28 (b) foi proposta,
considerando então a eliminação de NH3, com massa de 17 Da, e rearranjo do anel
aromático.
O íon “filho” m/z 160 possui fórmula molecular C10H10ON, com a diferença de
C2H3N em comparação com o íon principal, assim, ocorre a perda de 41 Da e
rearranjo. Zhao e colaboradores propuseram a estrutura do íon m/z 160 [M+H-41]+,
como apresentado na figura 28 (c) (ZHAO et al., 2012).
Harmalol #108-120 RT: 1.64-1.81 AV: 13 NL: 3.60E7T: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
160.07490
C 10 H10 O N = 160.07569
-4.96368 ppm
184.07480
C 12 H10 O N = 184.07569
-4.83421 ppm
201.10125
C 12 H13 O N2 = 201.10224
-4.91225 ppm
Figura 27 - Espectro CLUE-EM/EM, ionização eletrospray, modo positivo: fragmentação do [M+H]+ do alcaloide
harmalol.
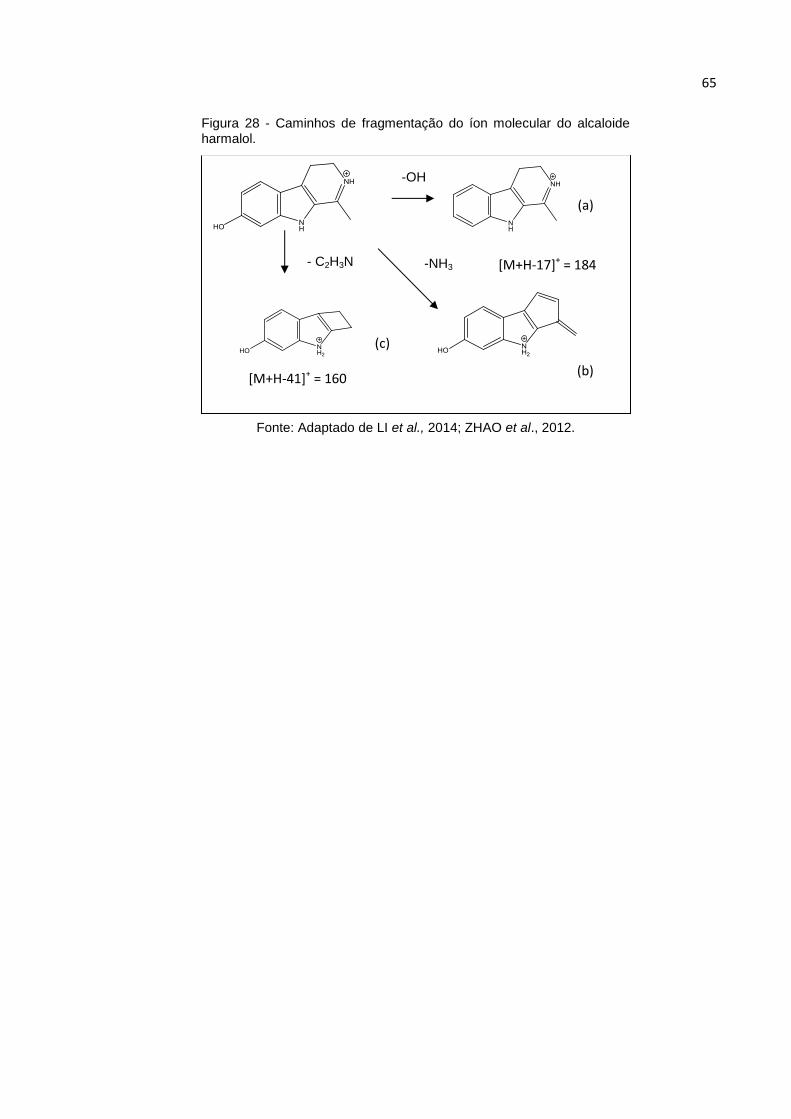
65
- C2H3N
[M+H-17]+ = 184
[M+H-41]+ = 160
-OH
-NH3
(a)
(b)
(c)
Fonte: Adaptado de LI et al., 2014; ZHAO et al., 2012.
Figura 28 - Caminhos de fragmentação do íon molecular do alcaloide harmalol.
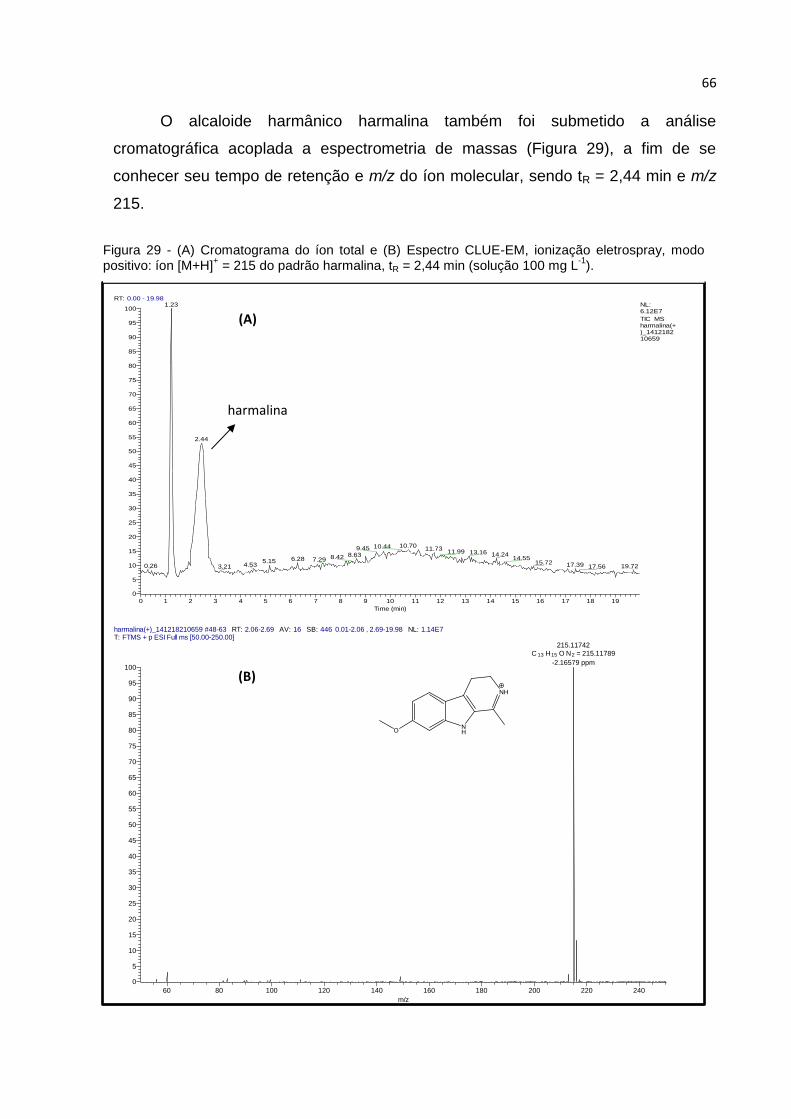
66
O alcaloide harmânico harmalina também foi submetido a análise
cromatográfica acoplada a espectrometria de massas (Figura 29), a fim de se
conhecer seu tempo de retenção e m/z do íon molecular, sendo tR = 2,44 min e m/z
215.
RT: 0.00 - 19.98
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lativ
e A
bu
nd
an
ce
1.23
2.44
10.7010.449.45 11.7311.99 13.16 14.248.638.42 14.556.28 7.295.15 15.72 17.394.530.26 19.723.21 17.56
NL:6.12E7
TIC MS harmalina(+)_141218210659
harmalina(+)_141218210659 #48-63 RT: 2.06-2.69 AV: 16 SB: 446 0.01-2.06 , 2.69-19.98 NL: 1.14E7T: FTMS + p ESI Full ms [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
215.11742
C 13 H15 O N2 = 215.11789
-2.16579 ppm
(A)
(B)
harmalina
Figura 29 - (A) Cromatograma do íon total e (B) Espectro CLUE-EM, ionização eletrospray, modo positivo: íon [M+H]
+ = 215 do padrão harmalina, tR = 2,44 min (solução 100 mg L
-1).
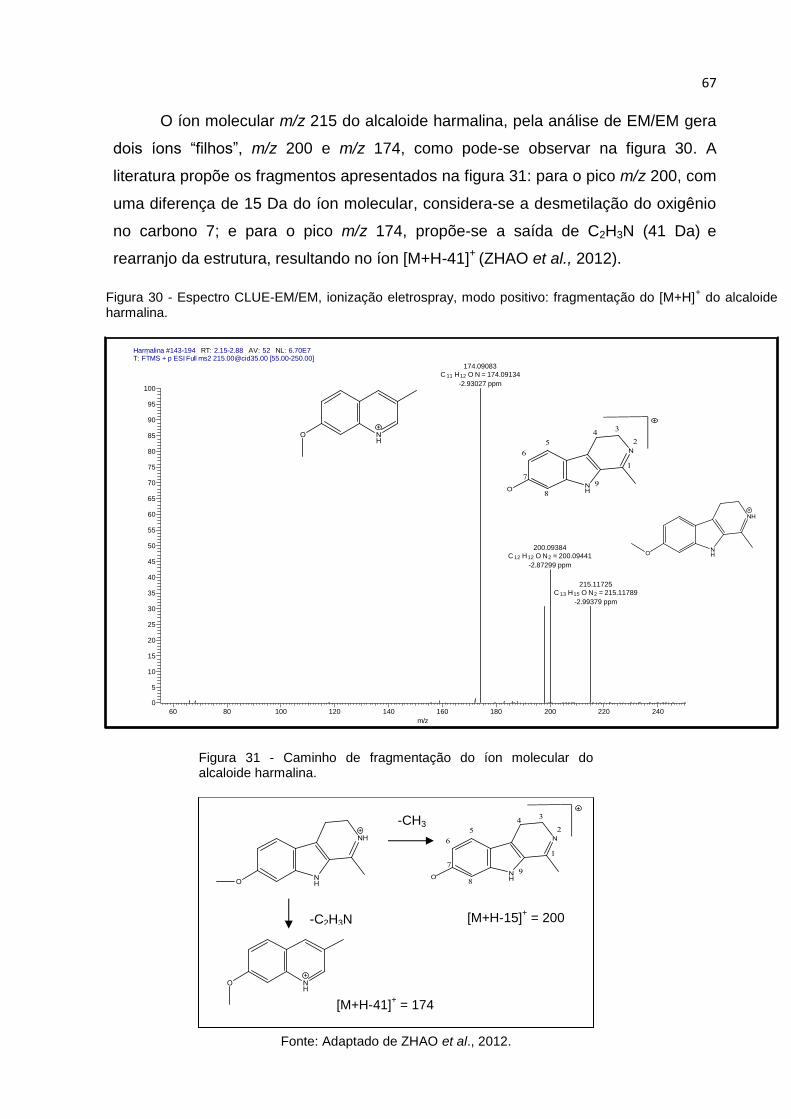
67
O íon molecular m/z 215 do alcaloide harmalina, pela análise de EM/EM gera
dois íons “filhos”, m/z 200 e m/z 174, como pode-se observar na figura 30. A
literatura propõe os fragmentos apresentados na figura 31: para o pico m/z 200, com
uma diferença de 15 Da do íon molecular, considera-se a desmetilação do oxigênio
no carbono 7; e para o pico m/z 174, propõe-se a saída de C2H3N (41 Da) e
rearranjo da estrutura, resultando no íon [M+H-41]+ (ZHAO et al., 2012).
Harmalina #143-194 RT: 2.15-2.88 AV: 52 NL: 6.70E7T: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
174.09083
C 11 H12 O N = 174.09134
-2.93027 ppm
200.09384
C 12 H12 O N2 = 200.09441
-2.87299 ppm
215.11725
C 13 H15 O N2 = 215.11789
-2.99379 ppm
[M+H-15]+ = 200
[M+H-41]+ = 174
-CH3
-C2H3N
Fonte: Adaptado de ZHAO et al., 2012.
Figura 30 - Espectro CLUE-EM/EM, ionização eletrospray, modo positivo: fragmentação do [M+H]+ do alcaloide
harmalina.
Figura 31 - Caminho de fragmentação do íon molecular do alcaloide harmalina.

68
4. 2. 1 Análise dos extratos de Passiflora alata e Passiflora edulis
O cromatograma do íon total e o espectro de massas do solvente (metanol)
utilizado para preparação das soluções de padrões e dos extratos, apresentados nas
figuras 32 e 33, foram obtidos nas mesmas condições CLUE-EM, para comparação
com os espectros dos padrões e extratos. Uma vez que os espectros dos extratos
apresentavam alguns picos desconhecidos, o espectro do branco (metanol) auxiliou
na detecção dos picos que pertenciam às amostras estudadas.

