
desenvolvimento de métodos e preparação de microesferas de ...

Universidade de São Paulo Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto Departamento de Química Programa de Pós-Graduação em Química
Microesferas Lipídicas encapsuladas com proteínas
antigênicas da membrana de Leishmania amazonensis
com potencial aplicação terapêutica.
Luiz Eduardo dos Reis Santos
Dissertação apresentada à Faculdade de Filosofia,
Ciências e Letras de Ribeirão Preto da Universidade
de São Paulo, como parte das exigências para a
obtenção do título de Mestre em Ciências, Área:
Química.
RIBEIRÃO PRETO -SP
2009

Universidade de São Paulo Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto Departamento de Química Programa de Pós-Graduação em Química
Microesferas Lipídicas encapsuladas com proteínas
antigênicas da membrana de Leishmania amazonensis
com potencial aplicação terapêutica.
Luiz Eduardo dos Reis Santos
Orientador: Prof. Dr. Pietro Ciancaglini
Dissertação apresentada à Faculdade de Filosofia,
Ciências e Letras de Ribeirão Preto da Universidade
de São Paulo, como parte das exigências para a
obtenção do título de Mestre em Ciências, Área:
Química.
RIBEIRÃO PRETO -SP
2009

FICHA CATALOGRÁFICA
Santos, Luiz Eduardo dos Reis.
Microesferas Lipídicas encapsuladas com proteínas antigênicas da membrana de Leishmania amazonensis com potencial aplicação terapêutica. Ribeirão Preto, 2009.
83 p.
Dissertação de Mestrado, apresentada à Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto/USP – Área de concentração: Química.
Orientador: Ciancaglini, Pietro.
1. Microesferas Lipídicas. 2. Leishmania amazonensis. 3. Delivery de Antígenos. 4. Proteínas Antigênicas. 5. Aplicação Terapêutica.

DDDEEEDDDIIICCCAAATTTÓÓÓRRRIIIAAA
Dedico este trabalho...
À minha Mãe. Obrigado por sempre acreditar em mim e por todo o incentivo,
amor, sacrifícios e pelas orações, aliás, foram muitas não é Mãe? A Senhora
dedicou a sua vida à minha educação, me ensinou que os problemas se tornam
pequenos quando temos fé e pessoas amadas ao nosso lado, esta dissertação é
uma vitória sua Mãe! Ah...Muitos créditos para minha Avó e minha Tia Luiza.
Ao meu Pai. Agradeço pelo investimento nos meus estudos, pelos conselhos e
pelas críticas. O Senhor me proporcionou a tranqüilidade para estudar, que
infelizmente, nem todas as pessoas tem.
Ao meu irmão. Obrigado por sempre estar perto nos momentos de alegrias e
tristezas e por sempre me incentivar e acreditar em mim. Mas do que um irmão
você é um grande amigo.
À Rafa. Dificilmente conseguirei expressar em palavras o quão importante você
foi. Você participou ativamente dos meus projetos, das minhas escolhas,
comemorou comigo minhas conquistas e também sofreu com meus fracassos.
“Pequena”, foi grande sua participação nesta conquista!

AAAGGGRRRAAADDDEEECCCIIIMMMEEENNNTTTOOOSSS EEESSSPPPEEECCCIIIAAAIIISSS
Primeiramente agradeço a Deus e a Nossa Senhora que durante toda essa
caminhada me guiaram de forma brilhante.
Em especial agradeço ao meu amigo e orientador,
Professor Dr. Pietro Ciancaglini, pela orientação excepcional, amizade, e pelo
exemplo de dedicação, competência e responsabilidade. Obrigado pelos
ensinamentos transmitidos nestes seis anos de convivência, seus conselhos e
sugestões foram indispensáveis para o meu amadurecimento e crescimento
profissional. Tenho orgulho de lhe ter como referência, pois você é um “verdadeiro
professor universitário”!
À Kátia e a Marcelle, que compartilharam comigo um pouco de seus
conhecimentos, e colaboraram de maneira direta neste trabalho.
À Associação Atlética Acadêmica Lucien Lison (Atlética Filô), pelos amigos e
experiências, que me proporcionou.
À Rep LOKS, agradeço por ter me acolhido nestes meus dois anos de mestrado,
foram anos extremamente valiosos. Agradeço a todos moradores, ex-moradores e
agregados, pelas conversas, festas, e conhecimentos compartilhados. Juma, Pala
e Flor, obrigado pelo carinho, companhia e por sempre me escutarem, sem pedir
nada em troca.

AAAGGGRRRAAADDDEEECCCIIIMMMEEENNNTTTOOOSSS
À CAPES pela bolsa concedida durante o desenvolvimento deste projeto.
À todos os meus amigos e amigas (ficaria difícil citar todos!) pela amizade, carinho e
apoio. A todos aqueles que, embora não nomeados, me brindaram com seus
inestimáveis apoios em inesquecíveis momentos, o meu reconhecido e carinhoso muito
obrigado!
Aos amigos do laboratório Marcelle, Simone, Carol, Ana Maria, Ju, Maytê, Rosângela e
Fernanda pela amizade, companheirismo, pelos momentos de alegria compartilhados e
acima de tudo obrigada por todo apoio e auxílio que me ofereceram durante todos esses
anos de convivência.
Aos alunos de iniciação científica Imaculada, Bruno, Thais, Ricardo, Andréia pelos
auxílios no laboratório.
Aos técnicos Dra. Ivana Aparecida Borin e Nilton Rosa Alves, sempre muito prestativos.
À Professora Maria Elisabete D. Zaniquelli, por permitir a utilização do equipamento Ultra
Turrax.
À Priscila Cerviglieri Ciancaglini, pela correção dos textos em inglês.
Aos docentes, técnicos e funcionários do Departamento de Química da Faculdade de
Filosofia, Ciências e Letras de Ribeirão Preto – USP.
À Maria Dolores (Tuca) e José Aparecido do Departamento de Morfologia da Faculdade
de Medicina de Ribeirão Preto – USP, pela ajuda e disponibilidade na obtenção de
imagens de microscopia eletrônica.
À FAPESP e CNPq pelos auxílios financeiros concedidos ao laboratório.
Enfim, agradeço a todos aqueles que contribuíram de alguma forma com a execução
deste trabalho.

“A Natureza faz do homem um ser natural; a
sociedade faz dele um ser social; somente o
homem é capaz de fazer de si um ser livre”.
Rudolf Steiner

“As pessoas estão sempre culpando as
circunstâncias pelo que elas são. Eu não
acredito em circunstâncias. As pessoas que
progridem neste mundo são as pessoas que
se levantam e procuram pelas circunstâncias
que elas querem, e, se elas não conseguem
encontrá-las, elas as fazem”.
George Bernard Shaw

SSSUUUMMMÁÁÁRRRIIIOOO
Lista de Siglas e Abreviaturas ............................................................................... I
Resumo .................................................................................................................. III
Abstract .................................................................................................................. IV
1. Introdução...................................................................................................... 1
1.1 – A Leishmania................................................................................................ 2
1.2 – Superfície celular da leishmania .................................................................. 3
1.3 – A doença Leishmaniose............................................................................... 6
1.4 - O desenvolvimento de vacinas contra leishmaniose.................................... 8
1.5 – O papel do adjuvante no desenvolvimento de vacinas ............................. 13
1.6 – Microesferas lipídicas................................................................................. 14
2. Objetivos ...................................................................................................... 19
2.1 – Objetivo geral ............................................................................................. 20
2.2 – Objetivos específicos ................................................................................. 20
3. Materiais e Métodos.................................................................................... 21
3.1 – Parasita ...................................................................................................... 22
3.2 – Obtenção da L. amazonensis .................................................................... 22
3.3 – Preparação do soro contendo os anticorpos contra os determinantes
imunogênicos de L. amazonensis ............................................................. 22
3.4 – Isolamento das membranas da L. amazonensis ....................................... 23
3.5 – Dosagem de proteína................................................................................. 23
3.6 – Solubilização das proteínas de membrana da L. amazonensis
com SDS.................................................................................................... 24
3.7 – Eletroforese em condição desnaturante (SDS-PAGE).............................. 24
3.8 – Avaliação da atividade antigênica das proteínas de membrana da
L. amazonensis.......................................................................................... 25
3.9 – Remoção do SDS do extrato protéico solubilizado ............................... 25
3.10 – Preparação das microesferas lipídicas .................................................... 26
3.10.1 – Protocolo 1 ......................................................................................... 26

3.10.2 – Protocolo 2 ......................................................................................... 26
3.10.3 – Protocolo 3 ......................................................................................... 27
3.10.4 – Protocolo 4 ......................................................................................... 27
3.10.5 – Protocolo 5 ......................................................................................... 28
3.11 – Marcação das proteínas antigênicas com isotiocianato de
fluoresceína (FITC).................................................................................... 29
3.12 – Microscopia eletrônica de varredura (MEV) ............................................ 30
3.13 – Microscopia de Fluorescência (MF)......................................................... 30
3.14 – Medidas de espalhamento de luz ............................................................ 30
3.15 – Centrifugação em gradiente de densidade de sacarose ......................... 31
3.16 – Dosagem de fosfato inorgânico ............................................................... 32
3.17 – Calorimetria diferencial de varredura (DSC)............................................ 33
3.18 – Quantificação das proteínas incorporadas nas ML ................................. 34
3.19 – Obtenção e o cultivo de macrófagos peritoneais murinos....................... 34
3.20 – Avaliação da viabilidade dos macrófagos peritoneais murinos............... 35
3.21 – Análise da produção de nitrito ................................................................. 35
3.22 – Análise estatística .................................................................................... 36
4. Resultados e Discussão............................................................................. 37
4.1 – Solubilização e identificação das proteínas antigênicas da
membrana da L. amazonensis .................................................................. 38
4.2 – Remoção do detergente do extrato bruto solubilizado .............................. 41
4.3 – Ensaios de preparação das microesferas lipídicas ................................... 43
4.4 – Caracterização das Microesferas Lipídicas ............................................... 55
4.5 – Quantificação das proteínas encapsuladas nas ML.................................. 62
4.6 - Análise da produção de NO e Teste de viabilidade (MTT) ........................ 65
5. Conclusões .................................................................................................. 69
6. Referências .................................................................................................. 72

I
LLLIIISSSTTTAAA DDDEEE SSSIIIGGGLLLAAASSS EEE AAABBBRRREEEVVVIIIAAATTTUUURRRAAASSS
APC Células apresentadoras de antígenos
C3b Componente C3 do sistema complemento
C3bi Componente C3 do sistema complemento
CMC Concentração micelar crítica
Cp Capacidade calorífica
DAB 3,3’ diaminobenzidina
DHFR-TS Dihidrofolato redutase-timidilato sintase
DNA Ácido desoxirribonucléico
DPPC Dipalmitoilfosfatidilcolina
DSC Calorimetria diferencial de varredura
ELISA Enzyme Linked Immunosorbent Assay
EPS Extrato de proteína solubilizada
FITC Isotiocianato de fluoresceína
FML Ligante de fucose e manose
fPPG Forma filamentosa das proteofosfoglicanas
GIPL Glicoinositolfosfolipídeos livres de GPI
GP42 Glicoproteína da superfície da Leishmania com massa de 42 KDa
GP46/M2 Glicoproteína da superfície da Leishmania com massa de 46 KDa
GP63 Glicoproteína da superfície da Leishmania com massa de 63 KDa
GPI Glicosilfosfatidilinositol
HEPES Ácido hidroxietil piperazina etanosulfúrico
IFN-γ Interferon-γ
IgG Imunoglobulina G
IL-12 interleucina-12
IL-2 interleucina-2
KO knock-out
LACK Leishmania homologue of receptors for activated C kinase
LPG Lipofosfoglicanas
LTA Leishmaniose Tegumentar Americana

II
MF Microscopia de fluorescência
MEV Microscopia eletrônica de varredura
ML Microesferas lipídicas
MLP Microesferas lipídicas com proteínas encapsuladas
MTT (3-(4,5-dimetil tiazol-2-il)-2,5-difenil brometo tetrazolina)
OS Óleo de soja
PBS Salina tamponada com fosfato
PPG Proteofosfoglicanas
PVA Álcool polivinílico
s.c subcutânea
Sch Schneider
SDS Dodecil sulfato de sódio
SDS-PAGE Eletroforese em condição desnaturante
SFB Soro fetal bovino
TC Tampão Carbonato
Th1 T helper 1
Th1 T helper 2
TLR4 Receptor Toll-like 4
Tm Temperatura de transição
Tris Tris-(hidroximetil)-aminometano
Tween 20 Polioxietileno sorbitan monolaurato

III
RRREEESSSUUUMMMOOO Microesferas lipídicas (ML) são excelentes sistemas de delivery de drogas e são
relativamente estáveis. O objetivo deste trabalho foi desenvolver um sistema capaz de
encapsular proteínas antigênicas da membrana de L. amazonensis. Proteínas de
membrana são importantes na formulação de vacinas, uma vez que estas proteínas são
os primeiros componentes celulares a entrarem em contato com a célula hospedeira,
provocando e mediando a resposta imune. Esta é uma ferramenta útil para evitar ou
inativar a invasão do parasita. As ML são constituídas por óleo de soja (OS),
dipalmitoilfosfatidilcolina (DPPC), colesterol e extrato de proteínas solubilizadas (EPS)
(previamente tratadas para remoção do detergente). Primeiramente foram ensaiadas
formulações de ML contendo álcool polivinílico (PVA). Estudos de microscopia eletrônica
de varredura (MEV) mostraram que as ML são formadas quando PVA 3% (p/v) é usado
na formulação. Além disso, foi feita a marcação das proteínas com isotiocianato de
fluoresceína (FITC) e a microscopia de fluorescência revelou a presença de estruturas
esféricas fluorescentes, o que indicou a encapsulação das proteínas na região lipofílica
das ML. A presença do PVA na formulação das ML acarretou em algumas limitações
cruciais para a continuidade desta pesquisa, levando a substituição deste pelo glicerol.
Com o objetivo de otimizar a formulação foram avaliadas cinco diferentes formulações
contendo glicerol 2,5% (p/v). Nestas formulações foram avaliadas as relações molares
DPPC:Colesterol:OS. As partículas formadas apresentaram um diâmetro médio de
200nm, baixa polidispersão, e estabilidade por um período de 30 dias, de acordo com
ensaios de espalhamento de luz dinâmico. Ensaios de gradiente de densidade de
sacarose das ML mostraram que proteínas e lipídios se encontram juntos no gradiente
de sacarose (5-50% p/v) sugerindo que a preparação de ML foi homogênea e que as
proteínas estão interagindo com este sistema lipídico. Termogramas obtidos por
calorimetria diferencial de varredura (DSC) corroboram com a formação de um sistema
de ML-proteína estável, além de fornecer indícios que as proteínas se encontram
localizadas no núcleo oleoso das ML. Os resultados mostraram que foram encapsulados
85% das proteínas do EPS nas microesferas, e que estas não perderam sua atividade
antigênica, mesmo após o complexo processo de preparação do sistema. Estudos de
viabilidade dos macrófagos peritoneais murinos e o ensaio de geração de nitrito
mostraram que o sistema ML associado às proteínas não apresenta efeito citotóxico para
os macrófagos e ainda estimula a produção de NO nos mesmos. Com isso, podemos
sugerir que ML contendo proteínas antigênicas de membrana de L. amazonensis
constituem um promissor sistema para a terapia da leishmaniose.

IV
AAABBBSSSTTTRRRAAACCCTTT Lipid microspheres (LM) are excellent drug delivery systems and they are also
relatively stable. The aim of this work was to develop a lipid-based system to encapsulate
antigenic membrane proteins from Leishmania amazonensis. Membrane proteins are
important for vaccine formulation because these proteins are the first ones to get in
contact with the host cell, triggering the cell mediated immune response. This is a useful
tool to avoid or to inactivate parasite invasion. LM are constituted by soybean oil (SO),
dipalmitoylphosphatidylcholine (DPPC), cholesterol and solubilized protein extract (SPE)
(previously treated to remove detergent). First, the LM formulations containing polyvinylic
alcohol (PVA) were assayed. Scanning electronic microscopy (SEM) studies have shown
that LM are formed when 3% PVA (w/v) is used in the formulation. Besides, proteins were
marked with fluorescein Isothiocyanate (FITC) and the fluorescence microscopy showed
the presence of fluorescent spherical structures, which indicated protein encapsulation in
the lipophilic region of the LM. The presence of PVA in the LM formulation has caused
some crucial limitations for the continuity of the research, leading to its substitution for
glycerol. In order to optimize the system, five different formulations containing 2.5%
glycerol (w/v) were evaluated for DPPC:Cholesterol:OS molar ratios. The particles formed
(LM-protein) presented an average diameter of 200 nm, low polydispersion and good
stability for a period of 30 days, according to dynamic light scattering assays. Isopycnic
density gradient centrifugation of LM-protein showed that proteins and lipids floated in the
sucrose gradient (5-50% w/v) suggesting that the LM preparation is homogeneous and
that the proteins are interacting with these systems. Thermograms obtained by differential
scanning calorimetry (DSC) corroborate with the formation of a stable LM system, besides
giving indication that proteins are located in the oily LM core. The results show that 85%
of the SPE proteins were encapsulated in the LM without loosely their antigenic activity
even after the system preparation process. Viability studies of the peritoneal macrophage
murines and the nitrate assay generation have shown that system does not have a
cytotoxic effect for the macrophages and it yet stimulate their NO production. Therefore,
we conclude that LM, encapsulated with L. amazonensis membrane antigenic proteins
seems to be a promising system for for therapy of Leishmaniasis.

