
Estudo e avaliação do uso de diferentes solventes no...
42
1 ESCOLA POLITÉCNICA DA UNIVERSIDADE DE SÃO PAULO Matheus Mlot Palma Tiago Tura Estudo e avaliação do uso de diferentes solventes no processo de precipitação durante a purificação de DNA plasmidial São Paulo 2014
Transcript of Estudo e avaliação do uso de diferentes solventes no...

1
ESCOLA POLITÉCNICA DA UNIVERSIDADE DE SÃO PAULO
Matheus Mlot Palma
Tiago Tura
Estudo e avaliação do uso de diferentes solventes no processo
de precipitação durante a purificação de DNA plasmidial
São Paulo
2014

2
Matheus Mlot Palma
Tiago Tura
Estudo e avaliação do uso de diferentes solventes no processo
de precipitação durante a purificação de DNA plasmidial
Trabalho de Conclusão de Curso apresentado à Graduação de Engehharia
Química da Escola Politécnica da USP como exigência para obtenção da
aprovação na matéria de TCC II.
Orientador: Prof. Dr. Adriano R. Azzoni
São Paulo
2014

3
AGRADECIMENTOS
“Primeiramente, gostaria de agradecer aos meus pais, Antonio e Esther,
que sempre colocaram minha educação à frente de tudo, me incentivando em
momentos difíceis e me mostrando que sempre vale a pena o esforço máximo e
a dedicação. Gostaria de agradecer ao Prof. Dr. Adriano Azzoni, por toda a
paciência e disponibilidade, estando sempre aberto ao diálogo e pronto para
cumprir a fantástica função que é ensinar, sendo um grande exemplo para mim.
Agradeço à Paula, por todo o incentivo e apoio nesses momentos finais do curso,
além do interesse pelo trabalho, que servia como mais uma motivação para
realizá-lo. Agradeço também a todos os amigos, tanto os que já existiam antes,
em especial Felipe, Guilherme e Khevin, quanto todos aqueles que eu conheci
durante esses cinco anos, que são tantos que dariam um novo Trabalho de
Conclusão de Curso se fosse enumerá-los. Obviamente, ao próprio Tiago, afinal,
sem o trabalho em equipe e a amizade, nada disso seria possível. Por fim,
agradeço à Associação de Engenharia por garantir tantos momentos essenciais
de descontração.”
Matheus Mlot Palma
“Agradeço primeiramente aos meus pais, Eduardo Tura e Rita Maria Silva
Tura que proporcionaram através do esforço de seu trabalho condições para que
eu tivesse uma boa educação. Ao Prof. Dr. Adriano Azzoni pela oportunidade e
pelo conhecimento fornecidos na área de engenharia bioquímica. À Andreza
Safiotti pela paciência e motivação essenciais para conclusão deste trabalho.
Aos meus amigos e companheiros politécnicos sem os quais essa jornada seria
extremamente difícil e monótona. E por fim, agradeço ao meu amigo Matheus
Mlot Palma, pela amizade e trabalho que propiciaram a realização deste
trabalho.”
TiagoTura

4
RESUMO
Os estudos baseados em vacinação gênica têm mostrado diversos avanços nos
últimos anos devido às suas inúmeras vantagens frente aos tratamentos
convencionais. Mais especificamente no caso do uso do DNA plasmidial para tal
fim, os principais benefícios são sua capacidade se replicar automaticamente,
sendo eficientes para carregar o gene terapêutico, além de serem muito mais
seguros do que vetores virais, por exemplo.
A produção do DNA plasmidial pode se dividir em cinco etapas: fermentação,
recuperação primária, recuperação intermediária, purificação final e
empacotamento do produto final. Geralmente, e no caso de interesse, o cultivo
desta molécula é realizada em células de Escherichia Coli, em que 3% em massa
do seu lisado neutralizado é composto de plasmídeos. Decorre disso a
necessidade de estudos de sua purificação. Mais especificamente, na etapa de
precipitação, soma-se o problema do comum uso do isopropanol, que é
agressivo ao meio ambiente e, portanto, há a necessidade do estudo de outras
vias e otimização da etapa.
Portanto, o trabalho realizado envolveu o uso de diferentes solventes a diferentes
concentrações visando uma análise de eficiência e seletividade da precipitação
do pDNA, sendo o RNA a principal impureza a ser eliminada. Foi, então,
realizado o cultivo da E. Coli, além da lise celular. Com esse lisado neutralizado,
precipitações foram realizadas com isopropanol, etanol e acetona e,
posteriormente, foram analisadas as absorbâncias de seus precipitados e
sobrenadantes, além de estudos qualitativos usando eletroforese em gel de
agarose.
Como resultados finais, chegou-se à conclusão de que, para o caso do
isopropanol, houve maior precipitação a 30% em volume de RNA e, a 80%, boa
precipitação de pDNA. No entanto, não pode-se notar boa seletividade ou
eficiência. Para o etanol, até 140% em volume, nota-se a precipitação da maior
parte dos ácidos nucleicos, havendo, portanto, alta eficiência, porém ainda com
baixa seletividade. No caso da acetona, com soluções até 100%, também há boa
eficiência e pouca seletividade, como no segundo caso. Apesar disso, não se
pode encontrar nenhuma condição ótima, apesar da preferência pelo uso de
etanol e acetona por sua eficiência. Além disso, esse solvente também é

5
recomendado devido aos seus custos associados, além das questões
ambientais.