69
RT: 0.00 - 19.99
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
0.86
16.86 16.959.59
16.6515.41
11.1310.329.424.08 11.86 19.397.031.33 6.35 16.358.40 17.986.26 18.535.11 7.42 14.0413.05
15.24 15.497.97 8.572.190.643.912.66
2.10
NL:4.58E9
TIC MS metanol(+)
metanol(+) #19-23 RT: 0.77-0.94 AV: 5 NL: 5.40E8T: FTMS + p ESI Full ms [50.00-250.00]
60 70 80 90 100 110 120 130 140 150 160 170 180 190 200
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
60.04399
C 2 H6 O N = 60.04439
-6.70806 ppm
90.50642
81.52007
111.01965
C 4 H3 O2 N2 = 111.01890
6.73513 ppm
99.51169
73.53134
125.98566
C 8 O N = 125.99744
-93.52048 ppm
184.98473
C 9 H O3 N2 = 184.99817
-72.63438 ppm
92.50284
156.98993
C 9 H O3 = 156.99202
-13.30062 ppm
Figura 32 - Cromatograma do íon total do branco (metanol), obtido por CLUE-EM, ionização eletrospray, modo positivo.
Figura 33 - Espectro CLUE-EM, ionização eletrospray, modo positivo, do branco (metanol).
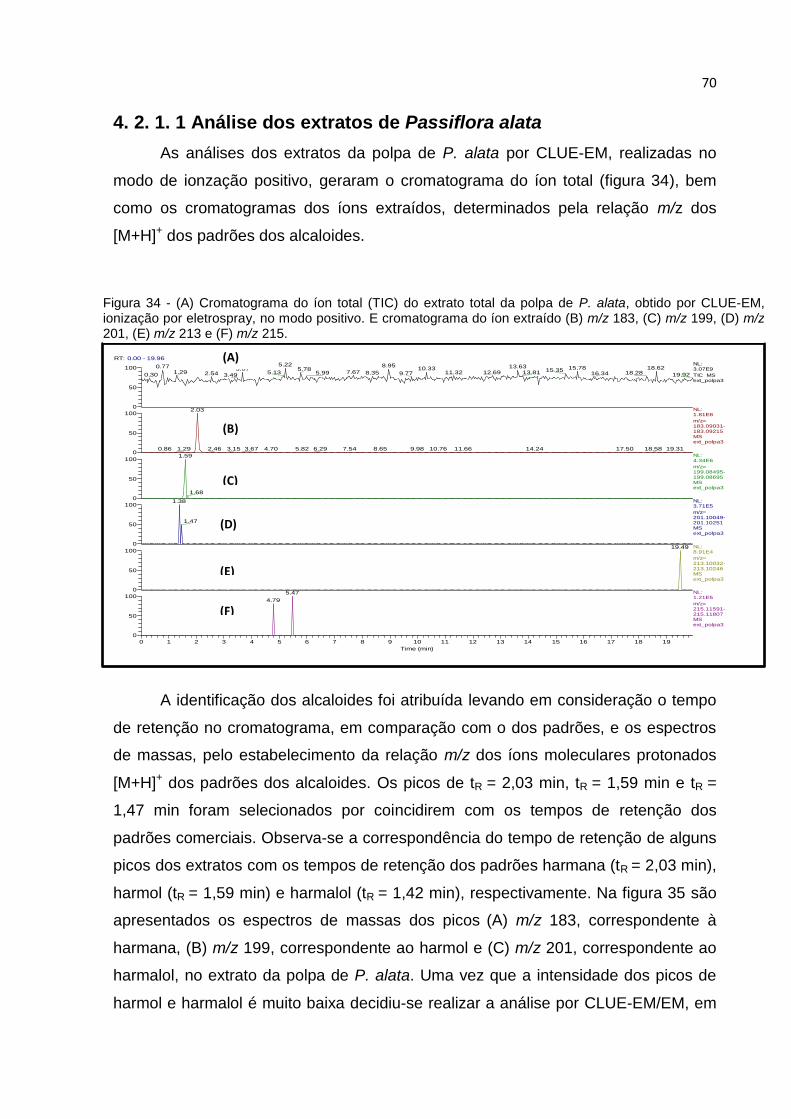
70
4. 2. 1. 1 Análise dos extratos de Passiflora alata
As análises dos extratos da polpa de P. alata por CLUE-EM, realizadas no
modo de ionzação positivo, geraram o cromatograma do íon total (figura 34), bem
como os cromatogramas dos íons extraídos, determinados pela relação m/z dos
[M+H]+ dos padrões dos alcaloides.
A identificação dos alcaloides foi atribuída levando em consideração o tempo
de retenção no cromatograma, em comparação com o dos padrões, e os espectros
de massas, pelo estabelecimento da relação m/z dos íons moleculares protonados
[M+H]+ dos padrões dos alcaloides. Os picos de tR = 2,03 min, tR = 1,59 min e tR =
1,47 min foram selecionados por coincidirem com os tempos de retenção dos
padrões comerciais. Observa-se a correspondência do tempo de retenção de alguns
picos dos extratos com os tempos de retenção dos padrões harmana (tR = 2,03 min),
harmol (tR = 1,59 min) e harmalol (tR = 1,42 min), respectivamente. Na figura 35 são
apresentados os espectros de massas dos picos (A) m/z 183, correspondente à
harmana, (B) m/z 199, correspondente ao harmol e (C) m/z 201, correspondente ao
harmalol, no extrato da polpa de P. alata. Uma vez que a intensidade dos picos de
harmol e harmalol é muito baixa decidiu-se realizar a análise por CLUE-EM/EM, em
RT: 0.00 - 19.96
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
50
100
0
50
100
0
50
100
0
50
100
Rel
ativ
e A
bund
ance
0
50
100
0
50
1005.22 8.95 13.630.77 15.78 18.6210.333.67 5.78 15.351.29 7.675.13 12.69 13.8111.325.99 8.35 18.2816.349.772.54 19.920.30 3.49
2.03
2.46 3.15 3.670.86 5.821.29 6.294.70 19.3110.768.65 11.66 14.24 17.507.54 9.98 18.58
1.59
1.68
1.38
1.47
19.49
5.47
4.79
NL:3.07E9
TIC MS ext_polpa3
NL:1.81E8
m/z= 183.09031-183.09215 MS ext_polpa3
NL:4.34E6
m/z= 199.08495-199.08695 MS ext_polpa3
NL:3.71E5
m/z= 201.10049-201.10251 MS ext_polpa3
NL:8.91E4
m/z= 213.10032-213.10246 MS ext_polpa3
NL:1.21E5
m/z= 215.11591-215.11807 MS ext_polpa3
(A)
(B)
(C)
(D)
(E)
(F)
Figura 34 - (A) Cromatograma do íon total (TIC) do extrato total da polpa de P. alata, obtido por CLUE-EM, ionização por eletrospray, no modo positivo. E cromatograma do íon extraído (B) m/z 183, (C) m/z 199, (D) m/z 201, (E) m/z 213 e (F) m/z 215.

71
que é possível a seleção do íon molecular que se deseja estudar e avaliar a sua
presença no espectro, bem como seus fragmentos (íons “filhos”).
ext_polpa3 #47-49 RT: 1.98-2.07 AV: 3 SB: 462 0.01-1.94 , 2.11-19.96 NL: 8.60E7T: FTMS + p ESI Full ms [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relat
ive A
bund
ance
183.09097
C 12 H11 N2 = 183.09167
-3.86420 ppm
81.52002
90.5062899.51161
73.53130
141.95790
C 8 O2 N = 141.99235
-242.73195 ppm
167.01203
C 11 H3 O2 = 167.01276
-4.37425 ppm
229.86616
C 14 O3 N = 229.98727
-526.87568 ppm
ext_polpa3 #37-39 RT: 1.55-1.64 AV: 3 SB: 459 0.01-1.51 , 1.68-19.83 NL: 1.81E6T: FTMS + p ESI Full ms [50.00-250.00]
170 175 180 185 190 195 200 205 210 215 220 225 230 235 240 245
m/z
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
360
380
400
420
440
460
480
500
520
540
Relat
ive Ab
unda
nce
182.98349
C 10 H O3 N = 182.99509
-63.41628 ppm
199.08563
C 12 H11 O N2 = 199.08659
-4.83542 ppm
192.10071
C 11 H14 O2 N = 192.10191
-6.20822 ppm172.97592
C 8 H O3 N2 = 172.99817
-128.60358 ppm209.74074
241.17605
C 15 H15 O2 N = 241.10973
274.97272 ppm225.59407215.67214
203.94852
C 13 O3 = 203.98420
-174.90446 ppm195.72285
ext_polpa3 #32-34 RT: 1.34-1.42 AV: 3 SB: 461 0.01-1.29 , 1.47-19.92 NL: 2.31E5T: FTMS + p ESI Full ms [50.00-250.00]
180 185 190 195 200 205 210 215 220 225 230 235
m/z
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700
1800
1900
2000
Relat
ive A
bund
ance
186.95541
C 13 H O N = 187.00527
-266.65613 ppm
201.10147
C 12 H13 O N2 = 201.10224
-3.81710 ppm
228.21945
C 14 H16 O N2 = 228.12571
410.73060 ppm
218.14416
C 13 H16 O2 N = 218.11756
121.95600 ppm208.54473
(B)
(A)
(C)
harmana
harmol
harmalol
[M+H]+
[M+H]+
[M+H]+
Figura 35 - Espectro CLUE-EM, ionização eletrospray, modo positivo: dos picos do íons (A) m/z 183, (B) m/z 199 e (C) m/z 201, presentes nos cromatogramas dos íons extraídos do extrato da polpa de P. alata figura 34.

72
As análises do extrato da polpa de P. alata por CLUE-EM/EM, realizadas no
modo de ionzação positivo, geraram os cromatogramas e os espectros de massas
de cada alcaloide harmânico presente no extrato. A identificação dos alcaloides foi
atribuída levando em consideração o tempo de retenção no cromatograma, em
comparação com o dos padrões, e as medidas de alta precisão das massas, por
comparação dos espectros de massas dos padrões e do extrato.
Pode-se observar a correspondência do tempo de retenção do pico do padrão
de harmana (tR = 2,09 min) (Figura 36 A) com o tempo de retenção (tR = 2,18 min)
do pico no cromatograma do extrato da polpa de P. alata (Figura 36 B).
RT: 0.00 - 20.00
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Time (min)
0
10
20
30
40
50
60
70
80
90
100
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
1.25
1.202.09
2.19
2.02
2.241.99
2.38
2.53 2.92 3.59 4.49 5.72 7.486.51 8.10 9.25 9.76 10.63 11.75 12.62 14.5614.12 15.07 16.48 17.08 18.15 18.73
2.18
2.15
2.26
2.32
2.36
2.40 19.671.71 14.562.77 7.78 11.90 18.8516.661.30 16.774.78 10.467.21 16.4212.806.38 7.83
NL:5.05E6
m/z= 183.08992-183.09176 MS harmana
NL:7.35E3
m/z= 183.08992-183.09176 MS palata_polpa_harmana
(A)
(B)
harmana
Figura 36 - Cromatogramas obtidos através das análises realizadas por CLUE-EM/EM do padrão de harmana e do extrato da polpa de P. alata. (A) solução padrão de harmana m/z 183, tR = 2,05 min e (B) extrato da polpa de P. alata.

