
PRESENÇA DE MARCADORES DE NEURODEGENERAÇÃO E...
81
UNIVERSIDADE DO SUL DE SANTA CATARINA SAMANTHA PEREIRA MIGUEL PRESENÇA DE MARCADORES DE NEURODEGENERAÇÃO E ATIVAÇÃO DE RAGE NO CÉREBRO DE RATOS SOBREVIVENTES DE SEPSE Tubarão 2011
Transcript of PRESENÇA DE MARCADORES DE NEURODEGENERAÇÃO E...

UNIVERSIDADE DO SUL DE SANTA CATARINA
SAMANTHA PEREIRA MIGUEL
PRESENÇA DE MARCADORES DE NEURODEGENERAÇÃO E ATIVAÇÃO DE
RAGE NO CÉREBRO DE RATOS SOBREVIVENTES DE SEPSE
Tubarão
2011

SAMANTHA PEREIRA MIGUEL
PRESENÇA DE MARCADORES DE NEURODEGENERAÇÃO E ATIVAÇÃO DE
RAGE NO CÉREBRO DE RATOS SOBREVIVENTES DE SEPSE
Dissertação apresentada ao curso de Mestrado em
Ciências da Saúde, da Universidade do Sul de Santa
Catarina, como requisito para obtenção do título de
Mestre em Ciências da Saúde.
Orientadora: Profª. Fabricia Petronilho, Dra.
Tubarão
2011

SAMANTHA PEREIRA MIGUEL
PRESENÇA DE MARCADORES DE NEURODEGENERAÇÃO E ATIVAÇÃO DE
RAGE NO CÉREBRO DE RATOS SOBREVIVENTES DE SEPSE
Esta Dissertação foi julgada adequada à obtenção do
título de Mestre em Ciências da Saúde e aprovado em
sua forma final pelo Curso de Ciência de Saúde, da
Universidade do Sul de Santa Catarina.
___________________________, ______ de ____________________ de 2011
Local dia mês ano
__________________________________________________________
Prof. e orientadora Fabricia Cardoso Petronilho, Dra.
Universidade do Sul de Santa Catarina - UNISUL
__________________________________________________________
Prof. Gislaine Tezza Rezin, Dra.
Universidade do Sul de Santa Catarina – UNISUL
__________________________________________________________
Prof. Gustavo da Costa Ferreira, Dr.
Universidade do Extremo Sul Catarinense

Dedico este trabalho e todo meu esforço a
minha mãe e ao meu esposo. Vocês são meus
amores... estarão sempre comigo!

AGRADECIMENTOS
São muitas as pessoas que fizeram parte desta minha etapa de estudo e a muitas
devo meus agradecimentos.
Gostaria então de iniciar meus agradecimentos à minha querida e doce
orientadora, Fabricia Petronilho. Foi ela que, assim que chegou à Universidade, aceitou meu
convite de orientação. Fa, você não foi apenas uma orientadora, foi amiga, companheira e
muito paciente, além de ter acreditado nos meus esforços e estar do meu lado durante todos os
momentos que precisei. Dessa forma, com todo carinho o meu muito obrigada.
Ao professor Gustavo da Costa que foi o primeiro professor que tive contato
assim que entrei no mestrado. Além de fazer parte da minha entrevista de seleção, foi quem
me ensinou os primeiros passos da pesquisa e me indicou à professora Fabricia. Muito
obrigada Gustavo, pela sua atenção e pela indicação.
A Lucinéia, minha ex-aluna e colega de pesquisa, que desde o início foi uma
adorável companheira e muito atenciosa, muito obrigada!
Ao meu esposo Andersson, que teve muita paciência, companheirismo e carinho
nos momentos em que pensei que não seria capaz... Você é meu amor e meu melhor amigo!
A minha mãe Albertina, minha grande incentivadora em todas as etapas de estudo
da minha vida. Mãe, certamente estou realizando um sonho que também é seu. És minha
estrela guia, eternamente vou te amar.
A minha irmã Sabrina, que me socorreu em vários momentos da pesquisa,
inclusive me mostrando caminhos que eu desconhecia...
Aos meus familiares, que sempre me deram incentivo aos estudos e de alguma
forma contribuíram para que eu alcançasse esse título.
Agradeço de coração cada um.

RESUMO
Sepse pode ser definida como a resposta inflamatória sistêmica frente como resposta à
infecção, denotando um processo progressivo de dano tecidual, onde a disfunção orgânica
múltipla representa sua expressão mais grave. Estudos de mortalidade indicam que a sepse é
uma das maiores causas de mortalidade em Unidade de Terapia Intensiva. No Brasil, a
mortalidade de pacientes com sepse grave chega a 43,6% e no mundo 23,9%. Já em pacientes
com choque séptico no Brasil a mortalidade chega a 65,7% enquanto no mundo, 37,4%.
Sobreviventes de sepse apresentam déficit cognitivo associados à diminuição da qualidade de
vida, aumentando a mortalidade a longo prazo. Certos aspectos dessas alterações resultam de
mecanismos fisiopatológicos de neurodegeneração, visto que encefalopatia séptica como uma
entidade que não possa ser explicada pela disfunção, hipotensão ou pela hipóxia hepática ou
renal, é relativamente nova, porém já está claro que a sepse e suas reações podem ser
associadas a danos e disfunções cerebrais. Nesse contexto, estudos mostram que o Receptor
de Produtos de Glicação Avançada (RAGE) parece ser importante na progressão da
neurodegeneração. Assim, nosso objetivo foi determinar a presença de marcadores de
neurodegeneração no cérebro de ratos sobreviventes de sepse e o possível envolvimento de
RAGE nessas alterações. Para tal, ratos Wistar machos (220-350g) foram sujeitos a sepse por
ligação e perfuração cecal (CLP) e trinta dias após, foram mortos por decaptação e foram
isolados o hipocampo e córtex pré-frontal para determinação das proteínas: β-amilóide (Aβ),
α-sinucleína, tau-DA, S100B, RAGE e a ativação de ERK 1/2 por técnica de imunoblotting.
Dessa forma, nossos resultados mostraram que o cérebro de animais sobreviventes à sepse
apresentaram vários marcadores de neurodegeneração, visto que houve aumento significativo
na expressão das proteínas Aβ, α-sinucleína e tau-DA no hipocampo, porém no córtex pré
frontal apenas a α-sinucleína apresentou-se aumentada de forma significativa. Houve aumento
na expressão de RAGE no hipocampo e no córtex pré-frontal e a ativação de ERK1/2
observou-se apenas no hipocampo. Em conclusão, os presentes resultados indicam que a via
Aβ-RAGE-MAPK pode ser a principal via responsável pelo déficit cognitivo a longo prazo
em sobreviventes de sepse.
Palavras-chave: Sepse. Neurodegeneração. RAGE. Disfunção cerebral.

ABSTRACT
Sepsis can be defined as the systemic inflammatory response in response to infection,
indicating a progressive process of tissue injury, where the multiple organ dysfunction
represents its more serious expression. Mortality studies indicate that sepsis is one of the
major causes of death in Intensive Care Unit. In Brazil, the mortality of patients with
severe sepsis reach 43,6%, while in the world it reaches 23,9% and patients with septic
shock as cause of the death in Brazil reached 65,7% while in the world, 37,4%. Survivors
of sepsis have cognitive decline associated with the decrease of the quality of life,
increasing the long-term mortality. Certain aspects of these changes are result of
pathophysiological mechanisms of neurodegeneration, seen asseptic encephalopathy as an
entity that cannot be explained by dysfunction, hypotension, and liver or kidney hypoxia
is relatively new, but it is already clear that sepsis and its reactions can be associated with
brain damage and impairment. In this context, some studies show that the Receptor for
Advanced Glycation Endproducts (RAGE) is important in the progression of
neurodegeneration. Thus, our objective was to determine the presence of
neurodegeneration markers in the brain of rats survivors of sepsis and possible
involvement of RAGE in these changes. So , male Wistar rats (220-350g) were subjected
to sepsis by cecal connection and perfuration (CLP) and, thirty days after that, they were
killed by decapitation and isolated the hippocampus and prefrontal cortex for the
determination of proteins: β-amyloid (Aβ), α-synuclein, AD-tau, S100B, RAGE and the
activation of Extracellular Signal-Regulated Kinase (ERK1/2) by the technique of
immunoblotting. So, our results showed that the brain of sepsis survivor animals showed
several markers of neurodegeneration, as there was a significant increase in expression of
Aβ, α-synuclein and tau-AD proteins in the hippocampus, but in the prefrontal cortex only
α-synuclein presented significantly. There was an increase in the expression of RAGE in
the hippocampus and in the prefrontal cortex and the activation of ERK 1/2 was observed
only in the hippocampus. In conclusion, the present results indicate that the Aβ -RAGE-
MAPK pathway may be the main responsable for the long-term cognitive deficit on sepsis
survivors.
Key-Words: neurodegeneration, RAGE, brain dysfunction.

LISTA DE ILUSTRAÇÕES
Figura 1 – Imunoconteúdo de Aβ, α-sinucleína e tau-DA no hipocampo dos animais
sobreviventes a sepse comparado ao grupo controle (sham)..... ............................................. 355
Figura 2 – Imunoconteúdo de Aβ, α-sinucleína e tau-DA no córtex pré-frontal dos animais
sobreviventes a sepse comparado ao grupo controle (sham). ................................................. 366
Figura 3 – Imunoconteúdo de RAGE, S100B e ERK1/2 fosforilada no hipocampo dos animais
sobreviventes sepse comparado ao grupo controle (sham).. .................................................. 377
Figura 4 – Imunoconteúdo de RAGE, S100B e ERK1/2 fosforilada no cortex pré-frontal dos
animais sobreviventes sepse comparado ao grupo controle (sham).. ..................................... 388

LISTA DE SIGLAS
OH- – Radical hidroxil
1O2 - Oxigênio singlet
AGE - Produtos finais de glicação avançada
Aβ – β-amilóide
BHE – Barreira Hematoencefálica
CLP – Ligação e perfuração cecal
DA – Doença de Alzheimer
DP – Doença de Parkinson
ERK1/2 - Quinase regulada por sinais extracelulares 1/2
ERN – Espécies reativas de nitrogênio
ERO – Espécies reativas de oxigênio
GSK-3β - Glicogênio Sintase Quinase 3β
HO-1 - heme oxigenase -1
IL-1 – Interleucina 1
IL-2 – Interleucina 2
IL-6 – Interleucina 6
IL-8 – Interleucina 8
ILAS – Instituto Latino Americano de Sepse
iNOS - Óxido nítrico sintase induzível
JA - Junções apertadas
JEA - Junções endoteliais aderentes
JNK - Quinase n-terminal c-Jun
LBP - Proteínas ligadoras de LPS
LPS - Lipopolissacarídeo
LTA - Ácido lipoteicóico
MAPK - Proteína quinase ativada por mitógeno
MD-2 - Proteína de diferenciação mielóide - 2
MyD88 - Fator de diferenciação mieloide humano 88
NF-kB - Fator nuclear-κB
NO• - Óxido nítrico

O2•- - Radical superóxido
ONOO- - Peroxinitrito
PAMPs - Padrões Moleculares Associados a Patógenos
PPR - Receptor de reconhecimento de padrão
RAGE - Receptor de Produtos de Glicação Avançada
SIRS – Síndrome da resposta inflamatória sistêmica
SNC – Sistema Nervoso Central
TIR - Domínio intracelular Toll/interleucina -1
TIRAP - Proteina adaptadora contendo o domínio TIR
TLR - Receptor toll-like
TLR-4 - Receptor toll-like-4
TNF-α - Fator de necrose tumoral – α
TRAM - Molécula adaptadora relacionada ao TRIF
TRIF - Domínio TIR contendo adaptador do indutor de interferon β
UTI – Unidade de Terapia Intensiva

SUMÁRIO
1 INTRODUÇÃO .................................................................................................................. 12
1.1 SEPSE: DEFINIÇÃO E ASPECTOS EPIDEMIOLÓGICOS .......................................... 12
1.2 FISIOPATOLOGIA DA SEPSE ....................................................................................... 14
1.2.1 Interação patógeno-hospedeiro .................................................................................. 144
1.2.2 Resposta Celular ........................................................................................................... 16
1.2.3 Radicais livres na progressão da sepse ........................................................................ 19
1.2.4 Barreira-hematoencefálica (BHE) ............................................................................... 20
1.3 ENCEFALOPATIA SÉPTICA ....................................................................................... 211
1.4 PROTEÍNAS RELACIONADAS A NEURODEGENERAÇÃO .................................... 23
1.4.1 Proteína Tau Fosforilada ............................................................................................ 233
1.4.2 Proteína β-amilóide (Aβ) .............................................................................................. 24
1.4.3 Proteína α-sinucleína .................................................................................................... 25
1.4.4 Proteína S100B ............................................................................................................ 266
1.5 NEURODEGENERAÇÃO E ATIVAÇÃO DE RAGE .................................................. 277
2 JUSTIFICATIVA ............................................................................................................ 300
3 OBJETIVOS ..................................................................................................................... 311
3.1 OBJETIVO GERAL ........................................................................................................ 311
3.2 OBJETIVOS ESPECÍFICOS .......................................................................................... 311
4 MATERIAL E MÉTODOS ............................................................................................ 322
4.1 TIPO DE ESTUDO ......................................................................................................... 322
4.1.1 Bioética ......................................................................................................................... 322
4.1.2 Animais ........................................................................................................................ 322
4.1.3 Indução de sepse .......................................................................................................... 333
4.1.4 Immunoblotting ........................................................................................................... 333
4.2 ANÁLISE ESTATÍSTICA .............................................................................................. 344
5 RESULTADOS ................................................................................................................ 355
6 DISCUSSÕES..................................................................................................................... 39
7 CONCLUSÕES ................................................................................................................ 433
8 PERSPECTIVAS ............................................................................................................. 444
REFERÊNCIAS ................................................................................................................... 455
APÊNDICE ........................................................................................................................... 600

APÊNDICE A – Artigo da Dissertação .............................................................................. 611

12
1 INTRODUÇÃO
1.1 SEPSE: DEFINIÇÃO E ASPECTOS EPIDEMIOLÓGICOS
Todo processo infeccioso desencadeia uma resposta inflamatória do hospedeiro,
cuja magnitude pode variar de indivíduo para indivíduo. A interação complexa entre o
organismo e o agente causador resulta no processo fisiopatológico que antigamente era
definido como septicemia e hoje é denominado como sepse (VICENT; KORKUT, 2008).
Louis Pasteur, em 1879-80, encontrou pela primeira vez, bactérias no sangue de
uma paciente que sobreviveu à sepse e assim as diferentes etapas desse processo foram
inicialmente definidas por Willian Osler em 1892 (BARON; BARON; PERRELLA, 2006).
Após a observação feita por Pasteur, aproximadamente um século depois é que finalmente se
chegou a um consenso sobre a definição de sepse. A relativa demora se deve principalmente à
dificuldade de identificação desta síndrome por clínicos e pesquisadores, uma vez que os
sinais e sintomas clínicos variam enormemente dependendo do estágio em que o paciente se
encontra (ANNANE; BELLISSANT; CAVAILLON, 2005).
Em 1991, na Conferência de Consenso feita pela American College of Chest
Physicians and the Society of Critical Care Medicine foram estabelecidos critérios para a
classificação de sepse e doenças similares (BONE et al., 1992). Optou-se por enquadrá-las
como Síndrome da Resposta Inflamatória Sistêmica (SIRS). Além disso, ficou estabelecido
também que o termo sepse deveria ser utilizado nos casos onde a infecção é documentada,
pois a SIRS pode ser causada por diversos outros insultos (além da infecção por bactérias,
vírus e fungos), considerada como “estéreis”, tais como trauma, queimaduras, choque
hemorrágico e pancreatite aguda (BEISHUIZEN; VERMES; HAANEN, 1999).
Esta medida foi necessária para adequação do tratamento à fase de evolução da
síndrome em que o paciente se encontra. Esses parâmetros vêm sendo reavaliados nas
subsequentes conferências e hoje compõe as diretrizes da “Surviving Sepsis Campaign” para
diagnóstico precoce e tratamento adequado em cada etapa do processo fisiopatológico da
sepse, com o objetivo único de diminuir a taxa de mortalidade (DELLINGER et al., 2008;
VICENT; KORHUT, 2008).

13
O desenvolvimento de alterações clínicas, hematológicas, bioquímicas e
imunológicas associadas à infecção caracteriza a sepse, a qual quando complicada pela
disfunção orgânica é denominada sepse severa. O choque séptico se refere a um estado de
falência circulatória aguda com hipotensão arterial apesar da reposição volêmica, fazendo-se
necessário a terapia vasopressora para manutenção de uma pressão arterial a valores
aceitáveis (VICENT; KORHUT, 2008).
O mau funcionamento dos mecanismos de regulação durante a sepse pode resultar
em uma perda de controle da inflamação, assim a fase precoce da sepse que é causada pela
excessiva ativação do sistema de reconhecimento do hospedeiro - patógeno pelos danos
teciduais em grande escala e/ou infecção grave, leva a uma desregulação de vários sistemas
do corpo. Atualmente, há evidências de que a sepse é uma condição que afeta não só o
sistema imunológico, mas também outros sistemas biológicos (RITTIRSCH; FLIERL;
WARD, 2008).
A ocorrência de falência orgânica segue um padrão comum: a disfunção pulmonar
ocorre quase sempre e precoce, persiste durante o choque que também ocorre precocemente, e
se resolve rapidamente ou é fatal. Sérias anormalidades da função hepática, coagulação e
manifestações neurológicas tendem a ocorrer horas ou dias após o início da sepse e persistem
por tempo indeterminado (BONE, 1996; HOTCHKISS; KARL, 2003; NATHENS;
MARSHALL, 1996; WARREN, 1997). O número de falências orgânicas, além da gravidade
destas, afeta o prognóstico do paciente, pois cada órgão adicional em falência acrescenta de
15-20% a taxa de mortalidade (BONE, 1992; FRIEDMAN; SILVA; VICENT, 1998;
HOTCHKISS; KARL, 2003; WARREN, 1997).
A incidência da sepse tem aumentado desde os anos 30 (DOMBROVSKIY et al.,
2007) e todas as recentes evidências sugerem que este aumento persiste. As razões para este
aumento sustentado são várias: aumento do uso de procedimentos invasivos, uso amplo de
imunossupressores e agentes citotóxicos, aumento na sobrevida de pacientes crônicos (como
por exemplo, diabetes) e aumento na infecção por organismos multiresistentes.
Dados de mortalidade mais recentes indicam (desde 2003) que a sepse está entre
as 10 causas mais comuns de morte nos Estados Unidos (KUNG et al., 2008), indicando que
ocorrem aproximadamente 751.000 casos de sepse por ano (REMICK, 2007) e se relata que o
choque séptico é a causa de 6 - 15% das internações em Unidades de Terapia Intensiva (UTI)
(ANTONELLI et al., 2007); a taxa de mortalidade anual é de 32,2% para sepse grave e 54,1%
para choque séptico (VINCENT et al., 2006), sendo que os índices aumentam continuamente
nos pacientes de maior idade (MARTIN; MANNINO; MOSS, 2006), isso ocorre devido à

