
GILENO VIEIRA LACERDA JÚNIOR -...
82
UNIVERSIDADE ESTADUAL DE SANTA CRUZ PRÓ-REITORIA DE PESQUISA E PÓS-GRADUAÇÃO Programa de Pós-graduação em Genética e Biologia Molecular GILENO VIEIRA LACERDA JÚNIOR Análise das alterações na comunidade microbiana e no metaproteoma de organismos saudáveis e doentes da esponja Cliona Varians Orientador (a): Profa. Dra. Rachel Passos Rezende ILHÉUS - BAHIA - BRASIL Agosto, 2012
Transcript of GILENO VIEIRA LACERDA JÚNIOR -...

UNIVERSIDADE ESTADUAL DE SANTA CRUZ
PRÓ-REITORIA DE PESQUISA E PÓS-GRADUAÇÃO
Programa de Pós-graduação em Genética e Biologia Molecular
GILENO VIEIRA LACERDA JÚNIOR
Análise das alterações na comunidade microbiana e no metaproteoma de
organismos saudáveis e doentes da esponja Cliona Varians
Orientador (a): Profa. Dra. Rachel Passos Rezende
ILHÉUS - BAHIA - BRASIL
Agosto, 2012

Análise das alterações na comunidade microbiana e no metaproteoma de organismos
saudáveis e doentes da esponja Cliona varians
Gileno Vieira Lacerda Júnior
Dissertação apresentada à Universidade
Estadual de Santa Cruz como parte das
exigências para obtenção do título de Mestre
em Genética e Biologia Molecular.
Orientador (a): Profa. Dra. Rachel Passos Rezende
ILHÉUS - BAHIA - BRASIL
Agosto, 2012

GILENO VIEIRA LACERDA JÚNIOR
Análise das alterações na comunidade microbiana e no metaproteoma de
organismos saudáveis e doentes da esponja Cliona Varians
Dissertação apresentada à
Universidade Estadual de Santa
Cruz, como parte das exigências
para obtenção do título de Mestre
Genética e Biologia Molecular.
BANCA EXAMINADORA
Profa. Dra. Helena Costa Profa. Profa. Dra. Idjane Santana de Oliveira (UESC) (UFPE) Profa. Dra. Bianca Mendes Maciel Profa. Dra. Rachel Passos Rezende (UESC) (UESC–Orientadora)
ILHÉUS - BAHIA - BRASIL
Agosto, 2012

DEDICATÓRIA
Aos meus pais e minha irmã que me incentivaram durante toda a jornada. A toda
família e amigos que me ajudaram de alguma forma a contornar as dificuldades
durante a minha caminhada, dedico.

AGRADECIMENTOS
A todos os meus familiares, em especial aos meus pais e a minha irmã Vivian,
por estarem sempre ao meu lado, apoiando e mostrando o melhor caminho a ser
seguido, nunca medindo esforços para o meu sucesso pessoal.
A minha namorada Lorena, por todo apoio emocional e por suportar os meus
dias difíceis.
Ao programa de Pós-Graduação em Genética e Biologia Molecular, ao Centro
de Aperfeiçoamento de Pessoal de Nível Superior (Capes) e a Fapesb pela
oportunidade e apoio financeiro durante a realização do curso de mestrado.
À Professora Drª Rachel Passos Rezende, que assumiu minha comissão de
orientação nos últimos momentos, sendo essencial para a finalização de todo esse
esforço.
Ao ex-orientador Drº Júlio Cézar de Mattos Cascardo (em memória), guardo
grandes recordações e muita admiração.
Ao ex-orientador Drº João Carlos Teixeira Dias, pela orientação e contribuição
inicial para o desenvolvimento do trabalho.
Aos amigos Sanderson, Ricardo, Eric e Salatiel pelas horas de companheirismo e
ajuda na execução dos experimentos laboratoriais.
À amiga e colega de trabalho Flamélia Carla que contribuiu muito na execução
desse trabalho e a Elizabeth Duarte pela amizade construída e companheirismo.
Ao Prof. Dr. Juliano de carvalho Cury pelas contribuições nas análises de
bioinformática.
Aos amigos e professores do Centro de Biotecnologia e Genética pelos
conselhos que me ajudaram a desenvolver este trabalho.
A todos que, direta ou indiretamente, colaboraram para a execução desse
trabalho.

i
ÍNDICE
LISTA DE FIGURA ............................................................................................... III
LISTA DE TABELA .............................................................................................. IV
RESUMO................................................................................................................ V
ABSTRACT ........................................................................................................... VI
1. INTRODUÇÃO ................................................................................................... 1
2. REVISÃO BIBLIOGRÁFICA .............................................................................. 3
2.1. Esponjas marinhas....................................................................................................... 3 2.2. Simbiose microbiana em esponjas marinhas .............................................................. 4 2.3. Esponjas e microrganismos associados: uma fonte de produtos naturais ................. 9 2.4. Doenças de esponjas: efeitos das alterações ambientais ......................................... 10 2.5.Métodos moleculares para acessar a diversidade microbiana: o uso do gene 16S rDNA ................................................................................................................................. 14 2.6. Metaproteômica ....................................................................................................... 16 2.7. Esponjas Cliona varians(Duchassaing e Micheootti, 1864) ...................................... 19
3. OBJETIVOGERAL ........................................................................................... 21
3.1. objetivos específicos ................................................................................................. 21
4. MATERIAL E MÉTODOS ................................................................................ 22
4.1. Local de estudo e coleta de esponjas ....................................................................... 22 4.2. Extração de DNA total ............................................................................................... 26 4.3. Amplificação do gene 16S rDNA ............................................................................... 26 4.4. Eletroforese em gel de gradiente desnaturante ....................................................... 28 4.5. Construção de biblioteca do gene 16S rDNA ............................................................ 29 4.6. Extração de DNA plasmidial e sequenciamento ....................................................... 30 4.7. Classificação das sequências ..................................................................................... 32 4.8. Curva de rarefação, análise de riqueza e diversidade .............................................. 32 4.9. Identificação filogenética .......................................................................................... 32 4.10. Extração de proteínas ............................................................................................. 33 4.11. Eletroforese 1D e 2D ............................................................................................... 34 4.12. Digestão tríptica das proteínas ............................................................................... 35 4.13. Espectrometria de massa (MS) ............................................................................... 36 4.14. Identificação das proteínas ..................................................................................... 37
5. RESULTADOS ................................................................................................ 38
5.1. Extração de DNA metagenômico .............................................................................. 38 5.2. Amplificação e purificação do gene 16S rDNA .......................................................... 39 5.3. Construção da biblioteca do gene 16S rDNA ............................................................ 41 5.4. Análise do perfil da comunidade microbiana por DGGE .......................................... 42 5.5. Comparação da diversidade bacteriana em esponjas saudáveis e doentes ............ 45 5.6. Análise da curva de rarefação, índices de diversidade e riqueza de espécies ......... 48 5.7. Separação dos extratos protéicos totais por 1D-PAGE ............................................. 49 5.8. Análise do perfil de expressão por 2D-PAGE ............................................................ 50 5.9. Identificação de proteínas por espetrometria de massa .......................................... 52

ii
6. DISCUSSÃO .................................................................................................... 55
6.1. Alteração na comunidade bacteriana de esponjas doentes ..................................... 55 6.2.Comparação da diversidade bacteriana em esponjas saudáveis e doentes ............. 57 6.3. análise em 1D e 2D-PAGE do metaproteoma ........................................................... 58 6.4. Identificação de proteínas ......................................................................................... 59
7. CONCLUSÃO .................................................................................................. 62
8. REFERÊNCIAS BIBLIOGRÁFICAS ................................................................ 63

iii
LISTA DE FIGURAS
Figura 1 - Imagens subaquáticas de esponjas marinhas demonstrando a diversidade de
cores, formas e tamanhos. .................................................................................................... 4
Figura 2- Um esquema geral do plano corporal básico de uma esponja. ............................. 6
Figura 3- Analise filogenética baseada no gene 16S rDNA de toda a microbiota
associada a esponja de acordo com uma meta-análise ........................................................ 8
Figura 4- Espécies de esponjas sendo afetadas por doenças atualmente. ......................... 11
Figura 5- Um modelo do desenvolvimento de doenças nas esponjas ................................ 13
Figura 6- Esponja Cliona varians perfurando substrato calcário ........................................ 19
Figura 7 - Mapa da Costa do descobrimento brasileiro, sul da Bahia, Brasil ...................... 24
Figura 8 - Vista panorâmica do recife de Coroa Vermelha e foto do platô recifal durante
período de baixamar ........................................................................................................... 25
Figura 9 - Mapa do vetor pGEM-T Easy .............................................................................. 30
Figura 10 - Análise em gel de agorose 0.8% do DNA total extraído de esponja saudável
(a) e doente (b). M: Marcador de peso molecular 1 Kb (Fermentas). ................................ 38
Figura 11 - Análise em gel de agarose 0.8% dos produtos do PCR utilizados para
construção da biblioteca 16S rDNA. .................................................................................... 39
Figura 12 - Análise em gel de agarose 0.8% dos produtos do PCR utilizados para análise
por DGGE do domínio Bactéria (1) e Archaea (2).. .............................................................. 40
Figura 13 - Perfil eletroforético em gel de agarose 0.8% do DNA plasmidial de uma
amostra de clones da biblioteca 16S rDNA ......................................................................... 41
Figura 14-Perfil das bandas do DGGE utilizando sequências 16S rDNA-V3 de Bactérias
(a) e Archaea (b). ................................................................................................................. 43
Figura 15 - Perfil das proteínas totais separadas em 1D-SDS-PAGE. .................................. 47
Figura 16 -Comparação do perfil metaproteômico das esponjas afetadas (a) e não
afetadas (b) pela doença. .................................................................................................... 48
Figura 17 - Classificação das sequências parciais do gene de 16S rRNA bacteriano das
esponjas doentes e saudáveis usando RDP classifier (limiar de 50%). ............................... 50
Figura 18 - Curva de rarefação gerada pela análise das sequências 16S rDNA da
biblioteca ............................................................................................................................. 51

iv
LISTA DE TABELAS
Tabela 1- Oligonucleotídeos utilizados no trabalho. ........................................................... 27
Tabela 2- Identificação das sequências 16S rDNA-V3 recuperados das bandas de DGGE
através do BLAST contra o banco de dados GenBank.. ....................................................... 44
Tabela 3-Identificação das sequências de Archaea recuperados das bandas de DGGE
através do BLAST contra o banco de dados GenBank ......................................................... 45
Tabela 4- Índices de diversidade e riqueza calculados ....................................................... 49
Tabela5- Identificação de proteínas separadas por 1D-SDS-PAGE utilizando Mascot
contra o banco de dados NCBI (nr) ..................................................................................... 54
Tabela 6- Identificação de proteínas separadas por 2D-SDS-PAGE utilizando Mascot
contra o banco de dados NCBI (nr) ..................................................................................... 54

v
RESUMO
Este trabalho acessou pela primeira vez a composição da comunidade microbiana e o
perfil de proteínas expressas (metaproteômica) para investigar o papel dos
microrganismos no processo de doença na esponja Cliona varians. Para isto, foram
analisados o perfil de bandas gerados por DGGE e o sequenciamento de bibliotecas de
clones do gene 16S rDNA a partir do DNA metagenômico extraído diretamente de
indivíduos sadios e doentes. Foi verificada uma alteração na comunidade bacteriana
em esponjas apresentando a doença. O perfil do DGGE apresentou múltiplas bandas
exclusivas na esponja doente, que foram relacionadas a sequências de esponjas
submetidas a estresse térmico e isoladas de esgoto. Os clones das bibliotecas foram
sequenciados e analisados taxonomicamente utilizando o classificador do Ribossomal
Database Project II (RDP). Observou-se predominância de ribotipos de Proteobacéria
(84%) no tecido saudável, já o tecido doente apresentou maior diversidade,
apresentando ribotipos de Proteobactérias (58%), Bacteroidetes (22%) e Firmicutes
(5%). Para análise funcional, proteínas foram extraídas e separadas por 1D e 2D-PAGE,
posteriormente analisados em espectrometria de massa (MS). As proteínas foram
identificadas através do “fingerprint” do MS contra o banco de dados do NCBI (nr). A
separação das proteínas por 1D e 2D-PAGE revelou um padrão de expressão proteica
diferenciado entre as amostras saudáveis e doentes de esponja. Foram identificadas
no total 12 proteínas a partir das bandas de 1D-PAGE e 10 proteínas a partir dos spots
de 2D-PAGE. Considerando todas as proteínas identificadas, 8 (36%) foram proteínas
com função desconhecida.
PALAVRAS-CHAVE: Esponjas marinhas, doença, comunidades microbianas, Cliona
Varians, metaproteômica.

vi
ABSTRACT
This work has accessed, for the first time, the microbial community and profile of
expressed proteins (metaproteomics) to investigate the role of microorganisms in the
disease process in the sponge Cliona varians. For this purpose, it were analyzed the bands
profile generated from DGGE and the sequencing of clone libraries of the 16S rRNA from
metagenomic DNA extracted directly from sick and healthy individuals. It was observed a
big change in the structure of the bacterial community in sponges presenting the disease.
The DGGE profile showed multiple bands in the sponge sick that were related to
sequences from sponges subjected to thermal stress and isolated from wastewater. The
clones from the libraries were sequenced and analyzed taxonomically using the Ribosomal
Database Project classifier II (RDP). Proteobacteria (84%) predominated in healthy tissue.
Diseased tissue showed greater diversity, with ribotypes of Proteobacteria (58%),
Bacteroidetes (22%) and Firmicutes (5%). For functional analysis, proteins were extracted
and separated by 1D-2D-PAGE PAGE and subsequently analyzed by mass spectrometry.
The spectra obtained were processed for protein identification attempts through the
"fingerprint" of MS against the NCBI database (nr). The separation of proteins through 1D-
2D-PAGE revealed a distinctive pattern of protein expression between the samples. Using
the database NCBInr it was identified a total of 12 protein from 1D-PAGE and 10 proteins
from spots. Considering all the proteins identified 8 (36%) were proteins with unknown
function.
Keywords: Marine sponges, microbial communities, disease, Cliona varians,
metaproteomic.

