
UNIVERSIDADE FEDERAL DE OURO PRETO ANÁLISE … · Crz1p - Fator de transcrição que ativa genes...
98
UNIVERSIDADE FEDERAL DE OURO PRETO NÚCLEO DE PESQUISAS EM CIÊNCIAS BIOLÓGICAS - NUPEB PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS BIOLÓGICAS ANÁLISE COMPARATIVA DA EXPRESSÃO GÊNICA NAS CEPAS SELVAGEM W303 E pkc1Δ DE Saccharomyces cerevisiae DURANTE O PROCESSO DE DESREPRESSÃO METABÓLICA AUTOR: ANTÔNIO HELVÉCIO TÓTOLA ORIENTADOR: PROF.DR. ROGELIO LOPES BRANDÃO Tese apresentada ao Programa de Pós-Graduação do Núcleo de Pesquisas em Ciências Biológicas da Universidade Federal de Ouro Preto, como parte integrante dos requisitos para obtenção do Título de Doutor em Ciências . Ouro Preto, Outubro de 2007.
Transcript of UNIVERSIDADE FEDERAL DE OURO PRETO ANÁLISE … · Crz1p - Fator de transcrição que ativa genes...

UNIVERSIDADE FEDERAL DE OURO PRETO NÚCLEO DE PESQUISAS EM CIÊNCIAS BIOLÓGICAS - NUPEB
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS BIOLÓGICAS
ANÁLISE COMPARATIVA DA EXPRESSÃO GÊNICA NAS CEPAS SELVAGEM W303 E pkc1∆ DE Saccharomyces cerevisiae DURANTE O
PROCESSO DE DESREPRESSÃO METABÓLICA AUTOR: ANTÔNIO HELVÉCIO TÓTOLA
ORIENTADOR: PROF.DR. ROGELIO LOPES BRANDÃO Tese apresentada ao Programa de Pós-Graduação do Núcleo de Pesquisas em Ciências Biológicas da Universidade Federal de Ouro Preto, como parte integrante dos requisitos para obtenção do Título de Doutor em Ciências .
Ouro Preto, Outubro de 2007.

Trabalho desenvolvido no Laboratório de Biologia celular e Molecular do Núcleo de
Pesquisas em Ciências Biológicas da Universidade Federal de Ouro Preto (NUPEB), sob
a orientação do professor Dr. Rogelio Lopes Brandão e com o auxílio financeiro da
Coordenadoria de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES), Fundação
de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) e Universidade Federal de
Ouro Preto.
II

“Cantar e cantar e cantar, A beleza de ser um eterno aprendiz”.
Dedico este trabalho Ao meu pai, grande pescador! “In memorian”.
A minha mãe, que sempre me incentivou e me serviu de exemplo! A Cristiane, minha companheira, amiga e mulher que sempre esteve ao meu lado mesmo quando era difícil. Seu amor e paciência me trouxeram até aqui. Te amo, sempre!
A minha filha Maria Clara, luz da minha vida!
III

AGRADECIMENTOS
Ao meu orientador professor Dr. Rogelio Lopes Brandão, pela confiança e pelo
exemplo.
Ao meu amigo professor Dr. Ieso de Miranda Castro, pelo apoio e suporte em
todos os momentos.
Ao amigo professor Dr.Luciano Gomes Fietto pela amizade e disponibilidade
para discutir as idéias, os resultados e as divagações.
A amiga professora Dra. Juliana Rangel Fietto, pela ajuda e pela amizade.
A Zezé, grande microbiologista do LBCM, sempre pronta a ajudar e sempre
trazendo alegria ao laboratório.
Ao Eduardo e demais colegas do LIP, pela ajuda e pelo papo sempre alegre
(principalmente quando o Cruzeiro ganhava ou o Galo perdia).
Aos amigos e colegas do LBCM, que sempre se mostraram dispostos a ajudar e
colaborar, mesmos nos momentos mais difíceis.
À Cida, sempre alegre e disposta a ajudar no que for necessário.
Aos professores, funcionários e alunos dos laboratórios do NUPEB, pela
convivência e amizade.
Aos alunos de graduação, pelo aprendizado e convivência. Ao
curso de pós-graduação pela oportunidade.
A minha família pelo apoio e incentivo. IV

RESUMO
A levedura Saccharomyces cerevisiae possue uma via MAP quinase de
transdução de sinais composta por Pkc1p, Bck1p, Mkk1p e Mkk2p, e Mpk1p. Já foi
descrito que esta via está envolvida no controle da integridade da parede celular, na
resposta ao estresse osmótico, no crescimento de pseudo-hifas e no controle do
metabolismo de carbono. Neste trabalho investigamos a participação de Pkc1p no
controle da expressão de genes envolvidos no metabolismo de carbono.Para isto, fizemos
uma analise da expressão gênica global através de microarranjos de DNA comparando a
resposta global sob condições de repressão e desrepressão metabólica no mutante a pkc1
∆ e na cepa selvagem correspondente W303. Os resultados obtidos indicaram uma
diferença significativa na expressão global dos genes nas duas cepas analisadas. Vinte e
oito genes envolvidos no metabolismo de carbono foram selecionados para validação por
RT-PCR e os resultados confirmaram que sete genes foram mais expressos na cepa W303
quando comparada com a cepa e quatro genes foram mais expressos na cepa pkc1 ∆
quando comparada com a cepa selvagem. Análises “In silico” apontaram três fatores de
transcrição Sok2p, Crz1p and Gcr1p, que podem estar envolvidos no controle da
expressão gênica por Pkc1p. Experimentos utilizando plasmídeos contendo o gene da β-
galactosidase sob controle dos promotores dos genes CYC1, HXT1 e SUC2 mostraram
que a expressão destes genes se apresentava diminuída na cepa pkc1 ∆ para o gene
HXT1, aumentada para o gene CYC1 e igual para o gene SUC2 quando comparada com a
cepa selvagem W303. A atividade de Invertase apresenta-se significativamente diminuída
na cepa pkc1 ∆ quando comparada com a cepa selvagem W303.
V

Nossos resultados confirmam a participação da proteína Pkc1p na regulação da expressão
gênica durante o processo de desrepressão metabólica.
VI

ÍNDICE
RESUMO... ........................................................................................................ V ABSTRACT... ..................................................................................................VII LISTA DE ABREVIAÇÕES............................................................................... X LISTA DE TABELAS... ................................................................................. XIV LISTA DE FIGURAS... ................................................................................... XV
1. Introdução... ........................................................................................................ 2 1.1. O modelo experimental: a levedura Saccharomyces cerevisiae... ........................... 2 1.2. O genoma da levedura Saccharomyces cerevisiae... .............................................. 4 1.3. O metabolismo de fontes de carbono na levedura Saccharomyces
cerevisiae... .......................................................................................................... 6 1.4. A transdução de sinais na levedura Saccharomyces cerevisiae... .......................... 10 1.4.1. As vias MAPK (Proteínas quinases ativadas por mitógenos) na levedura
Saccharomyces cerevisiae... ............................................................................... 11 1.4.2. A transdução de sinais na conjugação em leveduras: resposta aos
ferormônios... .................................................................................................... 12 1.4.3. A transdução de sinais envolvidos na resposta ao estresse osmótico..................... 14 1.4.4. A via HOG (High Osmolarity Glycerol) em resposta ao estresse
hipertônico... ...................................................................................................... 15 1.4.5. A via de integridade celular em resposta ao estresse hipotônico... ........................ 17
2. Objetivos... ........................................................................................................ 22 2.1. Objetivo geral... ................................................................................................. 23 2.2. Objetivos específicos... ...................................................................................... 23
3. Materiais e Métodos... ........................................................................................ 25 3.1. Microorganismos utilizados nos experimentos... ................................................. 25 3.1.1. Cepas de Saccharomyces cerevisiae... ................................................................. 25 3.1.2. Procedência das cepas de Saccharomyces cerevisiae... ........................................ 25 3.1.3. Cepa de bactéria... .............................................................................................. 25 3.2. Plasmídeos LacZ repórter... ................................................................................ 25 3.3. Meios de cultura e condições de crescimento... ................................................... 26 3.3.1. Meios de cultura... ............................................................................................. 26 3.3.1.1. Meio LB (Luria - Bertaine)................................................................................. 26 3.3.1.2. Meio YP... ......................................................................................................... 26 3.3.1.3. Meio mínimo SD... ............................................................................................ 26 3.3.2. Condições de crescimento... ............................................................................... 27 3.4. Transformação de bactérias e leveduras... ........................................................... 27 3.4.1. Preparação de células de bactérias competentes................................................... 27 3.4.2. Transformação de bactérias... ............................................................................. 27 3.4.3. Mini preparação de DNA plasmidial de bactéria... .............................................. 28 3.4.4. Preparo de células de levedura competentes... ..................................................... 28 3.4.5. Transformação de leveduras... ............................................................................ 29 3.5. Determinação da atividade enzimática da Invertase e da β -Galactosidase.. 30
VIII

3.5.1. Inóculo e crescimento das células.............................................................................. 30 3.5.2. Preparo de extratos celulares para a determinação da atividade enzimática
da Invertase e β - Galactosidase... ............................................................................. 30 3.5.3. Dosagem da Invertase... ............................................................................................. 30 3.5.4. Dosagem da β-Galactosidase..................................................................................... 31 3.6. Dosagem de proteína... .............................................................................................. 31 3.7. Extração do RNA Total... .......................................................................................... 31 3.7.1. Inóculo e crescimento das células.............................................................................. 31 3.7.2. Extração do RNA... .................................................................................................... 31 3.7.3. Eletroforese em gel de agarose... ............................................................................... 32 3.7.3.1. Preparação das amostras de RNA para corrida no gel... ........................................... 32 3.7.3.2. Montagem do gel para corrida de RNA... ................................................................. 32 3.8. Ensaios de microarranjos de DNA... ......................................................................... 33 3.8.1. Micro arranjos de DNA... .......................................................................................... 33 3.8.2. Marcação do cDNA para Micro arranjos... ............................................................... 34 3.8.3. Degradação do mRNA (Hidrólise alcalina)... ........................................................... 35 3.8.4. Purificação da sonda de cDNA... ............................................................................... 35 3.8.5. Reação de marcação com CyDye... ........................................................................... 36 3.8.6. Quantificação da sonda marcada... ............................................................................ 36 3.8.7. Hibridização manual... ............................................................................................... 37 3.8.8. Lavagens... ................................................................................................................. 37 3.8.9. Leitura das lâminas... ................................................................................................. 38 3.8.10. Análise estatística... .................................................................................................... 38 3.9. Reação em cadeia da polimerase em tempo real... .................................................... 39 3.9.1. Introdução... ............................................................................................................... 39 3.9.2. Obtenção dos cDNAs... ............................................................................................. 39 3.9.3. Desenho dos Iniciadores... ......................................................................................... 39 3.9.4. Reação da polimerase em cadeia... ............................................................................ 40 3.9.5. Análise dos resultados... ............................................................................................ 40
4. Resultados e discussão... ............................................................................................ 42 4.1. Análise dos experimentos de microarranjos de cDNA... .......................................... 42 4.2. Reação em cadeia da polimerase em tempo real - (RT-PCR)... ................................ 53 4.3. Análise “in silico” dos genes diferencialmente expressos... ..................................... 58 4.4. Análise da atividade de ß-galactosidase das construções CYC1-ß-Gal,
HXT1-ß-Gal e SUC2- ß-Gal...................................................................................... 64
5. Conclusões... .............................................................................................................. 70
6. Perspectivas... ............................................................................................................. 73
7. Referências Bibliográficas……………………………………………….. 75
IX

LISTA DE ABREVIAÇÕES
ARG82 - Gene que codifica para uma Inositol polifosfato multiquinase (IPMK), fosforila Ins
(1,4,5)P3 formando Ins(1,3,4,5,6)P5;
BEM2 - Gene que codifica para uma proteína Rho GTPase envolvida no controle da
organização do citoesqueleto e da morfogênese celular
Cdc24 - Gene que codifica para um fator de troca de guanina para Cdc42p
CLN1 - Gene que codifica para uma ciclina envolvida na regulação do ciclo celular; ativa a
quinase Cdc28p e promove a transição da fase G1 para S.
CLN2 - Gene que codifica para uma ciclina envolvida na regulação do ciclo celular; ativa a
quinase Cdc28p e promove a transição da fase G1 para S
COX14 - Gene que codifica para uma proteína da membrana mitocondrial, necessária à
montagem da citocromo oxidase.
Crz1p - Fator de transcrição que ativa genes envolvidos na resposta ao estresse.
CSD2 - Gene que codifica para uma quitina sintase que catalisa a transferência de N-
acetilglicosamina para a quitina.
CYB2 - Gene que codifica para o Citocromo b2; reprimido por glicose e condições
anaeróbicas.
CYC1 - Gene que codifica para o Citocromo c, isoforma 1; transportador de elétrons do
espaço intermembranas da mitocôndria.
DLD1 - Gene que codifica para a D-lactato desidrogenase, oxida D-lactato a piruvato;
reprimida por glicose.
FKS1 - Gene que codifica para uma subunidade catalítica do complexo 1,3-beta-D-
glucano sintase; envolvida na sintese e manutenção da parede celular.
FKS2 - Gene que codifica para uma subunidade catalítica do complexo 1,3-beta-D-
glucano sintase; envolvida na formação da camada interna da parede celular.
FUM1 - Gene que codifica para a Fumarase; converte acido fumárico a acido L-málico no
ciclo dos ácidos tricarboxílicos.
Gcr1p - Fator de transcrição envolvido na regulação da glicólise
GLC3 - Gene que codifica para uma enzima ramificadora do glicogênio
Gpa1p - Proteína G de membrana envolvida na conjugação de leveduras
X

GPT2 - Gene que codifica para a Glicerol-3-fosfato/dihidroxiacetona fosfato
aciltransferase envolvida na acilação de glicerol-3-fosfato e dihidroxiacetona fosfato
durante a síntese de lipídeos.
GRE1 - Gene que codifica para uma Hidrofilina de função desconhecida, induzida por
estresse; regulada pela via HOG.
GUT1 - Gene que codifica para a Glicerol quinase, converte glicerol a glicerol-3-fosfato
GUT2 - Gene que codifica para am Glicerol-3-fosfato desidrogenase mitocondrial;
reprimida por glicose e cAMP
Hog1p - Proteína quinase ativada por mitógenos envolvida na síntese de glicerol
Hxk2p - Proteína Hexoquinase P2, que catalisa a fosforilação de glicose no citosol.
ICL2 - Gene que codifica para uma 2-metilisocitrato liase da matriz mitocondrial, catalisa a
conversão de 2-metilisocitrato a succinato e piruvato.
JEN1 - Gene que codifica para um transportador de lactato, necessário à captação de
lactato e piruvato; reprimido por glicose.
MDH2 - Gene que codifica para a Malato desidrogenase citoplasmática; catalisa a
interconversão de malato e oxalacetato; interage com Pck1p e Fbp1p
Mid2p - Proteína da membrana plasmática que atua como sensor da via de integridade
celular; interage com Rom2p e com a proteína Zeo1p
Mig1p - Proteína repressora estimulada por glicose
MNN1 - Gene que codifica para uma Alfa-1,3-manosiltransferase do complexo de Golgi
responsável pela formação de ligações alfa1,3-manose em oligossacarídeos N e O
ligados.
Msb2p -Membro da família das mucinas, participa na via de crescimento filamentoso
dependente de Cdc42p e da via de MAP quinases.
Msn5p - Exportina envolvida na translocação do fator Mig1 p
Pak1p - Serina-Treonina quinase do complexo SNF1;parcialmente redundante a Elm1p e
Tos3p;
PCL1 - Gene que codifica para uma Ciclina envolvida na entrada do ciclo mitótico e na
regulação da morfogênese
PCL2 - Gene que codifica para uma Ciclina envolvida na entrada do ciclo mitótico e na
regulação da morfogênese
XI

PDC1 - Gene que codifica para a Piruvato descarboxilase, enzima envolvida na
fermentação alcoólica; descarboxila piruvato a acetaldeído
PFK2 - Gene que codifica para a Fosfofrutoquinase 2, envolvida na glicólise e
indispensável para o crescimento em anaerobiose; ativada por frutose 2,6-bisfosfato e
AMP.
PLC1 - Gene que codifica para a Fosfolipase C, hidrolisa fosfatidilinositol 4,5-bifosfato
(PIP2) para gerar inositol 1,4,5-trifosfato (IP3) e 1,2-diailglicerol (DAG)
PYC1 - Gene que codifica para a Piruvato carboxilase, enzima citoplasmática que
converte piruvato a oxalacetato
Ras - CDC42 - Proteína ativadora da enzima adenilato ciclase
Reg1p-Glc7p - Subunidades regulatórias envolvidas na regulação negativa de genes
reprimidos por glicose.
Rga1p - Proteína ativadora de GTPase
RHR2 - Gene que codifica para uma isoforma constitutivamente expressa da
DLglicerol-3-fosfatase; envolvida na biossíntese do glicerol; induzida em condições
anaeróbicas e por estresse osmótico.
Rlm1p - Regulador gênico envolvido na síntese da parede celular
ROM1 - Gene que codifica para uma proteína que atua como fator de troca GDP/GTP
ROM2 - Gene que codifica para uma proteína que atua como fator de troca GDP/GTP
para Rho1p e Rho2p
RSF2 - Gene que codifica para uma proteína do tipo dedo de zinco, envolvida no
controle transcricional de genes mitocondriais e nucleares necessários ao crescimento em
glicerol.
SAC7 - Gene que codifica para uma proteína ativadora da GTPase para Rho1p, envolvida
com o citoesqueleto de actina.
SDH1 - Gene que codifica para uma Flavoproteína do complexo da succinato
desidrogenase, participa na transferência de elétrons para a ubiquinona
Sho1p - Sensor osmótico transmembrana, participa na ativação de Cdc42p, do
crescimento filamentoso dependente da via de MAP quinases e da via HOG.
Slg1p -Sensor da via de resposta ao estresse ativada por PKC1-MPK1;envolvida na
manutenção da integridade da parede celular e na organização do citoesqueleto.
XII

SLN1 - Gene que codifica para um sensor osmótico que regula a cascata das MAP
quinases
Slt2p - Serina-treonina Map quinase envolvida na regulação da manutenção da parede
celular e na progressão do ciclo celular. Regulada pela via de PKC1.
Snf1p - Proteína quinase envolvida na via principal de repressão por glicose
Snf4p - Subunidade ativada da proteína Snf1;ativa genes reprimidos por glicose e reprime
genes induzidos por glicose.
Sok2p - Proteina nuclear que desempenha um papel regulatório na via de PKA
dependente de AMP cíclico; regula negativamente o crescimento de pseudo-hifas
Ssk2p - Gene que codifica para uma MAPKKK envolvida na síntese de glicerol; interage
com Ssk1p, induzindo a ativação de Ssk2p.
Ssk22p - Gene que codifica para uma MAPKKK envolvida na síntese de glicerol;
homologo a Ssk2p.
Ssn6p-Tup 1p - Co-repressores transcricionais do complexo que recruta SWI/SNF e
SAGA
Ste12p - Fator de transcrição ativado pela via das MAP quinases, ativa genes envolvidos na
conjugação e no crescimento de pseudohifas.
Ste11p Proteína quinase quinase quinase envolvida na conjugação de leveduras
STE2 - Gene que codifica para o receptor do fator α de acasalamento
Ste20p - Proteína quinase envolvida na conjugação de leveduras
STE3 - Gene que codifica para o receptor do fator a de acasalamento
SUC2 -Gene que codifica para a invertase
Swi4p-Swi6p - Componentes do complexo SBF de ligação ao DNA; ativador
transcricional que regula a transcrição de genes necessários à síntese e reparo do DNA.
TKL2 - Gene que codifica para uma transcetolase, similar a Tkl1p; catalisa a conversão de
xilulose-5-fosfato e ribose-5-fosfato a sedoheptulose-7-fosfato e gliceraldeído-3-
fosfato na via das pentoses fosfato.
TOR2 - Gene que codifica para uma proteína quinase do complexo TORC1, que regula o
crescimento em resposta a nutrientes; envolvida na meiose.
YDR 019c - Gene que codifica para uma subunidade do complexo da glicina
descarboxilase mitocondrial
XIII

LISTA DE TABELAS
Tabela 01 Classificação taxonômica da levedura Saccharomyces cerevisiae
Tabela 02 Marcos histórico da utilização da levedura Saccharomyces cerevisiae
Tabela 03 Processos e produtos obtidos com o emprego da levedura Saccharomyces cerevisiae
Tabela 04 Receptores e transportadores funcionais de glicose na levedura Saccharomyces cerevisiae
Tabela 05 Cepas de Saccharomyces cerevisiae utilizadas nos experimentos Tabela 06 Plasmídeos LacZ repórter
Tabela 07 Iniciadores utilizados nos experimentos de RT-PCR para validação dos experimentos de microarranjos de DNA
Tabela 08 Predição de sítios de fosforilação quinase específicos na proteína Sok2p
Tabela 09 Predição de sítios de fosforilação quinase específicos na proteína Crz1p
Tabela 10 Predição de sítios de fosforilação quinase específicos na proteína Gcr1p XIV