14
tendência epidemiológica nessa faixa etária onde há um aumento de pacientes cronicamente
doentes (O'BRIEN; ALINA; ABEREGG, 2007).
Em abril de 2011, o ILAS – Latim American Sepsis Institute, em sua campanha
sobrevivendo a sepse (Surviving Sepsis Campaign), apresentou em seu relatório trimestral os
dados de mortalidade de pacientes sépticos no Brasil e no mundo. No Brasil, a mortalidade de
pacientes com sepse grave é de 43,6% e no mundo 23,9%. Já pacientes com choque séptico
no Brasil a mortalidade chega a 65,7% enquanto no mundo, 37,4%. (LATIM AMERICAN
SEPSIS INSTITUTE, 2011).
No Brasil, através de estudos do Brazilian Sepsis Epidemiologic Study (BASES)
mostrou que cerca de 25% dos pacientes internados nas UTIs (Unidade de Terapia Intensiva)
apresentaram diagnósticos de sepse severa e choque séptico, com uma mortalidade de 34,7%
para sepse, 47,3% para sepse severa e 52,2% para choque séptico (SILVA et al., 2004).
Enquanto que a taxa de mortalidade em outro estudo foi de 21,4% para sepse, 30,9% para
sepse severa e 47,7% para choque séptico em UTIs (FERNANDES, 2008). A taxa de
mortalidade por sepse e suas complicações vem apresentando uma discreta redução nas
últimas décadas, provavelmente devido a melhor definição da síndrome e a incrementos nas
medidas de suporte a órgãos-alvos e prevenção de complicações, mesmo na ausência de uma
terapêutica específica com impacto considerável na mortalidade (FRIEDMAN; SILVA;
VICENT , 1998)
Apesar dos avanços significativos na terapia intensiva e uso de antibióticos, a
mortalidade global na sepse está associada a um custo anual de cuidados de saúde de cerca de
17 bilhões de dólares (ANGUS; WAX, 2001; FRIEDMAN; SILVA; VICENT, 1998;
MUNFORD; PUGIN, 2001). A possibilidade de diminuir a mortalidade por sepse e reduzir os
gastos com internação em UTIs, além das sequelas tardias destes pacientes justificam a
necessidade de maior investimento no estudo desta doença.
1.2 FISIOPATOLOGIA DA SEPSE
1.2.1 Interação patógeno-hospedeiro

15
A inflamação é uma resposta normal do hospedeiro contra agentes infecciosos.
Sepse e SIRS são caracterizadas pela produção excessiva de mediadores inflamatórios e pela
excessiva ativação de células inflamatórias (BONE, 1991). Os mecanismos de
reconhecimento específico, componentes da resposta inata, foram caracterizados como uma
via de controle da imunidade adquirida. Esses mecanismos são deflagrados por receptores de
membrana celular, que, por sua vez, são ativados com o reconhecimento do microorganismo
através de estruturas conservadas, constitutivamente expressas na superfície dos patógenos,
denominadas Padrões Moleculares Associados à Patógenos (PAMPs) (CINEL; DELLINGER,
2007). Assim, os fatores desencadeadores da ativação celular e da cascata de eventos
plasmáticos são principalmente os componentes da parede celular dos microorganismos,
como o ácido lipoteicóico (LTA) e peptideoglicanos, derivados de bactérias Gram-positivas
(exotoxinas), ou o lipopolissacarídeo (LPS), no caso de bactérias Gram-negativas
(endotoxinas).
O LPS e as exotoxinas são liberados normalmente durante a replicação da bactéria
e/ou como consequência de sua morte, devido a lise da parede celular. Diversos receptores
foram encontrados nos últimos anos, capazes de reconhecer essas moléculas e ativar a
resposta imune inata (TRIANTAFILOU; TRIANTAFILOU, 2002). Assim, os PAMPs são
reconhecidos por receptores encontrados em células do sistema imune inato como neutrófilos,
polimorfonucleares, macrófagos e células dendríticas. Esses receptores são denominados de
Receptores de Reconhecimento de Padrões (PPRs).
Entre os membros mais importantes dos PPRs destacam-se os receptores Toll-like
(TLRs). Tais receptores são uma família de receptores transmembrana do tipo 1, codificados
em linhagem germinativa e não clonais, que são caracterizados por domínios extracelulares
repetitivos ricos em leucina e um domínio citoplasmático homólogo ao receptor de
interleucina-1 (IL-1) (WEIGHARDT; HOLZMANN, 2007). Atualmente, 11 homólogos
humanos dos TLRs foram identificados e, se não todos os maioria envolvida no
reconhecimento dos principais padrões microbianos (HOPKINS; SRISKANDA, 2005)
existindo a evidência de que o polimorfismo de proteínas dos TLRs pode explicar em parte a
grande variabilidade de respostas individuais aos estímulos infecciosos (LORENZ et al.,
2000).
Logo, a ativação de macrófagos estimulados por LPS é dependente da presença de
proteínas ligadoras de LPS (LBP) e da proteína de membrana CD-14 (COHEN, 2002; LIEW
et al, 2005). A LBP é uma proteína de fase aguda que catalisa a transferência do LPS para
CD-14 potencializando a ativação de macrófagos induzida por LPS de 100 a 1.000 vezes.

16
Nesse sentido, o CD-14 em conjunto com a proteína de diferenciação mielóide-2 (MD-2),
uma pequena molécula sem região transmembrana, participa da apresentação de LPS para o
receptor similar ao Toll e Poltorak e colaboradores (1998), mostraram que a sinalização pelo
LPS é transmitida pelo receptor Toll-like-4 (TLR-4), o primeiro membro da família a ser
caracterizado em mamíferos.
TLR-4 transmite um sinal de ativação onde o domínio intracelular
Toll/Interleucina-1 (TIR) interage com quatro moléculas adaptação do domínio TIR. São elas
o fator de diferenciação mielóide humano 88 (MyD88), a proteína adaptadora contendo o
domínio TIR (TIRAP), a domínio TIR contendo o adaptador do indutor de interferon-β
(TRIF) e a molécula adaptadora relacionada ao TRIF (TRAM). Atualmente está claro que a
MyD88 e TRIF promovem uma plataforma central para a propagação de sinais complexos dos
TLRs via associação direta com quinases e fatores de transcrição (HONDA et al, 2004;
YAMAMOTO et al., 2003;). Nesse sentido com a ativação de quinases situadas na seqüência
do processo de sinalização, ocorre a liberação do fator nuclear-kB (NF-kB) que transloca para
o núcleo e aumenta a expressão gênica de citocinas inflamatórias determinando, por fim, uma
resposta pró-inflamatória (AKIRA; TAKEDA, 2004).
Assim, de um modo geral, após a interação inicial PAMPs-PPRs acontece
ativação da resposta imune inata com a finalidade de coordenar uma resposta defensiva
envolvendo componentes humorais e celulares. Nesse contexto, com tal interação, células
residentes têm um papel chave, liberando uma grande variedade de moléculas sinalizadoras
como prostaglandinas, leucotrienos, citocinas e quimiocinas que desencadeiam a resposta
inflamatória, culminando no recrutamento e ativação de leucócitos para o local da infecção,
representando uma das funções mais importantes da imunidade inata (JANEWAY, 2001,
2002).
1.2.2 Resposta Celular
Conforme descrito anteriormente, o recrutamento leucocitário para o local de
injúria celular é uma das etapas essenciais da defesa do organismo contra um agente agressor.
Nos estágios iniciais de variados processos infecciosos, incluindo infecções por fungos, vírus
e bactérias, o leucócito predominante é o neutrófilo, permanecendo em geral de 12-24 horas

17
no local do dano. Após esse período, o neutrófilo inicia um processo de morte programada
(apoptose) sendo, em seguida, fagocitado por macrófagos. A partir da décima hora surgem
progressivamente os eosinófilos, macrófagos e linfócitos, permanecendo por cerca de uma
semana no local, isto se o agente agressor for removido, caso contrário, ocorre a cronificação
do processo (GALLIN; SNYDERMAN, 1999).
A migração dessas células do compartimento intravascular para o extravascular
ocorre predominantemente nas vênulas pós-capilares, sendo mediadas por uma combinação
de processos mecânicos, químicos e moleculares. Tais eventos distintos estão ligados em uma
sequência temporal (SEELY; PASCUAL, 2003). Inicia-se com a marginação ou movimento
dos neutrófilos do fluxo central para a periferia do vaso. Esse mecanismo ocorre através da
interação molecular entre a superfície celular de neutrófilos e células endoteliais, resultando
no rolamento de neutrófilos ao longo da parede do vaso. Tal interação é dependente de forças
físicas e moleculares sendo mediado por selectinas e seus ligantes (MENG et al., 2004).
Durante o rolamento, os leucócitos passam a interagir com quimiocinas como
interleucina 8 (IL-8) e as quimiocinas da família GRO, que se encontram ancoradas na
superfície das células endoteliais, resultando na ativação dos leucócitos e promovendo
alterações conformacionais nas integrinas presentes nos leucócitos em rolamento, que
aumentam a sua capacidade adesiva. Consequentemente o rolamento é interrompido e os
leucócitos aderem firmemente ao endotélio vascular (HUTTENLOCHER; SANDBORG;
HORVITZ,1995; MENG et al., 2004). O estágio final do recrutamento de neutrófilos é a
passagem através da parede endotelial para o tecido inflamado (HUTTENLOCHER;
SANDBORG; HORVITZ, 1995).
Todos esses eventos que ocorrem são responsáveis, portanto, pelo papel
fundamental dos neutrófilos na circunscrição e controle do foco infeccioso. Uma vez no foco
infeccioso, são capazes de engolfar os patógenos por meio de um processo conhecido como
fagocitose, resultando na formação de vesículas citoplasmáticas formadas pela fusão dos
fagossomas e dos lisossomas (JANEWAY; MEDZHITOV, 2002). Através dessa fusão os
neutrófilos iniciam uma potente defesa antimicrobiana onde pode ser dividida em um
processo dependente e independente de oxigênio (HUTTENLOCHER; SANDBORG;
HORVITZ, 1995).
Os efetores da defesa antimicrobiana de neutrófilos independentes de oxigênio
estão contidos em grânulos no citoplasma dos neutrófilos conhecidos como grânulos
azurófilos ou primários (FAURSCHOU; BORREGAARD, 2003). Essas estruturas contêm

18
uma variedade de proteínas pré-formadas que são liberadas no fagossomo durante o processo
de morte microbiana como defensinas, elastase e lactoferrinas (MARSHALL, 2005)
Mecanismos dependentes de oxigênio envolvem a transformação de oxigênio
molecular em uma família de Espécies Reativas de Oxigênio (EROs) que são liberadas nos
fagossomos ou no ambiente extracelular (HAMPTON; KETTLE; WINTERBOURN, 1998).
Esses intermediários são altamente reativos com moléculas biológicas importantes causando
peroxidação de lipídios, modificações estruturais de proteínas e dano ao DNA, levando a
morte do microorganismo (MARSHALL, 2005).
Portanto, os neutrófilos são conhecidos por exercer um importante papel na
resposta inflamatória por uma série de funções efetoras que representam um mecanismo
central de imunidade contra infecções (APPELBERG, 2007; NATHAN, 2006). Da mesma
forma, estudos mostram que em modelo animal de sepse severa por ligação e perfuração cecal
(CLP) encontra-se a falência da migração de neutrófilos para o foco infeccioso, associado ao
número de bactérias aumentado no exsudato peritoneal e sangue, seguido pela inflamação
sistêmica caracterizada pelo aumento dos níveis de citocinas e quimiocinas circulantes e o
seqüestro de neutrófilos para o pulmão e redução da taxa de sobrevida (ALVES-FILHO et al.,
2006).
Diferentes mecanismos estão envolvidos no desequilíbrio desse sistema. Entre as
investigações no entendimento de tais mecanismos, verifica-se a redução da expressão do
receptor CXCR-2 um dos receptores da quimiocina IL-8 (CHISHTI et al., 2004) a produção
excessiva de citocinas e quimiocinas circulantes, proteínas de fase aguda, aumento da
atividade de óxido nítrico sintase induzível (iNOS), heme oxigenase-1 (HO-1) e a ativação
sistêmica de TLRs são associados a esse fenômeno (ALVES-FILHO et al., 2008). Conforme
mencionado, TLRs são componentes essenciais na resposta imune inata contra a infecção, no
entanto, diferentes evidências indicam que podem desempenhar um papel importante na
fisiopatologia da sepse (MENG et al., 2004). Durante uma infecção polimicrobiana onde
componentes estruturais de diferentes bactérias levam a ativação de vários TLRs, a ativação
de um deles é suficiente para o desencadeamento de uma eficiente resposta inflamatória local.
Por outro lado, a sinalização excessiva de todos os receptores leva à geração de uma resposta
inflamatória sistêmica que é caracterizada pela excessiva produção e liberação de citocinas e
quimiocinas na circulação (ALVES-FILHO et al., 2008).
Nesse contexto, a interação de vários componentes como infecciosos,
imunológicos, hemodinâmicos, bioquímico e até mesmo genéticos (DE MAIO; TORRES;
REEVES, 2005; REMICK, 2007), podem levar a uma resposta exacerbada do organismo com

19
produção excessiva de EROs e mediadores inflamatórios e consequentes alterações
fisiológicas (ANNANE; BELLISSANT; CAVAILLON, 2005; ROCHA; OLIVEIRA;
FARIAS - CORRÊA, 2006).
1.2.3 Radicais livres na progressão da sepse
Estudos indicam que os radicais livres possuem mecanismos coadjuvantes de
dano celular e progressão da sepse além dos citados até o momento (DAL PIZZOL et al,
2010). Um radical livre é qualquer espécie química capaz de existir de forma independente e
que contenha um ou mais elétrons desemparelhados (HALLIWELL, 2006, HALLIWELL,
2001; HALLIWELL; GUTTERIDGE, 2007; SOUTHORN; POWIS, 1988). Como em sua
maioria, são derivados do metabolismo do O2, o termo genérico “Espécies Reativas de
Oxigênio” é usado para incluir não só os radicais formados pela redução de O2, o radical
superóxido (O2•-
) e o radical hidroxil (•OH), mas também alguns não-radicais derivados do
oxigênio, como H2O2, o oxigênio singlet (1O2). Além dessas, existem ainda as espécies
reativas de nitrogênio (ERN), sendo o óxido nítrico (NO•) e o peroxinitrito (ONOO
-) os
principais representantes. (HALLIWELL; GUTTERIDGE, 2007)
Fisiologicamente, essas espécies reativas apresentam diversas funções
(BERGENDI et al, 1999). Assim, um aumento da liberação local de radicais livres pode ser
benéfico, como é o caso da liberação de espécies tóxicas oxidantes pelos neutrófilos, que
podem atuar na defesa do hospedeiro contra uma infecção (DELANTY; DICHTER, 1998;
HALLIWELL; GUTTERIDGE, 2007).
Com relação aos efeitos prejudiciais, quando formadas em excesso, essas espécies
altamente reativas têm potencial de oxidar moléculas (MAXWELL, 1995). Os radicais livres
podem promover lipoperoxidação, causar a oxidação de lipoproteínas de baixa densidade
(LDL), reagir com proteínas levando à sua inativação e consequente alteração de sua função e
reagir com o DNA e RNA, levando a mutações somáticas e a distúrbios de transcrição
(DELANTY; DICHTER, 1998; HALLIWELL; WHITEMAN, 2004).
Assim, usamos o termo estresse oxidativo para referir à situação na qual a geração
de ERO ultrapassa a capacidade de defesas antioxidantes disponíveis. Estudos mostram que o
estresse oxidativo pode resultar tanto de uma diminuição das defesas antioxidantes quanto de

20
uma produção aumentada de oxidantes, bem como da liberação de metais de transição ou a
combinação de quaisquer desses fatores (HALLIWELL, 2006).
O estresse oxidativo tem atraído muita atenção da comunidade científica, uma vez
que tem sido relatado por participar no mecanismo de muitas doenças incluindo o câncer,
aterosclerose, envelhecimento, diabetes tipo-2, de doenças neurodegenerativas como Doença
Alzheimer (DA) e Doença Parkinson (DP) (JEZEK; HLAVATÁ, 2005 apud DAL-PIZZOL et
al., 2010, p. 2). Muitos estudos com modelos animais e humanos já haviam mostrado
elevações nos parâmetros oxidativos durante o curso da sepse e também que o estresse
oxidativo é um dos fatores que levam a dano celular, disfunção orgânica e morte.
(SALVEMINI; CUZZOCREA, 2002; ZIMMERMANN, 1995).
Mais recentemente, têm-se verificado o papel dos radicais livres no Sistema
Nervoso Central (SNC) associado à disfunção do metabolismo energético celular bem como
mediadores inflamatórios em ratos sobreviventes de sepse (COMIM et al., 2011). Tais
mecanismos foram sugeridos por estarem associados a diminuição da memória e aprendizado
em ratos submetidos a sepse. (BARICHELLO et al., 2007b; BARICHELLO et al., 2007a;
COMIM et al., 2010).
1.2.4 Barreira-hematoencefálica (BHE)
Mediadores inflamatórios liberados por leucócitos na sepse podem ter profundos
efeitos sobre as células endoteliais e astrócitos e, danos a essas células, resulta em deficiência
na função neuronal (PAPADOPOULOS et al., 2000). O cérebro é pensado para ser um órgão
privilegiado, uma vez que é anatomicamente isolado do sistema imunológico pela
Barreira Hematoencefálica (BHE), não tem um sistema linfático e tem baixa expressão de
antígenos de histocompatibilidade complexa em seu parênquima celular (SHARSHAR et al.,
2004). Assim BHE é um importante componente da rede de comunicação conectando o SNC
e os tecidos periféricos, além de funcionar como uma interface que limita e regula a troca de
substâncias entre o sangue e o SNC (BANKS, 2010 apud ROJAS; RITTER; DAL PIZZOL,
2011, p. 224).
Além disso, a BHE expressa um número elevado de canais iônicos e
transportadores, tem uma baixa taxa de pinocitose e formas intercelular de complexos multi-

21
protéicos, como as junções apertadas (JA) e junções endoteliais aderentes (JEA) que limitam
a permeabilidade celular (HAWKINS; DAVIS, 2005). É formada pela presença das junções
endoteliais que controlam a abertura e fechamento coordenada das junções célula-célula
(STAMATOVIC; KEEP; ANDJELKOVIC, 2008). As células endoteliais cerebrais são
apoiadas sobre uma lamina basal que contém moléculas da matriz extracelular cobrindo mais
de 90% da superfície das células endoteliais, também estando envolvida na permeabilidade da
BHE (BAUER et al., 2010; ZLOKOVIC, 2008 apud ROJAS; RITTER; DAL PIZZOL, 2011,
p. 223).
Além das funções de permeabilidade seletiva, a BHE possui aspectos importantes
como funções neuroimune, incluindo a secreção de citocinas, prostaglandinas e óxido nítrico.
A BHE pode receber o estimulo de um compartimento (como por exemplo, o sistêmico) e,
simultaneamente, responder com secreções para o outro (por exemplo, SNC), sendo esta
função de papel central na resposta neuroimune (ROJAS; RITTER; DAL PIZZOL, 2011).
A disfunção da BHE, foi descrita há muito tempo como um elemento-chave da
progressão de várias doenças do SNC (WEISS et al., 2009), isso por que alguns estudos vem
demonstrando que o estresse oxidativo e aumento da produção de citocinas e interleucina-2
(IL-2); o fator de necrose tumoral-α (TNF-α) parecem ter um efeito neurotóxico sobre o
cérebro ou estão envolvidos em apoptose neuronal. Assim, o TNF-α aumenta a
permeabilidade de células endoteliais cerebrais em humanos, promovendo pinocitose,
afetando a JA. O TNF-α também reduz o consumo de oxigênio cerebral e fluxo sanguíneo
cerebral e eleva a pressão intracraniana e cérebro-espinhal de lactato de fluidos, como visto
em um modelo de coelho após a injeção subaracnóidea deste mediador. (TUREEN, 1995)
Um outro estudo sugere que interações recíprocas entre o sistema imunológico e
SNC são consideradas como principais componentes da resposta do hospedeiro em estado de
choque séptico (DAL PIZZOL et al., 2010). Além disso, a lesão cerebral parece ocorrer
durante o desenvolvimento sepse e vários mecanismos propostos para a disfunção do SNC
induzida por sepse incluem alteração na BHE, a interrupção de aminoácidos e isquemia
cerebral resultante de uma redução global ou regional no fluxo sanguíneo cerebral (FREUND
et al., 1985; POLLAND et al., 1997 apud DAL PIZZOL et al, 2010, p. 2).
1.3 ENCEFALOPATIA SÉPTICA