73
Ainda, há correspondência do íon molecular presente no espectro de EM/EM
do extrato, m/z 183 (Figura 37 B), com o pico do íon molecular apresentado pelo
espectro do padrão (Figura 37 A).
A presença dos picos m/z 141 e m/z 165 nos espectros do extrato da polpa de
P. alata foi questionada como possível fragmentação do íon molecular. Contudo, os
dados da literatura mostraram que não há semelhança entre os picos do extrato e os
identificados em outros trabalhos: Li e colaboradores realizaram CLUE-EM/EM para
avaliação do comportamento da harmana. O método utilizado (ESI, no modo positivo
de ionização e EM/EM) resultou no [M+H]+ com m/z 183,0955 e os fragmentos de
m/z 168,0719 ([M+H-15]+), m/z 142,0679 ([M+H-41]+) e m/z 115,0570 ([M+H-68]+).
O pico m/z 141,95766 do espectro de massas do extrato da polpa apresenta um erro
de -107 ppm com relação à fórmula molecular proposta (C10H8N+), sendo assim não
foi possível a determinação da origem dos outros picos presentes no espectro do
extrato da polpa de P. alata, pela aplicação de CLUE-EM/EM (Li et al., 2014).
60 80 100 120 140 160 180 200 220 240
m/z
0
10
20
30
40
50
60
70
80
90
100
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
183.09122
C 12 H11 N2 = 183.09167
-2.50878 ppm
66.10370
141.95750
183.09027
66.10421165.13734
97.96790
NL: 2.51E6
harmana#138-156 RT: 2.07-2.32 AV: 19 T: FTMS + p ESI Full ms2 [email protected] [50.00-250.00]
NL: 1.39E4
palata_polpa_harmana#132-148 RT: 2.07-2.32 AV: 17 T: FTMS + p ESI Full ms2 [email protected] [50.00-250.00]
(A)
(B)
[M+H]+
[M+H]+
Figura 37 - Espectros de massas da análise CLUE-EM/EM do íon [M+H]+ de m/z 183, tR = 2,05
min: (A) do padrão comercial de harmana e (B) extrato da polpa de P. alata.
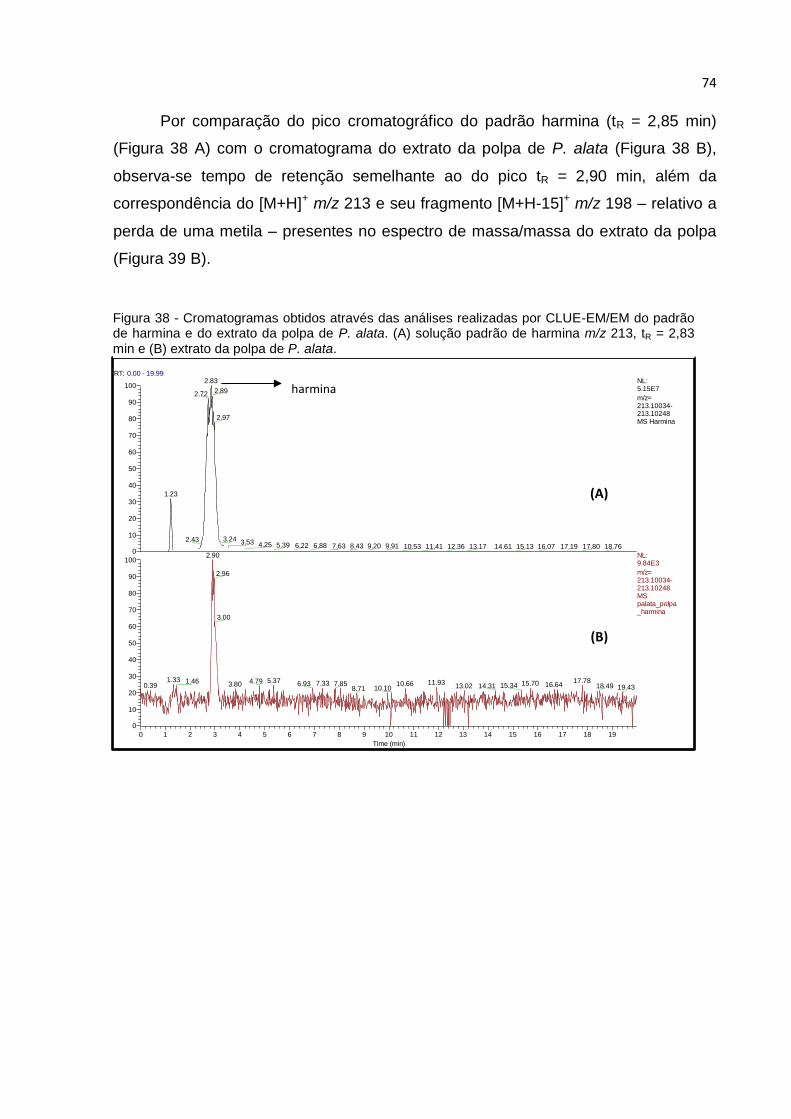
74
Por comparação do pico cromatográfico do padrão harmina (tR = 2,85 min)
(Figura 38 A) com o cromatograma do extrato da polpa de P. alata (Figura 38 B),
observa-se tempo de retenção semelhante ao do pico tR = 2,90 min, além da
correspondência do [M+H]+ m/z 213 e seu fragmento [M+H-15]+ m/z 198 – relativo a
perda de uma metila – presentes no espectro de massa/massa do extrato da polpa
(Figura 39 B).
RT: 0.00 - 19.99
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
10
20
30
40
50
60
70
80
90
100
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
2.83
2.892.72
2.97
1.23
3.242.43 3.53 4.25 5.39 6.22 6.88 7.63 8.43 9.20 9.91 10.53 11.41 12.36 13.17 14.61 15.13 16.07 17.19 17.80 18.76
2.90
2.96
3.00
1.33 17.785.374.791.46 11.937.33 15.707.856.93 10.663.80 16.640.39 15.3413.02 18.4914.31 19.4310.108.71
NL:5.15E7
m/z= 213.10034-213.10248 MS Harmina
NL:9.84E3
m/z= 213.10034-213.10248 MS palata_polpa_harmina
(B)
(A)
harmina
Figura 38 - Cromatogramas obtidos através das análises realizadas por CLUE-EM/EM do padrão de harmina e do extrato da polpa de P. alata. (A) solução padrão de harmina m/z 213, tR = 2,83 min e (B) extrato da polpa de P. alata.
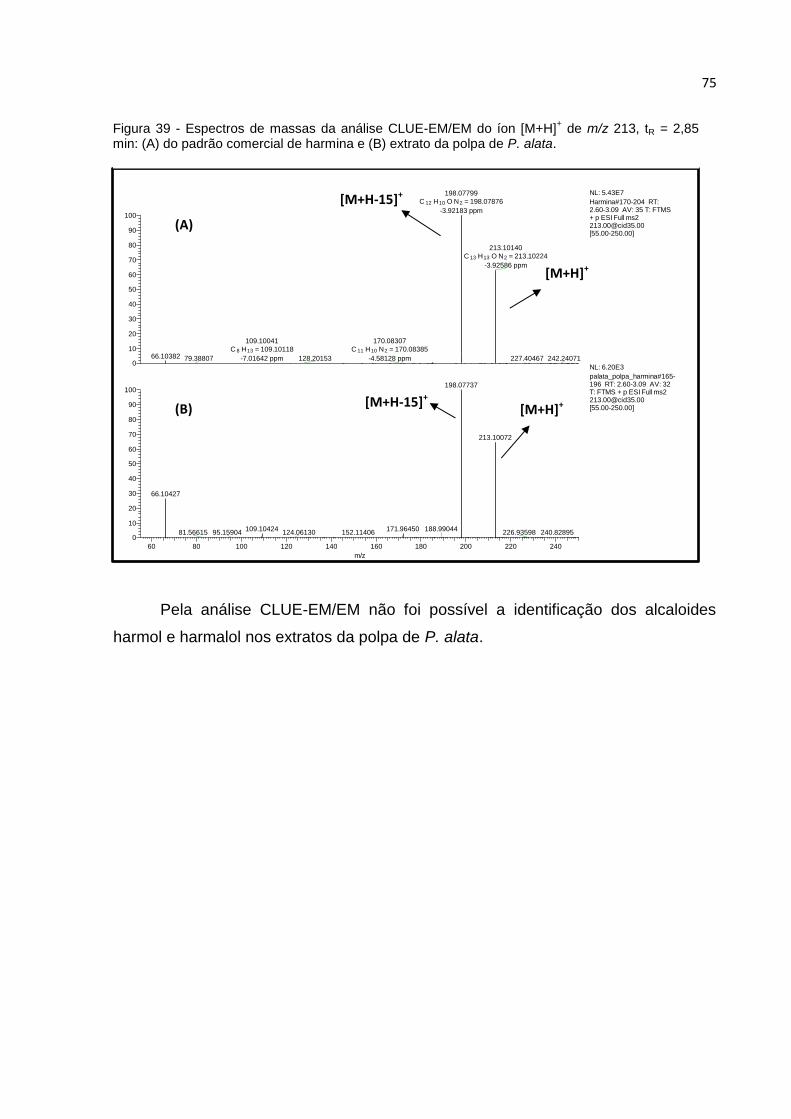
75
Pela análise CLUE-EM/EM não foi possível a identificação dos alcaloides
harmol e harmalol nos extratos da polpa de P. alata.
60 80 100 120 140 160 180 200 220 240
m/z
0
10
20
30
40
50
60
70
80
90
100
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
198.07799
C 12 H10 O N2 = 198.07876
-3.92183 ppm
213.10140
C 13 H13 O N2 = 213.10224
-3.92586 ppm
66.10382
170.08307
C 11 H10 N2 = 170.08385
-4.58128 ppm
109.10041
C 8 H13 = 109.10118
-7.01642 ppm 128.2015379.38807 227.40467 242.24071
198.07737
213.10072
66.10427
171.96450109.1042495.15904 152.11406124.0613081.56615 240.82895226.93598
188.99044
NL: 5.43E7
Harmina#170-204 RT: 2.60-3.09 AV: 35 T: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
NL: 6.20E3
palata_polpa_harmina#165-196 RT: 2.60-3.09 AV: 32 T: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
(A)
(B)
[M+H]+
[M+H-15]+
[M+H]+ [M+H-15]+
Figura 39 - Espectros de massas da análise CLUE-EM/EM do íon [M+H]+ de m/z 213, tR = 2,85
min: (A) do padrão comercial de harmina e (B) extrato da polpa de P. alata.
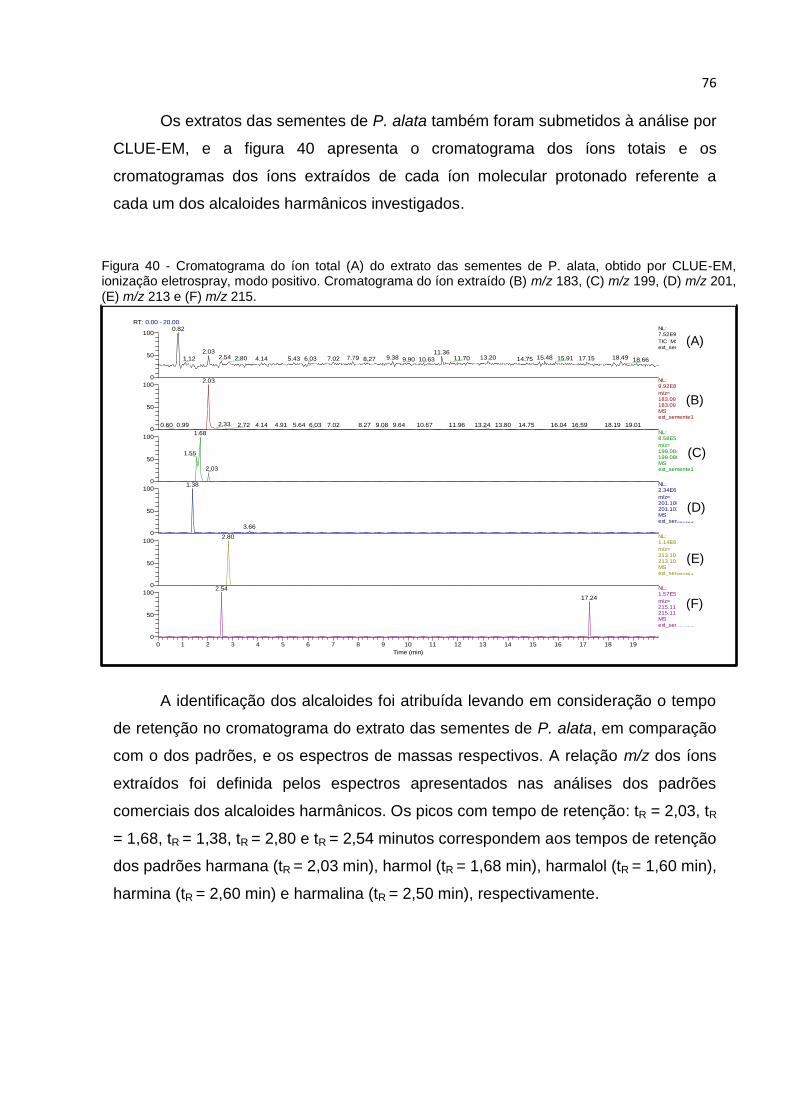
76
Os extratos das sementes de P. alata também foram submetidos à análise por
CLUE-EM, e a figura 40 apresenta o cromatograma dos íons totais e os
cromatogramas dos íons extraídos de cada íon molecular protonado referente a
cada um dos alcaloides harmânicos investigados.
A identificação dos alcaloides foi atribuída levando em consideração o tempo
de retenção no cromatograma do extrato das sementes de P. alata, em comparação
com o dos padrões, e os espectros de massas respectivos. A relação m/z dos íons
extraídos foi definida pelos espectros apresentados nas análises dos padrões
comerciais dos alcaloides harmânicos. Os picos com tempo de retenção: tR = 2,03, tR
= 1,68, tR = 1,38, tR = 2,80 e tR = 2,54 minutos correspondem aos tempos de retenção
dos padrões harmana (tR = 2,03 min), harmol (tR = 1,68 min), harmalol (tR = 1,60 min),
harmina (tR = 2,60 min) e harmalina (tR = 2,50 min), respectivamente.
RT: 0.00 - 20.00
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
50
100
0
50
100
0
50
100
0
50
100
Re
lativ
e A
bu
nd
an
ce
0
50
100
0
50
1000.82
2.03 11.362.54 13.20 15.48 18.499.387.79 11.70 15.91 17.152.80 7.021.12 5.434.14 6.03 14.758.27 9.90 10.63 18.66
2.03
2.33 2.72 4.14 5.644.91 6.03 8.27 13.807.02 14.759.64 16.0411.9610.679.08 19.0113.240.99 18.190.60 16.59
1.68
1.55
2.03
1.38
3.66
2.80
2.54
17.24
NL:7.52E9
TIC MS ext_semente1
NL:9.92E8
m/z= 183.09031-183.09215 MS ext_semente1
NL:8.58E5
m/z= 199.08495-199.08695 MS ext_semente1
NL:2.34E6
m/z= 201.10049-201.10251 MS ext_semente1
NL:1.14E6
m/z= 213.10032-213.10246 MS ext_semente1
NL:1.57E5
m/z= 215.11591-215.11807 MS ext_semente1
(A)
(B)
(C)
(D)
(E)
(F)
Figura 40 - Cromatograma do íon total (A) do extrato das sementes de P. alata, obtido por CLUE-EM, ionização eletrospray, modo positivo. Cromatograma do íon extraído (B) m/z 183, (C) m/z 199, (D) m/z 201, (E) m/z 213 e (F) m/z 215.
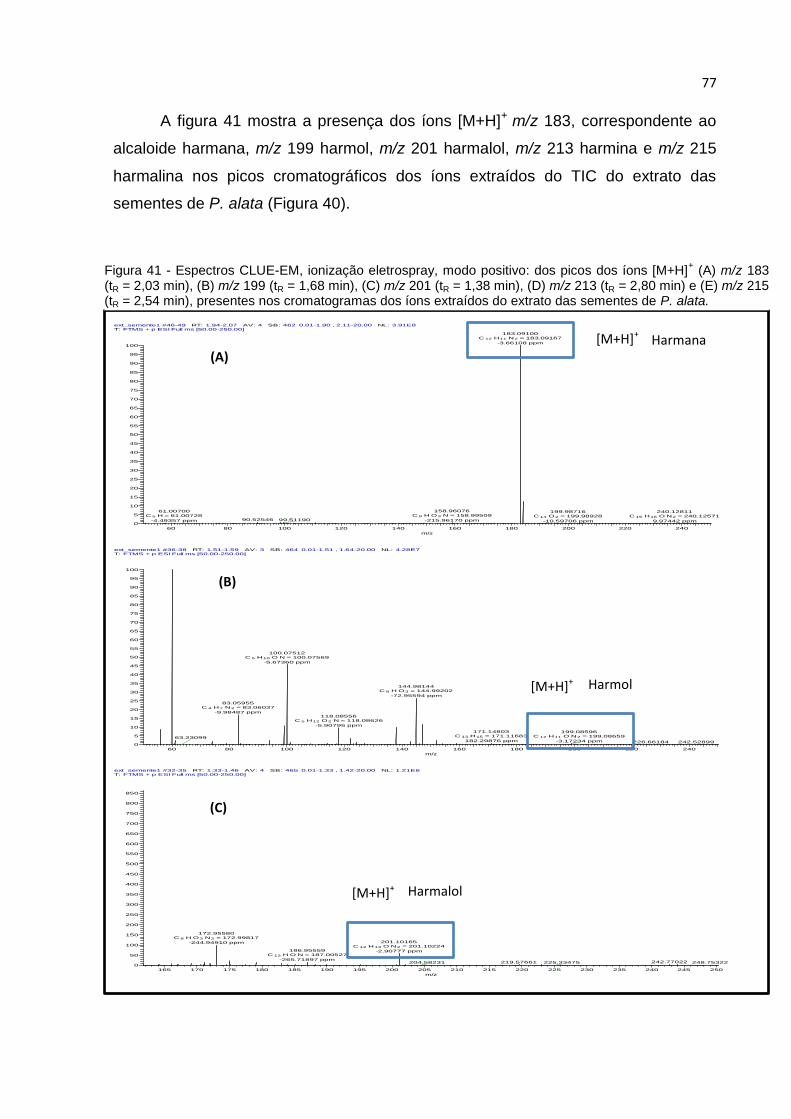
77
A figura 41 mostra a presença dos íons [M+H]+ m/z 183, correspondente ao
alcaloide harmana, m/z 199 harmol, m/z 201 harmalol, m/z 213 harmina e m/z 215
harmalina nos picos cromatográficos dos íons extraídos do TIC do extrato das
sementes de P. alata (Figura 40).
ext_semente1 #46-49 RT: 1.94-2.07 AV: 4 SB: 462 0.01-1.90 , 2.11-20.00 NL: 3.91E8T: FTMS + p ESI Full ms [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relat
ive Ab
unda
nce
183.09100
C 12 H11 N2 = 183.09167
-3.66108 ppm
90.52546 99.51190
158.96076
C 8 H O3 N = 158.99509
-215.96170 ppm
61.00700
C 5 H = 61.00728
-4.49357 ppm
199.98716
C 14 O2 = 199.98928
-10.59706 ppm
240.12811
C 15 H16 O N2 = 240.12571
9.97442 ppm
ext_semente1 #36-38 RT: 1.51-1.59 AV: 3 SB: 464 0.01-1.51 , 1.64-20.00 NL: 4.28E7T: FTMS + p ESI Full ms [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relat
ive Ab
unda
nce
100.07512
C 5 H10 O N = 100.07569
-5.67360 ppm
144.98144
C 8 H O3 = 144.99202
-72.95594 ppm
83.05955
C 4 H7 N2 = 83.06037
-9.98487 ppm118.08556
C 5 H12 O2 N = 118.08626
-5.90795 ppm
171.14803
C 13 H15 = 171.11683
182.29876 ppm
199.08596
C 12 H11 O N2 = 199.08659
-3.17234 ppm63.23099
242.52899226.66184
ext_semente1 #32-35 RT: 1.33-1.46 AV: 4 SB: 465 0.01-1.33 , 1.42-20.00 NL: 1.21E6T: FTMS + p ESI Full ms [50.00-250.00]
165 170 175 180 185 190 195 200 205 210 215 220 225 230 235 240 245 250
m/z
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
850
Relat
ive Ab
unda
nce
172.95580
C 8 H O3 N2 = 172.99817
-244.94910 ppm 201.10165
C 12 H13 O N2 = 201.10224
-2.90777 ppm186.95559
C 13 H O N = 187.00527
-265.71897 ppm242.77022219.57661204.58231 225.33475 248.75322
(A)
(C)
(B)
Harmana
Harmol
Harmalol
[M+H]+
[M+H]+
[M+H]+
Figura 41 - Espectros CLUE-EM, ionização eletrospray, modo positivo: dos picos dos íons [M+H]+ (A) m/z 183
(tR = 2,03 min), (B) m/z 199 (tR = 1,68 min), (C) m/z 201 (tR = 1,38 min), (D) m/z 213 (tR = 2,80 min) e (E) m/z 215 (tR = 2,54 min), presentes nos cromatogramas dos íons extraídos do extrato das sementes de P. alata.
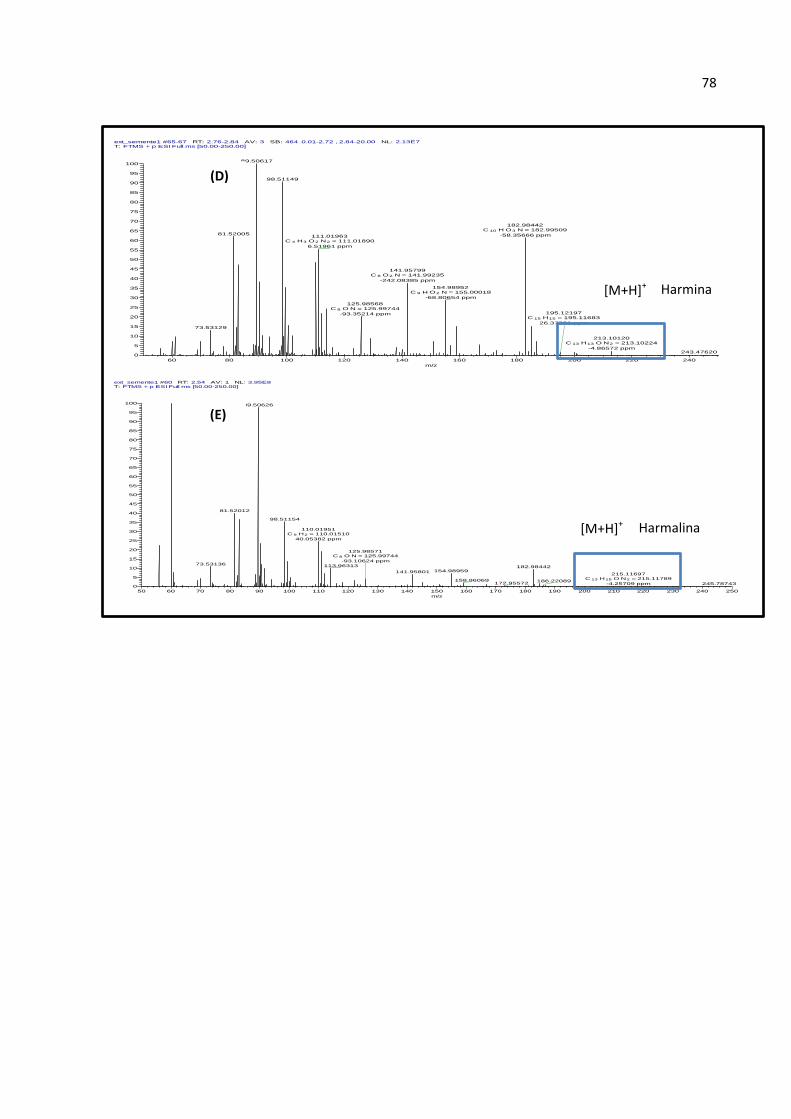
78
ext_semente1 #65-67 RT: 2.76-2.84 AV: 3 SB: 464 0.01-2.72 , 2.84-20.00 NL: 2.13E7T: FTMS + p ESI Full ms [50.00-250.00]
60 80 100 120 140 160 180 200 220 240
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relat
ive A
bund
ance
89.50617
98.51149
81.52005
182.98442
C 10 H O3 N = 182.99509
-58.35666 ppm111.01963
C 4 H3 O2 N2 = 111.01890
6.51961 ppm
141.95799
C 8 O2 N = 141.99235
-242.08385 ppm
154.98952
C 9 H O2 N = 155.00018
-68.80654 ppm
125.98568
C 8 O N = 125.99744
-93.35214 ppm
73.53129
213.10120
C 13 H13 O N2 = 213.10224
-4.86572 ppm
195.12197
C 15 H15 = 195.11683
26.37291 ppm
243.47620
ext_semente1 #60 RT: 2.54 AV: 1 NL: 3.95E8T: FTMS + p ESI Full ms [50.00-250.00]
50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relat
ive A
bund
ance
89.50626
81.52012
98.51154
110.01951
C 9 H2 = 110.01510
40.05382 ppm
73.53136 113.96313 182.98442154.98959141.95801
125.98571
C 8 O N = 125.99744
-93.10624 ppm
158.96069 186.22089172.95572
215.11697
C 13 H15 O N2 = 215.11789
-4.25709 ppm 245.78743
(E)
(D)
Harmalina
Harmina [M+H]+
[M+H]+
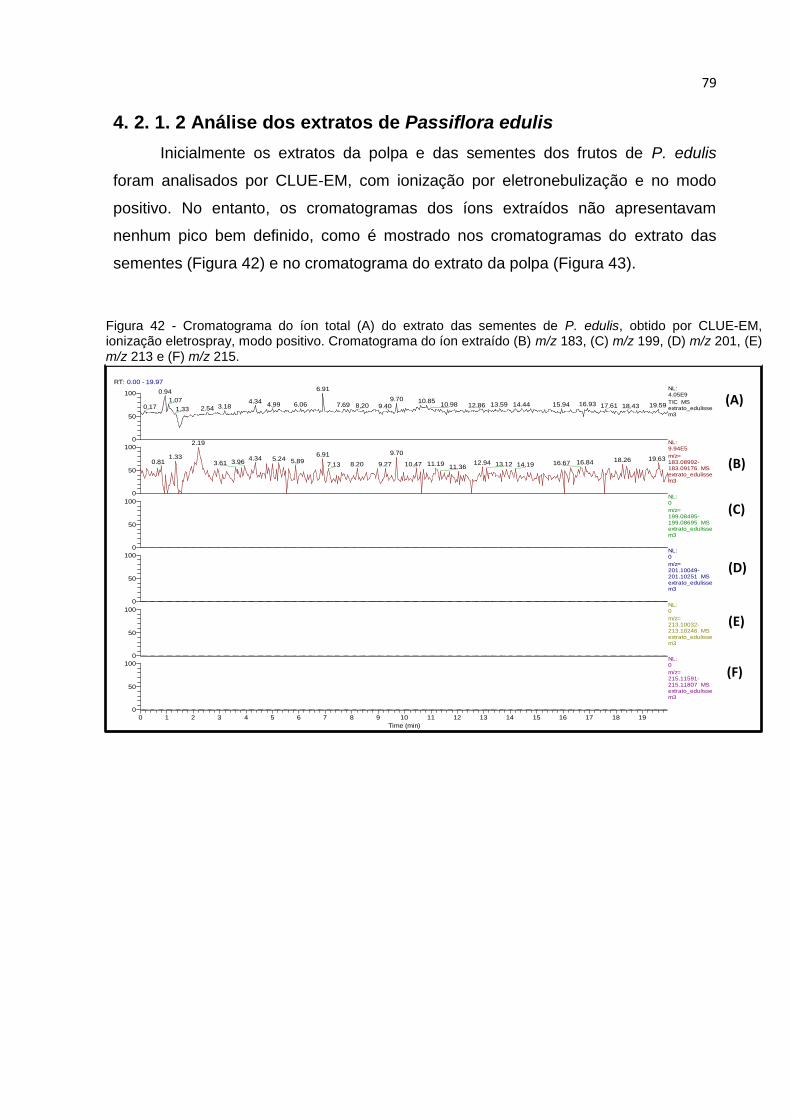
79
4. 2. 1. 2 Análise dos extratos de Passiflora edulis
Inicialmente os extratos da polpa e das sementes dos frutos de P. edulis
foram analisados por CLUE-EM, com ionização por eletronebulização e no modo
positivo. No entanto, os cromatogramas dos íons extraídos não apresentavam
nenhum pico bem definido, como é mostrado nos cromatogramas do extrato das
sementes (Figura 42) e no cromatograma do extrato da polpa (Figura 43).
RT: 0.00 - 19.97
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
50
100
0
50
100
0
50
100
0
50
100
Re
lativ
e A
bu
nd
an
ce
0
50
100
0
50
1006.910.94
9.701.07 10.854.34 16.9314.4410.98 13.596.064.99 15.947.69 12.86 19.598.20 17.61 18.439.403.180.17 2.541.33
2.19
9.706.911.33 4.34 19.635.24 18.265.893.960.81 16.8412.943.61 16.6711.199.278.20 13.1210.47 14.197.13 11.36
NL:4.05E9
TIC MS extrato_edulissem3
NL:9.94E5
m/z= 183.08992-183.09176 MS extrato_edulissem3
NL:0
m/z= 199.08495-199.08695 MS extrato_edulissem3
NL:0
m/z= 201.10049-201.10251 MS extrato_edulissem3
NL:0
m/z= 213.10032-213.10246 MS extrato_edulissem3
NL:0
m/z= 215.11591-215.11807 MS extrato_edulissem3
(E)
(D)
(C)
(F)
(B)
(A)
Figura 42 - Cromatograma do íon total (A) do extrato das sementes de P. edulis, obtido por CLUE-EM, ionização eletrospray, modo positivo. Cromatograma do íon extraído (B) m/z 183, (C) m/z 199, (D) m/z 201, (E) m/z 213 e (F) m/z 215.