1. INTRODUÇÃO

Introdução
2
1.1 – A Leishmania
Em 1903, Leishman e Donovan, separadamente, descreveram na Índia, um
protozoário presente em tecidos esplênicos de pacientes. A doença passou a ser
chamada de leishmaniose visceral e o parasita denominado Leishmania donovani
(Herwaltd, 1999).
Nos hospedeiros mamíferos, representados na natureza por várias ordens
e espécies, a Leishmania assume a forma amastigota, arredondada e imóvel, que
se multiplica obrigatoriamente dentro de células do sistema monocítico fagocitário.
À medida que as formas amastigotas vão se multiplicando, os macrófagos se
rompem liberando parasitas que são fagocitados por outros macrófagos (Gontijo
& Carvalho, 2003). Todas as espécies do gênero são transmitidas pela picada de
fêmeas infectadas de dípteros da subfamília Phlebotominae, pertencentes aos
gêneros Lutzomyia – no Novo Mundo, e Phlebotomus – no Velho Mundo. Nos
flebotomíneos as leishmanias vivem no meio extracelular, na luz do trato
digestivo. Ali, as formas amastigotas, ingeridas durante o repasto sangüíneo, se
diferenciam em formas promastigotas, flageladas, morfológicas e
bioquimicamente distintas das amastigotas (Gontijo & Carvalho, 2003). Esta forma
é alongada e apresenta um longo flagelo livre. No sistema digestivo de seus
vetores, as formas promastigotas multiplicam-se por aparente divisão simples e
assexuada e migram para a probóscíde do inseto após aproximadamente 4 a 5
dias. Neste ponto, bloqueiam o proventrículo, de onde podem ser inoculadas na
pele do hospedeiro vertebrado, junto com a saliva (Marzochi, 1992).
Há aproximadamente 21 espécies de Leishmania, transmitidas por cerca
de 30 espécies de flebotomíneos. Leishmania tem desenvolvido uma variedade
de mecanismos adaptativos não somente para viver dentro do vetor, mas também

Introdução
3
para escapar da resposta imune do hospedeiro vertebrado, inclusive dentro do
macrófago (Cunningham, 2002). Vários receptores da superfície celular dos
macrófagos, moléculas da superfície do parasita e fatores derivados do soro são
responsáveis pelas interações das formas promastigotas com a célula hospedeira
garantindo assim a sobrevivência, multiplicação, patogênese ou mesmo a morte
do parasita no hospedeiro (Mosser & Rosenthal, 1993; Garg et al., 2005).
1.2 – Superfície celular da leishmania
Considerando a estratégia de invasão da Leishmania, proteínas presentes
na superfície celular desses parasitas são de extrema importância, sendo
responsáveis pela interação com a célula do hospedeiro garantindo a
sobrevivência, multiplicação e patogênese do parasita no mesmo (Garg et al.,
2005).
A superfície celular de todos os tripanossomatídeos é dominada por
proteínas ancoradas por glicosilfosfatidilinositol (GPI), formando uma cobertura
que protege a superfície do parasita e medeia interações essenciais parasita-
hospedeiro. Acredita-se que essas proteínas ancoradas por GPI formem clusters
em microdomínios da membrana celular, geralmente chamados de lipids rafts, em
associação com glicoesfingolipidios, esfingomielina, colesterol e outras proteínas
sinalizadoras. No parasita Leishmania essas proteínas formam uma efetiva
barreira de difusão macromolecular efetiva que protege as formas promastigotas
de processos microbicidas tais como lise mediada pelo sistema complemento,
radicais oxigênio e hidrolases dos hospedeiros vertebrados e invertebrados
(Descoteaux &Turco, 1999; Ilgoutz & McConville, 2001).
As formas promastigotas de Leishmania são cobertas por um grande

Introdução
4
número de glicoproteínas ancoradas por GPI, como por exemplo, GP63, GP42
denominados de proteofosfoglicanas (PPG), um complexo lipofosfoglicano (LPG)
que possui uma âncora estruturalmente distinta da GPI (McConville & Ferguson,
1993) e uma abundante família de GPIs livre (aproximadamente 10 vezes mais
abundante do que proteínas ancoradas por GPI e LPG), denominada
glicoinositolfosfolipideos (GIPL) (McConville et al., 2002). Ao contrário, as formas
amastigotas de Leishmania, que proliferam dentro do fagolisossomo do
macrófago do hospedeiro, são recobertas por um mínimo glicocálix que é quase
exclusivamente composto por GIPLs e glicolipídeos derivado da célula do
hospedeiro, e a expressão das proteínas ancoradas por GPI e LPG é
dramaticamente baixo-regulada neste estágio (McConville & Blackwell, 1991;
Winter et al., 1994).
A glicoproteína ancorada por GPI mais predominante da superfície de
Leishmania é a zinco-metaloprotease chamada GP63 com peso molecular de 63
KDa (Bordier, 1987; Button & McMaster, 1988). Essa proteína é expressa na
superfície de promastigotas de diferentes espécies de Leishmania e em ambas
formas promastigotas e amastigotas de L. major e L. mexicana (Frommel et al.,
1990). Cada promastigota é coberta com aproximadamente 5x105 cópias de
GP63 o que corresponde a aproximadamente 1% das proteínas totais (Ilgoutz &
McConville, 2001). Essa glicoproteína apresenta um papel importante na
leishmaniose por agir com aceptor para o componente C3 do complemento (C3b
e C3bi) mediando a ligação da forma promastigota ao macrófago (Brittingham et
al., 1995; Russell et al., 1989). Outros estudos também mostraram que GP63 é o
ligante para múltiplos receptores de macrófagos, incluindo Mac-1 (CD11b/CD18)
e receptor celular para fibronectina (Brittingham et al., 1999). Ainda, GP63 pode

Introdução
5
ter um papel importante na proteção contra degradação dentro do fagolisossomo
do macrófago (Chaudhuri et al., 1989). Essa proteína exibiu atividade ótima sob o
ambiente ácido do fagolisossomo, e têm mostrado degradar as enzimas
lisossomais presentes neste ambiente (Cunningham, 2002).
Outras proteínas ancoradas também mostraram ter participação na
resposta imune como, por exemplo, as PPGs que constituem uma classe distinta
de moléculas e que são sintetizadas por ambas formas promastigotas e
amastigotas do parasita. Essas proteofosfoglicanas podem estar ancoradas por
GPI na superfície celular ou serem secretadas (forma filamentosa fPPG) (Ilgoutz
& McConville, 2001) respondendo pela virulência do parasita tanto no hospedeiro
vertebrado quanto no hospedeiro invertebrado (Ilg et al., 1999).
As proteínas antigênicas GP42 e GP46/M2 também parecem estar
envolvidas na resposta imunológica do hospedeiro vertebrado. A GP42 mostrou
induzir alta resposta proliferativa, assim como a produção de interleucina-2 (IL-2)
e interferon-γ (IFN-γ) por linfócitos humanos de pacientes com leishmaniose
(Burns et al., 1991).
Um outro composto (não protéico) importante na interação da superfície do
parasita e que participa da interação parasita-hospedeiro é o lipofosfoglicano
(LPG). Este glicolipídeo é um dos mais abundantes na superfície de
promastigotas de Leishmania, forma um denso glicocálix ao redor do parasita.
Este composto possui uma âncora estruturalmente distinta das GPIs, constituída
por unidades repetidas de Galβ1,4Manα-PO4 ligada a membrana de
promastigotas via uma ancora lipídica específica; 1-Ο-alquil-2-liso-
fosfatidil(mio)inositol com uma cadeia não usual longa de ácido graxo saturado de
24-26 carbonos (Turco & Descoteaux, 1992). A LPG, assim como outros

Introdução
6
glicoconjugados, têm uma participação importante na interação Leishmania-
complemento e ainda, se ligam e interagem com outras proteínas do soro para
promover a entrada na célula hospedeira. A LPG pode contribuir para a habilidade
da Leishmania em resistir ou escapar dos efeitos microbicidas dos macrófagos
(Descoteaux & Turco, 2002). Dentro dos macrófagos, as LPGs apresentam vários
efeitos como, por exemplo, inibição da maturação do fagossomo, inibição da
proteína C quinase e modulação da produção de óxido nítrico (NO) (Winberg et
al., 2009; Descoteaux & Turco, 2002).
1.3 – A doença Leishmaniose
Entre todas as doenças emergentes, aquelas causadas por protozoários são de
grande importância por apresentarem um elevado nível de morbidade e mortalidade. Este é
o caso da leishmaniose que é uma doença infecto-parasitária que acomete o homem,
causada por várias espécies de protozoários tripanossomatideos do gênero Leishmania
(Santos et al., 2008). Esta doença afeta aproximadamente 12 milhões de pessoas e está
presente em 88 países, principalmente em áreas tropicais e subtropicais. A incidência anual
é de aproximadamente 2 milhões de novos casos e cerca de 350 milhões de pessoas vivem
em áreas endêmicas, revelando assim a importância dessa doença negligenciada
(http://www.who.int/).
A leishmaniose pode causar um amplo espectro de manifestações clínicas. Na pele,
essas manifestações variam de úlceras cutâneas localizadas a lesões faciais mutilantes
(leishmaniose cutânea), lesões mucocutâneas (leishmaniose mucocutânea), e a
leishmaniose cutânea difusa e visceral que pode muitas vezes ser fatal (Teixeira, 2006).
Estas manifestações vão depender da espécie de Leishmania envolvida e da relação do
parasita com seu hospedeiro (Gontijo & Carvalho, 2003). A variedade de manifestações

Introdução
7
clínicas presentes na leishmaniose cutânea está diretamente relacionada ao estado
imunológico do paciente e a espécie de Leishmania envolvida. As diferentes formas de
leishmaniose cutânea constituem um importante problema de saúde pública principalmente
pela dificuldade de sua prevenção (MS/FUNASA, 2000).
Lesões causadas por Leishmania amazonensis, utilizada em nossos estudos,
usualmente apresentam uma lesão única ulcerada que é muito suscetível ao tratamento e
pode curar-se espontaneamente. Contudo, a diversidade de apresentação clínica desta
espécie parece variar de lugar, possivelmente devido a diferenças na sua virulência
(Weigle & Saravia, 1996).
A leishmaniose tegumentar americana, lesões cutâneas e muco cutâneas, (LTA) é
uma doença que acompanha o homem desde a antiguidade, existindo relatos e descrições
encontrados na literatura desde o séc. I d.C (Laison, 1997; Camargo & Barcinski, 2003). O
primeiro a observar o parasita do gênero Leishmania foi Cunningham, em 1885, na Índia,
em casos de leishmaniose visceral. No Brasil, Cerqueira, em 1855, observou a existência
da moléstia da pele, identificando-a clinicamente como “botão de Biskra”. Em 1895, na
Itália, Breda, descreveu a moléstia em italianos provenientes de São Paulo (Pessoa, 1982).
Entretanto, no Brasil, a natureza leishmaniótica das lesões cutâneas e nasofaríngeas só foi
confirmada, pela primeira vez, em 1909, por Lindenberg, que encontrou formas de
Leishmania, idênticas a Leishmania tropica da leishmaniose do Velho Mundo, em lesões
cutâneas de indivíduos que trabalhavam nas matas do interior do Estado de São Paulo
(Pessoa, 1982).
A leishmaniose tegumentar tem aumentando nos últimos 20 anos em quase todos os
estados brasileiros, e surtos epidêmicos tem ocorrido no Sudeste, Centro-Oeste, Nordeste,
e mais recentemente, na região Amazônica, devido ao processo predatório de colonização
(Delorenzi et al., 2001; Gontijo & Melo, 2004).

Introdução
8
O controle das leishmanioses esbarra no fato dela ser uma zoonose, sendo os
animais silvestres e domésticos potenciais reservatórios do parasita. O crescimento urbano
desordenado tem gerado muitas dificuldades no combate ao flebótomo (Handman, 2000).
1.4 - O desenvolvimento de vacinas contra leishmaniose
O controle da leishmaniose, tanto em saúde pública como individualmente,
é pouco eficiente, não existindo medidas simples e eficazes de proteção e
consiste principalmente em tratamentos quimioterápicos, pois infelizmente, ainda
não há uma vacina efetiva contra o parasita (Silva & Camargo-Neves, 2004).
Atualmente, o tratamento da leishmaniose primeiramente usa da
quimioterapia, com algumas tentativas no uso de imunoterapias (Ghosh et al.,
2003; Santos et al., 2003; Borja-Cabrera et al., 2004). A primeira linha de
tratamento é predominantemente baseada em antimoniais pentavalentes (ex.
Glucantime), um tratamento clássico na maioria das áreas endêmicas, contudo, a
sua utilidade tem sido comprometida pela acentuada resistência e efeitos
colaterais (Herwaldt, 1999). A segunda linha de tratamento inclui drogas tais como
anfotericina B e pentamidina, que são caracterizadas pela alta eficiência, mas são
relativamente caras e apresentam vários efeitos colaterais (Berman, 2003; Davis
e Kedzierski, 2005).
Novas drogas, tais como formulações lipídicas de anfotericina B, têm sido
efetivas no tratamento da leishmaniose visceral, mas o custo dessas formulações
é certamente inviável para a maioria dos pacientes (Berman et al., 1998; Murray,
2004). Recentemente, miltefosine tem mostrado ser um tratamento oral efetivo
para leishmaniose visceral na Índia (Sundar et al., 2002), e para leishmaniose
cutânea na América do Sul (Soto et al., 2001).

Introdução
9
Com a restrita oferta de drogas utilizadas para o tratamento de infecções
por Leishmania ssp. faz-se necessário a busca de alternativas profiláticas para
esta infecção. Devido a facilidade na utilização dos modelos in vivo e in vitro de
infecção com Leishmania, a eficiência de vários compostos e protocolos de
vacinação estão sendo testados (Handman et al., 2000).
A vacinação contra leishmaniose cutânea humana tem sido praticada por
séculos. A primeira vacina contra leishmaniose foi desenvolvida por Adler da
Universidade Hebrew de Jerusalém. Ele observou que mães libanesas expunham
os braços de seus filhos a picadas pelo inseto vetor, uma vez que elas
intuitivamente sabiam que o desenvolvimento de uma primeira e única lesão, que
se auto curava, poderia proteger seus filhos de uma forma mais severa da doença
no futuro. Por essa razão, práticas antigas inoculavam em indivíduos não
infectados o material infeccioso de lesões, em regiões do corpo onde a cicatriz
pudesse ser escondida (Palatnik-de-Souza, 2008). Conhecida como
leishmanização, esta prática utilizava amostras de lesões humanas ou de outros
animais. Posteriormente estas amostras foram substituídas pelo uso de formas
promastigotas de L. major, obtidas de culturas (Greenblatt, 1980; Senekji &
Beattie, 1941). A leishmanização foi usada em Israel e no Irã nas décadas de 70 e
80 respectivamente (Nadim et al., 1983), como uma vacina profilática. Esta prática
também foi adotada pelo Governo iraniano durante a guerra entre o Irã e o Iraque,
um conflito que foi de 1980 a 1988, este programa abrangeu cerca de 2 milhões
de pessoas. A variabilidade no tamanho e na duração das lesões resultantes da
inoculação de cepas virulentas fez com que a técnica fosse abandonada no Irã,
após a guerra. Assim, vacinas de primeira geração compostas de parasitas
mortos foram gradualmente substituindo a leishmanização (Modabber, 1995).