22
As interações recíprocas entre o SNC e sistema imunológico são considerados como
os componetes principais da resposta inflamatória à sepse, o que acaba causando alterações
nos sistemas neuroendócrino, autonômico (CHROUSOS, 1995), comportamental (GORDON
et al., 2004) e distúrbios em quaisquer funções adaptáveis como as resposta imuno-
inflamatórias e hemodinâmica (SHARSHAR et al., 2004; SAPER; BREDER, 1994; SPYER,
1989).
Nesse contexto, recentemente foi descrito que sobreviventes de sepse apresentam
déficit cognitivo associado com a diminuição da qualidade de vida e um aumento na
morbidade em longo prazo (WINTERS et al, 2010). Mesmo tal fato ser importante na
fisiopatologia da disfunção do SNC durante a sepse, essas alterações ainda foram pouco
estudadas. A encefalopatia séptica é considerada uma disfunção cerebral devido à sepse
podendo ocorrer em 8-70% dos pacientes sépticos, dependendo dos critérios de inclusão
empregados (SPRUNG et al., 1990; YOUNG et al., 1990), sendo a encefalopatia mais comum
nas UTIs (BLECK et al., 1993).
O conceito de encefalopatia séptica como uma entidade que não possa ser
explicada pela disfunção, hipotensão, ou pela hipóxia hepática ou renal, é relativamente nova,
porém já está claro que a sepse e suas reações podem ser associadas a um largo espectro de
danos e disfunções cerebrais (PAPADOPOULOS et al., 2000). Apesar de a fisiopatologia da
encefalopatia séptica não ser bem determinada, provavelmente deva ter origem multifatorial.
Diversos fatores de risco foram descritos e podem ser categorizados como: a) condição pré-
existente do paciente (como ex. idade, história de depressão, doença hepática, doença renal);
b) condição aguda do paciente (como ex. overdose de drogas, febre), e c) fatores iatrogênicos
ou ambientais (como ex. uso de sedativos, alimentação enteral, cateter venoso central)
(EBERSOLDT; SHARSHAR; ANNANE, 2007).
Em casos fatais, têm-se demonstrado proliferação de astrócitos e microglia no
córtex, infartos cerebrais, púrpura cerebral, múltiplas hemorragias pequenas na substância
branca e disseminação de microabsessos (JACKSON et al., 1985). Nos sobreviventes se tem
demonstrado redução do fluxo sanguíneo cerebral, disfunção da BHE e alteração de
neurotransmissores (BOWTON et al., 1989; DESCAMPS et al., 2003).
Além destas manifestações neurológicas agudas observadas em pacientes
criticamente enfermos, recentemente tem se demonstrado que sobreviventes de UTI,
incluindo pacientes sépticos, apresentam disfunções cognitivas em longo prazo (ANGUS;
WAX, 2001; HOPKINS et al., 1999, 2005; HOUGH; CURTIS, 2005). Assim como para a
encefalopatia, os mecanismos associados a estas alterações não são bem entendidos, mas

23
podem incluir inflamação, estresse oxidativo e disfunção energética cerebral (MESSARIS;
MEMOS; CHATZIGIANNI, 2004).
Semmler e colaboradores (2007) demonstraram em animais sobreviventes de
sepse perda neuronal no hipocampo e sub-regiões do córtex pré-frontal e inervação
colinérgica reduzida de áreas corticais, resultando em relevantes consequências funcionais,
tais como, a perda de memória, uma alteração semelhante à observado em pacientes com DA
(RITTER et al., 2004). Esta similaridade também foi demonstrada por outro grupo usando
rivastigmina para reverter déficit cognitivo a longo-prazo em animais sobreviventes de sepse,
sendo que a rivastigmina tem efeito inibitório de longa duração e é seletiva para a
acetilcolinesterase cerebral e da butirilcolinesterase predominantemente no hipocampo e no
córtex cerebral, áreas associadas à disfunção colinérgicas e habituação de memória (COMIM
et al, 2009b), características que podem ser verificadas em doenças neurodegenerativas
(COMIM et al, 2009a).
1.4 PROTEÍNAS RELACIONADAS A NEURODEGENERAÇÃO
Doenças neurodegenerativas possuem mecanismos comuns que estão ligados à
agregação patológica de proteínas deformadas que se acumulam como depósitos de proteínas
amilóide fibrilares em regiões seletivamente vulneráveis do SNC (TILLEMENT ; LECANU;
PAPADOPOULOS, 2010) como na de DA o acúmulo de filamentos de tau anormal e
depósitos de beta-amiloide (Aβ) (HAAPASALO et al, 2010) e, na DP, demência com corpos
de Lewy, alfa-sinucleína (α-sinucleína) (TAN; WONG; LIM, 2009). Além disso, a proteína
S100B que regula a fosforilação de proteínas que constituem o citoesqueleto e proteínas
associadas ao microtúbulo (DONATO, 2011)
1.4.1 Proteína Tau Fosforilada
Emaranhados neurofibrilares são constituídos primariamente de proteínas tau
associadas à microtúbulos. Em neurônios, tau é normalmente encontrada em axônios, porém

24
em certas taupatias ela se redistribui para o corpo celular e dendritos, em função de seu
desprendimento dos microtúbulos. As proteínas tau estabilizam os microtúbulos do
citoesqueleto neural, sendo esta função regulada por um processo de fosforilação e
desfosforilação. Nos neurônios que sofrem degeneração, as proteínas tau associadas a
microtúbulos tornam-se anormalmente hiperfosforiladas e se acumulam na forma de
filamentos emaranhados helicoidais pareados (NAGY et al., 1995).
O citoesqueleto possui a função chave de manter a forma e a polaridade estrutural
dos neurônios, essenciais para a fisiologia neuronal. Porém, em algumas doenças
neurodegenerativas, como a DA, um desequilíbrio nas atividades cinase/fosfatase parece levar
a hiperfosforilação da proteína tau, a qual nesse estado se desprende dos microtúbulos,
acumula-se no soma, forma filamentos helicoidais pareados e leva à desorganização do
citoesqueleto celular e comprometimento do transporte axonal. A formação dos emaranhados
neurofibrilares, desencadeado pelo acúmulo de peptídeo Aβ1-42 e deposição em placas senis,
podem acelerar a agregação da tau e formação dos emaranhados (LI et al., 2005; GOLDE,
2002). Por sua vez, as placas senis são constituídas da deposição de fragmentos amilóides no
parênquima cerebral.
1.4.2 Proteína β-amilóide (Aβ)
A proteína Aβ é um fragmento proteolítico formado a partir de uma glicoproteína
transmembrânica maior, a Proteína Precursora Amilóide (APP), que sofre a ação das enzimas
proteolíticas β e γ-secretases, resultando na secreção da Aβ em fragmentos amilóides de 38 a
43 aminoácidos (HAASS, 2004). O peptídeo Aβ mais produzido durante esse procedimento é
o peptídeo contendo 40 aminoácidos (Aβ1-40) que não é associado à formação das placas,
enquanto que uma pequena porção contém 42 aminoácidos (Aβ1-42). O peptídeo Aβ1-42 é a
forma mais hidrofóbica e mais propensa a formar fibrilas, além de ser o principal responsável
pela formação das placas senis (LAFERLA; GREEN; ODDO, 2007)
Os mecanismos responsáveis pela neurotoxicidade da Aβ são complexos, mas
parecem envolver a quebra da homeostase intracelular do cálcio e potássio, indução de
estresse oxidativo e ativação do processo de morte celular. Além disso, transtornos da

25
transmissão da acetilcolina e acetiltransferases ocorrem frequentemente nos indivíduos
afetados com DA. (HAASS, 2004; MATTSON, 2004).
Estudos indicam que o aumento dos níveis de Aβ é acompanhado por alterações
na atividade de cinases e fosfatases, levando a hiperfosforilação da proteína tau e formação
dos emaralhados neurofibrilares com consequente dano no transporte axonal. Desse modo, os
eventos da cascata amilóide causam disfunção sináptica e neuronal generalizada, perda
seletiva de neurônios e importante déficit de neurotransmissores que culminam em demência
com placas senis e emaranhados neurofibrilares (HAASS; SELKOE, 2007; HARDY;
SELKOE, 2002; MATTSON; CHAN, 2003)
Em doenças neurodegenerativas, como DA, tem sido proposto que uma fagocitose
ineficiente da proteína Aβ pela microglia, a consequente ativação microglial e liberação de
mediadores inflamatórios e fatores neurotóxicos, contribuiria de maneira decisiva na
progressão da doença, visto que uma vez ativa, a microglia pode também recruta os astrócitos
que ativamente aumentam a resposta inflamatória aos depósitos de Aβ (AKIYAMA et al.,
2000)
1.4.3 Proteína α-sinucleína
A α-sinucleína é outra importante proteína relacionada a progressão da
neurodegeneração pois interage com uma variedade de proteínas dependentes de microtúbulos
(LEE et al., 2006) e, na sua forma agregada, está relacionada à patogênese da DP. A primeira
evidência favorável à associação de um componente genético à etiologia da DP ocorreu a
partir da descoberta, em pacientes com parkinsonismo de herança autossômica dominante, da
mutação A53T no gene da α-sinucleína, a qual é o principal constituinte do corpúsculo de
Lewy. Uma vez que os corpos de Lewy são identificados em formas familiares e esporádicas
da DP, é possível que anormalidades da α-sinucleína façam parte da patogênese desta
enfermidade (POLYMEROPOULOS et al.,1997).
A função exata da α-sinucleína ainda não está bem estabelecida, mas acredita-se
que possa ter algum papel na proteção contra injúria neuronal, uma vez que se associa a uma
chaperona em terminais nervosos, prevenindo a neurodegeneração (KITADA, et al, 1998).

26
Experimentos in vitro demonstraram que tipos selvagem ou mutante da α-
sinucléina são capazes de formar protofibrilas (oligoméricas) ou assumir conformações
fibrilares, mas não conseguiram estabelecer que tipo de formação seria neurotóxica. Algumas
evidências apontam para as formações protofibrilares como principais responsáveis pela
neurotoxicidade. As mutações A53T e A30P na α-sinucleína promovem a formação de
protofibrilas, mas apenas a A53T leva à formação de fibrilas, enquanto a A30P inibe a
formação destas. A formação de protofibrilas é incrementada e estabilizada por quinonas de
dopamina (produtos de oxidação da dopamina), e talvez esta seja a causa de toxicidade maior
da α-sinucleína na substância negra. (GODEIRO JUNIOR, et al., 2007).
Além disso, a disfunção da α-sinucleína tem uma ação indireta e significativa na
função mitocondrial neuronal. O aumento de expressão da α-sinucleína sensibiliza os
neurônios ao estresse oxidativo e ao dano por toxinas como MPP+ e 6- hidroxidopamina e
talves isso seja uma forte evidência associando parkinsonismo e disfunção mitocondrial, com
base em influência genética. (GODEIRO JUNIOR, et al., 2007).
Estudos in vitro e in vivo demonstraram que a proteína promotora da
polimerização da tubulina (TPPP/p25) interage com sistema tubulina/microtúbulo.
(HLAVANDA et al., 2002). A co-agregação α-sinucleína/tubulina parece preceder a
degeneração neurítica (LEE et al., 2006). O microtúbulo é um alvo comum de toxinas e
parkina. Acúmulo de α-sinucleína interfere com parkina e com a solubilidade e distribuição de
α-tubulina, provocando alterações de citoesqueleto e disfunção neuronais (FENG, 2006).
A superexpressão de α-sinucleína reduz a α-tubulina ubiquitinada e isso poderia
provocar o acúmulo de α-tubulina insolúvel. A parkina protege neurônios dopaminérgicos
contra toxinas despolimerizantes de microtúbulos através da atenuação da ativação da
proteína-cinase associada a microtúbulo (REN et al., 2009). A despolarização de microtúbulo
exerce maior toxicidade em neurônios dopaminérgicos, neurônios que têm longos axônios que
se projetam ao estriado (LEE et al., 2006).
1.4.4 Proteína S100B
A proteína S100B é outra importante proteína que foi implicada no
desenvolvimento e manutenção do sistema nervoso. É pertencente a uma família de proteínas
ligantes de cálcio e de baixo peso molecular, possui atividade neurotrófica e gliotrófica e está

27
localizada principalmente no SNC em astrócitos e células de Schawnn, mas pode ser
detectada em menores concentrações em outras células, como melanócitos e adipócitos. De
acordo com experimentos “in vitro”, a proteína S100B foi estudada tanto no que se refere a
suas propriedades funcionais intracelulares quanto extracelulares. Intracelularmente, ela foi
relacionada a funções biológicas, tais como regular a fosforilação de proteínas constituintes
do citoesqueleto (DONATO, 2001; ZIMMER et al., 1995)
Estudos com o objetivo de esclarecer o potencial envolvimento da S100B em
processos neurodegenerativos e os mecanismos pelos quais ela exerce seus efeitos foram
tratados com grande interesse nos últimos anos (AZMITIA et al., 1992; DONATO, 2001;
ZIMMER et al., 1995). O aumento dos níveis cerebrais desta proteína foi implicado na
fisiopatologia das alterações cerebrais presentes na DA e na Síndrome de Down, sendo que
camundongos transgênicos que super expressam a proteína S100B apresentam alterações
morfológicas e comportamentais semelhantes àquelas que ocorrem na DA e síndrome de
Down (AZMITIA et al., 1992; DONATO, 2001; ZIMMER et al., 1995).
Devido aos seus efeitos extracelulares, a proteína S100B é também considerada
uma citocina, e, desta forma, ela pode estar não somente na fisiopatologia das alterações
presentes na DA e síndrome de Down, como também na resposta inflamatória ao dano
cerebral (GRIFFIN et al., 1996).
O efeito neurotóxico da proteína S100B pode ser atribuído à estimulação de uma
enzima iNOS de astrócitos, resultando na morte celular por apoptose, tanto dos neurônios
quanto de astrócitos (DONATO, 2001; HU; VAN ELDIK, 1996; HU; FERREIRA; VAN
ELDIK, 1997) ou também mediados via interação com Receptor de Produtos de Glicação
Avançada (RAGE), levando a um aumento na produção de ERO, liberação de citocromo C e
ativação da cascata das caspases, com posterior indução de apoptose (DONATO, 2001).
1.5 NEURODEGENERAÇÃO E ATIVAÇÃO DE RAGE
O RAGE é expresso em baixa concentração em vários tipos celulares, como
células endoteliais vasculares, musculares lisas, glomerulares, macrófagos e monócitos
(BRETT et al., 1993). Em situações de acúmulo dos seus ligantes, bem como em estresse,
desenvolvimento normal, diabetes, insuficiência renal, DA (HUDSON et al., 2002;

28
NISHIKAWA; EDELSTEIN; BROWNLEE, 2000; SCHMIDT; STERN, 2000; YAN et al.,
1996) ou processos inflamatórios como a sepse (HATTORI et al., 2010). Uma das
peculiaridades da proteína RAGE é a sua capacidade de reconhecer vários ligantes,
permitindo a sua participação em um amplo espectro de eventos fisiopatológicos,
relacionados principalmente à propagação de disfunção celular (SCHMIDT; STERN, 2000).
Os ligantes da proteína RAGE mais conhecidos são os produtos finais de glicação
avançada (AGEs), que constituem um grupo heterogêneo de compostos responsáveis por
efeitos adversos, incluindo redução de atividade enzimática, danos a ácidos nucléicos,
formação de ligações cruzadas (cross-linking) entre proteínas, além de indução de vias
citotóxicas (BROWNLEE, 1994). A formação de AGEs causa alterações fisiopatológicas,
entre elas podemos citar a interação de AGEs com receptores específicos, ativando vias pró-
inflamatórias e pró-trombóticas através de estresse oxidativo e da ativação de fatores de
transcrição, como é o caso da interação AGEs-RAGE (BROWNLEE, 1995).
Além das AGEs, na sepse tem-se verificado que ocorre a ativação de RAGE e a
progressão da resposta inflamatória pela ligação com a proteína HMGB1 secretada por
macrófagos, células dendríticas, tumores, células endoteliais e também liberada com o
processo de necrose sendo detectada no soro de pacientes com sepse em estágios tardios, e
com concentração plasmática inversamente proporcional com a sobrevivência (KARLSSON
et al, 2008).
Outros ligantes de RAGE incluem os polipeptídeos S100/calgranulinas e as
anfoterinas, com efeitos ainda pouco elucidados após a interação com RAGE. No entanto,
sabe-se que ambas desempenham papel durante o desenvolvimento normal e em inflamação
do tecido nervoso (HUDSON et al., 2002). Selkoe (1994) mostrou que a proteína Aβ, formada
através da proteólise da proteína amilóide, também é um dos ligantes de RAGE, sugerindo o
envolvimento deste receptor na DA. Neste mesmo estudo foi demonstrado que RAGE atua
como mediador do transporte da proteína Aβ através da barreira sanguínea do cérebro
contribuindo para o seu depósito na membrana basal de vasos sangüíneos.
Através da ativação de RAGE uma diversificada cascata de sinalização pode ser
estimulada. Por exemplo, MAPK (Proteína quinase ativada por mitógeno) incluindo ERK1/2
(Quinase regulada por sinais extracelulares 1/2), p38 e JNK (Quinase n-terminal c-Jun), vem
sendo mostrado por ser alvo do ligante RAGE em diversos tipos de células, tal como células
tumorais, neurônios, células endoteliais, células epiteliais Caco-2, podócitos e células
microgliais. (ALESHIN et al., 2008; CHANG et al., 2008; TAGUCHI et al., 2000).

29
As MAPKs abrangem um grupo de proteínas sinalizadores utilizadas para
diversos acontecimentos celulares, através de eventos de fosforilação. Elas estão presentes em
todos os eucariotos e controlam processos de proliferação, expressões gênicas, diferenciação e
apoptose (CANO; MAHADEVAM, 1995; COHEN, 1997). Os três grupos principais de
MAPKs: ERK1/2 (ativada principalmente por estímulos mitogênicos), p38 e JNK, são
ativadas principalmente por estímulos de estresse e citocinas inflamatórias (DAVIS, 2000;
KYRIAKIS; AVRUCH, 2001). Quando ativado, ERK1/2 é responsável pela ativação de
fatores de transcrição, como Ativador de Proteina 1 (AP-1), Glicogênio sintase quinase 3β
(GSK-3β) e NF-kB (CAMPBELL et al., 1998; WEINSTEIN-OPPENHEIMER et al., 2000).
Nesse contexto, RAGE pode ser considerado como um mediador da transdução de
sinal. A ativação e translocação nuclear de NF-kB aumentam a expressão de proteínas
envolvidas na adesão de leucócitos ao endotélio, um processo implicado com o início de
lesões ateroscleróticas e de outros distúrbios vasculares (CHAVAKIS; BIERHAUS;
NAWROTH, 2004; COLLINS, 1993). Assim através dessa via, tem se verificado que a
ativação do receptor RAGE gera o aumento nas concentrações de citocinas de resposta pró-
inflamatória, como as interleucinas IL-1β e IL-6 e o TNFα entre outros (HOFMANN et al.,
1999). Logo, a liberação de TNFα promovida pela amilóide sérica, em neutrófilos, requer a
participação de sinalizadores intracelulares como p38, ERK1/2, PI3K e da ativação de fator de
transcrição, NF-kB.(HATANAKA et al., 2004).
Tal aumento da resposta inflamatória no SNC pela liberação adicional de
citocinas pró-inflamatórias com ativação de RAGE pode potencialmente estar associado à
progressão do dano cerebral causado pela quebra da BHE visto que estudos mostram que na
sepse e na SIRS há alteração na permeabilidade da barreira cerebral sanguínea e o processo
inflamatório acaba afetando os sistemas de controle do SNC, causando alterações nas funções
fisiológicas cruciais à homeostase levando a encefalopatia (CHROUSOS; GOLD, 1992)
associado à modificações do débito sanguíneo cerebral e a sua auto-regulação (BOWIE;
O’CONNOR; MAHAJAN, 2003) que são implicadas na patogênese e a hipotensão
relacionada significativamente ao desenvolvimento de dano isquêmico cerebral (BOOKE et
al., 2003).
Nesse sentido, marcadores bioquímicos presentes no cérebro associados a
fisiopatologia de doenças neurodegenerativas podem estar também ligados ao
comprometimento cognitivo a longo prazo observado em modelo animal de sepse ou déficits
cognitivos observados em pacientes que se recuperam de sepse.

