
Sessão Temática 2 Análise Bayesiana Utilizando a abordagem Bayesiana no mapeamento de QTL´s...
35
Sessão Temática 2 Sessão Temática 2 Análise Bayesiana Análise Bayesiana Utilizando a abordagem Bayesiana no mapeamento de QTL´s Roseli Aparecida Leandro ESALQ/USP 11 o SEAGRO / 50ª RBRAS Londrina, Paraná 04 a 08 de Julho de 2005
-
date post
19-Dec-2015 -
Category
Documents
-
view
213 -
download
0
Transcript of Sessão Temática 2 Análise Bayesiana Utilizando a abordagem Bayesiana no mapeamento de QTL´s...

Sessão Temática 2Sessão Temática 2Análise BayesianaAnálise Bayesiana
Utilizando a abordagem Bayesiana no mapeamento de QTL´s
Roseli Aparecida LeandroESALQ/USP
11o SEAGRO / 50ª RBRAS Londrina, Paraná 04 a 08 de Julho de 2005

Colaboradores Colaboradores
Prof. Dr. Cláudio Lopes Souza Jr. Prof. Dr. Antônio Augusto Franco Garcia (Departamento de Genética ESALQ/USP)
Elisabeth Regina de Toledo (PPG Estatística e Experimentação
Agronômica, ESALQ/USP)

Qualitative traitQualitative trait
Mendelian gene
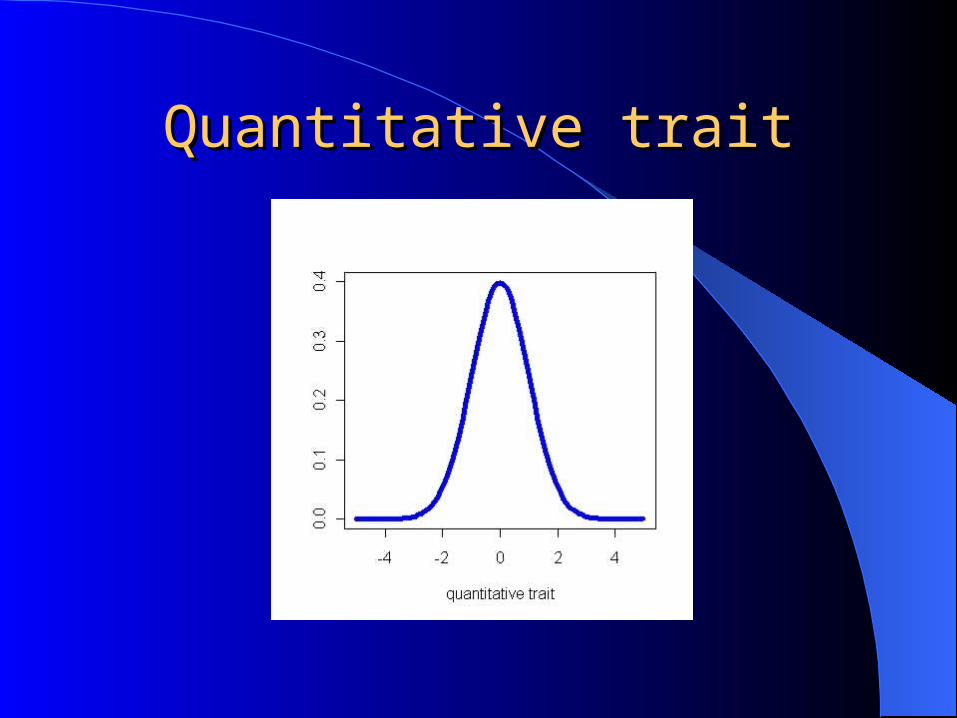
Quantitative traitQuantitative trait