Introdução
10
Em geral, as vacinas em desenvolvimento contra a leishmaniose, assim
como contra outras doenças infecciosas, podem ser divididas em três categorias:
(i) Vacinas vivas, incluindo também as construções geneticamente modificadas;
(ii) Vacinas de primeira geração, que são aquelas produzidas com frações do
agente patogênico ou ainda com o patógeno inteiro e morto, com ou sem
adjuvante e (iii) Vacinas de segunda e terceira geração, produzidas com proteínas
recombinantes ou DNA (ácido desoxirribonucléico) e vetores microbianos (vírus e
bactérias) expressando antígenos do parasita.
No Brasil, o uso de parasitas mortos na tentativa de se induzir uma
proteção contra a leishmaniose teve início na década de 40. A partir de 1970,
Wilson Mayrink e seus colaboradores na Universidade Federal de Minas Gerais
(UFMG), desenvolveram uma vacina morta composta de quatro espécies
diferentes de Leishmania (Genaro et al., 1996). Essa vacina acabou sendo
simplificada pelo uso de uma única espécie e está sendo usada aqui no Brasil
como um adjuvante durante o tratamento contra a leishmaniose (Machado-Pinto
et al., 2002) após ter sido aprovada pelo Ministério da Saúde. Além disso, essa
vacina também foi testada na Colômbia (Vélez et al., 2000; Vélez et al., 2005) e
no Equador (Armijos et al., 2004).
As vacinas de primeira geração, produzidas com frações do parasita,
também já foram empregadas. Conhecida como Leishmune® está sendo
produzida e comercializada no Brasil, desde que foi autorizada pelo Ministério da
Agricultura em junho de 2003, esta é uma vacina de subunidade contra a
leishmaniose visceral canina, constituída pelo ligante de fucose e manose (FML)
da L. donovani com saponina, induz cerca de 92 a 97% de proteção contra a
leishmaniose visceral zoonótica (Saraiva et al., 2006). A Leishmune®, apesar de

Introdução
11
eficaz na proteção canina, traz riscos para a população, uma vez que dificulta a
distinção entre animais vacinados e os animais contaminados assintomáticos.
Outra abordagem na busca de uma vacina contra a leishmaniose baseia-se
na manipulação gênica e criação de parasitas geneticamente modificados. Nessa
estratégia, aqueles genes que são essenciais para a sobrevivência da Leishmania
no seu hospedeiro são bloqueados, removidos ou mesmo substituídos por outros
genes. A primeira construção que gerou um mutante deficiente para um gene deu
origem a uma cepa de L. major knock-out (KO) para a enzima dihidrofolato
redutase-timidilato sintase (DHFR-TS) (Cruz et al., 1991). Os camundongos
inoculados com esses mutantes apresentaram uma proteção significativa, mas
temporária, após o desafio com os parasitas virulentos (Veras et al., 1999; Titus et
al., 1995).
Na construção das vacinas de segunda geração, muitos antígenos de
Leishmania já foram utilizados. A região codificadora para a parte protéica da
glicoproteína da superfície da Leishmania, a GP63, foi à primeira vacina de
plasmídeo produzida contra a leishmaniose (Xu & Liew, 1994). Ainda, outro
antígeno utilizado é o LACK (Leishmania homologue of receptors for activated C
kinase), contudo, resultados conflitantes foram observados. Enquanto cães
vacinados com vetores do vírus da vaccinia expressando esse antígeno
apresentaram 60% de proteção contra a leishmaniose visceral causada pela L.
infantum (Ramiro et al., 2003), camundongos BALB/c vacinados com DNA do
LACK não foram protegidos contra a infecção pela L. chagasi (Marques-da-Silva
et al., 2005).
Recentemente, o uso de substâncias com estruturas moleculares bem
definidas em vacinas de segunda e terceira geração contra leishmaniose estão

Introdução
12
emergindo (Coler & Reed, 2005). Embora o desenvolvimento de vacinas de
primeira geração seja mais barato e não necessite equipamentos sofisticados e
avanços tecnológicos, o desenvolvimento de uma vacina de antígenos bem
definidos tem vantagens na reprodutibilidade sobre vacinas com o extrato bruto
de Leishmania. Muitos dos antígenos de Leishmania que estão sendo testados
em modelos animais mostraram proteger quando usados com um adjuvante
apropriado (Afonso et al., 1994; Aebischer et al., 2000; Campos-Neto et al., 2001).
A inclusão de adjuvantes nas vacinas para leishmaniose é crítica para a
eficácia contra leishmaniose experimental murina. Pesquisas recentes têm focado
na identificação de adjuvantes que estimulam imunidade celular, e aqueles que
agem através do receptor Toll-like 4 (TLR4) (Coler & Reed, 2005; Cluff et al.,
2005), como por exemplo, ONO-4007 que tem capacidade para aumentar a
imunidade do tipo 1 e induzir produção de óxido nítrico, que pode ter um potente
efeito leishmanicida. Esse composto, sozinho, mostrou inibir a proliferação
intracelular de amastigotas de L. amazonensis e L. major dentro de macrófagos
(Calvopina et al., 2006).
Assim, vacinas preventivas são reconhecidas como as de melhor proteção
contra patógenos, a da Leishmania não é uma exceção. Contudo, o
desenvolvimento de vacinas para leishmaniose tem demonstrado ser uma tarefa
difícil e desafiadora, provavelmente devido a conhecimentos insuficientes da
patogênese do parasita e a complexidade da resposta imune necessária para
proteção. Vários novos antígenos estão sendo avaliados e com a conclusão do
projeto genoma de L. major e L. infantum serão, sem dúvida, descobertos mais
candidatos promissores (Kedzierski et al., 2006).

Introdução
13
1.5 – O papel do adjuvante no desenvolvimento de vacinas
Adjuvantes imunológicos foram originalmente descritos por Ramon, em
1924 como “substâncias usadas em combinação com um antígeno específico,
levando a uma resposta imune mais robusta do que o antígeno sozinho”. Estes
podem ser utilizados para melhorar a resposta imune de vacinas, de várias
formas diferentes: (1) aumentando a imunogenicidade de antígenos fracos; (2)
aumentando a velocidade e duração da resposta imune; (3) modulando a avidez
do anticorpo, especificidade, isotipo ou distribuição de subclasses; (4)
estimulando linfócitos T citotóxicos; (5) promovendo a indução de imunidade das
mucosas; (6) aumentando a resposta imune em indivíduos imunologicamente
imaturos; (7) decrescendo a dose do antígeno na vacina para reduzir o custo ou
(8) auxiliando a derrotar a competição entre os antígenos em vacinas combinadas
(O´Hagan & Singh, 2001).
O mecanismo de ação da maioria dos adjuvantes ainda permanece
parcialmente entendido, uma vez que a imunização freqüentemente ativa uma
cascata complexa de respostas que dificulta entender o efeito primário do
adjuvante. Contudo, se admitirmos o conceito local da reatividade imune, no qual
os antígenos que não alcançam os nódulos linfáticos locais não induzem resposta
imune (Zinckernagel et al., 1997), se torna mais fácil propor interpretações a
respeito do mecanismo de alguns adjuvantes, particularmente aqueles baseados
em um mecanismo de delivery. Se antígenos, que não alcançam os nódulos
linfáticos, não induzem resposta imune, então algum adjuvante, que aumente o
delivery de antígeno nas células que traficam para os nódulos linfáticos, pode
aumentar a resposta. Existem fortes indicações para supor que uma subclasse de
células dendríticas constituam as “células chaves” que circulam em tecidos

Introdução
14
periféricos e agem como “sentinelas”, sendo responsáveis pela captura de
antígenos e sua transferência para os nódulos linfáticos, onde eles são então,
apresentados para as células T (O´Hagan & Singh, 2001).
Os adjuvantes podem ser separados em duas classes, baseados em seus
principais mecanismos de ação: Sistemas de delivery e adjuvantes
imunoestimulatórios. Os sistemas de delivery dos antígenos são geralmente
particulados, por exemplo, emulsões, micropartículas e lipossomos. Estes
funcionam principalmente como agentes que direcionam os antígenos as células
apresentadoras de antígenos (APC). Em contraste, adjuvantes
imunoestimulatórios são predominantemente derivados de patógenos e
freqüentemente representam padrões moleculares associados, como por
exemplo, lipopolissacarídeos, e DNA, os quais ativam o sistema imune inato. Uma
vez ativado o sistema imune inato, é ativado o sistema imune adquirido (O´Hagan
& Singh, 2001).
Os aspectos práticos importantes para o desenvolvimento de adjuvantes e
a escolha de qual sistema usar vão depender da aplicação e do perfil do produto
de interesse. Devem ser levados em consideração: a via de administração, o
custo e a aplicabilidade para uma vasta gama de vacinas, estabilidade, facilidade
de manufatura, condições de uso e armazenamento (Boyd, 2008).
1.6 – Microesferas lipídicas
Microesferas lipídicas (ML) são também chamadas de emulsões lipídicas,
onde um dos dois líquidos que não dissolve homogeneamente se transforma em
micropartículas, as quais são dispersas no outro líquido. Quando o óleo está

Introdução
15
presente na micropartícula, esta é do tipo óleo-em-água, ou emulsão O/W,
todavia, no caso oposto temos emulsões tipo água-em-óleo, ou emulsão W/O.
Geralmente, as microesferas lipídicas estão na emulsão O/W. Um emulsificador é
requerido para tornar a emulsão estável e, para isto, surfactantes são geralmente
utilizados (Yamagushi & Mizushima, 1994). Microesferas lipídicas são produzidas
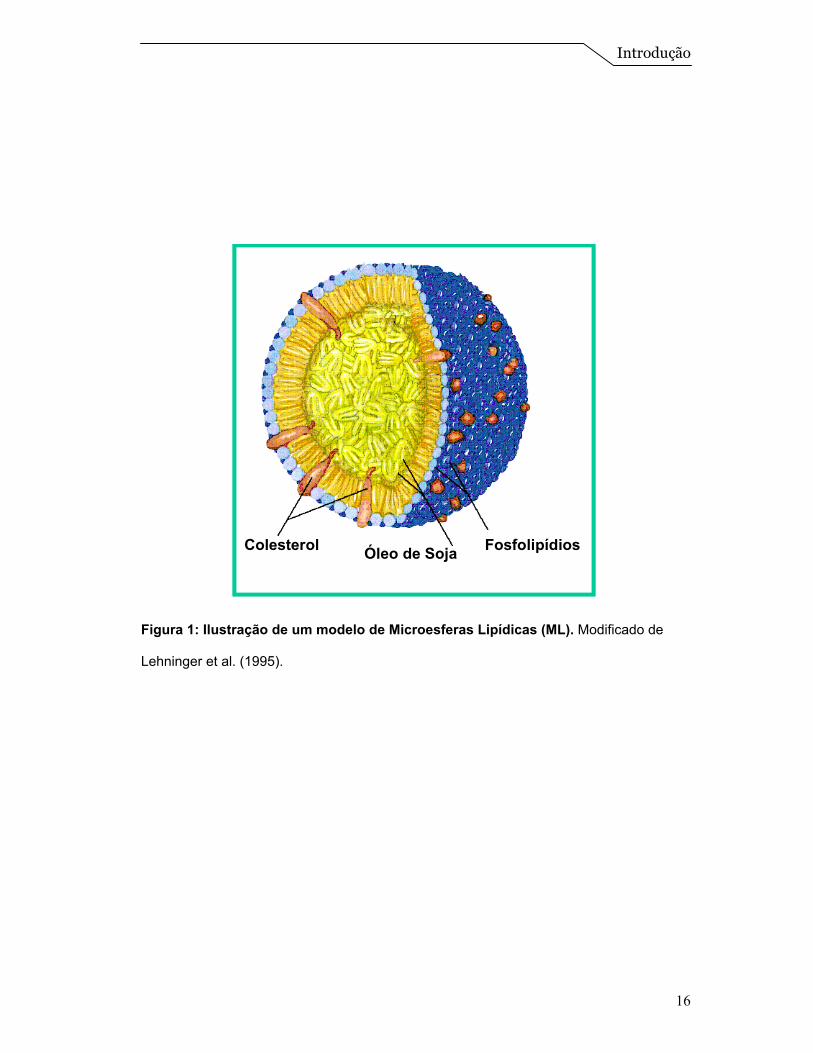
com óleo de soja e lecitina de gema de ovo (Figura 1) e comumente
administradas por via parenteral intravenosa. Podem ser usadas como
suprimentos calóricos em pacientes que são incapazes de obter nutrientes
adequados através de uma dieta normal. Seu uso clínico tem sido bem aceito,
como é o caso da Intralipid® e Lipofundin®. Nestas formulações o núcleo oleoso é
usado como um carreador de substâncias lipofílicas, para realçar sua ação
terapêutica (Igarashi et al., 1996; Nasirideen, 1998). Dentre os princípios ativos
encapsulados nesses carreadores podemos citar antiinflamatórios esteroidais e
não esteroidais, prostaglandinas bronco e vasodilatadoras, agentes
imunossupressores, citostáticos, etc. (Mizushima, 1996; Takenaga, 1996).
Introdução
16
Colesterol FosfolipídiosÓleo de SojaColesterol FosfolipídiosÓleo de Soja
Figura 1: Ilustração de um modelo de Microesferas Lipídicas (ML). Modificado de
Lehninger et al. (1995).

Introdução
17
Sua utilidade como delivery tem como base sua habilidade de incorporar
drogas com pouca solubilidade em água dentro da fase dispersa (Muller et al.,
2004). Assim, o contato direto da droga com fluidos sanguíneos e tecidos pode
ser evitado (reduzindo irritação) e a droga é liberada por um período de tempo
prolongado (liberação sustentada), o qual pode ser marcado por reduzir os efeitos
colaterais (Bock & Müller, 1994). A ML é um promissor veículo alternativo para
administração parenteral de drogas. Além disso, reduz a hidrólise, aumenta a
biodisponibilidade, bem como a atividade específica do composto carreado em
comparação com a administração em forma de solução (Nasirideen, 1998).
As ML são muito estáveis e podem ser estocadas por mais de dois anos a
temperatura ambiente (Lixin et al., 2006). Estas podem ser preparadas pelo
processo de sonicação, por métodos de agitação ou trituração. Dentre estes
métodos, o da trituração é o mais frequentemente usado até o momento, e é
chamado geralmente de método da emulsificação sob alta pressão. O princípio
deste método consiste no líquido receber uma força de trituração e emulsificar
quando dois líquidos passam através de um capilar sob alta pressão (Yamagushi
& Mizushima, 1994).
Em particular, emulsões lipídicas são consideradas sistemas superiores, devido ao
fato de poderem ser produzidas em escala industrial, ser estáveis durante armazenamento e
altamente biocompatíveis (Yamaguchi & Mizushima, 1994; Forssen & Willis, 1998).
Como, antígenos sozinhos são geralmente fracos imunógenos, estes requerem um
adjuvante para induzir imunidade protetora. As ML não somente protegem as proteínas da
degradação extracelular, como apresentam as mesmas às células relevantes do sistema
imune.

Introdução
18
No plasma sanguíneo, o transporte de lipídios é regulado por lipoproteínas
constituídas de apolipoproteínas específicas, receptores de lipoproteínas,
enzimas lipolíticas e proteínas de transferência que atuam para manter a
homeostase do colesterol e dos triacilglicerídeos (Kawakami et al. 2000). As
microesferas lipídicas são parecidas com as lipoproteínas, mas não possuem
apolipoproteínas específicas em sua superfície, enquanto sabe-se que algumas
emulsões lipídicas adquirem diversas apolipoproteínas do plasma, imediatamente
após a injeção intravenosa (Kawakami et al. 2000). As interações entre emulsões
lipídicas e apolipoproteínas desempenham um importante papel no
prolongamento do tempo de meia-vida desta emulsão na circulação lipídica. Esta
interação é controlada pelo tamanho e composição das emulsões lipídicas
(Kawakami et al. 2000).

2. OBJETIVOS

Objetivos
20
2.1 – Objetivo geral
Desenvolver uma metodologia para a preparação de microesferas lipídicas
estáveis, a fim de incorporar proteínas antigênicas da membrana de Leishmania
amazonensis, e avaliar sua viabilidade terapêutica no tratamento da leishmaniose.
2.2 – Objetivos específicos
I. Obter o extrato bruto solubilizado das proteínas de membrana da L. amazonensis e
avaliar a antigenicidade das mesmas;
II. Desenvolver um protocolo experimental para a preparação das microesferas lipídicas
na presença e na ausência das proteínas da membrana de L. amazonensis;
III. Caracterizar o tamanho das partículas e a sua estabilidade;
IV. Avaliar a interação entre as proteínas e o sistema de ML;
V. Quantificar a incorporação das proteínas pelas ML;
VI. Analisar a produção de NO e a citotoxicidade, em culturas de macrófagos
peritoneais murinos, estimulados com as proteínas de membrana e com o sistema de
ML.

3. MATERIAIS E MÉTODOS

4. RESULTADOS E DISCUSSÃO

Resultados e Discussão
38
4.1 – Solubilização e identificação das proteínas antigênicas da membrana
de L. amazonensis
O isolamento das proteínas de membrana de agentes patogênicos e a sua
posterior reconstituição em sistemas de delivery, tem sido empregado com
sucesso na tentativa de obter-se sistemas profiláticos para certas doenças
(Daghastanli et al., 2004; Santos et al., 2006; Jaafari et al., 2007; Migliaccio et al.,
2008).
Há aproximadamente trinta anos, o emprego de detergentes tem alcançado
grande sucesso na solubilização de proteínas de membranas (Silvius, 1992). Uma
solubilização efetiva destas proteínas envolve tanto a seleção do detergente
apropriado quanto a determinação de condições apropriadas para a solubilização.
Deve-se considerar a compatibilidade do detergente com métodos preparativos e
analíticos, bem como a possível remoção do mesmo na sucessiva aplicação das
proteínas de interesse (Hjelmeland, 1980; Santos & Ciancaglini, 2000).
A obtenção do parasita L. amazonensis foi realizada conforme descrito em
3.2 de materiais e métodos. Aproximadamente 5mL do pellet obtido (forma
promastigota do parasita) foi utilizado para se obter a fração de membrana,
conforme descrito em 3.4 de Materiais e Métodos.
Uma preparação típica das frações de membrana da L. amazonensis
utilizando cerca de 5mL de parasita produziu cerca de 35 mg de proteínas totais
de membrana. A eletroforese (SDS-PAGE) destas frações de membrana revelou
uma série de bandas protéicas, dentre as quais as mais intensas foram as de
massas moleculares em torno de 97; 87; 73; 66; 63; 46; 42 e 34 e 26kDa. (Figura
2A coluna 2).