LISTA DE FIGURAS
Figura 01 Principais laboratórios envolvidos no projeto genoma da levedura Saccharomyces cerevisiae.
Figura 02 Proteínas da membrana plasmática de células eucarióticas envolvidas na
detecção de nutrientes
Figura 03 Vias de MAPK presentes em Saccharomyces cerevisiae
Figura 04 Via de resposta aos hormônios de acasalamento
Figura 05 Mecanismos de adaptação aos choques hipertônico e hipotônico em Saccharomyces cerevisiae
Figura 06 Via HOG (High Osmolarity Glycerol) in Saccharomyces cerevisiae
Figura 07 Distribuição esquemática dos domínios na proteína Pkc1p
Figura 08 Representação esquemática dos principais componentes da via de Pkc1p de Saccharomyces cerevisiae
Figura 09 Análise do background das lâminas obtidas a partir da hibridização com os cDNAs da cepa W303.
Figura 10 Análise do background das lâminas obtidas a partir da hibridização com os cDNAs da cepa Pkc1Δ.
Figura 11 Normalização da intensidade de fluorescência das lâminas obtidas após hibridização com os cDNAs obtidos a partir da cepa W303
Figura 12 Normalização da intensidade de fluorescência das lâminas obtidas após hibridização com os cDNAs obtidos a partir da cepa Pkc1Δ
Figura 13 Box Plot apresentando a normalização por “Lowess fitting” das lâminas
obtidas após hibridização com os cDNAs obtidos a partir da cepa W303 Figura 14 Box Plot apresentando a normalização por “Lowess fitting” das lâminas
obtidas após hibridização com os cDNAs obtidos a partir da cepa Pkc1Δ Figura 15 Hibridização das lâminas com cDNAs produzidos a partir de mRNAs da
cepa selvagem W303
Figura 16 Hibridização das lâminas com cDNAs produzidos a partir de mRNAs da cepa pkc1 ∆.
XV

Figura 17 Expressão dos genes envolvidos com o metabolismo de carbono nas cepas W303 e Pkc1Δ.
Figura 18 Expressão dos genes envolvidos com o metabolismo de carbono selecionados através dos ensaios de microarranjos de DNA para validação por RT-PCR
Figura 19 Resultado da análise por RT-PCR da expressão de genes envolvidos no metabolismo de carbono nas cepas W303 e Pkc1Δ
Figura 20 Curvas de dissociação das reações de RT-PCR
Figura 21 Atividade de ß-galactosidase (em nmoles de ONP/min/mg de proteína) das cepas W303 e pkc1 Δ transformadas com a construção CYC1-ß- Galactosidase
Figura 22 Atividade de ß -galactosidase (em nmoles de ONP/min/mg de proteína) das cepas W303 e pkc1 Δ transformadas com a construção HXT1 - ß- Galactosidase
Figura 23 Atividade de ß -galactosidase (em nmoles de ONP/min/mg de proteína) das cepas W303 e pkc1 Δ transformadas com a construção SUC2 - ß- Galactosidase
Figura 24 Atividade de invertase (em nmoles de Glicose/min/mg de proteína) das cepas W303 e pkc1 Δ transformadas com a construção SUC2 - ß- Galactosidase
XVI

1
INTRODUÇÃO

2
1-Introdução
1.1-O modelo experimental: a levedura Saccharomyces cerevisiae
A levedura Saccharomyces cerevisiae é um fungo unicelular, pertencente ao filo
Ascomycota (Tabela 01) com formato redondo ou oval, medindo aproximadamente de 5
a 10 µm de diâmetro, sendo um dos modelos de organismo eucariótico mais estudados
em biologia celular e molecular.
Os estudos empregando S. cerevisiae como modelo experimental receberam um
forte impulso no final da década de 70 com o desenvolvimento da técnica de
transformação de fungos (Hinnen e cols., 1978). Algumas das propriedades que tornam
as leveduras tão interessantes como modelo experimental incluem a fácil obtenção, o
rápido crescimento na forma de células dispersas, um sistema genético bem definido, a
facilidade de transformação, replicação e isolamento de mutantes, seu genoma
completamente seqüenciado (Dujon, 1996) (Goffeau e cols., 1996), ser relativamente
estável tanto no estado haplóide quanto diplóide, além de não ser patogênico, dentre
outras.
Classificação Taxonômica Reino Fungi Filo Ascomycota Subfilo Saccharomycotina Classe Saccharomycetes Ordem Saccharomycetales Kudrjanzev, 1960 Família Saccharomycetaceae G. Winter, 1881 Gênero Saccharomyces Meyen Espécie Saccharomyces cerevisiae Hansen, 1883. Taxonomic Serial Nº 194157
Tabela 01: Classificação taxonômica da levedura Saccharomyces cerevisiae
Fonte: Integrated taxonomic information system (http://www.iti S. usda.gov)
Outro fator que deve ser levado em consideração é o fato de que uma vasta gama
de processos biológicos e funções celulares são altamente conservados e compartilhados
entre os fungos e organismos eucarióticos superiores, permitindo a utilização daqueles
como modelo no estudo comparativo desses processos. A possibilidade de

3
complementação com genes heterólogos (Atrian e cols., 1990; Smith e cols., 1990;
Wyckoff & Hsieh, 1988; Hensing e cols., 1995) e a facilidade de manipulação genética
dos fungos fazem dos mesmos um excelente sistema para o entendimento das funções
biológicas de produtos gênicos de outros organismos eucarióticos.
Além dos fatores descritos acima, deve-se levar em consideração a utilização de
Saccharomyces cerevisiae em uma vasta gama de processos industriais e
biotecnológicos (Ro e cols., 2006; Ostergaard e cols., 2000; Weber & Barth, 1988;
Maraz, 2002), além de constituir-se num dos mais antigos organismos domesticados
pelo ser humano, tendo sido empregada desde os primórdios da civilização (Tabela 02).
Tabela 02: Marcos histórico da utilização da levedura Saccharomyces cerevisiae.
Nos dias atuais, S. cerevisiae é utilizada na indústria alimentícia (Moreno-
Arribas & Polo, 2005; Chae e cols., 2001; Doores, 1983; Rose, 1981), na obtenção de
etanol (Bayrock & Michael, 2001; Taylor e cols., 2000; Taylor e cols., 1995), na
indústria farmacêutica para a produção de uma vasta gama de produtos (Maraz, 2002;
Masimirembwa e cols., 1999; Rombaut & Jore, 1997; Gill & Zaworski, 1991), em
pesquisas ambientais (Schofield e cols., 2007; Radhika e cols., 2007; Radhika e cols.,
2005)e no estudo bioquímico e biológico conforme descrito acima.
Cronologia Marcos históricos 6000-2000 AC Fermentação (Suméria, Babilônia) 1680 Levedura sob o microscópio (Van Leeuwenhoek) 1835 Fermentação alcoólica associada com a levedura 1837 Nome (S. cerevisiae) criado para as leveduras observadas no malte 1839 Açúcar com fonte de crescimento para leveduras 1857 Fermentação correlacionada com metabolismo (Pasteur) 1877 Introdução do termo Enzima (na levedura) por Kuhne 1880 Isolamento de cepas puras de levedura (Hansen) 1883 Produção de álcool e CO2 em extrato livre de células (Buchner) 1915 Produção de Glicerol 1949 Primeiro mapa genético (Lindegren); Sistema de conjugação e
acasalamento. 1930-1960 Taxonomia de leveduras por Kluyver 1960 Primeira estrutura de tRNA de levedura 1978 Primeira transformação em leveduras (Finck e cols..) 1990-1994 Primeiro produto farmacêutico a partir de levedura (vacina da hepatite B) 1990-1996 Seqüenciamento completo do genoma de S. cerevisiae

4
Na Tabela 03, podemos observar alguns dos processos e produtos obtidos com a
utilização de S. cerevisiae.
. Área Processos e produtos Indústria Química e de alimentos Enzimas
Fermento Acidulantes Redutores químicos
Indústria de fermentação Etanol Glicerol Vinho, cerveja, aguardentes. Panificação
Biomedicina Pesquisas em: Câncer, AIDS. Metabolismo de drogas Testes de genotoxicidade Desordens genéticas humanas
Meio ambiente Bioremediação Processos de reciclagem Bioadsorção de metais
Indústria Farmacêutica Probióticos Vacinas Hormônios Fatores de coagulação Fatores de crescimento Outros
Pesquisas biológicas Biologia celular Biologia molecular Genética Bioquímica Outros
Tabela 03: Processos e produtos obtidos com o emprego da levedura Saccharomyces
cerevisiae
1.2-O genoma da levedura Saccharomyces cerevisiae
A levedura S. cerevisiae contem um conjunto haplóide de 16 cromossomos com
tamanho variando entre 200 e 2200 KB. A seqüência completa do DNA cromossomal,
constituída de 12.052 KB foi descrita em 1996 (Dujon,1996;Goffeau e cols.,1996).
Contrastando com o genoma de organismos multicelulares, o genoma da levedura é
altamente compactado, onde os genes representam aproximadamente 72% da seqüência
total. O tamanho médio dos genes é de 1.45Kb ou 483 códons. Aproximadamente 3,8%

5
das janelas abertas de leitura contêm íntrons. Das 6.183 janelas abertas de leitura
descritas, compostas por mais que 100 aminoácidos, cerca de 5.800 correspondem a
possíveis genes que codificam para proteínas. Destas, cerca de 30% já foram
caracterizadas experimentalmente e 35% correspondem a genes que codificam para
proteínas que são relacionadas estruturalmente com produtos gênicos já caracterizados
em outros fungos ou organismos.
O RNA ribossômico é composto por aproximadamente 120 cópias arranjadas em
seqüência no cromossomo XII e os tRNAs são codificados por 262 genes, dos quais 80
possuem íntrons. O DNA de S. cerevisiae contém ainda uma série de elementos móveis
ou retro-transposons, que variam em quantidade e posição de acordo com as diferentes
cepas de levedura. O DNA mitocondrial tem aproximadamente 85Kb e contem os genes
que codificam para as subunidades I, II e III da citocromo C oxidase, apocitocromo b,
subunidades 6, 8 e 9 da ATPase e uma proteína associada ao ribossomo. Codifica ainda
para as unidades pequena e grande do RNA ribossomal, 24 RNAs transportadores e o
RNA 9 S. É caracterizado por um baixo conteúdo de GC (17%), possuindo uma grande
porção de regiões intergênicas representando 62% do mtDNA (Langkjaer e cols., 2003).
A Figura 01 representa um diagrama esquemático com os laboratórios
envolvidos no projeto de seqüenciamento do genoma da S. cerevisiae, publicado em
1996, com os 16 cromossomos em ordem decrescente de tamanho. As unidades
repetitivas estão representadas por boxes brancos nos cromossomos XII, IV e VIII
respectivamente.

6
Figura 1: Principais laboratórios envolvidos no projeto genoma da levedura Saccharomyces cerevisiae.
Fonte – Feldmann, H. in: Yeast molecular biology – A short compendium on basic features and novel aspects; 2005.
1.3-O metabolismo de fontes de carbono na levedura Saccharomyces cerevisiae.
As leveduras são organismos bem adaptados às mais variadas mudanças nas
condições ambientais, tendo desenvolvido uma série de mecanismos para responder às
mesmas. Uma vez que a disponibilidade de nutrientes no meio nunca é constante, as
leveduras devem se adaptar a estas variações e responder a elas modulando seu
metabolismo e seu crescimento (Holsbeeks e cols., 2004a) (Newcomb e cols., 2003).
Embora seja capaz de utilizar uma grande variedade de fontes de carbono a
levedura S. cerevisiae utiliza a glicose como sua fonte preferencial de carbono e energia

7
(Gancedo, 1998a; Mashego e cols., 2005). O metabolismo de glicose nas leveduras é
realizado principalmente através da fermentação alcoólica, mesmo sob condições de
aerobiose (Holm e cols., 2001). Embora a fermentação produza uma menor quantidade
de ATP por mol de açúcar, ela se processa a uma velocidade bem maior, trazendo uma
grande vantagem competitiva à levedura, principalmente porque o etanol produzido
durante a fermentação inibe o crescimento de outros microrganismos capazes de
competir com a levedura pelas fontes de carbono disponíveis. O etanol produzido pode
ainda ser utilizado através do metabolismo aeróbico, resultando na utilização completa
de todo o carbono presente no meio.
A capacidade que a levedura tem de detectar a flutuação dos níveis de glicose no
meio externo pode ser explicada pela presença de receptores de glicose na sua superfície
celular (Tabela 04).
Nome do gene ORF Descrição HXT1 YHR094C Transportador de hexose de baixa afinidade HXT2 YMR011W Transportador de hexose com baixa afinidade por
glicose HXT3 YDR345C Transportador de hexose de baixa afinidade HXT4 YHR092C Transportador de hexose de afinidade baixa a moderada HXT5 YHR096C Proteína da família Hxt com atividade transportadora
intrínseca HXT6 YDR343C Transportador de hexose de alta afinidade HXT7 YDR342C Transportador de hexose de alta afinidade HXT14 YNL318C Proteína da família de transportadores de açúcar capaz
de suportar o crescimento em galactose GPR1 YDL035C Receptor para glicose e sacarose acoplado à proteína G SNF3 YDL194W Sensor de glicose em baixas concentrações, semelhante
aos transportadore S. RGT2 YDL138W Sensor de glicose em altas concentrações, semelhante
aos transportadore S.
Tabela 04: Receptores e transportadores funcionais de glicose na levedura Saccharomyces cerevisiae. Fonte – SGD (Saccharomyces genome Database) http://www.yeastgenome.org
Além dos diversos transportadores de glicose, as células de S. cerevisiae
possuem dois tipos de sensores que detectam a presença de glicose, sendo um de baixa
(Rgt2p) e outro de alta afinidade (Snf3p) (Johnston & Kim, 2005) (Ozcan e cols., 1998).
Estes sensores são membros da família de transportadores de glicose com similaridade
parcial com os outros membros, possuindo longas caudas C - terminais citoplasmáticas.
Rgt2p e Snf3p não são capazes de atuar diretamente como transportadores de glicose;

8
entretanto, agem como sensores extracelulares de glicose envolvidos no controle da
expressão dos genes dos transportadores, através do fator de transcrição Rgt1. Rgt2 é
necessário para a máxima indução da expressão do transportador HXT1 em altas
concentrações de glicose, enquanto Snf3p é requerido para a indução da expressão dos
genes que codificam para Hxt2p, Hxt3p e Hxt4p em baixas concentrações de glicose
(Kaniak e cols., 2004) (Ozcan & Johnston, 1999). RGT2 é expresso constitutivamente,
ao passo que SNF3 é reprimido por altas concentrações de glicose. As caudas
citoplasmáticas de Rgt2p e Snf3p parecem ser responsáveis pela habilidade que estas
moléculas têm de traduzir a presença de glicose no meio externo para o interior a célula.
A fusão da cauda de Snf3p com os transportadores Hxt1p e Hxt2p foi capaz de
converter estes transportadores em sensores funcionais de glicose, capazes de gerar o
sinal necessário à indução da expressão dos genes dos transportadores funcionais de
glicose (Ozcan e cols., 1998;Tropia e cols., 2006)
Nas leveduras, uma das vias metabólicas mais estudadas e conhecidas,
envolvidas na regulação da utilização de glicose é a via principal de repressão por
glicose. A presença de glicose no meio desencadeia uma série de processos regulatórios
direcionados à utilização mais eficiente da mesma. A glicólise é ativada levando à
conversão da glicose em etanol e CO2, enquanto a gliconeogênese é inibida. Ocorre um
aumento acentuado na taxa de crescimento celular, precedido do aumento na
concentração de RNA ribossomal e da síntese de proteínas. Os genes envolvidos no
metabolismo de outras fontes de carbono e na resposta ao estresse são rapidamente
reprimidos e os carboidratos de reserva são utilizados (Gancedo, 1998b).
Os componentes centrais da via principal de repressão por glicose são o
complexo repressor Mig1p, o complexo da proteína quinase Snf1p e a proteína fosfatase
I – Reg1-Glc7 (Gelade e cols., 2003; Schuller, 2003). Mig1p é uma proteína com
domínios do tipo dedos de zinco Cys2–His2, que na presença de altas concentrações de
glicose recruta as proteínas co-repressoras Ssn6p e Tup1p, levando à repressão de
diversas famílias de genes e de seus respectivos indutores transcricionais (Treitel &
Carlson, 1995). A atividade de Mig1p como repressor é regulada por sua localização
celular, o que ocorre através da fosforilação ou defosforilação do mesmo. Em altas
concentrações de glicose, Mig1p é desfosforilado pela proteína fosfatase I e concentra-
se no núcleo da célula, onde se liga aos promotores dos genes reprimidos por glicose.
Quando a concentração de glicose decresce, Mig1p é fosforilado pela quinase Snf1p,
promovendo a interação de Mig1p com a exportina Msn5p, o que faz com que seja

9
transportado rapidamente para o citoplasma, aliviando a repressão gênica(Elbing e cols.,
2004; Kim e cols., 2006)
Snf1p é uma proteína serina/treonina quinase que apresenta dois domínios, um
regulatório na região carboxi-terminal e outro catalítico na região amino-terminal e
encontra-se associada a uma subunidade ativadora (Snf4p) e a três proteínas de
estruturação do complexo de alta massa molecular (subunidades Sip1p, Sip2p e
Gal83p). A subunidade Snf4p é necessária para a atividade de Snf1p, ao passo que
Sip1p, Sip2p e Gal83p ajudam a manter a associação entre as duas e determinam a
localização celular do complexo (Papamichos-Chronakis e cols., 2004).
A ativação de Snf1p parece estar relacionada a uma mudança conformacional da
mesma. Na presença de glicose, o domínio regulatório carboxi-terminal promove a
auto-inibição do complexo quinase, associando-se ao domínio catalítico. Na ausência de
glicose, ocorre a fosforilação de Snf1p, através das quinases Pak1p, Tos3p e Elm1p
(Nath e cols., 2003), ocasionando uma mudança conformacional que permite a interação
de Snf4p com o domínio regulatório de Snf1p, dissociando o mesmo do domínio
catalítico e promovendo a ativação do complexo, que fosforila Mig1p, promovendo sua
translocação do núcleo para o citoplasma. Quando a glicose é novamente disponível, o
complexo da proteína fosfatase I Glc7-Reg1p defosforila Snf1p no resíduo treonina 210,
fazendo com que a mesma retorne a sua conformação auto-inibitória. Ao lado disso, foi
proposta a participação da Hxk2p no processo de repressão por glicose, através da
interação com Mig1p, formando um complexo repressor localizado no núcleo celular da
levedura S. cerevisiae. Este complexo se ligaria aos promotores de diversos genes
reprimidos por glicose, reprimindo a transcrição dos mesmos (Ahuatzi e cols., 2004) .
Na ausência de glicose, outros carboidratos também podem ser utilizados por S.
cerevisiae como fonte de carbono e energia. Dentre eles podemos citar a frutose, a
manose, a galactose, a maltose, a sacarose entre outros. Frutose e manose são utilizadas
de modo semelhante à glicose, sendo que a galactose não é uma fonte usual de energia
para S. cerevisiae. Os genes envolvidos no metabolismo de galactose encontram-se
normalmente reprimidos. Na ausência de glicose e presença de galactose estes genes
são ativados cerca de 1.000 vezes, sendo um dos poucos genes neste organismo que
respondem da maneira tudo ou nada. A maltose e a sacarose são convertidas em seus
respectivos monossacarídeos através da ação da maltase e da invertase. As leveduras
podem ainda utilizar o glicerol após sua captação e conversão em glicerol-3-fosfato
através da enzima glicerol quinase.

10
1.4-A transdução de sinais na levedura Saccharomyces cerevisiae.
Na natureza, as leveduras lidam constantemente com flutuações nos níveis de
nutrientes disponíveis. A capacidade de detectar estas flutuações e de se adaptar a elas
é um fator preponderante para garantir a sobrevivência destes organismos. Durante a
evolução, as leveduras desenvolveram vários mecanismos que permitiram sua
adaptação a estas flutuações. Estes mecanismos envolvem tanto controle positivo como
negativo na regulação do metabolismo celular. Estes controles podem ser realizados
tanto a nível transcricional através da repressão ou indução de genes, quanto a nível pós
-transcricional, controlando a estabilidade e a tradução dos mRNAs, e pós-tradução,
através do controle da atividade enzimática por ativação ou inibição alostérica ou da
estabilidade das proteínas. A maioria desses processos é afetada direta ou indiretamente
pelos mecanismos de detecção e sinalização celular, através de vias de transdução de
sinais presentes nas leveduras.
A membrana plasmática das leveduras não se constitui somente numa barreira
celular onde moléculas que controlam o metabolismo e a proliferação celular são
detectadas, mas também numa barreira através da qual os nutrientes que garantem o
suprimento de energia e o crescimento celular são transportados (Holsbeeks e cols.,
2004b). Nela estão presentes diversos receptores adaptados a responder às condições
ambientais e a transformar os estímulos externos em respostas celulares específicas
através da ativação ou repressão transcricional, possibilitando às leveduras adaptar seu
metabolismo (Figura 02). A presença de um determinado tipo de molécula é detectada
pelo receptor apropriado, promovendo a ativação do mesmo e desencadeando uma série
de eventos celulares, geralmente através de uma cascata de reações enzimáticas.
Diversas vias de transdução de sinais foram identificadas em leveduras; dentre elas,
podemos citar aquelas envolvidas com a detecção da presença de hormônios e fatores de
crescimento; com as oscilações na temperatura e pH; com a presença de nutrientes e
com a variação da osmolaridade dentre outras.