Bayesian mapping of QTLBayesian mapping of QTL
Geneticists are often interested in locating regions in the chromosome contributing to phenotypic variation of a quantitative trait.
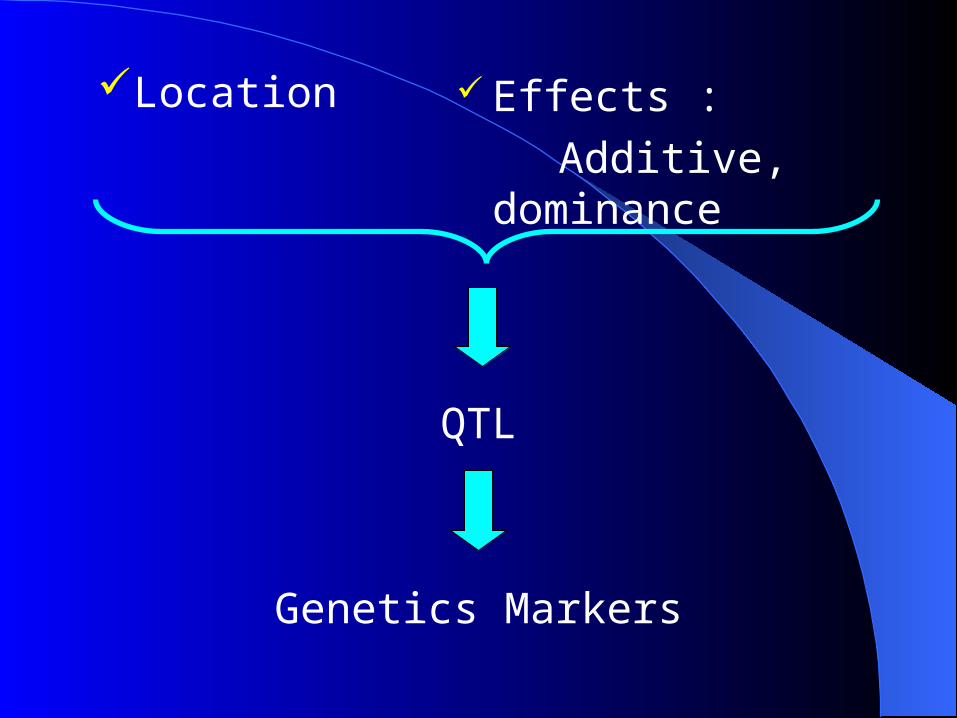
Effects :
Additive, dominance
Location
QTL
Genetics Markers
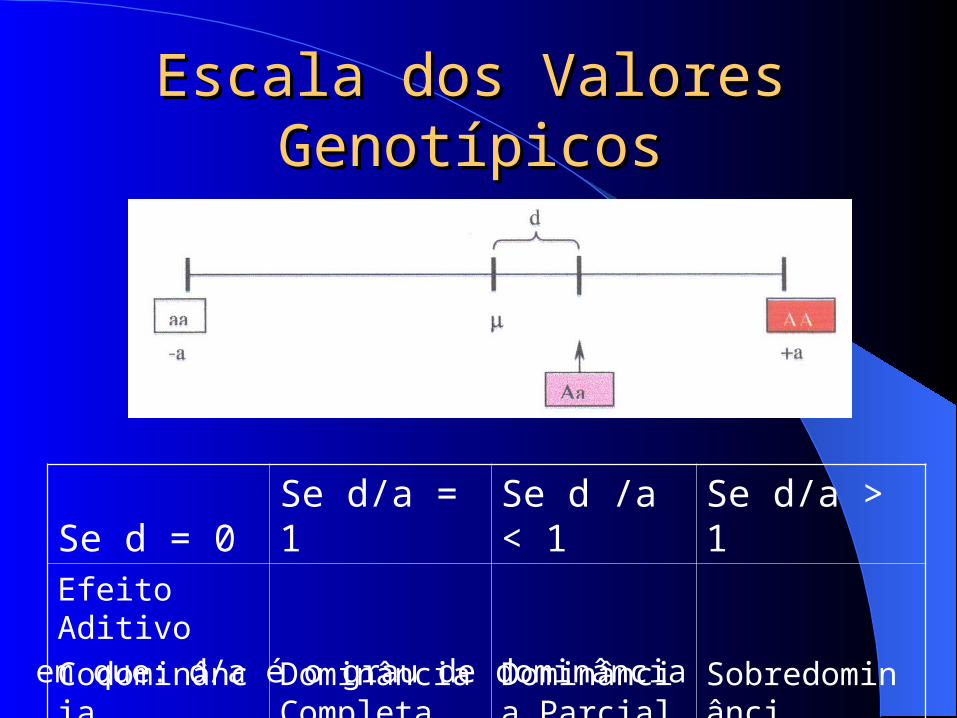
Escala dos Valores Escala dos Valores GenotípicosGenotípicos
Se d = 0 Se d/a = 1 Se d /a < 1 Se d/a > 1Efeito Aditivo
CodominânciaDominância Completa
Dominância Parcial Sobredominânci
em que: d/a é o grau de dominância

Chromosomal regions of known location
Do not have a physiological causal association to the trait under study

Genetics MarkersGenetics Markers

By studying the joint pattern of inheritance of the markers and trait
Inferences can be made about the number, location and effects of the QTL affecting trait.

