
Mecanismo Molecular de la Activación de la Síntesis de ...
117
Mecanismo Molecular de la Activación de la Síntesis de Fosfolípidos por la Proto-oncoproteína c-Fos Tesis para optar por el título de Doctor en Ciencias Químicas Lic. en Química Andrés M. Cardozo Gizzi Directora de Tesis Dra. Beatriz L. Caputto Centro de Investigaciones en Química Biológica de Córdoba (CIQUIBIC) – CONICET Depto. De Química Biológica – Facultad de Ciencias Químicas Universidad Nacional de Córdoba Córdoba, Junio de 2015
Transcript of Mecanismo Molecular de la Activación de la Síntesis de ...

Mecanismo Molecular de la Activación de la Síntesis de Fosfolípidos por la
Proto-oncoproteína c-Fos
Tesis para optar por el título de Doctor en Ciencias Químicas
Lic. en Química Andrés M. Cardozo Gizzi
Directora de Tesis Dra. Beatriz L. Caputto
Centro de Investigaciones en Química Biológica de Córdoba
(CIQUIBIC) – CONICET
Depto. De Química Biológica – Facultad de Ciencias Químicas
Universidad Nacional de Córdoba
Córdoba, Junio de 2015

Comisión de Tesis
Dr. Alfredo Cáceres
Dra. Graciela Panzetta
Dr. Marcos Villarreal
Evaluador Externo
Dr. Diego De Mendoza

Agradecimientos
En primer lugar, quisiera agradecer a las Instituciones involucradas: el CONICET, que
me brindó el apoyo económico; la Facultad de Ciencias Químicas, donde me formé como
estudiante de grado y postgrado; la Universidad Nacional de Córdoba, bajo cuyo paraguas
protector nos encontramos y a la Agencia (ANPCyT) por la financiación recibida. En segundo
lugar, al CIQUIBIC (entendido como un todo), por dejarme ser parte de esta gran familia.
Porque en todo momento supe que se podía contar con lo que hiciera falta, que siempre iba
a haber una mano amiga dispuesta a ayudar. Una mención especial merecen los
responsables de la sala de Microscopía; Cecilia, Carlos y Marcelo, que siempre me apoyaron
en lo que requerí. En tercer lugar, a mi comisión de Tesis, por seguirme estos años y
brindarme sus consejos.
A mi directora de Tesis, que confió (y aún lo hace) plenamente en mí, por apoyar cada
proyecto, cada idea, jugándosela cada vez que lo necesité. Por sus consejos y afecto. Porque
para ella no hay imposibles. A mis compañeros de laboratorio, porque nuestra relación
transciende lo laboral, porque me llevo muchos recuerdos, muchos lindos momentos
compartidos. Porque me soportan todos los días, y hacen que mis días malos sean más
soportables también. En especial a Gabriel, que fue quien me formó cuando empecé, porque
es un docente de alma que me tuvo paciencia y sabe mucho pero mucho.
A mi familia, que me hizo la persona que hoy soy. A mi papá y mis hermanos, por
estar siempre. A mi mamá que ya no puede verme. Sé que hubiera estado orgullosa. A mi
familia política por todo su cariño. A mis amigos, por su incondicionalidad, por tantas risas y
algunas lágrimas… y por todas las que vendrán.
Al último pero no la última, a Luli, por su permanente apoyo, amor y por nuestros
proyectos compartidos. Porque es mi asesora principal. Por todo lo que significa para mí.
A todos Uds., eternamente gracias.

“Termina siempre así, con la muerte. Pero antes, hubo vida. Escondido
debajo del bla, bla, bla, bla. Y todo sedimentado bajo los murmullos y el
ruido. El silencio y el sentimiento, la emoción y el miedo. Los demacrados,
caprichosos destellos de belleza. […] Más allá, está el más allá. Yo no me
ocupo del más allá. Por tanto, que esta novela dé comienzo. En el fondo, es
sólo un truco. Sí, es sólo un truco.”
Jep Gambardella – La grande bellezza

A la memoria de mi madre.

Contenido
ABREVIATURAS UTILIZADAS
RESUMEN
INTRODUCCIÓN 1
Membranas biológicas 2
Vías de síntesis de fosfolípidos 3
Lipinas 5
CDS – Vía del CDP-DAG 7
PIS 8
Biosíntesis de PIPs 9
PI4Ks tipo II 11
PI4Ks tipo III 12
c-Fos es un factor de transcripción tipo AP-1 con una función no genómica adicional 13
c-Fos como activador de la síntesis lipídica 14
Regulación de la función de c-Fos como activador de la síntesis de fosfolípidos 16
c-Fos en el crecimiento tumoral 16
c-Fos y un estado activado de la síntesis de lípidos 17
OBJETIVOS E HIPÓTESIS DE TRABAJO 18
RESULTADOS 21
Efecto de c-Fos en la síntesis de PIPs in vivo 22
Las actividades CDS y PIK pero no la de PIS son activadas por c-Fos 22
Las actividades de CDS1 y PI4KIIα son claves para la activación del marcado
radioactivo de PtdInsP y PtdInsP2 promovido por FBS, no así la de PI4KIIβ 29
CDS1 y PI4KIIα pero no PI4KIIβ participan en una asociación física con c-Fos 32

El domino BD sería el responsable de la activación de la síntesis de PIPs 35
La región N-terminal es la responsable de la asociación con CDS1 37
La región N-terminal es capaz de competir con c-Fos salvaje por su unión a la enzima
PI4KIIα 39
Activación de la actividad PAP por c-Fos 45
El BD de c-Fos sería el responsable de la actividad incrementada de Lipin 1 48
Fra-1, miembro de la familia Fos, a diferencia de c-Jun es capaz de activar a Lipin 1 49
Lipin 1 interacciona físicamente con c-Fos y Fra-1 pero no con c-Jun 50
c-Fos modifica la actividad de Lipin 1 al incrementar su capacidad catalítica 53
El dominio N-terminal de c-Fos es responsable de la asociación a Lipin 1 56
La región aa: 47-90 dentro del N-terminal de c-Fos estaría involucrado en la
asociación Lipin 1/c-Fos 58
DISCUSIÓN 60
La activación de la síntesis de fosfolípidos por c-Fos es un fenómeno no genómico 61
c-Fos es capaz de activar tanto a CDS1 como a Lipin 1 62
¿Para qué son necesarios los lípidos cuya síntesis c-Fos incrementa? 65
Un mecanismo compartido para la activación de la síntesis 67
La naturaleza intrínsecamente desestructurada de c-Fos y su asociación a múltiples
enzimas blanco 69
Fra-1 también incrementa la síntesis de lípidos 75
Conclusiones finales 77
MATERIALES Y MÉTODOS 78
BIBLIOGRAFÍA 93

ABREVIATURAS UTILIZADAS
aa: Aminoácidos
AP-1: Proteína Activadora-1
ASO: Oligonucleótido Anti-Sentido específico para c-Fos
BD: Dominio Básico de proteínas tipo AP-1
bZip: Dominio funcional de proteínas tipo AP-1 que comprende a BD y LZ
CDS: CDP-Diacilglicerol Sintasa
CDP-DAG: Citidina Difosfato – Diacilglicerol
CFP: Proteína Fluorescente Cyan
DAG: Diacilglicerol
DGK: Diacilglicerol Quinasa
FBS: Suero Fetal Bovino
FLIM: Fluorescence Lifetime Imaging Microscopy
FRET: Transferencia de Energía Resonante de Förster
G3P: Glicerol-3-Fosfato
GlcCerS: Glucosilceramida Sintasa
IDPs: Proteínas Intrínsecamente Desestructuradas
IPTG: isopropil-β-D-1-tiogalactopiranósido
LZ: Dominio cierre de leucinas de proteínas tipo AP-1
NGF: Factor de Crecimiento Nervioso
PA: Ácido Fosfatídico
PAP: Fosfatasas de Ácido Fosfatídico
PBD: Dominio Polibásico de Lipin 1
PI4K: Fosfatidilinositol-4-quinasa
PIPs: Polifosfoinosítidos
PIS: Fosfatidilinositol Sintasa
PtdCho: Fosfatidilcolina
PtdEth: Fosfatidiletanolamina
PtdIns: Fosfatidilinositol
PtdInsP2: Fosfatidilinositol bifosfato
PtdSer: Fosfatidilserina
RE: Retículo Endoplásmico
TAG: Triacilglicerol
YFP: Proteína Fluorescente Amarilla

RESUMEN

La proto-oncoproteína c-Fos es un factor de transcripción tipo AP-1 cuya función fue
descripta hace más de 25 años (Curran & Morgan 1987). En su función canónica, c-Fos forma
heterodímeros funcionales con proteínas de las familias Jun, ATF o JDP, que se unen a
regiones específicas del ADN y promueven la transcripción de múltiples genes blanco (Hess et
al. 2004). Esta actividad AP-1 ha sido involucrada en la transformación de señales de
crecimiento de corta duración en cambios de larga duración al regular la expresión de genes
involucrados en el crecimiento celular. Evidencias previas del laboratorio han demostrado
que la proteína c-Fos, de manera independiente de su actividad como factor de transcripción
tipo AP-1, activa la síntesis de fosfolípidos al asociarse al retículo endoplásmico (Caputto et
al. 2014). El objetivo principal de esta Tesis fue estudiar la actividad no genómica de c-Fos
por la es capaz de activar la síntesis de lípidos, y en particular el mecanismo molecular por el
cual c-Fos ejerce este efecto.
Encontramos que para alcanzar el incremento en el marcado radioactivo de todas las
especies de fosfolípidos analizadas, c-Fos afecta positivamente solo etapas metabólicas
específicas. Utilizando homogeneizados de células NIH3T3 quiescentes, encontramos que c-
Fos recombinante activa in vitro la actividad de CDP-diacilglicerol sintasa, Lipin 1 y
fosfatidilinositol 4-quinasa tipo II pero no fosfatidilinositol sintasa. La constante cinética
Vmax se incrementó para todas las enzimas analizadas mientras que no se observó ningún
efecto en Km, una medida de la afinidad de la enzima por su sustrato.
Más aún, hemos probado la activación in vitro en un sistema purificado que contiene
sólo a c-Fos, Lipin 1 y membranas modelo compuestas de micelas mixtas de Triton X-100/PA.
En este sistema altamente simplificado, encontramos que c-Fos incrementa la actividad
enzimática de Lipin1 sin modificar la afinidad de esta por las membranas modelo.
En orden de explicar el mecanismo de activación enzimática, realizamos ensayos de
interacción proteína-proteína. Mediante ensayos de co-inmunoprecipitación y de
microscopía FRET establecimos una interacción física entre c-Fos y las enzimas que activa
mientras que esta no se produce con aquellas enzimas cuya actividad c-Fos no regula.
El empleo de mutantes de deleción permitió determinar que la región involucrada en
la activación enzimática es diferente de la región responsable de la unión a enzimas

específicas, y que estas regiones de asociación/activación son comunes a todas las enzimas
estudiadas. Mientras que la región de activación es el dominio básico de c-Fos, la región de
asociación se encuentra en el N-terminal de la proteína.
Los resultados apoyan nuestra hipótesis de un mecanismo compartido en la
activación de la síntesis de fosfolípidos en el que c-Fos se asocia físicamente con las enzimas
responsables de la síntesis de fosfolípidos que es capaz de activar modificando su eficiencia
catalítica sin alterar la afinidad de la enzima blanco por su sustrato. A su vez, refuerzan el
concepto de una proteína que posee dos funciones diferenciales como lo son la de su
actividad como factor de transcripción y la capacidad de incrementar actividades enzimáticas
claves per se, para de esta forma sostener eventos de proliferación o diferenciación celular.

CAPITULO I
INTRODUCCIÓN

Introducción
2
Membranas biológicas
Las membranas de células eucariotas son una compleja mezcla de glicerolípidos,
esfingolípidos, proteínas y colesterol (Singer & Nicolson 1972) cuya síntesis requiere la
actividad sincronizada de múltiples pasos metabólicos que son coordinados en varios niveles.
Los lípidos son las especies moleculares cuantitativamente más importantes de las
membranas celulares (Spector & Yorek 1985). Entre la vasta diversidad de lípidos existentes,
los más abundantes son los glicerofosfolípidos [fosfatidilcolina (PtdCho),
fosfatidiletanolamina (PtdCho), fosfatidilinositol (PtdIns), fosfatidilserina (PtdSer) y
cardiolipina] (van Meer et al. 2008). La estructura de todos ellos deriva del glicerol-3-fosfato
(G3P), que se esterifica en las posiciones sn-1 y sn-2 con dos ácidos grasos, para dar ácido
fosfatídico (PA), y en el grupo fosforilo con alcoholes de bajo peso molecular como colina o
etanolamina (Fig. 1). Aunque los glicerofosfolípidos son clasificados de acuerdo a la
estructura de su cabeza polar (por ejemplo colina o inositol), cada clase de fosfolípido en
realidad consiste en numerosas especies moleculares que contienen la misma cabeza polar
pero se diferencian por las cadenas de los ácidos grasos que cada uno contiene (Wenk 2005).
En consecuencia, las membranas celulares son formadas por cientos de moléculas de
glicerofosfolípidos diferentes. Para alcanzar esta complejidad, las células eucariotas
necesitan invertir importantes recursos: aproximadamente 5% de sus genes están
involucrados en la síntesis de lípidos (Sud et al. 2007). La mayoría de los glicerofosfolípidos
están presentes en todas las membranas celulares, pero la abundancia relativa varía
significativamente de una organela a la otra. La síntesis de lípidos estructurales está limitada
espacialmente, por lo que el metabolismo lipídico local es el primer determinante de la
composición única de las distintas organelas (van Meer et al. 2008).
La biogénesis de membrana es un proceso complejo, fundamental para el
crecimiento, la proliferación y la diferenciación celular. Estos procesos acoplan señales de
crecimiento ambientales con respuestas nucleares para generar cambios funcionales y
morfológicos en la célula (Hermansson et al. 2011), que en conjunto necesitan de lípidos para
la expansión de membrana. Los lípidos requeridos son provistos por el sistema de

Introducción
3
endomembranas; en particular el retículo endoplásmico (RE), el principal sitio de síntesis de
la mayoría de los fosfolípidos, y el complejo de Golgi (Fagone & Jackowski 2009).
Fosfolípidos, junto con colesterol y proteínas integrales de membrana son
sintetizados en el RE e incorporados a membrana pre-existente. Membranas nacientes
surgen desde los sitios de salida del RE y se mueven por transporte vesicular hacia la
membrana plasmática pasando a través del complejo de Golgi, donde ocurren una serie de
modificaciones post-traduccionales en la carga y las proteínas asociadas a membrana. La
composición lipídica también es ajustada en el complejo de Golgi, especializado en la síntesis
de esfingomielina, glucosilceramida y glicoesfingolípidos complejos (Futerman & Riezman
2005). Adicionalmente, el transporte no vesicular tiene un rol relevante en la distribución y
tráfico intracelular lipídico. Los intercambios de monómeros lipídicos, tanto espontáneos
(lentos) como mediados por proteínas que transfieren lípidos (mucho más rápidos), son
incrementados enormemente por sitios de contacto directo con membranas, definidos como
pequeñas intersecciones entre el RE y prácticamente todas las otras organelas celulares (Lev
2010). La biogénesis de membrana es también sostenida con la inactivación de ciertas
fosfolipasas frente a determinados estímulos, volviendo más lento el recambio permanente
de un abundante rango de fosfolípidos (Manguikian & Barbour 2004, Bao et al. 2006).
Vías de síntesis de fosfolípidos
Los glicerofosfolípidos constituyen las unidades estructurales mayoritarias de las
membranas biológicas. Serán el principal objeto de estudio de esta Tesis, por lo que nos
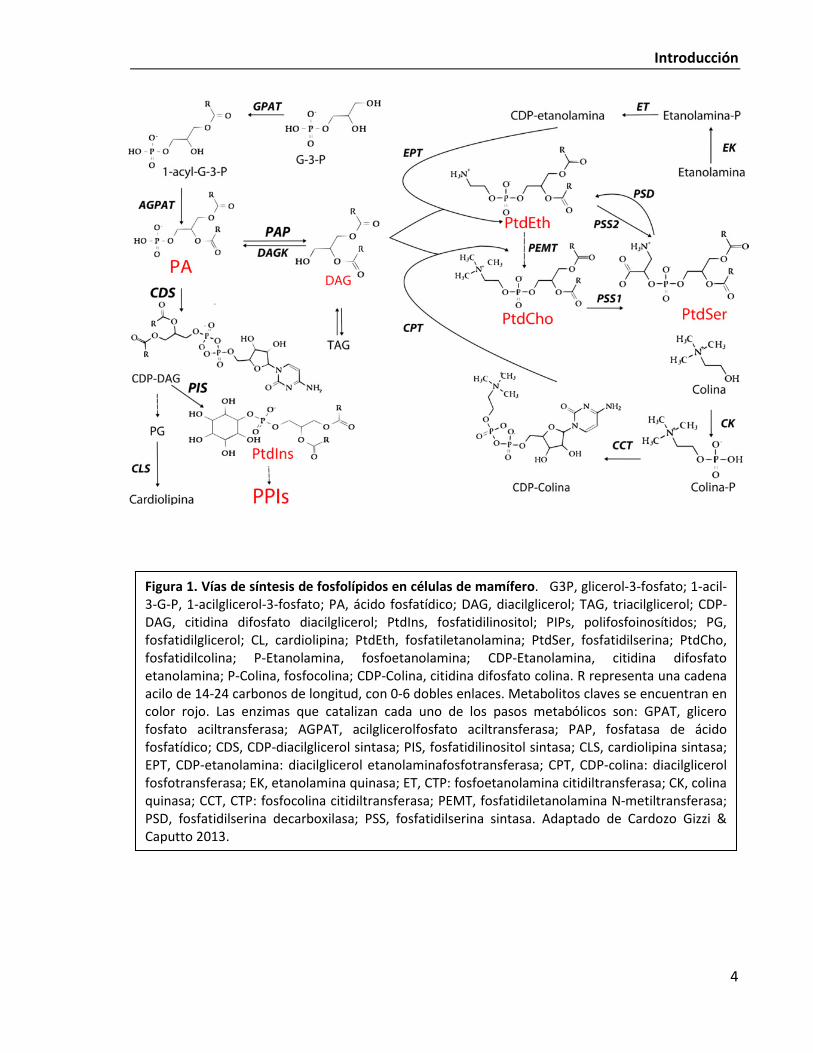
centraremos en la descripción de sus vías de síntesis (Fig. 1). El PA es un precursor central
para la biogénesis de membrana (Brindley 1984, Carman & Han 2009). Como puede
observarse en la Fig. 1, el PA se encuentra en un punto único de ramificación: puede ser
transformado por una fosfatasa a diacilglicerol (DAG), un precursor común a toda la vía de
Kennedy, o alternativamente puede ser derivado a la vía de síntesis del fosfatidilinositol y sus
derivados fosforilados por la enzima limitante de la vía, CDP-diacilglicerol sintasa (CDS)
(Athenstaedt & Daum 1999).
Introducción
4
Figura 1. Vías de síntesis de fosfolípidos en células de mamífero. G3P, glicerol-3-fosfato; 1-acil-3-G-P, 1-acilglicerol-3-fosfato; PA, ácido fosfatídico; DAG, diacilglicerol; TAG, triacilglicerol; CDP-DAG, citidina difosfato diacilglicerol; PtdIns, fosfatidilinositol; PIPs, polifosfoinosítidos; PG, fosfatidilglicerol; CL, cardiolipina; PtdEth, fosfatiletanolamina; PtdSer, fosfatidilserina; PtdCho, fosfatidilcolina; P-Etanolamina, fosfoetanolamina; CDP-Etanolamina, citidina difosfato etanolamina; P-Colina, fosfocolina; CDP-Colina, citidina difosfato colina. R representa una cadena acilo de 14-24 carbonos de longitud, con 0-6 dobles enlaces. Metabolitos claves se encuentran en color rojo. Las enzimas que catalizan cada uno de los pasos metabólicos son: GPAT, glicero fosfato aciltransferasa; AGPAT, acilglicerolfosfato aciltransferasa; PAP, fosfatasa de ácido fosfatídico; CDS, CDP-diacilglicerol sintasa; PIS, fosfatidilinositol sintasa; CLS, cardiolipina sintasa; EPT, CDP-etanolamina: diacilglicerol etanolaminafosfotransferasa; CPT, CDP-colina: diacilglicerol fosfotransferasa; EK, etanolamina quinasa; ET, CTP: fosfoetanolamina citidiltransferasa; CK, colina quinasa; CCT, CTP: fosfocolina citidiltransferasa; PEMT, fosfatidiletanolamina N-metiltransferasa; PSD, fosfatidilserina decarboxilasa; PSS, fosfatidilserina sintasa. Adaptado de Cardozo Gizzi & Caputto 2013.

Introducción
5
En comparación con sus precursores y sus metabolitos, los niveles de PA son bajos en
células en crecimiento a pesar del considerable flujo metabólico a través de PA requerido
para sostener la síntesis de fosfolípidos. Además de su rol biosintético, tanto PA como DAG
son también importantes en cascadas de transducción de señales (Stace & Ktistakis 2006). El
PA se encuentra implicado en regulación de la transcripción, activación del crecimiento
celular, biogénesis de membrana, secreción extracelular y transporte vesicular mientras que
el DAG se encuentra principalmente involucrado en la activación de proteína quinasa C (PKC),
aunque también se ha descripto la activación de otras quinasas también importantes en
señalización celular como por ejemplo proteína quinasa D o quinasas de DAG β and γ (Brose
et al. 2004, Wang et al. 2006, Carrasco & Merida 2007).
Inicialmente se caracterizaron bioquímicamente dos grupos distintos de fosfatasas de
PA (PAP), PAP-1 y PAP-2. Estas últimas se denominaron posteriormente LPP (lipid phosphate
phosphases o fosfatasas del fosfato de lípidos) debido a que no son específicas para PA. Su
actividad es independiente de Mg+2 y sus principales funciones están relacionadas a la
transducción de señalas a nivel de la membrana plasmática (Pascual & Carman 2013). En
cambio, las enzimas con actividad PAP-1 fueron clonadas y denominadas Lipinas.
Lipinas
Las Lipinas son enzimas PAP dependientes de Mg+2, que están involucradas en la
síntesis de novo de fosfolípidos y lípidos neutros (Harris & Finck 2011). A diferencia de la
mayoría de las enzimas de la vía de Kennedy, las Lipinas no son proteínas integrales de
membrana sino que se unen a membrana de forma periférica, por lo que necesitan
translocar desde el citosol hacia el RE para participar en la síntesis de glicerolípidos (Coleman
& Lee 2004, Csaki & Reue 2010).
El dominio polibásico de Lipin 1 (PBD) es un segmento compuesto por nueve residuos
básicos consecutivos que está involucrado en la unión selectiva a PA, lo que promueve la
translocación de la enzima a membranas. El dominio PBD de Lipin 1 es también su principal
señal de localización nuclear. Ha sido propuesto que el rol dual de PBD es regulado a partir
de los niveles de PA: un contenido elevado de PA en las membranas citoplásmicas antagoniza

Introducción
6
la localización nuclear de Lipin 1 (Ren et al. 2010). Existen evidencias de que la Lipinas dentro
del núcleo juegan un rol en la regulación directa de directa de la transcripción de genes
involucrados en la oxidación de ácidos grasos (Harris & Finck 2011). Por ejemplo, se ha
demostrado que Lipin 1 interacciona directamente con el receptor PPARα, un factor de
transcripción clave para el metabolismo de lípidos en el hígado, y con su co-activador PGC-1α
(Finck et al. 2006). Debido a que Lipin 1 no poseen una secuencia de unión a ADN
reconocible, la evidencia indica que Lipin 1 incrementaría la actividad de estos complejos de
factores de transcripción al reclutar otras proteínas co-activadoras con la habilidad de
modificar enzimáticamente la cromatina (Siniossoglou 2013).
En los últimos años las Lipinas han sido intensamente estudiadas dado su rol clave en
la síntesis de fosfolípidos (Pascual & Carman 2013). Su actividad se encuentra sujeta a
estrictos mecanismos de regulación, dados principalmente por múltiples fosforilaciones.
Estas son mediadas por varias quinasas, por ejemplo quinasas dependientes de ciclinas
(Grimsey et al. 2008) y mTOR (Huffman et al. 2002, Harris et al. 2007), que inducen su
disociación de la membrana. La fosforilación de las Lipinas altera su localización subcelular y
las proteínas con las que interactúa, cambiando indirectamente la actividad PAP al impedir
que se encuentre con el sustrato y/o directamente al disminuir el reconocimiento o la
afinidad por el sustrato (Harris & Finck 2011, Eaton et al. 2013). El reconocimiento por el
dominio PBD del PA di-aniónico promueve la asociación a membrana, mientras que la
fosforilación mediada por mTOR inhibe dicho reconocimiento (Eaton et al. 2013). La Lipin 1
hiper-fosforilada se asocia a las proteínas citosólicas 14-3-3 lo que facilita su retención en el
citosol (Peterfy et al. 2010). Por otro lado, el complejo transmembrana CTDNEP1-NEP1R1
(que es el ortólogo del Nem1p–Spo7p de levaduras) es capaz de defosforilar a Lipin y
consecuentemente actúa en la vía de activación de Lipin. Sin embargo, aún no está
esclarecido si para ser desfosforilada por el complejo CTDNEP1-NEP1R1, la Lipin que se
encuentra citosólica debe llegar a la región perinuclear (donde reside el complejo) o si Lipin 1
puede ser también defosforilada en el citosol por fosfatasas desconocidas (Han et al. 2012).
En conclusión, Lipin 1 es capaz de regular la síntesis de lípidos en múltiples niveles. Al
regular el destino de PA a través de su actividad PAP, puede controlar directamente la

Introducción
7
síntesis de fosfolípidos, mientras que puede hacerlo indirectamente al modular la actividad
de factores de transcripción relevantes para el metabolismo de lípidos. Ambas actividades
poseen claras separaciones espaciales: mientras que la actividad PAP de las Lipinas se ejerce
en las membranas nucleares/reticulares, la actividad como activador co-transcripcional
ocurre dentro del núcleo.
CDS – Vía del CDP-DAG
Como se mencionó anteriormente, el PA puede ser sustrato de la familia de Lipinas
que lo convierten en DAG o, en cambio, ser sustrato de la enzima CDP-DAG sintasa (CDS)
que cataliza la conversión PA a CDP-DAG (Heacock & Agranoff 1997). El CDP-DAG es el
precursor de PtdIns y sus derivados fosforilados, además de fosfatidilglicerol o cardiolipina.
La biosíntesis y metabolismo de PtdIns es de considerable interés debido a la participación de
este lípido y sus derivados fosforilados en la traducción de señales (Di Paolo & De Camilli
2006). Esta enzima tiene un rol clave entonces en la síntesis de fosfolípidos en general y en la
de polifosfoinosítidos (PIPs) en particular. La enzima es integral de membrana y es la
limitante de la vía (Heacock & Agranoff 1997). Recientemente se cristalizó el homólogo de
CDS del organismo procariota Thermotoga maritima (Liu et al. 2014).
Dos isoformas han sido identificadas en mamíferos, CDS1 y CDS2, las cuales han sido
descriptas localizando en el RE (Saito et al. 1997, Volta et al. 1999, Inglis-Broadgate et al.
2005). El ARNm de CDS2 muestra un patrón de expresión ubicua en ratón, en tanto el ARNm
de CDS1 se ha encontrado en cerebro, ojo, músculo liso, riñón y testículo. En ojo, CDS1 está
altamente expresada en la capa de fotorreceptores de retinas de adulto, lo que sugiere un rol
de CDS1 en la fototransducción de señales a través de la formación de PIP2 en mamíferos
(Inglis-Broadgate et al. 2005). Este rol fue también probado en invertebrados: en la
fototransducción en Drosophila se requiere de la enzima CDS para la regeneración de
PtdInsP2; los animales mutantes para esta enzima sufren una degeneración retinal mediada
por luz (Volta et al. 1999). En otro reporte, la mutación de CDS2 en pez cebra produce
defectos específicos en angiogénesis y en la formación de vasos sanguíneos, que fueron
atribuidos a la depleción de los niveles de PtdIns(4,5)P2 en células endoteliales estimuladas
por el factor de crecimiento VEGF. En estos estudios se concluyó que la actividad de la

Introducción
8
enzima CDS era necesaria para mantener una reserva de PtdIns(4,5)P2 para señalización
celular (Pan et al. 2012).
Ambas isoformas de CDS son inhibidas tanto por PtdInsP como por PtdInsP2, en un
mecanismo clásico de inhibición por un producto final de la vía (Saito et al. 1997, D'Souza et
al. 2014). Más allá de estos reportes, no hay otras pruebas de mecanismos regulatorios
endógenos para esta enzima. Existe un estudio que mostró cómo drogas antidepresivas
inducen la actividad de CDS, incrementando así la disponibilidad de CDP-DAG y facilitando la
síntesis de PtdIns (Aboukhatwa & Undieh 2010), y que postuló entonces la importancia de
esta enzima en cascadas de señalización mediadas por este lípido y sus derivados fosforilados
en células neurales en humanos.
PIS
La segunda enzima de la vía de síntesis de PIPs se denomina CDP-diacilglicerol:myo-
inositol 3-fosfatidil-transferasa o fosfatidilinositol sintasa (PIS). Cataliza la condensación de
una molécula de myo-inositol y una de CDP-DAG para formar PtdIns. Un único gen que
codifica para esta enzima fue clonado y caracterizado en mamíferos (Tanaka et al. 1996,
Lykidis et al. 1997). PIS es una proteína de 213 aminoácidos (aa) que se expresa
uniformemente en todos los tejidos humanos analizados. Estos estudios confirmaron además
que PIS es responsable tanto de la actividad de síntesis de PtdIns como así también de la de
recambio de myo-inositol en una molécula de PtdIns ya formada (Lykidis et al. 1997).
Estudios en pez cebra con una mutante para este gen indican que la falta de síntesis
de novo de PtdIns genera estrés crónico del RE lo que conduce por ejemplo a esteatosis
hepática (Thakur et al. 2011) y a daño e inflamación de la mucosa intestinal (Thakur et al.
2014). A su vez, esta mutante posee defectos estructurales del cristalino ocular y un reducido
número de fotorreceptores (Murphy et al. 2011).
Un trabajo reciente mostró que PIS se localiza no solo en el RE, donde co-localiza con
CDS1 y CDS2, sino también en un compartimento vesicular altamente móvil en donde estas
últimas enzimas quedan excluidas. Estas vesículas emergen desde la red membranosa del RE
en forma dependiente de la GTPasa pequeña Sar1 y es donde se concentra la mayor

Introducción
9
actividad PIS. Es importante remarcar que estas vesículas que contienen PIS no son
simplemente vesículas de transporte que se dirigen al aparato de Golgi, ya que no se
observa fusión de las mismas con esta organela. En cambio, se postula que este
compartimento hace contactos con una gran cantidad de membranas permitiendo así el
intercambio directo de lípidos (Kim et al. 2011). En un estudio posterior de otro grupo se
confirmó la existencia de estas estructuras altamente móviles que contienen PIS pero
también CEPT (la última enzima en la vía de síntesis de PtdCho) y dependen de otra GTPasa
pequeña llamada Rab10 para formar este compartimiento que es o bien un extremo que
crece o que ha emergido desde el RE (English & Voeltz 2013).
Biosíntesis de PIPs
Los PIPs son molecularmente diversos debido a la fosforilación variable de sus grupos
hidroxilo del anillo inositol (Fig. 2). La configuración de las siete especies de PIPs conocidos es
controlada rápida y reversiblemente por una batería de quinasas y fosfatasas que
interconvierten PIPs y que debido a ello resultan críticas para la localización y función de
cada uno de los distintos isómeros (Sasaki et al. 2009). Asimismo, se han identificado un gran
número de módulos de proteínas capaces de reconocer a isómeros específicos de PIPs (Czech
2003, Wenk & De Camilli 2004). Esto ha definido un paradigma en la regulación mediada por
PIPs en donde los mismos controlan una variedad de fenómenos de señalización y tráfico
celular al reclutar proteínas reguladoras hacia complejos de señalización ubicados en
membranas celulares (Balla & Balla 2006). De esta forma, los PIPs están involucrados en el
transporte vesicular, secreción y endocitosis (Balla et al. 2009), la organización de
citoesqueleto (Janmey 1994, Gilmore & Burridge 1996) y la estimulación de las cascadas de
proteína quinasas (Noh et al. 1995, Franke et al. 1997, Downward 2004). Las distintas clases
de PIPs son además importantes para definir la identidad de las organelas (van Meer et al.
2008).

Introducción
10
PI4Ks
Las fosfatidilinositol-4-quinasas (PI4Ks) fosforilan al anillo de inositol en la posición D4
para producir PtdIns4P. A la fecha, en base a la estructura de sus dominios y la sensibilidad a
inhibidores, se han identificado cuatro PI4K en mamíferos que se clasifican como tipo II:
PI4IIα y PI4IIβ; y tipo III: PI4IIIα y PI4IIIβ (Balla & Balla 2006). Las PI4K tipo III tienen un
dominio quinasa C-terminal similar a PI3Ks y son sensibles a los mismos inhibidores como por
ejemplo wortmanina (Balla 1998). Por otro lado, la región catalítica de las PI4K tipo II se
diferencian sustancialmente de las otras, son insensibles a wortmanina pero en cambio son
sensibles a adenosina. Cada isoenzima localiza en un compartimiento celular específico en un
tejido específico, sugiriendo que cada una de ellas produce PtdIns4P requerido en una
función celular diferente.
Figura 2. Estructura y formación de PIPs. El fosfatidilinositol está formado por un esqueleto de glicerol (recuadro azul), dos cadenas acilo en las posiciones sn1 y sn2 (recuadros en rojo) y una molécula de myo-inositol (recuadro verde). Los grupos hidroxilo pueden ser fosforilados en las posiciones 3,4 y 5, lo que permite la formación de hasta siete especies distintas de PIPs. La producción de cada una de estas especies depende de las acciones muy reguladas de las quinasas y fosfatasas de estos lípidos. Las distintas especies pueden ser interconvertidas según se indica con las flechas. Adaptado de D'Souza & Epand 2014.