1
1- INTRODUÇÃO
Esponjas marinhas são consideradas os animais multicelulares mais simples e
primitivos, possuindo importantes funções ecológicas em ecossistemas de recifes de
corais, sendo fonte de numerosos metabólitos com atividades farmacológicas (LOVE et
al. 2009; LAPORT, et al., 2009). As esponjas são conhecidas por sua interação com vários
tipos de microrganismos (bactérias, archaeas, fungos, protozoários e algas unicelulares,
incluindo os vírus) que podem compor até 40% da sua biomassa corporal (WEBSTER E
TAYLOR, 2012). Este consórcio microbiano simbionte é apontado como o responsável
pela nutrição, fixação de nitrogênio e mecanismos de defesa do organismo hospedeiro
(TAYLOR et al., 2007).
Perturbações na integridade desta relação simbiótica estão relacionadas a
fatores ambientais, tais como escoamento de esgotos e mudanças climáticas globais,
levando ao desenvolvimento de doenças acometidas por patógenos oportunistas
(WEBSTER et al., 2008). Nos últimos anos tem sido relatada uma ocorrência global de
doenças em esponjas marinhas nos recifes de corais localizados em várias regiões
geográficas (WEBSTER, 2007). Muitos estudos que reportam doenças de corais e
esponjas detectam uma mudança na comunidade bacteriana simbiótica em indivíduos
afetados, mas não conseguem atribuir esta mudança a um patógeno específico
(WEBSTER et al., 2008; ANGERMEIER et al., 2011; ANGERMEIER et al., 2012). Além
disso, os mecanismos funcionais e aspectos fisiológicos dessa interação ainda são muito
insipientes. Uma das alternativas propostas para este problema seria o uso da
metaproteômica visando um melhor conhecimento da interação das comunidades
microbianas com o hospedeiro através da análise de expressão de proteínas extraídas

2
diretamente do ambiente em estudo (RODRIGUEZ-VALERA, 2004). Essa abordagem
permite analisar a composição funcional de uma comunidade microbiana complexa.
Entretanto, é necessário sanar entraves relacionados à complexidade da amostra e a
identificação de proteínas desconhecidas (HETTICHet al., 2012).
A esponja Cliona varians está distribuída de forma endêmica por toda costa
americana tropical Atlântica Ocidental, sendo encontrada no Golfo do México, Caribe e
Brasil (Rio Grande do Norte, Fernando de Noronha, Atol das Rocas, Pernambuco,
Alagoas, Bahia e Espírito Santo). Recentemente, espécimes dessa esponja foram
reportados com sinais de necrose tecidual no litoral baiano de Santa Cruz de Cabrália,
Bahia, Brazil. Diante desta problemática, o objetivo desse trabalho foi examinar, pela
primeira vez, mudanças nos perfis de expressão proteica e na diversidade da
comunidade microbiana associadas a indivíduos doentes da espécie C. varians.

3
2- REVISÃO BIBLIOGRÁFICA
2-1 Esponjas marinhas
Esponjas marinhas são considerados os animais multicelulares mais simples e
primitivos, com registros fósseis datando cerca de 630 milhões de anos atrás (LOVE et
al., 2009). Este grupo de animais apresenta uma alta diversidade com aproximadamente
15.000 espécies descritas em três classes: Calcarea, Hexactinellida e Demospongiae
(Figura 1). Habitam vários tipos de ambientes marinhos, desde mares tropicais e
temperados, a regiões polares (HOOPER e VAN SOEST, 2002), representando um
significante componente da comunidade bentônica em recifes de corais, tanto em
termos de biomassa como importância ecológica. Como o próprio nome do filo sugere
(Porífera do latim porus = poro, e ferre = portador), a estrutura corporal das esponjas é
porosa, apresentando morfologia simples e com baixo grau de organização, sem a
formação de tecidos verdadeiros e com ampla diversidade de cores e tamanhos (Figura
1). As esponjas filtram seu alimento através do bombeamento da água por um complexo
sistema de canais que intercala seu corpo (chamado sistema aquífero) (REISWIG, 1974).
A água do mar penetra na esponja através de pequenas aberturas inalantes (óstio)
através de impulsos gerados pelo movimento de células flageladas chamadas
coanócitos, que capturam partículas orgânicas e microrganismos. O alimento é
fagocitado e distribuído para o mesênquima (matriz extracelular) por meio dos
amebócitos. Posteriormente, a água sai das esponjas através de um poro exalante
denominado ósculo (Figura 2).

4
Figura 1. Imagens subaquáticas de esponjas marinhas demonstrando a diversidade de
cores, formas e tamanhos. Mycale laxissima (A), Amphimedon queenslandica (B),
Ancorina alata (C), Rhopaloeides odorabile (D), Xestospongia muta (E), Cymbastela
concentrica (F), Aplysina aerophoba (G), Theonella swinhoei (H) e Ircinia felix (I). Fonte:
HEALNTSCHEL et al. (2012).
2.2- Simbiose microbiana em esponjas marinhas
Além de uma população transitória de bactérias da água do mar que são
utilizadas como fonte de alimento, as esponjas abrigam uma microbiota diversificada
que pode residir permanentemente em uma estreita interação de simbiose (LEE et al.,
2001; HENTSCHEL et al., 2002), incluindo vírus, bactérias, archeas, fungos, protozoários
e algas unicelulares, que podem compor 40% da sua biomassa corporal (WEBSTER e

5
TAYLOR, 2012). A maioria dos microrganismos associado às esponjas habitam o
mesênquima, uma matriz extracelular que é preenchida por células de esponja e
constitui a maior parte do corpo do hospedeiro (Figura 2). Embora alguns simbiontes
também sejam encontrados no meio intracelular, como evidenciado pelos primeiros
estudos de microscopia eletrônica (VACELET e DONADEY, 1977). Portanto, o
mesênquima é tanto o sítio da digestão de microrganismos filtrados (que podem ser
partículas de alimentos), como o habitat de extensas comunidades de microrganismos
simbióticos (Figura 2).
Figura 2. Esquema geral do plano corporal básico de uma esponja (a) e ampliação da
estrutura interna (b). (Fonte: HENTSCHEL et al., 2012)
Esponjas podem discriminar bactérias normalmente encontradas na água (que
são consideradas como alimento) e bactérias simbiontes através de compostos químicos
(como cápsulas de proteção) em torno da superfície bacteriana que mascaram e evitam
o reconhecimento de células fagocitárias da esponja (WILKINSONet al., 1984). Além

6
disso, possuem um sistema imune inato bem desenvolvido (WIENSet al., 2007) e
produzem uma ampla gama de compostos antimicrobianos (LAPORT et al., 2009), um
fator que aumenta a sua complexidade como um habitat para os microrganismos.
O processo de simbiose entre microrganismos e esponjas é estabelecido através
da absorção seletiva do simbionte específico, ou por transmissão vertical pelos estágios
reprodutivos como embriões e larvas (WEBSTER et al., 2010). A aplicação de técnicas
moleculares, incluindo eletroforese em gel de gradiente desnaturante (DGGE) e
sequenciamento do gene 16S rRNA permitiram identificação filogenética de bactérias
presentes em esponjas adultas e suas larvas (LEE et al., 2009; WEBSTER et al., 2010),
confirmando essa hipótese.
Microrganismos associados são determinantes para a biologia das esponjas. As
cianobactérias simbiontes, por exemplo, podem fornecer até 50% do requerimento
energético de certas esponjas tropicais (WILKINSON, 1983). Outros simbiontes podem
contribuir para a defesa química através da produção de metabólitos biologicamente
ativos, assim como suplementação alimentar pela biossíntese de vitaminas (THOMAS et
al., 2010; HENTSCHEL et al., 2012).
Por outro lado, simbiontes microbianos desfrutam de alguns benefícios. Os
nutrientes filtrados pelo hospedeiro, assim como compostos nitrogenados excretados
em forma de amônia pela esponja tornam-se um nicho particularmente atraente para
esses microrganismos (TAYLOR et al., 2007).
A partir de bibliotecas de genes 16S rDNA, foram reconhecidos
aproximadamente 14 filos bacterianos presente em esponjas (TAYLOR et al., 2007).

7
Recentemente estudos com pirosequenciamento de “amplicons” do gene 16S rDNA de
32 espécies de esponjas aumentaram esta riqueza taxonômica para 25 filos bacterianos
(SCHMITT, et al., 2012). Vários filos adicionais de bacterias, archeaes, fungos e outros
eucariotos foram detectados a partir de estudos utilizando o pirosequenciamento. Com
base numa meta-análise (método que combina os resultados de vários estudos) baseada
no gene 16S rDNA, Hentschel e colaboradores (2012) realizaram a construção de uma
análise filogenética mais rebuscada com todos os filos da microbiota associada as
esponjas (Figura 3). Independente da técnica utilizada, os filos dominantes de bactérias
associadas a esponjas são: Proteobactéria (principalmente as classes Alpha, Gamma e
Deltaproteobactéria), Spirochaetes, Chloroflexi, Actinoacteria, Acidobacteria,
Nitrospirae e Poribacteria (Figura 3). Este último, descoberto recentemente, existindo
quase que exclusivamente nas esponjas marinhas (FIESELERet al., 2004). A notável
diversidade química e microbiana da associação microrganismos-esponjas, juntamente
com o tempo dessa relação, faz delas um importante sistema modelo para o estudo dos
consórcios microbianos simbióticos na evolução dos metazoários, bem como fonte de
produtos naturais de interesse biotecnológico.

8
Figura 3. Analise filogenética baseada no gene 16S rDNA de toda a microbiota associada
a esponja de acordo com uma meta-análise (HENTSCHEL et al., 2012).
2.3- Esponjas e microrganismos associados: uma fonte de produtos naturais
Uma das propriedades mais notáveis de esponjas marinhas é sua capacidade de
produzir uma ampla variedade de produtos naturais. As esponjas estão entre as mais
ricas fontes de metabólitos secundários biologicamente ativos e produzem mais
compostos do que qualquer outro grupo de organismos marinhos (BLUNTet al., 2011).
Este repertório químico de substâncias desempenha importante função ecológica como
agentes de proteção contra predadores, epibiontes e microrganismos patogênicos,

9
contribuindo para o sucesso evolutivo das esponjas (THOMS e SCHUPP, 2007). Muitos
desses compostos são provenientes de microrganismos marinhos que vivem em
simbiose com as esponjas desempenhando um papel importante na defesa química do
hospedeiro (PIEL, 2006 e PIEL, 2009). Estes compostos englobam uma ampla variedade
de classes químicas (terpenóides, alcalóides, peptídeos e poliquetídeos) com grande
variedade de propriedades farmacológicas relevantes, como anticancerígenos
(SIMMONS et al., 2005), antimicrobianos (LAPORT et al., 2009) e antivirais (SAGAR et al.,
2010). Por exemplo, eribulina, um análogo sintético da halichondrina B isolado das
esponjas Halichondria e Lissodendoryxs pp., foi aprovado recentemente como um
medicamento para o tratamento do câncer de mama (HUYCK et al., 2011).
Muitas vezes os produtos naturais biologicamente ativos são produzidos em
quantidades relativamente pequenas e por animais raros, cuja população natural não
pode sustentar as extensas quantidades necessárias para os ensaios clínicos. Portanto,
meios alternativos são necessários para a produção em larga escala de metabólitos,
como a síntese química, embora a complexidade estrutural das moléculas possam
representar dificuldades para este tipo de método (AICHERet al., 1992).
Em face desses obstáculos, outras abordagens estão disponíveis para acessar
produtos naturais biologicamente ativos de esponjas e os microrganismos associados, e
estão divididos em três estratégias: cultivo de microrganismos produtores (PIEL, 2006),
cultivo de esponjas (DUCKWORTH e BATTERSHILL, 2003) e utilização de métodos
moleculares, como a metagenômica (HANDELSMAN, 2004). Esta última refere-se à
clonagem de fragmentos do genoma de uma complexa comunidade microbiana em
hospedeiros como a E. coli, possibilitando produção em larga escala e sustentável de

10
metabólitos bioativos, incluindo os produzidos por microrganismos não cultiváveis
(GURGUI e PIEL, 2010). A melhoria e combinação desses métodos fornecerão ótimas
chances de sucesso na descoberta de novas moléculas de interesses farmacêutico
produzido por esponjas.
2.4- Doenças de esponjas: efeito das alterações ambientais
Nas últimas décadas, tem se observado um aumento global de doenças em
organismos marinhos (LAFFERTYet al., 2004; PRZESLAWSKI et al., 2008). Fatores
ambientais como elevação da temperatura da água do mar assim como aumento da
poluição antropogênica, do enriquecimento de nutrientes e de espécies introduzidas
têm sido associadas a doenças em esponjas e outros organismos marinhos. É
importante ressaltar que as doenças afetam tanto espécies de vertebrados
quantoinvertebrados, incluindo ostras, vieiras, abalone e amêijoas, ouriços, esponjas e
corais (HARVELL et al., 1999).
As doenças de corais têm recebido enorme atenção devido ao importante papel
dos recifes de coral na biodiversidade, bem como o ritmo alarmante em que estes têm
sido dizimados no contexto do aquecimento global (BOURNEet al., 2009). Em
comparação, muito menos esforço foi realizado para investigar doenças de esponjas
marinhas. Epidemias de doenças podem causar drástica redução das populações de
esponjas, com efeitos negativos sobre a ecologia de recifes de corais (WEBSTER, 2007).
Eventos de doença é um fenômeno global, ocorrendo em diversas áreas geográficas.
Muitas espécies de esponja são acometidas, apresentando variados sintomas
fisiológicos, que geralmente começam com o aparecimento de manchas brancas ou

11
coloridas, seguidas de necrose do tecido, deixando o esqueleto exposto, podendo se
espalhar por todo o corpo dentro de semanas a meses (Figura 4).
Figura 4. Espécies de esponjas sendo afetadas por doenças atualmente. Phakellia
flabellata da grande barreira de corais (A) Ircinia fasciculata do Mediterrâneo Ocidental
(B) Aplysina aerophoba da Eslovênia (C). (Fonte: WEBSTER e TAYLOR, 2012)
Os fatores que causam doenças em esponjas são, em grande parte, ainda
desconhecidos. Se os surtos de doenças são devido a novos agentes patogênicos ou
novas condições ambientais é um tema de debate atual (WEBSTER e TAYLOR, 2012).
Mesmo com a dificuldade para identificar as causas exatas, o estresse ambiental é uma
causa admissível uma vez que é observada a diminuição do fitness das esponjas e sua
microbiota simbionte, tornando-a mais suscetível à doença (WEBSTER, 2007). Estudos
demonstram que em condições fisiológicas estressantes (alta temperatura,
enriquecimento de nutrientes e fluxo de água reduzido) pode ocorrer um incremento na

12
virulência de patógenos, causando desequilíbrio na simbiose e proliferação de bactérias
do ambiente. As esponjas podem ser incapazes de controlar a proliferação bacteriana
(VACELET et al., 1994), ocorrendo degeneração do tecido quando bactérias exógenas
substituem a população associada (Figura 5). Vários estudos têm relatado a perda de
simbiontes microbianos devido à exposição ao estresse térmico (LOPEZ-LEGENTIL et al.,
2008; WEBSTER et al., 2008a; PANTILE et al., 2011). Portanto, mudanças climáticas
globais tem efeito significativo em microrganismos marinhos e seus hospedeiros
invertebrados. Como está previsto pelo Painel Intergovernamental sobre Mudanças
Climáticas (IPCC) um aumento de 1,8 a 4°C na temperatura global na superfície do mar
até 2100, novos eventos de mortalidade em massa tornam-se extremamente
susceptíveis de ocorrer durante as próximas décadas.
Apesar das devastadoras epidemias, foram realizados poucos estudos
microbiológicos e os agentes etiológicos ou fatores responsáveis para as taxas de
mortalidade de esponja continuam pouco conhecidos, devido à dificuldade de cultivo
dos microrganismos causadores de doenças e determinação das doses patogênicas dos
mesmos. Muitos estudos que reportam doenças de corais e esponjas detectam uma
mudança na comunidade bacteriana simbiótica em indivíduos afetados, mas não
conseguem atribuir esta mudança a um patógeno específico (WEBSTER et al., 2008b;
ANGERMEIER et al., 2011; ANGERMEIER et al., 2012). Apenas um caso foi confirmado
ligando um microrganismo como agente causador de uma doença específica (WEBSTER
et al., 2002). Uma Alphaproteobactéria patogênica (denominada linhagem NW4327)
isolada de indivíduos infectados da esponja Rhopaloeidesodorabile causou necrose
tecidual através da degradação das fibras de colágeno, após experimentos de
inoculação. Em contraste, Luter e colaboradores (2010) não detectaram nenhuma