Experimental DesignExperimental Design
Offspring data: Divergent inbred lines
Backcross ( code 0=aa, 1=Aa )
(Recessive)
F2 (code –1=aa, 0=Aa, 1=AA)
Reason: maximize linkage desiquilibrium
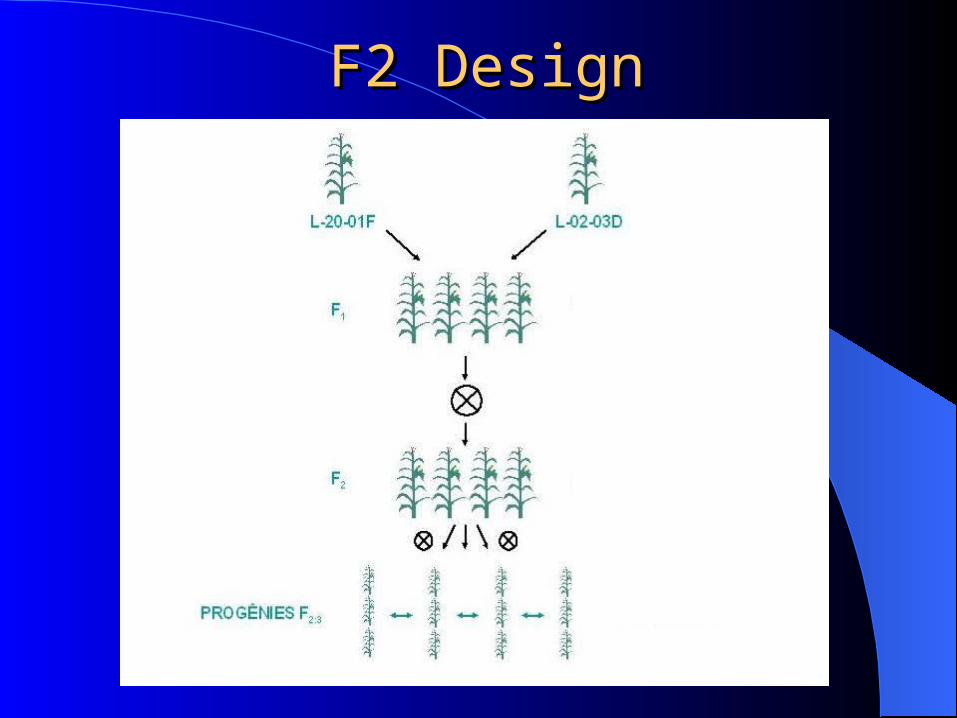
F2 DesignF2 Design
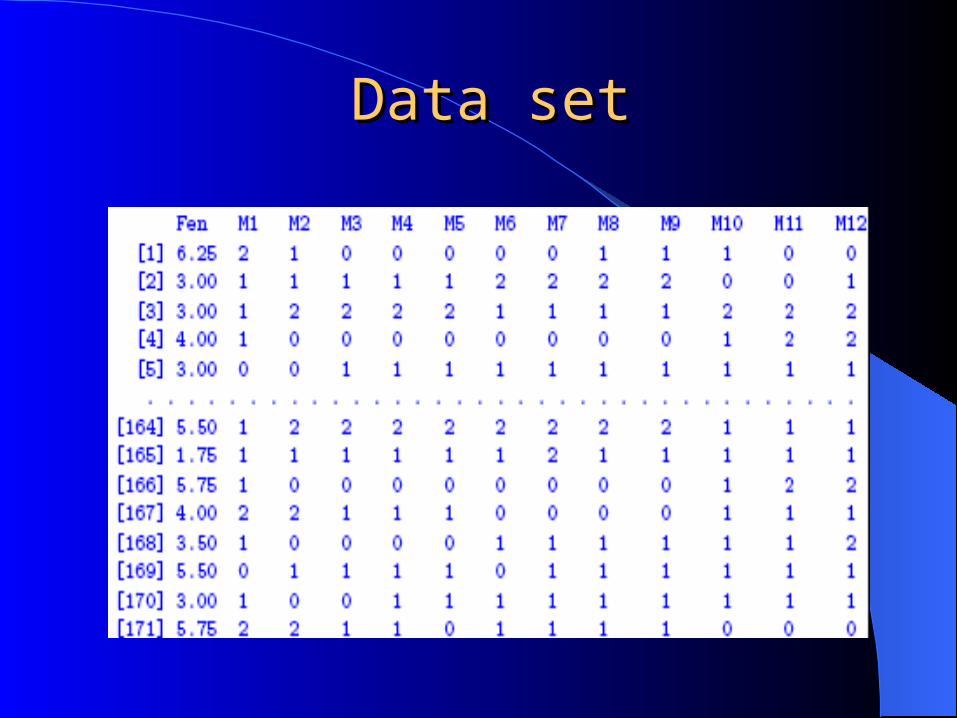
Data setData set