30
2 JUSTIFICATIVA
A sepse é a principal causa de mortalidade em UTIs não cardiológicas em todo
mundo, especialmente em decorrência da disfunção de múltiplos órgãos. Alterações
neurológicas precoces e tardias associam-se a a esses desfechos e embora existam grupos de
pesquisadores estudando o dano ao sistema nervoso central associado à sepse, comparando
com a disfunção em outros órgãos, o dano neurológico é um campo pouco explorado e de
extrema importância visto que ocorre a disfunção cognitiva em longo prazo como a perda da
memória e aprendizado comprometendo a capacidade produtiva do paciente diminuindo a sua
qualidade de vida.
Nesse contexto, estudos nos dão suporte de que a disfunção cerebral verificada na
sepse pode possuir mecanismos comuns ligados a fisiopatologia de doenças
neurodegenerativas. Tais mecanismos estão associados ao receptor RAGE que tem sido cada
vez mais implicado na progressão da morte neuronal em muitas doenças neurodegenerativas
como a DA, sendo também um mediador inflamatório importante visto que pode ser ativado
por mediadores inflamatórios em fluidos extracelulares durante a sepse. Sendo assim,
analisamos a presença de proteínas associadas as doenças neurodegenerativas e o seu possível
envolvimento com RAGE em cérebro de ratos sobreviventes de sepse.
Essa abordagem, portanto, poderá nos proporcionar resultados que poderão ser
potencialmente úteis para o melhor entendimento dos mecanismos fisiopatológicos dos danos
cerebrais em pacientes sépticos.

31
3 OBJETIVOS
3.1 OBJETIVO GERAL
Avaliar a presença de marcadores de neurodegeneração e ativação de RAGE no
cérebro de ratos sobreviventes de sepse.
3.2 OBJETIVOS ESPECÍFICOS
- Avaliar o imunoconteúdo das proteínas β-amilóide, isoforma tau de DA, α-sinucleína e
S100B no hipocampo e córtex pré-frontal em ratos 30 dias após a indução de sepse.
- Avaliar o imunoconteúdo protéico do receptor RAGE no hipocampo e córtex pré-frontal
em ratos 30 dias após a indução de sepse.
- Avaliar o imunoconteúdo protéico da quinase ERK 1/2 no hipocampo e córtex pré-
frontal em ratos 30 dias após a indução de sepse.

32
4 MATERIAL E MÉTODOS
4.1 TIPO DE ESTUDO
Estudo experimental com animais.
4.1.1 Bioética
O presente estudo foi aprovado pela Comissão de Ética no Uso de Animais
(CEUA) da Universidade do Extremo Sul Catarinense – UNESC, sob protocolo 56/2011 e
obedeceu aos Princípios de Cuidados de Animais de Laboratório (Principles of Laboratory
Animal Care, Instituto Nacional de Saúde dos Estados Unidos da América, NIH, publicação
número 85-23, revisada em 1996).
4.1.2 Animais
Ratos Wistar machos, entre 2 e 3 meses de idade, pesando aproximadamente 220
a 350g procedentes do Biotério da Universidade do Extremo Sul Catarinense foram divididos
em 2 grupos (1) sham; (2) CLP e mantidos em temperatura controlada (23°C ± 1) e ciclos de
luz artificial (12 horas claro/escuro). Receberam ração comercial padronizada para ratos de
laboratório e água ad libitum. Tiveram os níveis de imunoconteúdo avaliados 30 dias após a
indução da sepse, com n mínimo necessário para dosagem de cada parâmetro, totalizando 20
animais (n = 10 por grupo).

33
4.1.3 Indução de sepse
Sepse intra-abdominal foi produzida usando a técnica de CLP conforme
previamente descrito (RITTER, 2004). Brevemente, os ratos foram anestesiados com
cetamina (80 mg/kg) e xilazina (10 mg/kg), sendo submetidos à laparotomia com incisão
mediana abdominal. O ceco foi ligado logo abaixo da junção íleo-cecal com fio seda 3-0,
mantendo assim a continuidade intestinal. O ceco foi perfurado com uma agulha número 14
na face antimesentérica do ceco, e foi gentilmente comprimido até a extrusão de conteúdo
fecal. Os planos cirúrgicos foram fechados e os ratos foram observados em caixa de
recuperação por 2 horas. Como controle utilizamos animais submetidos a laparotomia, com
manipulação do ceco, mas sem ligação ou perfuração (sham). Os grupos sepse e sham
receberam reposição volêmica, realizada com salina, 50 mL/kg, imediatamente e 12 horas
após a cirurgia e ceftriaxona 30 mg / kg e 25 clindamicina mg / kg por via subcutânea a cada
6 horas por 3 dias. Ainda, após o procedimento cirúrgico os animais receberam 80 mg/kg de
dipirona sódica (i.m) para analgesia (PRADO; PONTES, 2002).
Trinta dias após a cirurgia os animais foram mortos por decaptação e isolados o
hipocampo w córtex pré-frontal. Os tecidos cerebrais foram congelados em freezer a -80°C
para que não ocorrece alterações.
4.1.4 Immunoblotting
Os tecidos cerebrais, hipocampo e córtex pré-frontal foram macerados picados e
imediatamente homogeneizados em tampão de extração. O extrato foi centrifugado a 11.000
rpm e 4°C por 40 min para remover o material insolúvel e o sobrenadante foi utilizado para a
quantificação da proteína, utilizando o método de Bradford (BRADFORD, 1976). As
proteínas foram desnaturadas por fervura (LAEMMLI, 1970) e o tampão contendo a amostra
foi submetido a corrida eletroforética em dodecil sulfato de sódio (SDS-PAGE) e transferidas
para membrana de nitrocelulose. A membrana foi bloqueada, submetida à sonda, conforme
método descrito anteriormente (DE SOUZA et al., 2005). Os anticorpos utilizados para o
immunoblotting foram (anti-RAGE, R5278; anti-isoforma tau de DA, A8855; comercializada

34
por Sigma; anti-S100B, ab41548; anti-α-sinucleína, ab6162; comercializada por Abcam; anti-
β-amilóide, No 2454 e anti-ERK 1/2, No 4376, comercializada por Cell Signaling
Technology) e anti-β-actina como controle. A intensidade das bandas foi quantificada por
densitometria óptica em autorradiografias. .
4.2 ANÁLISE ESTATÍSTICA
Os resultados foram expressos como média ± desvio padrão e o p<0.05 foi
considerado significativo. As diferenças entre os grupos foram determinadas por teste t
Student. Todas as análises estatísticas foram realizadas com o software SPSS 19.0 (SPSS,
Chicago, IL).
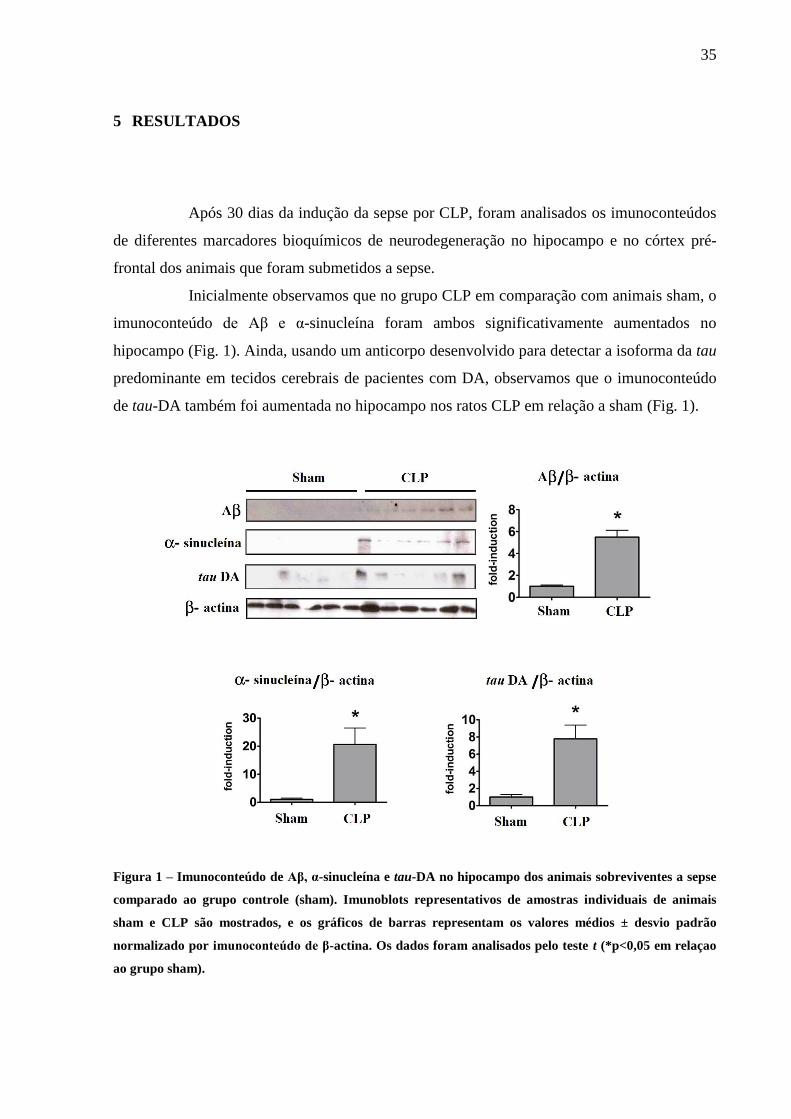
35
5 RESULTADOS
Após 30 dias da indução da sepse por CLP, foram analisados os imunoconteúdos
de diferentes marcadores bioquímicos de neurodegeneração no hipocampo e no córtex pré-
frontal dos animais que foram submetidos a sepse.
Inicialmente observamos que no grupo CLP em comparação com animais sham, o
imunoconteúdo de Aβ e α-sinucleína foram ambos significativamente aumentados no
hipocampo (Fig. 1). Ainda, usando um anticorpo desenvolvido para detectar a isoforma da tau
predominante em tecidos cerebrais de pacientes com DA, observamos que o imunoconteúdo
de tau-DA também foi aumentada no hipocampo nos ratos CLP em relação a sham (Fig. 1).
Figura 1 – Imunoconteúdo de Aβ, α-sinucleína e tau-DA no hipocampo dos animais sobreviventes a sepse
comparado ao grupo controle (sham). Imunoblots representativos de amostras individuais de animais
sham e CLP são mostrados, e os gráficos de barras representam os valores médios ± desvio padrão
normalizado por imunoconteúdo de β-actina. Os dados foram analisados pelo teste t (*p<0,05 em relaçao
ao grupo sham).
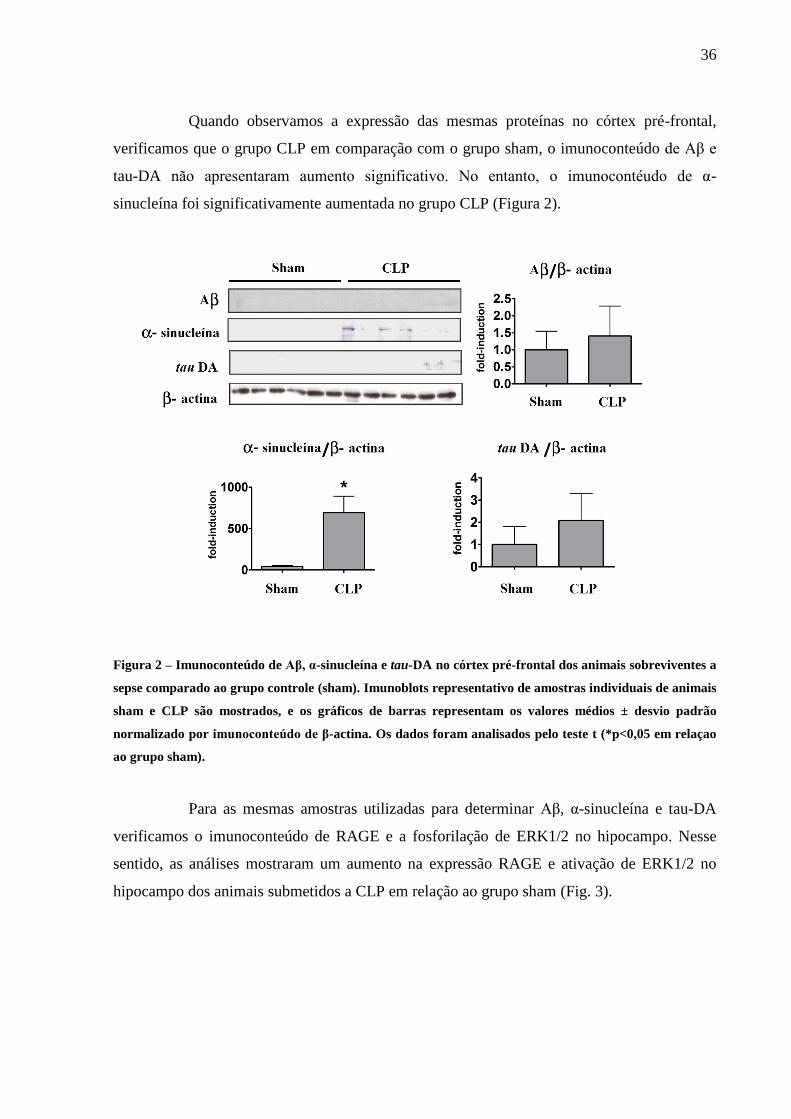
36
Quando observamos a expressão das mesmas proteínas no córtex pré-frontal,
verificamos que o grupo CLP em comparação com o grupo sham, o imunoconteúdo de Aβ e
tau-DA não apresentaram aumento significativo. No entanto, o imunocontéudo de α-
sinucleína foi significativamente aumentada no grupo CLP (Figura 2).
Figura 2 – Imunoconteúdo de Aβ, α-sinucleína e tau-DA no córtex pré-frontal dos animais sobreviventes a
sepse comparado ao grupo controle (sham). Imunoblots representativo de amostras individuais de animais
sham e CLP são mostrados, e os gráficos de barras representam os valores médios ± desvio padrão
normalizado por imunoconteúdo de β-actina. Os dados foram analisados pelo teste t (*p<0,05 em relaçao
ao grupo sham).
Para as mesmas amostras utilizadas para determinar Aβ, α-sinucleína e tau-DA
verificamos o imunoconteúdo de RAGE e a fosforilação de ERK1/2 no hipocampo. Nesse
sentido, as análises mostraram um aumento na expressão RAGE e ativação de ERK1/2 no
hipocampo dos animais submetidos a CLP em relação ao grupo sham (Fig. 3).
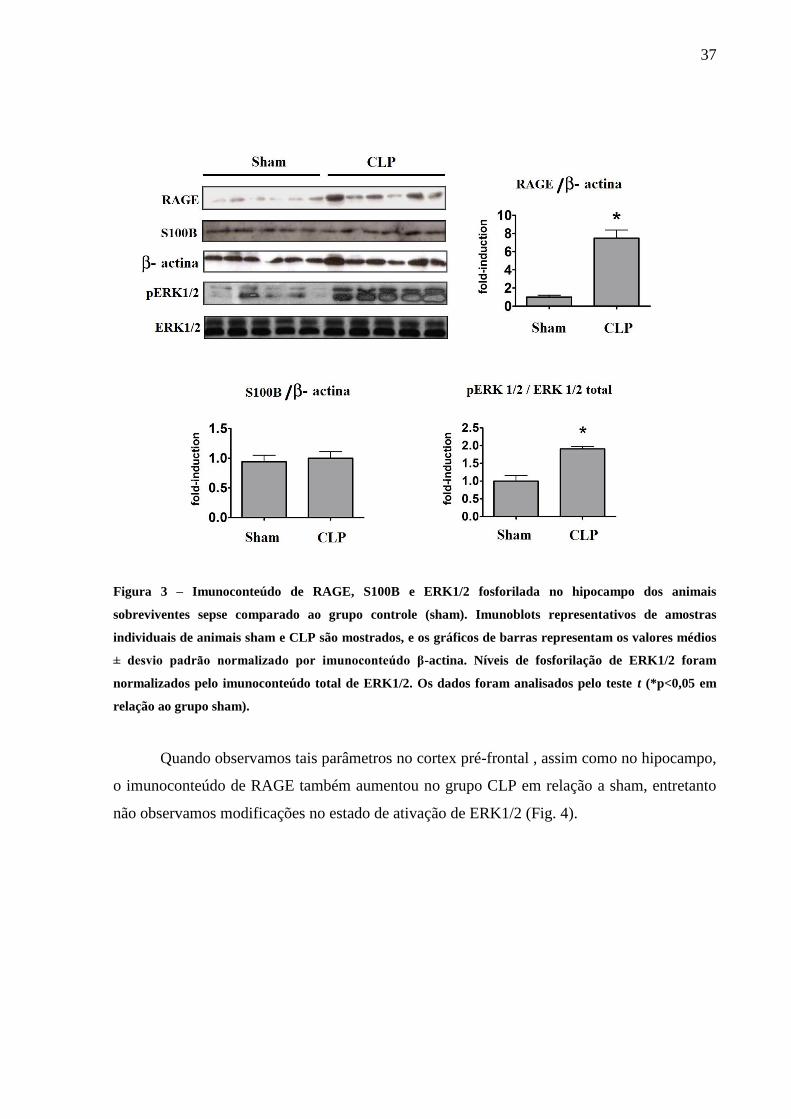
37
Figura 3 – Imunoconteúdo de RAGE, S100B e ERK1/2 fosforilada no hipocampo dos animais
sobreviventes sepse comparado ao grupo controle (sham). Imunoblots representativos de amostras
individuais de animais sham e CLP são mostrados, e os gráficos de barras representam os valores médios
± desvio padrão normalizado por imunoconteúdo β-actina. Níveis de fosforilação de ERK1/2 foram
normalizados pelo imunoconteúdo total de ERK1/2. Os dados foram analisados pelo teste t (*p<0,05 em
relação ao grupo sham).
Quando observamos tais parâmetros no cortex pré-frontal , assim como no hipocampo,
o imunoconteúdo de RAGE também aumentou no grupo CLP em relação a sham, entretanto
não observamos modificações no estado de ativação de ERK1/2 (Fig. 4).
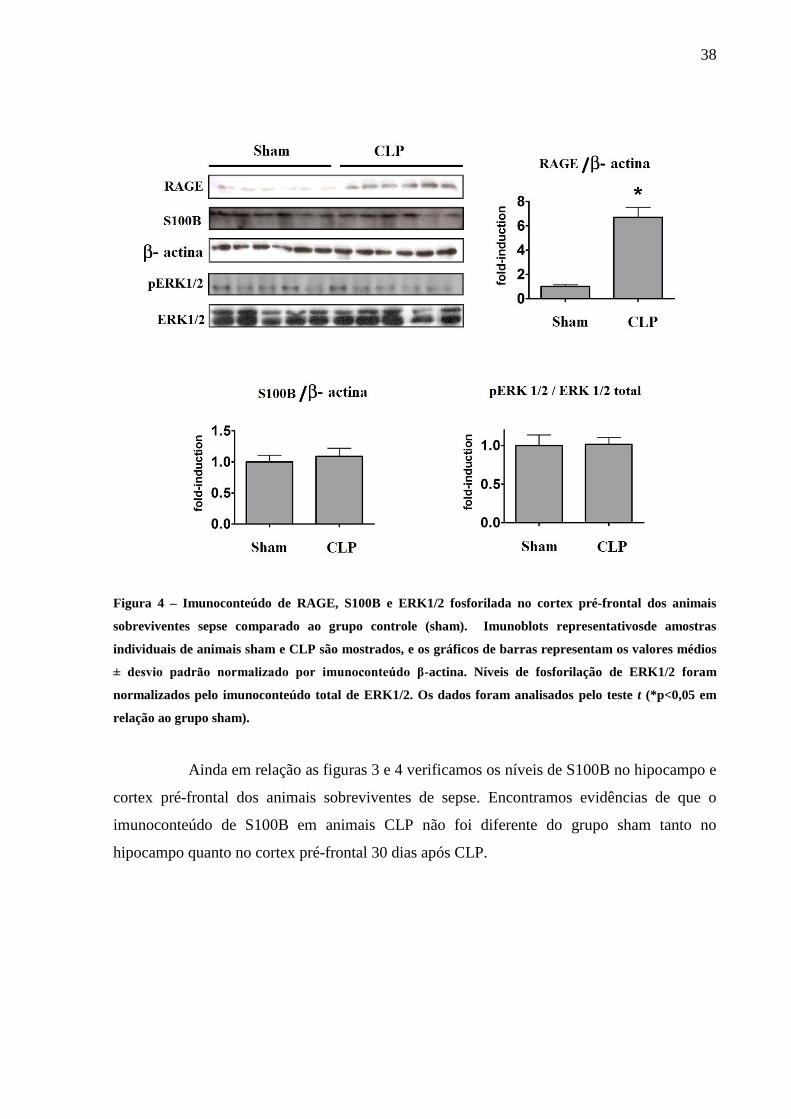
38
Figura 4 – Imunoconteúdo de RAGE, S100B e ERK1/2 fosforilada no cortex pré-frontal dos animais
sobreviventes sepse comparado ao grupo controle (sham). Imunoblots representativosde amostras
individuais de animais sham e CLP são mostrados, e os gráficos de barras representam os valores médios
± desvio padrão normalizado por imunoconteúdo β-actina. Níveis de fosforilação de ERK1/2 foram
normalizados pelo imunoconteúdo total de ERK1/2. Os dados foram analisados pelo teste t (*p<0,05 em
relação ao grupo sham).
Ainda em relação as figuras 3 e 4 verificamos os níveis de S100B no hipocampo e
cortex pré-frontal dos animais sobreviventes de sepse. Encontramos evidências de que o
imunoconteúdo de S100B em animais CLP não foi diferente do grupo sham tanto no
hipocampo quanto no cortex pré-frontal 30 dias após CLP.

