UNIVERSIDADE DE SÃO PAULO - USP · antifúngicos azólicos e também encontra-se envolvida na...
146
UNIVERSIDADE DE SÃO PAULO FACULDADE DE CIÊNCIAS FARMACÊUTICAS Programa de Pós-Graduação em Tecnologia Bioquímico-Farmacêutica Área de Tecnologia Químico-Farmacêutica Estudos de identificação de possíveis alvos para nitro-compostos azometínicos ou oxadiazolínicos com atividade antifúngica e anti-T. cruzi Ieda Yuriko Sonehara Tese para obtenção do grau de DOUTOR Orientador: Prof. Assoc. Leoberto Costa Tavares SÃO PAULO 2009
Transcript of UNIVERSIDADE DE SÃO PAULO - USP · antifúngicos azólicos e também encontra-se envolvida na...

UNIVERSIDADE DE SÃO PAULO
FACULDADE DE CIÊNCIAS FARMACÊUTICAS
Programa de Pós-Graduação em Tecnologia Bioquímico-Farmacêutica
Área de Tecnologia Químico-Farmacêutica
Estudos de identificação de possíveis alvos para
nitro-compostos azometínicos ou oxadiazolínicos com
atividade antifúngica e anti-T. cruzi
Ieda Yuriko Sonehara
Tese para obtenção do grau de
DOUTOR
Orientador:
Prof. Assoc. Leoberto Costa Tavares
SÃO PAULO
2009

Ieda Yuriko Sonehara
Estudos de identificação de possíveis alvos para
nitro-compostos azometínicos ou oxadiazolínicos com
atividade antifúngica e anti-T. cruzi
Comissão Julgadora da
Tese para obtenção do grau de Doutor
Prof. Assoc. Leoberto Costa Tavares orientador/presidente
_______________________________ 1o. examinador
_______________________________ 2o. examinador
_______________________________ 3o. examinador
_______________________________ 4o. examinador
São Paulo, __________ de 2009.

A Meus PaisA Meus PaisA Meus PaisA Meus Pais

AGRADECIMENTOS
Ao Programa de Pós-Graduação em Tecnologia Bioquímico-Farmacêutica da
Faculdade de Ciências Farmacêuticas da Universidade de São Paulo, pela
oportunidade e apoio na realização do presente trabalho.
Ao Professor Leoberto Costa Tavares, pela orientação deste trabalho, constante
apoio, incentivo e confiança, além da preciosa amizade e valiosos conselhos ao
longo destes anos de convivência.
Ao Dr. Ferran Sanz e ao Dr. Jordi Mestres, do Research Unit on Biomedical
Informatics do Institut Municipal d’Investigació Mèdica e Universitat Pompeu Fabra
(GRIB/IMIM/UPF), e colegas do Chemogenomics Laboratory/GRIB, pela
oportunidade oferecida, solicitude e meses de agradável convivência durante bolsa-
sandwich em Barcelona, Espanha.
Ao colega e amigo Salomão Dória Jorge, sempre presente e solícito, pelas
discussões e sugestões, por compartilhar seus conhecimentos e pelo auxílio
inestimável na realização deste trabalho.
À Maria das Graças Baptista Santos e Fanny Palace-Berl pela amizade, constante
troca de informações e auxílio e solicitude na preparação e realização dos ensaios
biológicos.
Aos colegas Alex de Oliveira, Fávero Paula e Marina Ishii, pelo auxílio constante,
horas de conversas, e pela convivência e companheirismo.
Ao amigos e colegas do curso de Ciências Farmacêuticas da Faculdade de
Medicina do ABC, em especial José Chisté Jr., Maria Lucila Teves e Hugo de
Alencar Isabel, e aos coordenadores Dra. Registila Beltrame e Marcelo Guimarães,
pelo apoio e compreensão durante estes anos, sem os quais não teria sido possível
dedicar as horas necessárias para a realização deste trabalho.
Aos amigos Victoria Bauld, Law Wilson, Erik Heeren e Gilberto Marques, pelas
longas conversas e apoio constantes.
Ao Jorge e Elaine, da Secretaria de Pós-Graduação, e Mirian e Elza, da Secretaria
do Departamento de Tecnologia Bioquímico-Farmacêutica, pela atenção e
solicitude.
Ao CNPq, pela bolsa de estudos no país (processo 142954/2006-3), apoio
financeiro e bolsa-sandwich (processo 201508/2007-9), sem os quais não teria sido
possível a realização deste trabalho.
A todos aqueles que de alguma forma, direta ou indiretamente, contríbuíram para a
realização deste trabalho.

i
SUMÁRIO
RESUMO.................................................................................................................................. 1
ABSTRACT.............................................................................................................................. 2
1. INTRODUÇÃO................................................................................................................. 3
2. FUNDAMENTAÇÃO BIBLIOGRÁFICA.......................................................................... 8
2.1. MÉTODOS DE PLANEJAMENTO DE FÁRMACOS AUXILIADOS POR
COMPUTADOR, CADD................................................................................................... 9
2.1.1 ANÁLISE DAS RELAÇÕES QUANTITATIVAS ENTRE ESTRUTURA
TRIDIMENSIONAL E ATIVIDADE, QSAR 3D............................................................... 15
2.2. 14α-DESMETILASE (CYP51): ESTRUTURA E FUNÇÃO ........................................... 26
2.2.1 CARACTERÍSTICAS DE COMPOSTOS INIBIDORES DE CYP51 ................. 38
3. OBJETIVOS .................................................................................................................. 44
4. EXPERIMENTAL........................................................................................................... 47
4.1. CÁLCULO DE DESCRITORES TOPOLÓGICOS......................................................... 49
4.2. ESTUDOS DE IDENTIFICAÇÃO DE POSSÍVEIS ALVOS POR COMPARAÇÃO DE
PERFIS MOLECULARES, VIRTUAL PROFILING........................................................ 53
4.3. CARACTERIZAÇÃO DA CAVIDADE CATALÍTICA DE CYP51.................................... 54
4.3.1 ESTUDOS DE INTERAÇÃO BASEADOS EM CARACTERÍSTICAS DAS
SUPERFÍCIES CATALÍTICAS DE PROTEÍNAS........................................................... 56
4.4. ESTUDOS DE ANCORAMENTO DO LIGANTE, DOCKING ........................................ 57
4.5. ANÁLISE DE PERFIL DE SOLUBILIDADE................................................................... 58
4.6. SÍNTESE........................................................................................................................ 59
4.7. DETERMINAÇÃO DE ATIVIDADE BIOLÓGICA........................................................... 60
4.7.1 ATIVIDADE ANTIFÚNGICA ............................................................................. 60
4.7.2 ATIVIDADE ANTI-T. CRUZI ............................................................................. 64
5. RESULTADOS E DISCUSSÃO .................................................................................... 66
6. CONCLUSÕES............................................................................................................ 108
7. PERSPECTIVAS FUTURAS....................................................................................... 111
8. REFERÊNCIAS BIBLIOGRÁFICAS........................................................................... 113
ANEXOS .............................................................................................................................. 131

ii
LISTA DE FIGURAS
Figura 1. Interrelação entre disciplinas relacionadas ao planejamento de fármacos in silico............................................................................................ 9
Figura 2. Métodos de QSAR ........................................................................ 10
Figura 3. Determinação de distância interatômica entre pares de átomos.. 14
Figura 4. Fluxo de trabalho para CoMFA. .................................................... 17
Figura 5. Processo de validação cruzada por leave-N-out........................... 23
Figura 6. Algumas reações catalizadas por enzimas da família P450. ........ 28
Figura 7. Dobramento comum de P450 mamífero ....................................... 29
Figura 8. Conservação de sequência primária em P450 ............................. 30
Figura 9. Reação da CYP51. ....................................................................... 32
Figura 10. Identidade da sequência de aminoácidos em 71 sequências da família CYP51. ............................................................................................. 32
Figura 11. Estrutura de CYP51 de M. tuberculosis em representação de Richardson. .................................................................................................. 36
Figura 12. Estruturas de alguns fármacos antifúngicos imidazólicos e triazólicos ..................................................................................................... 39
Figura 13. Interação de fluconazol no sítio catalítico de CYP51 de M.
tuberculosis. ................................................................................................. 43
Figura 14. Estrutura química geral das séries estudadas ............................ 48
Figura 15. Procedimento para cálculo de SHED.......................................... 52
Figura 16. Cavidade catalítica de CYP51 de Mycobacterium tuberculosis com CPTs em representação de superfície ................................................. 56
Figura 17. Síntese dos compostos azometínicos e oxadiazolínicos ............ 60
Figura 18. Inibição de crescimento* de formas epimastigotas de T. cruzi, cepa Y, em presença de compostos das séries azometínica e oxadiazolínica70
Figura 19. Equivalência dos átomos no sistema utilizado pelo programa Sybyl ............................................................................................................ 71
Figura 20. Estruturas dos antifúngicos azólicos selecionados para estudo . 72
Figura 21. Estruturas de compostos anti-T. cruzi ......................................... 72

iii
Figura 22. Perfis SHED das séries estudadas ............................................. 76
Figura 23. Estruturas de compostos responsáveis pelas anotações para alvos identificados em virtual profiling .......................................................... 77
Figura 24. Orientação de interação sugerida para COX-2 ........................... 78
Figura 25. Superposição de estruturas de análogo ao celecoxib (verde) e composto OxaS-CF3 (R) (azul) em orientação sugerida para interação com COX-2 .......................................................................................................... 79
Figura 26. Perfis SHED de compostos azometínicos selecionados............. 80
Figura 27. Perfis SHED de compostos oxadiazolínicos selecionados. ........ 81
Figura 28. Representação de CYP51 de M. tuberculosis............................. 83
Figura 29. Características doadoras de ligações de hidrogênio................... 85
Figura 30. Cavidade catalítica de CYP51..................................................... 88
Figura 31. Orientação de interação sugerida do composto OxaS-Cl (R) com CYP51.......................................................................................................... 89
Figura 32. Fragmentos presentes nos compostos das séries azometínica e oxadiazolínica, com os CPTs associados quando em interação com diversas proteínas*..................................................................................................... 89
Figura 33. Resultados de análise de PLS-DA ilustrando os escores das duas primeiras componentes principais para conjunto de 8 compostos*.............. 92
Figura 34. Resultados de análise de PLS-DA de conjunto de 8 compostos*: pesos............................................................................................................ 93
Figura 35. Resultados de análise de PLS-DA ilustrando os escores das duas primeiras componentes principais para conjunto de 13 compostos*............ 94
Figura 36. Resultados de análise de PLS-DA de conjunto de 13 compostos*: pesos............................................................................................................ 95
Figura 37. Perfil de descritores Volsurf para composto oxadiazolínico AzoO-CH3............................................................................................................... 96
Figura 38. Perfil de descritores Volsurf para composto oxadiazolínico OxaS-CH3 (R)......................................................................................................... 97
Figura 39. Perfil de descritores Volsurf para composto oxadiazolínico OxaS-OC4H8 (R)..................................................................................................... 98

iv
Figura 40. Mapas de contorno representando propriedades hidrofílicas (azul, sonda OH2) e lipofílicas (verde, sonda DRY) de compostos oxadiazolínicos.100
Figura 41. Volumes de contorno de compostos tiofilidênicos..................... 101
Figura 42. Orientação das conformações de interação de compostos antifúngicos em CYP51 de Cryptococcus neoformans. ............................. 102
Figura 43. Mapas de contorno representando propriedades hidrofílicas (azul, sonda OH2) e lipofílicas (verde, sonda DRY) do composto azometínico AzoO-CH3, obtidas com o programa Volsurf. ............................................. 102
Figura 44. Ligação de derivado 2-aminotetralínico no sítio ativo de CYP51 de C. albicans. ................................................................................................ 103
Figura 45. Interação de lanosterol (roxo) e fluconazol (verde) no sítio ativo de CYP51 de M. tuberculosis.......................................................................... 104
Figura 46. Fragmentos extraídos de banco de dados e respectivas regiões de CYP51 com possibilidade de interação................................................. 105
Figura 47. Proposição de características necessárias para interação de compostos oxadiazolínicos com sítio catalítico de CYP51......................... 107

v
LISTA DE TABELAS
Tabela 1. Alguns tipos de átomos utilizados pelo programa Sybyl .............. 49
Tabela 2. Alguns tipos de átomos utilizados pelo programa Sybyl e respectivas características farmacofóricas................................................... 50
Tabela 3. Proteínas selecionadas para estudo de docking .......................... 57
Tabela 4. Parâmetros de minimização utilizados no programa Sybyl 8.0 .... 58
Tabela 5. Concentração inibitória mínima, MIC, frente à cepa 537Y de C.
albicans ........................................................................................................ 67
Tabela 6. Concentração inibitória mínima, MIC, frente a cepa 537Y de C.
albicans, convertidas em potência. .............................................................. 68
Tabela 7. Inibição de crescimento de formas epimastigotas de T. cruzi, cepa Y, em presença de compostos das séries azometínica e oxadiazolínica..... 69
Tabela 8. Perfis SHED de compostos azometínicos.................................... 73
Tabela 9. Perfis SHED de compostos oxadiazolínicos ................................ 74
Tabela 10. Perfis SHED de compostos antifúngicos e anti-T.cruzi selecionados ................................................................................................ 75
Tabela 11. Resultados principais de virtual profiling .................................... 77
Tabela 12. Alguns descritores utizados pelo programa Volsurf ................... 90

vi
LISTA DE TERMOS* E ABREVIAÇÕES UTILIZADOS
AMINOÁCIDOS:
Alanina Ala A Asparagina Asn N Cisteína Cys C Prolina Pro P Ácido aspártico Asp D Glutamina Gln Q Ácido glutâmico Glu E Arginina Arg R Fenilalanina Phe F Serina Ser S Glicina Gly G Treonina Thr T Histidina His H Valina Val V Isoleucina Ile I Triptofano Trp W Lisina Lys L Tirosina Tyr Y Metionina Met M
CADD: Planejamento de Fármacos Auxiliado por Computador (Computer-
Aided Drug Design)
CoMFA: Análise Comparativa de Campos Moleculares (Comparative
Molecular Field Analysis)
CPT: Ponto Crítico (Critical Point)
Docking: exploração de possíveis modos de interação entre ligante e
biomacromolécula-alvo (estudos de ancoramento)
GRIND: Descritores Independentes de Grade (Grid Independent Descriptors)
In silico: métodos de estudo baseados em computador
LBDD: Planejamento de Fármacos Baseado Em Ligante (ligand-based drug
design)
MIF: Campos de Interação Moleculares (Molecular Interaction Fields)
MIC: Concentração Inibitória Mínima (Minimal Inhibitory Concentration)
PC: Componente Principal (Principal Component)
PCA: Análise de Componentes Principais (Principal Component Analysis)
PLS: Mínimos Quadrados Parciais (Partial Least Squares)
PLS-DA: Análise de Discriminante por Mínimos Quadrados Parciais (Partial
Least Squares Discriminant Analysis)

vii
QSAR 3D: Relações Quantitativas Tridimensionais Estrutura-Atividade
(Tridimensional Quantitative Struture-Activity Relationships)
PDB: Banco de Dados de Proteínas Brookhaven (Brookhaven Protein Data
Bank)
RMN: Ressonância Magnética Nuclear
SAR: Relações Estrutura-Atividade (Structure-Activity Relationships)
Scaffold: padrão molecular ou estrutura comum presente em compostos
químicos
SBDD: Planejamento de Fármacos Baseado em Estrutura (Structure-based
drug design)
SHED: Descritores de Entropia de Shannon (Shannon Entropy Descriptors)
Virtual profiling: procedimento de comparação de perfis moleculares para
identificação de possíveis alvos de compostos bioativos
WOMBAT™: World of Molecular Bioactivity™, banco de dados de estruturas
químicas
*Baseados em: SANT’ANNA, C.M.R. Glossário de termos usados no planejamento de fármacos (recomendações da IUPAC para 1997). Quim. Nova, São Paulo, v.25, n.3, p.505-512, 2002.

FBT/FCF/USP
Ieda Yuriko Sonehara
1
RESUMO
Este trabalho teve como objetivo a verificação de possíveis alvos para
compostos 5-nitro-heterocílicos de estrutura azometínica e oxadiazolínica
com bioatividade dual antifúngica e anti-T. cruzi. Os compostos estudados
pertencem a quatro séries intimamente relacionadas, a saber: Série AzoO:
5-nitro-2-furfurilideno benzidrazidas 4-substituídas; Série AzoS: 5-nitro-2-
tiofilideno benzidrazidas 4-substituídas; Série OxaO: 3-acetil-oxadiazolina
2,5-furfurilideno benzidrazidas 4-substituídas e Série OxaS: 3-acetil-
oxadiazolina 2,5-tiofilideno benzidrazidas 4-substituídas.
A determinação de atividade biológica demonstrou ação antifúngica
de alguns dos compostos contra Candida albicans, ATCC 537Y, com maior
atividade do composto OxaS-CH3 (MIC 3,28 µg/mL). Foi também
demonstrada, pelo grupo de trabalho, ação inibitória de crescimento de
formas epimastigotas de Trypanosoma cruzi, cepa Y.
A análise de resultados de virtual profiling apontou entre os possíveis
alvos as enzimas CYP19A (aromatase) e CYP3A4, que apresentam também
interação com compostos azólicos de ação antifúngica cujo alvo é CYP51
(14α-desmetilase). A inibição desta enzima é responsável pela ação de
antifúngicos azólicos e também encontra-se envolvida na ação anti-T. cruzi
de compostos azólicos, e foi portanto investigada como possível alvo
responsável pela ação dual dos compostos estudados.
A caracterização da cavidade catalítica de CYP51, tanto em termos
de descritores de propriedades físico-químicas como de características
estéricas, aliada a estudos de docking, confirmou a possibilidade de
interação da série oxadiazolínica com a CYP51 em orientação semelhante à
dos antifúngicos azólicos. A série azometínica não possui conformação
adequada para a interação da forma proposta, não excluindo porém a
possibilidade de interação no sítio catalítico de forma diferente à dos
compostos oxadiazolínicos. Os resultados apresentam-se coerentes com a
maior atividade antifúngica detectada em compostos pertencentes à série
oxadiazolínica.

FBT/FCF/USP
Ieda Yuriko Sonehara
2
ABSTRACT
This study had as objective the identification of potential targets for 5-
nitro-heterocyclic compounds with azomethynic or oxadiazolynic structures
presenting dual antifungal and anti-T. cruzi bioactivity. The studied
compounds belong to four closely related series: AzoO series: 4-substituted
5-nitro-2-furfurylidene benzhydrazides; AzoS Series: 4-substituted 5-nitro-2-
thiophylidene benzhydrazides; OxaO Series: 4-substituted 3-acetyl-
oxadiazolyne 2,5-furfurylidene benzhydrazides; and OxaS Series:
4-substituted 3-acetyl-oxadiazolyne 2,5-thiophilydene benzhydrazides.
The compounds showed antifungal activity against Candida albicans,
ATCC 537Y strain, with highest activity presented by the compound
OxaS-CH3 (MIC 3,28 µg/mL). It was also demonstrated that the compounds
present a growth inhibition action on epimastigote forms of Trypanosoma
cruzi, Y strain.
Analysis of virtual profiling results pointed among the possible targets
the P450 enzymes CYP19A (aromatase) and CYP3A4, which also interact
with azole antifungal compounds whose target is CYP51 (14α-demethylase).
The inhibition of this enzyme is responsible for the antifungal and anti-T. cruzi
activity of azole compounds, and was thus investigated as a possible target
responsible for the dual action of the studied compounds.
Characterization studies of CYP51 catalytic cavity considering not only
physicochemical properties descriptors but also steric characteristics, allied
to docking studies, confirmed the possibility of interaction of compounds from
the oxadiazolynic series with CYP51, in an orientation similar to that of azole
antifungals. The azomethynic series do not present an adequate
conformation for the interaction as proposed; however, the interaction of
compounds in a different way from that of oxadiazolynic compounds cannot
be excluded. The results are also coherent with the higher antifungal activity
detected in compounds from the oxadiazolinic series.

FBT/FCF/USP
Ieda Yuriko Sonehara
3
1. INTRODUÇÃO

FBT/FCF/USP
Ieda Yuriko Sonehara
4
Nos últimos anos houve aumento significativo na ocorrência de
infecções fúngicas graves devido, principalmente, ao aumento da população
suscetível. Fungos são, caracteristicamente, patógenos oportunistas, tendo
como alvo pacientes cujo sistema imunológico é incapaz de fazer frente a seu
ataque; assim, avanços na medicina tais como procedimentos de transplante
e regimes intensivos de quimioterapia acabaram por levar ao aumento no
número de pessoas expostas à infecção fúngica na medida em que tais
procedimentos tornaram-se mais comuns e/ou acessíveis, causando aumento
da população imunodeprimida [CHIH-CHENG et al, 2008]. Ao mesmo tempo,
a melhoria no acesso e qualidade do atendimento médico, aliado a um efetivo
controle da maioria das infecções bacterianas, resulta em aumento de
sobrevida em pacientes críticos, tendo como resultado um maior risco de
ocorrência de infecções fúngicas oportunistas [HOF, 2008; RICHARDSON,
LASS-FLÖRL, 2008]. A síndrome de imunodeficiência adquirida, (Acquired
Immunodeficiency Syndrome, AIDS), também possui papel importante nos
índices de morbidade e mortalidade relacionados à infecções fúngicas; em
países subdesenvolvidos ou em desenvolvimento apresentando epidemias de
AIDS, estas infecções ainda são uma importante causa de morbidade e
mortalidade [HOF, 2008].
Atualmente, apesar do aumento no número de fármacos antifúngicos
disponíveis no mercado, fatores como toxicidade, biodisponibilidade variável,
interações medicamentosas e surgimento de resistência aos diferentes
antifúngicos continuam a limitar as opções de tratamento; determinando a
necessidade constante de busca de novos compostos com atividade
antifúngica, visando à disponibilização de fármacos mais eficazes, com menor
toxicidade, maior espectro de ação e com menor tendência a causar
resistência [PASQUALOTTO, DENNING, 2008].
Em relação ao desenvolvimento de novos antifúngicos, compostos com
atividade potencial podem ser descobertos através do uso de diversos
instrumentos, seja pelos tradicionais métodos de triagem ou pela aplicação de
novas técnicas baseadas em genômica. O uso de diversas metodologias de
modificação e modelagem molecular permite o desenvolvimento de derivados

FBT/FCF/USP
Ieda Yuriko Sonehara
5
e análogos de compostos já conhecidos, proporcionando um melhor
entendimento dos alvos e mecanismos de ação envolvidos tanto na
patogênese como na atividade antifúngica. Desta forma, é facilitado o
desenvolvimento de fármacos mais específicos, com menor toxicidade e com
melhores perspectivas em relação ao problema de desenvolvimento de
resistência fúngica [CHAPMAN, SULLIVAN, CLEARY, 2008].
A enzima 14α-desmetilase, CYP51, pertencente ao complexo do
citocromo P450, é um dos alvos da terapia antifúngica. A CYP51, parte do
caminho biossintético de esteróis, é considerada a enzima do complexo P450
mais conservada entre os reinos, estando presente em micro-organismos,
plantas e animais [LEPESHEVA, WATERMAN, 2007; LEPESHEVA et al,
2008; DEBELJAK, FINK, ROZMAN, 2003]. Os compostos azólicos, classe a
qual pertencem os antifúngicos fluconazol e cetoconazol, entre outros, atuam
inibindo a ação desta enzima através de interação com o átomo de ferro do
grupo prostético heme presente no sítio catalítico da enzima [SHENG et al,
2009; SHENG et al, 2006; JI et al, 2000]. Entre os micro-organismos, esta
enzima é considerada como alvo de interesse no desenvolvimento de novos
compostos antifúngicos, anti-T. cruzi e tuberculostáticos [LEPESHEVA et al,
2008; LEPESHEVA et al, 2007; MATSUURA et al, 2005; BUCKNER et al,
2003; VANDEN BOSSCHE, KOYMANS, 1998].
A doença de Chagas, causada pelo Trypanosoma cruzi, protozoário
flagelado pertencente à ordem Kinetoplastida, família Trypanosomatidae, é
parte do grupo das chamadas doenças negligenciadas, com estimativa de 10
a 20 milhões de pessoas afetadas, tornando-a uma das doenças parasíticas
de maior importância nas Américas [REITHINGER et al, 2009; MONCAYO,
ORTIZ YANINE, 2006]. Esta infecção é a causa mais comum de
cardiomiopatia nas Américas Central e do Sul e é a principal responsável por
mortes devido a causas cardiovasculares em áreas onde a doença é
endêmica. Em indivíduos imunodeficientes, a doença de Chagas pode ser
reativada após alcançar o estágio crônico, com um grande número de

FBT/FCF/USP
Ieda Yuriko Sonehara
6
parasitas sendo encontrados no sangue e tecidos e levando frequentemente à
meningoencefalite severa, potencialmente fatal [REITHINGER et al, 2009].
O ciclo de vida do T. cruzi alterna hospedeiros vertebrados e insetos,
com diferentes etapas de desenvolvimento ocorrendo em cada hospedeiro
[DEVELOUS et al, 2009; MANOEL-CARNEIRO, SILVA, 2007]. A transmissão
por vetores ocorre somente nas Américas, porém a doença vem se
propagando no mundo devido à migração, estando presente nos Estados
Unidos, Europa e Japão [BERN, MONTGOMERY 2009; MILEI et al, 2009].
A eliminação da doença de Chagas implica em tratamento efetivo de
milhões de pessoas infectadas pelo T. cruzi no continente americano, assim
como em áreas não-endêmicas. Atualmente, os únicos fármacos disponíveis
para o tratamento desta doença são o nifurtimox e o benznidazol, geralmente
aceitos como eficazes no tratamento da fase aguda e crônica inicial. No
entanto, seu uso na fase crônica é ainda alvo de controvérsias e ambos os
fármacos apresentam efeitos adversos severos devido à sua toxicidade
[REITHINGER et al, 2009; JANNIN, VILLA, 2007]. O nifurtimox foi retirado do
mercado em vários países da América do Sul, sendo atualmente produzido e
utilizado principalmente na América Central [COURA, 2009]. Embora existam
avanços em estudos pré-clínicos, há mais de uma década não ocorre a
introdução de novos candidatos a fármacos em fases de estudo clínico
[REITHINGER et al, 2009].
É perceptível a necessidade da introdução de novos fármacos, tanto
antifúngicos como aqueles com ação anti-T. cruzi, de maior seletividade e
potência, menor toxicidade e capazes de atuar em casos de infecções por
micro-organismos resistentes. No caso específico da doença de Chagas, é
premente a descoberta de novos compostos que possuam menor toxicidade e
também ação na fase crônica da doença. Dentro deste cenário, compostos
nitro-heterocíclicos como os derivados 5-nitrofurfurilidênicos e 5-
nitrotiofilidênicos tem despertado grande interesse na comunidade científica
preocupada com a introdução de novos fármacos eficazes no tratamento de

FBT/FCF/USP
Ieda Yuriko Sonehara
7
doenças infecciosas [AGUIRRE et al, 2004; CARVALHO et al, 2004;
STEWART et al, 2004].
Os compostos nitrofurânicos têm sido utilizados desde a década de 40
para o tratamento de infecções, sendo os primeiros fármacos nitro-
heterocíclicos introduzidos em quimioterapia. Estudos realizados por Dodd e
colaboradores em 1944 resultaram na descoberta do nitrofural, 5-nitro-2-
furaldeído-semicarbazona, primeiro composto 5-nitrofurânico a ser utilizado
como fármaco. A síntese e ensaio de vários compostos análogos mostrou que
a maioria apresentava amplo espectro de ação, agindo não apenas sobre
bactérias, mas também sobre protozoários. Atualmente, os derivados
5-nitrofurânicos utilizados na prática médica, humana e veterinária, são
compostos sintéticos que se caracterizam estruturalmente pela presença de
anel furânico substituído na posição 5 pelo grupo nitro, sendo esta a
subestrutura mínima necessária para a atividade antimicrobiana ou
antiparasitária destes compostos [BEALE, 2004].
Sabendo-se que a atividade de um fármaco é função de interações
físico-químicas estabelecidas entre a estrutura molecular do fármaco e seu
receptor no sistema biológico e que alterações na estrutura química do
fármaco levam a modificações das propriedades físico-químicas e, por
extensão, da atividade biológica [WATERBEEMD, ROSE, 2008; TAVARES,
2004], é possível trabalhar com variações na estrutura molecular de
derivados 5-nitro-heterocíclicos, a fim de identificar análogos com melhor
atividade intrínseca e/ou melhor perfil farmacológico. Desta forma, com
interesse na otimização do perfil farmacológico de compostos 5-nitro-
heterocíclicos, análogos à nifuroxazida, 5-nitro-2-furfurilideno
4-hidroxibenzidrazida, sintetizados anteriormente e com atividade
antibacteriana e anti-T. cruzi relatadas [PAULA et al, 2009; ALMEIDA, 2009;
MASUNARI, TAVARES, 2007; MASUNARI, TAVARES, 2006; TAVARES et al,
1999; TAVARES, PENNA, AMARAL, 1997; TAVARES, 1993], foram
selecionados para estudo quanto à atividade antifúngica e determinação dos
possíveis alvos no sistema biológico.