QTL phenotype modelQTL phenotype model
One QTL

Multiple QTL phenotype Multiple QTL phenotype Model Model

Our aim is to make joint inference about the number of QTL, their positions (loci) and the sizes of their effects.
Assume that a linkage map has been developed for the genome.
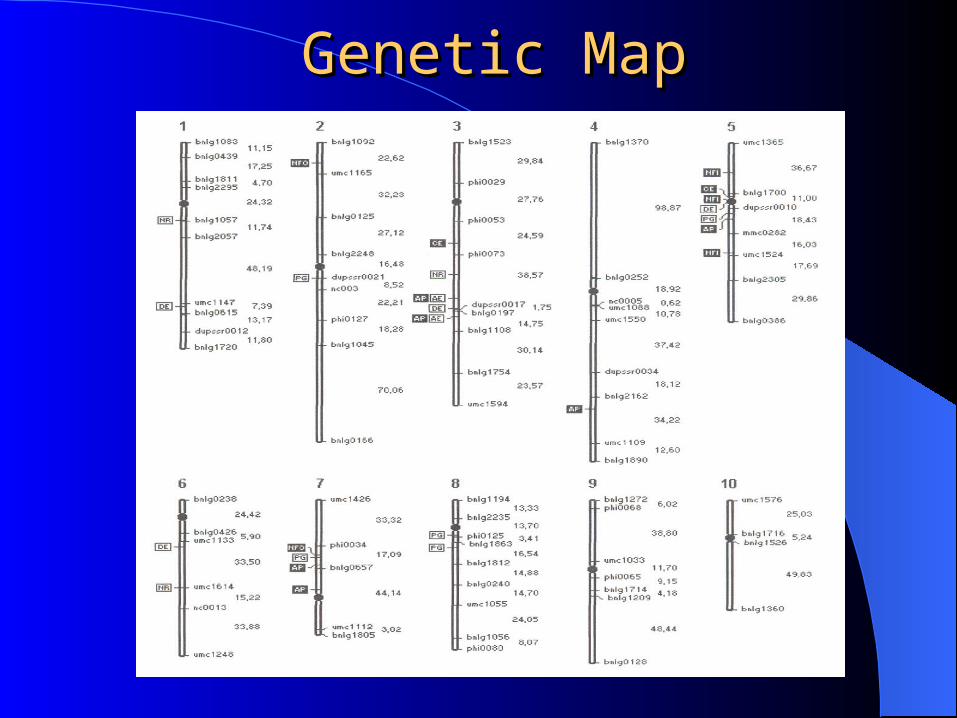
Genetic MapGenetic Map
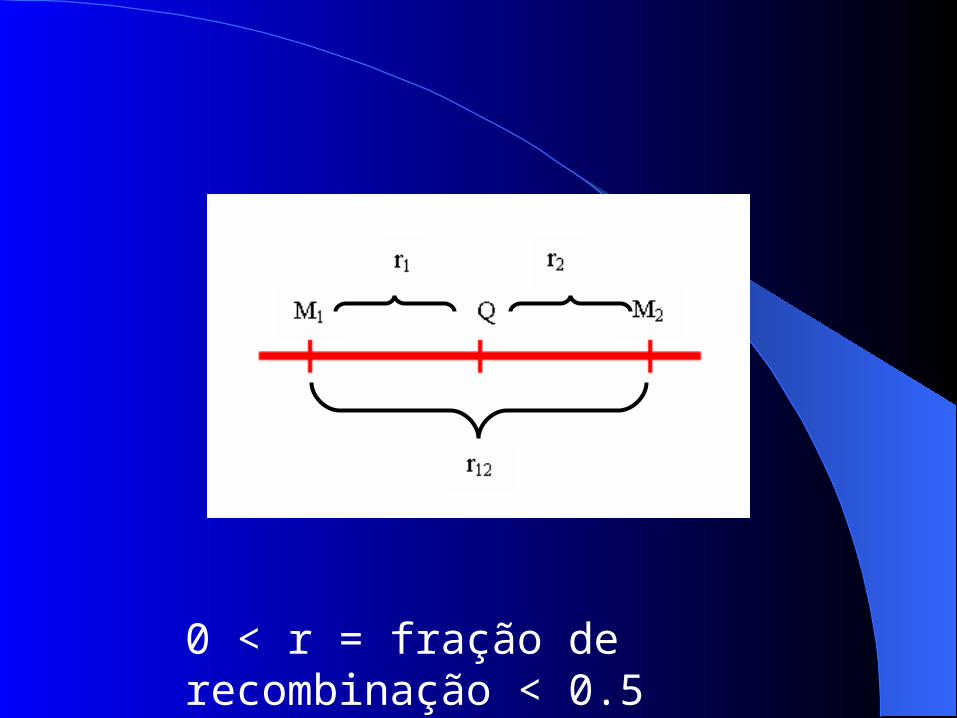
0 < r = fração de recombinação < 0.5