80
Então, a fim de obter melhores resultados para a identificação dos alcaloides
harmânicos nos extratos da polpa e das sementes de P. edulis, foram realizadas
análises por CLUE-EM/EM, realizadas no modo de ionzação positivo, que geraram o
cromatograma e o espectro de massas de cada alcaloide harmânico presente no
extrato referente a cada íon molecular [M+H]+ com a relação m/z dos padrões dos
alcaloides.
RT: 0.00 - 19.99
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
50
100
0
50
100
0
50
100
0
50
100
Re
lative
Ab
un
da
nce
0
50
100
0
50
1003.73
1.671.200.94 2.66 5.83 6.51 7.92 9.24 10.914.76 8.39 12.4510.14 16.906.68 14.8913.60 17.6715.57 18.53 19.00
2.23
2.15 3.86
16.901.46 2.40 17.673.99 6.510.60 14.169.54 19.525.53 18.449.03 10.317.54 14.7611.55 16.5111.85
9.76
19.43
NL:6.16E9
TIC MS extrato_edulispol2
NL:1.35E6
m/z= 183.08992-183.09176 MS extrato_edulispol2
NL:1.30E5
m/z= 199.08495-199.08695 MS extrato_edulispol2
NL:0
m/z= 201.10049-201.10251 MS extrato_edulispol2
NL:1.02E5
m/z= 213.10032-213.10246 MS extrato_edulispol2
NL:0
m/z= 215.11591-215.11807 MS extrato_edulispol2
(A)
(B)
(C)
(D)
(E)
(F)
Figura 43 - Cromatograma do íon total (A) do extrato da polpa de P. edulis, obtido por CLUE-EM, ionização eletrospray, modo positivo. Cromatograma do íon extraído (B) m/z 183, (C) m/z 199, (D) m/z 201, (E) m/z 213 e (F) m/z 215.
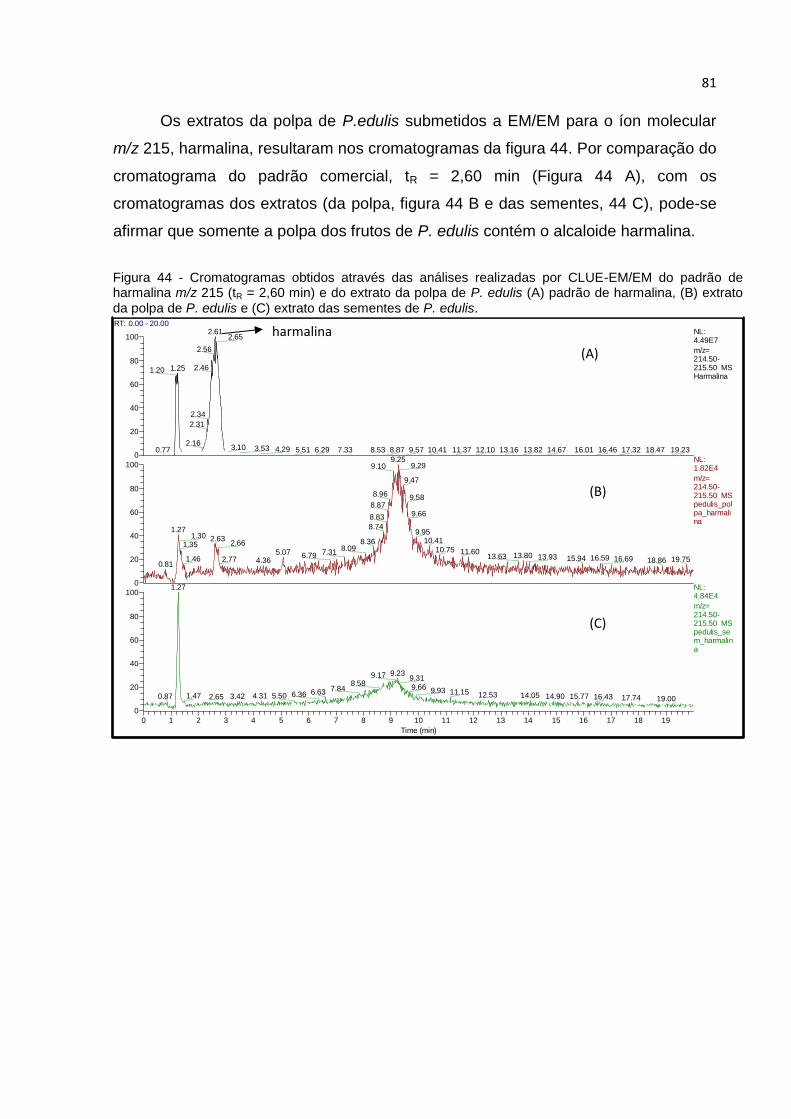
81
Os extratos da polpa de P.edulis submetidos a EM/EM para o íon molecular
m/z 215, harmalina, resultaram nos cromatogramas da figura 44. Por comparação do
cromatograma do padrão comercial, tR = 2,60 min (Figura 44 A), com os
cromatogramas dos extratos (da polpa, figura 44 B e das sementes, 44 C), pode-se
afirmar que somente a polpa dos frutos de P. edulis contém o alcaloide harmalina.
RT: 0.00 - 20.00
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
20
40
60
80
100
0
20
40
60
80
100
Re
lative
Ab
un
da
nce
0
20
40
60
80
1002.61
2.65
2.56
2.461.251.20
2.34
2.31
2.163.10 3.53 4.29 5.51 6.29 8.537.33 8.87 9.57 10.41 11.37 12.10 13.16 13.82 14.67 16.01 16.46 17.32 18.47 19.230.77
9.259.299.10
9.47
8.96 9.588.87
9.668.83
8.741.27 9.951.30 2.63 10.418.362.661.35
8.09 10.75 11.605.07 7.31 13.806.79 13.63 13.93 16.5915.94 16.691.46 2.77 19.7518.864.360.81
1.27
9.239.17 9.318.58
9.667.84 9.936.63 11.156.36 12.53 14.055.501.47 4.31 14.903.420.87 15.772.65 16.43 17.74 19.00
NL:4.49E7
m/z= 214.50-215.50 MS Harmalina
NL:1.82E4
m/z= 214.50-215.50 MS pedulis_polpa_harmalina
NL:4.84E4
m/z= 214.50-215.50 MS pedulis_sem_harmalina
(A)
(B)
(C)
harmalina
Figura 44 - Cromatogramas obtidos através das análises realizadas por CLUE-EM/EM do padrão de harmalina m/z 215 (tR = 2,60 min) e do extrato da polpa de P. edulis (A) padrão de harmalina, (B) extrato da polpa de P. edulis e (C) extrato das sementes de P. edulis.

