
INTRODUÇÃO AO ESTUDO DOS MATERIAISdemahp/ES242.doc · Web viewNa tabela I.1 são comparadas...
205
Introdução 1 Materiais de Engenharia ES242 Prof. Dr. Rubens Caram Departamento de Engenharia de Materiais Faculdade de Engenharia Mecânica Universidade Estadual de Campinas - Campinas SP -
Transcript of INTRODUÇÃO AO ESTUDO DOS MATERIAISdemahp/ES242.doc · Web viewNa tabela I.1 são comparadas...

Introdução 1
Materiais de Engenharia
ES242
Prof. Dr. Rubens CaramDepartamento de Engenharia de Materiais
Faculdade de Engenharia MecânicaUniversidade Estadual de Campinas
- Campinas SP -
1 9 9 8

Introdução 2
INTRODUÇÃO AO ESTUDO DOS MATERIAISI.1. INTRODUÇÃO
Os materiais estão intimamente ligados à existência e à evolução da espécie
humana. Desde o início da civilização, os materiais e a energia são usados com o
objetivo de melhorar o nível de vida do ser humano. Dentre os materiais mais comuns,
pode-se citar: madeira, cimento, pedra, aço, plástico, vidro, borracha, alumínio, cobre
e papel. Existem muitos outros tipos de materiais e, para se notar tal fato, basta
observar a constituição dos objetos ao nosso redor.
A produção e transformação de materiais em bens acabados, constitui uma das
mais importantes atividades de uma economia moderna. Um produto, para ser
manufaturado, requer uma etapa de planejamento de seu processo de produção.
Nesta etapa são selecionados diversos materiais, de acordo com custos e,
principalmente, com as necessidades técnicas exigidas. A elaboração dessa etapa
exige que o responsável pela mesma tenha noção das estruturas internas dos
materiais, pois o conhecimento das mesmas, aos níveis submicroscópicos, permite
prever o comportamento do material em serviço, bem como possibilita programar e
controlar suas propriedades e características.
Os materiais são analisados e desenvolvidos dentro do ramo do conhecimento
denominado de "Ciência e Engenharia de Materiais", o qual tem, como meta principal
a geração e emprego de conceitos envolvendo composição química, arranjo atômico e
processamento dos materiais com suas características e empregos.
A ciência dos materiais está associada à geração de conhecimento básico
sobre a estrutura interna, propriedades e processamento de materiais. Ela tem ainda
como objetivo, compreender a natureza dos materiais, estabelecendo conceitos e
teorias que permitam relacionar a estrutura dos materiais com suas propriedades e
comportamento. A engenharia dos materiais está principalmente ligada ao emprego
de conceitos fundamentais e empíricos dos materiais, na conversão dos mesmos em
produtos finais.
I.2. CLASSIFICAÇÃO DOS MATERIAISPor conveniência, a maioria dos materiais de engenharia é classificada em três
classes principais, quais sejam: materiais metálicos, materiais poliméricos (plásticos) e
materiais cerâmicos. Em adição a estes três tipos, um estudo mais abrangente deve

Introdução 3
incluir um outro tipo, que exibe, atualmente, grande importância tecnológica: os
materiais compósitos ou conjugados.
I.2.a. MATERIAIS METÁLICOSOs materiais metálicos são substâncias inorgânicas compostas por um ou mais
elementos metálicos e podem, também, conter elementos não-metálicos. Exemplos de
materiais metálicos: aço, cobre, alumínio, níquel e titânio. Elementos não-metálicos
como carbono, nitrogênio e oxigênio podem estar contidos em materiais metálicos.
Os metais tem uma estrutura cristalina, na qual os átomos estão arranjados de
maneira ordenada. Eles, em geral, são bons condutores térmicos e elétricos. Quase
todos os metais são mecanicamente resistentes, dúcteis e muitos mantém esta
resistência mesmo em altas temperaturas.
I.2.b. MATERIAIS POLIMÉRICOS (PLÁSTICOS)A maioria dos materiais poliméricos consiste de cadeias moleculares orgânicas
(carbono) de longa extensão. Estruturalmente, a maioria destes materiais não é
cristalina, porém alguns exibem uma mistura de regiões cristalinas e não-cristalinas. A
resistência mecânica e ductilidade dos materiais poliméricos varia enormemente.
Devido à natureza da estrutura interna, a maioria dos plásticos conduzem eletricidade
e calor de maneira extremamente precária. Isto permite que os mesmos sejam
freqüentemente utilizados como isolantes, tendo grande importância na confecção de
dispositivos e equipamentos eletrônicos. Em geral, os materiais poliméricos têm baixo
peso específico e apresentam temperatura de decomposição relativamente baixa.
I.2.c. MATERIAIS CERÂMICOS
Os cerâmicos são materiais inorgânicos constituídos por elementos metálicos e
não-metálicos unidos por meio de ligações químicas. Estes materiais podem ser
cristalinos, não-cristalinos ou uma mistura de ambos. A maioria dos cerâmicos
apresenta alta dureza e elevada resistência mecânica, mesmo em altas temperaturas.
Entretanto, tais materiais são, normalmente, bastante frágeis. Uma gama bastante
ampla de novos materiais cerâmicos está sendo desenvolvida, tendo como objetivo
diversas aplicações, como é o caso de peças para motores de combustão interna.

Introdução 4
Neste caso, estes materiais têm a vantagem do baixo peso, resistência e dureza
elevadas, ótima resistência ao calor e propriedades isolantes.
O fato de ser um bom isolante térmico, bem como ser resistente ao calor,
permite que os materiais cerâmicos tenham importante papel na construção de fornos
usados na indústria metalúrgica. Uma aplicação recente, que retrata com fidelidade o
potencial dos materiais cerâmicos, é o uso dos mesmos na construção do ônibus
espacial americano. A estrutura deste veículo é de alumínio revestida por milhares de
pastilhas cerâmicas. Estas pastilhas dão proteção térmica ao ônibus durante a subida
e por ocasião da reentrada do mesmo na atmosfera.
I.2.d. MATERIAIS COMPÓSITOSOs materiais compósitos ou conjugados são combinações de dois ou mais
materiais. A maioria destes materiais consiste de um elemento de reforço envolvido
por uma matriz, constituída de resina colante, com o objetivo de obter características
específicas e propriedades desejadas. Geralmente, os componentes não se dissolvem
um no outro e podem ser identificados, fisicamente, por uma interface entre os
mesmos, bem definida.
Os materiais compósitos podem ser de vários tipos e os mais comuns são os
fibrosos (fibras envolvidas por uma matriz) e os particulados (partículas envolvidas por
uma matriz). Existe uma infinidade de tipos de elementos de reforços, bem como
matrizes usadas industrialmente. Dois tipos notáveis de materiais compósitos, usados
intensamente na indústria, são: fibra de vidro em matriz de epóxi e fibra de carbono
também em matriz de epóxi. Um exemplo bastante familiar de material compósito é o
concreto armado que, nada mais é, que uma matriz de concreto (cimento, areia e
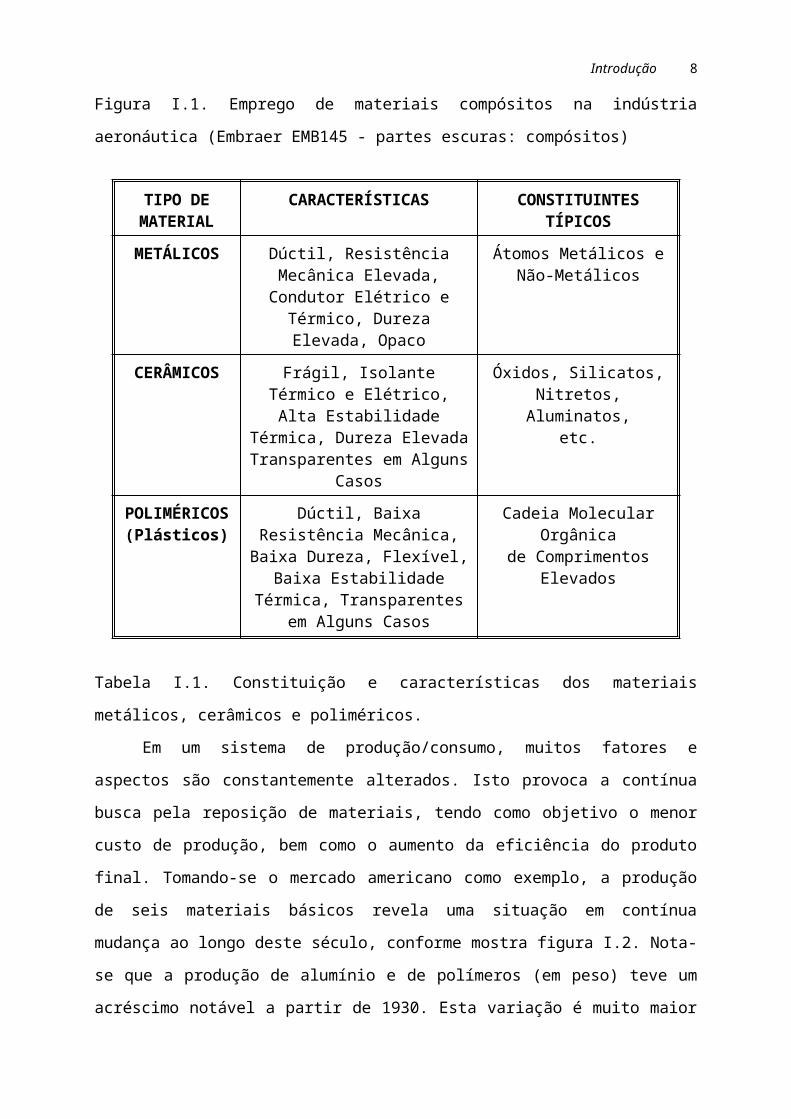
pedra) envolvendo o elemento de reforço, representado por barras de aço. A figura I.1
mostra o uso de materiais conjugados na indústria aeronáutica.
Na tabela I.1 são comparadas algumas propriedades dos materiais metálicos,
cerâmicos e poliméricos.
I.3. SUBSTITUIÇÃO E COMPETIÇÃO DE MATERIAISA competição entre diferentes tipos de materiais é um processo contínuo que
ocorre desde os primórdios da civilização, à medida que, em função de suas

Introdução 5
necessidades, o homem iniciou a transformação de materiais em ferramentas e
utensílios.
Figura I.1. Emprego de materiais compósitos na indústria aeronáutica (Embraer
EMB145 - partes escuras: compósitos)
TIPO DE MATERIAL
CARACTERÍSTICAS CONSTITUINTES TÍPICOS
METÁLICOS Dúctil, Resistência Mecânica Elevada, Condutor Elétrico e
Térmico, Dureza Elevada, Opaco
Átomos Metálicos e Não-Metálicos
CERÂMICOS Frágil, Isolante Térmico e Elétrico, Alta Estabilidade Térmica, Dureza ElevadaTransparentes em Alguns
Casos
Óxidos, Silicatos, Nitretos, Aluminatos,
etc.
POLIMÉRICOS(Plásticos)
Dúctil, Baixa Resistência Mecânica, Baixa Dureza,
Flexível, Baixa Estabilidade Térmica, Transparentes em
Alguns Casos
Cadeia Molecular Orgânica
de Comprimentos Elevados
Tabela I.1. Constituição e características dos materiais metálicos, cerâmicos e
poliméricos.

Introdução 6
Em um sistema de produção/consumo, muitos fatores e aspectos são
constantemente alterados. Isto provoca a contínua busca pela reposição de materiais,
tendo como objetivo o menor custo de produção, bem como o aumento da eficiência
do produto final. Tomando-se o mercado americano como exemplo, a produção de
seis materiais básicos revela uma situação em contínua mudança ao longo deste
século, conforme mostra figura I.2. Nota-se que a produção de alumínio e de
polímeros (em peso) teve um acréscimo notável a partir de 1930. Esta variação é
muito maior ainda se o volume produzido for considerado, pois estes dois materiais
exibem baixo peso específico.
Figura I.2. Produção americana de madeira, aço, cimento, plásticos, alumínio e cobre,
entre 1910 e 1990.
Um outro exemplo de alteração no perfil de consumo de materiais é o caso da
indústria automobilística. Em 1978, um carro médio americano pesava 1.800kg e
exibia 60% desse valor em ferro e aço, 10 a 20% em plásticos e 3 a 5% em alumínio.
Em 1985 o carro médio americano pesava 1.400 kg, sendo que 50 a 60% desse valor
em ferro e aço, 10 a 20% em plásticos e 5 a 10% em alumínio. Neste período a

Introdução 7
percentagem de aço decresceu, a quantidade de alumínio aumentou e a de plásticos
continuou estável. Durante a década de 1990, o carro médio americano deverá atingir
um peso próximo de 1.130 kg, e, deste valor, os plástico deverão representar 30%. Na
figura I.3. são mostrados alguns dos materiais utilizados na fabricação de automóveis.
Figura I.3. Materiais encontrados na fabricação de um automóvel moderno.
Em algumas aplicações, apenas determinados materiais apresentam os
requisitos técnicos necessários e, nestes casos, quase sempre tais materiais são
relativamente custosos. Por exemplo, a produção das turbinas para aviões requer o
emprego de superligas à base de níquel. O custo deste material é bastante elevado e
nenhum substituto de menor custo pode ser encontrado. Isto mostra que apesar do
custo ser um parâmetro importante na substituição de materiais, as características
técnicas podem decidir a escolha. A substituição de um material por outro é um
processo contínuo pois novos materiais são constantemente descobertos, assim como
novos processos são constantemente desenvolvidos.
I.4. ESTRUTURA E PROPRIEDADES DOS MATERIAISO emprego de materiais na forma de produtos acabados envolve, geralmente,
etapas de processamento onde algumas de suas características podem ser
significativamente alteradas, o que, normalmente, resulta em modificações na
RUBENS CARAM, 03/01/-1,
Introdução 8
estrutura interna do material. Por exemplo, a modificação da forma geométrica de um
material metálico, ou seja, a conformação plástica do mesmo, acarreta em alterações
no estado de tensão da estrutura atômica, bem como pode modificar a estrutura ao
nível atômico. As condições encontradas durante tal processamento exercem
influência decisiva no arranjo final dos átomos do material. Um exemplo típico é a
produção de uma peça metálica obtida pelo processo de fundição de metais, como é o
caso de um pistão de automóvel. Neste caso, um molde, geralmente metálico, com
uma cavidade com a mesma forma geométrica do pistão é preenchido por um volume
de metal líquido. Após a solidificação deste metal, a peça é desmoldada e a fundição
do pistão é concluída. Se a velocidade de solidificação do metal líquido foi alta ou
baixa, a estrutura interna do material será afetada em relação a defeitos nos arranjos
atômicos e, assim, influenciando as propriedades da peça.
Concluindo, um material para ser aplicado em engenharia necessita apresentar
dados sobre suas características básicas e também sobre a maneira com que o
mesmo foi processado até o momento de ser empregado. Uma chapa de aço, que é,
na verdade, uma liga de ferro e carbono, laminada "a frio" apresenta características
distintas de uma outra laminada "a quente". A figura I.4 relaciona estruturas,
propriedades e processamento dos materiais.
Figura I.4. Estruturas, propriedades e processos de modificação de propriedades dos
materiais.

Introdução 9
A natureza e comportamento dos materiais em serviço estão basicamente
associadas aos tipos de átomos envolvidos e aos arranjos dos mesmos. Um material
pode ser constituído por um ou mais tipos de elementos químicos. Entretanto, a forma
com que tais elementos se arranjam no espaço determinará as características do
material. A estrutura dos materiais pode ser estudada de acordo com quatro níveis: o
subatômico, atômico, microscópico e macroscópico. Para se ter uma visão global das
dimensões envolvidas, a figura I.5 compara o tamanho de diversas estruturas, desde a
muralha da China até as partículas elementares ou subatômicas.
Figura I.5. Comparação entre o tamanho de diversas estruturas.
O nível subatômico está relacionado à análise do átomo individual e o
comportamento de seu núcleo, e os elétrons de suas camadas periféricas. Existe um
compromisso bastante sólido entre o comportamento do átomo e suas partículas
subatômicas e propriedades elétricas, térmicas e magnéticas.
O nível atômico está ligado à análise do comportamento de um átomo em
relação a outro átomo, ou seja, a interação entre átomos e ligações entre os mesmos
e a formação de moléculas. As ligações interatômicas dependem do comportamento
do átomo ao nível subatômico. Em função do tipo e intensidade dessas ligações, um

Introdução 10
dado material, em uma determinada condição, pode apresentar-se como sólido,
líquido ou gasoso.
O nível microscópico relaciona-se à análise do arranjo dos átomos ou suas
moléculas no espaço. Um arranjo atômico pode resultar em três tipos estruturais,
quais sejam: arranjo cristalino, molecular e amorfo. O arranjo estrutural apresentado
por um material influencia diretamente as propriedades e características do mesmo.
O nível macroscópico relaciona-se às características e propriedades dos
materiais em serviço, que estão diretamente ligadas à natureza do comportamento
atômico nos três níveis anteriores, e à maneira com que o material foi processado.
EXERCÍCIOSI.1. Quais são as principais classes de materiais usados em engenharia ?
I.2. Liste alguns materiais normalmente encontrados em engenharia de materiais.
I.3. Defina o que são materiais conjugados ou compósitos.
I.4. Dê exemplos de materiais que foram substituídos por outros em determinadas
aplicações industriais. Explique as razões de tais substituições.
I.5. Atualmente, diversos componentes de motores de combustão interna são
confeccionados a partir de materiais cerâmicos. Qual a principal vantagem do
emprego destes materiais neste caso?
I.6. Considere uma aeronave moderna. Pesquise os novos materiais envolvidos na
construção da mesma.
I.7. Considere um automóvel moderno. Liste alguns materiais não-tradicionais
envolvidos na construção do mesmo.

Ligações Atômicas 11
LIGAÇÕES ATÔMICASII.1. INTRODUÇÃO
O comportamento de um material pode ser eficientemente previsto a partir da
análise do mesmo aos níveis subatômico, atômico e microscópico. Assim, torna-se
necessário examinar o mesmo, no tocante aos átomos que constituem o material, bem
como o comportamento eletrônico dos mesmos. A estrutura de qualquer material é
diretamente dependente dos tipos de átomo envolvidos e das ligações atômicas que
eles formam.
II.2. INTERAÇÕES ATÔMICASA base de qualquer unidade estrutural em ciência e engenharia de materiais é o
átomo. O átomo consiste basicamente de três partículas subatômicas: prótons,
elétrons e nêutrons. No centro do átomo localiza-se o núcleo, que tem diâmetro
próximo a 10-14 m. Este núcleo é envolvido por uma nuvem de elétrons de densidade
variável, que resulta em um diâmetro atômico final de 10-10m. No núcleo, onde residem
prótons e nêutrons, está a quase totalidade da massa atômica. A massa de um próton
é igual a 1,673x10-24g e sua carga elétrica é de +1,602x10-19 coulombs (C). O nêutron
é pouco mais pesado que o próton e tem massa igual a 1,675x10-24g porém, é
eletricamente neutro. O elétron tem massa de 9,109x10-28g e carga igual a -1,602x10-19
coulombs. Portanto, a quase totalidade do volume atômico concentra-se na nuvem de
elétrons, porém, esta colabora com apenas uma pequena parte da massa final do
átomo. Os elétrons, particularmente os mais externos, determinam a maioria das
características elétricas, mecânicas, químicas e térmicas do átomos e assim, o
conhecimento básico do mesmo é necessário no estudo dos materiais.
A estrutura interna dos materiais é resultado da agregação de átomos obtida
através de forças de ligação interatômicas. Esta agregação, em função das
características de tais ligações, pode resultar nos estados sólido, líquido e gasoso.
Basicamente, os átomos podem atingir uma configuração denominada de
estável a partir de três maneiras, quais sejam: ganho de elétrons, perda de elétrons ou
compartilhamento de elétrons. A facilidade em ganhar elétrons caracteriza o átomo
como elemento eletronegativo; a facilidade em perder elétrons o caracteriza como
sendo um elemento eletropositivo. Existem também os átomos que não apresentam
facilidade em perder ou ganhar elétrons. Estas características atômicas resultam na

Ligações Atômicas 12
existência de três tipos de ligações atômicas, denominadas como primárias ou fortes,
que são mostradas na tabela II.1
ELEMENTO ELETROPOSITIVO+
ELEMENTO ELETRONEGATIVOLIGAÇÃO IÔNICA
ELEMENTO ELETROPOSITIVO+
ELEMENTO ELETROPOSITIVOLIGAÇÃO METÁLICA
ELEMENTO ELETRONEGATIVO+
ELEMENTO ELETRONEGATIVOLIGAÇÃO COVALENTE
Tabela II.1. Relação entre características atômicas e ligações resultantes.
II.3. LIGAÇÕES IÔNICASÉ o resultado da interação entre íons positivos (cátions) e negativos (anions).
Um exemplo que pode ser considerado clássico de ligação iônica ocorre na formação
do NaCl (sal de cozinha). A estrutura formada pelo NaCl é exibida na figura II.1.
Figura II.1. Estrutura formada pelo NaCl.
O sódio possui as duas primeiras camadas eletrônicas completas e a terceira
com apenas um elétron. Isto mostra que o Na tem facilidade em perder um elétron
(eletropositivo) para adquirir a configuração eletrônica estável. Por outro lado, o cloro
apresenta em sua camada mais externa sete átomos ou seja, ele tem facilidade em

Ligações Atômicas 13
receber um elétron (eletronegativo) e tornar-se eletronicamente estável. Quando o Na
e Cl reagem, os elétrons externos dos átomos de sódio transferem-se para os átomos
de cloro, produzindo íons-sódio Na+ e os íons-cloretos Cl-, que são mantidos juntos
pela atração eletrostática de suas cargas opostas, formando o NaCl
II.4. LIGAÇÕES COVALENTESQuando dois elementos eletronegativos reagem entre si, não é formada uma
ligação iônica, pois os dois átomos têm facilidade em receber elétrons. Neste caso, a
configuração estável dos dois elementos ocorre por compartilhamento de elétrons.
Como exemplo, pode-se citar a formação da molécula de cloro Cl2. Cada átomo de
cloro compartilha um de seus elétrons com outro átomo. Dessa forma, um par
eletrônico pode ser compartilhado igualmente por dois átomos e, cada átomo, tem na
sua camada mais externa, seis elétrons originalmente dele e um par compartilhado.
Isto torna cada átomo eletronicamente estável, e o mesmo atinge a configuração do
gás nobre argônio. Da mesma forma, a formação da molécula de oxigênio envolve o
compartilhamento de quatro elétrons. Ligações covalentes podem ser observadas no
Si, como mostra a figura II.2.
Figura II.2. Ligações covalentes encontradas em um cristal de Silício.
II.5. LIGAÇÃO METÁLICAEsse tipo de ligação é normalmente encontrado em metais e envolve a
interação de elementos eletropositivos. A ligação metálica é resultado da ação entre

Ligações Atômicas 14
elétrons livres (nuvem eletrônica) e íons positivos. Estes elétrons livres são originários
da última camada de valência, fracamente presos ao átomo, e que estão livres dentro
da estrutura metálica. Através de tais elétrons pode-se explicar as altas condutividades
elétrica e térmica dos metais. A figura II.3. Mostra as ligações metálicas observadas
em metais.
Figura II.3. Ligações metálicas (nuvem de elétrons) encontradas nos metais.
Os três tipos de ligações primárias mencionados raramente ocorrem
individualmente. Na verdade, um mesmo material pode exibir uma combinação destes
tipos, formando materiais com ligações mistas. Um exemplo é o NaNO3 (nitrato de
sódio), que apresenta ligações covalente no radical nitrato NO3- e ligações iônicas
entre os íons Na+ e NO3-.
Uma outra classe de ligações, denominadas de ligações fracas, pode ser
encontrada em algumas substâncias. Estas ligações contribuem para a atração entre
átomos e são classificadas como forças de Van Der Walls. Tal classe de ligações
permite explicar a condensação dos gases nobres (He, Ne, Ar, Kr, Xe e Ra). Estes
elementos apresentam orbitais perfeitamente completos (8 e 2 elétrons na última
camada) e dessa forma tais átomos deveriam permanecer monoatômicos em qualquer
temperatura. Entretanto, em temperaturas extremamente baixas, a existência das
forças de Van Der Walls pode provocar a união dos elementos nobres.
II.6. DISTÂNCIAS INTERATÔMICASOs átomos constituintes de um material sólido encontram-se em um estado de
constante movimento vibratório ao redor de suas posições de equilíbrio. A intensidade
de tal movimento vibratório depende da temperatura em que se encontra o material.
Nos estudos das estruturas dos materiais sólidos, tal movimento pode ser considerado

Ligações Atômicas 15
desprezível ao se considerar que tal material é um agregado estático de átomos
interligados e localizados em pontos de equilíbrio de tais movimentos.
Independente do tipo de ligação existente entre dois átomos do agregado
atômico em questão, seja ela iônica, metálica ou ainda covalente, os pontos de
equilíbrio resultam da interação de dois tipos de força. O primeiro tipo é a força de
atração, que é resultante da ligação existente (iônica, metálica ou covalente) e é
responsável pela agregação atômica. O outro tipo de força a ser considerado é o de
repulsão, que resulta da proximidade acentuada de nuvens eletrônicas dos átomos.
Esta força permite explicar a existência de "espaços vazios" no volume em torno de
um núcleo atômico. Tais vazios comprovam o movimento de nêutrons dentro de
determinados materiais utilizados na confecção de reatores nucleares. Os nêutrons
caminham através de muitos átomos até colidirem com os núcleos. Este espaço
interatômico é resultado da interação entre forças de repulsão com as de atração
dando origem a uma distância de equilíbrio entre os átomos, que é o ponto onde
ambas as forças são iguais. Em termos de energia, a distância de equilíbrio entre os
átomos será aquela em que a energia potencial tem valor mínimo ou quando a força
de repulsão apresentar valor igual a de atração.
Para ilustrar a interação de forças pode-se tomar o caso de uma ligação iônica,
onde tais forças podem ser determinadas mais facilmente. A força de atração (FA)
neste caso é dada pela ação de duas cargas pontuais:
FA
Z e Z e
a 1 2
4 02
(II.1)
onde Z1 e Z2 são números de elétrons removidos ou adicionados aos átomos na
formação do íon, "e" é a carga do elétron, 0 é a permissividade do espaço vazio
(8,85x10-12C2/Nm2) e "a" é a distância interatômica.
A força de repulsão (FR) em uma ligação iônica é encontrada
experimentalmente como sendo inversamente proporcional à distância de separação
entre os íons e é dada pela equação:
RF = -nbn+1a
(II.2)
onde b e n são constante, sendo que n vale entre 7 e 9 para a ligação iônica no NaCl.
Assim, a força resultante (FTotal) é dada pela soma das forças de atração e repulsão:

Ligações Atômicas 16
TotalF = -( 1Z e)( 2Z e)
4 a -
nbn+1a0
2 (II.3)
A resistência mecânica de um material está relacionada com a FTotal envolvida
nas ligações entre os átomos do mesmo. A força resultante está associada à tensão
necessária para separar dois átomos. Como conseqüência deste fato, forças
interatômicas elevadas apresentam energias de ligação absolutas também elevadas
no ponto de equilíbrio, o que corresponde a materiais geralmente duros, como o
diamante ou o silício. Da mesma forma, o módulo de elasticidade do material, que é a
medida de rigidez do mesmo, pode ser obtido pela derivação de FTotal em relação à
distância, em posições próximas ao ponto de equilíbrio.
A energia (E) associada a uma ligação iônica, como a encontrada no NaCl, é
dada pela soma das energias envolvidas com a atração e repulsão dos íons. Esta
energia de ligação pode ser obtida pelo produto "força x distância", ou:
E = a
-( 1Z e)( 2Z e)
4 a -
nb
a n da
0
2 1 (II.4)
que resulta em:
E = +( 1Z 2Z 2e )
4 a +
bna0
(II.5)
O termo relativo à energia de atração corresponde à energia liberada quando
os íons aproximam-se e é negativa devido ao produto de uma carga positiva por uma
negativa (+Z1 . -Z2). O termo ligado à energia de repulsão representa a energia
absorvida quando os íons aproximam-se e é positiva. A soma destas duas energias
tem seu ponto mínimo quando os íons apresentam distância de separação de
equilíbrio. A figura II.4 apresenta um diagrama esquemático das variações da força
total com a distância de separação entre átomos.
As ligações atômicas permitem que os átomos exibam o estado de agregação
sólido. Em função da natureza dessas ligações atômicas e da forma com que os
átomos são arranjados no espaço, é possível prever propriedades, características e o
comportamento do material. Basicamente, pode-se classificar os arranjos atômicos
dos materiais sólidos em três tipos de estruturas atômicas: estrutura cristalina,
estrutura amorfa e estrutura molecular.

Ligações Atômicas 17
Figura II.4. Diagrama esquemático da interação entre forças de atração e repulsão no
ponto de equilíbrio.
A estrutura cristalina caracteriza-se por arranjos atômicos ordenados
espacialmente. É a estrutura típica dos metais. A estrutura amorfa caracteriza-se por
apresentar arranjos atômicos desordenados e aleatórios, semelhante à estrutura do
estado líquido. Entretanto, a estrutura amorfa pode exibir regiões isoladas de
ordenação atômica, sendo estas de curto alcance. A estrutura molecular se caracteriza
pela existência de moléculas como unidade estrutural. Tais moléculas são formadas
por átomos arranjados de forma ordenada e pré-determinada. É o tipo de estrutura
observada nos plásticos.
EXERCÍCIOSII.1. Descreva sucintamente as ligações atômicas primárias iônica, covalente e
metálica.
II.2. Após a ionização, por que um íon de sódio se torna menor que um átomo de
sódio?
II.3. Após a ionização, por que um íon de cloro se torna maior que um átomo de cloro?
II.4. Por que o diamante tem dureza muito elevada?

Ligações Atômicas 18
II.5. Por que os metais são bons condutores de calor e de eletricidade?
II.6. Calcule a força de atração entre o par iônico Na+ e F-, quando os mesmos estão
se tocando. Assuma que o raio iônico do Na+ é 0,095nm e do F- é 0,136nm.