6
SUMÁRIO
AGRADECIMENTOS ................................................................................................................. 3
RESUMO ..................................................................................................................................... 4
LISTA DE FIGURAS ................................................................................................................. 7
1. INTRODUÇÃO .................................................................................................................... 9
2. OBJETIVO ......................................................................................................................... 11
3. REVISÃO BIBLIOGRÁFICA .......................................................................................... 12
3.1. DNA Plasmidial ....................................................................................................... 12
3.2. Terapia Gênica ........................................................................................................ 13
3.3. Produção de pDNA através do cultivo de E. Coli .......................................... 14
3.4. Lise celular ............................................................................................................... 16
3.4.1 Características do lisado neutralizado ..................................................... 18
3.5.1. Clarificação e concentração plasmidial ................................................... 20
3.5.2. Processo Cromatográfico ............................................................................ 20
4. MATERIAIS E MÉTODOS .............................................................................................. 25
4.1. Materiais e Equipamentos .................................................................................... 25
4.2. Metodologia ............................................................................................................. 26
5. RESULTADOS E DISCUSSÃO ..................................................................................... 29
5.1. Dados de cultivo celular ....................................................................................... 29
5.2. Dados relativos às precipitações ....................................................................... 30
7. SUGESTÕES PARA TRABALHOS FUTUROS ......................................................... 40
8. REFERÊNCIAS BIBLIOGRÁFICAS ............................................................................. 41

7
LISTA DE FIGURAS
Figura 1 - Os estágios de desenvolvimento do pDNA. Retirado de Ferreira et. al, 2000
..................................................................................................................................................... 10
Figura 2 - Estruturas topológicas do pDNA. Adaptado de Prazeres et al., 1999 ........... 13
Figura 3 - Fluxograma da produção, concentração e purificação de pDNA. MF:
microfiltração; UF: ultrafiltração; DF: diafiltração e TFF: filtração tangencial. Adaptado
de Ferreira et al., 2000. ........................................................................................................... 16
Figura 4 - Representação da estrutura de endotoxina. Adaptado de Nemalia, 2011. .. 19
Figura 5 - SEC na presença de diferentes sais a diferentes concentrações. Retirado de
Stadler et al., 2004. .................................................................................................................. 22
Figura 6 - Gráfico da absorbância em função do tempo do cultivo de E. Coli. .............. 30
Figura 7 - Absorbância do precipitado A medida pelo espectrofotômetro ...................... 31
Figura 8 - Absorbância do sobrenadante A pelo espectrofotômetro ................................ 31
Figura 9 - Foto da eletroforese realizada com o precipitado de isopropanol .................. 32
Figura 10 - Absorbância do precipitado B pelo espectrofotômetro .................................. 33
Figura 11 - Absorbância do sobrenadante C pelo espectrofotômetro ............................. 34
Figura 12 - Foto da eletroforese realizada com o precipitado de etanol ......................... 34
Figura 13 - Absorbância do precipitado pelo espectrofotômetro ...................................... 36
Figura 14 - Absorbância do sobrenadante pelo espectrofotômetro ................................. 36
Figura 15 - Foto da eletroforese de amostra de precipitado de acetona ......................... 37

8
LISTA DE TABELAS
Tabela 1 - Constituição do lisadao neutralizado, Adaptado de Diogo et al., 2005. ....... 18
Tabela 2 - Quantidades e proporções volumétricas adicionadas de solventes. ............ 27
Tabela 3 - Dados de bsorbância em função do tempo para o cultivo de E. Coli. ......... 29
Tabela 4 - Resultados obtidos pela medição no espectrofotômetro de amostras de
isopropanol ................................................................................................................................ 31
Tabela 5 - Resultados obtidos pela medição no espectrofotômetro de amostras de
etanol .......................................................................................................................................... 33
Tabela 6 - Resultados obtidos pela medição no espectrofotômetro de amostras de
acetona....................................................................................................................................... 35

9
1. INTRODUÇÃO
O interesse nos estudos em terapia e vacinação gênica, ou seja, baseada
no uso de DNA tem crescido notavelmente nos últimos anos. As possibilidades
são inúmeras, visto que através deste novo tratamento, genes inteiros podem
ser inseridos e transcritos em células humana ou não-humanas, obtendo-se
assim a proteína terapêutica desejada. Assim, por exemplo, poderia-se repor
uma proteína com defeito, iniciar o sistema imunológico de modo a eliminar
célular de um tumor ou então imunizar indivíduos de certas doenças.
Além disso, somam-se as vantagens do uso do DNA plasmidial (pDNA)
nesse estudo. Tais moléculas são carregadores extra-cromossômicos de
informação genética e tem a habilidade de se replicar automaticamente e, assim,
são extremamente eficientes para entregar o gene terapêutico na célula alvo, se
destacando como uma alternativa mais fácil e segura de ser produzida se
comparada aos vetores virais, por exemplo.
Mas existem, obviamente, desvantagens no uso do pDNA que não devem
ser deixadas de ser consideradas. Para tal, são necessárias quantidades da
ordem de miligramas, por ser menos efetivo na introdução do ácido nucleico na
célula (estima-se que apenas uma em cada mil moléculas de plasmídio
entregues à célula alcança o núcleo e é, então, expressa) e, portanto, é
necessário o desenvolvimento de um processo em larga escala de manufatura.
Além disso, durante a separação do RNA e do pDNA, etapa importante no
contexto, utiliza-se comumente álcool isopropílico, ambientalmente degradante.
Levando em conta estes pontos negativos, tem-se a motivação deste trabalho,
que busca alternativas no uso do solvente, testando outras possibilidades para
melhorar esta separação, tendo em mente sempre as limitações econômicas e
ambientais.
De forma resumida, podemos descrever a produção e purificação de
pDNA dividindo-a em cinco etapas distintas: fermentação, recuperação primária,
recuperação intermediária, purificação final e empacotamento do produto final.
Todas estas serão posteriormente explicadas com mais detalhes na seção 3 do
trabalho. A figura 1, que se encontra a seguir, ilustra de maneira resumida o
processo que foi estudado.
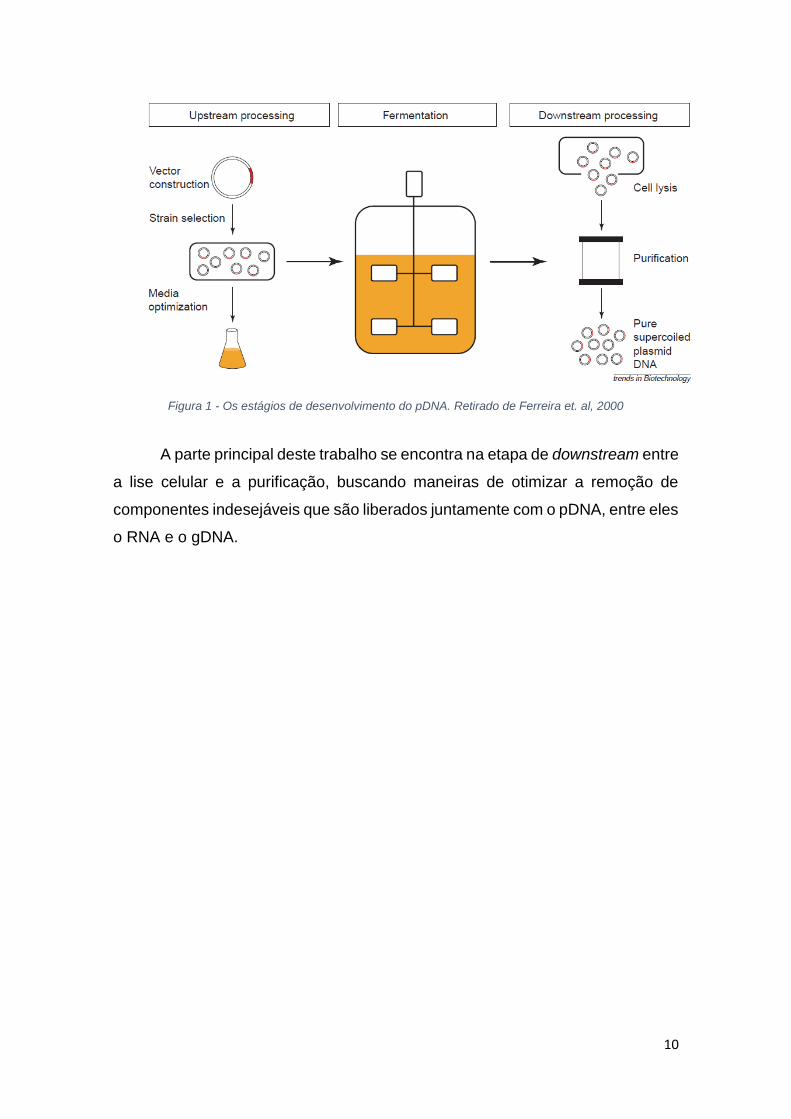
10
Figura 1 - Os estágios de desenvolvimento do pDNA. Retirado de Ferreira et. al, 2000
A parte principal deste trabalho se encontra na etapa de downstream entre
a lise celular e a purificação, buscando maneiras de otimizar a remoção de
componentes indesejáveis que são liberados juntamente com o pDNA, entre eles
o RNA e o gDNA.