82
Na figura 45, é possível observar que os espectros de massas do padrão
(Figura 45 A) e do extrato (Figura 45 B), no tempo de retenção do alcaloide (tR =
2,60 min), são semelhantes, onde podem ser observados os íons de m/z 215
([M+H]+), o fragmento iônico m/z 200, uma quebra simples com a saída de uma
metila ([M+H-15]+), e o fragmento iônico m/z 174, eliminação de C2H3N ([M+H-41]+).
60 80 100 120 140 160 180 200 220 240
m/z
0
10
20
30
40
50
60
70
80
90
100
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
174.09083
C 11 H12 O N = 174.09134
-2.90702 ppm
200.09384
C 12 H12 O N2 = 200.09441
-2.85638 ppm
66.10384
159.06734
C 10 H9 O N = 159.06787
-3.27322 ppm
109.10044
C 8 H13 = 109.10118
-6.74496 ppm83.59897
237.06279
C 14 H9 O2 N2 = 237.06585
-12.91051 ppm
174.09005
C 11 H12 O N = 174.09134
-7.40582 ppm
200.09293
C 12 H12 O N2 = 200.09441
-7.43666 ppm
66.10415 109.10316
C 8 H13 = 109.10118
18.14746 ppm 181.4139986.4564973.59283
145.89498 159.80556 224.83105
NL: 9.72E7
harmalina#157-184 RT: 2.35-2.73 AV: 28 F: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
NL: 1.15E4
Pedulis_polpa_harmalina#160-182 RT: 2.52-2.87 AV: 23 F: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
[M+H]+
[M+H-15]+
[M+H-41]+
[M+H]+
[M+H-15]+
[M+H-41]+
(A)
(B)
Figura 45 - Espectros de massas da análise CLUE-EM/EM do íon [M+H]+ de m/z 215, tR = 2,60 min: (A) do
padrão comercial de harmalina e (B) extrato da polpa de P. edulis.
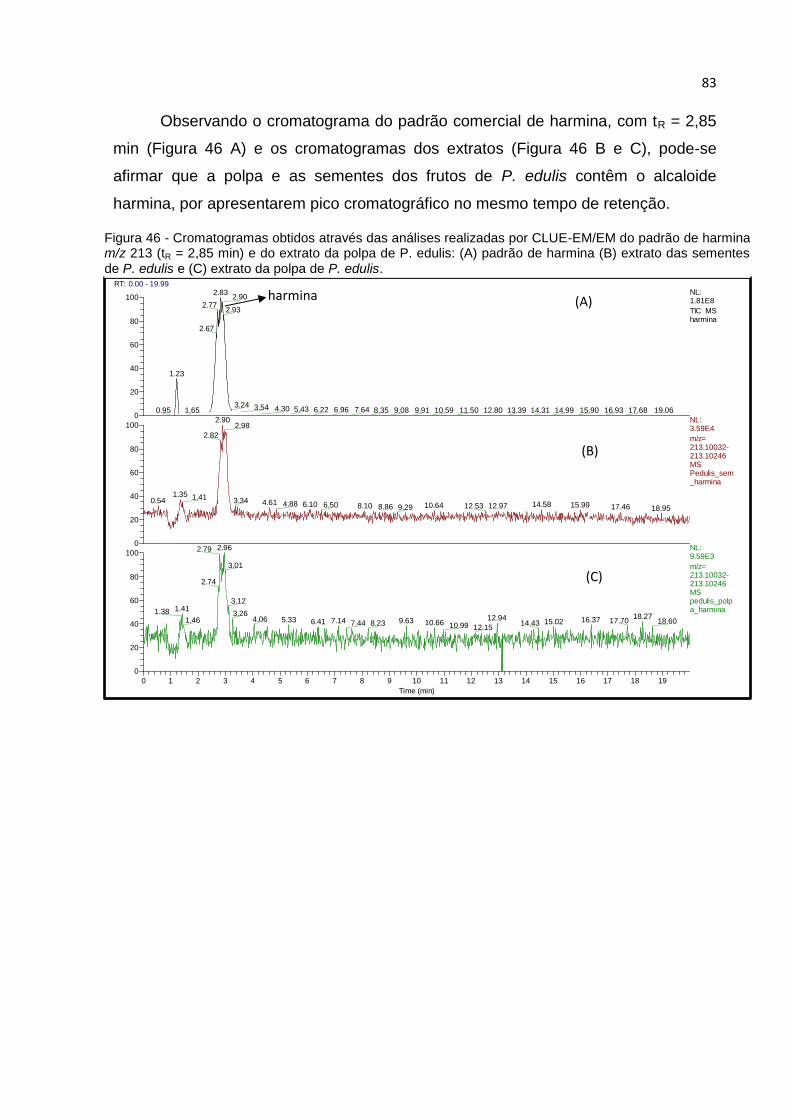
83
Observando o cromatograma do padrão comercial de harmina, com tR = 2,85
min (Figura 46 A) e os cromatogramas dos extratos (Figura 46 B e C), pode-se
afirmar que a polpa e as sementes dos frutos de P. edulis contêm o alcaloide
harmina, por apresentarem pico cromatográfico no mesmo tempo de retenção.
RT: 0.00 - 19.99
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Time (min)
0
20
40
60
80
100
0
20
40
60
80
100
Re
lative
Ab
un
da
nce
0
20
40
60
80
1002.83
2.902.77
2.93
2.67
1.23
3.24 3.54 4.30 5.43 6.22 6.96 7.64 8.35 9.08 9.91 10.59 11.50 12.80 13.39 14.31 14.99 15.90 16.93 17.68 19.061.650.95
2.902.98
2.82
1.35 1.410.54 3.34 4.61 4.88 6.10 14.58 15.996.50 10.64 12.9712.538.10 8.86 17.469.29 18.95
2.962.79
3.01
2.74
3.121.411.38 3.26 18.2712.944.06 5.33 16.371.46 7.14 9.63 17.70 18.606.41 15.0210.66 14.437.44 8.23 10.99 12.15
NL:1.81E8
TIC MS harmina
NL:3.59E4
m/z= 213.10032-213.10246 MS Pedulis_sem_harmina
NL:9.59E3
m/z= 213.10032-213.10246 MS pedulis_polpa_harmina
harmina (A)
(B)
(C)
Figura 46 - Cromatogramas obtidos através das análises realizadas por CLUE-EM/EM do padrão de harmina m/z 213 (tR = 2,85 min) e do extrato da polpa de P. edulis: (A) padrão de harmina (B) extrato das sementes de P. edulis e (C) extrato da polpa de P. edulis.

84
A comparação dos espectros de massas do padrão e dos extratos (Figura
47), no tempo de retenção do alcaloide (tR = 2,85 min), indica que estão presentes os
sinais referentes ao [M+H]+ (m/z 213) e o fragmento referente à perda de uma
metila, [M+H-15]+ (m/z 198), confirmando a presença do alcaloide harmina no extrato
da polpa e das sementes de P. edulis.
170 180 190 200 210 220 230 240
m/z
0
20
40
60
80
100
0
20
40
60
80
100
Re
lative
Ab
un
da
nce
0
20
40
60
80
100
198.07799
C 12 H10 O N2 = 198.07876
-3.91186 ppm
213.10140
C 13 H13 O N2 = 213.10224
-3.91692 ppm
185.10652
C 12 H13 N2 = 185.10732
-4.37359 ppm
170.08307
C 11 H10 N2 = 170.08385
-4.57022 ppm 227.40467 232.74769 242.24071221.82328
214.70690
198.07743
213.10080
171.96445 237.79539215.48345190.34061 224.60124201.62182180.79494 210.69831194.25286
198.07772
213.10111
171.96470 185.10627 224.31945194.72389 231.60981215.33904 241.51216202.01908 209.29978
NL: 4.85E7
harmina#175-210 RT: 2.67-3.17 AV: 36 T: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
NL: 8.99E3
pedulis_polpa_harmina#169-201 RT: 2.66-3.17 AV: 33 T: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
NL: 3.32E4
Pedulis_sem_harmina#169-201 RT: 2.66-3.17 AV: 33 T: FTMS + p ESI Full ms2 [email protected] [55.00-250.00]
(A)
(B)
(C)
[M+H]+
[M+H-15]+
[M+H-15]+
[M+H-15]+
[M+H]+
[M+H]+
Figura 47 - Espectros de massas da análise CLUE-EM/EM do íon [M+H]+ de m/z 213, tR = 2,85 min: (A)
padrão comercial de harmina, (B) extrato da polpa de P. edulis e (C) extrato das sementes de P. edulis.
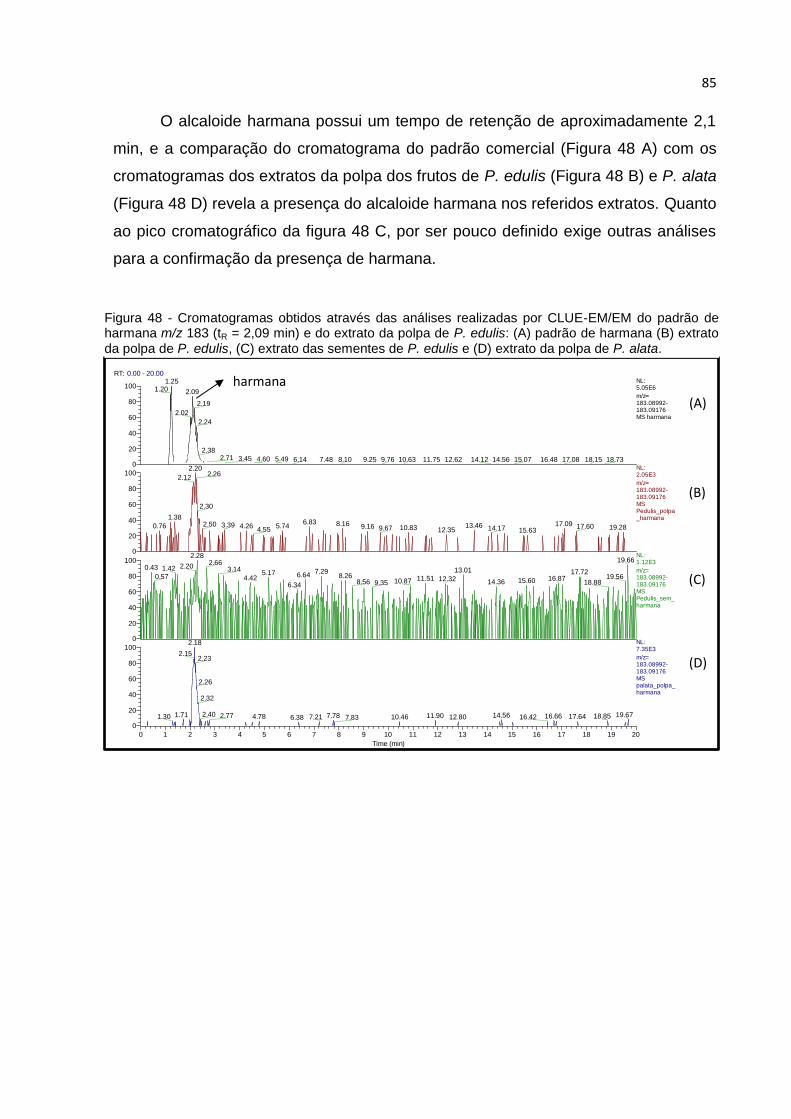
85
O alcaloide harmana possui um tempo de retenção de aproximadamente 2,1
min, e a comparação do cromatograma do padrão comercial (Figura 48 A) com os
cromatogramas dos extratos da polpa dos frutos de P. edulis (Figura 48 B) e P. alata
(Figura 48 D) revela a presença do alcaloide harmana nos referidos extratos. Quanto
ao pico cromatográfico da figura 48 C, por ser pouco definido exige outras análises
para a confirmação da presença de harmana.
RT: 0.00 - 20.00
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Time (min)
0
20
40
60
80
100
0
20
40
60
80
100
0
20
40
60
80
100
Re
lative
Ab
un
da
nce
0
20
40
60
80
1001.25
1.20 2.09
2.19
2.02
2.24
2.382.71 3.45 4.60 5.49 6.14 7.48 8.10 9.25 9.76 10.63 11.75 12.62 14.5614.12 15.07 16.48 17.08 18.15 18.73
2.202.26
2.12
2.30
1.386.83 17.098.162.50 3.39 13.465.744.260.76 17.609.16 19.2810.839.67 14.174.55 12.35 15.63
2.2819.662.66
2.200.43 1.42 3.14 13.017.29 17.725.17 6.64 8.260.57 19.564.42 16.8711.51 12.32 15.6010.878.56 14.36 18.889.356.34
2.18
2.152.23
2.26
2.32
2.40 19.671.71 14.562.77 7.78 11.90 18.8516.661.30 4.78 17.6410.467.21 16.4212.806.38 7.83
NL:5.05E6
m/z= 183.08992-183.09176 MS harmana
NL:2.05E3
m/z= 183.08992-183.09176 MS Pedulis_polpa_harmana
NL:1.12E3
m/z= 183.08992-183.09176 MS Pedulis_sem_harmana
NL:7.35E3
m/z= 183.08992-183.09176 MS palata_polpa_harmana
(A)
(B)
(C)
(D)
harmana
Figura 48 - Cromatogramas obtidos através das análises realizadas por CLUE-EM/EM do padrão de harmana m/z 183 (tR = 2,09 min) e do extrato da polpa de P. edulis: (A) padrão de harmana (B) extrato da polpa de P. edulis, (C) extrato das sementes de P. edulis e (D) extrato da polpa de P. alata.

86
O espectro de massa/massa do padrão de harmana no tR = 2,10 min (Figura
49 A) mostra o pico do íon [M+H]+ m/z 183. Comparado com os espectros EM/EM
dos extratos (Figura 49 B, C e D), estes também possuem o pico m/z 183, referente
ao íon [M+H]+. Como nas análises do extrato da polpa de P. alata discutidas
anteriormente neste trabalho (página 73), não foi possível a identificação dos picos
m/z 141, m/z 137 e m/z 165, presentes nos espectros dos extratos das sementes e
da polpa de P. edulis.
120 130 140 150 160 170 180 190 200
m/z
0
20
40
60
80
100
0
20
40
60
80
100
0
20
40
60
80
100
Re
lative
Ab
un
da
nce
0
20
40
60
80
100
183.09120
C 12 H11 N2 = 183.09167
-2.58422 ppm
186.42517
155.03662
C 10 H5 O N = 155.03657
0.35955 ppm118.49960 200.35004177.54761136.49132125.17021
141.95766
137.96324183.09050165.13757
141.95772
137.96330165.13759 183.09067
141.95749
183.09026137.96306 165.13734
NL: 1.46E6
harmana#152 RT: 2.26 AV: 1 T: FTMS + p ESI Full ms2 [email protected] [50.00-250.00]
NL: 1.59E4
Pedulis_polpa_harmana#144 RT: 2.26 AV: 1 T: FTMS + p ESI Full ms2 [email protected] [50.00-250.00]
NL: 1.47E4
Pedulis_sem_harmana#144 RT: 2.26 AV: 1 T: FTMS + p ESI Full ms2 [email protected] [50.00-250.00]
NL: 1.41E4
palata_polpa_harmana#144 RT: 2.26 AV: 1 T: FTMS + p ESI Full ms2 [email protected] [50.00-250.00]
(A)
(B)
(C)
(D)
[M+H]+
[M+H]+
[M+H]+
[M+H]+
Figura 49 - Espectros de massas da análise CLUE-EM/EM do íon [M+H]+ de m/z 183, tR = 2,05
min: (A) padrão comercial de harmana, (B) extrato da polpa de P. edulis e (C) extrato das sementes de P. edulis e (D) extrato da polpa de P. alata.