11
Figura 2 – Proteínas da membrana plasmática de células eucarióticas envolvidas na
detecção de nutrientes. Os receptores apresentam-se divididos em três grandes grupos: o
grupo de receptores não transportadores, envolvidos na detecção de sinais, mas não
acoplados ao transporte (Non-transporting transceptor), os receptores transportadores
não acoplados a proteínas G (transporting transceptors) e os transportadores
propriamente ditos acoplados a proteínas G (G-protein coupled receptors).
Fonte: (Holsbeeks e cols., 2004c)
1.4.1 - As vias MAPK (Proteínas quinases ativadas por mitógenos) na levedura
Saccharomyces cerevisiae.
As vias de proteínas quinases ativadas por mitógenos são vias de transdução de
sinais altamente conservadas nos organismos eucarióticos superiores, podendo ser
ativadas tanto por sinais externos como internos. Estas vias controlam uma série de
processos em leveduras como o crescimento celular, a morfogênese, a proliferação
celular, a resposta aos estresses, dentre outras. Geralmente, estas vias apresentam como
ponto comum, três proteínas quinases: uma MAP quinase (MAPK), uma MAP quinase
quinase (MAPKK) e uma MAP quinase quinase quinase (MAPKKK), atuando em
cascata. A ativação dessas vias se dá por sucessivos passos de fosforilação, culminando

12
com a regulação e modulação de fatores transcricionais. A regulação dessas vias
também se faz através da ação de diversas proteínas fosfatases que atuam normalmente
ao nível da MAPKK ou MAPK quinases. A ativação dessas vias normalmente ocorre
com a participação de uma proteína quinase inicial, associada geralmente a pequenas
proteínas G.
Em leveduras, são conhecidas seis vias de MAPK diferentes: a via de
conjugação em resposta aos ferormônios, a via de desenvolvimento de pseudo-hifas, a
via HOG (High Osmolarity Glycerol), a via de integridade celular, a via de montagem
da parede de esporos e a via de crescimento vegetativo(Chen & Thorner, 2007). Na
Figura 03, temos as cinco vias mais conhecidas, mostrando o compartilhamento de
algumas proteínas, o que contribui para a grande complexidade dos processos
envolvendo a ativação das vias de MAPK quinases.
Figura 3: Vias de MAPK presentes em Saccharomyces cerevisiae.
Fonte: Feldmann, H. in: Yeast molecular biology – A short compendium on basic
features and novel aspects; 2005.
1.4.2-A transdução de sinais na conjugação em leveduras: resposta aos
ferormônios
Células de leveduras haplóides com fatores de acasalamento opostos (a ou α)
podem se cruzar gerando células diplóides. As células MATα produzem o fator α e
possuem receptores para o fator a, codificado pelo gene STE2. As células haplóides

13
MATa produzem o fator a e possuem receptores para o fator α, codificado pelo gene
STE3. Após a ligação do fator de acasalamento ao seu respectivo receptor, ocorre a
ativação de Gpa1p, uma proteína G heterotrimérica. Em seguida, o sinal é transmitido
para uma proteína ligante de GTP da família das proteínas relacionadas a Ras (Cdc42p),
a qual está associada a uma proteína ativadora de GTPase (Rga1p) e ao fator
estimulador de troca GTP/GDP (Cdc24p). Ocorre então a ativação de Ste20p, uma
proteína do complexo das MAP quinases, com ativação subseqüente desta via. Os genes
específicos de acasalamento são por fim ativados pelo fator de transcrição Ste12p, que é
capaz de se ligar aos elementos de resposta a ferormônios (PRE) (Breitkreutz & Tyers,
2002)(Figura 04).
Figura 4: Via de resposta aos hormônios de acasalamento
Fonte: Feldmann, H. in: Yeast molecular biology – A short compendium on basic
features and novel aspects; 2005

14
1.4.3-A transdução de sinais envolvidos na resposta ao estresse osmótico.
A maioria dos organismos unicelulares encontrados na natureza como as
leveduras, freqüentemente estão expostos a grandes variações nas condições ambientais.
A capacidade de responder a estas mudanças eficientemente é de vital importância para
garantir a sobrevivência dos mesmos. Mudanças no ambiente podem ser de natureza
física ou química, como temperatura, pressão, radiação, concentração de solutos e água,
presença de determinados íons, agentes tóxicos, pH e disponibilidade de nutrientes.
Freqüentemente, as leveduras lidam com flutuações em mais de um dos fatores acima
descritos.
Na natureza, as células de leveduras estão expostas a variações dramáticas e
muito rápidas na osmolaridade, ou seja, na concentração de água disponível. Uma
levedura crescendo em um fruto seco pode estar exposta a altas concentrações de
açúcar, ao passo que imediatamente após uma chuva a mesma pode estar sujeita a altas
concentrações de água disponível. Durante o metabolismo fermentativo, ocorre um
aumento acentuado na concentração de álcool produzido como subproduto do
metabolismo, o que também diminui consideravelmente a água disponível (Tamas e
cols., 2000). O estresse osmótico é causado pela mudança nas concentrações de solutos
no meio onde a levedura está presente, alterando a disponibilidade de água. Em
membranas semipermeáveis, a água sempre flui na direção do compartimento com
maior concentração osmótica. Uma vez que a maioria das membranas biológicas é mais
permeável à água do que à maioria dos solutos, as células dos organismos na maioria
das vezes são afetadas pelas mudanças na osmolaridade. Um aumento da concentração
osmótica no meio (estresse hiperosmótico) fará com que a água tenha uma tendência a
fluir do interior da célula para o meio externo, enquanto uma diminuição da
concentração de solutos no meio (estresse hiposmótico) irá causar um aumento na
entrada de água para a célula (Figura 05).

15
Figura 5 – Mecanismos de adaptação aos choques hipertônico e hipotônico em
Saccharomyces cerevisiae.
Uma série de vias de sinalização permitem às leveduras detectar as variações
osmóticas no meio ambiente. As mais conhecidas são a via HOG (High Osmolarity
Glycerol) em resposta ao estresse hipertônico e a via de integridade celular ou via das
MAPK (Mitogen Activated Protein Kinase ou proteínas quinases ativadas por
mitógenos) em resposta ao estresse hipotônico.
1.4.4 - A via HOG (High Osmolarity Glycerol) em resposta ao estresse hipertônico. O glicerol é um álcool produzido durante o processo fermentativo em leveduras,
podendo ser utilizado como fonte de carbono e energia, sendo ainda importante na
biossíntese de fosfolipídios. Além disso, o glicerol é utilizado pela levedura como
regulador osmótico, através da regulação de sua concentração no interior da célula. Na
presença de estresse hipertônico, as leveduras acumulam o glicerol, contrabalançando as
altas concentrações de soluto no meio externo. O acúmulo do glicerol se dá pelo
aumento de sua captação, pelo aumento na produção do mesmo ou pelo controle da
abertura e fechamento dos canais de glicerol. A entrada do glicerol nas células de
leveduras pode acontecer por mecanismos diferentes: difusão passiva, difusão facilitada
e transporte ativo.
Segundos ou minutos
Entrada de água – Aumento da pressão interna. Inchamento da célula.
Saída de glicerol
Acúmulo de glicerol
Saída de água Célula murcha
Adaptação – Captação passiva de água, capacidade de proliferação, reassume forma regular
Adaptação – Saída passiva de água, diminuição pressão interna
Aumento da Osmolaridade
Choque hipertônico
Horas
Choque hipotônico
Diminuição da Osmolaridade

16
Normalmente, a difusão passiva de glicerol através da membrana celular de
leveduras é baixa. Em condições de estresse hiperosmótico a difusão passiva encontra-
se bastante diminuída. Isto parece ser devido ao fato de que as alterações na membrana
celular durante o estresse dificultam ainda mais a difusão.
A difusão facilitada de glicerol em leveduras é mediada pelo canal Fps1p. O
transporte de glicerol pelo mesmo pode se dar tanto num sentido quanto em outro. O
canal Fps1p desempenha um papel fundamental no controle dos níveis intracelulares de
glicerol em leveduras. O mecanismo de transporte ativo ainda não está bem estabelecido
em leveduras.
Em S. cerevisiae, foram identificadas três vias de sinalização capazes de ativar a
via HOG, através da fosforilação e ativação de Hog1p (Figura 06) (Van e cols., 2000)
(Saito & Tatebayashi, 2004) (Westfall e cols., 2004). Através de Sho1p, uma proteína
com quatro domínios transmembranares ou através de Msb2p, uma proteína semelhante
à mucina. Estas duas vias de sinalização estão conectadas à proteína Rho trifosfatase, a
Cdc42p, à proteína Pak1p, à proteína Ste20p e à proteína MAPKK quinase, Ste11p. A
outra via de sinalização que ocorre através de SLN1 normalmente é ativada em
condições de estresse hiperosmótico moderado, levando à ativação de duas MAPKKKs
redundantes, a Ssk2p e Ssk22p (O'Rourke e cols., 2002; O'Rourke & Herskowitz,
2004).
As vias de ativação via Sho1p e Msb2p ativam a via HOG através da ação de
Ste11p e a via de sinalização em resposta ao estresse moderado é ativada a partir de
Sln1p.
A ativação dessa vias leva a fosforilação de Hog1p, o ponto central da ativação da
resposta ao estresse hiperosmótico. A fosforilação de Hog1p resulta na sua translocação
do citoplasma para o núcleo, onde ela interage com uma grande variedade de proteínas
nucleares, incluindo fatores de transcrição e histona deacetilases, promovendo a
transcrição de vários genes envolvidos na resposta ao estresse. Quando a célula
restabelece o equilíbrio osmótico, Hog1p é defosforilada e exportada para o citoplasma,
cessando a resposta.

17
Figura 6 – Via HOG (High Osmolarity Glycerol) in Saccharomyces cerevisiae Fonte:http:stke.sciencemag.org/cgi .
1.4.5 - A via de integridade celular em resposta ao estresse hipotônico.
A via de integridade celular ou via PKC MAP quinases é uma via de transdução
de sinais envolvida em uma série de eventos celulares, como resposta a variações de
temperatura, resposta a ferormônios, ciclo celular e baixa osmolaridade. O principal
papel desta via parece ser o de manter a integridade celular, através do controle da
síntese da parede celular durante o crescimento, o desenvolvimento ou após eventos que
levam a expansão da célula ou perturbações da parede celular (Hohmann, 2002).
Mutantes nesta via de sinalização apresentam defeitos na formação da parede celular,
necessitando a adição de estabilizadores osmóticos (NaCl ou Sorbitol) para seu
crescimento e proliferação.

18
A proteína Pkc1p, codificada pelo gene PKC1 (Levin e cols., 1990) é a única
proteína quinase C em leveduras homóloga das proteínas quinases C de mamíferos. Ela
contém uma série de domínios funcionais similares aos encontrados nas proteínas
homólogas de mamíferos como duas regiões do tipo C1, que são seqüências do tipo
dedo de zinco com sítios de ligação para diacilglicerol, um domínio tipo C2 envolvido
na ligação de fosfolipídios dependente de cálcio, o domínio quinase, um sítio para
ligação do pseudosubstrato, a região V5 na porção C-terminal e dois domínios HR1 na
região N-terminal que ligam pequenas proteínas G (Figura 07). Apesar de possuir os
sítios para ligação de diacilglicerol e o domínio C2 dependente de cálcio, dados
experimentais obtidos até então demonstram que a Pkc1p de leveduras não é ativada por
esses sinais (Schmitz & Heinisch, 2003).
Figura 07 - Distribuição esquemática dos domínios na proteína Pkc1p.
Fonte: Denis,V. e cols.; 2005
Como descrito acima, a via da Pkc1p é ativada por perturbações ao nível da
parede celular. A capacidade de detectar essas perturbações e ativar essa via parece
estar associada à presença se sensores na superfície da célula, capazes de detectar e
transmitir os sinais para o interior da célula. Dois sensores foram identificados como
sendo possíveis componentes da via da Pkc1p. O primeiro deles é a proteína Slg1p (ou
Hcs77p ou ainda Wsc1p), que foi descoberta através de estudos que demonstraram sua
participação na ativação da fosforilação de Slt2p durante o estresse térmico (Gray e
cols., 1997). O outro é a proteína Mid2p, codificada pelo gene MID2, que participa no
controle da viabilidade celular durante o acasalamento e na manutenção da homeostase
do cálcio (Ono e cols., 1994). Sua seqüência de aminoácidos deduzida apresenta
similaridades com Slg1p. Ambas as proteínas apresentam um domínio rico em serina e
treonina, indicativo de que sejam proteínas glicosiladas localizadas na superfície
celular. Não se sabe como ocorre a transmissão do sinal entre esses possíveis sensores e
a via da Pkc1p.
O ponto central da via de Pkc1p é a cascata de quinases constituída por uma
MAPquinase quinase quinase (Bck1p), duas MAPquinase quinase redundantes (Mkk1p
e Mkk2p) e uma MAPquinase (Slt2p) (Figura 08). Esta cascata é ativada através da

19
fosforilação de resíduos de serina e treonina presentes em Bck1p pela ação da Pkc1p, o
que provoca uma seqüência de fosforilações nos componentes seguintes Mkk1p/Mkk2p
e Slt2p. A ativação de Pkc1p se dá através da ação de Rho1p, uma proteína da família
das proteínas ligantes de GTP. Rho1p é ativada através da ação do fator de troca
GDP/GTP (GEF), codificado pelos genes ROM1 e ROM2, sendo que Rom2 p parece ter
um papel preponderante neste processo. Por outro lado, as proteínas ativadoras de
GTPase, codificadas pelos genes BEM2 e SAC7, parecem ser as responsáveis pela
regulação negativa, inibindo a atividade de Rho1p (Schmidt e cols., 2002). O gene
TOR2 codifica para a proteína Tor2p que é responsável pela ativação de Rom2p
(Helliwell e cols., 1998b). Essa proteína possui uma grande homologia com as
fosfatidilinositol quinases de mamíferos, o que pode sugerir uma ligação entre o
metabolismo de lipídeos, importante na integridade da parede celular, e a via de Pkc1p.
Rho1p também participa diretamente na ativação do complexo da glucano sintase
(Mazur & Baginsky, 1996).
Como dissemos anteriormente, a via da Pkc1p está envolvida na manutenção da
integridade da parede celular e isso se dá através do controle da expressão de genes
envolvidos na síntese da mesma. Experimentos realizados por Igual(Igual e cols., 1996)
demonstraram que os genes FKS1 e FKS2, que codificam para as subunidades do
complexo β-1,3-glucano sintase, o gene MNN1 que codifica para uma α-1,3-
manosiltransferase e o gene CSD2 que codifica para uma quitina sintase, são
dependentes da ativação de Slt2p para sua expressão. Um dos fatores transcricionais
ativados por Slt2p é Rlm1p, um membro da família de proteínas MAD-Box. Estudos de
duplo híbrido demonstraram a interação de Slt2p com Rlm1p (Watanabe e cols., 1997)
e que Rlm1p ativa a transcrição de genes somente após a sua fosforilação (Watanabe e
cols., 1995). Outro alvo da fosforilação por Slt2p é o fator de transcrição SBF, um
heterodímero composto por Swi4p e Swi6p, envolvido na regulação do ciclo celular e
do crescimento polarizado em leveduras, principalmente durante a fase de transição de
G1 para a fase S, ativando a transcrição de genes como CLN1, CLN2, PCL1 e PCL2
(Madden e cols., 1997).

20
Figura 8 – Representação esquemática dos principais componentes da via de Pkc1p de Saccharomyces cerevisiae
Fonte: (Heinisch e cols., 1999b).
Estudos realizados por Levin (Levin e cols., 1994) com mutantes na via de PKC
MAP quinase demonstraram que mutações no gene PKC originaram um fenótipo mais
severo de osmosensibilidade quando comparados com mutações nos genes de outros
componentes desta via como BCK1 (MAPquinase quinase quinase), MKK1/MKK2
(MAPquinase quinase) e SLT2 (MAPquinase), o que sugere que Pkc1p possa estar
envolvida em outras vias de sinalização nas leveduras, além da via de MAPK quinases.
Outras vias envolvidas com Pkc1p já descritas (Heinisch e cols., 1999a); (Gustin
e cols., 1998) incluem a via de calcineurina (Mazur e cols., 1995), a via envolvida na
detecção de nutrientes, a via HOG, a via de fosfatidil inositol (Yoshida e cols., 1994;
Schmidt e cols., 1997), a via TOR (Barbet e cols., 1996), a via de controle do ciclo
celular dependente de Cdc-28 (Marini e cols., 1996), a organização do citoesqueleto
(Chai e cols., 2002) (Helliwell e cols., 1998a) e a via de conjugação celular (Zarzov e
cols., 1996). Valentini demonstrou, através da análise de supressores multicópia do
fator de iniciação eIF5A, a participação de Pkc1p nos mecanismos de controle da
estabilidade de RNAs mensageiros (Valentini e cols., 2002) .

21
Estudos realizados em nosso laboratório (Brandao e cols., 2002)) demonstraram
que uma cepa mutante pkc1∆ apresenta uma primeira fase de crescimento mais lenta e
um consumo de etanol menor do que as cepas selvagem e um mutante bck1∆. Foi
demonstrado ainda que o mutante pkc1∆ apresentava uma diminuição na expressão de
transportadores de glicose quando comparada com as cepas selvagem e bck1∆. Os
experimentos demonstraram ainda que o mutante pkc1∆ não crescia na presença de
glicerol como única fonte de carbono e que a desrepressão do gene SUC2 que codifica
para invertase estava bastante diminuída em comparação com a cepa selvagem. A
atividade enzimática de invertase também foi analisada e os resultados demonstraram
que a cepa pkc1∆ apresentava uma atividade invertásica bastante diminuída quando
comparada com as cepas selvagem, bck1∆ e mpk1∆. Posteriormente, Salgado
demonstrou que Pkc1p estava envolvida na desrepressão de outras enzimas como a
álcool desidrogenase e que o mecanismo envolvido nesse processo parece ser o controle
da localização subcelular do repressor Mig1p pela Pkc1p (Salgado e cols., 2002). Por
sua vez, Gomes (Gomes e cols., 2005) demonstrou que a cepa pkc1∆ não cresce em
glicerol por não ser capaz de desreprimir o gene GUT1 que codifica para a glicerol
quinase, a enzima que catalisa o primeiro passo do metabolismo do glicerol e que o
transporte de glicerol também estava afetado na cepa pkc1∆.
Os dados até então disponíveis, sugerem a participação da proteína Pkc1 p nos
processos de repressão e desrepressão por glicose; entretanto, como as vias de
transdução de sinais em leveduras apresentam uma grande complexidade, com
elementos comuns a várias vias, torna-se difícil avaliar quais são os componentes que
participam efetivamente nos processos de repressão e desrepressão por glicose que estão
associados à atividade de Pkc1p.
Os avanços relacionados à análise da expressão global de genes, através da
utilização de microarranjos de DNA, possibilitaram a utilização desta técnica na
elucidação do padrão de expressão gênica de diversos organismos submetidos a
condições pré-determinadas.
Decidimos então empregar a técnica de microarranjos de DNA, uma vez que a
mesma nos permite avaliar a expressão global dos genes de Saccharomyces cerevisiae
em condições de repressão e desrepressão, comparando as células selvagens com os
mutantes da cepa pkc1∆.

22
OBJETIVOS

23
2-Objetivos
2.1 - Objetivo geral
Tendo em vista os resultados anteriores obtidos em nosso laboratório, que
apontam para a participação da Pkc1p em processos metabólicos não relacionados
diretamente à via das Map quinases durante a desrepressão metabólica, decidimos
analisar comparativamente a expressão global de genes nas cepas selvagem W303 e
mutante pkc1∆ em condição de repressão e desrepressão metabólica.
2.2 - Objetivos específicos
2.2.1 - Analisar, através de ensaios de microarranjos de DNA, a expressão de todos os
genes da levedura Saccharomyces cerevisiae das cepas selvagem e mutante pkc1∆
durante o processo de desrepressão metabólica e selecionar os genes que apresentam
diferenças significativas de expressão entre as duas cepas.
2.2.2 – Validar os resultados obtidos nos ensaios de microarranjos de DNA para alguns
dos genes selecionados, através da reação em cadeia da polimerase em tempo real (RT-
PCR).
2.2.3 – Analisar o padrão da transcrição de alguns genes das cepas selvagem W303 e
pkc1∆ durante o processo de desrepressão metabólica empregando plasmídeos
contendo os promotores destes genes associados ao gene da β-galactosidase.