Estrutura Cristalina 19
ESTRUTURA CRISTALINAIII.1. INTRODUÇÃO
A estrutura física dos materiais sólidos depende fundamentalmente do arranjo
estrutural de seus átomos, íons ou moléculas. A grande maioria dos materiais
comumente utilizados em engenharia, particularmente os metálicos, exibe um arranjo
geométrico de seus átomos bem definido, constituindo uma estrutura cristalina. Um
material cristalino, independente do tipo de ligação encontrada no mesmo, caracteriza-
se por apresentar um agrupamento ordenado de seus átomos, íons ou moléculas, que
se repete nas três dimensões. Os arranjos atômicos em um sólido cristalino podem ser
descritos usando, como referência, os pontos de intersecção de uma rede de linhas
nas três dimensões. Em um cristal ideal, o arranjo destes pontos em torno de um
ponto particular deve ser igual ao arranjo em torno de qualquer outro ponto da rede
cristalina. Dessa maneira, é possível descrever um conjunto de pontos ou posições
atômicas repetitivo, denominado de célula unitária. Uma célula unitária é também
definida como a menor porção do cristal que ainda conserva as propriedades originais
do mesmo. Através da adoção de valores específicos, como parâmetros axiais e
ângulos interaxiais, pode-se obter células unitárias de diversas naturezas. O estudo da
estrutura interna dos materiais necessita da utilização de 7 arranjos atômicos básicos,
que podem representar as estruturas de todas as substâncias cristalinas conhecidas.
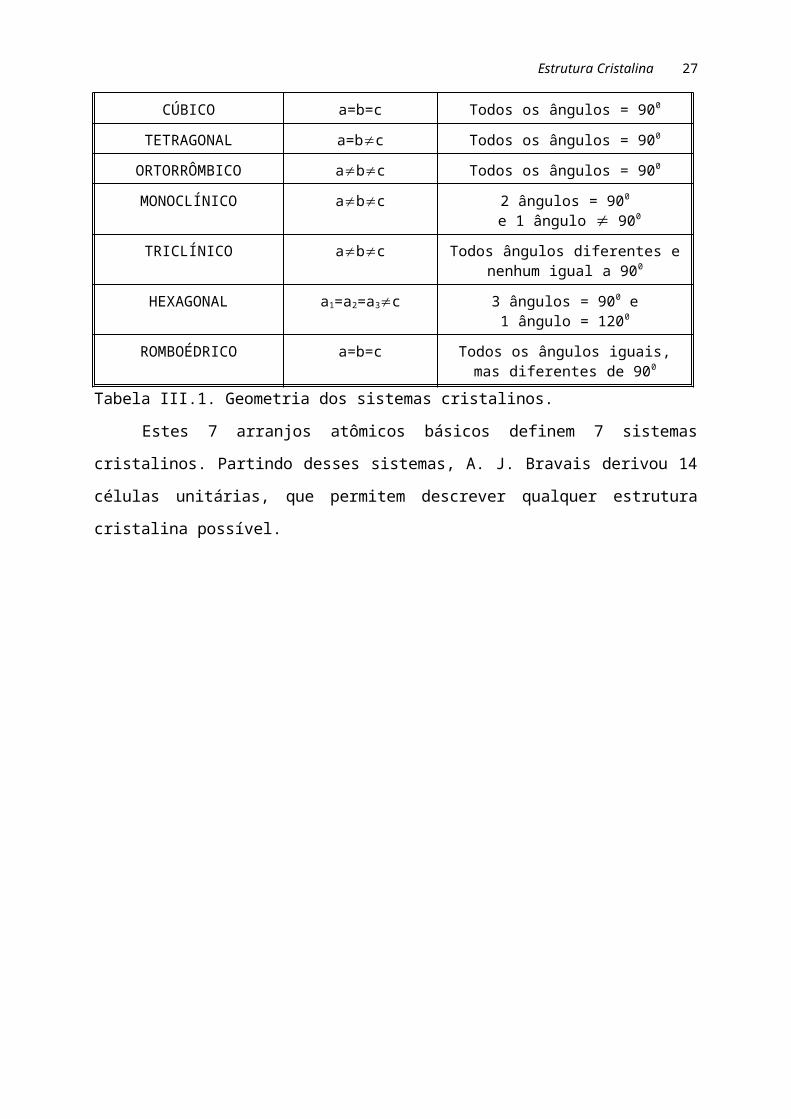
Na tabela III.1 e figura III.1 são mostradas características desses arranjos.
SISTEMAS EIXOS ÂNGULOS AXIAIS
CÚBICO a=b=c Todos os ângulos = 900
TETRAGONAL a=b¹c Todos os ângulos = 900
ORTORRÔMBICO a¹b¹c Todos os ângulos = 900
MONOCLÍNICO a¹b¹c 2 ângulos = 900
e 1 ângulo ¹ 900
TRICLÍNICO a¹b¹c Todos ângulos diferentes enenhum igual a 900
HEXAGONAL a1=a2=a3¹c 3 ângulos = 900 e1 ângulo = 1200
ROMBOÉDRICO a=b=c Todos os ângulos iguais, mas diferentes de 900
Tabela III.1. Geometria dos sistemas cristalinos.

Estrutura Cristalina 20
Estes 7 arranjos atômicos básicos definem 7 sistemas cristalinos. Partindo
desses sistemas, A. J. Bravais derivou 14 células unitárias, que permitem descrever
qualquer estrutura cristalina possível.
Figura III.1. Arranjos atômicos de Bravais.

Estrutura Cristalina 21
III.2. PRINCIPAIS ESTRUTURAS CRISTALINASA maioria dos elementos metálicos (em torno de 90%) transforma-se de líquido
para sólido assumindo estruturas altamente densas, quais sejam: cúbica de corpo
centrado (CCC), cúbica de face centrada (CFC) e hexagonal compacta (HC), que são
mostradas na figura III.2. A estrutura hexagonal compacta é na verdade uma
modificação da estrutura hexagonal simples, já mostrada anteriormente. A maioria dos
metais cristalizam-se seguindo estes arranjos compactos, pois a energia é liberada
com a aproximação dos átomos. Assim, uma estrutura densa apresenta nível de
energia mais baixo e portanto, mais estável. As dimensões de células unitárias
metálicas são extremamente pequenas. Como exemplo, a aresta do cubo de uma
célula unitária de ferro (CCC), na temperatura ambiente, mede 0,287x10-9m. Portanto,
se diversas células unitárias são colocadas em fila, em 1mm existiriam 3,48 x 106
células.
Figura III.2. Células unitárias de maior importância em estudos cristalográficos: CCC,
CFC e HC.
III.3. CRISTAIS CÚBICOSA estrutura cúbica é uma das que ocorrem com maior freqüência nas
substâncias cristalinas e é considerada a de maior importância. Dependendo da

Estrutura Cristalina 22
posição que os átomos ocupam na estrutura cúbica, a mesma pode ser classificada
em cúbica simples (CS), cúbica de corpo centrado (CCC) e cúbica de face centrada
(CFC).
III.3.a. CÉLULA CÚBICA SIMPLES (CS)Neste arranjo atômico existe um átomo em cada vértice de um cubo. Um
parâmetro de grande importância no estudo das estruturas cristalinas é o parâmetro
de rede (a). No caso da estrutura CS, o parâmetro de rede é dado pelo tamanho da
aresta deste cubo, ou seja, a=2R, onde R é o raio atômico. A figura III.3 mostra a
representação esquemática de tal célula cristalina.
Figura III.3. Representação esquemática de uma célula unitária CS.
Como forma de classificar o nível de ocupação por átomos em uma estrutura
cristalina, define-se o "fator de empacotamento (F.E.)", que é dado por:
F.E. = N V
VA
C(III.1)
onde: N = Número de átomos que efetivamente ocupam a célula;
VA = Volume do átomo (4/3..R3);
R = Raio do átomo;

Estrutura Cristalina 23
VC = Volume da célula unitária.
Na célula cúbica simples, o fator de empacotamento seria:
N = 18
8 = 1
A3V =
43
R
C3 3 3V = a = (2R ) = 8 R
F.E. = 1
43
R
8 R = 0,52
3
3
(III.2)
ou seja, apenas 52% desta célula unitária é preenchida por átomos. Devido ao baixo
índice de ocupação desta célula, os metais não apresentam este tipo de arranjo. Uma
única exceção é o polônio (Po).
III.3.b. CÉLULA CÚBICA DE CORPO CENTRADO (CCC)Neste arranjo estrutural existe um átomo em cada vértice de um cubo e um
outro átomo no centro do mesmo, como mostra a figura III.4. Esta estrutura pode ser
encontrada no tungstênio, tântalo, bário, nióbio, lítio, potássio, vanádio, cromo, etc.
Nesse caso, o fator de empacotamento pode ser calculado seguindo o mesmo
procedimento anterior:
N = 18
8 + 1 = 2
A3V =
43
R
O parâmetro de rede neste caso é calculado a partir do valor da diagonal
principal do cubo e de uma das face do mesmo. Assim,
2 2 2a + a 2 = (4R ) a = 4R
3 (III.3)
C3
3 3
V = a = 4R
3 =
64 R3 3

Estrutura Cristalina 24
F. E. = 2 4
3 R
64 R3 3
= 0,683
3
(III.4)
Figura III.4. Representação esquemática de uma célula unitária CCC.
III.3.c. CÉLULA CÚBICA DE FACE CENTRADA (CFC)Este arranjo caracteriza-se por exibir os mesmos átomos nos vértices
encontrados nos outros dois arranjos cúbicos e mais 1 átomo em cada face do cubo. A
estrutura cúbica de face centrada é a estrutura do alumínio, cálcio, níquel, cobre,
prata, ouro, platina, chumbo, etc. Neste caso existe um total de quatro átomos no
interior da célula unitária. A figura III.5 apresenta um diagrama esquemático desta
estrutura.
O parâmetro de rede no caso da estrutura CFC pode ser obtido através da
diagonal da face, ou:

Estrutura Cristalina 25
2 2 2a + a = (4R ) a = 2 2R (III.5)
C3 3 3V = a = 2R 2 = 16R 2
F.E. = 4
43
R
16 2 R = 0,74
3
3
(III.6)
Figura III.5. Representação esquemática de uma célula unitária CFC.
III.4. CRISTAIS HEXAGONAISAs estruturas cristalinas hexagonais, juntamente com as estruturas cúbicas,
formam os arranjos atômicos dos principais cristais elementares ou aqueles formados
por 1 único átomo. Destes cristais, aproximadamente 52% apresentam estrutura
cúbica, 28% exibem estrutura hexagonal e os 20% restantes estão distribuídos entre
os outros 5 tipos estruturais. Isto faz com que a estrutura hexagonal tenha grande
importância em cristalografia, o que torna necessário o estudo da mesma. Existem
dois tipos de arranjo hexagonal, quais sejam: hexagonal simples e hexagonal
compacto.
III.4.a. CÉLULA HEXAGONAL SIMPLES (HS)

Estrutura Cristalina 26
Esta estrutura é formada por dois hexágonos sobrepostos, e em cada vértice
destes hexágonos, existe um átomo. Um outro átomo localiza-se no centro de cada
hexágono. A estrutura cristalina hexagonal simples pode ser representada pelo arranjo
hexagonal mostrado na figura III.6. Neste caso, o parâmetro "a" é igual ao parâmetro
"c". Os ângulos basais são de 1200 e os verticais de 900. Esta estrutura cristalina pode
ser encontrada no selênio e no telúrio. O número de átomos existentes no interior de
uma célula hexagonal simples é 3. O fator de empacotamento de cristais hexagonais
simples é calculado da mesma forma feita anteriormente, sendo novamente
necessário determinar o volume de uma célula unitária desta estrutura. Tal volume é
dado por:
C3V = 12 R 3
O F.E. resulta em:
F. E. = 3
43
R
12 R 3 = 0,60
3
3
(III.7)
Figura III.6. Representação esquemática de uma célula unitária HS.
III.4.b. CÉLULA HEXAGONAL COMPACTA (HC)A estrutura hexagonal compacta é formada por dois hexágonos sobrepostos e
um plano intermediário de 3 átomos. Nos hexágonos, novamente, existem 6 átomos
nos vértices e um outro no centro.
Estrutura Cristalina 27
A estrutura cristalina hexagonal compacta pode ser observada na figura III.7.
Neste caso, o parâmetro "a" é diferente do parâmetro "c". Os ângulos basais são
novamente iguais a 1200 e os verticais de 900. A estrutura HC pode ser observada no
berílio, berquélio, lítio, magnésio, cádmio, cobalto, etc. O número de átomos que
efetivamente encontram-se dentro de uma célula unitária HC é igual a 6.
O fator de empacotamento é calculado da mesma maneira efetuada
anteriormente, e o volume da célula unitária é igual a:
C3V = 24 R 2
que resulta em:
F. E. = 6
43
R
24 R 2 = 0,74
3
3
(III.8)
Figura III.7. Representação esquemática de uma célula unitária HC.
III.5. SEQÜÊNCIA DE EMPILHAMENTOA estrutura cúbica de face centrada tem o mesmo fator de empacotamento da
estrutura hexagonal compacta (0,74). Este fato não é apenas uma coincidência, mas

Estrutura Cristalina 28
resultado da natureza dos planos cristalinos que constituem estas duas estruturas.
Observando a seqüência de empilhamento de planos cristalinos na direção da
diagonal do cubo da estrutura CFC e na direção perpendicular à base no caso da
hexagonal compacta, nota-se que os arranjos atômicos, em ambos os casos, são de
mesma natureza. A diferença entre as duas estruturas concentra-se no
posicionamento dos átomos destes planos em relação a um ponto de referência.
Enquanto os planos do cristal HC apresentam apenas duas variações de
posicionamento e assim, seguem uma seqüência do tipo "ABABAB...", os cristais CFC
apresentam três posicionamentos e exibem a seqüência "ABCABCABC..." . A figura
III.8 apresenta detalhes sobre o empilhamento de planos de tais estruturas.
Figura III.8. Empilhamento de planos compactos das estruturas CFC e HC.
III.6. ALOTROPIADiversos elementos, bem como compostos químicos apresentam mais de uma
forma cristalina, dependendo de condições como pressão e temperatura envolvidas.

Estrutura Cristalina 29
Este fenômeno é denominado de alotropia ou polimorfismo. Metais de grande
importância industrial como o ferro, o titânio e o cobalto apresentam transformações
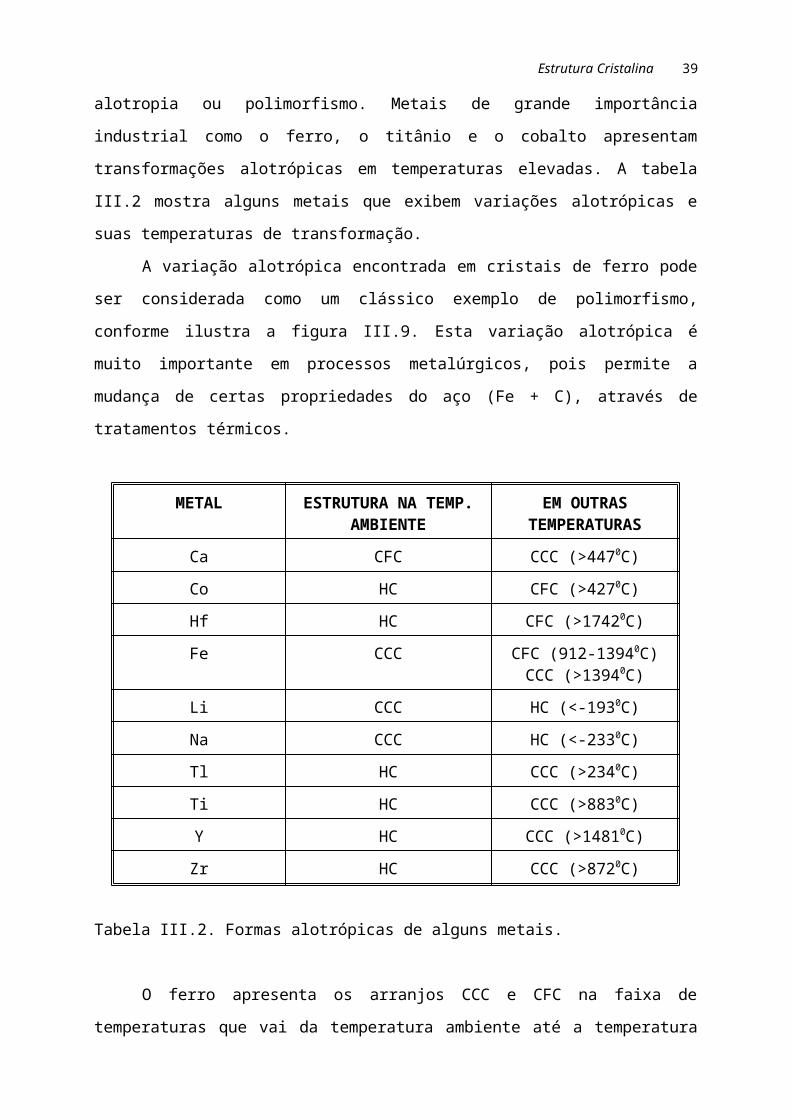
alotrópicas em temperaturas elevadas. A tabela III.2 mostra alguns metais que exibem
variações alotrópicas e suas temperaturas de transformação.
A variação alotrópica encontrada em cristais de ferro pode ser considerada
como um clássico exemplo de polimorfismo, conforme ilustra a figura III.9. Esta
variação alotrópica é muito importante em processos metalúrgicos, pois permite a
mudança de certas propriedades do aço (Fe + C), através de tratamentos térmicos.
METAL ESTRUTURA NA TEMP. AMBIENTE
EM OUTRAS TEMPERATURAS
Ca CFC CCC (>4470C)
Co HC CFC (>4270C)
Hf HC CFC (>17420C)
Fe CCC CFC (912-13940C)CCC (>13940C)
Li CCC HC (<-1930C)
Na CCC HC (<-2330C)
Tl HC CCC (>2340C)
Ti HC CCC (>8830C)
Y HC CCC (>14810C)
Zr HC CCC (>8720C)
Tabela III.2. Formas alotrópicas de alguns metais.
O ferro apresenta os arranjos CCC e CFC na faixa de temperaturas que vai da
temperatura ambiente até a temperatura de fusão do mesmo (15390C). O ferro a
existe de -273 a 9120C e tem estrutura cristalina CCC. Entre 768 e 9120C, o ferro a
deixa de ser magnético e, algumas vezes, é chamado de ferro b. O ferro g existe de
912 a 13940C e tem estrutura CFC. O ferro d existe de 1394 a 15390C, apresentando,
novamente, estrutura CCC. A diferença entre as estruturas CCC do ferro a e do ferro d
reside no valor do parâmetro de rede dos dois casos. Na faixa de temperaturas mais
baixa, o parâmetro de rede é menor.

Estrutura Cristalina 30
Figura III.9. Variação alotrópica do Ferro.
Um outro exemplo clássico de polimorfismo é a variação alotrópica do carbono.
Este elemento é encontrado como diamante, que é o material mais duro na natureza e
no grafite, um material de baixíssima dureza, que pode ser usado como lubrificante. O
diamante é duro porque todas as suas ligações são covalentes. Por outro lado, o
grafite tem ligações covalentes apenas em alguns planos. Estes planos são agregados
a outros planos através de forças secundárias e assim, é fácil provocar o deslizamento
dos mesmos. A figura III.10 apresenta as estruturas do diamante e do grafite.

Estrutura Cristalina 31
(a) Diamante (b) Grafite
Figura III.10. Estruturas cristalinas do carbono nas variações alotrópicas "diamante" e
"grafite".
III.7. POSIÇÕES, DIREÇÕES E PLANOS EM CRISTAISFreqüentemente, é necessário identificar posições, direções e planos em um
cristal. Isto é particularmente importante no caso de metais e suas ligas, que
apresentam propriedades que variam com a orientação cristalográfica. Por exemplo, a
existência de determinados conjuntos de planos e direções definidos como compactos,
desempenham importante papel durante a deformação plástica de metais. A
existência de propriedades dependentes da orientação cristalográfica resulta na
necessidade de se determinar posições, direções e planos em um cristal.
III.7.a. POSIÇÕES EM CRISTAIS CÚBICOSA localização de posições atômicas em uma célula unitária de um cristal cúbico
é obtida pelo uso de um sistema de eixos cartesiano. Em cristalografia, o eixo x é a
direção perpendicular ao plano do papel, o eixo y é a direção a direita do papel e o
eixo z é a direção para cima, como mostra a figura III.11. As direções negativas destes
eixos são opostas às direções mencionadas. As posições atômicas em uma célula
unitária são definidas pelo uso de unidades de distâncias ao longo dos eixos x, y e z,
como mostra a figura III.11.a. Por exemplo, as coordenadas das posições atômicas em
uma célula CCC são mostradas na figura III.11.b. As posições dos átomos nos oito
vértices do cubo têm as coordenadas: (0,0,0), (1,0,0), (0,1,0), (0,0,1), (1,1,1), (1,1,0),
(1,0,1) e (0,1,1). O átomo central desta estrutura ocupa a posição (½,½,½).

Estrutura Cristalina 32
Figura III.11. Posições atômicas em uma célula unitária cúbica.
III.7.b. DIREÇÕES EM CRISTAIS CÚBICOSNo sistema cúbico, as direções cristalográficas são obtidas a partir das
componentes da direção em questão, tomadas nos três eixos cartesianos. Para indicar
esquematicamente uma direção em uma célula unitária, desenha-se um vetor que
parte da origem e atinge a posição definida pelas coordenadas consideradas. Assim,
para se obter uma direção em um cristal, deve-se observar que:
a. Os eixos cristalinos são utilizados como direções básicas;
b. As coordenadas de um ponto são medidas em relação ao parâmetro de cada
eixo e assim, não representam valores reais de distância;
c. A direção [222] é idêntica à direção [111] e dessa forma, a combinação dos
menores números inteiros deve ser utilizada;
d. As direções negativas são indicadas com um traço sobre o índice;
e. Uma direção é representada por índices entre colchetes.
Por exemplo, considerando a figura III.12, as coordenadas do vetor OR, que
passa pela origem são (1,0,0). Assim, a direção do mesmo passa a ser [100]. As
coordenadas do vetor OT são (1,1,1) e sua direção é dada por [111]. As coordenadas
do vetor OM são (1,½,0) e sua direção é representada por [210]. As coordenadas do
vetor ON são (-1,-1,0). Como uma direção negativa é representada por um traço sobre
o índice, a direção deste vetor é dada por 1 1 0 . É importante notar que a origem

Estrutura Cristalina 33
neste último caso, foi deslocada. Genericamente, as letras u, v e w são usadas para
indicar os índices das direções x, y e z respectivamente, o que resulta em [uvw].
Figura III.12. Algumas direções em células unitárias cúbicas.
Algumas vezes, é necessário expressar um conjunto de direções que
representam determinadas direções como a diagonal da face do cubo. Existem 12
direções, neste caso, e uma representação geral de todas elas é dada pela
representação <110>. Esta representação é chamada de família de direções. A
notação <100> é usada para indicar o conjunto de arestas do cubo, assim como as
direções das diagonais do cubo são representadas por <111>.
III.7.c. PLANOS EM CRISTAIS CÚBICOSEm determinadas situações é necessário definir planos atômicos dentro de uma
estrutura cristalina. Para identificar planos cristalinos em cristais cúbicos, o sistema de
notação dos índices de Miller deve ser utilizado. Os índices de Miller de um plano
cristalino são definidos como sendo os inversos das coordenadas de interceptação do
plano com os eixos x, y e z. O procedimento básico para determinar os índices de
Miller para um cristal cúbico são:
a. Escolha de um plano que não passe pela origem (0,0,0);
b. Determinação dos pontos de interceptação do plano com os eixos x, y e z;
c. Obtenção dos inversos das interceptações;
d. Obtenção dos menores números inteiros para representar o plano;
e. Apresentação dos índices obtidos entre parênteses.
Genericamente, as letras h, k e l são usadas para indicar os índices de Miller de
um plano, o que resulta em (hkl). A figura III.13 mostra três dos mais importantes

Estrutura Cristalina 34
planos das estruturas cúbicas. Considerando o plano indicado na figura III.13.a, nota-
se que o mesmo intercepta x em 1, y em e z também em . Tomando os inversos
pode-se obter os índices de Miller, que resulta em (100). Como estes índices não
envolvem frações, os mesmos são usados para representar o referido plano.
Considerando o plano da figura III.13.b., observa-se que o mesmo intercepta os eixos
em 1, 1 e . Os inversos fornecem 1, 1 e 0, que resultam em (110). Finalmente, o
plano apresentado pela figura III.13.c intercepta os eixos em 1, 1 e 1, que resulta nos
índices de Miller (111).
Quando é necessário representar um conjunto de planos equivalentes dentro
de um cristal, são utilizadas as famílias de planos. Uma família de planos, como a dos
que passam pelas faces do cubo ou (100), (010), (001), etc, é representada pela
notação {100}. Da mesma forma, a família de planos que dividem o cubo em duas
partes iguais é dada por {110}.
Figura III.13. Planos em células unitárias cúbicas.
III.7.d. DIREÇÕES EM CRISTAIS HEXAGONAISDireções em cristais hexagonais são geralmente indicadas por quatro índices u,
v, t e w, apresentados entre colchetes. Os índices u, v e t são relativos aos eixos a1, a2
e a3, respectivamente e o índice w é relativo ao eixo c. Basicamente, o procedimento a
ser seguido no uso destes índices para a identificação de planos no sistema HC,
envolve a obtenção dos menores inteiros que representem a direção e que satisfaça a
relação u+v=-t. A figura III.14 apresenta algumas direções na célula hexagonal
compacta. Este método de identificação de planos no sistema HC não é considerado
uma técnica eficiente e prática.

Estrutura Cristalina 35
Figura III.14. Direções importantes em uma célula unitária hexagonal compacta.
III.7.e. PLANOS EM CRISTAIS HEXAGONAISOs planos em cristais hexagonais são identificados também pelo uso de
quatros eixos. Os índices empregados neste caso são denominados como índices de
Miller-Bravais e são representados pelas letras h, k, i e l, apresentadas como no caso
anterior entre parênteses ou (hkil). Estes índices são baseados em um sistema de
coordenadas com quatro eixos, conforme mostrado na figura III.15.
Figura III.15. Os quatro eixos usados como referência em um sistema hexagonal.
O plano basal nesta estrutura é considerado um plano muito importante. Como
o plano basal superior é paralelo aos eixos a1, a2 e a3, então o plano interceptará tais
eixos no infinito. Por outro lado, pode-se afirmar que tal plano intercepta o eixo c em 1.
Assim, a representação dos planos basais é dada por (0001), como mostra figura
III.16.a. Usando o mesmo método, os pontos onde o plano ABCD da figura III.16.b
intercepta os eixos são a1=+1, a2=, a3=-1, e c=. Isto permite afirmar que tal plano é
representado por (1010). Os outros planos das faces da célula unitária hexagonal
compacta são determinados seguindo o mesmo procedimento.

Estrutura Cristalina 36
Figura III.16. Índices de Miller de alguns planos no sistema hexagonal compacto.
III.8. DENSIDADES ATÔMICA EM CRISTAISDentre os planos e direções de um cristal, alguns revelam ser mais compactos,
ou seja, possuem mais átomos por unidade de comprimento ou de área. Assim no
sistema CS, as direções mais compactas são as da família <100> e os planos mais
compactos são os da família {100}.
A definição de uma direção compacta envolve a definição de densidade linear
de átomos. Esta densidade é obtida determinando o número de átomos que
efetivamente estão contidos em um determinado comprimento. Assim, a densidade
linear da família de direções <100>, no sistema CS (figura III.17) é igual a:
linear
o
D = n de atomoscomprimento
=
12
+ 12
a =
1a
(III.9)
Da mesma forma, um plano compacto é determinado calculando-se o número
de átomos que efetivamente ocupam uma certa área. Assim, a densidade planar de
átomos da família de planos {100}, no sistema CS (figura III.17) é igual a:
planar
o
2 2D = n de atomos
area =
14
+ 14
+ 14
+ 14
a =
1a
(III.10)

Estrutura Cristalina 37
Figura III.17. Plano (100) e direção [100] no sistema CS.
Os planos e direções compactos são importantes porque desempenham papel
significativo no estudo da deformação plástica de metais. Os átomos de um cristal
solicitado mecanicamente escorregam ao longo de planos compactos, seguindo
direções compactas.
III.9. ANÁLISE DE ESTRUTURAS CRISTALINASGrande parte do conhecimento adquirido sobre estruturas cristalinas é
resultado da utilização de técnicas de difração de raio-X. Estas técnicas permitem
obter informações detalhadas sobre dimensões, presença de defeitos e orientação
da rede cristalina. O uso do raio-X no estudo de cristais deve-se ao fato de que esta
radiação tem comprimento de onda próximo aos valores de distâncias entre planos
cristalinos.
A utilização de raio-X iniciou-se logo em seguida a sua descoberta em 1895,
por Roentgen. Apesar de, naquela época, a natureza desta radiação não ser
conhecida em detalhes (razão do nome "raio-X"), o raio-X foi então, aplicado em
estudos da estrutura interna de materiais opacos (radiografia) devido ao seu alto
poder de penetração. Desde aquela época, esta radiação era conhecida por
propagar-se em linha reta, sensibilizar filmes fotográficos e apresentar velocidade
de propagação definida. Os raios-X empregados em técnicas de difração são ondas
eletromagnéticas com comprimento de onda na faixa de 0,05 a 0,25nm (0,5 a 2,5
Å). Como comparação, o comprimento de onda da luz visível é da ordem de 600nm
(6.000 Å).