Introducción
11
PI4Ks tipo II
Se discutirán las características de ambas isoformas, α y β, en forma conjunta. Tanto
PI4KII α como β se encuentran fuertemente unidas a membranas, debido a la palmitoilación
de un segmento conservado de cisteínas que se encuentra dentro del dominio catalítico. A
pesar de tener una secuencia de palmitoilación muy similar, una fracción más importante de
PI4KIIβ respecto de PI4KIIα no se encuentra palmitoilada y consecuentemente se encuentra
en el citoplasma (Jung et al. 2008). PI4KIIβ es más soluble y menos activa que PI4KIIα, pero
puede asimismo ser movilizada y activada al haber un estímulo como por ejemplo el del
factor de crecimiento PDGF (Wei et al. 2002).
Tanto PI4KIIα como PI4KIIβ se expresan en forma ubicua en tejidos humanos.
Northern blot de tejidos humanos han mostrado que el contenido de ARNm de PI4KIIβ es
mayor en hígado mientras que para PI4KIIα es mayor en el cerebro (Minogue et al. 2001,
Balla et al. 2002). A nivel subcelular, análisis inmunoquímicos y de fraccionamiento
subcelular han mostrado que PI4KIIα en su forma palmitoilada se ubica en las membranas del
RE, trans-Golgi y endosomas (Wang et al. 2003, Waugh et al. 2003).
PI4KIIα y PI4KIIβ son proteínas de un peso molecular cercano a 55 kDa. La
comparación de secuencias entre las dos isoformas revela un alto grado de similitud en el
dominio catalítico C-terminal con una homología significativamente menor en la región N-
terminal, que comprende los primeros 100 residuos aminoacídicos. PI4KIIα en el N-terminal
posee una región rica en prolinas, mientras que PI4KIIβ no la tiene y en cambio muestra una
región acídica. El N-terminal de PI4KIIα es necesario para enviarla hacia membranas
específicas, incluyendo vesículas positivas para GLUT4 (Xu et al. 2006). Aún no está
esclarecido el rol del N-terminal de PI4KIIβ, si bien se estableció que sufre en esta región una
fosforilación, esta no influye en su asociación a membranas o actividad. Más aún, los últimos
160 aa en el C-terminal parecen ser los determinantes en la diferencia de solubilidades,
estado de palmitoilación o redistribución dependiente de estímulo (Jung et al. 2008). En
cambio, se ha sugerido que la región N-terminal divergente media interacciones proteína-
proteína específicas.

Introducción
12
Respecto de las funciones biológicas atribuidas a estas enzimas, ha sido descripto
que PI4KIIα es necesaria para la producción de PtdIns4P en el aparato de Golgi y la red trans-
Golgi y que esto regula el reclutamiento a membranas de adaptadores de clatrina como por
ejemplo los complejos proteicos adaptadores tipo I (Wang et al. 2003, Wang et al. 2007) y
tipo III (Craige et al. 2008). Otro estudio demostró que la mayor parte de la actividad PI4K en
el cerebro proviene de PI4KIIα, donde además se la encontró asociada a vesículas sinápticas y
se la relacionó con la neurotransmisión (Guo et al. 2003). En cambio, PI4KIIβ podría estar
asociada a señalización intracelular, ya que se la encontró asociada in vivo al complejo de
membrana TCR-CD3 (Srivastava et al. 2006).
PI4Ks tipo III
Existen dos tipos de quinasas tipo III: PI4KIIIα (~230 kDa) y PI4KIIIβ (~92 kDa)
(Nakagawa et al. 1996, Nakagawa et al. 1996). Ambas contienen un dominio quinasa C-
terminal similar al de las PI3Ks, y ambas son sensibles a wortmanina.
La enzima PI4KIIIα es la principal responsable de la generación del PtdIns4P de
membrana plasmática (Balla et al. 2005, Balla et al. 2008), a pesar de que su localización
inicialmente había sido descripta en la interface RE/Golgi (Nakagawa et al. 1996). A diferencia
de la isoforma β, que se expresa en forma ubicua, la isoforma α se encuentra expresada
principalmente en cerebro, aunque se detectaron niveles bajos en varios otros órganos
(Zolyomi et al. 2000).
La información disponible al presente permite inferir que PI4KIIIβ regula, mediante la
formación de PtdIns4P, el tráfico desde Golgi hacia membrana plasmática (Bruns et al. 2002,
Godi et al. 2004). Esta enzima se localiza principalmente en el Golgi (Godi et al. 1999),
aunque también se ha descripto que transloca desde el citosol al núcleo, si bien se desconoce
la función precisa que allí ejerce (de Graaf et al. 2002).

Introducción
13
c-Fos es un factor de transcripción tipo AP-1 con una función no genómica adicional
Células que se encuentran en un proceso activo de proliferación o en eventos de
expansión de la membrana demandan la biogénesis de membrana en forma masiva, por lo
que es razonable esperar que la homeostasis de las organelas sea diferente de aquellas
células que no se están dividiendo o creciendo activamente. Sin embargo, la naturaleza de
los eventos que regulan estos procesos todavía es pobremente entendida. Desde un punto
de vista simplista los mecanismos que regulan la síntesis lipídica pueden ser divididos en dos:
una al nivel de la transcripción y la traducción de las enzimas involucradas (regulación génica)
y otra al nivel post-transcripcional (regulación no-genómica). En mamíferos, la familia de
factores de transcripción SREBPs, actúa a nivel central controlando la expresión de
receptores de lipoproteínas de baja densidad junto con los genes para la síntesis de novo de
colesterol y ácidos grasos (Nohturfft & Zhang 2009).
En cambio, diversos mecanismos no-genómicos existen y aparecen como respuestas
rápidas frente a cambios en la necesidad de síntesis de fosfolípidos (Cardozo Gizzi & Caputto
2013). Dentro de estas respuestas no genómicas a la demanda de síntesis de lípidos, nos
concentraremos en aquellas producidas por la proteína de interés central para esta tesis: c-
Fos. Esta proteína fue descripta como un miembro de la familia de factores de transcripción
inducibles denominado Proteína Activadora 1 (AP-1) hace más de 25 años (Curran & Morgan
1987). El contenido celular de c-Fos se encuentra estrictamente regulado: mientras que en
células quiescentes se encuentra al límite de detección, su expresión es rápida y
transitoriamente inducido frente a diversos estímulos como por ejemplo factores de
crecimiento, estimulación sensorial o liberación de neurotransmisores (Kovary & Bravo 1992,
Ginty et al. 1994, Hughes & Dragunow 1995, Caputto & Guido 2000, Maggiolini et al. 2004) .
Un trabajo pionero de Hunt y colaboradores mostró como después de la estimulación
sensorial se inducía rápidamente la expresión de c-Fos en neuronas espinales de rata (Hunt
et al. 1987). A partir de allí, múltiples estudios publicados proveyeron evidencia de que
condiciones tanto fisiológicas como patológicas son capaces de inducir la expresión de c-Fos
y otros miembros de su familia de factores de transcripción en el sistema nervioso central
junto con otros tipos celulares.

Introducción
14
AP-1 es un factor de transcripción homo- o heterodimérico compuesto por proteínas
que pertenecen a la familias de Fos, Jun, ATF o JDP (Shaulian & Karin 2001). Todos los
miembros AP-1 poseen un dominio bZip, que consiste en un dominio básico (BD) adyacente a
un motivo de cierre de leucinas. El BD está implicado en la unión secuencia-específica del AP-
1 al ADN mientras que los residuos conservados de leucinas permiten la dimerización y
consecuentemente la formación de los dímeros AP-1 transcripcionalmente activos (Neuberg
et al. 1989). Mientras que la familia Jun existe como homo o heterodímeros, la familia Fos,
que no puede homodimerizar, forma heterodímeros estables principalmente con las
proteínas Jun. (Hess et al. 2004). Debido a que la dimerización es un pre-requisito para la
unión al ADN, c-Fos no se asocia al ADN por sí mismo (Halazonetis et al. 1988).
La actividad AP-1 ha sido involucrada en la transformación de señales de crecimiento
de corta duración en cambios de larga duración al regular la expresión de genes blanco
involucrados en el crecimiento celular como por ejemplo colagenasa (Angel et al. 1987),
metaloproteasa-3 (Kerr et al. 1990), metalotioneina IIA (Lee et al. 1987) o de genes críticos
para la re-entrada al ciclo celular como ciclina D1 (Brown et al. 1998). Cuando el
correspondiente estimulo cesa, c-Fos se degrada rápidamente, con una vida media que va
desde el minuto a la hora, dependiendo del tipo celular (Basbous et al. 2008, Adler et al.
2010).
c-Fos como activador de la síntesis lipídica
c-Fos ejerce un rol relevante en la regulación del crecimiento, diferenciación así como
también en procesos de transformación celular (Curran & Franza 1988, Cohen & Curran
1989). Estudios de nuestro laboratorio han establecido que c-Fos tiene dos funciones que lo
sitúan como capaz de regular el crecimiento celular no solo con su actividad como factor de
transcripción sino también al actuar como un activador de la biosíntesis de lípidos en
procesos celulares tanto normales como patológicos que demandan altas tasas de biogénesis
de membrana. La activación de la síntesis de lípidos ha sido observada en diferentes tipos
celulares: in vivo en células ganglionares retinales y fotorreceptoras estimuladas por luz
(Guido et al. 1996, Bussolino et al. 1998), en fibroblastos que se encuentran proliferando

Introducción
15
(Bussolino et al. 2001), en células PC12 inducidas a diferenciar (Gil et al. 2004, Crespo et al.
2008), en tumores del sistema nervioso central y periférico (Silvestre et al. 2010, Gil et al.
2012) y en tumores mamarios humanos malignos (Motrich et al. 2013).
En la retina, se observó que ante la estimulación lumínica de pollos se produce un
incremento tanto en expresión de c-Fos como en la síntesis de lípidos en células ganglionares
de la retina (Guido et al. 1996, Bussolino et al. 1998). Al bloquear específicamente la
expresión de c-Fos se bloquean las modificaciones inducidas por luz en la síntesis de
fosfolípidos. Debido a que la liberación de neurotransmisores ocurre en luz en células
ganglionares, se interpretó que el pico en la expresión de c-Fos responde a la necesidad de la
célula de incrementar la tasa de síntesis de membrana para restablecer el reciclado de
vesículas sinápticas ante la liberación de neurotransmisores (de Arriba Zerpa et al. 1999,
Caputto & Guido 2000).
La biogénesis de membrana requiere del suministro coordinado de todos sus
componentes integrales. En este sentido, fue notable como en células ganglionares el
marcado isotópico realizado tanto in vivo (3H-glicerol o 32P-ortofosfato) como in vitro (32P-γ-
ATP) mostró consistentemente que la marcación de todos los fosfolípidos analizados se
incrementó de manera similar, de un modo dependiente de c-Fos, en luz con respecto a
oscuridad. Resultados concordantes fueron encontrados en células PC12 inducidas a
diferenciar con factor de crecimiento nervioso (NGF) a un fenotipo similar a neuronas
simpáticas. c-Fos activa el marcado metabólico general tanto de fosfolípidos (Gil et al. 2004)
como de glicolípidos (Crespo et al. 2008). Al bloquearse la expresión de c-Fos se detiene
tanto la activación de síntesis de lípidos como así también la neuritogénesis. Análisis por TLC
de los extractos lipídicos luego del marcado isotópico de estas células con 14C-Galactosa o
con 32P-ortofosfato mostraron un incremento del 50-60% en todos los glicolípidos marcados
con 14C y de PtdCho, que también se marca en este ensayo. Asimismo, todos los fosfolípidos
marcados con 32P también mostraron resultados similares. Estos resultados son compatibles
con una estimulación global de la maquinaria de síntesis de lípidos en la respuesta a NGF
mediada por c-Fos.

Introducción
16
Regulación de la función de c-Fos como activador de la síntesis de fosfolípidos
La capacidad de c-Fos para activar la síntesis lipídica en el citoplasma requiere de la
asociación de c-Fos al RE (Bussolino et al. 2001), el sitio de síntesis cuantitativamente más
importante de la célula. Esta asociación al RE está regulada por el estado de fosforilación de
los residuos de tirosina 10 y 30 de c-Fos. Células quiescentes tienes cantidades muy
pequeñas de c-Fos que, además, se encuentra fosforilado en dichos residuos y por tanto
disociado del RE. Al inducir a las células a proliferar, se promueve la expresión de c-Fos
conjuntamente con la desfosforilación de esta proteína, lo que resulta en su asociación a las
membranas del RE y en la activación de la síntesis de lípidos (Portal et al. 2007).
c-Src fue identificada como la primer quinasa y TC45-PTP como la primer fosfatasa
que actúan sobre los residuos de tirosina de c-Fos (Ferrero et al. 2012). Estudios de
fraccionamiento subcelular y de marcado con 32P-ATP evidenciaron que c-Fos fosforilado es
incapaz de unirse a membranas y en consecuencia no activa la síntesis de fosfolípidos. La
regulación de esta modificación post-traduccional reversible se encuentra en la etapa de
desfosforilación. La inducción de las células con mitógenos promueve la concomitante
translocación de TC45-PTP desde el núcleo hacia el citoplasma, su activación y la formación
del complejo transitorio c-Fos/TC45 que resulta en la desfosforilación de c-Fos (Ferrero et al.
2012). Diferentes estudios han demostrado la activación de c-Src con el estímulo celular,
(revisado en Hunter 2009). Sin embargo, en el modelo celular empleado, no se observó un
incremento en la fosforilación de c-Fos frente al estímulo, lo que indicaría que la actividad de
c-Src basal es suficiente para mantener las pequeñas cantidades de c-Fos presente en células
quiescentes en su estado fosforilado. Es importante remarcar que la fosforilación en tirosina
tiene un efecto represor de la actividad no genómica de c-Fos mientras que la fosforilación
en serinas/treoninas tiene un efecto trans-activador de su actividad como AP-1.
c-Fos en el crecimiento tumoral
Existe abundante información acerca de los eventos genómicos que se encuentran
detrás del crecimiento exacerbado característico de células tumorales (Hanahan & Weinberg
2011). En cambio, son escasos los estudios respecto de los cambios pleiotrópicos que

Introducción
17
necesariamente acompañan el crecimiento tumoral. En este contexto, altas tasas de
proliferación correlacionan estrictamente con una elevada expresión de c-Fos junto con altas
tasas de síntesis de fosfolípidos en modelos tumorales. Bloqueando específicamente la
síntesis de fosfolípidos dependiente de la expresión de c-Fos se reduce significativamente la
proliferación en cultivo de células T98G, que derivan de un glioblastoma multiforme humano.
Más aún, al tratar ratones atímicos xeno-transplantados con estas células en el lugar de la
lesión con un oligonucleótido ARNm antisentido (ASO) específico para c-Fos, se impide el
crecimiento tumoral que de otro modo se desarrolla en el 90% de los animales control (Gil et
al. 2012). Resultados concordantes fueron encontrados en ratones NPcis, un modelo animal
de la enfermedad humana Neurofibromatosis tipo I, que a los 5-6 meses de edad generan
espontáneamente tumores del sistema nervioso central y periférico con una penetrancia del
100%. Los tumores ya formados crecen significativamente más lento que en los controles
cuando los ratones NPcis son tratados con ASO para c-Fos. Finalmente, ratones NPcis
homocigotas nulos para c-Fos no desarrollan tumores, en contraste con el 71.4% de sus
hermanos de camada Fos +/+ que si lo hacen (Silvestre et al. 2010).
c-Fos y un estado activado de la síntesis de lípidos
No solo patologías asociadas a tumores muestran c-Fos asociado al RE, en la médula
espinal de ratas sensibilizadas para desarrollar encefalomielitis alérgica en la que se
promueve gliosis, se induce la expresión de c-Fos y este se encuentra asociada al RE (Cammer
et al. 1989). La actividad no genómica de c-Fos promueve un estado activado de síntesis de
fosfolípidos que se ha probado como necesario en modelos de crecimiento, proliferación y/o
diferenciación celular para sostener estos procesos. En un reporte reciente, se estudiaron
cambios globales en la transcripción inducidos por c-Fos de una manera independiente a su
actividad AP-1. Se encontró que estos eran la consecuencia de la síntesis nuclear
dependiente de c-Fos de PtdIns(4,5)P2 (Ferrero et al. 2014), un lípido involucrado en la
remodelación de cromatina (Viiri et al. 2012). De esta manera, c-Fos integra ambas
funciones, tanto como i) activador de la síntesis lipídica (Caputto et al. 2014) como ii) un
modulador transcripcional (Angel & Karin 1991).

CAPITULO II
OBJETIVOS E HIPÓTESIS DE TRABAJO

Objetivos e Hipótesis de Trabajo
19
Los ejemplos planteados hasta aquí apuntan a la existencia de un mecanismo
compartido que permite un aumento en el abastecimiento de lípidos y nos permite formular
la siguiente hipótesis causa-consecuencia: 1) La expresión de c-Fos es rápidamente inducida
por factores externos, que llevan a 2) un incremento tanto en la actividad AP-1 como en la
activación de la síntesis de fosfolípidos que en consecuencia sostienen 3) proliferación o
diferenciación celular. El tiempo y la localización de estas dos funciones dentro de la célula
son distintos. c-Fos nuclear es necesario en etapas iniciales para desencadenar los
correspondientes programas génicos, participando entonces en preparar a las células para
crecer, mientras que c-Fos citoplásmico es necesario para mantener una tasa elevada de
síntesis lipídica tanto en etapas del crecimiento iniciales como tardías (Gil et al. 2004, Ferrero
et al. 2012, Gil et al. 2012, Cardozo Gizzi & Caputto 2013).
Distintos reportes del grupo han ido avanzando en el conocimiento de los fenómenos
dependientes de c-Fos. El objetivo principal de esta Tesis fue estudiar el mecanismo
molecular por el cual c-Fos es capaz de activar la síntesis de lípidos: la manera en que
consigue incrementar el marcado radioactivo de todas las especies de fosfolípidos analizadas.
En fibroblastos NIH 3T3 quiescentes estimulados a reingresar al ciclo celular, se
produce la rápida inducción transcripcional de c-Fos, en el orden de minutos (Greenberg &
Ziff 1984). Se producen dos ondas de expresión de c-Fos que son necesarias para que genere
concomitantemente la estimulación en la incorporación de 32P-ortofosfato a fosfolípidos in
vivo (Bussolino et al. 2001). La primera onda de marcado radioactivo incrementado de lípidos
tiene un pico a 7,5 minutos post-estímulo y regresa a niveles del control a los 15 minutos; la
segunda onda empieza a los 30 minutos y se mantiene elevada al menos hasta 120 minutos,
el tiempo más largo examinado. Los lípidos que incorporan 32P durante la primera onda son
PIPs, predominantemente segundos mensajeros ubicuos, mientras que en la segunda onda
los productos radioactivos mayoritarios son los principales lípidos constituyentes de
membranas. La vida media del ARNm de c-Fos es muy corta en la primer onda, de solo 10
minutos, en tanto es de alrededor de 85 minutos en la segunda onda (Bussolino et al. 2001).
Este antecedente fue el disparador de esta tesis doctoral, ya que nos propusimos
estudiar en detalle este fenómeno. El modelo de estudio empleado a lo largo de la tesis será

Objetivos e Hipótesis de Trabajo
20
entonces la mencionada línea celular y particularmente la transición G0/G1 inducida por el
agregado de suero fetal bovino (FBS) al 20% a células que se encuentran quiescentes luego de
permanecer 48 horas deprivadas de factores de crecimiento. Se utilizará este sistema como
modelo para estudiar la respuesta celular a un rápido requerimiento de síntesis de lípidos
frente a un estímulo externo. Se pretende resolver estos interrogantes mediante un enfoque
conjunto de técnicas bioquímicas clásicas junto con microscopía confocal de fluorescencia.
En otros modelos celulares ya se había observado que c-Fos activa el marcado
radioactivo de todos los lípidos analizados. Sin embargo, no se conocía si este efecto estaba
dado por la activación de todas las vías metabólicas involucradas o si en cambio c-Fos actuaba
sobre etapas específicas. Uno de los objetivos específicos fue el de establecer qué etapas
metabólicas c-Fos está afectando a partir del estudio de las mismas in vitro. La hipótesis es
que c-Fos sólo afecta etapas claves, ¿pero cómo lo hace? Nos propusimos estudiar las
posibles interacciones directas con las enzimas cuyos pasos metabólicos c-Fos incrementa.
Para hacerlo, la idea fue emplear co-inmunoprecipitaciones por su robustez pero también
avanzar en el empleo de microscopia de fluorescencia para obtener información espacial y
temporal del fenómeno.
Otro de los objetivos planteados fue el de establecer la región de c-Fos involucrada en
la función no genómica, conociendo por resultados anteriores que la región básica,
involucrada en la unión al ADN, también parecía clave en la función no genómica. El enfoque
empleado fue genético, generando mutantes de deleción de c-Fos y estudiar la pérdida de
función de las mismas en la activación de la síntesis lipídica.
Por último, queda planteado el interrogante de cómo se activan vías metabólicas para
dar como producto lípidos distintos. El PA es un intermediario clave en la síntesis de
glicerofosfolípidos ya que puede derivado tanto a la vía de PIPs como a la vía de Kennedy que
dará lugar finalmente a PtdCho, PtdEth y PtdSer. En el mencionado reporte (Bussolino et al.
2001), se indicó que existen las dos ondas de activación del marcado radioactivo de lípidos
que depende de c-Fos en donde los lípidos que incorporan 32P durante la primera onda son
diferentes a los de la segunda. Esto despertó un interés por parte mía para continuar
estudiando el destino final de PA en este sistema y su dependencia en la expresión de c-Fos.

CAPITULO III
RESULTADOS

Resultados
22
Efecto de c-Fos en la síntesis de PIPs in vivo
Cuando ingresé al laboratorio, la Dra. Marianne Renner había comenzado durante el
desarrollo de su tesis doctoral a dilucidar algunos aspectos de esta activación dependiente de
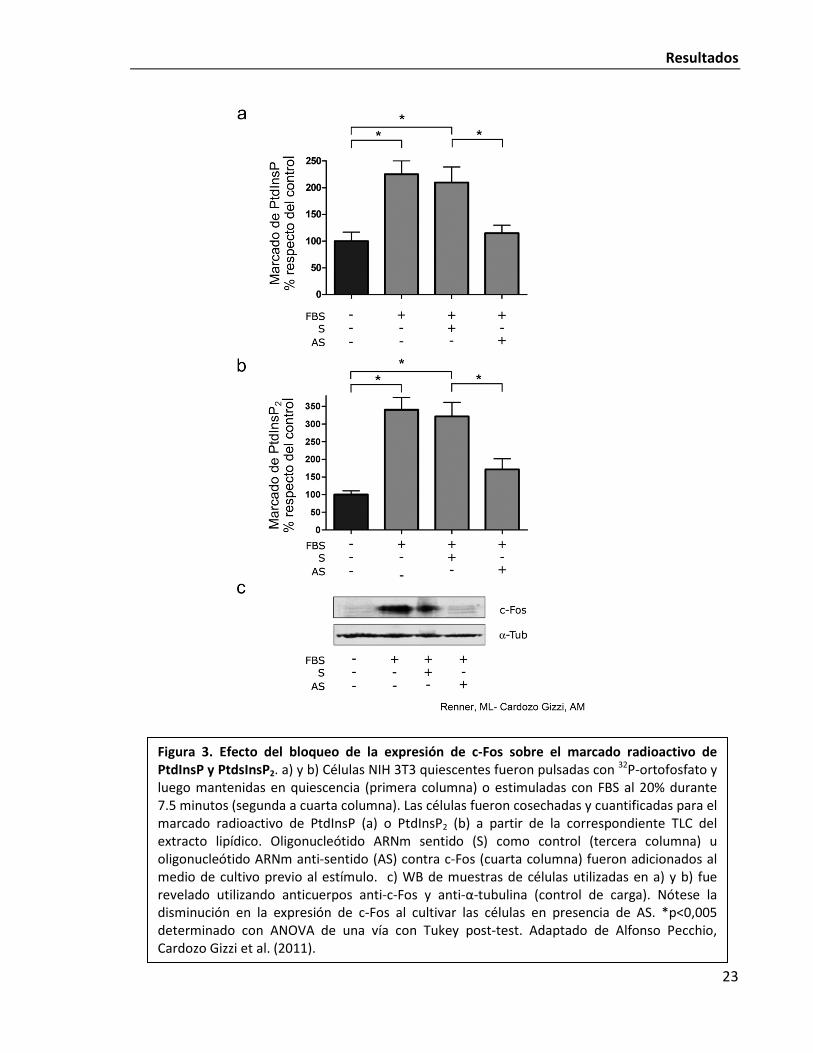
c-Fos. En la Fig. 3 se puede ver como se produce un acentuado incremento en el marcaje de
PtdIns y PtdInsP2 en las células luego de sólo 7,5 minutos de estímulo. Dicho incremento se
ve abolido frente al agregado a las células de un ASO que impide la traducción de la proteína
c-Fos. El experimento demuestra la relevancia biológica del fenómeno de la activación
dependiente de c-Fos para que la célula ajuste la síntesis de PIPs. Es concordante también
con estudios que indican que la expresión de c-Fos es necesaria para que células quiescentes
reingresen al ciclo celular (Robbins et al. 1990).
En la mencionada Fig. 3 se observa en la esquina inferior derecha el nombre de
Renner, ML reconociendo su trabajo. En próximas figuras, se continuará aclarando si el
trabajo fue hecho por otro miembro del laboratorio, en conjunto conmigo o si algunos
resultados fueron luego confirmados por mí. En los casos en los que no se coloque ningún
nombre, se trata de experimentos realizados íntegramente por mí.
Las actividades CDS y PIK pero no la de PIS son activadas por c-Fos
El incremento en el marcaje radioactivo de PIPs implica una más activa tasa de
síntesis o de recambio por sobre la de degradación de estos productos lipídicos. Nos
propusimos estudiar los distintos pasos metabólicos que conducen a la formación de los
mismos. En particular, se midieron las actividades enzimáticas en homogenatos celulares
totales de los tres primeros pasos de síntesis de PIPs utilizando precursores específicos de
cada etapa metabólica marcados radioactivamente, en presencia o ausencia de c-Fos
purificado obtenido en forma recombinante en E. Coli. Los homogenatos totales utilizados
como fuente de enzimas son de células NIH 3T3 quiescentes, que poseen niveles no
detectables c-Fos endógeno.
Resultados
23
Figura 3. Efecto del bloqueo de la expresión de c-Fos sobre el marcado radioactivo de PtdInsP y PtdsInsP2. a) y b) Células NIH 3T3 quiescentes fueron pulsadas con 32P-ortofosfato y luego mantenidas en quiescencia (primera columna) o estimuladas con FBS al 20% durante 7.5 minutos (segunda a cuarta columna). Las células fueron cosechadas y cuantificadas para el marcado radioactivo de PtdInsP (a) o PtdInsP2 (b) a partir de la correspondiente TLC del extracto lipídico. Oligonucleótido ARNm sentido (S) como control (tercera columna) u oligonucleótido ARNm anti-sentido (AS) contra c-Fos (cuarta columna) fueron adicionados al medio de cultivo previo al estímulo. c) WB de muestras de células utilizadas en a) y b) fue revelado utilizando anticuerpos anti-c-Fos y anti-α-tubulina (control de carga). Nótese la disminución en la expresión de c-Fos al cultivar las células en presencia de AS. *p<0,005 determinado con ANOVA de una vía con Tukey post-test. Adaptado de Alfonso Pecchio, Cardozo Gizzi et al. (2011).

Resultados
24
Luego de establecer las condiciones de linealidad de las distintas enzimas, se
realizaron ensayos con agregados crecientes de c-Fos purificado para establecer cuál o cuáles
actividad/es esta proteína es capaz de modificar. Como puede observarse en la Fig. 4, con el
agregado de 0,5 ng c-Fos/µg proteína total de homogeneizado, CDS tiene un pico en su
actividad de alrededor del 60% por encima del control que contiene solo el vehículo. Este
aumento se ve disminuido hasta alrededor del 30% por encima del control con mayores
cantidades de c-Fos. La actividad PIK requiere de mayores concentraciones de c-Fos (1 ng c-
Fos/µg proteína total) para alcanzar una activación del 60%. Sin embargo, hasta una
concentración de 3 ng c-Fos/µg proteína total (la más alta concentración de c-Fos ensayada)
sigue incrementándose el porcentaje de activación. En tanto, no se observó un incremento
en la actividad PIS a ninguna de las concentraciones de c-Fos ensayadas.
Figura 4. Curvas de activación dependiente de c-Fos para las actividades enzimáticas de CDS, PIS y PIK. Actividad in vitro de las enzimas CDS (círculos abiertos), PIS (cuadrados cerrados) y PIK (triángulos cerrados) en homogenatos totales de células NIH 3T3 quiescentes en función de la cantidad de c-Fos recombinante adicionado al medio de ensayo. Las actividades enzimáticas fueron determinadas según se describe en Materiales y Métodos, incubando los tubos durante 20 minutos para el caso de la actividad CDS y 10 minutos para los otros dos. Los Resultados son el promedio ± DE de un experimento representativo realizado en duplicado de un total de tres experimentos realizados. Adaptado de Alfonso Pecchio, Cardozo Gizzi et al. (2011).

Resultados
25
Es realmente interesante que a concentraciones inferiores de c-Fos se active CDS,
sugiriendo que los requerimientos para la activación del primer paso metabólico son
distintos del tercero.
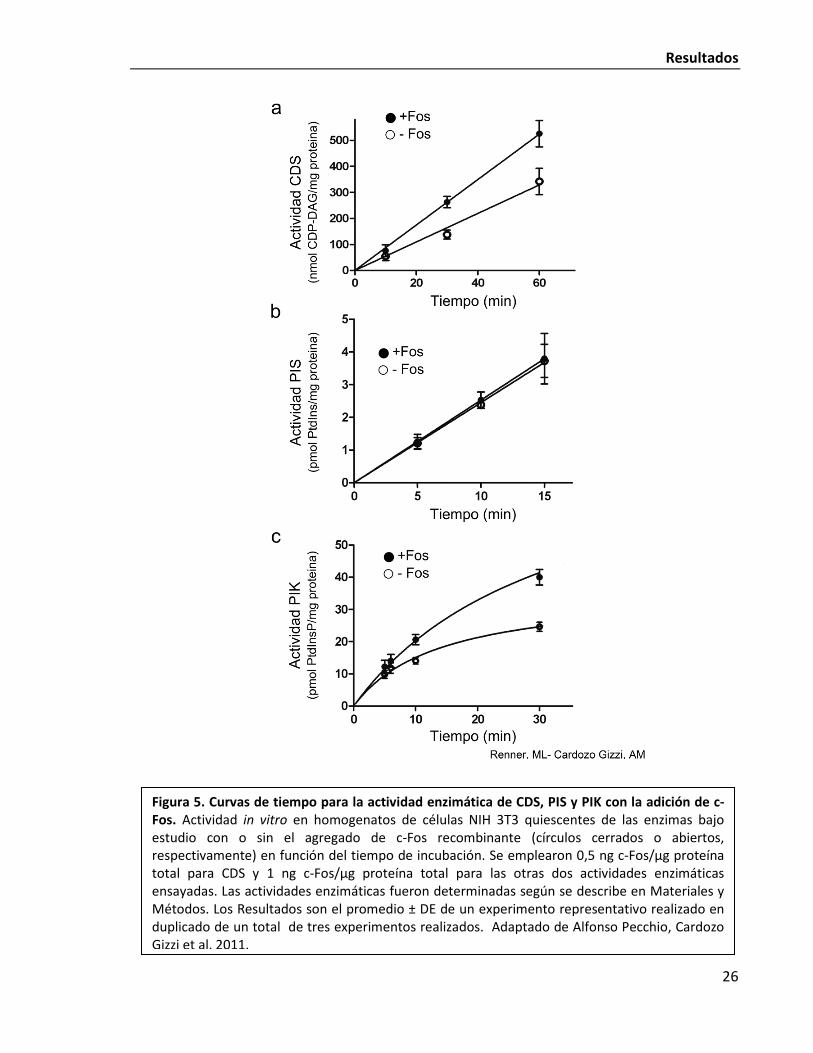
A continuación, se realizaron curvas de tiempo de las actividades enzimáticas a medir,
lo cual consiste en determinar la formación del producto de cada paso metabólico en
incubaciones a distintos tiempos; con o sin el agregado de c-Fos recombinante, usando 0,5
ng c-Fos/µg proteína total para CDS y 1 ng c-Fos/µg proteína total para las otras dos
actividades enzimáticas ensayadas. La actividad de CDS fue lineal hasta 60 minutos de
incubación (Fig. 5a). La actividad PIS fue lineal hasta 15 minutos, el tiempo más largo
ensayado (Fig. 5b). La formación de PtdInsP (actividad PIK) presenta una rápida cinética, por
lo que aún determinando la actividad a 25 °C no se obtuvo una clara zona de linealidad (Fig.
5c). En todos los tiempos analizados, se produjeron cantidades incrementadas de CDP-
DAG cercanas al 50% con el agregado de c-Fos. Esencialmente el mismo resultado se obtuvo
para PIK. En contraste, no se obtuvieron diferencias significativas en la actividad PIS en los
distintos tiempos analizados ni en las distintas concentraciones de c-Fos recombinante
ensayadas. Estos experimentos establecieron claramente que c-Fos es capaz de promover
una activación general de la vía al incrementar la actividad del primer y tercer paso
metabólico involucrado. Asimismo, la no activación de PIS estaría indicando la especificidad
de la activación enzimática.
Resultados
26
Figura 5. Curvas de tiempo para la actividad enzimática de CDS, PIS y PIK con la adición de c-Fos. Actividad in vitro en homogenatos de células NIH 3T3 quiescentes de las enzimas bajo estudio con o sin el agregado de c-Fos recombinante (círculos cerrados o abiertos, respectivamente) en función del tiempo de incubación. Se emplearon 0,5 ng c-Fos/µg proteína total para CDS y 1 ng c-Fos/µg proteína total para las otras dos actividades enzimáticas ensayadas. Las actividades enzimáticas fueron determinadas según se describe en Materiales y Métodos. Los Resultados son el promedio ± DE de un experimento representativo realizado en duplicado de un total de tres experimentos realizados. Adaptado de Alfonso Pecchio, Cardozo Gizzi et al. 2011.