13
diferença em comunidades microbianas da esponja Lanthella basta saudável e doente,
sugerindo que os microrganismos não são responsáveis pelos sintomas da doença nesta
espécie.
Figura 5. Modelo do desenvolvimento de doenças nas esponjas. Esponjas mantêm
simbiose e sistema de defesa bioquímico ativo. Os alimentos (bactérias) são filtrados e
digeridos (A). Mudanças nas condições ambientais causam desequilíbrio na simbiose (B)
e proliferação de bactérias do ambiente devido a falha no sistema de defesa química da
esponja (C), resultando nos sintomas da doença e posterior necrose do tecido. (Fonte:
WEBSTER, 2007)
Simbiontes microbianos são componentes importantes para a saúde das
esponjas. É provável que perturbações na simbiose como resultado das mudanças
climáticas e estresse ambiental, sejam os responsáveis por eventos de doenças. A
elucidação das causas e mecanismos no desenvolvimento de doenças em esponjas e
outros invertebrados marinhos representa um grande avanço para preservação e

14
sustentabilidade dos recursos marinhos como fonte de substâncias bioativas e
manutenção da biodiversidade do planeta.
2.5- Métodos moleculares para acessar a diversidade microbiana: o uso do gene 16S
rDNA
Técnicas baseadas em cultivo são insuficientes para estudar a diversidade
bacteriana a partir de amostras ambientais. Microrganismos isolados por métodos
tradicionais que utilizam meios de cultura não representam fielmente a comunidade
microbiana. Portanto, a aplicação de técnicas moleculares baseadas na análise de ácidos
nucléicos extraído diretamente do ambiente é essencial para descrever a diversidade
microbiana em ambientes naturais (AMANN et al., 1995). Estudos de biologia molecular,
baseada no gene 16S rRNA identificam um número maior de espécies de bactérias do
que as técnicas de cultura padrão, indicando que apenas uma pequena proporção de
espécies bacterianas são cultiváveis. Pace e colaboradores (1985), sugeriram
pioneiramente o gene 16S rDNA como um marcador molecular em estudos de
diversidade microbiana em amostras ambientais. O gene 16S rDNA possui vantagens na
identificação e análise filogenética, uma vez que é estável ao longo do tempo, altamente
conservado, presente em todos os organismos. Possui longas regiões conservadas
intercaladas com sequências de nucleotídeos variáveis úteis para determinar relações
filogenéticas próximas, aparentemente não sofrem transferência genética lateral e
possui tamanho considerado satisfatório, de cerca de 1500 nucleotídeos, para estudos
filogenéticos (AMANN, 1995). Portanto, métodos moleculares baseados no gene 16S
rRNA tornam possível descrever filogeneticamente comunidades complexas de
microrganismos não cultiváveis (HUGENHOLTZ et al., 1998).

15
A construção e análise de bibliotecas de genes ribossomais é uma ferramenta
altamente sensível para estudar ecologia microbiana ambiental (OLSENet al., 1986),
permitindo a comparação de sequências de diferentes amostras, com resolução em
diferentes níveis taxonômicos As sequências de 16S rDNA geradas a partir do
sequenciamento dos clones são comparadas com outras já depositadas em bancos de
dados públicos (GenBank e Ribossomal Database Project II). A análise da diversidade é
baseada no grau de similaridade dessas sequências que são agrupadas em unidades
taxonômicas operacionais (UTO`s). Sequências com similaridades maiores que 97% são
consideradas da mesma espécie, maiores que 95% do mesmo gênero, e maiores que
80% do mesmo filo (SCHLOSS e HANDELSMAN, 2005).
Outras técnicas moleculares baseadas em “fingerprinting” fornecem um perfil da
diversidade microbiana em uma amostra ambiental, como a amplificação por PCR do
gene 16S rDNA, com subsequente separação dos “amplicons” por DGGE (eletroforese
em gel com gradiente de desnaturação) tem sido utilizado para avaliação da diversidade
microbiana (MUYZER et al., 1993). Esta técnica é baseada no princípio da mobilidade
eletroforética diferencial de fragmentos de DNA em função de um gradiente de
desnaturação. O DNA é extraído das amostras biológicas e o gene 16S rDNA é
amplificado com oligonucleotídeos que flanqueiam regiões conservadas. Em seguida,
toda a mistura de produtos de PCR é separada em um gel de poliacrilamida contendo
gradiente linear de agentes desnaturantes (MUYZER e SMALLA, 1998). Sequências iguais
cessam a migração no mesmo ponto de gradiente do gel, enquanto amplicons com
mesmo tamanho, mas com composição diferente, em pelo menos um par de bases,
migram para posições diferentes no gel, gerando assim um perfil genotípico da
comunidade caracterizado por um padrão de bandas separadas. Para determinar as

16
identidades filogenéticas, as bandas podem ser excisadas do gel, amplificadas e
sequenciadas. Nos últimos anos, a aplicação de técnicas moleculares incluindo
eletroforese em gel de gradiente desnaturante (DGGE) e sequenciamento de biblioteca
do gene 16S rDNA permitiram explorar a diversidade filogenética de microrganismos
presentes em várias espécies de esponjas marinhas (LI et al., 2007; HARDOIM et al.,
2009; LEE et al., 2009; WEBSTER e TAYLOR, 2012).
2.6- Metaproteômica
Microrganismos representam o maior reservatório de diversidade genética na
terra. Durante a maior parte do passado geológico da terra, microrganismos tiveram um
papel essencial na formação das condições ambientais que existem hoje. Eles são os
principais modeladores da biogeoquímica, dos ciclos de nutrientes e responsáveis pela
decomposição de resíduos naturais ou antropogênicos do planeta (MADSEN, 2011).
Estudos independentes de cultivo microbiano que foram executadas nos últimos anos
forneceram conhecimento sobre a vastidão da diversidade microbiana. Em particular, a
metagenômica (análise genômica de comunidades microbianas diretamente do
ambiente natural) tem emergido como uma ferramenta poderosa para investigar
propriedades estruturais, evolutivas e metabólicas de amostras ambientais complexas
(HANDELSMAN et al., 1998). No entanto, estes métodos baseados em ácidos nucléicos
não são suficientes para a obtenção de informações sobre a função do componente
microbiano nos ecossistemas. A função ecológica exata que a sequência de DNA prediz é
quase impossível de se conhecer sem a informação das proteínas que são sintetizadas
em condições específicas (TORSVIK et al., 2002; RODRIGUEZ-VALERA, 2004). Graças ao
recente progresso nas tecnologias de sequenciamento (METZKER, 2010), as informações

17
de sequências metagenômicas continuam a crescer exponencialmente, e assim vai
oferecer uma base sólida para análises pós-genômica (GODZIK, 2011; SIMON e DANIEL,
2011; SHOKRALLA et al., 2012). A proteômica, uma abordagem experimental que
fornece medições qualitativas e quantitativas dos produtos finais dos genes (ou seja,
proteínas) pode fornecer informações das interações funcionais entre os membros de
uma comunidade microbiana no ambiente, permitindo a identificação de novas funções
envolvidas em complexas rotas metabólicas (MARON et al., 2007).
A maioria dos trabalhos realizados no campo da proteômica tem focado em
espécies únicas submetidas a diferentes condições, tornando as comunidades
microbianas uma área pouco explorada. Rodriguez-Valera (2004) propôs o termo
"metaproteômica" para descrever as proteínas expressas em amostras ambientais em
um determinado momento. O termo é derivado da metagenômica. Nesta abordagem,
uma cultura mista pode ser vista como um meta-organismo, em que mudanças na
população e no metaproteoma são formas de respostas funcionais. Estes estudos
ajudarão a alcançar um objetivo principal de microbiologia ambiental: a capacidade de
relacionar a espécie microbiana e a sua função no ambiente (LACERDA et al., 2009).
Os primeiros trabalhos com proteômica ambiental envolveram ambientes de
baixa complexidade (devido à dominância de uma ou algumas espécies), como a análise
de comunidades microbianas envolvidas na remoção biológica de fósforo em estações
de tratamento de esgotos (WILMES e BOND, 2004) e o consórcio microbiano de uma
drenagem de minas ácidas (RAM et al., 2005). Recentes abordagens de metaproteômica
foram aplicadas em ecossistemas aquático (SOWELL et al., 2011), terrestre, e em
microbiomas de hospedeiros eucarióticos, considerados sistemas de moderada ou alta

18
complexidade (que contém uma densa, complexa e diversificada microbiota). Em
ecossistemas terrestres, os trabalhos focaram no entendimento do fluxo de
carbono/nitrogênio e remediação de contaminantes, como metais pesados (LACERDA et
al., 2007) e xenobióticos (DESAI et al., 2010) em solos. Metaproteômica também tem
sido usada para investigar fatores que influenciam nas interações entre plantas e
microrganismos (KNIEF et al., 2011), ou nas comunidades que habitam a rizosfera (KNIEF
et al., 2011). E com o recente avanço das pesquisas do microbioma humano, análises
das comunidades do intestino humano também são alvos destes estudos
(VERBERKMOES et al.,2009).
Os estudos proteômicos e metaproteômicos estão baseados em técnicas de
separações de proteínas por meio de géis de eletroforese de duas dimensões e/ou pela
separação de peptídeos por cromatografia multidimensional acoplado a espectrômetro
de massas. A identificação das proteínas pode ser realizada com base na massa (obtida
por MALDI-TOF-MS) ou pela sequência dos peptídeos (LC-ESI-MS/MS) (AEBERSOLD e
MANN, 2003). Entretanto, a ausência de métodos padronizados e reproduzíveis de
extração, purificação e quantificação de proteínas que resultam em material favorável
para posteriores análises de metaproteômica como cromatografia líquida seguida por
espectrometria de massa, tem dificultado o progresso dessa área de estudo (HETTICHet
al., 2012). O aprimoramento destas técnicas permitirá a utilização da metaproteômica
para produzir novas informações funcionais e metabólicas para uma gama de
importantes ecossistemas ambientais.

19
2.7- Cliona varians (Duchassainge Michelotti, 1864)
A esponja Cliona varians (Figura 6) (Classe Demospongiae, Ordem Hadromerida,
Família Clionaidae) foi identificada primeiramente por Duchassainge Michelloti em
1864. Possui coloração marrom esverdeada na parte externa e a interna entre cinza e o
bege. Superfície regular, lisa, consistência firme, porém quebradiça. Ósculos circulares
dispersos, com 1-5 mm de diâmetro. Forma perfurante com 0,5 - 1,5 cm de espessura e
até 150 x 50 cm de largura (MORAES, 2011).
Figura 6. Esponja Cliona varians perfurando substrato calcário (Foto: Guilherme Muricy,
disponível em Moraes, 2011).
Essas esponjas estão distribuídas de forma endêmica na costa americana tropical
Atlântica Ocidental, sendo encontradas no Golfo do México, Caribe e Brasil (Rio Grande
do Norte, Fernando de Noronha, Atol das Rocas, Pernambuco, Alagoas, Bahia e Espírito
Santo). Na Bahia especificamente são encontradas em Salvador, Maraú, Abrolhos, Porto
Seguro e Santa Cruz de Cabrália (HAJDU et al., 2011). A espécie é relativamente comum
no mesolitoral (região sujeita às flutuações da maré, submersa durante a maré alta e
exposta durante a maré baixa) e no infra litoral (região permanentemente submersa) da

20
Bahia recobrindo substratos areno-calcários consolidados. A forma incrustante desta
espécie possui uma importante função ecológica, atuando no processo de erosão de
substratos carbonáticos em recifes de corais (RUTZLER, 2002). Espécimes desta esponja
com necroses teciduais foram reportadas no litoral baiano de Santa Cruz de Cabrália. A
partir de então, pela primeira vez, estudos foram realizados demonstrando a grande
diferença na diversidade microbiana e de expressão de proteínas (metaproteômica)
entre indivíduos saudáveis e doentes dessa espécie de esponja.

21
3- OBJETIVO GERAL
Examinar alterações no metaproteoma e na comunidade bacteriana e de archeae
associadas a indivíduos doentes da espécie Cliona varians, amplamente distribuída no
recife de corais de Coroa Vermelha (Santa Cruz de Cabrália, Bahia, Brazil).
3.1- OBJETIVOS ESPECÍFICOS
Testar e padronizar protocolo para extração de DNA metagenômico das
esponjas.
Amplificação do gene 16S rDNA a partir do DNA metagenômico utilizando
primers específicos do Domínio Bactéria e Archaea.
Avaliar diferenças na comunidade microbiana a partir do padrão de bandas
gerado por DGGE (eletroforese em gel de gradiente desnaturante).
Sequenciamento e identificação taxonômica das bandas do DGGE.
Comparar a diversidade microbiana através da construção e sequenciamento de
bibliotecas do gene 16S rDNA das esponjas sadias e doentes .
Testar protocolo de extração de proteínas diretamente do tecido de esponjas
sadias e infectadas.
Comparar o perfil de expressão proteica (metaproteoma) das esponjas por
eletroforese uni e bidimensional e identificar proteínas por espectrometria de
massa.

22
4- MATERIAL E MÉTODOS
4.1- Local de estudo e Coleta de esponjas
O Distrito de Coroa Vermelha, município de Santa Cruz Cabrália, está situado no
extremo sul baiano em uma região conhecida como "Costa do Descobrimento" (Figura
7A). A região é considerada patrimônio da humanidade pela UNESCO e é um dos
principais roteiros turísticos do Estado da Bahia devido às suas belezas naturais e ainda
pelo relevante papel dentro da história do país, por ter sido o local onde foi realizada a
primeira missa do Brasil. Devido ao valioso patrimônio ambiental (presença de
ambientes de restinga, remanescentes de Mata Atlântica, manguezais e recifes de
corais), ao valor histórico que reveste o local como marco do Descobrimento do Brasil,
realçado pela existência da comunidade indígena Pataxó, foi criada em sete de junho de
1993, através do decreto federal n° 2.184, a Área de Preservação Ambiental (APA) Coroa
Vermelha (Figura 7b), abrangendo 4.100ha. de extensão (SEMARH, 2012).
Dentre os recifes de corais localizados na APA (Figura 7), Coroa Vermelha
apresenta a concentração de nutrientes mais elevada, devido principalmente a fontes
antropogênicas como descargas de esgoto não tratado e águas residuais de áreas
urbanas próximas. Não existe tratamento de esgoto e o uso generalizado de fossas
sépticas em assentamentos urbanos ao longo da costa também aumenta as
concentrações de nutrientes das águas subterrâneas por contaminação via lençol
freático (COSTA JÚNIOR et al., 2000; COSTA JUNIOR et al., 2006; COSTA JÚNIOR et al.,
2007). O sistema recifal de Coroa Vermelha é formado por estruturas descontínuas
paralelas à costa. O platô recifal, quando exposto no período de baixamar, forma poças
de maré rasas com fundo arenoso. Estes ambientes agem como sistemas de incubações

23
“in situ”, o que permite estudo sobre processos metabólicos dos organismos e suas
interações com o ambiente (SOUZA et al., 2002). Fragmentos de esponjas Cliona varians
saudáveis e doentes foram coletadas aleatoriamente durante o mês de Outubro de
2010 em poças de maré de 0,30-0,60 metros na borda externa do platô recifal de Coroa
Vermelha durante a baixamar, momento de fluxo nulo entre as águas recifais e
oceânicas (Figura 8A). A amostragem de cinco pontos foi realizada próxima à costa
(Figura 8D). Em seguida, foram colocadas em sacos plásticos estéreis, mantidas
refrigeradas a quatro °C em caixa térmica isolante e encaminhadas ao laboratório de
Biotecnologia Microbiana da Universidade Estadual de Santa Cruz para armazenamento
em Ultra-freezer (-80°C). A identificação taxonômica foi realizada pelo grupo de
pesquisa em Poríferos da Universidade Federal da Bahia.