FBT/FCF/USP
Ieda Yuriko Sonehara
8
2. FUNDAMENTAÇÃO
BIBLIOGRÁFICA

FBT/FCF/USP
Ieda Yuriko Sonehara
9
2.1. MÉTODOS DE PLANEJAMENTO DE FÁRMACOS AUXILIADOS POR
COMPUTADOR, CADD
O planejamento de fármacos auxiliados por computador, CADD,
envolve disciplinas cujos limites encontram-se diluídos e frequentemente
sobrepostos [LANGER, BRYANT, 2008] (Figura 1). Entre os campos de
estudo diretamente relacionadas ao planejamento de fármacos, a modelagem
molecular utiliza um conjunto de ferramentas computacionais para a
construção, edição e visualização tridimensional de moléculas, com cálculo de
propriedades relevantes e compreensão de sua importância na interação com
biomacromoléculas-alvo. Um segundo campo envolve o estudo das relações
quantitativas entre estrutura química e atividade biológica (Quantitative
Structure-Activity Relationships, QSAR), buscando a obtenção de modelos
que permitam a previsão de propriedades relacionadas aos fármacos, com
uso de descritores frequentemente calculados por métodos de modelagem
molecular; além disso, no caso específico de QSAR 3D, a análise
conformacional de moléculas, técnica relacionada ao campo de modelagem
molecular, é uma etapa crítica uma vez que a conformação das moléculas
envolvidas é importante para o estudo da interação de ligantes com
biomacromolécula-alvo.
Figura 1. Interrelação entre disciplinas relacionadas ao planejamento de fármacos in silico

FBT/FCF/USP
Ieda Yuriko Sonehara
10
O QSAR procura determinar a capacidade de uma molécula para
interagir com um alvo macromolecular no organismo, representada por
modelos matemáticos relacionando à características estruturais mais
importantes de um composto químico, representadas por variáveis que
descrevem alterações estruturais (descritores moleculares), com sua
respectiva influência sobre a atividade biológica (Figura 2).
Figura 2. Métodos de QSAR
Utilizam propriedades físico-químicas e dados de atividade biológica na construção de modelos capazes de aumentar a informação e compreensão existentes acerca de compostos bioativos, além da previsão de propriedades físico-químicas e de bioatividade [adaptado de WATERBEEMD, ROSE, 2008]
A quimioinformática, por sua vez, é responsável pela codificação de
compostos químicos, permitindo seu armazenamento e busca em banco de
dados; através do uso de critérios de busca como descritores calculados por
métodos de modelagem molecular ou descritores de QSAR, sendo possível a
recuperação de informações para posterior análise.
A caracterização tridimensional dos alvos e o entendimento do
mecanismo de ação são considerados essenciais para o desenvolvimento de

FBT/FCF/USP
Ieda Yuriko Sonehara
11
novos fármacos, tornando possível o planejamento de compostos baseados
na estrutura do alvo biológico (structure-based drug design, SBDD).
Interações fortes e seletivas entre fármaco e biomacromolécula-alvo
dependem de complementaridade estrutural e físico-química, tornando
necessária e de grande importância a existência de informações confiáveis
sobre a estrutura das moléculas. Além disso, a determinação da estrutura
tridimensional de proteínas permite o entendimento de sua reatividade
química possibilitando, desta forma, o planejamento de ligantes
potencialmente eficazes. Técnicas computacionais permitem o descobrimento
de novos compostos-líderes baseados na recuperação de informações
detalhadas sobre o biorreceptor através de busca em banco de dados com
possibilidade de geração de estruturas químicas favoráveis à interação com o
mesmo. É comum a seleção de compostos baseada na complementaridade
estrutural com biorreceptores cujos ligantes são conhecidos, através de
técnicas de ancoramento (docking) visando à identificação de substâncias
com atividade semelhante a estes ligantes. É também possível o
planejamento de um ligante baseado nas características conhecidas do sítio-
alvo, no chamado planejamento de novo [RUGE, KORTING, BORELLI, 2005].
As metodologias in silico permitem também o planejamento de
fármacos baseados na estrutura de ligantes conhecidos (ligand-based drug
design, LBDD). Modelos de pequenas moléculas podem ser definidos com o
auxílio de técnicas envolvendo mecânica molecular, dinâmica molecular,
mecânica quântica e análise conformacional, enquanto propriedades
estruturais e físico-químicas de compostos ativos e inativos, além de relações
estrutura-atividade, podem ser utilizadas para caracterizar topograficamente
as enzimas, no procedimento conhecido como mapeamento de biorreceptores
ou identificação de farmacóforos. Em outro exemplo, modelos obtidos com
aplicação de técnicas de comparação de campos moleculares ou índices de
similaridade molecular de diferentes ligantes podem ser utilizados no
mapeamento das propriedades de receptores desconhecidos, na construção
de hipóteses de interação ligante-receptor, na otimização de estruturas-
protótipo, no planejamento de novos compostos ativos e na previsão de

FBT/FCF/USP
Ieda Yuriko Sonehara
12
atividades biológicas [EKINS, MESTRES, TESTA, 2007; RUGE, KORTING,
BORELLI, 2005; MESTRES, 2004].
Para uma navegação eficiente no espaço dos ligantes (espaço químico)
com objetivo de identificação de possíveis compostos-líderes na busca de
novos fármacos é necessária a descrição dos compostos usando
propriedades adequadas (descritores) e, em seguida, a utilização de uma
equação-mestre para medir a distância entre dois compostos (similaridade
métrica). Descritores podem ser uni-, bi-, tri-, ou quadridimensionais; os mais
simples e de maior facilidade de cálculo são os descritores unidimensionais,
que descrevem propriedades globais (peso molecular, contagem de ligações e
átomos, por exemplo) que podem ser derivadas a partir da fórmula química.
Estes descritores são capazes de refletir de forma adequada o tamanho,
forma e lipofilicidade das moléculas. Por outro lado, a maior parte dos
descritores de ligantes são bidimensionais, ou seja, descritores topológicos
onde a tabela de conectividade (lista de átomos e ligações) codifica as
propriedades de átomos e ligações ao mesmo tempo. O exemplo mais
simples deste tipo de descritor é a representação 2-D da estrutura química,
que permite a busca, em uma biblioteca de ligantes, de compostos partilhando
um padrão bidimensional (fragmento, subestrutura) em particular. Os modelos
derivados com a aplicação destes descritores são habitualmente referidos
como QSAR-2D, metodologia reconhecidamente capaz de predizer
propriedades físico-químicas e estimar quantitativamente diversos efeitos
biológicos. Contrastando com os descritores bidimensionais, descritores
tridimensionais codificam propriedades relacionadas à conformação
(coordenadas atômicas, farmacóforos 3-D, campos, potenciais, espectro), e
geralmente necessitam de um alinhamento comum entre as moléculas para
permitir a comparação no mesmo espaço cartesiano, e de uma amostragem
do espaço conformacional ocupado pelas moléculas individuais. Modelos
utilizando estes descritores são conhecidos como modelos de QSAR-3D
[EKINS, MESTRES, TESTA, 2007; ROGNAN, 2007; ENGEL, 2006;
MESTRES, 2004].

FBT/FCF/USP
Ieda Yuriko Sonehara
13
Com a finalidade de se evitar algumas das limitações características
dos estudos de QSAR 3D, notadamente os problemas de determinação de
conformação ativa e alinhamento adequado entre as moléculas, foi
desenvolvido o QSAR 4D, que faz uso dos conceitos da Teoria de Ensembles
da termodinâmica [LARANJEIRAS, CHIAPPIN, 2008; CRAMER, 2004]. Nesta
técnica, o conjunto total de possíveis conformações de uma molécula
(ensemble conformacional) é gerado por dinâmica molecular e utilizado para
gerar perfis que fornecem a base para a escolha de descritores e elaboração
de modelos. A quarta dimensão de uma análise de QSAR 4D é a “dimensão”
da amostragem de ensemble [HOPFINGER et al, 1997]. Os descritores
quadridimensionais, utilizados em QSAR 4D, incorporam desta forma a
variedade espacial das moléculas ao representar cada composto em
diferentes conformações, orientações, tautômeros, estereoisômeros ou
estados de protonação, com o modo de interação real ou conformação
bioativa identificada através do uso de algoritmos próprios [LILL, 2007].
Os descritores bidimensionais mais comuns utilizados na
caracterização de moléculas são as “impressões digitais” (fingerprints)
topológicas, que codificam a presença de fragmentos subestruturais em
compostos químicos [STANTON, 2008; EKINS, MESTRES, TESTA, 2007;
ESTRADA, URIARTE, 2001]. Em contraste com estes, existem os descritores
baseados na representação espacial das estruturas de moléculas; um
exemplo são os descritores baseados na superposição flexível de moléculas
sobre uma ou mais conformações bioativas de um ligante de referência.
Dentro da classe de descritores topológicos, o uso de distribuições de pares
de características centrados no átomo (atom-centered feature pairs) tem-se
mostrado eficaz em várias aplicações [EKINS, MESTRES, TESTA, 2007;
GREGORI-PUIGJANÉ, MESTRES, 2006; NETTLES et al, 2006; SCHNEIDER
et al, 1999; WILLETT, 1998].
Pares de átomos são subestruturas dentro de uma estrutura química,
consistindo de dois átomos, diferentes de H e não necessariamente
conectados entre si, e sua separação interatômica. Esta separação é medida
como a menor distância, expressa em número de ligações, conectando ambos

FBT/FCF/USP
Ieda Yuriko Sonehara
14
os átomos do par escolhido (Figura 3) [GREGORI-PUIGJANÉ, MESTRES,
2006; WILLETT, 1998; CARHART, 1985]. A codificação computacional dos
descritores baseados em pares de átomos tenta capturar sua distribuição
geral em uma molécula armazenando a ocorrência das características
centradas nos pares de átomos a diferentes distâncias, seja em nível
topológico ou geométrico, formando uma representação molecular por
fingerprint compartimentalizado (binned fingerprint) [GREGORI-PUIGJANÉ,
MESTRES, 2006].
Deve-se lembrar que funções baseadas em descritores de pares de
átomos em nível geométrico requerem coordenadas tridimensionais e, desta
forma, fornecem representações dependentes da conformação da molécula; a
transformação de coordenadas atômicas bidimensionais em tridimensionais
tem a potencialidade de gerar múltiplos confôrmeros de cada molécula, sendo
que nem todas são adequadas à interação com o alvo. Por outro lado,
descritores de pares de átomos em nível topológico podem não capturar todas
as informações essenciais presentes quando se utilizam coordenadas
tridimensionais [EKINS, MESTRES, TESTA, 2007; WILLETT, 1998;
SHERIDAN et al, 1996].
distância entre par de carbonos carbonílicos = 3 distância entre par de carbonos -CO-CH2CO = 2
Figura 3. Determinação de distância interatômica entre pares de átomos

FBT/FCF/USP
Ieda Yuriko Sonehara
15
2.1.1 Análise das Relações Quantitativas entre Estrutura Tridimensional e
Atividade, QSAR 3D
O QSAR tradicional, também chamado de QSAR 2D ou Análise de
Hansch [TAVARES, 2004; DEBNATH, 2001], é capaz de prever de forma
precisa a potência de compostos adicionais aos estudados inicialmente,
identificando, através das propriedades físico-químicas, quais variações
estruturais influenciam de forma mais importante as propriedades biológicas
analisadas [LANGER, BRYANT, 2008; TAVARES, 2004; DEBNATH, 2001].
Esta técnica, no entanto, apresenta limitações pelo fato de permitir aplicação
somente a conjuntos de moléculas que possuam um núcleo estrutural comum
e, principalmente, por não considerar o equilíbrio conformacional ou a
estrutura tridimensional, conhecidos fatores críticos para a interação com
biomacromoléculas e, portanto, para a determinação de atividade biológica
[LANGER, BRYANT, 2008; CRUCIANI, CAROSATI, CLEMENTI, 2003].
O QSAR 3D, por outro lado, pode ser considerado uma relação de
QSAR na qual os descritores moleculares possuem natureza tridimensional,
sendo geralmente calculados com auxílio de técnicas de modelagem
molecular. A premissa inicial deste método considera que as características
mais importantes na determinação da atividade de compostos bioativos são
forma, tamanho e propriedades eletrônicas. De modo análogo ao QSAR
tradicional ou Análise de Hansch, no QSAR 3D diversos compostos são
estudados simultaneamente em um modelo de regressão, com o objetivo de
identificar quais características moleculares influenciam de modo significativo
a resposta biológica de uma série de compostos [BLOCK, 2004; CRUCIANI,
CAROSATI, CLEMENTI, 2003].
O maior impulso no campo do QSAR 3D ocorreu com a introdução, por
Cramer, Patterson e Bunce em 1988, da técnica de Análise Comparativa de
Campos Moleculares (Comparative Molecular Field Analysis), CoMFA,
baseada em estudos anteriores desenvolvidos por Goodford [1985]. Esta
técnica, assim como ocorre em grande número das metodologias de QSAR
3D, analisa superfícies tridimensionais que representam a distribuição de

FBT/FCF/USP
Ieda Yuriko Sonehara
16
propriedades físico-químicas no espaço ao redor de uma molécula; a
informação extraída deste estudo é alimentada em um protocolo de regressão
multivariada com a finalidade de obter correlações entre as características
físico-químicas e a atividade biológica desempenhada pelos compostos
estudados [BARBANY et al, 2004; CRAMER, PATTERSON, BUNCE, 1988]. O
resultado de uma análise de QSAR 3D é tipicamente apresentado como
mapas de contorno superpostos a uma molécula representativa da série de
compostos estudada, permitindo a fácil visualização das regiões do espaço
onde determinadas características físico-químicas, como por exemplo
hidrofilicidade, lipofilicidade, ou potencial elestrostático, podem ser
importantes para a interação com o receptor.
Os campos de interação molecular (Molecular Interaction Fields, MIF),
também chamados potenciais de interação molecular (Molecular Interaction
Potentials, MIP), identificam regiões onde determinados grupos químicos
podem interagir favoravelmente e são propriedades moleculares que
representam as energias de interação entre os compostos de interesse e
sondas químicas (probes) que fazem o papel de fragmentos moleculares
importantes para o reconhecimento inter-molecular. Além da aplicação em
estudos de QSAR 3D, os campos de interação molecular podem ser utilizados
em SBDD, docking de biomacromoléculas, e previsão de propriedades
farmacocinéticas [LANGER, BRYANT, 2008; SIPPL , 2002; CRUCIANI,
PASTOR, GUBA, 2000; CRUCIANI et al, 2000; CRAMER, PATTERSON,
BUNCE, 1988].
Para a determinação de MIF, os compostos em estudo são alinhados
em uma grade (grid) tridimensional virtual, com as energias de interação
composto-sonda sendo calculadas nos vértices da grade e representadas
como superfícies isopotenciais [BARBANY et al, 2004; PASTOR et al, 2004;
RODRIGO et al, 2002]. Métodos de regressão multivariada, como por exemplo
Análise de Componentes Principais (Principal Component Analysis, PCA) e
Mínimos Quadrados Parciais (Partial Least Squares, PLS), relacionam a
atividade biológica de séries de compostos com descritores baseados em MIF

FBT/FCF/USP
Ieda Yuriko Sonehara
17
e são obtidos com a utilização de programas computacionais como CoMFA
[CRAMER, PATTERSON, BUNCE, 1988], GRID/GOLPE [PASTOR et al,
2000], Volsurf [CRUCIANI, PASTOR, GUBA, 2000; CRUCIANI et al, 2000] e
ALMOND [PASTOR et al, 2000] (Figura 4).
Figura 4. Fluxo de trabalho para CoMFA.
[LANGER, BRYANT, 2008]
A maior dificuldade apresentada na aplicação de métodos de QSAR 3D
dependentes de alinhamento, como CoMFA e similares, é a necessidade da
determinação da orientação espacial relativa dos ligantes, que representa
diferenças nas geometrias de ligação entre o composto e seu sítio de ligação.
Esta análise é reconhecidamente uma das etapas mais complexas e
demoradas de uma análise de campos moleculares, e determina seu sucesso
[PASTOR et al, 2000; CRAMER, PATTERSON, BUNCE, 1988]. Descritores
independentes da posição e orientação das estruturas moleculares no espaço,
como GRIND (GRid-INdependend Descriptors) [PASTOR et al, 2000], utilizado
pelo ALMOND, e os descritores utilizados pelo Volsurf foram desenvolvidos
para permitir estas análises sem a necessidade de alinhamento de moléculas.
A não-dependência do alinhamento de estruturas permite seu uso em outros
campos além do QSAR 3D, como, por exemplo, a pesquisa tridimensional,
identificação de farmacóforos e relações estrutura-metabolismo [FONTAINE et
al, 2003; PASTOR et al, 2000]. Outros descritores que procuram aproximar os

FBT/FCF/USP
Ieda Yuriko Sonehara
18
métodos dependentes de alinhamento com os independentes têm sido
desenvolvidos, como é o caso de Anchor-GRIND [FONTAINE et al, 2005].
Um estudo de QSAR 3D, tipicamente CoMFA, compreende as
seguintes etapas [LANGER, BRYANT, 2008; DEBNATH, 2001]:
1. Seleção de conjunto de treinamento e conjunto de avaliação;
2. Construção tridimensional dos ligantes;
3. Alinhamento e superposição moleculares;
4. Cálculos dos descritores de campo molecular tridimensionais;
5. Geração de modelo utilizando métodos de regressão multivariada;
6. Validação cruzada do modelo (métodos de leave-one-out e leave-N-out);
7. Previsão.
A qualidade e confiabilidade de um modelo de QSAR 3D são
dependentes da análise cuidadosa de cada etapa do estudo, em especial a
determinação de conformação e superposição moleculares e os cálculos de
descritores, capazes de influenciar significativamente os resultados, mesmo
quando um mesmo conjunto inicial de compostos é utilizado na geração do
modelo [LANGER, BRYANT, 2008; DEBNATH, 2001; KLEBE, ABRAHAM,
1993]. Além disso, assim como nos métodos de QSAR tradicionais, deve-se
considerar cuidadosamente a qualidade dos resultados de atividade biológica;
idealmente, estes devem ter sido obtidos no mesmo laboratório e em
condições de ensaio idênticas, e os resultados devem abranger pelo menos
três ordens de magnitude em potência. Todos os compostos testados devem
ter o mesmo mecanismo de interação com o alvo e sua estrutura
tridimensional deve ser bem definida. Compostos nos quais a estereoquímica
é desconhecida devem ser excluídos dos estudos [LANGER, BRYANT, 2008].
Em uma análise típica de QSAR 3D é necessário o alinhamento dos
compostos de acordo com sua orientação quando ligados ao alvo, ou seja, em
sua conformação bioativa, com a superposição de todas as estruturas
tridimensionais servindo como entrada de dados (input) para os cálculos de
campo molecular. No entanto, diversos fatores colaboram para que esta
situação ideal não seja a mais frequentemente encontrada. Em primeiro lugar,

FBT/FCF/USP
Ieda Yuriko Sonehara
19
mesmo que a estrutura do sítio de ligação do alvo seja conhecida, diferentes
ligantes podem interagir em diferentes orientações, sendo necessária uma
escolha que seja representativa da orientação de ligação. E, em segundo
lugar, se a estrutura do alvo não é conhecida, é necessário trabalhar com uma
orientação hipotética do ligante, com a superposição sendo realizada de
acordo com a hipótese considerada.
Deve-se lembrar que na maioria dos casos, tanto a conformação
bioativa de um composto como seu modo de ligação ao alvo não são
conhecidas. Além disso, uma desvantagem dos descritores moleculares
tridimensionais que deve ser considerada é sua sensibilidade à posição e
orientação das estruturas moleculares no espaço, resultando em diferentes
modelos gerados para um mesmo conjunto de dados em orientações
diferentes [LANGER, BRYANT, 2008]. Quando os ligantes são flexíveis a
superposição torna-se bastante complexa e, em muitos casos, completamente
subjetiva.
Uma vez que nenhum programa estatístico é capaz de detectar um mal
alinhamento, a validação do modelo de forma independente da qualidade de
superposição dos compostos é inviável; a única validação possível neste caso
é a interpretação do modelo ligada a uma análise cuidadosa dos resultados
experimentais [LANGER, BRYANT, 2008]. É compreensível, assim, o esforço
no desenvolvimento de métodos que permitam a obtenção de descritores
moleculares tridimensionais independentes de alinhamento.
Campos moleculares são definidos como sendo a área de influência de
uma determinada propriedade molecular sobre o espaço que circunda a
molécula, espaço este definido por uma grade de pontos tridimensional de
resolução conhecida, dependente do usuário, e que circunda todos os
compostos, superpostos, da série estudada [LANGER, BRYANT, 2008;
CRAMER, PATTERSON, BUNCE, 1988]. Existem várias propriedades de uma
molécula que podem ser medidas como campos, sendo as mais comuns as
propriedades estéricas e eletrostáticas [LANGER, BRYANT, 2008; CRUCIANI
et al, 2000].

FBT/FCF/USP
Ieda Yuriko Sonehara
20
Para a determinação dos campos moleculares, sondas químicas
adequadas percorrem os pontos da grade virtual determinada ao redor das
moléculas; as energias de interação entre sonda e a molécula são medidas
em cada um destes nodos, produzindo ao final uma matriz de valores de
energia de interação. A função mais simples que é utilizada em campos de
força para representar a combinação das energias de dispersão e repulsão
define o potencial de Lennard-Jones (U):
612)(
AB
AB
AB
ABAB
r
b
r
arU −=
onde a e b são constantes específicas aos átomos A e B, separados por uma
distância r [CRAMER, 2004].
O potencial eletrostático molecular (Molecular Electrostatic Potential,
MEP) permite calcular o grau de atração ou repulsão entre carga pontual
unitária e molécula, sendo particularmente útil quando visualizada em
superfícies ou regiões do espaço, pois fornece informações sobre a
polaridade local. Para uma posição r, o MEP pode ser calculado por
[CRAMER, 2004]:
∑ ∫ Ψ−
Ψ−−
=núcleo
k k
kMEP drr
rrr
rr
ZrV ')'(
|'|
1)'(
||)(
onde Zk representa a carga nuclear no átomo k e Ψ é o operador
Hamiltoniano.
Os átomos ou fragmentos utilizados como sondas são aqueles típicos
de serem encontrados em uma molécula de fármaco; átomos tipicamente
utilizados são C, H, N e O. Fragmentos são comumente C=O, CO2-, NH, entre
outros [LANGER, BRYANT, 2008]. De acordo com o procedimento
computacional utilizado e da natureza da sonda, o campo de energias de
interação obtido pode representar, entre outros, campos estéricos ou
eletrostáticos, interações hidrofóbicas ou densidades eletrônicas. Estes
campos podem ser utilizados como descritores pontuais tanto da estrutura

FBT/FCF/USP
Ieda Yuriko Sonehara
21
molecular tridimensional como do comportamento físico-químico da molécula,
uma vez que a maioria das propriedades relacionadas a interações
moleculares podem ser representadas por MIF. De forma geral, não se pode
dizer que existe um conjunto de descritores ideal para a obtenção de um
QSAR 3D; a escolha do conjunto a ser utilizado depende principalmente do
objetivo do estudo.
A etapa final de toda análise de QSAR 3D consiste em uma validação
estatística completa [LANGER, BRYANT, 2008], de forma a avaliar a
significância do modelo e, portanto, sua habilidade em prever atividades
biológicas de novos compostos. No entanto, análises de correlação clássicas
tais como métodos de regressão linear múltipla não são adequados em casos
de matrizes de dados não-simétricas contendo mais variáveis do que dados
observados, como é o caso dos modelos de QSAR 3D, em que existem
centenas de descritores de campos moleculares em relação ao número de
valores de atividade biológica. A validação estatística destes modelos utiliza
ferramentas não-clássicas, como PLS e PCA, que permitem a redução de
complexidade e simplificação de dados.
PCA não considera diretamente a atividade biológica, e condensa a
informação total contida nas variáveis descritivas em duas matrizes menores
em que descritores correlacionados encontram-se agrupados em uma nova
variável, chamada de componente principal. As componentes principais não
são correlacionadas e representam combinações lineares dos descritores
originais, sendo definidas em ordem decrescente da quantidade de variância
que são capazes de explicar. PLS permite a otimização das componentes
principais para descrever a relação entre descritores e atividades, através da
definição de variáveis latentes [FERREIRA, 2002; FERREIRA, MONTANARI,
GAUDIO, 2002].
O método mais comum de validação é o chamado método de validação
cruzada leave-one-out (LOO), em que um objeto é retirado do conjunto de
dados a cada vez, e previsto pelo modelo gerado em sua ausência. Após
aplicação para todos os compostos, são obtidos os parâmetros estatísticos

FBT/FCF/USP
Ieda Yuriko Sonehara
22
para validação do modelo, coeficiente de correlação de validação cruzada q2 e
desvio-padrão do erro de previsão (standard deviation of error of prediction,
SDEP), que são considerados os melhores critérios de robustez e capacidade
preditiva do modelo (Figura 5). Uma variação deste método que permite a
obtenção de modelo mais robusto é a validação cruzada por leave-N-out,
onde de 20 a 30% dos objetos é retirado do conjunto de dados, devendo ser
prevista pelo modelo gerado [LANGER, BRYANT, 2008].
Deve-se lembrar que métodos de validação cruzada avaliam um
modelo pela sua capacidade preditiva e, portanto, podem levar à exclusão de
compostos que trazem informações de SAR úteis, mas que não contribuem
para a capacidade preditiva do modelo em geral [CRAMER, WENDT, 2007].
Isto pode ocorrer, por exemplo, caso um único composto possua atividade
biológica desejável devido a características também únicas; ao ser retirado do
modelo durante a análise, não é capaz de ser previsto pelo modelo gerado,
levando a um valor de q2 artificialmente diminuído e influenciando as
conclusões sobre a existência de QSAR significante. Este composto único
pode, no entanto, representar um potencial composto-líder se adequadamente
tratado.

FBT/FCF/USP
Ieda Yuriko Sonehara
23
Figura 5. Processo de validação cruzada por leave-N-out. *SDEP = desvio-padrão do erro de previsão (Standard Deviation of Error of Prediction) [Adaptado de BLOCK, 2004]
A. Técnicas de QSAR 3D independentes de alinhamento
Com o objetivo de se evitar os problemas causados por erros durante o
procedimento de alinhamento das moléculas novos métodos para cálculo de
descritores derivados de MIFs foram desenvolvidos, sendo o GRIND e o
Volsurf os mais conhecidos.
Uma molécula pode ser caracterizada em termos de seu potencial de
formar ligações polares, hidrofóbicas, eletrostáticas e de hidrogênio no espaço
tridimensional. O Volsurf [CRUCIANI, PASTOR, GUBA, 2000] é um
procedimento computacional capaz de converter o tamanho e a distribuição

FBT/FCF/USP
Ieda Yuriko Sonehara
24
espacial dos contornos destas interações moleculares em descritores
bidimensionais de significado químico claro, sem necessidade de alinhamento
das moléculas. A idéia básica é a compressão de informação presente nos
mapas tridimensionais em alguns poucos descritores numéricos
bidimensionais, facilitando o entendimento e interpretação.
Os descritores Volsurf, por codificarem propriedades físico-químicas,
são planejados especialmente para a otimização in silico de propriedades
farmacocinéticas, que são por sua vez ligadas a propriedades físico-químicas.
A metodologia utilizada pelo Volsurf, apesar de ter sido desenvolvida com foco
em relações estrutura-propriedade (Quantitative Structure-Property
Relationthips, QSPR), pode ser também aplicada a relações estrutura-
atividade e estudos de diversidade molecular baseados em propriedades da
superfície molecular [CRUCIANI, PASTOR, GUBA, 2000].
No procedimento básico, campos de interação determinados com
sonda aquosa (OH2) e hidrofóbica (DRY) são calculados para todas as
moléculas do conjunto de dados. Os descritores moleculares obtidos referem-
se a tamanho e forma moleculares, tamanho e forma de regiões hidrofílicas e
hidrofóbicas, momentos anfifílicos e parâmetros de empacotamento crítico,
entre outros.
Os descritores GRIND [PASTOR et al, 2000], utilizados no programa
ALMOND, são também derivados diretamente de MIF. Com o uso de
diferentes sondas é possível determinar um conjunto de regiões em um
ligante em que grupos de um receptor em potencial apresentam a
possibilidade de interação favorável; estas posições constituem o chamado
sítio receptor virtual (Virtual Receptor Site, VRS), que define um sítio
complementar ideal para um composto químico e representa sua habilidade
potencial de interação com uma biomacromolécula. O conjunto de variáveis
representando a relação espacial entre regiões relevantes do VRS define o
GRIND.
Uma variação do GRIND é o chamado Anchor-GRIND [FONTAINE et
al, 2005], uma aplicação particular para casos onde há pelo menos um ponto

FBT/FCF/USP
Ieda Yuriko Sonehara
25
na estrutura comum dos compostos que pode ser considerado comum a todos
os elementos da série estudada, por razões químicas ou biológicas. Este
ponto comum é então utilizado como ponto de referência na determinação dos
descritores, resultando em uma descrição espacial mais precisa das regiões
MIF. Uma restrição existente desta aproximação, no entanto, é a necessidade
de uma estrutura química básica em comum para toda a série.
As características mais importantes do GRIND são sua independência
de alinhamento, rapidez de cálculo e relevância na descrição de propriedades
biológicas. A maior limitação desta metodologia é a sensibilidade à
conformação das estruturas, um problema comum nas análises de QSAR 3D;
idealmente o VRS deve ser obtido a partir da conformação bioativa do ligante,
com as sondas representando grupos químicos presentes no sítio ativo.
B. Vantagens e desvantagens dos métodos de QSAR 3D
Os métodos de QSAR 3D apresentam facilidade de visualização de
resultados finais, pois as interações favoráveis e desfavoráveis são
representadas graficamente por contornos tridimensionais ao redor de
molécula representativa da série estudada. Enquanto o QSAR tradicional não
sugere diretamente quais novos compostos podem apresentar atividade
biológica favorável, a análise das áreas onde são detectadas interações
favoráveis e desfavoráveis no QSAR 3D permite o planejamento de novas
estruturas [PATRICK, 2009].
Ao contrário dos métodos tradicionais, no QSAR 3D não existe
restrição quanto à presença de substituintes incomuns ou grau de similaridade
estrutural dentro da série de compostos a ser analisada, pois os descritores
são calculados individualmente por programas computacionais específicos; no
entanto, as moléculas devem necessariamente possuir o mesmo farmacóforo
e o mesmo mecanismo de interação com o receptor [PATRICK, 2009;
LANGER, BRYANT, 2008].