11
2. OBJETIVO
Este trabalho tem como principal objetivo estudar os diferentes
comportamentos de precipitação de DNA plasmidial, etapa importante no
processo de purificação deste material e que ocorre após a lise alcalina. Para
tal, foram utilizados diferentes solventes em proporções volumétricas
determinadas e, posteriormente, realizaram-se medidas de espectrofotometria e
eletroforese em gel de modo a permitir análises quantitativas e qualitativas e
avaliação de resultados.

12
3. REVISÃO BIBLIOGRÁFICA
3.1. DNA Plasmidial
Os plasmídeos são moléculas de dupla-fita de DNA, extracromossômicas,
com tamanhos que podem variar de entre 200 a mais de 100.000 pares de base
e são encontradas tanto em células eucarióticas quanto procarióticas. Uma de
suas principais características é a capacidade de se replicar independentemente
do cromossomo de seu hospedeiro e gerarem uma variedade de funções que,
mesmo não sendo essenciais para este organismo, conferem a ele propriedades
de alta adaptabilidade em ambientes distintos. Além disso, há um importante
controle na sua replicação nas bactérias em que está presente para manter uma
coexistência estável com seu hospedeiro e também são igualmente segregados
quando há divisão celular (Schmidt et al., 2001).
Os plasmídeos de interesse (no caso, para aplicação em terapia gênica)
são produzidos geralmente com Escherichia coli como hospedeiro, através de
fermentação, processo que será detalhado na seção 3.3 deste trabalho. É
importante destacar desde já, no entanto, que a maioria dos plasmídeos isolados
desse procarioto são negativamente superenovelados (ccc), com uma estrutura
fina, longa e ramificada, o que garante uma melhor adaptação num papel ativo
na fisiologia celular. Além desse, uma fração menor pode ser encontrada em
uma forma circular aberta (oc), sem enovelamento ou dupla hélice (Prazeres et
al., 1998). Este primeiro modelo, o ccc, apresenta vantagens em relação aos
outros por promover o desenrolamento e separação de fitas complementares
durante a transcrição (Nemailla, 2011). Um esquema simplificado das
possibilidades das estruturas topológicas dos plasmídeos pode ser encontrada
na figura 2. Vale ressaltar que esta preferência por essa forma de enovelamento
ainda não foi completamente justificada ou aceita. Caso esta necessidade seja
provada como irrelevante, um enorme impacto, tanto na operação quanto na
eficiência do processo seria esperado (Prazeres et al., 1999).

13
Figura 2 - Estruturas topológicas do pDNA. Adaptado de Prazeres et al., 1999
.
Os plasmídeos em questão, para a finalidade da vacina gênica, devem
ser produzidos em processos cuja reproducibilidade atenda diversos aspectos
de pureza, eficácia e segurança. Além disso, no caso da Escherichia coli devem
estar separados do DNA genômico (gDNA) (a relação entre os dois é estimada
em <10 ng gDNA/mg pDNA), proteínas deste hospedeiro, RNA e endotoxinas.
Adicionalmente, testes de esterilidade devem ser realizados, eliminando
qualquer presença de fungos e bactérias, que podem interferir nos resultados da
terapia gênica (Diogo et al., 1999).
3.2. Terapia Gênica
A terapia gênica vem apresentando interessantes promessas para
prevenção, tratamento e cura de diversas doenças, tais como cânce e AIDS
(Prazeres et al., 1998). Além disso, vem mostrando interessantes resultados
para veterinária, com alguns biofarmacêuticos já licenciados, como vacinas para
tratamento de melanoma maligno em cães (Gonçalves et al., 2014).
Consequentemente, a demanda por produção de DNA plasmidial, ponto-chave