24
MATERIAIS E MÉTODOS

25
3-Materiais e Métodos
3.1 - Microorganismos utilizados nos experimentos
3.1.1 - Cepas de Saccharomyces cerevisiae
As cepas de Saccharomyces cerevisiae utilizadas neste trabalho estão descritas na tabela
abaixo:
Nome Genótipo
aW303 Mat a ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 GAL mal SUC2
bYSH850 Mat a pkc1∆::HIS3 (W303-1A)
∗ Letras a-b, procedência das cepas de S. cerevisiae ítem 3.1.2
Tabela 05: Cepas de Saccharomyces cerevisiae utilizadas nos experimentos
3.1.2 - Procedência das cepas de Saccharomyces cerevisiae :
a- Johan Thevelein, Laboratorium voor Moleculaire Celbiologie, Katholieke
Universiteit Leuveun, Belgium.
b- Stefan Hohmann, CMB/Microbiology, Göteborg Univerty, Sweden.
3.1.3-Cepa de bactéria
A cepa de Escherichia coli utilizada para a amplificação do DNA plasmidial foi
a TOP10F’: F’, mcr A, ∆ (mrr-hadRMS – mcrBC) Ø 80∆lacZ ∆M15, ∆lacx74, deoR,
rec∆1, araD139,∆(ara,leu), 7697, galIU, galK, λ-,rs1p,end ∆1,nupG.
3.2-Plasmídeos LacZ repórter
Os plasmídeos contendo os promotores dos genes associados ao gene da β-
galactosidase estão descritos na tabela abaixo:
Nome Construção Gene do promotor associado
pBM2636 HXT1::lacZ em YEP 357R (URA3) YFL011W
pBM1942 pLG669-Z from L.Guarente (URA3)
= CYC-LacZ ( with no lexO sites)
YJR048W
pBM2773 SUC2::lacZ em YEP 356 (URA3) YIL162W
Tabela 06: Plasmídeos LacZ repórter

26
Todos os plasmídeos foram fornecidos pelo Dr. Mark Johnston, Washington University
School of Medicine, St. Louis, Missouri.
3.3-Meios de cultura e condições de crescimento
3.3.1-Meios de cultura
3.3.1.1-Meio LB (Luria – Bertaine)
O meio LB é composto por triptona 1% (p/v), extrato de levedura 0,5% (p/v) e cloreto
de sódio 0,5% (p/v), pH 7,5 ajustado com NaOH. Ao meio sólido acrescenta-se ágar
1,5% (p/v).
3.3.1.2-Meio YP
O meio YP é composto por extrato de levedura 1% (pv), bacto-peptona 2%
(p/v). O meio sólido é acrescido de ágar 1,5% (p/v). Em todos os experimentos o meio
YP foi adicionado de 1M de Sorbitol. As fontes de carbono utilizadas foram as seguintes:
glicose 2% ou 4% ou rafinose 2%. As fontes de carbono foram autoclavadas
separadamente e acrescentadas ao meio no fluxo laminar.
3.3.1.3-Meio mínimo SD
O meio SD é composto por 6.7 g/l de base nitrogenada de levedura sem
aminoácidos, suplementado com os aminoácidos arginina 20 mg/l, ácido glutâmico 100
mg/l, ácido aspártico 100 mg/l, fenilanina 50 mg/l, histidina 100 mg/l, isoleucina 30 mg/l,
lisina 30 mg/l, leucina 250 mg/l, metionina 20 mg/l, serina 375 mg/l, triptofano 100 mg/l,
tirosina 30 mg/l, treonina 200 mg/l, valina 150 mg/l e as bases adenina 50 mg/l e uracil
50 mg/l. Em todos os experimentos o meio SD foi adicionado de 1M de Sorbitol O meio
sólido foi acrescido de ágar 1,5% (p/v). O pH do meio SD foi acertado com KOH para o
valor 6,5. As fontes de carbono usadas foram: glicose 2% (p/v) ou rafinose 2% (p/v).

27
3.3.2-Condições de crescimento
As células de Saccharomyces cerevisiae foram crescidas a 30°C sob agitação a
200 rpm (Incubabor Rotatório New Brunswick Model G25) em meio YP suplementado
com a fonte de carbono adequada. Para as cepas transformadas portadoras de
plasmídeos, foi utilizado o meio seletivo SD-URA. As células de E. coli foram
crescidas a 37°C sob agitação de 200 rpm (incubador rotatório New Brunswick Model
G25) em meio LB.
3.4-Transformação de bactérias e leveduras
3.4.1-Preparação de células de bactérias competentes
A bactéria Escherichia coli Top 10 F’, estocada à - 70°C com 25% glicerol, foi
crescida em meio LB suplementado com 15 µg/ml de tetraciclina e 20 µg/ml de
estreptomicina a 37°C durante 16 horas. Uma colônia dessa cultura foi inoculada em 3
ml de meio LB e incubada sob agitação de 200 rpm (Incubador Rotatório New
Brunswick Model G25) nas mesmas condições descritas acima. Após este período,
utilizou-se uma alíquota de 1 ml dessa cultura para inocular 100 ml de meio LB,
mantidos a 37 °C sob agitação de 200 rpm (Incubador Rotatório New Brunswick Model
G25) até atingir a DO600nm próxima de 0.8 nm (Du-68 Spectrophtometer-Beckman).
Após o crescimento, as bactérias foram coletadas por centrifugação a 1.000g, por 10
minutos, a 4°C. O sedimento celular foi suspenso em 40 ml de CaCl2 0.1 M e mantido
em gelo por 1 hora. Após centrifugação, as células foram suspensas em 1 ml de CaCl2
1M. As células competentes foram aliquotadas e usadas imediatamente ou estocadas a -
70°C em presença de glicerol em uma concentração final de 25%.
3.4.2 - Transformação de bactérias
Cerca de 10 ng de mistura de ligação ou 2 µl de plasmídeo foram adicionados a
um tubo Eppendorf de 1,8 ml contendo 100 µl de bactéria TOP 10F’competentes (item
3.9). Este material foi mantido em gelo por 30 minutos. Após este período, as células

28
foram submetidas a um choque térmico a 42°C por 2 minutos. Em seguida, as células
foram mantidas em 1 ml de meio LB, a 37°C, por 50 minutos. Após centrifugação a
6.000 rpm (Heraeus Sepatech, Biofure 13) por 1 minuto, o sobrenadante foi descartado.
As células foram suspensas em 100 µl de LB, plaqueadas com auxílio de pérolas de
vidro em meio líquido LB-ampicilina (100 µg/ml) e crescidas a 37°C sob agitação 200
rpm (incubador rotatório New Brunswick Model G25), por 16 horas.
3.4.3 - Mini preparação de DNA plasmidial de bactéria
As colônias obtidas no procedimento anterior foram inoculadas em 5 ml de meio
LB-ampicilina100 µg/ml a 37° C por 12 horas. Após o crescimento, a suspensão de
células foi transferida para tubos de microcentrífuga de 1,8 ml e centrifugadas a 3500
rpm (Heraeus Sepatech, Biofuge 13) por 10 minutos. O sobrenadante foi descartado e o
sedimento celular ressuspendido em 100 µl da solução I (dextrose 0,5M, EDTA 7,5
mM, Tris-HCl pH 8, 25mM e Ribonuclease A 0,15mg/ml) . Em seguida, foram
adicionados 200µl de solução de lise (SDS 1% e NaOH 0,2 M). As células foram
homogeneizadas por inversão e incubadas a 4º C por 20 minutos. Após este período, o
material foi centrifugado a 13000 rpm (Heraeus Sepatech, Biofuge 13) por 20 minutos.
O sobrenadante foi transferido para outro tubo de microcentrífuga de 1,8 ml, adicionado
de 300 µl de Isopropanol e centrifugado a 13000 rpm por 20 minutos O sobrenadante
foi descartado e o DNA plasmidial foi lavado com 800 µl de etanol 70%. Após uma
nova centrifugação nas mesmas condições, o sobrenadante foi eliminado e o DNA seco
a vácuo (speedvac Savant AS 290). O DNA plamidial foi ressuspendido em 25 µl de
água milli-Q (Sistema Milli-Q Millipore).
3.4.4-Preparo de células de levedura competentes
A cepa de Sacharomyces cerevisiae a ser transformada foi crescida a 30°C, 200
rpm (Incubabor Rotatório New Brunswick Model G25), durante 24 horas, em 5 ml de
YPD com sorbitol 1M. Este pré-inóculo foi utilizado para inocular 100 ml do mesmo
meio e foi incubado nas mesmas condições de temperatura e agitação descritas acima,
até que a cultura atingisse um valor de densidade ótica de 0,6. Após o crescimento, as
células foram coletadas por centrifugação a 1.000g por 4 minutos. O sobrenadante da

29
cultura foi descartado e as células foram lavadas com 20 ml de TE (10mM de Tris-HCl
pH 8,0, 1mM EDTA) e suspensas em 5 ml de TE acrescidos de 250 µL de uma solução
de acetato de lítio 2,5 M. A suspensão de células foi incubada a 30°C sob agitação de
200 rpm durante no mínimo 1 hora. As células de levedura, tornadas competentes pelo
tratamento descrito acima, foram aliquotadas em frações de 300µl e usadas
imediatamente.
3.4.5-Transformação de leveduras
A transformação de leveduras foi realizada de acordo com a técnica descrita por
Ito e colaboradores (Ito e cols.., 1983) com pequenas modificações. Em um tubo de
microcentrífuga contendo 300 µl de leveduras competentes, adicionaram-se 5 µg do
DNA plasmidial de interesse e 10 µl de DNA de esperma de salmão (10mg/ml),
previamente desnaturado a 95°C (Thermomixer 5436). Após incubação por 30 minutos
a 30°C, foram adicionados 700µL de PEG 4000 (50%), procedeu-se à homogeneização
do conteúdo delicadamente e incubou-se novamente por 1 hora nas mesmas condições
anteriores. Em seguida, as células foram submetidas a choque térmico a 42°C, por 10
minutos, e logo após, foram centrifugadas por 10 minutos a 4.000 rpm (Heraeus
Sepatech, Biofure 13). Após essa nova centrifugação nas mesmas condições anteriores,
o sobrenadante foi descartado, e as células foram ressuspensas no sobrenadante residual
e plaqueadas com pérolas de vidro em meio sólido seletivo (SD-URA). As placas foram
incubadas a 30°C, por 48 a 72 horas. As colônias crescidas nos meios seletivos foram
inoculadas em meio líquido YPD com Sorbitol 1M para posterior confirmação dos
marcadores genéticos com o objetivo de confirmar se a transformação foi efetiva
Como controle negativo do experimento, utilizamos 300 µl de leveduras
competentes e 10 µl de DNA de esperma de salmão (10 mg/ml) previamente
desnaturado a 95°C.

30
3.5-Determinação da atividade enzimática da Invertase e da β - Galactosidase
3.5.1 - Inóculo e crescimento das células
As células de Saccharomyces cerevisiae foram crescidas em 20 ml do meio SD-
URA acrescido de glicose 4% ou rafinose 2%, durante 24 horas, a 30°C, sob agitação a
200 rpm em incubador rotatório New Brunswick Model G25.
3.5.2-Preparo de extratos celulares para a determinação da atividade enzimática
da invertase e β - Galactosidase.
As células de leveduras foram coletadas em diferentes intervalos de tempo e
centrifugadas a 1000 g, a 4°C, por 5 minutos. Em seguida as células foram lavadas uma
vez com tampão de extração (Na2PO4 460mM, NaH2PO4 40mM, KCl 10mM e MgSO4
1mM, pH 7,0) e ressuspensas em 500µl do mesmo tampão. Após este procedimento,
adicionou-se, em cada tubo, 0,5 g de pérolas de vidro (0,5 mm de diâmetro) e as células
foram lisadas por agitação em vortex (três vezes, 60segundos) com repouso em gelo nos
intervalos de agitação. Em seguida, os extratos foram centrifugados (3000g por 5 min) e
o sobrenadante foi coletado e congelado a – 70º C para posterior dosagem no aparelho
bioquímico COBAS-FARA (Roche).
3.5.3-Dosagem da invertase
A dosagem da enzima invertase foi realizada no aparelho de dosagem
bioquímica COBAS-FARA (Roche), tendo sido utilizado como substrato para a reação
uma solução de sacarose 0,3 M em acetato de potássio 100 mM pH 5,1. Após 5 minutos
de incubação, a 30°C, a reação foi interrompida pela adição de NaOH 1 M. Em seguida
um volume igual de HCl 1M foi adicionado para neutralizar o pH da reação. A glicose
liberada foi determinada pela reação clássica de glicose – oxidase / peroxidase, tendo a
ortodianisidina como reativo de cor. A leitura da absorbância foi feita a 560 nm. A
atividade da enzima invertase foi expressa em nmoles de glicose /min /mg proteína.

31
3.5.4 - Dosagem da β-Galactosidase
Os ensaios para determinação da atividade de β-galactosidase foram realizados
com extratos de células de leveduras pré-incubados por 5 minutos a 30º C. Depois disso,
0,44 mM de ONPG foi acrescentado e uma outra incubação de 25 minutos foi realizada.
A reação foi interrompida pela adição de Na2CO3. Procedeu-se então a leitura da
absorbância a 420nm e os resultados foram expressos em nmoles ONP min /mg
proteína. Todas as reações foram realizadas pelo aparelho bioquímico COBAS-FARA
(Roche).
3.6 - Dosagem de proteínas
A dosagem de proteínas foi realizada segundo o método de Lowry (LOWRY e
cols., 1951).
3.7-Extração do RNA Total
3.7.1 - Inóculo e crescimento das células
As leveduras foram inoculadas em 10 ml de meio YPD 4% e incubadas a 30°C,
sob agitação a 200 rpm em incubador rotatório (New Brunswick Model G25) até
atingirem e a densidade ótica entre 0,8 e 1,0 a 600 nm (DU-68 Spectrophotometer-
Beckman). Em seguida, as células foram coletadas por centrifugação (3000 g por 5
min), lavadas em 1M de sorbitol e ressuspensas em meio YPD 4% e em meio
YPRafinose 2%.As células foram então incubadas sob agitação a 200rpm a uma
temperatura de 30º C por 2 horas, para que ocorresse a desrepressão das células
crescidas em rafinose.
3.7.2-Extração do RNA
As células foram coletadas por centrifugação (3000 g por 10 min, 4º C)
descartando-se o sobrenadante e em seguida foram lavadas com 2 ml de sorbitol 1M,
4ºC. Após nova centrifugação (3000 g por 10 min, 4º C), o sobrenadante foi descartado
e o conteúdo restante foi ressuspenso em 2 ml de fenol / água 3,75:1 e 2 ml de TES
(10mM Tris:Cl; 10mM EDTA; 0,5% SDS; pH 7,5) seguido de agitação vigorosa em

32
Vortex. Os tubos foram incubados em banho Maria a 70º C por 1 hora, sendo agitados
vigorosamente em Vortex a intervalos de 5 minutos. Os tubos foram centrifugados
(3000g por 10 min a 4º C) e a fase superior coletada em tubos de microcentrífuga.
Foram adicionados 800 µl de fenol / água 3,75: 1, e após nova agitação os tubos foram
centrifugados (3000g por 10 min a 4º C). O sobrenadante foi coletado e foram
adicionados 600 µl de clorofórmio. Procedeu-se à nova agitação em vortex e em
seguida, os tubos foram centrifugados (3000g por 10 min a 4º C). O sobrenadante foi
coletado e foram adicionados acetato de sódio (50µl, 3M pH 5,3) e etanol gelado
100%(1 ml). Em seguida os tubos foram incubados a –70º C por 10 minutos. As
amostras foram centrifugadas (15000 rpm por 10 min, 4º C). O sobrenadante foi
descartado e o sedimento lavado com 500 µl de etanol 70%. As amostras foram
centrifugadas e o sobrenadante foi descartado. O RNA foi ressuspenso em 50 µl H2O
DEPC (200ml H2O milli Q estéril, 200µl DEPC) e armazenado a -70ºC até a utilização.
3.7.3 - Eletroforese em gel de agarose
3.7.3.1 - Preparação das amostras de RNA para corrida no gel
As amostras foram desnaturadas a 68º C por 5 minutos em aparelho termoblott.
Uma alíquota de 5µl de cada amostra foi misturada com 15µl de tampão de amostra (1x
volume NBC 20x, 3x volume formaldeído 37%, 2x volume corante, 10x volume
formamida) e 1µl de brometo de etídio 10mg/ml. As amostras foram então mantidas em
gelo por 5 minutos antes da aplicação.
3.7.3.2 - Montagem do gel para corrida de RNA Todo o material utilizado (cuba de eletroforese, pente e suporte para o gel) foi
previamente tratado com NaOH 0,1M por duas horas e depois enxaguado por várias
vezes com água milli Q. Para a eletroforese foi preparado um gel de agarose 1% em
NBC 1x e formaldeído 1%. Antes da aplicação, os géis foram percorridos por 30
minutos a 100V em tampão NBC 1X. Depois disto as amostras foram aplicadas e a
eletroforese realizada nas mesmas condições. Em seguida os géis foram visualizados no
transiluminador a 365 nm e fotografados (Sony-KAISER-RSI).

33
3.8 – Ensaios de microarranjos de DNA 3.8.1 – Microarranjos de DNA
Os avanços obtidos nos últimos anos, relativos ao seqüenciamento do genoma de
várias espécies, inclusive do genoma humano realizado pelo consórcio internacional
para seqüenciamento do genoma humano, trouxeram uma vasta gama de informações a
respeito da constituição do genoma destas espécies, mas independente dos esforços
realizados, muitas questões permanecem não elucidadas. Com relação ao ser humano,
não se sabe ainda quantos genes são realmente funcionais, sendo que as estimativas
realizadas através de análises computacionais variam entre 30.000 e 120.000 (Das e
cols., 2001).
Independentemente do número exato de genes funcionais necessários à
sobrevivência de um dado organismo, o desafio consiste em determinar quais genes são
necessários em determinada condição fisiológica ou estágio do desenvolvimento, ou
ainda quais genes são expressos em diferentes tecidos ou em determinadas situações
patológicas. Estas são questões abordadas por uma nova área do conhecimento,
denominada genômica funcional (Asnagli & Murphy, 2001).
Um aspecto crucial da genômica funcional é a descrição da expressão global dos
genes de um determinado organismo, abordando basicamente duas questões:
a) Quando, no organismo em questão, os genes são expressos ou reprimidos?
b)Como a expressão desses genes varia em função de mudanças fisiológicas ou
metabólicas ou ainda patológicas?
Durante muito tempo, essas questões foram abordadas com o emprego de
técnicas com baixa capacidade de resolução, como “Northern blot”, “RNAse
protection”, “Western blot”, permitindo avaliar um pequeno conjunto de genes de cada
vez. A disponibilidade de dados através dos projetos de análise de genomas e o
desenvolvimento da técnica de microarranjos de DNA, possibilitou a análise da
expressão de centenas ou milhares de genes em um único ensaio.
A técnica de microarranjos de DNA consiste na hibridização de múltiplas sondas
fluorescentes com um DNA alvo de determinada célula ou tecido. As células de
interesse são submetidas à extração do mRNA. O mRNA é transcrito em cDNA e
marcado com uma sonda fluorescente. Em seguida, o cDNA marcado é hibridizado com

34
sondas de DNA conhecidas (representando um genoma ou um conjunto de genes
específicos) afixadas a uma matriz sólida (um lamina de vidro recoberta com uma
matriz especifica para adesão das sondas). A hibridização ocorre através do pareamento
complementar de bases entre as sondas e os cDNAs, proporcionalmente à concentração
de moléculas de cDNA presentes. Em seguida, as laminas são submetidas à leitura de
fluorescência em comprimentos de onda específicos para cada corante e os dados são
coletados na forma de intensidade de fluorescência para cada ponto (cada sonda alvo),
sendo posteriormente analisados.
3.8.2 - Marcação do cDNA para Microarranjos Para a realização do experimento de marcação do cDNA obtido a partir dos
RNAs extraídos foi usado o CyScribe Post-Labelling kit. Os seguintes reagentes foram
descongelados em gelo: tampão CyScript, DTT, mix de nucleotídeos, AA-dUTP, H2O,
RNA total e iniciadores. Em seguida foi feita a reação de anelamento do RNA total
com os iniciadores de oligo-dT para a marcação direta. Foi preparado o Mix1 (um para
controle e o outro para o experimental), sendo utilizada uma quantidade de RNA total
equivalente a 20µg de amostra e como iniciador foi utilizado o oligo-dT.
Reagentes do Mix1 Amostra 1 Amostra 2
RNA total X µl (20µg) X µl (20µg)
Oligo-dT ancorado 4 µl(0,5µg/µl) 4 µl
Água (do kit) Y µl Y µl
TOTAL 11 µl 11 µl
As amostras foram misturadas e incubadas a 70º C por 5 minutos no
termociclador. Deixou-se esfriar por 10 minutos a temperatura ambiente e preparou-se o
Mix2.