Estrutura Cristalina 38
III.9.a. OBTENÇÃO DE RAIOS-X
A obtenção de raios-X para difração envolve a aplicação de tensões da
ordem de 35kV entre um catodo e um anodo, dentro de um sistema apresentando
alto vácuo. A figura III.18 mostra um diagrama esquemático de um equipamento de
raio-X.
Figura III.18. Diagrama esquemático de um sistema de geração de raios-X.
O funcionamento do mesmo é bastante simples:
- Ao ser aquecido, o filamento de tungstênio (catodo) libera elétrons por
emissão termo-iônica. Devido a elevada diferença de potencial (35kV), os elétrons
liberados são acelerados, ganham energia cinética e movimentam-se em direção ao
anodo (molibdênio). Ao colidirem com o anodo, tais elétrons provocam a emissão de
raio-X. Entretanto, em torno de 98% da energia cinética dos elétrons é transformada
em calor, o que torna necessário o emprego de um sistema de refrigeração do
anodo.
III.9.b. DIFRAÇÃO DE RAIOS-XEm 1912, a partir da teoria eletromagnética da luz, foi possível prever que o
raio-X podia ser difratado por estruturas cristalinas. Este fato foi inicialmente
Estrutura Cristalina 39
implementado experimentalmente na investigação da estrutura cristalina do NaCl,
KCl e KBr. Até aquela época, a estrutura cristalina de metais era desconhecida.
Se um feixe de raio-X monocromático (freqüência única) incide sobre um
átomo isolado, elétrons do mesmo são excitados e vibram com a mesma freqüência
do feixe incidente. Tais elétrons em vibração emitirão raio-X em todas direções com
a mesma freqüência do feixe incidente. Assim, o átomo isolado espalha o feixe
incidente em todas as direções. Entretanto, quando o mesmo feixe incide sobre um
conjunto de átomos ordenados, como é o caso da estrutura cristalina e se este feixe
monocromático tiver comprimento de onda com valor semelhante aos
espaçamentos entre tais átomos, então ocorrerá interferência construtiva em
algumas direções e destrutiva em outras. A figura III.19 ilustra casos de interferência
destrutiva (figura II.19.a) e construtiva (figura III.19.b).
Observando esta mesma ilustração (figura III.19.c), nota-se que a
interferência construtiva de dois raios monocromáticos (raio 1 e 2) ocorrerá quando
os mesmos permanecerem em fase. Isto acontecerá quando o raio 2 percorrer uma
distância extra MP+PN, equivalente a um número inteiro de comprimentos de ondas
(l). Então:
n = MP + PNl (IIII.11)
onde n=1,2,3,... e é chamado ordem de difração. Como MP e PN são iguais a
dhlksenq, onde dhlkl é a distância entre dois planos com índices (hkl), a condição
necessária para ocorrer interferência construtiva deverá ser:
n = 2d senhkll q (III.12)
Esta equação é conhecida como lei de Bragg e relaciona comprimento de
onda (l) e ângulo do feixe (q) de raio-x incidente e distância interplanar dhkl. Como
na maioria dos casos a ordem de difração é 1, a lei de Bragg torna-se igual a:
l q = 2 d senhkl (III.13)
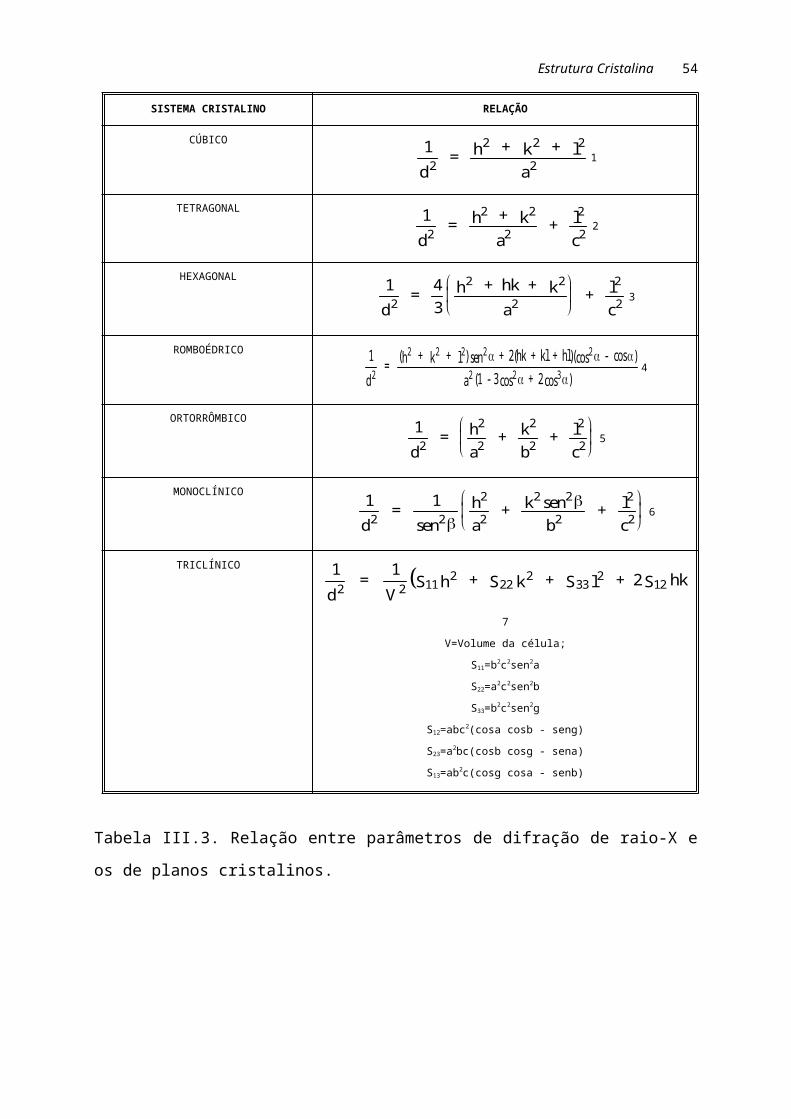
A tabela III.3 apresenta a relação entre espaçamento interplanar (dhkl),
parâmetros da célula unitária (a, b, c), ângulos a (entre os eixos y e z), b (entre os
eixos x e z) e g (entre os eixos x e y) e planos cristalinos (h k l).
SISTEMA CRISTALINO RELAÇÃO
CÚBICO 1d
= h + k + la2
2 2 2
21

Estrutura Cristalina 40
SISTEMA CRISTALINO RELAÇÃO
TETRAGONAL 1d
= h + ka
+ lc2
2 2
2
2
22
HEXAGONAL 1d
= 43
h + hk + ka
+ lc2
2 2
2
2
2
3
ROMBOÉDRICO 1
d =
(h + k + l ) sen + 2(hk + kl + hl)( - )
a (1 - 3 + 2 )2
2 2 2 2 2
2 2 3a a a
a acos cos
cos cos4
ORTORRÔMBICO1d
= ha
+ kb
+ lc2
2
2
2
2
2
2
5
MONOCLÍNICO1d
= 1sen
ha
+ k senb
+ lc2 2
2
2
2 2
2
2
2bb
6
TRICLÍNICO 1
d =
1
VS h + S k + S l + 2 S hk + 2 S kl + 2 S hl
2 2 112
222
332
12 23 13
7
V=Volume da célula;
S11=b2c2sen2a
S22=a2c2sen2b
S33=b2c2sen2g
S12=abc2(cosa cosb - seng)
S23=a2bc(cosb cosg - sena)
S13=ab2c(cosg cosa - senb)
Tabela III.3. Relação entre parâmetros de difração de raio-X e os de planos
cristalinos.

Estrutura Cristalina 41
Figura III.19. Reflexão de raios-X de natureza monocromática por planos de um
cristal.
III.9.c. ANÁLISE DE ESTRUTURAS CÚBICASUm ensaio de raio-X é executado com o emprego de um dispositivo
denominado de goniômetro, conforme mostra a figura III.20. Nesse equipamento, a
amostra é colocada no ponto O e é girada para que o ângulo de incidência do feixe
de raio-X (T) seja variado.
O feixe de raios-X difratados é medido através do detetor C. Em função das
características de um goniômetro, em geral, o ângulo de difração é medido como
2q. A figura III.21 apresenta um difratograma resultante de um ensaio de raio-X do
tungstênio. A intesidade de difração é maior para os planos de alta densidade de
átomos. Como, geralmente, a distância entre planos compactos é grande, a análise
da equação III.13, permite concluir que os planos de maior intensidade de difração
correspodem a baixos ângulos.
Na análise de estruturas cúbicas, apenas alguns planos podem provocar
difração. No caso das estruturas CCC, a difração é possível quando a soma dos
índices de Miller resulta em um número par. Para as estruturas CFC, a difração
ocorre quando todos os índices são pares ou todos são impares. A tabela III.4
mostra os planos de difração nas estruturas cúbicas.
Família (h2+k2+l2) Planos de DifraçãoCCC CFC
{100} 1
{110} 2 X
{111} 3 X
{200} 4 X X
{210} 5
{211} 6 X
{220} 8 X X
{221} 9
{310} 10 X
Tabela III.4. Família de planos em estruturas cúbicas que provocam difração.

Estrutura Cristalina 42
Figura. III.20. Goniômetro empregado em ensaios de difração de raios-X
A técnica de difração de raios-X pode ser facilmente empregada para
diferenciar estruturas CCC e CFC. Analisando a tabela III.3, observa-se que para as
estruturas cúbicas vale a relação:
12d
= 2h + 2k + 2l
2a(III.14)
Figura III.21. Difratograma de raio-X do tungstênio
Combinando as equações III.13 e III.14 e elevando ambos os lados ao
quadrado, pode-se obter:
sen2 = 2
4a22h + 2k + 2lq
l (III.15)

Estrutura Cristalina 43
Como l e a são constantes, então:
sensen
21
22
= 2h + 2k + 2l2h + 2k + 2l
1 1 1
2 2 2
(III.16)
Onde q1 e q2 estão associados aos principais planos de difração. A aplicação
da equação III.16 associada à tabela III.4 permite prever que os dois primeiros
planos de uma estrutura CCC resulta no valor sen2q1/sen2q2=0,5. No caso das
estruturas CFC, a relação sen2q1/sen2q2=0,75.
EXERCÍCIOSIII.1. Quais são as 14 células unitárias de Bravais ?
III.2. Quais são as estruturas cristalinas metálicas mais comuns ? Liste alguns
metais que apresentam estas estruturas.
III.3. Qual é o número de coordenação dos átomos de uma estrutura CCC ?
III.4. Qual é a relação entre tamanho da aresta "a" da célula CCC e raio atômico ?
III.5. O Nb, na temperatura ambiente tem estrutura CCC e apresenta raio atômico de
0,147 nm. Calcule o valor do parâmetro de rede "a" em nanometros.
III.6. Calcule o fator de empacotamento da estrutura CFC.
III.7. Quantos átomos por célula existem na estrutura HC ?
III.8. O Ni é CFC com uma densidade de 8,9 Mg/m3 e tem sua M.A. é igual a 58,71.
a. Qual é o volume por célula unitária baseado no valor da densidade ? b. Calcule o
raio atômico do Ni a partir de sua resposta na parte (a).
III.9. O Titânio é CCC em alta temperatura. Seu raio aumenta em 2% durante sua
transformação de CCC para HC no resfriamento. Qual a variação percentual de
volume que ocorre nesta transformação ?

Estrutura Cristalina 44
III.10. Liste as coordenadas das posições atômicas dos 8 átomos nos vértices e as
dos 6 nas faces de uma estrutura CFC.
III.11. Desenhe as seguintes direções cristalográficas em uma célula CCC e em
outra tetragonal com a/c=3:
a. [001] b. [110] c. [111] d. [113] e. [223]
III.12. Qual é a família de planos {100} no sistema cúbico ?
III.13. Um plano no sistema cúbico intercepta os eixos em x=2/3, y=-1/2 e z=1/2.
Qual são os índices de Miller para este plano ?
III.14. Desenhe os seguintes planos cristalográficos na estrutura CCC e liste as
coordenadas dos átomos com centros nestes planos:
a. (100) b. (110) c. (111)
III.15. O Al é CFC e tem parâmetro de rede "a" igual a 0,3158 nm. Calcule a
densidade planar de átomos nos planos (100) e (111).
III.16. Considerando novamente o Al, calcule a densidade linear de átomos nas
direções [100] e [111].
III.17. Derive a lei de Bragg a partir de um caso onde um raio incidente sofre
difração pelos planos paralelos de um cristal.
III.18. Uma amostra de um metal CCC foi colocada em um difratômetro de raios-X
com l=0,1541nm. A difração obtida pela família de planos {220} apresentou
2q=82,5500. Calcule o valor do parâmetro de rede deste elemento. Assuma difração
de 1ª ordem.

Formação e Imperfeições da Estrutura Cristalina 45
FORMAÇÃO E IMPERFEIÇÕES DA ESTRUTURA CRISTALINAIV.1. INTRODUÇÃO
Durante a solidificação, os metais sofrem o rearranjo de seus átomos que
determina a estrutura cristalina dos mesmos. Dependendo do modo com que o líquido
transforma-se em sólido, podem ocorrer defeitos no empilhamento e organização dos
átomos, resultando em imperfeições estruturais. O tipo e a quantidade destas
imperfeições afetam decisivamente algumas propriedades e o comportamento dos
materiais cristalinos.
Com exceção de alguns poucos produtos conformados por sinterização
(metalurgia do pó), todos os produtos metálicos passam necessariamente pelo
processo de solidificação, em algum estágio de sua fabricação. Em geral, o processo
de solidificação pode ser dividido em duas etapas:
a. Formação de embriões de cristais estáveis dentro do líquido ou etapa de
nucleação, como mostra figura IV.1;
b. Transformação dos núcleos em cristais, ou etapa de crescimento.
Figura IV.1. Formação de embriões de cristais estáveis dentro do líquido ou etapa de
nucleação.
A transformação líquido/sólido e a conseqüente formação da estrutura cristalina
é observada na prática em duas situações diferentes, quais sejam: solidificação com
nucleação e crescimento controlados e solidificação com nucleação e crescimento
não-controlados. O primeiro caso envolve situações onde existe a necessidade de se

Formação e Imperfeições da Estrutura Cristalina 46
produzir um sólido, onde a característica principal do mesmo é a qualidade do arranjo
cristalino. Esta situação é geralmente encontrada na obtenção de insumos básicos
para microeletrônica, onde a necessidade de monocristais perfeitos de silício, arseneto
de gálio, etc, é fundamental. Os processos de crescimento a partir do líquido
(solidificação), com controle de seus parâmetros operacionais, basicamente são
derivações do processo de solidificação direcional. O processo de solidificação
direcional consiste em solidificar uma amostra na forma de uma barra e inicialmente
no estado líquido, a partir de uma das extremidades, como mostra a figura IV.2.
Efetuando esta operação com velocidades extremamente baixas (1,0 cm/h) e assim,
tendo controle sobre a direção e taxa de resfriamento do líquido, é possível obter um
sólido com alta perfeição cristalina.
Figura IV.2. Diagrama esquemático do processo de solidificação unidirecional, que
consiste em solidificar uma amostra na forma de uma barra e inicialmente no estado
líquido, a partir de uma das extremidades.
Três das mais conhecidas técnicas industriais para a obtenção de monocristais
são: o processo Czochralski, o processo Bridgman e o processo de Fusão Zonal
Flutuante. A figura IV.3 apresenta diagramas esquemáticos destes processos. A outra
classe de transformação líquido/sólido, ou seja, a solidificação sem controle rigoroso
de seus parâmetros operacionais, pode ser sintetizada nos casos encontrados na
industria metalúrgica-siderúrgica. Fazem parte deste caso, os processos de
lingotameto de metais (contínuo e estático) e fundição, etc. Neste caso, o líquido a ser
transformado em sólido é vazado em moldes e perde calor conforme a geometria e
parâmetros térmicos do sistema. Assim, o sólido obtido exibe estrutura cristalina com a
presença acentuada de defeitos. Como, geralmente, a direção de crescimento não é
única, esta classe de processos caracteriza-se por apresentar um sólido com diversas

Formação e Imperfeições da Estrutura Cristalina 47
orientações cristalográficas, que resulta em um material definido como policristalino. A
figura IV.4 apresenta diagramas esquemáticos dos processos de fundição e
lingotamento de metais.
Figura IV.3. Processos industriais para a obtenção de monocristais: (a) Czochralski; (b)
Bridgman e (c) Fusão Zonal Flutuante.

Formação e Imperfeições da Estrutura Cristalina 48
Figura IV.4. Diagramas esquemáticos dos processos: (a) Fundição e (b) Lingotamento
contínuo de metais.

Formação e Imperfeições da Estrutura Cristalina 49
IV.2. IMPERFEIÇÕES ESTRUTURAISAs estruturas cristalinas analisadas até aqui apresentam como característica
básica, arranjos cristalinos muito bem definidos. Entretanto, os cristais observados na
prática nunca são totalmente perfeitos, exibindo defeitos de diversas naturezas. Tais
imperfeições afetam diretamente várias características dos materiais, como os
parâmetros envolvidos na deformação plástica, na condutividade elétrica de semi-
condutores, na corrosão em metais e em processos de difusão atômica. As
imperfeições presentes em estruturas cristalinas podem ser de três tipos básicos,
quais sejam: defeitos pontuais, defeitos em linha e defeitos de superfície.
IV.2.a. DEFEITOS PONTUAISOs cristais podem apresentar defeitos em pontos isolados de sua estrutura,
dando lugar às imperfeições de ponto. Dentre as imperfeições pontuais, as mais
importantes são: as vacâncias ou vazios, os átomos intersticiais e os átomos
substitucionais.
O tipo de defeito mais simples é a vacância. As vacâncias são vazios pontuais
causados pela ausência de átomos em algumas posições da rede cristalina, como
mostra a figura IV.5. Este tipo de defeito pode ser produzido durante o processo de
solidificação, como resultado de perturbações locais no crescimento do cristal. Uma
outra causa destas imperfeições é o rearranjo atômico de um cristal já existente,
devido à mobilidade de seus átomos. Nos metais, a concentração de vacâncias
raramente passa de 1 para cada 104 átomos. As vacâncias podem ainda ser
resultantes da deformação plástica, do resfriamento rápido e do bombardeamento da
rede cristalina por partículas atômicas, como nêutrons.
Figura IV.5. Vacância ou vazio pontual causado pela ausência de átomos em algumas
posições da rede cristalina.

Formação e Imperfeições da Estrutura Cristalina 50
Em cristais iônicos, os defeitos pontuais exibem caráter mais complexo devido à
necessidade de manter a neutralidade elétrica do sistema. Mesmo assim, pode-se
observar defeitos estruturais, como o caso em que dois íons de cargas opostas
perdidos dentro da estrutura entram em contato, criando uma vacância dupla. Este tipo
de defeito é conhecido como imperfeição de Schottky. Quando um íon positivo move-
se para uma posição intersticial do cristal iônico, uma "vacância cátion" é criada,
conhecida como imperfeição de Frenkel. Os defeitos de Schottky e Frenkel são
mostrados na figura IV.6. A presença dos defeitos de Schottky e Frenkel em cristais
iônicos aumenta a condutividade elétrica dos mesmos.
Uma outra classe de defeitos são os átomos substitucionais e intersticiais
estranhos à rede cristalina. Os átomos intersticiais são imperfeições causadas pela
presença de átomos estranhos nos interstícios da rede cristalina e os átomos
substitucionais são defeitos provocados pela existência de átomos estranhos nos
próprios vértices da rede cristalina, em substituição aos átomos que alí deveriam estar
se não existissem vacâncias. Defeitos dessa natureza podem modificar o
comportamento de certas propriedades. Por exemplo, a presença de uma quantidade
muito pequena de átomos estranhos à rede cristalina do silício pode afetar, de modo
significativo, a condutividade elétrica do mesmo. Estes dois tipos de defeitos pontuais
são frequentemente observados durante a formação das ligas metálicas, na forma de
soluções sólidas.
Figura IV.6. Defeitos de Frenkel e Schottky.
Na maioria das aplicações de engenharia, a necessidade de propriedades
específicas, faz com que o uso de materiais metálicos nem sempre esteja restrito aos
metais puro. Na verdade, apenas em um número bastante limitado de aplicações, os

Formação e Imperfeições da Estrutura Cristalina 51
metais podem ser encontrados na forma pura ou quase pura. Por exemplo, o cobre de
alta pureza (99,99%) é usado na confecção de fios elétricos devido a sua elevada
condutividade elétrica. O alumínio super-puro (99,99%) é usado na fabricação de
objetos decorativos, pois o mesmo permite obter uma superfície melhor acabada. Por
outro lado, a maioria dos materiais metálicos usados em engenharia, estão
combinados com outros metais ou não-metais. Estas combinações, denominadas de
ligas metálicas, têm o objetivo de aumentar a resistência mecânica, a resistência à
corrosão ou melhorar outras propriedades.
Uma liga metálica, ou simplesmente uma liga, é a mistura de dois ou mais
metais ou metais e não-metais. Estas ligas podem ter estruturas relativamente
simples, como a de uma peça de bronze. O bronze é essencialmente uma liga binária
(dois metais), contendo 70% em peso de Cu e 30% em peso de Zn. Por outro lado,
certas ligas podem ser extremamente complexas como as superligas à base de
níquel, denominadas comercialmente de Inconel 718 e usadas na confecção de peças
de motores a jato. Estas ligas contém nominalmente em torno de 10 elementos. Um
outro exemplo de liga metálica pode ser observado no aço, onde as estruturas CFC e
CCC do ferro abrigam átomos de carbono. Esta combinação permite obter um material
extremamente versátil, com aplicações bastante diversificadas.
O tipo mais simples de liga metálica é aquele que forma uma solução sólida.
Uma solução sólida é um sólido que consiste de dois ou mais elementos
atomicamente dispersos em uma estrutura monofásica. Em geral existem dois tipos de
soluções sólidas: substitucional e intersticial.
Nas soluções sólidas substitucionais formadas por dois elementos, os átomos
do soluto podem ser substitutos dos átomos do solvente na rede cristalina. Na figura
IV.7 é mostrado um plano (111) de um cristal CFC contendo átomos do soluto
substituindo os átomos do solvente. Neste caso, a estrutura do solvente não é
alterada, sendo comum a distorção da rede cristalina, já que os átomos do soluto nem
sempre exibem o mesmo diâmetro atômico dos átomos do solvente.
A fração de átomos de um elemento que pode ser dissolvida em outro, é
definida como solubilidade. O termo solubilidade significa a quantidade de um certo
material A (soluto) que pode ser dissolvido em outro B (solvente) e varia de um valor
muito pequeno, próximo de zero, até 100%. A solubilidade é dada em "% peso" e "%
atômica".

Formação e Imperfeições da Estrutura Cristalina 52
Assim, uma liga cobre-zinco com 20% em peso de zinco, possui, em 100
gramas da liga, 20 gramas de zinco e 80 gramas de cobre. Do mesmo modo, uma liga
com 20% em átomos de zinco apresenta em cada 100 átomos de liga, 20 átomos de
zinco e 80 átomos de cobre. Como as densidades dos materiais são diferentes, é
evidente que 20% de zinco em peso não correspondem a 20% de zinco em átomos.
Para o caso de formação de uma solução sólida substitucional, a solubilidade de um
elemento em outro será elevada, desde que as seguintes condições sejam satisfeitas:
a. Os raios dos átomos dos dois elementos não devem diferir em mais de 15%;
b. A estrutura cristalina dos dois elementos deve ser a mesma;
c. Não deve existir diferença significativa entre a eletronegatividade dos dois
elementos, assim compostos não serão formados;
d. Os dois elementos devem ter a mesma valência.
Na tabela IV.1 observa-se que a facilidade de um elemento dissolver-se em
outro, é maior se o seu diâmetro for próximo do diâmetro do solvente, no caso o cobre.
SOLUTO SOLVENTE RELAÇÃO DE RAIOS SOLUBILIDADE
% PESO % ATÔMICA
Ni (CFC) Cu (CFC) 1,246/1,278=0,98 100 100
Al (CFC) Cu (CFC) 1,431/1,278=1,12 9 19
Ag (CFC) Cu (CFC) 1,444/1,278=1,14 8 6
Pb (CFC) Cu (CFC) 1,750/1,278=1,37 » 0 » 0
Tabela IV.1. Solubilidade de elementos CFC no cobre, em função de seus raios
atômicos.
Figura IV.7. Plano (111) de um cristal CFC com o soluto substituindo o solvente.

Formação e Imperfeições da Estrutura Cristalina 53
O soluto intersticial é o que fica nos "vãos" da matriz. Estes vãos ou vazios são
chamados de interstícios. As soluções sólidas intersticiais são formadas quando um
átomo é muito maior que o outro. Por exemplo, o ferro a 10000C apresenta estrutura
CFC com o maior vão de diâmetro igual a 1,0 Å. Assim estes "buracos" abrigam
facilmente o hidrogênio (d=0,9 Å), o boro (d=0,92 Å) e com certa dificuldade, o
carbono (d=1,5 Å). Entretanto, apesar dessa diferença, um máximo de 2,08 % em
peso de carbono pode ser dissolvido intersticialmente no ferro a 11480C. A figura IV.8
ilustra esquematicamente a distorção da rede cristalina do ferro quando o carbono
ocupa posições intersticiais na mesma. Na figura IV.9 são apresentadas as estruturas
CFC e CCC do ferro, com os interstícios tetraédricos e octaédricos. Apesar da célula
unitária CCC apresentar diversos vãos, a solubilidade de carbono no Fe é maior em
células CFC, pois as mesmas concentram o espaço vazio da célula, nos vãos
octaédricos.
Figura IV.8. Diagrama esquemático de uma solução sólida de carbono em ferro CFC,
mostrando o plano (100). Note a distorção da rede cristalina do ferro.
Figura IV.9. Interstícios da estrutura do Fe CFC e CCC. (+) octaédrico e (·)
tetraédrico.
Formação e Imperfeições da Estrutura Cristalina 54
IV.3. DEFEITOS LINEARES (DISCORDÂNCIAS)Os cristais podem apresentar defeitos alinhados e contínuos em sua estrutura,
dando origem às imperfeições de linha. Os defeitos de linha, também chamados de
discordâncias são defeitos que causam a distorção da rede cristalina em torno de uma
linha e caracterizam-se por envolver um plano extra de átomos. Estas imperfeições
podem ser produzidas durante a solidificação, na deformação plástica de sólidos
cristalinos ou ainda como resultado da concentração de vacâncias.
Os três principais tipos de defeitos em linha são conhecidos como: discordância
em cunha, discordância em hélice e discordância mista.
IV.3.a. DISCORDÂNCIA EM CUNHAOcorre pela interrupção de um plano atômico como mostra a figura IV.10.
A distância de deslocamento dos átomos ao redor da discordância é
denominada de vetor de Burgers (b) e neste caso esse vetor é perpendicular à linha
de discordância.
Figura IV.10. Ilustração de uma discordância em cunha, que ocorre pela interrupção
de um plano atômico. A letra b corresponde ao vetor de Burgers.
IV.3.b. DISCORDÂNCIA EM HÉLICEUma discordância é helicoidal quando o empilhamento é feito como se fôsse
uma mola. Neste caso, o vetor de Burgers é paralelo à linha de discordância, conforme
mostra a figura IV.11.

Formação e Imperfeições da Estrutura Cristalina 55
Figura IV.11. Discordância em hélice. O vetor de Burgers é paralelo à linha de
discordância.
IV.3.c. DISCORDÂNCIA MISTA (CUNHA + HÉLICE)As discordâncias são produzidas durante solidificação do material ou quando é
aplicada uma tensão cisalhante sobre o mesmo, como mostra a figura IV.12. A
discordância mista é formada por uma discordância em cunha associada a uma
discordância em hélice. Neste caso, as duas discordâncias apresentam uma única
linha de discordância.
Figura IV.12. Discordância mista, que é produzida durante a solidificação do material
ou quando aplica-se uma tensão cisalhante sobre o mesmo. A discordância mista é
formada por uma discordância em cunha associada a uma discordância em hélice.
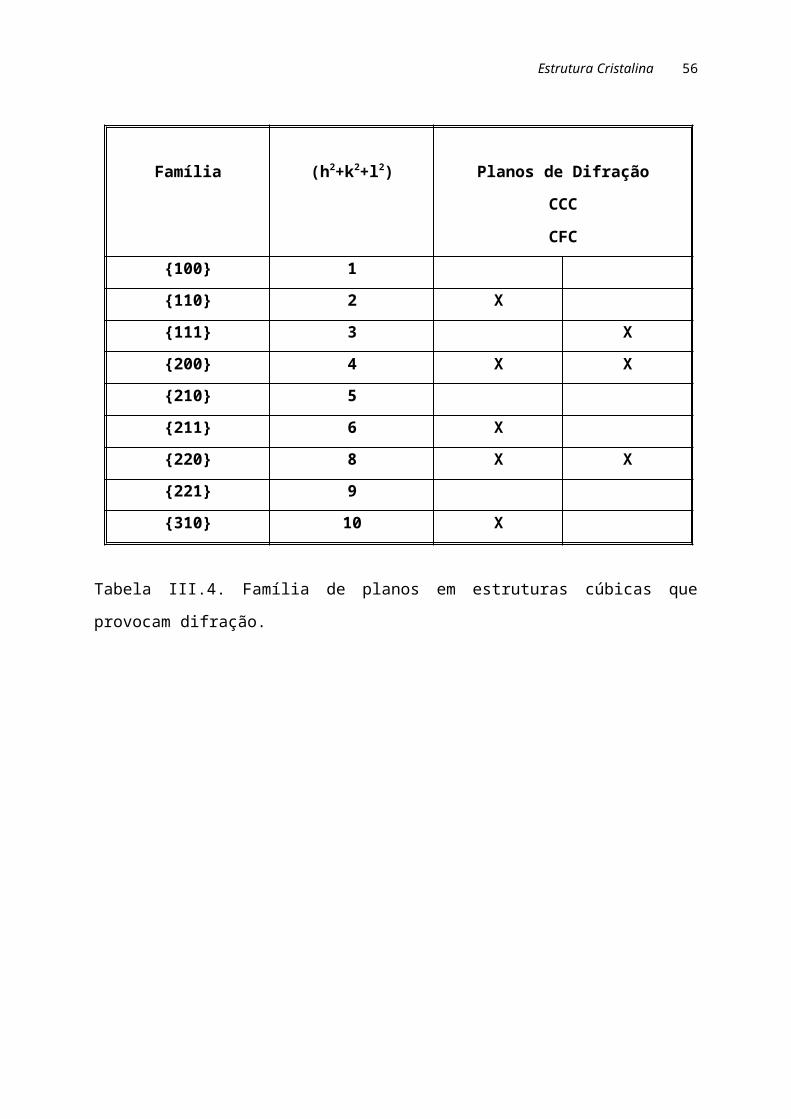
Formação e Imperfeições da Estrutura Cristalina 56
IV.4. DEFEITOS DE SUPERFÍCIESOs cristais também apresentam defeitos que se estendem ao longo de sua
estrutura, formando superfícies e denominados de imperfeições de superfície. Esse
tipo de imperfeição cristalina pode ser de três tipos: superfícies livres, contornos de
grão e maclas.
IV.4.a. SUPERFÍCIES LIVRESApesar de serem consideradas o término da estrutura cristalina, as superfícies
externas de um cristal são consideradas defeitos cristalinos, já que o número de
vizinhos de um átomo superficial não é o mesmo de um átomo no interior do cristal
(figura IV.13). Os átomos superficiais possuem vizinhos apenas de um lado, tem maior
energia e assim, estão ligados aos átomos internos mais fragilmente.
Figura IV.13. Apesar de ser considerada o término da estrutura cristalina, as
superfícies externas de um cristal são consideradas defeitos cristalinos, já que o
número de vizinhos de um átomo superficial não é o mesmo de um átomo no interior
do cristal.
IV.4.b. CONTORNOS DE GRÃOUma barra de cobre puro, embora contenha um único elemento, possui vários
grãos, ou seja, regiões onde a estrutura cristalina tem a mesma orientação. Durante a
solidificação, vários núcleos sólidos surgem no interior do líquido, como é apresentado
na figura IV.14.
Numa fase seguinte, denominada de crescimento, estes núcleos crescem e se
juntam, formando nestas "juntas", uma região conhecida como contorno de grão.
Como os diversos grãos não apresentam necessariamente a mesma orientação

Formação e Imperfeições da Estrutura Cristalina 57
cristalográfica, como pode ser visto na figura IV.15, o encontro dos mesmo cria
surperfícies de contato dentro do cristal, como é observado na figura IV.16.
Figura IV.14. Formação de um material policristalino: (a) Etapa de nucleação; (b)
Etapa de crescimento; (c) Material policristalino.
Figura IV.15. Os grãos de um material policristalino não apresentam uma mesma
orientação cristalográfica.
Figura IV.16. Superfícies de contato associadas aos contornos de grão.

Formação e Imperfeições da Estrutura Cristalina 58
O tamanho de grão de um material policristalino é importante ser conhecido, já
que o número de grãos tem papel significativo em muitas propriedades dos materiais,
especialmente na resistência mecânica. Em baixas temperaturas, até metade da
temperatura de fusão do material, os contornos de grão aumentam a resistência do
material através da limitação do movimento de discordâncias. Em altas temperaturas
pode ocorrer o escorregamento de contornos de grão ou seja o mecanismo de
deformação plástica nestas temperaturas é o de fratura intergranular. Este mecanismo
é um dos responsáveis pela queda da resistência mecânica do material em
temperaturas elevadas. Na produção de peças submetidas a temperaturas mais
próximas a de fusão do material, como é o caso de turbinas de avião, são utilizados
processos de fundição com crescimento direcional e controlado. Isto permite obter um
sólido com um número de grãos pequeno e indicado para temperaturas de trabalho
elevadas. A tabela IV.2 apresenta a padronização do tamanho de grão cristalino
segundo a ASTM. Nesta tabela, o número do tamanho de grão pode ser determinado
pela equação:
N = 2n-1 (IV.1)
onde n é um número inteiro definido como o número do tamanho de grão da ASTM
(American Society for Testing and Materials) e N é o número de grãos por pol2, em um
material polido, atacado quimicamente e observado com o aumento de 100X. A figura
IV.17 mostra micrografias de aço baixo carbono, atacado com NITAL (H2NO3 e álcool)
e o respectivo tamanho de grão segundo a ASTM.
Figura IV.17. Micrografias de aço baixo carbono, atacado com Nital (H2NO3 + álcool) e
o respectivo tamanho de grão segundo a ASTM (X 100): (a) nº7; (b) nº8; (c) nº9.