14
no processo, tem crescido rapidamente em resposta aos avanços alcançados
sobre o tema.
A principal estratégia da terapia gênica é inserir ácidos nucleicos nas
cpelulas de interesse de modo a alterar seu repertório, seja inibindo,
intensificando, iniciando ou reiniciando uma função bioquímica (Ferreira et al.,
2000). Essa entrega gênica pode ser feita através de vetores virais ou não-virais,
sendo a segunda alternativa a de interesse deste trabalho. Os problemas
apresentados pelo uso do vírus como vetor são questões de segurança e,
portanto, os plasmídeos tem se mostrado uma alternativa válida, podendo ser
usados como um produto farmacêutico comum, como por exemplo, através de
injeções musculares (Prazeres et al., 1999). No caso estudado, o DNA plasmidial
contendo o antígeno com as sequêncais codificadas são introduzidas por
diferentes técnicas que permitam a sua expressão in vivo (Reimann et al., 2001).
Em contrapartida aos vetores virais, a entrega gênica realizada por genes
não-virais apresenta aspectos interessantes. Podemos ressaltar, entre eles, a
abrangência na dimensão das moléculas que serão transportadas, em que
diferentes tamanhos de moléculas poderão ser utilizadas em relação aos virais;
eles podem gerados por alguns poucos componentes; além do baixo custo da
produção dos plasmídeos em grande escala (Adler et al., 2010).
No entanto, a produção do pDNA para terapias gênicas apresenta
também suas desvantagens e problemas. Primeiramente, faz-se necessária uma
grande quantidade de plasmídeo em um grau farmacêutico para sua aplicação,
sendo a purificação, portanto, o principal obstáculo do processo, já que o
tratamento provavelmente deverá ser realizado diversas vezes, por causa da
baixa eficiência e duração da expressão do gene no alvo. Acredita-se que entre
0,1 a 0,001% das partículas chegam num estado interessante ao processo, no
núcleo da célula (Adler et al., 2010). Há também riscos envolvendo questões de
pureza, uma vez que certos contaminantes podem induzir a respostas
imunológicas e biológicas (Prazeres et al., 1999).
3.3. Produção de pDNA através do cultivo de E. Coli
Como dito anteriormente, para a produção de pDNA, comumente utiliza-
se a Escherichia Coli transformada com o plasmídio de interesse. Através de

15
um processo fermentativo, a E. Coli é multiplicada até uma concentração celular
de interesse, e o pDNA é replicado autonomamente no meio intracelular
(Nemailla, 2011). As temperaturas geralmente utilizadas para o crescimento
giram em torno de 37 °C. Entretanto, para alguns tipos de pDNA de alto número
de cópias, como os da família pUC, pode-se obter maior quantidade final de
plasmídeos apenas mudando a temperatura de crescimento de 37 °C para 42-
45 °C (Prazeres et al., 1999). Essa mudança de faixa de temperatura favorece a
replicação do pDNA ao mesmo tempo que reduz a taxa de crescimento do
hospedeiro, oque diminui a contaminação, após a lise, por RNA e gDNA
(Prazeres et al., 1999). Realizado o cultivo, as células com os plasmídeos devem
ser recuperadas, por centrifugação ou filtração, etapa esta chamada de
isolamento primário. Após esta primeira separação, são realizadas operações
unitárias com o objetivo de obter plasmídeos que atendam as especificações de
qualidade.
A primeira dessas etapas é a clarificação e concentração. A clarificação
consiste em uma etapa de pré-purificação geralmente conduzida através de
precipitação com sais/solventes de forma a remover debris celulares e impurezas
como proteínas e ácidos nucleicos de baixo peso molecular, ao mesmo tempo
que promove a concentração de pDNA para futuras etapas de purificação
(Ferreira et al., 2000). A etapa de purificação consiste geralmente na utilização
de colunas de cromatografia que exploram as diferenças de propriedades físico-
químicas entre pDNA superenovelados e pDNA desnaturados, DNA genômico,
RNA de alto peso melecular e endotoxinas (Ferreira et al., 2000).
A grande maioria dos plasmídeos obtidos a partir do isolamento de
procariotos como E. Coli encontram-se em uma forma negativamente
superenovelada (sc) que é melhor adaptada às atividades fisiológicas da célula,
havendo também variações de pDNA: forma circular aberta, linear e desnaturada
(Prazeres et al., 1998). A dificuldade na purificação de pDNA está na separação
dos debris celulares, impurezas e de variações de pDNA que podem ser
ineficazes na transferência de genes para a célula (Prazeres et al., 1998).
O plasmídio final deve atender às especificações de DNA genômico (<10
ng gDNA/MG pDNA), proteínas da célula hospedeira (não detectável por BCA
ou em gel de coloração-prata), RNA (não observável em 0,8% em gel de
agarose), e endotoxinas (<0,1 EU/mg pDNA) (Horn et al., 1995). Além disso,

16
testes finais não devem verificar a presença de bactérias e fungos. Para melhor
entendimento do fluxo de processo de pDNA em E. Coli segue a figura 3.
Figura 3 - Fluxograma da produção, concentração e purificação de pDNA. MF: microfiltração; UF:
ultrafiltração; DF: diafiltração e TFF: filtração tangencial. Adaptado de Ferreira et al., 2000.
3.4. Lise celular
Na etapa de lise celular, o método mais utilizado é a de lise alcalina
(Birboim e Doly, 1979) ou variações dela. Este processo foi desenvolvido para
romper as células ao mesmo tempo que causa a desnaturação de gDNA e
proteínas que são posteriormente precipitadas junto com debris celulares e
outras impurezas (Diogo et al., 1999). O sobrenadante desta solução é
precipitado com a ajuda de solventes como isopropanol e etanol, etapa esta que
será focada neste trabalho, e depois resuspendido em solução tampão
apropriada (Diogo et al., 1999).