39
6 DISCUSSÕES
Na amplitude das manifestações clínicas da sepse, a encefalopatia séptica é uma
das mais comuns, embora pouco diagnosticada, além de ser a causa de maior taxa de
mortalidade. Em modelo animal de sepse polimicrobiana, ocorre a encefalopatia aguda e os
sobreviventes apresentam danos cognitivos no SNC. Além disso, sobreviventes de UTIs,
incluindo pacientes sépticos, podem ter morbidade associada ao cérebro incluindo déficit
cognitivo e desenvolvimento de distúrbios psiquiátricos (DAL PIZZOL et al., 2010).
Nesse sentido, os resultados encontrados no presente estudo sugerem pela
primeira vez, que o estímulo inflamatório sistêmico agudo está associado ao aparecimento de
marcadores bioquímicos de neurodegeneração no cérebro, tais como, Aβ, tau-DA, α-
sinucleína e RAGE que se assemelha a diversos tipos de doenças cerebrais associadas a idade.
Nosso estudo propõe que a ativação da via Aβ-RAGE-ERK1/2 pode ser
importante para o comprometimento cognitivo a longo prazo observado em modelo animal
de sepse, e também pode estar implicado no déficit cognitivo persistente observado em alguns
pacientes que se recuperam de sepse.
Há um número crescente de evidências que relatam que a inflamação e
neurodegeneração são eventos relacionados (SEMMLER et al.; 2005). Procalcitonina (PCT)
no líquido cefalorraquidiano foi encontrado em níveis significativamente aumentados em
pacientes com DA, demência vascular, demência com corpos de Lewy e a demência
frontotemporal, semelhante ao observado em estados agudos neuroinflamatórios, como na
encefalite e meningite (ERNST et al., 2007). Isto indica que a inflamação central pode ter um
papel no desenvolvimento de doenças neurodegenerativas crônicas.
Neurodegeneração também está relacionada à inflamação sistêmica, o que é
exemplificado pela infecção pelo HIV. A patologia geralmente atribuída a DA foi visto em
pacientes HIV+, incluindo deposição aumentada de Aβ no cérebro, formação de placas
amilóides extracelulares e níveis de Aβ diminuído no líquido cefalorraquidiano. A DP pode
ser outra doença particularmente potencial para pacientes idosos HIV+, dada a importância
dos núcleos da base e vias dopaminérgicas em ambas as doenças (VALCOUR; PAUL, 2006).
Além disso, eventos inflamatórios sistêmicos agudos foram associados com um
aumento de 2 vezes na taxa de declínio cognitivo ao longo de um período de 6 meses em
pacientes DA (HOLMES et al., 2009). Neste contexto, foi demonstrado, em modelo animal,

40
que a administração intracerebroventricular de proteína C-reativa está associada à perda de
memória e fase inicial da patogênese da DA (LIN et al., 2009).
Até o momento, consequências a longo prazo da encefalopatia séptica sobre a
função cerebral são pouco compreendidas. Foi demonstrado em modelo animal que o
comprometimento comportamental foi independente dos níveis de glicose cerebral, mas
dependente do gene iNOS, sugerindo uma relação com a ativação microglial sustentada
(WEBERPALS et al., 2009). Além disso, temos relatado anteriormente que as fases iniciais
da sepse são caracterizadas pelo aumento na produção de ERO levando ao estresse oxidativo
(BARICHELLO et al, 2006) e que o tratamento antioxidante durante a disfunção cerebral na
fase aguda induzida por CLP, preveniu o desenvolvimento de déficit cognitivo tardio
(BARICHELLO et al, 2007b).
Uma vez que o estresse oxidativo e a inflamação são eventos relacionados em
doenças neurodegenerativas e na sepse, é possível que tanto o estresse oxidativo e ativação da
microglia colaboram para o dano cognitivo a longo prazo observado em sobreviventes de
sepse. Microglias são fontes comuns de estresse oxidativo e mediadores pró-inflamatório em
DA como mostrado por observações consistentes de resposta inflamatória alto-regulada em
regiões cerebrais que expoem altos níveis de marcadores patológicos na DA, tais como, o
córtex frontal e região límbica (CRAS et al., 1990 e GOOD et al., 1996). Por outro lado, esses
marcadores de inflamação ou estresse oxidativo estão ausentes ou mínimos em regiões do
cérebro que normalmente são menos suscetíveis a eventos neurotóxicos relacionados à
patologia, como o cerebelo (AKIYAMA et al, 2000).
Foi sugerido que tanto a inflamação como estresse oxidativo aumentam antes de
um diagnóstico da DA (ou pelo menos no início de DA), devido à super-ativação de certas
respostas pró-inflamatórias por fagócitos mononucleares, reforçando a nossa sugestão de que
a inflamação cerebral precoce e estresse oxidativo pode estar relacionado com a ocorrência de
marcadores tardios de neurodegeneração (FAGIOLO et al., 1993; NUNOMURA et al., 2001).
Além disso, mediadores inflamatórios liberados por leucócitos resultam na deficiência
neuronal (PAPADOPOULOS et al., 2000), causando a disfunção da BHE que aumenta a
progressão de várias doenças do SNC (WEISS et al., 2009).
Os receptores TLRs desempenham um papel importante na resposta inflamatória
inata na sepse (FULOP et al., 2004). Esses receptores podem estar associados com o
envelhecimento normal e doenças neurodegenerativas, interligando a inflamação com
patologia cerebral. A expressão de TLR4 é encontrada em neutrófilos não estimulados de
indivíduos idosos, quando comparados com indivíduos jovens, sugerindo uma inflamação de

41
baixo grau no envelhecimento humano (FULOP et al., 2004). Contudo, foi demonstrado que o
polimorfismo pró-inflamatório do gene TLR4 é um fator de risco independente para o
desenvolvimento de DA (MINORETTI et al., 2006).
Observamos também que há evidências farmacológicas ligadas ao dano cognitivo
a longo prazo associado a sepse e doenças neurodegenerativas (CASSOL JUNIOR et al.,
2010; COMIM et al., 2009a; COMIM et al., 2009b). Essas mudanças também são
temporalmente correlacionadas com a ocorrência de déficits cognitivos em tarefas
dependentes dos circuitos hipocampais (TUON et al., 2008), sugerindo uma relação entre
esses dois fenômenos. Portanto, uma vez que essas modificações são comumente associados a
doenças neurodegenerativas e/ou processos neurotóxicos agudos, nossos resultados indicaram
uma alteração nos níveis de proteínas no cérebro associadas a neurodegeneração em sepse.
Dessa forma, como a Aβ desempenha um papel central nos eventos neurotóxicos
celulares relacionados a doenças neurodegenerativas, como AD, nós também investigamos
uma via relacionada com a neurotoxicidade de Aβ nestes animais. Nossos resultados
apresentaram que a Aβ está aumentada no hipocampo de ratos sobreviventes a sepse, isso
porque RAGE atua como um sitio de ligação na superfície celular para Aβ, que resulta na
ativação de vias controladas por MAPK associadas à apoptose neuronal, produção de espécies
reativas dependentes de NADPH oxidase, disfunção sináptica e declínio cognitivo, bem como
o desenvolvimento de respostas inflamatórias em DA (ARANCIO et al., 2004 e LECLERC et
al., 2009).
Além disso, um dos principais alvos intracelulares da ativação do RAGE é o fator
de transcrição NF-kB, que induz a expressão de fator estimulador de colônia de macrófagos
que ativa a microglia , perpetuando a resposta inflamatória cerebral e o estresse oxidativo (DU
YAN et al., 1997). Como o gene RAGE também contém um elemento de resposta para NF-
kB (p65), a ligação da Aβ a RAGE resulta em um ciclo de feedback positivo de indução de
RAGE, que por sua vez aumenta ainda mais a inflamação no cérebro, o estresse oxidativo e a
produção Aβ (CREAGHT-BROWN et al., 2010; CREAGH-BROWN; QUINLAN; EVANS,
2010).
Nesse sentido, encontramos no presente estudo, um aumento dos níveis de RAGE
nas duas estruturas estudadas, mostrando a relação de ativação de RAGE em sobreviventes de
sepse. Nossos resultados no entanto não demonstraram relação da alta expressão de RAGE
com níveis de S100B visto que trata-se de outro ligante de RAGE, provavelmente por sua
superexpressão se apresentar na fase aguda do desenvolvimento de sepse (LIPCSEY et al.,
2010).

42
Como alvo da ativação de RAGE temos como resultado a ativação das MAPKs
que podem ser expressas nas células neuronais no SNC durante estados dinâmicos em
resposta de vários estímulos externos, tais como, fatores de crescimento, o glutamato e
hormônios, fatores de estresse (por exemplo, estresse oxidativo e hipóxia) e agentes
patogênicos (ORIGLIA et al., 2009). Entre elas, as ERK1/2 são frequentemente descritas
como alvos primários da ativação do RAGE entre as proteínas quinases intracelulares (YANG
et al., 2008) . A ERK1 e ERK 2 são moléculas que desempenham um papel importante na
cascata de sinalização intracelular, nas quais a fosforilação é induzida como parte da resposta
celular a vários estímulos (THOMAS; HUGANIR, 2004). Estudos recentes sobre retardo
mental humano sugerem que ERK1/2 desempenham um papel prejudicial na aprendizagem
(COSTA; SILVA, 2002).
Além disso, resultados tem mostrado que ERKs são envolvidas em diversas
formas de plasticidade sináptica no hipocampo e em outras áreas do cérebro que envolvem o
processo de aprendizagem e memória (THOMAS; HUGANIR, 2004) o que pode estar
associado aos nossos achados com aumento significativo da fosforilação de ERK 1/2 no
hipocampo.
Ainda, recentemente foi demonstrado que a ativação do RAGE induz
hiperfosforilação de tau pela ativação de GSK-3 em modelos animais e celulares, e o bloqueio
desse efeito em ratos restaura o comprometimento do potencial a longo prazo, a diminuição
de proteínas sinápticas e deterioração da memória (LI et al., 2010). Anormalmente tau
fosforilada é o primeiro passo na formação de emaranhados neurofibrilares, causando
deterioração neurítica e formando depósitos de proteínas hidrofóbicas anormais observadas
em AD e outras doenças neurodegenerativas. (LI et al., 2005)
O polimorfismo RAGE G82S (associado com afinidade de ligantes de RAGE
aumentado) também foi associado com o início precoce em DA, e com aumento significativo
na imunorreatividade de RAGE (LI et al., 2010). Além desta quantidade cada vez maior de
dados apontando para este receptor, como um dos principais mediadores da sinalização
neurotóxica no cérebro, também foi demonstrado um papel significativo para RAGE na
progressão da neurodegeneração, como esta proteína foi identificado para mediar o transporte
de Aβ através da BHE, sendo assim a principal causa de translocação de Aβ sistêmica para o
SNC (CANDELA et al., 2010).
Nesse sentido, por tudo o que foi mencionado até o momento, nossos achados
sugerem que o cérebro de sobreviventes de sepse é caracterizado por um aumento de
marcadores potenciais de neurodegeneração associados a alteração do receptor RAGE.

43
7 CONCLUSÕES
O hipocampo dos ratos sobreviventes de sepse apresentou níveis aumentados de
Aβ, α-sinucleína e tau-DA enquanto o córtex pré-frontal apresentou resultados aumentados
para α-sinucleína, ambas estruturas parecem estar associados a alterações de vias secundárias
a ativação de RAGE.

44
8 PERSPECTIVAS
Nosso propósito como continuidade para o estudo até agora apresentado, está
relacionado à investigação da ação de RAGE em sobreviventes de sepse sob modelo animal
de CLP. Portanto pretendemos verificar o dano cerebral a longo prazo em camundongos
knockot de RAGE além da ação de inibidores de RAGE em animais submetidos ao modelo de
sepse por CLP.
Essa abordagem nos proporcionará resultados mais refinados que poderão ser
potencialmente úteis para o desenvolvimento de novas estratégias terapêuticas para a sepse.

45
REFERÊNCIAS
AKIRA, S.; TAKEDA, K. Toll-like receptor signalling. Nat Rev Immunol, v. 4, p. 499-511,
2004.
AKIYAMA, H. et al. Inflammation and Alzheimer's disease. Neurobiol Aging, v. 21, p. 383-
421, 2000.
ALESHIN, A. et al. RAGE modulates myocardial injury consequent to LAD infarction via
impact on JNK and Stat signaling in a murine model. Am J Physiol Heart Circ Physiol, v.
294, p. H1823–H1832, 2008.
ALKIYAMA, H. et al. Inflammation and Alzheimer’s disease. Neurobiol Aging, v. 21, p.
383-421, 2000.
ALVES-FILHO, J. C. et al. Neutrophil function in severe sepsis. Endocr Metab Immune
Disord Drug Targets, v. 6, p. 151-8, 2006.
ALVES-FILHO, J. C. et al. The role neutrophils in severe sepsis. Shock, v. 1, p. 3-9, 2008.
ANGUS, D.; WAX, R. S. Epidemiology of sepsis: an update. Crit Care Med, v. 29, n. 7, p.
S109–S116, 2001.
ANNANE, D.; BELLISSANT, E.; CAVAILLON, J. Septic shock. The Lancet, v. 365, p. 63-
78, 2005.
ANTONELLI M. et al. Hemodynamic monitoring in shock and implications for management:
International Consensus Conference, Paris, France, 27-28 April 2006. Int Care Med, v. 33, p.
575-590, 2007.
APPELBERG, R. Neutrophils and intracellular pathogens: beyond phagocytosis and killing.
Trends Microbiol, v. 15, p. 87-92, 2007.
ARANCIO, O. et al. RAGE potentiates Abeta-induced perturbation of neuronal function in
transgenic mice. EMBO J, v. 23, p. 4096-4105, 2004.

46
AZMITIA, E.C. S100 beta and serotonin: a possible astrocytic-neuronal link to
neuropathology of Alzheimer’s disease. Prog Brain Res, v. 94, p. 459-473, 1992.
BARICHELLO, T. et al. Antioxidant treatment prevented late memory impairment in an
animal model of sepsis. Crit Care Med, v. 35, p. 2186-2190, 2007b.
BARICHELLO, T. et al. Behavioral deficits in sepsis-surviving rats induced by cecal ligation
and perforation. Braz J Med Biol Res, v. 40, n.6, p. 831-7, 2007a.
BARICHELLO, T. et al. Oxidative variables in the rat brain after sepsis induced by cecal
ligation and perforation. Crit Care Med, v. 34, p. 886-889, 2006.
BARON, R. M.; BARON, M. J.; PERRELLA, M. A. Pathobiology of sepsis: are we still
asking the same questions? Am J Respir Cell Mol Biol, v. 34, n. 2, p. 129-34, 2006.
BEISHUIZEN, A.; VERMES, I.; HAANEN, C. Endogenous mediators in sepsis and septic
shock. Adv Clin Chem, v. 33, p. 55-131, 1999.
BERGENDI, L. et al. Chemistry, physiology and pathology of free radicals. Life Sci, v. 65, p.
1865-74, 1999.
BLECK, T.P. et al. Neurologic complications of critical medical illnesses. Crit Care Med, v.
21, p. 98–103, 1993.
BONE, R.C. et al. Definitions for sepsis and organ failure and guidelines for the use of
innovative therapies in Sepsis. The ACCP-SCCM consensus conference on sepsis and organ
failure. Chest, v. 101, p. 1481-1483, 1992.
BONE, R.C. Immunologic dissonance: a continuing evolution in our understanding in our
understanding of the systemic inflammatory response syndrome (SIRS) and the multiple
organ dysfunction syndrome (MODS). Ann Intern Med, v. 125, n. 8, p. 680-7, 1996.
BONE, R.C. The pathogenesis of sepsis. Ann Intern Med, v. 115, p. 457-469, 1991.
BOOKE, M. et al. Cerebral blood flow is not altered in sheep qith Pseudomonas aeruginosa
sepsis treated with norepinephrine or nitric oxide synthase inhibit. Anesth Anlg, v. 96,
p.1122-8, 2003.

47
BOWIE, RA.; O’CONNOR, P.J.; MAHAJAN, R.P. Cerebrovascular reactivity to carbon
dioxide in sepse syndrome. Anaesthesia, v. 58, p. 261-79, 2003.
BOWTON, D.L. et al. Cerebral blood flow is reduced in patients with sepsis syndrome. Crit
Care Med, v. 17, p. 399–403, 1989.
BRADFORD, M.M. A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, v. 72, p.
248– 254, 1976.
BRETT, J. et al. Survey of the distribution of a newly characterized receptor for advanced
glycation end products in tissues. Am J Pathol, v. 143, p. 1699-1712, 1993.
BROWNLEE, M. Advanced Protein Glycosilation in Diabetes and Aging. Annu Rev Med, v.
46, p. 223-234, 1995.
BROWNLEE, M. Lilly Lecture 1993. Glycation and diabetic complications. Diabetes, v. 43,
p. 836-41, 1994.
CAMPBELL, S.L. et al. Increasing complexity of Ras signaling. Oncogene, v. 17, p. 1395-
1413, 1998.
CANDELA, P. et al. Apical-to-basolateral transport of amyloid-beta peptides through blood-
brain barrier cells is mediated by the receptor for advanced glycation end-products and is
restricted by P-glycoprotein. J Alzheimers Dis, v. 22, p. 849-859, 2010.
CANO, E.; MAHADEVAN, L.C. Parallel signal processing among mammalian MAPKs.
Trends Biochem Sci, v. 20, p. 117-122, 1995.
CASSOL JUNIOR, O.J. et al. Low dose dexamethasone reverses depressive-like parameters
and memory impairment in rats submitted to sepsis. Neurosci Lett, v. 473, p. 126-130, 2010.
CHANG, J. S. et al. Oxygen deprivation triggers upregulation of early growth response-1 by
the receptor for advanced glycation endproducts. Circ Res, v. 102, p. 905–913, 2008.
CHAVAKIS, T.; BIERHAUS, A.; NAWROTH, P. P. RAGE (receptor for advanced glycation
end products): a central player in the inflammatory response. Microbes Infect, v. 6, p. 1219–
1225, 2004.