24
A.
B.
Figura 7. (A) Mapa da Costa do descobrimento brasileiro, sul da Bahia, Brasil. Em
destaque a localização geográfica do recife de Coroa Vermelha. (B) (MAPES, 2012).

25
D
Figura 8. Vista panorâmica do recife de Coroa Vermelha e foto do platô recifal durante
período de baixamar (A). Amostras de indivíduos de esponjas saudáveis (B) e doentes
(C). Seta em vermelho estágio primário da doença e seta preta estágio avançado com
necrose do tecido. Pontos de coleta e as respectivas coordenadas geográficas (D).

26
4.2- Extração de DNA total
A estratégia de extração do DNA total utilizada foi o método “direto”, lise das
células microbianas junto com as células da esponja (KENNEDY et al., 2008 –
modificado). Para extração do DNA, 2 g de esponja foram macerados em nitrogênio
líquido. O macerado foi misturado em 5 ml de tampão de lise (EDTA 100 mM, Tris
100mM, CTAB 1%, SDS 2% e NaCl 1,5 M, pH 8,0) e incubado a 70 °C por 30 min. A
mistura foi centrifugada por 10 min a 10.000 g. Então o sobrenadante foi transferido
para um novo tubo e adicionado o mesmo volume de solução de
fenol/clorofórmio/álcool isoamílico (25:24:1). Transferiu-se a fase aquosa para um novo
tubo e o DNA foi precipitado acrescentado 0.7V de isopropanol e 0.1V de acetato de
sódio (3M, pH 7.4) por 16 ha -20 °C. Então a mistura foi centrifugada por 10 min a
10.000 g, o sobrenadante foi descartado e o precipitado lavado duas vezes com etanol
(70%). Em seguida, o DNA foi suspenso em 100 µL de água ultrapura. A quantidade e
qualidade do DNA extraído foram avaliadas em espectrofotômetro (Gene Quant ®) na
razão 260/280 e por eletroforese em gel de agarose (1%) corado com brometo de etídio
e visualizado em luz ultravioleta.
4.3- Amplificação do gene 16S rDNA
Após extração de DNA, a PCR foi realizada com os primers 341F (5’-
CCTACGGGAGGCAGCAG-3’) e 518R (5’-ATTACCGCGGCTGCTGG-3’) (Muyzer et al., 1993)
para amplificar a região V3 do gene 16S rDNA bacteriano (correspondente a posição
341-534 pb em E. coli). Para amplificação de fragmentos específicos de Archaea foram
utilizados os primers 1100F (5’-AGTCAGGTAACGAGCGAG-3’) e 1400R (5’-
GTGCAAGGAGCAGGGAC-3’) (Kudo et al., 1997). Um grampo-GC de 40 pb foi incluído na

27
extremidade 5’ dos primers Forward para separação das bandas em DGGE (Muyzer et
al., 1993). Para a construção da biblioteca foi utilizado um conjunto de primers
universais 27F (5’-AGAGTTTGATCCTGGCTCAG-3’) e 1525R (5’-AAGGAGGTGWTCCARCC-
3’) para o domínio Bacteria, que gera fragmento de DNA de aproximadamente 1500
pares de bases. Os primers utilizados estão listados na tabela 1. Foram realizadas
reações de 25 µl contendo 3 mM de MgCl2, 1X de tampão de PCR, 0,2 mM de cada dNTP
(Promega), 5 pmol de cada primer, 0,05 U/µl de Taq DNA polimerase (Promega) e 1,5 µl
de DNA (aproximadamente 75 ng/µl).
Tabela 1.Oligonucleotídeos utilizados no trabalho
Gene Nome do primer
Sequências (5'-3') Tamanho
(pb) Referências
16SrDNA
27F AGAGTTTGATCACTGGCTCAG ~1500
Lane (1991) 1525R AAGGAGGTGATCCAGCC
Região V3 (16S rDNA)
341F-GC CCTACGGGAGGCAGCAG
~200
Muyzer et al. (1993) 518R ATTACCGCGGCTGCTGG
16SrDNA (Archaea)
1100F-GC AGTCAGGTAACGAGCGAG
~300
Kudo et al. (1997) 1400R GTGCAAGGAGCAGGGAC
A reação foi realizada em um termociclador (Mastercycler Gradiente,
Eppendorf), nas seguintes condições: (i) temperatura inicial de desnaturação 94 °C por 5
minutos; (ii) 34 ciclos de amplificação à 94 °C por 1 minuto; 55 °C (Bactéria) - 57 °C
(Archaea) por 1 minuto; 72 °C por 2 minutos e (iii) temperatura de extensão final de 72
°C por 10 minutos.

28
4.4- Eletroforese em gel de gradiente desnaturante (DGGE)
Os produtos da amplificação utilizando os primers com grampo GC para Bacteria
e Archaea foram separados através do sistema de eletroforese em gel de poliacrilamida
com gradiente de um agente desnaturante (uréia e formamida) conforme Muyzer et al.
(1993). O procedimento foi conduzido aplicando-se 15µl da reação de PCR realizada
previamente em gel vertical de poliacrilamida a 8% Acrilamda/Bis-acrilamida (37,5:1)
contendo gradiente de desnaturação de 15-55 % (Bactéria) e 40-55 % (Archaea), sendo
que a solução 100% desnaturante utilizada contém 40 % (V/V) de formamida (VETEC) e
7 M de Uréia (Promega), e a solução 0% não possui agentes desnaturantes. A
eletroforese foi realizada a 60°C em um sistema para análise de mutação CDC 20x20 cm
(BioAgency Biotecnologia LTDA.) seguindo os seguintes passos: 15 min a 60 V, seguido
por 5 horas a 200 V em temperatura constante de 60 °C. Posteriormente o gel foi corado
com Nitrato de Prata para visualização das bandas.
4.5- Identificação filogenética das bandas do DGGE
As bandas do DGGE foram excisadas assepticamente e estocadas em água
ultrapura estéril a -20°C até a reamplificação dos fragmentos. Os amplicons foram
clonados usando TA cloning kit® (Invitrogen) de acordo com as recomendações do
fabricante. As colônias transformadas foram amplificadas com os primers M13F (5’-GTT
TTC CCA GTC ACG AC-3’) e M13R (5’-CAG GAA ACA GCT ATG ACC-3’). As reações de PCR
continham 3mM de MgCl2, 1X de PCR buffer, 0,2 mM de cada dNTP (Promega), 5 pmol
de cada primer, 0,05 U/µL de Taq DNA polymerase (Promega) e 1,5 µL de DNA molde. As
condições da PCR foram as seguintes: 30 ciclos de 94 °C por 20 s, 55 °C por 30 s e 72 °C
por 60 s. Os amplicons foram sequenciados, processados e analisados com o software

29
Phrap/Phred/Cross Match (ERWING & GREEN, 1998) e foram comparadas contra o
banco de dados do GenBank (NCBInt) pela ferramenta de busca BLASTn (ALTSCHUL et
al., 1990).
4.6- Construção de biblioteca do gene 16S rDNA
Para uma comparação bacteriana mais detalhada, o gene 16S rRNA de cinco
amostras de esponjas doentes e saudáveis foram amplificadas por PCR com os primers
universais 27F (5’-AGAGTTTGATCCTGGCTCAG-3’) e 1525R (5′-AAGGAGGTGATCCAGCC-
3′) (Lane, 1991) específicos para o domíno Bacteria (Tabela 1). Os produtos obtidos da
PCR das cinco amostras de esponjas saudáveis e doentes foram agrupados
separadamente e purificados com o Kit GFX PCR DNA and Gel Band Purification Kit (GE
Healthcare) para posterior clonegam em vetores pGEM-T (pGEM-T Easy vector system,
Promega) (Figura 9), seguindo especificações do fabricante. Após a ligação, foi feito o
procedimento de eletro-transformação, utilizando 20 µL das células de E. coli DH10β
eletrocompetentes (Invitrogen) e 4 µL da reação de ligação. A amostra foi submetida à
eletroporação utilizando o GenePulser II (Biorad). Foi adicionado 1 ml de meio LB (Luria-
Bertani) a suspensão celular e incubado sob agitação a 37 °C por 1 hora. Para seleção
visual direta (seleção branca/azul) das colônias, as células transformadas foram
plaqueadas em meio LB suplementado com ampicilina (100 mg/ml), IPTG (Isopropyl β-D-
1-thiogalactopyranoside) 200 µg/ml e X-gal (5-bromo-4-cloro-3indolil-β-D-
galactopiranosideo) 20 µg/ml. Em seguida as placas foram incubadas a 37 °C por 16 h.
Os clones com inserto (colônias brancas) foram transferidos para placas de polietileno
de 96 poços. As biblioteca de 16S rDNA foram estocadas a -80 °C com glicerol 15 %. A
confirmação da clonagem dos insertos de 16S rDNA nos clones da biblioteca foi

30
realizada utilizando uma amplificação por PCR com os primers M13F (5’ - GTA AAA CGA
CGG CCA GT - 3’) e M13R (5’ - CAG GAA ACA GCT ATG AC - 3’) complementares aos sítios
de clonagem do vetor pGEM-T Easy.
Figura 9. Mapa do vetor de clonagem pGEM-T Easy.
4.7- Extração de DNA plasmidial e sequenciamento
A purificação de DNA plasmidial foi realizada pelo método de lise alcalina, de
acordo com Sambrook et al. (2001). Os clones foram transferidos para placas de 96
poços (Deep well) com 1,2 ml de meio LB suplementado com ampicilina 100 µg/ml e
foram incubados em agitador horizontal com agitação (240 rpm) por 18 h a 37 °C. Após
o crescimento, os clones foram centrifugados durante 6 minutos, a 10.000 x g, sendo o
sobrenadante descartado e a placa invertida em papéis absorventes durante 5 minutos.
Foi adicionado 240 µL de solução GET (Glicose 50 mM; Tris- HCl 25 mM (pH 8); EDTA 10

31
mM) em cada poço. A placa foi agitada vigorosamente e as amostras ressuspendidas.
Centrifugou-se a 10.000 x g por 6 minutos, a 20 °C. Novamente os sobrenadantes foram
descartados e a placa colocada invertida em papel para a secagem. A cada poço foram
adicionados uma solução de GTE/RNAse composta por 80 µL de solução GET e 5 µg de
Ribonuclease A e as amostras ressuspensas por agitação vigorosa. Transferiu-se 60µL do
ressuspenso para uma microplaca de 250 µL e foi adicionado 60 µL de solução de lise
(NaOH 0,2 N; SDS 1%). A placa foi selada e invertida 10 vezes, incubada por 10 minutos a
temperatura ambiente e centrifugada por 30 segundos a 250 x g. Foi adicionado 60 µL
de acetado de potássio (3M, pH 5,2), misturando-as por inversão. Novamente
centrifugou-se a placa a 250 x g por 30 segundos, para a deposição do material (restos
celulares) e em seguida incubou-se por 10 minutos a temperatura ambiente. Após esse
período, o adesivo foi removido e a placa colocada em uma estufa numa temperatura
de 90 °C, durante exatamente 30 minutos. Logo depois, a placa foi resfriada sobre o
gelo, durante 10 minutos, e centrifugada a 3000 x g por 6 minutos a 20°C. O
sobrenadante coletado foi colocado em um filtro (PVDF 0,2 µm, Millipore) fixado no
topo de uma microplaca de fundo em “V” de 250 µL de polipropileno, e centrifugado a
3000 x g por 6 minutos, a 20°C. Ao filtrado foi adicionado 110 µL de isopropanol
absoluto. O material foi centrifugado a 3000 x g por 45 minutos e o sobrenadante
descartado. O pellet foi lavado com 200 µL de etanol 70 % gelado. Os sobrenadantes
foram descartados e a placa foi deixada durante 1 hora em temperatura ambiente e em
seguida cada amostra de DNA foi ressuspensas em 40 µL de água ultrapura estéril. O
sequenciamento parcial do gene 16S rDNA dos clones foi realizado pelo sequenciador
automático (Megabace 1000TM, Amersham Bioscience), cuja reação de amplificação foi

32
realizada com o primer M13 F (5’-GTAAAACGACGGCCAGT-3’), complementares aos
sítios de clonagem do vetor.
4.8- Classificações das sequências
Foi realizada a classificação das sequências 16S rDNA das bibliotecas ao nível de
filo e classe utilizando a comparação feita pelo programa Classifier com o banco de
dados de genes ribossomais do RDP II (Ribosomal Database Project II), disponível em
http://rdp.cme.msu.edu. Este programa classifica taxonomicamente as sequências de
acordo com o padrão do manual de Sistemática bacteriana de Bergey.
4.9- Curva de rarefação, análise de riqueza e diversidade.
Depois de alinhadas, as sequências foram em seguida agrupadas em unidades
taxonômicas operacionais (OTU) a um nível de similaridade de 97% (cut-off 0.03)
utilizando o software MOTHUR (SCHLOSS et al., 2009). Neste programa também foram
calculadas as curvas de rarefação, índices de diversidade Shannon e Simpson, além dos
estimadores de riqueza Chao-1. A curva de rarefação representa uma parcela do
número de espécies em função do número de sequências amostradas. As curvas
normalmente se desenvolvem rapidamente no princípio, onde as espécies mais comuns
são encontradas, e começam a atingir o platô quando apenas espécies raras continuam
a ser amostradas.