Classic approachClassic approach
Interval mapping (Lander & Botstein,1989)
Least squares method (Haley & Knott,1992)
Composite interval mapping
(Jansen, 1993; Jansen and Stam, 1994; Zeng 1993, 1994)

Bayesian approachBayesian approach
Satagopan et al. (1996)
Satagopan & Yandell (1998)
Sillanpää & Arjas (1998)

The joint posterior distribution of all unknowns (s, , Q, ) is proportional to

In practice, we observe the phenotypic trait . and the marker genotypes but NOT the QTL genotypes .
For convenience consider only one linkage group with ordered markers {1,2,...,m}. Assume that genotypes:
The markers are assumed to be at known distances

The conditional distribution
* assuming the loci segregate independently
** under Haldane assumption of independent recombination

The marginal likelihood of the parameters s, and for the ith individual may be obtained from the joint distribution of traits and QTL genotypes.
by summing over the set of all possible QTL genotypes for the ith individual,

Therefore,
When the data Y are n independent observations, the marginal likelihood for the trait data is the product over individuals, a familiar misture model likelihood,

The joint likelihood is a mixture of densities, and hence, is difficult to evaluate when there are multiple QTL.
The joint posterior distribution of all unknowns (s, , Q, ) is proportional to

A Bayesian approach combined with reversible jump MCMC is well suited for QTL studies