Resultados e Discussão
39
Figura 2: SDS-PAGE e Western Bloting das proteínas da membrana de L.
amazonensis. (A) proteínas coradas com prata e (B) Western Blotting de: (1)
extrato de proteína solubilizada; (2) fração de membrana. SDS-PAGE e Western
Blotting foram realizados conforme descrito nos itens 3.7 e 3.8, respectivamente,
de Materiais e Métodos.
205 kDa
116 kDa
66 kDa
45 kDa
29 kDa
97,4 kDa
1 2 1 2205 kDa
116 kDa
66 kDa
45 KDa
29 kDa
97,4 kDa
A B

Resultados e Discussão
40
Posteriormente a eletroforese, foi avaliada a atividade antigênica das
proteínas presentes na fração de membrana, para isso foi realizado ensaio de
Western Blotting. A figura 2B coluna 2 mostra que muitas das bandas protéicas
presentes na fração de membrana foram reconhecidas pelo anticorpo contra
antígenos totais de L. amazonensis, obtido conforme descrito em 3.3 de materiais
e métodos.
A solubilização das proteínas da fração de membrana da L. amazonensis
foi realizada incubando-se esta na concentração de 1,0 mg/mL de proteína com
SDS 0,1% (p/v) por 30 minutos sob agitação, conforme descrito no item 3.6 de
materiais de métodos. A dosagem de proteína do material solubilizado, separado
por ultracentrifugação, revelou um rendimento de solubilização de
aproximadamente 85% das proteínas presentes nas frações de membrana.
O perfil eletroforético da fração de membrana e do extrato de proteínas
solubilizadas (EPS) foi muito semelhante, mostrando assim a eficiência do
processo de solubilização (Figura 2A coluna1). Foi feita também uma
investigação da atividade antigênica das proteínas presentes no EPS. Após o
ensaio de Western Blotting pode-se observar que bandas protéicas do EPS foram
reconhecidas pelo anticorpo contra antígenos totais de L. amazonensis assim
como na fração de membrana (Figura 2B coluna 1).
O processo de solubilização utilizado neste trabalho teve como principal
objetivo, o de preservar as características estruturais das proteínas de membrana
desejáveis para sua antigenicidade. Através da Figura 2B coluna 1, pudemos
observar que não houve perda da antigenicidade das proteínas solubilizadas, em
comparação a membrana do parasita (Figura 2B coluna 2). Processos de
solubilização de proteínas de membrana de L. amazonensis, similares ao utilizado

Resultados e Discussão
41
neste trabalho, foram descritos com êxito por Colhone et al. (2009) e Santos et al.
(2006).
Assim, uma vez obtido este extrato de proteínas solubilizadas rico em
proteínas de membrana de L. amazonensis, e sabendo que se conseguiu a
manutenção da atividade antigênica destas, efetuamos estudos sistemáticos para
definir a melhor condição para a incorporação dessas proteínas em sistemas de
microesferas lipídicas.
Vale a pena ressaltar que até o presente momento não é descrito na
literatura a utilização de ML como sistemas de delivery de um pool de proteínas
antigênicas de membrana.
4.2 – Remoção do detergente do extrato bruto solubilizado
Muitos métodos têm sido empregados para a remoção completa ou parcial
do detegente a partir de misturas de detergente-proteína ou de detergente-lipídio-
proteína (Furth, 1980; Furth et al. 1984). Dentre estes métodos tem-se: uma
simples diluição (Racker et al., 1979), diálise (Mimms et al., 1981; McCormick et
al., 1984), ultracentrifugação (Jones et al., 1988), precipitação seletiva e
ultrafiltração (Furth et al. 1984) que removem anfifílicos na forma de monômeros
(ou pequenos agregados), entretanto estes métodos são usados somente para
detergentes com alto valor de Concentração Micelar Crítica (CMC). Em casos de
detergentes com baixas CMC, como é o caso do SDS, detergente usado neste
trabalho, utiliza-se a adsorção seletiva do detergente em resinas hidrofóbicas
(Haaker e Racker, 1979; Levy et al., 1990a; Silvius, 1992). Atualmente, algumas
resinas como BioBeads® e Calbiosorb® têm sido empregadas. Este método
remove seletivamente o detergente na sua forma monomérica a qual é rápida e

Resultados e Discussão
42
eficientemente adsorvido pela resina hidrofóbica quando esta entra em contato
com a amostra detergente-proteína. Na prática, entretanto, uma significativa
quantidade de proteína pode ser adsorvida pela resina durante o processo de
remoção de detergente. Este problema, algumas vezes, pode ser reduzido por
ajustes no raio das partículas das resinas, pelos detergentes utilizados ou pela
exposição da amostra à resina por curtos períodos de incubação (Levy et al.,
1990b; Silvius, 1992; Rigaud et al., 1995; Rigaud et al., 1998; Daghastanli et al.,
2004).
Considerando que o objetivo deste trabalho é a incorporação das proteínas
da membrana de L. amazonensis no núcleo oleoso das ML, torna-se necessário à
obtenção de uma solução protéica livre de detergente. Para isso padronizamos
uma metodologia para a remoção do detergente empregado na solubilização das
proteínas da fração de membrana do parasita.
Para tal, foram realizados testes de remoção do SDS utilizando-se a resina
Calbiosorb, uma vez que já se conhece a sua eficiência na remoção do
detergente durante a formação dos proteolipossomos pelo método da co-
solubilização conforme descrito em Daghastanli et al. (2004).
Assim, foram realizados dois testes, no primeiro utilizamos uma proporção
de 200mg/mL de resina, uma vez que esta quantidade de resina é indicada para
obtenção de proteolipossomos. Os resultados obtidos revelaram uma remoção
final de 99,4% do detergente após 4 horas de tratamento, com uma troca de
resina após 2 horas, e com uma recuperação final de 76% de proteína. No
segundo teste, a quantidade de resina foi ajustada proporcionalmente a
concentração de detergente a ser removida, para eliminar o excesso de resina e
maximizar a recuperação de proteína. Assim, utilizamos 6,2mg de Calbiosorb

Resultados e Discussão
43
para cada 1mL de EPS. Ao final das 2 primeiras horas de tratamento obteve-se
uma remoção de 76,3% do SDS e nenhuma perda de proteína. Neste ponto, a
resina foi trocada e a amostra tratada por mais 2 horas, como resultado final,
observou-se uma remoção de 98,7% e uma recuperação de 92% de proteína.
Este extrato de proteína solubilizada livre de detergente foi então utilizado
na formulação das ML. É importante ressaltar que este procedimento de remoção
de detergente foi realizado sempre no momento do uso efetivo deste extrato
protéico, para evitar ao máximo o processo de agregação das proteínas
solubilizadas.
4.3 – Ensaios de preparação das microesferas lipídicas
Embora não exista nenhuma vacina comercial disponível baseada em
emulsões lipídicas, existem vários artigos na literatura documentando o grande
potencial deste sistema de transporte guiado. As possíveis formas de uso deste
sistema estão relacionadas à maneira de encapsular um antígeno viável em uma
pequena partícula, tal que este se mantenha estável e que possa ser
seguramente liberado para sítios apropriados no hospedeiro. A adequação do
veiculo a vacina pode ser obtida pela seleção de diferentes materiais e técnicas
(Kawakami et al., 2000).
O método mais usado para produção de ML encapsuladas com algum
“principio ativo” é dissolver este componente na fase oleosa e depois misturar
com os componentes aquosos sobre forte agitação, promovendo a emulsificação
do sistema (Lixin et al., 2006; Yu et al., 2006). Entretanto, para proteínas de
membrana, esta não é uma opção praticável porque estas se encontram em fase
aquosa, ou seja, não são diretamente inseridas na fase oleosa.

Resultados e Discussão
44
De acordo com a literatura os três fatores principais que influenciam a
estabilidade física de ML são: (a) Fase oleosa apropriada, (b) emulsificante
apropriado, (c) bom controle de homogeneização (Zhang et al., 2007).
Neste trabalho foram estudadas diferentes formulações de ML com o
objetivo de ajustar estes três fatores que influenciam em sua estabilidade física.
O primeiro ensaio para a preparação das microesferas lipídicas foi
realizado conforme descrito no Protocolo 1 do item 3.10.1 de Materiais e
Métodos. A metodologia empregada aqui é uma adaptação daquela descrita por
Yamaguchi e Mizushima (1994).
A suspensão resultante desta preparação, após a centrifugação, mostrou-
se não homogênea, apresentando uma fase oleosa de cor branca na superfície
da amostra o que sugere a presença de uma fase emulsionada.
Uma alíquota desta emulsão foi tomada e observada em microscópio ótico
onde foi visualizada a presença de gotículas translúcidas que são características
de microgotas de óleo e não a formação de microesferas lipídicas as quais
aparecem como partículas opacas.
Com o intuito de minimizar a agregação destes sistemas passamos a
empregar o Protocolo 2, o qual introduz na preparação o álcool polivinílico (PVA),
uma vez que é descrito que este polímero é capaz de revestir este sistema
proporcionando assim uma maior estabilidade das partículas (Hilbert et al., 1999).
Também para o Protocolo 2 foi observada a fase emulsionada na
superfície da amostra, indicando mais uma vez que esta metodologia também
não é a mais adequada para a formação das microesferas lipídicas.
Uma vez que não obtivemos êxito na separação da fase emulsionada por
centrifugação, modificamos a metodologia para a filtração a vácuo em

Resultados e Discussão
45
membranas Amicon YM-3 (Protocolo 3). Porém, durante o processo a fase oleosa
adsorveu na membrana obstruindo seus poros e impedindo assim a filtração da
amostra.
Visto que os protocolos anteriores não resultaram nas microesferas
lipídicas, elaboramos outra metodologia de montagem das microesferas
(Protocolo 4), desta vez unindo e adaptando 2 métodos descritos na literatura
(Asai & Watanabe, 1999; Hilbert et al., 1999). As amostras resultantes deste
protocolo apresentaram-se homogêneas, com aparência turva, característica de
uma suspensão de micropartículas. Devido a isto, foi adotado este protocolo para
melhor investigarmos os componentes da suspensão.
Inicialmente, foram realizadas montagens de sistemas de microesferas na
ausência de proteínas antigênicas, para a adequação das condições
experimentais.
Foram realizados estudos relacionados a concentração de PVA mais
adequada para a obtenção das microesferas lipídicas com a colaboração da
Profa. Dra. Juliana Maldonado Marchetti da FCFRP-USP e da Dra. Kátia R.P.
Dhagastanli do IQ-USP. Dentre as diversas concentrações de PVA testadas, a
formulação que resultou em uma suspensão característica de microesferas foi
quando utilizamos para a preparação da segunda emulsão uma solução de PVA
3% (p/v).
Esta preparação foi centrifugada a 2.000xg, conforme descrito no item
3.10.4 de Materiais e Métodos, e foram realizados ensaios de MEV tanto no
sobrenadante quanto no pellet obtido.
A imagem obtida para o sobrenadante (Figura 3A) revela a presença de
estruturas esféricas bem definidas, características das microesferas lipídicas

Resultados e Discussão
46
recobertas por PVA (Hilbert et al., 1999; Dinarvand et al., 2002; Bouissou et al.,
2004), e a ausência das tramas poliméricas observadas nas imagens de
microscopia das formulações com menores concentrações de PVA (0,01, 0,05 e
0,1%) como pode ser observado nas figuras 3B, 3C, 3D, respectivamente.
Quando utilizamos concentrações de PVA de 0,01; 0,05 e 0,1%, não havia
quantidade suficiente de polímero na formulação, ou seja, não havia um balanço
adequado na relação lipídio:polímero para que pudéssemos alcançar a formação
de ML. Devido a isto, obtínhamos apenas tramas de PVA ao acaso e não uma
estrutura organizada, referente as microesferas. De fato, as imagens de
microscopia eletrônica (Figuras 3B, 3C, 3D) indicam que não houve formação de
micropartículas para as preparações realizadas com menores concentrações de
PVA (Hu et al., 2002).
Já a imagem obtida do pellet, mostra a presença de microesferas que,
neste caso, estão associadas sob o excesso de PVA sedimentado pela
centrifugação (Figura 4), formando uma espécie de “tapete” de PVA.

Resultados e Discussão
47
Figura 3: Microscopia eletrônica de varredura das formulações obtidas pelo
Protocolo 4, conforme descrito no item 3.10.4 de Materiais e Métodos. (A)
sobrenadante da preparação utilizando 3% (p/v) de PVA; (B) 0,01% (p/v) de PVA;
(C) 0,05% (p/v) de PVA; (D) 0,1% de PVA.

Resultados e Discussão
48
Figura 4: Microscopia eletrônica de varredura do pellet da formulação
obtida pelo Protocolo 4, utilizando 3% PVA (p/v), conforme descrito no item
3.10.4 de Materiais e Métodos.

Resultados e Discussão
49
Estes estudos referentes à concentração de PVA indicam que para
alcançar o balanço lipídio:proteína ideal para a estabilização das ML é necessário
o uso de PVA 3 % (p/v) na formulação, no entanto, a alta viscosidade do sistema
conferida pelo uso deste, torna ineficiente a separação efetiva das ML por
centrifugação, acarretando assim a presença das ML tanto no pellet quanto no
sobrenadante.
Para a obtenção das microesferas lipídicas na presença das proteínas
antigênicas (MLP), foi utilizada a relação proteína:lipídios 1:10 (p/p), portanto
durante a preparação do sistema, o Tampão Tris-HCl 5 mM foi substituído pelo
extrato de proteínas solubilizadas (EPS) livre de detergente, em quantidade
suficiente de proteína para obtermos a mesma relação proteína:lipídio desejada.
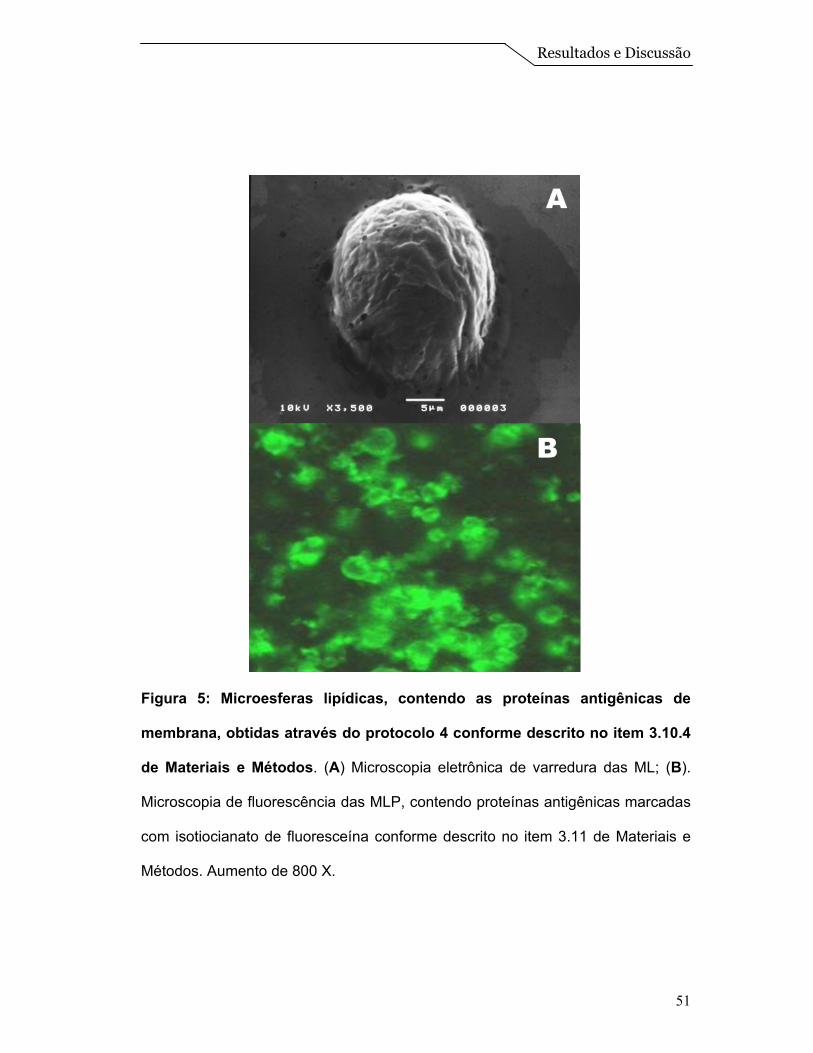
A micrografia obtida pela microscopia eletrônica de varredura para a
preparação de microesferas na presença de proteínas está mostrada na Figura
5A. Podemos observar nesta imagem a formação de sistemas esféricos
referentes à ML, semelhantes àquelas obtidas na ausência de proteína (Figura
3A).
A fim de determinarmos se as proteínas antigênicas estavam encapsuladas
nas microesferas, submetemos o EPS livre de detergente ao processo de
marcação com o fluoróforo isotiocianato de fluoresceína (FITC), o qual tem a
propriedade de se ligar aos grupamentos amino dos resíduos laterais e terminais
presentes nas proteínas.
A marcação das proteínas com FITC foi realizada conforme descrito no
item 3.11 de Materiais e Métodos. Para disponibilizar os grupos amino não
acessíveis das proteínas e então proporcionar um maior contato do fluoróforo
com a amostra de EPS na ausência de detergente, foi feita a adição de uréia

Resultados e Discussão
50
7mM. De fato, este tratamento possibilitou a marcação das proteínas as quais
ficaram visíveis no topo do gel de eletroforese sob condições desnaturantes. A
agregação das proteínas no gel pode ser devido a um novo estado
conformacional resultante do tratamento com a uréia ou ainda a presença de
uréia residual não removida durante o processo de diálise (resultados não
mostrados).
Quando estas proteínas marcadas com FITC foram adicionadas a
formulação de microesferas foi possível verificar por microscopia de fluorescência
a presença de esferas fluorescentes (Figura 5B) indicando êxito na encapsulação
das proteínas antigênicas no interior das ML.
Resultados semelhantes foram encontrados por Sun et al. (2003) para
proteínas marcadas com FITC encapsuladas em microesferas poliméricas.
Resultados e Discussão
51
Figura 5: Microesferas lipídicas, contendo as proteínas antigênicas de
membrana, obtidas através do protocolo 4 conforme descrito no item 3.10.4
de Materiais e Métodos. (A) Microscopia eletrônica de varredura das ML; (B).
Microscopia de fluorescência das MLP, contendo proteínas antigênicas marcadas
com isotiocianato de fluoresceína conforme descrito no item 3.11 de Materiais e
Métodos. Aumento de 800 X.
B