FBT/FCF/USP
Ieda Yuriko Sonehara
26
Existem alguns problemas potenciais que estão intrinsecamente ligados
às metodologias de QSAR 3D; o maior deles sem dúvida envolve a
necessidade de conhecimento da conformação ativa dos compostos
estudados e, em métodos como CoMFA, o fato de ser imprescindível o
alinhamento e superposição corretos das moléculas para obtenção de um
modelo válido. Relacionado a estes dois fatores, existe também a
necessidade de similaridade na interação com o receptor, ou seja, os
compostos devem interagir com o receptor em orientação semelhante
[PATRICK, 2009; LANGER, BRYANT, 2008].
Outra desvantagem dos métodos de QSAR 3D é a limitação no
tamanho do conjunto de moléculas estudadas, não maior que algumas
centenas de compostos, impedindo seu uso em aplicações de ensaios
robotizados em grande escala, high-throughput screening, HTS, que permitem
o estudo de centenas de compostos em um período de tempo mínimo
[LANGER, BRYANT, 2008]. No entanto, Cramer e Wendt [2007]
mencionaram, em estudo recente, o uso de protocolos de alinhamento por
topômeros na obtenção de QSAR 3D que possibilita a triagem de bancos de
dados; neste tipo de protocolo, o estado conformacional inicial das moléculas
é ignorado e o alinhamento ocorre através de regras que determinam valores
de valência e geometria de anéis, com posterior padronização de torções e
quiralidade. Este tipo de protocolo considera a molécula localmente, sem
considerar presença de outros ligantes similares ou interação com cavidade
de bioreceptores.
2.2. 14α-DESMETILASE (CYP51): ESTRUTURA E FUNÇÃO
A família do citocromo P450 (CYP), existente em todos os reinos
biológicos, compreende várias enzimas contendo grupo prostético heme, cuja
função primária é a oxidação de diversos substratos hidrofóbicos. Suas
funções principais são o metabolismo de compostos exógenos em preparação
para a sua excreção, e biossíntese de moléculas sinalizadoras essenciais na

FBT/FCF/USP
Ieda Yuriko Sonehara
27
homeostase e desenvolvimento dos organismos. Em mamíferos, as enzimas
do complexo P450 metabolizam fármacos e xenobióticos e estão envolvidas
na biossíntese de compostos de baixo peso molecular que atuam como
reguladores em diferentes processos no organismo [BROWN, REISFELD,
MAYENO, 2008; LAMB, WATERMAN, KELLY, 2007; OTYEPKA et al, 2007;
MESTRES, 2005; ANZENBACHER, ANZENBACHEROVÁ, 2001].
O mecanismo de ação do P450 é uma cascata complexa envolvendo
interação com outras proteínas durante o processo redox, utilizando NAD(P)H
como fonte de elétrons. As proteínas mais frequentemente envolvidas na
reação são, em eucariotas, citocromo P450 redutase e, em procariotas,
ferredoxina/ferredoxina redutase. Entre as reações enzimáticas básicas
catalizadas por CYPs encontram-se C-hidroxilação, oxigenação e
dealquilação de heteroátomos, formação de epóxido e migração de grupos.
Reações mais complexas incluem oxigenação de cloro, dealogenação
aromática, formação de dímeros, migração oxidativa de grupos arílicos e
fusão, contração, e formação de anéis [BROWN, REISFELD, MAYENO, 2008;
LAMB et al, 2007; OTYEPKA et al, 2007; DENISOV et al, 2005] (Figura 6).

FBT/FCF/USP
Ieda Yuriko Sonehara
28
Figura 6. Algumas reações catalizadas por enzimas da família P450.
Extraído de LAMB et al, 2007.
Em todos os CYPs, um grupo prostético heme é ligado à proteína
através da interação do átomo de ferro em seu estado férrico com um ânion
tiolato formado pelo átomo de enxofre de uma cisteína proximal
absolutamente conservada, encontrando-se inserido entre as α-hélices I distal
e L proximal. Este ligante tiolato origina a absorbância de Soret característica
em 450 nm. A sequência primária das enzimas do complexo P450 é
diversificada, fato compreensível ao considerar-se que esta superfamília de
enzimas necessita acomodar uma grande variedade de substratos de
diferentes características tridimensionais. Embora a identidade de sequência
entre as enzimas P450 seja menor que 20%, o dobramento como um todo
permaneceu inalterado através de toda a evolução, assim como um núcleo
estrutural ligado ao grupo heme. A Figura 7 mostra o dobramento comum
encontrado em CYPs, constituído principalmente de α-hélices de arquitetura
ortogonal segundo a classificação CATH [ORENGO et al, 1997], com o

FBT/FCF/USP
Ieda Yuriko Sonehara
29
segmento F/G (α-hélice F, alça F/G e α-hélice G) perpendicular à α-hélice I
altamente conservada em região muito próxima ao grupo heme. A α-hélice I
possui uma curvatura na região próxima ao grupo heme e estende-se através
de toda a estrutura do CYP [ANZENBACHER et al, 2008; OTYEPA et al,
2007; MESTRES, 2005].
Figura 7. Dobramento comum de P450 mamífero CYP2C9, código pdb 1og2 (resolução 2,6 Å), representado em cartoon. Heme em representação CPK [ANZENBACHER et al, 2008],
As regiões mais conservadas nas CYPs de mamíferos são α-hélice E,
porção C-terminal da α-hélice I, α-hélices J e K, folhas β6-1 e β1-3, α-hélices
K’ e K”, bolso cisteínico, α-hélice L, e folha β3-2 [OTYEPA et al, 2007;
MESTRES, 2005] (Figura 8). Existe ainda semelhança estrutural entre P450s
de mamíferos e micro-organismos, diferindo em detalhes que são
responsáveis por diferentes características de ligação de substrato. A maior
diferença ocorre no posicionamento das α-hélices F e G e da alça F/G, que
formam o teto da cavidade de ligação do substrato em conjunto com a alça
B/C [OTYEPA et al, 2007; PODUST, POULOS, WATERMAN, 2001].

FBT/FCF/USP
Ieda Yuriko Sonehara
30
Figura 8. Conservação de sequência primária em P450
Conservação representada em valores de RMSD (desvio quadrático médio, Root Mean Square Deviation) entre estruturas de 9 citocromos P450. Variação representada em escala de cores de vermelho (mínima) a azul (máxima). Representação em cartoon de CYP101 (cód. PDB 1phc, resolução 1,6 Å). Heme em representação palito (vermelho), átomo de ferro central representado como esfera. [MESTRES, 2005].
Os CYPs são considerados de interesse no desenvolvimento de
fármacos tanto por seu papel na metabolização e processos de destoxificação
de substâncias exógenas, como pelo seu potencial como alvos para novos
fármacos quando responsáveis por processos regulatórios no metabolismo.
O combate a infecções causadas por patógenos microbianos tem
encontrado dois problemas principais, a emergência de novas espécies
patogênicas e o crescente desenvolvimento de resistência de micro-
organismos já conhecidos aos quimioterápicos tradicionalmente utilizados. A
identificação de novos alvos é uma necessidade constante dentro da busca
por fármacos quimioterápicos mais eficazes e, embora somente alguns
poucos micro-organismos tenham revelado a presença de genes codificando

FBT/FCF/USP
Ieda Yuriko Sonehara
31
CYPs, a importância desta família de enzimas para o metabolismo e
desenvolvimento de organismos leva a um crescente interesse no seu uso
como alvo potencial para fármacos quimioterápicos [LAMB et al, 2007].
Os CYPs catalizam ainda uma grande parte das reações mais
complexas na biossíntese de diversos produtos naturais utilizados na prática
médica, como por exemplo vancomicina, anfotericina B, artemisina e taxol
entre muitos outros. Na maioria dos casos, as enzimas do P450 possuem
papel essencial nos processos oxidativos que levam à biossíntese final destes
compostos. A manipulação genética destes sistemas enzimáticos, por
exemplo através de deleção ou mutação de aminoácidos na sequência
proteica da enzima, pode permitir a obtenção de compostos modificados com
finalidade de melhoria de atividade ou diminuição de toxicidade [LAMB et al,
2007].
Entre os CYPs humanos somente o CYP19A1, aromatase, enzima
envolvida na biosíntese de estrogênio, é um alvo estabelecido. Inibidores de
aromatase são utilizados no tratamento de tumores estrogênio-dependentes.
O CYP5A1, tromboxana sintase, permanece como alvo em potencial para
compostos com atividade anticoagulante. Em micro-organismos, a CYP51,
14α-desmetilase, é alvo de antifúngicos azólicos, além de representar um
possível alvo na terapia anti-T. cruzi e anti-leishmania [LAMB et al, 2007;
LEPESHEVA et al, 2007]. O CYP121 é um alvo válido para compostos
azólicos com potencial atividade frente a micobactérias [LAMB et al, 2007;
SEWARD, et al, 2006; MUNRO et al, 2003].
A CYP51 é parte do caminho biossintético de esteróis, sendo notável
por ser a única da família P450 encontrada em todos os reinos biológicos,
estando presente em mais de 80 organismos entre bactérias, eucariotas
inferiores, plantas, fungos e mamíferos [JACKSON et al, 2008; LEPESHEVA,
WATERMAN, 2007]; além disso, diversas plantas e fungos apresentam
múltiplos genes CYP51, de forma que o número de sequências conhecidas
ultrapassa a 100. A 14α-desmetilase de todos os filos é classificada como
uma única família de P450 devido à manutenção estrita de sua função,

FBT/FCF/USP
Ieda Yuriko Sonehara
32
catalisando uma reação regio- e estereoespecífica de remoção oxidativa do
grupo 14α-metil de precursores de esteróis formados a partir da ciclização do
esqualeno-2,3-epóxido (Figura 9). A identidade da sequência total de
aminoácidos na família da CYP51 é de cerca de 30%, variando conforme a
proximidade evolutiva das espécies, sendo em média de 23% – 34% entre os
reinos biológicos [LEPESHEVA, WATERMAN, 2007] (Figura 10)
Figura 9. Reação da CYP51. Grupo 14α-metil oxidado sequencialmente a álcool e aldeído, seguido de remoção de ácido fórmico e introdução de ligação dupla ∆14,15. Lanosterol (L), 24,25-diidrolanosterol (D) e 24-metilenodiidro-lanosterol (M) são substratos fisiológicos de CYP51 de mamíferos e fungos; obtusifoliol (O) é substrato fisiológico de plantas; norlanosterol (N), substrato fisiológico de Trypanosoma brucei. [LEPESHEVA et al, 2006]
Figura 10. Identidade da sequência de aminoácidos em 71 sequências da família CYP51. *número de sequências no alinhamento/porcentagem de identidade [LEPESHEVA, WATERMAN, 2007]

FBT/FCF/USP
Ieda Yuriko Sonehara
33
Desde sua descoberta, a CYP51 de Mycobacterium tuberculosis,
ortólogo solúvel de seus equivalentes eucarióticos ligados à membrana, serve
de modelo de homologia e modelagem tridimensional de interações CYP51-
inibidores em patógenos microbianos [EDDINE et al, 2008]. Este CYP51
procariótico possui, no entanto, estrutura substancialmente diferente das
isoformas eucarióticas, principalmente na região que define o canal de acesso
ao substrato, embora apresente in vitro a atividade específica de 14α-
desmetilase; níveis de esteróis mais baixos que em fungos foram detectados
em M. smegmatis, embora não se saiba ainda qual o papel exato dos esteróis
em procariotas [JACKSON et al, 2000; EDDINE et al, 2008]. Embora
inibidores de CYP51, especificamente azóis, exerçam atividade tóxica sobre
M. tuberculosis, esta atividade não é suficiente ainda para ser de utilidade
terapêutica [JACKSON et al, 2008].
A reação catalizada pela CYP51 envolve a remoção da metila na
posição C14 de precursores 14α-metil-esteroidais e ocorre em três etapas,
cada uma necessitando de oxigênio e NADPH, através da conversão do grupo
14α-metil sucessivamente em 14α-carboxiálcool e 14α-carboxialdeído, com
este grupo sendo liberado na última etapa como ácido fórmico, com formação
de uma dupla ∆14.15 no núcleo esteroidal (Figura 9, p. 32) [LEPESHEVA,
WATERMAN, 2007; PODUST, POULOS, WATERMAN, 2001]. Os produtos
14α-desmetilados são intermediários em caminhos biossintéticos, resultando
em colesterol em animais, ergosterol em fungos e diversos esteróis olefinados
e 24-alquilados em plantas, algas e protozoários. Normalmente as reações
ocorrem no retículo endoplasmático, porém existem evidências de que em
kinetoplastídeos a biossíntese de esteróis possa ocorrer na mitocôndria
[LEPESHEVA, WATERMAN, 2007]. Esteróis volumosos como colesterol,
ergosterol e sitosterol (plantas) são componentes da membrana plasmática,
tendo papel estrutural importante na regulação da fluidez e permeabilidade de
membrana, além de modular indiretamente a atividade e distribuição de
proteínas integrais de membrana, incluindo enzimas, canais iônicos e
componentes dos caminhos de transdução de sinais [LEPESHEVA,

FBT/FCF/USP
Ieda Yuriko Sonehara
34
WATERMAN, 2007]. Além disso, esteróis servem como precursores de
moléculas bioativas como hormônios esteroidais em mamíferos e hormônios
brassinoesteroidais em plantas. Em mamíferos, a CYP51 também converte
lanosterol em esteróis ativadores de meiose (Meiosis-Activating Sterols, MAS),
precursores de colesterol que modulam o desenvolvimento de céulas
germinativas masculinas e femininas [LEPESHEVA, WATERMAN, 2007;
ZARN, BRÜSCHWEILER, SCHLATTER, 2003]
Além de seu papel na biossíntese de esteróis, a CYP51 também possui
atividade regulatória; em mamíferos um dos intermediários da reação
(derivado 14α-carboxialdeído da desmetilação do lanosterol) regula a
produção de colesterol por atuar como supressor de HMG-CoA redutase (3-
hidróxi-metilglutaril coenzima A redutase). O papel regulatório da CYP51 pode
ser de especial importância em tripanosomatídeos, onde o intermediário
aldeídico do lanosterol pode ser utilizado pelo parasita para alternar o
caminho biossintético da produção de esterol endógeno para o consumo de
precursores exógenos presentes no sangue do hospedeiro mamífero, em
processo energeticamente mais favorável [LEPESHEVA, WATERMAN, 2007].
Ao contrário dos P450s voltados para a metabolização de fármacos,
que são capazes de acomodar uma grande variedade de estruturas, a CYP51
tem uma faixa de especificidade de produto muito estreita. O conjunto de
substratos é limitado aos cinco 14α-metil-esteróis naturais (lanosterol, 24, 25-
diidrolanosterol, 24-metilenodiidro-lanosterol, obtusifoliol e 4β-desmetil-
lanosterol (norlanosterol) (Figura 9, p. 32) [LEPESHEVA, WATERMAN, 2007].
O complexo mecanismo da reação catalizada pela CYP51 sugere que um
arranjo espacial específico do substrato é necessário para assegurar sua
orientação apropriada no sítio catalítico durante as três etapas da catálise, o
que torna necessária certa conservação de aminoácidos apesar da baixa
identidade de sequência nesta família [LEPESHEVA, WATERMAN, 2007].
Estudos indicam que o substrato é estabilizado no sítio ativo
predominantemente através de interações hidrofóbicas e ligações de H, com a
cadeia lateral em C17 do substrato posicionada em um canal hidrofóbico

FBT/FCF/USP
Ieda Yuriko Sonehara
35
estreito; ligações de H envolvendo o grupo 3-OH do substrato auxiliam em seu
correto posicionamento no sítio catalítico da enzima [JI et al, 2000].
Sete resíduos de glicina aparentemente são essenciais para assegurar
a flexibilidade conformacional da CYP51, separando elementos secundários
de sua estrutura. A maioria dos demais resíduos conservados nesta enzima
estão aglomerados em seis regiões, que representam as cinco regiões de
reconhecimento de substrato (Substrate Recognition Sites, SRS) e a região ao
redor do resíduo de cisteína que coordena com o grupo heme. Embora a
conservação nesta região de ligação do heme seja geral entre as famílias do
P450, a alta conservação das SRS é uma característica específica de CYP51
[LEPESHEVA, WATERMAN, 2007]. Os resíduos variáveis presentes no sítio
catalítico incluem F78, M79, K97, M99, H101, F255, S252, I323 e V434,
revelando diferenças consideráveis entre os reinos e sugerindo que são
resíduos que contribuem para a especificidade da CYP51 em relação ao
substrato em diferentes espécies [PODUST, POULOS, WATERMAN, 2001].
Estudos da estrutura cristalina de CYP51 de Mycobacterium
tuberculosis [PODUST et al, 2004; PODUST, POULOS, WATERMAN, 2001]
revelaram a presença de uma α-hélice I cuja porção N-terminal afasta-se do
núcleo estrutural da enzima em um ângulo de 145° com a porção C-terminal.
Este afastamento em relação ao grupo heme libera a alça BC de sua
conformação fechada, aumentando o espaço disponível para ligação de
substrato ou inibidores; como consequência do reposicionamento da α-hélice
I, as regiões adjacentes – α-hélices H e G e alças intermediárias – também
exibem diferenças topológicas significativas em relação a outros P450s. A
posição aberta da alça BC forma, em conjunto com a α-hélice I deslocada,
uma abertura de grandes dimensões que conecta a superfície à cavidade
catalítica e ao grupo heme; este canal (canal 1) corre aproximadamente
paralelo ao plano do grupo heme, formando o canal de acesso do substrato
(Figura 11). Este canal de acesso difere daqueles encontrados em outros
P450 (canal 2), que se encontra perpendicular ao plano do grupo heme. O
canal 2 também é aparente na estrutura da CYP51, porém sua entrada na

FBT/FCF/USP
Ieda Yuriko Sonehara
36
superfície encontra-se fechada pela interação entre a α-hélice A’ e a alça FG;
no entanto, a rotação das α-hélices F e G poderia permitir um movimento de
abertura/fechamento da alça FG, abrindo o acesso ao canal 2. Na CYP51, a
abertura do canal por este mecanismo torna necessário o fechamento do
canal 1; este cenário leva a uma imagem dinâmica e sincronizada de catálise
onde enquanto o canal 1 encontra-se aberto, o canal 2 encontra-se fechado.
Esta sincronização poderia possibilitar ao substrato a entrada por um canal,
com o produto saindo por outro. Considerando as etapas de oxidação
múltiplas necessárias na atividade da CYP51, este movimento poderia ser
necessário para o posicionamento de resíduos-chave nas diversas etapas do
caminho catalítico. Acredita-se que o substrato hidrofóbico é reconhecido na
superfície da proteína através de uma região hidrofóbica de resíduos de
aminoácidos adjacente ao canal 1 de acesso de substrato, correspondendo a
resíduos na região N-terminal da α-hélice I na alça BC; após o
reconhecimento, o substrato penetra o canal de acesso e é particionado para
o sítio ativo através de interações hidrofóbicas [SHENG et al, 2009; PODUST,
POULOS, WATERMAN, 2001; JI et al, 2000].
Figura 11. Estrutura de CYP51 de M. tuberculosis em representação de Richardson.
Heme em representação de bolas (vermelho), ligante 4-fenilimidazol em representação CPK (rosa). α-hélice I em verde [PODUST et al, 2001].

FBT/FCF/USP
Ieda Yuriko Sonehara
37
A maioria das mutações identificadas em isolados de C. albicans
resistentes aos antifúngicos azólicos encontram-se distribuídas ao redor das
regiões altamente dinâmicas responsáveis pela movimentação da alça FG e
consequente abertura/fechamento dos canais de acesso 1 e 2 [EDDINE et al,
2008; PODUST et al, 2004], indicando um possível envolvimento direto ou
indireto com a interação de ligantes. No entanto, considerando o alto nível de
conservação dos resíduos envolvidos na interação do substrato com a enzima
dentro de cada filo, e sua importância para o reconhecimento de substrato, é
improvável que ocorram mutações importantes nestes resíduos no que se
refere a resistência a fármacos [PODUST et al, 2004]. Existe, porém, a
possibilidade de mutações em resíduos não envolvidos diretamente na
interação com o substrato, mas que modulam a abertura e o fechamento dos
canais de acesso ao sítio catalítico; desta forma, pode ser possível a
manutenção do canal aberto mesmo com a ligação do inibidor, o que leva a
um estado de baixa afinidade do mesmo e permite seu deslocamento pelo
substrato. Este mecanismo não permite a diminuição da afinidade abaixo de
um valor mínimo definido pela área de interação do inibidor e,
consequentemente, no planejamento de novos fármacos pode-se aumentar o
tamanho da molécula inibidora de forma a permitir que ocupe uma área mais
extensa quando ligado ao sítio catalítico, procurando assim manter a afinidade
com a enzima em níveis adequados para a inibição enzimática efetiva
[PODUST et al, 2004; KELLY et al, 2001; PODUST et al, 2001]. Esta
estratégia foi aplicada no desenvolvimento de antifúngicos como itraconazol e
posaconazol, que possuem estruturas maiores que fluconazol. Existe, no
entanto, limitação na utilidade desta estratégia devido ao próprio tamanho da
região de interação [PODUST et al, 2004].
A importância da CYP51 como possível alvo para fármacos deve-se ao
efeito causado por sua inibição, que leva ao bloqueio da síntese de esteróis,
letal para organismos unicelulares por sua incapacidade de assimilação de
esterol exógeno; a inibição de CYP51 também influencia o crescimento e
processos de desenvolvimento em plantas, e diminui a produção de colesterol
endógeno em animais. Embora inibidores de CYP51 estejam sendo

FBT/FCF/USP
Ieda Yuriko Sonehara
38
estudados como herbicidas e fármacos hipocolesterolêmicos, capazes de
diminuir os níveis de colesterol [KOROSEC et al, 2008; LEPESHEVA,
WATERMAN, 2007], estes compostos são mais comumente utilizados como
fungicidas no tratamento de micoses e na prevenção de infecções em
agricultura. A necessidade de inibidores de CYP51 aumenta continuamente,
devido ao aumento mundial da ocorrência de infecções fúngicas oportunistas
como consequência do aumento de hospedeiros imunodeprimidos (resultado
de infeções por HIV, quimioterapia antineoplásica, transplantes de órgãos e
de medula entre outras causas) e pacientes com infecções primárias como
tuberculose. Um novo aspecto para inibidores de CYP51 é a descoberta de
sua utilidade no tratamento de infecções causadas por Trypanosoma e
Leishmania [EDDINE et al, 2008; LEPESHEVA et al, 2008; LEPESHEVA,
WATERMAN, 2007; ALRAJHI et al, 2002; BERMAN, 1981], doenças
parasitárias negligenciadas que afetam milhões de pessoas na América Latina
[REITHINGER et al, 2009; MONCAYO, ORTIZ YANINE, 2006].
2.2.1 Características de compostos inibidores de CYP51
Os inibidores de CYP51 mais conhecidos são os compostos azólicos
(imidazóis e triazóis) (Figura 12, p. 39). Estes compostos ligam-se à sexta
posição de coordenação do ferro do grupo heme, impedindo assim a ligação e
metabolização do substrato; a especificidade pela CYP51 fúngico está
relacionada aos grupos substituintes ligados ao N1 do grupo azólico [SHENG
et al, 2009; SHENG et al, 2006; JI et al, 2000], que também determinam a
potência através de interações com a apoproteína [LAMB et al, 2000]. Os
compostos azólicos têm papel importante no tratamento de micoses
sistêmicas e superficiais, tendo também apresentado eficácia em modelos de
leishmaniose e doença de Chagas [URBINA et al, 2003; ALRAJHI el al, 2002;
MOLINA et al, 2000; McCABE, REMINGTON, ARAUJO, 1986; BERMAN,
1981].

FBT/FCF/USP
Ieda Yuriko Sonehara
39
miconazol fluconazol
cetoconazol voriconazol
itraconazol
pozaconazol
Figura 12. Estruturas de alguns fármacos antifúngicos imidazólicos e triazólicos
Os azóis apresentam menor toxicidade em relação a outros fármacos
antifúngicos e anti-trypanosoma e são de menor custo e mais facilmente
disponíveis, porém apresentam diversas desvantagens. Entre elas destacam-
se que o seu uso a longo prazo pode levar à inibição de outras enzimas do
complexo P450, entre elas várias envolvidas na metabolização de
xenobióticos tais como CYP3A4, CYP2A6, CYP2C9, CYP2C19, CYP2D16 e
CYP2B6 [ITOKAWA et al, 2007; ZHANG, et al, 2002; LAMB et al, 2000],
causando efeitos indesejáveis devido a interações medicamentosas com
outros fármacos metabolizados por estas enzimas. Em relação à inibição da
CYP3A4, parece haver influência das características do grupo N-substituído

FBT/FCF/USP
Ieda Yuriko Sonehara
40
na porção imidazólica dos compostos, assim como da lipofilicidade das
cadeias laterais; estas características parecem estender-se a outras enzimas
P450, embora a capacidade inibidora dos compostos dependa das
características tridimensionais do sítio catalítico que pode ou não permitir que
o grupo N-substituinte facilite a coordenação do átomo de nitrogênio do anel
azólico com o grupo heme da enzima [ITOKAWA, YAMAUCHI, CHUMAN,
2009; ITOKAWA et al, 2007; ; ZHANG, et al, 2002; LAMB et al, 2000].
O uso contínuo de antifúngicos azólicos também leva ao surgimento de
resistência, permitindo no patógeno o desenvolvimento de tolerância ao
fármaco. Entre as possíveis causas da resistência, encontram-se mutação na
CYP51 fúngica, aumento na expressão do gene CYP51, e aumento no efluxo
de azóis ou diminuição de permeabilidade [COWEN, 2008; LEPESHEVA,
WATERMAN, 2007; AKINS, 2005]. Uma alternativa para contornar a
resistência a azóis é a inibição da CYP51 por análogos do substrato [EDDINE
et al, 2008; LEPESHEVA et al, 2008; LEPESHEVA, WATERMAN, 2007], idéia
que tem sido explorada na busca por novos agentes hipocolesterolêmicos. Os
análogos ao substrato apresentam como vantagens a especificidade pela
CYP51, melhor permeabilidade celular e maior tempo de meia-vida em
soluções aquosas quando comparadas a diversos azóis; além disso, possuem
baixa probabilidade de causar resistência devido à sua semelhança com
esteróis endógenos. No entanto, estes tipos de inibidores apresentam como
grande problema a própria especificidade da enzima, que reconhece seu
substrato baseado em requisitos bastante rígidos; desta forma, alterações
mínimas na estrutura do esterol podem afetar a qualidade da ligação e a
potência de inibição [LEPESHEVA et al, 2008].
A CYP51 encontra-se presente também nos tripanosomatídeos,
agentes causadores da doença de Chagas (Trypanosoma cruzi) e doença do
sono ou tripanosomíase africana (Trypanosoma brucei). Ambas espécies de
protozoários possuem ciclos de vida complexos que incluem estágios em
insetos e mamíferos, porém diferem quanto aos vetores, hospedeiros

FBT/FCF/USP
Ieda Yuriko Sonehara
41
mamíferos preferidos, localização no organismo do hospedeiro e
manifestação sintomática [LEPESHEVA et al, 2008].
Do ponto de vista de planejamento de fármacos, a diferença mais
importante refere-se às necessidades desses parasitas em relação aos
esteróis. O T. cruzi é dependente da biossíntese de esteróis endógenos em
todos os estágios de vida, enquanto que o T. brucei é capaz de assimilar
colesterol de hospedeiros mamíferos durante seu período de vida na corrente
sanguínea. Enquanto que a inibição da CYP51 em T. cruzi causa depleção de
esteróis estruturais e consequente dano às funções de membrana, no T.
brucei a mesma inibição interfere na síntese de esteróis funcionais, ou seja,
aqueles produzidos em níveis hormonais e importantes no desenvolvimento
do parasito e regulação de seu ciclo de vida. Existem também diferenças
entre estas espécies em relação à especificidade por substrato, sendo 24-
metilenodiidrolanosterol substrato para T. cruzi, e esteróis C4-monometilados,
possivelmente 31-norlanosterol, para T. brucei (Figura 9, p.32) [LEPESHEVA
et al, 2008]. Esta especificidade em relação ao substrato relaciona-se com
diferenças em um resíduo filo-específico na α-hélice B’ da CYP51 (F105 em T.
brucei, I105 em T. cruzi) [LEPESHEVA et al, 2006].
Nas enzimas pertencentes à família do citocromo P450, o átomo de
ferro do grupo prostético heme possui em seu estado de repouso uma
molécula de água ligada à sexta posição de coordenação. Na interação do
substrato, ocorre deslocamento desta molécula de água, com alteração de
spin do átomo de ferro seguido por redução, ligação de oxigênio molecular,
nova redução e duas protonações, resultando, ao final, em espécie ativa oxo-
ferro [BALDING et al, 2008; DENISOV et al, 2005; FRIESNER, GUALLAR,
2005]. Os inibidores azólicos ocupam o espaço destinado aos substratos do
citocromo P450, ligando-se diretamente ao grupo heme e tornando-o inativo
em relação a reações com oxigênio. A molécula do azol aproxima-se do grupo
heme, atraída pela da aceitação de uma ligação de H da molécula de água
presente no estado de repouso; a uma distância adequada, a molécula de
água é liberada do grupo heme e é substituída pelo grupo azólico, que desta

FBT/FCF/USP
Ieda Yuriko Sonehara
42
forma bloqueia a ligação de oxigênio molecular e inativa o centro heme.
Modelos quânticos de teoria funcional de densidade (Density Functional
Theory, DFT) mostram que o processo é sutil e induzido pela presença de
grupos doadores e aceptores de ligações de H presentes no sítio ativo,
levando à liberação da molécula de água ligada ao grupo heme. Interações
via ligações de H com a molécula de água do estado de repouso ou com a
molécula do inibidor podem levar à maior estabilização do estado de repouso
ou do complexo ligado ao inibidor [BALDING et al, 2008].
A Figura 13 ilustra o modo de interação do fluconazol com o sítio ativo
da CYP51; o anel triazólico posiciona-se perpendicularmente ao plano da
porfirina, permitindo interação de um átomo de N do anel com o átomo de Fe
do grupo heme, enquanto que o grupo difluorofenil direciona-se para o canal
de acesso de substrato 1 (alça BC), com o segundo anel triazólico
apresentando orientação preferida em direção ao canal de acesso de
substrato 2 (alça FG) [SHENG et al, 2009; PODUST, POULOS, WATERMAN,
2001]. O fluconazol também interage com pelo menos três moléculas de água
presentes no sítio ativo [PODUST, POULOS, WATERMAN, 2001], que podem
estar mediando interações entre o grupo hidroxila e o resíduo H320, altamente
conservado na família da CYP51 [SHENG et al, 2009]. Estruturalmente, a
maior diferença entre o fluconazol e outros antifúngicos triazólicos encontra-se
na cadeia lateral ligada ao C3, que permite a formação de ligações
hidrofóbicas adicionais com consequente aumento da afinidade pela enzima
[PODUST et al, 2004], como é o caso de itraconazol e posaconazol em que as
longas cadeias laterais permitem a formação de interações hidrofóbicas e de
van der Waals adicionais no sítio catalítico [SHENG et al, 2009].