Resultados
27
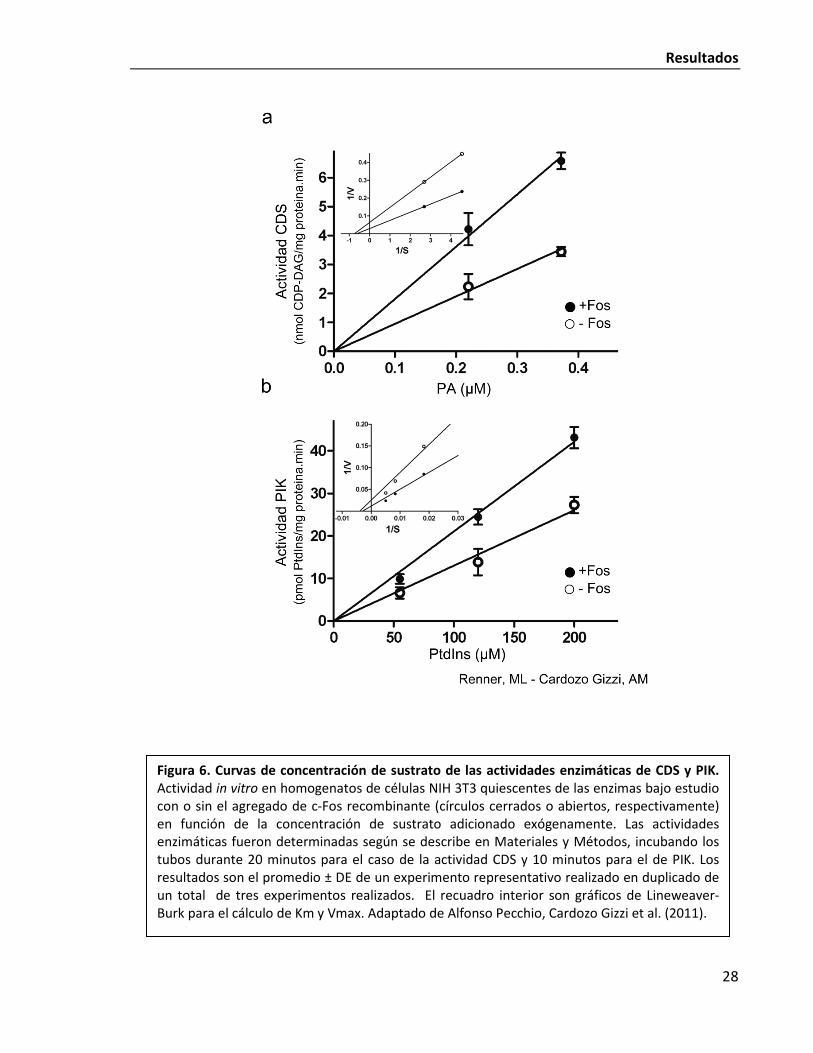
Finalmente, se realizaron curvas de concentración de sustrato de las enzimas que son
activadas por c-Fos (Fig. 6) con el objetivo de establecer los parámetros cinéticos, Km y
Vmax, de las enzimas en presencia y ausencia de c-Fos. El ajuste de los datos a curvas de
Michaelis-Menten permitió determinar los Km y Vmax de CDS y PIK, cuyos resultados se
encuentran en la Tabla 1. Estos estudios cinéticos son nuestro primer acercamiento al
mecanismo molecular mediante el cual c-Fos consigue el efecto de activación. En los dos
casos analizados, se observó el mismo comportamiento: el agregado de c-Fos recombinante
produjo incrementos en la velocidad máxima de la reacción de alrededor del 100% por
encima del control, sin que se produzcan cambios estadísticamente significativos en los Km.
Esto indica: i) que c-Fos incrementa la capacidad catalítica de las enzimas involucradas en la
activación sin que ello implique un cambio en la afinidad de la enzima por su sustrato y ii) son
las primeras evidencias de que existe un mecanismo compartido en la activación de las
distintas enzimas involucradas. Los pasos metabólicos estudiados catalizan reacciones
totalmente distintas: CDS transfiere un nucleótido (CTP) hacia el grupo fosfato del PA
mientras que PI4KIIα transfiere un fosfato desde ATP hacia el grupo alcohol del anillo de
inositol.
Es importante destacar que la mayor concentración de c-Fos exógena adicionada (1
ng/µg proteína total) es comparable a una concentración cercana a 105 moléculas de c-Fos
por célula. Esta concentración es la que se encuentra en fibroblastos cuando la expresión de
c-Fos es inducida, según fuera calculada por Kovary & Bravo (1991).
Resultados
28
Figura 6. Curvas de concentración de sustrato de las actividades enzimáticas de CDS y PIK. Actividad in vitro en homogenatos de células NIH 3T3 quiescentes de las enzimas bajo estudio con o sin el agregado de c-Fos recombinante (círculos cerrados o abiertos, respectivamente) en función de la concentración de sustrato adicionado exógenamente. Las actividades enzimáticas fueron determinadas según se describe en Materiales y Métodos, incubando los tubos durante 20 minutos para el caso de la actividad CDS y 10 minutos para el de PIK. Los resultados son el promedio ± DE de un experimento representativo realizado en duplicado de un total de tres experimentos realizados. El recuadro interior son gráficos de Lineweaver-Burk para el cálculo de Km y Vmax. Adaptado de Alfonso Pecchio, Cardozo Gizzi et al. (2011).

Resultados
29
Tabla 1. Parámetros cinéticos de las enzimas de la vía de síntesis de PIPs que son activadas por el agregado de c-Fos recombinante, calculados a partir de los datos de la Fig. 6.
Constantes cinéticas de la enzima CDS
Constantes cinéticas de la enzima PIK
Km (µM) Vmax (nmol producto/
mg proteina total min)
Km (µM) Vmax (pmol producto/
mg proteina total min)
Control 1,3±0,2 15,8±0,7 254±30 4,0±0,5
+c-Fos 1,6±0,3 34,3±1,6 331±57 8,5±0,6
Las actividades de CDS1 y PI4KIIα son claves para la activación del marcado radioactivo de
PtdInsP y PtdInsP2 promovido por FBS, no así la de PI4KIIβ
En esta instancia, se había establecido que al estimular fibroblastos a reingresar al
ciclo celular se produce un rápido incremento en el marcaje radioactivo de PIPs que requiere
de la expresión de c-Fos. La hipótesis inicial es que c-Fos media este efecto al activar etapas
metabólicas específicas, la primera y la tercera para el caso de la vía de síntesis de PIPs. Para
confirmar el rol de enzimas particulares en la síntesis de PtdInsP y PtdInsP2 in vivo, decidimos
estudiar el marcado de estos lípidos en células en cultivo previamente tratadas con
pequeños ARN de interferencia (siRNA) para deprimir la expresión de las enzimas en estudio.
Respecto de la formación de PtdInsP a partir del PtdIns, se ha observado que en
homogenatos totales de células NIH 3T3 se puede abolir prácticamente en su totalidad la
actividad PI4K con el inhibidor adenosina, que es específico para PI4K tipo II (de Graaf et al.
2002). Aún más, la vía predominante de fosforilación de PtdIns es iniciada por este tipo de
quinasas en muchas células de mamíferos (Fruman et al. 1998) y en particular la enzima
PI4KIIα se ha descripto como la más activa (Balla et al. 2002). Por tanto, se decidió utilizar las
dos isoformas (α y β) de esta familia de enzimas como blanco para disminuir su expresión. En
concreto, se trató a las células con siRNA contra CDS1, PI4KIIα y PI4KIIβ.
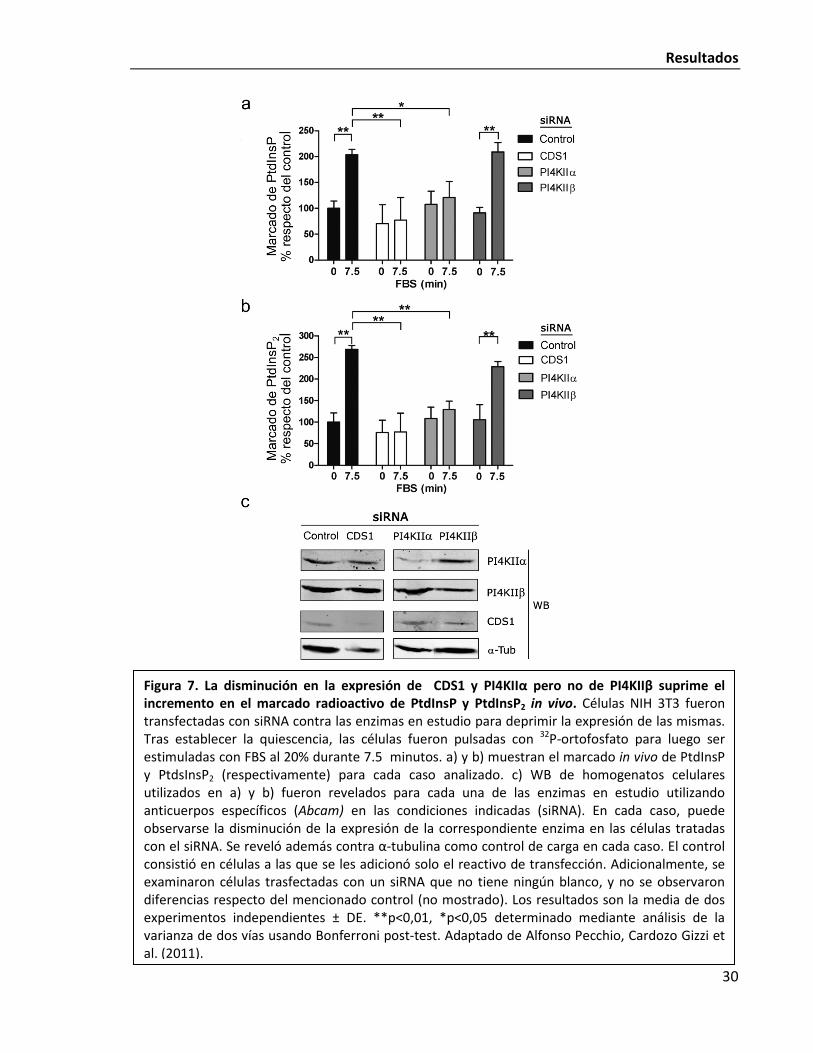
Resultados
30
Figura 7. La disminución en la expresión de CDS1 y PI4KIIα pero no de PI4KIIβ suprime el incremento en el marcado radioactivo de PtdInsP y PtdInsP2 in vivo. Células NIH 3T3 fueron transfectadas con siRNA contra las enzimas en estudio para deprimir la expresión de las mismas. Tras establecer la quiescencia, las células fueron pulsadas con 32P-ortofosfato para luego ser estimuladas con FBS al 20% durante 7.5 minutos. a) y b) muestran el marcado in vivo de PtdInsP y PtdsInsP2 (respectivamente) para cada caso analizado. c) WB de homogenatos celulares utilizados en a) y b) fueron revelados para cada una de las enzimas en estudio utilizando anticuerpos específicos (Abcam) en las condiciones indicadas (siRNA). En cada caso, puede observarse la disminución de la expresión de la correspondiente enzima en las células tratadas con el siRNA. Se reveló además contra α-tubulina como control de carga en cada caso. El control consistió en células a las que se les adicionó solo el reactivo de transfección. Adicionalmente, se examinaron células trasfectadas con un siRNA que no tiene ningún blanco, y no se observaron diferencias respecto del mencionado control (no mostrado). Los resultados son la media de dos experimentos independientes ± DE. **p<0,01, *p<0,05 determinado mediante análisis de la varianza de dos vías usando Bonferroni post-test. Adaptado de Alfonso Pecchio, Cardozo Gizzi et al. (2011).

Resultados
31
Al estimular las células con FBS, se observa el esperando incremento en el marcado
de PtdInsP y PtdInsP2 (Fig. 7). Este incremento se ve suprimido en células tratadas con siRNA
contra CDS1 o PI4KIIα. En cambio, disminuir la expresión de PI4KIIβ no tiene efecto alguno en
el fenómeno. Estos resultados ponen de manifiesto la importancia de CDS1 y PIKIIα en la
activación de la síntesis de PIPs a tiempos cortos en células NIH 3T3 que reingresan al ciclo
celular. Es importante remarcar en este punto que según se observa en la Fig. 3, dicha
activación es dependiente de la expresión de c-Fos.
Es interesante destacar que en un reporte reciente se vio que mientras CDS1 no
muestra una especificidad o preferencia por las cadenas laterales del sustrato (posiciones sn-
1 y sn-2 de PA), CDS2, la segunda isoforma de esta enzima, tiene una marcada preferencia
por 1-estearoil-2-araquidonil PA como sustrato, así como también una inhibición de su
actividad por parte del PtdInsP2 con estas cadenas de los ácidos grasos en particular
(D'Souza et al. 2014). Estudios de otros laboratorios demuestran que el PtdIns formado a
partir de la vía de síntesis de novo contiene principalmente cadenas del resto acilo saturadas
o mono-insaturadas (Holub & Kuksis 1971, Luthra & Sheltawy 1976). En cambio, el ciclo de
PtdIns involucra la ruptura de PtdIns(4,5)P2 en la membrana plasmática y la re-síntesis de
PIPs por parte de enzimas, entre ellas DGKε (Lung et al. 2009) o PtdIns4P-5-quinasa (Shulga
et al. 2012), que tienen una importante especificidad por el sustrato, lo cual resulta en un
enriquecimiento cíclico del PtdIns de cadenas acilo particulares, siendo las principales 1-
estearoil-2-araquidonil (18:0 sn−1/20:4 sn−2). Esto último induce a pensar que CDS2 estaría
involucrada principalmente en el ciclo del PtdIns mientras que CDS1, que no exhibe ninguna
especificidad por la cadena lateral, en la síntesis de novo de PtdIns (D'Souza & Epand 2014).
La enzima CDS cuya actividad incrementada depende de la expresión de c-Fos y que además
al deprimir la expresión de la misma se impide el incremento en la marcación de PtdInsP2,
sería entonces la involucrada en la síntesis de novo.

Resultados
32
CDS1 y PI4KIIα pero no PI4KIIβ participan en una asociación física con c-Fos
Los experimentos mostrados hasta aquí plantearon el interrogante sobre la forma en
que c-Fos logra la activación de enzimas específicas. Un posible mecanismo es mediante una
interacción directa con las enzimas que activa. Para estudiar esta alternativa, inicialmente se
realizaron experimentos de coinmunoprecipitacion en homogenatos totales de fibroblastos
NIH 3T3 transfectados para expresar las enzimas bajo estudio con un rótulo que permita
dirigir un anticuerpo especifico contra el mismo. Concordantemente con lo observado en la
activación de enzimas particulares (Figs. 4 y 5), c-Fos coinmunoprecipitó con CDS1 y PI4KIIα
pero no con PIS1 o PI4KIIβ (Fig. 8). Estos estudios sugirieron que c-Fos está modulando la
actividad de estas enzimas a través de una interacción física, lo cual permitiría la activación
específica de las mismas.
La técnica de coinmunoprecipitación es muy robusta ya que si al bajar una proteína
con un anticuerpo especifico y luego revelar la existencia en el inmuno-complejo de una
segunda proteína, se está probando que ambas participan en un complejo. Sin embargo, esta
Figura 8. CDS1 y PI4KIIα pero no PI4KIIβ coinmunoprecipitan con c-Fos. Células NIH 3T3 fueron transfectadas para expresar PIS1-myc, PI4KIIα-myc, CDS1-YFP ó PIKIIβ-YFP y estimuladas con FBS al 20% durante 1 hora para inducir la expresión de c-Fos endógeno. Lisados de estas células fueron inmunoprecipitados usando anti-GFP (Roche) o anti-myc (Sigma), ambos producidos en ratón. Se muestra en la parte superior el WB revelado utilizando anti-c-Fos (Santa Cruz,
producido en conejo) y en la parte inferior los inmunoprecipitados obtenidos para cada enzima revelados usando anti-GFP (Sigma) o anti-myc (Santa Cruz) ambos producidos en conejo. Adaptado de Alfonso Pecchio, Cardozo Gizzi et al. (2011).

Resultados
33
técnica tiene sus limitaciones. En primer lugar, al tratarse de homogenatos totales, no se
brinda ninguna información espacial del fenómeno. Más aún, la disrupción del entorno
natural en que se encuentran las proteínas de interés puede promover interacciones que no
están presentes en una célula intacta. Por último, que ambas proteínas se encuentren en el
mismo inmuno-complejo no descarta la posibilidad de terceras proteínas que actúen como
proteínas puente.
Para superar estas limitaciones, decidimos estudiar la interacción proteína-proteína
usando microscopía FRET por emisión sensibilizada (Elangovan et al. 2003). Dicha técnica se
basa en la transferencia de energía desde un donor fluorescente excitado hacia un aceptor
(fluorescente o no) por medio de un fenómeno no radiativo. Este fenómeno, denominado
FRET o transferencia de energía de Förster, por quien estableciera los principios que lo rigen,
depende fuertemente de la distancia. En términos prácticos, donor y aceptor deben
encontrarse a no más de 10 nm de distancia para que ocurra el fenómeno de FRET, para el
caso de la utilización de proteínas fluorescentes como par aceptor-donor. Esta distancia es
típica de una interacción directa entre dos proteínas, por lo que esta técnica permite medir
interacciones proteína-proteína (Vogel et al. 2006). El FRET agota la población del estado
excitado del donor, disminuyendo su emisión mientras que causa la emisión (sensibilizada)
del aceptor. La forma de medir el fenómeno consiste en fusionar las proteínas de interés a
proteínas fluorescentes, usando un par o combinación que permita la transferencia de
energía. En este caso en particular empleamos a la proteína fluorescente cyan (CFP) como
donor y a la proteína fluorescente amarilla (YFP) como aceptor (Piston & Kremers 2007). Para
realizar el experimento de FRET, se transfectaron las células con los plásmidos
correspondientes al tiempo que se deprivaban a estas de factores de crecimiento. Luego de
establecerse la quiescencia, se indujeron las células durante 7,5 minutos con FBS para
generar la transición G0/G1 y situarnos en el momento de más alta síntesis de PIPs (Bussolino
et al. 2001). Este estímulo es necesario para desencadenar los fenómenos celulares que
aseguran la interacción de ambas proteínas. Sin embargo, el estímulo también promueve la
rápida traducción de c-Fos endógeno, que podría competir con el c-Fos fusionado a CFP por
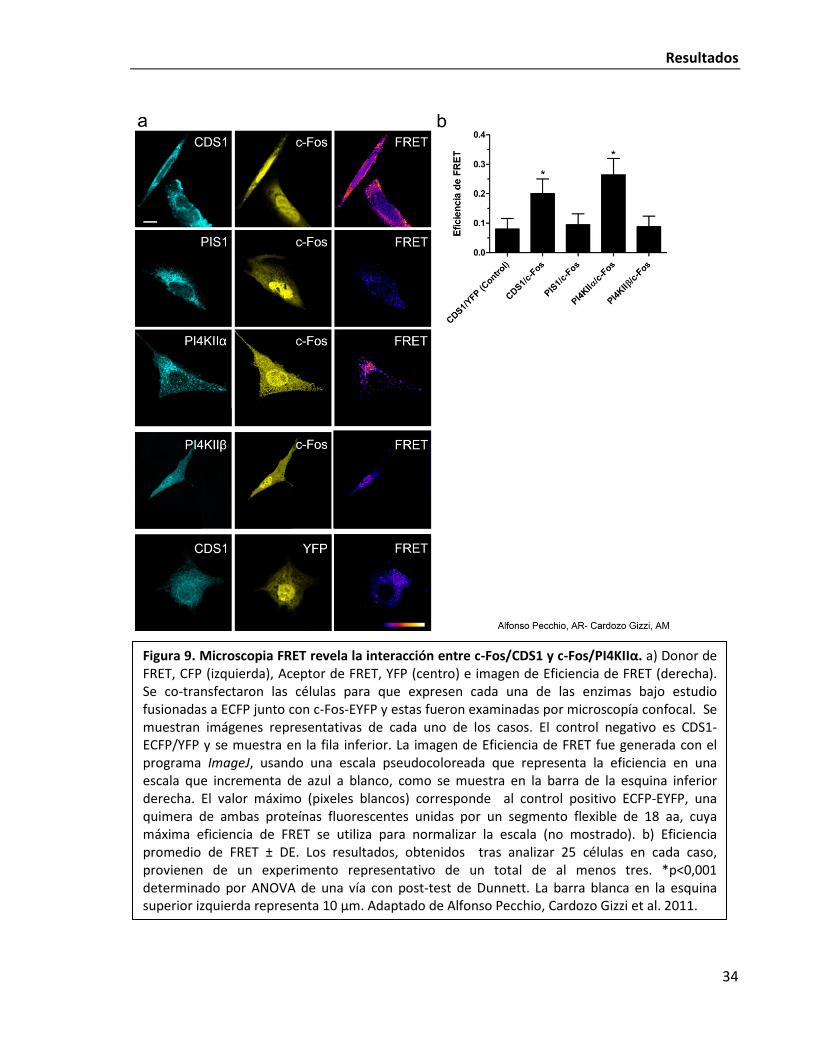
Resultados
34
Figura 9. Microscopia FRET revela la interacción entre c-Fos/CDS1 y c-Fos/PI4KIIα. a) Donor de FRET, CFP (izquierda), Aceptor de FRET, YFP (centro) e imagen de Eficiencia de FRET (derecha). Se co-transfectaron las células para que expresen cada una de las enzimas bajo estudio fusionadas a ECFP junto con c-Fos-EYFP y estas fueron examinadas por microscopía confocal. Se muestran imágenes representativas de cada uno de los casos. El control negativo es CDS1-ECFP/YFP y se muestra en la fila inferior. La imagen de Eficiencia de FRET fue generada con el programa ImageJ, usando una escala pseudocoloreada que representa la eficiencia en una escala que incrementa de azul a blanco, como se muestra en la barra de la esquina inferior derecha. El valor máximo (pixeles blancos) corresponde al control positivo ECFP-EYFP, una quimera de ambas proteínas fluorescentes unidas por un segmento flexible de 18 aa, cuya máxima eficiencia de FRET se utiliza para normalizar la escala (no mostrado). b) Eficiencia promedio de FRET ± DE. Los resultados, obtenidos tras analizar 25 células en cada caso, provienen de un experimento representativo de un total de al menos tres. *p<0,001 determinado por ANOVA de una vía con post-test de Dunnett. La barra blanca en la esquina superior izquierda representa 10 µm. Adaptado de Alfonso Pecchio, Cardozo Gizzi et al. 2011.

Resultados
35
la unión de las enzimas y disminuir los valores de FRET. Para contrarrestar esto, se incubó a
las células durante el estímulo (y desde 45 minutos antes) con cicloheximida a una
concentración de 50 µg/mL, que impide la traducción al nivel del ribosoma. De esta forma, se
permiten los eventos post-traduccionales necesarios para la activación de algunas enzimas al
tiempo que se inhibe la síntesis de c-Fos endógeno.
Las condiciones de FRET indican que las proteínas de interés se encuentran a una
distancia menor a 10 nm, actuando entonces como una regla molecular; en tanto que al ser
una técnica de microscopía, permite obtener información respecto de la localización
subcelular de la interacción. Los resultados correlacionan con lo observado anteriormente en
las coinmunoprecipitaciones: tanto CDS1 como PI4KIIα pero no PI4KIIβ participan de una
interacción física con c-Fos (Fig. 9). Si bien se observa un acumulamiento de c-Fos dentro del
núcleo, la asociación ocurre peri-nuclearmente, aparentemente asociado al RE según puede
verse en las imágenes de eficiencia de FRET (Fig. 9). Es importante mencionar que c-Fos
experimenta un transporte activo entre el núcleo y el citoplasma, y que esta proteína es
capaz de experimentar ciclos de entrada y salida al núcleo (Malnou et al. 2007). El RE es,
como se dijo anteriormente, el principal sitio de síntesis de fosfolípidos y el sitio subcelular
donde se ha encontrado previamente a c-Fos asociado (Bussolino et al. 2001) por lo que el
sitio de asociación enzima/c-Fos se corresponde con la hipótesis de que c-Fos interacciona
directamente con enzimas específicas para activar la síntesis metabólica de fosfolípidos en el
RE.
El domino BD sería el responsable de la activación de la síntesis de PIPs
Para determinar cuál o cuáles son los dominios de c-Fos requeridos para esta función
no genómica, construimos inicialmente mutantes de deleción de la proteína y ensayamos su
capacidad para activar la síntesis de PIPs in vitro. Puede observarse una representación
esquemática de las mutantes empleadas en la Fig. 10c. La mutante NB (aa: 1-160) es capaz
de activar la síntesis de PtdInsP y PtdInsP2 mientras que NA (aa: 1-138) no lo hace (Figs 10 a y
b). La diferencia entre ambas mutantes son únicamente los 21 aa del dominio básico (BD)
que NB incluye y NA no. Por otro lado, la mutante de deleción ΔBD, a la que solo le faltan los
21 aa correspondientes al BD respecto de c-Fos salvaje, es incapaz de activar la síntesis de
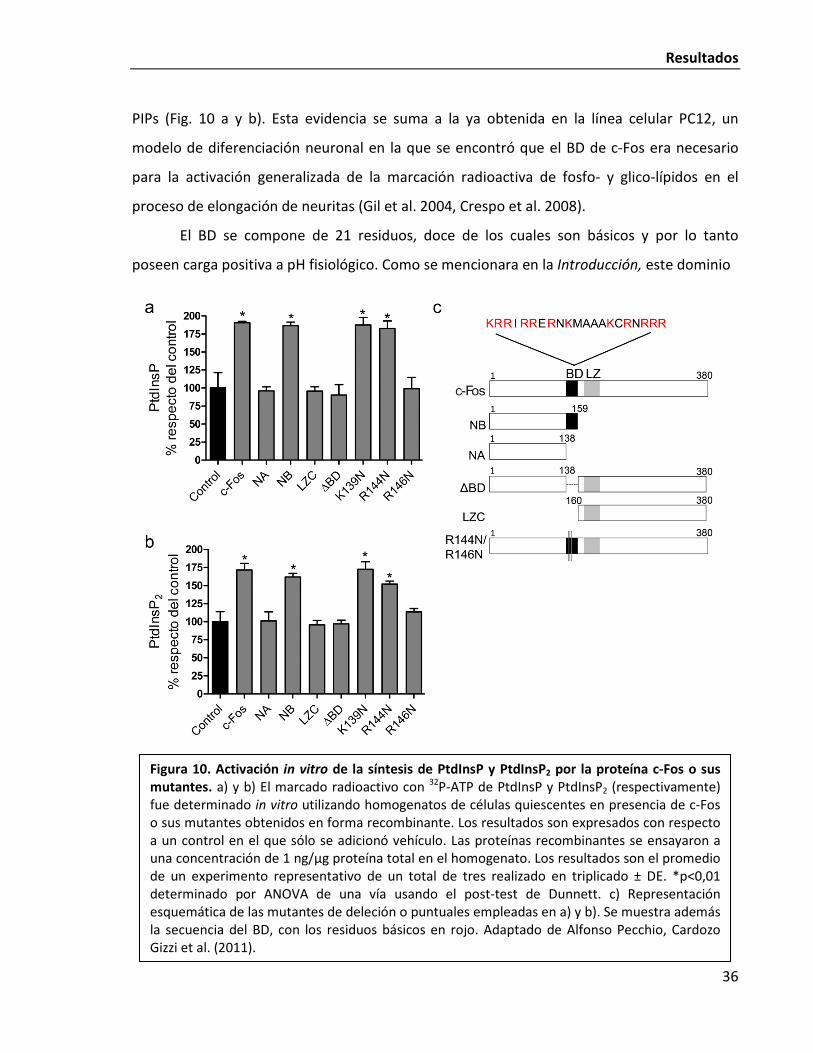
Resultados
36
Figura 10. Activación in vitro de la síntesis de PtdInsP y PtdInsP2 por la proteína c-Fos o sus mutantes. a) y b) El marcado radioactivo con 32P-ATP de PtdInsP y PtdInsP2 (respectivamente) fue determinado in vitro utilizando homogenatos de células quiescentes en presencia de c-Fos o sus mutantes obtenidos en forma recombinante. Los resultados son expresados con respecto a un control en el que sólo se adicionó vehículo. Las proteínas recombinantes se ensayaron a una concentración de 1 ng/µg proteína total en el homogenato. Los resultados son el promedio de un experimento representativo de un total de tres realizado en triplicado ± DE. *p<0,01 determinado por ANOVA de una vía usando el post-test de Dunnett. c) Representación esquemática de las mutantes de deleción o puntuales empleadas en a) y b). Se muestra además la secuencia del BD, con los residuos básicos en rojo. Adaptado de Alfonso Pecchio, Cardozo Gizzi et al. (2011).
PIPs (Fig. 10 a y b). Esta evidencia se suma a la ya obtenida en la línea celular PC12, un
modelo de diferenciación neuronal en la que se encontró que el BD de c-Fos era necesario
para la activación generalizada de la marcación radioactiva de fosfo- y glico-lípidos en el
proceso de elongación de neuritas (Gil et al. 2004, Crespo et al. 2008).
El BD se compone de 21 residuos, doce de los cuales son básicos y por lo tanto
poseen carga positiva a pH fisiológico. Como se mencionara en la Introducción, este dominio

Resultados
37
está directamente involucrado en la unión al ADN, que por sus fosfatos posee una fuerte
carga negativa. Sin embargo, la unión al ADN es altamente específica y los heterodímeros AP-
1 solo interaccionan con secuencias definidas. En la Fig. 10c puede verse la secuencia de este
dominio donde se han resaltado los residuos básicos en rojo. Decidimos realizar mutantes
puntuales sobre estos residuos básicos, en particular sobre lisina-139, arginina-144 y
arginina-146, reemplazándolas por asparagina, un aminoácido polar que tiene un grupo
carboxamida como su cadena lateral. Los resultados indican que la arginina-146 es
indispensable para conseguir la activación de la síntesis lipídica, ya que al mutarla se suprime
el efecto. Las otras dos mutantes ensayadas, K139N y R144N, son capaces de incrementar el
marcado radioactivo de lípidos a niveles comparables con c-Fos salvaje a pesar de tener una
carga positiva menos (Fig. 10 a y b), lo que indicaría que el efecto no es únicamente una
cuestión de carga.
La región N-terminal es la responsable de la asociación con CDS1
Habiendo establecido el dominio involucrado en la activación de la síntesis de PIPs,
fue de interés establecer cuál era el dominio de asociación con la enzima CDS1, elegida como
modelo de estudio. Para ello se fusionaron las mismas mutantes esquematizadas en la Fig.
10c a la proteína fluorescente CFP y se analizó por microscopía FRET la transferencia de
energía a CDS1-YFP. Según puede observarse en la Fig. 11, tanto la mutante NA como NB
retienen la capacidad de unión a CDS1 a pesar de no contar con la porción C-terminal de la
proteína c-Fos salvaje. Ello, sumado a que la mutante LZC (160-380) es incapaz de unirse a la
enzima, indicaría que la región C-terminal de c-Fos no está involucrada en el fenómeno. Por
otro lado, la mutante ΔBD, a pesar de no poseer capacidad como activadora de la síntesis de
PIPs, sigue asociándose a CDS1, lo cual descartaría que el BD esté involucrado en esta
asociación. Aún más, el hecho que tanto NA como NB se asocien a la enzima, apuntaría a la
región N-terminal (aa: 1-138) como la implicada en la interacción, ya que es la región mínima
compartida. En conclusión, la interpretación más sencilla de los resultados obtenidos hasta
aquí es que el dominio N-terminal de c-Fos interacciona con CDS1 mientras que el dominio
de activación es el BD, lo que indica que, sorpresivamente, la unión y la activación de CDS1 se
deben a regiones diferentes de c-Fos.

Resultados
38
Figura 11. El dominio N-terminal es el responsable de la asociación de c-Fos a la enzima CDS1. a) Donor de FRET, CFP (izquierda), Aceptor de FRET, YFP (centro) e imagen de Eficiencia de FRET (derecha). Se co-transfectaron las células para que expresen cada una de las enzimas bajo estudio fusionadas a ECFP junto con c-Fos-EYFP y estas fueron examinadas por microscopía confocal. Se muestran imágenes representativas de cada uno de los casos. El control negativo es CDS1-ECFP/YFP (no mostrado). La imagen de Eficiencia de FRET fue generada con el programa ImageJ, usando una escala pseudocoloreada que representa la eficiencia en una escala que incrementa de azul a blanco, como se muestra en la barra de la esquina inferior derecha. El valor máximo (pixeles blancos) corresponde al control positivo ECFP-EYFP, una quimera de ambas proteínas fluorescentes unidas por un segmento flexible de 18 aa, cuya máxima eficiencia de FRET se utiliza para normalizar la escala (no mostrado). b) Eficiencia promedio de FRET ± DE. Los resultados, obtenidos tras analizar 25 células en cada caso, provienen de un experimento representativo de un total de al menos tres. *p<0,001 determinado por ANOVA de una vía con post-test de Dunnett. La barra blanca en la esquina superior izquierda representa 10 µm. Adaptado de Alfonso Pecchio, Cardozo Gizzi et al. (2011).