Formação e Imperfeições da Estrutura Cristalina 59
Como os contornos de grão são regiões onde os átomos estão fragilmente
ligados uns aos outros, a ação de um ataque químico permite revelar o mesmo, pois
nestes pontos é mais fácil "arrancar" os átomos, em comparação com regiões no
interior do grão, como mostra a figura IV.18. A região do contorno de grão aparece
mais escura no microscópio devido a menor capacidade de reflexão de luz da mesma
(figura IV.19).
Número do Tamanho de Quantidade Média de Grãos
Grão (n) Por mm2 X 1 Por pol2 X 100
1 15,5 1,0
2 31,0 2,0
4 124 8,0
6 496 32,0
10 7940 512
Tabela IV.2. Tamanho de grão segundo a ASTM.
(a) (b)
Figura IV.18. (a) Amostra só polida e (b) atacada quimicamente.

Formação e Imperfeições da Estrutura Cristalina 60
O contorno grão tem átomos fragilmente interligados e assim, em tal local é
mais fácil "arrancar" os átomos da estrutura cristalina em comparação com o interior
do grão.
Figura IV.19. A região do contorno de grão aparece mais escura no microscópio
devido à menor capacidade de reflexão de luz da mesma.
IV.4.c. MACLASAs maclas constituem um outro tipo de defeito de superfície e podem surgir a
partir de tensões térmicas ou mecânicas. Tal defeito de superfície ocorre quando parte
da rede cristalina é deformada, de modo que a mesma forme uma imagem especular
da parte não deformada (figura III.20).
O plano cristalográfico de simetria entre as regiões deformadas e não
deformada, é chamado de plano de maclação. A maclação, ocorre em direções
específicas chamadas de direções de maclação.
Figura IV.20. Diagrama esquemático do defeito de maclação

Formação e Imperfeições da Estrutura Cristalina 61
EXERCÍCIOSIV.1. Uma liga contém 85% em peso de cobre e 15% em peso de estanho. Calcule a
percentagem atômica de cada elemento.
IV.2. Uma liga contém 80% em peso de Ni e 20% em peso de Cu. O Cu apresenta-se
como átomo substitucional na estrutura CFC do Ni. Supondo que a estrutura atômica
do Ni não é deformada e seu raio atômico é de 0,1246 nm, calcule a densidade desta
liga. M.A. do Ni=58,71 / M.A. do Cu=63,54.
IV.3. Calcule o raio do maior átomo que pode se situar no interstício octaédrico do Fe
CFC, sem deformar a estrutura.
IV.4. Uma liga Al-Mg contém 5% em átomos de Mg. Calule a percentagem em peso de
magnésio.
IV.5. Uma solução sólida intersticial de carbono em ferro CCC apresenta 1 átomo de
carbono para cada 500 células unitários de ferro. Calcule a percentagem em peso do
carbono.
IV.6. Por que a solubilidade total entre dois componentes de uma solução sólida pode
ocorrer caso a mesma seja substitucional, mas não ocorre no caso intersticial ?
IV.7. Quais são os tipos de defeitos em linha de uma estrutura cristalina ?

Estrutura Cristalina em Materiais Cerâmicos 62
ESTRUTURA CRISTALINA EM MATERIAIS CERÂMICOSV.1. INTRODUÇÃO
A origem da palavra cerâmica está intimamente ligada ao processo de
transformação envolvido na produção dos materiais cerâmicos: “keramikos” - palavra
grega que significa “algo queimado”. Na fabricação dos materiais cerâmicos, os
insumos são submetidos a altas temperaturas, o que resulta em reações
termoquímicas que produzem as ligações atômicas no material. Os materiais
cerâmicos ou cerâmicas são conhecidos por representarem uma classe de materiais
de elevada dureza, alta fragilidade e resistentes a temperaturas elevadas, conforme
apresenta a tabela V.1.
Composto Cerâmico Temperatura de Fusão, oC
HfC 4.150
TiC 3.120
WC 2.850
MgO 2.798
SiC 2.500
B4C 2.450
Al2O3 2.050
SiO2 1.715
Si3N4 1.900
Tabela V.1. Temperatura de fusão de alguns materiais cerâmicos.
Tais características estão diretamente ligadas à natureza das ligações e
arranjos que os átomos dos materiais cerâmicos exibem. Estes materiais, que são
definidos como substâncias inorgânicas e não-metálicas, são constituídos por
elementos metálicos e não-metálicos, unidos através de ligações iônicas ou
covalentes.
Quando comparados aos materiais metálicos, observa-se que os materiais
cerâmicos transmitem calor e eletricidade de forma precária, e são química e
termicamente mais estáveis que os metais. Enquanto os metais têm melhor
resistência à tração, as cerâmicas são resistentes à compressão. Os materiais

Estrutura Cristalina em Materiais Cerâmicos 63
metálicos exibem microestruturas monofásicas ou bifásicas, enquanto os materiais
cerâmicos exibem número elevado de fases. Sem dúvida, quando as cerâmicas são
comparadas aos materiais metálicos, a principal diferença está ligada à fragilidade
das mesmas. Enquanto o deslizamento de planos atômicos é um fenômeno
relativamente fácil de ocorrer em metais, nas cerâmicas tal processo é mais difícil.
Tal fato permite explicar a razão da alta fragilidade das cerâmicas e da boa
ductilidade dos metais.
Por outro lado, quando comparados aos materiais poliméricos, nota-se que
os cerâmicos têm estabilidade térmica superior e resistência mecânica muito maior
que a dos poliméricos. Além disso, ambos os materiais (cerâmicos e poliméricos)
não conduzem bem calor e eletricidade, exibem processo de cristalização difícil,
resultado das complexidades estruturais nesses dois tipos de material.
Há mais ou menos quatro décadas, os materiais cerâmicos deixaram de ser
produzidos de maneira rudimentar. Com a introdução de novas tecnologias uma
nova geração de materiais cerâmicos surgiu, a qual tem despertado o interesse,
tanto de pesquisadores, como da indústria. Essa nova classe de cerâmicas
provocou a divisão dos materiais cerâmicos em cerâmicas tradicionais e cerâmicas
avançadas. A tabela V.2 apresenta comparações entre os materiais cerâmicos
tradicionais e os avançados.
Característica Avançadas Tradicionais
Matéria-prima Sintética Natural
Preparação do pó Muito controlada Pouco controlada
Conformação Muito controlada Pouco controlada
Análise microestrutural Microscopia eletrônica Microscopia óptica
Resistência relativa 10 - 1.000 1
Preço relativo 10 - 10.000 1
Tabela V.2. Características dos materiais cerâmicos avançados comparados aos
tradicionais.

Estrutura Cristalina em Materiais Cerâmicos 64
Os materiais cerâmicos avançados resultam de processos de transformação
altamente controlados de matérias-primas sintéticas. Tais materiais são
empregados em indústrias com alta densidade tecnológica, como a nuclear, a
aeroespacial e a eletrônica. Por exemplo, na indústria automobilística, materiais
cerâmicos são empregados em componentes de motores submetidos a
temperaturas elevadas.
Por outro lado, os materiais cerâmicos tradicionais envolvem materiais
empregados na fabricação de objetos e utensílios domésticos, tais como tijolos,
copos e porcelana. Tais materiais envolvem processos de transformação com pouco
controle de seus parâmetros operacionais, utilizando, quase sempre, matéria-prima
natural.
V.2. ESTRUTURA DOS MATERIAIS CERÂMICOSA estrutura dos materiais cerâmicos pode ser extremamente complexa à
medida que um número elevado de átomos, com diferentes funções, pode formar a
mesma. Tal estrutura, como de outros materiais (metálicos e poliméricos) é
determinada pela natureza das ligações atômicas presentes, bem como das
características dos elementos envolvidos em tais ligações. Na maioria dos materiais
cerâmicos, a estrutura é resultado da quantidade relativa de ligações iônicas e
covalentes presentes. As parcelas iônica e covalente dependem basicamente da
eletronegatividade dos átomos envolvidos. De acordo com o critério de Pauling, o
caráter iônico de um composto tipo AB é dado pela equação:
% Carater Ionico e
X XA B
1 100%
2
4 (V.1)
onde XA e XB são as eletronegatividades dos átomos A e B, respectivamente. A tabela
V.3 mostra o caráter de alguns compostos.
O caráter iônico ou covalente define, em parte, o tipo de estrutura que o
composto cerâmico exibe. Como na maioria dos compostos cerâmicos o caráter iônico
é predominante, a estrutura dos mesmos é determinada por dois fatores
fundamentais. No caso de compostos iônicos simples, do tipo AB, o arranjo dos íons é
definido pelos seguintes fatores:
a. A relação entre os raios do cátion e do ânion;

Estrutura Cristalina em Materiais Cerâmicos 65
b. A necessidade de existir um balanço de cargas no sólido iônico.
Composto Átomos
Diferença de
eletronegatividade
% de caráter
iônico
% de caráter
covalente
MgO Mg-O 2,3 73 27
Al2O3 Al-O 2,0 63 37
SiO2 Si-O 1,7 51 49
Si3N4 Si-N 1,2 30 70
SiC Si-C 0,7 11 89
Tabela V.3. Caráter das ligações em alguns compostos cerâmicos.
Como os sólidos iônicos exibem tendência a formar estruturas altamente
compactas, o limite de tal compactação é dado pela relação entre raios iônicos e pelo
balanço eletrostático dos íons envolvidos. Além disso, para que a ligação iônica ocorra
é necessário que os cátions e ânions estejam em contato.
No caso de um composto iônico, o número de ânions em contato com um
cátion é definido como número de coordenação (N.C.). Genericamente, o N.C. pode
ainda representar o número de átomos que estão em contato com outro átomo, ou o
número de vizinhos mais próximos de um átomo. Como citado, para os materiais com
ligações iônicas, o fator que exerce influência fundamental, além do eletroquímico,
está ligado às relações geométricas entre os íons envolvidos. Assim, para o caso de
uma estrutura onde os íons são iguais, é fácil perceber que o número de
coordenação será igual a 12 (estruturas CFC ou HC). Se os íons são diferentes, o
N.C. dependerá da relação entre seus raios r/R, onde r é o raio do cátion e R do
ânion. Existe uma relação (r/R) ideal, onde o ajuste geométrico é perfeito, como mostra
a tabela V.4. Quando as dimensões dos íons são comparadas, observa-se que os
ânions são, geralmente, maiores que os cátions. Este fato está relacionado à força que
o núcleo exerce em relação à eletrosfera. Com a perda de elétrons (gerando cátions),
os elétrons restantes são atraídos em direção ao núcleo de maneira mais forte, o que
reduz o raio iônico. O fenômeno oposto, ou seja, o aumento do raio iônico ocorre com
o ganho de elétrons e a formação de ânions. A tabela V.5 exibe valores do raio iônico
de alguns cátions e ânions formadores de estruturas cerâmicas simples.

Estrutura Cristalina em Materiais Cerâmicos 66
Cátion Raio Iônico (nm) Ânion Raio Iônico
Cs+ 0,170 Br- 0,196
K+ 0,138 Cl- 0,181
Na+ 0,098 F- 0,133
Ni2+ 0,069 I- 0,220
Mg2+ 0,072 S2- 0,184
Mn2+ 0,067 O2- 0,140
Tabela V.4. Raios iônicos de alguns cátions e ânions.
Número de
Coordenação
Relação (r/R)ideal
3 0,155
4 0,225
6 0,414
8 0,732
12 1,00
Tabela V.4. Número de coordenação para as relações entre raios metálicos ou
iônicos.
A estrutura dos materiais cerâmicos pode ser altamente complexa. Entretanto,
no caso de compostos simples, a estrutura dos mesmos pode ser prevista de forma
relativamente simples.
V.3. ESTRUTURA DOS COMPOSTOS CERÂMICOS AXConforme citado, a estrutura cerâmica exibe tendência a alta compactação, o
que significa que um cátion tem tendência a exibir o maior N.C. possível.
Fundamentando-se nas principais estruturas cristalinas dos metais, nota-se que as
mesmas são ocupadas parcialmente por átomos, o que é evidente aos se observar os
fatores de empacotamento das estruturas CS, CCC, CFC e HC. Tais arranjos

Estrutura Cristalina em Materiais Cerâmicos 67
poderiam ser assumidos por compostos iônicos, desde que os ânions, de maior
tamanho, estivessem situados nas posições originais da rede e os cátions, de menor
tamanho, nos seus interstícios. Em função de tal forma de ocupação da estrutura, os
compostos cerâmicos simples, do tipo AX, onde A representa um cátion e X um ânion,
podem apresentar as seguintes estruturas: estrutura do NaCl, do CsCl e do ZnS.
a. Estrutura do NaCl
Neste tipo de estrutura existe um número equivalente de cátions e ânions. O
número de coordenação, que é obtido da relação r/R e resulta no valor de 0,564
conforme dados obtidos na tabela V.5, é igual a 6. Como o número de cátions é igual
ao de ânions, o número de coordenação 6 é igual para ambos os íons. A estrutura
desse composto é gerada a partir de um arranjo CFC dos ânions, tendo em seus
interstícios, os cátions, como mostra a figura V.1. Além do NaCl, o MgO, o MnS e o LiF
também apresentam este tipo de arranjo estrutural.
b. Estrutura do CsCl
Semelhante ao NaCl, a estrutura do CsCl é formada por um número
equivalente de cátions e ânions. O número de coordenação nesse caso, onde
r=0,170nm e R=0,181, produz relação r/R relacionada a N.C.=8. A estrutura desse
composto é gerada a partir de um arranjo CS dos ânions, tendo em seus interstícios,
os cátions, como mostra a figura V.2. A troca de posições dos ânions e dos cátions
não conduz a qualquer alteração do arranjo iônico.
Figura V.1. Célula unitária do NaCl.

Estrutura Cristalina em Materiais Cerâmicos 68
Figura V.2. Célula unitária do CsCl.
c. Estrutura do ZnS
Nesta estrutura, o composto ZnS tem estrutura formada a partir de um arranjo
CFC do enxofre e o Zn ocupando interstícios tetraédricos. O caráter das ligações é
altamente covalente. Além do ZnS, os compostos ZnS, ZnTe e o SiC também exibem
este tipo de arranjo. A estrutura do composto ZnS é mostrada na figura V.3.
Figura V.3. Célula unitária do ZnS.
V.4. ESTRUTURA DOS COMPOSTOS CERÂMICOS AnXm
Além dos cristais simples do tipo AX, alguns cristais do tipo AnXm podem ser
facilmente previstos. Dos cristais AnXm , os mais simples são do tipo AX2. O número de
cátions e ânions é diferente devido à necessidade de um balanço de cargas, o que
resulta em dois ânions para cada cátion.
a. Estrutura do CaF2
Esse composto forma uma estrutura relativamente simples onde os ânions
exibem o arranjo CS e os cátions ocupam posições intersticiais. O número de
coordenação é agora diferente para o cátion e para o ânion. Para os íon do Ca, o N.C.

Estrutura Cristalina em Materiais Cerâmicos 69
é igual a 8, enquanto para os íons F, é de 4. A estrutura do composto CaF 2 é
mostrada na figura V.4.
Figura V.4. Célula unitária do CaF2.
EXERCÍCIOS
V.1. Determine a relação (r/R)ideal para número de coordenação igual a 3.
V.2. Determine a relação (r/R)ideal para número de coordenação igual a 4.
V.3. Determine a relação (r/R)ideal para número de coordenação igual a 6.
V.4. Determine a relação (r/R)ideal para número de coordenação igual a 8.
V.5. Determine a estrutura cristalina do FeO.
V.6. Calcule a densidade do NiO sabendo-se que a M.A. do Ni é de 58,7 g/mol e a do
O é igual a 16 g/mol.
V.7. Calcule o fator de empacotamento do NaCl.

Estrutura Moleculares 70
ESTRUTURAS MOLECULARESVI.1. INTRODUÇÃO
A estrutura molecular é formada pelo agrupamento de diversas moléculas
constituídas, por sua vez, por um número limitado de átomos fortemente ligados
entre si, através de ligações covalentes. Em alguns casos, é possível constatar a
presença, também, de ligações iônicas. Em qualquer destes casos, as moléculas
apresentam um grupo de átomos eletricamente neutro. A formação da estrutura
molecular é resultado da interação entre diversas moléculas por meio de ligações
fracas, do tipo de Van der Walls.
Nos materiais de estrutura molecular, a natureza das ligações
intermoleculares e intramoleculares exerce influência decisiva em suas
características e propriedades. Características como pontos de fusão e ebulição,
resistência mecânica e dureza dependem do tipo de tais ligações.
Dentre os muitos compostos moleculares, pode-se citar a água (H2O), o gás
carbônico (CO2), o metano (CH4), o oxigênio (O2) e o hidrogênio (H2). A figura VI.1
exibe diagramas esquemáticos da molécula do etano.
Figura VI.1. Diagrama esquemático da molécula de etano: (a) convencional; (b) com
par eletrônico covalente e (c) tridimensional.
VI.2. NATUREZA DAS LIGAÇÕES EM ESTRUTURAS MOLECULARES
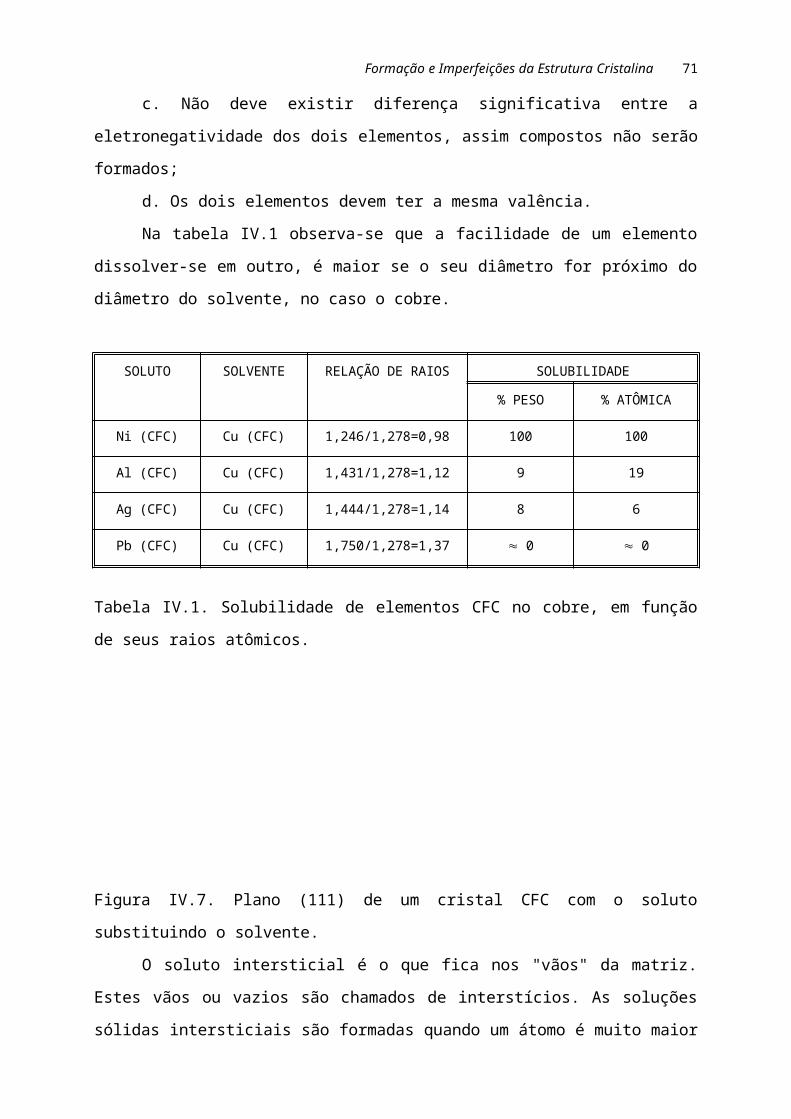
Estrutura Moleculares 71
No estudo das estruturas moleculares, o número de ligações entre os
átomos, o comprimento e a intensidade das mesmas e finalmente, o ângulo formado
por tais ligações são parâmetros básicos para o perfeito entendimento destes
arranjos atômicos.
Para os materiais com ligações covalentes, o número de ligações de um átomo
depende do número de elétrons de valência do mesmo. Assim, os elementos do grupo
VII da tabela periódica, exibem apenas uma ligação e, portanto, têm número de
coordenação, N.C., igual a um. Seguindo o mesmo raciocínio, os elementos do grupo
VI podem ter um número de coordenação máximo igual a dois e da mesma forma, os
elementos do grupo V têm número máximo de vizinhos igual a três. Finalmente, os
elementos do grupo IV têm número de coordenação máximo igual a quatro. O número
de coordenação neste caso, com exceção das ligações do hidrogênio, é obtido pela
equação:
LN = 8 - G (VI.1)
onde NL é o número máximo de ligações e G é o grupo da tabela periódica. Na tabela
VI.1 são apresentados grupos da tabela periódica onde as ligações covalente ocorrem
com maior freqüência.
GRUPO DA TABELA PERIÓDICA
4A 5A 6A 7A
C N O F
Si P S Cl
Ge Br
I
Tabela VI.1. Elementos químicos de grupos onde as ligações covalente ocorrem
com maior freqüência.
As características dos átomos envolvidos em um par covalente, bem como o
número de ligações entre o mesmo, exercem influência fundamental no
comprimento e na intensidade de tais ligações. Se o número de ligações atômicas
existente entre o par covalente é elevado, os átomos envolvidos na ligação estão
relativamente mais próximos e assim, o comprimento da ligação é menor. Quanto

Estrutura Moleculares 72
mais próximos estão tais átomo, mais forte é a ligação, ou seja, é necessário utilizar
uma quantidade de energia maior para romper a mesma. Por exemplo, um par
atômico apresentando ligações dupla ou tripla apresenta comprimento de ligação
menor que aquele com ligação simples e portanto, requer mais energia para romper
as mesmas. A tabela VI.2 apresenta valores de comprimentos e energia de ligação
para algumas ligações covalentes.
TIPO DE COMPRIMENTO ENERGIA DE LIGAÇÃO*
LIGAÇÃO DA LIGAÇÃO (10-9 m) kcal/mol kJ/mol
C - C 0,154 88 370
C = C 0,13 162 680
C º C 0,12 213 890
C - H 0,11 104 435
C - N 0,15 73 305
C - O 0,14 86 360
C = O 0,12 128 535
C - F 0,14 108 450
C - Cl 0,18 81 340
O - H 0,10 119 500
O - O 0,15 52 220
O - Si 0,16 90 375
N - O 0,12 60 250
N - H 0,10 103 430
F - F 0,14 38 160
H - H 0,074 104 435
* Estes valores podem variar em função da natureza das ligações adjacentes.
Tabela VI.2. Comprimento e energia de ligação de algumas ligações covalentes.
A natureza da ligação covalente faz com que os átomos se associem com
outros em apenas algumas direções. Isto resulta na formação de moléculas com
características tridimensionais bem definidas. Por exemplo, na molécula de água, o
ângulo entre os átomos de hidrogênio é 1050 como pode ser visto na figura VI.2. Na
molécula de metano, o ângulo entre os átomos de hidrogênio é de 109,5 0, como é

Estrutura Moleculares 73
visto na figura VI.3.a. Para ligações entre átomos de carbono (por exemplo: -C-C-
C- ), o ângulo entre o primeiro átomo e o terceiro é de 109,50, como pode ser
observado na figura VI.3.b.
Figura VI.2. Representação esquemática de uma molécula de água.
(a) Metano (b) Butano
Figura VI.3. Moléculas do metano e do butano.
VI.3. HIDROCARBONETOSUma classe importante de materiais com estrutura molecular é aquela
formada pelos hidrocarbonetos. Este compostos são constituídos essencialmente
por átomos de carbono ligados a átomos de hidrogênio. Um dos exemplos mais
comuns de hidrocarboneto, além de ser o menor deles, é o metano (CH4).

Estrutura Moleculares 74
Os hidrocarbonetos podem ser classificados como saturados e insaturados.
O conceito de saturação pode ser obtido pela análise de uma molécula de metano,
tomada aqui como unidade de uma estrutura. Se átomos de hidrogênio e carbono
são adicionados a esta "molécula unidade", pode-se obter uma molécula de
tamanho teoricamente infinito. Essas moléculas, de formula geral CnH2n+2 são
denominadas "parafinas". Uma molécula é considerada parafínica se todas as suas
ligações são simples. Assim, cada átomo de carbono dentro da cadeia tem número
de coordenação igual a 4 e não existe possibilidade de novos átomos serem
adicionados a esta molécula, que é considerada "saturada".
As moléculas saturadas apresentam ligações intramoleculares fortes
(covalentes) e intermoleculares fracas (Van der Walls). Quando o número de
átomos de uma molécula é elevado, a intensidade das forças de Van der Walls é
também elevada, pois existe um número maior de posições ao longo da molécula, o
que permite que forças secundárias do tipo dipolo-dipolo e efeito de dispersão
ocorram com maior probabilidade. Por exemplo, a parafina contém em torno de 30
átomos de carbono por molécula e se funde à temperatura ambiente. Já o plástico
polietileno, que é um hidrocarboneto com milhares de átomos de carbono por
molécula, tem temperatura de fusão em torno de 145oC.
Por outro lado, as moléculas consideradas "insaturadas" apresentam átomos
de carbono com ligações duplas e triplas. A quebra dessas ligações é possível e
permite a adição de novos átomos à molécula. Em geral, qualquer molécula com
ligações carbono-carbono múltiplas, é considerada insaturada. Tais moléculas são
bastante importantes industrialmente, já que permitem a polimerização de pequenas
moléculas em uma única molécula, de tamanho bem maior, como mostra figura
VI.4.
Os hidrocarbonetos dão origem aos materiais poliméricos. Os materiais
poliméricos englobam os plásticos, as borrachas sintéticas, as borrachas naturais e
os materiais biológicos, como couro, lã e celulose. Por outro lado, em função da
origem dos materiais poliméricos, os mesmos são classificados em naturais
(madeira, borracha natural), artificiais, que são preparados a partir de matéria-prima
natural (acetato de celulose) e sintéticos, que são obtidos de matéria-prima artificial.

Estrutura Moleculares 75
Figura VI.4. Polimerização do etileno: (a) Monômeros de etileno e (b) Polímero
contendo muitas unidade de C2H4 (meros).
VI.4. PROCESSOS DE POLIMERIZAÇÃOA polimerização é a etapa básica na formação dos materiais poliméricos. Tal
processo consiste na reação de monômeros, que formarão os polímeros. Os
monômeros, que são o insumo fundamental em tal processo, são definidos como
substâncias constituídas por pequenas moléculas com ligações covalentes. Para
formar um polímero, os monômeros devem apresentar pelo menos dois pontos
reativos em cada molécula. Tais pontos reativos estão relacionados a ligações
insaturadas entre átomos de carbono e grupos funcionais oxigenados e nitrogenados.
O processo de polimerização pode ocorrer de duas formas principais: polimerização
por adição e polimerização por condensação. Na polimerização por adição, os pontos
reativos dos monômeros são gerados pela quebra de ligações duplas (C=C),
resultando na possibilidade de duas novas ligações à molécula. Em tal forma de
polimerização, não ocorre a geração de subprodutos. A polimerização por adição
ocorre em três etapas:
a. Iniciação
Ocorre quando, pela aplicação de calor, luz, pressão ou catalisador, as ligações
duplas podem ser rompidas.
b. Preparação
É a etapa onde as moléculas do monômero, com pontos reativos iniciam o
processo de formação de cadeias poliméricas.
c. Término

Estrutura Moleculares 76
Nessa última etapa, os pontos reativos são eliminados, o que encerra o
processo de polimerização. A figura VI.5 apresenta um diagrama do processo de
polimerização por adição.
Figura VI.5. Diagrama esquemático do processo de polimerização por adição.
Na polimerização por condensação, o processo é iniciado pela reação de duas
ou mais substâncias diferentes. A polimerização ocorre com a eliminação, geralmente,
de água e HCl. No caso da reação envolver apenas compostos bifuncionais (apenas
dois pontos reativos), as cadeias poliméricas resultantes serão lineares. No caso da
reação envolver compostos trifuncionais, é possível a obtenção de retículos
tridimensionais. A figura VI.6. mostra um diagrama do processo de polimerização por
condensação.

Estrutura Moleculares 77
(a) (b)
Figura VI.6. Diagrama esquemático do processo de polimerização por condensação:
(a) cadeias lineares e (b) cadeias não-lineares.
Além dos dois processos de polimerização citados, existe ainda o processo de
copolimerização, que envolve a polimerização por adição de dois ou mais monômeros
de natureza distinta, denominados de comonômeros.
VI.5. ESTRUTURAS DOS MATERIAIS POLIMÉRICOSA forma das cadeias poliméricas exerce influência significativa nas
propriedades do material polimérico. As cadeias poliméricas podem ser dos tipos

Estrutura Moleculares 78
linear, ramificada e com ligações cruzadas. A lineares são formadas por monômeros
bifuncionais. Nesse caso, as moléculas adjacentes são unidas por forças secundárias,
o que permite o escorregamento intermolecular. Além disso, em função do arranjo da
cadeia, a plasticidade aumenta com a temperatura. Em geral, os polímeros de cadeia
linear podem ser submetidos a elevados níveis de deformações em altas temperaturas
e quando resfriados voltam a exibir as características originais.
Os polímeros de cadeias ramificadas são obtidos quando cadeias lineares
formam ligações paralelas no corpo do monômero. Tais ramificações permitem o
entrelaçamento das cadeias, limitando o movimento das mesmas. Os materiais
poliméricos de ligações cruzadas são formados quando uma ligação ocorre entre duas
cadeias lineares. Um exemplo clássico de polímero com ligações cruzadas é
encontrado na borracha vulcanizada, onde átomos de enxofre permitem a união de
duas cadeias lineares. A figura VI.7 mostra um diagrama diferenciando as cadeias
lineares, ramificadas e com ligações cruzadas.
A estrutura dos materiais poliméricos é caracterizada pela presença de cadeias
de átomos bastante longas ligadas entre si por ligações covalentes. A natureza das
ligações possíveis em polímeros, no tocante à tetravalência do carbono, bem como
aos diversos tipos de monômeros existentes, permite combinações quase que
ilimitadas de arranjos estruturais. Um exemplo de tal fato é a natureza da cadeia
principal do polímero. A figura VI.8 mostra as cadeias do tipo carbônica e do tipo
heterogênea, o que é definido pelos átomos presentes na mesma.
A natureza da estrutura dos materiais poliméricos define o comportamento dos
mesmo quando submetidos a altas temperaturas. O comportamento de um material
polimérico em relação a ação de calor, permite classificar os polímeros como
termoplásticos e termofixos. Os polímeros termoplásticos, geralmente de cadeias
lineares, podem ser amolecidos sob ação de calor, deformados sob ação de tensões e
após o resfriamento, recuperam a natureza sólida. Este processo pode ser repetido e
exemplos de materiais dessa classe envolvem o polietileno e o pvc.
Por outro lado, os polímeros termofixos, de cadeias mais complexas, quando
submetidos a altas temperaturas podem ser amolecidos e conformados sob ação de
tensões. Entretanto, após o resfriamento e recuperação da natureza sólida, o processo
não pode ser repetido. Exemplos de polímeros termofixos: silicones, poliuretano e
epóxi.