33
4.10- Extração de proteína
As proteínas foram extraídas de acordo com o protocolo desenvolvido por
Pirovani et al. (2008) com algumas modificações. Amostras compostas por pequenos
pedaços (2 cm3) de três indivíduos diferentes de esponjas doentes e saudáveis foram
pulverizados em nitrogênio líquido. Posteriormente, 200 mg desse material foi suspenso
em acetona 100% contendo β-mercaptoetanol (0,07%) e EDTA (ácido
etilenodiaminotetracético) 5 mmol.L-1, e sonicado (amplitude de 70%, 3 pulsos de 10
segundos com intervalos de 5 segundos) no gelo. Após cada sonicação, os tubos foram
centrifugados por 10 minutos a 10.000 x g. Em seguida o pellet foi ressuspenso em
solução contendo 10% de ácido tricloroacético (TCA), 0,07% de β-mercaptoetanol e
acetona absoluta, sonicado de acordo com o programa do passo anterior e centrifugado
por 12 min a 10.000 x g. Esta etapa foi repetida 4 vezes até a coloração amarelada do
pellet ser removida. O pellet remanescente foi ressuspenso em solução contendo 10%
de TCA e 0,07% de β-mercaptoetanol solubilizados em água, sonicado1 vez por 7
segundos “on” e 7 segundos “off” a 50% de amplitude e centrifugado por 12 min a
10.000 x g. Por fim, o pellet foi lavado mais uma vez em solução contendo 80% de
acetona e 0,07% de β-mercaptoetanol solubilizados em água, agitado em vortex,
centrifugado por 12 min a 10.000 x g e secado à vácuo por 3 minutos. Em seguida foi
adicionado 800 µl de SDS-Denso (contendo 100 mmol.L-1 Tris-HCl pH 8,0, 30% de
sacarose, 2% de Dodecil Sulfato de Sódio (SDS) e 5% de β-mercaptoetanol em água),
sonicado 1 vez por 3 segundos a 50% de amplitude e incubado por 10 minutos em gelo.
Foi acrescentado 800 µl de fenol tamponado (pH 8,0), agitado suavemente por 40
minutos e centrifugado por 12 minutos a 10.000 x g. A fase fenólica foi coletada e
transferida para um tubo novo de 15 ml no qual se seguiu a precipitação das proteínas

34
em 5 volumes de solução contendo 0,1 M de acetato de amônio em metanol, e
incubada durante 12 horas a -20°C. Por fim, o precipitado foi lavado 3 vezes com a
solução anterior, 2 vezes com acetona gelada e 1 vez com etanol a 70%.
O precipitado foi tradado com o kit 2D Clean up (GE Healthcare), de acordo com
as instruções do fabricante, para a remoção de compostos interferentes que podem
atrapalhar na migração das proteínas no gel e ressuspensas em tampão de reidratação
(uréia 7 mol.L-1, tiouréia 2 mol.L-1, CHAPS 2% e azul de bromofenol 0,002 %). As
proteínas foram quantificadas utilizando o kit 2D Quant (GE Healthcare), de acordo com
as instruções do fabricante, para realizações das focalizações e posterior migração em
SDS-PAGE.
4.11- Eletroforese 1D e 2D
A eletroforese 1D (SDS-PAGE) foi realizada em géis de 8 x 10 cm contendo 12,5%
de poliacrilamida no gel de separação e 5 % no gel de empilhamento, de acordo com
Laemmli (1970). Após desnaturação a 95°C por 3 min, 30 µg de proteínas foram
aplicadas no gel e submetidas a 150 v, 280 ma/60wem minicubas de eletroforese
(Biorad) durante 1h e 30 min. Após corrida, o gel foi corado por 4 horas com Coomassie
Blue (Brilliant Blue 0.1%, metanol 25%, ácido acético 5%) e descorado com ácido acético
7% para visualização das proteínas.
Na 2D-E (eletroforese bidimensional), a separação das proteínas de acordo com
o ponto isoelétrico (primeira dimensão) foi realizada utilizando o sistema Ethan IPGphor
III (GE Healthcare). Amostras de 350 µg de proteína diluída em 250 µl de solução de
reidratação (7 MUréia, 2 M tiuréia, 10 mM DTT, 0,4% Triton X-100, 4% Chaps, 0,5%
anfólito e 0,005% de azul de bromofenol) foram aplicadas em “strips” (tiras de gel) de

35
13 cm de comprimento, com gradiente de pH imobilizado não linear de 3-10 (Amersham
Biosciences, Immobiline™ Dry-Strip) e condicionadas em sarcófagos por 12h. Após
reidratação o sistema foi submetido a focalização isoelétrica com os seguintes as
seguintes condições: 01:00 h a 500 Vh, 01:40 h a 1000 Vh, 2:30 h a 2.200 Vh e 04:35 h a
8000 Vh. A corrente foi limitada a 50 µA para cada strip e a temperatura mantida a 20 °C
em todos os passos da focalização. As “strips” foram tratadas com 7mL de tampão de
equilíbrio (Tris 50 mM, uréia 6 M, glicerol 30% e SDS 2%) contendo DTT 1% w/v, por 15
min em agitação lenta. Em seguida as mesmas foram transferidas para o tampão de
equilíbrio contendo iodoacetamida a 2,5% por mais 15 min. Após, as strips foram
lavadas com tampão de corrida (Tris 0,025 mol.L-1 pH 8.3, Glicina 0,19 mol.L-1, SDS
0.1%.) por 15 min. Para a segunda dimensão, as “strips” foram transferidas para gel SDS-
PAGE 12,5% no sistema de eletroforese vertical Ruby SE600 (GE Healthcare), sendo
aplicada uma corrente elétrica inicial de 15 mA/gel por 15 min, seguida de 50 mA/gel
por 3h e 30min. Após eletroforese os géis foram corados com Coomassie G-250 e
descorado com ácido acético 7%. Para obtenção das imagens, os géis foram escaneados
(Image Scanner, GE) em duplicata utilizando o software Lab Scan (GE Healthcare). E a
análise dos spots com o software Image Master 2D Platinum 7.0 (GE Healthcare).
4.12- Digestão tríptica das proteínas
Todos os spots do gel 2D e 15 bandas diferenciais entre as esponjas doente e
saudáveis foram excisadas com uso de bisturi, cortados em pedaços menores e
colocados em microtubos, em seguida foram descorados em 200 µL de bicarbonato de
amônio (NH4HCO3) contendo acetonitrila 50%, sendo o sobrenadante descartado e os
fragmentos de gel desidratados em 100 µL de acetonitrila 100 % por 5 minutos e secos a

36
vácuo no Concentrator 5301 (Eppendorf) por 10 minutos. Foram adicionados 4 µL de
tripsina Gold (Promega) 25 ng/µL, mantidos a 4 °C por 10 minutos para absorção da
solução nos fragmentos de gel. Posteriormente foi adicionado NH4HCO3 até cobrir os
fragmentos e incubados a 37 °C por 16 horas para ação da tripsina. O sobrenadante foi
coletado e transferido para um novo tubo. Os peptídeos foram recuperados dos
fragmentos de gel por duas eluições com 50 µL de acetonitrila à 50 % contendo ácido
fórmico 0,1 %, sendo agitados por 15 minutos no vórtex em cada lavagem. Em seguida,
as amostras foram concentradas a vácuo até atingirem o volume entre 10 e 15 µL. Os
peptídeos extraídos foram analisados por espectrometria de massa.
4.13- Espectrometria de massa (MS)
Os peptídeos foram resolvidos por cromatografia de fase-reversa no nano
Acquity UPLC (WATERS) em duas colunas C18, sendo a primeira uma coluna “trapping”
de 5 µm, 180 µm x 20 mm e a segunda de 1,7 µm 100 µm x 100 mm, sob um fluxo de 0,6
µL.min-1 em uma corrida de 50 minutos, onde foram coletados 4 µL de cada amostra.
Os peptídeos foram separados de acordo com gradiente de acetonitrila, sendo 1 % até 1
minuto, de 1 % a 50 % em 40 minutos, de 50 % a 85 % em 5 minutos, mantendo-se
nessa concentração por mais 2 minutos, voltando à concentração de 1 % em um minuto
e permanecendo nessa condição por 2 minutos, totalizando 50 minutos de corrida. Os
peptídeos separados foram ionizados em capilar sob voltagem de 3000 V (Micromass Q-
TOF micro), fragmentados no modo positivo com seleção da intensidade relativa mínima
de 10 counts, sendo analisados os 3 íons mais intensos por cada varredura de 1
segundo, com energia de colisão variando entre 20 e 95 eV de acordo com a relação
massa/carga (m/z) dos peptídeos.

37
4.14- Identificação das proteínas
Os espectros processados foram analisados com a ferramenta MASCOT MS/MS Ion
Search (www.matrixsciesce.com) para a comparação contra o banco de proteínas não
redundantes do NCBI, configurados para digestão tríptica, com 1 sítio de clivagem
perdida, cisteínas modificadas por carboxamidometilação e as possíveis modificações,
como oxidação de metionina, erro tolerante de 30 ppm e tolerância para erro de massa
igual a 0,3 Da. De acordo com a probabilidade de análise do MASCOT apenas os “hits”
significativos (p<0.05) foram aceitos.

38
5-RESULTADOS
5.1- Extração de DNA metagenômico
Através do método de extração direta do DNA de espécimes da esponja C.
varians saudáveis e doentes foram obtidos concentrações de 446 ng/µL e 494 ng/µL,
respectivamente. Apesar de rastros de degradação terem sido detectados em gel de
agarose, uma grande quantidade de DNA de alto peso molecular (acima de 10.000 pb)
foi observada , considerando adequado para amplificação do gene 16S rDNA pela
técnica da PCR (Figura 10). Em relação a qualidade, o DNA extraído apresentou leitura
média de 1,7 na razão 260/230 nm em espectrofotômetro Gene Quant (Amershan
Biosciences), revelando um DNA livre de contaminantes, em especial ácidos húmicos. O
método utilizado para extração de DNA metagenômico total de esponjas foi satisfatório,
obtendo-se DNA de quantidade e qualidade satisfatório para posteriores reações
enzimáticas.
Figura 10. Análise em gel de agorose 0.8% do DNA total extraído de esponja saudável (a)
e doente (b). M: Marcador de peso molecular 1 Kb (Fermentas).
39
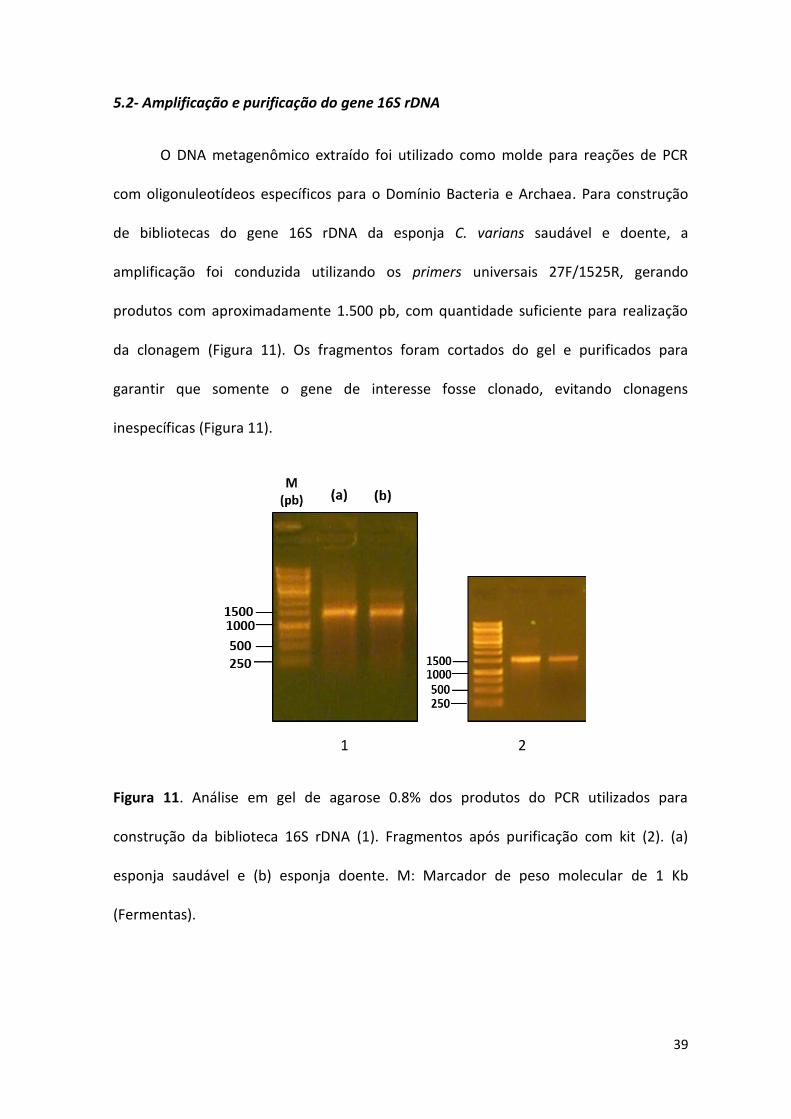
5.2- Amplificação e purificação do gene 16S rDNA
O DNA metagenômico extraído foi utilizado como molde para reações de PCR
com oligonuleotídeos específicos para o Domínio Bacteria e Archaea. Para construção
de bibliotecas do gene 16S rDNA da esponja C. varians saudável e doente, a
amplificação foi conduzida utilizando os primers universais 27F/1525R, gerando
produtos com aproximadamente 1.500 pb, com quantidade suficiente para realização
da clonagem (Figura 11). Os fragmentos foram cortados do gel e purificados para
garantir que somente o gene de interesse fosse clonado, evitando clonagens
inespecíficas (Figura 11).
1 2
Figura 11. Análise em gel de agarose 0.8% dos produtos do PCR utilizados para
construção da biblioteca 16S rDNA (1). Fragmentos após purificação com kit (2). (a)
esponja saudável e (b) esponja doente. M: Marcador de peso molecular de 1 Kb
(Fermentas).

40
Como resultado das amplificações com os iniciadores 1100F/1400R para o
Domíno Archae e 341F/518R pra amplifcar a região V3 do gene 16S rDNA (Domínio
Bacteria), foram gerados com sucesso fragmentos de aproximadamente 200pb e 400pb,
respectivamente, com era esperado para essas regiões do gene (Figura 12).
1 2
Figura 12. Análise em gel de agarose 0.8% dos produtos do PCR utilizados para análise
por DGGE do domínio Bactéria (1) e Archaea (2).Em (a) esponja saudável e (b) esponja
doente. M: Marcador de peso molecular 1 Kb (Fermentas).

41
5.3- Construção da biblioteca do gene 16S rDNA
O DNA plasmidial extraído dos clones das duas bibliotecas foi analisado em gel
de agarose. A maioria dos clones apresentou padrão de bandeamento característico
para DNA plasmidial (Figura 13). Os clones foram sequenciados com primers universais
que anelam na região terminal do vetor. Após eliminação das sequências de baixa
qualidade e retirada de fragmentos de vetor, 37 sequências da esponja doente e 48 da
saudável foram selecionadas para posteriores análises.
Figura 13. Perfil eletroforético em gel de agarose 0.8% do DNA plasmidial de uma
amostra de clones da biblioteca 16S rDNA.

42
5.4- Análise do perfil da comunidade microbiana por DGGE
A análise do perfil de bandas do DGGE foi utilizada para explorar as alterações na
comunidade de bacteria e archeae entre amostras saudáveis e doentes de C. varians. O
DGGE revelou uma diversa comunidade microbiana em âmbos tecidos doentes e
saudáveis. Entretanto, uma clara diferença foi observada no padrão de bandas entre as
amostras (Figura 14). Apenas uma banda em comum, sendo dominante no tecido não
afetado pela doença. Todas as outras bandas foram exclusivas nas lesões. Este padrão
revela grande alterações na microbiota normal na composição de archae e bactérias.
Para obter um detalhamento filogenético, as sequências do gene 16S rDNA foram
recuperadas das bandas do DGGE de Archaea e Bacteria para clonagem e
sequenciamento. Dessas, 11 foram sequenciadas com sucesso para o domínio Bacteria
(Tabela 2) e cinco para o domínio Archaea (Tabela 3).

43
Figura 14. Perfil das bandas do DGGE utilizando sequências 16S rDNA-V3 de Bactérias (a)
e Archaea (b). As setas indicam as bandas isoladas. As letras representam a identificação
do grupo taxonômico após sequenciamento. Poço 1 – esponja saudável. Poço 2 –
esponja doente.