Resultados
39
La región N-terminal es capaz de competir con c-Fos salvaje por su unión a la enzima
PI4KIIα
Dado el hallazgo de que la región N-terminal es la responsable de la asociación a la
enzima CDS1 pero no de la activación de la misma (ya que el dominio de activación se localiza
en el BD) se decidió utilizarlo para una posible intervencion en el metabolismo celular. La
idea consiste en establecer si la región N-terminal por si misma puede competir por el sitio
de unión de la enzima con c-Fos y de esta forma, al impedir la asociación, evitar el
incremento en la actividad enzimática. El objetivo último de esta intervención en el
metabolismo es establecer una posible estrategia para detener la proliferación celular que,
como se dijo, depende del estado activado de síntesis de fosfolípidos. A partir de la
evidencia obtenida hasta aquí, diseñamos un experimento de competencia, utilizando a la
enzima PI4KIIα para esta prueba de concepto. Estos experimentos se realizaron en conjunto
con el Dr. Cesar Prucca, que comenzaba entonces una estancia post-doctoral en el
laboratorio con el objetivo de desarrollar esta estrategia anti-tumoral mediante el bloqueo
específico de la actividad no genómica de c-Fos.
Para hacer estos experimentos de competencia utilizamos otra forma de determinar
la ocurrencia de FRET llamado FLIM (Fluorescence Lifetime Imaging Microscopy o
Microscopía de tiempo de vida media de fluorescencia). El método de emisión sensibilizada
se basa en la intensidad de la imagen para poder cuantificar una eficiencia “aparente” en la
transferencia de energía y por lo tanto depende de la concentración de los fluoróforos, que
debe ser tenida en cuenta para normalizar la señal. En cambio, la técnica de FLIM mide el
tiempo de vida media de fluorescencia (τ), el cual no depende de la concentración de
fluoróforos, el microscopio empleado o las condiciones de adquisición. Este método se
encuentra entre los mas robustos y confiables para medir FRET, ya que se basa en un
parámetro físico cuantificable directamente (Sun et al. 2013). Toda población de fluoróforos
que es excitada por un pulso de luz reside en el estado electrónico excitado un tiempo
determinado antes de decaer al estado basal. Para decaer, emite energía en forma radiativa
o bien puede disipar esa energía en forma no radiativa. En presencia de un aceptor de FRET,
la población de fluoróforos excitados posee una nueva vía para transmitir o disipar la

Resultados
40
energía. En consecuencia, el tiempo de vida media en el estado excitado es menor respecto
de una población de fluoróforos que no tienen disponible un aceptor. En otras palabras, el
tiempo de vida media del fluoróforo emisor disminuye ante la ocurrencia de FRET. El cambio
en el tiempo de vida media con o sin el aceptor (o el delta τ) indica la eficiencia de FRET en
forma cuantitativa, siendo este un parámetro muy robusto para establecer una interacción
proteína-proteína. Una de las principales desventajas de este método es que la adquisición
de datos confiables depende de colectar múltiples imágenes sucesivas para así tener los
fotones necesarios para hacer un análisis, y por lo tanto es mas lento que la técnica de
emisión sensibilizada. La otra desventaja es que requiere instrumentación especializada y
costosa. Para mas detalles, ver la sección Materiales y Métodos.
El experimento se desarrolla de la siguiente manera: se toman las imágenes de FLIM
para un campo en particular y a continuación se obtienen las imágenes para los canales de
CFP, YFP y λ660 (el fluoróforo con el que se marcó a NA-myc). Las imágenes de FLIM se
toman excitando únicamente al donor de FRET, CFP, utilizando un láser modulado y un
detector para el mismo, ya que se estudia el delta τ en la fluorescencia del donor. Las
imágenes confocales de los tres canales en cambio permiten tener la información espacial
para las tres proteínas, para luego hacer análisis de ubicación subcelular y co-localización.
Analizamos entonces la ocurrencia de FRET como una disminución en el tiempo de
vida media de fluorescencia del donor utilizando un enfoque novedoso, el análisis de fasor
(Digman et al. 2008). En este enfoque, la información de FLIM se representa de forma que el
decamiento de fluorescencia de cada pixel de la imagen genera un punto en un gráfico de
fasores (o phasor plot) (Fig. 12). El gráfico de fasores es un gráfico en el que se representan
las coordinadas s y g de cada fasor, un vector que representa el tiempo de vida media. Existe
un fasor por cada pixel en la imagen que representa entonces el tiempo de vida media de ese
pixel (ver Materiales y Métodos para la descripción de un fasor). Se utiliza un cursor en el
gráfico de fasores, de forma que al seleccionar los fasores que están dentro de este cursor, se
“pintan” los pixeles en la imagen que se corresponden con estos fasores (Fig. 13, en este caso
se “pintan” de violeta los pixeles que se encuentran en el cursor negro redondo del gráfico
de fasores). El fasor que finalmente se asocia con una célula es el fasor promedio (o el que
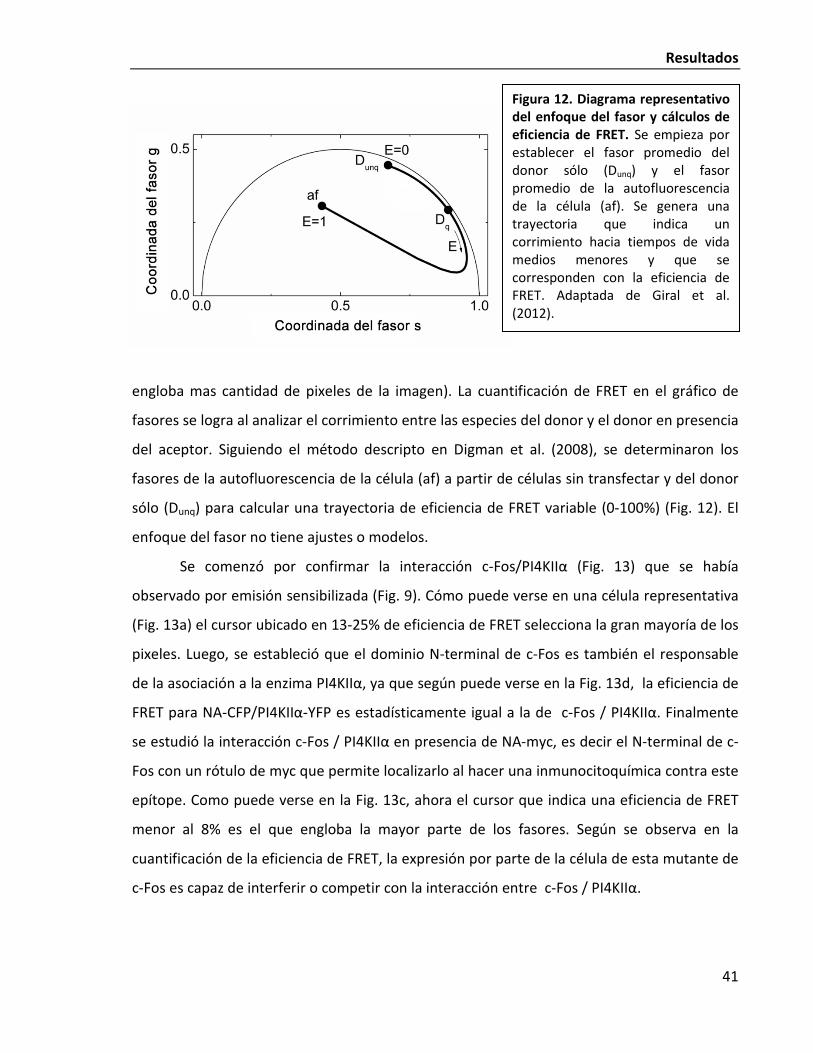
Resultados
41
engloba mas cantidad de pixeles de la imagen). La cuantificación de FRET en el gráfico de
fasores se logra al analizar el corrimiento entre las especies del donor y el donor en presencia
del aceptor. Siguiendo el método descripto en Digman et al. (2008), se determinaron los
fasores de la autofluorescencia de la célula (af) a partir de células sin transfectar y del donor
sólo (Dunq) para calcular una trayectoria de eficiencia de FRET variable (0-100%) (Fig. 12). El
enfoque del fasor no tiene ajustes o modelos.
Se comenzó por confirmar la interacción c-Fos/PI4KIIα (Fig. 13) que se había
observado por emisión sensibilizada (Fig. 9). Cómo puede verse en una célula representativa
(Fig. 13a) el cursor ubicado en 13-25% de eficiencia de FRET selecciona la gran mayoría de los
pixeles. Luego, se estableció que el dominio N-terminal de c-Fos es también el responsable
de la asociación a la enzima PI4KIIα, ya que según puede verse en la Fig. 13d, la eficiencia de
FRET para NA-CFP/PI4KIIα-YFP es estadísticamente igual a la de c-Fos / PI4KIIα. Finalmente
se estudió la interacción c-Fos / PI4KIIα en presencia de NA-myc, es decir el N-terminal de c-
Fos con un rótulo de myc que permite localizarlo al hacer una inmunocitoquímica contra este
epítope. Como puede verse en la Fig. 13c, ahora el cursor que indica una eficiencia de FRET
menor al 8% es el que engloba la mayor parte de los fasores. Según se observa en la
cuantificación de la eficiencia de FRET, la expresión por parte de la célula de esta mutante de
c-Fos es capaz de interferir o competir con la interacción entre c-Fos / PI4KIIα.
Figura 12. Diagrama representativo del enfoque del fasor y cálculos de eficiencia de FRET. Se empieza por establecer el fasor promedio del donor sólo (Dunq) y el fasor promedio de la autofluorescencia de la célula (af). Se genera una trayectoria que indica un corrimiento hacia tiempos de vida medios menores y que se corresponden con la eficiencia de FRET. Adaptada de Giral et al. (2012).
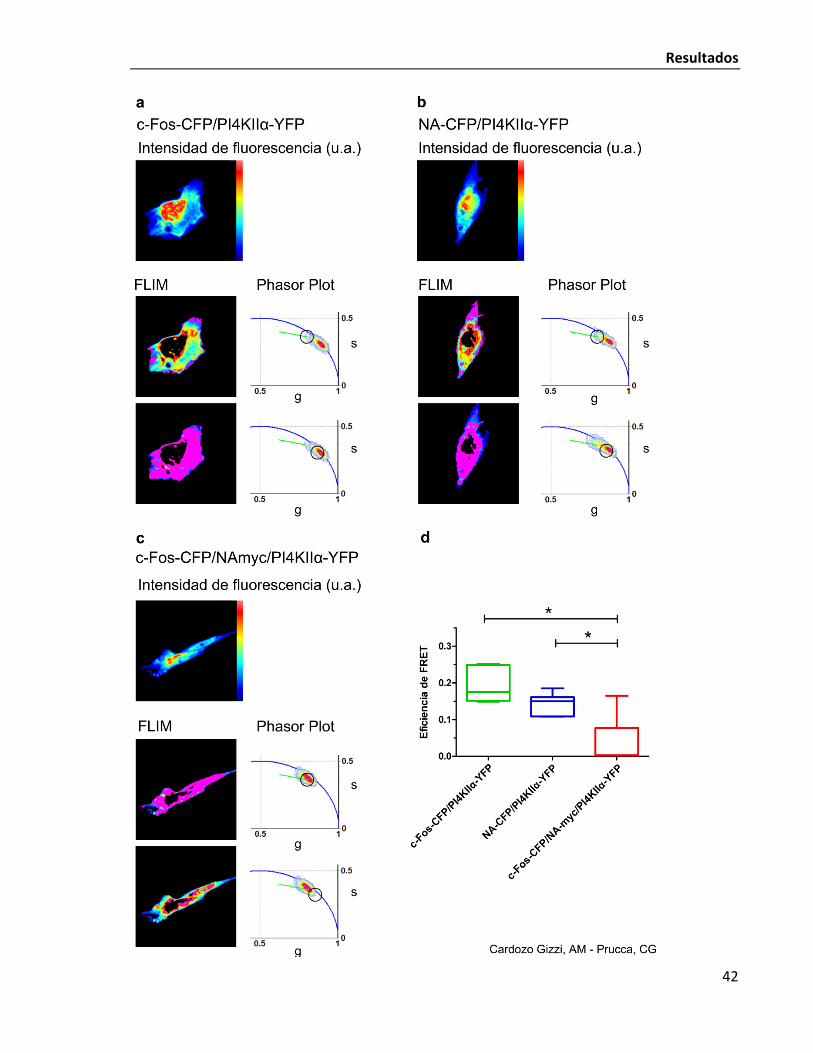
Resultados
42

Resultados
43
El análisis posterior de la fluorescencia en los tres canales aportó nueva información
(Fig. 14). La enzima PI4KIIα se observa teniendo una amplia distribución extranuclear y se
encuentra asociada a membranas de distintas organelas/vesículas en forma particulada
concordantemente con la bibliografía que la señala asociada a RE, Golgi y endosomas
(Waugh et al. 2003, Minogue et al. 2006). Por otro lado, c-Fos-CFP se observa tanto dentro
como fuera del núcleo, a diferencia de NA-myc que aparece concentrado en forma peri-
nuclear, excluido del núcleo y asociado aparentemente a membranas. Es posible que la
fusión de c-Fos a CFP genere una acumulación de esta quimera en el núcleo, que ya no se
observa con el pequeño rótulo de myc para el caso de la mutante NA. Precisamente en la
estructura donde localiza NAmyc es donde se da una triple co-localización de las proteínas en
estudio. Es de esperar que allí sea donde se produzca la competencia por la unión de PI4KIIα.
Sin embargo, en las imágenes de FLIM, la ocurrencia de FRET se da en toda la región
extranuclear, que es la que marca el cursor (Fig. 13) y no en esta región en particular. Dos
posibles explicaciones a este resultado surgen rápidamente: 1) la resolución espacial del FLIM
no es suficiente para observar una región precisa o 2) la interacción c-Fos/PI4KIIα se produce
en multiples localizaciones citoplásmicas más allá de la acumulación en alguna región
particular.
Figura 13. NA-myc compite con la unión de c-Fos salvaje con PI4KIIα. (a, b y c) Un experimento de FLIM-estrategia del fasor en células NIH 3T3 muestra la imagen de intensidad de fluorescencia, el gráfico del fasor y la localización en la imagen de FLIM (máscara violeta) de los pixeles que se encuentran dentro del área de selección (circulo negro en el gráfico de fasores). Se observan dos subconjuntos de fasores, y los correspondientes pixeles se seleccionan en la máscara de la imagen de FLIM como pixeles violetas: pixeles en el rango <8% y 13-25% de eficiencia de FRET. En a y b) el cursor de selección 13-25% de eficiencia de FRET selecciona al menos el 80% de los pixeles totales, en tanto en c) el cursor de eficiencia de FRET <8% selecciona al menos el 80% de los pixeles totales. Es importante destacar que se dispuso un umbral de intensidad que descarta los pixeles dentro del núcleo, para así excluirlos del análisis. d) Cuantificación de la eficiencia de FRET tomando un total de 15 células para cada caso. *p< 0,01 usando ANOVA y post-test de Dunnett. Nótese como NAmyc es capaz de disminuir la eficiencia de FRET y por lo tanto la interacción c-Fos/PI4KIIα.
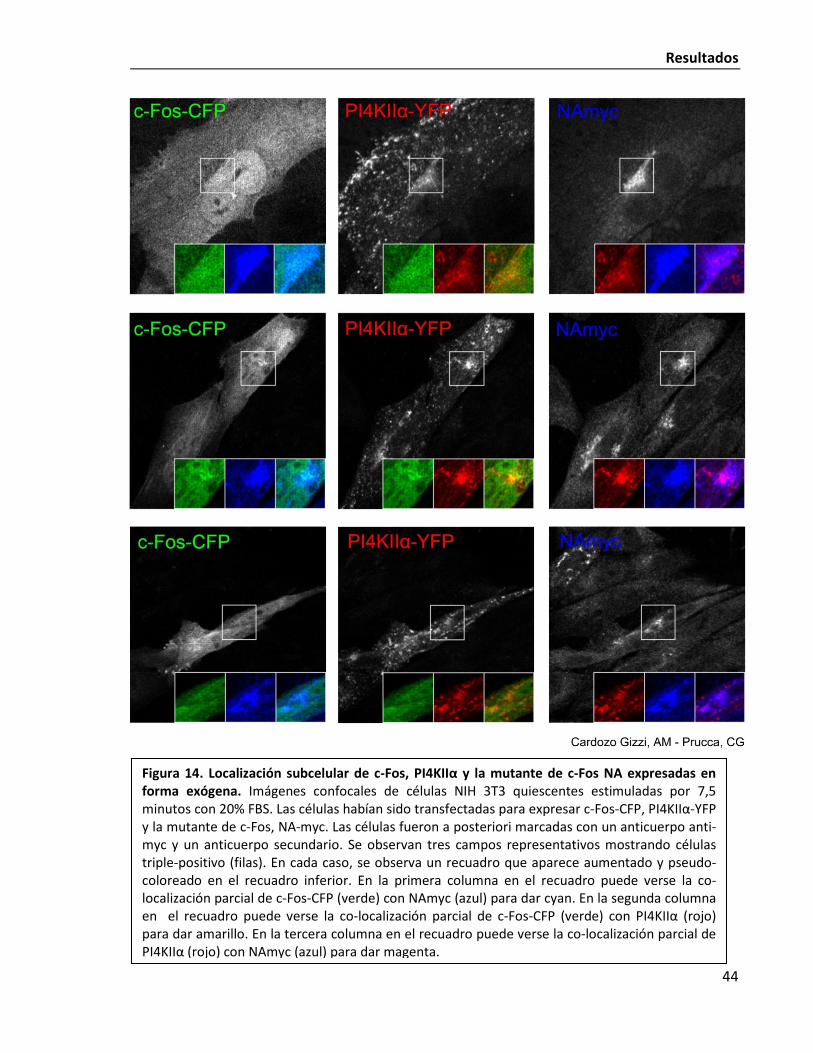
Resultados
44
Figura 14. Localización subcelular de c-Fos, PI4KIIα y la mutante de c-Fos NA expresadas en forma exógena. Imágenes confocales de células NIH 3T3 quiescentes estimuladas por 7,5 minutos con 20% FBS. Las células habían sido transfectadas para expresar c-Fos-CFP, PI4KIIα-YFP y la mutante de c-Fos, NA-myc. Las células fueron a posteriori marcadas con un anticuerpo anti-myc y un anticuerpo secundario. Se observan tres campos representativos mostrando células triple-positivo (filas). En cada caso, se observa un recuadro que aparece aumentado y pseudo-coloreado en el recuadro inferior. En la primera columna en el recuadro puede verse la co-localización parcial de c-Fos-CFP (verde) con NAmyc (azul) para dar cyan. En la segunda columna en el recuadro puede verse la co-localización parcial de c-Fos-CFP (verde) con PI4KIIα (rojo) para dar amarillo. En la tercera columna en el recuadro puede verse la co-localización parcial de PI4KIIα (rojo) con NAmyc (azul) para dar magenta.

Resultados
45
Es interesante destacar que a partir de estas observaciones, el Dr. César Prucca
continuó con experimentos de proliferación de células tumorales en cultivo sobreexpresando
la mutante NA y observó que se limita la proliferación, dando los primeros indicios de que
esta estrategia podría funcionar para controlar la proliferación exarcerbada de las células
tumorales. En particular, se utilizaron tres sistemas: 1) células transfectadas para expresar
NAmyc, 2) líneas celulares estables que expresan NA al ser inducidas con mifepristona y 3)
profección (adición de proteínas en lípidos catiónicos a las células) de la mutante NA. En
todos los casos, la presencia de NA disminuyó la tasa de proliferación de las células
tumorales respecto de los controles (Resultados no mostrados). Las consecuencias de esto se
analizarán en la sección Discusión.
Activación de la actividad PAP por c-Fos
El PA es un intermediario clave en la síntesis de glicerofosfolípidos ya que puede ser
convertido por la enzima CDS en CDP-DAG para continuar hacia la vía de PIPs y cardiolipina o
alternativamente ser convertido por una enzima PAP a DAG, intermediario común a toda la
vía de Kennedy que dará lugar finalmente a PtdCho, PtdEth y PtdSer. Hasta aquí hemos
establecido que c-Fos logra una activación general de la vía de síntesis de PIPs mediante una
interacción física directa y la consecuente activación de CDS1 y PI4KIIα. Resultados previos de
la Dra. Marianne Renner indican que c-Fos también es capaz de incrementar la actividad PAP
en homogenatos totales de células NIH 3T3 quiescentes, derivando PA entonces hacia la vía
de Kennedy al formar DAG, intermediario común de todos los productos lipídicos de esta vía
(Fig. 15a). Esto último podría explicar los cambios en las especies marcadas radioactivamente
en la segunda onda de la transición G0/G1. La actividad PAP que se incrementa por c-Fos en
dichos homogenatos corresponde a PAP I, ya que no se detectó la formación de producto
bajo condiciones de reacción adecuadas para medir PAP II (Tesis Doctoral Marianne Renner,
2003).
La activación de PAP I dependiente de la expresión de c-Fos también había sido
descripta en células ganglionares de la retina de pollo. Al suprimir la expresión de c-Fos
mediante un ARN antisentido contra esta proteína inyectado en la retina de los animales, se

Resultados
46
cancela la activación de PAP I estimulada por luz (de Arriba Zerpa et al. 1999). Si bien los
resultados mostraron claramente que la activación PAP I es dependiente de c-Fos, dado la
complejidad del sistema experimental in vivo utilizado, resultaba muy difícil avanzar en los
estudios del mecanismo molecular involucrado en esta activación.
Dada una reciente publicación en donde se reporta la purificación de la enzima Lipin 1
(Han & Carman 2010), con actividad PAP I, se decidió poner a punto un sistema de ensayo in
vitro con componentes purificados donde analizar el rol de c-Fos sobre la actividad de esta
enzima. En el sistema empleado la membrana modelo está formada por micelas del
detergente no iónico Tritón X-100. Este detergente forma micelas de tamaños relativamente
homogéneas de un radio cercano a 6 nm para micelas hidratadas (Lichtenberg et al. 1983,
Thomas et al. 1999). A dichas micelas se les adiciona PA de forma de obtener un máximo de
10 mol% de PA, de esta manera no se alteran las características propias de las micelas de
Tritón X-100 (Lin & Carman 1990). Las micelas mixtas de Tritón X-100/PA resultantes permite
analizar la actividad PAP en un ambiente que simula la superficie de membranas celulares
(Carman et al. 1995).
El gen Lpn1 da lugar a tres transcritos distintos (Lipin 1α, Lipin 1β y Lipin 1γ) por
procesamiento alternativo del ARN. Cada una de las isoformas posee una localización
subcelular particular así como una actividad PAP diferenciada. Lipin 1α aparece
predominantemente nuclear mientras que Lipin 1β (que posee 33 residuos extra) reside
mayormente en el citoplasma/ER (Peterfy et al. 2005). En cambio, Lipin 1γ se encuentra
asociada a gotas lipídicas a través de un dominio hidrofóbico específico de esta isoforma
(Wang et al. 2011). Se escogió para el estudio a la enzima Lipin 1β dada su localización
subcelular. De aquí en adelante, Lipin 1β será simplemente Lipin 1.
Se purificó hasta homogeneidad la enzima Lipin 1 expresada recombinante en E. Coli,
y se obtuvo una banda del peso molecular aparente esperado de 110 kDa en SDS-PAGE. Se
ensayó su actividad en micelas mixtas de Tritón X-100/PA con el agregado de
concentraciones crecientes de c-Fos purificado (Fig. 15b). En este sistema purificado, el
agregado de c-Fos recombinante incrementa la actividad PAP de Lipin 1 hasta cerca del 40%
por encima del control. Este ensayo presenta claras ventajas respecto del realizado

Resultados
47
ensayando la actividad PAP en homogenatos totales, en donde existen una vasta cantidad de
proteínas celulares conjuntamente con la compleja composición de membranas biológicas.
Aunque no son directamente comparables debido, entre otros motivos, a que las condiciones
del ensayo son diferentes (Pasquare et al. 2004) se pueden establecer algunas conclusiones
parciales. Por ejemplo en el ensayo de micelas mixtas los únicos componentes presentes son
c-Fos, Lipin 1 y las membranas modelo compuestas por micelas mixtas de Tritón-100/PA, lo
cual descarta que otras proteínas puedan estar involucradas en la activación de la enzima por
c-Fos.
Figura 15. Efecto de la adición de c-Fos sobre la actividad PAP. a) Actividad PAP in vitro en homogenatos celulares totales de células NIH 3T3 quiescentes en función del agregado de c-Fos recombinante. b) Actividad PAP de Lipin 1 purificada sobre micelas mixtas de Tritón X-100/PA conteniendo 9.1 mol% y 1 mM de PA en función del agregado de c-Fos recombinante. La actividad enzimática está expresada con respecto al control (referenciado como el 100%) que contiene vehículo en lugar de c-Fos. Los resultados son el promedio ± DE de un experimento representativo hecho en triplicado de al menos tres realizados.
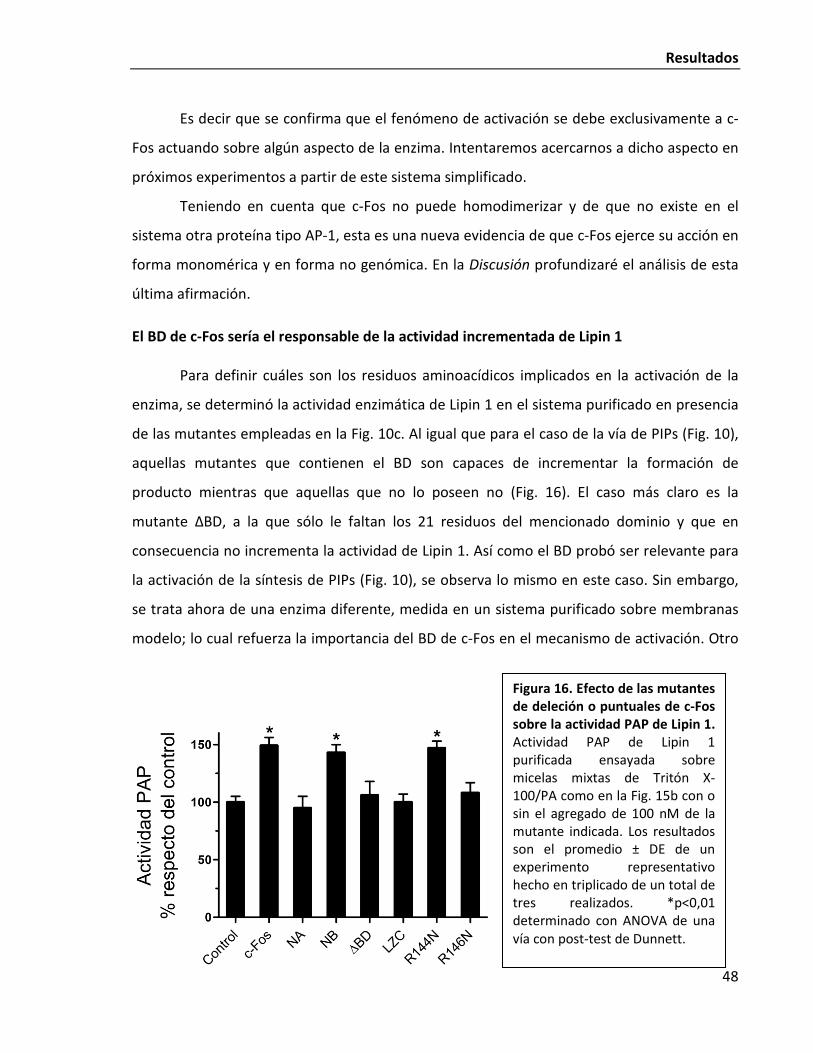
Resultados
48
Es decir que se confirma que el fenómeno de activación se debe exclusivamente a c-
Fos actuando sobre algún aspecto de la enzima. Intentaremos acercarnos a dicho aspecto en
próximos experimentos a partir de este sistema simplificado.
Teniendo en cuenta que c-Fos no puede homodimerizar y de que no existe en el
sistema otra proteína tipo AP-1, esta es una nueva evidencia de que c-Fos ejerce su acción en
forma monomérica y en forma no genómica. En la Discusión profundizaré el análisis de esta
última afirmación.
El BD de c-Fos sería el responsable de la actividad incrementada de Lipin 1
Para definir cuáles son los residuos aminoacídicos implicados en la activación de la
enzima, se determinó la actividad enzimática de Lipin 1 en el sistema purificado en presencia
de las mutantes empleadas en la Fig. 10c. Al igual que para el caso de la vía de PIPs (Fig. 10),
aquellas mutantes que contienen el BD son capaces de incrementar la formación de
producto mientras que aquellas que no lo poseen no (Fig. 16). El caso más claro es la
mutante ΔBD, a la que sólo le faltan los 21 residuos del mencionado dominio y que en
consecuencia no incrementa la actividad de Lipin 1. Así como el BD probó ser relevante para
la activación de la síntesis de PIPs (Fig. 10), se observa lo mismo en este caso. Sin embargo,
se trata ahora de una enzima diferente, medida en un sistema purificado sobre membranas
modelo; lo cual refuerza la importancia del BD de c-Fos en el mecanismo de activación. Otro
Figura 16. Efecto de las mutantes de deleción o puntuales de c-Fos sobre la actividad PAP de Lipin 1. Actividad PAP de Lipin 1 purificada ensayada sobre micelas mixtas de Tritón X-100/PA como en la Fig. 15b con o sin el agregado de 100 nM de la mutante indicada. Los resultados son el promedio ± DE de un experimento representativo hecho en triplicado de un total de tres realizados. *p<0,01 determinado con ANOVA de una vía con post-test de Dunnett.

Resultados
49
punto a considerar es que al tratarse de un sistema purificado, puede descartarse que el BD
esté interactuando con otra proteína o actuando como puente hacia una tercera proteína
para generar el efecto de activación, lo que indicaría de que se trata de un efecto del BD por
sí mismo. Es importante remarcar además que el agregado de la mutante R146N no
incrementa la actividad respecto del control (Fig. 16), por lo que la significancia de este
residuo no es exclusiva en la activación de la vía de síntesis de PIPs.
Fra-1, miembro de la familia Fos, a diferencia de c-Jun es capaz de activar a Lipin 1
Dado el rol del BD de c-Fos en la activación enzimática de Lipin 1, decidimos estudiar
si otras dos proteínas que forman complejos de tipo AP-1 y que comparten un dominio BD, c-
Jun y Fra-1, también promueven la activación. Las proteínas AP-1 poseen un dominio BD,
responsable de la asociación al ADN, junto a un dominio llamado “cierre de leucinas”,
responsable de la heterodimerización, que en conjunto se denomina “bZip”. La Fig. 17
muestra un esquema de las tres onco-proteínas con sus dominios bZip correspondientes,
junto con un alineamiento de las secuencias de sus BD. Fra-1 es un miembro de la familia Fos
que posee un BD altamente conservado con respecto a c-Fos aunque la homología total de
las dos proteínas es inferior al 44%. Por el contrario, c-Jun tiene una secuencia muy
divergente, y esa divergencia de secuencia incluye al domino bZip en su totalidad. Cuando se
adicionó Fra-1 obtenido en forma recombinante en lugar de c-Fos al sistema micelar, se
incrementó la actividad PAP de Lipin 1 en una medida similar al aumento provocado por el
agregado de c-Fos (Fig. 17c). La similitud de ambos BD refuerza la idea de la participación de
este dominio no solo en su unión secuencia-especifica al ADN sino también en la activación
enzimática. En tanto c-Jun, que tiene un BD con baja homología respecto de las otras dos
proteínas (e incluso una densidad de carga inferior), no promueve un incremento en la
actividad de Lipin 1 cuando es adicionada al sistema purificado en lugar de c-Fos (Fig. 17c).

Resultados
50
Lipin 1 interacciona físicamente con c-Fos y Fra-1 pero no con c-Jun
Hasta aquí, hemos establecido que c-Fos incrementa la actividad Lipin 1 a través de su
dominio BD, en homogenatos totales pero también en un sistema purificado diseñado
específicamente para ensayar la actividad de esta enzima. Esto planteó el interrogante
respecto a si Lipin 1 y c-Fos participan en una asociación física como forma de alcanzar el
estado activado de la enzima. Para responderlo, se analizó por microscopía FRET por emisión
sensibilizada la posible interacción utilizando ahora el par de FRET mTurquoise2/ SYFP2. En la
bibliografía se encuentran descriptas las mutaciones puntuales de CFP y YFP que resultan en
proteínas fluorescentes que resultan más brillantes y resistentes al fotoblanqueo (Kremers et
al. 2006, Goedhart et al. 2012), denominadas mTurquoise2 y SYFP2, y que son entonces
mejores herramientas para microscopía de fluorescencia.
Figura 17. Efecto de c-Fos, Fra-1 y c-Jun en la actividad PAP de Lipin 1. a) Representación esquemática de Fra-1, c-Fos y c-Jun. Se indica en negro el BD y en gris el cierre de leucinas de las tres proteínas que se encuentran alineadas. El bZip se conforma por el BD y el cierre de leucinas. El cierre de leucinas permite la dimerización para formar dímeros AP-1 activos y el BD funciona asociándose a secuencias blanco de ADN pero también activando a enzimas blanco. b) BDs de las tres proto-oncoproteínas. La línea indica “identidad” y los dos puntos indican una substitución conservativa. Notese la alta homología entre los BDs de Fra-1 y c-Fos pero no así con el BD de c-Jun. c) Actividad PAP de Lipin 1 purificada sobre micelas mixtas de Tritón X-100/PA como se indica en la leyenda de la Fig. 15b con el agregado de c-Fos o Fra-1 o c-Jun purificados.

Resultados
51
Se fusionaron c-Fos, c-Jun o Fra-1 a la proteína fluorescente mTurquoise2 y se estudió
una posible interacción con Lipin 1-SYFP2 en células NIH 3T3 quiescentes luego de ser
estimuladas durante 1 hora con 20% FBS en presencia de cicloheximida, un inhibidor de la
síntesis de proteínas a nivel ribosomal. Se eligió este tiempo de estímulo porque así como a
7,5 minutos post-estímulo se produce el pico en la síntesis de PIPs, a 1 hora post-estímulo el
análisis indica que los lípidos marcados son mayoritariamente los principales constituyentes
de membrana (Bussolino et al. 2001). Por lo tanto, al estimular las células por 1 hora, nos
situamos en las condiciones a priori más favorables para observar el fenómeno de
interacción. Como un control positivo, se utilizó c-Fos/ PI4KIIα, una interacción que ya se
había establecido (Figuras 9 y 13), si bien en ese caso la células habían sido estimuladas
durante 7,5 minutos. Se encontró que tanto c-Fos como Fra-1 interaccionan físicamente con
Lipin 1 mientras que c-Jun no lo hace (Fig. 18); lo cual correlaciona con nuestra hipótesis de
que las onco-proteínas que activan a la enzima son capaces de interaccionar con ella.
La interacción aparece localizada fuera del núcleo, si bien tanto c-Fos como Lipin 1
localizan también dentro del mismo. Las enzimas analizadas en la vía de síntesis de PIPs son
extra-nucleares, en cambio según bibliografía, Lipin 1 se ubica tanto en RE como en núcleo,
donde cumple funciones diferentes. Su actividad PAP 1 se encuentra asociada al RE mientras
que su función como co-activador transcripcional se produce en el núcleo (Siniossoglou
2013). c-Fos ejerce su actividad genómica en el núcleo, mediante su actividad como factor de
transcripción de tipo AP-1, pero también de manera independiente a esta al incrementar la
síntesis de PtdInsP2 y promoviendo de esta forma cambios transcripcionales globales (Ferrero
et al. 2014). Sin embargo, c-Fos ejerce su efecto no genómico principalmente en el
citoplasma, asociado al RE (Caputto et al. 2014). Según se observa en la Fig. 18, la interacción
c-Fos/Lipin 1 se produce peri-nuclearmente, probablemente asociado al RE. Por lo tanto, si
bien las dos proteínas localizan en ambos compartimientos, la asociación y
consecuentemente la activación parecen ocurrir únicamente a nivel del RE.
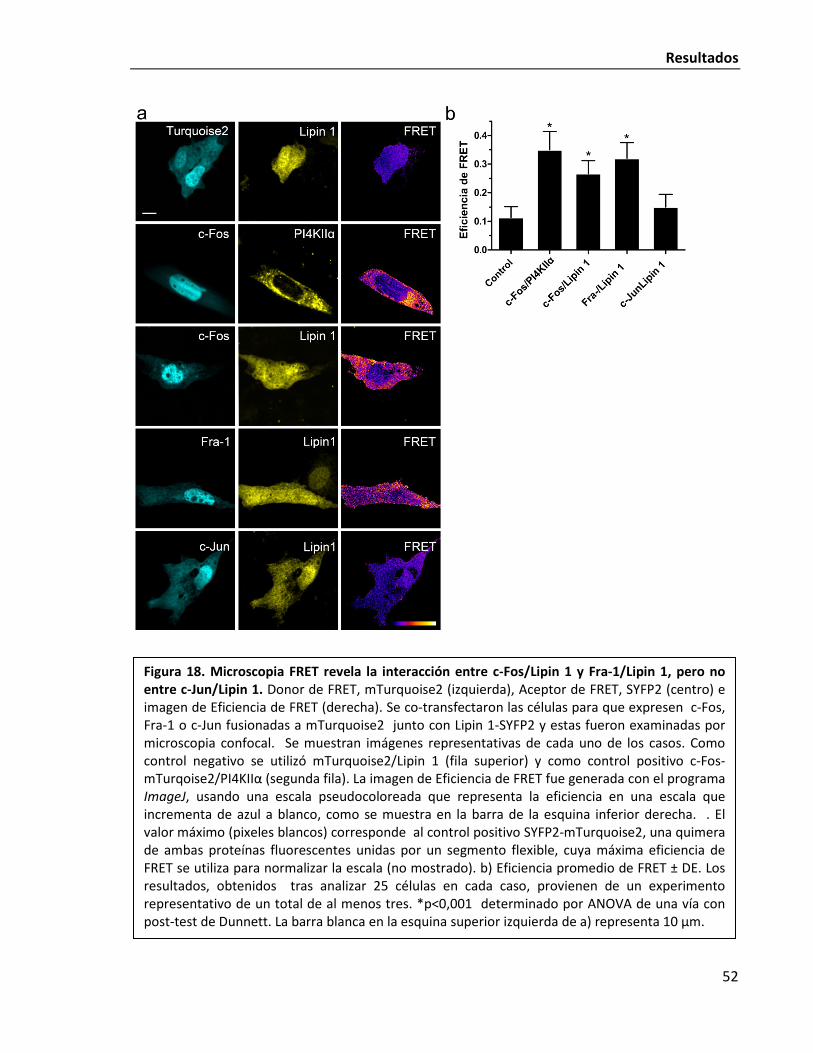
Resultados
52
Figura 18. Microscopia FRET revela la interacción entre c-Fos/Lipin 1 y Fra-1/Lipin 1, pero no entre c-Jun/Lipin 1. Donor de FRET, mTurquoise2 (izquierda), Aceptor de FRET, SYFP2 (centro) e imagen de Eficiencia de FRET (derecha). Se co-transfectaron las células para que expresen c-Fos, Fra-1 o c-Jun fusionadas a mTurquoise2 junto con Lipin 1-SYFP2 y estas fueron examinadas por microscopia confocal. Se muestran imágenes representativas de cada uno de los casos. Como control negativo se utilizó mTurquoise2/Lipin 1 (fila superior) y como control positivo c-Fos-mTurqoise2/PI4KIIα (segunda fila). La imagen de Eficiencia de FRET fue generada con el programa ImageJ, usando una escala pseudocoloreada que representa la eficiencia en una escala que incrementa de azul a blanco, como se muestra en la barra de la esquina inferior derecha. . El valor máximo (pixeles blancos) corresponde al control positivo SYFP2-mTurquoise2, una quimera de ambas proteínas fluorescentes unidas por un segmento flexible, cuya máxima eficiencia de FRET se utiliza para normalizar la escala (no mostrado). b) Eficiencia promedio de FRET ± DE. Los resultados, obtenidos tras analizar 25 células en cada caso, provienen de un experimento representativo de un total de al menos tres. *p<0,001 determinado por ANOVA de una vía con post-test de Dunnett. La barra blanca en la esquina superior izquierda de a) representa 10 µm.