Resultados e Discussão
52
A preparação oriunda do protocolo 4 embora tenha apresentado bons
resultados, quando analisada por MEV e microscopia de fluorescência,
apresentou algumas limitações cruciais para a continuidade da pesquisa. A
preparação utilizando uma solução de PVA 3% resultou em uma solução
extremamente viscosa, o que inviabiliza estudos de espalhamento de luz
dinâmico e estudos de interação lipídio:proteína; também tivemos problemas para
quantificação das proteínas encapsuladas, uma vez que o PVA interfere em todos
métodos de quantificação disponíveis em nosso laboratório.
Diante das dificuldades encontradas para posteriores estudos, devido
principalmente a presença de PVA na formulação, optamos por substituir o PVA
por glicerol, esta mudança foi fundamentada em trabalhos descritos na literatura,
(Lixin et al., 2006; Yu et al., 2006).
Com o objetivo de otimizar a formulação de ML foram avaliadas cinco
diferentes formulações (protocolo 5), conforme descrito no item 3.10.5 de
Materiais e Métodos, (Tabela 1), estas foram preparadas e caracterizadas
separadamente, podendo-se assim ajustar a quantidade de cada componente na
formulação.
Nas formulações número 1 e 2 foram feitas principalmente investigações
quanto a proporção molar DPPC:Óleo de Soja(OS) a ser utilizada. Na primeira foi
utilizada uma proporção de aproximadamente 1:1, enquanto na segunda
formulação utilizou-se uma relação de 1:7. A avaliação feita por ensaios de
espalhamento de luz dinâmico revelou um alto índice de polidispersão na
formulação número 1, evidenciando a baixa homogeneidade da preparação, uma
vez que, quanto maior este índice, mais ampla a distribuição de tamanhos de
partículas no sistema (Nele & Pinto, 2002). Esta formulação também apresentou

Resultados e Discussão
53
baixa estabilidade, fato que ficou evidenciado nos resultados de espalhamento,
que registrou grandes valores de diâmetro médio, oriundos de uma provável
agregação no sistema.
Já na formulação de número 2 obteve-se bom índice de polidispersão,
porém assim como na primeira, houve agregação em poucos dias. Nas
formulações 3 e 4 tomou-se como base a relação DPPC:óleo de soja da
formulação número 2 e adicionou-se colesterol na proporção molar de 1:3
(colesterol:DPPC), nestas formulações foi variada a concentração de glicerol no
meio. O colesterol diminui a permeabilidade da monocamada lipídica
(Brockerhoff,1974) enquanto o glicerol é descrito como estabilizador de emulsões
(Zeringue, 1964), ambos evitam a coalescência entre as partículas estabilizando
assim as ML. Os resultados de espalhamento de luz obtidos para as formulações
número 3 e 4 mostraram a maior estabilidade da formulação de número 3.
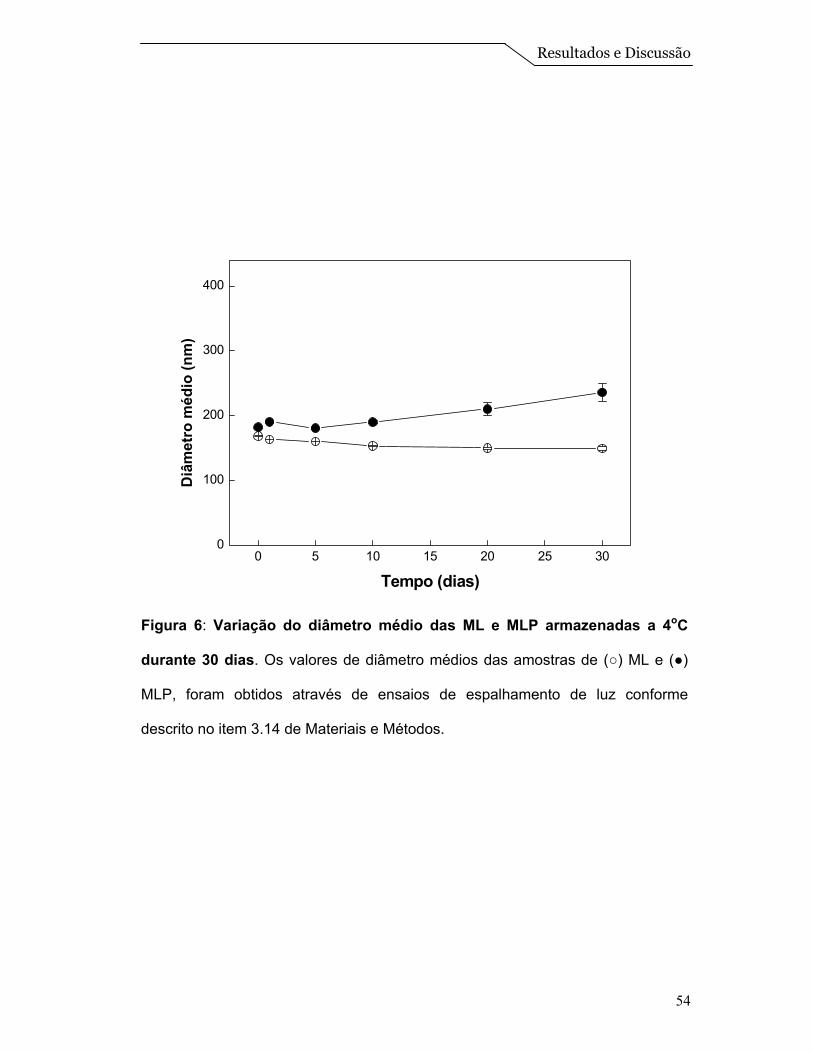
Com isso, acompanhamos por medidas de espalhamento a formulação de
número 3 por um período de 30 dias, a fim de se obter informações sobre a
estabilidade do tamanho das partículas (Figura 6). Nesta Figura pode-se observar
que as partículas se mantêm estáveis no período estudado. As ML apresentaram
diâmetro médio de 160 nm, enquanto as MLP apresentaram 200 nm de diâmetro
médio. Houve um aumento de 25% no tamanho médio das ML com a
incorporação das proteínas, e também foi observado uma possível agregação nas
MLP em torno do 15º dia, porém as partículas se mantiveram com tamanho
próximos a 200 nm. Este aumento nas partículas devido à incorporação das
proteínas também foi observado em sistemas de proteolipossomos, descritos por
Daghastanli et al. (2002).
Resultados e Discussão
54
0 5 10 15 20 25 300
100
200
300
400
Diâ
met
ro m
édio
(nm
)
Tempo (dias)
Figura 6: Variação do diâmetro médio das ML e MLP armazenadas a 4oC
durante 30 dias. Os valores de diâmetro médios das amostras de (○) ML e (●)
MLP, foram obtidos através de ensaios de espalhamento de luz conforme
descrito no item 3.14 de Materiais e Métodos.

Resultados e Discussão
55
Após o estudo de tamanho de partícula, iniciou-se estudos de interação
lipídeo:proteína, neste ponto nos deparamos com outro problema da formulação,
a concentração de lipídeos e proteínas atingidos no final da preparação estavam
abaixo do limite de detecção das nossa metodologias de dosagem, além de não
apresentarem sinal quando submetidas a ensaios prévios de calorimetria
diferencial de varredura. Para resolvermos este problema, montamos a
formulação número 5, onde apenas aumentamos em cinco vezes a quantidade de
cada componente da formulação, mantendo somente a concentração de glicerol
em 2,5% (p/v). Foi usada a formulação de número 5 para os ensaios de
caracterização da microesferas lipídicas.
4.4 – Caracterização das Microesferas Lipídicas
Para avaliar a interação entre as proteínas e o sistema de ML, foi feito um
ensaio de centrifugação em gradiente de densidade de sacarose. Amostras de
EPS, ML e MLP foram submetidas ao gradiente, conforme descrito no item 3.15
de Materiais e Métodos.
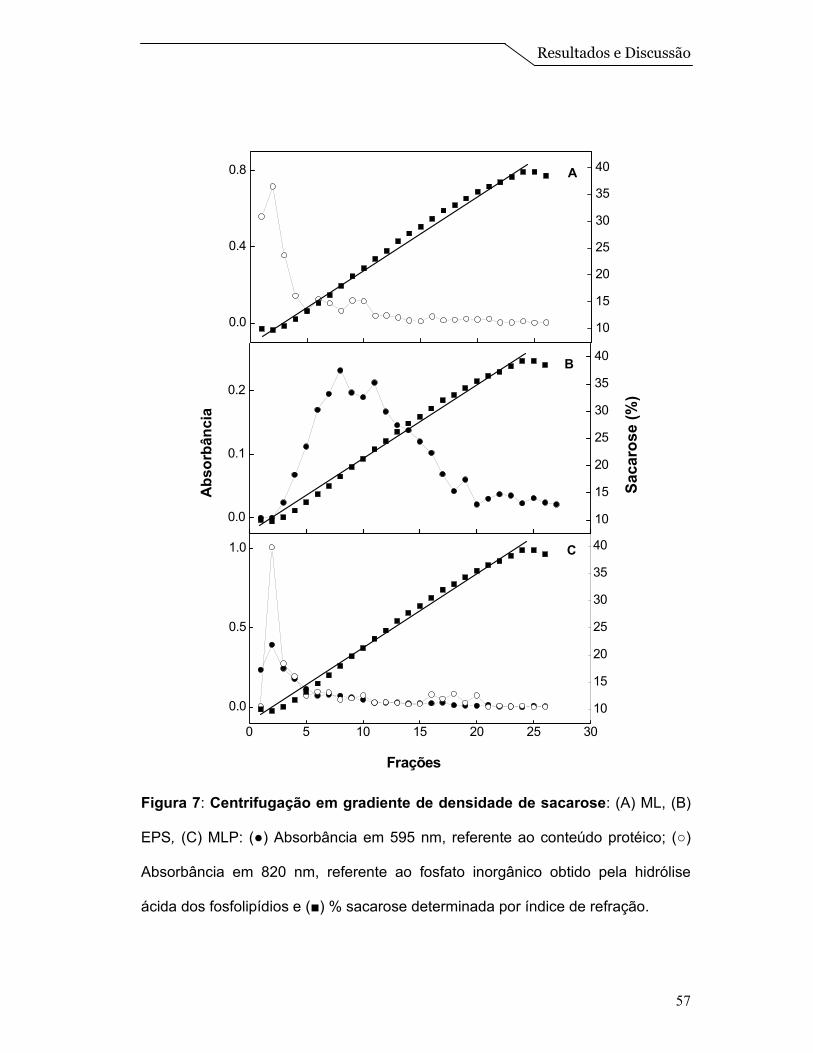
A centrifugação em gradiente de densidade de sacarose, do extrato
solubilizado de proteínas, mostrado na Figura 7B, revelou a presença de proteína
em concentrações de sacarose na faixa de 10 a 25 % (p/v), mostrando que a
amostra do EPS aparece espalhada na primeira metade do gradiente. Quando o
experimento foi realizado com ML (Figura 7A), observou-se a presença de um
único pico em uma concentração de sacarose ao redor de 13% (p/v) que é
identificado pela dosagem de fosfato inorgânico oriundo da hidrólise ácida dos
fosfolipídios, conforme descrito por Chen et al. (1956). Finalmente a centrifugação
em gradiente de densidade de sacarose das MLP, revelou a presença de um

Resultados e Discussão
56
único pico em concentrações de sacarose ao redor de 13% (p/v) (o mesmo
acontece com as ML) onde, tanto as proteínas quanto o lipídio se equilibram na
mesma densidade, sugerindo, portanto, a associação das proteínas às
microesferas lipídicas (Figura 7C).
O fato das proteínas de membrana quando livres equilibrarem-se numa
faixa de densidade de sacarose entre 10 a 25% e ao redor de 13% quando
colocadas juntas na preparação das ML sugerem que estas proteínas estão
interagindo com o sistema de ML e não simplesmente coexistindo na mesma
solução. Resultados semelhantes foram obtidos por Daghastanli et al. (2004) na
caracterização da interação de proteínas de membrana com sistemas de
lipossomos.
Resultados e Discussão
57
0 5 10 15 20 25 30
0.0
0.5
1.0
Saca
rose
(%)
C
Frações
0.0
0.1
0.2
Abs
orbâ
ncia
B
0.0
0.4
0.8
A
10
15
20
25
30
35
40
10
15
20
25
30
35
40
10
15
20
25
30
35
40
Figura 7: Centrifugação em gradiente de densidade de sacarose: (A) ML, (B)
EPS, (C) MLP: (●) Absorbância em 595 nm, referente ao conteúdo protéico; (○)
Absorbância em 820 nm, referente ao fosfato inorgânico obtido pela hidrólise
ácida dos fosfolipídios e (■) % sacarose determinada por índice de refração.

Resultados e Discussão
58
Posteriormente, foi realizado um ensaio de calorimetria diferencial de
varredura (DSC). Esta é uma técnica de grande importância na caracterização
termodinâmica de modelos de membranas e sistemas lipídicos estruturados. Na
Figura 8 pode-se visualizar os termogramas obtidos para as diferentes amostras
analisadas. Nota-se que os termogramas referentes às ML e as MLP apresentam
um comportamento muito similar com temperatura de transição ao redor de 38,1 e
37,4ºC, respectivamente. Já a amostra que continha somente uma mistura física
dos componentes das ML, apresentou uma transição na temperatura de 43,0ºC.
Cabe destacar que com esta técnica, na faixa de temperatura estudada, é
possível determinar diretamente o calor absorvido de modo a determinar, por
exemplo, a transição de fase gel para fase líquido-cristalino (fluido) do DPPC. Os
termogramas revelam picos de capacidade calorífica (Cp) numa faixa estreita de
temperatura, dos quais se obtém facilmente a temperatura de transição (Tm) para
a qual a transição está concluída em 50% (Tristam-Nagle & Nagle, 2004). Pelo
fato das ML serem um sistema mais complexo, não abordamos os parâmetros
termodinâmicos, mas apenas a temperatura de transição do DPPC que está
localizado na forma de uma monocamada na superfície das esferas (Figura 1).
Assim, a transição apresentada na “mistura física” é característica de
sistemas constituídos somente por DPPC, que apresenta uma temperatura de
transição ao redor de 42,0ºC (Tristam-Nagle & Nagle, 2004). Entretanto, observa-
se um deslocamento desta temperatura de transição para valores menores nos
sistemas de ML e MLP, que reflete a um comportamento de um sistema
estruturado de DPPC:Colesterol. Além disso, a presença da proteína não está
afetando significativamente a temperatura de transição do sistema
(DPPC:Colesterol), e isso pode ser um indicio que as mesmas estariam mais

Resultados e Discussão
59
localizadas na região interna da ML (Figura 1).
Os resultados obtidos nos ensaios de DSC (Figura 8), juntamente com os
obtidos no gradiente de densidade de sacarose (Figura 7), confirmam que houve
a formação de um sistema de microesferas lipídicas estáveis, bem como um
sistema de LM encapsuladas com proteínas de membrana da L. amazonensis.
Como as técnicas utilizadas para isolar e purificar as proteínas de
membranas, bem como as técnicas de reconstituição destas em sistemas de
delivery são bastante drásticas, podendo causar a desorganização da estrutura
nativa da proteína, e consequentemente a perda de sua função, foi feito um
ensaio de eletroforese e western blotting para as amostras de EPS sem
detergente (pós sonicação) e das MLP. A Figura 9 mostra que mesmo após o
processamento destas proteínas (obtenção das frações de membrana,
solubilização, remoção de detergente e incorporação em ML), aparentemente não
há o aparecimento de novas bandas protéicas de menor massa molecular
oriundas de proteólise, nem a perda de atividade imunogênica das proteínas,
quando reveladas no western blotting.
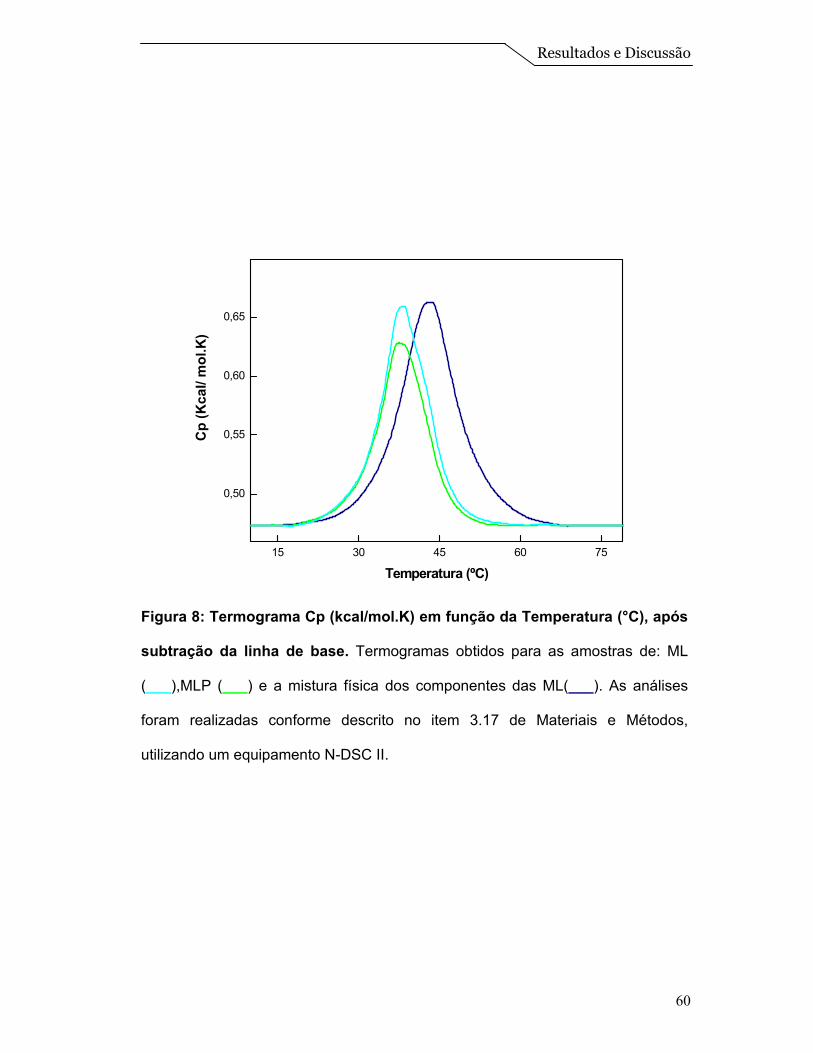
Resultados e Discussão
60
15 30 45 60 75
0,50
0,55
0,60
0,65
Cp
(Kca
l/ m
ol.K
)
Temperatura (ºC)
Figura 8: Termograma Cp (kcal/mol.K) em função da Temperatura (°C), após
subtração da linha de base. Termogramas obtidos para as amostras de: ML
(___),MLP (___) e a mistura física dos componentes das ML(___). As análises
foram realizadas conforme descrito no item 3.17 de Materiais e Métodos,
utilizando um equipamento N-DSC II.
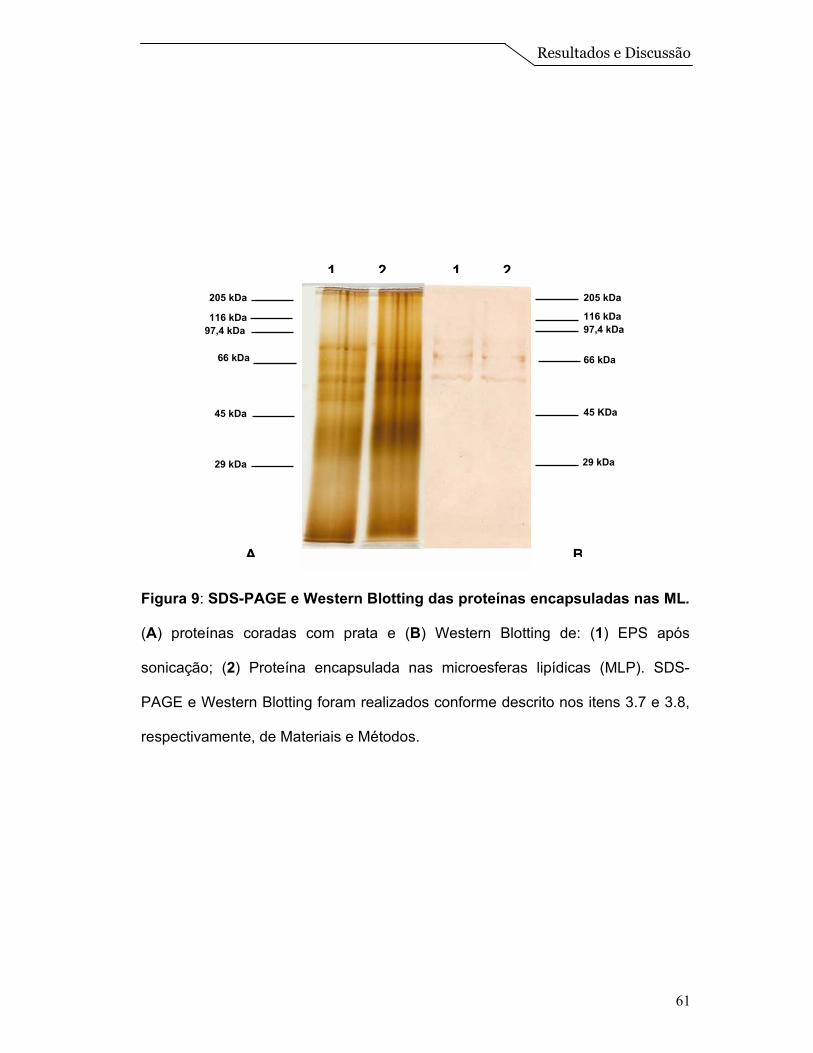
Resultados e Discussão
61
Figura 9: SDS-PAGE e Western Blotting das proteínas encapsuladas nas ML.
(A) proteínas coradas com prata e (B) Western Blotting de: (1) EPS após
sonicação; (2) Proteína encapsulada nas microesferas lipídicas (MLP). SDS-
PAGE e Western Blotting foram realizados conforme descrito nos itens 3.7 e 3.8,
respectivamente, de Materiais e Métodos.
205 kDa
116 kDa
66 kDa
45 kDa
29 kDa
97,4 kDa
1 2 1 2205 kDa
116 kDa
66 kDa
45 KDa
29 kDa
97,4 kDa
A B