17
O método da lise alcalina consiste na ruptura das células através do uso
de solução tampão de dodecil sulfato de sódio (SDS) com NaOH em altos
valores de pH. A lise celular libera os plasmídeos e também todos os
componentes da célula hospedeira: parede celular, proteínas, DNA genômico,
RNA, etc. O pDNA liberado sofre desnaturação reversível por conta do alto valor
de ph do meio (Nemailla, 2011). Rompida a célula pela lise alcalina, segue-se a
etapa de neutralização em que uma solução tampão de acetato de potássio é
adicionada causando a precipitação de SDS, componentes celulares e DNA
genômico, que se encontra em fita simples por conta da desnaturação sofrida na
etapa de lise alcalina (Diogo et al., 2005). Esses componentes precipitados
devem ser retirados da solução através de métodos que minimizem as forças de
cisalhamento como a centrifugação em rotor de ângulo fixo, operação esta mais
comumente utilizada em escala laboratorial (Ferreira et al., 2000). Durante as
etapas de lise e neutralização é importante evitar movimentos bruscos de forma
a impedir a clivagem do DNA genômico por cisalhamento pois dessa forma
podem ser gerados fragmentos de gDNA do tamanho do pDNA o que dificultaria
a separação posterior (Freitas, 2007).
A liberação e recuperação de grandes quantidades de pDNA (sc) nesta
etapa são cruciais para obtenção de altas eficiências do processo como um todo
(Ferreira et al., 2000). Um grande desafio para o engenheiro bioquímico é a
extração de grandes quantidades de pDNA da célula sem qualquer danos. A
grande sensibilidade ao cisalhamento do pDNA e do gDNA, bem como a alta
viscosidade do lisado são de grande preocupação durante a ruptura das células
(Ferreira et al., 2000). Estudos sobre a ruptura mecânica de E. Coli revelaram
que apenas dois processos são capazes de gerar pDNA intactos:
microfluidização e moinho de esferas. O estudo mostra que a recuperação de
pDNA atingiu 35% para 65% das células rompidas pelo processo de
microfluidização e 74% de recuperação para 50% das células rompidas
utilizando o moinho de esferas. Do total de pDNA liberado, 80% e 94% estavam
na forma superenovelada (sc) no processo de microfluidização e no de moinho
de esferas respectivamente (Ferreira et al., 2000).
18
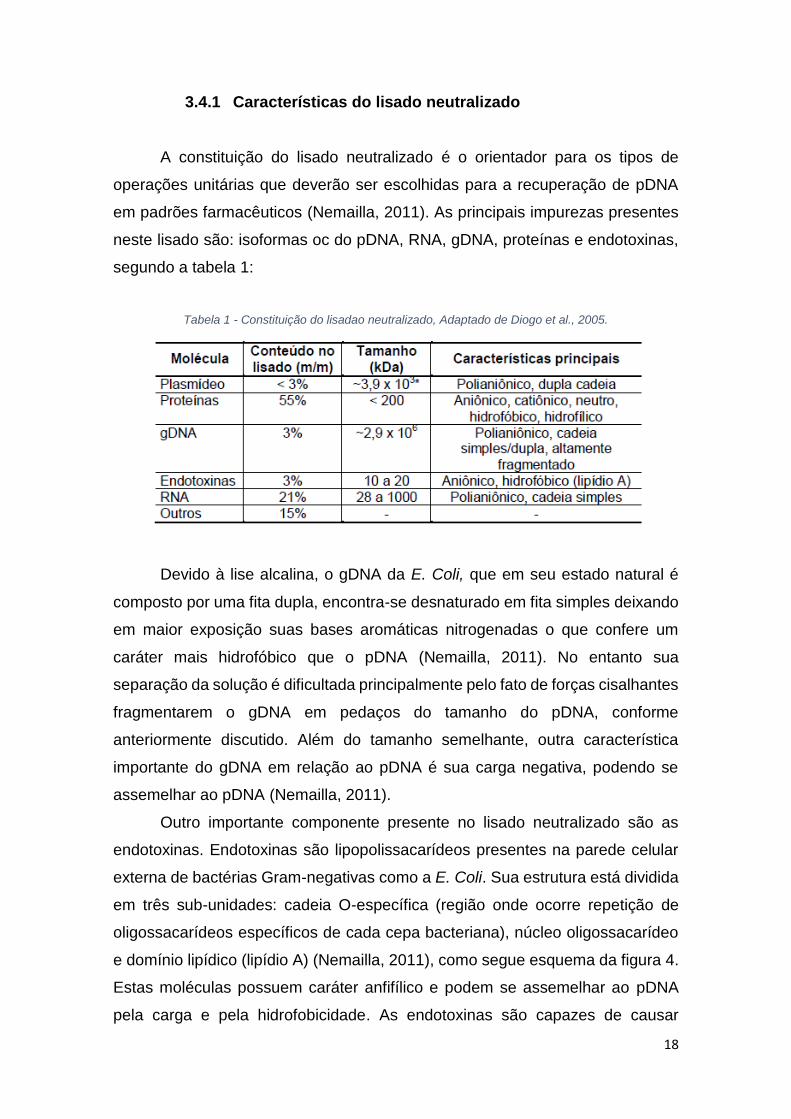
3.4.1 Características do lisado neutralizado
A constituição do lisado neutralizado é o orientador para os tipos de
operações unitárias que deverão ser escolhidas para a recuperação de pDNA
em padrões farmacêuticos (Nemailla, 2011). As principais impurezas presentes
neste lisado são: isoformas oc do pDNA, RNA, gDNA, proteínas e endotoxinas,
segundo a tabela 1:
Tabela 1 - Constituição do lisadao neutralizado, Adaptado de Diogo et al., 2005.
Devido à lise alcalina, o gDNA da E. Coli, que em seu estado natural é
composto por uma fita dupla, encontra-se desnaturado em fita simples deixando
em maior exposição suas bases aromáticas nitrogenadas o que confere um
caráter mais hidrofóbico que o pDNA (Nemailla, 2011). No entanto sua
separação da solução é dificultada principalmente pelo fato de forças cisalhantes
fragmentarem o gDNA em pedaços do tamanho do pDNA, conforme
anteriormente discutido. Além do tamanho semelhante, outra característica
importante do gDNA em relação ao pDNA é sua carga negativa, podendo se
assemelhar ao pDNA (Nemailla, 2011).
Outro importante componente presente no lisado neutralizado são as
endotoxinas. Endotoxinas são lipopolissacarídeos presentes na parede celular
externa de bactérias Gram-negativas como a E. Coli. Sua estrutura está dividida
em três sub-unidades: cadeia O-específica (região onde ocorre repetição de
oligossacarídeos específicos de cada cepa bacteriana), núcleo oligossacarídeo
e domínio lipídico (lipídio A) (Nemailla, 2011), como segue esquema da figura 4.
Estas moléculas possuem caráter anfifílico e podem se assemelhar ao pDNA
pela carga e pela hidrofobicidade. As endotoxinas são capazes de causar