48
CHISHTI, A. D. et al. Neutrophil chemotaxis and receptor expression in clinical septic shock.
Intensive Care Med, v. 30, p. 605-611, 2004.
CHROUSOS, G.P.; GOLD, P.W. The concepts of stress and stress system disorders.
Overview of physical and behavorial homeostasis. JAMA, v. 267, p. 1244-1252, 1992.
CHROUSOS, G. P. The hypothalamic-pituitary-adrenal-axis and immune-mediated
inflammation. N Engl J Med, v. 332, p. 1351-1362, 1995.
CINEL, I.; DELLINGER, R.P. Advances in pathogenesis and management of sepsis. Curr
Opin Infect Dis, v. 20, p. 345-352, 2007.
COHEN, J. The immunopathogenesis of sepsis. Nature, v. 420, n. 6917, p. 885-891, 2002.
COHEN, P. The search for physiological substrate of MAP and SAP kinases in mammalian
cells. Trends Biochem Sci, v. 7, p. 353-361, 1997.
COLLINS, T. Endothelial nuclear factor-kappa B and the initiation of the atherosclerotic
lesion. Lab Invest, v. 68, p. 499–508, 1993.
COMIM, C. M. et al. Effects of acute treatment with amphetamine in locomotor activity in
sepsis survivor rats. J Neuroimmunol, v. 212, p. 145-147, 2009a.
COMIM, C. M. et al. Rivastigmine reverses habituation memory impairment observed in
sepsis survivor rats. Shock, v. 32, p. 270-271, 2009b.
COMIM, C. M. et al. Alterations in inflammatory mediators, oxidative stress parameters and
energetic metabolism in the brain of sepsis survivor rats. Neurochem Res, v. 36, n.2, p. 304-
11, 2011.
COSTA, R. M.; SILVA, A. J. Molecular and cellular mechanisms underlying the cognitive
deficits associated with neurofibromatosis 1. J Child Neurol, v. 17, n. 8, p. 622-651. 2002.
CRAS, P. et al. Neuronal and microglial involvement in beta-amyloid protein deposition in
Alzheimer's disease. Am J Pathol, v. 137, p. 241-246, 1990.

49
CREAGH-BROWN, B. C. et al. The RAGE axis in systemic inflammation, acute lung injury
and myocardial dysfunction: an important therapeutic target? Intensive Care Med, v. 36, p.
1644-1656, 2010.
CREAGH-BROWN, B. C.; QUINLAN, G. J.; EVANS, T. W. RAGE inhibition: healthy or
harmful? Crit Care Med, v. 38, p. 1487-1490, 2010.
DAL-PIZZOL, F. et al. Oxidative Mechanisms of Brain Dysfunction During Sepsis.
Neurochem Res, v. 35, p. 1–12, 2010.
DAVIS, R.J. Signal transduction by the JNK group of MAP kinases. Cell, v. 103, p. 239-252,
2000.
DE MAIO, A.; TORRES, M.B.; REEVES, R.H. Genetic determinants influencing the
response to injury, inflammation, and sepsis. Shock, v. 23, p. 11-17, 2005.
DE SOUZA, C.T. et al. Consumption of a fat-rich diet activates a proinflammatory response
and induces insulin resistance in the hypothalamus. Endocrinology, v. 146, p. 4189–4191,
2005.
DELANTY, N.; DICHTER, M. A. Oxidative injury in the nervous system. Acta Neurol
Scand Sep, v. 98, p. 145-53, 1998.
DELLINGER. R.P. et al. Surviving Sepsis Campaing: international guidelines for
management of severe sepsis and septic shock. Crit Care Med, v. 36, p. 296-327, 2008.
DESCAMPS, L. et al. Protective effect of glial cells against lipopolysaccharide-mediated
blood barrier injury. Glia, v. 42, p. 46–58, 2003.
DOMBROVSKIY, V.Y. et al. Rapid increase in hospitalization and mortality rates for severe
sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med, v. 35, p.
1244-1250, 2007.
DONATO, R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type
with intracellular and extracellular functional roles. Int J Biochem Cell Biol, v. 33, p. 637-
668, 2001.
DU YAN, S. et al. Amyloid-beta peptide-receptor for advanced glycation endproduct
interaction elicits neuronal expression of macrophage-colony stimulating factor: a

50
proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci USA, v. 94, p. 5296-
5301; 1997.
EBERSOLDT, M.; SHARSHAR, T.; ANNANE, D. Sepsis-associated delirium. Int Care
Med, v. 33, p. 941–950, 2007.
ERNST, A. et al. Procalcitonin is elevated in the cerebrospinal fluid of patients with dementia
and acute neuroinflammation. J Neuroimmunol, v. 189, p. 169-174, 2007.
FAGIOLO, U. et al. Increased cytokine production in mononuclear cells of healthy elderly
people. Eur J Immunol, v. 23, p. 2375-2378, 1993.
FAURSCHOU, M.; BORREGAARD, N. Neutrophil granules and secretory vesicles in
inflammation. Microbes Infect, v. 5, n. 14, p. 1317-27, 2003.
FENG, J. Microtubule: a common target for parkin and Parkinson’s disease toxins.
Neuroscientist, v. 12, p. 469-476, 2006.
FERNANDES V.J. Avaliação dos critérios de definição de sepse baseados no center for
diseases control na unidade de terapia intensiva de adultos do hospital de clínicas da
universidade federal de Uberlândia. Hor Ci, v. 1, p. 1-14, 2008.
FRIEDMAN, G.; SILVA, E.; VICENT, J.L. Has the mortality of septic shock change with
time? Crit Care Med, v. 26, p. 2078-2086, 1998.
FULOP, T. et al. Signal transduction and functional changes in neutrophils with aging. Aging
Cell, v. 3, p. 217-226, 2004.
GALLIN, J. I.; SNYDERMAN, R. Overview, in Inflammation: Basic Principles and Clinical
Correlates. Lippincott Willians & Wilkins, 1999.
GODEIRO JUNIOR, C. O. et al. Mitocôndria e doença de Parkinson: contribuições da
genética no conhecimento do processo patogênico. Einstein, v. 5, n. 2, p. 177-181, 2007.
GOLDE, T. Inflammation takes on Alzheimer’s disease. Nat Med, v. 8, p. 936-938. 2002.
GOOD, P. F. et al. Evidence of neuronal oxidative damage in Alzheimer's disease. Am J
Pathol, v. 149, p. 21-28, 1996.

51
GORDON, S. M. et al. Clinical identification of cognitive impairment in ICU survivors:
insights for intensivists. Intensive Care Med, v. 30, p. 1997-2008, 2004.
GRIFFIN, S.W.T.; SHENG, J.G.; MRAK, R.E. Inflamatory pathways. Implications. In:
EASCO, W.; TANZI R.E. (Edts). Alzheimer’s disease: molecular mechanism of dementia,
human press. Totowa: NJ, 1996. p. 169-176.
HAAPASALO, A. et al. Emerging role of Alzheimer's disease-associated ubiquilin-1 in
protein aggregation. Biochem Soc Trans, v. 38, p. 150-155, 2010.
HAASS, C.; SELKOE, D.J. Soluble protein oligomers in neurodegeneration: lessons from the
Alzheimer’s amyloid β-peptide. Nature, v. 8, p. 101-112, 2007.
HAASS; C. Take five--BACE and the gamma-secretase quartet conduct Alzheimer's amyloid
beta-peptide generation. EMBO J, v. 23, p. 483-488, 2004.
HALLIWELL, B.; GUTTERIDGE, J. M. C. Free Radicals in Biology and Medicine.
Oxford: Oxford University Press, 614-677, 2007.
HALLIWELL, B. Reactive species and antioxidants. Redox biology is a fundamental theme
of aerobic life. Plant Physiol, v. 141, p. 312-22, 2006.
HALLIWELL, B. Role of free radicals in the neurodegenerative diseases: therapeutic
implications for antioxidant treatment. Drugs Aging, v. 18, p. 685-716, 2001.
HALLIWELL, B.; WHITEMAN, M. Measuring reactive species and oxidative damage in
vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol,
v. 142, p. 231-55, 2004.
HAMPTON, M.B.; KETTLE, A.J.; WINTERBOURN, C.C. Inside the neutrophil
phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood, v. 92, p. 3007–3017,
1998.
HARDY, J.; SELKOE, D.J. The amyloid hypothesis of Alzheimer’s disease: progress and
problems on the Road to therapeutic. Science, v. 297, p. 353-356, 2002.
HATANAKA, E. et al. Serum amyloid A induced mRNA expression and release of tumor
necrosis factor-alpha (TNFα) in human neutrophils. Immunol Lett. v. 91, n.1, p. 33-7, 2004.

52
HATTORI, Y. et al. Insigths Into Sepsis Therapeutic Design Based on the Apoptotic Death
Pathway. J Pharmacol Sci, v. 114, p. 354-365, 2010
HAWKINS, B. T.; DAVIS, T. P. The blood brain barrier/neurovascular unit in health and
disease. Pharmacol Rev, v. 57, p. 173–85, 2005
HLAVANDA, E. et al. Brain-specific p25 protein binds to tubulin and microtubules and
induces aberrant microtubule assemblies at substoichimetric concentrations. Biochemistry, v.
41, p. 8657-8664, 2002.
HOFMANN, M. A. et al. RAGE mediates a novel proinflammatory axis: a central cell surface
receptor for S100/calgranulin polypeptides. Cell, v. 97, p. 889-901, 1999.
HOLMES, C. et al. Systemic inflammation and disease progression in Alzheimer disease.
Neurology, v. 73, p. 768-774, 2009.
HONDA, K. et al. Role of a transductional-transcriptional processor complex involving
MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natt Acad Sci USA, v. 101, n. 43, p.
15416-21, 2004.
HOPKINS, P. A.; SRISKANDA, S. Mammalian Toll-like receptors: to immunity and beyond.
Clin Exp Immunol, v. 140, p. 395-407, 2005.
HOPKINS, R.O. et al. Neuropsychological sequelae and impaired health status in survivors of
severe acute respiratory distress syndrome. Am J Respir Crit Care Med, v. 160, p. 50–56,
1999.
HOTCHKISS, R.S.; KARL, I.E. The pathophysiology and treatment of sepsis. New Engl J
Med, v. 348, p. 138-150, 2003.
HOUGH, C.L.; CURTIS, J.R. Long-term sequelae of critical illness: memories and health-
related quality of life. Crit Care, v. 9, p. 145-146, 2005.
HU, J.; FERREIRA, A.; VAN ELDIK, L.J. S100β induces neuronal cell death through nitric
oxide release from astrocytes. J Neurochem, n. 69, p. 2294-2301, 1997.
HU, J.; VAN ELDIK, L.J. S100β induces apoptotic cell death in cultured astrocytes via a
nitric oxide-dependet pathway. Biochim Biophys Acta, n. 13, n. 13, p. 239-245, 1996.

53
HUDSON, B. I. et al. Glycation and diabetes: The RAGE connection. Current Science, v.
83, p. 1515-1520, 2002.
HUTTENLOCHER, A.; SANDBORG, R. R.; HORVITZ, A. F. Adhesion in cell migration.
Curr Opin Cell Biol, v. 7, n. 5, p. 697-706, 1995.
JACKSON, A.C. et al. The encephalopathy of sepsis. Can J Neurol Sci, v. 12, p. 303–307,
1985.
JANEWAY, C. A. JUNIOR. How the immune system protects the host from infection.
Microbes Infect, v. 3, n. 13, p. 1167-71, 2001.
JANEWAY, C. A. JUNIOR. Medzhitov R. Innate immune recognition. Annu Rev
Immunol, v. 20, p. 197-216, 2002.
KARLSSON, S. et al. HMGB1 as a predictor of organ dysfunction and outcome in patients
with severe sepsis. Intensive Care Med, v. 34, n. 6, p. 1046-53, 2008.
KITADA, T. Mutations in parkin gene cause autossomal recessive juvenile parkinsonism.
Nature, v. 392, n. 6676, p. 605-8, 1998.
KUNG H.C. et al. Deaths: final data for 2005. Natl Vital Stat Rep, v. 56, p. 1–120, 2008.
KYRIAKIS, J.; AVRUCH, J. Mammalian mitogen-activated protein kinase signal
transduction pathways activated by stress and inflammation. Physiol Rev, v. 81, p. 807–869,
2001.
LAEMMLI, U.K. Cleavage of structural proteins during the assembly of the of bacteriophage
T4. Nature, v. 227, p. 680–685, 1970.
LAFERLA, F.M.; GREEN, K. N.; ODDO, S. Intracellular amyloid-β in Alzheimer’s disease.
Nat Rev Neurosci, v. 8, p. 499-509, 2007.
LATIM AMERICAN SEPSIS INSTITUTE. Campanha sobrevivendo a sepse. Disponível em:
<http://www.sepsisnet.org/PDF/Relatorio%20Nacional%20SSC.pdf>. Acesso em: 15 out.
2011.

54
LECLERC, E. et al. Crosstalk between calcium, amyloid beta and the receptor for advanced
glycation endproducts in Alzheimer's disease. Rev Neurosci, v. 20, p. 95-110, 2009.
LEE, H-J. et al. Impairment of microtubule-dependent trafficking by overexpression of α-
sinuclein. Eur J Neurosci, v. 24, p. 3153-3162, 2006.
LI, K. et al. Association between the RAGE G82S polymorphism and Alzheimer's disease. J
Neural Transm, v. 117, p. 97-104, 2010.
LI, X.C. et al. Effect of melatonin on calyculin A-induced tau hyperphosphorylation. Eur J
Pharmacol, v. 510, p. 25-30, 2005.
LIEW, F. et al. Negative regulation of toll-like receptor-mediated immune responses. Nat
Rev Immunol, v. 5, n. 6, p. 446-58, 2005.
LIN, H. B. et al. Memory deficits and neurochemical changes induced by C-reactive protein
in rats: implication in Alzheimer's disease. Psychopharmacology, v. 204, p.705-714, 2009.
LIPCSEY, M. et al. The brain is a source of S100B increase during endotoxemia in the pig.
Anesth Analg, v. 110, p.174-180, 2010.
LORENZ, E. et al. A novel polymorphism in the toll-like receptor 2 gene and its potential
association with staphylococcal infection. Infect Immun, v. 68, p. 6398-401, 2000.
MARSHALL, J.C. Neutrophils in the pathogenesis of sepsis. Crit Care Med, v. 33, p. S502-
S505, 2005.
MARTIN, G. S; MANNINO, D. M; MOSS, M. The effect of age on the development and
outcome of adult sepsis. Crit Care Med, v. 34, p. 15–21, 2006.
MATTSON, M. P.; CHAN, S. L. Neuronal and calcium signalin in Alzheimer’s disease. Cell
Calcium, v. 34, p. 385-397, 2003.
MATTSON, M.P. Pathways towards and away from Alzheimer's disease. Nature, v. 430, p.
631-639, 2004.
MAXWELL, S.R. Prospects for the use of antioxidant therapies. Drugs, v. 49, p. 345-61,
1995.

55
MENG, G. et al. Antagonistic antibody prevents toll-like receptor-2-driven lethal shock-like
syndromes. J Clin Invest, v. 113 p. 1473-1481, 2004.
MESSARIS, E.; MEMOS, N.; CHATZIGIANNI, E. Time-dependent mitochondrial-mediated
programmed neuronal cell death prolongs survival in sepsis. Crit Care Med, v. 32, p. 1764–
1770, 2004.
MINORETTI, P. e al. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism
on susceptibility to late-onset Alzheimer's disease. Neurosci Lett, v. 391, p. 147-149, 2006.
MUNFORD, R. S.; PUGIN, J. Normal responses to injury prevent systemic inflammation and
can be immunosuppressive. AM J Respir Crit Care Med, 163, 316–321, 2001.
NAGY Z. et al. Relative roles of plaques and tangles in the dementia of Alzheimer’s disease:
correlations using three sets of neuropathological criteria. Dementia, v. 6, p. 21-31. 1995.
NATHAN, C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol,
v. 6, n. 3, p. 173-82, 2006.
NATHENS, A.B.; MARSHALL, J.C. Sepsis, SIRS, and MODS: wat´s in a name? World J
Surg, v. 20, p. 386-391, 1996.
NISHIKAWA, T.; EDELSTEIN, D.; BROWNLEE, M. The missing link: a single unifying
mechanism for diabetic complications. Kidney Int Suppl, v. 77, p. S26-30, 2000.
NUNOMURA, A. et al. Oxidative damage is the earliest event in Alzheimer disease. J
Neuropathol Exp Neurol, v. 60, p. 759-767, 2001.
O'BRIEN, J. M; ALINA, JUNIOR; ABEREGG, S. K. Sepsis. Am J Med, v. 120, p. 1012-22,
2007.
ORIGLIA, N. et al. MAPK, beta-amyloid and synaptic dysfunction: the role of RAGE.
Expert Rev Neurother, v. 9, n. 11, p. 1635-45, 2009.
PAPADOPOULOS, M.C. et al. Pathophysiology of septic encephalopathy: a review. Crit
Care Med, v. 28, p. 3019-3024, 2000.

56
POLTORAK, A. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice:
mutations in Tlr4 gene. Science, v. 282, n. 5396, p. 2085-8, 1998.
POLYMEROPOULOS, M. H. et al. Mutation in the alfa-synuclein gene identified in families
with Parkinson’s disease. Science, v. 276, n. 5321, p. 2045-7, 1997.
PRADO, W. A.; PONTES, R. M. Presurgical ketoprofen, but not morphine, dipyrone,
diclofenac or tenoxicam, preempts post-incisional mechanical allodynia in rats. Braz J Med
Biol Res, v. 35, p. 111-119, 2002.
REMICK, D.G. Pathophysiology of sepsis. Am J Pathol, v. 170, p. 1435-1444, 2007.
REN, Y. et al. Parkin protects dopaminergic neurons against microtubule-depolymerizing
toxins by attenuating microtubule-associated protein kinase activation. J Biol Chem, v. 6, n.
284, p. 4009-4017, 2009.
RITTER, C. et al. Drug intervention trials in sepsis. Lancet, p. 364-498, 2004.
RITTIRSCH, D.; FLIERL, M. A.; WARD, P.A. Harmful molecular mechanisms in sepsis
Nat Rev Immunol, v.10, p. 776-87, 2008.
ROCHA, M.J.A.; OLIVEIRA, G.R.; FARIAS-CORRÊA, P.B. Neurohypophyseal homone
secretion during septic shock. In: CHEN, F.J. (Ed.), New Trends in Brain Research. New
York: Nova Science Publishers, 2006. p. 75-94.
ROJAS, H., RITTER, C., DAL PIZZOL, F. Mecanismos de disfunção da barreira
hematoencefálica no paciente criticamente enfermo: ênfase no papel das metaloproteinases de
matriz. Ver Bras Ter Intensiva, v. 23, n. 2, p. 222-227, 2011.
SALVEMINI, D; CUZZOCREA, S. Oxidative stress in sepstic shock and disseminated
intravascular coagulation. Free Radic Biol Med, v. 33, p. 1173-1185, 2002.
SAPER, C. B; BREDER, C. D. The neurologic basis of fever. N Engl J Med, v. 330, p. 1880-
1886, 1994.
SCHMIDT, A. M.; STERN, D. M. RAGE: A new target for the prevention and treatment of
the vascular and inflammatory complications of diabetes. Trends in Endocrinology and
Metabolism, v. 11, p. 368-375, 2000.