Resultados
53
c-Fos modifica la actividad de Lipin 1 al incrementar su capacidad catalítica
Considerando que Lipin 1 interacciona con c-Fos y es activada por esta proteína,
decidimos examinar la cinética de su activación en el sistema simplificado como estrategia
para obtener información acerca del mecanismo molecular de esta activación. La capacidad
catalítica incrementada de Lipin 1 en presencia de c-Fos puede deberse a un incremento en
la capacidad para unirse a membranas, un incremento en la eficiencia catalítica de la enzima
(eficiencia del sitio activo) o una combinación de ambos factores. Para resolver este
interrogante, se realizaron ensayos enzimáticos utilizando micelas mixtas de Tritón X-100/PA
(Fig. 19).
Como se mencionó anteriormente, Lipin 1 es una enzima soluble que primero debe
asociarse a membranas para, una vez unida, buscar sobre la superficie a su sustrato PA para
así catalizar la reacción como fosfatasa. Este proceso depende entonces de interacciones en
solución con las membranas y de interacciones de superficie con los lípidos presentes sobre
la membrana. Un modelo cinético que describe esta situación ha sido formulado y se
denomina “modelo de dilución de superficie” (Carman et al. 1995). Usando este modelo, se
realizaron dos tipos de experimentos: i) manteniendo la concentración de superficie de PA
constante a 9.1 mol% mientras se incrementa su concentración molar y ii) manteniendo la
concentración molar de PA fija a 1 mM mientras se modifica su concentración de superficie
(o mol%).
Los experimentos de tipo i) permiten determinar la primera etapa en donde la enzima
que se encuentra soluble se une a las membranas modelo en una transición desde la solución
hacia la superficie para formar el complejo enzima-micela mixta. Esta asociación depende de
la concentración de solución (o molar) del lípido (Lin & Carman 1990). Debido a que la
concentración de superficie de PA está fija, al incrementar la concentración molar de PA es
necesario incrementar la concentración molar de Tritón X-100 en la misma proporción. En
consecuencia, se está incrementando el número total de micelas, que además tienen la
misma composición. Los experimentos de tipo ii) en cambio permiten el estudio de cómo la
enzima unida a la membrana modelo encuentra PA en la superficie de esta membrana para
poder llevar a cabo la reacción enzimática.
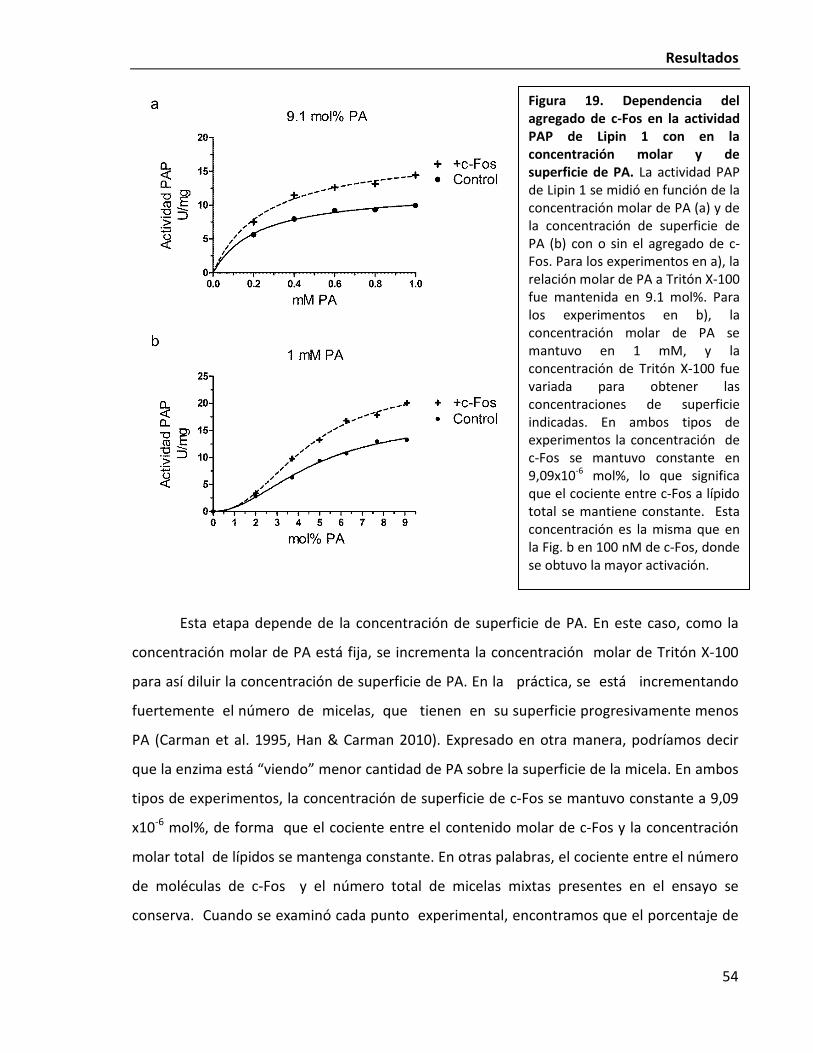
Resultados
54
Esta etapa depende de la concentración de superficie de PA. En este caso, como la
concentración molar de PA está fija, se incrementa la concentración molar de Tritón X-100
para así diluir la concentración de superficie de PA. En la práctica, se está incrementando
fuertemente el número de micelas, que tienen en su superficie progresivamente menos
PA (Carman et al. 1995, Han & Carman 2010). Expresado en otra manera, podríamos decir
que la enzima está “viendo” menor cantidad de PA sobre la superficie de la micela. En ambos
tipos de experimentos, la concentración de superficie de c-Fos se mantuvo constante a 9,09
x10-6 mol%, de forma que el cociente entre el contenido molar de c-Fos y la concentración
molar total de lípidos se mantenga constante. En otras palabras, el cociente entre el número
de moléculas de c-Fos y el número total de micelas mixtas presentes en el ensayo se
conserva. Cuando se examinó cada punto experimental, encontramos que el porcentaje de
Figura 19. Dependencia del agregado de c-Fos en la actividad PAP de Lipin 1 con en la concentración molar y de superficie de PA. La actividad PAP de Lipin 1 se midió en función de la concentración molar de PA (a) y de la concentración de superficie de PA (b) con o sin el agregado de c-Fos. Para los experimentos en a), la relación molar de PA a Tritón X-100 fue mantenida en 9.1 mol%. Para los experimentos en b), la concentración molar de PA se mantuvo en 1 mM, y la concentración de Tritón X-100 fue variada para obtener las concentraciones de superficie indicadas. En ambos tipos de experimentos la concentración de c-Fos se mantuvo constante en 9,09x10-6 mol%, lo que significa que el cociente entre c-Fos a lípido total se mantiene constante. Esta concentración es la misma que en la Fig. b en 100 nM de c-Fos, donde se obtuvo la mayor activación.

Resultados
55
activación con respecto al control es constante en alrededor del 40% en todas las
condiciones ensayadas (Fig. 19).
Bajo las condiciones de tipo i), se observó una cinética de tipo Michaelis-Menten; en
tanto en los experimentos de tipo ii) se observó un comportamiento cinético cooperativo de
tipo alostérico (ecuación de Hill). Utilizando regresiones no lineales utilizando estos modelos,
fue posible determinar los parámetros cinéticos a partir de los datos de la Fig. 19, y se
encuentran mostrados en la Tabla 2. Se encontró en los experimentos de tipo i) que, a pesar
de que la constante kcat se incrementa cerca del 50% (Fig. 19a), c-Fos no promovió una
unión más eficiente de Lipin 1 a las membranas modelo, dado que las diferencias en Ks no
fueron estadísticamente significativas (Tabla 2). El parámetro Ks es una medida de la afinidad
de la enzima por las membranas, es decir una medida de las interacciones de la enzima en
solución con las micelas. Análisis de los datos a partir de la ecuación de Hill de los
experimentos de tipo ii) se encontró que el parámetro Km no es modificado
significativamente frente a la adición de c-Fos (Fig. 19b). El parámetro Km puede ser
considerado como la afinidad de enzima por su sustrato. La probada cooperatividad de Lipin
1 (Han & Carman 2010), reflejada en el número de Hill, no se ve alterada tampoco. En
cambio, la kcat de la reacción se ve positivamente modificada por c-Fos, e incrementa
alrededor de 40%. Los experimentos realizados revelaron que la eficiencia catalítica de Lipin
1 es modificada por c-Fos sin que ello implique alterar la asociación de Lipin a las micelas,
reflejada en Ks, o su interacción específica con PA, reflejada en Km.
Los resultados son concordantes con otros obtenidos previamente para otras enzimas
Tabla 2. Parámetros cinéticos de Lipin 1 empleando micelas mixtas de Triton X-100/PA con o sin el agregado de c-Fos recombinante, calculados con el programa GraphPad Prism 5 a partir de los datos de la Fig. 19a y b. Se usaron las ecuaciónes de Michelis-Menten o de Hill, respectivamente, para ajustar los datos.
Constantes cinéticas con respecto a la concentración molar de PA
Constantes cinéticas con respecto a la concentración de superficie de PA
Ks (mM) kcat(s-1) Km (mol%) kcat(s
-1) No. Hill
Control 0,22±0,03 20,3±0,7 4,7±0,6 28,7±3,0 2,0±0,3
+c-Fos 0,26±0,04 29,9±1,6 4,4±0,5 39,4±2,5 2,2±0,2

Resultados
56
activadas por c-Fos y que muestran que se produce un incremento en la capacidad catalítica
sin modificar la afinidad por el sustrato. Pero además aquí la utilización del sistema
purificado permite su análisis en un sistema muy simple en donde es posible establecer
conclusiones en forma directa.
En los últimos años, múltiples reportes han descripto nuevas proteínas que
interaccionan con Lipin 1, alterando su distribución subcelular a través de modificaciones
post-traduccionales y/o al asociarse directamente con la misma, regulando así sus funciones
celulares (Huffman et al. 2002, Harris et al. 2007, Grimsey et al. 2008, Liu & Gerace 2009). c-
Fos interacciona con Lipin 1 (Fig. 18), pero la co-transfección con c-Fos no cambia la
localización subcelular de Lipin 1 respecto a la transfección sólo de la enzima (Resultados no
mostrados), lo cual indicaría que c-Fos no cambia la localización de la enzima sino que logra
activarla donde esta localiza. Asimismo, c-Fos tampoco modifica su afinidad por la membrana
modelo estudiada, ya que no cambia los parámetros Ks o Km de la enzima. Hasta donde llega
nuestro conocimiento, este es el primer estudio que reporta una proteína que interacciona
con Lipin 1 que modifica su actividad directamente, y no su localización subcelular o afinidad
por membranas, aunque el mecanismo preciso de esta activación no está esclarecido
completamente.
El dominio N-terminal de c-Fos es responsable de la asociación a Lipin 1
Los experimentos en la Fig. 16 nos indican que el BD de c-Fos es el responsable de la
activación promovida sobre Lipin 1. En cambio, el domino responsable de la asociación a
Lipin 1 es desconocido en este punto. Para explorar este interrogante, se utilizaron las
diferentes mutantes de deleción que se esquematizaron previamente en la Fig. 10,
fusionadas a mTurqouise2 para estudiar una posible interacción con Lipin 1 mediante
microscopía FRET. Las mutantes NA (aa: 1-138, sin el BD) y ΔBD (solo le faltan los aa: 139-
160) mantienen la asociación a Lipin 1, descartando de este modo que el BD sea el
responsable de esta. La mutante LZC, que comprende el C-terminal de c-Fos, no se une a la
enzima, sugiriendo que esta porción no participa en el fenómeno. La región mínima
compartida por las mutantes capaces de unirse a Lipin 1 es nuevamente el N-terminal de c-
Fos.
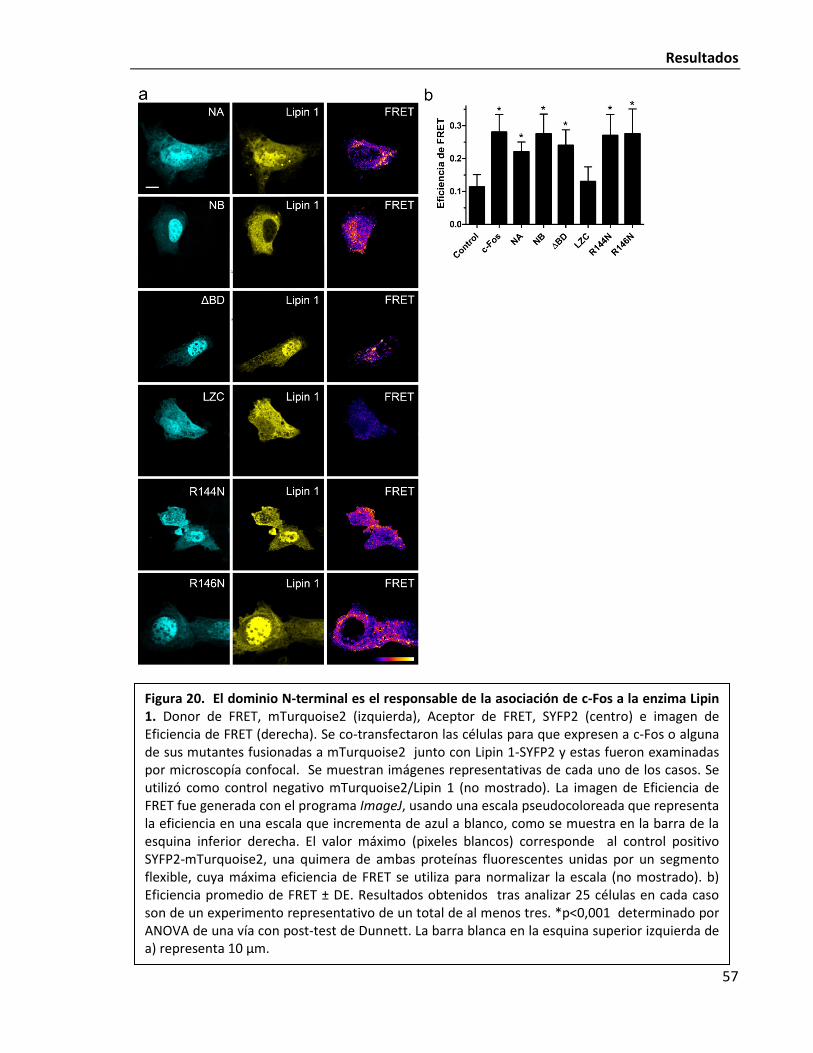
Resultados
57
Figura 20. El dominio N-terminal es el responsable de la asociación de c-Fos a la enzima Lipin 1. Donor de FRET, mTurquoise2 (izquierda), Aceptor de FRET, SYFP2 (centro) e imagen de Eficiencia de FRET (derecha). Se co-transfectaron las células para que expresen a c-Fos o alguna de sus mutantes fusionadas a mTurquoise2 junto con Lipin 1-SYFP2 y estas fueron examinadas por microscopía confocal. Se muestran imágenes representativas de cada uno de los casos. Se utilizó como control negativo mTurquoise2/Lipin 1 (no mostrado). La imagen de Eficiencia de FRET fue generada con el programa ImageJ, usando una escala pseudocoloreada que representa la eficiencia en una escala que incrementa de azul a blanco, como se muestra en la barra de la esquina inferior derecha. El valor máximo (pixeles blancos) corresponde al control positivo SYFP2-mTurquoise2, una quimera de ambas proteínas fluorescentes unidas por un segmento flexible, cuya máxima eficiencia de FRET se utiliza para normalizar la escala (no mostrado). b) Eficiencia promedio de FRET ± DE. Resultados obtenidos tras analizar 25 células en cada caso son de un experimento representativo de un total de al menos tres. *p<0,001 determinado por ANOVA de una vía con post-test de Dunnett. La barra blanca en la esquina superior izquierda de a) representa 10 µm.

Resultados
58
La región aa: 47-90 dentro del N-terminal de c-Fos estaría involucrado en la asociación Lipin
1/c-Fos
Dado que la región N-terminal de c-Fos se asocia pero no activa a Lipin 1 (Figuras 20 y
16, respectivamente), decidimos estudiar la porción mínima de esta región que conserva la
capacidad para unirse a esta enzima. El Dr. César Prucca gentilmente cedió las mutantes
conteniendo diferentes porciones de los primeros 138 residuos de c-Fos fusionadas a la
proteína fluorescente mTurquoise2 y que utilizamos para establecer la región mínima
necesaria para la asociación. Estas mutantes se encuentran representadas en la Fig. 21c. Las
tres mutantes pequeñas, de 46 residuos de longitud (NA 1-46, NA 47-92, NA 93-138), no
mostraron asociación a Lipin 1 (Fig. 21b). Sorpresivamente, tanto los primeros 92 residuos
como la región aa: 47-138 alcanzaron eficiencias de FRET comparables con la de c-Fos
salvaje. Una posible explicación a este resultado es que la región aa: 47-92 (la región mínima
compartida por las dos mutantes capaces de interaccionar con Lipin 1) esté involucrada en el
fenómeno de asociación pero que esta región no sea suficiente por sí misma para asegurar la
unión a la enzima. Otra explicación posible es que la región aa: 47-92 sea suficiente para la
asociación, pero el impedimento estérico debido al gran volumen de la proteína fluorescente
que se encuentra a continuación del péptido de solo 46 residuos de longitud impida la
asociación a la enzima. El perfil hidrofóbico de c-Fos muestra dos segmentos que poseen los
valores más altos de hidrofobicidad de toda la proteína entre los residuos 50-68 y 76-86 (Ver
Discusión, Fig. 23). La hidrofobicidad parece tener un rol de importante para la asociación.
Dado que tanto los primeros como los últimos 46 residuos (aa: 1-46 y aa: 93-138,
respectivamente) son también requeridos para la asociación, podría ser que estos residuos
actúan simplemente como espaciadores o conectores flexibles que permiten aliviar la
tensión estérica entre las secciones hidrofóbicas asociadas a la enzima y la proteína
fluorescente citoplásmica. Alternativamente, estas regiones de c-Fos (aa: 1-46 y aa: 93-138)
podrían poseer características particulares, lo que será discutido en la próxima sección
conjuntamente con el hecho de que todas las enzimas activadas interactúan específicamente
con la región N-terminal.
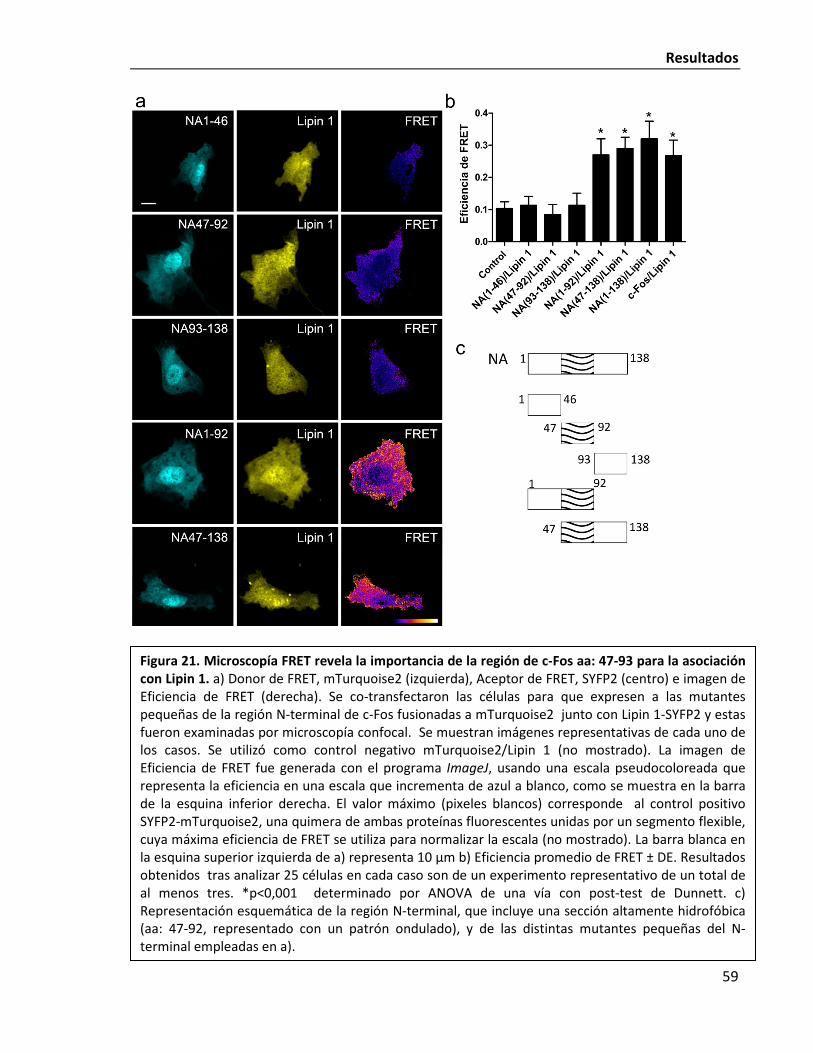
Resultados
59
Figura 21. Microscopía FRET revela la importancia de la región de c-Fos aa: 47-93 para la asociación con Lipin 1. a) Donor de FRET, mTurquoise2 (izquierda), Aceptor de FRET, SYFP2 (centro) e imagen de Eficiencia de FRET (derecha). Se co-transfectaron las células para que expresen a las mutantes pequeñas de la región N-terminal de c-Fos fusionadas a mTurquoise2 junto con Lipin 1-SYFP2 y estas fueron examinadas por microscopía confocal. Se muestran imágenes representativas de cada uno de los casos. Se utilizó como control negativo mTurquoise2/Lipin 1 (no mostrado). La imagen de Eficiencia de FRET fue generada con el programa ImageJ, usando una escala pseudocoloreada que representa la eficiencia en una escala que incrementa de azul a blanco, como se muestra en la barra de la esquina inferior derecha. El valor máximo (pixeles blancos) corresponde al control positivo SYFP2-mTurquoise2, una quimera de ambas proteínas fluorescentes unidas por un segmento flexible, cuya máxima eficiencia de FRET se utiliza para normalizar la escala (no mostrado). La barra blanca en la esquina superior izquierda de a) representa 10 µm b) Eficiencia promedio de FRET ± DE. Resultados obtenidos tras analizar 25 células en cada caso son de un experimento representativo de un total de al menos tres. *p<0,001 determinado por ANOVA de una vía con post-test de Dunnett. c) Representación esquemática de la región N-terminal, que incluye una sección altamente hidrofóbica (aa: 47-92, representado con un patrón ondulado), y de las distintas mutantes pequeñas del N-terminal empleadas en a).

CAPITULO IV
DISCUSIÓN

Discusión
61
La activación de la síntesis de fosfolípidos por c-Fos es un fenómeno no genómico
La activación de la síntesis de fosfolípidos podría ser en principio una consecuencia de
la actividad de c-Fos como factor de transcripción tipo AP-1, al incrementar por ejemplo la
expresión de enzimas de síntesis de los mismos. Los primeros experimentos de marcación de
fosfolípidos totales en células de la retina de pollo indicaron la dependencia de la expresión
de c-Fos para el incremento en el marcado radioactivo en respuesta a la luz (Guido et al.
1996, Bussolino et al. 1998). Es importante discutir en este punto las evidencias
experimentales encontradas fundamentalmente en esta Tesis y en otros trabajos del grupo
que muestran como c-Fos realiza esta función no canónica en un nivel no genómico.
La primera evidencia directa de esto fueron los experimentos realizados in vitro en los
cuales se midió la incorporación de 32P desde 32P-ATP a fosfolípidos totales (Gil et al. 2004) o
en particular a PtdInsP y PtdInsP2 (esta Tesis, Fig. 10) utilizando como fuente de enzima
homogenatos totales u homogenatos libres de la fracción nuclear de células quiescentes que
contienen niveles despreciables de c-Fos endógeno. En estos experimentos, se adicionó en
forma exógena 32P-γ-ATP junto con c-Fos purificado, para que luego las enzimas presentes en
el homogenato celular incorporaran fosfato radioactivo a fosfolípidos, que se extrajeron y
cuantificaron. El agregado de c-Fos recombinante incrementa el marcado radioactivo de
fosfolípidos in vitro alrededor del 100% por encima del control. Lo que es clave en los
experimentos de marcación radioactiva in vitro es que, en células quiescentes, los niveles
endógenos de los otros miembros AP-1 tampoco son detectables. c-Fos es incapaz de
estimular la transcripción en ausencia de c-Jun (Chiu et al. 1988), lo que descarta una
activación de la síntesis lipídica a partir de mecanismos regulatorios genómicos en este
sistema. Por otro lado, c-Jun purificado ensayado en los experimentos antes mencionados no
tuvo un efecto en la tasa de síntesis de fosfolípidos (Gil et al. 2004) o en particular en la
activación de Lipin 1 en un sistema purificado (Fig. 17), apuntando hacia la especificidad del
fenómeno de activación.
La segunda línea de evidencia de que el efecto que estudiamos se produce de manera
no genómica se obtuvo a partir de la utilización de mutantes de deleción de c-Fos en los

Discusión
62
mencionados ensayos de marcación de fosfolípidos in vitro, donde el ejemplo más claro
surge de la mutante NB (aa: 1-160). Esta mutante es capaz de activar la síntesis de
fosfolípidos y en particular de PIPs (Fig. 10), así como de activar a Lipin 1 (Fig. 16) a niveles
comparables con la proteína c-Fos completa. Sin embargo, la mutante NB no contiene
dominio de cierre de leucinas, indispensable para la heterodimerización y consecuente
formación de un dímero AP-1. Dado entonces que NB no puede actuar como factor de
transcripción, pero aun así activa la síntesis de fosfolípidos, ello confirma la función no
genómica de c-Fos. Más aún, la mutante NB ha sido capaz de sostener in vivo la
diferenciación o la proliferación celular en células deprivadas de factores de crecimiento (Gil
et al. 2004, Gil et al. 2012). La utilización de mutantes ha establecido también que la función
anti-apoptótica de c-Jun, antagonizando a MEKK en células PC12, está dada de forma no
genómica ya que no depende de su dimerización o unión al ADN (Leppa et al. 2001), lo cual
indica que las funciones no genómicas de los miembros AP-1 no son exclusivas de c-Fos.
La confirmación última de que la activación de la síntesis lipídica se trata de un efecto
independiente de la transcripción ha sido descripta en esta Tesis doctoral, en el ensayo de la
actividad PAP de Lipin 1 en micelas mixtas. En este sistema altamente simplificado, sin más
componentes que las membranas modelo, Lipin 1 y la propia onco-proteína, c-Fos es capaz
por sí mismo de generar un efecto sobre la actividad catalítica de la enzima (Fig. 15b). En
definitiva, las distintas líneas de evidencia convergen en establecer que este fenómeno de
activación es claramente no genómico.
c-Fos es capaz de activar tanto a CDS1 como a Lipin 1
Al comenzar esta Tesis, se había determinado previamente que c-Fos es capaz de
incrementar el marcado radioactivo de todas las especies de fosfolípidos analizadas
(Bussolino et al. 2001, Gil et al. 2004), si bien los pasos metabólicos particulares permanecían
sin examinar. Para profundizar entonces sobre las etapas metabólicas que pudieran ser
afectadas por c-Fos utilizamos como modelo de estudio la inducción de células quiescentes a
re-ingresar al ciclo de celular mediante el agregado de FBS al 20% al medio de cultivo, lo cual
dispara una serie de eventos celulares que estudiamos en la ventana de la primera hora post-

Discusión
63
estímulo. En particular, estudiamos el marcado radioactivo de las dos ondas consecutivas de
síntesis de lípidos en esta ventana temporal. Estas ondas poseen características distintas ya
que mientras que en la primera se marcan fundamentalmente PIPs en la segunda, en cambio,
se marcan mayoritariamente los principales constituyentes de membrana, como por ejemplo
PtdEth (Bussolino et al. 2001). En este sentido, en la primera parte de esta Tesis se estableció
que para incrementar el marcado de PIPs c-Fos actúa sobre la enzima CDS1 (la primera de la
vía) y sobre PI4KIIα (que cataliza el tercer paso metabólico). El fenómeno se probó de
relevancia biológica, ya que al deprimir la expresión de c-Fos, se inhibe el incremento en el
marcado (Fig. 3). Más aún, se estableció que las mencionadas enzimas están necesariamente
implicadas en el proceso, ya que al deprimir la expresión de CDS1 o de PI4KIIα se anulaba el
efecto de activación de la síntesis al estimular a las células (Fig. 7).
Claramente, CDS1 tiene un rol destacado en la síntesis de fosfolípidos y en la
regulación de este metabolismo. La enzima CDS de Drosophila (que posee un único gen para
la enzima) ha sido descripta como clave para coordinar el crecimiento celular y el acúmulo de
grasas ya que al aumentar la disponibilidad de PtdIns, incrementa la actividad de la vía de la
insulina, lo que induce el crecimiento celular (Liu et al. 2014). La vía de la insulina es una vía
de señalización conservada desde mamíferos hasta Drosophila que depende del suministro
de PtdIns y regula numerosos procesos fisiológicos, incluyendo el metabolismo lipídico
(Teleman 2010). Las moscas que son mutantes nulos para CDS poseen un menor tamaño de
células y órganos conjuntamente con un acumulamiento de lípidos neutros (Liu et al. 2014).
Esto viene de la mano de otros reportes, donde en el mencionado organismo se involucra a
CDS en vías de señalización celular vinculadas a PIPs (Wu et al. 1995) o incluso en células de
mamífero donde afecta las respuestas de cascadas de señalización inducidas por citoquinas
pro-inflamatorias específicas (Weeks et al. 1997). Por otro lado, numerosos estudios
genéticos y bioquímicos llevados a cabo en levaduras y células de mamíferos señalan a Lipin
1 como un regulador crucial para el metabolismo lipídico y la fisiología celular (Csaki & Reue
2010, Harris & Finck 2011, Pascual & Carman 2013, Siniossoglou 2013). Por ejemplo, un
estudio implicó a Lipin 1 en la síntesis de fosfolípidos que acompaña la expansión de
membranas del RE en la diferenciación de linfocitos B (Fagone et al. 2007).
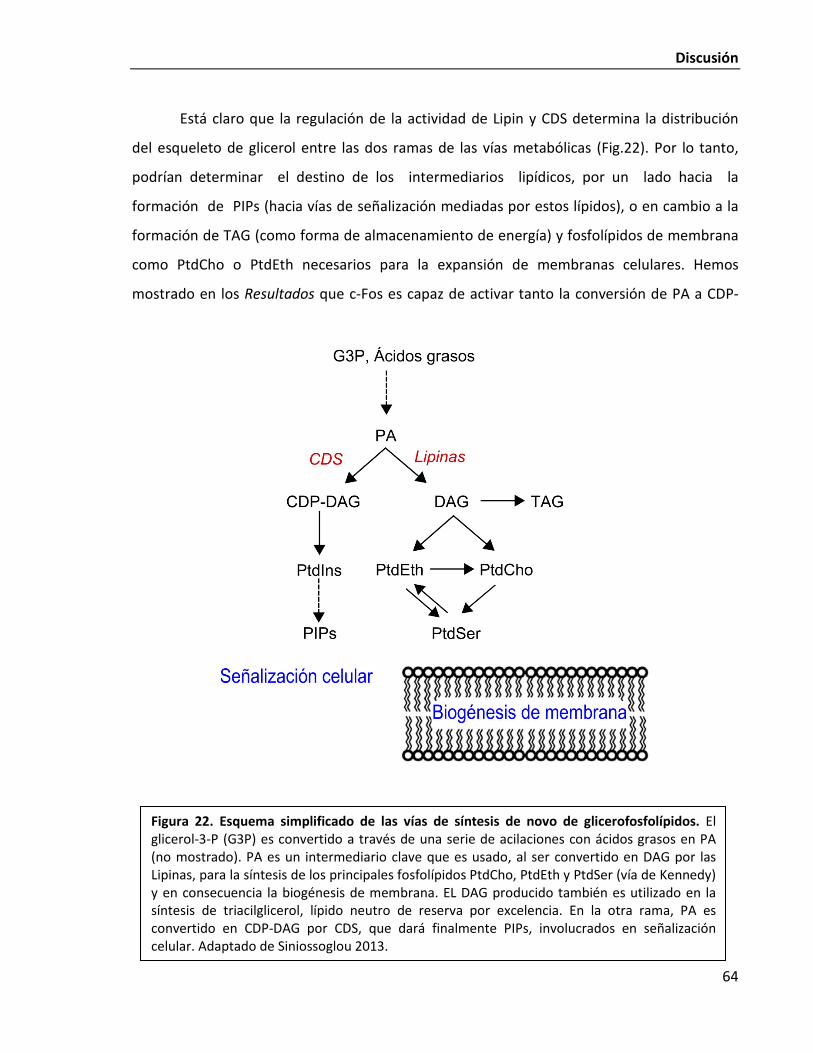
Discusión
64
Está claro que la regulación de la actividad de Lipin y CDS determina la distribución
del esqueleto de glicerol entre las dos ramas de las vías metabólicas (Fig.22). Por lo tanto,
podrían determinar el destino de los intermediarios lipídicos, por un lado hacia la
formación de PIPs (hacia vías de señalización mediadas por estos lípidos), o en cambio a la
formación de TAG (como forma de almacenamiento de energía) y fosfolípidos de membrana
como PtdCho o PtdEth necesarios para la expansión de membranas celulares. Hemos
mostrado en los Resultados que c-Fos es capaz de activar tanto la conversión de PA a CDP-
Figura 22. Esquema simplificado de las vías de síntesis de novo de glicerofosfolípidos. El glicerol-3-P (G3P) es convertido a través de una serie de acilaciones con ácidos grasos en PA (no mostrado). PA es un intermediario clave que es usado, al ser convertido en DAG por las Lipinas, para la síntesis de los principales fosfolípidos PtdCho, PtdEth y PtdSer (vía de Kennedy) y en consecuencia la biogénesis de membrana. EL DAG producido también es utilizado en la síntesis de triacilglicerol, lípido neutro de reserva por excelencia. En la otra rama, PA es convertido en CDP-DAG por CDS, que dará finalmente PIPs, involucrados en señalización celular. Adaptado de Siniossoglou 2013.