19
respostas pirogênicas em humanos quando em pequenas quantidades e até
choque séptico quando em grandes quantidades, portanto sua remoção a fim de
que seja atendida as especificações farmacêuticas, já discutidas anteriormente,
é crucial (Nemailla, 2011).
A principal impureza do lisado neutralizado são moléculas de RNA, que
são polianiônicas de fita simples. Devido a maior exposição de bases
nitrogenadas em relação ao pDNA, as moléculas de RNA são mais hidrofóbicas
que as de pDNA, mas ainda assim se assemelham bastante em relação à carga
(Nemailla, 2011).
Por último, as proteínas, os compostos mais heterogêneos do lisado
neutralizado, podendo em algumas técnicas de purificação de pDNA em colunas
cromatográficas competir por sítios de adsorção na resina, diminuindo
consideravelmente a capacidade da resina de adsorver a molécula alvo
(Nemaila, 2011).
Figura 4 - Representação da estrutura de endotoxina. Adaptado de Nemalia, 2011.

20
3.5. Purificação do pDNA
3.5.1. Clarificação e concentração plasmidial
O DNA plasmidial representa apenas 2% do total de ácidos nucleicos
presentes no lisado celular obtido e, portanto, grandes quantidades de RNA e
proteínas remanescentes devem ser removidas antes dos próximos passos de
purificação, sendo a primeira substância citada a de principal interesse na
separação durante a clarificação. (Ferreira et al., 2001).
Diversas operações unitárias são conhecidas para tal função, como
precipitações (principal motivação deste trabalho e a mais comumente utilizada)
e filtração tangencial (Nemailla, 2011). Apesar destas operações poderem sofrer
bypass, existem comprovações de que são importantes para uma melhora nos
processos cromatográficos (Prazeres et al., 1999). Os principais solventes
utilizados são isopropanol e etanol, porém outros agentes podem ser usados
para passos específicos de cromatografia (Diogo et al., 1999).
Para a concentração, geralmente é utilizada a precipitação com polietileno
glicol (PEG), com a função de remover pequenos ácidos nucleicos e reduzir o
volume das correntes de processo, etapa necessária para as próximas etapas
cromatográficas (Ferreira et al., 2001).
3.5.2. Processo Cromatográfico
A purificação do DNA plasmidial tradicionalmente utiliza a cromatografia
como técnica escolhida para tal etapa. Nessa parte do processo, a maior parte
das impurezas consiste de RNA, fragmentos de gDNA, endotoxinas e variantes
do plasmídio desejado e devem ser separadas usando a técnica citada. Algumas
características do ácido nucleico envolvido, tais como seu tamanho, carga e
hidrofobicidade são estudadas para aperfeiçoar a interação e seletividade entre
o DNA e a fase sólida. Portanto, as cromatografias por exclusão de tamanho
(Size-exclusion chromatography, SEC), a cromatografia de troca iônica são
alguns dos casos mais utilizados (Ferreira et al., 2001).
A cromatografia de exclusão de tamanho vem apresentando resultados
extremamente satisfatórios para a separação de RNA, endotoxinas e proteínas

21
do plasmídio (Ferreira et al., 2001). Basicamente, nesta técnica, ácidos nucleicos
de maior tamanho ficam retidos por menos tempo na coluna, devido a sua
dificuldade de penetração na fase estacionária se comparada com moléculas
menores (Ferreira et al., 2000). No entanto, etapas de clarificação e
concentração pré-SEC precisam ser realizadas para aumentar a seletividade da
operação (Ferreira et al., 2001).
Um dos parâmetros operacionais mais importantes no que diz respeito ao
controle da SEC é a vazão. Tanto fluxos muito altos quanto muito baixos podem
levar a baixas resoluções de macromoléculas. Apesar de muitas vezes esta
variável ter um valor sugerida pelos fornecedores, é de extrema importância
realizar uma otimização própria para o caso em estudo (Ferreira et al., 2001).
A presença de sais a diferentes concentrações também influencia
drasticamente a separação de pDNA em função do RNA na SEC. A figura 5 a
seguir apresenta três casos diferentes de uso desses sais. Temos,
respectivamente, o uso de 0,15M e 0,50M de NaCl e também presença de 2M
de (NH4)2SO4, mostrando os diferentes comportamentos de separação para
cada caso.
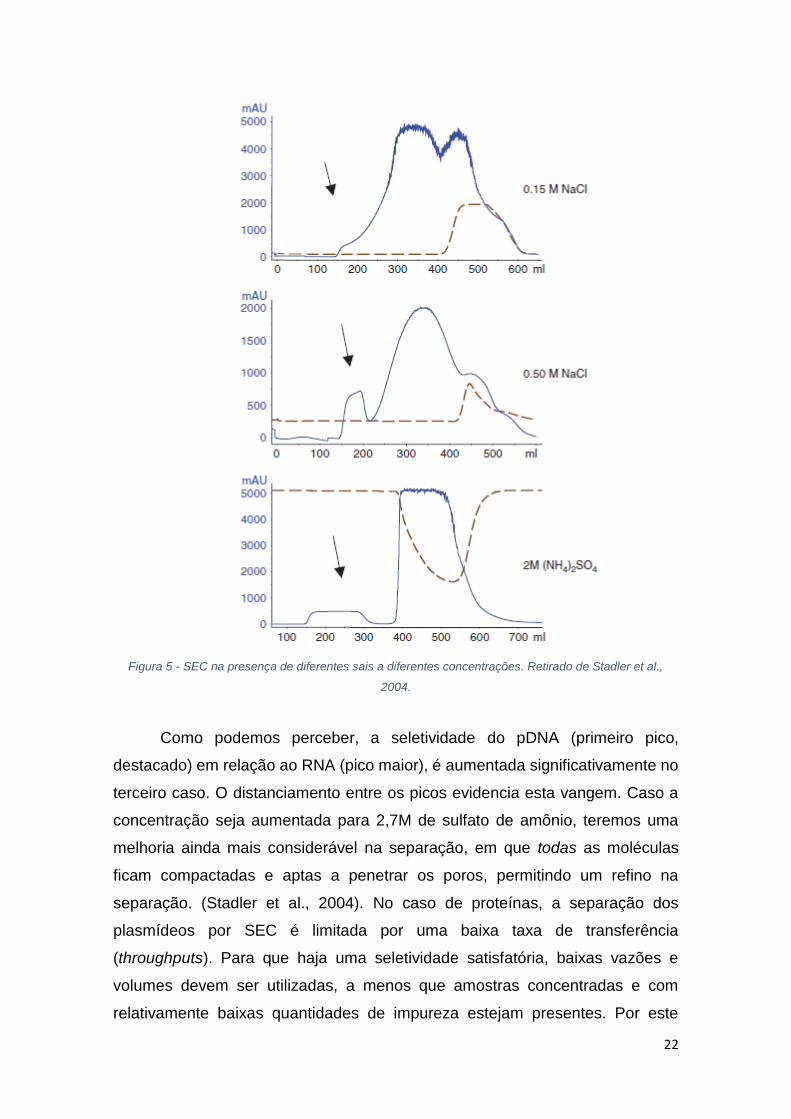
22
Figura 5 - SEC na presença de diferentes sais a diferentes concentrações. Retirado de Stadler et al.,
2004.
Como podemos perceber, a seletividade do pDNA (primeiro pico,
destacado) em relação ao RNA (pico maior), é aumentada significativamente no
terceiro caso. O distanciamento entre os picos evidencia esta vangem. Caso a
concentração seja aumentada para 2,7M de sulfato de amônio, teremos uma
melhoria ainda mais considerável na separação, em que todas as moléculas
ficam compactadas e aptas a penetrar os poros, permitindo um refino na
separação. (Stadler et al., 2004). No caso de proteínas, a separação dos
plasmídeos por SEC é limitada por uma baixa taxa de transferência
(throughputs). Para que haja uma seletividade satisfatória, baixas vazões e
volumes devem ser utilizadas, a menos que amostras concentradas e com
relativamente baixas quantidades de impureza estejam presentes. Por este