35
Reagentes do Mix2 x 1
5x tampão CyScript 4 µl
DTT 0,1M 2 µl
Mix de nucleotídeos 1 µl
AA-dUTP 1 µl
Transcriptase reversa CyScript 1 µl
TOTAL 9 µl
Foram transferidos 9µl dos tubos de reação do Mix2 para os quatro tubos
contendo os 11 µl do Mix1. Os tubos de reação foram misturados bem com a pipeta e
quando necessário, submetidos a uma rápida centrifugação para homogeneização da
mistura. A reação foi incubada 10 minutos a temperatura ambiente e, em seguida a 42ºC
por 1,5 h. Nesta fase, os tubos de reação foram colocados em gelo para purificação
posterior.
3.8.3 - Degradação do mRNA (Hidrólise alcalina)
Foram adicionados aos tubos de reação 2µl de 2,5M NaOH. Misturou-se bem e
em seguida centrifugou-se durante 30 segundos. Os tubos foram incubados no
termociclador a 37º C por 15 minutos. Aos tubos foram adicionados 10µl de 2M
HEPES “free acid”. Novamente, misturou-se bem e centrifugou-se por 30 segundos.
Neste momento, pôde-se congelar a -20º C ou prosseguir o experimento fazendo a
purificação da sonda de cDNA.
3.8.4 - Purificação da sonda de cDNA
Foram adicionados 120µl de tampão de ligação a cada reação e todo o volume
foi transferido para um poço da placa de purificação de 96 cavidades (Millipore
Multiscreen filter plate). Centrifugou-se a 1000xg por 1 minuto a 25ºC e descartou-se o
filtrado. Cada poço do filtro foi lavado 4x usando 200µl de Etanol 80%, sendo que a
última lavagem foi realizada centifugando-se a 3500rpm por 5 minutos. O filtrado foi
descartado e a placa de coleta foi trocada por uma nova. As amostras foram eluídas 2x
com 45µl de 10mM Tris pH 8,0 e a cada eluição foram centrifugadas a 3000rpm por 5
minutos. Juntou-se os 85µl de sonda marcada em um tubo âmbar. As amostras foram

36
secas no speed-vac por aproximadamente 40 a 60 minutos, até secar (não em excesso).
Neste ponto pôde-se parar a marcação e as amostras puderam ser congeladas secas por
vários dias.
3.8.5 - Reação de marcação com CyDye
O cDNA amino allyl purificado foi seco e ressuspendido em 15µl em água
DEPC. Também foram ressuspendidas uma alíquota de Cy3 (corante verde) e outra de
Cy5 (corante vermelho) do kit em 15µl de 0,1M Bicarbonato de Sódio pH 9,0.
Adicionou-se imediatamente uma alíquota de CyDye (corante Cy) a uma alíquota de
cDNA. A reação foi misturada por agitação e incubada a temperatura ambiente, no
escuro, por uma hora. Foram adicionados 15µl de Hidroxilamina 4M a cada reação.
Novamente, a mistura foi homogeneizada e incubada à temperatura ambiente, no
escuro, por 15 minutos. Em seguida, realizou-se a purificação como descrito
anteriormente, sem secar e sem incidência direta de luz. O volume eluído foi medido e a
marcação foi dosada em espectrofotômetro. A etapa de marcação dos cDNAs com os
corantes fluorescentes foi realizada sem a incidência de luz direta.
3.8.6 - Quantificação da sonda marcada
Foram utilizadas cubetas de quartzo de 50µl reservadas para cada Corante. As
cubetas foram lavadas cerca de 5 vezes com água milli-Q, tendo sido adicionados cerca
de 70µl de Tris-HCl pH 8,0 para zerar o espectrofotômetro. Apos a retirada da solução
de Tris da cubeta, colocou-se todo o volume de sonda marcada (não diluída) na cubeta.
A leitura foi feita nos seguintes comprimentos de onda: 550 nm para Cy3, 650 nm para
Cy5 e 260 nm para quantificação de cDNA. Foi medido o volume final de cada uma das
reações. O total de corante incorporado foi determinado de acordo com a seguinte
fórmula:
(OD x fator de diluição x volume) / (E x 10-6) = total pmol de Cy
Volume = volume de sonda purificada
OD = densidade ótica, unidade de medida: nanômetros.
E 550 coeficiente de extinção molar Cy3 = 150000M
E 650 coeficiente de extinção molar Cy5 = 250000M

37
A fórmula também pode ser dada resumidamente como:
Para Cy3 → (ABS / 0,15) x vol = pmol
Para Cy5 → (ABS / 0,25) x vol = pmol
ABS = absorbância
Esta etapa, tal como a anterior, foi feita sem a incidência de luz direta.
3.8.7 - Hibridização manual
As amostras foram secas totalmente no aparelho Speed-vac (Savant), à 30º C por
aproximadamente 40 a 60 minutos. Cada uma foi ressuspensa com 6,75 µl água DEPC.
As sondas foram misturadas Cy3 e Cy5 (total 13,5 µl) e então foram acrescentados 13,5
µl de tampão de hibridização e 27 µl de formamida. Incubou-se a 92º C por 2 minutos e
colocou-se no gelo. Os 54µl da amostra foram aplicados em uma das extremidades da
lâmina contendo os microarranjos (oligos de DNA) (EuroGentec), sem encostar a
ponteira na lâmina, fazendo um arraste de solução. Com cuidado para não fazer bolhas,
colocou-se por cima uma lamínula muito limpa com o auxílio de uma pinça. A lamínula
foi colocada dentro de um tubo de centrífuga de 50 ml (preso em suporte de ferro) com
um filme de água, na horizontal. O suporte foi colocado no banho a 42º C por 16 h.
3.8.8 - Lavagens
As duas primeiras soluções foram aquecidas em banho a 55º C. Foram feitas 5
lavagens, como descrito a seguir, sob leve agitação e protegendo as lâminas da luz:
1. 1x SSC, 0,2% SDS → 10min 55º C
2. 0,1x SSC, 0,2% SDS → 10min 55º C
3. 0,1x SSC, 0,2% SDS → 10min 55º C
4. 0,1x SSC → 1min temperature ambiente
5. 4 vezes em água milli-Q
Após mergulhar em água, as lâminas foram secas imediatamente com nitrogênio gasoso
e as imagens e fluorescência foram captadas em seguida.

38
3.8.9 – Leitura das laminas.
Ao final da hibridização, a fluorescência nas lâminas foi captada com o auxilio do
aparelho scanner utilizando os parâmetros de intensidade de fluorescência relacionados
abaixo. Estes parâmetros são padronizados de acordo com a intensidade de
fluorescência emitida por cada corante, sendo menores para Cy5 uma vez que a emissão
de fluorescência deste corante é maior que a emissão de Cy3.
PMT Cy3 – 900
PMT Cy5 – 650
3.8.10 – Análise estatística
Os experimentos de hibridização foram realizados no mínimo 3 vezes e os
resultados foram analisados utilizando-se dos métodos estatísticos descritos por Yang,
Y.H. e colaboradores. (Yang e cols., 2002). Foram considerados como genes
diferencialmente expressos apenas aqueles que apresentaram sinal de fluorescência
igual ou maior que oito vezes o valor do sinal de fundo (background) e que
apresentaram intensidade de fluorescência maior que 1x log 2 em relação ao gene
controle. Uma segunda analise foi realizada utilizando o pacote estatístico R, usando o
critério de Fold Variation de acordo com Gentleman e colaboradores(Gentleman R &
Ihaka R, 1997). Foram selecionados os genes que apresentaram valores superiores a
dois desvios da média.

39
3.9 – Reação em cadeia da polimerase em tempo real.
3.9.1 – Introdução
Os experimentos de microarranjos de DNA são capazes de fornecer um conjunto
muito grande de dados referentes à expressão gênica global de determinado organismo.
Estes dados, apesar de fornecerem informações qualitativas muito importantes, não são
precisos do ponto de vista quantitativo, uma vez que as condições empregadas nos
experimentos não são as ideais para cada gene analisado. Para validar os dados obtidos
nos experimentos de microarranjos, devemos empregar um método capaz de fornecer
dados quantitativos mais precisos. Dentre os vários métodos disponíveis, um dos mais
empregados é a reação em cadeia da polimerase em tempo real (RT-PCR), o qual
permite, através da reação de amplificação dos cDNAs obtidos a partir dos RNAs
mensageiros, quantificar exatamente o número de cópias de cada RNA mensageiro
presente nas amostras, através da utilização de corantes ou sondas fluorescentes. Os
métodos que empregam os corantes fluorescentes se baseiam na propriedade que estes
corantes apresentam de se ligarem à dupla fita de DNA tão logo ela tenha sido
sintetizada, permitindo a quantificação dos mesmos. A metodologia que emprega as
sondas fluorescentes utiliza sondas associadas a um reagente fluorescente e a uma
substância inibidora de fluorescência. Quando a sonda hibridiza com a seqüência alvo e
ocorre a amplificação, ocorre a remoção da substância inibidora de fluorescência e a
emissão de fluorescência proporcional ao número de cópias das seqüências alvo.
3.9.2 – Obtenção dos cDNAs
A partir de 5µg de RNAs extraídos das cepas selvagem W303 e pkc1∆ extraídos
nas mesmas condições descritas para os experimentos de microarranjos de DNA, foram
sintetizados os cDNAs através do mesmo protocolo descrito em material e métodos.
Estes cDNAs foram armazenados a -80º C até a sua utilização.
3.9.3 – Desenho dos iniciadores
Os iniciadores para os experimentos de PCR em tempo real foram desenhados
com o auxílio do programa Primer Express da Applied Biosystems. Foram escolhidos
os pares de iniciadores com a TM mais próxima e com menor conteúdo de GC,

40
principalmente nas extremidades. A tabela 07 apresenta a seqüência dos iniciadores
utilizados.
3.9.4 – Reação da polimerase em cadeia
Para a reação de polimerase em cadeia em tempo real, foram utilizados os
reagentes e procedimentos abaixo descritos:
Adicionamos a um tubo de PCR:
5 µl cDNA 5 µl iniciadores (direto e reverso) (150 µg/ml or 20 pMol cada) 4 µl 5 mM 4dNTP 10 µl 10× tampão de amplificação 70.5 µl H2O.
A mistura foi aquecida por 2 min a 94° C e rapidamente centrifugada.
Foram adicionados 0.5 µl (2.5 U) de Taq DNA polimerase, a solução foi gentilmente
misturada, e rapidamente centrifugada.
Os tubos de reação foram colocados no aparelho e os ciclos da reação estão descritos abaixo:
Numero de Ciclos Tempo por ciclo Temperatura 39 ciclos: 2 min 55° C
2 min 72° C 1 min 94° C
1 ciclo: 2 min 55° C 7 min 72° C
Para cada conjunto de cDNAs utilizado foi feita uma reação com o gene normalizador
(Actina).Para cada produto de amplificação foi feita uma curva de dissociação para
checar a especificidade de cada reação.
3.9.5 – Análise dos resultados
Para a análise dos resultados, foi utilizado o método de quantificação relativa,
utilizando o gene da actina como gene normalizador, conforme descrito por Livak and
Schmittgen ((Livak & Schmittgen, 2001) .

41
RESULTADOS E DISCUSSÃO

42
4 - Resultados e discussão 4.1. - Análise dos experimentos de microarranjos de cDNA.
Para os experimentos de análise da expressão global de genes na cepa pkc1∆,
foram utilizadas lâminas comerciais fornecidas pela Eurogentec, contendo todas as 5803
ORFs conhecidas de Saccharomyces cerevisiae, cepa 288c em duplicata, perfazendo um
total de 11.606 dados por lâmina. A hibridação e a análise de dados foram realizadas
segundo o protocolo seguido pelo CAGE (Cooperação para a análise dos genes e
genomas do departamento de Bioquímica, Instituto de Química da Universidade de São
Paulo) e o pacote estatístico R. Cada experimento foi realizado no mínimo três vezes e
os resultados foram analisados conforme descrito em material e métodos. Foram
hibridizadas sete lâminas, sendo quatro com cDNAs provenientes de mRNAs da cepa
pkc1∆ e três com cDNAs provenientes de mRNAs da cepa W303, incubadas na
presença de glicose ou rafinose como fonte de carbono.
Os dados obtidos foram filtrados e selecionados e de um total de 81.242 pontos
experimentais iniciais, um primeiro tratamento foi realizado visando eliminar os dados
que não apresentavam confiabilidade, excluindo aqueles cujo valor de intensidade de
fluorescência apresentaram discrepâncias estatísticas entre as duplicatas, pontos com
interferência na fluorescência (sujeira ou falha na impressão) e dados cujos valores de
fluorescência foram iguais ao “background” da lâmina. Utilizando o pacote estatístico
R, foi feita uma análise do background das lâminas e os resultados estão apresentados
nas figuras 9 e 10. Em seguida, foi feita uma normalização das intensidades de
fluorescência para correção dos desvios da média dos valores de fluorescência para cada
um dos corantes. O resultados das normalizações para todas as lâminas são apresentados
nas figuras 11 e 12.

43
Figura 9– Análise do background das lâminas obtidas a partir da hibridização com os
cDNAs da cepa W303. Background verde representa o corante Cy3 e background
vermelho o corante Cy5.
Figura 10 – Análise do background das lâminas obtidas a partir da hibridização com os
cDNAs da cepa pkc1∆. Background verde representa o corante Cy3 e background
vermelho o corante Cy5.

44
Antes da normalização Após a normalização
Figura 11 – Normalização da intensidade de fluorescência das lâminas obtidas após
hibridização com os cDNAs obtidos a partir da cepa W303. Estão representadas as três
lâminas hibridizadas com os cDNAs da cepa W303. Em verde está representada a
fluorescência do corante Cy3 e em vermelho a fluorescência do corante Cy5.
Lâmina 01
Lâmina 02
Lâmina 03

45
Antes da normalização Após a normalização
Figura 12 – Normalização da intensidade de fluorescência das lâminas obtidas após
hibridização com os cDNAs obtidos a partir da cepa pkc1∆. Estão representadas as
quatro lâminas hibridizadas com os cDNAs da cepa pkc1∆. Em verde está representada
a fluorescência do corante Cy3 e em vermelho a fluorescência do corante Cy5.
Lâmina 03
Lâmina 04
Lâmina 01
Lâmina 02

46
Após a normalização da intensidade de fluorescência, foi realizada a
normalização dos dados globais por “Lowess fitting” para cada uma das lâminas obtidas
com os cDNAs da cepa W303 e da cepa pkc1∆.Estes resultados são apresentados nas
figuras 13 e 14, que apresenta um Box plot antes e após a normalização dos dados.
Antes da normalização Após a normalização
Figura 13 – Box Plot apresentando a normalização por “Lowess fitting” das lâminas obtidas após hibridização com os cDNAs obtidos a partir da cepa W303, onde M = log2
(Cy5 / Cy3).
Antes da normalização Após a normalização
Figura14 – Box Plot apresentando a normalização por “Lowess fitting” das lâminas obtidas após hibridização com os cDNAs obtidos a partir da cepa pkc1∆, onde M = log2
(Cy5 / Cy3).
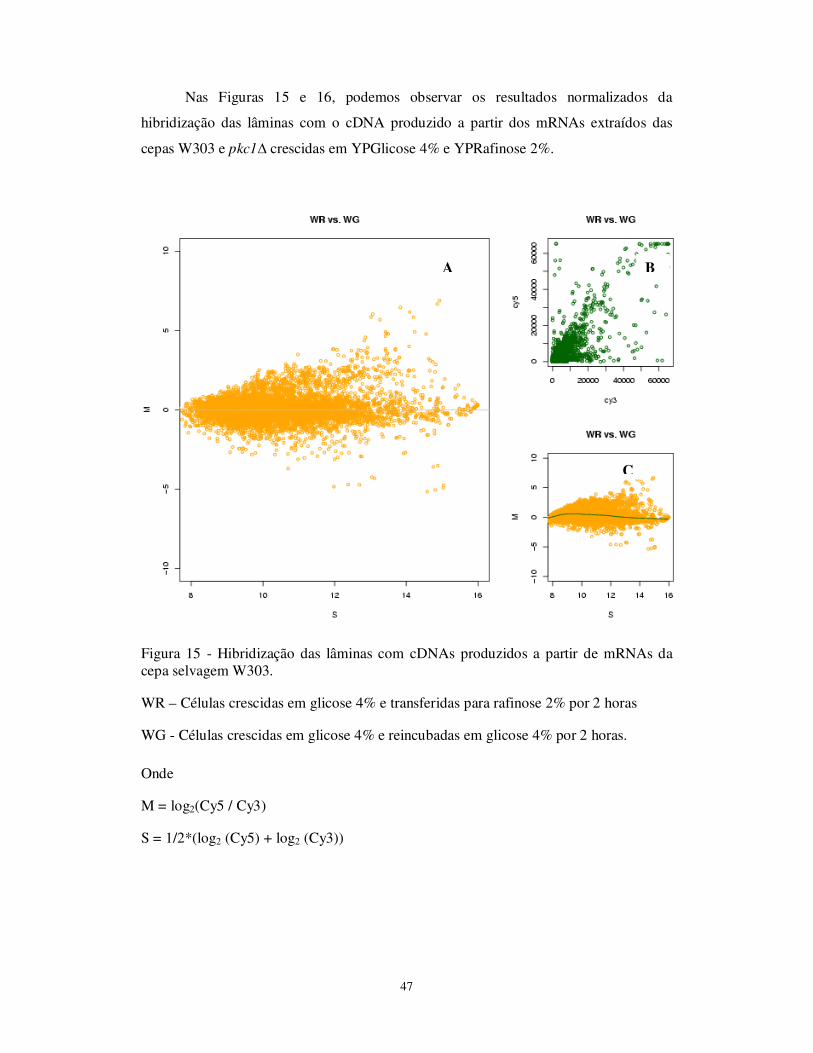
47
Nas Figuras 15 e 16, podemos observar os resultados normalizados da
hibridização das lâminas com o cDNA produzido a partir dos mRNAs extraídos das
cepas W303 e pkc1∆ crescidas em YPGlicose 4% e YPRafinose 2%.
Figura 15 - Hibridização das lâminas com cDNAs produzidos a partir de mRNAs da cepa selvagem W303.
WR – Células crescidas em glicose 4% e transferidas para rafinose 2% por 2 horas
WG - Células crescidas em glicose 4% e reincubadas em glicose 4% por 2 horas.
Onde
M = log2(Cy5 / Cy3)
S = 1/2*(log2 (Cy5) + log2 (Cy3))
B A
C

48
Figura 16 - Hibridização das lâminas com cDNAs produzidos a partir de mRNAs da cepa Pkc1∆.
PR – Células crescidas em glicose 4% e transferidas para rafinose 2% por 2 horas
PG - Células crescidas em glicose 4% e reincubadas em glicose 4% por 2 horas
Onde
M = log2(Cy5 / Cy3)
S = 1/2*(log2 (Cy5) + log2 (Cy3))
As figuras 15 A e 16 A apresentam em um gráfico de M x S, o resultado da
fluorescência emitida a partir da hibridização dos cDNAs obtidos a partir das cepas
incubadas em rafinose sobre a fluorescência emitida a partir da hibridização dos cDNAs
obtidos das cepas incubadas em glicose. O valor de M indica a razão de log2 de rafinose
sobre glicose, onde cada ponto corresponde a um gene. Os valores de M maiores que
zero indicam os genes que foram mais expressos na presença de rafinose e os genes
abaixo do eixo M (menores que zero) indicam os genes que foram mais expressos em
A B
C

49
glicose. Os valores de S indicam a intensidade de fluorescência quando comparados
com o background da lâmina. Foram considerados apenas os genes que apresentaram
intensidade de fluorescência (S) maior que oito vezes o valor do background. As figuras
15B e 16B indicam a intensidade de marcação pelos corantes Cy3 versus Cy5. As
figuras 15C e 16C representam os resultados de fluorescência obtidos em A após a
normalização por lowess fitting dos resultados de hibridização das lâminas.
A primeira observação que podemos fazer é que embora tenhamos empregado a
mesma quantidade de mRNA inicial e a mesma quantidade de cDNA marcado nas
hibridizações. houve uma marcação muito mais acentuada na cepa W303 do que na
cepa pkc1∆.
Podemos ainda observar uma marcação muito mais numerosa de genes quando
as cepas W303 e pkc1∆ foram incubadas em rafinose, em comparação com as mesmas
cepas incubadas em glicose. Isto pode ser explicado pela inibição da expressão de um
grande número de genes nas leveduras quando as mesmas estão em presença de glicose,
através da via principal de repressão por glicose, discutida anteriormente.
Utilizando o banco de dados de ontologia gênica do SGD, classificamos os
genes de acordo com a ontologia gênica e separamos os mesmos em grupos diversos.
Selecionamos então os genes envolvidos no metabolismo de carbono para uma análise
mais específica dos dados.
Os resultados obtidos estão apresentados na figura 17, onde estão representados
todos os 178 genes envolvidos com o metabolismo de carbono na levedura S.
cerevisiae, de acordo com a ontologia gênica do SGD. Cada coluna representa as
médias ( Log2 fold variation)dos experimentos de microarranjos de DNA para as cepas
W303 e pkc1∆ .Cada bloco representa a média dos resultados para um gene isolado,
comparando as condições de desrepressão versus repressão (rafinose/glicose). O grau de
saturação de cor indica a magnitude da expressão (rafinose/glicose) de acordo com a
escala apresentada.

50
Figura 17– Expressão dos genes envolvidos com o metabolismo de carbono nas cepas
W303 e pkc1∆. As colunas representam a expressão dos genes nas cepas W303 e pkc1∆
. Cada bloco representa o Log2 (Fold variation) para cada gene, comparando a expressão
em condição de desrepressão versus a condição de repressão (rafinose/glicose). A
saturação de cor representa a magnitude da razão da expressão de rafinose sobre glicose
de acordo com a escala indicada na figura.

51
Apesar de alguns genes se apresentarem mais expressos na cepa pkc1∆ , como
por exemplo o gene YDR019c, podemos observar um maior número de genes cuja
expressão é maior na cepa W303 quando comparada com a cepa pkc1∆.
A partir da análise da expressão dos genes envolvidos com o metabolismo de
carbono, selecionamos um grupo de genes cuja expressão estava aumentada na cepa
W303 em relação à cepa pkc1∆ em condição de desrepressão metabólica. Na figura 18
estão representados os genes selecionados e os resultados da expressão dos mesmos
obtidos a partir dos experimentos de microarranjos de DNA.

52
-3
-2
-1
0
1
2
3
4
5
6
7
ARG82 COX14 CYB2 DLD1 FUM1 GLC3 GPT2 GRE1 GTT1 GUT1 GUT2 HXT2 HXT3 HXT5 ICL2 JEN1 MDH2 PDC1 PFK2 PLC1 PYC1 RHR2 RSF2 SDH1 SUC2 TKL2
Fo
ld v
ari
ati
on
WR/WG
PR/PG
Figura 18 – Expressão dos genes envolvidos com o metabolismo de carbono selecionados através dos ensaios de microarranjos de DNA para
validação por RT-PCR. Os valores estão expressos em Log2 (Fold variation).