Discusión
65
DAG (activando CDS1, hacia la vía de PIPs, Figs. 4,5 y 6) como a DAG (activando Lipin 1, hacia
la vía de Kennedy, Figs. 15 y 16). Los cambios en la activación de estas dos enzimas pueden
en principio explicar las diferencias en las especies marcadas en las dos ondas. No existe una
manera fácil de determinar cómo c-Fos podría incrementar una etapa metabólica en favor de
la otra para generar un flujo metabólico hacia una vía en particular. No está claro como c-Fos
podría generar este efecto, pero sí que la interrelación entre CDS, Lipin 1 y c-Fos está
involucrada en el fenómeno, según se desprende de las vías de síntesis de fosfolípidos
presentados en la Fig. 22.
¿Se activa primero una enzima y luego otra? Un punto que quedó pendiente fue el
de establecer la dinámica de la activación mediante FRET en células estimuladas,
siguiéndolas en el tiempo. ¿Es una cuestión temporal, de localización o una combinación de
ambas? Mientras que CDS es una proteína integral de membrana, Lipin 1 debe translocar
hacia las membranas para catalizar la reacción enzimática. Ello ya impone una limitación
temporal. Por otro lado, en los ensayos in vitro, CDS requiere menores concentraciones de c-
Fos respecto de Lipin 1 para activarse (Figs. 4 y 15a, respectivamente). Si bien ambos ensayos
no son directamente comparables, por las condiciones diferentes del ensayo, podrían estar
indicando una tendencia. Si la afinidad de CDS por c-Fos fuera mayor que la de Lipin, se
podría activar a menores concentraciones de c-Fos, y por lo tanto a tiempos cortos post-
estímulo cuando la concentración de c-Fos es menor. A la hora post-estimulo, la mayor
concentración de c-Fos permitiría la activación de Lipin. Esta es sólo una de las posibles
explicaciones, teniendo en cuenta la cuestión temporal y de afinidad, sin embargo, hay
muchas otras consideraciones a tener en cuenta y que seguramente serán interesantes
explorar.
¿Para qué son necesarios los lípidos cuya síntesis c-Fos incrementa?
Es demasiado simplista a esta altura pensar que el aumento en la síntesis lipídica
sólo puede ser para generar nueva membrana que directamente se utilice para la expansión
de membranas necesaria para el crecimiento celular. Si bien el ensayo in vitro de marcación
de fosfolípidos a partir de 32P-γ-ATP no distingue entre síntesis de novo o recambio parcial,

Discusión
66
también se hicieron experimentos in vivo utilizando como precursor 3H-glicerol y analizando
los fosfolípidos marcados, y este ensayo da cuenta exclusivamente de la síntesis de novo, en
donde se mostró la dependencia de la expresión de c-Fos para su activación (de Arriba Zerpa
et al. 1999). Entonces c-Fos activa la síntesis de novo de los principales fosfolípidos, pero esto
no implica la producción neta de membranas. Por ejemplo, las altas tasas de síntesis de
PtdCho en células en G1 del ciclo celular se ven acompañadas de altas tasas de degradación
del mencionado lípido mediado por la fosfolipasa independiente de calcio A2 (iPLA2)
(Manguikian & Barbour 2004) y la esterasa diana de neuropatía (Zaccheo et al. 2004, Glynn
2005), generando un balance durante esta etapa. La acumulación neta de PtdCho se da al
llegar a la fase S, donde una reducción en el recambio lo permite (Jackowski 1994). Sin
embargo, esta fase de alto recambio es necesaria para la proliferación celular. De hecho, la
inhibición de iPLA2 en células de cáncer de ovario produce un arresto de la proliferación en la
fase S, que puede ser rescatado con el agregado exógeno de ácido lisofosfatídico (LPA),
producto de la degradación de PtdCho por esta enzima (Song et al. 2007). LPA ha probado
tener funciones de crecimiento y supervivencia en este tipo particular de células (Fang et al.
2002).
En el caso de c-Fos, hemos probado cómo aumenta la síntesis de fosfolípidos
mediando etapas claves. Pero, ¿podría c-Fos también incrementar la actividad de
fosfolipasas? Evidencias encontradas en monocapas como modelo de membranas indican
que c-Fos modula la actividad de tres clases de fosfolipasas (Borioli et al. 2002), al menos en
este sistema simplificado. Interesantemente, varios lípidos bioactivos que se producen
durante el metabolismo de glicerofosfolípidos como PA, DAG, PtdIns(4,5)P2 y ácido
araquidónico, pueden regular vías de señalización celular (Oude Weernink et al. 2007). El
metabolismo lipídico es muy complejo y aún no se entiende acabadamente como funciona
pero claramente la regulación del mismo tiene consecuencias sobre la crecimiento celular,
proliferación, supervivencia y apoptosis al actuar sobre vías de señalización (Huang & Freter
2015) y que van más allá de la simple acumulación de membrana. A pesar de no comprender
todavía el fin último de los fosfolípidos cuya síntesis c-Fos induce, las consecuencias
celulares de c-Fos han sido puestas de manifiesto en numerosos estudios, ya que al deprimir

Discusión
67
la expresión de c-Fos se reduce el marcado radiactivo de fosfolípidos y ello conlleva a la
detención de la diferenciación (Gil et al. 2004) y de la proliferación (Gil et al. 2012). Hemos
estudiado la forma en que c-Fos consigue la activación de la síntesis, pero queda abierta la
pregunta de los mecanismos que estos lípidos desencadenan para sostener finalmente la
proliferación.
Un mecanismo compartido para la activación de la síntesis
Un aspecto que emerge del cuerpo de resultados de esta Tesis es el mecanismo con el
que c-Fos activa en forma general la síntesis de glicerofosfo- y glico- lípidos. En todos los
casos estudiados, la activación de enzimas particulares tiene puntos en común: un aumento
en la Vmax sin modificar el Km. Este fue el caso para CDS1, PI4KIIα, Lipin 1, pero también para
glucosilceramida sintasa (GlcCerS), la primera enzima en la vía de síntesis de glicolípidos
(Crespo et al. 2008). Mientras que algunas enzimas son activadas, otras no lo son, y en todos
los casos estudiados, la activación involucra una asociación física directa entre c-Fos y la
enzima activada. Los resultados obtenidos sostienen la existencia de un mecanismo común
para la activación de la síntesis de lípidos mediada por la proto-oncoproteína c-Fos.
El mecanismo común comienza por la localización subcelular de los participantes del
fenómeno en estudio, así como c-Fos se encuentra asociado a las membranas del RE
(Bussolino et al. 2001), también lo están las enzimas que es capaz de activar. Es para destacar
que el RE es la principal organela donde transcurre la biosíntesis de lípidos (Bell et al. 1981,
van Meer et al. 2008) y aparece como un sitio de activación en común para todas las
enzimas con las que c-Fos interactúa. Está claro que CDS1 localiza en el RE (Inglis-Broadgate
et al. 2005), así como Lipin 1 (Peterfy et al. 2005, Grimsey et al. 2008). En cambio la enzima
PI4KIIα se encuentra en múltiples compartimientos, se localiza en el trans-Golgi (Wang et al.
2003), membranas del sistema endosomal (Salazar et al. 2005) así como intermediarios de
tráfico especializados como por ejemplo vesículas sinápticas (Guo et al. 2003). Sin embargo,
un reporte muestra sólida evidencia bioquímica de que existe una fracción minoritaria pero
muy activa enzimáticamente que localiza en el RE (Waugh et al. 2003), donde podría ser
activada por c-Fos (Clayton et al. 2013). En esta última revisión, se cita de este modo al

Discusión
68
reporte producto de los resultados de la primera parte de la presente Tesis doctoral que
fueran publicados en la revista Mol. Biol. Cell (Alfonso Pecchio, Cardozo Gizzi et al. 2011).
Por otro lado, aunque la enzima GlcCerS normalmente se clasifica como una enzima
residente en Golgi, también se la ha detectado en membranas de pre-Golgi/RE (Futerman &
Pagano 1991, Kohyama-Koganeya et al. 2004). Más aún, GlcCerS es única entre las enzimas
glicolípido glicosiltransferasa en el sentido de que el sitio catalítico se orienta hacia la cara
citoplásmica en vez de la cara luminal del RE (Kohyama-Koganeya et al. 2004). En conclusión,
la activación de la síntesis de lípidos por c-Fos se logra al incrementar la actividad de enzimas
particulares que translocan o son componentes integrales del retículo endoplásmico. La
única excepción encontrada hasta aquí es PI4P5K (PtdIns4P-5-quinasa), que se ubica
principalmente en el núcleo (Boronenkov et al. 1998, Mellman et al. 2008). Se mostró que c-
Fos activa a esta enzima en matrices nucleares, promoviendo la formación de PtdIns(4,5)P2 y
consecuentemente cambios transcripcionales globales independientes de la actividad AP-1
de c-Fos (Ferrero et al. 2014).
Una pregunta que queda abierta en la presente Tesis es si existen determinantes en
las enzimas que permitan su interacción con c-Fos, ya que no hemos apuntado a establecer
las regiones involucradas en las enzimas activadas. Todavía es incierta la región de Lipin 1β
involucrada en la asociación con c-Fos. Existen descriptos en Lipin 1 una organización en
forma de dominios conservados que comprende al amino terminal (N-LIP) con una función
desconocida y un dominio C-terminal (C-LIP) que contiene el dominio catalítico, una
secuencia tipo HAD. Sumados a estos dominios, se encuentra además el dominio PBD
involucrado en la unión de PA y algunas otras regiones más de alta homología con los otros
miembros de la familia, junto con extensivas regiones no conservadas. Es por lo tanto de
interés estudiar a futuro si la región de interacción es un dominio conservado en Lipin 2 y 3.
¿Se trata de una activación PAP I general o está restringida a Lipin 1? En la vía de síntesis de
PIPs encontramos que la isoforma α pero no la β de PI4KII se asocia y es activada por c-Fos
(Figs. 8 y 9). La comparación de secuencias entre estas isoformas muestra, como se
mencionó en la Introducción, una región catalítica C-terminal muy conservada mientras que
en la región N-terminal (los primeros 100 residuos) existe una muy baja homología e incluso

Discusión
69
se ha postulado que esta región interviene en asociaciones proteína-proteína específicas.
Podría entonces tratarse de una región que la isoforma α posee pero no la β o, por otro lado,
podría tratarse simplemente de que estas dos proteínas no localizan en los mismos
compartimientos subcelulares. Para el caso de Lipin 1 y Lipin 2, se estableció que ambas no
sólo tienen un patrón de localización diferencial sino también un patrón de expresión
temporal diferente (Grimsey et al. 2008). Estos ejemplos ponen de manifiesto que la
existencia de más de una enzima que cataliza los mismos pasos metabólicos no significa que
sus funciones sean redundantes.
La naturaleza intrínsecamente desestructurada de c-Fos y su asociación a múltiples enzimas
blanco
¿Cómo es que c-Fos es capaz de activar enzimas que no comparten similitudes
estructurales obvias entre ellas? La respuesta puede encontrarse en la naturaleza
intrínsecamente desordenada de c-Fos (Campbell et al. 2000, Gaggiotti et al. 2009).
Pertenece a la familia de proteínas intrínsecamente desestructuradas (IDPs), proteínas que
contienen extensas regiones que no poseen una estructura 3D definida en su estado nativo
(Dyson & Wright 2005). Este tipo de proteínas tienen una plasticidad conformacional que les
permite el reconocimiento y unión a múltiples proteínas blanco con una combinación única
de interacciones de alta especificidad y baja afinidad (Dyson & Wright 2005, Wright & Dyson
2009, Liu & Huang 2014). La existencia de IDPs está cambiando el paradigma de secuencia-
estructura-función. Las IDPs no poseen una única estructura y se adaptan a diferentes roles,
si bien se ha encontrado que las proteínas relacionadas a señalización celular y en particular
a cáncer se encuentran enriquecidas en regiones desordenadas (Iakoucheva et al. 2002).
El orden o desorden de una proteína está definido al nivel de la secuencia de sus
aminoácidos, lo que ha permitido el desarrollo de predictores de desorden basados en la
secuencia. Podría decirse en base a la bibliografía que estos predictores poseen una
exactitud general cercana al 80% al comparar los resultados de probabilidad de desorden con
la información experimental (He et al. 2009).

Discusión
70
Figura 23. Perfil de hidrofobicidad y de probabilidad de desorden de c-Fos. Representación esquemática de c-Fos. BD (aa: 139-159, en negro) es el segmento compuesto por 21 residuos responsable de la unión secuencia-especifica al ADN y de la activación de enzimas blanco. LZ (aa: 165-193, en gris) es el dominio que permite la dimerización y consecuentemente la formación de AP-1 activos. El N-terminal (primeros 138 residuos) es el responsable de la interacción proteína-proteína con enzimas de síntesis de fosfolípidos. El dominio aa: 47-92, tiene un rol importante en esta función y está representado con patrón ondulado. En la parte inferior se observa el resultado de predictores de probabilidad de desorden y de hidrofobicidad basados en análisis de la secuencia. En rojo se observa la representación de la probabilidad del desorden en función del número de residuo obtenido con PONDR-FIT (Xue et al. 2010). El valor de corte es 0,5 lo que significa que residuos por encima de ese valor son desordenados. En azul se observa la representación de hidrofobicidad en función del número de residuo obtenida a partir de la escala Kyte & Doolittle. Nótese que en la región aa: 47-92 existen dos segmentos altamente hidrofóbicos que solapan con una región ordenada, según se discute en el texto.

Discusión
71
En la sección Resultados, hemos mostrado como el N-terminal de c-Fos es
responsable de la asociación específica pero no la activación de CDS1, PI4KIIα y Lipin 1. Para
encontrar nuevas perspectivas, hemos ingresado la secuencia de c-Fos en diferentes
predictores de desorden. Para todos los predictores utilizados, se encontró
consistentemente un perfil similar al mostrado en la Fig. 23 para la región N-terminal. Si bien
toda la proteína c-Fos es una IDP, y esto incluye su N-terminal, hemos observado que la
probabilidad de desorden cae significativamente en la región alrededor de aa: 45-80 (Fig. 23),
punto que discutiremos a continuación.
Algunos de los predictores que utilizamos son clasificadores que han sido
entrenados con secuencias ordenadas y desordenadas de bases de datos como PONDR-VLXT
(Romero et al. 2001); otros, como como IUPred, se basan en propiedades físico-químicas
tales como la capacidad de polipéptidos de formar contactos estabilizadores (Dosztanyi et al.
2005). Los valores de probabilidad se calculan residuo por residuo, tomando una ventana o
rango de aminoácidos vecinos. La Fig. 23 muestra una representación de probabilidad de
desorden utilizando PONDR-FIT (Xue et al. 2010) junto con un perfil hidrofóbico utilizando la
escala de Kyte y Doolite (1982). PONDR-FIT utiliza una red neuronal artificial para predecir
cuales residuos son desordenados. La idea detrás de la utilización de redes neurales es dar
diferente peso y combinar de manera no lineal distintos tipos de atributos como la
composición general de aminoácidos, la complejidad de la secuencia o características de los
mismos aminoácidos como carga o hidropatía (He et al. 2009).
Mientras que la repulsión de cargas favorece el desplegado, el incremento en la
hidrofobicidad favorece el plegado, según las presupuestos del primer modelo predictor
(Uversky et al. 2000). En otras palabras, regiones hidrofóbicas tienden a ser ordenadas
mientras que regiones con cargas netas difícilmente podrán compactarse y establecer una
estructura única, sino que por lo contrario tenderán a tener múltiples configuraciones.
Gráficos de carga neta total en función de hidropatía permiten distinguir proteínas
desordenadas de aquellas que no lo son. También se desarrolló un algoritmo para predecir
residuo por residuo la probabilidad de desorden a partir de esta idea (Prilusky et al. 2005).

Discusión
72
Este simple modelo funciona notoriamente bien aun teniendo en cuenta los desarrollos
actuales (Uversky 2013).
La alta hidrofobicidad de la región de c-Fos aa: 47-92 ya ha sido señalada en la sección
Resultados, donde se la vinculó a la asociación con Lipin 1 (Fig. 21). Puede verse en la Fig. 23
como la mencionada región forma un segmento hidrofóbico que según el predictor tiene
mayor tendencia al orden. Las regiones con mayor propensión al orden han sido apuntadas
como dominios funcionales dentro de IDPs, y se piensa que participan en el reconocimiento
molecular de los múltiples blancos de unión (Hsu et al. 2013). Según los resultados
obtenidos, la región intermedia del N-terminal (aa: 47-92) está involucrada de una manera
compleja con la asociación a Lipin 1 (Fig. 21) pero también a PI4KIIα, según indican resultados
preliminares del Dr. César Prucca. Nuevos experimentos utilizando mutantes de esta región
podrían esclarecer su rol y a la vez brindar información acerca de si es un fenómeno
secuencia-especifica o si, como suponemos, se trata de un efecto debido a las propiedades
globales del segmento en cuestión.
Fue establecido que el dominio BD de c-Fos experimenta una transición desorden a
orden al unirse al ADN como un complejo AP-1, formando una alfa hélice que interacciona
directamente con el ADN (Patel et al. 1990, Glover & Harrison 1995). La idea detrás de esto
es que proteínas que son desestructuradas en su estado nativo pueden adoptar una
estructura definida bajo ciertas condiciones, que incluyen la presencia de un ligando, un
cambio en el pH o la presencia de cationes o sales específicos (Wright & Dyson 2009).
Aunque todavía resta por establecerse si existe una transición “desorden a orden” acoplada a
la asociación del N-terminal de c-Fos a las enzimas blanco. Es posible pensar que el plegado
de c-Fos al unirse a su enzima blanco podría ser relevante para la formación del complejo c-
Fos/enzima. Sin embargo, el plegado en respuesta una unión no es universal y existen
ejemplos de IDPs que preservan un alto nivel de desorden aún unidas a su blanco, un
fenómeno denominado “fuzziness” o “estructuras difusas” (Hazy & Tompa 2009).
En cualquier caso, está definido que la interacción con distintas proteínas puede
involucrar una misma región, como sería el caso del N-terminal para la unión de c-Fos a
múltiples enzimas blanco para ser activadas. En cambio, la región BD tiene una función

Discusión
73
directa tanto en la actividad AP-1 como en la activación en si misma de enzimas puntuales,
por lo que c-Fos utiliza la misma región para realizar acciones muy distintas. Proponemos a c-
Fos como una proteína IDP capaz de múltiples funciones (o moonlighting por su
denominación en inglés). La manifestación última de la naturaleza IDP de c-Fos es su
promiscuidad para unirse a múltiples blancos, como c-Jun o Lipin 1 para citar solo dos
ejemplos, y así poder ejercer como un proteína moonlighting (Tompa et al. 2005, Jeffery
2009).
Existen diversos mecanismos para que una proteína multi-tarea pase de una función a
otra, tales como una regulación en su localización subcelular o capacidad para unirse a
ligandos. En este sentido, vale la pena mencionar que una fosforilación en los residuos de
tirosina 10 y 30 vuelve a c-Fos incapaz de asociarse al RE y consecuentemente de activar la
síntesis de lípidos (Ferrero et al. 2012). La región aa: 47-92 se encuentra flanqueada por dos
regiones flexibles desestructuradas (Fig. 23). En base a los Resultados obtenidos, parece
razonable pensar que la fosforilación en las tirosinas 10 y 30 afecta de alguna manera la
región aa: 47-92 que posiblemente es la responsable de interaccionar con las enzimas
blanco. Se ha señalado que regiones desordenadas dentro de una proteína facilitan la
fosforilación (Liu & Huang 2014) dado que la fosforilación se da predominantemente en estas
regiones (Iakoucheva et al. 2004). Más aún, los niveles de fosforilación son mucho más
dinámicos durante el ciclo celular para IDPs (Tyanova et al. 2013), regulando de esta forma el
accionar de proteínas claves para la progresión del ciclo. La fosforilación en tirosinas es sólo
una de las formas de regular las actividades de c-Fos, otro ejemplo resulta de la acción de las
quinasas de serinas/treoninas ERK y RSK que fosforilan a c-Fos luego de estímulos
mitogénicos en su dominio de trans-activación C-terminal promoviendo un incremento en su
estabilidad y capacidad como factor de transcripción (Murphy et al. 2002, Murphy & Blenis
2006). Es importante resaltar que las distintas fosforilaciones generan efectos opuestos,
mientras que la fosforilación en tirosinas reprime su actividad como activador de la síntesis
lipídicas, la fosforilación en ser/treo incrementa su actividad AP-1.
Otra característica determinada por la naturaleza IDP de c-Fos es su comportamiento
interfacial. c-Fos es una proteína anfitrópica que a partir de su naturaleza flexible ejerce una

Discusión
74
actividad de superficie. Estudios biofísicos han mostrado que c-Fos tiene una gran afinidad
por membranas y una marcada capacidad para detectar y responder a cambios en la
composición y estado de fase de lípidos en una monocapa (Borioli et al. 2001, Gaggiotti et al.
2009). Todo ello apoya la idea de que c-Fos tiene las características moleculares (alta
actividad de superficie, capacidad para detectar y responder a lípidos y desorden intrínseco
que permite cierta promiscuidad en sus asociaciones) (Borioli 2011) para llevar a cabo un rol
regulatorio en la síntesis de glicerolípidos. En un contexto celular, c-Fos puede ser imaginado
detectando la composición lipídica, asociándose específicamente a membranas del RE y
encontrando su camino hacia enzimas blanco para finalmente activarlas in situ (Cardozo Gizzi
& Caputto 2013).
La región N-terminal de c-Fos media la asociación con CDS1, PI4KIIα y Lipin 1. En la
Fig. 14 se observa una localización de NA-myc (mutante que comprende todo el N-terminal,
aa: 1-138) exclusivamente peri-nuclear, en tanto en la Fig. 13 se probó como NA-myc es
capaz de competir por la unión de PI4KIIα con c-Fos. Es posible que la sobre-expresión de NA
bloquee la interacción c-Fos/enzima para todos los casos en donde se observó que la región
N-terminal es la involucrada en la unión. Nuestra hipótesis es que c-Fos activa a las enzimas
exclusivamente en esta localización subcelular, en el RE, por lo que la presencia de la
mutante NA allí sería capaz de competir y generar el efecto de detener la activación general
de la síntesis de fosfolípidos. En relación a lo anterior, la sobre-expresión de la mutante NA
puede detener la proliferación de células T98G en cultivo, resultados obtenidos por el Dr.
Cesar Prucca en su proyecto post-doctoral que se continuó a partir de los Resultados de la
primera parte de esta Tesis. Esto último es una prueba más de que la activación de lípidos
dependiente de c-Fos es requerida por la célula para sostener la proliferación, pero además
indica que el N-terminal de c-Fos es clave para ello y aparece como un posible blanco
terapéutico. Anteriormente ya se utilizaron estrategias para disminuir los efectos de c-Fos al
impedir su expresión mediante el uso de ARN antisentido (Mercola et al. 1987, Edwards et al.
1988, Ledwith et al. 1990). Sin embargo, la mutante NA aparece como atractiva por bloquear
únicamente el rol no genómico de c-Fos, compitiendo con la proteína endógena por la unión
de sus enzimas blanco.

Discusión
75
Fra-1 también incrementa la síntesis de lípidos
Ya se ha establecido que Fra-1 está involucrado en la proliferación de células de
cáncer de mama al incrementar el marcado radioactivo de fosfolípidos (Motrich et al. 2013),
si bien el mecanismo por el cual lo hace es desconocido. En esta Tesis hemos encontrado
que, al menos para la actividad PAP-1, Fra-1 es capaz de incrementar la actividad enzimática
en la misma medida que c-Fos. Ya se discutió la homología del BD de ambas proteínas y de la
relevancia de este dominio a partir de experimentos con mutantes de deleción. Por otro
lado, también se encontró una unión directa entre Fra-1 y Lipin 1, por lo que es razonable
pensar en que el mecanismo de activación de Fra-1 posee características comunes con el de
c-Fos. Esto nos lleva a especular acerca de las regiones de Fra-1 que podrían mediar la
interacción con la mencionada enzima.
En términos generales, existen dos regiones altamente conservadas entre c-Fos y Fra-
1: el dominio bZip (que comprende el BD) y los últimos 70 residuos en C-terminal de ambas
proteínas. Sin embargo, existe una tercera región de similitud entre la secuencia primaria de
ambas proteínas, los aa: 54-84 de c-Fos y los aa: 31-61 de Fra-1 (Fig. 24). Esta región
comprende para el caso de c-Fos en forma casi completa el dominio que se ha identificado
como necesario para la asociación a Lipin 1 (Fig. 21), la región comprendida por aa: 47-90 de
la que se ha venido discutiendo su función. Por lo tanto, la tercera región de homología entre
c-Fos y Fra-1 se trataría de la que está involucrada, al menos en el caso de c-Fos, en la
interacción con las enzimas blanco. Predictores de desorden e hidropatía también indican
que esta región de Fra-1 comparte las características de la región equivalente de c-Fos. Es un
segmento altamente hidrofóbico, ligeramente más corto que el de c-Fos, en donde PONDR-
FIT predice una región ordenada flanqueada por regiones desordenadas. Es interesante
remarcar que si bien no existe una conservación a nivel de secuencia, la región ordenada aa:
31-61 de Fra-1 tiene a cada lado regiones desestructuradas como se da en c-Fos. Estudios a
futuro podrían confirmar si este es efectivamente el dominio de interacción, y si existe un
mecanismo de activación compartido por las dos proto-oncoproteínas.

Discusión
76
Más importante aún sería determinar las consecuencias celulares de ello. Podemos
especular que la expresión de c-Fos, Fra-1 o de ambas proteínas en un modelo celular en
particular podría determinar el resultado final en la maquinaria de biogénesis de membrana.
Por ejemplo, el mencionado rol de Fra-1 en la proliferación de células de cáncer de mama se
debe a un incremento en la expresión en este tipo celular que ha sido descripto
extensivamente (Belguise et al. 2005, Song et al. 2006, Logullo et al. 2011). En nuestro
laboratorio se encontró que más del 95% de las muestras de carcinoma de mama ductal
expresan Fra-1 pero también c-Fos, a diferencia de lo que ocurre en tejido normal donde no
se encuentran ninguna de las dos proteínas (Motrich et al. 2013).
Figura 24. Alineamiento las secuencias de c-Fos y Fra-1. Se destacan tres regiones conservadas: 1) N-terminal (recuadro rojo), bZip (recuadro azul) y C-terminal (recuadro violeta). * indica aminoácido conservado, : alta homología, . baja homología. La escala que en el recuadro inferior derecho indica grado de conservación y va desde verde (no conservado) a rosa fuerte (máxima conservación).

Discusión
77
Conclusiones finales
La regulación de la síntesis lipídica para las diferentes etapas del crecimiento y la
proliferación celular es un proceso clave que debe ser finamente regulado debido a que
involucra la actividad sincronizada de múltiples pasos metabólicos que se orquestan en
varios niveles. Sin duda, un mecanismo clave para alcanzar la regulación global de la
biogénesis de membrana es a través de factores de transcripción que actúan en forma
coordinada para regular la expresión de enzimas responsables de la síntesis lipídica como las
que ocurren durante la división celular (Nohturfft & Zhang 2009). Sin embargo, dado que la
biosíntesis de lípidos involucra numerosas etapas metabólicas, la regulación de la síntesis
requeriría de complejos y variados mecanismos regulatorios. A pesar de ello, a la luz de los
resultados de muchos laboratorios, se observa que al regular únicamente etapas limitantes
se altera toda la vía.
En este sentido, la evidencia obtenida en esta Tesis doctoral indica que al incrementar
la capacidad catalítica de enzimas clave, c-Fos incrementa la disponibilidad de los principales
lípidos. Esto apunta a un posible mecanismo molecular de activación compartido por las
distintas enzimas a las que c-Fos se asocia físicamente. Hemos avanzado en el conocimiento
del cómo es que c-Fos actúa a nivel no genómico, pero se abre el interrogante del “para
qué”. No está claro en este punto el fin de la activación de la síntesis de lípidos, pero si sus
consecuencias últimas: el sostenimiento del crecimiento o la proliferación celular. Quizás
estudios en esta dirección nos indiquen en un futuro como limitar la proliferación irrestricta
de células tumorales, al interferir con la activación lipídica que es promovida por c-Fos.

CAPITULO V
MATERIALES Y MÉTODOS

Materiales y Métodos
79
Materiales
Los reactivos radioactivos son de PerkinElmer. Los lípidos son de Avanti Polar Lipids.
La Lipofectamina 2000, los medios y reactivos de cultivo son de Invitrogen. Los reactivos de
biología molecular, kits, enzimas de restricción, etc. son de Promega. Los demás reactivos
son de máxima pureza adquiridos en Sigma, Merck o equivalente. Se harán aclaraciones
cuando sea pertinente.
Cultivos celulares
Todos los experimentos fueron realizados con la línea celular derivada de
fibroblastos de ratón NIH 3T3 adquiridas al American Type Culture Collection, USA. Esta línea
fue crecida en cápsulas de Petri descartables de diferentes tamaños según cada experimento
(multicelda de 24, cápsulas de 35, 60 o 100 mm de diámetro). Las células fueron crecidas
hasta alcanzar más del 90% de confluencia en cada caso. Para la adquisición de imágenes en
experimentos de microscopía fueron crecidas sobre cubreobjetos de vidrio redondos
previamente tratados con ácido y enjuagados exhaustivamente. Los cultivos se sembraron a
baja densidad y se dejaron proliferando a 37°C durante 48 horas en presencia de 5% de CO2
en DMEM suplementado con 10% v/v suero de ternero bovino (Gibco). Una vez alcanzada la
confluencia, las células se arrestaban al reemplazar el medio de cultivo con DMEM. Se
dejaron las células con este medio durante 48 horas más. En los experimentos de
estimulación se adicionó FBS para llevar al 20 % final durante los tiempos especificados en
cada experimento.
Marcado metabólico de PtdInsP y PtdInsP2
Células quiescentes fueron pulsadas con 25 µCi/mL de 32P-ortofosfato 15 minutos
antes de cosecharlas. Para el experimento del bloqueo específico de la expresión de c-Fos al
producirse el estímulo, ARNm anti-sentido (Morpholino oligonucleótidos, Gene Tools) contra
c-Fos (5´-TGC-GTT-GAA-GCC-CGA-GAA-3´) (Bussolino et al. 1998) o un oligonucleótido de

Materiales y Métodos
80
secuencia aleatoria que no tiene blanco en células de ratón fueron adicionados al medio a
una concentración de 1 µg/mL medio cultivo, 30 minutos antes del pulsado radioactivo.
En cambio, para deprimir la expresión de las enzimas blanco se utilizaron dúplex de
ARN pequeño de interferencia (Dharmacon). En todos los casos, 240 pmol de ARN total
fueron transfectados a cada pocillo en una placa multicelda de 6 con Lipofectamina de
acuerdo al protocolo del fabricante. Después de 24 horas, las células fueron transfectadas
una segunda vez de la misma forma y fueron cultivadas por 48 horas más en medio libre de
suero antes de realizar el marcado metabólico. Para corregir por posibles diferencias en el
número de células en los distintos pocillos en los ensayos con ARN pequeño de interferencia,
se corrió en paralelo un pocillo que no se marcó radioactivamente y que se utilizó para WB.
El mencionado WB se reveló contra las distintas enzimas para así poner de manifiesto si
había funcionado el tratamiento con ARN pequeño de interferencia. Por densitometría se
estableció además la intensidad de la banda de α-tubulina, utilizada como control de carga
para normalizar las cuentas radioactivas obtenidas con la proteína total sembrada.
Luego del pulsado, se estimuló a las células durante 7,5 minutos con FBS al 20%.
Luego del estímulo, se enjuagó cuidadosamente con dos lavados de PBS frío antes de
cosechar las células en agua. Los ensayos se hicieron en duplicado en un total de tres
experimentos independientes como mínimo.
Marcado de PtdInsP y PtdInsP2 in vitro
La capacidad de marcado radioactivo de 32P-PtdInsP y 32P-PtdInsP2 de células
quiescentes fue determinado in vitro según Gil et al. (2004) en tubos de Khan de vidrio. El
buffer de reacción contenía HEPES 64 mM (pH 7,4), NaCl 140 mM, KCl 4,5 mM, MgCl2 0,5
mM, glucosa 5,6 mM, 3 µCi de 32P-ATP (actividad específica 3000 Ci/mmol). Se determinó la
marcación en presencia de c-Fos o sus mutantes de deleción obtenidos en forma
recombinante y purificados, utilizando 1 ng c-Fos recombinante/µg proteína total
resuspendido en 3 µL de buffer de elusión, que contiene imidazol 500 mM /urea 8 M. A los
incubados control se les adicionó en cambio 3 µL de buffer de elusión. Las reacciones, que
tenían un volumen final de 80 µL, fueron iniciadas con la adición de 100 µg de homogenato

Materiales y Métodos
81
celular total al medio de ensayo, incubadas por 1 hora a 37 °C y terminadas por la adición de
20 volúmenes de cloroformo/metanol 2:1. Los ensayos se hicieron por triplicado en al menos
tres experimentos independientes.
Purificación de PtdInsP y PtdInsP2 y cromatografía en capa delgada
Para la obtención de los mencionados fosfolípidos tanto desde el marcado metabólico
o el marcado in vitro, se utilizó el método de Folch (Folch et al. 1957). Tras la adición de 20
volúmenes de cloroformo/metanol 2:1, la fase orgánica fue particionada por el agregado de
agua (0,2 volúmenes). Luego de vorterear por 3 segundos, se centrifugó durante 2 minutos a
1.500 rpm para separar las fases y se lavó la fase inferior (orgánica) 5 veces con 1 mL de fase
superior teórica (metanol/agua/cloroformo 48:47:3, v/v/v) para eliminar el precursor
radioactivo soluble. Finalmente, se extrajo cuidadosamente la fase inferior mediante pipeta
Pasteur y se transfirió a un nuevo tubo de vidrio. Se secó esta fase bajo flujo de nitrógeno
para luego resuspender en 50 µL de cloroformo/metanol 2:1.
Los extractos lipídicos fueron separados en placas cromatográficas delgadas de Sílica
Gel 60 utilizando la metodología descripta en Catz & Sterin-Speziale (1996). Las placas de
sílica fueron tratadas con una solución de oxalato de potasio al 1% en metanol/agua 2:3 y
calentadas posteriormente a 110 °C durante al menos 30 minutos previo a ser utilizadas para
remover humedad residual. Las placas fueron desarrolladas en dos corridas consecutivas en
un sistema monodimensional utilizando cloroformo/metanol/ácido acético/agua 25:15:8:2
para la primera corrida, luego de la cual las placas fueron secadas a temperatura ambiente y
desarrolladas en el mismo sistema de solventes pero en las proporciones 25:15:16:2.
Fosfolípidos de referencia fueron sembrados en carriles junto a las muestras y
visualizadas con vapores de iodo en una cuba dispuesta para este fin, para de esta forma
identificar los productos obtenidos. La radioactividad de los lípidos individuales fue
cuantificada por exposición de la placa de sílica a una pantalla de fósforo y posteriormente
procesada para su visualización por PhosPhorImager (Molecular Dinamycs) utilizando el
scanner especifico FLA-3000 (Fijifilm).