FBT/FCF/USP
Ieda Yuriko Sonehara
43
Figura 13. Interação de fluconazol no sítio catalítico de CYP51 de M. tuberculosis.
α-hélices representadas em fita. Fluconazol (FLU, verde), grupo heme (vermelho) e resíduos de aminoácidos (cinza) representados em palito. [PODUST, POULOS, WATERMAN, 2001]
Estudos de inibição enzimática mostram que a presença de grupo
aromático volumoso em α ou β em relação ao anel imidazólico é essencial
para uma interação eficiente com a enzima; grupos α-fenílicos são facilmente
deslocados pelo substrato, porém o deslocamento do grupo fenílico para a
posição β resulta em inibidores muito mais potentes, chegando à ação
irreversível [LEPESHEVA et al, 2007].
Existem ainda estudos em que se relatam diferentes modos de
interação de outros compostos inibidores, de características não-azólicas, com
o sítio catalítico da CYP51. Neste caso, interações hidrofóbicas e ligações de
hidrogênio são responsáveis pela interação do inibidor com a enzima, sem
envolvimento do grupo heme [YAO et al, 2007; ZHU et al, 2006; JI et al, 2000].

FBT/FCF/USP
Ieda Yuriko Sonehara
44
3. OBJETIVOS

FBT/FCF/USP
Ieda Yuriko Sonehara
45
A utilização de forma integrada de ferramentas pertencentes às áreas
de química farmacêutica, quimioinformática e bioinformática permite a
caracterização de cavidades catalíticas de proteínas e a identificação das
propriedades físico-químicas consideradas essenciais para a interação
adequada entre ligante e biomacromolécula-alvo. Por outro lado, permitem
também a identificação de alvos a partir da comparação de estruturas
químicas, com as propriedades físico-químicas associadas, entre ligantes
conhecidos e possíveis alvos; este procedimento de comparação de perfis
moleculares, conhecido como virtual profiling, parte do princípio de que
moléculas semelhantes a ligantes conhecidos de proteínas têm o potencial de
interagir com essa mesma proteína, onde o grau de semelhança é
determinado não somente pela estrutura química, mas também pela
distribuição de características físico-químicas. As ferramentas utilizadas em
estudos in silico incluem também análise de descritores topológicos que
permitem a caracterização de propriedades físico-químicas considerando sua
distribuição espacial em relação à estrutura química de uma dada molécula. O
uso destes descritores permite a determinação do grau de semelhança entre
moléculas ou partes de moléculas (fragmentos moleculares), e encontra-se na
base das metodologias de triagem e de caracterização de sítios catalíticos de
proteínas.
No presente trabalho, propõe-se um estudo das características mais
importantes que possam influenciar a atividade antifúngica e anti-T. cruzi de
nitro-compostos com estrutura azometínica ou oxadiazolínica, com formulação
de hipótese sobre possíveis biomacromoléculas-alvo. Esta identificação de
alvos teve como objetivo principal o fornecimento de bases sólidas para
estudos posteriores das relações entre a estrutura química e a atividade,
principalmente no aspecto quantitativo, proporcionando melhor entendimento
do mecanismo de ação em nível molecular e possibilitando o planejamento
mais eficiente de compostos químicos candidatos a fármacos ou compostos-
líderes para estudos de modificação molecular.

FBT/FCF/USP
Ieda Yuriko Sonehara
46
Para tanto, métodos de CADD foram aplicados, com uso de descritores
topológicos como ferramentas para identificação de possíveis biomacro-
moléculas-alvo através de comparação de perfis moleculares com ligantes
anotados para alvos conhecidos, no procedimento conhecido como virtual
profiling. A caracterização físico-química do possível sítio de interação dos
compostos auxiliou na formulação de hipóteses sobre o possível alvo, assim
como estudos de perfis de solubilidade com uso do programa Volsurf.

FBT/FCF/USP
Ieda Yuriko Sonehara
47
4. EXPERIMENTAL

FBT/FCF/USP
Ieda Yuriko Sonehara
48
As séries de compostos analisadas encontram-se representadas na
Figura 14. As séries AzoO e AzoS são séries azometínicas, diferindo apenas
no anel contendo o grupo nitro; estas séries foram sintetizadas pelo grupo de
pesquisa do Laboratório de Planejamento e Desenvolvimento de Fármacos,
LPDF, da FCF/USP, sob coordenação do Prof. Leoberto Costa Tavares,
orientador do presente trabalho, seguindo descrição em literatura
[MASUNARI, TAVARES, 2007; MASUNARI, TAVARES, 2006; TAVARES et
al, 1999; TAVARES, PENNA, AMARAL, 1997; TAVARES, 1993]. As séries
OxaO e OxaS são séries oxadiazolínicas, sendo que a série OxaO é apenas
teórica, sem compostos sintetizados no momento. A série OxaS foi sintetizada
também pelo mesmo grupo de pesquisa [ALMEIDA, 2009].
Estabeleceu-se, como regra, que os compostos individuais são
identificados por um código, correspondente à série à qual pertence, seguida
pelo o seu substituinte. Assim, o composto AzoO-OH refere-se ao composto
4-hidroxi-substituído da série AzoO, ou seja, 5-nitro-2-furfurilideno-4-
hidroxibenzidrazida (nifuroxazida).
SÉRIES AZOMETÍNICAS SÉRIES OXADIAZOLÍNICAS
Série AzoO: 5-nitro-2-furfurilideno benzidrazidas 4-substituídas (X=O)
Série AzoS: 5-nitro-2-tiofilideno benzidrazidas 4-substituídas (X=S)
Série OxaO: 3-acetil-oxadiazolina 2,5-furfurilideno benzidrazidas 4-substituídas (X=O)
Série OxaS: 3-acetil-oxadiazolina 2,5-tiofilideno benzidrazidas 4-substituídas (X=S)
substituintes R para todas as séries: -H, -CH3, -C2H5, -OCH3, -OC2H5, -Cl, -Br, -I, -F, -CF3, -COCH3, -NH2, -N(CH3)2, -NO2, -OH (composto AzoO-OH = nifuroxazida)
Figura 14. Estrutura química geral das séries estudadas

FBT/FCF/USP
Ieda Yuriko Sonehara
49
4.1. CÁLCULO DE DESCRITORES TOPOLÓGICOS
Com o objetivo de encontrar um equilíbrio entre métodos geométricos
e topológicos para a determinação de fingerprints moleculares, GREGORI-
PUIGJANÉ e MESTRES desenvolveram, em 2006, um conjunto de
descritores baseados no conceito de entropia de Shannon, denominados
Descritores de Entropia de Shannon, SHED (Shannon Entropy Descriptors);
estes descritores representam um meio de se quantificar a variabilidade
demonstrada pelas distribuições topológicas de pares de características
centrados no átomo nas diferentes moléculas.
Para a determinação do SHED, cada átomo de uma estrutura química é
primeiro mapeado para seu equivalente utilizado pelo programa Sybyl (Tripos,
Inc.) [GREGORI-PUIGJANÉ, MESTRES, 2006] (Tabela 1). Em um segundo
passo, determina-se que cada tipo de átomo possui uma ou mais das
seguintes quatro características centradas no átomo: hidrofobicidade (H),
aromaticidade (R), capacidade receptora (A) ou doadora (D) de ligações de
hidrogênio (Tabela 2).
Tabela 1 Alguns tipos de átomos utilizados pelo programa Sybyl
Código Descrição Código Descrição
C.3 carbono sp3 S.3 enxofre sp3
C.2 carbono sp2 S.2 enxofre sp2
C.1 carbono sp S.O enxofre em sulfóxido
C.ar carbono aromático S.O2 enxofre em sulfona
N.3 nitrogênio sp3 F flúor
N.2 nitrogênio sp2 Cl cloro
N.1 nitrogênio sp Br bromo
N.ar nitrogênio aromático I iodo
N.am nitrogênio amínico O.3 oxigênio sp3
N.pl3 nitrogênio trigonal planar O.2 oxigênio sp2
N.4 nitrogênio sp3 carregado
positivamente
O.co2 oxigênio em grupos
carboxilato e fosfato
H hidrogênio P.3 fósforo sp3

FBT/FCF/USP
Ieda Yuriko Sonehara
50
Tabela 2 Alguns tipos de átomos utilizados pelo programa
Sybyl e respectivas características farmacofóricas
Extraído de GREGORI-PUIGJANÉ, MESTRES, 2006
É derivado, em seguida, o caminho de menor distância, contado como
número de ligações, entre estas características de pares de átomos; sua
ocorrência em diferentes distâncias é armazenada para criar-se uma
distribuição de pares de características. Para a determinação desta
distribuição é utilizada uma distância máxima de 20 ligações; pares de
características a distâncias maiores são acumuladas no último compartimento.
O conceito de entropia de Shannon é então aplicado para a determinação da

FBT/FCF/USP
Ieda Yuriko Sonehara
51
variabilidade das distribuições dos pares de características; dentro desta
sistemática, a entropia S de uma população P, distribuída em certo número de
compartimentos (representando neste caso as diferentes distâncias) N=20, é
fornecida por
onde ρi e pi são, respectivamente, a probabilidade e a população em cada
compartimento i da distribuição. Os valores de S variam de 0, refletindo a
situação em que a população inteira concentra-se em um único
compartimento, a um valor máximo Smax = ln N, refletindo a situação de uma
população distribuída uniformemente entre todos os compartimentos.
Uma medida mais intuitiva que pode ser relacionada linearmente à
situação de ocupação completamente uniforme é dada pela transformação
dos valores de entropia em valores de entropia projetada, E=eS. Para qualquer
população P > 0, os valores de E variam de 1 (entropia zero, população
totalmente concentrada em um único compartimento) a N (entropia máxima,
população uniformemente distribuída entre todos os compartimentos). No
caso limite em que P=0, considera-se E=0. Este valor E corresponde ao
SHED para o par de características considerado. O conjunto de valores SHED
obtido para os 10 pares de características possíveis constituem o perfil SHED
da molécula. Este perfil SHED é normalmente representado utilizando-se
gráfico em radar, como pode ser visualizado na Figura 22 (p. 76), com os
dados correspondentes na Tabela 8 e Tabela 9 (p. 73 e 74). A Figura 15
esquematiza o procedimento geral para obtenção de perfil SHED.
∑=
−=N
i
iiS1
ln ρρ Ppii /=ρ

FBT/FCF/USP
Ieda Yuriko Sonehara
52
Figura 15. Procedimento para cálculo de SHED.
Extraído de GREGORI-PUIGJANÉ, MESTRES, 2006.
O perfil SHED considera implicitamente o tamanho das moléculas,
particularmente em valores SHED correspondentes a pares de características
envolvendo centros hidrofóbicos e aromáticos; à medida em que as moléculas
tornam-se maiores e mais complexas em termos de combinação de
características, a área ocupada por seus perfis SHED tende a se tornar
também maior.
Perfis SHED semelhantes refletem a possibilidade de moléculas
realizarem interações análogas em bolsos semelhantes em seus alvos
respectivos; no entanto, deve-se lembrar que, dependendo do tamanho da
cavidade de ligação da proteína, sua flexibilidade e grau de exposição ao
solvente, ligantes são capazes de interagir em diferentes bolsos, explorar
diferentes interações e apresentar diferentes grupos funcionais expostos ao
solvente. Cada perfil SHED representa uma distribuição particular de
características permitidas em um composto ativo para um determinado alvo.
Ao mesmo tempo, os perfis SHED são capazes de reconhecer a presença de
características semelhantes arranjadas de modo semelhante ao redor de
padrões estruturais (scaffolds) significativamente diferentes, uma habilidade
comumente referida como scaffold hopping.

FBT/FCF/USP
Ieda Yuriko Sonehara
53
Os perfis SHED apresentados neste trabalho foram calculados com uso
de script preparado por Gregori-Puigjané e Mestres, durante a vigência de
bolsa-sandwich junto ao Chemogenomics Laboratory (CGL), pertencente ao
Research Unit on Biomedical Informatics (GRIB/IMIM/UPF), Barcelona,
Espanha, durante o ano de 2008.
4.2. ESTUDOS DE IDENTIFICAÇÃO DE POSSÍVEIS ALVOS POR
COMPARAÇÃO DE PERFIS MOLECULARES, VIRTUAL PROFILING
Considerando-se que as principais famílias de alvos podem ser
reconhecidas pelo conjunto das propriedades físico-químicas (massa
molecular, log P, contagem de doadores e receptores de ligações de
hidrogênio) de seus ligantes, pode-se dizer que o uso de descritores mais
específicos pode permitir a previsão de um perfil global do alvo com o qual a
interação de um determinado composto é possível, desde que o alvo a ser
previsto se encontre descrito de forma suficiente por ligantes já identificados.
Esta técnica, conhecida também como target fishing, possui três componentes
básicos, e são eles: 1- conjunto de compostos de referência para o qual
descritores 2-D ou 3-D estejam disponíveis em um banco de dados; 2-
procedimento de triagem; e 3- conjunto de compostos a serem testados para
identificação de novos compostos provavelmente ativos nos mesmos alvos
dos compostos de referência [ROGNAN, 2007].
Neste trabalho, optou-se por utilizar perfis SHED como descritores para
a realização do virtual profiling. Dessa forma, perfis SHED foram calculados
para cada composto das séries e comparados com os perfis SHED de
compostos, anotados para diversos alvos, pesquisados no banco de dados
Wombat™ (World of Molecular Bioactivity) [BALAKIN, 2006; Sunset Molecular
Discovery] versão 2007.1 (datado de 30/11/2007) e filtrados para selecionar
aqueles com atividade inibitória em concentrações não superiores a 10 µM. O
critério de classificação por similaridade foi definido considerando o menor

FBT/FCF/USP
Ieda Yuriko Sonehara
54
valor de todas as distâncias euclidianas calculadas entre os compostos de
referência e os compostos testados.
O protocolo de virtual profiling utilizado no presente trabalho foi
desenvolvido por Gregori-Puigjané e Mestres, e utilizado durante vigência de
bolsa-sandwich junto ao Chemogenomics Laboratory (CGL), pertencente ao
Research Unit on Biomedical Informatics (GRIB/IMIM/UPF), Barcelona,
Espanha, durante o ano de 2008.
4.3. CARACTERIZAÇÃO DA CAVIDADE CATALÍTICA DE CYP51
À medida em que ocorre o avanço da ciência e das técnicas
experimentais, há aumento progressivo no número de proteínas para as quais
se conhece a estrutura, seja por determinação experimental (cristalografia de
raios-X, RMN) ou através de modelos de homologia obtidos por modelagem
comparativa. A disponibilidade destas estruturas abre novas perspectivas no
campo do desenvolvimento de novos fármacos na medida em que revela
possíveis novos alvos terapêuticos. No entanto, são também necessárias
novas ferramentas que permitam o uso destas informações de forma racional
e com o mínimo dispêndio de recursos e tempo. Dentro deste raciocínio é
altamente desejável um método que torne possível prever funções da proteína
diretamente a partir de sua estrutura tridimensional; no entanto, embora a
geometria de uma proteína carregue informações sobre sua função
bioquímica em nível molecular, a função em si não é restrita a um dobramento
particular e não é visível através de sequenciamento. Por outro lado, sabe-se
que padrões de reconhecimento molecular podem encontrar-se preservados
ao longo de cavidades de ligação de proteínas com funções semelhantes,
uma vez que enzimas apresentam alta especificidade em relação ao
substrato, com requisitos bem definidos quanto ao arranjo espacial das
moléculas de substrato. Assim, foram desenvolvidos métodos que permitem a
detecção e extração automáticas de sítios de ligação putativos de proteínas,
com a constituição destes sítios sendo traduzidas em termos de descritores

FBT/FCF/USP
Ieda Yuriko Sonehara
55
moleculares associados a propriedades físico-químicas que podem ser
utilizados para a comparação mútua de diferentes sítios catalíticos [SCHMITT,
KUHN, KLEBE, 2002].
A caracterização do sítio catalítico de CYP51 foi realizada durante
vigência de bolsa-sandwich com uso de metodologia desenvolvida pelo grupo
do Chemogenomics Laboratory, pertencente ao Research Unit on Biomedical
Informatics (GRIB/IMIM/UPF), Barcelona, Espanha, durante o ano de 2008.
As estruturas de CYP51 em resolução abaixo de 2,5 Å foram obtidas
em formato pdb a partir do RCSB Protein Data Bank [KIRCHMAIR et al, 2008;
BERMAN et al, 2002; RCSB Protein Data Bank]. A superposição das
estruturas assim obtidas foi realizada utilizando-se o programa GAPS
(Gaussian-based Alignment of Protein Structures) [MESTRES, 2000]. Na
determinação das características da cavidade catalítica foram usados
protocolos internos do laboratório [JALENCAS, 2008] que se baseiam na
distribuição de características físico-químicas assinaladas para cada átomo.
Assim, a cada átomo diferente de H e pertencente à superfície da cavidade
catalítica da proteína foram associadas determinadas características físico-
químicas, a saber: aromática (R), hidrofóbica (H), doadora (D) e receptora (A)
de ligações de hidrogênio, além de descritores indicando a existência de
carga positiva (P) e negativa (N). Estas características são mapeadas e
representadas graficamente como esferas de diferentes cores. Estes pontos
críticos para interação ligante-proteína (Critical Points, CPTs) representam as
características físico-químicas mais importantes na caracterização da
superfície da cavidade catalítica e suas coordenadas. A Figura 16 ilustra um
resultado de cálculo de CPTs, superpostos à estrutura da proteína.

FBT/FCF/USP
Ieda Yuriko Sonehara
56
Figura 16. Cavidade catalítica de CYP51 de Mycobacterium tuberculosis com CPTs em representação de superfície
H=ciano, R=laranja, A=vermelho, D=azul Fluconazol em verde, grupo heme em rosa escuro. Átomo Fe como esfera laranja. Código de acesso PDB 1ea1 (resolução 2,21 Å).
4.3.1 Estudos de interação baseados em características das superfícies
catalíticas de proteínas
Em uma segunda etapa, a estrutura de uma proteína foi adotada como
referência para a família e sua superfície catalítica mapeada como descrito no
item anterior. Em um processo similar ao virtual profiling, um banco de dados
contendo fragmentos de ligantes de proteínas foi pesquisado para extração
daqueles fragmentos que se ligam a regiões com características presentes na
proteína de referência. Este banco de dados é composto por fragmentos de
ligantes, originalmente co-cristalizados com as respectivas proteínas-alvo, e
os CPTs correspondentes pertencentes à região de interação deste fragmento
na cavidade catalítica da proteína-alvo; desta forma, a triagem detecta CPTs
comuns à proteína de referência e ao fragmento de ligante, extraindo este,
com os CPTs associados, para posterior análise. Esta etapa possibilita a
identificação de regiões dos compostos estudados (séries azometínica e
oxadiazolínica) passíveis de interação com uma biomacromolécula-alvo. Para
ilustração, veja-se a Figura 46 (p. 105).

FBT/FCF/USP
Ieda Yuriko Sonehara
57
Estudo semelhante ao descrito acima, desta vez separando as
estruturas dos compostos considerados em fragmentos, foi realizado com o
objetivo de detectar que tipo de região da proteína, caracterizada pelos CPTs,
é favorável à interação destes fragmentos. Assim, os diferentes fragmentos
pertencentes aos compostos estudados foram extraídos do mesmo banco de
dados citado acima, permitindo a análise de quais CPTs permitem a interação
dos fragmentos com quais regiões típicas de uma proteína-alvo. A Figura 32
(p. 89) ilustra alguns fragmentos moleculares originários dos compostos e a
que tipo de regiões eles podem interagir.
4.4. ESTUDOS DE ANCORAMENTO DO LIGANTE, DOCKING
A escolha dos alvos a serem estudados por docking foi realizada após
análise de perfis SHED dos compostos e estudos preliminares de atividade
biológica. As estruturas de cristalografia de raios-X das biomacromoléculas-
alvo selecionadas foram obtidas do Brookhaven Protein Data Bank
[KIRCHMAIR et al, 2008; BERMAN et al, 2002; RCSB Protein Data Bank],
com códigos de acesso e resoluções indicados na Tabela 3.
Tabela 3 Proteínas selecionadas para estudo de docking
ALVO código PDB
resolução (Å) Descrição da estrutura*
CYP51 1ea1 2,21 14-α-esterol desmetilase associada ao citocromo P450 (CYP51) de Mycobacterium tuberculosis complexado com fluconazol
COX2 1cx2 3,0 ciclo-oxigenase-2 (prostaglandina sintase-2) complexada com inibidor seletivo SC-558.
*estruturas de proteínas obtidas do RCSB Protein Data Bank, conforme código de acesso PDB citado
Os estudos de docking foram realizados pela Chemotargets
(www.chemotargets.com). O processo de docking foi realizado com uso do

FBT/FCF/USP
Ieda Yuriko Sonehara
58
programa AutoDock 3.0 [Scripps Research Institute; SOTRIFFER et al, 2000;
GOODSELL, MORRIS, OLSON, 1996]. A minimização posterior do ligante em
interação com o sítio ativo, mantendo os átomos-alvo estacionários, foi
realizada utilizando o programa Moloc [Gerber Molecular Design] e campo de
força MAB [GERBER, MÜLLER, 1995].
4.5. ANÁLISE DE PERFIL DE SOLUBILIDADE
As análises foram realizados com utilização dos programas Sybyl 8.0
(Tripos, Inc) e Volsurf 4.0 (Molecular Discovery, Ltd), considerando-se quando
pertinente a atividade antifúngica dos compostos estudados.
Os compostos selecionados foram aqueles para os quais foi possível
determinar a concentração inibitória mínima (Minimal Inhibitory Concentration,
MIC) para C. albicans, conforme dados da Tabela 6 (p. 68). As estruturas dos
compostos foram desenhadas no programa Sybyl 8.0, e minimizadas no
mesmo programa com uso de campo de força MMFF94 (Merck Molecular
Force Field) [HALGREN, 1996] e cargas parciais MMFF. Os demais
parâmetros utilizados pelo programa encontram-se na Tabela 4.
Tabela 4 Parâmetros de minimização utilizados no programa Sybyl 8.0
Campo de Força MMFF94 Cargas MMFF
NB cutoff 8 Constante Dielétrica 1,0
Método Powell Otimização inicial / nº de iterações
Simplex /
20 Gradiente de terminação
0,05 kcal/(mol*Å)
Iterações máximas 3000
As atividades biológicas foram introduzidas na forma de potência de
MIC (pMIC = log (1/MIC)), com MIC expresso em concentração molar (M).

FBT/FCF/USP
Ieda Yuriko Sonehara
59
Para os compostos oxadiazolínicos (série OxaS), que possuem centro quiral
porém não tiveram configuração determinada, existem três possibilidades na
presença de mistura de ambos isômeros: atividade idêntica dos isômeros;
presença de um isômero inativo; ou ambos isômeros apresentando atividade,
porém em grau diferente. Seguindo uma opção apresentada por Sheng e
colaboradores [2006], foi considerado como pMIC para cada configuração R/S
um valor equivalente ao dobro daquele determinado experimentalmente,
baseando-se na hipótese de MIC ter sido determinado em mistura de
isômeros apresentando diferentes atividades.
4.6. SÍNTESE
A síntese dos compostos foi realizada pelo grupo de pesquisa do
Laboratório de Pesquisa e Desenvolvimento de Fármacos do Departamento
de Tecnologia Bioquímico-Farmacêutica da Faculdade de Ciências
Farmacêuticas da Universidade de São Paulo, LPDF/FBT/FCF/USP, sob
coordenação do Prof. Leoberto Costa Tavares, orientador de presente
trabalho, dentro da programação de diversos projetos de iniciação científica,
mestrado e doutorado com foco no estudo das atividades biológicas e
análises de QSAR das séries azometínicas e oxadiazolínicas.
A síntese dos compostos foi realizada em quatro etapas. A saber:
esterificação dos ácidos benzóicos 4-substituídos; obtenção das
benzidrazidas a partir dos respectivos benzoatos de metila; obtenção de
bases de Schiff e ciclização da ponte azometínica [ALMEIDA, 2009;
MASUNARI, TAVARES, 2007; MASUNARI, TAVARES, 2006; TAVARES et al,
1999; TAVARES, PENNA, AMARAL, 1997; TAVARES, 1993], conforme
Figura 17.
A identificação estrutural dos compostos e de seus intermediários foi
realizada por meio de análise espectrométrica IV, RMN-1H, RMN-13C, análise
elementar de CHN e faixa de fusão.

FBT/FCF/USP
Ieda Yuriko Sonehara
60
Figura 17. Síntese dos compostos azometínicos e oxadiazolínicos
4.7. DETERMINAÇÃO DE ATIVIDADE BIOLÓGICA
4.7.1 Atividade antifúngica
A. Materiais
• Dimetilsulfóxido PA (Labsynth Produtos Químicos para Laboratórios)
• Meio de cultura RPMI-1640 com L-Glutamina, sem bicarbonato de
sódio (Sigma-Aldrich)
• ácido 3-(N-morfolino)propanosulfônico, MOPS (Sigma-Aldrich)
• Meio de cultura Ágar Sabouraud-dextrose (Fluka)
• solução fisiológica estéril (cloreto de sódio 0,85%)
B. Métodos
A determinação de atividade antifúngica foi realizada no
LPDF/FBT/FCF/USP através da determinação de concentração inibitória

FBT/FCF/USP
Ieda Yuriko Sonehara
61
mínima, MIC (Minimal Inhibitory Concentration), por método de microdiluição
em microplaca de acordo com metodologia padronizada pelo Clinical and
Laboratory Standards Institute (CLSI, anteriormente NCCLS – National
Committee on Clinical Laboratory Standards) [NCCLS M27-A2], utilizando-se
cepa ATCC 537Y de Candida albicans.
A determinação dos valores de MIC envolveu duas fases. Na fase I, a
atividade antifúngica é determinada quantitativamente com uso da técnica de
microdiluição padronizada pelo CLSI [NCCLS M27-A2]. Em uma fase
seguinte, fase II, a sensibilidade do ensaio é aumentada através da adaptação
de metodologia aplicada para determinação de MIC fase II em bactérias
[JORGE et al, 2009].
Para a preparação do meio de cultura, 10,4 g de meio RPMI-1640 em
pó foram dissolvidos em 900 mL de água destilada, acrescentando-se em
seguida 34,53 g de tampão MOPS (ácido 3-(N-morfolino)propanosulfônico),
agitando-se até dissolução. O pH foi ajustado para 7,0 a 25 C utilizando-se
hidróxido de sódio 1M. Completou-se o volume para 1000 mL com água
destilada, e o meio esterilizado por filtração em membrana, sendo
armazenado a 4 °C até o momento da utilização.
Para preparação do inóculo foram escolhidas cinco colônias, com
diâmetro aproximado de 1 mm, de cultura de 24 horas de Candida albicans
em ágar Sabouraud-dextrose inclinado, que foram colhidas com uso de swab
estéril e suspendidas em 5 mL de solução salina estéril 0,85%. A suspensão
foi colocada em agitador de vórtex por 15 segundos e sua densidade celular
ajustada com espectrofotômetro UV/VIS por acréscimo de solução salina
estéril a 0,85% suficiente para obter-se transmitância equivalente a uma
solução-padrão da escala de McFarland 0,5, em comprimento de onda de 530
nm.
A solução de sulfato de bário utilizada como padrão de turbidez
McFarland 0,5 é preparada acrescentando-se 0,5 mL de solução de cloreto de
bário (BaCl2 . 2 H2O) 0,048 mol/L a 99,5 mL de ácido sulfúrico 0,18 mol/L. A
densidade é corrigida utilizando-se espectrofotômetro com via de luz de 1cm e

FBT/FCF/USP
Ieda Yuriko Sonehara
62
cubeta correspondente, com absorbância em 625 nm situando-se entre 0,08 e
0,10. Esta solução foi armazenada ao abrigo da luz e em temperatura
ambiente, sendo agitada vigorosamente em vórtex antes de ser utilizada. A
solução foi substituída a cada três meses após o preparo.
Após ajuste da turbidez, a suspensão de levedura sofreu diluição 1:50
em solução salina estéril 0,85%, seguida por agitação por 15 minutos em
frasco com agitador e pérolas de vidro. Foi realizada diluição 1:20 em meio
RPMI-1640 estéril, seguida por nova agitação por 15 minutos. Esta suspensão
de trabalho fornece inóculo com concentração de 1 x 103 a 5 x 103 unidades
formadoras de colônia (UFC) por mL, confirmadas por contagem de UFC em
placas de ágar Sabouraud-dextrose.
Para cada composto a ser testado na fase I, prepararam-se soluções-
estoque em meio RPMI contendo 20% de DMSO, em concentrações que
variaram de 20 a 30 µg/mL dependendo da solubilidade do composto. Estas
soluções foram obtidas pela diluição 1:9 em DMSO de uma solução-mãe em
DMSO de composto em concentração adequada, seguida por diluição 1:4 em
meio RPMI para fornecer a solução-estoque.
O teste de microdiluição em fase I foi realizado em placas de
microdiluição estéreis, descartáveis, com 96 poços em formato de U.
DispeNsaram-se 100 µL de meio RPMI em todas as colunas exceto a coluna
1. Dispensaram-se a seguir 100 µL da solução-estoque com uso de pipeta
multicanal nos poços das colunas 1, 2, e 11. Procedeu-se à diluição seriada
1:2 da coluna 2 à coluna 10; para tanto, 100 µL foram transferidos da coluna 2
para a coluna 3. Após homogeneização, 100 µL foram novamente transferidos
da coluna 3 para a 4, e assim sucessivamente até a coluna 10, que tem
descartados 100 µL.
Todos os poços exceto aqueles da coluna 11 (controle de esterilidade)
foram inoculados com 100 µL de suspensão de C. albicans, preparada como
descrito acima, sendo a coluna 12 o controle de crescimento. As microplacas

FBT/FCF/USP
Ieda Yuriko Sonehara
63
foram incubadas a 35 °C por 48 horas. Todos os ensaios foram realizados em
triplicata.
Nesta fase, com exceção da coluna 1, as concentrações de DMSO
durante o ensaio foram iguais ou menores que 5%, concentração esta abaixo
do limite de tolerância de 6% determinado através de testes para C. albicans
frente a este solvente.
A leitura considerou a concentração inibitória mínima (MIC) dos
compostos como sendo a menor concentração em que se observou redução
proeminente de crescimento em relação ao controle. Para tanto, os poços de
microdiluição receberam uma pontuação de 0 a 4, de acordo com o
crescimento em comparação com o controle de crescimento, sendo:
0: opticamente claro (inibição completa)
1: crescimento indefinido
2: redução proeminente de crescimento (definido como o valor de MIC)
3: ligeira redução do crescimento
4: nenhuma inibição de crescimento (crescimento equivalente ao
controle)
Uma fase II de ensaios foi realizada com a finalidade de estreitar-se a
faixa de valores MIC obtidos na fase I. Adaptando-se metodologia aplicada
para determinação de MIC fase II em bactérias [JORGE et al, 2009], 1 mL de
soluções-estoque foram preparadas em concentrações de composto
equivalentes ao dobro do valor de MIC determinado na fase I, em meio RPMI
contendo 10% de DMSO. Estas soluções foram obtidas pela diluição 1:9 em
DMSO de uma solução-mãe em DMSO de composto em concentração
adequada, seguida por diluição 1:9 em meio RPMI para fornecer a solução-
estoque.
Distribuíram-se 100 µL da solução-estoque nos poços da coluna 1 da
microplaca. Adicionaram-se 100 µL de meio RPMI 1640 à solução-estoque
inicial, desta forma diluindo-a em 10%. Após homogeneização, 100 µL desta

FBT/FCF/USP
Ieda Yuriko Sonehara
64
nova solução-estoque foram distribuídos na coluna 2 da microplaca.
Procedeu-se a nova diluição de 10% da solução-estoque pela adição de 100
µL de RPMI 1640, com distribuição nos poços da coluna 3. Este procedimento
foi repetido sucessivamente até a coluna 11, que recebeu adicionalmente 100
µL de meio (controle de esterilidade). A coluna 12 recebeu 100 µL de meio
(controle de crescimento). Todos os poços, à exceção daqueles da coluna 11,
foram inoculados com 100 µL de suspensão de C. albicans e incubados a
35 °C por 48 horas. Todos os ensaios foram realizados em triplicata. A leitura
do MIC de fase II seguiu o mesmo procedimento adotado para a fase I.
4.7.2 Atividade anti-T. cruzi
A avaliação da atividade anti-T.cruzi dos compostos foi realizada pelo
grupo de pesquisa, frente a formas epimastigotas de Trypanosoma cruzi, cepa
Y [SILVA E NUSSENSZWEIG, 1953], cultivadas a 28 ºC, em meio LIT (liver
infusion tryptose), suplementado com 10% de soro fetal bovino v/v. Os testes
foram realizados durante a fase log de crescimento parasitário.
Os parasitas na fase logarítmica (cultura parasitária de três dias) foram
inoculados em 50 mL de meio LIT (concentração inicial de 1,0 x 107
parasitas/mL). Os parasitas foram incubados com os compostos
(concentração de 10 µg/mL) diluídos em DMSO (concentraçao final de 1%)
durante 24, 48, 72 e 96 horas [POZAS, 2005]. A viabilidade e o crescimento
parasitário foram verificados em intervalos de 24 horas em Câmara de
Neubauer e espectrofotômetro de UV/VIS (leituras realizadas em 580 nm). A
porcentagem de inibição de crescimento (%IC) foi calculada de acordo com a
seguinte equação:
%IC = {1-[(AP – A0P)/(AC – A0C)]} x 100
onde AP= absorbância em 580nm (A580) da cultura contendo composto em 24,
48, 72 e 96 horas; A0P = A580 da cultura contendo o composto logo após

FBT/FCF/USP
Ieda Yuriko Sonehara
65
adição de inóculo (dia 0); AC = A580 da cultura na ausência de qualquer
composto (controle positivo); A0C = A580 da cultura na ausência de qualquer
composto em 24, 48, 72 e 96 horas [BOIANI et al, 2008; AGUIRRE et al,
2005; ARAN et al, 2005; CERECETTO et al, 1999].