87
A Tabela 8 apresenta, de forma resumida, a identificação dos alcaloides
estudados neste trabalho. Os alcaloides harmalol e harmol só foram identificados
nos extratos das sementes de P. alata analisadas por SBSE/CLUE-EM, não sendo
detectados nos outros extratos, analisados por SBSE/CLUE-EM/EM. A harmina está
presente nas sementes e na polpa de ambas as espécies estudadas, enquanto a
harmana foi identificada nos extratos da polpa e das sementes de P. alata e nos
extratos da polpa de P. edulis. O alcaloide harmalina foi encontrado nos extratos da
polpa de P. edulis e nas sementes de P. alata.
Ainda, os extratos das sementes de P. edulis necessitam de futuras análises,
pois suspeita-se da presença da harmana, pois o cromatograma da análise CLUE-
EM/EM apresentou um pico cromatográfico pouco definido no tR = 2,09 min,
referente ao alcaloide, ainda que o espectro de EM/EM mostre a presença do sinal
do íon [M+H]+ de m/z 183.
Tabela 8 - Avaliação da identificação dos alcaloides harmânicos nos extratos da polpa e das sementes de P. alata e P. edulis.
Extratos P. edulis (sementes) P. edulis (polpa) P. alata (sementes) P. alata (polpa)
Alcaloides
Harmana Sim Sim Sim Sim
Harmina Sim Sim Sim Sim
Harmol Não Não Sim Não
Harmalol Não Não Sim Não
Harmalina Não Sim Sim Não

88
4. 3 Extração de alcaloides harmânicos por SBSE-EG Silicone.
O planejamento experimental é considerado uma ferramenta conveniente
para determinar as condições ótimas de procedimentos envolvendo um número
razoável de análises. Uma vez que as barras para SBSE recobertas com EG-
Silicone não haviam sido utilizadas por outros pesquisadores para a extração de
alcaloides harmânicos, o processo de planejamento fatorial fracionário foi realizado,
a fim de se avaliar os fatores mais relevantes, e assim definir os valores dos seus
níveis correspondentes às melhores condições de extração e dessorção.
O planejamento fatorial fracionário 25-1 foi escolhido com base no trabalho de
Pereira e colaboradores, para a extração de alcaloides da polpa e das sementes de
maracujá azedo, utilizando barras SBSE revestidas de PDMS. Os mesmos fatores
foram mantidos, uma vez que são cinco as variáveis que podem afetar a eficiência
de uma extração por SBSE: quantidade de NaCl, pH, tempo de extração, tempo de
dessorção e porcentagem de metanol como solvente de dessorção (PEREIRA, et al.
2014).
Após os experimentos do planejamento fatorial fracionário, foram utilizadas as
áreas dos picos cromatográficos de cada alcaloide como resposta, e os resultados
das análises quimiométricas podem ser observados nos gráficos de Pareto e dos
Efeitos Principais nas figuras 50 e 51. O Gráfico de Pareto é representado por barras
horizontais, e o comprimento dessas são proporcionais ao valor absoluto do seu
efeito estimado associado ou do efeito padronizado. A linha vertical, que corta as
barras, indica a significância estatística e que, neste estudo, corresponde ao limite
de 95%. Desta forma, os fatores avaliados são significativos se a barra
correspondente cruzar a linha vertical. Para o alcaloide harmana (Figura 50 a), todos
as variáveis são importantes, e em seus valores máximos. O Gráfico de Pareto
referente ao alcaloide harmina (Figura 50 b), observa-se que somente a
porcentagem de metanol é significante, no nível máximo e no nível de confiança de
95%.
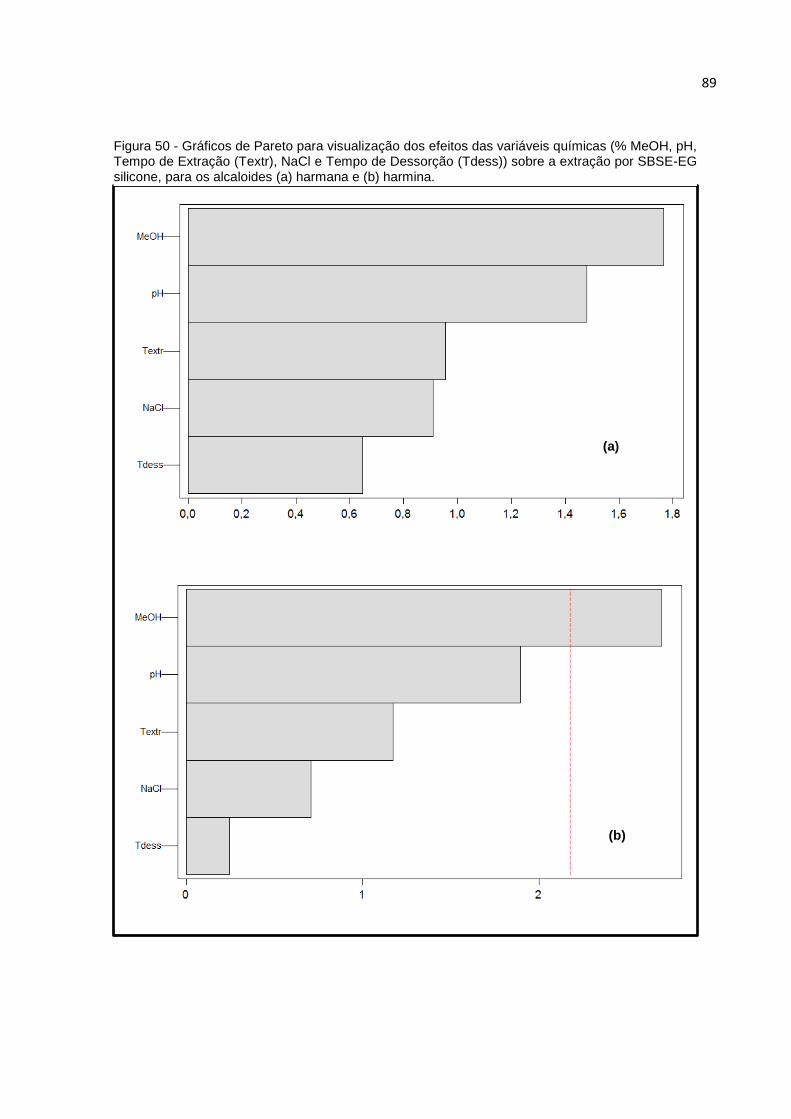
89
(a)
(b)
Figura 50 - Gráficos de Pareto para visualização dos efeitos das variáveis químicas (% MeOH, pH, Tempo de Extração (Textr), NaCl e Tempo de Dessorção (Tdess)) sobre a extração por SBSE-EG silicone, para os alcaloides (a) harmana e (b) harmina.
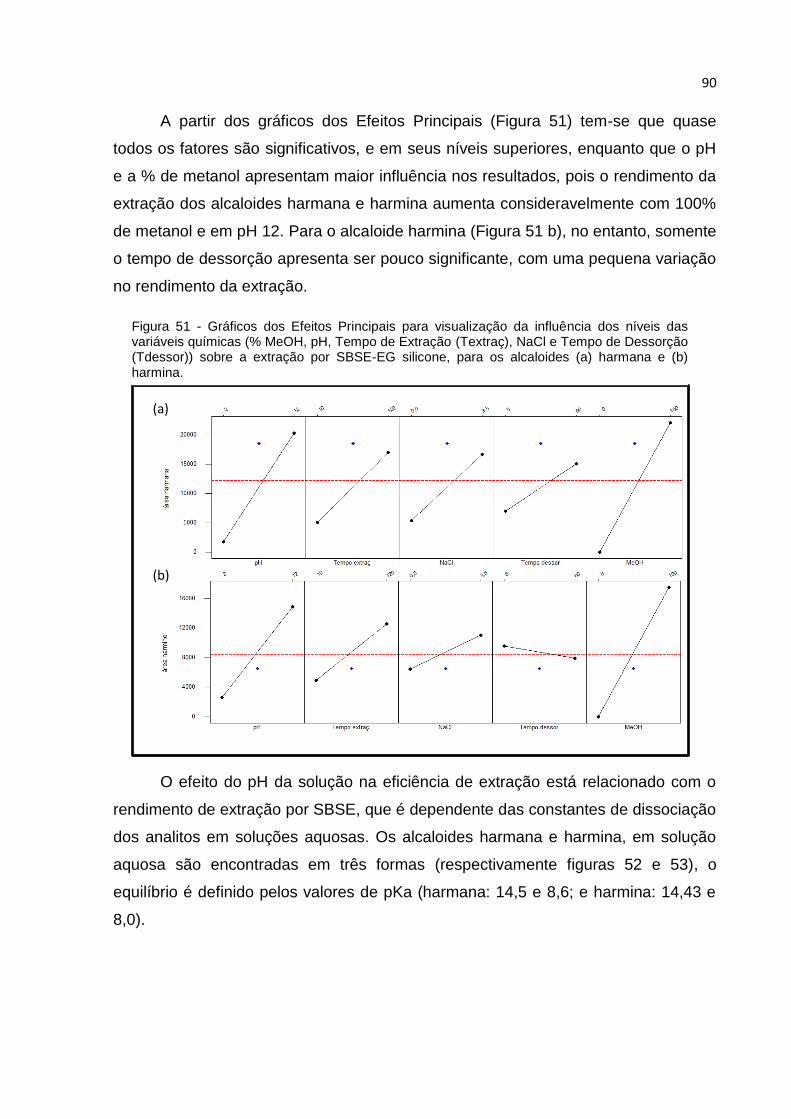
90
A partir dos gráficos dos Efeitos Principais (Figura 51) tem-se que quase
todos os fatores são significativos, e em seus níveis superiores, enquanto que o pH
e a % de metanol apresentam maior influência nos resultados, pois o rendimento da
extração dos alcaloides harmana e harmina aumenta consideravelmente com 100%
de metanol e em pH 12. Para o alcaloide harmina (Figura 51 b), no entanto, somente
o tempo de dessorção apresenta ser pouco significante, com uma pequena variação
no rendimento da extração.
O efeito do pH da solução na eficiência de extração está relacionado com o
rendimento de extração por SBSE, que é dependente das constantes de dissociação
dos analitos em soluções aquosas. Os alcaloides harmana e harmina, em solução
aquosa são encontradas em três formas (respectivamente figuras 52 e 53), o
equilíbrio é definido pelos valores de pKa (harmana: 14,5 e 8,6; e harmina: 14,43 e
8,0).
(a)
(b)
Figura 51 - Gráficos dos Efeitos Principais para visualização da influência dos níveis das variáveis químicas (% MeOH, pH, Tempo de Extração (Textraç), NaCl e Tempo de Dessorção (Tdessor)) sobre a extração por SBSE-EG silicone, para os alcaloides (a) harmana e (b) harmina.
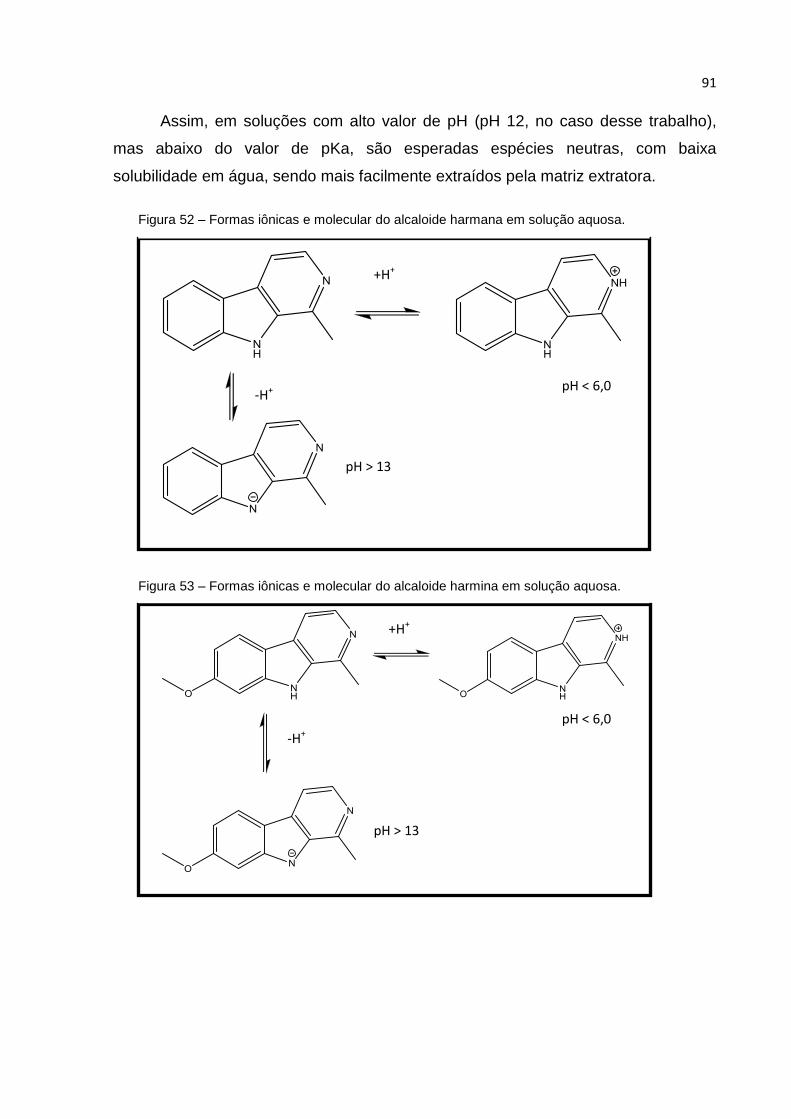
91
Assim, em soluções com alto valor de pH (pH 12, no caso desse trabalho),
mas abaixo do valor de pKa, são esperadas espécies neutras, com baixa
solubilidade em água, sendo mais facilmente extraídos pela matriz extratora.
pH < 6,0
+H+
pH > 13
-H+
+H+
pH < 6,0
-H+
pH > 13
Figura 52 – Formas iônicas e molecular do alcaloide harmana em solução aquosa.
Figura 53 – Formas iônicas e molecular do alcaloide harmina em solução aquosa.

92
Os resultados significativos obtidos para a quantidade de NaCl adicionado,
correspondentes ao nível superior, estão relacionados à força iônica do meio de
extração, que devido ao efeito de salting-out e as interações eletrostáticas entre
moléculas polares e os íons de NaCl aumenta a quantidade de analitos extraídos.
Para a determinação do melhor tempo de extração e de dessorção, foi
realizado um estudo da cinética do processo de extração e de dessorção, mantendo
o pH 12, 3,5 g de NaCl e com 100% de metanol como solvente de dessorção. Os
resultados avaliados foram o aumento da área do pico cromatográfico referente ao
alcaloide extraído por SBSE usando as barras recobertas com EG-Silicone, com o
aumento do tempo de extração (Figura 54) e dessorção (Figura 55).
O comportamento dos alcaloides harmana e harmina frente aos processos de
extração e dessorção é semelhante, e com base nos resultados dos estudos
cinéticos, utilizando as barras recobertas com EG-Silicone, o melhor tempo de
extração são 30 minutos sob agitação, com o tempo de dessorção de 15 minutos no
ultrassom. A partir destes tempos o ganho de massa tende a estabilizar e o
aumento não é considerável em relação ao aumento do tempo de extração e
dessorção.