44
Tabela 2. Identificação das sequências 16S rDNA-V3 recuperados das bandas de DGGE através do BLAST
contra o banco de dados GenBank
Banda Sequência mais relacionada Similaridade
(%) Fonte
Saudável
F11 Uncultured bacterium clone Ma2E10 100% Coral Mussismiliasp. (Brasil)
F12 Uncultured alpha proteobacterium clone MPWIC_C07
97% Esponja Clathria prolifera
F10 Uncultured Chloroflexi bacterium clone 289R 92% Esponja Corallistessp.
G1 Uncultured alpha proteobacterium clone MPWIC_C07
96% EsponjaClathriaprolifera
G3 Uncultured bacterium isolate DGGE gel band B7-2 96% EsponjaIrciniafelix
Doente
G5 Uncultured bacterium clone TV10-3-7_C9 97% Esponja Ircinia variabilis
G8 Uncultured bacterium clone Mfav_M07 98% Coral Montastraea faveolata
G9 Uncultured bacterium partial clone i118 97% Esponja Aplysina fulva (Brasil)
G10 Uncultured bacterium partial clone i118 97% Lodo de esgoto
G12 Uncultured bacterium isolate DGGE gel band SAC11-2
96% Esponja Aplysina fulva (Brasil)
H2 Uncultured Gemmatimonadetes bacterium 100% Esponja exposta a alta temperatura
H2 Uncultured sponge bacterium clone JZ66-1 100% Esponja do mar vermelho

45
Tabela 3. Identificação das sequências de Archaea recuperados das bandas de DGGE através do
BLAST contra o banco de dados GenBank
Banda Sequência mais relacionada Similaridade
(%) Fonte
Saudável
H04 Uncultured marine group 1 crenarchaeote clone
95% Sedimento marinho
H05 Uncultured marine archaeal group 1 crenarchaeote
99% Esponja Geodia Media
H06 Uncultured Nitrosopumilalesarchaeon 99% Esponja Geodia Media
Doente
H07 Unculturedcrenarchaeote 98% Esponja Mycale armata
H08 Uncultured marine group I thaumarchaeote 92% Fontes hidrotermais
5.5- Comparação da diversidade bacteriana em esponjas saudáveis e doentes
Após verificação da qualidade e formação de quimeras, um total de 48 e 37
sequências foram obtidas a partir das bibliotecas 16S rDNA das esponjas saudáveis e
doentes, respectivamente. Para classificar as sequências filogeneticamente e comparar
a composição bacteriana das bibliotecas, foi utilizado o programa Classifier do
“RibossomalDatabase Project II” (RDP), com um limite de confiança de 50%. A maioria
das sequências pôde ser atribuída a filos bacterianos conhecidos, as outras sequências
foram classificadas como não caracterizadas. Em ambas as bibliotecas da esponja C.
varians o filo Proteobacteria foi o mais abundante. No tecido saudável observou-se
baixa diversidade de filos com a predominância de Proteobacéria (84%), e bactérias
ainda não caracterizadas (16%). Já o tecido doente apresentou maior diversidade de

46
filos, sendo as sequências classificadas em Proteobactérias (58%), Bacteroidetes (22%),
Firmicutes (5%) e bactérias ainda não caracterizadas (15%) (Figura 15A).
Quando o filo Proteobacteria foi analisado ao nível de classe, observou-se, para a
classe Alphaproteobacteria, uma diminuição de 91% de ribotipos no tecido saudável
para 8% de no tecido doente. Com a perda em Alphaproteobacteria (91% para 8%), um
aumento das classes Gammaproteobacteria (8%) e Epsilonproteobacteria (56%) foi
observado, sendo encontradas apenas nos tecidos afetados (Figura 15B).
O filo Bacteroidetes foi encontrado apenas em amostras de C. varians doente,
representando 29% do total. Este filo apresentou alta variabilidade composta pelas
classes Sphingobacteria, Flavobacteria, Bacteroidia (Figura 15C). 22% das sequências
foram incluídas no filo Bacteroidetes, mas não foram classificadas dentro dessas classes.
Como evidências adicionais não podem justificar a classificação em uma das classes
conhecidas ou em uma nova classe, estes ribotipos foram agrupados em Bacteroidetes
incertae sedis (do latim “posicionamento incerto”) (Figura 15C).

47
Figura 15.Classificação das sequências parciais do gene de 16S rRNA bacteriano das
esponjas doentes e saudáveis usando RDP classifier (limiar de 50%). Comparação da
diversidade com base na distribuição ao nível de filos (A), ao nível de classes do filo
Proteobacteria (B) e ao nível de classe do filo Bacteroidetes (C).

48
5.6- Análise de rarefação, índices de diversidade e riqueza de espécies
As curvas de rarefação foram obtidas pela relação entre o numero de filotipos
(OTU`s) agrupados a 97% de similaridade, e o numero de sequências analisados. As
curvas geradas mostraram que o número de clones sequenciados não foi suficiente para
representar toda a diversidade de bactérias nas esponjas saudável e doente, indicando
que mais amostras de ambas as bibliotecas de clone teria revelado diversidade
adicional. Apesar disso, essa amostragem foi suficiente para acessar os grupos mais
abundantes. Com base na análise comparativa entre as duas bibliotecas, as curvas
demonstram uma maior diversidade na esponja doente, pois com um menor número de
sequências amostradas, observou-se uma maior representatividade de unidades
taxonômicas operacionais (UTO’s) (Figura 16).
Figura 16. Curva de rarefação gerada pela análise das sequências 16S rDNA da
biblioteca.

49
Índices de diversidade e riqueza também foram estimados com um nível de
confiança de 97% para determinação das OTU’s (Tabela 4). Os resultados indicam que
existe uma maior diversidade e riqueza de espécies bacterianas nas esponjas doentes,
corroborando com a curva de rarefação. Isto também pode ser observado quando
relacionamos o número de OTU’s formadas e o número de sequências amostradas.
Apesar de menor número de sequências na esponja doente, foi possível formar a
mesma quantidade de OTU’s que a saudável.
Tabela x. Índices de diversidade e riqueza calculados
Amostra Label N OTU's Chao Shannon Simpson
Doente 0.3 37 32 265.500 3.423 0.01
Saudável 0.3 48 32 120.750 3.235 0.039
OTU's - unidades taxonômicas operacionais
Label - Nivel de significância para atribuir OTU's
N - Número de sequências amostradas
5.7- Separação dos extratos proteicos totais por 1D-SDS-PAGE
No presente estudo, avaliamos as mudanças nos padrões de expressão proteica
entre metaproteomas de esponjas marinhas sadias e doentes, com o intuito de melhor
compreender a interação entre microrganismos e esponjas em resposta a doença. Para
verificar a integridade da amostra proteica obtida e a eficiência do método de extração,
30 µg de proteínas totais foram separadas em SDS-PAGE 12,5%, produzindo um padrão
muito distinto de bandas com baixa intensidade que se distribui entre 14,4 a mais de 97
kDa (Figura 17). Poucas bandas foram compartilhadas nas duas amostras, além de um

50
perfil de arraste heterogêneo localizado entre 27 a 40 kDa. Apesar de ambos os extratos
de esponjas saudáveis e doentes apresentarem várias bandas exclusivas, um maior
número de bandas únicas foi encontradas na amostra dos tecidos lesionados (Figura 17).
Figura 17. Perfil das proteínas totais em 1D-SDS-PAGE. M - Marcador de massa
molecular, cujos valores de massa em kDa estão indicados à esquerda. Na coluna 1
encontra-se o extrato proteico da esponja doente. E na coluna 2, extrato proteico da
esponjas sadia. As setas correspondem a bandas exclusivas. À direita as 15 faixas de
bandas que foram analisadas em MS.
5.8- Análise do perfil de expressão por 2D-PAGE
Após separação das proteínas em primeira e segunda dimensão (focalização
isoelétrica e eletroforese 2-D, respectivamente), os géis foram digitalizados, e as
análises das imagens realizadas no software Image Master 2D Platinum para avaliar a
presença de spots foram realizadas, para posteriormente comparadas entre as

51
amostras. Os géis foram avaliados quanto à distribuição dos spots em relação ponto
isoelétrico (pI) e massa molecular. Verifica-se que a maioria das proteínas que compõem
os metaproteomas de tecido doente encontram-se distribuídas na faixa de 25 a 65 kDa e
pontos isoelétricos aproximadamente entre 4 a 7. Nas esponjas saudáveis não foi
possível observar um padrão de distribuição em relação a massa molecular devido a
pequena quantidade de spots observados, mas os spots detectados também
apresentaram-se entre estes pontos isoelétricos (pI) (Figura 18).
Figura 18. Comparação do perfil metaproteômico das esponjas não afetadas (a) e
afetadas (b) pela doença. As amostras foram focalizadas em tiras de 13 cm com
gradiente de pH 3-10 não linear (NL).
Após a análise dos géis, todos os spots foram excisados e analisados por
espectrometria de massas do tipo Nano-ESI-Q-TOF.

52
5.9- Identificação de proteínas por espectrometria de massa
Para identificação de proteínas a partir do 1D-PAGE, cada coluna do gel das
esponjas foi cortado em 15 bandas (Figura 17). E para identificação a partir do 2D-PAGE,
um total de 89 spots foram excisadas das esponjas saudáveis e 32 da doente. Após
digestão tríptica, os peptídeos extraídos foram submetidos a análise por espectrometria
de massa (nanoLC-ESI-MS/MS). A procura por similaridade dos espectros de massa
("MS/MS ionsearch") no banco de dados foram conduzidas pela ferramenta de interface
web Mascot (http://www.matrixscience.com/), contra todas as proteínas não
redundantes do NCBInr (GenBank;http://www.ncbi.nlm.nih.gov/ index.html). De acordo
com a probabilidade de análise do MASCOT apenas os “hits” significativos (p<0.05)
foram aceitos. Todos os spots que tiveram as suas identidades conhecidas estão nas
tabelas 5 e 6, classificadas de acordo com suas funções. Utilizando o banco de dados do
NCBInr foram identificadas um total de 12 proteínas a partir das bandas de 1D-PAGE e
10 proteínas a partir dos spots. Considerando todas as proteínas identificadas, 8 (36%)
foram proteínas hipotéticas (com função desconhecida).

53
Tabela 5. Identificação de proteínas separadas por 1D-SDS-PAGE utilizando Mascot contra o banco de dados
NCBI (nr)
Banda* Proteína identificada N Nº de acesso Score Taxonomia
M_A Hypothetical protein 1 gi|262371871 41 Proteobacteria
M_E serine protease-like protein 1 gi|385805376 32 Archaea/Crenarchaeota
M_L Hypothetical protein 1 gi|345564129 29 Ascomycota
M_G Cytoplasmic actin CyII 7 gi|2304965 81 Echinodermata
M_K Hypothetical protein 1 gi|124002932 19 Bacteroidetes
M_P Hypothetical protein 1 gi|156403682 28 Cnidaria
MD_B Hypothetical protein 1 gi|255088677 14 Viridiplantae
MD_C Hypothetical protein 1 gi|340966885 20 Fungi/Dikarya
MD_H TetR Family transcriptional
regulator 1 gi|334564404 17 Actinobacteria
MD_J putative dead/deah box helicase 1 gi|223039658 43 Proteobacteria
MD_K Hypothetical protein 1 gi|166030969 22 Firmicutes/Clostridia
MD_L Hypothetical protein 1 gi|126641907 35 Proteobacteria
*Código das bandas: M = Saudável, MD = Doente N corresponde ao número de peptídeos sequenciados

54
Tabela 6. Identificação de proteínas separadas por 2D-SDS-PAGE utilizando Mascot contra o banco de dados NCBI (nr)
Spot* Proteína identificada N Nº de acesso Score Taxonomia
M_3 Coactosin like protein 1 gi|340367995 67 Porifera
MD_06 trpC gene product 1 gi|127513182 55 Proteobacteria
MD_12 Splicing factor 3A subunit 1 1 gi|358339810 59 Metazoa
MD_16 putative septation ring formation regulator EzrA 1 gi|293376359 30 Firmicutes
MD_24 Hypothetical protein 2 gi|46107788 62 Fungi
MD_46 Cytochrome b dubiquinol oxidase subunit I 1 gi|220923257 38 Proteobacteria
MD_46 gelsolin-likeprotein 2-like 1 gi|115891439 129 Metazoa
MD_68 Heat shock protein 70 4 gi|195964871 218 Metazoa
MD_89 glucose-1-phosphate adenylyltransferase 1 gi|152966845 50 Actinobacteria
MD_89 acyl-CoA dehydrogenase 1 gi|301057558 26 Proteobacteria
*Código dos spots: M = Saudável, MD = Doente
N corresponde ao número de peptídeos sequenciados

55
6-DISCUSSÃO
Vários sintomas patológicos de doenças têm sido usados para descrever uma
série de síndromes em esponjas marinhas (WEBSTER, 2007). Este é o primeiro estudo
da literatura que avalia o impacto na microbiota simbionte e no metaproteoma
provados por doença acometendo a espécie C. varians, endêmica da costa atlântica
ocidental, e o primeiro estudo das interações microbianas de esponjas doentes em
recifes de corais da costa brasileira. As esponjas desse estudo apresentaram como
sintomas patológicos da doença manchas pretas na superfície, caracterizando o
estágio inicial da doença. A evolução caracterizou-se por grandes áreas de tecido
necrosado com coloração branca, levando à exposição das fibras esqueléticas da
esponja (Figura 8C). Sintomas patológicos similares a estes foram encontradas na
esponja Aplysina aerophoba (WEBSTER et al., 2008b), coletada no mar Adriático,
durante surtos da doença denominada “Aplysina Black Patch Syndrome” (Figura 4).
Devido às similaridades nos padrões de sintomas apresentados nomeamos esta
doença como “Cliona Black Patch Syndrome”.
6.1- Alterações na composição da comunidade microbiana simbionte
Este estudo revela uma grande mudança na composição da comunidade
microbiana entre esponjas C.varians saudável e doente, detectada tanto por DGGE,
quanto pela análise de sequências de bibliotecas do gene 16S rDNA.
Na análise comparativa da classificação taxonômica realizada pelo RDP pode-se
observar aumento do número de sequências de deltaproteobacteria (2% para 9%) e
epsilonproteobacteria (0% para 60%) em amostras doentes, aproximando-se com os
padrões previamente observados nos corais Montastrea annularis e Diploria strigosa

56
afetada por Black Band Disease (BBD) (FRIAS-LOPEZ et al., 2002), e a esponja Aplysina
aerophoba afetada por “Aplysina Black Patch Syndrome” (WEBSTER et al., 2008b).
Todas as sequências de Alphaproteobacteria da biblioteca são correlacionadas a
linhagens de Arcobacter spp. Essa espécie foi previamente relacionada a doenças de
corais (FRIAS-LOPEZ et al., 2002; SWEET e BYTHELL, 2012) e é frequentemente
encontrada em águas contaminadas por esgotos (COLLADO et al., 2010), sendo um
candidato potencial na patogênese da doença.. Um incremento do filo Bacteroidetes
foi detectado (0% para 22%) em amostras doentes, apresentando uma grande
variabilidade distribuídas em 3 classes, sendo totalmente ausente no tecido não
afetado (Figura 15A,C). Este aumento na comunidade de Bacteroidetes foi
previamente relatado em inúmeras espécies (FRIAS-LOPEZ et al., 2002; WEBSTER et al.,
2008b). Em contrapartida, o número de Alphaproteobacterias, filo em maior
abundância nas esponjas saudáveis, diminuiu drasticamente (90% para 8%) (Figura
15B). Isto evidencia um desbalanço da microbiota simbiótica das esponjas. Esse
desequilíbrio no consórcio microbiano natural das esponjas parece ser o marco em
doenças de esponjas e corais. E suas causas são muitas vezes atribuídas ao estresse
ambiental (WEBSTER, 2007). Neste trabalho não foi investigado diretamente as causas,
mas algumas evidências sugerem a poluição antrópica dos recifes por escoamento de
esgotos e o aumento da temperatura no contexto do aquecimento global.
A banda MD10 do DGGE (tabela 2), exclusiva nas esponjas doentes está
estritamente relacionada (com 100% de similaridade) a sequências 16S rDNA da
esponja Rhopaloeides odorabile exposta a estresse térmico. A esponja Amphimedon
compressa apresentando branqueamento também apresentou similaridade a estas
sequências (ANGERMEIER et al., 2012). E a banda MD6 (tabela 2), apresentou