Resultados e Discussão
62
Trabalhos de Colhone et al. (2009), Migliaccio et al. (2008), Santos et al.
(2006) e Daghastanli et al. (2004), onde foram reconstituídas proteínas de
membrana de patógenos de L. amazonensis (promastigota), T. cruzi, L.
amazonensis (amastigota) e Pasteurella multocida, respectivamente, em
sistemas de lipossomos, mostraram resultados similares aos obtidos no presente
trabalho onde, as frações de proteínas de membrana que foram solubilizadas e
reconstituídas em sistemas de proteolipossomos, mantiveram suas respectivas
atividades antigênicas.
É descrito que ML possuem uma atividade farmacológica mais potente
quando comparada com administrações de princípios ativos na forma de solução
(Nasirideen, 1998). Por exemplo, já estão disponíveis no mercado ML contendo
diazepam, etomidato e propofol com boas relações custo beneficio (Lixin et al.
2006). Assim, seguindo a mesma linha de raciocínio, apesar de ainda não haver
dados suficientes disponíveis na literatura, é possível que a veiculação das
proteínas em ML possa levar a uma resposta imunogênica mais eficiente.
4.5 – Quantificação das proteínas encapsuladas nas ML
A quantificação das proteínas encapsuladas nas ML foi estimada através
da dosagem de proteína nas frações cromatográficas onde se encontravam as
ML, ou seja, as frações que apresentaram maior leitura de fosfato inorgânico
(Figura 10). Primeiramente foi aplicada na coluna, uma amostra de ML, após a
dosagem de fosfato inorgânico das frações coletadas, pode-se identificar o
volume de eluição das ML. A quantidade total de proteínas encapsuladas nas ML
foi estimada após a filtração em gel, que foi feita, para garantir a total separação
das proteínas não encapsuladas. Assim, a quantificação de proteínas foi avaliada

Resultados e Discussão
63
especificamente para as frações que continham as ML (maior leitura de fosfato
inorgânico) (Figura 10).
A quantidade total de proteína encontrada nestas frações, quando
comparada à quantidade total de proteína no sistema, revelou uma incorporação
de 85% das proteínas de membrana da L. amazonensis nas microesferas
lipídicas. Santos et al. (2006) trabalhando com sistemas de lipossomos obteve
uma incorporação de 60% de proteínas solubilizadas de membrana de L.
amazonensis (amastigotas), e em ensaios de imunização utilizando
camundongos BALB/c obteve cerca de 50% de redução da lesão provocada pelo
parasita, quando o animal foi imunizado com 40µg de proteolipossomo.
Portanto, conseguiu-se uma maior incorporação das proteínas nos
sistemas de ML, quando comparados com sistemas de lipossomos descritos na
literatura (Santos et al. 2006), afirmando assim o grande potencial que este
sistema possui na incorporação de proteínas de membrana.
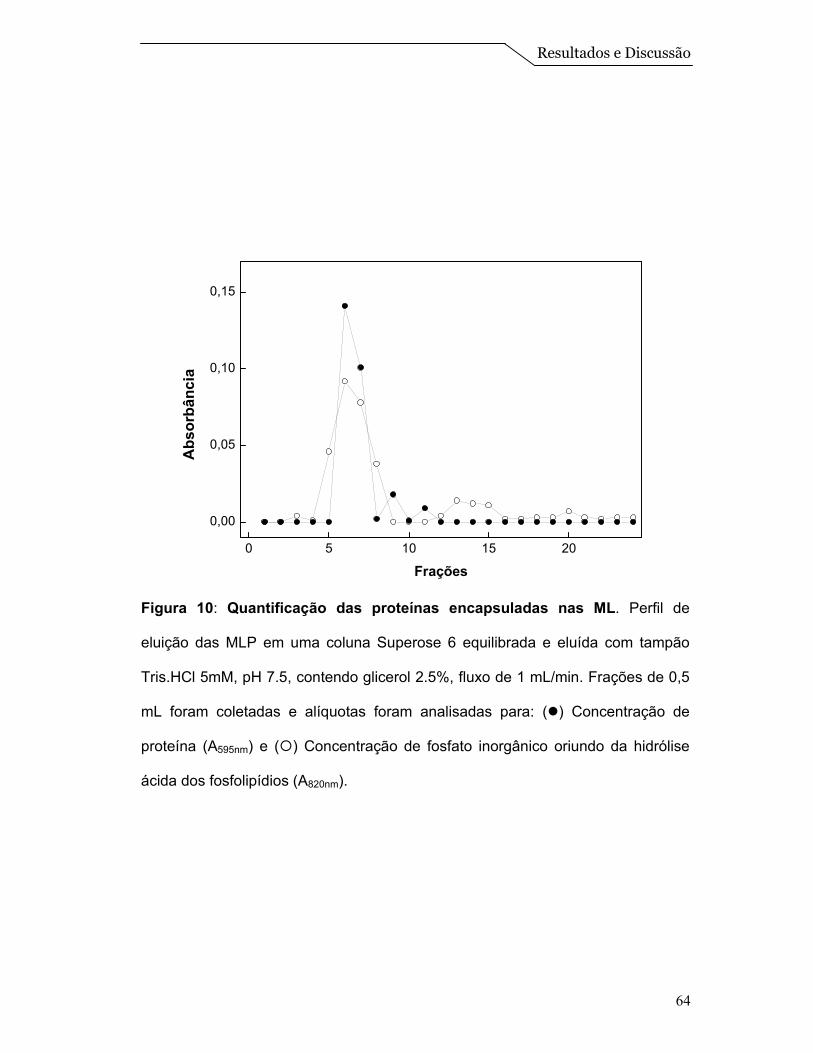
Resultados e Discussão
64
0 5 10 15 20
0,00
0,05
0,10
0,15
Abs
orbâ
ncia
Frações
Figura 10: Quantificação das proteínas encapsuladas nas ML. Perfil de
eluição das MLP em uma coluna Superose 6 equilibrada e eluída com tampão
Tris.HCl 5mM, pH 7.5, contendo glicerol 2.5%, fluxo de 1 mL/min. Frações de 0,5
mL foram coletadas e alíquotas foram analisadas para: ( ) Concentração de
proteína (A595nm) e ( ) Concentração de fosfato inorgânico oriundo da hidrólise
ácida dos fosfolipídios (A820nm).

Resultados e Discussão
65
4.6 - Análise da produção de NO e Teste de viabilidade (MTT)
Atualmente existe uma gama de evidências clínicas e experimentais sobre
a via de controle da leishmaniose cutânea: macrófagos ativados produzem IL-12
que conduz a diferenciação e a proliferação de células Th1. As células Th1
produzem IFN-γ que ativa macrófagos para matar o parasita Leishmania através
da produção de NO. É bem documentado que a produção de NO é a etapa chave
na atividade leishmanicida de macrófagos murinos (Liew et al., 1997; Ropert and
Gazzinelli, 2000; Balaraman et al., 2004). Consequentemente foi avaliada a
produção de NO por macrófagos peritoneais murinos em resposta aos diferentes
estímulos com os sistemas de microesferas lipídicas.
Como se pode observar na Figura 11, maiores níveis de NO são
produzidos quando as culturas são estimuladas com 150 µL/mL de ML e 150
µL/mL de MLP (0,42 mg/mL) quando comparados as culturas controle que não
receberam nenhum estímulo. Apesar da literatura não apresentar dados que
possibilitem a comparação dos nossos resultados, aumentos na produção de NO
em culturas estimuladas com antígenos de Leishmania incorporados em outros
sistemas constituídos de lipídios tais como, lipossomos, também foram
observados por outros autores (Mazumdar et al., 2004; Bhowmick et al., 2008).
Também, Afrin et al. (2002) observaram que estimulações com GP63 livres ou
incorporadas em lipossomos ativaram macrófagos peritoneais, impedindo assim a
multiplicação de amastigotas de L. donovani in vitro. Em conclusão, o aumento na
produção de nitrito observado nos sobrenadantes das culturas que foram
estimuladas com os diferentes tratamentos (Figura 11) sugere que esses
estímulos podem estar ativando as células, o que pode conduzir a um maior efeito
leishmanicida dos macrófagos.

Resultados e Discussão
66
Ainda, para avaliar se as microesferas, na presença ou ausência das
proteínas, poderiam exercer algum efeito tóxico nos macrófagos estimulados por
esses diferentes tratamentos, verificamos a viabilidade celular através do teste do
MTT. Esse teste tem muitas vantagens, como, por exemplo, é rápido, permite
analisar muitas amostras de uma só vez e o substrato não interfere na medida do
produto. Segundo Mosmann (1983) através do teste do MTT é possível
diferenciar células vivas de células mortas, sendo assim de ampla aplicabilidade
para medidas de sobrevivência e proliferação de várias células. Pode-se observar
na Figura 12 que macrófagos peritoneais murinos estimulados com 70 µL/mL de
EPS (0,85 mg/mL), 150 µL/mL de ML e 150 µL/mL de MLP (0,42 mg/mL) por 48
horas, não mostram diferenças significativas na viabilidade celular quando
comparados as culturas controles que não foram estimuladas. Assim, estes
resultados demonstram que os macrófagos não sofrem efeitos tóxicos extremos
por ação de tais estímulos. Embora a literatura não ofereça dados que permitam
uma análise comparativa destes resultados, foi observado que o sistema de ML e
MLP nas concentrações testadas, não apresentam efeitos citotóxicos.
Portanto, todos esses resultados indicam que as microesferas lipídicas
podem estar ativando as células a produzirem maiores níveis de NO e ainda
mantêm as funções mitocondriais normais dos macrófagos na presença dos
diferentes estímulos.
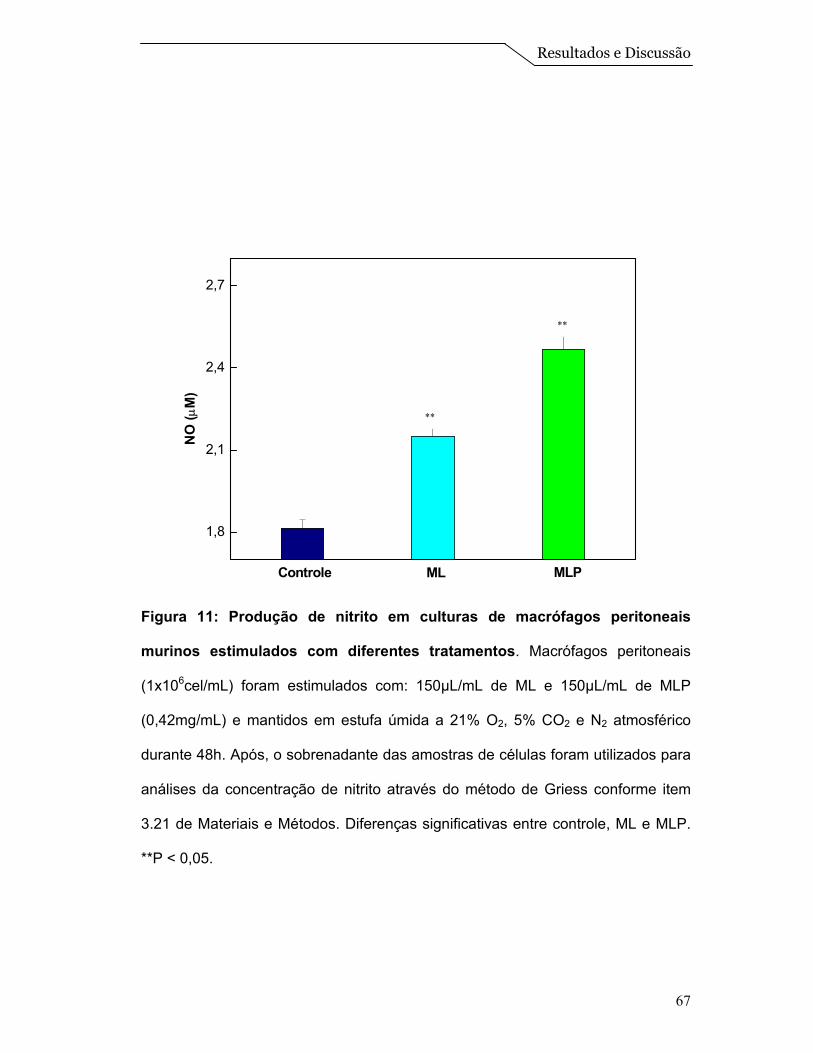
Resultados e Discussão
67
1,8
2,1
2,4
2,7
*
*
MLPMLControle
NO
( µM
)
Figura 11: Produção de nitrito em culturas de macrófagos peritoneais
murinos estimulados com diferentes tratamentos. Macrófagos peritoneais
(1x106cel/mL) foram estimulados com: 150µL/mL de ML e 150µL/mL de MLP
(0,42mg/mL) e mantidos em estufa úmida a 21% O2, 5% CO2 e N2 atmosférico
durante 48h. Após, o sobrenadante das amostras de células foram utilizados para
análises da concentração de nitrito através do método de Griess conforme item
3.21 de Materiais e Métodos. Diferenças significativas entre controle, ML e MLP.
**P < 0,05.
**
**
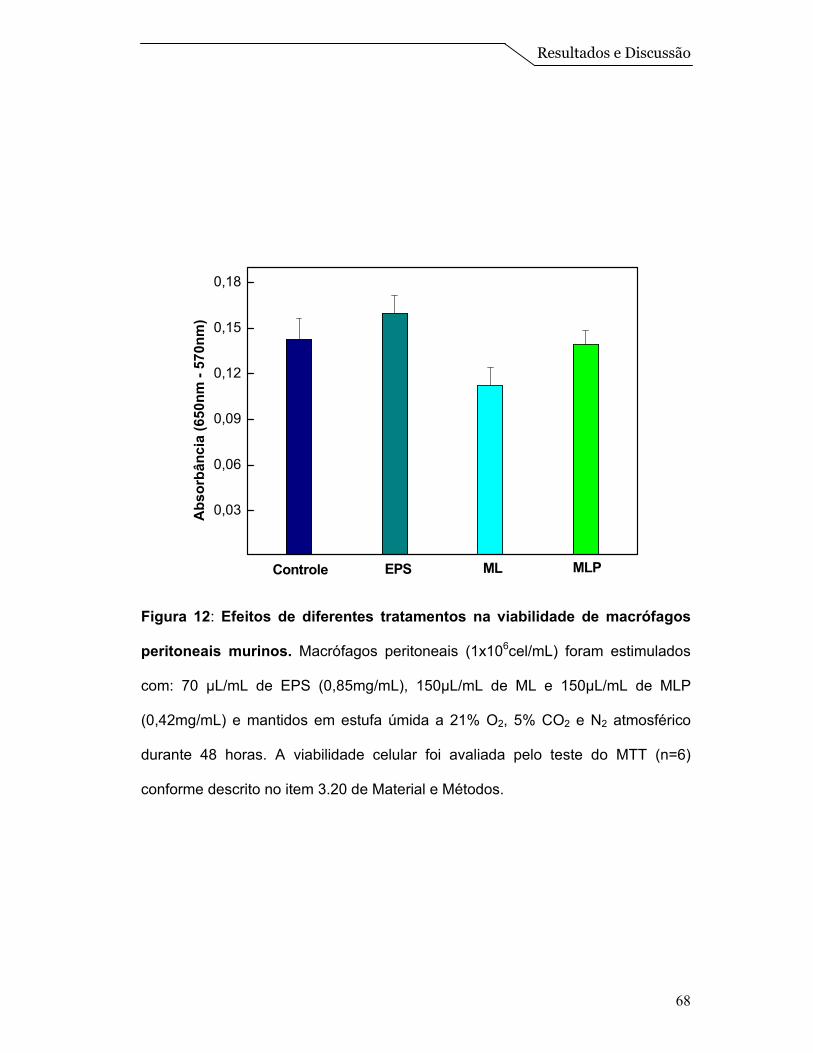
Resultados e Discussão
68
0,03
0,06
0,09
0,12
0,15
0,18
MLPMLEPSControle
Abs
orbâ
ncia
(650
nm -
570n
m)
Figura 12: Efeitos de diferentes tratamentos na viabilidade de macrófagos
peritoneais murinos. Macrófagos peritoneais (1x106cel/mL) foram estimulados
com: 70 µL/mL de EPS (0,85mg/mL), 150µL/mL de ML e 150µL/mL de MLP
(0,42mg/mL) e mantidos em estufa úmida a 21% O2, 5% CO2 e N2 atmosférico
durante 48 horas. A viabilidade celular foi avaliada pelo teste do MTT (n=6)
conforme descrito no item 3.20 de Material e Métodos.