FBT/FCF/USP
Ieda Yuriko Sonehara
66
5. RESULTADOS E DISCUSSÃO

FBT/FCF/USP
Ieda Yuriko Sonehara
67
Estudos de atividade biológica mostraram atividade frente a C. albicans
em compostos das séries azometínica e oxadiazolínica (Tabela 5). Os
compostos da série azometínica contendo anel tiofilidênico (5-nitro-2-
tiofilideno benzidrazidas 4-substituídas) apresentaram problemas de
solubilidade no procedimento para determinação de atividade antifúngica, com
leitura de MIC prejudicada pela precipitação de compostos e foram, por este
motivo, excluídos do estudo. A Tabela 6 traz os resultados de MIC convertidos
para potência.
Tabela 5 Concentração inibitória mínima, MIC, frente à cepa 537Y de C. albicans
SÉRIE AZOMETÍNICA (AzoO) 5-nitro-2-furfurilideno benzidrazidas
4-substituídas
SÉRIE OXADIAZOLÍNICA (OxaS) 3-acetil-oxadiazolina 2,5-tiofilideno
benzidrazidas 4-substituídas
R código MIC (µµµµg/mL) R código MIC (µµµµg/mL)
-Cl AzoO-Cl 7,50 – 7,20 -Cl OxaS-Cl 4,72 – 4,25
-CH3 AzoO-CH3 7,50 – 7,20 -CH3 OxaS-CH3 3,28 – 2,95
-OCH3 AzoO-OCH3 7,50 – 7,20 -OC2H5 OxaS-OC2H5 6,48 – 5,83
-CF3 OxaS-CF3 8,0 – 7,20
-OC4H8 OxaS-OC4H8 7,20 – 6,48
CETOCONAZOL < 0,0293
FLUCONAZOL 0,1172 – 0,0586
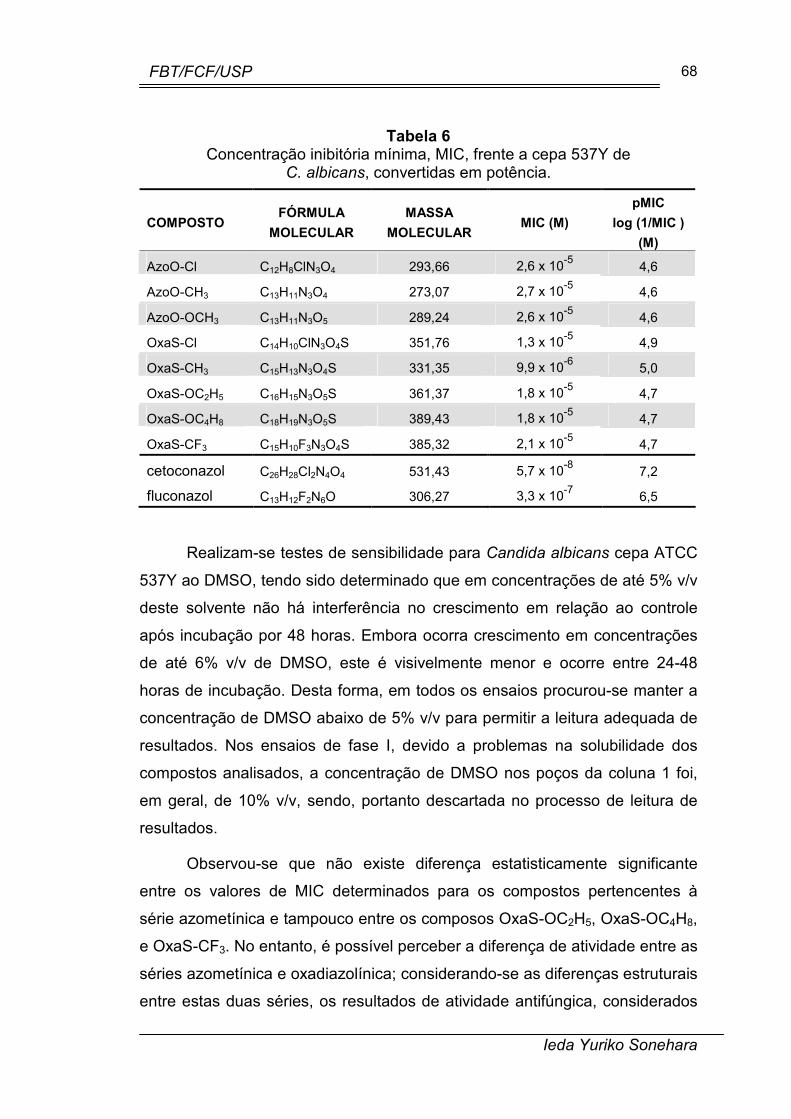
FBT/FCF/USP
Ieda Yuriko Sonehara
68
Tabela 6 Concentração inibitória mínima, MIC, frente a cepa 537Y de
C. albicans, convertidas em potência.
COMPOSTO FÓRMULA
MOLECULAR
MASSA
MOLECULAR MIC (M)
pMIC
log (1/MIC )
(M)
AzoO-Cl C12H8ClN3O4 293,66 2,6 x 10-5
4,6
AzoO-CH3 C13H11N3O4 273,07 2,7 x 10-5
4,6
AzoO-OCH3 C13H11N3O5 289,24 2,6 x 10-5
4,6
OxaS-Cl C14H10ClN3O4S 351,76 1,3 x 10-5
4,9
OxaS-CH3 C15H13N3O4S 331,35 9,9 x 10-6
5,0
OxaS-OC2H5 C16H15N3O5S 361,37 1,8 x 10-5
4,7
OxaS-OC4H8 C18H19N3O5S 389,43 1,8 x 10-5
4,7
OxaS-CF3 C15H10F3N3O4S 385,32 2,1 x 10-5
4,7
cetoconazol C26H28Cl2N4O4 531,43 5,7 x 10-8
7,2
fluconazol C13H12F2N6O 306,27 3,3 x 10-7
6,5
Realizam-se testes de sensibilidade para Candida albicans cepa ATCC
537Y ao DMSO, tendo sido determinado que em concentrações de até 5% v/v
deste solvente não há interferência no crescimento em relação ao controle
após incubação por 48 horas. Embora ocorra crescimento em concentrações
de até 6% v/v de DMSO, este é visivelmente menor e ocorre entre 24-48
horas de incubação. Desta forma, em todos os ensaios procurou-se manter a
concentração de DMSO abaixo de 5% v/v para permitir a leitura adequada de
resultados. Nos ensaios de fase I, devido a problemas na solubilidade dos
compostos analisados, a concentração de DMSO nos poços da coluna 1 foi,
em geral, de 10% v/v, sendo, portanto descartada no processo de leitura de
resultados.
Observou-se que não existe diferença estatisticamente significante
entre os valores de MIC determinados para os compostos pertencentes à
série azometínica e tampouco entre os composos OxaS-OC2H5, OxaS-OC4H8,
e OxaS-CF3. No entanto, é possível perceber a diferença de atividade entre as
séries azometínica e oxadiazolínica; considerando-se as diferenças estruturais
entre estas duas séries, os resultados de atividade antifúngica, considerados

FBT/FCF/USP
Ieda Yuriko Sonehara
69
apenas em seu aspecto qualitativo, podem indicar uma diferença de
mecanismo de ação ou modo de interação com a biomacromolécula-alvo.
A Tabela 7 e Figura 18 mostram a atividade inibitória de crescimento de
compostos das séries azometínica e oxadiazolínica sobre formas
epimastigotas de T. cruzi, como determinados pelo grupo de pesquisa do
Laboratório de Desenvolvimento e Planejamento de Fármacos,
FBT/FCF/USP. Observa-se, também neste caso, uma diferença qualitativa
entre as duas séries de compostos.
Tabela 7 Inibição de crescimento de formas epimastigotas de T. cruzi, cepa Y, em presença de compostos das séries azometínica e oxadiazolínica
INIBIÇÃO DE CRESCIMENTO RELATIVO (%)* COMPOSTO
24 horas 48 horas 72 horas 96 horas
AzoS-Cl 100,6 115,1 100,6 100,4
AzoS-CH3 22,8 48,8 55,7 52,4
AzoS-OC2H5 74,7 80,1 68,5 58,1
AzoS-CF3 84,2 94,5 97,3 98,7
OxaS-Cl 25,3 18,3 15,2 11,7
OxaS-CH3 15,7 15,1 20,3 18,5
OxaS-OC2H5 30,7 15,9 12,2 7,7
OxaS-OC4H8 113,8 19,5 21,9 23,9
OxaS-CF3 16,8 20,1 11,4 9,4
benznidazol 33,5 19,6 17,6 12,8
* em relação ao controle positivo (controle de crescimento) Todos compostos em concentração de 10µM

FBT/FCF/USP
Ieda Yuriko Sonehara
70
Figura 18. Inibição de crescimento* de formas epimastigotas de T. cruzi, cepa Y, em presença de compostos das séries azometínica e oxadiazolínica
* em relação ao controle positivo (controle de crescimento). Todos os compostos em concentração de 10µM
Na determinação de perfil SHED das séries estudadas, constatou-se
que devido à metodologia empregada para a determinação destes descritores
não há diferença entre compostos de mesma estrutura que diferem apenas na
presença do anel tiofilidênico ou furfurilidênico, uma vez que os átomos de
oxigênio e enxofre possuem as mesmas propriedades para efeito da
determinação do SHED (Tabela 2, p. 50). Desta forma, para clareza na
apresentação dos resultados, serão consideradas apenas as séries contendo
anel tiofilidênico, ou seja, séries AzoS (compostos azometínicos) e OxaS
(compostos oxadiazolínicos). As conclusões são válidas para as séries
correspondentes em que X=O. A Figura 19 ilustra um exemplo de
equivalência dos átomos das séries estudadas para os tipos de átomos
utilizados pelo programa Sybyl.

FBT/FCF/USP
Ieda Yuriko Sonehara
71
a
b
Figura 19. Equivalência dos átomos no sistema utilizado pelo programa Sybyl
a) AzoO-CH3. b) OxaS-CH3
Foram também calculados perfis SHED de compostos antifúngicos
selecionados (Figura 20) e compostos anti-T. cruzi (Figura 21) com a
finalidade de possibilitar estudos comparativos e formulação de hipóteses
quanto a alvos e mecanismos de ação.

FBT/FCF/USP
Ieda Yuriko Sonehara
72
fluconazol voriconazol
miconazol cetoconazol
itraconazol
Figura 20. Estruturas dos antifúngicos azólicos selecionados para estudo
benznidazol nifurtimox
Figura 21. Estruturas de compostos anti-T. cruzi
A Tabela 8, Tabela 9 e Tabela 10, e as Figura 22 (p.76), Figura 26,
(p. 80) e Figura 27 (p. 81) trazem os perfis SHED para as séries azometínica,
oxadiazolínica, compostos antifúngicos e compostos anti-T. cruzi.

73
Tabela
8
Pe
rfis
SH
ED
de
com
post
os
azo
me
tínic
os
HH
HR
HA
HD
RR
RA
RD
AA
AD
DD
Azo
S-B
r 4,8
130
5,5
926
5,4
641
2,6
644
5,2
717
5,3
889
3,2
734
1,8
899
1,4
142
0,0
000
Azo
S-C
2H5
5,2
873
5,9
565
5,9
214
3,0
446
5,2
717
5,3
889
3,2
734
1,8
899
1,4
142
0,0
000
Azo
S-C
F3
4,8
130
5,7
830
5,4
341
2,6
644
6,1
504
6,0
090
3,2
132
1,8
899
1,4
142
0,0
000
Azo
S-C
H3
4,8
130
5,5
926
5,4
341
2,6
644
5,2
717
5,3
889
3,2
734
1,8
899
1,4
142
0,0
000
Azo
S-C
l 4,8
130
5,5
926
5,4
341
2,6
644
5,2
717
6,3
889
3,2
734
1,8
899
1,4
142
0,0
000
Azo
S-C
OC
H3
5,2
873
5,9
921
5,9
908
3,0
446
5,9
283
6,2
934
3,4
128
3,2
988
1,2
932
0,0
000
Azo
S-F
4,8
130
5,5
926
5,5
206
2,6
644
5,2
717
5,8
719
3,2
734
2,7
253
1,8
946
0,0
000
Azo
S-H
4,3
959
5,2
676
4,9
486
2,2
969
5,2
717
5,3
889
3,2
734
1,8
899
1,4
142
0,0
000
Azo
S-I
4,8
130
5,5
926
5,4
341
2,6
644
5,2
717
5,3
889
3,2
734
1,8
899
1,4
142
0,0
000
Azo
S-N
(CH
3)2
5,4
821
6,1
646
6,0
818
3,1
506
5,5
801
5,7
694
3,3
877
1,8
899
1,4
142
0,0
000
Azo
S-N
H2
4,3
959
5,3
148
4,9
486
3,8
940
5,5
801
5,7
694
5,6
734
1,8
899
2,2
278
0,5
000
Azo
S-N
O2
4,3
959
5,5
220
5,0
072
2,2
969
6,1
261
6,4
780
3,3
885
3,2
646
1,1
906
0,0
000
Azo
S-O
C2H
5 5,7
459
6,3
622
6,4
101
3,4
358
5,2
717
5,3
889
3,2
734
1,8
899
1,4
142
0,0
000
Azo
S-O
CH
3 5,2
873
5,9
565
5,9
214
3,0
446
5,2
717
5,3
889
3,2
734
1,8
899
1,4
142
0,0
000
Azo
S-O
H
4,3
959
5,2
676
5,0
597
3,8
940
5,2
717
5,8
719
5,6
734
2,7
253
2,2
929
0,5
000
As
colu
nas
corr
esp
onde
m
aos
pare
s de
cara
cterí
stic
as
consi
dera
dos
na
medid
a
de
dis
tânci
as;
H
=h
idro
fobic
ida
de,
R=
aro
matic
idade,
D=
capa
cidad
e d
oadora
de
ligaçõ
es
de h
idro
gên
io,
A=
cap
aci
da
de a
cepto
ra d
e li
gaçõ
es
de
hid
rogê
nio

74
Tabela
9
Pe
rfis
SH
ED
de
com
post
os
oxa
dia
zolín
ico
s
HH
HR
HA
HD
RR
RA
RD
AA
AD
DD
Oxa
S-B
r 4,3
638
4,9
348
5,4
018
0,0
000
4,8
379
5,0
854
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-C
2H5
4,7
474
5,2
617
5,7
915
0,0
000
4,8
379
5,0
854
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-C
F3
4,3
638
5,2
900
5,4
018
0,0
000
5,7
568
5,4
803
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-C
H3
4,3
638
4,9
348
5,4
018
0,0
000
4,8
379
5,0
854
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-C
l 4,3
638
4,9
348
5,4
018
0,0
000
4,8
379
5,0
854
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-C
OC
H3
4,7
474
5,3
791
5,8
405
0,0
000
5,5
142
5,9
926
0,0
000
1,3
747
0,0
000
0,0
000
Oxa
S-F
4,9
348
5,4
156
0,0
000
4,8
379
5,5
439
0,0
000
1,8
899
0,0
000
0,0
000
0,0
000
Oxa
S-H
4,0
057
4,6
443
5,0
142
0,0
000
4,8
379
5,0
854
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-I
4,3
638
4,9
348
5,4
018
0,0
000
4,8
379
5,0
854
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-N
(CH
3)2
4,9
541
5,4
908
5,8
978
0,0
000
5,1
564
5,5
057
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-N
H2
4,0
057
4,7
640
5,0
142
4,5
000
5,1
564
5,5
057
5,5
456
0,9
449
0,9
449
0,0
000
Oxa
S-N
O2
4,0
057
5,0
581
5,0
678
0,0
000
5,7
236
6,1
175
0,0
000
1,4
359
0,0
000
0,0
000
Oxa
S-O
C2H
5 5,1
431
5,6
236
6,1
468
0,0
000
4,8
379
5,0
854
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-O
CH
3 4,7
474
5,2
617
5,7
915
0,0
000
4,8
379
5,0
854
0,0
000
0,9
449
0,0
000
0,0
000
Oxa
S-O
H
4,0
057
4,6
443
5,0
766
4,5
000
4,8
379
5,5
439
5,5
456
1,8
899
0,9
449
0,0
000
Oxa
S-O
C4H
8 5,9
413
6,4
207
6,9
489
0,0
00
4,8
379
5,1
085
0,0
000
1,8
899
0,0
000
0,0
000
As
colu
nas
corr
esp
onde
m
aos
pare
s de
cara
cterí
stic
as
consi
dera
dos
na
medid
a
de
dis
tânci
as;
H
=hid
rofo
bic
ida
de,
R=
aro
matic
idade,
D=
capa
cidad
e d
oadora
de
ligaçõ
es
de h
idro
gên
io,
A=
cap
aci
da
de a
cepto
ra d
e li
gaçõ
es
de
hid
rogê
nio

75
Tabela
10
Pe
rfis
SH
ED
de
com
post
os
an
tifú
ngi
cos
e a
nti-T.cruzi
sele
cion
ado
s
HH
HR
HA
HD
RR
RA
RD
AA
AD
DD
ceto
conazo
l 8,3
824
8,7
021
9,3
711
0,0
000
8,4
918
7,0
324
0,0
000
0,5
000
0,0
000
0,0
000
itraco
nazo
l 9,2
324
9,1
346
7,7
751
0,0
000
8,4
538
6,3
690
0,0
000
1,1
906
0,0
000
0,0
000
mic
onazo
l 4,5
551
4,5
305
4,3
361
0,0
000
3,9
545
3,1
506
0,0
000
0,0
000
0,0
000
0,0
000
vorico
nazo
l 4,0
521
4,2
581
4,3
932
2,5
363
3,9
545
4,4
219
2,0
302
4,0
631
1,4
709
0,0
000
fluco
nazo
l 3,9
208
4,1
134
4,2
097
2,5
929
3,5
967
4,1
783
1,7
472
3,3
342
1,3
747
0,0
000
benzn
ida
zol
4,3
954
4,9
537
5,0
674
2,3
912
5,0
525
5,0
960
2,7
374
1,8
899
1,4
142
0,0
000
nifu
rtim
ox
3,6
489
4,5
356
4,1
101
0,0
000
4,9
631
5,2
918
0,0
000
1,8
946
0,0
000
0,0
000
As
colu
nas
corr
esp
on
de
m
aos
pare
s de
cara
cterí
stic
as
consi
dera
dos
na
medid
a
de
dis
tânci
as;
H
=hid
rofo
bic
idad
e,
R=
aro
matic
idade,
D=
capa
cidad
e d
oadora
de
ligaçõ
es
de h
idro
gên
io,
A=
cap
aci
da
de a
cepto
ra d
e li
gaçõ
es
de
hid
rogê
nio

FBT/FCF/USP
Ieda Yuriko Sonehara
76
a b
Figura 22. Perfis SHED das séries estudadas a) compostos azometínicos. b) compostos oxadiazolínicos
Partindo-se da premissa que compostos com perfis SHED
semelhantes possuem capacidade potencial de interação com os mesmos
alvos biológicos [GREGORI-PUIGJANÉ, MESTRES, 2006], optou-se por
analisar os perfis das séries estudadas em comparação a fármacos
antifúngicos e anti-T. cruzi. Os fármacos antifúngicos de estrutura azólica
(imidazóis e triazóis) foram escolhidos devido ao fato de atuarem sobre a
CYP51 [VANDEN BOSSCHE, KOYMANS, 1998], também alvo para
compostos anti-T. cruzi [LEPESHEVA et al, 2007; BUCKNER et al, 2003] e
que poderia explicar a ação dual apresentada pelos compostos das séries
estudadas.
A análise dos resultados de virtual profiling (Tabela 11), que tem como
objetivo identificar possíveis alvos a partir da análise de similaridade
topográfica entre ligantes conhecidos, traz como principais alvos possíveis
para os compostos oxadiazolínicos enzimas pertencentes ao complexo
P450, a saber CYP19A (aromatase), CYP3A4, CYP3A5 e CYP3A7. Sabe-se
que alguns compostos azólicos com atividade antifúngica, atuando sobre
CYP51, também possuem atividade inibitória de CYP19A [CASTELLANO et
al, 2008; TRÖSKEN et al, 2004; ZARN, BRÜSCHWEILER, SCHLATTER,
2003] e CYP3A4 [ITOKAWA et al, 2007; LAMB et al, 2000]. Estes resultados

FBT/FCF/USP
Ieda Yuriko Sonehara
77
corroboram a escolha da CYP51 como possível alvo a ser investigado. A
Figura 23 ilustra os compostos responsáveis pelas principais anotações.
Tabela 11 Resultados principais de virtual profiling
ALVO número de anotações*
distância euclidiana mínima / código do
composto
aromatase (CYP19A) 8 0,1432 / OxaS-H
CYP3A4 5 0,1432 / OxaS-H
CYP3A5 5 0,1432 / OxaS-H
CYP3A7 5 0,1432 / OxaS-H
glutationa dissulfito redutase 7 0,1814 / OxaS-H
prostaglandina endoperóxido sintase (COX-2) 11 0,2069 / OxaS-C2H5
* O número de anotações refere-se ao número de compostos analisados anotados para um determinado alvo
a b c
Figura 23. Estruturas de compostos responsáveis pelas anotações para alvos identificados em virtual profiling
a) Letrozol (inibidor de CYP19A, CYP3A4, CYP3A5, CYP3A7). b) Inibidor de glutationa dissulfito redutase. c) Inibidor de COX.
Além da CYP51, o virtual profiling aponta também a COX-2 (ciclo-
oxigenase 2) como alvo possível para os compostos da série oxadiazolínica;
considerando-se a semelhança estrutural entre esta série e compostos anti-

FBT/FCF/USP
Ieda Yuriko Sonehara
78
inflamatórios seletivos para COX-2, a anotação é compreensível e serve
também como validação para o processo. Estudos de docking indicam a
possibilidade de interação da série oxadiazolínica com COX-2, tanto em
conformação R como S (Figura 24), com Ki esperado, determinado pelo
programa Autodock, variando de 1 a 50 µM.
a b
Figura 24. Orientação de interação sugerida para COX-2 a) OxaS-CF3 (R), Ki Est. = 1 µM b) OxaS-OC2H5 (S), Ki Est. = 1 µM
Estes compostos mostram possibilidade de interação com COX-2 de
forma coerente com a orientação de ligação determinada experimentalmente
do composto SC-558, um composto análogo ao celecoxib (Figura 25) e que
possui IC50 experimental de 9 nM. Deve-se, no entanto, ressaltar que os
compostos oxadiazolínicos não apresentam grupo sulfonamídico ou
equivalente, considerado como essencial nos inibidores seletivos de COX-2
para proporcionar a estabilização do composto enzima-inibidor [MICHAUX,
CHARLIER, 2004; POUPLANA et al, 2002]; desta forma, a hipótese de
interação dos compostos oxadiazolínicos com COX-2 deve ser verificada
através de estudos mais aprofundados. Além disso, estudos de docking
isolados podem ser considerados indicativos de possibilidades a serem
estudadas de forma mais aprofundada, devendo ser validadas, por exemplo,
através de procedimentos de dinâmica molecular.

FBT/FCF/USP
Ieda Yuriko Sonehara
79
Figura 25. Superposição de estruturas de análogo ao celecoxib (verde) e composto OxaS-CF3 (R) (azul) em orientação sugerida para interação com COX-2
Corroborando a validade da suposição de atividade dual inibidora de
COX-2 e CYP51, existem relatos na literatura de atividade anti-inflamatória
variável em compostos antifúngicos azólicos [LIEBEL et al, 2006; KÖFELER
et al, 2000], que apresentam basicamente o mesmo perfil SHED de
compostos oxadiazolínicos.
O virtual profiling apresenta como primeiro alvo anotado para
compostos da série azometínica o Receptor Nuclear 1B (Nuclear Receptor
1B, NR1B), em distância euclidiana de 0,4353 (composto AzoO-OC2H5),
seguido de tromboxana sintase a uma distância euclidiana de 0,4820
(composto AzoO-C2H5). Em ambos os casos, considerou-se que os alvos
anotados não auxiliam na elucidação do possível mecanismo de ação
antifúngica ou anti-T. cruzi, embora seja sugestivo o fato de que estes
compostos não sejam anotados para os mesmos alvos da série
oxadiazolínica.
A comparação de perfis revela a semelhança entre compostos
azometínicos e benznidazol, fármaco anti-T. cruzi (Figura 26b), e entre
compostos oxadiazolínicos e os antifúngicos cetoconazol, itraconazol e
miconazol, e o fármaco anti-T. cruzi nifurtimox (Figura 27). Esta semelhança
permite, em princípio, supor que existam alvos em comum entre estes

FBT/FCF/USP
Ieda Yuriko Sonehara
80
compostos, de forma que os compostos oxadiazolínicos possuem o
potencial de interagir com CYP51. Assim, uma consequência interessante é
a possibilidade de nifurtimox também possuir certa atividade antifúngica,
embora esta hipótese não tenha sido verificada devido à falta de
disponibilidade deste composto para testes. Por outro lado, não existe um
consenso quanto ao mecanismo de ação de benznidazol e nifurtimox, sendo
que as hipóteses consideram em sua maior parte a redução do grupo nitro
gerando espécies reativas que causam dano à célula por estresse oxidativo,
porém sem especificar o mecanismo exato pelo qual esta redução
ocorre [GERPE et al, 2008; KRAUTH-SIEGEL, INHOFF, 2003; MAYA et al,
2003; VIODÉ et al, 1999; MAYA et al, 1997; HENDERSON et al, 1988].
a b
Figura 26. Perfis SHED de compostos azometínicos selecionados a) Comparados a antifúngicos selecionados. b) Comparados ao benznidazol

FBT/FCF/USP
Ieda Yuriko Sonehara
81
a b
Figura 27. Perfis SHED de compostos oxadiazolínicos selecionados. a) Comparados a antifúngicos selecionados. b) Comparados ao nifurtimox
Uma das hipóteses para a ação anti-T. cruzi de derivados
nitrofurânicos, como nifurtimox, é a interferência com a enzima tripanotiona
redutase [IRIBARNE et al, 2007; VEGA-TEIJIDO, CARACELLI, ZUKERMAN-
SCHPECTOR, 2006; MAYA et al, 2003; BLUMENSTIEL et al, 1999], parte
do sistema de proteção do micro-organismo contra o estresse oxidativo. Os
derivados nitrofurânicos possuem a capacidade de atuar como substratos
subversivos desta enzima, produzindo, ao serem metabolizados, espécies
de oxigênio reativo que causam dano celular por estresse oxidativo
[KRAUTH-SIEGEL, INHOFF, 2003; HENDERSON et al, 1988]. Esta hipótese
é coerente com o resultado de virtual profiling para os compostos estudados
(Tabela 11, p.77), que aponta como um possível alvo a glutationa redutase,
enzima humana correspondente à tripanotiona redutase e com a qual possui
40% de homologia de sequência total, compartilhando mecanismo catalítico
e substratos subversivos (inibidores não-competitivos) [IRIBARNE et al,
2007]. Sabendo-se que tanto o nifurtimox como a nifuroxazida, protótipo da
série azometínica, são capazes de inibir, em certo grau, a glutationa
redutase (37% e 85%, respectivamente) e a tripanotiona redutase (62% e
75%) [IRIBARNE et al, 2007], não se pode descartar a possibilidade de que
a inibição desta enzima seja responsável pelo menos em parte pela

FBT/FCF/USP
Ieda Yuriko Sonehara
82
atividade anti-T. cruzi dos compostos estudados, em especial da série
azometínica, levando-se em consideração a ação inibitória relatada para a
nifuroxazida (composto AzoO-OH). Um estudo envolvendo derivados
5-nitrofurânicos e 5-nitrotiofênicos de semicarbazonas mostra também a
possibilidade de interação não-competitiva destes compostos com a
tripanotiona redutase [VEGA-TEIJIDO, CARACELLI, ZUKERMAN-
SCHPECTOR, 2006]. Deve-se lembrar, porém, que o sítio de interação de
substratos subversivos tanto da tripanotiona redutase como da glutationa
redutase possui diferentes resíduos carregados cuja interação com o ligante
é importante, tanto para interação quanto para seletividade [IRIBARNE et al,
2007].
É possível também a existência de um mecanismo de ação dual dos
compostos em sua ação anti-T. cruzi, atuando por estresse oxidativo e
inibição de CYP51, à semelhança de outros compostos que possuem a
capacidade de provocar estresse oxidativo e inibição de outras enzimas no
caminho metabólico da biossíntese de esteróis [GERPE et al, 2008].
Observe-se que, mesmo que o virtual profiling não tenha revelado
anotação importante para a glutationa redutase/tripanotiona redutase em
compostos azometínicos, este procedimento é um instrumento útil porém
dependente de coleta de dados e atualização dos mesmos; o banco de
dados Wombat, utilizado no método aplicado, possui dados capturados de
periódicos selecionados (Journal of Medicinal Chemistry, Bioorganic and
Medicinal Chemistry, Bioorganic and Medicinal Chemistry Letters, European
Journal of Medicinal Chemistry) [Sunset Molecular Discovery, 13/11/2009]
durante determinado período de tempo; a não-anotação para determinados
alvos pode ser apenas um reflexo da cobertura do banco de dados.
Compostos antifúngicos azólicos têm como alvo principal a enzima
CYP51, 14α-desmetilase, pertencente ao complexo do citocromo P450. A
Figura 28 mostra o fluconazol ligado ao sítio ativo de CYP51 de M.
tuberculosis (código de acesso PDB 1ea1, resolução 2,21 Å).