Estrutura Moleculares 79
Figura VI.7. Diagrama esquemático mostrando as diferenças entre cadeias lineares,
ramificadas e com ligações cruzadas.
Cadeia Carbônica Cadeia Heterogênea
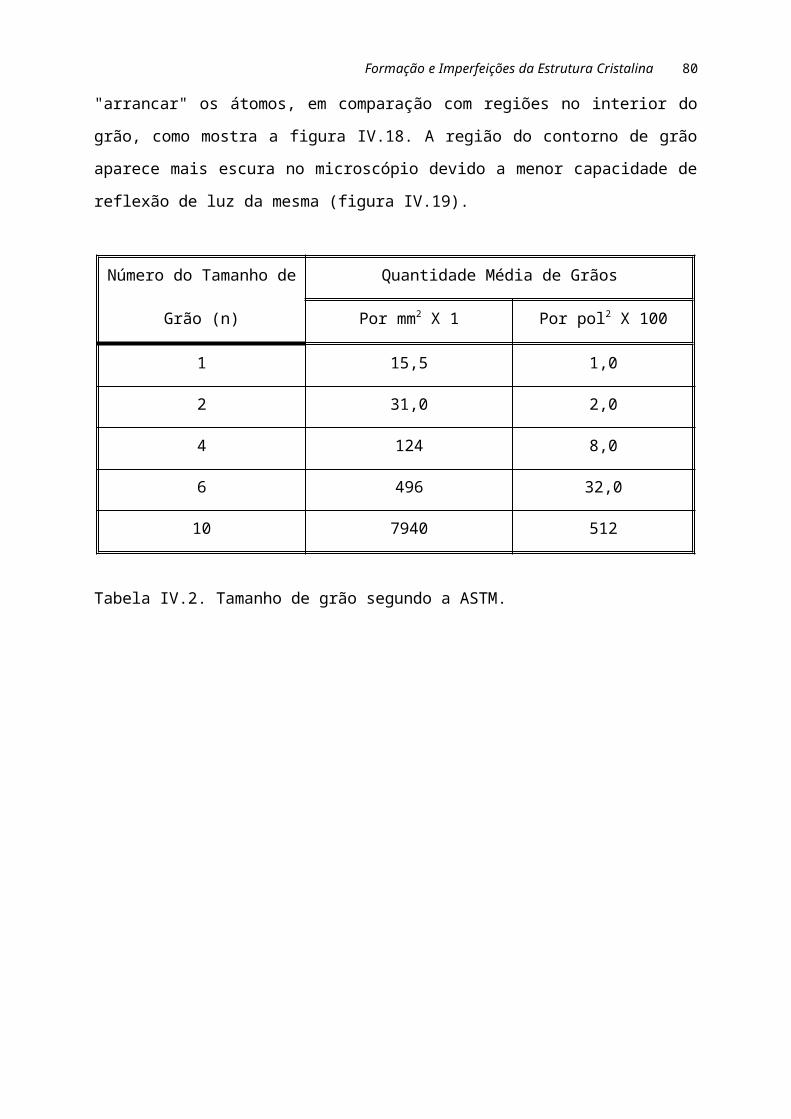
Estrutura Moleculares 80
Figura VI.8. Cadeias do tipo carbônica e do tipo heterogênea.
EXERCÍCIOS
VI.1. Descreva as diferenças básicas dos materiais poliméricos quando comparados
ao materiais metálicos e cerâmicos.
VI.2. Descreva os dois processos básicos de polimerização.
VI.3. Como são formados os materiais poliméricos?
VI.4. É possível formar um material polimérico a partir de monômeros com apenas um
ponto reativo? Explique.
VI.5. Como é realizado o processo de vulcanização da borracha?
VI.6. Descreva qual é a diferença entre termoplásticos e termofixos.

Estrutura Moleculares 81

Estrutura Amorfas 82
ESTRUTURAS AMORFASVII.1. INTRODUÇÃO
As estruturas amorfas, também chamadas de estruturas vítreas são formadas
por arranjos atômicos aleatórios e sem simetria ou ordenação de longo alcance.
Este tipo de estrutura pode ser encontrado em gases, em líquidos e em certos
sólidos, representados basicamente pelos vidros.
Por definição, um material apresenta estrutura amorfa quando o mesmo é
resfriado a partir do líquido e exibe um aumento contínuo de sua viscosidade.
Quando o material atinge uma determinada temperatura, definida como temperatura
de transição vítrea, TV, o valor da viscosidade é da ordem de 1012 a 1013 Poise (1
Poise=0,1 N.s/m2). Este valor de viscosidade é semelhante a aqueles de materiais
no estado sólido. Nesta situação, devido ao limitado movimento atômico, o rearranjo
dos átomos não é possível e a cristalização do material não ocorre. Por outro lado,
se durante tal resfriamento, a cristalização do material ocorre, a viscosidade do
mesmo é abruptamente alterada, atingindo valores próximos de 1012 Poise. A figura
VII.1 apresenta um diagrama esquemático da variação da viscosidade em função da
temperatura, para materiais cristalinos e amorfos.
Figura VII.1. Variação do viscosidade com a temperatura para materiais vítreos e
cristalinos.

Estrutura Amorfas 83
Uma outra forma de diferenciar o processo de formação da estrutura
cristalina (cristalização) da estrutura amorfa (vitrificação) é a medida de variação de
volume durante o resfriamento do material líquido. Quando o material cristaliza-se,
ocorre uma rápida variação de volume em uma determinada temperatura, definida
como a de fusão (Tf). Este fenômeno é resultante da reorganização dos átomos
para formar um cristal do material. Se o material apresenta o processo de
vitrificação durante o resfriamento, a variação de volume é contínua, já que a
ordenação dos átomos não ocorre totalmente. A figura VII.2 mostra a variação do
volume específico com a temperatura para materiais vítreos e para materiais
cristalinos. Resumindo, é possível afirmar que o vidro tem estrutura de um líquido
"congelado". Este "congelamento" da estrutura do líquido se dá à temperatura de
vitrificação, que é inferior à temperatura de cristalização.
A estrutura amorfa é geralmente observada em materiais que poderiam
apresentar estrutura cristalina se solidificados sob condições especiais. Alguns
compostos cerâmicos à base de óxidos, silicatos, boratos e aluminatos formam
estruturas vítreas em condições normais de solidificação.
Figura VII.2. Variação do volume específico com a temperatura para materiais
vítreos e cristalinos.

Estrutura Amorfas 84
A sílica (SiO2) é o exemplo clássico de material que em condições especiais
pode exibir o processo de cristalização e formar o quartzo. Por outro lado, se o
resfriamento da sílica, a partir do líquido, ocorre em condições normais, a estrutura
resultante é a amorfa. A figura VII.3 mostra as duas estruturas observada na sílica:
estrutura cristalina e estrutura vítrea.
Figura VII.3. Diagrama bidimensional das estruturas cristalina e vítrea da sílica.
Além dos vidros, uma classe de materiais sólidos que apresenta estrutura
amorfa e destaca-se pelo interesse tecnológico que desperta são os "metais
amorfos", também chamados de "vidros metálicos". Os vidros metálicos
representam uma nova classe de materiais que começou a ser desenvolvida com
sucesso na década de 60 e são obtidos a partir do estado líquido, por resfriamento
ultra-rápido, ou seja, taxas de resfriamento próximas de um milhão de graus por
segundo.
Os metais amorfos foram obtidos pela primeira vez da solidificação rápida da
liga Au-25%Si, em 1960 no Caltec (California Institute of Technology, EUA). Tal feito
provocou interesse imediato da comunidade científica, pois até aquela data sempre
se associava a um sólido metálico, estrutura atômica perfeitamente organizada
(cristal). Este desenvolvimento motivou uma intensa corrida ao desenvolvimento dos

Estrutura Amorfas 85
metais amorfos e atualmente, este material já é utilizado comercialmente em
diversos campos.
Embora os metais amorfos apresentem estrutura semelhante a dos vidros
tradicionais, eles exibem algumas características bastante diferenciadas destes.
Como os metais amorfos são constituídos por elementos metálicos, ligados entre si
por ligações metálicas, eles apresentam elevada condutividade elétrica e térmica,
assim como são dúcteis. Por outro lado, os metais amorfos não são transparentes
como os vidros à base de óxidos, nem são frágeis. Geralmente, os metais vítreos
mostram qualidades particulares como a facilidade de magnetização, elevada
dureza, alta tenacidade, resistência à corrosão e expansão térmica reduzida.
Os metais vítreos apresentam propriedades mecânicas bastante
interessantes, como elevada resistência mecânica, podendo ser empregados como
elemento de reforço em concreto, plástico e borracha. A estrutura amorfa apresenta
um arranjo estrutural em que, a rigor, não exibe as mesmas imperfeições
observadas em materiais cristalinos. Assim, os mecanismos de deformação plástica
em cristais não são observados nos vidros metálicos. A resistência mecânica destes
materiais é bastante elevada, chegando próximas do valor teórico (monocristais sem
discordâncias).
Com relação às propriedades químicas vale destacar que a isenção de
defeitos estruturais, como contornos de grão, discordâncias, precipitados e
segregações, resultam em um material com comportamento químico bastante
diferenciado dos metais cristalinos. Quando constituídos por elementos adequados,
como o cromo, os metais vítreos apresentam resistência à corrosão ideal. Dentre as
possíveis aplicações dos metais vítreos, com relação a características químicas,
pode-se destacar o uso em lâminas de barbear, cutelaria, bio-implantes, eletrodo
para células eletrolíticas e vasos de reatores químicos.
Como a resistividade elétrica de um material está relacionada com a
desordem de seus átomos, em metais vítreos este parâmetro é elevado quando
comparado aos cristalinos e pouco dependente da temperatura. Isto significa que os
metais amorfos exibem baixo valor do coeficiente de variação de resistividade com a
temperatura, podendo ser usados como resistências de precisão, ou ainda como
sensores de campos magnéticos.
Como os metais vítreos possuem alta permeabilidade magnética, alta
resistividade elétrica em relação aos cristalinos, eles podem ser utilizados na
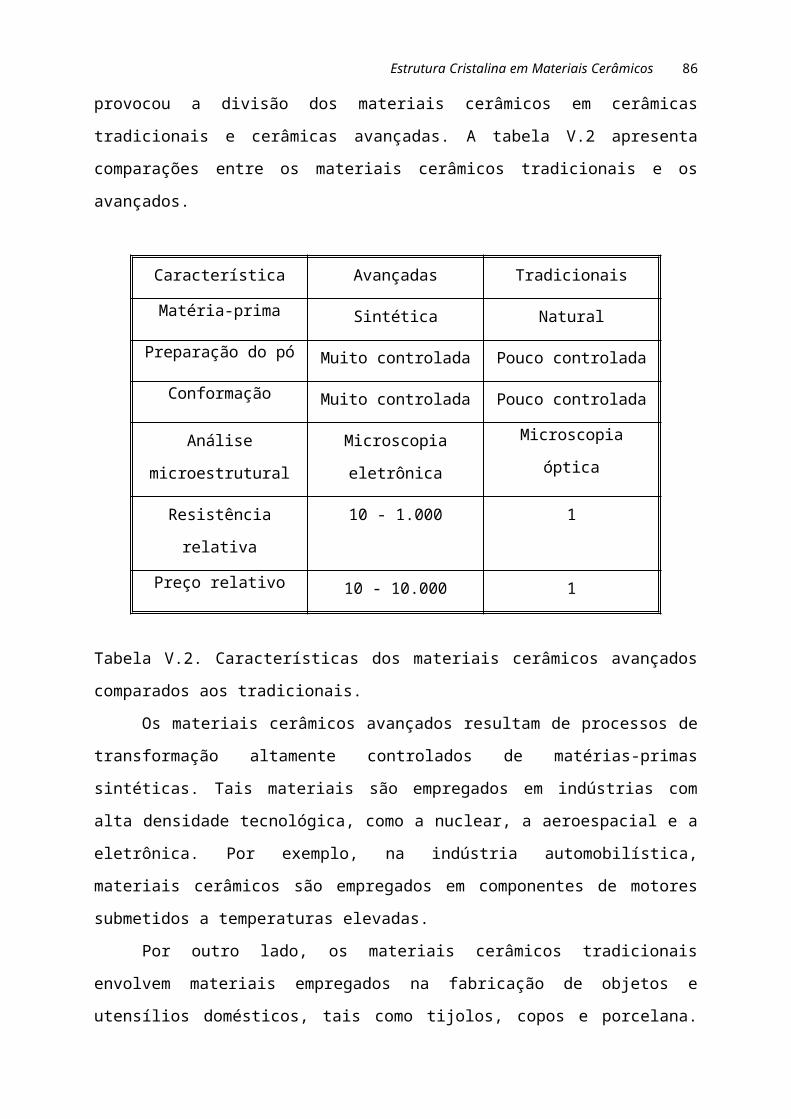
Estrutura Amorfas 86
fabricação de transformadores elétricos, cabeçotes de gravadores e transdutores
magnéticos. Na tabela VII.1 observa-se um quadro das características e
propriedades dos metais vítreos comparados ao metais tradicionais.
Característica/
Propriedade
Metais
Tradicionais
Vidros
Tradicionais
Metais
Vítreos
Estrutura Cristalina Amorfa Amorfa
Ligação Metálica Covalente Metálica
Tensão de
Escoamento
Não Ideal Quase Ideal Quase Ideal
Trabalhabilidade Boa, Dúctil Pobre, Frágil Boa, Dúctil
Dureza Baixa / Alta Muito Alta Muito Alta
Resistência
Mecânica
Baixa / Alta Baixa Alta / Muito Alta
Característica
Ótica
Opaca Transparente Opaca
Condutividade
Elet. Térm.
Muito Boa Pobre Boa
Resistência à
Corrosão
Pobre / Boa Muito Boa Muito Boa
Propriedades
Magnéticas
Diversas Não Existe Diversas
Tabela VII.1. Comparação de características e propriedades de metais tradicionais,
vidros tradicionais e metais vítreos.
EXERCÍCIOSVII.1. Descreva a evolução da viscosidade com a diminuição de temperatura de um
material cristalino e de outro amorfo.
VII.2. Descreva a evolução do volume específico com o aumento de temperatura de
um material cristalino e de outro amorfo.

Estrutura Amorfas 87
VII.3. Quando uma liga metálica de estrutura cristalina complexa é resfriada
rapidamente, existe a possibilidade de se obter estruturas amorfas. Cite uma razão
para tal tendência
VII.4. Dê exemplos de possíveis aplicações de metais amorfos ou vítreos na indústria.
VII.5. Os metais vítreos, quando comparado aos seus respectivos no estado cristalino,
podem exibir resistência mecânica bastante superior. Cite uma razão para tal fato.
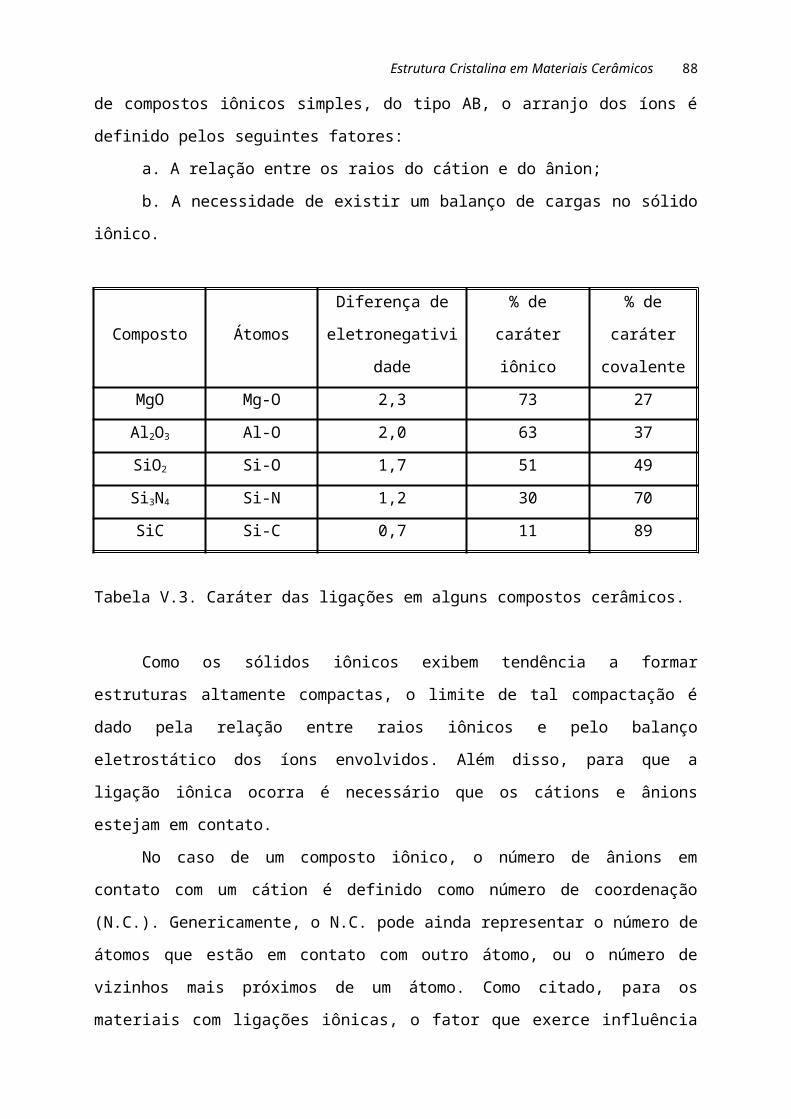
Difusão Atômica 88
DIFUSÃO ATÔMICAVIII.1. INTRODUÇÃO
A difusão atômica pode ser definida como um mecanismo pelo qual a matéria
é transportada através da matéria. Os átomos em gases, líquidos e sólidos estão
em constante movimento. O movimento atômico em gases é relativamente rápido. O
movimento atômico em líquidos é, em geral, mais lento que em gases, como pode
ser observado durante o movimento de um corante em água. O movimento atômico
em sólidos é bastante restrito, já que as forças de ligação atômicas são elevadas e
também, devido à existência de posições de equilíbrio bem definidas. Entretanto,
vibrações atômicas de origem térmica existentes em sólidos permitem movimentos
atômicos limitados. A difusão atômica em metais e ligas é particularmente
importante, pois a maioria das reações de estado sólido, que são fundamentais em
metalurgia, envolve movimentos atômicos. Exemplos de reações de estado sólido
são obtidos na nucleação e crescimento de novas fases em sólidos cristalinos, no
tratamento térmico de aços, na produção de circuitos eletrônicos, etc.
VIII.2. MECANISMOS DO MOVIMENTO ATÔMICOOs átomos apenas estão em repouso absoluto quando a temperatura é igual
a zero absoluto (-2730C). Acima desta temperatura os átomos começam a vibrar e
saem de suas posições originais. À medida que a temperatura aumenta, esse
movimento atômico torna-se mais intenso. Existem dois mecanismos básicos de
difusão de átomos em um sólido cristalino, quais sejam: mecanismo substitucional
ou de vazios e mecanismo intersticial. Além desses dois, o movimento atômico
pode-se dar através do mecanismo de anel, que é de ocorrência mais difícil.
VIII.2.a. MECANISMO SUBSTITUCIONAL OU DE VAZIOSOs átomos podem mover-se no interior de um cristal, de uma posição
atômica para outra se os mesmos apresentam energia de vibração suficiente e se
existem posições atômicas vazias ou defeitos cristalinos na estrutura atômica. Esta
energia de vibração é resultante da energia térmica dos átomos. Os vazios ou
vacâncias em metais e ligas são defeitos de equilíbrio e assim, estão sempre
presentes para permitir o movimento atômico pelo mecanismo substitucional. Com o
aumento da temperatura em metais, mais vacâncias podem ser observadas e mais
energia térmica estará disponível. Assim, a taxa de difusão atômica aumentará com
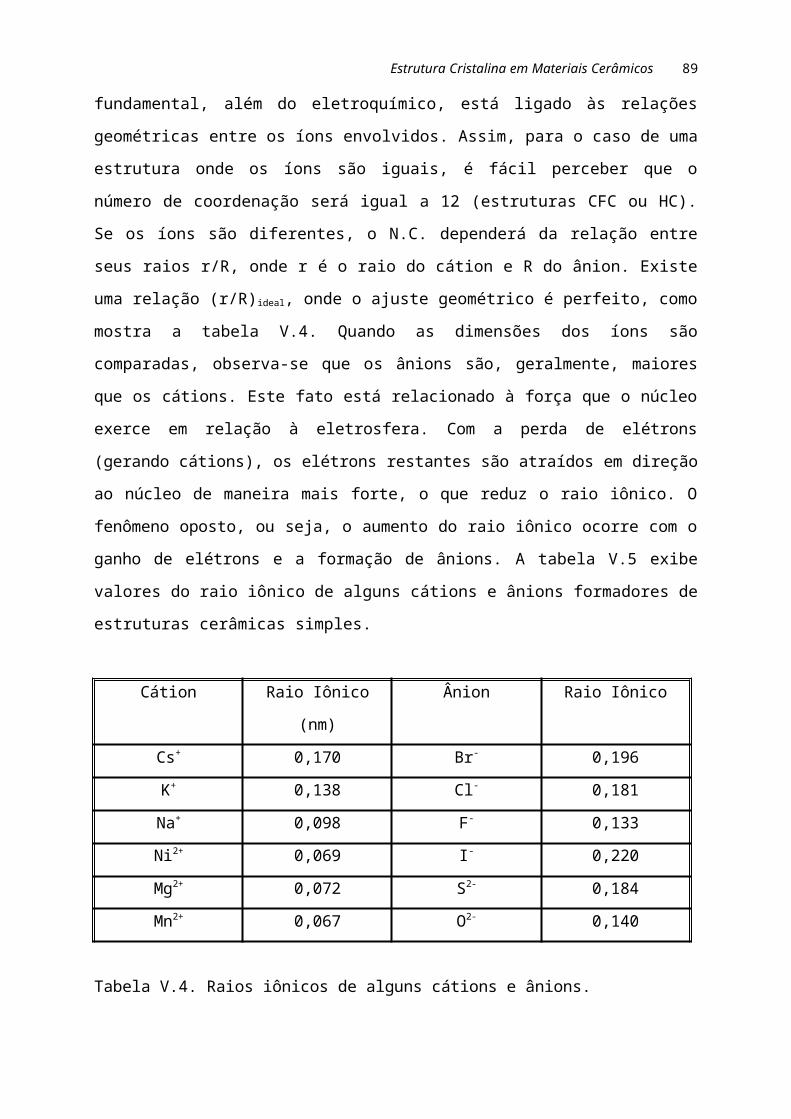
Difusão Atômica 89
a temperatura. Considere o processo de difusão apresentado na figura VIII.1.
Se um átomo próximo à vacância tem energia suficiente, ele poderá mover-se até a
posição vazia. As diferenças de tamanho atômico e energias de ligação são fatores
que afetam a taxa de difusão atômica através de vazios.
Figura VIII.1. Mecanismo de difusão atômica de vazios ou substitucional
VIII.2.b. MECANISMO INTERSTICIALA difusão de átomos intersticiais em um sólido cristalino ocorre quando um
átomo se move de uma posição intersticial para outra posição vizinha intersticial,
sem que exista deslocamento de átomos da matriz cristalina, como mostra a figura
VIII.2. Para que o mecanismo de difusão intersticial seja ativo, o tamanho do átomo
em difusão deve ser relativamente pequeno quando comparado com os átomos da
matriz. Pequenos átomos como o hidrogênio, oxigênio, nitrogênio e carbono podem
apresentar difusão intersticial em alguns sólidos cristalinos. Por exemplo, o carbono
pode difundir-se intersticialmente no ferro a e no ferro g. Na difusão intersticial de
carbono em ferro, os átomos de carbono são comprimidos entre a matriz atômica do
ferro.

Difusão Atômica 90
Figura VIII.2. Mecanismo de difusão atômica intersticial
VIII.2.c. MECANISMO DE ANELEste mecanismo é mais raro devido a suas particularidades. A difusão
atômica através deste mecanismo envolve a rotação de três ou quatro átomos
simultaneamente, como mostra a figura VIII.3.
(a) (b)
Figura VIII.3. Mecanismo de difusão atômica de anel de 3 átomos e de quatro
átomos.
VIII.3. DISTRIBUIÇÃO DE ENERGIA TÉRMICAOs átomos dentro de um material, em uma determinada temperatura,
apresentam diferentes níveis de energia, sendo esta uma distribuição estatística,
como mostra a figura VIII.4.

Difusão Atômica 91
Figura VIII.4. Distribuição de energia dos átomos de um material.
Nesta distribuição, nota-se que poucos átomos possuem energia de ativação
suficiente para "saltar" fora de sua posições originais, ou em outras palavras, mover-
se na rede cristalina. Porém, aumentando-se a temperatura do sistema, a energia
de cada átomo aumenta e assim, alguns átomos que não podiam saltar de suas
posições, podem agora fazê-lo. Isto significa que a energia dos mesmos é maior
que a energia de ativação, como mostra a figura VIII.5.
Fazendo uso de análise estatística é possível determinar a parcela de átomos
com energia suficiente para apresentar movimento atômico. Boltzmann estudou o
efeito da temperatura na energia das moléculas em um gás. Usando os
fundamentos estatísticos empregados por Boltzmann, pode-se calcular o número de
átomos com energia maior que a energia de ativação:
n = aN -EkT
Aexp (VIII.1)
onde k=1,38x10-23 [Joule/átomo.K], n é o número de átomos com energia maior que
a de ativação, N é o número total de átomos do sólido, a é uma constante típica do
sistema, EA é a energia de ativação e T é a temperatura absoluta.

Difusão Atômica 92
Figura VIII.5. Distribuição de energia dos átomos de um material para duas
temperaturas diferentes.
VIII.4. COEFICIENTE DE DIFUSÃOA análise estatística de Boltzmann aplicada ao movimento atômico permite
estabelecer a intensidade de difusão atômica em materiais. A difusão de um
material A (soluto) dentro de um outro material B (solvente) é representada pelo
coeficiente de difusão (D), definido como:
D = D e0-
QRT
(VIII.2)
onde D é o coeficiente de difusão, D0 é constante do sistema soluto/solvente, Q é a
energia de ativação e R é a constante molar dos gases (8,314 J/mol.K ou 1,987
cal/mol.K).
Pela equação VIII.2, observa-se que o coeficiente de difusão depende da
temperatura, aumentando quando a mesma aumenta. Na tabela VIII.1 são
apresentados os valores de Q e D0 para alguns sistemas em difusão.
Soluto Matriz D0 (10-5m2/s) Q (10-19J/átomo)
Carbono Ferro CFC 2,0 2,4
Carbono Ferro CCC 22,0 2,0
Ferro Ferro CFC 2,2 4,5
Ferro Ferro CCC 20,0 4,0
Níquel Ferro CFC 7,7 4,6

Difusão Atômica 93
Zinco Cobre 3,4 3,2
Cobre Cobre 2,0 3,3
Prata Prata (Cristal) 4,0 3,1
Prata Prata (Cont.Grão.) 1,4 1,5
Silício Silício 320 6,8
Fósforo Silício 3,9 5,0
Boro Silício 140 5,9
Tabela VIII.1. Coeficientes de difusão para diversos sistemas solvente/soluto.
VIII.5. PRIMEIRA LEI DE FICKO fenômeno de difusão atômica pode ser analisado considerando o
movimento de átomos entre duas regiões em contato, como mostra a figura VIII.6.
Assumindo que as concentrações de átomos de soluto nas regiões 1 e 2 não sofrem
alterações com o tempo, o sistema pode ser considerado como em regime
permanente ou estacionário. A figura VIII.7 mostra um processo de difusão em
regime permanente, provocado pelo gradiente de concentração (C2-C1)/(x2-x1). Um
caso semelhante é observado quando um gás difunde-se através de uma folha fina
de metal, como é o caso do hidrogênio difundindo-se por uma folha fina de Paládio.
O movimento de átomos por difusão atômica ocorre devido à vibração
térmica do átomo. Tal vibração faz com que cada átomo permaneça "saltando" de
uma posição a outra. O equacionamento do fluxo atômico em regime permanente,
que é dado pela 1ª lei de Fick, é implementado pela definição das seguintes
variáveis:
Dx - Espessura das regiões 1 e 2;
A - Área de contato entre as regiões 1 e 2;
f - Freqüência de saltos dos átomos (saltos/s), igual em todas as direções;
C1 - Concentração de átomos de soluto na região 1 (át./cm3);
C2 - Concentração de átomos de soluto na região 2 (át./cm3).
J - Fluxo de átomos entre as regiões 1 e 2 (át./cm2.s)

Difusão Atômica 94
Figura VIII.6. Diagrama esquemático do fluxo de átomos entre duas regiões, em
contato, de concentrações diferente.
Figura VIII.7. Difusão atômica em regime estacionário provocada pelo gradiente de
concentração (C2-C1)/(x2-x1).
Considerando o movimento atômico espacial, um átomo tem a possibilidade
de saltar em seis diferentes direções. Assim, entre as regiões 1 e 2, a freqüência de
saltos pode ser dada por f/6 e conseqüentemente, em um intervalo de tempo Dt, o
número de átomos saltando da região 1 para a região 2 é proporcional aos valores
de C1, de Dt, de f, e do volume da região 1, que pode ser representado por sua
espessura, Dx, pois a área de contato é igual para as duas regiões:

Difusão Atômica 95
12 1N = K Cf6
t xD D (VIII.3)
onde K é uma constante.
O fluxo de átomos entre as regiões 1 e 2 é dado pela diferença entre os
átomos que saltam da região 1 para a região 2 e aqueles que fazem o caminho
inverso. Assim,
J t = N - N = K Cf6
t x - K Cf6
t x12 21 1 2D D D D D (VIII.4)
ou
J = K C Cf6
x1 2 D (VIII.5)
Desta equação é possível prever que se as concentrações das regiões 1 e 2
são iguais, o fluxo de massa entre elas será nulo. Por outro lado, se existe um
gradiente de concentração de átomos de soluto, o fluxo de átomos será diferente de
zero. Uma relação entre as concentrações C1 e C2 pode ser obtida se a
concentração é contínua ao longo da direção x (paralelo ao fluxo de átomos), ou
seja:
2 1t
C = C + xCx
D
(VIII.6)
Substituindo o valor de C2, o fluxo de átomos entre as regiões 1 e 2 torna-se
igual a:
J = - K xf6
Cx
2
tD
(VIII.7)
Esta equação é conhecida como a 1ª lei de Fick e o coeficiente de difusão
atômica, D, é dado por:
D = K xf6
2D (VIII.8)
Se no sistema em difusão considerado não ocorrem reações químicas entre
os átomos do soluto e os do solvente, a diferença de concentração entre as regiões
1 e 2 resultará em um fluxo atômico que vai do ponto de maior para o de menor
concentração. O fluxo de átomos neste tipo de sistema pode ser representado pela
equação:
J = - DCx
(VIIII.9)