93
(B)
(A)
Figura 54 - Gráficos da variação da área do pico cromatográfico dos alcaloides (A) harmina e (B) harmana extraídos por SBSE-EG Silicone, em função dos diferentes tempos de extração sob agitação.
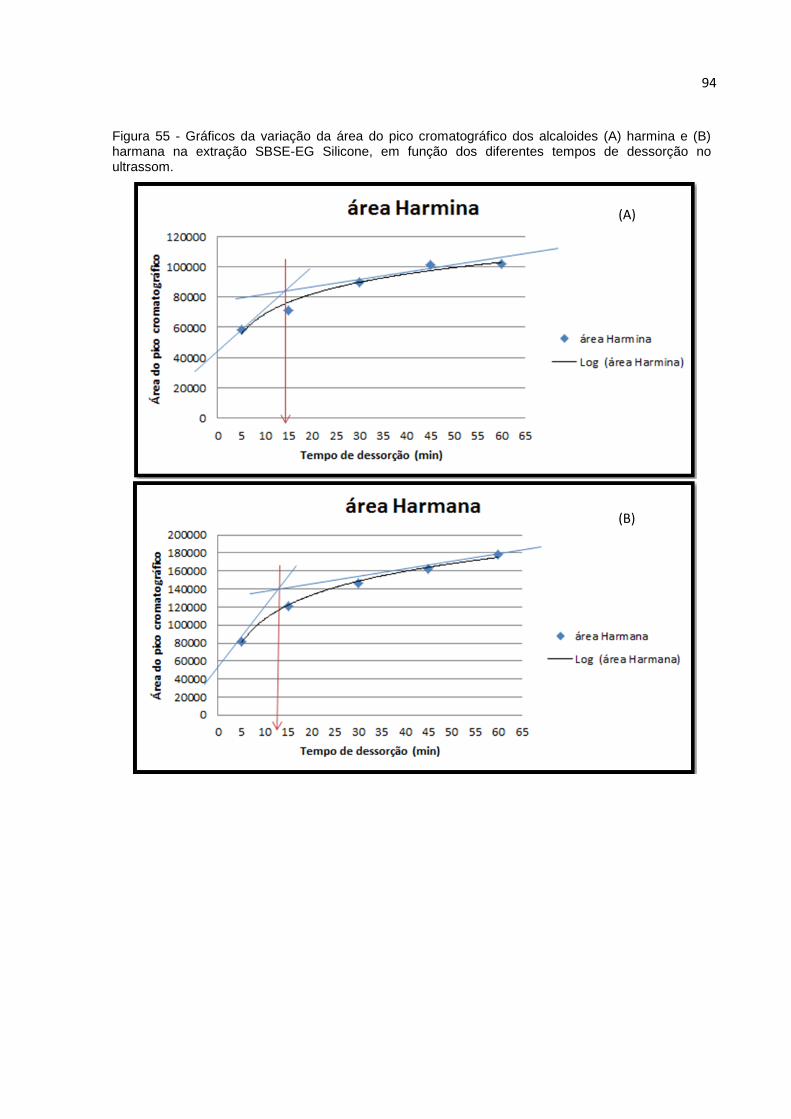
94
(A)
(B)
Figura 55 - Gráficos da variação da área do pico cromatográfico dos alcaloides (A) harmina e (B) harmana na extração SBSE-EG Silicone, em função dos diferentes tempos de dessorção no ultrassom.

95
O estudo da recuperação do procedimento de extração e dessorção definido
pelo planejamento experimental e pelo estudo cinético foi realizado a partir da
análise prévia, por CLAE-Flu, de alíquotas da amostra a ser submetida ao processo
de extração por SBSE com as barras recobertas por EG-Silicone, determinando-se a
porcentagem inicial dos alcaloides contidos em cada solução. A área dos picos
cromatográficos de cada um dos alcaloides foi considerada como 100% para o
cálculo da recuperação. Após a dessorção das barras magnéticas, os alcaloides
dessorvidos foram analisados por CLAE-Flu, obtendo-se a área do pico
cromatográfico referente à porcentagem final dos alcaloides presentes.
O valor da área do pico que corresponde a 100% de recuperação foi
calculado a partir dos resultados obtidos pela análise das alíquotas da amostra antes
de ser submetida ao processo de extração:
Área1 (obtida antes da extração) ..................10 mL
X (áreateórica correspondente a extração) .....150 µL
Xárea teórica = Área1 x 10 mL / 150 µL
Então, a porcentagem de recuperação real foi calculada, após a extração dos
alcaloides por SBSE/CLAE-Flu:
[Área2 (obtida após extração por SBSE/CLAE-Flu)/ Xárea teórica] x 100%
As concentrações das soluções dos padrões dos alcaloides harmana e
harmina utilizados para o estudo da recuperação foram: 20, 250, 500, 1000, 1500 e
2000 ng L-1. O perfil da curva da porcentagem de recuperação em função da
concentração pode ser observado nas figuras 56 e 57.
A curva de recuperação para a harmana (Figura 56 A) mostra inicialmente um
comportamento crescente, onde a recuperação aumenta com o aumento da
concentração de alcaloides na solução extraída. Uma concentração maior que 500
ng L-1 leva a uma recuperação porcentual menor, que diminui gradativamente. No
intervalo entre 20, 250 e 500 ng L-1, a recuperação do alcaloide harmana é linear
(Figura 56 B).
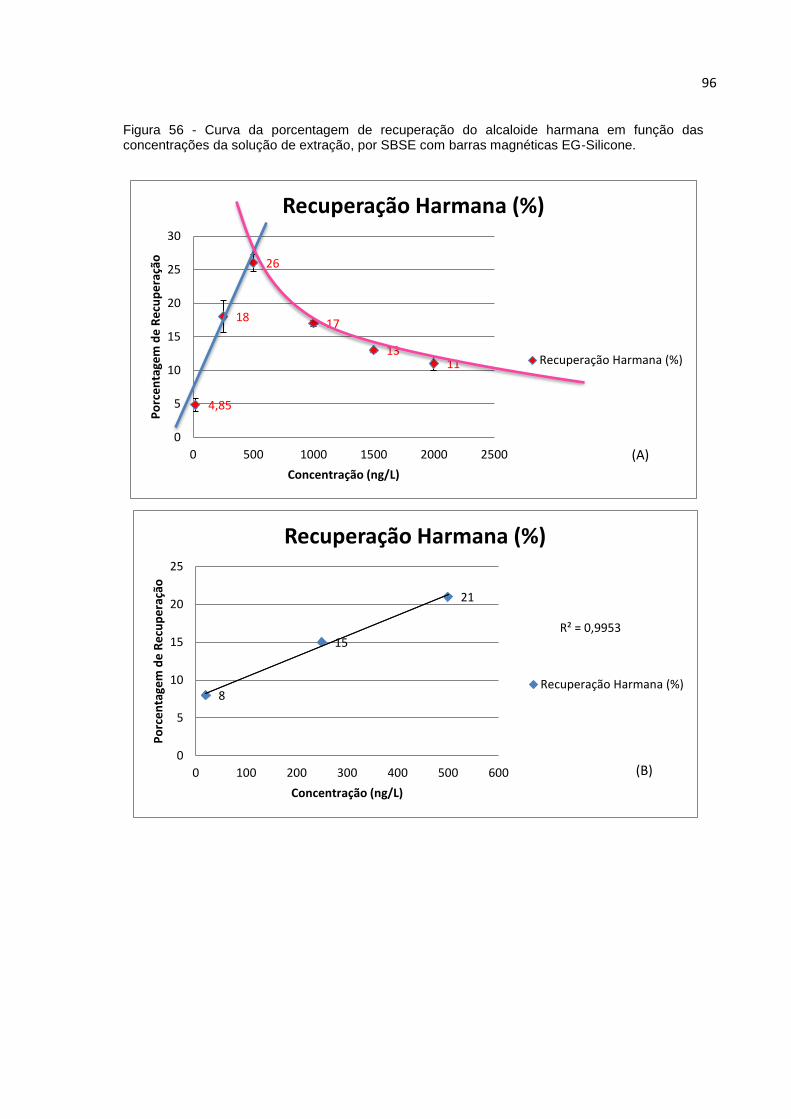
96
4,85
18
26
17
13 11
0
5
10
15
20
25
30
0 500 1000 1500 2000 2500
Po
rce
nta
gem
de
Re
cup
era
ção
Concentração (ng/L)
Recuperação Harmana (%)
Recuperação Harmana (%)
8
15
21
R² = 0,9953
0
5
10
15
20
25
0 100 200 300 400 500 600
Po
rce
nta
gem
de
Re
cup
era
ção
Concentração (ng/L)
Recuperação Harmana (%)
Recuperação Harmana (%)
(A)
(B)
Figura 56 - Curva da porcentagem de recuperação do alcaloide harmana em função das concentrações da solução de extração, por SBSE com barras magnéticas EG-Silicone.
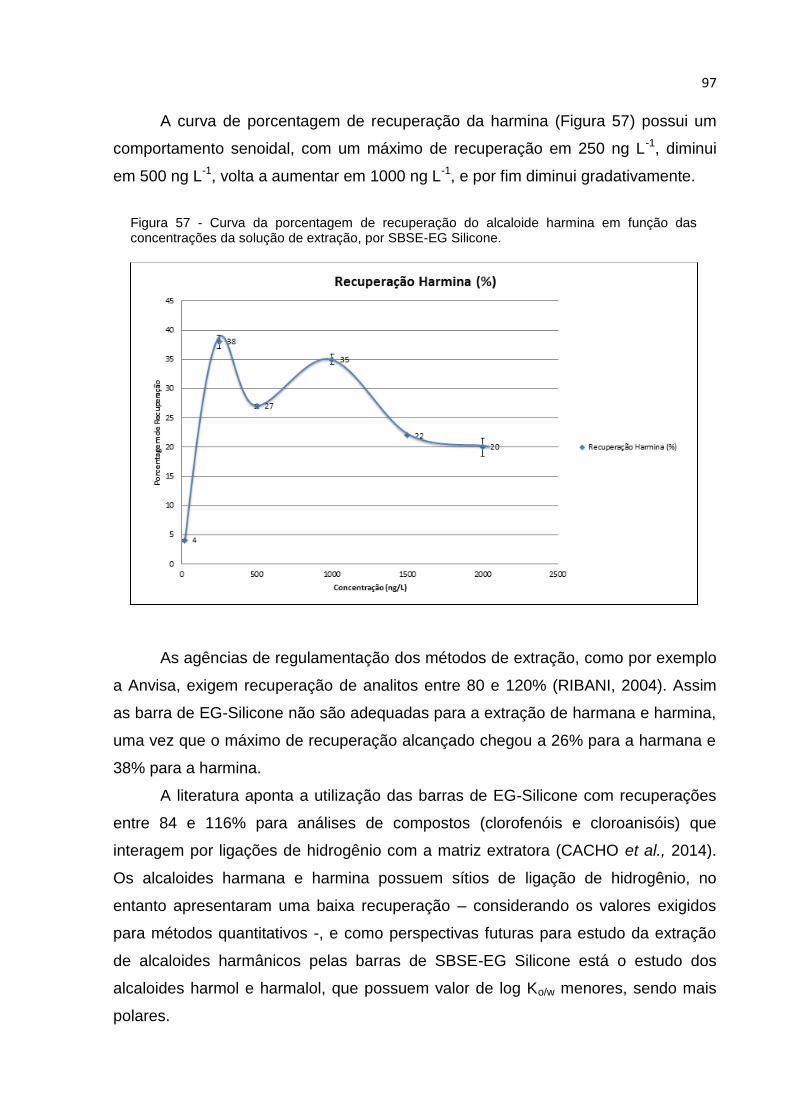
97
A curva de porcentagem de recuperação da harmina (Figura 57) possui um
comportamento senoidal, com um máximo de recuperação em 250 ng L-1, diminui
em 500 ng L-1, volta a aumentar em 1000 ng L-1, e por fim diminui gradativamente.
As agências de regulamentação dos métodos de extração, como por exemplo
a Anvisa, exigem recuperação de analitos entre 80 e 120% (RIBANI, 2004). Assim
as barra de EG-Silicone não são adequadas para a extração de harmana e harmina,
uma vez que o máximo de recuperação alcançado chegou a 26% para a harmana e
38% para a harmina.
A literatura aponta a utilização das barras de EG-Silicone com recuperações
entre 84 e 116% para análises de compostos (clorofenóis e cloroanisóis) que
interagem por ligações de hidrogênio com a matriz extratora (CACHO et al., 2014).
Os alcaloides harmana e harmina possuem sítios de ligação de hidrogênio, no
entanto apresentaram uma baixa recuperação – considerando os valores exigidos
para métodos quantitativos -, e como perspectivas futuras para estudo da extração
de alcaloides harmânicos pelas barras de SBSE-EG Silicone está o estudo dos
alcaloides harmol e harmalol, que possuem valor de log Ko/w menores, sendo mais
polares.
Figura 57 - Curva da porcentagem de recuperação do alcaloide harmina em função das concentrações da solução de extração, por SBSE-EG Silicone.

98
5 CONCLUSÕES
Pereira e colaboradores e Rodrigues analisaram os alcaloides harmana e
harmina por SBSE/CLAE-Flu dos extratos da polpa e das sementes de P. edulis,
determinando as quantidades de cada um nestes extratos. Nos estudos deste
trabalho verificou-se que a quantidade dos alcaloides harmana e harmina
encontrada nos extratos da polpa de P. alata, por SBSE/CLAE-Flu é pequena, em
comparação com a polpa de P. edulis, de acordo com o trabalho de Pereira e
colaboradores (PEREIRA et al., 2014; RODRIGUES, 2013). Para complementar o
estudo dos alcaloides harmana e harmina em maracujá doce é necessário realizar a
quantificação nos extratos das sementes desta espécie.
A técnica de extração por SBSE-PDMS aplicada no estudo dos alcaloides
harmânicos nos extratos da polpa e das sementes de P. alata e P.edulis por
SBSE/CLUE-EM/EM mostrou-se vantajosa por ser rápida, fácil de realizar, e exige
menor quantidade de amostra e solvente, quando comparada a técnica de extração
líquido-líquido, por exemplo.
Os resultados obtidos neste trabalho contribuem para o conhecimento da
composição fitoquímica da polpa e das sementes das espécies de maracujá
estudados, quanto aos alcaloides harmânicos, especialmente nas sementes do
maracujá doce (P. alata), em que foram identificados os alcaloides harmana,
harmina, harmol, harmalol e harmalina, sem outros relatos na literatura sobre estes
alcaloides nesta espécie. Os alcaloides identificados em P. edulis, harmana, harmina
e harmalina foram detectados nos extratos das folhas desta espécie, por
cromatografia em papel, utilizando o reagente de Dragendorff (LUTOMONSKI et al.,
1975). Este trabalho utilizou técnicas mais modernas e confirma a presença destes
alcaloides.
A extração dos alcaloides harmana e harmina com as barras de SBSE-EG
Silicone mostrou uma baixa recuperação, em torno de 30 %, sugerindo que a
interação dessas moléculas com a fase extratora, dentro das condições estudadas,
não foi significativa. Assim, outros alcaloides harmânicos precisam ser testados, e/ou
a metodologia deve ser estudada, para otimizações.