57
similaridade de 97% com sequências oriundas de estações de tratamento de esgoto.
Dentre os recifes de corais localizados na APA, Coroa Vermelha apresenta a
concentração de nutrientes mais elevada, devido principalmente a fontes
antropogênicas como descargas de esgoto não tratado e águas residuais de áreas
urbanas próximas (COSTA JUNIOR et al., 2006; COSTA JÚNIOR et al., 2007). Estes dados
corroboram que a temperatura e enriquecimento por nutrientes (como descarga de
esgoto) realmente podem desencadear desequilíbrio da microbiota simbionte dessas
esponjas (WEBSTER et al., 2008), embora mais estudos com o monitoramento dessas
espécies submetida a influências ambientais e antropogênicas serão requeridos para
responder estas questões.
6.2- Comparação da diversidade bacteriana em esponjas saudáveis e doentes
Alta diversidade de bactérias foi detectada em esponjas doentes de C. varians
(Figura 15 e tabela 4). Resultado como este foi demonstrado em várias doenças ou
organismos marinhos estressados como corais Acropora palmata e Montastrea
anullaris acometidos por branqueamento (PANTOS E BYTHELL, 2006; PANTOS et al.,
2003), Diploria strigosa e Montastrea anullaris infectado por BBD (COONEY et al.,
2002). Além de esponjas Rhopaloeides odorabile estressadas por temperatura
(WEBSTER et al., 2008) e infectadas por doença (WEBSTER et al., 2008b).
A maior diversidade bacteriana detectada em animais doentes pode ser
explicada pela formação de diferentes micronichos para habitação (ex., tecido íntegro,
tecido lesionado, expostos do esqueleto), e elevação da disponibilidade de nutrientes
oriundos da degradação do tecido, proporcionando uma variada fonte de compostos
que podem ser utilizados no metabolismo microbiano de diferentes grupos

58
taxonômicos. Quando doentes, a produção de metabólitos pelos simbiontes como
mecanismos de defesa podem também tornar-se comprometida, facilitando o
estabelecimento de populações bacterianas oportunistas que normalmente não
poderiam sobreviver dentro do animal, como proposto por WEBSTER (2007).
6.3- Análise em 1D e 2D-PAGE do metaproteoma
A metaproteômica está sendo muito empregada para descrever o perfil de
proteínas expressas de comunidades microbianas para explorar melhor o potencial
genético oferecido por conjuntos de dados metagenômicos (LACERDA et al., 2009).
Entretanto, a detecção e identificação de proteínas produzidas por uma complexa
comunidade microbiana ambiental é uma tarefa difícil. Desafios para análise
metaproteômica incluem a distribuição desigual de espécies, a ampla gama de níveis
de expressão de proteína dentro de microrganismos, e a grande heterogeneidade
genética nas comunidades microbianas (SCHNEIDER E RIEDEL, 2011).
O sucesso na aplicação das técnicas baseadas em proteínas para caracterizar
estrutura funcional de comunidades microbianas complexas requer uma efetiva
recuperação de proteínas do ambiente, com eficiente lise celular e remoção de
compostos interferentes (ácidos húmicos, ácidos fenólicos, sais, dentre outros).
Entretanto amostras ambientais são bem conhecidas por conter substâncias
interferentes que dificultamas análises proteômicas desses ambientes (THOMPSON et
al., 2008). Embora vários métodos tenham sido descritos e utilizados com sucesso para
recuperação e caracterização de metaproteomas de solo, água, intestino humano,
nenhuma metodologia para extração de proteínas diretamente de esponjas marinhas
foi desenvolvida. Neste trabalho, um método desenvolvido por Pirovani e

59
colaboradores (2008) para extração proteica de tecidos de plantas recalcitrantes com
elevados níveis de polissacarídeos e polifenóis foi utilizado com sucesso em amostras
de esponjas marinhas (Figura 17). Esse protocolo combina métodos físicos e químicos,
incluindo fenol, tampão SDS denso e sonicação, além de sucessivas lavagens com
acetona para eliminação de contaminantes. Os passos de sonicação foram muito
importantes para quebra de polissacarídeos, separação da matrix do tecido e lise
celular, através do efeito das ondas de choque geradas a partir de ultrassom (TANG et
al., 2002). A falta de padrões de bandas nítidos em extratos proteicos complexos,
obtidos diretamente do ambiente tem sido observada anteriormente em extratos de
solos e de águas residuais (BENNDORF et al., 2009), bem como em amostras de
comunidade microbiana da água superficial da Baía de Chesapeake (KAN et al., 2005).
Em comparação com estes trabalhos, os resultados obtidos revelaram um padrão de
bandas satisfatório em gel 1D-SDS-PAGE para posteriores análises de espectrometria
de massa (MS).
6.4- Identificação de proteínas
Um pequeno número de proteínas foi identificado neste trabalho, sendo que
36% delas foram consideradas sem atribuição de função ou de natureza hipotética
(tabela 5 e 6). Isto sugere a presença de muitas funções ainda não reconhecidas na
comunidade microbiana simbiótica de esponja. Vários trabalhos fracassaram em inferir
função de microrganismos em seu ambiente através dessa abordagem. Essa
deficiência pode ser encontrada devido à baixa representatividade dos bancos de
dados disponíveis atualmente em sequências dos organismos presentes na amostra,

60
pois como visto nas análises de diversidade, muitos desses microrganismos são
desconhecidos e não cultiváveis.
Nenhum método de separação das células eucarióticas e microbiana foi utilizado. As
proteínas foram extraídas diretamente do tecido da esponja, portanto era esperado
encontrar proteínas relacionadas tanto a microrganismos como a animais
(Metazoários). A maioria das proteínas procarióticas foi relacionada ao filo
Proteobacteria, que também foi o mais abundante nas esponjas doentes e saudáveis
(Figura 15). O spot M_3 foi identificado como a proteína “coactosin-like protein” da
esponja Amphimedon queesland. Esta esponja teve seu genoma sequenciado
recentemente, sendo o único de esponjas depositas no banco de dados. Apesar disso,
somente essa proteína foi identificado, isso reflete a grande diversidade genética entre
diferentes espécies de esponjas. Essas proteínas se ligam a filamentos de actina
ativando processos como motilidade e fagocitose (Li et al., 2004), atividade realizada
pelos amebócitos, células responsáveis pela digestão e distribuição do alimento nas
esponjas. Outras proteínas, provavelmente componentes estruturais do citoesqueleto
das esponjas foram detectadas, como “cytoplasmic actin” e “gelsolin-likeprotein”.
A proteína “serine protease-like” identificada na esponja saudável, está envolvida em
vários processos fisiológico com metabolismo, sinalização celular e mecanismos de
defesa, importante para manutenção da homeostase. Já nas esponjas doentes foram
encontradas proteínas relacionadas a respostas de estresse oxidativo, como uma
Thioredoxina bacteriana e uma proteína de choque térmico (Heat Shock Protein). A
elevada expressão dessas proteínas tem sido reportada em corais submetidos a

61
estressores ambientais e antropogênico como elevação da temperatura, salinidade e
metis pesados (Fanget al., 1995; Downs et al., 2000).
Recentemente, estratégias são empregadas para obter um melhor sucesso na
identificação de proteínas a partir de metaproteomas. Predições de proteínas a partir
de sequências metagenômicas obtidas do ambiente em questão por métodos de
sequenciamento de larga escala podem ser utilizadas como banco de dados para busca
por homologia de metaproteomas (CANTARELet al., 2011). Utilizando essa abordagem,
Leary e colaboradores (2011) observaram um aumento do número de proteínas
identificadas quando as buscas eram feitas contra um banco de dados com sequências
metagenômicas da mesma amostra de biofilme, quando comparado com
identificações contra o banco de dados do SwissProt.
Apesar dessas limitações, a metaproteômica tem um enorme potencial de
vincular a diversidade genética de comunidades microbianas com o seu impacto no
funcionamento do ecossistema. Estes dados mostram que é possível obter proteína de
boa qualidade diretamente do tecido de esponjas marinhas. Essa abordagem poderá
ser usada para posteriores investigações da fisiologia da interação entre esponjas e
seus microrganismos simbióticos, bem como comunidades microbianas relacionadas a
doenças, podendo gerar um maior entendimento dos mecanismos envolvidos na
ruptura da simbiose envolvida nessa relação, até então pouco conhecidos.

62
7- CONCLUSÃO
A comunidade microbiana associada a amostras de C. varians doente e
saudável demonstra diferenças notáveis em vários níveis taxonômicos. O incremento
observado de Bacteroidetes, Epsilonproteobacteria e Deltaproteobacteria em esponjas
doentes caracteriza o desequilíbrio na microbiota simbionte, deixando clara a
importância destes para saúde das esponjas. Esta é a primeira tentativa em utilizar
abordagens metaproteômicas para tentar esclarecer a função que esta microbiota
alterada desempenha nestes organismos. Apesar do pouco sucesso em identificar
proteínas, a separação eletroforética das proteínas foi sensível o suficiente para
mostrar uma alteração no padrão de expressão proteica em esponjas doentes,
revelando grande impacto na fisiologia da interação simbiótica esponja-
microrganismos. Este resultado justifica maiores esforços para determinar
mecanismos responsáveis por esse processo patológico na ruptura da simbiose.

63
8- REFERENCIAS BIBLIOGRÁFICAS
AEBERSOLD, R. e MANN, M. Mass spectrometry‐basedproteomics. Nature. v. 422, p.198-205, 2003.
AICHER, T.D., K.R. BUSZEK, F.G. FANG, C.J. FORSYTH, S.H. JUNG, Y. KISHI, M.C. MATELICH, P.M., SCOLA, SPERO, D.M E YOON, S.K. Total synthesis of halichondrin B and norhalichondrin. B. J. Am. Chem. Soc. 114:3162–3164, 1992.
ALTSCHUL, S.F., GISH, W., MILLER, W., MYERS, E.W., LIPMAN, D.J. Basic local alignment search tool. J. Mol. Biol., 215, 403–410, 1990.
AMANN, R.I., LUDWIG, W., SCHLEIFER, K.H: Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143-169. 1995.
ANGERMEIER, H., GLÖCKNER, V., PAWLIK, J.R., LINDQUIST, N.L., HENTSCHEL, U. Sponge white patch disease affecting the Caribbean sponge Amphimedon compressa. Dis. Aquat. Organ. Jun 13; 99(2):95-102, 2012.
ANGERMEIER, H., KAMKE, J., ABDELMOHSEN, U.R., KROHNE, G., PAWLIK, J.R., LINDQUIST, N.L. E HENTSCHEL, U. The pathology of sponge orange band disease affecting the Caribbean barrel sponge Xestospongia muta. FEMS Microbiol. Ecol. 75: 218-230, 2011.
BLUNT, J. W., COPP, B. R., MUNRO, M. H. G., NORTHCOTE, P. T. PRINSEP, M. R. Marine natural products. Nat. Prod. Rep. 28, 196-268, 2011.
BOURNE, D.G.; GARREN, M.; WORK, T.M.; ROSENBERG, E.; SMITH, G.W.; HARVELL, C.D. Microbial disease and the coral holobiont. Trends Microbiol. Dec; 17(12):554-62, 2009.
COONEY, R.P., PANTOS, O., LE TISSIER, M.D., BARER, M.R., O’DONNELL, A.G., AND BYTHELL, J.C. Characterization of the bacterial consortium associated with black band disease in coral using molecular microbiological techniques. Environ. Microbiol. 4: 401–413, 2002.
COSTA J.R., O. S..; LEÃO, Z.M.A.N.; NIMMO, M.; ATTRILL, M. Nutrification impacts on coral reefs from northern Bahia, Brazil. Hydrobiologia, v. 440, p. 307-315, 2000.
http://www.ncbi.nlm.nih.gov/pubmed?term=Angermeier%20H%5BAuthor%5D&cauthor=true&cauthor_uid=22691978

64
COSTA J.R., OZEAS S. Anthropogenic nutrient pollution of coral reefs in Southern Bahia, Brazil. Braz. J. Oceanogr. São Paulo, v. 55, n. 4, Dec. 2007.
COSTA JÚNIOR, O.S.; ATTRILL, M.J.; NIMMO, M. Seasonal and spatial controls on the delivery of excess nutrients to nearshore and offshore coral reefs of Brazil. J. Mar. Syst. v. 60, p. 63-74, 2006.
DESAI, C., PATHAK, H., MADAMWAR, D. Advances in molecular and ‘‘-omics’’ technologies to gauge microbial communities and bioremediation at xenobiotic/anthropogen contaminated sites. Bioresour. Technol. 101:1558-1569, 2010.
DUCKWORTH, A. R., AND C. N. BATTERSHILL. Developing farming structures for production of biologically active sponge metabolites. Aquaculture. 217:139-156. 2003.
DUCKWORTH, A., E BATTERSHILL C.N. Sponge aquaculture for the production of biologically active metabolites: the influence of farming protocols and environment. Aquaculture. 221:311-329. 2003.
FIESELER, L., HORN, M., WAGNER, M. E HENTSCHEL, U. Discovery of the novel candidate phylum “Poribacteria” in marine sponges. Appl. Environ. Microbiol. 70, 3724-3732, 2004.
GODZIK, A. Metagenomics and the protein universe. Curr. Opin. Struct. Biol. Jun; 21(3):398-403, 2011.
GURGUI, C., PIEL, J. Metagenomic approaches to identify and isolate bioactive natural products from microbiota of marine sponges. Methods. Mol. Biol. 668:247-64. 2010
HAJDU, E.; PEIXINHO, S. E FERNANDEZ, J.C.C. Esponjas Marinhas da Bahia - Guia de Campo e Laboratório. Série Livros 45. Museu Nacional/UFRJ, Rio de Janeiro. 276 pags, 2011.
HANDELSMAN, J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68: 669–685, 2004.
HANDELSMAN, J., RONDON, M.R., BRADY, S.F., CLARDY, J., GOODMAN, R.M. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chemistry e Biology 5, 245-249, 1998.