Difusão Atômica 96
onde J é o fluxo de átomos, D é o coeficiente de difusão e C/x é o gradiente de
concentração. Tomando a direção x como referência, o sinal negativo mostra que o
fluxo de massa tem sentido contrário ao aumento da concentração e é usado
porque o fluxo de átomos vai da maior para a menor concentração e o mesmo tende
a anular o gradiente de concentração.
A equação VIII.9 é denominada de 1ª lei de Fick e define que para condições
estacionárias ou permanentes (concentrações constantes com o tempo), o fluxo de
átomos por difusão atômica é igual à difusividade D multiplicada pelo gradiente de
concentração. No sistema SI, esta equação é dada por:
J atomos
m .s = D m
s
Cx
atomos
mx
1m2
2
3
(VIII.10)
A tabela VIII.2 lista valores de difusividade atômica para alguns sistemas. A
difusividade atômica depende de diversos fatores, sendo que os mais importantes
são:
a. Tipo de mecanismo de difusão (substitucional ou intersticial) - Dependendo
dos tamanhos atômicos envolvidos, o mecanismo de difusão influencia a
intensidade de difusão. Átomos de tamanhos próximos tem difusão elevada quando
o mecanismo é substitucional. Quando os átomos apresentam tamanhos muito
diferentes, o mecanismo apropriado é o intersticial;
b. Temperatura na qual a difusão ocorre - A temperatura aumenta a difusão;
c. Tipo de estrutura cristalina do solvente - Estruturas compactas (CFC, HC)
dificultam a difusão atômica;
d. Tipo e quantidade de imperfeições presentes na rede cristalina - Defeitos
como discordâncias e vazios aumentam a intensidade de difusão.
Soluto Solvente Coeficiente de Difusão (m2/s)
5000C 10000C
Carbono Ferro CFC 5x10-15(metaest.) 3x10-11
Carbono Ferro CCC 10-12 2x10-9(metaest.)
Ferro Ferro CFC 2x10-23(metaest.) 2x10-16
Ferro Ferro CCC 10-20 3x10-14(metaest.)
Níquel Ferro CFC 10-23(metaest.) 2x10-16
Manganês Ferro CFC 3x10-24(metaest.) 10-16
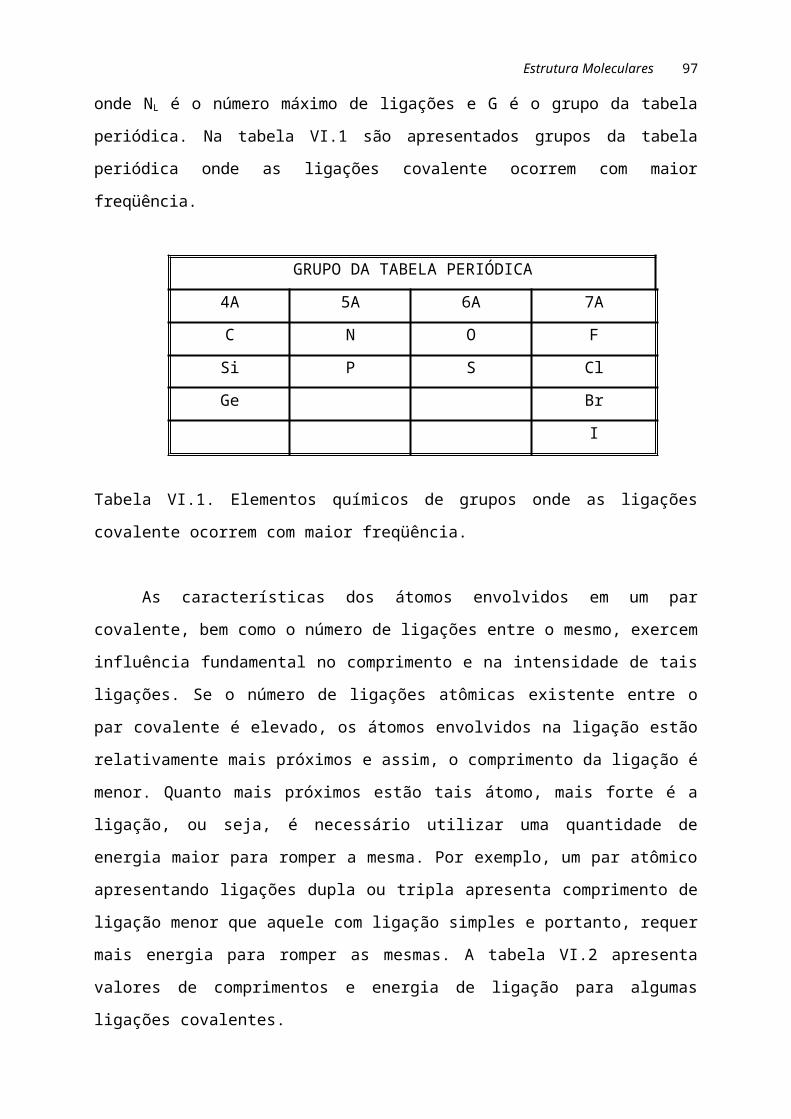
Difusão Atômica 97
Zinco Cobre 4x10-18 5x10-13
Cobre Alumínio 4x10-14 10-10(teórico)
Cobre Cobre 10-18 2x10-11
Prata Prata (Cristal) 10-17 10-12(teórico)
Prata Prata (Cont.Grão) 10-11 -
Carbono Titânio 3x10-16 2x10-11(metaest.)
Tabela 3.2. Coeficientes de difusão atômica para 5000C e 10000C.
VIII.6. SEGUNDA LEI DE FICKO movimento atômico em condições estacionárias não é comum em
engenharia de materiais. Na maioria dos casos, este movimento ocorre em regime
transitório ou em situações onde as concentrações mudam com o tempo. Por
exemplo, se o carbono está sendo difundido através da superfície de uma
engrenagem de aço para cementar a mesma, a concentração de carbono no interior
da peça será alterada à medida que o tempo de processamento aumenta, como
mostra a figura VIII.8. Nestes casos, onde o regime não é permanente, é
interessante determinar a evolução da variável composição em função do tempo de
processamento e da posição de um dado ponto a ser estudado.
Considere uma barra de um material qualquer de concentração C, exibindo
transporte de massa do soluto por difusão, como mostra o diagrama da figura VIII.9.
Considere também a existência de um elemento de volume de largura Dx e área da
secção transversal A. Suponha que em tal elemento está entrando fluxo de massa
J1 e deixando o mesmo, o fluxo de massa J2. Após um intervalo de tempo, Dt, a
variação na concentração de soluto em tal elemento é dada por:
1 2J A t - J A t = A x CD D D D (VIII.11)
onde:
J - Fluxo de átomos do soluto (át/cm2.s)
A - Área (cm2)
Dx - largura (cm)
Dt - Intervalo de tempo (s)
DC - Variação na concentração de soluto (át/cm3)
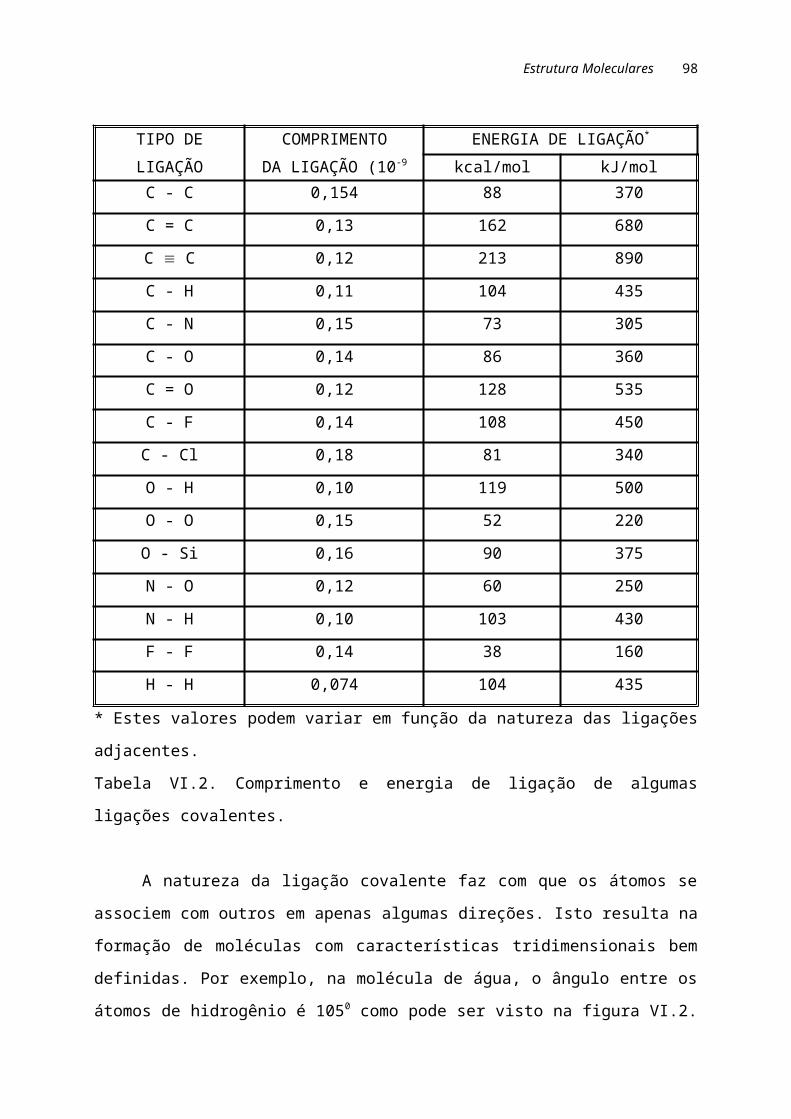
Difusão Atômica 98
Figura VIII.8. Modelo para aplicação da segunda lei de Fick.
Se o fluxo atômico é contínuo ao longo de x, pode-se escrever:
2 1J = J + Jx
x
D (VIII.12)
A substituição de J2 na equação VIII.11 permite obter:
-Jx
xA t = A x Ct
D D D D (VIII.13)
Fazendo Dt tender a zero,
-Jx
= Ctt x
(VIII.14)
O fluxo de átomos pode ser dado pela 1ª lei de Fick. Substituindo VIII.9 em
VIII.14, obtêm-se:
-Jx
= x
DCx
(VIII.15)
ou
Ct
= DC
x
2
2 (VIII.16)
Esta equação é denominada de 2ª lei de Fick e é aplicada a casos de difusão
atômica em regime transitório. Em função das condições de contorno do problema
tratado, esta equação apresenta vários tipos de solução. Em termos práticos, dois
tipos de problema podem ser abordados através da equação VIII.16: cementação de
aços e tratamento de homogeneização de peças fundidas.

Difusão Atômica 99
Figura VIII.9. Diagrama do fluxo de átomos na formulação da 2a lei de Fick
VIII.6.a. CEMENTAÇÃO DE AÇOSO tratamento da cementação de aços através das equações de difusão
envolve o emprego da "função erro". A figura VIII.10 exibe um diagrama
esquemático de um processo de cementação. Com a solução da equação VIII.16 é
possível descrever o perfil de concentração de carbono em aços durante o
processo. Esta solução é dada por:
C(x, t) = C - C - C erf x
2 D tS S 0
(VIII.17)
na qual C(x,t) é a concentração de carbono num determinado ponto "x", para um
certo tempo "t" de cementação (% em peso), C0 é a concentração inicial de carbono
(% em peso), CS é a concentração de carbono na superfície da peça, D é o
coeficiente de difusão do carbono em aço (m2/s), x é a distância a partir da
superfície (m) e t é o tempo de cementação (s). A função erro, "erf", é uma função
matemática com valores tabelados da mesma forma que funções trigonométricas.

Difusão Atômica 100
Figura VIII.10. Modelo de análise do processo de cementação de aços.
FUNÇÃO ERRO (erf) a. Definição:
erf(Z) = 2
eZ Z
0
Z-u
3 52 du = 2 Z -
3x1! +
5x2!...
(VIII.18)
b. Características:
erf(-Z) = -erf (Z)
erf(-) = -1
erf(0) = 0
erf(+) = +1
c. Derivação:
erf(Z)Z
= 2Z
exp - Z2 (VIII.19)
2
22erf(Z)
Z = -
4Z
Z exp Z
(VIII.20)
d. Função Erro complementar (erfc):
erfc(Z) = 1 - erf(Z) = 2Z
exp u duZ
2
(VIII.21)
e. Aproximações:
Z > 2 erf(Z) 1
Z < 0,2 erf(Z)2
Z
erf(Z) = 1 - uy + by + cy - Z2 3 2exp (VIII.22)
onde:

Difusão Atômica 101
y = 1
1 + Zd (Z > 0) (VIII.23)
sendo:
a = 0,348
b = -0,096
c = 0,748
d = 0,471
f. Representação gráfica esquemática:
g. Valores selecionados da função erro complementar
Z erfc(Z) Z erfc(Z)
1,0 0,15730 1,6 0,02365
1,1 0,11980 1,7 0,01621
1,2 0,08969 1,8 0,01091
1,3 0,06599 1,9 0,007210
1,4 0,04777 2,0 0,004678
1,5 0,03389
h. Valores selecionados da função erro

Difusão Atômica 102
VIII.6.b. TRATAMENTO DE HOMOGENEIZAÇÃO DE FUNDIDOSEsta abordagem é aplicada na minimização de microsegregação em peças
obtidas pelo processo de fundição. Em geral, a microsegregação de solutos ocorre
em torno de braços dendríticos, o que leva o perfil de concentração de solutos a
apresentar uma variação senoidal, conforme mostra a figura VIII.11. Assumindo que
a concentração de solutos, antes do tratamento de homogeneização, tem variação
do tipo:
C(x, t = 0) = C + senxl
b
(VIII.24)
A solução desse processo de difusão é dada por:
C(x, t) = C + senxl
.e-Dt
l
2
2b
(VIII.25)

Difusão Atômica 103
Figura VIII.11. Representação da variação de composição em torno de ramos
dendríticos.
VIII.7. DIFUSÃO ATÔMICA EM CONTORNOS DE GRÃO E DISCORDÂNCIASO movimento atômico considerado até agora envolveu difusão em cristais
perfeitos. No entanto, a existência de defeitos cristalográficos, como discordâncias e
contornos de grão na rede cristalina pode influir no transporte atômico por difusão.
Nos contornos de grão ocorre uma maior concentração de vazios, o que facilita o
movimento de átomos. Nas regiões onde existem discordâncias, a rede cristalina
sofre deformações (fica com a estrutura mais aberta), o que da mesma forma facilita
a difusão. Como conseqüência, os coeficientes de difusão em contornos de grão e
nas vizinhanças de discordâncias são bem maiores que o observado no cristal
perfeito. Porém, as regiões associadas a contornos de grão e às discordâncias são
pequenas comparadas com o volume de material, de modo que, em geral, a
contribuição dos contornos de grão e discordâncias é pequena.
Por exemplo, em altas temperaturas, o coeficiente de difusão é alto no cristal,
facilitando o movimento de átomos na rede, tornando desprezível a difusão em
contornos de grão. Por outro lado, se a temperatura é baixa, a difusão na rede
cristalina é difícil e a contribuição dos contornos de grão passa a ser significativa,
como é mostrado na figura VIII.12. Já no caso das discordâncias, essa contribuição
é, em geral, desprezível nos material recozidos (recozimento é o processo de
aquecimento e resfriamento para recuperar a ductilidade de um material), onde a
densidade desse defeito é, em geral, baixa. Em materiais com alto grau de
deformação plástica, a densidade de discordâncias é elevada e o efeito das

Difusão Atômica 104
mesmas na difusão atômica pode ser significativo.
Figura VIII.12. Evolução do coeficiente de difusão atômica na prata (autodifusão)
com a temperatura no cristal e em contornos de grão.
EXERCÍCIOS
VIII.1. Considere um tanque de aço contendo hidrogênio na pressão de 10 atm e
vácuo no lado externo. Sabendo-se que a solubilidade do H no aço a 10 atm é igual a
10-2 g/cm3 e o coeficiente de difusão do H no aço é igual a 10 -5 cm2/s, determinar o
fluxo de H através de uma parede de aço de 1mm, em g/cm2.s.
VIII.2. O coeficiente de difusão do carbono na austenita (ferro CFC) pode ser
aproximado pela equação:
C2D = 0,2 -33000cal / mol
RTcm / sexp
(a) Determine o valor do coeficiente de difusão a 9200C;
(b) Supondo um processo de cementação na temperatura dada acima, qual o
tempo necessário para que um ponto localizado a 1 mm da superfície mude sua
concentração de 0% de C para 0,1% de C, considerando que esta superfície contém
2% de C.
(c) Determine a temperatura necessária para diminuir este tempo à metade.

Difusão Atômica 105
VIII.3. O transporte atômico por difusão é governado pelas leis de Fick. A partir da 1ª
lei de Fick (regime permanente), derive a expressão que representa a 2ª lei (regime
transitório).
VIII.4. Uma liga Fe-C é colocada em uma atmosfera descarburizante, que mantém a
superfície da mesma na concentração de carbono igual a zero.
(a) Desenhe a concentração de carbono em função da posição, C(x), após um
determinado tempo finito.
(b) Em função de C(x), determine C/x versus x. Lembre-se que a
concentração da superfície é mantida zero.
(c) A partir de C/x, plote J(x) versus x.
VIII.5. Em um processo de cementação de um aço 1020, a superfície da peça
tratada é mantida à concentração de 1,5% de carbono e à temperatura de 950oC.
Calcular a composição da citada peça em um ponto situado a 0,1mm de sua
superfície, após 30 minutos de processamento.

Diagrama de Fases 106
DIAGRAMA DE FASESIX.1. INTRODUÇÃO
Da grande variedade de materiais metálicos utilizados em engenharia,
poucos são constituídos de metais puros. Em muitos casos, com o objetivo de
aprimorar propriedades, adiciona-se propositadamente ao metal original, um ou
mais elementos. Nesta situação, o material resultante é denominado de liga
metálica. As ligas metálicas podem ser classificadas como monofásicas e
polifásicas, dependendo do número de fases observadas em uma determinada
condição de composição, temperatura e pressão. Fases em materiais são definidas
como regiões que diferenciam-se de outras em termos de estrutura e/ou
composição. O estudo de um sistema de um, dois ou mais componentes, sendo
monofásico ou polifásico, pode ser feito a partir dos diagramas de fases. Os
diagramas de fases são representações gráficas das fases presentes em um
sistema em função da temperatura, pressão e composição. A maioria dos
diagramas de fases são obtidos em condições de equilíbrio e são usados para
entender e prever o comportamento dos materiais. Dentre algumas das informações
obtidas dos diagramas de fases, pode-se listar:
a. Fases presentes em diferentes condições de temperatura, pressão e
composição;
b. Solubilidade sólida de um elemento ou composto em outro;
c. Temperaturas ou faixas de temperatura de transformação de uma liga em
condições de equilíbrio.
O uso de diagramas de fases permite explicar situações curiosas como é o
caso onde se adiciona sal (NaCl) ao gelo acumulado em estradas de países frios. O
sal provoca um decréscimo na temperatura de transformação de fase do sistema
(H2O-NaCl) e assim, o gelo pode se transformar em líquido.
Em geral, dependendo de variáveis como temperatura, pressão e
composição, uma liga pode exibir microestrutura monofásica ou polifásica. Uma
microestrutura é monofásica quando existe completa solubilidade do soluto no
solvente, ou seja, não ocorre a formação de precipitado, que em outras palavras
seria uma segunda fase. Exemplos de ligas que podem exibir microestrutura
monofásica: Latão (cobre e zinco), bronze (cobre e estanho), etc.
Na figura IX.1 apresenta-se uma fotomicrografia da liga Al-4%Cu, com
microestrutura monofásica. Para tal composição, todo o cobre pode ser dissolvido

Diagrama de Fases 107
no alumínio, sem ocorrer precipitação de outra fase. Em geral, as propriedades das
ligas com microestrutura monofásica são diferentes das propriedades dos metais
componentes da mesma. Nas figuras IX.2 e IX.3 apresenta-se a influência da adição
de um segundo elemento em algumas propriedades físicas e mecânicas da liga Cu-
Zn (latão) e da liga Cu-Ni.
Figura IX.1. Liga metálica Al-4%Cu monofásica.
Quando não ocorre a completa solubilidade do soluto no solvente, ou seja,
quando o limite de solubilidade é ultrapassado, formam-se então, as ligas com
microestruturas polifásicas. Em uma liga com microestrutura polifásica pode-se
notar claramente a presença de dois ou mais compostos ou soluções sólidas, que
são chamados de fases. A fase é a região homogênea de uma mistura heterogênea.
Um exemplo de liga polifásica é encontrado na liga eutética Pb-Sn, muito usada em
soldagem de componentes semicondutores. Uma fotomicrografia dessa liga,
apresentada na figura IX.4.a, revela a existência de duas fases distintas com
estruturas e composições diferentes: estanho (HC) em solução sólida com chumbo
(soluto) e chumbo (CFC) em solução sólida com estanho (soluto). A figura IX.4.b
mostra a estrutura polifásica da liga Al-7%Si-2%Cu, utilizada na fundição de peças
automotivas.

Diagrama de Fases 108
Figura IX.2. Propriedades físicas e mecânicas de ligas Cu-Zn (latão).
Figura IX.3. Propriedades físicas e mecânicas de ligas Cu-Ni.

Diagrama de Fases 109
(a)
(b)
Figura IX.4. Estruturas polifásicas: (a) Liga Sn-Pb: Sn (HC) em solução sólida com
Pb (soluto) e Pb (CFC) em solução sólida com Sn (soluto); (b) liga Al-7%Si-2%Cu,
utilizada na fundição de peças automotivas.
IX.2. DIAGRAMA DE FASES DE SUBSTÂNCIAS PURASUma substância pura, como a água, pode existir nas fases sólida, líquida e
vapor, dependendo das condições de temperatura e pressão. Um exemplo bastante
familiar de uma substância pura com duas fases em equilíbrio é um copo de água
contendo cubos de gelo. Neste caso, a água nos estados líquido e sólido apresenta
duas fases distintas separadas por uma região interfacial, a superfície do gelo.
Durante a ebulição da água, esta substância nos estados líquido e vapor apresenta
duas fases em equilíbrio.
Uma representação gráfica das fases da água existentes em diferentes
condições de temperatura e pressão é mostrada na figura IX.5. Neste diagrama P-T
existe um ponto triplo em baixa pressão (4,6 torr) e baixa temperatura (0,00980C),
onde coexistem as fases sólida, líquida e vapor. As fases líquida e vapor coexistem
ao longo da linha de vaporização e as fases sólida e líquida ao longo da linha de

Diagrama de Fases 110
solidificação. Nestas duas linhas, estão em equilíbrio duas fases.
Figura IX.5. Diagrama de fases (pressão-temperatura) da água.
IX.3. DIAGRAMA DE FASES BINÁRIO
Na análise de sistemas de um único componente, as variáveis consideradas
eram temperatura e pressão. Quando os sistemas passam a ter mais componentes,
uma nova variável é introduzida, a composição. O tratamento de sistemas com dois
componentes é complexo por envolver figuras tridimensionais. O que se faz então, é
fixar uma das variáveis para transformar o sistema em bidimensional. No caso da
transformação de fase dos materiais metálicos, serão analisadas as transformações
líquido/sólido e transformações no estado sólido. Como as fases líquida e sólida
sofrem pouca influência da variável pressão e em geral, os processos metalúrgicos
são realizados à pressão atmosférica, esta variável (pressão) é fixada, permitindo
que os sistemas metálicos sejam estudados a partir de diagramas isobáricos, onde
as variáveis são temperatura e composição.
Os diagramas binários podem ser de dois tipos: isomorfos e anisomorfos. Os
diagramas isomorfos são sistemas cujos componentes têm a mesma estrutura
cristalina e são totalmente solúveis um no outro, em qualquer composição.
Considerando um sistema cristalino com dois componentes A e B, tal que A é o
solvente e B o soluto, dois tipos de solução sólida são possíveis: solução sólida
intersticial e solução sólida substitucional. Entretanto, apenas as soluções sólidas do
tipo substitucional permitem a solubilidade completa de B em A, qualquer que seja a

Diagrama de Fases 111
proporção de ambos. Na figura IX.6 apresenta-se um diagrama isomorfo, também
chamado de diagrama de solução sólida. Em diagramas de fases, as fases sólidas
são designadas por letras gregas: a, b, g, etc.
Em tal diagrama isomorfo, resfriando um líquido com qualquer composição
(diferente de 0 ou 100% de soluto) dentro desse diagrama, é possível obter:
Fase Líquida; Fase Líquida + Fase Sólida; Fase SólidaAs microestruturas hipotéticas dessa liga também estão apresentadas na
figura IX.6.
Figura IX.6. Diagrama de fases do tipo isomorfo.
Os sistemas isomorfos com solubilidade total são constituídos por
componentes que apresentam mesma estrutura cristalina e características físico-
químicas compatíveis. Entretanto, os casos práticos mais freqüente envolvem
componentes com diferentes estruturas cristalinas e outros fatores que resultam em
situações onde a solubilidade total não é possível, havendo um limite de solubilidade
de um constituinte no outro, além de surgirem fases intermediárias estáveis, o que
resulta em sistemas anisomorfos.
Os sistemas anisomorfos podem ser classificados de acordo com o tipo de
reação característica:
a. Sistema Eutético

Diagrama de Fases 112
O sistema eutético envolve a reação característica onde um líquido (L) transforma-
se em dois sólidos (a e b), durante o resfriamento.
L + a b (IX.1)
onde a e b são fases sólidas, podendo ser compostos ou soluções sólidas. A figura
IX.7 apresenta o diagrama de fases do sistema eutético Cu-Ag: (a) sem considerar a
formação da estrutura eutética e (b) considerando a formação da estrutura eutética.
Exemplos de sistemas eutéticos: Pb-Sn, Cu-Al e Al-Si.
(a)
(b)
Figura IX.7. Diagrama de fases Cu-Ag: (a) sem considerar a estrutura eutética; (b)
considerando a estrutura eutética.
b. Sistemas Semelhantes aos Eutéticos
São sistemas semelhantes ao sistema eutético aqueles que apresentam uma
reação onde uma fase transforma-se em duas outras, durante o resfriamento.
- Sistema Monotético (figura IX.8)
1 2L + L a (IX.2)
onde L1 e L2 são líquidos de características diferentes.

Diagrama de Fases 113
Figura IX.8. Diagrama de fases monotético.
- Sistema Metatético (figura IX.9)
b a L + (IX.3)
- Sistema Eutetóide (figura IX.10)
b a g + (IX.4)
c. Sistema Peritético
São sistemas onde existe uma reação onde um líquido (L) e um sólido (b)
transformam-se em um sólido (a), como mostra a figura IX.11.
L + b a (IX.5)
d. Sistemas Semelhantes aos Peritéticos
São considerados semelhantes aos sistemas peritéticos, os sistemas que
apresentam uma reação onde duas fases transformam-se numa terceira fase,
durante o resfriamento.
- Sistema Peritetóide (figura IX.12)
a g b + (IX.6)

Diagrama de Fases 114
Figura IX.9. Diagrama de fases metatético.
Figura IX.10. Diagrama de fases eutetóide.
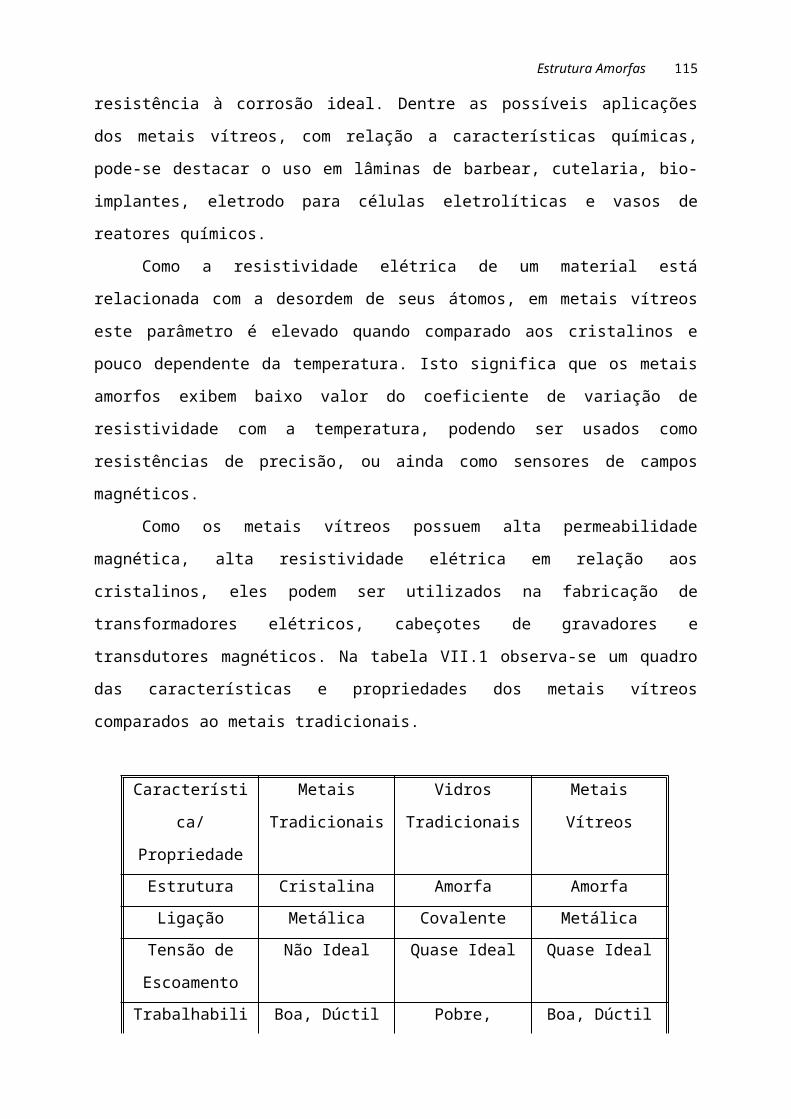
Diagrama de Fases 115
Figura IX.11. Diagrama de fases peritético
Figura IX.12. Diagrama de fases peritetóide
IX.4. REGRA DAS FASES DE GIBBSPartindo de conceitos termodinâmicos, J.W. Gibbs derivou uma equação que
define o número de fases que podem coexistir em um determinado sistema, em
condições particulares de pressão, temperatura e composição. Esta equação é
denominada de Regra das Fases de Gibbs e é dada pela relação:
P + F = C + 2 (IX.8)
onde P é o número de fases que coexistem no sistema, C é o número de
componentes do sistema e F é o grau de liberdade do sistema. Normalmente, um
componente do sistema pode ser um elemento, um composto ou ainda uma
solução. O grau de liberdade é o número de variáveis (pressão, temperatura e
composição da fase) que podem ser mudadas independentemente, sem alterar o
número de fases em equilíbrio neste sistema.
A análise do diagrama P-T da água pela regra de Gibss resulta em:
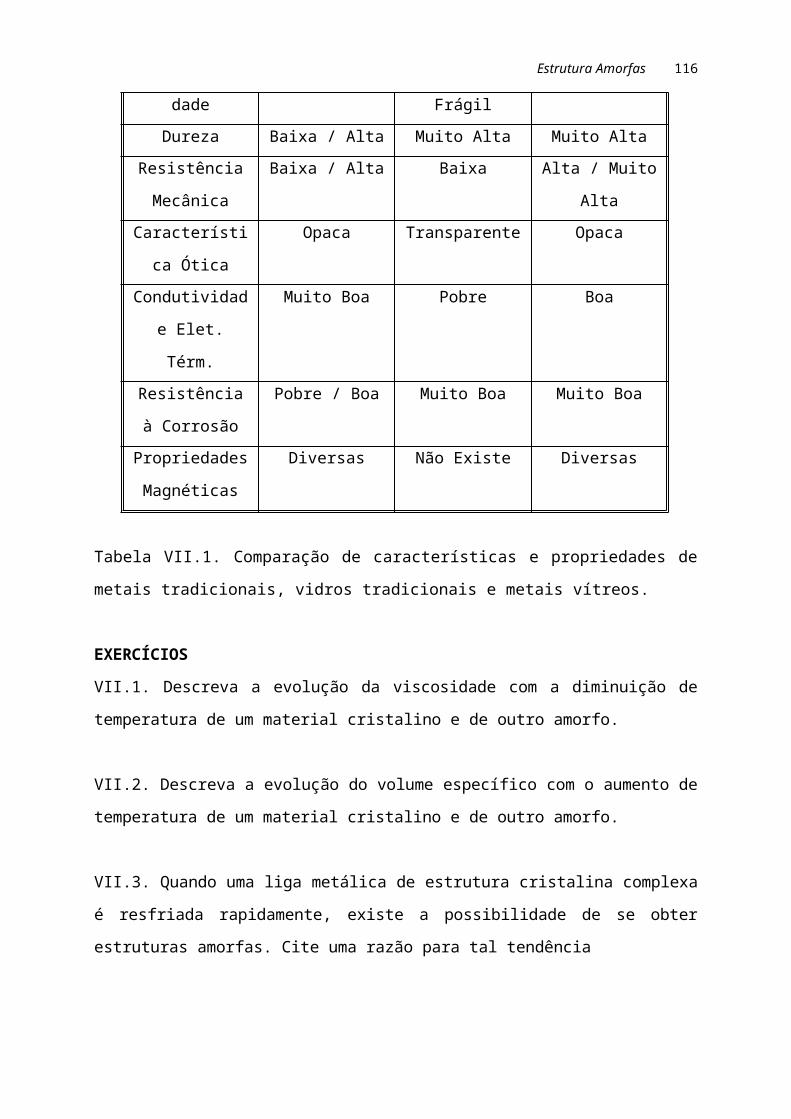
Diagrama de Fases 116
a. No ponto triplo coexistem três fases em equilíbrio e já que existe apenas
um componente (água pura), o grau de liberdade é dado por:
P + F = C + 2 (IX.9)
3 + F = 1 + 2
ou F=0 (nenhum grau de liberdade). Nenhuma variável (temperatura ou pressão)
pode ser mudada mantendo-se a existência das três fases, e assim, o ponto triplo é
chamado de ponto invariante.
b. Considerando um ponto ao longo da linha de solidificação. Em qualquer
ponto desta linha existirão duas fases. Assim,
2 + F = 1 + 2
ou F=1 (um grau de liberdade). Este resultado indica que, para manter a existência
das duas fases em equilíbrio, apenas uma das variáveis (temperatura ou pressão)
pode ser mudada, ficando a outra determinada. Assim, se uma pressão em
particular é especificada, existe apenas uma temperatura em que líquido e sólido
estão em equilíbrio.
c. Considerando um ponto dentro de uma fase, a regra das fases permite
obter:
1 + F = 1 + 2
ou F=2 (dois graus de liberdade). Este resultado indica que a temperatura ou
pressão podem ser mudadas independentemente sem comprometer a existência da
fase citada.
Na tabela IX.1 estão apresentadas as várias possibilidades para um sistema
binário. Observe que, para um sistema binário monofásico, há três graus de
liberdade. Normalmente, as variáveis consideradas são temperatura, pressão e a
composição da fase. No caso de duas fases em equilíbrio, em um sistema binário,
há dois graus de liberdade. Assim, escolhidas uma temperatura e uma pressão, nas
quais duas fases podem ser mantidas em equilíbrio, as composições das fases
estão univocamente determinadas. Naturalmente, é possível variar a temperatura e
a pressão, mas tais variações provocam alterações de composições das fases.
No caso de diagramas binários de ligas metálicas, já que a pressão é mantida
constante, a regra das fases de Gibbs é usada considerando apenas duas variáveis:
temperatura e composição. Assim a regra de Gibbs torna-se igual a:
P + F = C + 1 (IX.10)