Random-sweep Metropolis-Hastings Random-sweep Metropolis-Hastings algorithm for general state spaces algorithm for general state spaces
(Richardson and Green, 1997)(Richardson and Green, 1997)
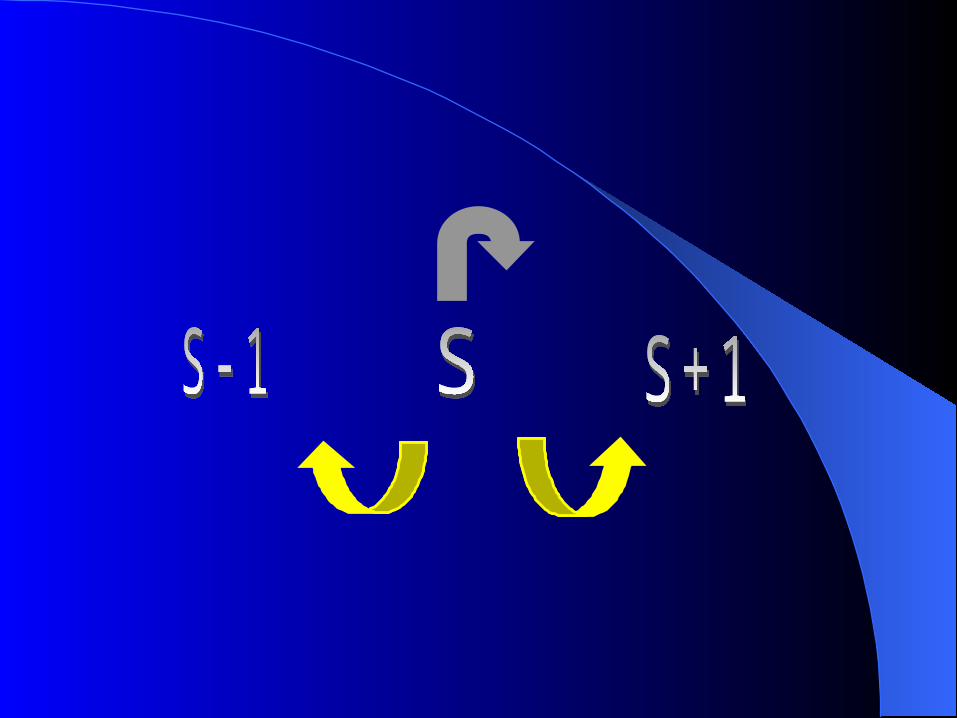
Suppose
current state of the chain indexed by s.

The chain can
(1) move to a “birth” step
(number of loci s s+1 )
(2) move to a “death” step
(number of loci s s-1 )
(3) continue with “current” number (s) of loci