53
Podemos observar nesta figura que o gene SUC2 apresentou uma expressão
maior na cepa pkc1∆ em relação à expressão na cepa selvagem W303. Também não
estão apresentados os valores para os genes HXT1, HXT4 e HXT6 que codificam para
transportadores de glicose. Estes genes não estão representados porque seus dados
foram excluídos durante os procedimentos de análise dos microarranjos de DNA. Para
os genes HXT1 e HXT4 o número de dados não foi suficiente, uma vez que em duas
lâminas da cepa W303 a leitura dos mesmos ficou comprometida. O gene HXT6
apresentou valores muito discrepantes entre as réplicas, não podendo ser analisado.
Apesar disto, estes genes foram incluídos nos experimentos de RT-PCR, uma
vez que dados anteriores obtidos em nosso laboratório indicavam que SUC2, HXT1 e
HXT4 apresentavam uma expressão diminuída na cepa pkc1∆ quando comparada com a
cepa W303. Incluímos o gene HXT6 porque assim teríamos a possibilidade de avaliar o
comportamento de todos os genes para transportadores funcionais de glicose em
Saccharomyces cerevisiae.
4.2. - Reação em cadeia da polimerase em tempo real – (RT-PCR).
Baseados nos resultados obtidos a partir dos experimentos de microarranjos de
DNA, selecionamos 28 genes envolvidos no metabolismo de carbono para validação
através de RT-PCR. Como descrito em material e métodos, utilizamos o gene da actina
como controle interno da análise da expressão dos genes selecionados.
Através do banco de dados disponível no SGD, obtivemos as seqüências das
regiões codificadoras de cada um dos genes e utilizamos estas seqüências para o
desenho dos iniciadores a serem utilizados nos experimentos de RT-PCR. Os
iniciadores foram desenhados com o auxílio do programa Primer Express da Applied
Biosystems. Foram escolhidos os pares de iniciadores com a TM mais próxima e com
menor conteúdo de GC, principalmente nas extremidades. Realizamos então os
experimentos de RT-PCR, a partir dos mRNAs extraídos das cepas W303 e pkc1∆,
comparando a expressão dos mesmos em condição de desrepressão e repressão
metabólica ( rafinose/glicose). Os resultados foram normalizados utilizando o gene da
actina, como descrito em material e métodos. Os resultados estão apresentados na figura
19, e são expressos em quantificação relativa. As figuras estão ordenadas pela
magnitude da expressão, ou seja, primeiro são apresentados os resultados dos genes que
apresentaram uma expressão relativa menor.

54
Gene Iniciador direto Iniciador reverso
Actina GCCGAAAGAATGCAAAAGGA TCTGGAGGAGCAATGATCTTGA
ARG82 CCGGAGCGATGGGAATTACTAA ACCAGTTGTTTTTTTGTCCTTTGG
COX14 GACGTTGGTTGGTGGTACGTT TTGTTCGTACTTCTTACCGTTCATG
DLD1 CCCTGAAGAACACGAAACCTGTAG AAATCGACGGGTGCTTCACCTA
CYB2 GGTGTTCAACGTACCGAAGATG CCACCATGATTGGATAGAACCA
FUM1 TTAGTCACCGCTTTGAACCCTAA TTCAGGAACAACCCATTCATCA
GLC3 GCTGTGGGATTCCCGTCTTT AAAATGCCAGGTTTGCTAATAAGAA
GPT2 TCGACCCGGCTTTGGTAAT CATTCTGGATCGGGACTTTCC
GRE1 TGCTCAAAGTAACCGCTACCAA CGTTTCCTGACCCAGACAGATC
GUT1 CCACGTTGAAGTTCCCCAAA TTGGACGACGTTCACCAGTAAT
GUT2 GCCTCGCTTGTGTACCATGA ACAGCCGTGATGGCTAAAGTG
HXT1 GCTTCCTGGGTTCCAGTATCC TGGTTGGTCATCATGCATTAGG
HXT2 GGCCCCAATTGCCTACGT CAATAGCCATAGCACGATTTTTGA
HXT3 CGAAGCAAGAGCATCTCTTTCC ACCCCATGATGCTGAACCA
HXT4 TGCTGCTTCCATGACAGCTT CTTCTTACCATTTGGCCACAATC
HXT5 CTGGTACCGCCGGTTACAA GCCTTCATGGGAAATGTAACTTG
HXT6 GCTGCATCCATGACTGCTTGT TCTTGACCATTTGGCCATAATCT
ICL2 GCCGGATTTTGGCGATT AATTGCTGCGCCTTGAATATTC
JEN1 CCTTACTGCACATCCACGAGTTT CCATGTCATTTTGCGCAGTT
MDH2 ACCGGAGCGTTACCCACAT GGCGGTAACACCGTTGATG
PDC1 CCAACTTTCGGTGCTAAGGACTAT CAAGTTTTGTGGAGCATCGAAGA
PFK2 GAAAACTTCAACGCTGATGACAA CCAGTGAATGACCTTTGGCATT
PLC1 GCAAACTAATGATATTGGACAGCAA TGGAATCATTTTCGCCTTTGTT
PYC1 TTACTAAATCCAAAGCAGACATGCA TTTCCATTTTCATGGCGCTTAA
RHR2 TGCCTTTGACCACAAAACCTTT GAATAACGTGTTCGGCATCGAA
RSF2 CAATAAACGCCAATCCAGCAAT CATAGCAAACTTCCTCCAAAGCTT
SDH1 TGATGAAGAAACTGGCGAAGAC CAGAAACACAAGCGGCTTCAC
SUC2 CTATGCCTTGCAAACTTTCTTCAA TTGAAGCCCAGGCAATACCT
TKL2 ATGGCACAGTTCTCCGACATT TCCACCTGGTCAACGGAAAG
Tabela 07 – Iniciadores utilizados nos experimentos de RT-PCR para validação dos
experimentos de microarranjos de DNA.

55
A
0
2
4
6
8
10
12
14
16
18
20
COX14 DLD1 FUM1 HXT1 HXT2 HXT3 HXT4 MDH2 PDC1 PFK2 PLC1 PYC1 RHR2 SDH1
Qu
an
tifi
ca
çã
o R
ela
tiv
a
WR/WG
PR/PG
B
0
10
20
30
40
50
ARG82 CYB GLC3 GPT2 RSF2 SUC2
Qu
an
tifi
ca
çã
o R
ela
tiv
a
WR/WG
PR/PG
C
0
20
40
60
80
100
GUT1 GUT2 HXT5 HXT6 ICL2
Qu
an
tifi
caç
ão
Re
lati
va
WR/WG
PR/PG
D
0
50
100
150
200
250
300
350
400
450
500
GRE1 JEN1 TKL2
Qu
anti
fica
çã
o R
ela
tiva
WR/WG
PR/PG
Figura 19 – Resultado da análise por RT-PCR da expressão de genes envolvidos no
metabolismo de carbono nas cepas W303 e pkc1∆. Os valores estão expressos em
quantificação relativa, usando o gene da actina como gene normalizador e representam a
media de três experimentos.

56
Analisando os resultados da figura 19, podemos observar, para nossa surpresa ,
que alguns genes apresentaram uma expressão maior na cepa pkc1∆ do que na cepa
W303 em condição de desrepressão metabólica. São eles o gene HXT4, que não havia
sido analisado nos experimentos de microarranjos de DNA e os genes MDH2 uma
enzima com atividade málica, RHR2 uma enzima com atividade de glicerol 1 fosfatase e
SDH1 uma enzima com atividade de succinato desidrogenase. Aparentemente estes
resultados contradizem aqueles encontrados nos experimentos de microarranjos.
Entretanto, é conveniente salientar que a validação dos experimentos de microarranjos
de DNA através do RT-PCR visa exatamente eliminar os desvios que podem acontecer
nos experimentos de microarranjos. Devemos lembrar que a hibridização dos cDNAs
nos experimentos de microarranjos não é otimizada para um determinado gene, uma vez
que a temperatura de hibridização não leva em consideração um único gene. Assim,
podemos estar trabalhando numa faixa de temperatura de hibridização que é boa para a
maior parte dos genes mas está longe da ideal para outros em particular. Nos
experimentos de RT-PCR, os iniciadores são desenhados e selecionados para uma faixa
de temperatura ótima para todos os genes, com desvios muito pequenos da temperatura
ideal. Nestas condições, podemos analisar com um grau de certeza maior a expressão
dos genes selecionados.
Voltando à analise dos resultados do RT-PCR, podemos observar que sete genes
apresentam uma expressão maior na cepa W303 quando comparada com a cepa pkc1∆
em condição de desrepressão metabólica. São eles ARG82 que codifica para uma
inositol muliquinase envolvida no metabolismo de inositois polifosfatos, HXT5 que
codifica para um transportador de glicose de afinidade moderada, ICL2 que codifica
para uma proteína envolvida no metabolismo de ácido propiônico, JEN1 que codifica
para um transportador de lactato, PLC1 que codifica para uma fosfolipase que hidroliza
Fosfatidilinositol -4,5-bifosfato gerando inositol 1,4,5 trifosfato e diacilglicerol, RSF2
que codifica para uma proteína do tipo dedo de zinco e está relacionada ao controle de
genes envolvidos no metabolismo respiratório e no crescimento em glicerol e TKL2 que
codifica para uma proteína envolvida no metabolismo de pentoses fosfato. Todos os
demais genes não apresentam uma expressão diferente quando comparadas as duas
cepas analisadas.
Para excluirmos a possibilidade da existência de artefatos nos experimentos de
RT-PCR como a formação de dímeros de iniciadores durante a reação, realizamos as
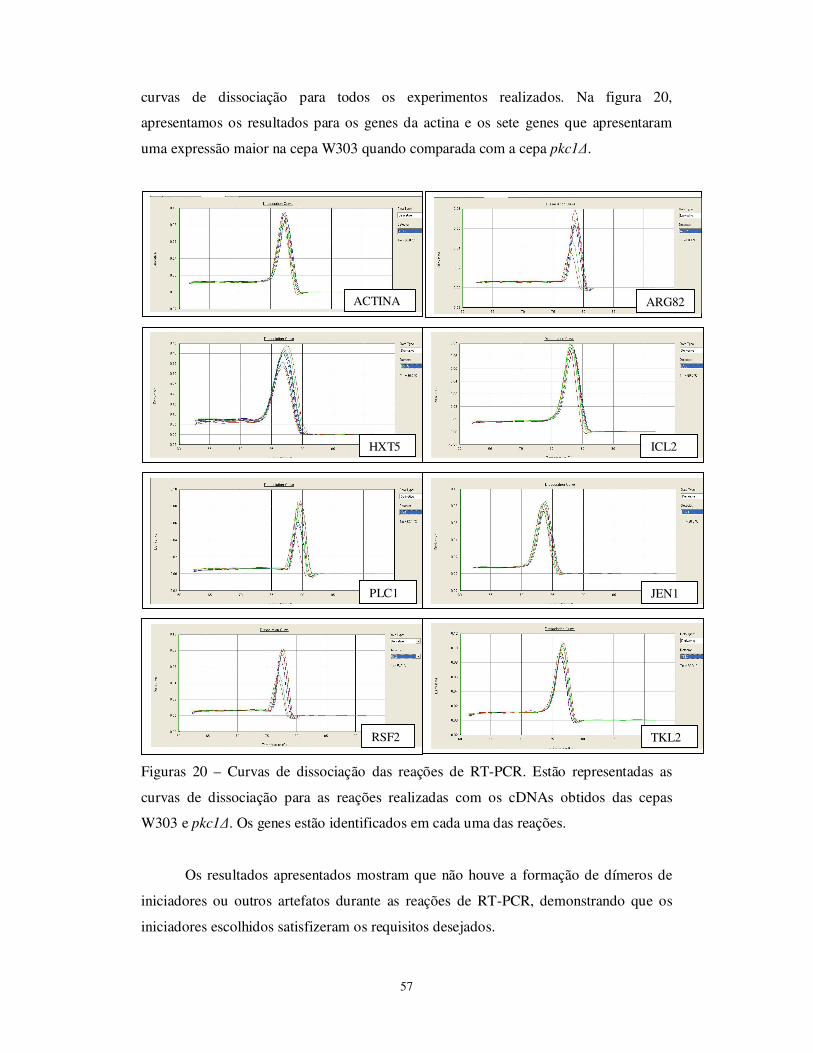
57
curvas de dissociação para todos os experimentos realizados. Na figura 20,
apresentamos os resultados para os genes da actina e os sete genes que apresentaram
uma expressão maior na cepa W303 quando comparada com a cepa pkc1∆.
Figuras 20 – Curvas de dissociação das reações de RT-PCR. Estão representadas as
curvas de dissociação para as reações realizadas com os cDNAs obtidos das cepas
W303 e pkc1∆. Os genes estão identificados em cada uma das reações.
Os resultados apresentados mostram que não houve a formação de dímeros de
iniciadores ou outros artefatos durante as reações de RT-PCR, demonstrando que os
iniciadores escolhidos satisfizeram os requisitos desejados.
ACTINA ARG82
HXT5 ICL2
PLC1 JEN1
RSF2 TKL2

58
4.3. – Análise “in silico” dos genes diferencialmente expressos.
Com base nestes resultados foi realizada uma analise “in silico “ tentando
encontrar algum fator que pudesse conectar os genes que apresentaram uma menor
expressão na cepa pkc1∆. Para isto, foi realizada uma busca por fatores de transcrição
relacionados com o controle da expressão dos genes acima relacionados, através de
pesquisa no site Yeastract (Teixeira e cols., 2006). Quatro dos sete genes (HXT5, ICL2,
JEN1 e RSF2) apresentaram um sitio de ligação para o fator de transcrição Sok2p, um
repressor transcricional dependente de glicose cuja atividade supressora depende da
fosforilação no resíduo T 598 por PKA (Shenhar & Kassir, 2001).Três genes (HXT5,
ICL2 e TKL2) possuem sítios de ligação para o fator de transcrição Crz1p, um ativador
transcricional dependente de calcineurina envolvido na ativação de FKS2, um
componente do complexo glucano sintase regulado por Pkc1p (Lu e cols., 2005a;
Stathopoulos & Cyert, 1997) (Boustany & Cyert, 2002) e no controle da expressao de
diversos genes em S.cerevisiae (Matheos e cols., 1997).
Buscamos então por possíveis sítios de fosforilação em Sok2p e Crz1p por
Pkc1p utilizando as ferramentas disponíveis no site NETPHOSK (Blom e cols., 2004)e
encontramos dezoito sítios de fosforilação por Pkc1p com “score” acima de 0,70 com o
maior “score” (0,91) na posição 132, indicando uma grande possibilidade de que Sok2p
possa ser fosforilado por Pkc1p. O resíduo T598 não aparece como possível sítio de
fosforilação por Pkc1p, indicando que caso Pkc1p atue sobre Sok2p isto aconteça em
outros resíduos. (Tabela 08)

59
Metodo: NetPhosK com filtro ESS – Corte - 0,70
Nome: Sok2p
Pos: AA: Kinase: Score: Positivo: Negativo:
Pos AA Kinase Score Positivo Negativo
14 S PKC 0.79 2 0
132 T PKC 0.906 2 0
133 T PKC 0.905 2 0
186 S PKC 0.818 2 0
220 T PKC 0.738 2 0
291 S PKC 0.772 2 0
377 T PKC 0.728 2 0
384 T PKC 0.834 2 0
385 T PKC 0.718 2 0
395 T PKC 0.724 2 0
515 S PKC 0.796 2 0
532 S PKC 0.705 2 0
533 T PKC 0.809 2 0
647 T PKC 0.726 2 0
695 S PKC 0.711 2 0
731 T PKC 0.7762 2 0
738 T PKC 0.708 2 0
780 T PKC 0.794 2 0
Tabela 08 – Predição de sítios de fosforilação quinase específicos na proteína Sok2p utilizando as ferramentas disponíveis no site of NetphosK. A predição dos sítios de fosforilação foi realizada utilizando uma faixa de corte de 0,70 e a ferramenta ESS (Evolutionary Stable Sites). Somente os sítios de fosforilação por Pkc1p estão representados.
Estes dados, juntamente com os resultados por nos obtidos, parecem apresentar
uma controvérsia, uma vez que Sok2p funciona como um repressor transcricional
dependente de glicose e que os genes controlados por Sok2p estão menos expressos em
Pkc1p durante o processo de desrepressão metabólica. Uma hipótese para explicar isto,
seria a de que Pkc1p possa regular negativamente a atividade de Sok2p através da
fosforilação em outros resíduos. Na ausência de Pkc1p, Sok2p poderia estar exercendo
seu efeito repressor sobre os demais genes por ele regulados, caracterizando o fenótipo
reprimido da cepa pkc1∆. Para confirmar esta hipótese, teríamos que analisar se Pkc1p
efetivamente fosforila Sok2p.É importante dizer que os níveis de expressão do gene
Sok2p não estão afetados na cepa de pkc1∆ quando comparada com a cepa selvagem
W303, durante o processo de desrepressão metabólica. Outra possibilidade, uma vez
que recentemente nosso laboratório adquiriu a coleção com todos os mutantes contendo
a deleção de todos os genes da cepa Saccharomyces cerevisiae da EuroScarf, seria
analisar se a dupla deleção de PKC1 e SOK2 reverteria o fenótipo de pkc1∆ para os
genes regulados por Sok2p.

60
A busca por sítios de fosforilação em Crz1p resultou em treze possíveis sítios de
fosforilação por Pkc1p com “score” acima de 0,70, com o maior “score” (0,92) na
posição 546, indicando a possibilidade de que Pkc1p fosforile Crz1p (tabela 09).
Metodo: NetPhosK com filtro ESS – Corte - 0,70
Nome: Crz1p
Pos: AA: Kinase: Score: Positivo: Negativo:
Pos AA Kinase Score Positivo Negativo
69 S PKC 0.84 2 0
143 T PKC 0.86 2 0
183 T PKC 0.89 2 0
318 S PKC 0.70 2 0
405 S PKC 0.85 2 0
418 T PKC 0.84 2 0
422 S PKC 0.83 2 0
490 T PKC 0.88 2 0
491 S PKC 0.72 2 0
544 T PKC 0.87 2 0
546 S PKC 0.91 2 0
620 T PKC 0.88 2 0
657 S PKC 0.74 2 0
Tabela 09 – Predição de sítios de fosforilação quinase específicos na proteína Crz1p utilizando as ferramentas disponíveis no site NetphosK. A predição dos sítios de fosforilação foi realizada utilizando uma faixa de corte de 0,70 e a ferramenta ESS (Evolutionary Stable Sites). Somente os sítios de fosforilação por Pkc1p estão representados.
Crz1p é um fator de transcrição envolvido na ativação da transcrição de FKS2
dependente de calcineurina em resposta ao Cálcio. Zhao e colaboradores (Zhao e cols.,
1998)demonstraram que a via de calcineurina e a via de integridade celular funcionam
em paralelo durante o crescimento em condições de altas temperaturas moderadas e que
a indução de FKS2 durante o crescimento celular em meio pobre em fontes de carbono
era dependente de SNF1 e MIG1. Resultados anteriores em nosso laboratório
demonstraram que PKC1 está envolvido na regulação da localização celular de Mig1p.
Zakrzewska e colaboradores (Zakrzewska e cols., 2005)demonstraram que Crz1p está envolvido
na resposta celular a quitosano, um composto formado por cadeias lineares de resíduos de β-1,4
glicoseaminas que age perturbando a membrana celular, mostrando que células
deletadas do gene CRZ1 são mais sensíveis ao quitosano. Garcia e colaboradores
mostraram que o uso de agentes que causam perturbações na parede celular como
vermelho congo e zimolase induziu um aumento da expressão de diversos genes
relacionados à via de integridade da parede celular e que Crz1p estava relacionado à

61
regulação da expressão de alguns desses genes.(Garcia e cols., 2004). Lagorce e
colaboradores (Lagorce e cols., 2003), usando mutantes da via de síntese da parede
celular, demonstraram que a via de integridade celular e a via de Ca++/Calcineurina
atuavam em conjunto regulando a expressão de FKS2 através da ação de Crz1p.
Analisando estes dados em conjunto, podemos estabelecer uma conexão entre
Pkc1p e Crz1p, atuando conjuntamente na manutenção da integridade celular. A
ausência da Pkc1p e conseqüentemente mudanças estruturais ao nível da parede celular,
uma vez que Pkc1p está diretamente envolvida no metabolismo da parede celular
através da ativação da via das MAP quinases e do complexo glucano sintase, pode se
refletir na composição da membrana celular numa tentativa de se manter a integridade
celular. Uma vez que um dos alvos da via de integridade celular é o gene FKS2, que
também é controlado por CRZ1, é possível pensar na hipótese de que outros genes
compartilhem um mecanismo de controle exercido por Pkc1p e Crz1p. Estudos mais
detalhados, com a construção de duplos mutantes pkc1∆:crz1∆ podem permitir uma
visão mais detalhada das possíveis interações de Pkc1p com Crz1p.
Voltando aos genes cuja expressão está diminuída na cepa pkc1∆ , é interessante
notar que dois deles codificam para transportadores presentes na membrana celular (
HXT5 e JEN1). Resultados anteriores obtidos em nosso laboratório mostram que outros
transportadores de glicose também estão menos expressos na cepa pkc1∆ quando
comparada com a cepa selvagem W303. Uma menor expressão destes transportadores
poderia explicar o crescimento mais lento de pkc1∆, uma vez que a captação de
nutrientes, essencial para o crescimento celular, é diretamente dependente da expressão
dos mesmos. Como discutido anteriormente, perturbações ao nível da parede celular
podem implicar em alterações ao nível da membrana celular, o que pode afetar a
organização e a funcionalidade das proteínas associadas à mesma.
Dois outros genes (ARG82 e PLC1) estão envolvidos no metabolismo de
inositois e na sinalização celular dependente de fosfolipídios. Embora em mamíferos
exista uma correlação direta entre PKC e PLC1 através da ação de Diacilglicerol e
Ca++, produtos finais da ação de Plc1p que atuam ativando a Pkc1p (O'Brian & Ward,
1989; Bell e cols., 1986), esta conexão não foi observada em leveduras. A menor
expressão de ARG82 e PLC1 pode estar relacionada com o metabolismo dos lipídeos
da membrana celular, uma vez que a cepa pkc1∆ apresenta problemas relacionados à
manutenção da integridade da parede celular. Outro fator a ser levado em conta é o fato
que produtos do metabolismo de fosfolipídeos como IP3, IP4, IP5 e IP6 e outros, estão

62
relacionados com vias de controle da expressão gênica e nos mecanismos envolvidos no
transporte de mRNAs a partir do núcleo (Odom e cols., 2000; York e cols., 1999; cazar-
Roman e cols., 2006; Resnick e cols., 2005; Auesukaree e cols., 2005; Chi e cols.,
2004). Uma menor expressão de ARG82 e PLC1 poderia afetar o metabolismo destes
fosfolipídeos, afetando a expressão de diversos genes ou mesmo afetando de uma
maneira geral o transporte de mRNAs a partir do núcleo.
O gene RSF2 é um gene cuja expressão é fundamental para o metabolismo
aeróbico e para o metabolismo do glicerol. Lu e colaboradores (Lu e cols.,
2005b)demonstraram que RSF2 controla uma série de genes envolvidos com a
utilização de glicerol como fonte de carbono e genes mitocondriais envolvidos na
respiração celular.Uma expressão diminuída de RSF2 pode explicar a ausência da
segunda fase de crescimento e a incapacidade que a cepa pkc1∆ tem de crescer em
glicerol como única fonte de carbono, conforme descrito anteriormente em nosso
laboratório.