57
SEELY, A.J.; PASCUAL, J. L.; CHRISTOU, N.V. Science review: Cell membrane
expression (connectivity) regulates neutrophil delivery, function and clearance. Crit Care, v.
7, p. 291-307, 2003.
SELKOE, D. J. Alzheimer's disease: a central role for amyloid. Journal of Neuropathology
and Experimental Neurology, v. 53, p. 438-447, 1994.
SEMMLER, A. et al. Long-term cognitive impairment, neuronal loss and reduced cortical
cholinergic innervation after recovery from sepsis in a rodent model. Exp Neurol, v. 204, p.
733-740, 2007.
SEMMLER, A. et al. Systemic inflammation induces apoptosis with variable vulnerability of
different brain regions. J Chem Neuroanat, v. 30, n. 2-3, p. 144-57, 2005.
SHARSHAR, T. et al. The neuropathology of septic shock. Brain Pathol, v. 14, p. 21–23,
2004.
SILVA, E. et al. Brazilian Sepsis Epidemiological Study (BASES Study). Crit Care Med, v.
8, p. R251-R260, 2004.
SOUTHORN, P.A.; POWIS, G. Free radicals in medicine. I. Chemical nature and biologic
reactions. Mayo Clin Proc, v. 63, p. 381-389, 1988.
SPRUNG, C.L. et al. Impact of encephalopathy on mortality in the sepsis syndrome. The
Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med, v. 18, p.
801–806, 1990.
SPYER, K. M. Neural mechanisms involved in cardiovascular control during affective
behavior. Trends Neurosci, v. 12, p. 506-513, 1989.
STAMATOVIC, S. M.; KEEP, R. F.; ANDJELKOVIC, A. V. Brain endothelial cell-cell
junctions: how to “open” the blood brain barrier. Curr Neuropharmacol, v. 6, n. 3, p. 179-
92, 2008.
TAGUCHI, A. et al. Blockade of RAGE-amphoterin signaling suppresses tumor growth and
metastases. Nature, v. 405, p. 354–360, 2000.
TAN, J.M.; WONG, E.S.; LIM, K.L. Protein misfolding and aggregation in Parkinson's
disease. Antioxid Redox Signal, v. 11, p. 2119-2134, 2009.

58
THOMAS, G. M.; HUGANIR, R. L. MAPK cascade signalling and synaptic plasticity. Nat.
Rev. Neurosci, v. 5, p. 173-183, 2004.
TILLEMENT, J.P.; LECANU, L.; PAPADOPOULOS, V. Amyloidosis and
neurodegenerative diseases: current treatments and new pharmacological options.
Pharmacology, v. 85, p. 1-17, 2010.
TRIANTAFILOU, M.; TRIANTAFILOU, K. Lipopolysaccharide recognition: CD14, TLRs
and the LPS-activation cluster. Trends Immunol, v. 23, p. 301-304, 2002.
TUON, L. et al. Memory-enhancing treatments reverse the impairment of inhibitory
avoidance retention in sepsis-surviving rats. Crit Care, v. 12, p. 133, 2008.
TUREEN, J. Effect of recombinant human tumor necrosis factor-alpha on cerebral oxygen
uptake, cerebrospinal fluid lactate, and cerebral blood flow in the rabbit: role of nitric oxide. J
Clin Invest, v. 95, p. 1086–1091, 1995.
VALCOUR, V.; PAUL, R. HIV infection and dementia in older adults. Clin Infect Dis, v. 42,
p. 1449-1454, 2006.
VICENT, J. L.; KORKUT, H.Á. Defining sepsis. Clin Chest Med, v. 29, p. 589-590, 2008.
VINCENT J. L. et al. Sepsis in European intensive care units: results of the SOAP study. Crit
Care Med, v. 34, p. 344–353, 2006.
WARREN, H.S. Strategies for the treatmente of sepsis. N Engl J Med, v. 336, p. 952-953,
1997.
WEBERPALS, M. et al. NOS2 gene deficiency protects from sepsis-induced long-term
cognitive deficits. J Neurosci, v. 29, p. 14177-14184, 2009.
WEIGHARDT, H.; HOLZMANN, B. Role of Toll-like receptor responses for sepsis
pathogenesis. Immunobiology, v. 212, p. 715-722, 2007.
WEINSTEIN-OPPENHEIMER, C.R. et al. The Raf signal transduction cascade as a target for
chemotherapeutic intervention in growth factor-responsive tumors. Pharmacol Ther, v. 88,
p. 22-279, 2000.

59
WEISS, N. et al. The bloodbrain barrier in brain homeostasis and neurological diseases.
Biochim Biophys Acta, v. 1788, p.842-57, 2009.
WINTERS, B.D. et al. Long-term mortality and quality of life in sepsis: a systematic review.
Crit Care Med, v. 38, p. 1276-1283, 2010.
YAMAMOTO, M. et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor
signaling pathway. Science, v. 301, n. 5633, p. 640-3, 2003.
YAN, S. D. et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease.
Nature, v. 382, p. 674-5, 1996.
YANG, S. et al. Common and distinct mechanisms of different redox-active carcinogens
involved in the transformation of mouse JB6P+ cells. Mol Carcinog, v. 47, p. 485-491, 2008.
YOUNG, G.B. et al. The encephalopathy associated with septic illness. Clin Invest Med, v.
13, p. 297–304, 1990.
ZIMMER, D. B. et al. The S100 protein family: history, function, and expression. Brain Res
Bull, v. 37, p. 417-429, 1995.
ZIMMERMANN, J. J. Defining the role of oxyradicals in the pathogenesis of sepsis. Crit
Care Med, v. 23, p. 616-617, 1995.
ZLOKOVIC, B. V. The blood-brain barrier in health and chronic neurodegenerative
disorders. Neuron, v. 57, p.178-201, 2008.

60
APÊNDICE

61
APÊNDICE A – Artigo da Dissertação
Brain markers of neurodegeneration in sepsis survivor rats
Samantha Pereira Miguel BSc; Matheus Pasquali MSc; Daniel Pens Gelain PhD; Francieli
Vuolo BSc, João Quevedo MD, PhD; José Cláudio Fonseca Moreira PhD Felipe Dal-Pizzol
MD, PhD; Fabricia Petronilho PhD
Artigo científico a submetido para publicação ao periódico
SHOCK: Injury, Inflammation, and Sepsis: Laboratory and Clinical Approaches

62
Brain markers of neurodegeneration in sepsis survivor rats
Samantha Pereira Miguel BSc1; Matheus Pasquali MSc
2; Daniel Pens Gelain PhD
2; Francieli
Vuolo BSc3, João Quevedo MD, PhD
4; José Cláudio Fonseca Moreira PhD
2 Felipe Dal-Pizzol
MD, PhD3; Fabricia Petronilho PhD
1
1 Postgraduate Program in Health Sciences, University of Southern Santa Catarina, Tubarão,
Santa Catarina, Brazil.
2Center of Oxidative Stress Research, Professor Tuiskon Dick Department of Biochemistry,
Institute of Health Basic Sciences, Federal University of the Rio Grande do Sul (UFRGS),
Porto Alegre, Rio Grande do Sul, Brazil.
3Experimental Physiopathology Laboratory and National Institute of Translational Science
and Technology in Medicine, Postgraduate Program in Health Sciences, University of the
Extreme-South Catarinense, Criciúma, Santa Catarina, Brazil.
4Laboratory of Neurosciences and National Institute of Translational Science and Technology
in Medicine, Postgraduate Program in Health Sciences, University of the Extreme-South
Catarinense, Criciúma, Santa Catarina, Brazil.
Address requests for reprints to: Prof. Dra. Fabricia Petronilho. Programa de Pós-graduaçao
em Ciências da Saúde, Universidade do Sul de Santa Catarina, Avenida José Acácio Moreira,
787, Bairro Dehon, Tubarão, SC, Brazil, 88704-900. E-mail address:
[email protected]. Phone / fax: 55 48 3621-3363
Running head: Neurodegeneration and sepsis

63
Abstract
Survivors from sepsis presented cognitive deficits associated with decreased quality of
life and increased long-term morbidity. Some of these alterations resembled the
pathophysiological mechanisms of neurodegenerative diseases. The receptor for advanced
glycation (RAGE) has been increasingly implicated in the progression of neuronal death in
many neurodegenerative diseases, such as Alzheimer’s Disease (AD), and is also an important
pro-inflammatory mediator. For this reason, we analyzed biochemical parameters related to
neurodegeneration in rats that survived sepsis, and their possible involvement with RAGE.
Rats were subjected to sepsis by cecal ligation and puncture (CLP), and thirty days after
surgery the hippocampus and pre-frontal cortex have been isolated. The immunocontent of β-
amyloid peptide (Aβ), α-synuclein and AD-like tau were analyzed by western blot. Besides,
we analyzed the immunocontent of RAGE, the activation state of its downstream signaling
kinase ERK 1/2 and the presence of the RAGE agonist S100B. Alpha-synuclein, Aβ and AD-
like tau were increased in the hippocampus, but not in the pre-frontal cortex. RAGE was
upregulated in both structures, but activated ERK 1/2 was observed only in hippocampus. The
immunocontent of S100B was not different from control at both structures. In conclusion,
brain from sepsis survivor animals presented several markers of neurodegeneration suggesting
that the RAGE-MAPK pathway could be responsible to long-term cognitive deficits in sepsis
survivors.
KEY WORDS: sepsis; neurodegeneration; RAGE; brain dysfunction; long-term deficits

64
INTRODUCTION
The brain may be one of the first organs affected in sepsis. In fact, sepsis-associated
delirium is a frequent feature associated with increased morbidity and mortality [1]. Recently,
cognitive deficits associated with decreased quality of life and increased long-term morbidity
were observed in sepsis survivors [2]. Despite its clinical impact, the pathophysiology of
central nervous system (CNS) dysfunction during sepsis remains poorly understood.
In animal models, acute encephalopathy is observed in the course of sepsis [3], and
survivors were described to present long-term cognitive impairment [4]. Such models
represent an important tool to study the mechanisms associated with CNS dysfunction during
and after sepsis. Central cholinergic deficiency is the leading hypothesized mechanism for
delirium [5], a finding that overlaps the pathophysiological mechanisms for delirium,
Alzheimer’s Disease (AD) and neurodegeneration [6]. Thus, delirium and dementia may
represent different points along a continuum of cognitive disorders. Recently, Semmler et al.
demonstrated neuronal loss in hippocampal subregions and in the prefrontal cortex, and
reduced cholinergic innervation of postrolandic cortical areas in sepsis survivor animals - a
feature similar to that observed in AD patients [7]. This similarity was also demonstrated by
our group using rivastigmine to reverse long-term cognitive deficits in sepsis survivor animals
[8].
Neurodegenerative disorders share common mechanisms directly associated to the
pathological aggregation of misfolded proteins. These accumulate as fibrillar amyloid
deposits in selectively vulnerable regions of the CNS [9]. In AD, abnormally phosphorylated
tau proteins form neurofibrillary tangles, and beta-amyloid peptide (Aβ) accumulates in the
extracellular environment and forms amyloid plaques, which cause neuronal cell death as

65
consequence of hydrophobic misfolded protein aggregation [10]. Beta-amyloid peptide is also
able to trigger the activation of the receptor for advanced glycation endproducts (RAGE).
RAGE activation induces the phosphorylation of MAP kinases (MAPKs), stimulates the NF-
kB-dependent production of pro-inflammatory cytokines and enhances the production of
reactive oxygen species by NADPH oxidase [11]. These are all important cellular events in
the progression of neurodegeneration [12]. Furthermore, RAGE may be activated by
HMGB1, which is increased in extracellular fluids during sepsis, and also by S100B, which is
a relevant protein to brain dysfunction [13].
These evidences point to a major role of RAGE in the mechanisms underlying the pro-
inflammatory-neurocytotoxic axis in both sepsis-induced encephalopathy and some
neurodegenereative diseases. Thus, the aim of the present work was to study the presence of
biochemical markers of neurodegeneration in the brain of sepsis survivor rats and their
possible relationship with the regulation of RAGE immunocontent.
MATERIALS AND METHODS
Animals
Adult male Wistar rats (220 to 300 g) were obtained from our breeding colony. They
were housed five to a cage with food and water available ad libitum and were maintained on a
12-hour light/dark cycle (lights on at 7 a.m.). All experimental procedures involving animals
were performed in accordance with the National Institutes of Health Guide for the Care and
Use of Laboratory Animals and approved by the local Ethic Board in Research.
Cecal ligation and perforation (CLP) surgery

66
Animals were subjected to CLP as described [14] with adaptations [15]. Briefly, rats
were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg) given
intraperitoneally. Under aseptic conditions, a 3-cm midline laparotomy was performed to
allow exposure of the cecum with the adjoining intestine. The cecum was tightly ligated with
a 3.0 silk suture at its base, below the ileocecal valve, and was perforated once with a 14-
gauge needle. The cecum was then gently squeezed to extrude a small amount of feces from
the perforation site returned to the peritoneal cavity, and the laparotomy was closed with 4.0
silk sutures. Animals were resuscitated with normal saline (50 mL/kg subcutaneously)
immediately and 12 hours after CLP. All animals were returned to their cages with free access
to food and water. In the sham-operated group, the rats were submitted to all surgical
procedures but the cecum was neither ligated nor perforated. After surgery, animals received
30 mg/kg ceftriaxone and 25 mg/kg clindamycin subcutaneously every 6 hours for a total of 3
days. Survival rates were 100% in the sham group and 43% in the sepsis group, which were in
accordance with our previous reports [15]. Animals were randomly distributed to sham and
CLP groups. Thirty days after surgery the animals were killed, and hippocampus and pre-
frontal cortex isolated. We choose this period of time since we previously demonstrated
several cognitive dysfunctions in sepsis survival animals 30 days after CLP [4, 16-19].
Western Blot Analyses
Hippocampus and frontal cortex samples were homogenized in Laemmli-sample
buffer (62.5mM Tris-HCl, pH 6.8, 1% (w/v) SDS, 10% (v/v) glycerol) and equal amounts of
cell protein (30 µg/well) were fractionated by SDS-PAGE and electro-blotted onto
nitrocellulose membranes. Protein loading and electro-blotting efficiency were verified
through Ponceau S staining, and the membrane was blocked in Tween-Tris buffered saline
(TTBS: 100mM Tris-HCl, pH 7.5, containing 0.9% NaCl and 0.1% Tween-20) containing 5%

67
albumin. Membranes were incubated overnight at 4ºC with primary antibody diluted at
1:1000 in TTBS (1:1000) (anti-RAGE, R5278; anti-Alzheimer’s disease isoform tau, A8855;
were purchased from Sigma; anti-S100B, ab41548; anti-α-synuclein, ab6162; were purchased
from Abcam; anti-Aβ, No 2454; anti-ERK1/2, No 4376 and anti-phospho-ERK1/2 N. 4370,
were purchased from Cell Signaling Technology) and then washed with TTBS. Anti-IgG
from mouse or rabbit (according the species that originated the primary antibody) linked to a
peroxidase was then incubated with the membrane for additional 2 h at room temperature
(1:5000 dilution range), the membrane was washed again and the immunoreactivity was
detected by enhanced chemiluminescence using Pierce WestPico Chemiluminescence kit.
Densitometric analysis of the films was performed with ImageQuant software. Blots were
developed to be linear in the range used for densitometry.
Statistical analyses
The results are expressed as means ± SE in all figures. Differences among groups were
compared by t-test. All statistical analysis were performed with SPSS® 12.0 for Windows and
statistical significance was assigned to P<0.05.
RESULTS
After 30 days of sepsis induction by CLP, we analyzed the immunocontent of different
biochemical markers of neurodegeneration in the hippocampus and prefrontal cortex of septic
animals. Compared to sham animals, the immunocontent of both Aβ and α-synuclein were
significantly increased in the hippocampus (Fig. 1A). Using an antibody developed to detect
the isoform of tau predominant in brain tissues of Alzheimer’s disease patients, we observed
that the immunocontent of this pathogenic tau (“AD-like tau”) was also enhanced (Fig. 1A).

68
In the prefrontal cortex, only α-synuclein immunocontent was increased (Fig. 1B). Since these
modifications are commonly associated to neurodegenerative conditions and/or acute
neurotoxic processes, this result indicates a persistent biochemical dysfunction in brain. These
changes are also temporally correlated with the occurrence of cognitive deficits in tasks
dependent on hippocampus circuits [20], suggesting a relationship between these two
phenomena. Since Aβ plays a central role in cellular neurotoxic events related to
neurodegenerative disorders such as AD, we also investigated a pathway related to Aβ
neurotoxicity in these animals.
Overexpression and activation of RAGE in CNS have been increasingly implicated in
the progression of neurodegeneration observed in AD and other neurodegenerative disorders
[11]. The neurotoxicity related to RAGE signaling in brain is believed to be related both to
mechanisms that enhance RAGE expression and/or the production and secretion of RAGE
ligands, such as Aβ, S100B and AGEs [21]. Once activated by its ligands, RAGE induces the
phosphorylation of MAPKs such as ERK1/2, which triggers a phosphorylation cascade that
results in increased expression of pro-inflammatory cytokines and NADPH oxidase [11].
These events, in turn, are described to induce free radical production, recruitment and
activation of microglia which enhance Aβ production and tau hyperphosphorylation, thus
leading to a positive feedback cycle of neurotoxicity. We then analyzed the immunocontent of
RAGE and the phosphorylation state (i.e. activation) of ERK1/2 in the hippocampus and pre-
frontal cortex of septic animals 30 days of CLP. We also analyzed the immunocontent of
S100B, which is another RAGE ligand and was previously demonstrated to be overexpressed
in the acute phase of sepsis development [22]. Western blot analysis of the same samples used
to determine Aβ, α-synuclein and phosphorylated tau showed increased RAGE expression
and ERK1/2 activation in hippocampus of septic animals (Fig 2A). The immunocontent of
RAGE was also increased in pre-frontal cortex, but there were no modifications in the

69
activation state of ERK1/2 (Fig. 2B). Finally, the immunocontent of S100B in septic animals
was not different from sham group in both structures (Fig. 2A and B).
DISCUSSION
The findings of the present study suggest, for the first time, that the acute systemic
inflammatory stimulus is associated to the appearance of biochemical markers of
neurodegeneration in the brain that resembles several kinds of age-related brain diseases, such
as Aβ, AD-like tau, α-synuclein and RAGE. We propose that the activation of Aβ-RAGE-
ERK1/2 pathway could be important to the long-term cognitive impairment observed in this
animal model, and it also may be implicated in the persistent cognitive deficits observed in
some patients that recover from sepsis.
There is increasing evidence that inflammation and neurodegeneration are related
events. Procalcitonin (PCT) in cerebrospinal fluid was found to be significantly increased in
patients with AD, vascular dementia, dementia with Lewy bodies and frontotemporal
dementia in pattern similar to that observed in acute neuroinflammatory states, such as in
encephalitis and meningitis [23]. This indicates that central inflammation could have a role in
the development of chronic neurodegenerative diseases. Neurodegeneration is also related to
systemic inflammation, which is exemplified by HIV infection. The pathology typically
attributed to AD has now been reported in HIV+ patients, including increased brain Aβ
deposition, extracellular amyloid plaques formation and decreased CSF Aβ levels [24] .
Parkinson disease may be another particularly potential disease for older HIV+ patients, given
the importance of the basal ganglia and dopaminergic pathways in both diseases [24]. In
addition, acute systemic inflammatory events were associated with a 2-fold increase in the