Diagrama de Fases 117
Número de Componentes,
C
Número de fases,
P
Graus de liberdade,
F
1 1 2 (T, P)
1 2 1 (T ou P)
1 3 0
2 1 3 ( T, P, Ca)
2 2 2 (T, P)
2 3 1 (T ou P)
2 4 0
* Composição das fases
Tabela IX.1. Número de fases e grau de liberdade em sistemas unitários e binários
(P+F=C+2).
X.5. REGRA DA ALAVANCA
Considere o diagrama de fases Cu-Ag mostrado na figura IX.7. O
resfriamento de líquidos com diversas composições exibe os seguintes resultados:
a. Liga com 28,1% de cobre (composição eutética)
Seqüência: Fase líquida; Fase a + Fase b
b. Liga com 95% de cobre
Seqüência: Fase líquida; Fase líquida + Fase b; Fase b; Fase a + Fase b
c. Liga com 50% de cobre
Seqüência: Fase líquida; Fase líquida + Fase b; Fase a + Fase b
d. Liga com 5% de cobre
Seqüência: Fase líquida; Fase líquida + Fase a; Fase a + Fase b
A figura IX.7 apresenta, também, as possíveis microestruturas, encontradas
nas várias regiões do diagrama. É importante observar que a estrutura obtida em
amostras com a composição eutética apresenta um arranjo estrutural bastante
particular. A formação de duas fases ao mesmo tempo, durante solidificação de um
líquido de composição eutética, pode resultar em uma estrutura onde placas das
duas fases são alternadamente sobrepostas.
Da mesma forma, é possível observar que fora da composição eutética, por
exemplo, uma amostra com 20% de cobre apresenta duas regiões distintas. Uma

Diagrama de Fases 118
exibe regiões de fase a-livre (escura) e a outra região com estrutura eutética
(lamelas das fases a e b). Do mesmo modo, a microestrutura de uma amostra com
50% de cobre apresenta regiões de fase b-livre e regiões lamelares.
Em algumas situações é interessante conhecer a quantidade relativa de uma
determinada fase, em uma dada condição de temperatura e composição. Neste
caso é necessário utilizar a regra da alavanca, que pode ser aplicada ao diagrama
de fases mostrado na figura IX.13.
Regra da Alavanca:
% = C - CC - C
.100% e % = C - CC - C
.100%0 0a b
b
b a
a
b a(IX.11)
onde:
C0 - Concentração da liga no ponto em questão;
Ca - Concentração da fase a;
Cb - Concentração da fase b;
%a - Percentagem da fase a;
%b - Percentagem da fase b.
Figura IX.13. Diagrama de aplicação da regra da alavanca.

Diagrama de Fases 119
IX.5.a. EXEMPLO DE APLICAÇÃO DA REGRA DA ALAVANCACalcular a quantidade relativa das fases existentes numa liga Ag-Cu com
40% de cobre, nas temperaturas:
a. 10000C
b. 779,40C + DT (ligeiramente acima de 779,40C)
c. 779,40C - DT (ligeiramente abaixo de 779,40C)
d. 4000C
Resolução:
a. A 10000C toda mistura é líquida - 100% de líquido.
b. %L=(92%-40%)/(92%-28,1%).100%=81,4%
%b=(40%-28,1%)/(92%-28,1%).100%=18,6%
c. %a=(92%-40%)/(92%-8,8%).100%=62,5%
%b=(40%-8,8%)/(92%-8,8%).100%=37,5%
d. %a=(100%-40%)/(100%-0%).100%=60%
%b=(40%-0%)/(100%-0%).100%=40%
Uma outra maneira de calcular as quantidades relativas é considerar a
estrutura eutética como sendo uma “fase". Assim,
%E=(92%-40%)/(92%-28,1%).100%=81,4%
%b=(40%-28,1%)/(92%-28,1%).100%=18,6%
Para cada fase do eutético:
%a=(92%-28,1%)/(92%-8,8%).100%=76,8%
%b=(28,1%-8,8%)/(92%-8,8%).100%=23,2%
IX.6. DIAGRAMA DE FASES TERNÁRIOSA maioria das ligas industriais contém um constituinte principal, outro em
concentração moderada e diversos em quantidade menor, que são resultantes de
adições acidentais ou propositais. Um diagrama de fases binário dificilmente permite
representar as reações e fases envolvidas em um sistema de 3 ou mais
constituintes. conseqüentemente, é necessária uma análise, mesmo elementar, dos
princípios que regem os diagramas ternários. Na figura IX.14, observa-se a
representação de sistemas com uma a três variáveis. Para o caso de uma variável,

Diagrama de Fases 120
a representação é uma linha; para duas variáveis utiliza-se um plano; finalmente,
para três variáveis é necessário o emprego de uma figura tridimensional. A
observação de sistemas com quatro variáveis não é simples como os diagramas
vistos anteriormente, exigindo para isto recursos especiais. Para se contornar tal
problema, uma das variáveis pode ser fixada, reduzindo o mesmo a um sistema de
três variáveis.
Os diagramas de fases de sistemas metálicos contendo três constituintes
apresentam quatro variáveis (temperatura e composição de três constituintes). A
análise detalhada dos mesmos deve utilizar figuras tridimensionais, o que,
geralmente envolve dificuldades elevadas. Uma maneira conveniente de abordar o
problema é fixar a variável temperatura, e conseqüentemente, transformar o
problema tridimensional (quatro variáveis) para o caso bidimensional (três
variáveis). Assim, considerando a figura IX.14.c, o emprego deste diagrama é
fundamentado no seguinte princípio de geometria plana: A soma das distâncias de
qualquer ponto interno de um triângulo equilátero aos lados do mesmo (ou seja,
numa linha perpendicular ao lado), é constante e igual à altura do triângulo.
Portanto, se a altura do triângulo, h, representa 100%, a distância X a representa a
percentagem do elemento A e assim sucessivamente para Xb e Xc.

Diagrama de Fases 121
Figura IX.14. Diagramas de fases com uma, duas, três e quatro variáveis
A figura IX.15 mostra a superfície liquidus de um diagrama ternário
simplificado, constituído por três sistemas eutéticos, sem solubilidade sólida. A
análise deste diagrama pode ser executada através de cortes isotérmicos do
mesmo, como mostra a figura IX.16. A temperatura escolhida para este corte está
situada abaixo do ponto de fusão do constituinte B. Na figura IX.16.b, apresenta-se
um corte isotérmico localizado abaixo do patamar eutético AB e a figura IX.16.c
mostra um corte logo acima do ponto eutético ternário. As linhas cheias nas figuras
IX.16 (a, b e c) representam a intersecção dos planos isotérmicos com as
superfícies liquidus. As linhas tracejadas indicam os contornos dos campos de três
fases.

Diagrama de Fases 122
Figura IX.15. Superfície liquidus de um diagrama ternário simplificado, constituído
por três sistemas eutéticos.
Para algumas aplicações pode ser mais útil projetar a superfície liquidus e
solidus sobre a base do triângulo. Outra forma interessante de usar um diagrama
ternário é a do corte pseudo-binário, no qual a concentração de um componente é
mantida constante e as proporções dos outros são alteradas (como em um sistema
binário).

Diagrama de Fases 123
Figura IX.16. Cortes isotérmicos para análise do diagrama mostrado na figura IX.15.
EXERCÍCIOS
IX.1. Contrua o diagrama Cu-Ni a partir dos seguintes dados:
% em peso de Ni Temperatura Liquidus (0C) Temperatura Solidus (0C)
0 1083 1083
20 1195 1135
40 1275 1205
60 1345 1290
80 1410 1375
100 1453 1453
IX.2. Dado o diagrama de fase abaixo, determine:
(a) A percentagem relativa das fases a e b em uma liga com 50% de B, logo
abaixo da temperatura eutética.
(b) A percentagem relativa das fases a e líquido numa liga com 20% de B, logo
acima da temperatura eutética.
(c) A percentagem relativa das fases a e b numa liga com 20% de B, logo
abaixo da temperatura eutética.
(d) A percentagem relativa da fase a-livre (fora da estrutura lamelar) numa liga
com 20% de B, logo abaixo da temperatura eutética.
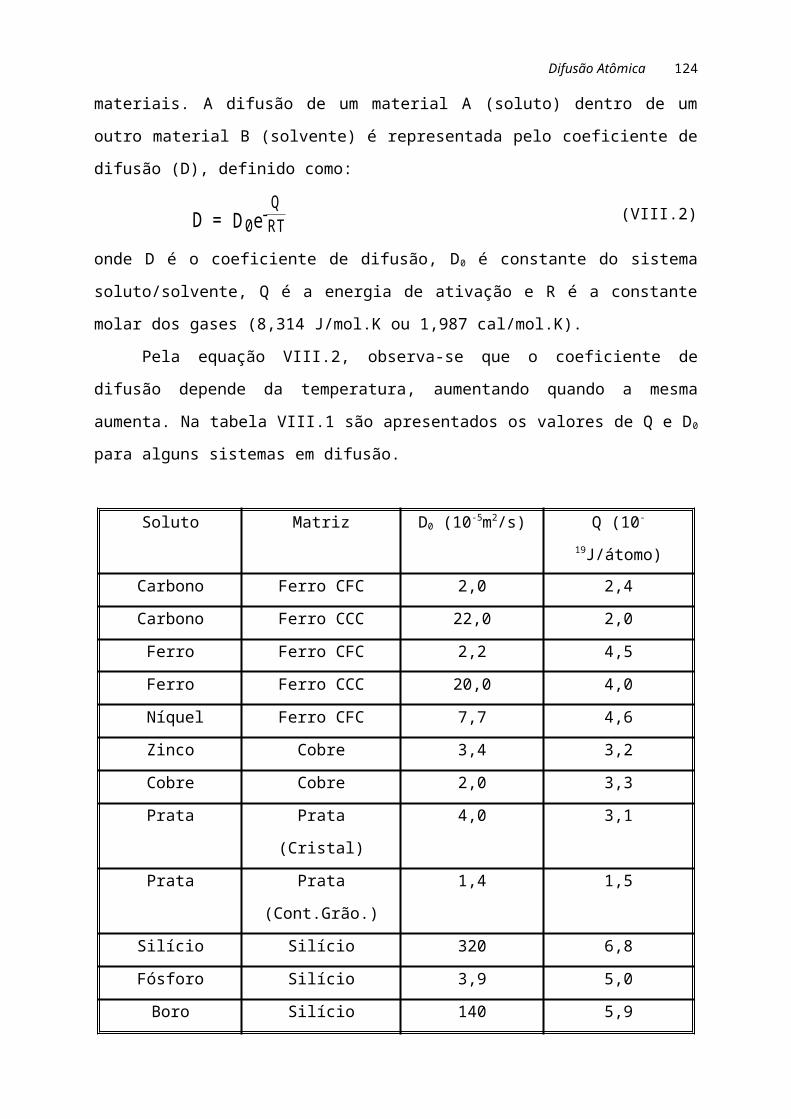
Diagrama de Fases 124
IX.3. No exercício anterior, determine o grau de liberdade para os seguintes pontos:
(a) No ponto eutético;
(b) Sobre a linha liquidus, em amostras ricas em A;
(c) Dentro do campo monofásico, onde a fase a existe;
(d) Sobre o patamar eutético.
IX.4. Denomine o tipo ou tipos de fases que existem nos campos indicados no
diagrama de fases da liga A-B mostrada abaixo.
IX.5. Determine para a liga 92%Mg-8%Al, a composição e quantidade relativa das
fases presentes nas seguintes temperaturas:
(a) 6500C;
(b) 5300C;
(c) 4200C;
(d) 3100C;
(e) 2000C.

Diagrama de Fases 125
IX.5. Descreva como as linhas liquidus e solidus de um diagrama isomorfo podem ser
determinadas experimentalmente.
IX.6. Considerando um diagrama de solubilidade total como referência, derive a regra
da alavanca para calcular as quantidades relativas (percentagem em peso) das fases
existentes na região bifásica do mesmo.

Comportamento dos Materiais Sob Tensão 126
COMPORTAMENTO DOS MATERIAIS SOB TENSÃOX.1. INTRODUÇÃO
O comportamento de um material sob solicitação mecânica é fundamental na
identificação de propriedades de interesse em engenharia mecânica. Tal
comportamento é função direta de três fatores básicos ligados às características do
material, ou seja: o tipo e a intensidade das ligações envolvendo átomos ou moléculas;
a natureza do arranjo dos átomos ou moléculas e a natureza e quantidade de defeitos
no arranjo dos átomos ou moléculas do material. Além desses três fatores, o
processamento a que o material foi submetido, determina intensamente a definição
das propriedades do mesmo.
Uma das características mais importantes dos materiais no estado sólido é a
capacidade dos mesmos em resistir ou transmitir tensões. A resposta desses materiais
sob tensão está intimamente relacionada com a propriedade do material em se
deformar elasticamente ou plasticamente. Quando um material é submetido a esforços
mecânicos, ele deforma-se de duas maneiras: elasticamente e plasticamente.
Considera-se que um material exibe comportamento elástico, quando o mesmo,
ao ser submetido a esforços mecânicos, apresenta deformações não-permanentes, ou
seja, ao se remover tais tensões, o material retorna as suas dimensões originais. Ao
nível atômico, a deformação elástica é observada quando as células unitárias alteram
suas dimensões, alongando, se o esforço for de tração ou comprimindo, se o esforço
for de compressão, como apresenta a figura X.1. Quando os esforços de tração ou
compressão cessam, as células cristalinas voltam às formas e dimensões originais.
O comportamento plástico é observado quando o mesmo material é submetido
a tensões mais elevadas e suas dimensões são alteradas permanentemente, ou seja,
cessados os esforços, o material não retorna as suas dimensões originais. Ao nível
atômico, a deformação plástica é principalmente observada quando planos atômicos
são deslizados uns sobre os outros, de tal maneira que ao se remover os esforços
mecânicos, o material não exibe suas dimensões originais.
A caracterização do comportamento mecânico de um material pode ser
implementada pelo emprego do ensaio de tração do mesmo (figura X.2). A aplicação
de uma força em um material provoca tensões e deformações (permanentes ou não)
no mesmo, como mostra a figura X.3.

Comportamento dos Materiais Sob Tensão 127
Figura X.1. Deformação elástica em cristais: (a) Cristal sem deformação; (b) Cristal
deformado por tração; (c) Cristal deformado por compressão.
A tensão, s, é definida como força por unidade de área, ou:
s = FA
(X.1)
onde:
s = Tensão (Pa = N/m2);
F = Força Aplicada (N);
A = Área do Plano (m2).
A deformação () é definida como o efeito da tensão em um material, relaciona-
se à alteração nas dimensões originais do material e é expressa como variação do
comprimento inicial, ou:
= LL0
D(X.2)
onde:
= Deformação (%);
DL = (L - L0) = Variação de comprimento (m);
L0 = Comprimento inicial (m);
L = Comprimento final (m).
O ensaio de tração revela duas fases distintas: a elástica e a plástica. Na fase
elástica um dos principais parâmetros que o ensaio de tração permite revelar é o
módulo de elasticidade do material.

Comportamento dos Materiais Sob Tensão 128
(a) (b)
Figura X.2. Diagrama esquemático de um ensaio de tração: (a) Sem deformação; (b)
Com deformação.
(a) (b) (c)Tensão limite de escoamento (sLE) - Tensão necessária para iniciar a deformação plástica;
Tensão limite de ruptura (sRU) - Final do comportamento plástico (ruptura);
Tensão limite de resistência (sRE)- Máxima carga suportada pelo material.
Figura X.3. Curvas tensão-deformação relativa: (a) Material não-dúctil sem
deformação plástica (ex.: ferro fundido); (b) Material dúctil com ponto de escoamento
definido (ex.: aço de baixo carbono); (c) Material dúctil sem ponto de escoamento
definido (ex.: alumínio).
X.2. DEFORMAÇÃO ELÁSTICASe um material apresenta comportamento elástico, o mesmo segue a lei de
Hook, que estabelece que sua deformação varia linearmente com a tensão aplicada. A

Comportamento dos Materiais Sob Tensão 129
relação entre tensão aplicada e deformação resultante é constante e denominada de
Módulo de Elasticidade (E), ou:
E = s
(X.3)
O módulo de elasticidade de um material é a medida de rigidez do mesmo. Se
um material exibe valor elevado desse parâmetro, isso siginifica que uma tensão
mecânica elevada será necessária para deformá-lo. Como visto no estudo das forças
interatômicas, o módulo de elasticidade está diretamente relacionado com a variação
de FTotal (equação II.3), em relação à distância interatômica ou,
dFda
= -d (Z e)(Z e)
4 a + nb
ada
Total
1 2
02 n+1
(X.4)
A temperatura influencia intensamente o módulo de elasticidade, e quanto mais
elevada for a mesma, menor será o módulo de elasticidade. Como o módulo de
elasticidade varia com a direção em um cristal (depende da densidade linear de
átomos), a anisotropia dos cristais permite que o mesmo varie intensamente com a
orientação do cristal. Como exemplo, o ferro tem um módulo de elasticidade médio de
cerca de 205 MPa. Porém, o módulo real de um cristal de ferro varia de 208 MPa na
direção [111], para apenas 125 GPa na direção [100]. A tabela X.1 apresenta valores
de módulo de elasticidade de diversos materiais.
Um outro parâmetro importante, relacionado ao comportamento mecânico dos
materiais, é o módulo de elasticidade transversal (G), que relaciona tensões e
deformações de cisalhamento dentro da fase elástica do material. Este parâmetro é
definido de maneira semelhante ao módulo de elasticidade (E), ou seja:
G = g (X.5)
onde:
= Tensão de cisalhamento
g = Deformação elástica por cisalhamento.
Conforme mostra figura X.4, a deformação elástica por cisalhamento, g, é
definida como a tangente do ângulo de cisalhamento a, ou g = tg a.
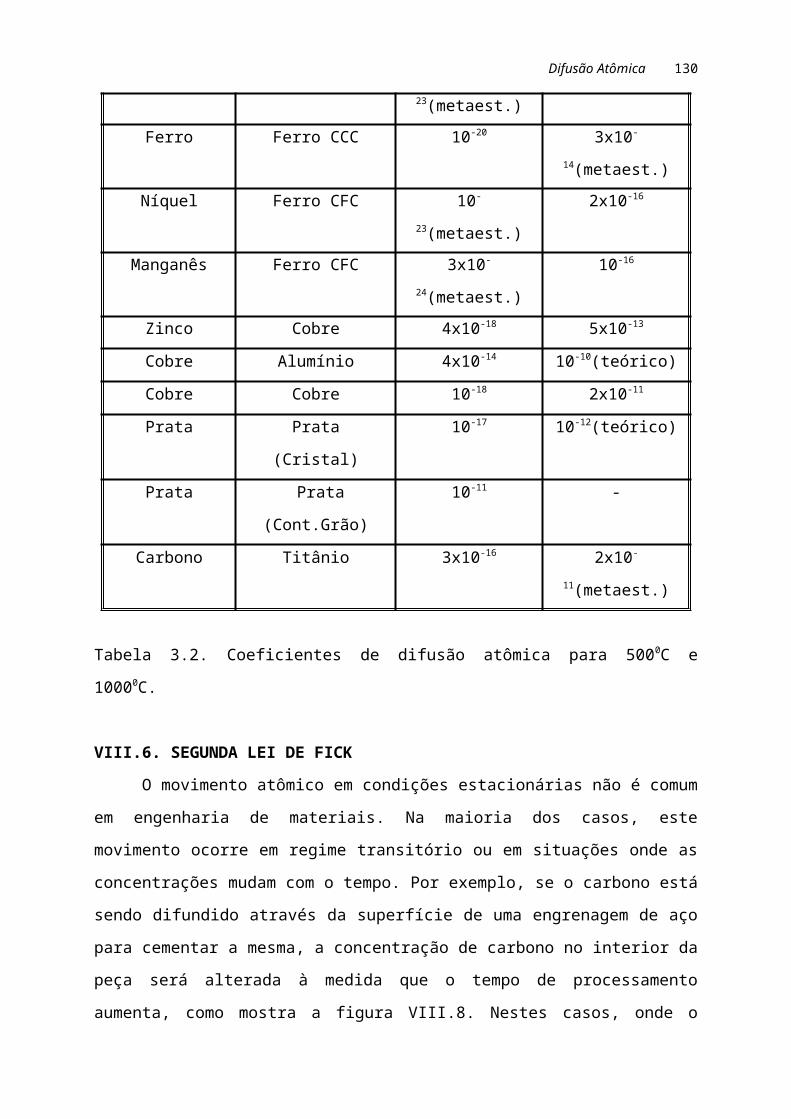
Comportamento dos Materiais Sob Tensão 130
X.3. DEFORMAÇÃO PLÁSTICA POR DESLIZAMENTONa fase plástica do material, ocorrem as deformações permanentes no mesmo.
Neste caso, planos atômicos do material são deslocados permanentemente de suas
posições originais. Existem dois mecanismos de deformação plástica, quais sejam: o
deslizamento de planos cristalinos e a maclação. Em ambos os casos, a deformação
ocorre devido às componentes de cisalhamento das tensões aplicadas. A deformação
permanente de um material submetido a ensaio de tração pode ser tratada como o
alongamento, que é a quantidade de deformação permanente observada antes da
ruptura ou estricção, que é a redução da área da secção transversal, observada antes
da ruptura.
Material Módulo de elasticidade (GPa)
Borracha Sintética 0,004 - 0,075
Nylon 2,8
Borracha Vulcanizada 3,5
Chumbo 14
Magnésio 45
Ligas de Alumínio 72,4
Cobre 110
Aço de Baixo Carbono 200
Aço Inoxidável 193
Titânio 117
Quartzo (SiO2) 310
Alumina (Al2O3) 350
Tungstênio 400
Tabela X.1. Valores do módulo de elasticidade de diversos materiais.

Comportamento dos Materiais Sob Tensão 131
Figura X.4. Diagrama esquemático da deformação elástica por cisalhamento.
O principal mecanismo de deformação plástica é o deslizamento de planos
atômicos e caracteriza-se pelo movimento de uma parte do cristal em relação a outra
(figura X.5). Nas estruturas cristalinas, estes deslizamentos acontecem em torno de
planos atômicos compactos (de alta densidade planar de átomos), seguindo direções
compactas (de alta densidade linear de átomos). A tabela X.2 apresenta planos e
direções de deslizamento das estruturas cristalinas mais comuns.
A tensão necessária para iniciar a deformação plástica em monocristais,
depende não apenas da tensão necessária para deslizar os planos cristalinos, mas
também da orientação da tensão aplicada em relação ao sistema de deslizamento.
Para que esta deformação plástica por deslizamento de planos atômicos ocorra, é
necessário que a componente de cisalhamento da força aplicada ao material, no plano
e na direção de deslizamento, atinja o limite de resistência ao cisalhamento do
material, também chamado de tensão crítica de cisalhamento. O cálculo desta tensão
necessária para deformar o cristal envolve a determinação da área de cisalhamento,
bem como da força na direção de cisalhamento.
Considere como exemplo um ensaio de tração. Assumindo que um corpo de
prova de geometria cilíndrica e secção transversal A, é solicitado mecanicamente
através da força de tração F, como mostra a figura X.6.

Comportamento dos Materiais Sob Tensão 132
Figura X.5. Deslizamento em um monocristal de zinco paralelo ao plano (0001): (a)
vista frontal - cristal real; (b) vista lateral - cristal real;(c) diagrama da vista lateral com
os planos de deslizamento; (d) célula HC mostrando os planos de deslizamento.
A aplicação da força F no corpo de prova provoca o aparecimento de uma
tensão de tração no mesmo, que é dada por:
s = FA
(X.6)
No plano de deslizamento, a componente desta força F, causa uma tensão de
cisalhamento , igual a:
= FA
S
S(X.7)
onde:
AS = Área da secção transversal no plano de deslizamento;
FS = Força aplicada no plano de deslizamento;
Estrutura Plano de
Deslizamento
Direção de
Deslizamento
Número de
Sistemas
Diagrama
CFC
(Cu, Al, Ni,
Pb, Au, Ag ...)
{111} <110> 4 X 3 = 12
CCC
(Fea, W, Mo)
{110} <111> 6 X 2 = 12

Comportamento dos Materiais Sob Tensão 133
CCC
(Fea, W, Mo)
{211} <111> 12 X 1 = 12
CCC
(Fea, K)
{321} <111> 24 X 1 = 24
HC
(Cd, Zn, Mg,
Ti, Be ...)
{0001} <1120> 1 X 3 = 3
HC
(Ti)
{1010} <1120> 3 X 1 = 3
HC
(Ti, Mg)
{1011} <1120> 6 X 1 = 6
Tabela X.2. Planos e direções preferenciais de deslizamento em alguns cristaias

Comportamento dos Materiais Sob Tensão 134
(a) (b)
Figura X.6. Tensão de cisalhamento em um cristal submetido a esforços de tração: (a)
Nível macroscópico; (b) Nível atômico, mostrando direções e planos compactos
(sistemas de deslizamento)
Supondo que a força F apresenta um ângulo f em relação à normal ao plano
de deslizamento, então:
SA = Acosf (X.8)
A força FS atuando no plano e na direção de deslizamento é dada por:
SF = Fcosl (X.9)
A tensão de cisalhamento no plano de deslizamento é igual a:
f = FA
S cos (X.10)
ou
f l s f l = FA
= cos cos cos cos (X.11)
Esta tensão atinge seu valor máximo quando os valores de f e l são iguais a
450. Neste caso tem-se:
maxs
= 2
(X.12)

Comportamento dos Materiais Sob Tensão 135
A tabela X.3. apresenta valores do limite de resistência ao cisalhamento de
diversos metais.
Na deformação plástica por deslizamento, assumindo um mecanismo
simplificado onde a deformação implica na ruptura de muitas ligações interatômica ao
mesmo tempo, nota-se que a resistência do material considerado será muito maior
que a encontrada experimentalmente.
Evidências experimentais sugerem um mecanismo de deformação envolvendo
movimentos de discordâncias (figura X.7). As discordâncias facilitam o movimento de
planos de átomos dentro de um material. Assim, a tensão de cisalhamento calculada
para o ferro é de 1010 N/m2, porém a obtida experimentalmente é de 108 N/m2, ou seja,
é 100 vezes menor. A razão disto reside no fato de que as ligações entre os átomos
são reajustadas passo a passo pelo movimento das discordâncias.
X.4. DEFORMAÇÃO PLÁSTICA POR MACLAÇÃOO segundo mecanismo de deformação plástica que pode ocorrer em metais é a
deformação por maclação. Neste processo, parte da rede cristalina é deformada de
modo que a mesma forme uma imagem especular da parte não deformada (figura
X.8).
O plano cristalográfico de simetria entre as regiões deformadas e não
deformada, é chamado de plano de maclação. A maclação, como o deslizamento,
ocorre em direções específicas chamadas de direções de maclação. Entretanto, no
deslizamento, os átomos em um lado do plano de deslizamento deslocam-se
igualmente. Na maclação, as distâncias de movimentação dos átomos são
proporcionais a suas distâncias do plano de maclação. A maclação envolve pequenas
frações do volume total da estrutura atômica e assim, a deformação resultante deste
mecanismo é mínima. Porém, um importante papel da maclação é a mudança de
orientação do cristal, permitindo a reorientação do mesmo segundo sistemas
favoráveis à deformação por deslizamento. A figura X.9 compara os dois tipos de
mecanismos de deformação plástica.

Comportamento dos Materiais Sob Tensão 136
Figura X.7. Ilustração do movimento de uma discordância em cunha produzindo um
degrau unitário com a aplicação de uma pequena tensão de cisalhamento: (a)
discordância em cunha; (b) tensão provoca mudança das ligações atômicas, para
libertar um novo plano intercalado; (c) repetição do processo faz com que a
discordância se mova através do cristal; (d) analogia do movimento de uma
discordância com o movimento de uma dobra em um tapete.