63
Uma busca por fatores de transcrição associados aos genes cuja expressão estava
aumentada em pkc1∆ através do programa Yeastract, nos permitiu identificar o fator de
transcrição Gcr1p, que possue sítios de ligação em três dos quatro genes pesquisados
(HXT4, RHR2 e SDH1).
Gcr1p é um fator transcricional ativador de genes envolvidos na glicólise,
atuando como regulador positivo da enolase e de genes da família das gliceraldeido -3 -
fosfato desidrogenases.
Uma busca por sítios de fosforilação por Pkc1p em Gcr1p resultou em nove
possíveis sítios de fosforilação com score superior a 0,70, indicando a possibilidade de
que Gcr1p seja fosforilado por Pkc1p ( Tabela 10).
Tabela 10 – Predição de sítios de fosforilação quinase específicos na proteína Gcr1p1p utilizando as ferramentas disponíveis no site NetphosK. A predição dos sítios de fosforilação foi realizada utilizando uma faixa de corte de 0,70 e a ferramenta ESS (Evolutionary Stable Sites). Somente os sítios de fosforilação por Pkc1p estão representados.
Zeng e colaboradores (Zeng e cols., 1997) demonstraram que Gcr1p é altamente
fosforilado em resíduos de serina e que esta fosforilação é dependente do fator Gcr2p.
Entretanto, não foram identificadas qual ou quais seriam as quinases responsáveis pela
fosforilação de Gcr1p e muito menos se a fosforilação do mesmo aumentava ou
diminuía sua atividade como ativador transcricional.
Para analisar se Pkc1p efetivamente fosforila Gcr1p, serão necessários ensaios
de fosforilação “in vitro” e “in vivo” para confirmação. Também seria interessante
analisar o fenótipo do duplo mutante pkc1∆:gcr1∆ e a expressão dos genes regulados
por Gcr1p durante o processo de desrepressão metabólica..
Metodo: NetPhosK com filtro ESS – Corte - 0,70
Nome: Gcr1p
Pos: AA: Kinase: Score: Positivo: Negativo:
20 S PKC 0.85 2 0
48 T PKC 0.8 2 0
132 S PKC 0.81 2 0
225 S PKC 0.78 2 0
479 S PKC 0.81 2 0
605 T PKC 0.77 2 0
607 T PKC 0.77 2 0
612 S PKC 0.8 2 0
687 T PKC 0.75 2 0

64
4.4. - Análise da atividade de ß-galactosidase das construções CYC1-ß-Gal, HXT1-
ß-Gal e SUC2- ß-Gal.
Resultados obtidos em nosso laboratório por Brandão e cols.. (2002)
demonstraram que uma cepa de S. cerevisiae com deleção do gene PKC1 apresentava
uma série de problemas na desrepressão metabólica. As células de pkc1∆ não eram
capazes de crescer em glicerol como única fonte de carbono, apresentavam uma
atividade invertásica bastante diminuída mesmo em condições de desrepressão e baixa
expressão do gene SUC2 que codifica para a invertase; além disso, tinham um
crescimento menor na segunda fase de crescimento e apresentava problemas de
expressão dos genes que codificam para os transportadores de glicose Hxt1, Hxt2 e
Hxt4.
Através da transformação das cepas pkc1∆ e W303 com os plasmídeos contendo
o gene da ß-galactosidase sob controle dos promotores dos genes HXT1, SUC2 e CYC1
foram obtidos diversos transformantes selecionados em meio SD-Uracila. Os
transformantes foram transferidos para meio líquido e testados de acordo com a
metodologia descrita.
Na Figura 21, apresentamos os resultados da incubação de células de pkc1∆ e
W303 transformadas com a construção CYC1-ß-galactosidase. O gene CYC1 codifica a
proteína citocromo C, isoforma 1, uma proteína envolvida no transporte de elétrons
durante o processo de respiração celular. Como podemos observar ambas as células
apresentaram atividade de ß-galactosidase quando transferidas para o meio contendo
rafinose como fonte de carbono. A cepa pkc1∆ apresentou uma atividade de ß-
galactosidase superior se comparada à cepa selvagem W303. Os dados representam a
média de três experimentos independentes, realizados com células de leveduras
transformadas isoladas de colônias diferentes.

65
CYC1-LacZ
0
2500
5000
7500
10000
12500
0 10 20 30
Tempo ( horas)
Ativ
idad
e β
-Gal
acto
sida
se n
mol
es
ON
P/m
in/m
g pr
otei
na
W303
pkc1
*
**
*
Figura 21 – Atividade de ß-galactosidase (em nmoles de ONP/min/mg de proteína) das
cepas W303 e pkc1∆ transformadas com a construção CYC1–ß-Galactosidase.(*)
p<0,05.
Estes resultados indicam que a indução da transcrição do gene da ß-
galactosidase associado ao promotor do gene CYC1 foi superior na cepa pkc1∆ quando
comparamos com a cepa selvagem W303. Isto indica que, apesar de não apresentar uma
segunda fase metabólica respiratória, este problema não se encontra associado à
ativação da transcrição do gene CYC1, devendo haver outros mecanismos envolvidos na
deficiência respiratória da cepa pkc1∆.
Na Figura 22 apresentamos os resultados referentes às células do mutante pkc1∆
e da cepa selvagem correspondente W303 transformadas com a construção HXT1-ß-
galactosidase transferidas de um meio SD-Uracila e rafinose2% para um meio SD-
Uracila e glicose 4% para desrepressão do gene HXT1. O gene HXT1 codifica para um
transportador de glicose de baixa afinidade.Como podemos observar, tanto as células
pkc1∆ quanto as células W303 apresentaram atividade da ß-galactosidase, compatível
com a indução do promotor do gene HXT1. Entretanto, os níveis de indução foram
significativamente menores na cepa pkc1∆ do que na cepa W303, indicando que a cepa
pkc1∆ apresenta problemas de desrepressão do gene HXT1.

66
HXT1
0
3000
6000
9000
12000
15000
0 10 20 30
Tempo ( Horas)
Ati
vida
de
-Gal
acto
sida
se n
mol
es
ON
P/m
in/m
g pr
otei
na
W303
pkc1
*
*
*
*
Figura 22 – Atividade de ß -galactosidase (em nmoles de ONP/min/mg de proteína) das
cepas W303 e pkc1∆ transformadas com a construção HXT1 – ß-Galactosidase. .(*)
p<0,05.
Estes dados corroboram aqueles apresentados por Brandão (2002), que
demonstraram que a cepa pkc1∆ apresentava problemas relacionados à expressão de
alguns transportadores de glicose, inclusive HXT1. O defeito apresentado pela cepa
pkc1∆ na expressão de transportadores de baixa e alta afinidade por glicose, pode ser
um dos fatores cruciais no que diz respeito ao crescimento mais lento que esta cepa
apresenta quando comparada à cepa selvagem, uma vez que a disponibilidade
intracelular de glicose pode estar afetada. Para confirmar esta hipótese, seria necessário
realizar experimentos de captação de glicose, comparando-se as cepas pkc1∆ e W303.
Os dados obtidos para este experimento não podem ser comparados com os
dados obtidos com os experimentos de microarranjos de DNA e RT-PCR, uma vez que
para este ensaio, as células foram crescidas em condição de desrepressão metabólica
(rafinose) e depois foram reincubadas em condições de repressão metabólica (glicose),
uma vez que a transcrição do gene HXT1 é ativada por altas concentrações de glicose. O
fato de não encontrarmos diferença na expressão do gene HXT1 nos experimentos de
microarranjos de DNA e RT-PCR, pode ser explicado pelo fato de termos incubado as
células em glicose e depois termos reincubado as mesmas em rafinose. O tempo em que
as células foram mantidas em condição de desrepressão metabólica (2 horas) pode não
ter sido suficiente para que os níveis de mRNA do gene HXT1 fossem alterados
significativamente nas duas cepas estudadas.

67
Na Figura 23, podemos ver o perfil de indução da transcrição da ß-galactosidase
associada ao promotor do gene SUC2 que codifica para a enzima invertase, após a
transferência das células de meio SD-Ura mais glicose 4% para um meio SD-Ura mais
rafinose 2%. Como podemos observar, ocorre a indução da transcrição do gene da ß-
galactosidase associado ao promotor do gene SUC2 tanto na cepa W303 quanto na cepa
Pkc ∆, não havendo diferença significativa entre as duas cepas.
SUC2-LacZ
0
250
500
750
1000
1250
1500
0 5 10 15 20 25
Tempo ( Horas)
Ati
vida
de
-Gal
acto
sida
se n
mol
es
ON
P/m
in/m
g pr
otei
na
W303
pkc1
*
Figura 23 – Atividade de ß -galactosidase (em nmoles de ONP/min/mg de proteína) das
cepas W303 e pkc1∆ transformadas com a construção SUC2 – ß-Galactosidase. .(*)
p<0,05.
Dados apresentados por Brandão (2002) indicavam que a atividade de invertase
estava bastante diminuída na cepa pkc1∆ quando comparada com a cepa selvagem
W303. Decidimos então analisar a atividade de invertase nas cepas transformadas com a
construção SUC2–ß-galactosidase. Os resultados são apresentados na Figura 24.

68
Invertase - SUC2
0
0,5
1
1,5
2
2,5
0 5 10 15 20 25Tempo (Horas)
nmol
Gli
cose
x m
in-1
x m
g pr
otei
na-1
W303
pkc1
*
*
**
Figura 24 – Atividade de invertase (em nmoles de glicose/min/mg de proteína) das
cepas W303 e pkc1∆ transformadas com a construção SUC2 – ß-Galactosidase. .(*)
p<0,05.
Como podemos observar, a atividade de invertase da cepa selvagem W303 é
muito maior que aquela apresentada pela cepa pkc1∆ em todo o intervalo de tempo
analisado. Os tempos correspondem ao tempo de transferência das células de um meio
YPglicose 4% para um meio YPrafinose 2%. Os dados encontrados corroboram aqueles
descritos por Brandão, exceto pelos resultados da atividade de ß-galactosidase das
construções de SUC2 – ß-galactosidase. Estes resultados, associados aos encontrados
nos experimentos de microarranjos de DNA e RT-PCR, indicam que a indução da
transcrição do gene SUC2 não apresenta problemas na cepa pkc1∆ quando comparada
com a cepa selvagem. Estes resultados sugerem que os baixos níveis de atividade
invertásica encontrados na cepa pkc1∆ não são devidos a problemas relacionados com a
transcrição do gene SUC2. Nossos dados apontam que a baixa atividade de invertase na
cepa pkc1∆ parece estar relacionada a problemas pós-transcricionais, podendo estar
relacionada com a estabilidade da proteína invertase por exemplo. Estudos mais
específicos devem ser realizados para avaliar esta hipótese.

69
CONCLUSÕES

70
5 - Conclusões
5.1 - Com base nos resultados das análises do padrão de expressão global dos
genes através de microarranjos de DNA e RT-PCR, podemos confirmar a participação
de Pkc1p nos mecanismos envolvidos na regulação da expressão de diversos genes
durante o processo de desrepressão metabólica.
5.1.1 - A cepa pkc1∆ de S. cerevisiae apresenta níveis diminuídos de expressão
dos genes ARG82, HXT5, ICL2, JEN1, PLC1, RSF2 e TKL2 quando comparada com a
cepa selvagem W303 durante o processo de desrepressão metabólica.
5.1.2 – Os genes HXT4, MDH2, RHR2 e SDH1 apresentam um nível de
expressão aumentado na cepa pkc1∆ quando comparada com a cepa selvagem W303
durante o processo de desrepressão metabólica.
5.1.3 – Os genes HXT1 e SUC2 não apresentaram expressão alterada nos
experimentos de RT-PCR realizados quando comparadas as duas cepas analisadas.
5.2 – A análise “in silico” dos sete genes cuja expressão está diminuída na cepa
pkc1∆ quando comparada com a cepa selvagem W303 encontraram dois fatores
transcricionais, Sok2p e Crz1p, possivelmente envolvidos na regulação desses genes.
5.2.1 – O fator Sok2p, um repressor transcricional dependente de glicose, possue
sítios de ligação em quatro genes HXT5, ICL2, JEN1 e RSF2.
5.2.2 – O fator Crz1p, um ativador transcricional dependente de calcineurina,
possue sítios de ligação em três genes HXT5,ICL2 eTKL2
5.2.3 – A análise “in silico” dos quatro genes cuja expressão está aumentada na
cepa pkc1∆ quando comparada com a cepa selvagem W303 encontrou um fator de
transcrição, Gcr1p, envolvido na regulação de três dos quatro genes analisados (HXT4,
RHR2 e SDH1).
.

71
5.2.4 – Os fatores de transcrição Sok2p, Crz1p e Gcr1p possuem prováveis sítios
de fosforilação por Pkc1p com scores superiores a 0,70. Sok2p apresenta dezoito sítios
de fosforilação por Pkc1p, Crz1p apresenta treze sítios de fosforilação e Gcr1p possue
nove sítios de fosforilação por Pkc1p.
5.3 – Os ensaios com as construções com os plasmídeos contendo o gene da ß-
galactosidase sob controle dos promotores dos genes CYC1, HXT1 e SUC2 mostraram
que:
5.3.1 - a indução da transcrição do gene da ß-galactosidase associado ao
promotor do gene CYC 1 foi superior na cepa pkc1∆ quando comparamos com a cepa
selvagem W303.
5.3.2 - tanto as células pkc1∆ quanto as células W303 apresentaram atividade da
ß-galactosidase, compatível com a indução do promotor do gene HXT1. Entretanto, os
níveis de indução foram significativamente menores na cepa pkc1∆ do que na cepa
W303, indicando que a cepa pkc1∆ apresenta diminuição da resposta de desrepressão
do gene HXT1.
5.3.3 - ocorre a indução da transcrição do gene da ß-galactosidase associado ao
promotor do gene SUC2 tanto na cepa W303 quanto na cepa pkc1∆, não havendo
diferença significativa entre as duas cepas. Entretanto, a atividade de invertase é
significativamente menor na cepa pkc1∆ quando comparada com a cepa selvagem
W303.

72
PERSPECTIVAS

73
6 - Perspectivas
6.1 – Analisar através de ensaios de fosforilação “in vitro” e “in vivo” se Pkc1p
fosforila os fatores de transcrição Sok2p, Crz1p e Gcr1p.
6.2 – Analisar a localização celular dos fatores de transcrição Sok2p, Crz1p e
Gcr1p nas cepas selvagem W303 e pkc1∆ durante o processo de desrepressão
metabólica.
6.3 – Analisar a expressão de outros genes controlados por Sok2p, Crz1p e
Gcr1p durante o processo de desrepressão metabólica nas cepas selvagem W303 e
pkc1∆.
6.4 – Analisar o fenótipo de cepas obtidas através da construção de duplos
mutantes pkc1∆:sok2∆; pkc1∆:crz1∆; pkc1∆:gcr1∆ durante o processo de desrepressão
metabólica e a expressão dos genes analisados nos ensaios de RT-PCR que
apresentaram uma expressão alterada nas cepas selvagem W303 e pkc1∆ relacionados a
estes fatores.
6.5 – Analisar os mecanismos pós-transcricionais envolvidos nos processos de
síntese e estabilidade de proteínas, com o objetivo de explicar porque os níveis de
atividade de invertase estão diminuídos na cepa pkc1∆ quando comparada com a cepa
selvagem W303.

74
REFERÊNCIAS BIBLIOGRÁFICAS

75
7 - Referências Bibliográficas
AHUATZI D., HERRERO P., DE LA C.T. & MORENO F. (2004) The glucose-regulated nuclear localization of hexokinase 2 in Saccharomyces cerevisiae is Mig1-dependent. J.Biol.Chem. 279, 14440-14446.
ASNAGLI H. & MURPHY K.M. (2001) The functional genomics experience (are you experienced?). Nat.Immunol. 2, 826-828.
ATRIAN S., GONZALEZ-DUARTE R. & FOTHERGILL-GILMORE L.A. (1990) Synthesis of Drosophila melanogaster alcohol dehydrogenase in yeast. Gene 93, 205-212.
AUESUKAREE C., TOCHIO H., SHIRAKAWA M., KANEKO Y. & HARASHIMA S. (2005) Plc1p, Arg82p, and Kcs1p, enzymes involved in inositol pyrophosphate synthesis, are essential for phosphate regulation and polyphosphate accumulation in Saccharomyces cerevisiae. J.Biol.Chem. 280, 25127-25133.
BARBET N.C., SCHNEIDER U., HELLIWELL S.B., STANSFIELD I., TUITE M.F. & HALL M.N. (1996) TOR controls translation initiation and early G1 progression in yeast. Mol.Biol.Cell 7, 25-42.
BAYROCK D.P. & MICHAEL I.W. (2001) Application of multistage continuous fermentation for production of fuel alcohol by very-high-gravity fermentation technology. J.Ind.Microbiol.Biotechnol. 27, 87-93.
BELL R.M., HANNUN Y.A. & LOOMIS C.R. (1986) Mechanism of regulation of protein kinase C by lipid second messengers. Symp.Fundam.Cancer Res. 39, 145-156.
BLOM N., SICHERITZ-PONTEN T., GUPTA R., GAMMELTOFT S. & BRUNAK S. (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 4, 1633-1649.
BOUSTANY L.M. & CYERT M.S. (2002) Calcineurin-dependent regulation of Crz1p nuclear export requires Msn5p and a conserved calcineurin docking site. Genes
Dev. 16, 608-619.
BRANDAO R.L., ETCHEBEHERE L., QUEIROZ C.C., TROPIA M.J., ERNANDES J.R., GONCALVES T., LOUREIRO-DIAS M.C., WINDERICKX J., THEVELEIN J.M., LEIPER F.C., CARLING D. & CASTRO I.M. (2002) Evidence for involvement of Saccharomyces cerevisiae protein kinase C in glucose induction of HXT genes and derepression of SUC2. FEMS Yeast Res. 2, 93-102.
BREITKREUTZ A. & TYERS M. (2002) MAPK signaling specificity: it takes two to tango. Trends Cell Biol. 12, 254-257.