Materiales y Métodos
82
Clonado de mutantes de c-Fos
Mutantes de deleción de c-Fos: Las diferentes mutantes se construyeron mediante el diseño
de oligonucleótidos específicos que abarcaron a los diferentes dominios de c-Fos salvaje, y
con estos oligonucleótidos se realizaron reacciones de PCR y clonado direccional a partir de
la secuencia de un vector que codifica para c-Fos de rata con un rótulo de 6 x histidinas con
codones optimizados para procariotas (Abate et al. 1991). Los productos de PCR obtenidos se
verificaron en geles de agarosa, se purificaron con columnas de purificación y se ligaron al
vector de expresión procariota pET15b (Novagen) utilizando los sitios de restricción NdeI y
BamHI.
Mutagénesis sitio dirigida: La generación de mutantes puntuales de c-Fos se realizó mediante
procedimientos diferentes. Estas secuencias se construyeron por una estrategia denominada
“Superposición Extendida” u “OE-PCR” (Higuchi et al. 1988). Brevemente, consiste en la
utilización de 4 oligonucleótidos, 2 conteniendo la mutación puntual deseada y que además
son 100% complementarios entre sí (“mutagénicos”), y los otros 2 restantes que se unen a
los extremos de la secuencia de c-Fos salvaje (“No mutagénicos”). Los oligonucleótidos
mutagénicos son usados en una primera reacción de PCR con su correspondiente
oligonucleótido no mutagénico, para obtener 2 productos de PCR conteniendo cada uno de
ellos la mutación deseada. Estos productos de PCR se corren en un gel de agarosa y se
purifican adecuadamente y debido a que estos complementan perfectamente sus extremos,
se mezclan y se realiza una tercera reacción de PCR, con los oligonucleótidos no mutagénicos
como cebadores de la reacción. Finalmente se obtiene amplificada a la secuencia completa
de c-Fos pero ahora con la mutación introducida. Las etapas finales son las mismas que para
los clonados anteriores.
Expresión y purificación de c-Fos, Fra-1, c-Jun y sus mutantes
Se siguió el protocolo descripto en Borioli et al. (2001). Una colonia de BL21 (DE3)
conteniendo el plásmido fue inoculada a 5 mL de medio LB suplementado con ampicilina
(100 µg/mL), y se incubó durante la noche a 37 °C. Dos mL del cultivo saturado se inocularon

Materiales y Métodos
83
a 100 mL de LB que se creció con agitación constante de 250 rpm a 37 °C hasta que la
absorbancia a 600 nm alcanzó 0,5. Entonces se le agregó al cultivo 1 µM de concentración
final de isopropil-β-D-1-tiogalactopiranósido (IPTG) y se lo incubó por 4 horas más a 37 °C. El
cultivo inducido para la expresión de c-Fos fue cosechado por centrifugación a 5,000 x g por
10 minutos a 4 °C. El sedimento se re-suspendió en 2 mL de buffer conteniendo 20 mM de
Tris-HCl (pH 8,0), 500 mM NaCl y 5 mM imidazol (buffer de unión). Las bacterias
resuspendidas fueron incubadas con lisozima durante 15 minutos a temperatura ambiente y
luego lisadas adicionalmente con 5 pulsos de sonicación a la máxima potencia durante 1
minuto en hielo, para evitar que aumente la temperatura del lisado. El lisado fue
centrifugado a 12,000 x g por 15 minutos a 4 °C. El sedimento contiene a la proteína en
forma mayoritaria, que permanece insoluble en cuerpos de inclusión. Se resuspendió en
buffer de unión pero que ahora además contiene urea 6 M. Se lo incubó durante una hora a
4 °C por agitación constante favoreciendo la solubilización de los cuerpos de inclusión. Se
centrifugó nuevamente a 12,000 x g por 15 minutos a 4 °C. El sobrenadante fue pasado por
un filtro de 0,4 µm y luego a través de una columna de afinidad de Níquel-Sefarosa de 600 µL
(suspensión acuosa). Se procedió al lavado primero con 3 mL de buffer de unión y luego con
2 mL de buffer de lavado conteniendo 20 mM de Tris-HCl (pH 8,0), 500 mM NaCl, 20 mM
imidazol y urea 6 M. La proteína fue eluída en fracciones de 0,4 mL en un total de 2 mL con
buffer conteniendo 20 mM de Tris-HCl (pH 8,0), 500 mM NaCl, 500 mM imidazol y urea 6 M.
Se determinó la concentración de proteína por el método de Bradford (Bradford 1976). Las
preparaciones, que tenían una concentración cercana a 1 mg/mL, se guardaron a 4 °C por
hasta un mes.
Anticuerpos empleados
Se emplearon los siguientes anticuerpos: anti-myc (ratón, M4439), anti-tubulina (ratón,
DM1A), anti-GFP (conejo, G1544), son de Sigma. Anti-c-Fos (ratón, sc-52) y anti-myc (conejo,
sc-789) de Santa Cruz. Anti-CDS1 (conejo, Ab-84019), anti-PI4KIIα (conejo, Ab-71824) y β
(conejo, Ab-71823) son de Abcam. Anti-GFP de Roche (ratón, #1-814-460).

Materiales y Métodos
84
Inmunoprecipitaciones
Las inmunoprecipitaciónes se realizaron según se describió previamente (Bonifacino
& Dell'Angelica 2001). Brevemente, cultivos celulares de NIH 3T3 (aproximadamente 250 μg)
arrestados y transfectados con la proteína de interés, se estimularon (Ver Cultivos celulares)
y lavaron dos veces con PBS 10mM frío. Luego se agregó 500 μl de buffer de lisis (50 mM
TrisHCl pH: 7,5, 1% v/v Tritón X-100, 300 mM NaCl, cóctel inhibidor de proteasas Complete
(Roche) y 0,05% P/v azida sódica) y fueron dejados en hielo durante 20 minutos. Después de
este tiempo se recolectaron los lisados y se pusieron en tubos de microcentrífuga de 1,5 mL,
se agitaron con vortex al máximo durante 3 segundos y se dejaron en hielo durante 15
minutos más. Los lisados fueron pre-absorbidos con el agregado de 10 μl de proteína G-
Sepharosa (GE Healthcare) durante una hora en un rotor de 360º a 4ºC, y después de este
tiempo fueron nuevamente centrifugados a 13.200 rpm a 4ºC. El sobrenadante fue
cuidadosamente transvasado a un nuevo tubo. Se separó el 20% del volumen obtenido para
posterior análisis por WB (homogenato total) y el resto del lisado fue sometido a la co-
inmunoprecipitación. Se agregó 25 μl de proteína G-Sepharosa previamente conjugada con
aproximadamente 1 μg de anticuerpo policlonal correspondiente a cada caso (ver
Resultados). Las inmunoprecipitaciones se dejaron en un rotor 360º a 4ºC durante 4 horas.
Luego de este tiempo los inmunocomplejos fueron centrifugados 30 segundos a 4.200 rpm y
lavados con buffer de lavado (50 mM TrisHCl pH: 7,5, 0,1% v/v Tritón X-100, 300 mM NaCl,
cóctel inhibidor de proteasas Complete y 0,05% P/v azida sódica). Este procedimiento se
realizó 4 veces y después se hizo un lavado final con PBS para remover el detergente
remanente que puede interferir en la siguiente etapa de WB. Las muestras se calentaron a
100 C° durante 5 minutos y se sembraron para el posterior SDS-PAGE.
SDS-PAGE y Western Blot
Cápsulas de Petri conteniendo las células se lavaron cuidadosamente 3 veces con PBS
10 mM frío y se cosecharon con un volumen mínimo de agua milliQ (o Buffer de lisis, según lo
especifique algún procedimiento) conteniendo además cóctel inhibidor de proteasas
Complete. Después de cuantificar proteínas por Bradford, y dependiendo del anticuerpo y/o

Materiales y Métodos
85
proteína a detectar, una cantidad de proteínas que variaba entre 10 µg a 150 µg fue
calentada durante 5 minutos a 100 ºC en presencia de Buffer de siembra de proteínas
(sample buffer) conteniendo 20 % del agente reductor 2-β mercaptoetanol, para ser
posteriormente sometidas a electroforesis en gel de poliacrilamida SDS-PAGE. Todos los
geles SDS-PAGE se realizaron en concentración 12% final de acrilamida. Después de la
electroforesis y habiéndose separado las proteínas de diferente peso molecular, las mismas
fueron transferidas (70 minutos, 300 mA) a una membrana de nitrocelulosa de 0,2 μm de
poro e incubadas durante al menos una hora con buffer de bloqueo (5% leche descremada
en PBS). Luego de este tiempo las membranas estaban listas para ser incubadas con los
anticuerpos primarios correspondientes según cada caso. Como anticuerpos secundarios se
usaron IRDye 800 y 680 anti-ratón o anti-conejo (LI-COR), según corresponda. Para revelar las
bandas se utilizó un scanner fluorescente en infrarrojo Oddysey (LI-COR). El peso molecular
de cada banda fue calculada por extrapolación a un estándar de pesos moleculares sembrado
en cada gel (BioRad).
Expresión y purificación de Lipin 1
Se siguió el protocolo descripto en Han & Carman (2010). Una colonia de Rosetta2
(DE3) conteniendo el plásmido de la Lipin 1β humana fue inoculado a 5 mL de medio LB
suplementado con kanamicina (30 µg/mL) y cloroanfenicol (50 µg/mL), y se incubó durante la
noche a 37 °C. Dos mililitros del cultivo saturado se inocularon a 500 mL de medio fresco que
se creció con agitación constante de 250 rpm a 37 °C hasta que la absorbancia a 600 nm
alcanzó 0,5. Entonces se le agregó al cultivo 1 µM de concentración final de IPTG y se lo
incubó por 3 horas más a 37 °C. El cultivo inducido para la expresión de Lipin 1 fue cosechado
por centrifugación a 5,000 x g por 10 minutos a 4 °C. El sedimento se re-suspendió en 20 mL
de buffer conteniendo 20 mM de Tris-HCl (pH 8,0), 500 mM NaCl y 5 mM imidazol. Las
bacterias resuspendidas fueron lisadas a través de dos pasajes en un Emulsiflex a 15,000 p.s.i.
y el lisado centrifugado a 12,000 x g por 30 minutos a 4 °C. El sobrenadante fue pasado a
través de una columna de afinidad de Níquel-Sefarosa de 1 mL (suspensión acuosa). Se
procedió al lavado con 25 mL con buffer conteniendo 20 mM de Tris-HCl (pH 8,0), 500 mM

Materiales y Métodos
86
NaCl, 45 mM imidazol, 7 mM 2-mercaptoetanol y 10% v/v glicerol. La enzima, que posee un
rótulo de 6 histidinas en línea, fue eluída en fracciones de 0,5 mL en un total de 3 mL con
buffer conteniendo 20 mM de Tris-HCl (pH 8,0), 500 mM NaCl, 250 mM imidazol, 7 mM 2-
mercaptoetanol y 10% glicerol. Después de establecer cuáles eran las alícuotas más
concentradas, se las mezcló y se determinó la concentración de proteína por el método de
Bradford (Bradford 1976). Las preparaciones de enzimas, que tenían una concentración
cercana a 1 mg/mL, se guardaron en pequeñas alícuotas a -70 °C.
Preparación del reactivo de verde de malaquita-molibdato
Un reactivo coloreado para detectar fosfato inorgánico libre fue preparado según
Mahuren et al. (2001) con algunas modificaciones, y se compone de 3 volúmenes de verde
de malaquita 0,1 mM y un volumen de molibdato de amonio 34 mM en HCl 5 M. Luego de
mezclar los componentes, se deja en agitación por lo menos durante 30 minutos, se filtra y se
guarda protegido de la luz. El reactivo así preparado se usó dentro de una semana de
preparado.
Preparación de las micelas mixtas de Tritón X-100/PA
PA en cloroformo/metanol (2:1) fue transferido a un tubo de ensayo de vidrio, secado
bajo un flujo de N2 y finalmente mantenido in vacuo durante 1 hora. Tritón X-100 fue
adicionado al PA justo después del buffer de ensayo para preparar las micelas mixtas. El
vortereo aplicado extensivamente asegura la formación de micelas. El contenido mol% de PA
en las micelas mixta fue calculado de acuerdo a la fórmula:
mol%PA = 100 x [PA (molar)]/ ([PA (molar)]+ [Triton X-100(molar)])
En forma similar, el mol% de c-Fos recombinante fue calculado como:
mol%c-Fos = 100 x [c-Fos (molar)]/ ([PA (molar)]+ [Triton X-100(molar)])

Materiales y Métodos
87
La concentración total de PA en las micelas mixtas se mantuvo por debajo del 15% para
asegurar que la estructura de la misma sea similar a la de micelas de Tritón X-100 puras
Lichtenberg et al. (1983).
Actividades enzimáticas
En todos los casos se utilizó como fuente de enzima homogenatos totales de NIH 3T3.
Los incubados tuvieron 100 µg proteína total en un volumen final de 80 µL.
CDS
La reacción se llevó a cabo según Lykidis et al. (1997). El buffer de reacción contenía
Bis Tris-HCl (pH 6,5) 100 mM H3-CTP (actividad específica 14,5 Ci/mmol), PA 2 mM
(suspensión acuosa sonicada en buffer), MgCl2 10 mM. La reacción se comenzó con el
agregado de MgCl2. La reacción fue incubada a 37 °C durante los tiempos indicados.
PIS
La reacción se llevó a cabo según Lykidis et al. (1997). El buffer de reacción contenía
Tris-HCl (pH 8,0) 100 mM, MgCl2 50 mM, MnCl2 2 mM, CDP-DAG 1 µM sonicado en buffer y
3.3 mM myo-[H3]inositol (actividad específica 30 Ci/mmol). La reacción se desarrolló durante
tiempos variables a 37 °C.
PI4K
La reacción se llevó a cabo según Wong et al. (1997) con algunas modificaciones. El
buffer de reacción contenía Hepes (pH 7,5) 20 mM, MgCl2 20 mM, ATP 20 µM, PtdIns 70 µM
y PtdSer 35 µM sonicados en buffer y γ-32P-ATP 10 µCi (actividad específica 3000 Ci/mmol)
incubando la reacción a 25 °C durante tiempos variables.
PAP
La actividad PAP se midió según Pasquare et al. (2001) con algunas modificaciones. El
buffer de reacción contenía Tris-maleato (pH 6,5) 50 mM, L-α-dipalmitoil-C14-PA 0,75 µCi

Materiales y Métodos
88
(actividad específica 100-200 mCi/mmol) y dioleoil-PA 0,3 mM como dispersión en agua y
MgCl2 1 mM. La reacción se llevó a cabo a 37 °C durante tiempos variables.
Actividad de Lipin 1 en micelas mixtas de Tritón X-100/PA
La actividad PAP de Lipin 1 purificada fue medida según Han & Carman (2000)
mediante la determinación de la liberación de fosfato inorgánico proveniente del PA por la
acción enzimática en micelas mixtas preparadas como se indicó anteriormente. El buffer de
reacción contiene 50 mM Tris-HCl (pH 7,5), 0,5 mM MgCl2, 10 mM 2-mercaptoetanol, 1 mM
L-α-PA purificado de huevo de gallina, 10 mM Tritón X-10 y 50 ng enzima en un volumen
total de 0,1 mL. La mezcla de reacción fue incubada durante 20 minutos a 37 °C, momento en
que se terminó la reacción enzimática con el agregado de 0,5 mL HCl 0,1 M en metanol, 1 mL
de cloroformo y 1 mL de agua milliQ. Tras 3 segundos de agitación en vortex, se procedió a
separar las fases por centrifugación durante 2 minutos a 2,000 x g de los tubos de ensayo de
vidrio. Se tomaron 300 µL de la fase acuosa superior y se mezcló con 600 µL del reactivo de
verde de malaquita-molibdato, el reactivo pasa de amarillo a verde. Tras esperar 15 minutos,
se midió la absorbancia a 650 nm. Como blanco se utilizó 300 µL de agua milliQ, que no
muestra diferencias con la fase superior de un tubo en el que el incubado a 37 °C fue
omitido.
Transfecciones transitorias de ADN plasmídico
Los plásmidos utilizados se encuentran descriptos en la Tesis doctoral de Dr. Alfonso
Pecchio (2008). Para el caso de Lipin 1, se partió del plásmido cedido gentilmente por el Dr.
Carman y que fue utilizado en la publicación de Wang, H (2011). A partir de dicho plásmido,
se subclonó en el vector pSYFP2-N1 empleando los sitios Nhe y XhoI, dado que el vector de
origen (pEGFP-N1) y el de destino poseen los mismos sitios de clonado y marco de lectura.
La introducción de los diferentes plásmidos a las células para los experimentos de FRET
utilizadas se realizó mediante la utilización de lípidos catiónicos comerciales, Lipofectamina
2000. Las condiciones no difirieron significativamente del protocolo del fabricante, pero se

Materiales y Métodos
89
hicieron cambios considerables respecto a las cantidades de ADN utilizadas. Los diferentes
plásmidos usados necesitaron optimizarse individualmente para obtener niveles de
expresión aceptables. Para el caso de c-Fos y sus mutantes, se emplearon 0,8 µg de ADN por
pocillo en un multiwell de 24. En cambio, para las enzimas se utilizó la cantidad descripta en
la Tabla 3. Para las co-transfecciones, se adicionaron ambas cantidades de ADN en la mezcla
con Lipofectamina 2000.
Tabla 3. Cantidades de ADN plasmídico empleadas en la transfecciones transitorias para FRET
Enzima µg ADN/pocillo
CDS1 2
PIS1 1,4
PI4KIIα 1
PI4KIIβ 1
Lipin 1 1,6
Adquisición de imágenes confocales para FRET
La imágenes para los análisis de FRET fueron tomadas usando microscopios
confocales Olympus FV1000, Olympus FV300 o Zeiss Pascal equipados en todos los casos con
un láser de Argón multilínea (458, 488 y 514 nm). La fluorescencia de CFP (donor) fue
detectada utilizando excitación a 458 nm, y una ventana de detección de 465-495 nm. La
fluorescencia de YFP (aceptor) fue detectada utilizando excitación a 514 nm y una ventana de
detección de 530-560 nm. Las imágenes fueron adquiridas utilizando objetivos 60X apertura
numérica = 1,42 con un zoom digital de 3X, 512x512, ajustando el pinhole de forma de tomar
fetas ópticas de 0.8-1 µm. Las condiciones de adquisición se ajustaron para minimizar el
fotoblanqueo y tiempo de escaneo pero maximizando la relación señal/ruido en las distintas
condiciones. Una vez ajustadas las mismas, se procedió a tomar el conjunto de imágenes de
todo el experimento con las mismas condiciones en el mismo día.

Materiales y Métodos
90
FRET (Förster Resonance Energy Transfer) por Emisión Sensibilizada
Para el análisis de imágenes célula por célula realizada post-adquisición se utilizó el
programa de distribución libre ImageJ (http://rsbweb. nih.gov/ij/). Se utilizó el algoritmo
previamente descripto en Elangovan et al. (2003) para remover el sangrado espectral del
donor (SED) y la excitación directa del aceptor (EDA) que contaminan la señal de FRET. Con
ese propósito se usaron siete imágenes: dos imágenes de referencia marcadas sólo para el
donor y otras dos imágenes de referencia marcadas sólo para el aceptor (Dex/Dem y
Dex/Aem); y tres imágenes con doble marca proveniente de células co-transfectadas
(Aex/Aem, Dex/Dem, Dex/Aem). La imagen Dex/Dem muestra la fluorescencia apagada o
quenched del donor (qD) y corresponde al canal del donor (D), mientras que la imagen
Aex/Aem indica la fluorescencia del aceptor y corresponde al canal del aceptor (A). La
imagen Dex/Aem corresponde a la imagen de FRET no corregida (uFRET), que es procesada
segun el algoritmo de Elagonvan para generar la imagen de precisión de FRET (PFRET),
mostrando los niveles de transferencia de energía corregidos:
PFRET = uFRET - qD x SED – A x EDA (1)
La eficiencia aparente de FRET:
E = 1 – qD/uD (2)
se expresa como la fracción de fluorescencia del donor en la ausencia de FRET o unquenched
donor (uD). Este último parámetro se calcula como:
uD= qD +PRET (3)
como una aproximación de la Eq. B.19 en Elangovan et al. (2003). ImageJ fue usado para
calcular este parámetro. Luego de sustraer el ruido de fondo o background, se aplicó una
máscara independiente a las imágenes qD, uFRET y A para excluir del análisis pixeles

Materiales y Métodos
91
saturados o pixeles por debajo de un umbral determinado. Los valores de eficiencia de FRET
promedio fueron calculados desde regiones de interés de acuerdo a:
E = 1 −��
������(4)
en un análisis pixel por pixel. De esta forma, se puede lograr la separación de pixeles que
contienen señal significativa de aquellos que sólo representan mayoritariamente ruido de
fondo para el cálculo de FRET. La imágenes de E resultantes fueron pseudocoloreadas con
ImageJ en donde la intensidad del pixel se representa mediante una escala de color: blanco
es el valor máximo normalizado de pSYFP2-mTurquoise2 mientras que azul corresponde al
valor mínimo, 0.
FLIM (Fluorescence Lifetime Imaging Microscopy) en el dominio de la frecuencia
Un microscopio confocal Olympus FV-1000 equipado con un módulo de FastFlim
FlimBox (ISS) fue utilizado para los estudios de FLIM. FLIMBox es un hardware de frecuencia
digital capaz de análisis multi-armónico (Colyer et al. 2008). Un láser de diodo a 445 nm
modulado con un frecuencia de 20 MHz fue usado para la excitación de CFP (donor), con una
excitación cruzada muy baja de YFP (aceptor). La emisión de fluorescencia de CFP fue
colectado en una ventana espectral de 470-495 nm. Una solución de fluoresceína en
Cumarina 6 disuelta en etanol (pico de excitación y emisión: 460 y 505, respectivamente) que
tiene un tiempo de vida media de fluorescencia de 2,5 ns fue usada como estándar para
calibrar la respuesta instrumental. Se siguió el protocolo establecido en Sun et al. (2012).
Las imágenes fueron obtenidas en 256x256 con 20 µs/pixel y promediando 25-50
imágenes tomadas consecutivamente. La información de FLIMBox conteniendo la
información de intensidad y de tiempo de vida fueron adquiridas y procesadas utilizando el
programa Sim-FCS, desarrollado en el Laboratory for Fluorescence Dynamics. Se utilizó la
estrategia del fasor, una representación de vectores (Clayton et al. 2004, Digman et al. 2008),
para cuantificar el tiempo de vida del donor (Verveer et al. 2000, Caiolfa et al. 2007). El fasor

Materiales y Métodos
92
representa el decaimiento de fluorescencia en un espacio transformado de Fourier. La fase y
la modulación son medidas en cada pixel de una imagen y son usados para determinar las
coordinadas del fasor en ese pixel, para así establecer el tiempo de vida de fluorescencia del
donor. El fasor es entonces un vector, cuyas coordinadas polares son s y g, relacionadas
directamente con la fase y modulación, los parámetros físicos que se miden directamente. Es
importante destacar que la estrategia del fasor no requiere de un modelo de ajuste, sino que
expresa el decaimiento de fluorescencia en términos de las coordinadas polares. En los
experimentos utilizamos la componente del segundo armónico, que corresponde a 40 MHz.
El fasor asociado con una célula obtenida a través de este sistema se determinó
como un promedio de la imagen celular. El fasor de autofluorescencia fue determinado
tomando las imágenes de FLIM de células no transfectadas. Se determinó el fasor del donor
en ausencia y presencia del aceptor. Para cada experimento, se colectaron múltiples
imágenes del donor en ausencia del aceptor para tomar un valor fasor promedio, que fue
asignada a la posición del donor en ausencia del aceptor. El FLIM puede de esta forma medir
el acortamiento en el tiempo de vida media que resulta de FRET (Wouters & Bastiaens 1999).
La cuantificación de FRET en el gráfico del fasor se alcanza al medir el corrimiento del fasor
entre las dos especies: el donor en ausencia y en presencia del aceptor. Según se describe en
Digman et al. (2008) a partir de los fasores de autofluorescencia y del donor en ausencia del
aceptor se calcula la trayectoria de eficiencia variable de FRET (0-100%). Esto permite
determinar la eficiencia de FRET del fasor promedio de cada célula de donor en presencia del
aceptor.

BIBLIOGRAFÍA

Referencias bibliográficas
94
1. Abate, C., D. Luk, and T. Curran, Transcriptional regulation by Fos and Jun in vitro:
interaction among multiple activator and regulatory domains. Mol Cell Biol, 1991. 11(7): p. 3624-32.
2. Aboukhatwa, M.A. and A.S. Undieh, Antidepressant stimulation of CDP-diacylglycerol
synthesis does not require monoamine reuptake inhibition. BMC Neurosci, 2010. 11: p. 10.
3. Adler, J., N. Reuven, C. Kahana, and Y. Shaul, c-Fos proteasomal degradation is
activated by a default mechanism, and its regulation by NAD(P)H:quinone
oxidoreductase 1 determines c-Fos serum response kinetics. Mol Cell Biol, 2010. 30(15): p. 3767-78.
4. Alfonso Pecchio, A.R., A.M. Cardozo Gizzi, M.L. Renner, M. Molina-Calavita, and B.L. Caputto, c-Fos activates and physically interacts with specific enzymes of the pathway
of synthesis of polyphosphoinositides. Mol Biol Cell, 2011. 22(24): p. 4716-25. 5. Angel, P., et al., 12-O-tetradecanoyl-phorbol-13-acetate induction of the human
collagenase gene is mediated by an inducible enhancer element located in the 5'-
flanking region. Mol Cell Biol, 1987. 7(6): p. 2256-66. 6. Angel, P. and M. Karin, The role of Jun, Fos and the AP-1 complex in cell-proliferation
and transformation. Biochim Biophys Acta, 1991. 1072(2-3): p. 129-57. 7. Athenstaedt, K. and G. Daum, Phosphatidic acid, a key intermediate in lipid
metabolism. Eur J Biochem, 1999. 266(1): p. 1-16. 8. Balla, A. and T. Balla, Phosphatidylinositol 4-kinases: old enzymes with emerging
functions. Trends Cell Biol, 2006. 16(7): p. 351-61. 9. Balla, A., et al., Maintenance of hormone-sensitive phosphoinositide pools in the
plasma membrane requires phosphatidylinositol 4-kinase IIIalpha. Mol Biol Cell, 2008. 19(2): p. 711-21.
10. Balla, A., G. Tuymetova, M. Barshishat, M. Geiszt, and T. Balla, Characterization of
type II phosphatidylinositol 4-kinase isoforms reveals association of the enzymes with
endosomal vesicular compartments. J Biol Chem, 2002. 277(22): p. 20041-50. 11. Balla, A., G. Tuymetova, A. Tsiomenko, P. Varnai, and T. Balla, A plasma membrane
pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase
type-III alpha: studies with the PH domains of the oxysterol binding protein and
FAPP1. Mol Biol Cell, 2005. 16(3): p. 1282-95. 12. Balla, T., Phosphatidylinositol 4-kinases. Biochim Biophys Acta, 1998. 1436(1-2): p. 69-
85. 13. Balla, T., Z. Szentpetery, and Y.J. Kim, Phosphoinositide signaling: new tools and
insights. Physiology (Bethesda), 2009. 24: p. 231-44. 14. Bao, S., et al., Effects of stable suppression of Group VIA phospholipase A2 expression
on phospholipid content and composition, insulin secretion, and proliferation of INS-1
insulinoma cells. J Biol Chem, 2006. 281(1): p. 187-98. 15. Basbous, J., I. Jariel-Encontre, T. Gomard, G. Bossis, and M. Piechaczyk, Ubiquitin-
independent- versus ubiquitin-dependent proteasomal degradation of the c-Fos and
Fra-1 transcription factors: is there a unique answer? Biochimie, 2008. 90(2): p. 296-305.

Referencias bibliográficas
95
16. Belguise, K., N. Kersual, F. Galtier, and D. Chalbos, FRA-1 expression level regulates
proliferation and invasiveness of breast cancer cells. Oncogene, 2005. 24(8): p. 1434-44.
17. Bell, R.M., L.M. Ballas, and R.A. Coleman, Lipid topogenesis. J Lipid Res, 1981. 22(3): p. 391-403.
18. Bonifacino, J.S. and E.C. Dell'Angelica, Immunoprecipitation. Curr Protoc Cell Biol, 2001. Chapter 7: p. Unit 7 2.
19. Borioli, G.A., Immediate early proto-oncoproteins and membranes: not just an
innocent liaison. Curr Protein Pept Sci, 2011. 12(8): p. 685-90. 20. Borioli, G.A., B.L. Caputto, and B. Maggio, c-Fos is surface active and interacts
differentially with phospholipid monolayers. Biochem Biophys Res Commun, 2001. 280(1): p. 9-13.
21. Borioli, G.A., M.L. Fanani, B.L. Caputto, and B. Maggio, c-Fos is a surface pressure-
dependent diverter of phospholipase activity. Biochem Biophys Res Commun, 2002. 295(4): p. 964-9.
22. Boronenkov, I.V., J.C. Loijens, M. Umeda, and R.A. Anderson, Phosphoinositide
signaling pathways in nuclei are associated with nuclear speckles containing pre-
mRNA processing factors. Mol Biol Cell, 1998. 9(12): p. 3547-60. 23. Bradford, M.M., A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 1976. 72: p. 248-54.
24. Brindley, D.N., Intracellular translocation of phosphatidate phosphohydrolase and its
possible role in the control of glycerolipid synthesis. Prog Lipid Res, 1984. 23(3): p. 115-33.
25. Brose, N., A. Betz, and H. Wegmeyer, Divergent and convergent signaling by the
diacylglycerol second messenger pathway in mammals. Curr Opin Neurobiol, 2004. 14(3): p. 328-40.
26. Brown, J.R., et al., Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol, 1998. 18(9): p. 5609-19.
27. Bruns, J.R., M.A. Ellis, A. Jeromin, and O.A. Weisz, Multiple roles for
phosphatidylinositol 4-kinase in biosynthetic transport in polarized Madin-Darby
canine kidney cells. J Biol Chem, 2002. 277(3): p. 2012-8. 28. Bussolino, D.F., et al., Light affects c-fos expression and phospholipid synthesis in both
retinal ganglion cells and photoreceptor cells in an opposite way for each cell type. Brain Res Mol Brain Res, 1998. 58(1-2): p. 10-5.
29. Bussolino, D.F., et al., c-Fos associates with the endoplasmic reticulum and activates
phospholipid metabolism. FASEB J, 2001. 15(3): p. 556-8. 30. Caiolfa, V.R., et al., Monomer dimer dynamics and distribution of GPI-anchored uPAR
are determined by cell surface protein assemblies. J Cell Biol, 2007. 179(5): p. 1067-82. 31. Cammer, W., F.A. Tansey, and C.F. Brosnan, Gliosis in the spinal cords of rats with
experimental allergic encephalomyelitis: immunostaining of carbonic anhydrase and
vimentin in reactive astrocytes. Glia, 1989. 2(4): p. 223-30.

Referencias bibliográficas
96
32. Campbell, K.M., A.R. Terrell, P.J. Laybourn, and K.J. Lumb, Intrinsic structural disorder
of the C-terminal activation domain from the bZIP transcription factor Fos. Biochemistry, 2000. 39(10): p. 2708-13.
33. Caputto, B.L., A.M. Cardozo Gizzi, and G.A. Gil, c-Fos: an AP-1 transcription factor with
an additional cytoplasmic, non-genomic lipid synthesis activation capacity. Biochim Biophys Acta, 2014. 1841(9): p. 1241-6.
34. Caputto, B.L. and M.E. Guido, Immediate early gene expression within the visual
system: light and circadian regulation in the retina and the suprachiasmatic nucleus. Neurochem Res, 2000. 25(1): p. 153-62.
35. Cardozo Gizzi, A.M. and B.L. Caputto, Mechanistic insights into the nongenomic
regulation of phospholipid synthesizing enzymes. IUBMB Life, 2013. 65(7): p. 584-92. 36. Carman, G.M., R.A. Deems, and E.A. Dennis, Lipid signaling enzymes and surface
dilution kinetics. J Biol Chem, 1995. 270(32): p. 18711-4. 37. Carman, G.M. and G.S. Han, Phosphatidic acid phosphatase, a key enzyme in the
regulation of lipid synthesis. J Biol Chem, 2009. 284(5): p. 2593-7. 38. Carrasco, S. and I. Merida, Diacylglycerol, when simplicity becomes complex. Trends
Biochem Sci, 2007. 32(1): p. 27-36. 39. Catz, S.D. and N.B. Sterin-Speziale, Bradykinin stimulates phosphoinositide turnover
and phospholipase C but not phospholipase D and NADPH oxidase in human
neutrophils. J Leukoc Biol, 1996. 59(4): p. 591-7. 40. Clayton, A.H., Q.S. Hanley, and P.J. Verveer, Graphical representation and
multicomponent analysis of single-frequency fluorescence lifetime imaging
microscopy data. J Microsc, 2004. 213(Pt 1): p. 1-5. 41. Clayton, E.L., S. Minogue, and M.G. Waugh, Mammalian phosphatidylinositol 4-
kinases as modulators of membrane trafficking and lipid signaling networks. Prog Lipid Res, 2013. 52(3): p. 294-304.
42. Cohen, D.R. and T. Curran, The structure and function of the fos proto-oncogene. Crit Rev Oncog, 1989. 1(1): p. 65-88.
43. Coleman, R.A. and D.P. Lee, Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res, 2004. 43(2): p. 134-76.
44. Colyer, R.A., C. Lee, and E. Gratton, A novel fluorescence lifetime imaging system that
optimizes photon efficiency. Microsc Res Tech, 2008. 71(3): p. 201-13. 45. Craige, B., G. Salazar, and V. Faundez, Phosphatidylinositol-4-kinase type II alpha
contains an AP-3-sorting motif and a kinase domain that are both required for
endosome traffic. Mol Biol Cell, 2008. 19(4): p. 1415-26. 46. Crespo, P.M., et al., c-Fos activates glucosylceramide synthase and glycolipid synthesis
in PC12 cells. J Biol Chem, 2008. 283(45): p. 31163-71. 47. Csaki, L.S. and K. Reue, Lipins: multifunctional lipid metabolism proteins. Annu Rev
Nutr, 2010. 30: p. 257-72. 48. Curran, T. and B.R. Franza, Jr., Fos and Jun: the AP-1 connection. Cell, 1988. 55(3): p.
395-7. 49. Curran, T. and J.I. Morgan, Memories of fos. Bioessays, 1987. 7(6): p. 255-8.