FBT/FCF/USP
Ieda Yuriko Sonehara
83
a b
Figura 28. Representação de CYP51 de M. tuberculosis. a) Representação de superfície. b) Cavidade catalítica (interna) em representação de superfície. código PDB 1ea1 (resolução 2,21 Å). Heme em representação palito rosa escuro com átomo de Fe em esfera laranja. Fluconazol em representação palito, verde
As propriedades hidrofóbicas dos ligantes de CYP51 são
consideradas importantes para a interação com a enzima, uma vez que o
reconhecimento do substrato ocorre através de interações hidrofóbicas com
resíduos adjacentes à entrada do canal de acesso 1. Nos antifúngicos
azólicos, o grupo fenílico dissubstituído interage em canal estreito e
altamente hidrofóbico presente no sítio catalítico da enzima [JI et al, 2000];
naqueles compostos com presença de cadeia lateral longa, caso de
cetoconazol e itraconazol, ocorrem interações hidrofóbicas e de van der
Waals adicionais, possivelmente em direção ao canal de acesso de
substrato 2 [SHENG et al, 2009; SHENG et al, 2006; XIAO et al, 2004; JI et
al, 2000]. A presença de interações adicionais devido à maior extensão da
cadeia lateral presente em alguns compostos inibidores de CYP51 pode
representar uma melhor afinidade à enzima; existem suposições que
atribuem a esta maior afinidade a menor ocorrência de resistência em
presença de mutações em resíduos nas regiões próximas ao grupo heme
[XIAO et al, 2004].
Moléculas menores, como é o caso de fluconazol e voriconazol,
interagem também através de interações hidrofóbicas [SHENG et al, 2009;

FBT/FCF/USP
Ieda Yuriko Sonehara
84
PODUST, POULOS, WATERMAN, 2001]. O grupo hidroxila presente no C2
destas moléculas aparentemente não apresenta interações observáveis com
a enzima, apesar de influenciar significativamente a atividade biológica; uma
possível explicação é a interação através de moléculas de água presentes
no sítio catalítico que fariam o papel de ‘ponte’ entre o inibidor e a enzima
[SHENG et al, 2009; SHENG et al, 2006; JI et al, 2000]. O menor tamanho
da molécula com consequente menor número de pontos de interação com a
enzima é uma das hipóteses para explicar a menor atividade biológica do
fluconazol em relação à itraconazol e à cetoconazol [VANDEN BOSSCHE,
KOYMANS, 1998].
É importante ressaltar que o perfil SHED, ao ser representado
graficamente, também incorpora informação sobre o tamanho relativo dos
compostos, representada pela área ocupada no gráfico de radar [GREGORI-
PUIGJANÉ, MESTRES, 2006]; isso se deve ao fato de que, em moléculas
maiores, as distâncias entre centros apresentando as mesmas
características tendem também a ser maiores. Percebe-se esta diferença ao
se compararem os perfis de miconazol, cetoconazol e itraconazol (Figura
27a), onde o perfil do miconazol indica claramente uma estrutura de
tamanho menor que cetoconazol e itraconazol, de tamanhos equivalentes
(estruturas dos antifúngicos mostradas na Figura 20, p. 72). Esta diferença
deve-se basicamente ao tamanho da cadeia lateral presente nas moléculas
destes últimos compostos. É de se esperar que a interação dos compostos
oxadiazolínicos com a CYP51, caso ocorra, não seja tão eficiente quanto à
de itraconazol e de cetoconazol devido ao menor número de possíveis
pontos de interação em sua estrutura.
A diferente orientação de ligação ao sítio ativo destes dois grupos de
compostos antifúngicos é também aparente na análise de perfis SHED, em
que fluconazol e voriconazol apresentam perfis praticamente iguais e
claramente diferentes de moléculas maiores como itraconazol e cetoconazol.
A maior característica hidrofóbica destes últimos também é visível através da
magnitude dos parâmetros HA/HR/HH (envolvendo distâncias entre pares

FBT/FCF/USP
Ieda Yuriko Sonehara
85
com características hidrofóbicas, aromáticas e receptoras de ligações de
hidrogênio) quando comparados ao de fluconazol e voriconazol. Entre os
compostos antifúngicos azólicos, também pode-se considerar que existem
padrões estruturais diferentes em que apenas um deles apresenta presença
de grupo hidroxila. A presença deste grupo ligado ao carbono assimétrico no
fluconazol e voriconazol resulta em característica doadora de ligações de
hidrogênio não presente nos compostos semelhantes ao cetoconazol. Este
comportamento apresenta um certo paralelo em reação ao padrão estrutural
dos compostos estudados (Figura 20, p. 72 e Figura 29).
Figura 29. Características doadoras de ligações de hidrogênio
A interação de antifúngicos azólicos com CYP51 ocorre com
participação importante de moléculas de água presentes na cavidade
catalítica e que são responsáveis por diversas interações por ligação de H
com a molécula do inibidor. A aproximação da molécula do antifúngico ao
átomo de Fe do grupo heme, ao qual deve-se ligar para exercer ação
inibitória, ocorre com auxílio da molécula de água ligada ao próprio átomo de
Fe em seu estado de repouso, em processo influenciado pela distribuição de

FBT/FCF/USP
Ieda Yuriko Sonehara
86
grupos doadores e receptores de ligações de H no sítio catalítico da enzima
[BALDING et al, 2008]; o fluconazol, além disso, é capaz de interações com
moléculas de água presentes no sítio catalítico, podendo formar ligações de
H entre a hidroxila ligada ao seu carbono assimétrico e moléculas de água
que intermediam a interação com resíduo de histidina altamente conservado
presente no sítio catalítico [SHENG et al, 2009; PODUST, POULOS,
WATERMAN, 2001]. Existe ainda a possibilidade de que a presença de
pontos que tornam possível a formação de ligações de H adicionais
influencie tanto a dinâmica de aproximação da molécula ao grupo heme,
como a qualidade da interação com resíduos presentes na cavidade
catalítica, embora os fatores determinantes da afinidade com o receptor
sejam a capacidade de coordenação com o átomo de Fe do grupo heme e a
presença de grupos hidrofóbicos que permitam a interação com resíduos
lipofílicos presentes no canal de acesso ao substrato [SHENG et al, 2009].
Os compostos azometínicos e oxadiazolínicos diferem
estruturalmente em sua flexibilidade e na presença de regiões que
possibilitam interações por ligações de hidrogênio (Figura 29, p. 85). Os
compostos oxadiazolínicos apresentam estrutura mais rígida e com
presença de grupo acetila, que representa possibilidade de interações por
ligação de hidrogênio na região da carbonila e hidrofóbica através do grupo
metila, confirmada pelos mapas de distribuição de propriedades lipofílicas e
hidrofílicas obtidos pelo programa Volsurf (Figura 40, p.100). Além disso,
existe estudo demonstrando a interação de derivados de 1,3,4-oxadiazol-
2(3H)-onas com CYP51 de Mycobacterium tuberculosis, onde a rigidez do
anel oxadiazolínico proporciona condições para a interação das moléculas
com o átomo de ferro do heme ao mesmo tempo em que preenche a
cavidade catalítica através de duas ligações de hidrogênio mediadas por
moléculas de água envolvendo N4 e O do anel oxadiazolínico [ZAMPIERI et
al, 2009].
Observando-se o perfil SHED da série oxadiazolínica em comparação
com os compostos azólicos (Figura 27a, p. 80), percebe-se que a

FBT/FCF/USP
Ieda Yuriko Sonehara
87
distribuição de regiões hidrofóbicas pode levar ao reconhecimento dos
compostos pela enzima de forma a permitir sua passagem pelo canal de
entrada de substrato, e posterior orientação adequada no sítio catalítico.
Nota-se também que os compostos azometínicos (Figura 26a, p. 80)
possuem menor presença de características aromáticas (parâmetro AA), o
que pode interferir com a formação de interações π-π. Embora exista certa
semelhança de perfis entre os compostos azometínicos e os antifúngicos
fluconazol e voriconazol, não se pode concluir de forma definitiva que as
características topológicas são semelhantes o suficiente para levar a uma
interação com o mesmo receptor.
Por outro lado, a hipótese de interação dos compostos da série
oxadiazolínica com CYP51 é reforçada por estudos relatando a ação anti-T.
cruzi dos compostos antfúngicos azólicos itraconazol, posaconazol,
ravuconazol e albaconazol em modelos animais [GUEDES et al, 2004;
URBINA et al, 2003; MOLINA et al, 2000; McCABE, REMINGTON, ARAUJO,
1986].
O estudo de caracterização da cavidade catalítica de CYP51 usando
os mesmos parâmetros utilizados em SHED, (H, R, A, D) mostra a
distribuição destas características de acordo com os resíduos presentes
delimitando a cavidade catalítica. A Figura 30 traz os resultados superpostos
à cavidade da CYP51 de Mycobacterium tuberculosis com fluconazol co-
cristalizado como ligante (código de acesso PDB 1ea1, resolução 2,21 Å). A
cavidade catalítica encontra-se no interior da proteína (área sombreada na
Figura 30a); a Figura 30b traz os limites da cavidade catalítica (malha em
cinza), com a distribuição dos CPTs em sua superfície.

FBT/FCF/USP
Ieda Yuriko Sonehara
88
a b
Figura 30. Cavidade catalítica de CYP51 a) Cavidade catalítica (interna) de CYP51 em representação de superfície, com fluconazol (representação em palito, verde) ligado (cód. de acesso PDB 1ea1). b) CPTs, com cavidade catalítica delineada em cinza; H=ciano, R=laranja, A=vermelho, D=azul. Heme representado como palitos rosa escuro com átomo de Fe em esfera laranja
Na análise de CPTs, percebe-se a distribuição de características
hidrofóbicas principalmente em direção ao canal 2, com presença de
características doadoras de receptoras de ligações hidrogênio na região de
interação do anel fenílico dissubstituído do fluconazol, correspondente ao
canal 1. Comparativamente, resultados de docking identificaram uma
possível orientação de interação para compostos oxadiazolínicos em
conformação R, com o grupo nitro em coordenação com o átomo de ferro do
grupo heme (Figura 31). O Ki esperado, como determinado pelo programa
Autodock, variou de 15 a 130 nM. No modelo proposto por este docking,
percebe-se que o grupo acetila acomoda-se na região equivalente à
ocupada pelo anel fenílico dissubstituído em antifúngicos azólicos, podendo-
se supor que existam ligações de hidrogênio envolvidas nesta interação do
grupo acetila. O anel fenílico substituído dos compostos oxadiazolínicos, de
acordo com o docking, direciona-se para o canal 2, concordando com a
possibilidade de interação em regiões hidrofóbicas.

FBT/FCF/USP
Ieda Yuriko Sonehara
89
Figura 31. Orientação de interação sugerida do composto OxaS-Cl (R) com CYP51
Ki Est. = 15 nM
A análise de fragmentos realizada na base de dados de ligantes co-
cristralizados com proteínas sugere que o anel tiofilidênico interage em
regiões do receptor com características predominantemente hidrofóbicas,
com anel furfurilidênico apresentando interação também em regiões com
características doadoras ou receptoras de ligações hidrogênio (Figura 32).
a b c
Figura 32. Fragmentos presentes nos compostos das séries azometínica e oxadiazolínica, com os CPTs associados quando em interação com diversas proteínas*
a) fragmento R-C=N-N-R b) fragmento tiofilideno c) fragmento furfurilideno. Todos fragmentos representados em modelo palito. *cores indicam as características físico-químicas mapeadas; H=cinza, R=laranja, A=vermelho, D=azul. Os CPTs de diversas proteínas encontram-se superpostos em cada caso.

FBT/FCF/USP
Ieda Yuriko Sonehara
90
Estudos de perfis de solubilidade foram realizados em um conjunto de
13 compostos das séries azometínicas com anel nitrofurfurilidênico (série
AzoO) e oxadiazolínica com anel nitrotiofilidênico (série OxaS), conforme
Tabela 6 (p.68) com uso do programa Volsurf 4.0. A Tabela 12 apresenta
alguns dos descritores utilizados pelo programa Volsurf.
No caso dos compostos oxadiazolínicos, a presença de centro quiral
com possibilidade de configuração R ou S dos compostos levou ao uso de
ambas a configurações nos cálculos, uma vez que não se determinou a
configuração real dos compostos testados biologicamente. Partindo-se de
hipótese de que os compostos estão presentes em mistura, a atividade
biológica (em potência) teve seu valor dobrado para efeito de cálculos. Os
resultados da análise de discriminante por método de mínimos quadrados
(Partial Least Squares Discriminant Analysis, PLS-DA) [INDAHL, MARTENS,
NAES, 2007; NOCAIRI et al, 2005] deste conjunto encontram-se na Figura
35 (p.94) e Figura 36 (p. 95).
Tabela 12
Alguns descritores utizados pelo programa Volsurf
DESCRITOR SIGNIFICADO
CW1-CW8 Fator de capacidade (Capacity Factor)
razão entre regiões hidrofílicas e superfície molecular total
D1-D8 Regiões hidrofóbicas distribuiçãodas regiões hidrofóbicas
HL1-HL2 Balanço hidrofílico-lipofílico
razão entre regiões hidrofílicas e regiões hidrofóbicas; descreve qual efeito é dominante na molécula
ID1-ID8 Momento Integy (INTEraction enerGY) hidrofóbico
medida do desequilíbrio entre centro de massa e baricentro da região hidrofóbica
IW1-IW8 Momento Integy (INTEraction enerGY) hidrofílico
medida do desequilíbrio entre centro de massa e baricentro da região hidrofílica
W1-W8 Regiões hidrofílicas distribuição das regiões hidrofílicas
[CRUCIANI et al, 2000]

FBT/FCF/USP
Ieda Yuriko Sonehara
91
Realizou-se também análise utilizando-se conjunto de 8 compostos,
considerando-se entre os compostos oxadiazolínicos apenas aqueles em
configuração R. Esta escolha foi baseada nos resultados de docking
indicando a interação com CYP51 somente dos compostos nesta
configuração. Os resultados considerando-se um conjunto de 8 compostos
(3 compostos azometínicos, 5 compostos oxadiazolínicos em configuração
R) são apresentados na Figura 33 (escores) e Figura 34 (pesos).
A comparação de resultados entre este dois conjuntos mostrou que,
em princípio, não há diferença significativa entre eles; em ambos os casos,
as duas primeiras componentes principais (Principal Component, PC)
explicam a maior parte da variabilidade dos dados, 77,3% no conjunto de 8
dados e 84,5% no conjunto de 13 dados. Desta forma, optou-se pela análise
do conjunto com maior número de dados para finalidade de discussão de
resultados.

FBT/FCF/USP
Ieda Yuriko Sonehara
92
Figura 33. Resultados de análise de PLS-DA ilustrando os escores das duas primeiras componentes principais para conjunto de 8 compostos*.
*Compostos azometínicos: AzoO-CH3, AzoO-OCH3, AzoO-Cl. Compostos oxadiazolínicos em conformação R: OxaS-CH3, OxaS-OC2H5, OxaS-OC4H8, OxaS-Cl, OxaS-CF3

FBT/FCF/USP
Ieda Yuriko Sonehara
93
Figura 34. Resultados de análise de PLS-DA de conjunto de 8 compostos*: pesos
*Compostos azometínicos: AzoO-CH3, AzoO-OCH3, AzoO-Cl. Compostos oxadiazolínicos em conformação R: OxaS-CH3, OxaS-OC2H5, OxaS-OC4H8, OxaS-Cl, OxaS-CF3
A análise de resultados do conjunto de 13 compostos (3 compostos
azometínicos, 5 compostos oxadiazolínicos em configurações R e S) mostra
que existe uma influência importante das propriedades hidrofóbicas sobre a
atividade biológica na série oxadiazolínica, como pode ser evidenciado
através do gráfico de escores e do gráfico de pesos (loading plot). No gráfico
de escores (Figura 35) são visíveis claramente dois grupos, com duas
componentes principais respondendo por 84% da variabilidade detectada na
atividade biológica.

FBT/FCF/USP
Ieda Yuriko Sonehara
94
Figura 35. Resultados de análise de PLS-DA ilustrando os escores das duas primeiras componentes principais para conjunto de 13 compostos*.
*Compostos azometínicos: AzoO-CH3, AzoO-OCH3, AzoO-Cl. Compostos oxadiazolínicos em conformação R e S: OxaS-CH3, OxaS-OC2H5, OxaS-OC4H8, OxaS-Cl, OxaS-CF3
No gráfico de pesos (Figura 36), é possível visualizar a importância
dos descritores, lembrando que a maior distância em módulo em relação ao
eixo X representa maior influência sobre a atividade. Observa-se que há
concentração de descritores de regiões hidrofóbicas gerados pela sonda
DRY, como por exemplo D1-D8 e ID1-ID8, nos quadrantes superior e
inferior direito, onde se encontram os compostos oxadiazolínicos. A
influência de descritores de regiões hidrofílicas geradas pela sonda OH2,
como por exemplo W1-W8 e Cw1-Cw8, é maior nos quadrantes superior e
inferior esquerdo, onde se encontram os compostos azometínicos no gráfico
de escores.
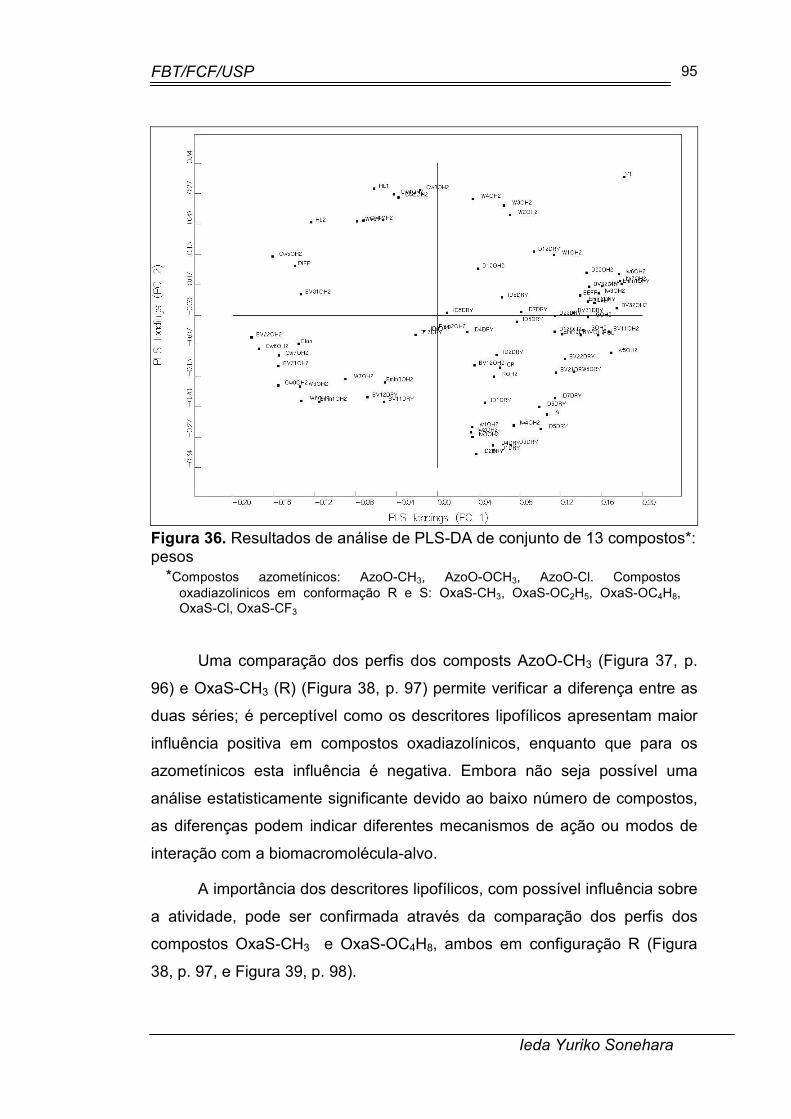
FBT/FCF/USP
Ieda Yuriko Sonehara
95
Figura 36. Resultados de análise de PLS-DA de conjunto de 13 compostos*: pesos
*Compostos azometínicos: AzoO-CH3, AzoO-OCH3, AzoO-Cl. Compostos oxadiazolínicos em conformação R e S: OxaS-CH3, OxaS-OC2H5, OxaS-OC4H8, OxaS-Cl, OxaS-CF3
Uma comparação dos perfis dos composts AzoO-CH3 (Figura 37, p.
96) e OxaS-CH3 (R) (Figura 38, p. 97) permite verificar a diferença entre as
duas séries; é perceptível como os descritores lipofílicos apresentam maior
influência positiva em compostos oxadiazolínicos, enquanto que para os
azometínicos esta influência é negativa. Embora não seja possível uma
análise estatisticamente significante devido ao baixo número de compostos,
as diferenças podem indicar diferentes mecanismos de ação ou modos de
interação com a biomacromolécula-alvo.
A importância dos descritores lipofílicos, com possível influência sobre
a atividade, pode ser confirmada através da comparação dos perfis dos
compostos OxaS-CH3 e OxaS-OC4H8, ambos em configuração R (Figura
38, p. 97, e Figura 39, p. 98).

FBT/FCF/USP
Ieda Yuriko Sonehara
96
Figura 37. Perfil de descritores Volsurf para composto oxadiazolínico AzoO-CH3
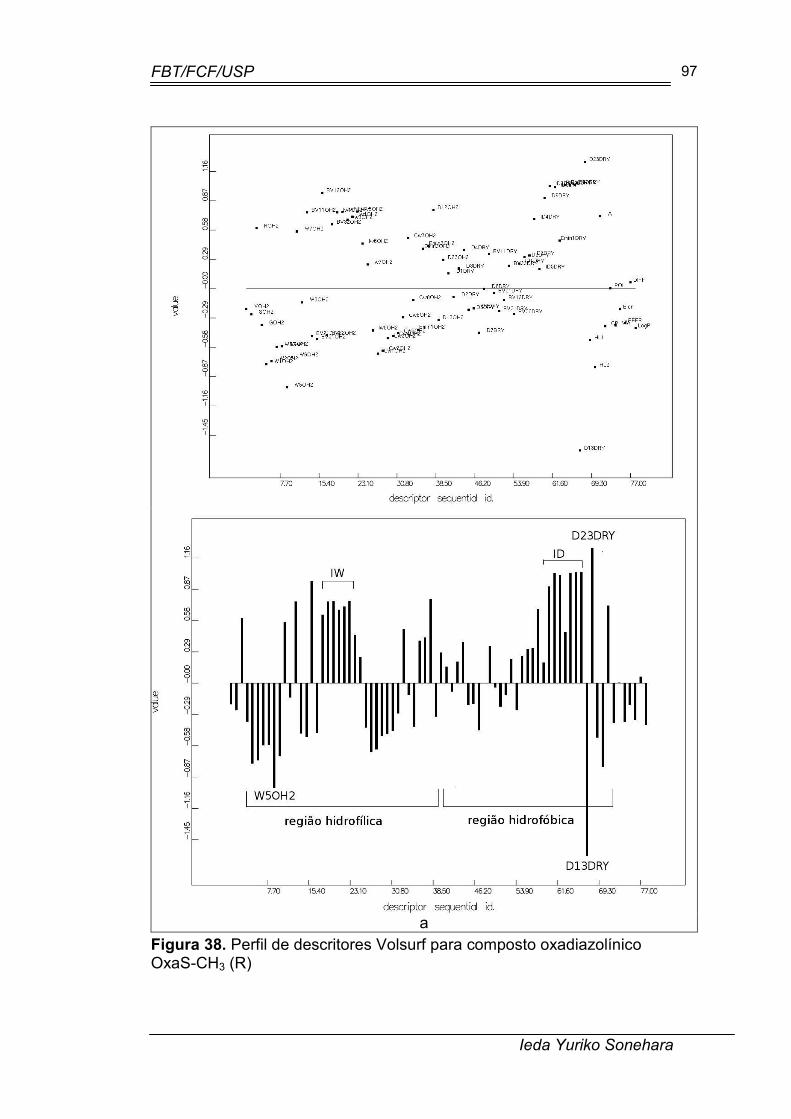
FBT/FCF/USP
Ieda Yuriko Sonehara
97
a Figura 38. Perfil de descritores Volsurf para composto oxadiazolínico OxaS-CH3 (R)
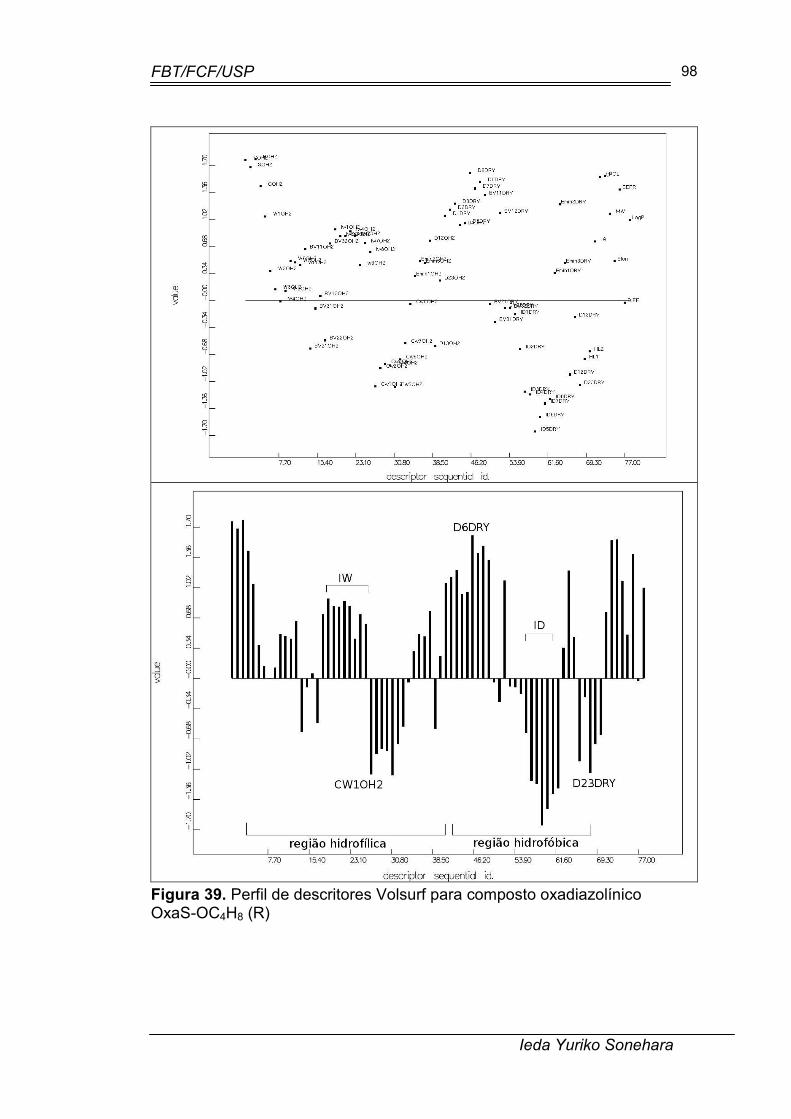
FBT/FCF/USP
Ieda Yuriko Sonehara
98
Figura 39. Perfil de descritores Volsurf para composto oxadiazolínico OxaS-OC4H8 (R)

FBT/FCF/USP
Ieda Yuriko Sonehara
99
Os resultados de atividade antifúngica (Tabela 6, p. 68) mostram
maior atividade anti-Candida do composto OxaS-CH3, com potência
decrescente nos compostos OxaS-OC2H5 e OxaS-OC4H8. Percebe-se como,
entre OxaS-CH3 e OxaS-OC4H8 (Figura 37 e Figura 39), há uma modificação
na direção da influência de vários parâmetros hidrofóbicos, com aumento da
influência positiva de alguns parâmetros hidrofílicos, evidenciando a
influência de substituinte alcoxílico sobre a atividade analisada, embora de
forma menos acentuada. Considerando-se a natureza hidrofóbica do canal
onde teoricamente estes substituintes acomodar-se-iam em uma interação
com CYP51, é compreensível a maior influência de parâmetros hidrofóbicos
sobre a atividade. A comparação dos mapas de contorno obtidos com uso
das sondas OH2 e DRY (Figura 40) também evidencia este fato, chamando
atenção à sutil diferença na conformação do composto OxaS-OC4H8 (R),
com um ângulo maior entre o anel fenílico substituído e o anel
nitrotiofilidênico que poderia ser suficiente para provocar uma interação
estereoquimicamente menos favorável com o receptor.
Em relação às atividades antifúngicas determinadas para os
compostos oxadiazolínicos (Tabela 6, p. 68), percebe-se que não há
diferença estatística nas potências observadas entre os compostos
OxaS-CH3 e OxaS-Cl; este fato pode ser devido à similaridade de volume e
lipofilicidade entre estes dois grupos substituintes, não afetando de forma
significativa a possível interação com a biomacromolécula-alvo. Da mesma
forma, a similaridade de potência entre OxaS-OC2H5, OxaS-OC4H8 e
OxaS-CF3, apesar das diferenças em relação a caracteristicas eletrônicas,
lipofílicas e estéricas, pode indicar que estas modificações foram realizadas
em regiões que não afetam significativamente a interação com o alvo.