76
CAZAR-ROMAN A.R., TRAN E.J., GUO S. & WENTE S.R. (2006) Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat.Cell Biol. 8, 711-716.
CHAE H.J., JOO H. & IN M.J. (2001) Utilization of brewer's yeast cells for the production of food-grade yeast extract. Part 1: Effects of different enzymatic treatments on solid and protein recovery and flavor characteristics. Bioresour.Technol. 76, 253-258.
CHAI B., HSU J.M., DU J. & LAURENT B.C. (2002) Yeast RSC function is required for organization of the cellular cytoskeleton via an alternative PKC1 pathway. Genetics 161, 575-584.
CHEN R.E. & THORNER J. (2007) Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim.Biophys.Acta 1773, 1311-1340.
CHI Z.M., LI J.F., WANG X.H. & YAO S.M. (2004) Inositol and phosphatidylinositol mediated mediated glucose derepression, gene expression and invertase secretion in yeasts. Acta Biochim.Biophys.Sin.(Shanghai) 36, 443-449.
DAS M., BURGE C.B., PARK E., COLINAS J. & PELLETIER J. (2001) Assessment of the total number of human transcription units. Genomics 77, 71-78.
DOORES S. (1983) The microbiology of apples and apple products. Crit Rev.Food
Sci.Nutr. 19, 133-149.
DUJON B. (1996) The yeast genome project: what did we learn? Trends Genet. 12, 263-270.
ELBING K., STAHLBERG A., HOHMANN S. & GUSTAFSSON L. (2004) Transcriptional responses to glucose at different glycolytic rates in Saccharomyces cerevisiae. Eur.J.Biochem. 271, 4855-4864.
GANCEDO J.M. (1998b) Yeast carbon catabolite repression. Microbiol.Mol.Biol.Rev. 62, 334-361.
GANCEDO J.M. (1998a) Yeast carbon catabolite repression. Microbiol.Mol.Biol.Rev. 62, 334-361.
GARCIA R., BERMEJO C., GRAU C., PEREZ R., RODRIGUEZ-PENA J.M., FRANCOIS J., NOMBELA C. & ARROYO J. (2004) The global transcriptional response to transient cell wall damage in Saccharomyces cerevisiae and its regulation by the cell integrity signaling pathway. J.Biol.Chem. 279, 15183-15195.
GELADE R., VAN d., V, VAN D.P. & THEVELEIN J.M. (2003) Multi-level response of the yeast genome to glucose. Genome Biol. 4, 233
GENTLEMAN R & IHAKA R (1997) The R language. In Proceedings of the 28th
Symposium on the Interface, L.Billard and N.Fisher Eds.The Interface
Foundation of North America

77
GILL G.S. & ZAWORSKI P.G. (1991) Use of yeasts in production and discovery of pharmaceuticals. Ann.N.Y.Acad.Sci. 646, 172-180.
GOFFEAU A., BARRELL B.G., BUSSEY H., DAVIS R.W., DUJON B., FELDMANN H., GALIBERT F., HOHEISEL J.D., JACQ C., JOHNSTON M., LOUIS E.J., MEWES H.W., MURAKAMI Y., PHILIPPSEN P., TETTELIN H. & OLIVER S.G. (1996) Life with 6000 genes. Science 274, 546, 563-546, 567.
GOMES K.N., FREITAS S.M., PAIS T.M., FIETTO J.L., TOTOLA A.H., ARANTES R.M., MARTINS A., LUCAS C., SCHULLER D., CASAL M., CASTRO I.M., FIETTO L.G. & BRANDAO R.L. (2005) Deficiency of Pkc1 activity affects glycerol metabolism in Saccharomyces cerevisiae. FEMS Yeast Res. 5, 767-776.
GRAY J.V., OGAS J.P., KAMADA Y., STONE M., LEVIN D.E. & HERSKOWITZ I. (1997) A role for the Pkc1 MAP kinase pathway of Saccharomyces cerevisiae in bud emergence and identification of a putative upstream regulator. EMBO J. 16, 4924-4937.
GUSTIN M.C., ALBERTYN J., ALEXANDER M. & DAVENPORT K. (1998) MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol.Mol.Biol.Rev. 62, 1264-1300.
HEINISCH J.J., LORBERG A., SCHMITZ H.P. & JACOBY J.J. (1999b) The protein kinase C-mediated MAP kinase pathway involved in the maintenance of cellular integrity in Saccharomyces cerevisiae. Mol.Microbiol. 32, 671-680.
HEINISCH J.J., LORBERG A., SCHMITZ H.P. & JACOBY J.J. (1999a) The protein kinase C-mediated MAP kinase pathway involved in the maintenance of cellular integrity in Saccharomyces cerevisiae. Mol.Microbiol. 32, 671-680.
HELLIWELL S.B., HOWALD I., BARBET N. & HALL M.N. (1998a) TOR2 is part of two related signaling pathways coordinating cell growth in Saccharomyces cerevisiae. Genetics 148, 99-112.
HELLIWELL S.B., SCHMIDT A., OHYA Y. & HALL M.N. (1998b) The Rho1 effector Pkc1, but not Bni1, mediates signalling from Tor2 to the actin cytoskeleton. Curr.Biol. 8, 1211-1214.
HENSING M.C., ROUWENHORST R.J., HEIJNEN J.J., VAN DIJKEN J.P. & PRONK J.T. (1995) Physiological and technological aspects of large-scale heterologous-protein production with yeasts. Antonie Van Leeuwenhoek 67, 261-279.
HINNEN A., HICKS J.B. & FINK G.R. (1978) Transformation of yeast. Proc.Natl.Acad.Sci.U.S.A 75, 1929-1933.
HOHMANN S. (2002) Osmotic stress signaling and osmoadaptation in yeasts. Microbiol.Mol.Biol.Rev. 66, 300-372.
HOLM H.E., NISSEN P., SOMMER P., NIELSEN J.C. & ARNEBORG N. (2001) The effect of oxygen on the survival of non-Saccharomyces yeasts during mixed

78
culture fermentations of grape juice with Saccharomyces cerevisiae. J.Appl.Microbiol. 91, 541-547.
HOLSBEEKS I., LAGATIE O., VAN N.A., VAN d., V & THEVELEIN J.M. (2004c) The eukaryotic plasma membrane as a nutrient-sensing device. Trends
Biochem.Sci. 29, 556-564.
HOLSBEEKS I., LAGATIE O., VAN N.A., VAN d., V & THEVELEIN J.M. (2004b) The eukaryotic plasma membrane as a nutrient-sensing device. Trends
Biochem.Sci. 29, 556-564.
HOLSBEEKS I., LAGATIE O., VAN N.A., VAN d., V & THEVELEIN J.M. (2004a) The eukaryotic plasma membrane as a nutrient-sensing device. Trends
Biochem.Sci. 29, 556-564.
IGUAL J.C., JOHNSON A.L. & JOHNSTON L.H. (1996) Coordinated regulation of gene expression by the cell cycle transcription factor Swi4 and the protein kinase C MAP kinase pathway for yeast cell integrity. EMBO J. 15, 5001-5013.
JOHNSTON M. & KIM J.H. (2005) Glucose as a hormone: receptor-mediated glucose sensing in the yeast Saccharomyces cerevisiae. Biochem.Soc.Trans. 33, 247-252.
KANIAK A., XUE Z., MACOOL D., KIM J.H. & JOHNSTON M. (2004) Regulatory network connecting two glucose signal transduction pathways in Saccharomyces cerevisiae. Eukaryot.Cell 3, 221-231.
KIM J.H., BRACHET V., MORIYA H. & JOHNSTON M. (2006) Integration of transcriptional and posttranslational regulation in a glucose signal transduction pathway in Saccharomyces cerevisiae. Eukaryot.Cell 5, 167-173.
LAGORCE A., HAUSER N.C., LABOURDETTE D., RODRIGUEZ C., MARTIN-YKEN H., ARROYO J., HOHEISEL J.D. & FRANCOIS J. (2003) Genome-wide analysis of the response to cell wall mutations in the yeast Saccharomyces cerevisiae. J.Biol.Chem. 278, 20345-20357.
LANGKJAER R.B., CASAREGOLA S., USSERY D.W., GAILLARDIN C. & PISKUR J. (2003) Sequence analysis of three mitochondrial DNA molecules reveals interesting differences among Saccharomyces yeasts. Nucleic Acids Res. 31, 3081-3091.
LEVIN D.E., BOWERS B., CHEN C.Y., KAMADA Y. & WATANABE M. (1994) Dissecting the protein kinase C/MAP kinase signalling pathway of Saccharomyces cerevisiae. Cell Mol.Biol.Res. 40, 229-239.
LEVIN D.E., FIELDS F.O., KUNISAWA R., BISHOP J.M. & THORNER J. (1990) A candidate protein kinase C gene, PKC1, is required for the S. cerevisiae cell cycle. Cell 62, 213-224.
LIVAK K.J. & SCHMITTGEN T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402-408.

79
LOWRY O.H., ROSEBROUGH N.J., FARR A.L. & RANDALL R.J. (1951) Protein measurement with the Folin phenol reagent. J.Biol.Chem. 193, 265-275.
LU L., ROBERTS G.G., OSZUST C. & HUDSON A.P. (2005a) The YJR127C/ZMS1 gene product is involved in glycerol-based respiratory growth of the yeast Saccharomyces cerevisiae. Curr.Genet. 48, 235-246.
LU L., ROBERTS G.G., OSZUST C. & HUDSON A.P. (2005b) The YJR127C/ZMS1 gene product is involved in glycerol-based respiratory growth of the yeast Saccharomyces cerevisiae. Curr.Genet. 48, 235-246.
MADDEN K., SHEU Y.J., BAETZ K., ANDREWS B. & SNYDER M. (1997) SBF cell cycle regulator as a target of the yeast PKC-MAP kinase pathway. Science 275, 1781-1784.
MARAZ A. (2002) From yeast genetics to biotechnology. Acta
Microbiol.Immunol.Hung. 49, 483-491.
MARINI N.J., MELDRUM E., BUEHRER B., HUBBERSTEY A.V., STONE D.E., TRAYNOR-KAPLAN A. & REED S.I. (1996) A pathway in the yeast cell division cycle linking protein kinase C (Pkc1) to activation of Cdc28 at START. EMBO J. 15, 3040-3052.
MASHEGO M.R., JANSEN M.L., VINKE J.L., VAN GULIK W.M. & HEIJNEN J.J. (2005) Changes in the metabolome of Saccharomyces cerevisiae associated with evolution in aerobic glucose-limited chemostats. FEMS Yeast Res. 5, 419-430.
MASIMIREMBWA C.M., OTTER C., BERG M., JONSSON M., LEIDVIK B., JONSSON E., JOHANSSON T., BACKMAN A., EDLUND A. & ANDERSSON T.B. (1999) Heterologous expression and kinetic characterization of human cytochromes P-450: validation of a pharmaceutical tool for drug metabolism research. Drug Metab Dispos. 27, 1117-1122.
MATHEOS D.P., KINGSBURY T.J., AHSAN U.S. & CUNNINGHAM K.W. (1997) Tcn1p/Crz1p, a calcineurin-dependent transcription factor that differentially regulates gene expression in Saccharomyces cerevisiae. Genes Dev. 11, 3445-3458.
MAZUR P. & BAGINSKY W. (1996) In vitro activity of 1,3-beta-D-glucan synthase requires the GTP-binding protein Rho1. J.Biol.Chem. 271, 14604-14609.
MAZUR P., MORIN N., BAGINSKY W., EL-SHERBEINI M., CLEMAS J.A., NIELSEN J.B. & FOOR F. (1995) Differential expression and function of two homologous subunits of yeast 1,3-beta-D-glucan synthase. Mol.Cell Biol. 15, 5671-5681.
MORENO-ARRIBAS M.V. & POLO M.C. (2005) Winemaking biochemistry and microbiology: current knowledge and future trends. Crit Rev.Food Sci.Nutr. 45, 265-286.
NATH N., MCCARTNEY R.R. & SCHMIDT M.C. (2003) Yeast Pak1 kinase associates with and activates Snf1. Mol.Cell Biol. 23, 3909-3917.

80
NEWCOMB L.L., DIDERICH J.A., SLATTERY M.G. & HEIDEMAN W. (2003) Glucose regulation of Saccharomyces cerevisiae cell cycle genes. Eukaryot.Cell 2, 143-149.
O'BRIAN C.A. & WARD N.E. (1989) Biology of the protein kinase C family. Cancer
Metastasis Rev. 8, 199-214.
O'ROURKE S.M. & HERSKOWITZ I. (2004) Unique and redundant roles for HOG MAPK pathway components as revealed by whole-genome expression analysis. Mol.Biol.Cell 15, 532-542.
O'ROURKE S.M., HERSKOWITZ I. & O'SHEA E.K. (2002) Yeast go the whole HOG for the hyperosmotic response. Trends Genet. 18, 405-412.
ODOM A.R., STAHLBERG A., WENTE S.R. & YORK J.D. (2000) A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science 287, 2026-2029.
ONO T., SUZUKI T., ANRAKU Y. & IIDA H. (1994) The MID2 gene encodes a putative integral membrane protein with a Ca(2+)-binding domain and shows mating pheromone-stimulated expression in Saccharomyces cerevisiae. Gene 151, 203-208.
OSTERGAARD S., OLSSON L. & NIELSEN J. (2000) Metabolic engineering of Saccharomyces cerevisiae. Microbiol.Mol.Biol.Rev. 64, 34-50.
OZCAN S., DOVER J. & JOHNSTON M. (1998) Glucose sensing and signaling by two glucose receptors in the yeast Saccharomyces cerevisiae. EMBO J. 17, 2566-2573.
OZCAN S. & JOHNSTON M. (1999) Function and regulation of yeast hexose transporters. Microbiol.Mol.Biol.Rev. 63, 554-569.
PAPAMICHOS-CHRONAKIS M., GLIGORIS T. & TZAMARIAS D. (2004) The Snf1 kinase controls glucose repression in yeast by modulating interactions between the Mig1 repressor and the Cyc8-Tup1 co-repressor. EMBO Rep. 5, 368-372.
RADHIKA V., MILKEVITCH M., AUDIGE V., PROIKAS-CEZANNE T. & DHANASEKARAN N. (2005) Engineered Saccharomyces cerevisiae strain BioS-1, for the detection of water-borne toxic metal contaminants. Biotechnol.Bioeng. 90, 29-35.
RADHIKA V., PROIKAS-CEZANNE T., JAYARAMAN M., ONESIME D., HA J.H. & DHANASEKARAN D.N. (2007) Chemical sensing of DNT by engineered olfactory yeast strain. Nat.Chem.Biol. 3, 325-330.
RESNICK A.C., SNOWMAN A.M., KANG B.N., HURT K.J., SNYDER S.H. & SAIARDI A. (2005) Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proc.Natl.Acad.Sci.U.S.A 102, 12783-12788.

81
RO D.K., PARADISE E.M., OUELLET M., FISHER K.J., NEWMAN K.L., NDUNGU J.M., HO K.A., EACHUS R.A., HAM T.S., KIRBY J., CHANG M.C., WITHERS S.T., SHIBA Y., SARPONG R. & KEASLING J.D. (2006) Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440, 940-943.
ROMBAUT B. & JORE J.P. (1997) Immunogenic, non-infectious polio subviral particles synthesized in Saccharomyces cerevisiae. J.Gen.Virol. 78 ( Pt 8), 1829-1832.
ROSE A.H. (1981) The microbiological production of food and drink. Sci.Am. 245, 126-34, 136, 138.
SAITO H. & TATEBAYASHI K. (2004) Regulation of the osmoregulatory HOG MAPK cascade in yeast. J.Biochem.(Tokyo) 136, 267-272.
SALGADO A.P., SCHULLER D., CASAL M., LEAO C., LEIPER F.C., CARLING D., FIETTO L.G., TROPIA M.J., CASTRO I.M. & BRANDAO R.L. (2002) Relationship between protein kinase C and derepression of different enzymes. FEBS Lett. 532, 324-332.
SCHMIDT A., BICKLE M., BECK T. & HALL M.N. (1997) The yeast phosphatidylinositol kinase homolog TOR2 activates RHO1 and RHO2 via the exchange factor ROM2. Cell 88, 531-542.
SCHMIDT A., SCHMELZLE T. & HALL M.N. (2002) The RHO1-GAPs SAC7, BEM2 and BAG7 control distinct RHO1 functions in Saccharomyces cerevisiae. Mol.Microbiol. 45, 1433-1441.
SCHMITZ H.P. & HEINISCH J.J. (2003) Evolution, biochemistry and genetics of protein kinase C in fungi. Curr.Genet. 43, 245-254.
SCHOFIELD D.A., WESTWATER C., BARTH J.L. & DINOVO A.A. (2007) Development of a yeast biosensor-biocatalyst for the detection and biodegradation of the organophosphate paraoxon. Appl.Microbiol.Biotechnol.
SCHULLER H.J. (2003) Transcriptional control of nonfermentative metabolism in the yeast Saccharomyces cerevisiae. Curr.Genet. 43, 139-160.
SHENHAR G. & KASSIR Y. (2001) A positive regulator of mitosis, Sok2, functions as a negative regulator of meiosis in Saccharomyces cerevisiae. Mol.Cell Biol. 21, 1603-1612.
SMITH H.A., GORMAN J.W., KOLTIN Y. & GORMAN J.A. (1990) Functional expression of the Candida albicans beta-tubulin gene in Saccharomyces cerevisiae. Gene 90, 115-123.
STATHOPOULOS A.M. & CYERT M.S. (1997) Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 11, 3432-3444.

82
TAMAS M.J., REP M., THEVELEIN J.M. & HOHMANN S. (2000) Stimulation of the yeast high osmolarity glycerol (HOG) pathway: evidence for a signal generated by a change in turgor rather than by water stress. FEBS Lett. 472, 159-165.
TAYLOR F., KURANTZ M.J., GOLDBERG N. & CRAIG J.C., Jr. (1995) Continuous fermentation and stripping of ethanol. Biotechnol.Prog. 11, 693-698.
TAYLOR F., KURANTZ M.J., GOLDBERG N., MCALOON A.J. & CRAIG J.C., Jr. (2000) Dry-grind process for fuel ethanol by continuous fermentation and stripping. Biotechnol.Prog. 16, 541-547.
TEIXEIRA M.C., MONTEIRO P., JAIN P., TENREIRO S., FERNANDES A.R., MIRA N.P., ALENQUER M., FREITAS A.T., OLIVEIRA A.L. & SA-CORREIA I. (2006) The YEASTRACT database: a tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic
Acids Res. 34, D446-D451
TREITEL M.A. & CARLSON M. (1995) Repression by SSN6-TUP1 is directed by MIG1, a repressor/activator protein. Proc.Natl.Acad.Sci.U.S.A 92, 3132-3136.
TRÓPIA MJ, CARDOSO AS, TISI R, FIETTO LG, FIETTO JL, MARTEGANI E, CASTRO IM, BRANDÃO RL. (2006) Calcium signaling and sugar-induced activation of plasma membrane H(+)-ATPase in Saccharomyces cerevisiae cells. Biochem Biophys Res Commun. 19;343(4):1234-43.
VALENTINI S.R., CASOLARI J.M., OLIVEIRA C.C., SILVER P.A. & MCBRIDE A.E. (2002) Genetic interactions of yeast eukaryotic translation initiation factor 5A (eIF5A) reveal connections to poly(A)-binding protein and protein kinase C signaling. Genetics 160, 393-405.
VAN W.O., REISER V., SIDERIUS M., KELDERS M.C., AMMERER G., RUIS H. & MAGER W.H. (2000) Response of Saccharomyces cerevisiae to severe osmotic stress: evidence for a novel activation mechanism of the HOG MAP kinase pathway. Mol.Microbiol. 37, 382-397.
WATANABE Y., IRIE K. & MATSUMOTO K. (1995) Yeast RLM1 encodes a serum response factor-like protein that may function downstream of the Mpk1 (Slt2) mitogen-activated protein kinase pathway. Mol.Cell Biol. 15, 5740-5749.
WATANABE Y., TAKAESU G., HAGIWARA M., IRIE K. & MATSUMOTO K. (1997) Characterization of a serum response factor-like protein in Saccharomyces cerevisiae, Rlm1, which has transcriptional activity regulated by the Mpk1 (Slt2) mitogen-activated protein kinase pathway. Mol.Cell Biol. 17, 2615-2623.
WEBER H. & BARTH G. (1988) Nonconventional yeasts: their genetics and biotechnological applications. Crit Rev.Biotechnol. 7, 281-337.
WESTFALL P.J., BALLON D.R. & THORNER J. (2004) When the stress of your environment makes you go HOG wild. Science 306, 1511-1512.

83
WYCKOFF E. & HSIEH T.S. (1988) Functional expression of a Drosophila gene in yeast: genetic complementation of DNA topoisomerase II. Proc.Natl.Acad.Sci.U.S.A 85, 6272-6276.
YANG Y.H., DUDOIT S., LUU P., LIN D.M., PENG V., NGAI J. & SPEED T.P. (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30, e15
YORK J.D., ODOM A.R., MURPHY R., IVES E.B. & WENTE S.R. (1999) A phospholipase C-dependent inositol polyphosphate kinase pathway required for efficient messenger RNA export. Science 285, 96-100.
YOSHIDA S., OHYA Y., NAKANO A. & ANRAKU Y. (1994) Genetic interactions among genes involved in the STT4-PKC1 pathway of Saccharomyces cerevisiae. Mol.Gen.Genet. 242, 631-640.
ZAKRZEWSKA A., BOORSMA A., BRUL S., HELLINGWERF K.J. & KLIS F.M. (2005) Transcriptional response of Saccharomyces cerevisiae to the plasma membrane-perturbing compound chitosan. Eukaryot.Cell 4, 703-715.
ZARZOV P., MAZZONI C. & MANN C. (1996) The SLT2(MPK1) MAP kinase is activated during periods of polarized cell growth in yeast. EMBO J. 15, 83-91.
ZENG X., DEMINOFF S.J. & SANTANGELO G.M. (1997) Specialized Rap1p/Gcr1p transcriptional activation through Gcr1p DNA contacts requires Gcr2p, as does hyperphosphorylation of Gcr1p. Genetics 147, 493-505.
ZHAO C., JUNG U.S., GARRETT-ENGELE P., ROE T., CYERT M.S. & LEVIN D.E. (1998) Temperature-induced expression of yeast FKS2 is under the dual control of protein kinase C and calcineurin. Mol.Cell Biol. 18, 1013-1022.


