99
REFERÊNCIAS
ABOURASHED, E. A., VANDERPLANK, J., KHAN, I. A. High-Speed Extraction and CLAE Fingerprinting of Medicinal Plants – II. Application to Harman Alkaloids of Genus Passiflora. Pharmaceutical Biology, v. 42, No. 2, pp. 100-106 (2003).
BRASIL. Ministério da Saúde. Agência Nacional de Vigilância Sanitária. Resolução RDC 48 de 16 de março de 2004. Dispõe sobre o registro de medicamentos fitoterápicos. D.O.U., Brasília, 18 mar. 2004.
BUSQUETS, R., PUIGNOU. L., GALCERAN, M. T., SKOG, K. Effect of Red Wine Marinades on the Formation of Heterocyclic Amines in Fried Chicken Breast. Journal of Agricultural and Food Chemistry, v. 54, pp. 8376-8384 (2006).
BICCHI, C., CORDERO, C., LIBERTO, E., RUBIOLO, P., SGORBINI, B., DAVID, F., SANDRA, P. Dual-Phase Twisters: A New Approach to Headspace Sorptive Extraction and Stir Bar Sorptive Extraction. Journal of Chromatography A, v. 1094, pp. 9-16 (2005).
CACHO, J. I., CAMPILLO, N., VIÑAS, P., CÓRDOBA, M. H. Stir Bar Sorptive Extraction with EG-Silicone Coating for Bisphenols Determination in Personal Care Products by GC-MS. Journal of Pharmaceutical and Biomedical Analysis, v. 78-79, pp 255-260 (2013).
CACHO, J. I., CAMPILLO, N., VIÑAS, P., CÓRDOBA, M. H. Stir Bar Sorptive Extraction Polar Coatings for the Determination of Chlorophenols and Chloroanisoles in Wines Using Gas Chromatography and Mass Spectrometry. Talanta, v. 118, pp. 30-36 (2014).
CAICEDO, N. H., KUMIRSKA, J., NEUMANN, J., STOLTE, S., THÖMING, J. Detection of Bioactive Exometabolites Produced by the Filamentous Marine Cyanobacterium Geitlerinema sp. Mar Biotechnology, v. 14, pp. 436-445 (2012).
CALDAS, S. S., GONÇALVES, F.F., PRIMEL, E. G., PRESTES, O. D., MARTINS, M. L., ZANELLA, R. Principais Técnicas de Preparo de Amostra pra a Determinação de Resíduos de Agrotóxicos em Água por Cromatografia Líquida com Detecção por Arranjo de Diodos e por Espectrometria de Massas. Química Nova, v. 34, No. 9, pp. 1604-1617 (2011).
CARERI, M., BIANCHI, F., CORRADINI, C. Recent Advances in the Application of Mass Spectrometry in Food-Related Analysis. Journal of Chromatography A, v. 970, pp. 3-64 (2002).

100
CASS, Q. B., BARREIRO, J. C. Os Avanços Tecnológicos na Química Analítica: Sucesso e Desafios. Ciência e Cultura, v. 63, No. 1, pp. 37-40 (2011).
CROTTI, A. E. M., VESSECCHI, R., LOPES, J. L. C., LOPES, N. P. Espectrometria de Massas com Ionização por “Electrospray”: Processos Químicos Envolvidos na Formação de Íons de Substâncias Orgânicas de Baixo Peso Molecular. Química Nova, v. 29, No. 2, pp. 287-292 (2006).
DAVID, F., SANDRA, P. Stir Bar Sorptive Extraction for Trace Analysis. Journal of Chromatography A, v. 1152, pp. 54-69 (2007).
DHAWAN, K., DHAWAN, S., SHARMA, A. Passiflora: A Review Update. Journal of Ethnopharmacology, v. 94, pp. 1-23 (2004).
EG Silicone Twister® GERSTEL GmbH & Co. KG Edição 11/2011.
GALCERAN, M. T., MOYANO, E., PUIGNOU, L., PAIS, P. Determination of heterocyclic amines by pneumatically assisted electrospray liquid chromatography-mass spectrometry. Journal of Chromatography A, v. 730, pp. 185-194 (1996).
GILART, N., MIRALLES, N., MARCÉ, R. M., BORRULL, F., FONTANALS, N. Novel Coatings for Stir Bar Sorptive Extraction to Determine Pharmaceuticals and Personal Care Products in Environmental Waters by Liquid Chromatography and Tandem Mass Spectrometry. Analytica Chimica Acta, v. 774, pp. 51-60 (2013).
HAJNOS, M. W. e SHERMA, J. High Performance Liquid Chromatography in Phytochemical Analysis. Chromatographic Science Series, v. 102. CRC Press Taylor and Francis Group (2011).
HERRAIZ, T. Analysis of the Bioactive Alkaloids Tetrahydro-β-Carboline and β-Carboline in Food. Journal of Chromatography A, v. 881, pp. 483-499 (2000).
LANÇAS, F. M. A Cromatografia Líquida Moderna e a Espectrometria de Massas: Finalmente “Compatíveis”? Scientia Chromatographica, v. 1, No. 2, pp 35-61 (2009).
LANÇAS, F. M. A Cromatografia Líquida Moderna e a Espectrometria de Massas: Finalmente “Compatíveis”? II A Escolha do Analisador de Massas. Scientia Chromatographica, v. 5, No. 1, pp. 27-46 (2013).

101
LI, S., LIU, W, TENG, L., CHENG, X., WANG, Z., WANG, C. Metabolites Identification of Harmane in vitro/in vivo in Rats by Ultra-Performance Liquid Chromatography Combined with Electrospray Ionization Quadrupole Time-of-Flight Tandem Mass Spectrometry. Journal of Pharmaceutical and Biomedical Analysis, v. 92, pp. 53-62 (2014).
LUMBRERAS, R. G., CONRADO, N. R., GONZÁLEZ, M. E. L., ARRIBAS, P., DÍEZ, L. M. P. Capillary Liquid Chromatography with Diode Array and Mass Spectrometry Detection for Heterocyclic Aromatic Amine Determination in Ready-to-Eat Food Treated with Electron-Beam Irradiation. Journal of Chromatography A, v. 1217, pp. 6778-6784 (2010). LUTOMSKI, J., MALEK, B., RYBACKA, L. Pharmacochemical Investigation of the Raw Materials from Passiflora genus.2. The Pharmacochemical estimation of Juices from the Fruits of Passiflora edulis and Passiflora edulis forma flavicarpa. Planta Medica, vol 27, pp. 112-121 (1975) apud ZERAIK, M. L., PEREIRA, C. A. M., ZUIN, V., G., YARIWAKE, J. H. Maracujá: Um Alimento Funcional? Revista Brasileira de Farmacognosia, v. 20, No. 3, pp. 459-471 (2010).
MACHADO, M. W., NETO, C. S., SALGADO, J., ZAFFARI, A. B., CAMPOS, F. R., CORILO, Y. E., EBERLIN, M. N., BIAVATTI, M. W. Search for Alkaloids on Callus Culture of Passiflora alata. Brazilian Archives of Biology and Technology, v. 53, No. 4, pp. 901-910 (2010).
MAGI, E., DI CARRO, M., SCAPOLLA, C., NGUYEN, K. Stir Bar Sorptive Extraction and LC/MS-MS for Trace Analysis of UV Filters in Different Water Matrices. Chromatographia, v. 75, pp 973-982 (2012).
MALDANER, L., JARDIM, I. C. S. F. O Estado da Arte da Cromatografia Líquida de Ultra Eficiência, Química Nova, v. 32, No. 1, pp. 214-222 (2009).
MARAKOV, A. Electrostatic Axially Harmonic Orbital Trapping: A High-Performance Technique of Mass Analysis. Analytical Chemistry, v. 72, pp. 1156-1162 (2000).
MCLLHENNY, E. H., RIBA, J., BARBANOJ, M. J., STRASSMAN, R., BARKER, S. A. Methodology for and the Determination of the Major Constituents and Metabolites of the Amazonian Botanical Medicine Ayahuasca in Human Urine. Biomedical Chromatography, v. 25, pp. 970-984 (2011).
NIE, Y., BENNE, E. K. Using Three Types of Twister Phases for Stir Bar Sorptive Extraction of Whisky, Wine and Fruit Juice. Global analytical Solutions – Gerstel, v. 3 (2011).

102
NOGUEIRA, J. M. F. Novel Sorption-Based Methodologies for Static Microextraction Analysis: A Review on SBSE and Related Techniques. Analytica Chimica Acta, v. 757, pp. 1-10 (2012).
ONISHI, Y., OISHI, K., KAWANO, Y., YAMAZAKI, Y. The Harmala Alkaloid Harmine is a Modulator of circadian Bmal1 Transcription. Bioscience Reports, v. 32, pp. 45-52 (2012).
PEREIRA, C. A. M. ; J.H. Yariwake . Quantification of harman alkaloids in passion fruit samples by dual SBSE-LC/Flu. In: 36th ISCC / 9th GC x GC Symposium, 2012, Riva del Garda, Italia. Proceedings 36th ISCC / 9th GC x GC Symposium, (2012).
PEREIRA, C. A. M., RODRIGUES, T. R., YARIWAKE, J. H. Quantification of Harman Alkaloids in Sour Passion Fruit Pulp and Seeds by a Novel Dual SBSE-LC/Flu (Stir Bar Sorptive Extraction-Liquid Chromatography with Fluorescence Detector) Method. Journal Brazilian Chemical Society, v. 25, No. 08, pp. 1472-1483 (2014).
Phamacognosy. Acesso em 15 de agosto de 2013: http://www.epharmacognosy.com/2012/07/simple-carboline-alkaloids.html
PLOTKA, J., TOBISZEWSKI, M., SULEJ, A. M., KUPSKA, M., GÓRECKI, T., NAMIESNIK, J. Green Chromatography. Journal of Chromatography A, v. 1307, pp. 1-20 (2013).
RIBANI, M., BOTTOLI, C. B. G., COLLINS, C. H., JARDIM, C. S. F., MELO, L. F. C. Validação em Métodos Cromatográficos e Eletroforéticos. Química Nova, v. 27, No. 5, pp. 771-780 (2004).
RIPA, F. A., NAHAR, L., HAQUE, M., ISLAM, M. M. Antibacterial, Cytotoxic, and Antioxidant Activity of Crude Extract of Marsilea Quadrifolia. European Journal of Scientific Research, v. 33, No. 1, pp. 123-129 (2009).
RODRIGUES, T. R. Estudo de Alcaloides Harmânicos em Sementes de Passiflora edulis Sims. F. flavicarpa Degener (Maracujá Azedo) por SBSE/CLAE-Flu dual. Dissertação de Mestrado. Instituto de Química de São Carlos – USP (2013).
RODRIGUEZ, J. A., ARTEAGA, K. A., DÍEZ, C., BARRADO, E. Recent Advances in the Extraction of Triazines from Water Samples. Intech – Herbicides: Advances in Research. Capítulo 13, pp. 255-276 (2013).

103
SHARMA, V., SARKAR, I. Leveraging Biodiversity Knowledge for Potential Phyto-Therapeutic Applications.Journal American Medicinal Informatics Association. v. 20, pp. 668-679 (2013).
SILVA, C., G. A., COLLINS, C. H., Aplicações de Cromatografia Líquida de Alta Eficiência para o Estudo de Poluentes Orgânicos Emergentes. Química Nova, v. 34, pp. 665-676 (2011).
SILVERSTEIN, R. M., WEBSTER, F. X., KIEMLE, D. J.Identificação Espectrométrica de Compostos Orgânicos; tradução de Ricardo Bicca de Alencastro. – [Reimpr.]. – Rio de Janeiro: LTC, 2010. Capítulo 1, pp.13-18 e 45.
TEOFILO, R. F., FERREIRA, M. M C. Quimiometria II: Planilhas Eletrônicas para Cálculos de Planejamentos Experimentais, um Tutorial. Química Nova, v. 29, No. 2. pp. 338-350 (2006).
THERMO SCIENTIFIC. Protein Methods Library. Overview of Mass Spectrometry. Rockford, IL USA, 2014. Disponível em: <
http://www.piercenet.com/method/overview-mass-spectrometry>. Acesso em: 22 nov. 2014.
VIÑAS, P., CAMPILLO, N., PÉREZ, M. H., CÓRDOBA, M. H. A Comparison of Solid-Phase Microextraction and Stir Bar Sorptive Extraction Coupled to Liquid Chromatography for the Rapid Analysis of Resveratrol Isomers in Wines, Musts and Fruit Juices. Analytica Chimica Acta, v. 611, pp. 119-125 (2008).
WATERS CORPORATION. HPLC Columns. Continuing the Legacy of HPLC Column Performance. Mildford, MA USA, 2014. Disponível em: <
http://www.waters.com/webassets/cms/library/docs/720003995en.pdf> Acesso em: 02 dez. 2014.
WATERS CORPORATION. XP Columns. Extended Performance – Xbridge XSelect Columns. Mildford, MA USA, 2012. Disponível em: <http://www.waters.com/webassets/cms/library/docs/720004195en.pdf> Acesso em: 28 nov. 2014.
WU, H., GUO, J., CHEN, S., LIU, X., ZHOU, Y., ZHANG, X., XU, X. Recent Developments in Qualitative and Quantitative Analysis of Phytochemical Constituents and their Metabolites Using Liquid Chromatography-Mass Spectrometry. Journal of Pharmaceutical and Biomedical Analysis, v. 72, pp. 267-291 (2013).

104
XU, J., CHEN, B., HE, M., HU, B. Analysis of Preservatives with Different Polarities in Beverage Samples by Dual-Phase Dual Stir Bar Sorptive Extraction Combined with High-Performance Liquid Chromatography. Journal of Chromatography A, vol 1278, pp. 8-15 (2013).
YADAV, R. N. S., AGARWALA, M. Phytochemical Analysis of Some Medicinal Plants. Journal of Phytology, v. 12, No. 3, pp. 10-14 (2011).
YANG, M., SUN, J., LU, Z., CHEN, G., GUAN, S., LIU, X., JHANG, B., YE, M., GUO, D. Phytochemical Analysis of Traditional Chinese Medicine Using Liquid Chromatography Coupled with Mass Spectrometry. Journal of Chromatography A, v. 1216, pp. 2045-2062 (2009).
YOUDIM, K. A., SAUNDERS, K. C. A Review of LC-MS Techniques and High-Throughput Approaches Used to Investigate Drug Metabolism by Cytochrome P450s. Journal of Chromatography B, v. 878, pp. 1326-1336 (2010).
ZERAIK, M. L., PEREIRA, C. A. M., ZUIN, V., G., YARIWAKE, J. H. Maracujá: Um Alimento Funcional? Revista Brasileira de Farmacognosia, v. 20, No. 3, pp. 459-471 (2010).
ZERAIK, M. L. Estudo analítico dos flavonóides dos frutos do maracujá (Passiflora edulis Sims f. Flavicarpa Degener). Tese de doutorado. Instituto de Química de São Carlos – USP (2010).
ZHANG, Y., LIN, C., FANG, G., MEI, J., WANG, X., WANG, S. Tandem Solid Phase Extraction Coupled to LC-ESI-MS/MS for the Accurate Simultaneous Determination of Five Heterocyclic Aromatic Amines in Processed Meat Products. European Food Research Technology, v. 234, pp. 197-205 (2012).
ZHAO, T., ZHENG, S., ZHANG, B., LI, Y., BLIGH, S. W. A., WANG, C., WANG, Z. Metabolic Pathways of the Psychotropic-Carboline Alkaloids, Harmaline and Harmine, by Liquid Chromatography/Mass Spectrometry and NMR Spectroscopy. Food Chemistry, v. 134, pp. 1096-1105 (2012).
ZUCOLOTTO, S. M., FAGUNDES, C., REGINATTO, F. H., RAMOS, F. A., CASTELLANOS, L., DUQUE, C., SCHEKEL, E. P. Analysis of C-glycosyl Flavonoids from South America Passiflora Species by CLAE-DAD and CLAE-MS. Phytochemical Analysis, v. 23, pp. 232-239 (2012).

105
ZUIN, V. G., MONTERO, L., BAUER, C., POPP, P. Stir Bar Sorptive Extraction and High-Performance Liquid Chromatography-Fluorescence Detection for the Determination of Polycyclic Aromatic Hydrocarbons in Mate Teas. Journal of Chromatography A, v. 1091, pp. 2-10 (2005).
ZUIN, V. G., SCHELLIN, M., MONTERO, L., YARIWAKE, J. H., AUGUSTO, F., POPP, P. Comparison of Stir Bar Sorptive Extraction and Membrane-Assisted Solvent Extraction as Enrichment Techniques for the Determination of Pesticide and Benzo[a]pyrene Residues in Brazilian Sugarcane Juice. Journal of Chromatography A, v. 1114, pp. 180-187 (2006).
Top Related