FBT/FCF/USP
Ieda Yuriko Sonehara
100
a
b
c
Figura 40. Mapas de contorno representando propriedades hidrofílicas (azul, sonda OH2) e lipofílicas (verde, sonda DRY) de compostos oxadiazolínicos.
a) OxaS-CH3 (R) b) OxaS-OC2H5 (R) c) OxaS-OC4H8 (R). Todas as estruturas representadas em modelo palito. C = cinza, N= azul, O = vermelho, S = amarelo.

FBT/FCF/USP
Ieda Yuriko Sonehara
101
Os resultados de atividade antifúngica (Tabela 6, p. 68) também
indicam uma certa atividade anti-Candida para compostos da série
azometínica, embora menor em comparação com a série oxadiazolínica. No
entanto, resultados de docking levam a crer que a ação dos compostos
azometínicos, caso ocorra por ação inibitória de CYP51, não seja devida à
interação com CYP51 na forma proposta uma vez que não foi detectada
orientação da molécula adequada para esta interação.
A comparação dos volumes das estruturas minimizadas com uso do
programa Sybyl 8.0 (parâmetros de minimização apresentados na Tabela 4,
p. 58) mostra as diferenças de orientação e distribuição de volumes entre as
séries azometínica e oxadiazolínica (Figura 41). Comparados com dados
publicados em relação ao docking de fluconazol e itraconazol no sítio ativo
de CYP51 [SHENG et al, 2009] (Figura 42), percebe-se que os compostos
da série oxadiazolínica mostram possuir conformação mais favorável à
interação com a enzima, o que concorda com os resultados de docking que
indicam interação favorável para os compostos oxadiazolínicos em
configuração R (Figura 31, p.89). A comparação da distribuição de regiões
lipofílicas e hidrofílicas como determinado pelo Volsurf 4.0 também evidencia
a diferença na distribuição destas propriedades, como mostra a Figura 43.
a b
Figura 41. Volumes de contorno de compostos tiofilidênicos. a) composto AzoO-Cl. b) composto OxaS-Cl (R)

FBT/FCF/USP
Ieda Yuriko Sonehara
102
a b
Figura 42. Orientação das conformações de interação de compostos antifúngicos em CYP51 de Cryptococcus neoformans.
a) Fluconazol. b) Itraconazol. Heme em representação palito rosa. [SHENG et al, 2009]
Figura 43. Mapas de contorno representando propriedades hidrofílicas (azul, sonda OH2) e lipofílicas (verde, sonda DRY) do composto azometínico AzoO-CH3, obtidas com o programa Volsurf.
Composto AzoO-CH3 representado em modelo palito. C= cinza, N = azul, O = vermelho
Não se pode descartar completamente o CYP51 como alvo para a
série de compostos azometínicos, embora a conformação desta série não
seja favorável para a interação com CYP51 da forma descrita para
antifúngos azólicos. Existem estudos demonstrando a possível interação de

FBT/FCF/USP
Ieda Yuriko Sonehara
103
compostos não-azólicos com CYP51, porém sem envolver interação com o
átomo de ferro do grupo heme. Neste modelo, interações por ligações de
hidrogênio e hidrofóbicas com a apoproteína são responsáveis pela
manutenção do inibidor na cavidade catalítica da enzima, impedindo assim a
ligação do substrato. A interação proposta é mostrada na Figura 44, onde
existe uma interação por ligação de hidrogênio com resíduo de tirosina
(Y69), com a cadeia lateral hidrofóbica interagindo em canal estreito
hidrofóbico que corresponde ao possível local de interação da cadeia lateral
em C17 no substrato natural de CYP51 [YAO et al, 2007; ZHU et al, 2006; JI
et al, 2000]. A Figura 45 compara a interação de fluconazol com a possível
interação de lanosterol com CYP51.
Figura 44. Ligação de derivado 2-aminotetralínico no sítio ativo de CYP51 de C. albicans.
S1 (amarelo), região hidrofílica. S2 (azul), região hidrofóbica. S3 (rosa), região canal hidrofóbico estreito. S4 (vermelho), região de ligação de hidrogênio. Ligante representado em modelo palito, heme e resíduos em representação de linhas cinza [YAO et al, 2007].

FBT/FCF/USP
Ieda Yuriko Sonehara
104
Considerando-se este modo alternativo de interação é possível a
interação da série azometínica com CYP51 através da formação de ligações
de hidrogênio na região do anel nitrofurânico, com interações
predominantemente hidrofóbicas na região do anel fenílico substituído. A
distribuição de regiões hidrofílicas e lipofílicas como determinadas pelo
Volsurf (Figura 43, p. 102) reforça esta possibilidade.
Figura 45. Interação de lanosterol (roxo) e fluconazol (verde) no sítio ativo de CYP51 de M. tuberculosis.
Estruturas em representação de linhas. Grupo heme em preto. α-hélice em representação de fita azul . [Modificado de PODUST et al, 2001]
Outro fator interessante a ser considerado são os resultados da
análise de fragmentos (Figura 46); onde foi detectado um composto,
acenaftenoquinona, que possui parte de sua estrutura química em comum
com a série azometínica e, por análise de características de sua interação
com seu receptor, pôde-se detectar que o anel nitrofurânico, analisado como
dois fragmentos, grupo nitro e anel furânico, poderia interagir com regiões do
sítio catalítico de CYP51 que possuem características doadoras de ligação
H, com fragmento azometínico estando localizado em regiões com
características aromáticas.

FBT/FCF/USP
Ieda Yuriko Sonehara
105
a** b**
Figura 46. Fragmentos extraídos de banco de dados e respectivas regiões de CYP51 com possibilidade de interação
a) Três fragmentos da acenaftenoquinona, ligante da proteína de código de acesso PDB 1oay (Imunoglobulina E), comuns à série azometínica, superpostos no sítio catalítico (limites não visíveis) da enzima CYP51 (código PDB 1eae1, resolução 2,21 Å). Porção comum com série azometínica destacada em vermelho na estrutura. b) mesma imagem de A, em superposição aos CPTs da enzima CYP51. ** cores indicam as características físico-químicas mapeadas; H=ciano, R=laranja, A=vermelho, D=azul, P=rosa.
Por outro lado, embora a similaridade topográfica da série
azometínica com os antifúngicos azólicos possa ser questionada existe, sem
dúvida, similaridade significativa com o benznidazol. Embora o mecanismo
de ação deste fármaco não tenha sido elucidado, existe relato de atividade
inibitória sobre CYP51, embora com baixa afinidade [LEPESHEVA et al,
2008]. Uma hipótese é sua ação como inibidor irreversível após redução do
grupo nitro pela citocromo P450 redutase, ligando-se então de forma
covalente à enzima [LEPESHEVA et al, 2008].
A observação dos resultados de inibição de crescimento de T. cruzi
(Tabela 7 e Figura 18) indica atividade inibitória de crescimento menor da
série oxadiazolínica em relação aos compostos azometínicos, resultado este
oposto ao que seria esperado caso a atividade anti-T. cruzi fosse devida
exclusivamente à inibição de CYP51. Este resultado pode indicar uma
diferença no mecanismo de ação entre as duas séries, não se descartando
de imediato a ação inibitória de CYP51 da série oxadiazolínica uma vez que

FBT/FCF/USP
Ieda Yuriko Sonehara
106
estes compostos apresentam atividade inibitória de crescimento comparável
ao do benznidazol.
Considerando a hipótese de interação dos compostos da série
oxadiazolínica no sítio ativo de CYP51 seguindo uma orientação similar à
dos compostos antifúngicos azólicos, pode-se concluir que algumas
características estruturais e físico-químicas são necessárias para uma
interação adequada. Em primeiro lugar, é necessária a presença de um
grupo que seja capaz de coordenar-se com o átomo de Fe do grupo heme
de forma semelhante ao nitrogênio básico dos anéis imidazólico e triazólico;
no caso dos compostos oxadiazolínicos, este papel seria assumido pelo
grupo nitro ligado ao anel furfurilidênico ou tiofilidênico. É também
necessário um substituinte lipofílico de volume adequado para interação em
bolso hidrofóbico de tamanho restrito, na posição equivalente ao do anel
fenílico dissubstituído dos antifúngicos azólicos; nos compostos
oxadiazolínicos de configuração R, o grupo 3-acetil apresenta orientação
adequada para a interação com este bolso hidrofóbico. O anel oxadiazolínico
possivelmente mantém a relação de distância adequada entre estes dois
pontos hipotéticos de interação, grupo nitroheterocíclico e grupo acetila, e o
anel fenílico que se posicionaria em direção ao canal de acesso de substrato
2, de característica hidrofóbica. Substituintes adequados na posição 4 do
anel fenílico são possivelmente necessários para assegurar a melhor
interação com a enzima através de interações hidrofóbicas adicionais ao
longo do canal de acesso. A Figura 47 ilustra estas características.
Em relação aos compostos estudados pertencentes à série
oxadiazolínica, seguindo-se a proposição do modo de interação com CYP51
sugere-se para o futuro a síntese e ensaio de atividade antifúngica de
compostos com substituintes lipofílicos de maior volume porém não-
ramificados na posição 4 do anel fenílico, com o objetivo de melhorar a
interação com a enzima através da introdução de pontos adicionais de
interação hidrofóbica.

FBT/FCF/USP
Ieda Yuriko Sonehara
107
Figura 47. Proposição de características necessárias para interação de compostos oxadiazolínicos com sítio catalítico de CYP51.
Heme em representação palito rosa, átomo de Fe em esfera laranja. Cavidade catalítica delineada em cinza. Baseado em estrutura de CYP51, código PDB 1ea1 (resolução 2,21 Å).

FBT/FCF/USP
Ieda Yuriko Sonehara
108
6. CONCLUSÕES

FBT/FCF/USP
Ieda Yuriko Sonehara
109
• Os compostos das séries azometínica e oxadiazolínica apresentam
atividade antifúngica, com a série oxadiazolínica apresentando maior
atividade. O composto mais ativo entre aqueles testados foi OxaS-
CH3, com MIC entre 3,28 e 2,95 µg/mL.
• A análise de similaridade de perfis topológicos com uso de perfis SHED
indicou semelhança da série oxadiazolínica com compostos
antifúngicos azólicos, cujo sítio de ação é a CYP51.
• Estudos de virtual profiling sugeriram como possíveis alvos para a série
oxadiazolínica as enzimas CYP19 (aromatase), CYP3A4, CYP3A5 e
CYP3A7, enzimas estas que também são inibidas por compostos
azólicos. Estes estudos indicaram ainda possível ação inibitória de
COX-2, corroborados por estudos de docking.
• A distribuição de características físico-químicas na cavidade catalítica
de CYP51 e o perfil de solubilidade de compostos oxadiazolínicos,
obtido com Volsurf, sugerem a possibilidade de interação com o sítio
catalítico da CYP51. Estudos de docking indicam a possibilidade de
interação de compostos oxadiazolinicos em configuração R com este
sítio, com o grupo nitro do anel nitroheterocíclico coordenado ao átomo
de Ferro do grupo heme.
• Os compostos azometínicos não possuem distribuição de propriedades
físico-quimicas ou perfis SHED que indiquem possível interação com
CYP51 na orientação adotada pelos compostos azólicos. É possível
que a interação ocorra em orientação diferente daquela de compostos
azólicos, sem interação de coordenação com o átomo de Ferro do
grupo heme.
• Os compostos oxadiazolínicos são menos ativos em relação a
compostos azometínicos como inibidores de crescimento de formas
epigastigotas de T. cruzi, fato que pode indicar diferentes mecanismos
de ação contra este protozoário. Existe a possibilidade de que os
compostos azometínicos interajam com outro alvo, como por exemplo

FBT/FCF/USP
Ieda Yuriko Sonehara
110
tripanotiona redutase, ou que interfiram com o mecanismo de proteção
do micro-organismo contra estresse oxidativo devido à presença do
grupo nitro.
• Com estes estudos é possível sugerir alguns requisitos estruturais que
dever ser considerados no planejamento de novos análogos e que,
provavelmente, favorecem a atividade desses novos ligantes, de
estrutura oxadiazolínica, frente à CYP51. Assim, sugere-se que no
planejamento destes novos ligantes de CYP51 se considerem as
seguintes características:
o grupo ligado ao anel heterocíclico adequado para interação
com átomo de Ferro do grupo heme;
o substituinte na posição 3 do anel oxadiazolínico com
características hidrofóbicas e restrição de volume, permitindo
interação com bolso hidrofóbico presente na CYP51;
o presença de substituintes hidrofóbicos na posição 4 do anel
fenílico com características estéricas adequadas para a
interação no canal de acesso de substrato 2 de CYP51.

FBT/FCF/USP
Ieda Yuriko Sonehara
111
7. PERSPECTIVAS FUTURAS

FBT/FCF/USP
Ieda Yuriko Sonehara
112
O presente estudo traz indicações interessantes sobre o possível
mecanismo de ação dos compostos azometínicos e oxadiazolínicos
estudados. Assim, para etapas futuras, propõe-se:
• Síntese e determinação de atividade biológica de derivados com
substuintes mais lipofílicos na posição 4 do anel fenílico. Sugere-se,
em primeiro lugar, a síntese e ensaio dos compostos OxaS-propil e
OxaS-butil;
• Estudos de inibição enzimática, para confirmação da hipótese proposta
de atividade antifúngica e anti-T. cruzi dos compostos oxadiazolínicos
por inibição de CYP51;
• Estudos de docking validados por dinâmica molecular, possibilitando
um melhor entendimento dos resíduos e tipos de interação envolvidos
na ligação com a biomacromolécula-alvo;
• Estudos de docking e inibição enzimática dos compostos frente a
outras enzimas, como por exemplo tripanotiona redutase, verificando
hipótese alternativa para ação anti-T. cruzi.
• Determinação de possível padrão de substituição na estrutura dos
compostos oxadiazolínicos que possa levar a um melhor perfil de
atividade biológica e à introdução de novo composto-líder ou protótipo
para o desenvolvimento de novos fármacos antifúngicos e anti-T. cruzi.

FBT/FCF/USP
Ieda Yuriko Sonehara
113
8. REFERÊNCIAS BIBLIOGRÁFICAS

FBT/FCF/USP
Ieda Yuriko Sonehara
114
AGUIRRE, G., BOIANI, L., BOIANI, M., CERECETTO, H., DI MAIO, R., GONZÁLEZ, M., PORCAL, W., DENICOLA, A., PIRO, O.E., CASTELLANO, E.E., SANT’ANNA, C.M.R., BARREIRO, E.J. New potent 5-substitued benzofuroxans as inhibitors of Trypanosoma cruzi growth: Quantitative structure-activity relationship studies. Bioorg. Med. Chem., Oxford, v. 13, n. 23, p. 6336-6346, 2005
AGUIRRE, G., CABRERA, M.; CERECETTO, H.; DI MAIO, R.; GONZÁLEZ, M.; SEOANE, G.; DUFFAULT, A.; DENICOLA, AN.; GIL, M.J.; MARTÍNEZ-MERINO, V. Design, synthesis and biological evaluation of new potent 5-nitrofuryl derivatives as anti-Trypanosoma cruzi agents. Studies of trypanothione binding site of trypanothione reductase as target for rational design. Eur. J. Med. Chem., Paris, v. 39, n. 5, p. 421-431, 2004.
AKINS, R.A. An update on antifungal targets and mechanisms of resistance in Candida albicans. Med. Mycol., Oxford, v. 43, n. 4, p. 285-318, 2005.
ALMEIDA, L.V. Síntese e determinação da atividade antimicrobiana de 2-[5-nitro-tiofen-2-il]-3-acetil-5-[4-fenil-substituído]-2,3-diidro-1,3,4-oxadiazolinas frente à cepa ATCC 25923 de Staphylococcus aureus. 2009. 133p. Dissertação (Mestrado). Faculdade de Ciências Farmacêuticas da Universidade de São Paulo. São Paulo, 2009.
ALRAJHI, A.A.; IBRAHIM, E.A.; DE VOL, E.B.; KHAIRAT, M.; FARIS, R.; MAGUIRE, J.H. Fluconazole for the treatment of cutaneous leishmaniasis caused by Leishmania major. N. Engl. J. Med., Boston, v. 346, n. 12, p.892-895, 2002.
ANZENBACHER, P.; ANZENBACHEROVÁ, E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol. Life Sci., Basel, v. 58, n. 5-6, p. 737-747, 2001.
ANZENBACHER, P.; ANZENBACHEROVÁ, E.; LANGE, R.; SKOPALÍK, J.; OTYEPKA, M. active sites of cytochromes P450: what are they like? Acta Chim. Slov., Ljubljana, v. 55, n. 1, p. 63-66, 2008.
Referências bibliográficas de acordo com a norma NBR-6023/2002 da Associação Brasileira de Normas Técnicas (ABNT).

FBT/FCF/USP
Ieda Yuriko Sonehara
115
ARAN, V.J., OCHOA, C., BOIANI, L., BUCCINO, P., CERECETTO, H., GERPE, A., GONZALEZ, M., MONTERO, D., NOGAL J.J., GOMEZ-BARRIO, A., AZQUETA, A., CERAIN A.L., PIRO,O.E., CASTELLANO, E.E. Synthesis and biological properties of new 5-nitroimidazole derivatives. Bioorg. Med. Chem., Oxford, v. 13, n. 9, p. 3197–3207, 2005.
BALAKIN, K.V.; TKCHENKO, S.E.; KISELYOV, A.; SAVCHUK, N.P. Focused chemistry from annotated libraries. Drug Discov. Today: Technol., Kidlington, v. 3, n. 4, p. 397-403, 2006.
BALDING, P.R.; PORRO, C.S. ; MCLEAN, K.J.; SUTCLIFFE, M.J.; MARÉCHAL, J.D.; MUNRO, A.W.; DE VISSER, S.P. How do azoles inhibit cytochrome P450 enzyme? A density functional study. J. Phys. Chem. A, Washington, v. 112, n. 50, p. 12911-12918, 2008.
BARBANY, M.; GUTIÉRREZ-DE-TERÁN, H.; SANS, F.; VILLÀ-FREIXA, J. Towards a MIP-based alignment and docking in computer-aided drug design. Proteins, New York, v. 56, n. 3, p. 585-594, 2004.
BEALE JR, J.M. Anti-infective agents. In Block, J.H.; Beale, Jr, J.H., eds. Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry. 11th ed, Baltimore: Lippincot-Wiliams & Wilkins, 2004, p. 217-281.
BERMAN, H.M.; BATTISTUZ, T.; BHAT, T.N.; BLUHM, W.F.; BOURNE, P.E.; BURKHARDT, K.; FENG, Z.; GILLILAND, G.L.; IYPE, L.; JAIN, S.; FAGAN, P.; MARVIN, J.; PADILLA, D.; RAVICHANDRAN, V.; SCHNEIDER, B.; THANKI, N.; WEISSIG, H.; WESTBROOK, J.D.; ZARDECKI, C. The Protein Data Bank. Acta Crystallogr. D., Oxford, v. 58, n. 6, p. 899-907, 2002.
BERMAN, J.D. Activity of imidazoles against Leishmania tropica in human macrophage cultures. Am. J. Trop. Med. Hyg., Northbrook, v. 30, n.3, p. 566-569, 1981.
BERN, C.; MONTGOMERY, S.P. An estimate of the burden of Chagas disease in the United States. Clin. Infect. Dis., Chicago, v. 49, n. 5, e52-e54, 2009.

FBT/FCF/USP
Ieda Yuriko Sonehara
116
BLOCK, J.H. Physicochemical properties in relation to biological action. In BLOCK, J.H.; BEALE JR., J.H., ed. Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry. 11th ed. Baltimore: Lippincot-Wiliams & Wilkins; 2004, 3-42.
BLUMENSTIEL, K.; SCHONECK, R.; YARDLEY, V.; CROFT, S.L.; KRAUTH-SIEGEL, R.L. Nitrofuran drugs as common subversive substrates of Trypanosoma cruzi lipoamide dehydrogenase and trypanothione reductase. Biochem. Pharmacol., Oxford, v. 58, n. 11, p. 1791-1799, 1999.
BOIANI, L., GERPE, A., ARÁN, V.J., TORRES DE ORTIZ, S., SERNA, E., VERA DE BILBAO, N., SANABRIA, L., YALUFF, G., NAKAYAMA, H., ROJAS DE ARIAS, A., MAYA, J.D., MORELLO, J.A., CERECETTO, H., GONZÁLEZ, M. In vitro and in vivo antitrypanosomatid activity of 5-nitroimidazoles. Eur. J. Med. Chem., Paris, v. 44, n. 3, p. 1034-1040, 2008.
BROWN, C.M.; REISFELD, B.; MAYENO, A.N. Cytochromes P450: a structure-based summary of biotransformations using representative substrates. Drug Metab. Rev., London, v. 40, n. 1, p. 1-100, 2008.
BUCKNER, F.; YOKOYAMA, K.; LOCKMAN, J.; AIKENHEAD, K.; OHKANDA, J.; SADILEK, M.; SEBTI, S.; VAN VOORHIS, W.; HAMILTON, A.; GELB, M.H. A class of sterol 14-demethylase inhibitors as anti-Trypanosoma cruzi agents. Proc. Natl. Acad. Sci. USA, Washington, v.100, n. 25, p. 15149-15153, 2003.
CARHART, R.E.; SMITH, D.H.; VENKATARAGHAVAN, R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J. Chem. Inf. Comput. Sci., Washington, v. 25, n. 2, p. 64-73, 1985.
CARVALHO, S. A.; DA SILVA, E.F.; SANTA-RITA, R.M.; DE CASTRO, S.L.; FRAGA, C.A.M. Synthesis and antitrypanosomal profile of new functionalized 1,3,4-thiadiazole-2-arylhydrazone derivatives, designed as non-mutagenic megazol analogues. Bioorg. Med. Chem. Lett., Oxford, v. 14, n. 24, p. 5967-5970, 2004.
CASTELLANO, S.; STEFANCICH, G.; RAGNO, R.; SCHEWE, K.; SANTORIELLO, M.; CAROLI, A.; HARTMANN, R.W.; SBARDELLA, G. CYP19 (aromatase): exploring the scaffold flexibility for novel selective inhibitors. Bioorg. Med. Chem., Oxford, v.16, n. 18, p. 8349-8358, 2008.

FBT/FCF/USP
Ieda Yuriko Sonehara
117
CERECETTO, H., DI MAIO, R., GONZÁLEZ, M., RISSO, M., SAENZ, P., SEOANE, G., DENICOLA, A. PELUFFO, G., QUIJANO, C., OLEA-AZAR, C. 1,2,5-oxadizaole N-oxide derivatives and related compounds as potential antitrypanosomal drugs: structure-activity relationships. J. Med. Chem., Washington, v. 42, n. 11, p. 1941-1950, 1999.
CHAPMAN, S.W.; SULLIVAN, D.C.; CLEARY, J.D. In search of the Holy Grail of antifungal therapy. Trans. Am. Clin. Climatol. Assoc., Baltimore, v. 119, p. 197-216, 2008.
CHIH-CHENG, L.; CHE-KIM, T.; YU-TSUNG, H.; PEI-LAN, S.; PO-REN, H. Current challenges in the management of invasive fungal infections. J. Infect. Chemother., Tokyo, v. 14, n. 2, p. 77-85, 2008.
COURA, J.R. Present situation and new strategies for Chagas disease chemotherapy – a proposal. Mem. Inst. Oswaldo Cruz, Rio de Janeiro, v. 104, n. 4, p. 549-554, 2009.
COWEN, L.E. The evolution of fungal drug resistance: modulating the trajectory from genotype to phenotype. Nature Rev. Microbiol., London, v. 6, n. 3, p. 187-198, 2008.
CRAMER, C.J. Essentials of Computational Chemistry. 2nd ed. Chichester: John Wiley & Sons; 2004. 596p.
CRAMER, R.D.; PATTERSON, D.F.; BUNCE, J.D. Comparative Molecular Field Analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc., Washington, v. 110, n. 18, p. 5959-5967, 1988.
CRAMER, R.D.; WENDT, B. Pushing the boundaries of 3D-QSAR. J. Comput.-Aided Mol. Des., Amsterdam, v. 21, p. 23-32, n. 1-3, 2007.
CRUCIANI, G.; CRIVORI, P.; CARRUPT, P.-A.; TESTA, B. Molecular fields in quantitative structure-permeation relationships: the VolSurf approach. J. Mol. Struct. (TheoChem), Amsterdam, v. 503, n. 1-2, p. 17-30, 2000.
CRUCIANI, G.; PASTOR, M.; GUBA, W. VolSurf: a new tool for the pharmacokinetic optimization of lead compounds. Eur. J. Pharm. Sci., Paris, v. 11, Supl. 2, p. S29-S39, 2000.

FBT/FCF/USP
Ieda Yuriko Sonehara
118
CRUCIANI, G; CAROSATI, E.; CLEMENTI, S. Three-dimensional quantitative structure-property relationships. In: WERMUTH, C.G, ed. The Practice of Medicinal Chemistry. 2nd ed. Oxford:Academic Press; 2003. p. 69-89.
DEBELJAK, N.; FINK, M.; ROZMAN, D. Many facets of mammalian lanosterol 14α-demethylase from the evolutionarily conserved cytochrome P450 family CYP51. Arch. Biochem. Biophys., New York, v. 409, n. 1, p. 159-171, 2003.
DEBNATH, A.K. Quantitative Structure-Activity Relationship (QSAR) paradigm – Hansch Era to New Milllenium. Mini Rev. Med. Chem., Hilversum, v. 1, n. 2, p. 187-195, 2001.
DENISOV, I.G.; MAKRIS, T.M.; SLIGAR, S.G.; SCHLICHTING, I. Structure and chemistry of cytochrome P450. Chem. Rev., Washington, v. 105, n. 6, p. 2253-2277, 2005.
DEVELOUX, M.; LESCURE, F.-X.; LE LOUP, G.; PIALOUX, G. Maladie de Chagas. Rev. Medecine Int., Paris, v. 30, n. 8, p. 686-695, 2009.
DODD, M. C.; STILLMAN, W. B.; ROYS, M.; CROSBY, C. The in vitro bacteriostatic action of some simple furan derivatives J. Pharmacol. Exp. Ther., Bethesda, v. 82, n. 1, p. 11-18, 1944.
EDDINE, A.N.; VON KRIES, J.P.; PODUST, M.V.; WARRIER, T.; KAUFMANN, S.H.E.; PODUST, L.M. X-ray structure of 4,4’-dihydroxybenzophenone mimicking sterol substrate in the active site of sterol 14α-demethylase (CYP51). J. Biol. Chem., Baltimore, v. 283, n. 22, p. 15152-15159, 2008.
EKINS, S.; MESTRES, J.; TESTA, B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br. J. Pharmacol., London, v. 152, n. 1, p. 9–20, 2007.
ENGEL, T. Basic Overview of Chemoinformatics. J. Chem. Inf. Model., Washington, v. 46, n. 6, p. 2267-2277, 2006.
ESTRADA, E.; URIARTE, E. Recent advances on the role of topological indices in drug discovery research. Curr. Med. Chem., Schiphol, v. 8, n. 13, p. 1573-1588, 2001.

FBT/FCF/USP
Ieda Yuriko Sonehara
119
FERREIRA, M.M.C. Multivariate QSAR. J. Braz. chem. Soc., São Paulo, v. 13, n. 6, p. 742-753, 2002.
FERREIRA, M.M.C.; MONTANARI, C.A.; GAUDIO, A.C. Seleção de variáveis em QSAR. Quim. Nova, São Paulo, v. 25, n. 3, p. 439-448, 2002.
FONTAINE, F.; PASTOR, M.; GUTIÉRREZ-DE-TERÁN, H.; LOZANO, J.J.; SANZ, F. Use of alignment-free molecular descriptors in diversity analysis and optimal sampling of molecular libraries. Mol. Divers., Leiden, v. 6, n. 2., p. 135-147, 2003.
FONTAINE, F.; PASTOR, M.; ZAMORA, I.; SANZ F. Anchor-GRIND: Filling the gap between standard 3D QSAR and the GRid-INdependent Descriptors. J. Med. Chem., Washington, v. 48, n. 7, p. 2687-2694, 2005.
FRIESNER, R.A.; GUALLAR, V. Ab initio quantum chemical and mixed quantum mechanics/molecular mechanics (QM/MM) methods for studying enzymatic catalysis. Annu. Rev. Phys. Chem., Palo Alto, v. 56, p. 389-427, May 2005.
Gerber Molecular Design, http://www.moloc.ch/ (acessado em 10/11/2009)
GERBER, P.R.; MÜLLER, K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput.-Aided Mol. Des., Amsterdam, v. 9, n. 3, p. 251-268, 1995.
GERPE, A.; ODREMAN-NUÑEZ, I.; DRAPER, P.; BOIANI, L.; URBINA, J.A.; GONZÁLEZ, M.; CERECETTO, H. Hereroallyl-containing 5-nitrofuranes as new anti-Trypanosoma cruzi agents with a dual mechanism of action. Bioorg. Med. Chem., Oxford, v. 16, n. 1, p. 569-577, 2008.
GOODFORD, P. A computational procedure for determining energetically favourable binding sites on biologically important macromolecules. J. Med. Chem., Washington, v. 28, n. 7, p. 849-857, 1985.
GOODSELL, D.S.; MORRIS, M.M.; OLSON, A.J. Automated Docking of Flexible Ligands: Applications of AutoDock. J. Mol. Recog., Chichester, v. 9, n. 1, p. 1-5, 1996.