5. CONCLUSÕES

Conclusões
70
I. Foi padronizada uma metodologia rápida e eficiente para a obtenção de um
extrato de membrana de L. amazonensis rico em proteínas antigênicas.
II. As proteínas de membrana da L. amazonensis foram eficientemente
solubilizadas após tratamento com o SDS.
III. Foi adaptado e otimizado um protocolo para a remoção do detergente do EPS
com uma recuperação de 92% de proteína total.
IV. Foi possível obter ML revestidas por PVA encapsuladas com proteínas
antigênicas de membrana.
V. Microscopia eletrônica de varredura das ML com PVA revelou estruturas
esféricas revestidas pelo polímero.
VI. Microscopia de fluorescência das ML constituídas de proteínas antigênicas
marcadas com FITC indicou que este sistema foi capaz de encapsular as
proteínas antigênicas.
VII. A presença do PVA na formulação das ML implicou em elevada viscosidade e
interferência nos métodos de dosagem de proteína. Assim, o PVA foi
substituído por formulações contendo glicerol.
VIII. A condição ótima para a obtenção de ML foi: DPPC (56,50mg), OS (376,64mg);
Colesterol (10,00mg); Glicerol (2,5%p/v) e Água (q.s.p. 50mL). Esta formulação
apresentou maior estabilidade, baixa viscosidade e polidispersão.
IX. Uma encapsulação de 85% das proteínas do extrato protéico solubilizado de L.
amazonensis foi obtida nas ML.
X. ML e MLP são estáveis por um período de até 30 dias de armazenamento a
4ºC e possuem um diâmetro médio de 160 e 200 nm, respectivamente, quando
analisadas por espalhamento de luz dinâmico.
XI. Experimentos de gradiente de densidade de sacarose e DSC confirmam que se

Conclusões
71
conseguiu obter um sistema organizado onde as proteínas estão interagindo
com as ML. Ainda, pelos resultados de DSC podemos sugerir que as proteínas
estão localizadas no interior das ML.
XII. As proteínas da membrana L. amazonensis não perderam a atividade
antigênica, mesmo após os processos de solubilização, remoção do detergente
e posterior encapsulação.
XIII. Ensaios de viabilidade celular e de geração de NO em macrófagos peritoneais
murinos, revelaram que os sistemas de ML e MLP não apresentaram nenhum
efeito citotóxico e ativaram a produção de NO nas células.
XIV. Baseado nos resultados obtidos pode-se sugerir a utilização das microesferas
lipídicas encapsuladas com proteínas antigênicas da membrana de L.
amazonensis como um potencial sistema para terapia da leishmaniose.

6. REFERÊNCIAS

Referências
73
Aebischer, T.; Wolfram, M.; Patzer, S. I.; Ilg, T., Wiese, M. & Overath, P. (2000).
Subunit vaccination of mice against new world cutaneous leishmaniasis
comparison of three proteins expressed in amastigotas and six adjuvants. Infect.
Immun., 68: 1328-1336.
Afonso, L. C. C.; Scharton, T. M.; Vieira, L. W.; Wysocka, M.; Trinchieri, G.; Scott,
P. (1994). The adjuvant effect of interleukin-12 in a vaccine against Leishmania
major. Science, 263: 235-237.
Afrin, F.; Rajesh, R.; Anam, K.; Gopinath, M.; Pal, S. & Ali, N. (2002)
Characterization of Leishmania donovani antigens encapsulated in liposomes
that induce protective immunity in BALB/c mice. Infect. Immun., 70: 6697-6706.
Amons, R. & Schrier, P.I. (1981) Removal of sodium dodecyl sulfate from proteins
and peptides by gel filtration. Anal. Biochem. 116: 439-443.
Armijos, R. X.; Weigle, M. M.; Calvopina, M.; Hidalgo, A.; Cevallos, W. &.; Correa,
J. (2004). Safety, immunogenecity, and efficacy of an autoclaved Leishmania
amazonensis vaccine plus BCG adjuvant against New World cutaneous
leishmaniasis. Vaccine, 22: 1320-1326.
Asai, Y.; Watanabe, S. (1999) Interaction of sesame oil with soybean
phosphatidylcholine and their formation of samall dispersed particles. J.
Microencapsul. 16: 705-713.
Balaraman, S.; Tewary, P.; Singh, V.K. & Madhubala, R. (2004). Leishmania
donovani induces interferon regulatory factor in murine macrophages: a host
defense response. Biochem. Biophys. Res. Commun. 317: 639-647.
Berman, J. (2003). Current treatment approaches to leishmaniasis. Curr. Opin.
Infect. Dis., 16: 397-401.
Berman, J. D.; Badaro, R.; Thakur, C. P.; Wasunna, K. M.; Behbehani, K.;
Davidson, R.; Kuzoe, F.; Pang, L.; Weerasuriya, K. & Bryceson, A. D. (1998).
Efficacy and safety of liposomal amphotericin B (AmBiosome) for visceral
leishmaniasis in endemic developing countries. Bull. World Health Organ., 76:
25-32.
Bhowmick, S.; Ravindran, R. & Ali, N. (2008) Gp63 in stable cationic liposomes
confers sustained vaccine immunity to susceptible BALB/c mice infected with
Leishmania donovani. Infect. Immun., 76: 1003-1015.
Bock, T., Müller, B. W. (1994) A novel assay to determine the hemolytic activity of

Referências
74
drugs incorporated in colloidal carriers systems. Pharm. Res. 11, 589–591.
Bordier, C. (1987) The promastigote surface of Leishmania. Parasitol. Today 24:
73-79.
Borja-Cabrera, G. P.; Cruz Mendes, A.; Paraguai de Souza, E.; Hashimoto Okada,
L. Y.; de Atrivellato, F. A.; Kawasaki, J. K.; Costa, A. C.; Reis, A. B.; Genaro, O.;
Batista, L. M.; Palatnik, M. & Palatnik-de-Sousa, C. B. (2004). Effective
immunotherapy against canine visceral leishmaniasis with the FML-vaccine.
Vaccine, 2234-2243.
Bouissou, C.; Potter, U.; Altroff, H.; Mardon, H & van der Walle, C. (2004)
Controlled release of the fibronectin central cell binding domain from polymeric
microspheres. J. Control. Release 95: 557-566.
Boyd, B. J. (2008) Past and future evolution in colloidal drug delivery systems. Expert Opin. Drug Deliv. 5: 69-85.
Brittingham, A.; Chen, G.; McGwire, B. S.; Chang, K. P. & Mosser, D. M. (1999)
Interaction of Leishmania gp63 with cellular receptors for fibronectin. Infect.
Immun., 67: 4477-4484.
Brittingham, A.; Morrison, C.J.; Mcmaster, W.R.; Mcgwire, B.S.; Chang, K.P. &
Mosser, D.M. (1995) Role of the Leishmania surface protease gp63 in
complement fixation, cell adhesion, and resistance to complement-mediated
lysis. J. Immunol. 155: 3102-3111.
Brockerhoff, H. (1974) Model of interaction of polar lipids, cholesterol, and proteins
in biological membranes. Lipids. 9: 645 – 650.
Burns, J. M. Jr.; Scott, J. M.; Carvalho, E. M.; Russo, D. M.; March, C. J.; Van
Ness, K. P. & Reed, S. G. (1991) Characterization of a membrane antigen of
Leishmania amazonensis that stimulates human immune responses. J. Immunol.
146: 742-748.
Button, L. L. & Mcmaster, W. R. (1988) Molecular cloning of the major surface
antigen of Leishmania. J. Exp. Med. 167: 724-729.
Calvopina, M.; Barroso, P. A.; Marco, J. D.; Korenaga, M.; Cooper, P. J.; Nonaka,
S. & Hashiguchi, Y. (2006) Efficacy of vaccination with a combination of
Leishmania amastigote antigens and the lipid A-analogue ONO-4007 for
immunoprophylaxis and immunotherapy against Leishmania amazonensis
infection in a murine model of New World cutaneous leishmaniasis. Vaccine. 24:
5645-5652.

Referências
75
Camargo, L. M. A. & Barcinski, M. A. (2003). Leishmanioses, feridas bravas e
kalazar. Cienc. Cult., 1: 34-37.
Campos-Neto, A.; Porrozzi, R.; Greeson, K.; Coler, R. N.; Webb, J. R.; Seiky, Y.
A. W.; Reed, S. G. & Grimaldi, G. (2001). Protection against cutaneous
leishmaniasis induced by recombinant antigens in murine and non-human
primate models of the human disease. Infect. Immun., 69: 4103-4108.
Chaudhuri, G.; Chaudhuri, M.; Pan, A. & Gang, K. P. (1989). Surface acid
proteinase (gp63) of Leishmania mexicana. J. Biol. Chem., 264: 7483-7489.
Chaudhuri, G.; Chaudhuri, M.; Pan, A. & Gang, K.P. (1989). Surface acid
proteinase (gp63) of Leishmania mexicana. J. Biol. Chem. 264: 7483-7489.
Chen, P.S.Jr.; Toribara, T.Y & Warner, H. (1956). Microdetermination of
phosphorus. Anal. Chem. 28: 1756-1758.
Cluff, C. W.; BaldridgE, J. R.; Stover, A. G.; Evans, J. T.; Johnson, D. A.; Lacy, M.
J.; Clawson, V. G.; Yorgensen, V. M.; Johnson, C. L.; Livesay, M. T.; Hershberg,
R. M. & Persing, D. H. (2005). Synthetic toll-like receptor 4 agonists stimulate
innate resistance to infectious challenge. Infect. Immun. 73:3044-3052.
Coler, R. N. & Reed, S. G. (2005). Second-generation vaccines against
leishmaniasis. Trends. Parasitol. 21: 244-249.
Colhone, M.C.; Nobre, T.M.; Zaniquelli, M.E.D.; Stabeli, R.G; Ciancaglini, P.
(2009) Incorporation of antigenic GPI-proteins from Leishmania amazonensis to
membrane mimetic systems: influence of DPPC/cholesterol ratio. J. Colloid
Interf. Sci.. 333: 373–379.
Cunningham, A. C. (2002) Parasitic adaptive mechanisms in infection by
Leishmania. Exp. Mol. Pathol. 72: 132-141.
Daghastanli, K.R.P., Ferreira, R.B., Thedei Jr., G., Maggio, B. & Ciancaglini, P.
(2004). Lipid composition-dependent incorporation of multiple membrane
proteins into liposomes. Coll. Surf. B. 36: 127-137.
Davis, A. J. & Kedzierski, L. (2005). Recent advances in antileishmanial drug
development. Current Opinion in Investigational Drugs, 6: 163-169.
Delorenzi, J.C.; Attias, M.; Gattass, C.; Andrade, M.; Rezende, C.; Pinto, A.C.;
Henriques, A.T.; Bou-Habib, D.C. & Saraiva, E.M. (2001) Antileishmanial activity
of na índole alkaloid from Peschiera australis. Antimicrob. Agents. Chemother.,
45:1349–1354.
Descoteaux, A. & Turco, S. J. (1999). Glycoconjugates in Leishmania infectivity.

Referências
76
Biochim. Biophys. Acta., 1455: 341-352.
Descoteaux, A. & Turco, S. J. (2002). Functional aspects of the Leishmania
donovani lipophosphoglycan during macrophage infection. Microbes Infect., 4:
975-981.
Dinarvand, R.; Mirfattachi, S.; Atyabi, F. (2002) Preparation, characterization and
in vito drug release of isosorbide dinitrate microspheres. J. Microencapsul., 19:
73-81.
Forssen, E. & Willis, M. (1998). Ligand-targeted liposomes. Adv. Drug Deliv. Rev.,
29: 249–271
Frommel, T. O.; Button, L. L.; Fujikura, Y. & Mcmaster, W. R. (1990) The major
surface glycoprotein (GP63) is present in both life stages of Leishmania. Mol.
Biochem. Parasitol. 38: 25-32.
Furth, A.J. (1980). Removing unbound detergent from hydrophobic proteins. Anal.
Biochem. 109: 207-215.
Furth, A.J.; Bolton, H.; Potter, J. & Priddle, J.D. (1984) Separating detergent from
proteins. Meth. Enzymol. 104: 318-328.
Garg, R.; Srivastava, J. K.; Pal, A.; Naik, S. & Dube, A. (2005). Isolation of integral
membrane proteins of Leishmania promastigotes and evaluation of their
prophylactic potencial in hamsters against experimental visceral leishmaniasis.
Vaccine, 23: 1189-1196.
Genaro, O.; de Toledo, V. P.; da Costa, C. A.; Hermeto, M. V.; Afonso, L. C. &
Mayrink, W. (1996). Vaccine for prophylaxis and immunotherapy, Brazil. Clin.
Dermatol., 14: 503-512.
Ghosh, M.; Pal, C.; Ray, M.; Maitra, S.; Mandal, L. & Bandyopadhyay, S. (2003).
Dendritic cell-based immunotherapy combined with antimony-based
chemotherapy cures established murine visceral leishmaniasis. J. Immunol.,
170: 5625-5629.
Gontijo, B. & Carvalho, M. L. R. (2003) Leishmaniose tegumentar americana. Rev.
Soc. Bras. Med. Trop. 36(1): 71-80.
Gontijo, C.M.F. & Melo, M.N. (2004) Leishmaniose Visceral no Brasil: Quadro
Atual, Desafios e Perspectivas. Rev Bras Epidemiol 7 (3):338–349.
Green, S. J.; Meltzer, M. S.; Hibbs J. B. & Nacy, N. A. (1990) Activated
macrophages destroy intracellular Leishmania major amastigotes by an L-
arginine-dependent killing mechanism. J. Immunol., 144: 278-283.

Referências
77
Greenblatt, C. L. (1980). The present and future of vaccination for cutaneous
leishmaniasis. Prog. Clin. Biol. Res., 47: 259-285.
Haaker, H. & Racker, E. (1979). Purification and reconstitution of the Ca2+-ATPase
from plasma membrane of pig erythrocytes. J. Biol. Chem. 254: 6598-6602.
Handman, E. (2000) Cell biology of Leishmania. Adv. Parasitol., 44:1-39.
Handman, E.; Noormohammadi, A. H.; Curtis, J. M.; Baldwin, T. & Sjolander, A.
(2000) Therapy of murine cutaneous leishmaniasis by DNA vaccination. Vaccine.
18: 3011-3017.
Hartree, E.F. (1972) Determination of protein: a modification of the Lowry method
that gives a linear photometric response. Anal. Biochem. 48: 422-427.
Herwaldt, B. L. (1999) Leishmaniasis. Lancet 354: 1191-1199.
Hilbert, A.K.; Fritzsche, U.; Kissel, T. (1999) Biodegradable microspheres
containing influenza A vaccine: immune response in mice. Vaccine 17: 1065-
1073.
Hjelmeland, L.M. (1980). A nonderaturing zwitterionic detergent for membrane
biochemistry: design and synthesis. Proc. Natl. Acad. Sci. USA, 77: 6368-6370.
Hu, F.Q.; Yuan, H.; Zhang, H.H. & Fang, M. (2002) Preparation of solid lipid
nonoparticles with clobetasol proprionate by a novel solvent diffusion method in
aquous system and physicochemical characterization. Int. J. Pharm., 239: 121-
128.
Igarashi, R., Takenaga, M., Matsuda, T., 1996. Distribution of lipid microsphere
preparations. Adv. Drug. Deliv. Rev. 20, 147–154.
Ilg, T.; Handman, E. & Stierhof, Y. D. (1999) Proteophosphoglycan from
Leishmania promastigotes and amastigotes. Biochem. Soc. Trans. 27: 518-525.
Ilgoutz, S. C. & MCconville, M. J. (2001) Function and assembly of the Leishmania
surface coat. Intern. J. Parasitol. 31: 899-908.
Jaafari, M. R.; Badiee, A.; Khamesipour, A.; Samiei, A.; Soroush, D.; Kreiri, M. T.;
Barkhordari, F.; McMaster, W. R. & Mahboudi, F. (2007). The role of CpG ODN
in enhancement of immune response and protection in BALB/c mice immunized
with recombinant major surface glycoprotein of Leishmania (rgp63) encapsulated
in cationic liposome. Vaccine, 25: 6107-6117.