23
motivo, a cromatografia por exclusão de tamanho deve ser um dos últimos
passos de downstream da produção do plasmídio (Ferreira et al., 2000)
No caso da cromatografia de troca iônica, uma especificação deste
método é utilizado, em que a fase estacionária contem carga positiva e, deste
modo, retem as cargas negativas do produto a ser separado. Esse processo é
conhecido como cromatografia de troca aniônica e é amplamente utilizado para
a obtenção de plasmídeos (Ferreira et al., 2001, 1). A estrutura poliônica dos
ácidos nucleicos permite uma exploração do uso deste método (Ferreira et al.,
2000).
A princípio, a cromatografia de troca aniônica fornece boa separação
entre o pDNA e outros componentes, tais como RNA, proteínas residuais e
endotoxinas. No entanto, essa eficiência varia consideravelmente de acordo com
a composição da amostra que está sendo tratada, de acordo com sua origem e
outros tratamentos prévios (Stadler et al., 2004). Sugere-se que sejam realizadas
as etapas de clarificação e concentração discutidas na seção 3.5.1 deste
trabalho para aumentar a capacidade da coluna, apesar de que, se passarem
por bypass, ou seja, indo diretamente da lise celular para a cromatografia de
troca iônica, amostras de idêntica pureza podem ser alcançadas (Ferreira et al.,
2001).
A presença de uma carga negativa por base de ácido nucleico causa uma
interação com a fase móvel de modo que as maiores moléculas sejam eluídas a
partir de forças iônicas maiores (Ferreira et al., 2001). Do mesmo modo, apesar
de altos fluxos de fase móvel não interferirem na resolução da cromatografia, um
menor tempo de residência pode ser atribuído a tal situação, o qual é controlado
por difusividade, viscosidade da amostra e tamanho de partícula e a perda de
pDNA pode ser atribuída a essas elevadas vazões (Ferreira et al., 2000, 5). Outro
fator que pode influenciar nesta técnica são variações de cátions utilizados. Caso
seja mantido o cloreto como ânion, foi observado que cátions menores eluem os
ácidos nucleicos a menores forças iônicas (Ferreira et al., 2001).
Uma das maores limitações no processo de cromatografia de troca
aniônica é a baixa capacidade da grande maioria dos adsorbentes encontrados
comercialmente. Para que grandes quantidades de plasmídio sejam adsorvidas,
são necessárias maiores colunas com menores capacidades. Sabe-se também
que esta capacidade depende do tamanho da partícula do adsorvente utilizado,

24
como já foi estudado, por exemplo, o aumento deste parâmetro para Q-
sepharoes XL, alterando- ão
de 5,3 mg/ml para 3,4 mg/ml (Ferreira et al., 2001).

25
4. MATERIAIS E MÉTODOS
Nessa seção do trabalho serão detalhados todos os equipamentos e
substâncias utilizados, bem como suas purezas e fornecedores. Posteriormente
serão explicadas o modo como todas as técnicas foram utilizadas.
4.1. Materiais e Equipamentos
O meio utilizado para a fermentação das bactérias foi o meio LB (1 L)
composto por triptona (10 g/L), extrato de levedura (5 g/L) e NaCl (5 g/L).
Triptona e extrato de levedura do fornecedor Acumedia, sem especificação de
pureza. Para a medida das quantidades utilizaram-se duas balanças de
precisão: Mettler Toledo e Marte AS 2000. Para a esterilização, utilizou-se a
Autoclave Vertical Phoenix. Após esterilização, o processo fermentativo ocorreu
utilizando bactérias E. Coli DH5α já transformadas com o plasmídio pVAX1GFP
(3697 pb). O antibiótico usado no crescimento foi a canamicina. O cultivo das
células ocorreu na incubadora C24 incubator shaker da New Brunswick
Scientific. Durante o cultivo foi utilizado também para efeito de
acompanhamento, um espectrofotômetro analítico AJX-1000 da Micronal.
Para a lise celular e neutralização do lisado, foram realizados 100 mL de 3
soluções tampão: solução de tris (10 mmol/L) da Sigma e de pureza >99,9%,
glicose (50 mmol/L) da Merck, sem especificação de pureza, e EDTA (10
mmol/L) da Promega; solução de acetato de potássio (3 mol/L) com o acetato de
pureza >99,95 da Sigma e água mili-Q do equipamento de purificação de águas
Millipore; e solução de dodecil sulfato de sódio (SDS) a 1% com NaOH (200
mmol/L). Foi utilizado também ácido acético de pureza 100% da Emsure e uma
centrífuga para volumes maiores Z326K da Hermle. As medidas de ph das
soluções foi realizada com o phmetro digital da Quimis.
Para a parte das precipitações utilizou-se 3 solventes: Isopropanol (2-
propanol) de pureza >99,8% da Emsure; etanol de pureza >99,8% da Emsure e
acetona de pureza >99,5% da Spectrum. As concentrações utilizadas serão
discutidas na metodologia. Os equipamentos utilizados foram o
espectrofotômetro para pequenos volumes Nanodrop 2000 Spectrophotometer
e a centrífuga para pequenos volumes 5415 R da Eppendorf.