Comportamento dos Materiais Sob Tensão 137
Figura X.8. Diagrama esquemático do processo de maclação, mostrando planos e
direções de maclação.
Metal Estrutura
Cristalina
Plano de
Deslizamento
Direção de
Deslizamento
Resistência ao
Cisalhamento
(MPa)
Zn HC (0001) [1120] 0,18
Mg HC (0001) [1120] 0,77
Cd HC (0001) [1120] 0,58
Ti HC (1010) [1120] 13,7
Ag CFC (111) [110] 0,48
Cu CFC (111) [110] 0,65
Ni CFC (111) [110] 5,7
Fe CCC (110) [111] 27,5
Mo CCC (110) [111] 49,0
Tabela X.3. Valores de tensão limite de cisalhamento necessários para ocorrer o
deslizamento em monocristais de alguns metais.
Figura X.9. Diagrama esquemático de superfícies de um cristal deformado por
deslizamento (a) e por maclação (b).

Comportamento dos Materiais Sob Tensão 138
X.5. PROPRIEDADES MECÂNICASEm função do comportamento mecânico dos materiais, as seguintes
propriedades mecânicas podem ser definidas:
a. Elasticidade - Capacidade do material ser deformado elasticamente, sem
atingir o campo plástico. A relação entre tensão e deformação elástica (s/) é definida
como módulo de elasticidade (E).
b. Ductilidade - Capacidade do material ser deformado plasticamente, sem
atingir a ruptura. Pode ser obtida da análise do alongamento e da estricção.
c. Fragilidade - Comportamento oposto à ductilidade.
d. Fluência - Capacidade do material se deformar lentamente, quando
submetido a tensões menores que a de escoamento, sob temperaturas elevadas.
e. Tenacidade - Capacidade de um material em armazenar energia sem se
romper. Pode ser quantificada através do cálculo da área sob a curva
tensão/deformação.
f. Dureza - Capacidade de um material em resistir à penetração de sua
superfície e está intimamente relacionada com a tensão limite de resistência do
material.
EXERCÍCIOS
X.1. Por que a resistência mecânica de um metal monocristalino, calculada
teoricamente, é sempre maior que aquela obtida na prática ?
X.2. Quais são os 2 principais mecanismos de deformação plástica ?
X.3. Qual é a função do fenômeno de maclação em um processo de deformação
plástica?
X.4. Descreva o papel das discordância no processo de deformação plástica ?

Comportamento dos Materiais Sob Tensão 139
X.5. A tensão de cisalhamento crítica para os sistema <110>/{111} do Cu
monocristalino é de 1 MPa. a. Qual a tensão a ser aplicada na direção [001] para
produzir deslizamento na direção [011] e no plano (111)?

Introdução aos Materiais Compósitos 140
INTRODUÇÃO AOS MATERIAIS COMPÓSITOSXI.1. INTRODUÇÃO
Desde o inicio da civilização, o homem tem utilizado os materiais para garantir
sua sobrevivência e melhorar o seu nível de vida. Inicialmente, foram empregados os
materiais disponíveis até então, como ossos, pedras, madeira, barro, etc. A
curiosidade e principalmente, a inteligência humana deram origem a processos de
transformação de matérias-primas naturais em materiais sintéticos, que passaram a
suprir algumas necessidades. O desenvolvimento de técnicas de transformação
apropriadas resultou na produção de materiais com melhores propriedades e
desempenhos mais eficientes. Entretanto, à medida que as necessidades do homem
tornaram-se mais complexas, os processos de seleção de materiais tornaram-se mais
severos, pois o grau de dificuldade em encontrar materiais de características
diversificadas, elevou-se. Tal evento desencadeou um processo de conjugação de
diferentes materiais, em função de suas propriedades, com o objetivo de alcançar
características particulares. Embora tal processo de conjugação tenha ocorrido de
maneira mais intensa, só após a metade deste século, e devido ao desenvolvimento
da indústria aeroespacial, este tipo de alternativa já era usado desde os primórdios da
Humanidade.
Os materiais compósitos ou conjugados para aplicações sofisticadas e
envolvendo alta tecnologia só surgiram há não mais que 40 anos, com o aparecimento
das resinas poliméricas. Inicialmente, tais resinas foram reforçadas com fibras
naturais, como asbesto, madeira ou tecidos. Durante a 2a Guerra Mundial, devido às
necessidades militares, deu-se o desenvolvimento da fibra de vidro, o que permitiu a
produção dos materiais compósitos do tipo fibra de vidro/resina plástica. Isto resultou
em um impulso muito grande, que deu origem a atual era dos materiais compósitos
avançados. Com o término desta guerra, os materiais compósitos começaram a ser
empregados em diversas indústrias, como a aeroespacial, a automobilística e a naval.
Os materiais compósitos podem ser definidos como o resultado da combinação de
dois ou mais diferentes constituintes. A maioria destes materiais consiste de um
elemento de reforço envolvido por uma matriz, com o objetivo de obter características
específicas e propriedades desejadas. Geralmente, os componentes não se dissolvem
um no outro e podem ser identificados fisicamente, por uma interface bem definida
entre os mesmos.

Introdução aos Materiais Compósitos 141
Na prática, esta definição não pode ser aplicada de modo genérico. Ao nível
atômico, alguns materiais, como as ligas metálicas e os polímeros, podem ser
classificados como compósitos, já que resultam da associação de diferentes
elementos químicos. Ao nível microscópico (10-6 a 10-4 m), algumas ligas metálicas
apresentam diferentes fases, como é o caso dos aços, que em determinadas
condições de processamento apresentam as fases ferrita e cementita. Estas duas
fases podem ser facilmente identificadas em um microscópio e dessa forma, tais ligas
podem ser classificadas como materiais compósitos. Ao nível macroestrutural (>10-4
m), os plásticos reforçados com fibras de vidro podem ter seus constituintes
identificados sem auxílio de microscopia e assim, podem ser considerados como
materiais compósitos. Concluindo, a definição de um material compósito está
necessariamente associada ao nível de grandeza considerado. Em engenharia, a
definição adotada de materiais compósitos, refere-se ao estudo de materiais que
apresentam uma mistura ou combinação de dois ou mais micro ou macro-
constituintes, diferentes em forma e composição química e essencialmente insolúveis
um no outro. Ainda do ponto de vista de engenharia, um material compósito
representa a combinação de dois ou mais materiais para resultar em um material que
possui propriedades e características superiores as de seus constituintes.
XI.2. TIPOS DE MATERIAIS COMPÓSITOSOs diversos tipos de materiais compósitos podem ser classificados de acordo
com a composição química dos constituintes e com a geometria ou forma das fases
presentes. Materiais com características orgânicas podem ser conjugados com
aqueles de natureza inorgânica. Componentes na forma de fibras, longas ou curtas,
de laminados, de partículas, podem ser incorporados, proporcionando diferentes
estruturas e características ao compósito. De um modo geral, entre os materiais
compósitos mais comuns, encontram-se os plásticos reforçados. Além destes tipos de
materiais compósitos, existem outras classes de desenvolvimento mais recente, como
é o caso dos compósitos de matriz metálica e os de matriz cerâmica.
XI.3. PLÁSTICOS REFORÇADOSUm dos tipos mais comuns de material compósito refere-se aos plásticos
reforçados através de fibras. Neste caso, uma matriz plástica é reforçada por um outro
tipo de material na forma de fibras. São denominados de reforços aqueles

Introdução aos Materiais Compósitos 142
constituintes que modificam determinadas propriedades da matriz polimérica. Desde
simples cargas minerais, que são adicionadas aos polímeros com o objetivo de
diminuir custos, até fibras de alta resistência podem ser considerados como elementos
de reforço. As fibras, pela sua característica unidirecional, incrementam intensamente
grande parte das propriedades mecânicas em determinadas direções.
Os reforços podem ser classificados de acordo com a sua natureza ou com
relação as suas características geométricas. Os reforços podem ser: duros - para
aumentar a dureza e a resistência à abrasão; resistentes à ruptura - para elevar a
resistência à tração, à flexão e ao cisalhamento; rígidos - para elevar o módulo de
elasticidade; e finalmente, térmicamente resistentes - para elevar a estabilidade
térmica do compósito. Quanto às características geométricas, os reforços podem ser:
particulados, fibrosos ou laminados. Os particulados apresentam comprimento próximo
à espessura. Os fibrosos tem seu comprimento bastante superior à espessura e suas
fibras podem ser contínuas ou curtas. Em ambos os casos citados (particulados e
fibrosos), os reforços são envolvidos por uma matriz polimérica contínua. No caso de
reforços laminados, estes se apresentam na forma de uma fase contínua. Os reforços
particulados proporcionam reforço isotrópico, enquanto que os fibrosos favorecem o
reforço anisotrópico.
A maior parte dos reforços particulados tende a aumentar a rigidez da matriz
polimérica, podendo ou não aumentar a tenacidade ou a resistência à tração. Em
muitos casos, a tenacidade é substancialmente decrescida pelas partículas
isométricas. Por outro lado, mesmo fibras razoavelmente curtas conferem
combinações de elevados valores de módulo de elasticidade, resistência mecânica e
tenacidade.
Dentre os diversos tipos de fibras utilizadas como reforços em materiais
compósitos, destacam-se as fibras de vidro, de carbono e as arâmidas.
As fibras de vidro são as mais utilizadas, pois têm custo significativamente
reduzido. As fibras de carbono e de poliamidas aromáticas (arâmidas) apresentam
propriedades mecânicas bastante especiais, sendo ideais para aplicações envolvendo
alta tecnologia, como é o caso da industria aeronáutica e espacial, apesar do custo
das mesmas ser elevado. Estas três classes de fibras são responsáveis por quase
90% do reforço de matrizes poliméricas e quando longas e/ou contínuas permitem a
melhora de diversas propriedades.

Introdução aos Materiais Compósitos 143
As fibras de vidro permitem a obtenção de materiais compósitos com boa
estabilidade dimensional, elevada resistência mecânica, boa resistência ao calor, ao
frio, à umidade e à corrosão, boa isolação elétrica, facilidade de produção e baixo
custo.
Existem dois tipos básicos de vidro utilizados na obtenção dessas fibras: O tipo
E (elétrico) e o tipo S (alta resistência mecânica). O vidro do tipo E foi o primeiro a ser
desenvolvido especialmente para a produção de fibras contínuas, indicado
inicialmente para aplicações elétricas. Basicamente, este tipo é constituído de óxidos
de alumínio e boro, com uma quantidade de sódio e potássio bastante baixa. A
composição básica do vidro tipo E é de 52-56% de SiO2, 12-16% de Al2O3, 16-25% de
CaO e 8-13% de B2O3. A resistência à tração suportada por fibras de vidro do tipo E é
próxima de 3,44 GPa e o módulo de elasticidade deste material é de 72,5 GPa. O
vidro tipo S é mais resistente, porém de custo mais elevado e é usado principalmente
em aplicações aeroespaciais e militares. A resistência à tração suportada por este tipo
de vidro é próxima de 4,48 GPa e seu módulo de elasticidade é de 85,4 GPa. A
composição típica do mesmo é de 65% de SiO2, 25% de Al2O3 e 10% de MgO.
Quando comparadas com as fibras de carbono e de arâmidas, as fibras de
vidro apresentam resistência à tração e módulo de elasticidade inferiores. Porém,
como mostra a tabela XI.1, o alongamento é maior no caso das fibras de vidro. Em
termos de densidade, as fibras de vidro apresentam valores maiores que as de
carbono e as de arâmida. Entretanto, devido ao baixo custo de produção e
versatilidade das mesmas, estas fibras são as mais utilizadas industrialmente no
reforço de matrizes poliméricas.
Os materiais compósitos com fibras de carbono e matrizes à base de resinas
poliméricas, são caracterizados por exibir uma combinação de baixo peso, alta
resistência à tração e elevado módulo de elasticidade. Tais propriedades permitem
que tal material compósito seja intensamente utilizado na indústria aeroespacial.
Entretanto, o custo elevado para se produzir as fibras de carbono inibem o uso deste
tipo de compósito, em diversos tipos de indústrias, como é o caso da automobilística.
Um dos tipos de fibra de carbono mais comuns é o produzido a partir de PAN
(poliacrilonitrila). A produção deste tipo de fibra de carbono envolve três estágios: (a)
estabilização; (b) carbonização e (c) grafitização. No primeiro estágio ou estabilização,
as fibras PAN são esticadas e oxidadas em temperaturas próximas de 2000C,
enquanto tensionadas mecanicamente.

Introdução aos Materiais Compósitos 144
No estágio de carbonização, o objetivo principal é elevar a resistência mecânica
do material. Neste processo, as fibras PAN já estabilizadas, são aquecidas até que as
mesmas transformem-se em carbono, através da eliminação de oxigênio, hidrogênio e
nitrogênio. O tratamento térmico de carbonização é implementado em presença de
atmosfera inerte, em uma faixa de temperaturas entre 1.000 e 1.5000C. Durante esta
etapa, núcleos de grafite são formados dentro de cada fibra, o que resulta no aumento
na resistência mecânica do material.
Propriedade Fibra de Vidro
Tipo E (HTS)
Fibra de
Carbono (HT)
Arâmida
(Kevlar 49)
Resistência
Mecânica (MPa)
2.410 3.100 3617
Módulo de Elast.
(GPa)
69 220 124
Alongamento
Máximo (%)
3,5 1,4 2,5
Densidade
(g/cm3)
2,54 1,75 1,48
Tabela XI.1. Propriedades mecânicas das fibras de vidro, carbono e Kevlar.
O terceiro estágio ou grafitização é usado quando se deseja aumentar o
módulo de elasticidade da fibra de carbono já carbonizada. Durante a etapa de
grafitização, que é efetuada em temperaturas próximas de 1.8000C, a cristalinidade
dos núcleos de grafites, surgidos no estágio anterior, é intensificada.
As fibras de carbono obtidas a partir de PAN têm resistência à tração variando
de 3,1 a 4,45 GPa e módulo de elasticidade com valores que vão de 193 a 241 GPa.
Em geral, quando o módulo de elasticidade desta fibra é aumentado, sua resistência
mecânica cai e vice-versa. A densidade de uma fibra de carbono, após os processos
de carbonização e grafitização varia de 1,7 a 2,1 g/cm3, enquanto que seu diâmetro é
de 7 a 10 mm.
Fibras de arâmidas é o nome genérico dado às fibras do tipo poliamidas
aromáticas. A fibras de arâmidas foram introduzidas comercialmente em 1972 pela Du
Pont, sob o nome comercial de Kevlar. Atualmente, existem dois tipos comerciais de

Introdução aos Materiais Compósitos 145
Kevlar: o Kevlar 29 e o 49. O Kevlar 29 é uma classe de fibras que apresenta baixa
densidade, alta resistência mecânica e é empregada, por exemplo, em proteções
balísticas. O outro tipo, ou seja, o Kevlar 49 exibe baixa densidade e elevados valores
de resistência mecânica e módulo de elasticidade. As propriedades encontradas no
Kevlar 49 fazem suas fibras ideais na produção de plásticos reforçados para a
indústria aeroespacial, naval e automobilística.
Nesta estrutura, os átomos de hidrogênio ligam as cadeias poliméricas umas as
outras, na direção transversal. Assim, analisando globalmente, tais fibras tem elevada
resistência mecânica na direção longitudinal e reduzida resistência na direção
transversal. Os anéis aromáticos da estrutura dão alta rigidez à cadeia polimérica.
As fibras de Kevlar são utilizadas em materiais compósitos de elevado
desempenho, onde o baixo peso, a alta resistência mecânica, o alto módulo de
elasticidade e a elevada resistência à fadiga são fundamentais. Um tipo de material
compósito bastante interessante é o de resina epóxi com fibras de Kevlar, que é usado
na confecção de diversas partes do ônibus espacial americano.
A figura XI.1 compara curvas tensão-deformação de fibras de vidro, carbono e
arâmidas. Pode-se observar que a resistência destas fibras está entre 1.720 a 3.440
MPa, enquanto que a deformação máxima das mesmas vai de 0,4 a 4,0%. O módulo
de elasticidade destas fibras apresenta valores na faixa de 68,9 a 413 MPa. As fibras
de carbono exibem as melhores combinações de altos valores de resistência
mecânica e módulo de elasticidade e baixos valores de densidade, porém apresentam
baixos valores de alongamento. A fibra a base de Kevlar 49 tem uma combinação de
alta resistência mecânica, alto módulo de elasticidade (não tão alto quanto as fibras de
carbono), baixa densidade e alto alongamento. Quando comparadas às fibras de
carbono e arâmidas, as fibras de vidro têm baixos valores de resistência mecânica e
módulo de elasticidade e altos valores de densidade. Devido ao baixo custo de
produção, as fibras de vidro são as mais utilizadas.
A figura XI.2 apresenta valores de resistência mecânica específica e módulo de
elasticidade específico (resistência mecânica e módulo de elasticidade por unidade de
peso) para diversos tipos de fibra de reforço. Quando comparados com o alumínio e
aço, os valores de resistência mecânica específica e módulo de elasticidade específico
das fibras de carbono são excepcionais. Devido a tais vantagens, os materiais
compósitos à base de fibras de carbono e arâmidas estão substituindo, com sucesso,
os metais em diversas aplicações aeroespaciais. Com relação às matrizes de plásticos

Introdução aos Materiais Compósitos 146
reforçados, basicamente, existem dois tipos empregados na produção de materiais
compósitos: o poliester insaturado e as resinas epóxi.
Figura XI.1. Curvas tensão-deformação de materiais usados no reforço de plásticos.
A tabela XI.2 mostra propriedades das resinas epóxi e poliester sólidas, antes
de serem reforçadas com fibras. As resinas de poliester apresentam baixo custo de
produção, porém, não são tão resistentes quanto as resinas epóxi. As resinas de
poliester instaurados são largamente utilizadas como matrizes de plásticos reforçados.
Estes materiais são empregados na fabricação de embarcações, automóveis, aviões,
etc.
As resinas epóxi custam mais, porém apresentam vantagens, como melhores
propriedades mecânicas e menor índice de contração após a moldagem. As resinas
epóxi são geralmente usadas como matrizes na produção materiais compósitos de
fibras de carbono e arâmidas.

Introdução aos Materiais Compósitos 147
Figura XI.2. Resistência à tração e módulo de elasticidade específicos de diversos
materiais.
Propriedade Resina de Poliester Resina de Epóxi
Resistência à Tração (MPa) 40-90 55-130
Módulo de Elasticidade (GPa) 2,0-4,4 2,8-4,2
Resistência ao Impacto (J/m) 10,6-21,2 5,3-53
Densidade (g/cm3) 1,1-1,46 1,2-1,3
Tabela XI.2. Propriedades das resinas epóxi e poliester sólidas, antes de serem
reforçadas com fibras.
A resistência mecânica dos compósitos empregando resina de poliester/fibra de
vidro é principalmente dependente do tipo de vidro e do arranjo das fibras de vidro.
Geralmente, quanto maior a quantidade de vidro no material compósito, mais
resistente será o mesmo. Quando as fibras estão arranjadas paralelamente, a
quantidade de vidro no material compósito pode alcançar até 80% em peso, o que
resulta em valores de resistência mecânica bastante elevados.
Quando o arranjo unidirecional e paralelo das fibras é alterado, o material
compósito tem sua resistência mecânica reduzida. Por exemplo, os compósitos feitos
de tecidos de fibras de vidro e desta forma, não apresentam arranjo unidirecional,
exibem resistência mecânica inferior a aqueles de arranjo paralelo, como mostra

Introdução aos Materiais Compósitos 148
tabela XI.3. Se as fibras de um compósito são picotadas, ou seja, o comprimento das
mesmas é bastante reduzido, a orientação de tais fibras será aleatória. Isto implica na
redução da resistência mecânica em uma determinada direção, porém, a resistência
apresentada é igual em qualquer que seja a direção considerada.
Propriedade Compósitos de
Fibras Trançadas
(Tecido)
Compósitos de
Fibras
Picotadas
Compósitos
de Fibras
Laminadas
Resistência à Tração (MPa) 206-344 103-206 55-138
Módulo de Elasticidade (GPa) 103-310 55-138 374-1175
Resistência ao Impacto (J/m) 267-1600 107-1070 -----
Densidade (g/cm3) 1,1-1,46 1,2-1,3 1,65-2,0
Tabela XI.3. Propriedade de compósitos reforçados com tecido de fibra de vidro, fibra
de vidro picotada e fibra de vidro laminada.
Nos materiais compósitos a base de fibras de carbono, as fibras possibilitam a
melhoria nas propriedades mecânicas ligadas à tração, enquanto que a matriz
polimérica serve para elevar a resistência ao impacto.
As resinas epóxi são, sem dúvida, as mais utilizadas como matrizes de
compósitos reforçados com fibra de carbono, apesar da existência de outros tipos
como as de poliamidas e de polifenil sulfetos, que podem ser utilizadas em
determinadas aplicações.
A maior vantagem observada em materiais compósitos com fibras de carbono
refere-se aos elevados valores de resistência a tração e módulo de elasticidade,
combinados com baixa densidade. Por essa razão, tais materiais estão
gradativamente substituindo os metais em diversas aplicações aeroespaciais. A tabela XI.4 lista algumas propriedades mecânicas típicas de um compósito que apresenta 62% de
seu volume ocupado por fibras de carbono.
Propriedade Sentido Longitudinal Sentido Transversal
Resistência à Tração (MPa) 1.860 65
Módulo de Elasticidade (GPa) 145 9,4
Deformação Máxima (%) 1,2 0,70

Introdução aos Materiais Compósitos 149
Tabela XI.4. Propriedades mecânicas de um compósito reforçado com fibras de
carbono e resina de epóxi.
A figura XI.3 mostra as propriedades de fadiga de um material compósito de
fibras de carbono arranjadas longitudinalmente comparadas com outros materiais. Os
materiais compósitos empregados industrialmente geralmente são laminados. O
arranjos de tais placas pode ter diferentes orientações, de forma a apresentar
propriedades mecânicas desejadas.
Figura XI.3. Propriedades ligadas à fadiga de diversos materiais.
XI.4. MATERIAIS COMPÓSITOS DE MATRIZ METÁLICAOs compósitos do tipo matriz metálica (CMM) estão sendo pesquisados com
muita intensidade nos últimos anos e o resultado de tal fato pode ser constatado pelo
desenvolvimento de inúmeros materiais dessa natureza, com propriedades mecânicas
extremamente interessantes. A maioria dos compósitos com matriz metálica foi
desenvolvida para ser utilizada, principalmente, na industria aeroespacial. Além disso,
inúmeras partes de motores de automóveis fabricadas a partir de tais materiais, já são
atualmente utilizadas comercialmente. Em geral, existem três tipos de elementos de
reforço empregados nos compósitos de matriz metálica, quais sejam: reforços com
fibras contínuas, reforços com fibras descontínuas e reforços particulados.
Os conjugados de matriz metálica reforçados com fibras contínuas apresentam
os maiores valores de módulo de elasticidade e resistência à tração dentre os CMM.

Introdução aos Materiais Compósitos 150
Um dos primeiros CMM de fibras contínuas desenvolvidos foi o de matriz de ligas de
alumínio com reforço de fibras de boro. As fibras de boro, para este tipo de compósito,
são produzidas através de deposição química de boro, a partir do vapor, em um
substrato representado por um fio de tungstênio. O compósito alumínio/boro é obtido
da pressagem a quente das fibras de boro entre folhas de alumínio. Desta forma, o
alumínio é deformado em torno das fibras e permite a ligação do elemento de reforço
à matriz. Quando uma liga de alumínio é reforçada com adição de boro, em uma
proporção de 51% em volume, a resistência à tração da liga é aumenta de 310 para
1.417 MPa, enquanto que seu módulo de elasticidade passa de 69 para 231 GPa. Os
materiais compósitos do tipo Al-B possuem características tão particulares em termos
de propriedades mecânicas, que os mesmos são empregados na fabricação de
diversas partes estruturais do ônibus espacial americano.
Outros materiais do tipo fibra contínua, utilizados no reforço dos CMM são:
fibras de carboneto de silício, de grafite, de alumina e de tungstênio. Por exemplo,
fibras contínuas de SiC, utilizadas como reforço de ligas de alumínio, estão sendo
testadas como matéria-prima na construção de peças de aviões militares americanos
do tipo caça. Um dos materiais compósitos que apresentam boa perspectiva de uso é
o CMM com matriz metálica de titânio e reforço de fibras contínuas de SiC. Este
material deverá ser usado na produção de partes do avião hipersônico americano X-
30. Devido às velocidades hipersônicas que este avião atingirá (Mach 12 a Mach 25),
sua fuselagem deverá exibir temperaturas superiores as de fusão de diversas
superligas atualmente produzidas (>1.8000C). Existem diversos tipos de materiais
compósitos reforçados com fibras descontínuas e particulados. Tais materiais
apresentam diversas características notáveis, como elevados valores de resistência
mecânica, módulo de elasticidade e melhor estabilidade dimensional quando
comparados às ligas metálicas sem reforço. Os materiais compósitos CMM com
reforço de fibras descontínuas são produzidos, basicamente, através da metalurgia do
pó e por processos envolvendo infiltração de ligas fundidas. No processo envolvendo
metalurgia do pó, o carboneto de silício, na forma de agulhas com diâmetros entre 1 e
3 mm e comprimentos variando de 50 a 200 mm, é misturado ao metal pulverizado,
consolidado por prensagem a quente e extrudado ou forjado na forma final desejada.
A tabela XI.5 apresenta propriedades mecânicas de diversas ligas metálicas, com e
sem reforço. Observando esta tabela, nota-se que a resistência à tração da liga Al
6061 é aumentada de 310 para 480 MPa, com a adição de fibras descontínuas de

Introdução aos Materiais Compósitos 151
SiC, enquanto que seu módulo de elasticidade passa de 69 para 115 GPa. Apesar do
significativo aumento de resistência mecânica e módulo de elasticidade que pode ser
obtido neste caso, os processos de metalurgia do pó e de infiltração têm custo relativo
elevado. Os CMM reforçados com fibras descontínuas são aplicados na construção de
pistões de motores de combustão interna.
Tipo de Material Resistência à
Tração MPa
Módulo de
Elast. GPa
Deformação
Máxima %
CMM - Reforço com Fibras Contínuas
Al 20241 (45% B) (longitudinal) 1.458 220 0,81
Al 60612 (51% B) (longitudinal) 1.417 231 0,74
Al 6061 (47 % SiC) (longitudinal) 1.462 204 0,89
CMM - Reforço com Fibras Descontínuas
Al 2124 (20% SiC) 650 127 2,40
Al 6061 (20% SiC) 480 115 5,00
CMM - Reforço com Partículas
Al 2124 (20% SiC) 552 103 7,00
Al 6061 (20% SiC) 496 103 5,50
Sem Reforço
Al 2124 455 71 9,00
Al 6061 310310 68,9 12,0
Tabela XI.5. Propriedades mecânicas de materiais compósitos de matriz metálica.
Como exemplo de CMM de reforço particulado, pode-se citar a matriz metálica
de aluminídio de titânio reforçado com partículas de alumina e de carboneto de silício,
com diâmetros na faixa de 3 a 200 mm. As partículas, que em certos casos podem
sofrer um revestimento apropriado para melhorar a molhabilidade, são adicionadas à
liga de alumínio fundida e solidificada na forma de lingotes para uma futura refusão e
utilização.
Com a adição de partículas de SiC, a liga Al 6061 tem sua resistência à tração
aumentada de 310 para 496 MPa e o módulo de elasticidade passa de 68,9 para 103
1 ? Al 2024 - Composição: 4,4% Cu; 1,5% Mg; 0,6% Mn.
2 ? Al 6061 - Composição: 1,0% Mg; 0,6% Si; 0,27% Cu; 0,2% Cr.

Introdução aos Materiais Compósitos 152
GPa. O CMM (Al-SiC) é utilizado principalmente na fabricação de materiais esportivos
e partes de motores de automóveis.
XI.5. MATERIAIS COMPÓSITOS DE MATRIZ CERÂMICADa mesma forma que os CMM, o desenvolvimento dos materiais compósitos de
matriz cerâmica (CMC) é muito recente. Os CMC apresentam resistência à tração e
tenacidade superiores, quando comparados ao material da matriz (cerâmico) sem
reforço. Novamente, da mesma forma que os CMM, os compósitos CMC podem
apresentar três tipos básicos de reforço: fibras contínuas, fibras descontínuas e
particulados.
Os dois principais materiais empregados como reforços do tipo fibra contínua
são: o carboneto de silício e a alumina. Na fabricação de CMC com reforço através de
SiC, as fibras são encapsuladas por uma matriz de material cerâmico. Este tipo de
material compósito é utilizado principalmente na confecção de trocadores de calor, de
sistemas de proteção térmica e de equipamentos e dispositivos submetidos a
ambientes corrosivos.
As fibras descontínuas de SiC podem aumentar significativamente a tenacidade
à fratura de materiais cerâmicos. Como mostra a tabela XI.6, quando uma matriz de
alumina é reforçada com fibras de SiC, na proporção de 20% em volume, a tenacidade
à fratura é aumentada de 4,5 para 8,5 MPa.m½. Os compósitos CMC de fibra curta e
particulados têm a vantagem de serem produzidos por processos utilizados na
fabricação de cerâmicas comuns, como compactação a quente. Acredita-se que os
compósitos CMC têm suas propriedades mecânicas melhoradas a partir de três
mecanismos principais, que são resultados da interferência das fibras de reforço na
propagação de trincas na matriz cerâmica. Tais mecanismos são:
a. Desvio de Trincas
Quando a trinca atinge um elemento de reforço, a mesma é desviada, o que faz
com que o trajeto da mesma aumente, pois este adquire forma sinuosa.
Conseqüentemente, para propagar esta trinca, um esforço maior será necessário.
b. Ponte sobre Trincas
Durante a propagação de uma trinca, as fibras de reforço servem como ligação
entre as partes fraturadas e assim, o nível de solicitação mecânica necessário para
propagar a mesma deverá ser aumentado.
c. Deslizamento de Fibras

Introdução aos Materiais Compósitos 153
O atrito causado pelo deslizamento das fibras no interior da matriz, absorve
energia. Assim, um valor de tensão mais elevado é necessário para dar continuidade à
propagação da trinca. Desta forma, quanto melhor a qualidade da ligação interfacial
entre a matriz e a fibra, maior será a resistência mecânica do compósito. Também,
quanto menor a diferença entre os coeficientes de expansão térmica da matriz e das
fibras, maior será a resistência mecânica do material, principalmente quando as
temperaturas de trabalho são elevadas.
Matriz Fração de SiC
% em Volume
Tenacidade à Fratura
MPa m 9
Si3N4 0 5-7
10 6,5-9,5
30 7,5-10
Al2O3 0 4,5
10 7,1
20 7,5-9.0
Tabela XI.6. Tenacidade à fratura de compósitos CMC reforçados com fibras
descontínua de SiC, na temperatura ambiente.
EXERCÍCIOS
XI.1. Defina uma material compósito.
XI.2. Explique o processo de obtenção das fibras de carbono.
XI.3. Cite razões para o elevado consumo das fibras de vidro.
XI.4. Compare, em termos de propriedades mecânicas, as fibras de vidro, de carbono
e Kvelar.
XI.5. Descreva um material compósito de matriz metálica.
XI.6. Descreva um material compósito de matriz cerâmica.


