Referências
78
Jones, O.T.; Eubanks, J.H.; Earmest, J.P. & McNamee, M.G. (1988).
Reconstitution of the nicotinic acetylcholine receptor using a lipid substitution
technique. Biochim. Biophys. Acta, 944: 359-366.
Kawakami, S.; Yamashita, F & Hashida, M. (2000) Disposition characteristics of
emulsions and incorporated drugs after systemic or local injection. Adv. Drug.
Deliv. Rev, 45: 77–88
Kedzierski, L.; Zhu, Y. & Handman, E. (2006). Leishmania vaccines: progress and
problems. Parasitol., 133: S87-S112.
Laemmli, U.K. (1970) Cleavage of structural proteins during the assembly of the
head bacteriophage T4. Nature 227: 680-685.
Laison, R. (1997). Leishmania e leishmaniose, com particular referência à região
Amazônica do Brasil. Revista Paraense de Medicina, 11: 29-40.
Lehninger, A. L.; Nelson, D. L. & Cox, M. M. (1995). Princípios de bioquímica.
Tradução de W.R. Loodi, e A.A. Simões. São Paulo: Sarvier. 839 p. Tradução
de: Principles of biochemistry
Levy, D.; Bluzat, A.; Seigneuret, M. & Rigaud, J.L. (1990b). A systematic study of
liposome and proteolipossome reconstitution involving Bio-Bead-mediated triton
X-100 removal. Biochim. Biophys. Acta., 1025: 179-190.
Levy, D.; Gulik, A.; Seigneuret, M. & Rigaud, J.L. (1990a) Phospholipid vesicle
solubilization and reconstitution by detergents. Symmetrical analysis of the two
processes using octaethylene glycol mono-n-dodecyl ether. Biochem., 29: 9480-
9488.
Liew, F.Y.; Wei, X.Q.; Proudfoot, L. (1997). Cytocines and nitric oxide as effector
molecules against parasitic infections. Philos. Trans. R. Soc. Lond. B. Biol. Sci.
352: 1311-1315.
Lixin, W.; Haibing, H.; Xing, T.; Ruiying, S.; Dawei, C. (2006) A less irritant
norcantharidin lipid microspheres: Formulation and drug distribution. Int. J.
Pharm 323, 161 – 167.
Machado-Pinto, J.; Pinto, J.; da Costa, C. A.; Genaro, O.; Marques, M. J.;
Madabber, F.; Mayrink, W. (2002) Immunochemotherapy for cutaneous
leishmaniasis: a controlled trial using killed Leishmania (Leishmania)
amazonensis vaccine plus antimonial. Int. J. Dermatol. 41(2): 73-78.

Referências
79
Marques-da-Silva, E. A.; Coelho, E. A.; Gomes, D. C.; Vilela, M. C.; Masioli, C. Z.;
Tavares, C. A.; Fernandes, A. P.; Afonso, L. C. & Rezende, S. A. (2005).
Intramuscular immunization with p36(LACK) DNA vaccine induces IFN-gamma
production but does not protect BALB/c mice against Leishmania chagasi
intravenous challenge. Parasitol. Res., 98: 67-74.
Marzochi, M. C. A. (1992). Leishmanioses no Brasil (As leishmanioses
tegumentares). J. B. M., 63: 81-105.
Mazumdar, T.; Anam, K. & Ali, N. (2004) A mixed Th1/Th2 response elicited by a
liposomal formulation of Leishmania vaccine instructs Th1 responses and
resistance to Leishmania donovani in susceptible BALB/c mice. Vaccine. 22:
1162-1171.
McConville, M. J. & Ferguson, M. A. (1993). The structure, biosynthesis and
function of glycosylated phosphatidylinositols in the parasite protozoa and higher
eukaryotes. Biochem. J., 294: 305-324.
Mcconville, M.J. & Blackwell, J.M. (1991) Developmental changes in the
glycosylated phosphatidylinositols of Leishmania donovani. Characterization of
the promastigotes and Amastigote glycolipids. J. Biol. Chem. 266: 1510-1519.
MCconville, M.J.; Mullin, K.; Ilgoutz, S. & Teasdale, R.D. (2002) Secretory
pathaway of trypanosomatids parasites. Microbiol. Mol. Biol. Rev. 66: 122-154.
McCormick, J.I.; Tsang, D. & Jhonstone, R.M. (1984). A simple and efficient
method for reconstitution of amino acid and glucose transport systems from
Ehrlich ascites cells. Arch. Biochem. Biophys. 231: 355-265.
Migliaccio, V.; Santos, F.R.; Ciancaglini, P. & Ramalho-Pinto, F.J. (2008) The use
of proteoliposome as a vaccine against Trypanosoma cruzi in mice. Chem phys
lipids 152: 86-94.
Mimms, L.T.; Zampighi, G.; Nozaki, Y.; Tanford, C. & Reynolds, J.A. (1981).
Phospholipid vesicle formation and transmembrane protein incorporation using
octyl glucoside. Biochem., 20: 833-840.
Ministério da Saúde. Fundação Nacional de Saúde. Manual de Controle da
Leishmaniose Tegumentar Americana. Coordenação de Vigilância
Epidemiológica. 5ª edição, 2000.
Mizushima, Y. (1996) Adv. Drug Del. Rev 20, 113.
Modabber, F. (1995). Vaccines against leishmaniasis. Ann. Trop. Med. Parasitol.,
89: 83-88.

Referências
80
Mosmann, T. (1983) Rapid colorimetric assay for cellular growth and survival:
Application to proliferation and cytotoxicity assays. J. Immunol. Meth., 65: 55-63.
Mosser, D. M. & Rosenthal, L. A. (1993). Leishmania-macrophage interactions:
multiple receptors, multiple ligands and diverse cellular responses. Semin. Cell.
Biol., 4: 315-322.
Müller, R.H., Schmidt, S., Buttle, I., Akkar, A., Schmitt, J., Br¨omer, S., 2004.
Solemuls-novel technology for the formulation of i.v. emulsions with poorly
soluble drugs. Int. J. Pharm. 269, 293–302.
Murray, H. W. (2004). Progress in the treatment of a neglected infectious disease:
visceral leishmaniasis. Expert. Rev. Anti. Infect. Ther., 2: 279-292.
Nadim, A.; Javadian, E.; Tahvildar-Bidruni, G. & Ghorbani, M. (1983).
Effectiveness of leishmanization in the control of cutaneous leishmaniasis. Bull.
Soc. Pathol. Exot. Filiales, 76: 377-383.
Nasirideen, S. (1998). Naproxen incorporated lipid emulsion. I. Fomulation and
stability study. J. Clin. Pharm. Ther. 23, 57–65.
Nele, M. & Pinto,J.C. (2002). Molecular-Weight Multimodality of Multiple Flory
Distributions. Macromol. Theory Simul.. 11, 293 – 307.
O´Hagan, D.T.; Mac Kichan, M.L.; Singh, M. (2001) Recent developments in
adjuvants for vaccines against infectious diseases. Biomol. Eng., 18: 69-85
Palatnik-de-Souza, C. B. (2008). Vaccines for leishmaniasis in the fore coming 25
years. Vaccine, 26: 1709-1724.
Pessoa, S. M. (1982). Parasitologia Médica. Rio de Janeiro: Guanabara Koogan.
Racker, E.; Violand, B.; O’Neal, S.; Alfonzo, M. & Telford, J. (1979).
Reconstitution, a way of biochemical research, some new approaches to
membrane-bound enzymes. Arch. Biochem. Biophys., 198: 470-477.
Ramiro, M. J.; Zarate, J. J.; Hanke, T.; Rodriguez, D.; Rodriguez, J. R.; Esteban,
M.; Lucientes, L.; Castillo, J. A. & Larraga, V. (2003). Protection in dogs against
visceral leishmaniasis caused by Leishmania infantum is achieved by
immunization with a heterologous prime-boost regime using DNA and vaccinia
recombinant vectors expressing LACK. Vaccine, 21: 2474-2484.
Read, S. M. & Northcote, D. H. (1981) Minimization of variation in the response to
different proteins of the coomassie blue G dye-binding assay for protein. Anal.
Biochem., 116, 53 – 64.

Referências
81
Rigaud, J.L.; Levy, D.; Mosser, G. & Lamperd, O. (1998). Detergent removal by
non-polar polystyrene beads-applications to membrane. Protein Reconstitution
and two-dimensional crystallization. Eur. Biophys. J., 27: 3004-319.
Rigaud, J.L.; Pitard, B. & Levy, D. (1985). Reconstitution of membrane proteins
into lipossomes: application to energy-transducing membrane proteins. Biochim.
Biophys. Acta. 1231: 223-246.
Ropert, C. & Gazzinelli, R.T. (2000). Signalling of immune system cells by
glycosylphosphatidylinositol (GPI) anchor and related structures derived from
parasitic protozoa. Curr. Opin. Microbiol. 3: 395-403.
Russel, D.G.; Talamas-Rohana, P. & Zelechowski, J. (1989) Antibodies raised
against synthetic peptides from the Arg-Gly-Asp-containing region of the
Leishmania surface protein gp63 cross-react with human C3 and interfere with
pg63-mediated binding to macrophages. Infec. Immun. 57: 630-632.
Santos, D. O.; Coutinho, C. E. R.; Madeira, M. F.; Bottino, C. G.; Vieira, R. T.;
Nascimento, S. B.; Bernardino, A.; Bourguignon, S. C.; Corte-Real, S.; Pinho, R.
T.; Rodrigues, C. R. & Castro, H. C. (2008). Leishmaniasis treatment – a
challenge that remains: Rev. Parasitol. Res., 103: 1-10.
Santos, F. R.; Ferraz, D. B.; Daghastanli, K. R. P.; Ramalho-Pinto, F. J. &
Ciancaglini, P. (2006). Mimetic membrane system to carry multiple antigenic
proteins from Leishmania amazonensis. J. Membrane Biol., 210: 173-181.
Santos, H.L. & Ciancaglini, P. (2000). A practical approach to the choice of a
suitable detergent and optimal conditions to solubilize a membrane protein. J.
Biochem. Educ., 28: 178-182.
Santos, W. R.; Aguiar, I. A.; Paraguai de Souza, E.; de Lima, V. M.; Palatnik, M. &
Palatnik-de-Sousa, C. B. (2003). Immunotherapy against murine experimental
visceral leishmaniasis with the FML-vaccine. Vaccine, 21: 4668-4676.
Saraiva, E. M.; de Figueiredo Barbosa, A.; Santos, F. N.; Borja-Cabrera, G. P.;
Nico, D.; Souza, L. O.; de Oliveira Mendes-Aguiar, C.; de Souza, E. P.; Fampa,
P.; Parra, L. E.; Menz, I.; Dias Jr, J. G.; de Oliveira, S. M. & Palatnik-de-Sousa,
C. B. (2006). The FNL-vaccine (Leishmune®) against canine visceral
leishmaniasis: a transmission blocking vaccine. Vaccine, 24: 2423-2431.
Senekji, H. A. & Beattie, C. P. (1941). Artificial infection and immunization of man
with cultures of Leishmania tropica. Trans. R. Soc. Trop. Med. Hyg., 34: 415-
419.

Referências
82
Silva, L. J.; Camargo-Neves, V. L. F. (2004). As leishmanioses, uma visão para o
clínico. Prática Hospitalar. 36(VI): nov-dez. Disponível em:
<http://www.praticahospitalar.com.br/pratica2036/paginas/materia/2010-
36.html>. (10/07/2006).
Silvius, J.R. (1992). Solubilization and functional reconstitution of biomembrane
components. Annu. Rev. Bioph. Biol., 21: 323-348.
Soto, J.; Toledo, J.; Gutierrez, P.; Nicholls, R. S.; Padilha, J.; Engel, J.; Fischer,
C.; Voss, A. & Berman, J. (2001). Treatment of American cutaneous
leishmaniasis with miltefosine, an oral agent. Clin. Infect. Dis., 33: E57-61.
Sun, S.W.; Jeong, Y.I.; Jung, S.W. & Kim, S.H. (2003) Characterization of FITC-
albumin encapsulated poly(DL-lacco-glycolide) microspheres and its release
characteristics. J. microencapsul. 20: 479-488.
Sundar, S.; Jha, T. K.; Thakur, C. P.; Engel, J.; Sindermann, H.; Fischer, C.;
Junge, K.; Bryceson, A & Berman, J. (2002). Oral miltefosine for Indian visceral
leishmaniasis. N. Engl. J. Med., 347: 1739-1746.
Takenaga, M. (1996) Adv. Drug Del. Rev. 20, 209.
Teixeira, M. J.; Teixeira, C. R.; Andrade, B. B.; Barral-Netto, M.; Barral, A. (2006).
Chemokines in host-parasite interactions in leishmaniasis. Trends Parasitol., 22:
32-40.
Titus, R. G.; Gueiros-Filho, F. J.; de Freitas, L. A. & Beverley, S. M. (1995).
Development of a safe live Leishmania vaccine line by gene replacement. Proc.
Natl. Acad. Sci. USA, 92: 10267-10271.
Tristam-Nagle, S. & Nagle, J.F. (2004) Lipid bilayers:termodynamics, structure,
fluctuations and interactions. Chemistry and Physics of Lipids 127:3-14.
Turco, S. J. & Descoteaux, A. (1992). The lipophosphoglycan of Leishmania
parasites. Annu. Rev. Microbiol., 46: 65-94.
Vélez, I. D.; del Pilar Agudelo, S.; Arbelaez, M. P.; Gilchrist, K.; Robledo, S. M.;
Puerta, J. A.; Zicker, F.; Berman, J. & Modabber, F. (2000). Safety and
immunogenicity of a killed Leishmania (L.) amazonensis vaccine against
cutaneous leishmaniasis in Colombia: a randomized controlled trial. Trans. R.
Soc.Trop. Med. Hyg., 94: 698-703.
Vélez, I. D.; Gilchrist, K.; Arbelaez, M. P.; Rojas, C. A.; Puerta, J. A.; Antunes, C.
M.; Zicker, F. & Modabber, F. (2005). Failure of a killed Leishmania amazonensis
vaccine against American cutaneous leishmaniasis in Colombia. Trans. R. Soc.

Referências
83
Trop. Med. Hyg., 99: 593-598.
Veras, P.; Brodskyn, C.; Balestieri, F.; Freitas, L.; Ramos, A.; Queiroz, A.; Barral,
A.; Beverley, S. & Barral-Netto, M. (1999). A dhfr-ts- Leishmania major knock-out
mutant cross-protects against Leishmania amazonensis. Mem. Inst. Oswaldo
Cruz, 94: 491-496.
Weigle, K. & Saravia, N. G. (1996) Natural history, clinical evolution, and the host-
parasite interaction in New World cutaneous leishmaniasis. Clin. Dermatol. 14:
433-450.
Winberg, M. E.; Holm, A.; Sarndahl, E.; Vinet, A. F.; Descoteaux, A.; Magnusson,
K-E.; Rasmusson, B. & Lerm. (2009). Leishmania donovani lipophosphoglycan
inhibits phagosomal maturation via action on membrane rafts. Microbes Infec., 3:
1-8.
Winter, G.; Fuchs, M.; Mcconville, M.J.; Stierhof, Y.D. & Overath, P. (1994)
Surface antigens of Leishmania mexicana amastigotes-characterization of
glicoinositol phospholipids and a macrophage-derived glycosphingolipid. J. Cell.
Sci. 107: 2471-2482.
Xu, D. & Liew, F. Y. (1994). Genetic vaccination against leishmaniasis. Vaccine,
12: 1534-1536.
Yamaguchi, T.; Mizushima, Y. (1994) Lipid microspheres for drug delivery from the
pharmaceutical viewpoint. Crit. Rev. Ther. Drug Carrier Syst., 11: 215-229.
Yu, J.; He, H. B.; Tang, X. (2006) Formulation and evaluation of nimodipine-loaded
lipid microspheres. J. Pharm. Pharmacol., 58: 1429 – 1435.
Zeringue, H. J.; Brown, M. L. & Singleton, W. S. (1964) Chromatographically
homogeneous egg lecithin as stabilizer of emulsions for intravenous nutrition. J.
Amer. Oil Chem. Soc., 41: 688 – 691.
Zhang, H.Y.; Tang, X.; Li, H.Y. & Liu, X.L. (2008). A lipid microsphere vehicle for
vinorelbine: Stability, safety and pharmacokinetics. Int. J. Pharm., 348: 70–79.
Zinckernagel, R.M.; Ehl, S.; Aichele, P.; Oehen, S.; Kundig, T.; Hengartner, H.
(1997). Antigen Localisation regulates emmune responses in a dose and time-
dependent fashion: a geographical view of immune reactivity. Immunol. Rev.,
156: 199-209.

Referências
84


