70
rate of cognitive decline over a 6-month period in AD patients [25]. In this context, it was
demonstrated, in an animal model, that the intracerebroventricular administration of C-
reactive protein was associated to memory loss and early phase of AD pathogenesis [26].
Toll-like transmembrane receptors (TLRs) play an important role in innate
inflammatory response in sepsis [27]. These receptors could be associated with normal aging
and neurodegenerative disorders, linking inflammation to brain pathology. Expression of
TLR4 is enhanced in lipid rafts and non-raft membrane fractions in non-stimulated
neutrophils from elderly individuals compared to young subjects, suggesting a low-grade
inflammation in human ageing [27]. In addition, it was demonstrated that the pro-
inflammatory polymorphism of TLR4 gene is an independent risk factor for developing AD
[28].
To date, long-term consequences of septic encephalopathy on cerebral function are
poorly understood. It was demonstrated in a murine model that the behavioral impairment
was independent of the cerebral glucose levels, but dependent on the inducible nitric oxide
synthase gene, thus suggesting a relation with sustained microglial activation [29].
Furthermore, we have previously reported that early phases of sepsis are characterized by
enhanced reactive species production leading to oxidative stress [30]. Antioxidant treatment
during this CLP-induced acute phase of brain dysfunction prevented the development of late
cognitive deficits in an animal model of sepsis [31].
Since oxidative stress and inflammation are related events in both neurodegenerative
disorders and in sepsis, it is possible that both oxidative stress and microglia activation
collaborate to the long-term cognitive impairments observed in sepsis survivors. Microglia
are the common source of pro-inflammatory and oxidative stress in AD as shown by
consistent gross observations of upregulated inflammatory responses in brain regions that

71
exhibit high levels of AD pathological markers such as the frontal and limbic cortices [32,
33]. Conversely, these markers of inflammation or oxidative stress are absent or minimal in
brain regions which are typically less susceptible to neurotoxic events related to AD
pathology, such as cerebellum [34]. It was suggested that inflammation and oxidative stress
both increase prior to a diagnosis of AD (or at least early in AD) due to over-activation of
certain pro-inflammatory responses by mononuclear phagocytes, reinforcing our suggestion
that early brain inflammation and oxidative stress could be related to the occurrence of late
markers of neurodegeneration [35, 36]. We also observed that pharmacological evidences
linked sepsis-associated long-term cognitive impairment to neurodegenerative diseases [8, 19,
37, 38].
RAGE, a multiligand receptor in the immunoglobulin superfamily, acts as a cell
surface binding site for Aβ, which results in the activation of MAPK-controlled pathways
associated to neuronal apoptosis, NADPH oxidase-dependent reactive species production,
synaptic dysfunction and cognitive decline, as well as development of inflammatory
responses in AD [39, 40]. Besides, one of the main intracellular targets of RAGE activation is
the transcription factor NF-κB, which induces expression of macrophage-colony stimulating
factor that activates microglia, thus perpetuating the brain inflammatory response and
oxidative stress [41]. Since the RAGE gene also contains a response element for NF- κB
(p65), binding of Aβ to RAGE results in a positive feedback cycle of RAGE induction, which
in turn further increases brain inflammation, oxidative stress and Aβ production [42, 43].
Phosphorylation of MAPK is one of the main consequences of RAGE activation by
Aβ and other ligands, such as AGEs, HMGB1 and S100B. ERK1/2 are frequently described
as the primary targets of RAGE activation among intracellular protein kinases [44]. Recently,
it was demonstrated that RAGE activation induces tau hyperphosphorylation by activation of
GSK-3 in animal and cellular models, and blockade of this effect in rats restores the

72
impairment of LTP and memory deterioration [45]. Abnormally phosphorylated tau is the
first step in the formation of neurofibrillary tangles, causing neurite deterioration and forming
hydrophobic deposits of misfolded proteins observed in AD and other neurodegenerative
conditions.
It has been recently demonstrated that the RAGE G82S polymorphism (associated
with increased ligand affinity of RAGE) is associated with early onset AD , and significant
increase in RAGE immunoreactivity was observed in early AD-like pathology [46]. Besides
this increasingly amount of data pointing this receptor as one of the major mediators of
neurotoxic signaling in brain, it was also demonstrated a striking role for RAGE in the
progression of neurodegeneration, as this protein was identified to mediate the transport of Aβ
across the blood-brain barrier, thus being the major cause of translocation of systemic Aβ to
the CNS [47]. In this context, our results suggest that the brain of sepsis survivors are
characterized by a persistent increased of several potential mediators of neurodegeneration of
the RAGE pathway.
In conclusion, we demonstrated that brain from sepsis survivors animals presented
several markers of neurodegeneration and suggested that the Aβ-RAGE-MAPK pathway
could be a major pathway responsible to long-term cognitive deficits in sepsis survivors.
Further studies will address the relationship among the persistence of cognitive impairment in
sepsis survivors with RAGE expression, activation and the systemic and cerebral production
of RAGE ligands.
ACKNOWLEDGEMENTS
Supported by grants from the National Council for Scientific and Technological Development
(CNPq), FAPERGS PqG 2010 (1008860) and Universidade do Extremo Sul Catarinense.

73
REFERENCES
[1] Iacobone, E.; Bailly-Salin, J.; Polito, A.; Friedman, D.; Stevens, R. D.; Sharshar, T.
Sepsis-associated encephalopathy and its differential diagnosis. Crit Care Med 37:S331-336;
2009.
[2] Winters, B. D.; Eberlein, M.; Leung, J.; Needham, D. M.; Pronovost, P. J.; Sevransky,
J. E. Long-term mortality and quality of life in sepsis: a systematic review. Crit Care Med
38:1276-1283; 2010.
[3] Comim, C. M.; Rezin, G. T.; Scaini, G.; Di-Pietro, P. B.; Cardoso, M. R.; Petronilho,
F. C.; Ritter, C.; Streck, E. L.; Quevedo, J.; Dal-Pizzol, F. Mitochondrial respiratory chain
and creatine kinase activities in rat brain after sepsis induced by cecal ligation and perforation.
Mitochondrion 8:313-318; 2008.
[4] Barichello, T.; Martins, M. R.; Reinke, A.; Feier, G.; Ritter, C.; Quevedo, J.; Dal-
Pizzol, F. Long-term cognitive impairment in sepsis survivors. Crit Care Med 33:1671; 2005.
[5] van Gool, W. A.; van de Beek, D.; Eikelenboom, P. Systemic infection and delirium:
when cytokines and acetylcholine collide. Lancet 375:773-775; 2010.
[6] Buckingham, S. D.; Jones, A. K.; Brown, L. A.; Sattelle, D. B. Nicotinic acetylcholine
receptor signalling: roles in Alzheimer's disease and amyloid neuroprotection. Pharmacol Rev
61:39-61; 2009.
[7] Semmler, A.; Frisch, C.; Debeir, T.; Ramanathan, M.; Okulla, T.; Klockgether, T.;
Heneka, M. T. Long-term cognitive impairment, neuronal loss and reduced cortical
cholinergic innervation after recovery from sepsis in a rodent model. Exp Neurol 204:733-
740; 2007.

74
[8] Comim, C. M.; Pereira, J. G.; Steckert, A.; Petronilho, F.; Barichello, T.; Quevedo, J.;
Dal-Pizzol, F. Rivastigmine reverses habituation memory impairment observed in sepsis
survivor rats. Shock 32:270-271; 2009.
[9] Tillement, J. P.; Lecanu, L.; Papadopoulos, V. Amyloidosis and neurodegenerative
diseases: current treatments and new pharmacological options. Pharmacology 85:1-17; 2010.
[10] Haapasalo, A.; Viswanathan, J.; Bertram, L.; Soininen, H.; Tanzi, R. E.; Hiltunen, M.
Emerging role of Alzheimer's disease-associated ubiquilin-1 in protein aggregation. Biochem
Soc Trans 38:150-155; 2010.
[11] Yan, S. D.; Bierhaus, A.; Nawroth, P. P.; Stern, D. M. RAGE and Alzheimer's disease:
a progression factor for amyloid-beta-induced cellular perturbation? J Alzheimers Dis 16:833-
843; 2009.
[12] Tan, J. M.; Wong, E. S.; Lim, K. L. Protein misfolding and aggregation in Parkinson's
disease. Antioxid Redox Signal 11:2119-2134; 2009.
[13] Sims, G. P.; Rowe, D. C.; Rietdijk, S. T.; Herbst, R.; Coyle, A. J. HMGB1 and RAGE
in inflammation and cancer. Annu Rev Immunol 28:367-388; 2010.
[14] Wichterman, K. A.; Baue, A. E.; Chaudry, I. H. Sepsis and septic shock--a review of
laboratory models and a proposal. J Surg Res 29:189-201; 1980.
[15] Ritter, C.; Andrades, M. E.; Reinke, A.; Menna-Barreto, S.; Moreira, J. C.; Dal-Pizzol,
F. Treatment with N-acetylcysteine plus deferoxamine protects rats against oxidative stress
and improves survival in sepsis. Crit Care Med 32:342-349; 2004.
[16] Barichello, T.; Martins, M. R.; Reinke, A.; Feier, G.; Ritter, C.; Quevedo, J.; Dal-
Pizzol, F. Cognitive impairment in sepsis survivors from cecal ligation and perforation. Crit
Care Med 33:221-223; discussion 262-223; 2005.
[17] Barichello, T.; Martins, M. R.; Reinke, A.; Constantino, L. S.; Machado, R. A.;
Valvassori, S. S.; Moreira, J. C.; Quevedo, J.; Dal-Pizzol, F. Behavioral deficits in sepsis-

75
surviving rats induced by cecal ligation and perforation. Braz J Med Biol Res 40:831-837;
2007.
[18] Tuon, L.; Comim, C. M.; Antunes, M. M.; Constantino, L. S.; Machado, R. A.;
Izquierdo, I.; Quevedo, J.; Dal-Pizzol, F. Imipramine reverses the depressive symptoms in
sepsis survivor rats. Intensive Care Med 33:2165-2167; 2007.
[19] Tuon, L.; Comim, C. M.; Petronilho, F.; Barichello, T.; Izquierdo, I.; Quevedo, J.;
Dal-Pizzol, F. Time-dependent behavioral recovery after sepsis in rats. Intensive Care Med
34:1724-1731; 2008.
[20] Tuon, L.; Comim, C. M.; Petronilho, F.; Barichello, T.; Izquierdo, I.; Quevedo, J.;
Dal-Pizzol, F. Memory-enhancing treatments reverse the impairment of inhibitory avoidance
retention in sepsis-surviving rats. Crit Care 12:R133; 2008.
[21] Zhang, L.; Postina, R.; Wang, Y. Ectodomain shedding of the receptor for advanced
glycation end products: a novel therapeutic target for Alzheimer's disease. Cell Mol Life Sci
66:3923-3935; 2009.
[22] Lipcsey, M.; Olovsson, M.; Larsson, E.; Einarsson, R.; Qadhr, G. A.; Sjolin, J.;
Larsson, A. The brain is a source of S100B increase during endotoxemia in the pig. Anesth
Analg 110:174-180; 2010.
[23] Ernst, A.; Morgenthaler, N. G.; Buerger, K.; Dodel, R.; Noelker, C.; Sommer, N.;
Schwarz, M.; Koehrle, J.; Bergmann, A.; Hampel, H. Procalcitonin is elevated in the
cerebrospinal fluid of patients with dementia and acute neuroinflammation. J Neuroimmunol
189:169-174; 2007.
[24] Valcour, V.; Paul, R. HIV infection and dementia in older adults. Clin Infect Dis
42:1449-1454; 2006.

76
[25] Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford,
D.; Perry, V. H. Systemic inflammation and disease progression in Alzheimer disease.
Neurology 73:768-774; 2009.
[26] Lin, H. B.; Yang, X. M.; Li, T. J.; Cheng, Y. F.; Zhang, H. T.; Xu, J. P. Memory
deficits and neurochemical changes induced by C-reactive protein in rats: implication in
Alzheimer's disease. Psychopharmacology (Berl) 204:705-714; 2009.
[27] Fulop, T.; Larbi, A.; Douziech, N.; Fortin, C.; Guerard, K. P.; Lesur, O.; Khalil, A.;
Dupuis, G. Signal transduction and functional changes in neutrophils with aging. Aging Cell
3:217-226; 2004.
[28] Minoretti, P.; Gazzaruso, C.; Vito, C. D.; Emanuele, E.; Bianchi, M.; Coen, E.; Reino,
M.; Geroldi, D. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on
susceptibility to late-onset Alzheimer's disease. Neurosci Lett 391:147-149; 2006.
[29] Weberpals, M.; Hermes, M.; Hermann, S.; Kummer, M. P.; Terwel, D.; Semmler, A.;
Berger, M.; Schafers, M.; Heneka, M. T. NOS2 gene deficiency protects from sepsis-induced
long-term cognitive deficits. J Neurosci 29:14177-14184; 2009.
[30] Barichello, T.; Fortunato, J. J.; Vitali, A. M.; Feier, G.; Reinke, A.; Moreira, J. C.;
Quevedo, J.; Dal-Pizzol, F. Oxidative variables in the rat brain after sepsis induced by cecal
ligation and perforation. Crit Care Med 34:886-889; 2006.
[31] Barichello, T.; Machado, R. A.; Constantino, L.; Valvassori, S. S.; Reus, G. Z.;
Martins, M. R.; Petronilho, F.; Ritter, C.; Quevedo, J.; Dal-Pizzol, F. Antioxidant treatment
prevented late memory impairment in an animal model of sepsis. Crit Care Med 35:2186-
2190; 2007.
[32] Cras, P.; Kawai, M.; Siedlak, S.; Mulvihill, P.; Gambetti, P.; Lowery, D.; Gonzalez-
DeWhitt, P.; Greenberg, B.; Perry, G. Neuronal and microglial involvement in beta-amyloid
protein deposition in Alzheimer's disease. Am J Pathol 137:241-246; 1990.

77
[33] Good, P. F.; Werner, P.; Hsu, A.; Olanow, C. W.; Perl, D. P. Evidence of neuronal
oxidative damage in Alzheimer's disease. Am J Pathol 149:21-28; 1996.
[34] Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G. M.; Cooper, N. R.;
Eikelenboom, P.; Emmerling, M.; Fiebich, B. L.; Finch, C. E.; Frautschy, S.; Griffin, W. S.;
Hampel, H.; Hull, M.; Landreth, G.; Lue, L.; Mrak, R.; Mackenzie, I. R.; McGeer, P. L.;
O'Banion, M. K.; Pachter, J.; Pasinetti, G.; Plata-Salaman, C.; Rogers, J.; Rydel, R.; Shen, Y.;
Streit, W.; Strohmeyer, R.; Tooyoma, I.; Van Muiswinkel, F. L.; Veerhuis, R.; Walker, D.;
Webster, S.; Wegrzyniak, B.; Wenk, G.; Wyss-Coray, T. Inflammation and Alzheimer's
disease. Neurobiol Aging 21:383-421; 2000.
[35] Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E. K.; Jones, P. K.;
Ghanbari, H.; Wataya, T.; Shimohama, S.; Chiba, S.; Atwood, C. S.; Petersen, R. B.; Smith,
M. A. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol
60:759-767; 2001.
[36] Fagiolo, U.; Cossarizza, A.; Scala, E.; Fanales-Belasio, E.; Ortolani, C.; Cozzi, E.;
Monti, D.; Franceschi, C.; Paganelli, R. Increased cytokine production in mononuclear cells
of healthy elderly people. Eur J Immunol 23:2375-2378; 1993.
[37] Cassol-Jr, O. J.; Comim, C. M.; Petronilho, F.; Constantino, L. S.; Streck, E. L.;
Quevedo, J.; Dal-Pizzol, F. Low dose dexamethasone reverses depressive-like parameters and
memory impairment in rats submitted to sepsis. Neurosci Lett 473:126-130; 2010.
[38] Comim, C. M.; Constantino, L. S.; Petronilho, F.; de Souza, B.; Barichello, T.;
Quevedo, J.; Dal-Pizzol, F. Effects of acute treatment with amphetamine in locomotor activity
in sepsis survivor rats. J Neuroimmunol 212:145-147; 2009.
[39] Arancio, O.; Zhang, H. P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.; Hegde,
A.; Yan, S. F.; Stern, A.; Luddy, J. S.; Lue, L. F.; Walker, D. G.; Roher, A.; Buttini, M.;
Mucke, L.; Li, W.; Schmidt, A. M.; Kindy, M.; Hyslop, P. A.; Stern, D. M.; Du Yan, S. S.

78
RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice.
EMBO J 23:4096-4105; 2004.
[40] Leclerc, E.; Sturchler, E.; Vetter, S. W.; Heizmann, C. W. Crosstalk between calcium,
amyloid beta and the receptor for advanced glycation endproducts in Alzheimer's disease. Rev
Neurosci 20:95-110; 2009.
[41] Du Yan, S.; Zhu, H.; Fu, J.; Yan, S. F.; Roher, A.; Tourtellotte, W. W.; Rajavashisth,
T.; Chen, X.; Godman, G. C.; Stern, D.; Schmidt, A. M. Amyloid-beta peptide-receptor for
advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony
stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci U S
A 94:5296-5301; 1997.
[42] Creagh-Brown, B. C.; Quinlan, G. J.; Evans, T. W. RAGE inhibition: healthy or
harmful? Crit Care Med 38:1487-1490; 2010.
[43] Creagh-Brown, B. C.; Quinlan, G. J.; Evans, T. W.; Burke-Gaffney, A. The RAGE
axis in systemic inflammation, acute lung injury and myocardial dysfunction: an important
therapeutic target? Intensive Care Med 36:1644-1656; 2010.
[44] Yang, S.; Misner, B.; Chiu, R.; Meyskens, F. L., Jr. Common and distinct mechanisms
of different redox-active carcinogens involved in the transformation of mouse JB6P+ cells.
Mol Carcinog 47:485-491; 2008.
[45] Li, X. H.; Lv, B. L.; Xie, J. Z.; Liu, J.; Zhou, X. W.; Wang, J. Z. AGEs induce
Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation.
Neurobiol Aging; 2011.
[46] Li, K.; Dai, D.; Zhao, B.; Yao, L.; Yao, S.; Wang, B.; Yang, Z. Association between
the RAGE G82S polymorphism and Alzheimer's disease. J Neural Transm 117:97-104; 2010.
[47] Candela, P.; Gosselet, F.; Saint-Pol, J.; Sevin, E.; Boucau, M. C.; Boulanger, E.;
Cecchelli, R.; Fenart, L. Apical-to-basolateral transport of amyloid-beta peptides through

79
blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and
is restricted by P-glycoprotein. J Alzheimers Dis 22:849-859; 2010.
FIGURE LEGENDS
Figure 1 – Immunocontent of neurodegeneration markers in the hippocampus and pre-
frontal cortex of sepsis survivor animals compared to control (sham). Sepsis was induced
by CLP and after thirty days the imunnocontent of beta-amyloid peptide (Aβ), α-synuclein
and an isoform of tau related to Alzheimer’s disease (AD-like tau) was evaluated in samples
from the hippocampus (A) and pre-frontal cortex (B). Representative immunoblots of
individual samples from different animals from Sham and Sepsis groups are shown, and the
bar graphs represent mean values ± SE normalized by β–actin immunocontent. Data were
analyzed by t test and * denotes difference from sham group (p < 0.05).
Figure 2 – Immunocontent of RAGE, S100B and phosphorylated ERK1/2 in the
hippocampus and pre-frontal cortex of sepsis survivor animals compared to control
(sham). Sepsis was induced by CLP and after thirty days the imunnocontent of RAGE,
S100B and phosphorylated ERK1/2 was evaluated in samples from the hippocampus (A) and
pre-frontal cortex (B). Representative immunoblots of individual samples from different
animals from Sham and Sepsis groups are shown, and the bar graphs represent mean values ±
SE normalized by β–actin immunocontent. Levels of phosphorylated ERK1/2 were
normalized by the total immunocontent of ERK1/2. Data were analyzed by t test and *
denotes difference from sham group (p < 0.05).

80
Figure 1
Figure 2


![sepse [Modo de Compatibilidade] - INETI · • Sepse grave: sepse + disfunção (cardiovascular, respiratória, hematológica, renal, metabólica, hepática, neurológica) ... UTI](https://static.fdocumentos.tips/doc/165x107/5bfee81209d3f224168c6c90/sepse-modo-de-compatibilidade-sepse-grave-sepse-disfuncao-cardiovascular.jpg)