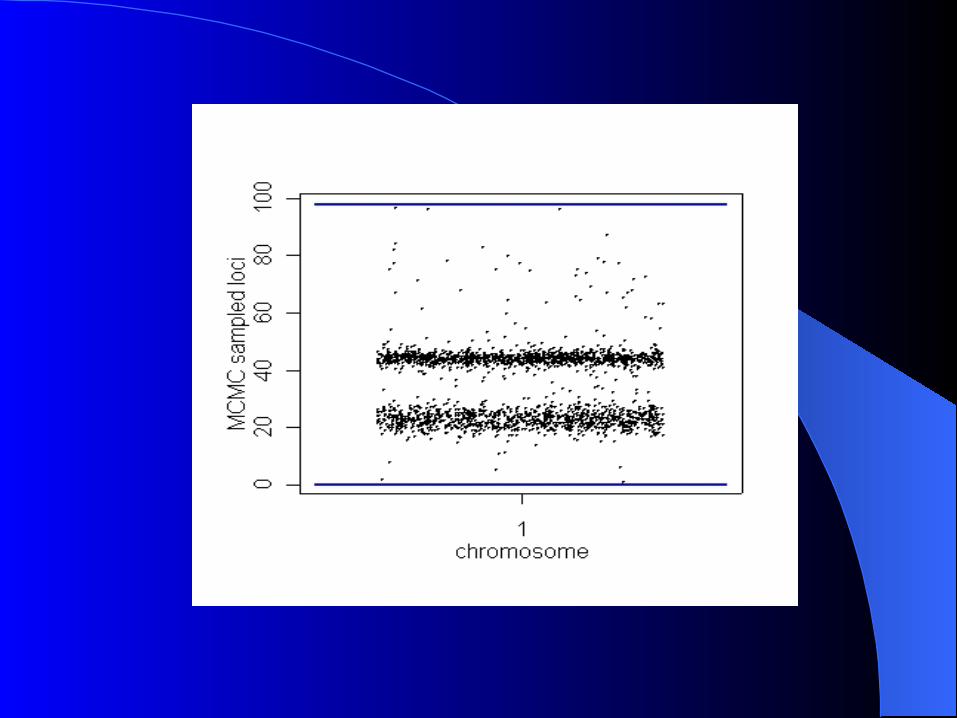
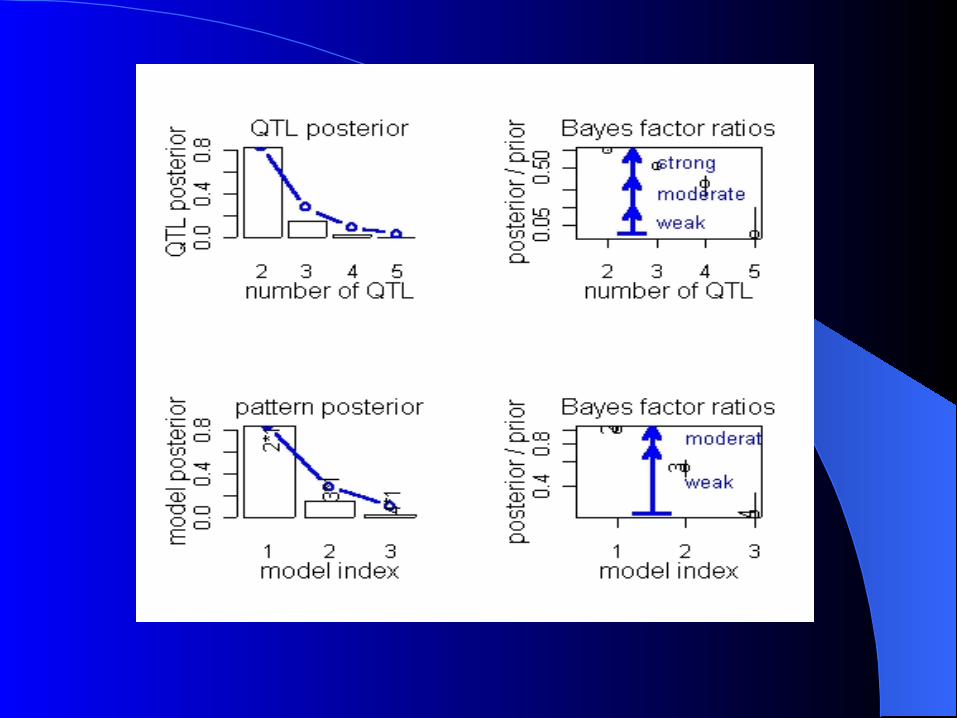
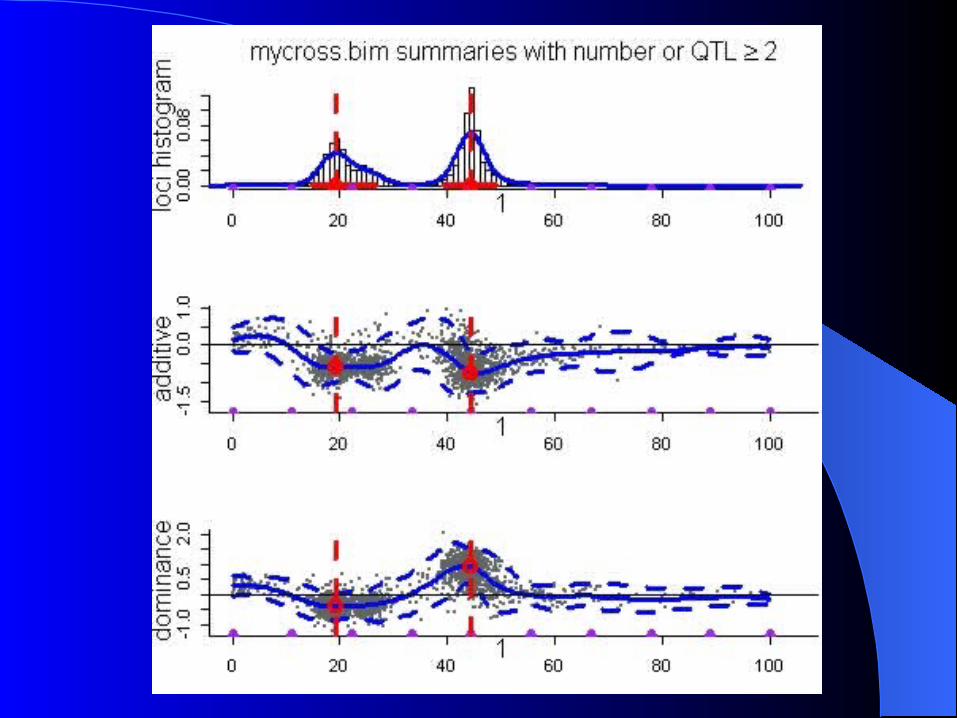
Simulation Simulation
Simulated F2 intercross n=250 1 cromossome 2 QTL

ReferênciasReferências
Satagopan, J. M.; Yandell, B. S. (1998)Bayesian model determination for
quantitative trait


