Referencias bibliográficas
97
50. Czech, M.P., Dynamics of phosphoinositides in membrane retrieval and insertion. Annu Rev Physiol, 2003. 65: p. 791-815.
51. Chiu, R., et al., The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of
AP-1 responsive genes. Cell, 1988. 54(4): p. 541-52. 52. D'Souza, K. and R.M. Epand, Enrichment of phosphatidylinositols with specific acyl
chains. Biochim Biophys Acta, 2014. 1838(6): p. 1501-8. 53. D'Souza, K., Y.J. Kim, T. Balla, and R.M. Epand, Distinct properties of the two isoforms
of CDP-diacylglycerol synthase. Biochemistry, 2014. 53(47): p. 7358-67. 54. de Arriba Zerpa, G.A., et al., Light exposure activates retina ganglion cell
lysophosphatidic acid acyl transferase and phosphatidic acid phosphatase by a c-fos-
dependent mechanism. J Neurochem, 1999. 73(3): p. 1228-35. 55. de Graaf, P., et al., Nuclear localization of phosphatidylinositol 4-kinase beta. J Cell Sci,
2002. 115(Pt 8): p. 1769-75. 56. Di Paolo, G. and P. De Camilli, Phosphoinositides in cell regulation and membrane
dynamics. Nature, 2006. 443(7112): p. 651-7. 57. Digman, M.A., V.R. Caiolfa, M. Zamai, and E. Gratton, The phasor approach to
fluorescence lifetime imaging analysis. Biophys J, 2008. 94(2): p. L14-6. 58. Dosztanyi, Z., V. Csizmok, P. Tompa, and I. Simon, IUPred: web server for the
prediction of intrinsically unstructured regions of proteins based on estimated energy
content. Bioinformatics, 2005. 21(16): p. 3433-4. 59. Downward, J., PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol, 2004. 15(2): p.
177-82. 60. Dyson, H.J. and P.E. Wright, Intrinsically unstructured proteins and their functions. Nat
Rev Mol Cell Biol, 2005. 6(3): p. 197-208. 61. Eaton, J.M., G.R. Mullins, D.N. Brindley, and T.E. Harris, Phosphorylation of lipin 1 and
charge on the phosphatidic acid head group control its phosphatidic acid phosphatase
activity and membrane association. J Biol Chem, 2013. 288(14): p. 9933-45. 62. Edwards, S.A., A.Y. Rundell, and E.D. Adamson, Expression of c-fos antisense RNA
inhibits the differentiation of F9 cells to parietal endoderm. Dev Biol, 1988. 129(1): p. 91-102.
63. Elangovan, M., et al., Characterization of one- and two-photon excitation fluorescence
resonance energy transfer microscopy. Methods, 2003. 29(1): p. 58-73. 64. English, A.R. and G.K. Voeltz, Rab10 GTPase regulates ER dynamics and morphology.
Nat Cell Biol, 2013. 15(2): p. 169-78. 65. Fagone, P. and S. Jackowski, Membrane phospholipid synthesis and endoplasmic
reticulum function. J Lipid Res, 2009. 50 Suppl: p. S311-6. 66. Fagone, P., et al., Phospholipid biosynthesis program underlying membrane expansion
during B-lymphocyte differentiation. J Biol Chem, 2007. 282(10): p. 7591-605. 67. Fang, X., et al., Lysophosphatidic acid is a bioactive mediator in ovarian cancer.
Biochim Biophys Acta, 2002. 1582(1-3): p. 257-64. 68. Ferrero, G.O., M.L. Renner, G.A. Gil, L. Rodriguez-Berdini, and B.L. Caputto, c-Fos-
activated synthesis of nuclear phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P(2)]
promotes global transcriptional changes. Biochem J, 2014. 461(3): p. 521-30.

Referencias bibliográficas
98
69. Ferrero, G.O., F.N. Velazquez, and B.L. Caputto, The kinase c-Src and the phosphatase
TC45 coordinately regulate c-Fos tyrosine phosphorylation and c-Fos phospholipid
synthesis activation capacity. Oncogene, 2012. 31(28): p. 3381-91. 70. Finck, B.N., et al., Lipin 1 is an inducible amplifier of the hepatic PGC-
1alpha/PPARalpha regulatory pathway. Cell Metab, 2006. 4(3): p. 199-210. 71. Folch, J., M. Lees, and G.H. Sloane Stanley, A simple method for the isolation and
purification of total lipides from animal tissues. J Biol Chem, 1957. 226(1): p. 497-509. 72. Franke, T.F., D.R. Kaplan, L.C. Cantley, and A. Toker, Direct regulation of the Akt proto-
oncogene product by phosphatidylinositol-3,4-bisphosphate. Science, 1997. 275(5300): p. 665-8.
73. Fruman, D.A., R.E. Meyers, and L.C. Cantley, Phosphoinositide kinases. Annu Rev Biochem, 1998. 67: p. 481-507.
74. Futerman, A.H. and R.E. Pagano, Determination of the intracellular sites and topology
of glucosylceramide synthesis in rat liver. Biochem J, 1991. 280 ( Pt 2): p. 295-302. 75. Futerman, A.H. and H. Riezman, The ins and outs of sphingolipid synthesis. Trends Cell
Biol, 2005. 15(6): p. 312-8. 76. Gaggiotti, M.C., M. Del Boca, G. Castro, B.L. Caputto, and G.A. Borioli, The immediate-
early oncoproteins Fra-1, c-Fos, and c-Jun have distinguishable surface behavior and
interactions with phospholipids. Biopolymers, 2009. 91(9): p. 710-8. 77. Gil, G.A., et al., c-Fos activated phospholipid synthesis is required for neurite
elongation in differentiating PC12 cells. Mol Biol Cell, 2004. 15(4): p. 1881-94. 78. Gil, G.A., D.C. Silvestre, N. Tomasini, D.F. Bussolino, and B.L. Caputto, Controlling
cytoplasmic c-Fos controls tumor growth in the peripheral and central nervous system. Neurochem Res, 2012. 37(6): p. 1364-71.
79. Gilmore, A.P. and K. Burridge, Regulation of vinculin binding to talin and actin by
phosphatidyl-inositol-4-5-bisphosphate. Nature, 1996. 381(6582): p. 531-5. 80. Ginty, D.D., A. Bonni, and M.E. Greenberg, Nerve growth factor activates a Ras-
dependent protein kinase that stimulates c-fos transcription via phosphorylation of
CREB. Cell, 1994. 77(5): p. 713-25. 81. Giral, H., et al., NHE3 regulatory factor 1 (NHERF1) modulates intestinal sodium-
dependent phosphate transporter (NaPi-2b) expression in apical microvilli. J Biol Chem, 2012. 287(42): p. 35047-56.
82. Glover, J.N. and S.C. Harrison, Crystal structure of the heterodimeric bZIP transcription
factor c-Fos-c-Jun bound to DNA. Nature, 1995. 373(6511): p. 257-61. 83. Glynn, P., Neuropathy target esterase and phospholipid deacylation. Biochim Biophys
Acta, 2005. 1736(2): p. 87-93. 84. Godi, A., et al., FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF
and PtdIns(4)P. Nat Cell Biol, 2004. 6(5): p. 393-404. 85. Godi, A., et al., ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates
synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol, 1999. 1(5): p. 280-7. 86. Goedhart, J., et al., Structure-guided evolution of cyan fluorescent proteins towards a
quantum yield of 93%. Nat Commun, 2012. 3: p. 751.

Referencias bibliográficas
99
87. Greenberg, M.E. and E.B. Ziff, Stimulation of 3T3 cells induces transcription of the c-
fos proto-oncogene. Nature, 1984. 311(5985): p. 433-8. 88. Grimsey, N., et al., Temporal and spatial regulation of the phosphatidate
phosphatases lipin 1 and 2. J Biol Chem, 2008. 283(43): p. 29166-74. 89. Guido, M.E., G.A. de Arriba Zerpa, D.F. Bussolino, and B.L. Caputto, Immediate early
gene c-fos regulates the synthesis of phospholipids but not of gangliosides. J Neurosci Res, 1996. 43(1): p. 93-8.
90. Guo, J., et al., Phosphatidylinositol 4-kinase type IIalpha is responsible for the
phosphatidylinositol 4-kinase activity associated with synaptic vesicles. Proc Natl Acad Sci U S A, 2003. 100(7): p. 3995-4000.
91. Halazonetis, T.D., K. Georgopoulos, M.E. Greenberg, and P. Leder, c-Jun dimerizes
with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell, 1988. 55(5): p. 917-24.
92. Han, G.S. and G.M. Carman, Characterization of the human LPIN1-encoded
phosphatidate phosphatase isoforms. J Biol Chem, 2010. 285(19): p. 14628-38. 93. Han, S., et al., Nuclear envelope phosphatase 1-regulatory subunit 1 (formerly
TMEM188) is the metazoan Spo7p ortholog and functions in the lipin activation
pathway. J Biol Chem, 2012. 287(5): p. 3123-37. 94. Hanahan, D. and R.A. Weinberg, Hallmarks of cancer: the next generation. Cell, 2011.
144(5): p. 646-74. 95. Harris, T.E. and B.N. Finck, Dual function lipin proteins and glycerolipid metabolism.
Trends in Endocrinology and Metabolism, 2011. 22(6): p. 226-33. 96. Harris, T.E. and B.N. Finck, Dual function lipin proteins and glycerolipid metabolism.
Trends Endocrinol Metab, 2011. 22(6): p. 226-33. 97. Harris, T.E., et al., Insulin controls subcellular localization and multisite
phosphorylation of the phosphatidic acid phosphatase, lipin 1. J Biol Chem, 2007. 282(1): p. 277-86.
98. Hazy, E. and P. Tompa, Limitations of induced folding in molecular recognition by
intrinsically disordered proteins. Chemphyschem, 2009. 10(9-10): p. 1415-9. 99. He, B., et al., Predicting intrinsic disorder in proteins: an overview. Cell Res, 2009.
19(8): p. 929-49. 100. Heacock, A.M. and B.W. Agranoff, CDP-diacylglycerol synthase from mammalian
tissues. Biochim Biophys Acta, 1997. 1348(1-2): p. 166-72. 101. Hermansson, M., K. Hokynar, and P. Somerharju, Mechanisms of glycerophospholipid
homeostasis in mammalian cells. Prog Lipid Res, 2011. 50(3): p. 240-57. 102. Hess, J., P. Angel, and M. Schorpp-Kistner, AP-1 subunits: quarrel and harmony among
siblings. J Cell Sci, 2004. 117(Pt 25): p. 5965-73. 103. Higuchi, R., B. Krummel, and R.K. Saiki, A general method of in vitro preparation and
specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res, 1988. 16(15): p. 7351-67.
104. Holub, B.J. and A. Kuksis, Differential distribution of orthophosphate- 32 P and
glycerol- 14 C among molecular species of phosphatidylinositols of rat liver in vivo. J Lipid Res, 1971. 12(6): p. 699-705.

Referencias bibliográficas
100
105. Hsu, W.L., et al., Exploring the binding diversity of intrinsically disordered proteins
involved in one-to-many binding. Protein Sci, 2013. 22(3): p. 258-73. 106. Huang, C. and C. Freter, Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci,
2015. 16(1): p. 924-49. 107. Huffman, T.A., I. Mothe-Satney, and J.C. Lawrence, Jr., Insulin-stimulated
phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc Natl Acad Sci U S A, 2002. 99(2): p. 1047-52.
108. Hughes, P. and M. Dragunow, Induction of immediate-early genes and the control of
neurotransmitter-regulated gene expression within the nervous system. Pharmacol Rev, 1995. 47(1): p. 133-78.
109. Hunt, S.P., A. Pini, and G. Evan, Induction of c-fos-like protein in spinal cord neurons
following sensory stimulation. Nature, 1987. 328(6131): p. 632-4. 110. Hunter, T., Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol,
2009. 21(2): p. 140-6. 111. Iakoucheva, L.M., C.J. Brown, J.D. Lawson, Z. Obradovic, and A.K. Dunker, Intrinsic
disorder in cell-signaling and cancer-associated proteins. J Mol Biol, 2002. 323(3): p. 573-84.
112. Iakoucheva, L.M., et al., The importance of intrinsic disorder for protein
phosphorylation. Nucleic Acids Res, 2004. 32(3): p. 1037-49. 113. Inglis-Broadgate, S.L., et al., Isolation and characterization of murine Cds (CDP-
diacylglycerol synthase) 1 and 2. Gene, 2005. 356: p. 19-31. 114. Jackowski, S., Coordination of membrane phospholipid synthesis with the cell cycle. J
Biol Chem, 1994. 269(5): p. 3858-67. 115. Janmey, P.A., Phosphoinositides and calcium as regulators of cellular actin assembly
and disassembly. Annu Rev Physiol, 1994. 56: p. 169-91. 116. Jeffery, C.J., Moonlighting proteins--an update. Mol Biosyst, 2009. 5(4): p. 345-50. 117. Jung, G., et al., Molecular determinants of activation and membrane targeting of
phosphoinositol 4-kinase IIbeta. Biochem J, 2008. 409(2): p. 501-9. 118. Kerr, L.D., D.B. Miller, and L.M. Matrisian, TGF-beta 1 inhibition of transin/stromelysin
gene expression is mediated through a Fos binding sequence. Cell, 1990. 61(2): p. 267-78.
119. Kim, Y.J., M.L. Guzman-Hernandez, and T. Balla, A highly dynamic ER-derived
phosphatidylinositol-synthesizing organelle supplies phosphoinositides to cellular
membranes. Dev Cell, 2011. 21(5): p. 813-24. 120. Kohyama-Koganeya, A., et al., Drosophila glucosylceramide synthase: a negative
regulator of cell death mediated by proapoptotic factors. J Biol Chem, 2004. 279(34): p. 35995-6002.
121. Kovary, K. and R. Bravo, Existence of different Fos/Jun complexes during the G0-to-G1
transition and during exponential growth in mouse fibroblasts: differential role of Fos
proteins. Mol Cell Biol, 1992. 12(11): p. 5015-23. 122. Kovary, K. and R. Bravo, The jun and fos protein families are both required for cell
cycle progression in fibroblasts. Mol Cell Biol, 1991. 11(9): p. 4466-72.

Referencias bibliográficas
101
123. Kremers, G.J., J. Goedhart, E.B. van Munster, and T.W. Gadella, Jr., Cyan and yellow
super fluorescent proteins with improved brightness, protein folding, and FRET Forster
radius. Biochemistry, 2006. 45(21): p. 6570-80. 124. Kyte, J. and R.F. Doolittle, A simple method for displaying the hydropathic character of
a protein. J Mol Biol, 1982. 157(1): p. 105-32. 125. Ledwith, B.J., S. Manam, A.R. Kraynak, W.W. Nichols, and M.O. Bradley, Antisense-fos
RNA causes partial reversion of the transformed phenotypes induced by the c-Ha-ras
oncogene. Mol Cell Biol, 1990. 10(4): p. 1545-55. 126. Lee, W., P. Mitchell, and R. Tjian, Purified transcription factor AP-1 interacts with TPA-
inducible enhancer elements. Cell, 1987. 49(6): p. 741-52. 127. Leppa, S., M. Eriksson, R. Saffrich, W. Ansorge, and D. Bohmann, Complex functions of
AP-1 transcription factors in differentiation and survival of PC12 cells. Mol Cell Biol, 2001. 21(13): p. 4369-78.
128. Lev, S., Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat Rev Mol Cell Biol, 2010. 11(10): p. 739-50.
129. Lichtenberg, D., R.J. Robson, and E.A. Dennis, Solubilization of phospholipids by
detergents. Structural and kinetic aspects. Biochim Biophys Acta, 1983. 737(2): p. 285-304.
130. Lin, Y.P. and G.M. Carman, Kinetic analysis of yeast phosphatidate phosphatase
toward Triton X-100/phosphatidate mixed micelles. J Biol Chem, 1990. 265(1): p. 166-70.
131. Liu, G.H. and L. Gerace, Sumoylation regulates nuclear localization of lipin-1alpha in
neuronal cells. PLoS One, 2009. 4(9): p. e7031. 132. Liu, X., Y. Yin, J. Wu, and Z. Liu, Structure and mechanism of an intramembrane
liponucleotide synthetase central for phospholipid biosynthesis. Nat Commun, 2014. 5: p. 4244.
133. Liu, Y., W. Wang, G. Shui, and X. Huang, CDP-diacylglycerol synthetase coordinates cell
growth and fat storage through phosphatidylinositol metabolism and the insulin
pathway. PLoS Genet, 2014. 10(3): p. e1004172. 134. Liu, Z. and Y. Huang, Advantages of proteins being disordered. Protein Sci, 2014. 23(5):
p. 539-50. 135. Logullo, A.F., et al., Role of Fos-related antigen 1 in the progression and prognosis of
ductal breast carcinoma. Histopathology, 2011. 58(4): p. 617-25. 136. Lung, M., et al., Diacylglycerol kinase epsilon is selective for both acyl chains of
phosphatidic acid or diacylglycerol. J Biol Chem, 2009. 284(45): p. 31062-73. 137. Luthra, M.G. and A. Sheltawy, The metabolic turnover of molecular species of
phosphatidylinositol and its precursor phosphatidic acid in guinea-pig cerebral
hemispheres. J Neurochem, 1976. 27(6): p. 1501-11. 138. Lykidis, A., P.D. Jackson, C.O. Rock, and S. Jackowski, The role of CDP-diacylglycerol
synthetase and phosphatidylinositol synthase activity levels in the regulation of
cellular phosphatidylinositol content. J Biol Chem, 1997. 272(52): p. 33402-9.

Referencias bibliográficas
102
139. Maggiolini, M., et al., The G protein-coupled receptor GPR30 mediates c-fos up-
regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J Biol Chem, 2004. 279(26): p. 27008-16.
140. Mahuren, J.D., S.P. Coburn, A. Slominski, and J. Wortsman, Microassay of phosphate
provides a general method for measuring the activity of phosphatases using
physiological, nonchromogenic substrates such as lysophosphatidic acid. Anal Biochem, 2001. 298(2): p. 241-5.
141. Malnou, C.E., et al., Heterodimerization with Jun family members regulates c-Fos
nucleocytoplasmic traffic. J Biol Chem, 2007. 282(42): p. 31046-59. 142. Manguikian, A.D. and S.E. Barbour, Cell cycle dependence of group VIA calcium-
independent phospholipase A2 activity. J Biol Chem, 2004. 279(51): p. 52881-92. 143. Mellman, D.L., et al., A PtdIns4,5P2-regulated nuclear poly(A) polymerase controls
expression of select mRNAs. Nature, 2008. 451(7181): p. 1013-7. 144. Mercola, D., A. Rundell, J. Westwick, and S.A. Edwards, Antisense RNA to the c-fos
gene: restoration of density-dependent growth arrest in a transformed cell line. Biochem Biophys Res Commun, 1987. 147(1): p. 288-94.
145. Minogue, S., et al., Cloning of a human type II phosphatidylinositol 4-kinase reveals a
novel lipid kinase family. J Biol Chem, 2001. 276(20): p. 16635-40. 146. Minogue, S., et al., Phosphatidylinositol 4-kinase is required for endosomal trafficking
and degradation of the EGF receptor. J Cell Sci, 2006. 119(Pt 3): p. 571-81. 147. Motrich, R.D., G.M. Castro, and B.L. Caputto, Old players with a newly defined
function: Fra-1 and c-Fos support growth of human malignant breast tumors by
activating membrane biogenesis at the cytoplasm. PLoS One, 2013. 8(1): p. e53211. 148. Murphy, L.O. and J. Blenis, MAPK signal specificity: the right place at the right time.
Trends Biochem Sci, 2006. 31(5): p. 268-75. 149. Murphy, L.O., S. Smith, R.H. Chen, D.C. Fingar, and J. Blenis, Molecular interpretation
of ERK signal duration by immediate early gene products. Nat Cell Biol, 2002. 4(8): p. 556-64.
150. Murphy, T.R., et al., Phosphatidylinositol synthase is required for lens structural
integrity and photoreceptor cell survival in the zebrafish eye. Exp Eye Res, 2011. 93(4): p. 460-74.
151. Nakagawa, T., K. Goto, and H. Kondo, Cloning and characterization of a 92 kDa soluble
phosphatidylinositol 4-kinase. Biochem J, 1996. 320 ( Pt 2): p. 643-9. 152. Nakagawa, T., K. Goto, and H. Kondo, Cloning, expression, and localization of 230-kDa
phosphatidylinositol 4-kinase. J Biol Chem, 1996. 271(20): p. 12088-94. 153. Neuberg, M., M. Schuermann, J.B. Hunter, and R. Muller, Two functionally different
regions in Fos are required for the sequence-specific DNA interaction of the Fos/Jun
protein complex. Nature, 1989. 338(6216): p. 589-90. 154. Noh, D.Y., S.H. Shin, and S.G. Rhee, Phosphoinositide-specific phospholipase C and
mitogenic signaling. Biochim Biophys Acta, 1995. 1242(2): p. 99-113. 155. Nohturfft, A. and S.C. Zhang, Coordination of lipid metabolism in membrane
biogenesis. Annu Rev Cell Dev Biol, 2009. 25: p. 539-66.

Referencias bibliográficas
103
156. Oude Weernink, P.A., L. Han, K.H. Jakobs, and M. Schmidt, Dynamic phospholipid
signaling by G protein-coupled receptors. Biochim Biophys Acta, 2007. 1768(4): p. 888-900.
157. Pan, W., et al., CDP-diacylglycerol synthetase-controlled phosphoinositide availability
limits VEGFA signaling and vascular morphogenesis. Blood, 2012. 120(2): p. 489-98. 158. Pascual, F. and G.M. Carman, Phosphatidate phosphatase, a key regulator of lipid
homeostasis. Biochim Biophys Acta, 2013. 1831(3): p. 514-22. 159. Pasquare, S.J., M.G. Ilincheta de Boschero, and N.M. Giusto, Aging promotes a
different phosphatidic acid utilization in cytosolic and microsomal fractions from brain
and liver. Exp Gerontol, 2001. 36(8): p. 1387-401. 160. Pasquare, S.J., G.A. Salvador, and N.M. Giusto, Phospholipase D and phosphatidate
phosphohydrolase activities in rat cerebellum during aging. Lipids, 2004. 39(6): p. 553-60.
161. Patel, L., C. Abate, and T. Curran, Altered protein conformation on DNA binding by Fos
and Jun. Nature, 1990. 347(6293): p. 572-5. 162. Peterfy, M., T.E. Harris, N. Fujita, and K. Reue, Insulin-stimulated interaction with 14-
3-3 promotes cytoplasmic localization of lipin-1 in adipocytes. J Biol Chem, 2010. 285(6): p. 3857-64.
163. Peterfy, M., J. Phan, and K. Reue, Alternatively spliced lipin isoforms exhibit distinct
expression pattern, subcellular localization, and role in adipogenesis. J Biol Chem, 2005. 280(38): p. 32883-9.
164. Piston, D.W. and G.J. Kremers, Fluorescent protein FRET: the good, the bad and the
ugly. Trends Biochem Sci, 2007. 32(9): p. 407-14. 165. Portal, M.M., G.O. Ferrero, and B.L. Caputto, N-Terminal c-Fos tyrosine
phosphorylation regulates c-Fos/ER association and c-Fos-dependent phospholipid
synthesis activation. Oncogene, 2007. 26(24): p. 3551-8. 166. Prilusky, J., et al., FoldIndex: a simple tool to predict whether a given protein sequence
is intrinsically unfolded. Bioinformatics, 2005. 21(16): p. 3435-8. 167. Ren, H., et al., A phosphatidic acid binding/nuclear localization motif determines lipin1
function in lipid metabolism and adipogenesis. Mol Biol Cell, 2010. 21(18): p. 3171-81. 168. Robbins, P.D., J.M. Horowitz, and R.C. Mulligan, Negative regulation of human c-fos
expression by the retinoblastoma gene product. Nature, 1990. 346(6285): p. 668-71. 169. Romero, P., et al., Sequence complexity of disordered protein. Proteins, 2001. 42(1): p.
38-48. 170. Saito, S., K. Goto, A. Tonosaki, and H. Kondo, Gene cloning and characterization of
CDP-diacylglycerol synthase from rat brain. J Biol Chem, 1997. 272(14): p. 9503-9. 171. Salazar, G., et al., Phosphatidylinositol-4-kinase type II alpha is a component of
adaptor protein-3-derived vesicles. Mol Biol Cell, 2005. 16(8): p. 3692-704. 172. Sasaki, T., et al., Mammalian phosphoinositide kinases and phosphatases. Prog Lipid
Res, 2009. 48(6): p. 307-43. 173. Shaulian, E. and M. Karin, AP-1 in cell proliferation and survival. Oncogene, 2001.
20(19): p. 2390-400.

Referencias bibliográficas
104
174. Shulga, Y.V., R.A. Anderson, M.K. Topham, and R.M. Epand, Phosphatidylinositol-4-
phosphate 5-kinase isoforms exhibit acyl chain selectivity for both substrate and lipid
activator. J Biol Chem, 2012. 287(43): p. 35953-63. 175. Silvestre, D.C., G.A. Gil, N. Tomasini, D.F. Bussolino, and B.L. Caputto, Growth of
peripheral and central nervous system tumors is supported by cytoplasmic c-Fos in
humans and mice. PLoS One, 2010. 5(3): p. e9544. 176. Singer, S.J. and G.L. Nicolson, The fluid mosaic model of the structure of cell
membranes. Science, 1972. 175(4023): p. 720-31. 177. Siniossoglou, S., Phospholipid metabolism and nuclear function: roles of the lipin
family of phosphatidic acid phosphatases. Biochim Biophys Acta, 2013. 1831(3): p. 575-81.
178. Song, Y., et al., An association of a simultaneous nuclear and cytoplasmic localization
of Fra-1 with breast malignancy. BMC Cancer, 2006. 6: p. 298. 179. Song, Y., et al., Inhibition of calcium-independent phospholipase A2 suppresses
proliferation and tumorigenicity of ovarian carcinoma cells. Biochem J, 2007. 406(3): p. 427-36.
180. Spector, A.A. and M.A. Yorek, Membrane lipid composition and cellular function. J Lipid Res, 1985. 26(9): p. 1015-35.
181. Srivastava, R., R.K. Sinha, and G. Subrahmanyam, Type II phosphatidylinositol 4-kinase
beta associates with TCR-CD3 zeta chain in Jurkat cells. Mol Immunol, 2006. 43(5): p. 454-63.
182. Stace, C.L. and N.T. Ktistakis, Phosphatidic acid- and phosphatidylserine-binding
proteins. Biochim Biophys Acta, 2006. 1761(8): p. 913-26. 183. Sud, M., et al., LMSD: LIPID MAPS structure database. Nucleic Acids Res, 2007.
35(Database issue): p. D527-32. 184. Sun, Y., N.M. Hays, A. Periasamy, M.W. Davidson, and R.N. Day, Monitoring protein
interactions in living cells with fluorescence lifetime imaging microscopy. Methods Enzymol, 2012. 504: p. 371-91.
185. Sun, Y., C. Rombola, V. Jyothikumar, and A. Periasamy, Forster resonance energy
transfer microscopy and spectroscopy for localizing protein-protein interactions in
living cells. Cytometry A, 2013. 83(9): p. 780-93. 186. Tanaka, S., J. Nikawa, H. Imai, S. Yamashita, and K. Hosaka, Molecular cloning of rat
phosphatidylinositol synthase cDNA by functional complementation of the yeast
Saccharomyces cerevisiae pis mutation. FEBS Lett, 1996. 393(1): p. 89-92. 187. Teleman, A.A., Molecular mechanisms of metabolic regulation by insulin in
Drosophila. Biochem J, 2010. 425(1): p. 13-26. 188. Thakur, P.C., J.M. Davison, C. Stuckenholz, L. Lu, and N. Bahary, Dysregulated
phosphatidylinositol signaling promotes endoplasmic-reticulum-stress-mediated
intestinal mucosal injury and inflammation in zebrafish. Dis Model Mech, 2014. 7(1): p. 93-106.
189. Thakur, P.C., et al., Lack of de novo phosphatidylinositol synthesis leads to
endoplasmic reticulum stress and hepatic steatosis in cdipt-deficient zebrafish. Hepatology, 2011. 54(2): p. 452-62.

Referencias bibliográficas
105
190. Thomas, M.J., et al., Lipid exchange between mixed micelles of phospholipid and triton
X-100. Biochim Biophys Acta, 1999. 1417(1): p. 144-56. 191. Tompa, P., C. Szasz, and L. Buday, Structural disorder throws new light on
moonlighting. Trends Biochem Sci, 2005. 30(9): p. 484-9. 192. Tyanova, S., J. Cox, J. Olsen, M. Mann, and D. Frishman, Phosphorylation variation
during the cell cycle scales with structural propensities of proteins. PLoS Comput Biol, 2013. 9(1): p. e1002842.
193. Uversky, V.N., A decade and a half of protein intrinsic disorder: biology still waits for
physics. Protein Sci, 2013. 22(6): p. 693-724. 194. Uversky, V.N., J.R. Gillespie, and A.L. Fink, Why are "natively unfolded" proteins
unstructured under physiologic conditions? Proteins, 2000. 41(3): p. 415-27. 195. van Meer, G., D.R. Voelker, and G.W. Feigenson, Membrane lipids: where they are and
how they behave. Nat Rev Mol Cell Biol, 2008. 9(2): p. 112-24. 196. Verveer, P.J., F.S. Wouters, A.R. Reynolds, and P.I. Bastiaens, Quantitative imaging of
lateral ErbB1 receptor signal propagation in the plasma membrane. Science, 2000. 290(5496): p. 1567-70.
197. Viiri, K., M. Maki, and O. Lohi, Phosphoinositides as regulators of protein-chromatin
interactions. Sci Signal, 2012. 5(222): p. pe19. 198. Vogel, S.S., C. Thaler, and S.V. Koushik, Fanciful FRET. Sci STKE, 2006. 2006(331): p.
re2. 199. Volta, M., et al., Identification and characterization of CDS2, a mammalian homolog of
the Drosophila CDP-diacylglycerol synthase gene. Genomics, 1999. 55(1): p. 68-77. 200. Wang, H., et al., Lipin-1gamma isoform is a novel lipid droplet-associated protein
highly expressed in the brain. FEBS Lett, 2011. 585(12): p. 1979-84. 201. Wang, J., et al., PI4P promotes the recruitment of the GGA adaptor proteins to the
trans-Golgi network and regulates their recognition of the ubiquitin sorting signal. Mol Biol Cell, 2007. 18(7): p. 2646-55.
202. Wang, X., S.P. Devaiah, W. Zhang, and R. Welti, Signaling functions of phosphatidic
acid. Prog Lipid Res, 2006. 45(3): p. 250-78. 203. Wang, Y.J., et al., Phosphatidylinositol 4 phosphate regulates targeting of clathrin
adaptor AP-1 complexes to the Golgi. Cell, 2003. 114(3): p. 299-310. 204. Waugh, M.G., et al., Localization of a highly active pool of type II phosphatidylinositol
4-kinase in a p97/valosin-containing-protein-rich fraction of the endoplasmic
reticulum. Biochem J, 2003. 373(Pt 1): p. 57-63. 205. Weeks, R., et al., Isolation and expression of an isoform of human CDP-diacylglycerol
synthase cDNA. DNA Cell Biol, 1997. 16(3): p. 281-9. 206. Wei, Y.J., et al., Type II phosphatidylinositol 4-kinase beta is a cytosolic and peripheral
membrane protein that is recruited to the plasma membrane and activated by Rac-
GTP. J Biol Chem, 2002. 277(48): p. 46586-93. 207. Wenk, M.R., The emerging field of lipidomics. Nat Rev Drug Discov, 2005. 4(7): p. 594-
610.

Referencias bibliográficas
106
208. Wenk, M.R. and P. De Camilli, Protein-lipid interactions and phosphoinositide
metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc Natl Acad Sci U S A, 2004. 101(22): p. 8262-9.
209. Wong, K., R. Meyers dd, and L.C. Cantley, Subcellular locations of phosphatidylinositol
4-kinase isoforms. J Biol Chem, 1997. 272(20): p. 13236-41. 210. Wouters, F.S. and P.I. Bastiaens, Fluorescence lifetime imaging of receptor tyrosine
kinase activity in cells. Curr Biol, 1999. 9(19): p. 1127-30. 211. Wright, P.E. and H.J. Dyson, Linking folding and binding. Curr Opin Struct Biol, 2009.
19(1): p. 31-8. 212. Wu, L., B. Niemeyer, N. Colley, M. Socolich, and C.S. Zuker, Regulation of PLC-
mediated signalling in vivo by CDP-diacylglycerol synthase. Nature, 1995. 373(6511): p. 216-22.
213. Xu, Z., G. Huang, and K.V. Kandror, Phosphatidylinositol 4-kinase type IIalpha is
targeted specifically to cellugyrin-positive glucose transporter 4 vesicles. Mol Endocrinol, 2006. 20(11): p. 2890-7.
214. Xue, B., R.L. Dunbrack, R.W. Williams, A.K. Dunker, and V.N. Uversky, PONDR-FIT: a
meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta, 2010. 1804(4): p. 996-1010.
215. Zaccheo, O., D. Dinsdale, P.A. Meacock, and P. Glynn, Neuropathy target esterase and
its yeast homologue degrade phosphatidylcholine to glycerophosphocholine in living
cells. J Biol Chem, 2004. 279(23): p. 24024-33. 216. Zolyomi, A., X. Zhao, G.J. Downing, and T. Balla, Localization of two distinct type III
phosphatidylinositol 4-kinase enzyme mRNAs in the rat. Am J Physiol Cell Physiol, 2000. 278(5): p. C914-20.


