65
HARDOIM, C.C., COSTA, R., ARAUJO, F.V., HAJDU, E., PEIXOTO, R. et al. Diversity of bacteria in the marine sponge Aplysina fulva in Brazilian coastal waters. Appl. Environ. Microbiol. 75, 3331-3343, 2009.
HARVELL, C.D.; KIM, K., BURKHOLDER, J.M., COLWELL, R.R., EPSTEIN, P.R., GRIMES, D.J., et al. Emerging marine diseases-climate links and anthropogenic factors. Science. 285: 1505-1510, 1999.
HENTSCHEL, U., J. HOPKE, M. HORN, A. B. FRIEDRICH, M. WAGNER, J. HACKER, E B. S. MOORE. Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 68: 4431-4440, 2002.
HENTSCHEL, U.; PIEL, J.; DEGNAN, S.M., TAYLOR, M.W. Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 30 July, 2012.
HETTICH, R.L., SHARMA, R., CHOUREY, K., GIANNONE, R.J. Microbial metaproteomics: identifying the repertoire of proteins that microorganisms use to compete and cooperate in complex environmental communities. Curr. Opin. Microbiol. Jun, 15(3):373-80, 2012.
HOOPER, J.N.A., VAN SOEST, R.W.M. Systema Porifera: a guide to the classification of sponges. Kluwer Academic/Plenum Publishers, New York. Vol. 2, 1756 p., 2002.
HUGENHOLTZ, P., GOEBEL, B.M., PACE, N.R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180:4765-4774, 1998.
HUYCK, T. K., GRADISHAR, W., MANUGUID, F. KIRKPATRICK, P. Eribulin mesylate. Nature Rev. Drug. Discov. 10, 173–174, 2011.
KENNEDY, J., CODLING, C.E., JONES, B.V., DOBSON, A.D.W., MARCHESI, J.R. Diversity of microbes associated with the marine sponge, Haliclona simulans, isolated from Irish waters and identification of polyketide synthase from the sponge metagenome. Environ. Microbiol. 10(7), 1888-1902, 2008.
KNIEF, C., DELMOTTE, N., VORHOLT J.A. Bacterial adaptation to life in association with plants - a proteomic perspective from culture to in situ conditions. Proteomics. 11:3086-3105. 2011.
KUDO, Y., NAKAJIMA, T., MIYAKI, T., OYAIZU, H. Methanogen flora of paddy soils in Japan. FEMS Microbiol. Ecol. 22: 39-42, 1997.

66
LACERDA, C.M.R., CHOE, L.H., REARDON, K.F. Metaproteomic analysis of a bacterial community response to cadmium exposure. J. Proteome Res. 6:1145-1152, 2007.
LACERDA, C. M., REARDON, K. F. Environmental proteomics: applications of proteome profiling in environmental microbiology and biotechnology. Brief. Funct. Genomic. Proteomic. 8, 75-87, 2009.
LAEMMLI, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227: 680-685. 1970.
LAFFERTY, K.D., PORTER, J.W., AND FORD, S.E. Are diseases increasing in the ocean? Annu. Rev. Ecol. Evol. Syst. 35:31-54, 2004.
LANE, D.J. 16S/23S rRNA sequencing. In: Stackebrandt E. and Goodfellow M. (eds), Nucleic Acid Techniques in Bacterial Systematics. John Wiley and Sons, Chichester, United Kingdom, pp. 115-175, 1991.
LAPORT, M.S., SANTOS, O.C., MURICY, G. Marine sponges: potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol. Jan; 10(1):86-105, 2009.
LAPORT, M.S.; SANTOS, O.C.; MURICY, G. Marine sponges: potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol. Jan: 10(1):86-105, 2009.
LEE, O.O., CHUI, P.Y., WONG, Y.H., PAWLIK, J.R., QIAN, P.-Y. Evidence for vertical transmission of bacterial symbionts from adult to embryo in the Caribbean sponge Svenzeazeai. Appl. Environ. Microbiol. 75: 6147-6156, 2009.
LEE, Y.K.; LEE, J.H.; LEE, H.K. Microbial Symbiosis in Marine Sponges. J. Microbiol. Vol. 39: 4 p.254-264, 2001.
LI Z, H.E.L, MIAO, X. Cultivable bacterial community from South China Sea sponge as revealed by DGGE fingerprinting and 16S rDNA phylogenetic analysis. Curr. Microbiol. Dec; 55(6):465-72, 2007.
LOPEZ-LEGENTIL, S., SONG, B., MCMURRAY, S. E. E PAWLIK, J. R. Bleaching and stress in coral reef ecosystems: hsp70 expression by the giant barrel sponge Xestospongia muta. Mol. Ecol. 17, 1840-1849, 2008.
LOVE G.D., GROSJEAN, E., STALVIES C., FIKEDA GROTZINGER, J.P., BRADLEY, A.S., KELLY, A.E., BATIAM, M., MEREDITH, W., SNAPE C.E., BOWRING S.A., CONDON D.J.,

67
SUMMONS R.E. Fossil steroids record the appearance of Demospongiae during the Cryogenian period. Nature. 457, 718-721, 2009.
LUTER, H.M., WHALAN, S., AND WEBSTER, N.S. Exploring the role of microorganisms in the disease-like syndrome affecting the sponge Ianthella basta. Appl. Environ. Microbiol. 76: 5736-5744, 2010.
MADSEN, E.L. Microorganisms and their roles in fundamental biogeochemical cycles. Curr. Opin. Biotechnol. Jun; 22(3):456-64, 2011.
MARON, P.A., RANJARD, L., MOUGEL, C., LEMANCEAU, P. Metaproteomics: a new approach for studing functional microbial ecology. Microbiol.Ecol., v.53, p.486‐493, 2007.
METZKER, M.L. Sequencing technologies - the next generation. Nat. Rev. Genet. 11(1): 31-46, 2010.
MORAES, F. Esponjas das ilhas oceânicas brasileiras. Série Livros 44, Museu Nacional/UFRJ. Rio de Janeiro. 252 pags, 2011.
MOURA, R.M., QUEIROZ, A.F., FOOK, J.M., DIAS, A.S., MONTEIRO, N.K., RIBEIRO, J.K., MOURA, G.E., MACEDO, L.L., SANTOS, E.A., SALES, M.P. CvL, a lectin from the marine sponge Cliona varians: Isolation, characterization and its effects on pathogenic bacteria and Leishmania promastigotes. Comparative Biochemistry and Physiology - Part A: Molecular e Integrative Physiology. Dec: 145(4): 517-23, 2006.
MUYZER, G., DE WAAL, E.C., e UITTERLINDEN, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695-700, 1993.
MUYZER, G., SMALLA, K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van Leeuwenhoek 73:127-141, 1998.
OLSEN, G.J; LANE, D.J.; GIOVANNONI, S.J.; PACE, N.R. Microbial ecology and evolution: a ribosomal RNA approach. Ann. Rev. Microbiol. 40: 337-365, 1986.
PACE, N.R., STAHL, D.A., LANE, D.J. e OLSEN, G.J. Analyzing natural microbial populations by rRNA sequences. ASM News, 51: 4-12, 1985.

68
PACE, N.R., STAHL, D.A., LANE, D.J. e OLSEN, G.J. The analysis of natural microbial populations by ribosomal RNA sequences. Adv. Microbial Ecol. 9: 1-55, 1986.
PANTILE, R. E WEBSTER, N. Strict thermal threshold identified by quantitative PCR in the sponge Rhopaloeides odorabile. Mar. Ecol. Prog. Ser. 431, 97-105, 2011.
PANTOS, O., AND BYTHELL, J.C. Bacterial community structure associated with white band disease in the elkhorn coral Acropora palmata determined using culture-independent 16S rRNA techniques. Dis. Aquat. Organ. 69: 79–88, 2006.
PANTOS, O., COONEY, R.P., LE TISSIER, M.D., BARER, M.R., O’DONNELL, A.G., AND BYTHELL, J.C. The bacterial ecology of a plague-like disease affecting the Caribbean coral Montastrea annularis. Environ. Microbiol. 5: 370-382, 2003.
PIEL, J. Bacterial symbionts: prospects for the sustainable production of invertebrate-derived pharmaceuticals. Curr. Med. Chem. 13, 39-50, 2006.
PIEL, J. Metabolites from symbiotic bacteria. Nat. Prod. Rep. 26: 338-362, 2009.
PIROVANI, C.P.; CARVALHO, H.A.; MACHADO, R.C.; GOMES, D.S.; ALVIM, F.C.; POMELLA, A.W.; GRAMACHO, K.P.; CASCARDO, J.C.; PEREIRA, G.A.; MICHELI, F. Protein extraction for proteome analysis from cacao leaves and meristems, organs infected by Moniliophthora perniciosa, the causal agent of the witches' broom disease. Electrophoresis. Vol. 11, pp 2391-401, 2008.
PRZESLAWSKI, R.; AHYONG, S.; BYRNE, M.; WORHEIDE, G.; HUTCHINGS, P. Beyond corals and fish: the effects of climate change on non-coral benthic invertebrates of tropical reefs. Global Change Biology. 14:1-23, 2008.
QUEIROZ, A.F., SILVA, R.A., MOURA, R.M., DREYFUSS, J.L., PAREDES-GAMERO, E.J., SOUZA, A.C., TERSARIOL, I.L., SANTOS, E.A., NADER, H.B., JUSTO, G.Z., DE SALES, M.P. Growth inhibitory activity of a novel lectin from Cliona varians against K562 human erythroleukemia cells. Can. Chem. Pharm. May; 63 (6):1023-33, 2009.
RAM, R.J., VERBERKMOES, N.C., THELEN, M.P., TYSON, G.W., BAKER, B.J., BLAKE, R.C. 2ND, SHAH, M., HETTICH, R.L., BANfiELD, J.F. Community proteomics of a natural microbial biofilm. Science. 308:1915-1920, 2005.
REISWIG, H. M. Water transport, respiration and energetics of three tropical marine sponges. J. Exp. Mar. Biol. Ecol. 14: 231-249, 1974.

69
RODRIGUEZ-VALERA, F. Environmental genomics, the big picture? FEMS Microbiol. Lett. 231:153-158, 2004.
RUTZLER, K. Impact of Crustose Clionid Sponges on Caribbean Reef Corals. Acta Geol. Hisp., v. 37, nº 1, p. 61-72, 2002.
SAGAR, S., KAUR, M., MINNEMAN, K.P. Antiviral lead compounds from marine sponges. Mar. Drugs Oct 11;8(10):2619-38, 2010.
SAMBROOK, J. e RUSSELL, D.W. Molecular cloning: A laboratory manual. New York, Cold Spring Harbor Laboratory Press, 2001.
SCHLOSS, P.D., HANDELSMAN, J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol., 71:1501-1506, 2005.
SCHMITT, S., TSAI, P., BELL, J., FROMONT, J., ILAN, M., LINDQUIST, N., PEREZ, T., RODRIGO, A., SCHUPP, P.J., VACELET, J., WEBSTER, N., HENTSCHEL, U., TAYLOR, M.W. Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6, 564-576, 2012.
SEMARH - Secretaria de Meio Ambiente e Recursos Hídricos do Estado da Bahia: Disponível em: http://www.semarh.ba.gov.br/DecretosUnidadesdeConservacao/DECRETO%20Nº%202.184%20DE%2007%20DE%20JUNHO%20DE%201993%20%20Coroa%20Vermelha.pdf.
SIMMONS, T.L., ANDRIANASOLO, K. MCPHAIL, P.M. FLATT, AND GERWICK, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 4:333–342, 2005.
SIMON, C., DANIEL, R. Metagenomic analyses: past and future trends. Appl. Environ. Microbiol. Feb: 77(4):1153-61, 2011.
SOWELL, S.M., ABRAHAM, P.E., SHAH, M., VERBERKMOES, N.C., SMITH, D.P., BAROFSKY, D.F., GIOVANNONI, S.J. Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J. 5:856-865, 2011.
TAMURA, K., D. PETERSON, N. PETERSON, G. STECHER, M. NEI AND S. KUMAR. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol., 28: 2731-2739, 2011.

70
TAYLOR, M.W., RADAX, R., STEGER, D., AND WAGNER, M. Sponge-associated microorganisms: evolution, ecology and biotechnological potential. Microbiol. Mol. Biol. Rev. 71:295-347, 2007.
THOMAS, T., RUSCH, D., DEMAERE, M.Z., YUNG, P.Y., LEWIS, M.,HALPERN, A., et al. Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME Journal 4: 1557-1567, 2010.
THOMPSON, J.D., D.G. HIGGINS AND T.J. GIBSON.CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22: 4673-4680, 1994.
THOMS, C.; SCHUPP, P. J. Chemical defense strategies in sponges: a review. Book: Porifera research: biodiversity, innovation and sustainability. p. 627-637, 2007.
TORSVIK, V.L, OVREAS, L. Microbial diversity and function in soil: from genes to ecosystems. Curr. Opin. Microbiol. 5: 240-245, 2002.
VACELET, J. DONADEY, C.Electron microscope study of the association between some sponges and bacteria.J. Exp. Mar. Biol. Ecol. 30, 301-314 1977.
VERBERKMOES, N.C., RUSSELL, A.L., SHAH, M., GODZIK, A., ROSENQUIST, M., HALFVARSON, J., LEFSRUD, M.G., APAJALAHTI, J., TYSK, C., HETTICH, R.L, JANSSON, J. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 3:179-189, 2009.
WANG, H.B., ZHANG, Z.X., LI, H., HE, H.B., FANG, C.X., ZHANG, A.J., LI, Q.S., CHEN, R.S., GUO, X.K., LIN, H.F et al. Characterization of metaproteomics in crop rhizospheric soil. J. Proteome Res., 10:932-940, 2011.
WEBSTER, N.S., TAYLOR, M.W., BEHNAM, F., LÜCKER, S., RATTEI, T., WHALAN, S., HORN, M., WAGNER, M. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070-2082, 2010.
WEBSTER, N. S., COBB, R. E. NEGRI, A. P. Temperature thresholds for bacterial symbiosis with a sponge. ISME J. 2, 830-842, 2008a.
WEBSTER, N.S., XAVIER, J.R., FRECKELTON, M., MOTTI, C.A., E COBB, R. Shifts in microbial and chemical patterns within the marine sponge Aplysina aerophoba during a disease outbreak. Environ. Microbiol. 10: 3366-3376, 2008b.

71
WEBSTER, N.S.; NEGRI, A.P.; WEBB, R.I. E HILL, R.T. A sponging-boring α-proteobacterium is the etiological agent of disease in the Great Barrier Reef sponge Rhopaloeides odorabile. Mar. Ecol. Prog. Ser. 232: 305-309, 2002.
WEBSTER, N.S.; TAYLOR, M.W. Marine sponges and their microbial symbionts: love and other relationships. Environ. Microbiol. Feb: 14(2):335-46, 2012.
WEBSTER, N.S. Sponge disease: a global threat? Environ Microbiol 9: 1363–1375, 2007.
WIENS, M., KORZHEV, M., PEROVIC-OTTSTADT, S., LUTHRINGER, B., BRANDT, D., KLEIN, S., MÜLLER, W.E. Toll-like receptors are part of the innate immune defense system of ponges (Demospongiae: Porifera). Mol. Biol. Evol. 24, 792-804, 2007.
WILKINSON, C. R., GARRONE, R. E VACELET, J. Marine sponges discriminate between food bacteria and bacterial symbionts: electron microscope radioautography and in situ evidence. Proc. R. Soc. Lond. B 220, 519-528, 1984.
WILMES, P., BOND, P.L. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms.Environ. Microbiol. 6:911-920, 2004.
WILMES, P., WEXLER, M. E BOND, P. L. Metaproteomics provides functional insight into activated sludge wastewater treatment. PLoS ONE 3, e1778, 2008.


