FBT/FCF/USP
Ieda Yuriko Sonehara
120
GREGORI-PUIGJANÉ, E.; MESTRES, J. SHED: Shannon Entropy Descriptors from Topological Feature Distributions. J. Chem. Inf. Model., Washington, v. 46, n. 4, p. 1615-1622, 2006.
GUEDES, P.M.M.; URBINA, J.A.; LANA, M.; AFONSO, L.C.C.; VELOSO, V.M.; TAFURI, W.L.; MACHADO-COELHO, G.L.L.; CHIARI, E.; BAHIA, M.T. Activity of the new triazole derivative albaconazole against Trypanosoma (Schizotrypanum) cruzi in dog hosts. Antimicrob. Agents Chemother., Washington, v. 48, n. 11, p. 4286-4292, 2004.
HALGREN, T.A. Merck molecular force field. I. Basis, scope, parameterization, and performance of MMFF94. J. Comput. Chem., New York, v. 17, n. 5-6, p. 490-519, 1996.
HENDERSON, G.B.; ULRICH, P.; FAIRLAMB, A.; ROSENBERG, I.; PEREIRA, M.; SELA, M.; CERAMI, A. “Subversive” substrates for the enzyme trypanothione disulfide reductase: Alternative approach to chemotherapy of Chagas disease. Proc. Natl. Acad. Sci. USA, Washington, v. 85, n. 15, p. 5374-5378, 1988.
HOF, H. Developments in the epidemiology of invasive fungal infections – implications for the empiric and targeted antifungal therapy. Mycoses, Berlin, v. 51, Supl. 1, p. 1–6, 2008.
HOPFINGER, A.J.; WANG, S.; TOKARSKI, J.S.; JIN, B.; ALBUQUERQUE, M.; MADHAV, P.J.; DURAISWAMI, C. Construction of 3D-QSAR using the 4D-QSAR analysis formalism. J. Am. Chem. Soc., Washington, v. 119, n. 43, p. 10509-10524, 1997.
INDAHL, U.G.; MARTENS, H.; NAES, T. From dummy regression to prior probabilities in PLS-DA. J. Chemom., Chichester, v. 21, n. 12, p. 529-536, 2007.
IRIBARNE, F.; GONZÁLEZ, M.; CERECETTO, H.; AGUILERA, S.; TAPIA, S.; PAULINO, M. Interaction energies of nitrofurans with trypanothione reductase and glutathione reductase studied by molecular docking. Theochem, Amsterdam, v. 818, n. 1-3, p. 7-22, 2007.
ITOKAWA, D.; NISHIOKA, T.; FUKUSHIMA, J.; YASUDA, T.; YAMAUCHI, A.; CHUMAN, H. Quantitative Structure-Activity Relationship study of binding affinity of azole compounds with CYP2B and CYP3A. QSAR Comb. Sci., Weinheim, v. 26, n. 7, p. 828-836, 2007.

FBT/FCF/USP
Ieda Yuriko Sonehara
121
ITOKAWA, D.; YAMAUCHI, AL; CHUMAN, H. Quantitative Structure-Activity Relationship for inhibition of CYP2B6 and CYP3A4 by azole compounds – comparison with their binding affinity. QSAR Comb. Sci., Weinheim, v. 28, n. 6-7, p.629-636, 2009.
JACKSON, C.J.; LAMB, D.C.; KELLY, D.E.; KELLY, S.L. Bactericidal and inhibitory effects of azole antifungal compounds on Mycobacterum smegmatis. FEMS Microbiol. Lett., Amsterdam. 192, n. 2, p. 159-162, 2000.
JACKSON, C.J.; LAMB, D.C.; WARRILOW, A.G.S.; PARKER, J.E.; MELO, N.R.; KELLY, D.E.; KELLY, S.L. P450s in microbial sterol biosynthesis and drug targets. Acta Chim. Slov., Ljubljana, v. 55, n. 1, p.58-62, 2008.
JALENCAS, X., comunicação pessoal, 2008.
JANNIN, J.; VILLA, L. An overview of Chagas disease treatment. Mem. Inst. Oswaldo Cruz, Rio de Janeiro, v. 102, Supl. 1, p. 95-97, 2007.
JI, H.; ZHANG, W.; ZHOU, Y.; ZHANG, M.; ZHU, J.; SONG, Y.; LU, J.; ZHU, J. A three-dimensional model of lanosterol 14α-demethylase of Candida albicans and its interaction with azole antifungals. J. Med. Chem., Washington, v. 43, n. 13, p.2493-2505, 2000.
JORGE, S.D.; MASUNARI, A.; RANGEL-YAGUI, C.O.; PASQUALOTO, K.F.M.; TAVARES, L.C. Design, synthesis, antimicrobial activity and molecular modeling studies of novel benzofuroxan derivatives against Staphylococcus aureus. Bioorg. Med. Chem., Oxford, v. 17, n. 8., p. 3028-3036, 2009.
KELLY, S.L.; LAMB, D.C.; CANNIEUX, M.; GREETHAM, D.; JACKSON, C.J.; MARCZYLO, T.; UGOCHUKWU, C.; KELLY, D.E. An old activity in the cytochrome P450 superfamily (CYP51) and a new story of drugs and resistance. Biochem. Soc. Trans., London, v. 29, part 2, p. 122-128, 2001.
KIRCHMAIR, J.; MARKT, P.; DISTINTO, S.; SCHUSTER, D.; SPITZER, G.M.; LIEDL, K.R.; LANGER, T.; WOLBER, G. The Protein Data Bank (PDB), its related services and software tools as key components for in silico guided drug discovery. J. Med. Chem., Washington, v. 51, n. 22, p. 7021-7040, 2008.

FBT/FCF/USP
Ieda Yuriko Sonehara
122
KLEBE, G.; ABRAHAM, U. On the prediction of binding properties of drug molecules by Comparative Field Analysis. J. Med. Chem., Washington, v. 36, n. 1, p. 70-80, 1993.
KÖFELER, H.C.; FAULER, G.; WINDISCHHOFER, W.; LEIS, H.J. Effect of cytochrome P-450 inhibitors econazole, bifonazole and clotrimazole on prostanoid formation. Br. J. Pharmacol., London, v. 130, n. 6, p. 1241-1246, 2000.
KOROSEC, T.; ACIMOVIC, J.; SELISKAR, M.; KOCJAN, D.; TACER, K.F.; ROZMAN, D.; URLEB, U. Novel cholesterol biosynthesis inhibitors targeting human lanosterol 14α-demethylase (CYP51). Bioorg. Med. Chem., Oxford, v. 16, n.1, p. 209-221, 2008.
KRAUTH-SIEGEL, R.L.; INHOFF, O. Parasite-specific trypanothione reductase as a drug target molecule. Parasitol. Res., Berlin, v. 90, Supl. 2., p. S77-S85, 2003.
LAMB, D.C.; KELLY, D.E.; BALDWIN, B.C.; KELLY, S.L. Differential inhibition of human CYP3A4 and Candida albicans CYP51 with azole antifungal agents. Chem. Biol. Interact., Amsterdam, v. 125, n. 3., p. 165-175, 2000.
LAMB, D.C.; WATERMAN, M.R.; KELLY, S.L.; GUENGERICH, F.P. Cytochromes P450 and drug discovery. Curr. Opin. Biotechnol., London, v. 18, n. 6, p. 504-512, 2007.
LANGER, T.; BRYANT, S.D. 3D Quantitative Structure-Property Relationships. In: WERMUTH, C.G., ed. The Practice of Medicinal Chemistry. 3rd ed. Oxford:Elsevier, 2008. p. 587-604.
LARANJEIRAS, C.C.; CHIAPPIN, J.R.N. A construção de uma teoria de ensembles: antecedentes em Maxwell e Boltzmann. Rev. Bras. Ens. Fis. São Paulo, v. 30, n.1, p. 1601-1611, 2008.
LEPESHEVA, G.I.; HARGROVE, T.Y.; KLESHCHENKO, Y.; NES, W.D.; VILLALTA, F.; WATERMAN, M. CYP51: A major drug target in the cytochrome P450 superfamily. Lipids, Berlin, v. 43, n. 12, p.1117-1125, 2008.

FBT/FCF/USP
Ieda Yuriko Sonehara
123
LEPESHEVA, G.I.; OTT, R.D.; HARGROVE, T.Y.; KLESHCHENKO, Y.Y.; SCHUSTER, I.; NES, W.D.; HILL, G.C.; VILLALTA, F.; WATERMAN, M.R. Sterol 14α-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem. Biol., St. Louis, v. 14, n. 11, p. 1283-1293, 2007.
LEPESHEVA, G.I.; WATERMAN, M.R. Sterol 14α-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta, Amsterdam, v. 1770, n. 3, p. 467-477, 2007.
LEPESHEVA, G.I.; ZAITSEVA, N.G.; NES, W.D.; ZHOU, W.; ARASE, M.; LIU, J.; HILL, G.D.; WATERMAN, M.R. CYP51 from Trypanosoma cruzi. A phyla-specific residue in the B’ helix defines substrate preferences of sterol 14α-demethylase. J. Biol. Chem., Baltimore, v.281, n. 6, p. 3577-3585, 2006.
LIEBEL, F.; LYTE, P.; GARAY, M.; BABAD, J.; SOUTHALL, M.D. Anti-inflammatory and anti-itch activity of sertaconazole nitrate. Arch. Dermatol. Res., Berlin, v. 298, n. 4, p. 191-199, 2006.
LILL, M.A. Multi-dimensional QSAR in drug design. Drug Discov. Today, Kidlington, v.12, n. 23/24, p. 1013-1017, 2007.
MANOEL-CARNEIRO, F.S.; SILVA, A.E. Implications of genetic variability of Trypanosoma cruzi for the pathogenesis of Chagas disease. Cad. Saude Publ., Rio de Janeiro, v. 23, n. 10, p. 2263-2274, 2007.
MASUNARI, A.; TAVARES, L.C. A new class of nifuroxazide analogues: synthesis of 5-nitrothiophene derivatives with antimicrobial activity against multidrug-resistant Staphylococcus aureus. Bioorg. Med. Chem., Oxford, v. 15, n. 12, p. 4229-4236, 2007.
MASUNARI, A.; TAVARES, L.C. Síntese e determinação da atividade antimicrobiana de derivados 5-nitro-2-tiofilidênicos frente a Staphylococcus aureus multi-resistente. Rev. Bras. Cienc. Farmac., São Paulo, v. 42, n. 3, p. 461-471, 2006.
MATSUURA, K.; YOSHIOKA, SH.; TOSHA, T.; HORI, H.; ISHIMORI, K.; KITAGAWA, T.; MORISHIMA, I.; KAGAWA, N.; WATERMAN, M.R. Structural diversity of active site in clinical azole-bound forms between sterol 14α-demethylases (CYP51s) from human and Mycobacterium tuberculosis. J. Biol. Chem., Baltimore, v. 280, n. 10, p.9088-9096, 2005.

FBT/FCF/USP
Ieda Yuriko Sonehara
124
MAYA, J.D.; BOLLO, S.; NUÑEZ-VERGARA, L.J.; SQUELLA, J.A.; REPETTO, Y.; MORELLO, A.; PÉRIÉ, J.; CHAUVIÈRE, G. Trypanosoma cruzi: effect and mode of action of nitroimidazole and nitrofuran derivatives. Biochem. Pharmacol., Oxford, v. 65, n. 6, p. 999-1006, 2003.
MAYA, J.D.; REPETTO, Y.; AGOSÍN, M.; OJEDA, J.M.; TELLEZ, R.; GAULE, C.; MORELLO, A. Effects of Nifurtimox and benznidazole upon glutathione and trypanothione content in epimastigote, trypomastigote and amastigote forms of Trypanosoma cruzi. Mol. Biochem. Parasitol., Amsterdam, v. 86, n. 1, p. 101–106, 1997
McCABE, R.E.; REMINGTON, J.S.; ARAUJO, F.G. In vitro and in vivo effects of itraconazole against Trypanosoma cruzi. Am. J. Trop. Med. Hyg., Northbrook, v. 35, n.2, p. 280-284, 1986.
MESTRES, J. Computational chemogenomics approaches to systematic knowledge-based drug discovery. Curr. Opin. Drug Disc. Devel., London, v. 7, n. 3, p. 304-313, 2004.
MESTRES, J. Gaussian-based Alignment of Protein Structure: deriving a consensus superposition when alternative solutions exist. J. Mol. Model., Berlin, v.6, n. 7-8, p.539-549, 2000.
MESTRES, J. Structure conservation in Cytochromes P450. Proteins, New York, v. 58, n.3, p. 596-609, 2005.
MICHAUX, C.; CHARLIER, C. Structural approach for COX-2 inhibition. Mini Rev. Med. Chem., Hilversum, v. 4, n. 6, p. 603-615, 2004.
MILEI, J.; GUERRI-GUTTENBERG, R.A.; GRANA, D.R.; STORINO, R. Prognostic impact of Chagas disease in the United States. Am. Heart J., St. Louis v. 157, n. 1, p. 22-29, 2009.
MOLINA, J.; MARTINS-FILHO, O.; BRENER, Z.; ROMANHA, A.J.; LOEBENBERG, D.; URBINA, J.A. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicr. Agents Chemother., Washington, v. 44, n. 1, p. 150-155, 2000.

FBT/FCF/USP
Ieda Yuriko Sonehara
125
MONCAYO, A.; ORTIZ YANINE, M.I.. An update on Chagas disease (human American trypanosomiasis). Ann. Trop. Med. Parasitol., Abingdon , v. 100, n. 8, p. 663-677, 2006.
MUNRO, A.W.; MCLEAN, K.J.; MARSHALL, K.R.; WARMAN, A.J.; LEWIS, G.; ROITEL, O.; SUTCLIFFE, M.J.; KEMP, C.A.; MODI, S.; SCRUTTON, N.S.; LEYS, D. Cytochromes P450: novel drug targets in the war against multidrug-resistant Mycobacterium tuberculosis. Biochem. Soc. Trans., London, v. 31, part 3, p. 625-630, 2003.
NCCLS. Método de Referência para Testes de Diluição em Caldo para a Determinação da Sensibilidade a Terapia Antifúngica das Leveduras; Norma Aprovada - Segunda Edição. NCCLS document M27-A2 [ISBN 1-56238-469-4]. NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 Estados Unidos, 2002.
NETTLES, J.H.; JENKINS, J.L.; BENDER, A.; DENG, Z.; DAVIES, J.W.; GLICK, M. Bridging chemical and biological space: “target fishing” using 2D and 3D molecular descriptors. J. Med. Chem., Washington, v. 49, n. 23, p. 6802-6810, 2006.
NOCAIRI, H.; QANNARI, E.M.; VIGNEAU, E.; BERTRAND, D. Discrimination on latent components with respect to patterns. Application to multicollinear data. Comput. Stat. Data Anal., Amsterdam, v. 48, n. 1, p. 139-147, 2005.
ORENGO, C.A.; MICHIE, A.D.; JONES, S.; JONES, D.T.; SWINDELLS, M.B.; THORNTON, J.M. CATH – a hierarchic classification of protein domain structures. Structure, London, v. 5, n. 8, p, 1093-1107, 1997.
OTYEPKA, M.; SKOPALIK, J.; ANZENBACHEROVÁ, E.; ANZENBACHER, P. What common structural features and variations of mammalian P450s are known to date? Biochim. Biophys. Acta, Amsterdam, v. 1770, n. 3, p. 376-389, 2007
PASQUALOTTO, A.C.; DENNING, D.W. New and emerging treatments for fungal infections. J. Antimicrob. Chemother., London, v. 61, Supl. 1, p. i19–i30, 2008
PASTOR, M.; CRUCIANI, G.; MCLAY, I.; PICKETT, S.; CLEMENT, S. GRid-INdependent Descriptors (GRIND): A novel class of alignment-independent three-dimensional molecular descriptors. J. Med. Chem., Washington, v. 43, n. 17, p. 3233-324, 2000.

FBT/FCF/USP
Ieda Yuriko Sonehara
126
PATRICK, G.L. An introduction to Medicinal Chemistry. 4th ed. Oxford: Oxford University Press; 2009, 377-402.
PAULA, F.R.; JORGE, S.D.; ALMEIDA, L.V.; PASQUALOTO, K.F.M.; TAVARES, L.C. Molecular modeling studies and in vitro bioactivity evaluation of a set of novel 5-nitro-heterocyclic derivatives as anti-T. cruzi agents. Bioorg. Med. Chem., Oxford, v. 17, n. 7 , p. 2673-2679, 2009.
PODUST, L.M.; POULOS, T.L.; WATERMAN, M.R. Crystal structure of cytochrome P450 14α-sterol demethylase (CYP51), from Mycobacterium tuberculosis in complex with azole inhibitors. Proc. Natl. Acad. Sci. USA, Washington, v. 98, n. 6, p. 3068-3073, 2001.
PODUST, L.M.; STOJAN, J.; POULOS, T.L.; WATERMAN, M.R. Substrate recognition sites in 14α-sterol demethylase from comparative analysis of amino acid sequences and X-ray structure of Mycobacterium tuberculosis CYP51. J. Inorg. Biochem., New York, v. 87, n. 4, p. 227-235, 2001.
PODUST, L.M.; YERMALITSKAYA, L.V.; LEPESHEVA, G.I.; PODUST, V.N.; DALMASSO, E.A.; WATERMAN, M.R. Estriol bound and ligand-free structures of sterol 14α-demethylase. Structure, London, v. 12, n. 11, p. 1937-1945, 2004.
POUPLANA, R.; LOZANO, J.J.; PÉREZ, C.; RUIZ, J. Structure-based QSAR study on differential inhibition of human prostaglandin endoperoxide H synthase-2 (COX-2) by nonsteroidal anti-inflammatory drugs. J. Comput.-Aided Mol. Des., Amsterdam, v. 16, n. 10, p. 683-709, 2002.
POZAS, R., CARBALLO, J., CASTRO, C., RUBIO, J. Synthesis and in vitro antitrypanosomal activity of novel Nifurtimox analogues. Bioorg. Med. Chem. Lett., Oxford, v. 15, n. 5, p. 1417–1421, 2005.
RCSB Protein Data Bank, http://www.rcsb.org. (acessado em 11/11/2009).
REITHINGER, R.; TARLETON, R.L.; URBINA, J.A.; KITRON, U.; GURTLER, R.E. Eliminating Chagas disease: challenges and a roadmap. BMJ v. 338:b1283, doi: 10.1136/bmj.b1283 (published 14 April 2009).
RICHARDSON, M.; LASS-FLÖRL, C. Changing epidemiology of systemic fungal infections. Clin. Microbiol. Infect., Oxford, v. 14, Suppl. 4, p. 5–24, 2008.

FBT/FCF/USP
Ieda Yuriko Sonehara
127
RODRIGO, J.; BARBANY, M.; GUTIÉRREZ-DE-TERÁN, H.; CENTENO, N.; DE-CÀCERES, M.; DEZI, C.; FONTAINE, F.; LOZANO, J.J.; PASTOR, M.; VILLÀ, J.; SANZ, F. Comparison of biomolecules on the basis of Molecular Interaction Potentials. J. Braz. Chem. Soc., São Paulo, v. 13, n. 6, p. 795-799, 2002.
ROGNAN, D. Chemogenomic approaches to rational drug design. Br. J. Pharmacol., London, v. 152, n. 1, p. 38-52, 2007.
RUGE, E.; KORTING, H.C.; BORELLI, C. Current state of three-dimensional characterisation of antifungal targets and its use for molecular modelling in drug design. Int. J. Antimicrob. Agents, Amsterdam, v. 26, n. 6, p. 427–441, 2005.
SALMON-CHEMIN, L.; BUISINE, E.; YARDLEY, V.; KOHLER, S.; DEBREU, M.A.; LANDRY, V.; SERGHERAERT, C.; CROFT, S.L.; DRAUTH-SIEGEL, R.L.; DAVIOUD-CHARVET, E. 2- and 3-substituted 1,4-naphthoquinone derivatives as subversive substrates of trypanothione reductase and lipoamide dehydrogenase from Trypanosoma cruzi: synthesis and correlation between redox cycling activities and in vitro cytotoxicity. J. Med. Chem., Washington, v. 44, n. 4, p. 548-565, 2001
SCHMITT, S.; KUHN, D.; KLEBE, G. A new method to detect related function among proteins independent of sequence and fold homology. J. Mol. Biol., London, v. 323, n. 2, p. 387-406, 2002.
SCHNEIDER, G.; NEIDHART, W.; GILLER, T.; SCHMID, G. “Scaffold-Hopping” by topological pharmacophore search: a contribution to virtual screening. Angew. Chem. Int., Weinhein, v. 38, n. 19, p. 2894-2896 , 1999.
SEWARD, H.E.; ROUJEINIKOVA, A.; MCLEAN, K.J.; MUNRO, A.W.; LEYS, D. Crystal structure of the Mycobacterium tuberculosis P450 CYP121-fluconazole complex reveals new azole drug-P450 binding mode. J. Biol. Chem., Baltimore, v. 281, p. 39437-39443, 2006.
SHENG, C.; MIAO, Z.; JI, H.; YAO, J.; WANT, W.; CHE, X; DONG, G.; LU, J.; GUO, W.; ZHANG, W. A three-dimensional model of lanosterol 14α-demethylase from Cryptococcus neoformans: active site characterization and insights into azoles binding. Antimicrob. Agents Chemother., Washington, v. 53, n. 8, p. 3487-3485, 2009.

FBT/FCF/USP
Ieda Yuriko Sonehara
128
SHENG, C.; ZHANG, W.; JI, H.; ZHANG, M.; SONG, Y.; XU, H.; ZHU, J.; MIAO, Z.; JIANG, Q.; YAO, J.; ZHOU, Y.; ZHU, J.; JIAGO, L. Structure-based optimization of azole antifungal agents by CoMFA, CoMSIA, and molecular docking. J. Med. Chem., Washington, v. 49, n. 8, p. 2512-2525, 2006.
SHERIDAN, R.P.; MILLER, M.D.; UNDERWOOD, D.J.; KEARSLEY, S.K. Chemical similarity using geometric atom pair descriptors. J. Chem. Inf. Comput. Sci., Washington, v. 36, n. 1, p. 128-136, 1996.
SILVA, L. H. P.; NUSSENSZWEIG, V. Sobre uma cepa de Trypanosoma cruzi altamente virulenta para o camundongo branco. Folia Clin. Biol., São Paulo, v. 20, p. 191, 1953.
SIPPL, W. Development of biologically active compounds by combining 3D QSAR and structure-based design methods. J. Aided. Mol. Des., Leiden, v. 16, n. 11, p. 825-830, 2002.
SOTRIFFER, C.A.; FLADER, W.F.; WINGER, R.H.; RODE, B.M.; LIEDL, K.R.; VARGA, J.M. Automated docking of ligands to antibodies: methods and applications. Methods, San Diego, v. 20, n. 3, p. 280-291, 2000.
STANTON, D.T. On the importance of topological descriptors in understanding structure-property relationships. J. Comput.-Aided. Mol. Des., Amsterdam, v. 22, n. 6-7, p. 441-460, 2008.
STEWART, M.L.; BUENO, G.J.; BALIANI, A.; KLENKE, B.; BRUN, R.; BROCK, J.M.; GILBER, I.H.; BARRETT, M.P. Trypanocidal activity of melamine-based nitroheterocycles. Antimicrob. Agents Chemother., Washington, v. 48, n. 5, p. 1733-1738, 2004.
Sunset Molecular Discovery, http://www.sunsetmolecular.com (acessado em 11/11/2009)
TAVARES, L.C. QSAR: A abordagem de Hansch. Quim. Nova, São Paulo, v. 27, n. 4, p. 631-639, 2004.
TAVARES, L.C. Relações Quantitativas Entre a Estrutura Química e a Atividade Antimicrobiana de Análogos à Nifuroxazida. Tese (Doutorado). Faculdade de Ciências Farmacêuticas da Universidade de São Paulo, São Paulo, 1993.

FBT/FCF/USP
Ieda Yuriko Sonehara
129
TAVARES, L.C.; CHISTÉ JÚNIOR, J., SANTOS, M.G.B., PENNA, T.C.V. Synthesis and biological activity of nifuroxazide and analogs. II. Boll. Quim. Farm., Milano, v. 138, n. 8, p. 432-6, 1999.
TAVARES, L.C.; PENNA, T.C.V.; AMARAL, A.T. Synthesis and biological activity of nifuroxazide and analogs. Boll. Quim. Farm., Milano, v. 136, n. 3, p. 244-249, 1997.
The Scripps Research Institute, http://autodock.scripps.edu/ (acessado em 11/11/2009)
TRÖSKEN, E.R.; SCHOLZ, K.; LUTZ, R.W.; VÖLKEL, W.; ZARN, J.A.; LUTZ, W.K. Comparative assessment of the inhibition of recombinant human CYP19 (aromatase) by azoles used in agriculture and as drugs for humans. Endocr. Res., London, v. 30, n. 3, p. 387-394, 2004.
URBINA, J.A.; PAYARES, G.; SANOJA, C.; LIRA, R.; ROMANHA, A.J. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int. J. Antimicrob. Agents, Amsterdam, v. 21, n. 1, p. 27-38, 2003.
VANDEN BOSSCHE, H.; KOYMANS, L. Cytochromes P450 in fungi. Mycoses, Berlin, v. 41, S1, p.32-38, 1998.
VEGA-TEIJIDO, M.; CARACELLI, I.; ZUKERMAN-SCHPECTOR, J. Conformational analyses and docking studies of a series of 5-nitrofuran- and 5-nitrothiophen-semicarbazone derivatives in three possible binding sites of trypanothione and glutathione reductases. J. Mol. Graph. Model. New York, v. 24, n.5, p. 349-355, 2006.
VIODÉ, C.; BETTACHE, N.; CENAS, N.; KRAUTH-SIEGEL, R.L.; CHAUVIÈRE, G.; BAKALARA, N.; PÉRIÉ, J. Enzymatic reduction studies of nitroheterocycles. Biochem. Pharmacol., Oxford, v. 57, n. 5, p. 549-557, 1999.
WATERBEEMD, H.; ROSE, S. Quantitative approaches to Structure-Activity relationships. . In: WERMUTH, C.G., ed. The Practice of Medicinal Chemistry. 3rd ed. Oxford:Elsevier, 2008. p. 491-513.
WILLETT, P. Chemical similarity searching. J. Chem. Inf. Comput. Sci., Washington, v. 38, n. 6, p. 983-996, 1998.

FBT/FCF/USP
Ieda Yuriko Sonehara
130
XIAO, L.; MADISON, V.; CHAU, A.S.; LOEBENBERG, D.; PALERMO, R.E.; MCNICHOLAS, P.M. Three-dimensional models of wild-type and mutated forms of Cytochrome P450 14α-demethylases from Aspergillus fumigatus and Candida albicans provide insights into posaconazole binding. Antimicrob. Agents Chemother., Washington, v. 48, n.2, p. 568-574, 2004.
YAO, B.; JI, H.; CAO, Y.; ZHOU, Y.; ZHU, J.; LÜ, J.; LI, Y.; CHE, J.; ZHENG, C.; JIANG, Y.; LIANT, R.; TANG, H. Synthesis and antifungal activities of novel 2-aminotetralin derivatives. J. Med. Chem., Washington, v. 50, n. 22, p. 5293-5300, 2007.
ZAMPIERI, D.; MAMOLO, M.G.; LAURINI, E.; FERMEGLIA, M.; POSOCCO, P.; PRICL, S.; BANFI, E.; SCIALINO, G.; VIO, L. Antimycobacterial activity of new 3,5-substituted 1,3,4-oxadiazol-2(3H)-one derivatives. Molecular modeling investigations. Bioorg. Med. Chem., Oxford, v. 17, n. 13, p. 4693-4707, 2009.
ZARN, J.A.; BRÜSCHWEILER, B.J.; SCHLATTER, R. Azole fungicides affect mammalian steroidogenesis by inhibiting sterol 14α-demethylase and aromatase. Environ. Health Perspect., Research Triangle Park, v. 111, n. 3, p. 255-261, 2003.
ZHANG, W.; RAMAMOORTHY, Y.; KILICARSLAN, T.; NOLTE, H.; TYNDALE, R.F.; SELLERS, E.M. Inhibiton of cytochromes P450 by antifungal imidazole derivatives. Drug Metab. Dispos., Baltimore, v. 30, n. 3, p. 314-318, 2002.
ZHU, J.; LU, J.; ZHOU, Y.; LI, Y.; CHENG, J.; ZHENG, C. Design, synthesis, and antifungal activities in vitro of novel tetrahydroisoquinoline compounds based on the structure of lanosterol 14α-desmethylase (CYP51) of fungi. Bioorg. Med. Chem. Lett., Oxford, v. 16, n. 20, p. 5285-5289, 2006.

FBT/FCF/USP
131
ANEXOS

UNIVERSIDADE DE SÃO PAULO
Faculdade de Ciências Farmacêuticas Secretaria de Pós-Graduação
Av. Prof. Lineu Prestes, 580, Bloco 13 A - Cidade Universitária - CEP 05508-900 - São Paulo - SP Fone: (11) 3091 3621 - Fax (11) 3091 3141 – e-mail: [email protected]
Informações para os Membros de Bancas Julgadoras de Mestrado/Doutorado
1. O candidato fará uma apresentação oral do seu trabalho, com duração máxima de trinta minutos.
2. Os membros da banca farão a argüição oral. Cada examinador
disporá, no máximo, de trinta minutos para argüir o candidato, exclusivamente sobre o tema do trabalho apresentado, e o candidato disporá de trinta minutos para sua resposta.
2.1 Com a devida anuência das partes (examinador e candidato),
é facultada a argüição na forma de diálogo em até sessenta minutos por examinador.
3. A sessão de defesa será aberta ao público. 4. Terminada a argüição por todos os membros da banca, a
mesma se reunirá reservadamente e expressará na ata (relatório de defesa) a aprovação ou reprovação do candidato, baseando-se no trabalho escrito e na argüição.
4.1 Caso algum membro da banca reprove o candidato, a
Comissão Julgadora deverá emitir um parecer a ser escrito em campo exclusivamente indicado na ata.
4.2 Será considerado aprovado o aluno que obtiver aprovação por unanimidade ou pela maioria da banca.
5. Dúvidas poderão ser esclarecidas junto à Secretaria de Pós-
Graduação: [email protected], (11) 3091 3621.
São Paulo, 18 de março de 2005.
Profa. Dra. Bernadette D. G. M. Franco Presidente da CPG/FCF/USP