26
Para a eletroforese, foi utilizado um gel de agarose da Invitrogen (0,4 g)
dissolvido em 50 ml de solução TAE 1X. Para dissolução, utilizou-se microondas
da Brastemp. A eletroforese foi conduzida, com auxílio do corante Gel loading
DYE blue (6x) da Biolabs, em suporte HE33 mini horizontal submarine unit da
Amesham. O tratamento do gel após a eletroforese foi realizado com solução
SYBR Safe DNA Gel Stain (10.000x concentrada em DMSO) da Invotrogen. O
gel com as bandas de DNA foi analisado no equipamento Gel Logic 212 PRO
Carestream da Analítica utilizando o software específico do aparelho.
4.2. Metodologia
Preparado o meio de cultura LB com os componentes já especificados na
seção de materiais (triptona, extrato de levedura e NaCl) o mesmo foi esterilizado
em autoclave a 121 °C por período de tempo suficiente, segundo especificações
do equipamento. Esterilizado o meio, o mesmo foi armazenado em refrigerador
à 4°C. Para o inóculo, utilizou-se 40 μL de solução de células E. Coli juntamente
com 5 mL da solução do meio LB em um frasco pequeno que foi resfriado a 4 °C
pelo período de 16h, a fim de reduzir a fase LAG do crescimento das células.
No dia seguinte, o inóculo foi adicionado a 0,5 L da solução de meio LB
juntamente com o antibiótico canamicina a uma concentração de 30 mg/mL. O
crescimento foi realizado por 8h na incubadora à 37 °C e à uma agitação de 250
rpm. A cada meia hora, foram retiradas amostras do cultivo a fim de se obter a
curva de crescimento das células, identificando o fim da fase exponencial onde
o cultivo seria finalizado, através da medida de absorbância pelo
espectrofotômetro AJX-1000 (λ = 600 nm). Finalizado o cultivo, as células foram
recuperadas por centrifugação à 6000g e 8 °C por 10 min. Os dados de
crescimento celular, comprovando o atingimento do final da fase de exponencial,
se encontram na seção de resultados deste trabalho.
A etapa de lise celular foi conduzida ressuspendendo o pellet de células
em 16 ml de solução tampão de Tris-HCl seguida da adição de 16 ml de solução
tampão de SDS 1% e NaOH. A solução foi deixada parada em temperatura
ambiente por 10 min ocorrendo neste momento a primeira precipitação. Após
esse períodode tempo, o lisado que se encontra alcalino, foi neutralizado com a
adição de 16 ml de solução tampão de acetato de potássio a 3 M e o pH corrigido
27
para 5,5 com a adição de ácido acético glacial previamente resfriado a 0 °C. A
solução foi mantida por 10 min a 4 °C. Seguiu-se com a centrifugação a 12.000g
a 4 °C por 50min. O sobrenadante foi armazenado a -70 °C e o precipitado
obtido, descartado.
Para as precipitações com solventes (iso-propanol, etanol, acetona) foram
utilizadas 6 amostras do lisado neutralizado, cada uma contendo 0,5 ml. Para
cada amostra foi adicionada uma quantidade de solvente proporcional ao volume
de 0,5 ml. Seguem as tabelas das quantidades adicionadas de cada tipo de
solvente abaixo:
Tabela 2 - Quantidades e proporções volumétricas adicionadas de solventes.
Solução 30% 50% 60% 70% 80% 100%
Vol. Isop.
(mL)
0,15 0,25 0,3 0,35 0,4 0,5
Solução 100% 120% 140% 160% 180% 200%
Vol. EtOH
(mL)
0,5 0,6 0,7 0,8 0,9 1
Solução 40% 70% 100% 130% 160% 200%
Vol. Acet.
(mL)
0,2 0,35 0,5 0,65 0,8 1
Após a adição de cada tipo de solventes às amostras, esperou-se por 1h
para que ocorresse a precipitação do pDNA, teoricamente. Precipitado o
plasmídio, seguiu-se a etapa de centrifugação, na centrifuga pequena 5415 R, a
12.000g e 4 °C por 10 min. O sobrenadante foi separado em outros 6 frascos
pequenos e o precipitado ressuspendido em 100 μL de água mili-Q. Assim, para
cada solvente, foram obtidos 12 frascos de amostra, 6 com sobrenadante e 6
com o precipitado que a princípio seria o pDNA. Para as 12 amostras, foi
analisado a absorbância no espectrofotômetro de DNA Nanodrop 2000 em
comprimentos de onda de λ= 260 nm e de λ= 280 nm. Para calibração do
instrumento foi utilizado como “branco” água destilada. Os resultados obtidos, de
concentração de ácidos nucleicos, RNA, etc. serão apresentados e discutidos na
próxima seção.
A etapa seguinte foi a de eletroforese dos precipitados de cada solvente.
Para a formação do gel, utilizou-se 0,4 g para 50 mL de solução TAE 1x. Para

28
que toda a agarose fosse dissolvida utilizou-se o microondas, aquecendoa
amostra durante 1,5 min e parando a cada 15 segunda para agitação e para
evitar aquecimento excessivo. O resfriamento foi feito em água corrente de forma
suave a fim de evitar a precipitação da agarose. Parte do volume da amostra é
perdido devido ao aquecimento, sendo assim, foi completado o volume perdido
com água mili-Q. A solução foi deixada no molde por 20 min para resfriar e formar
o gel.
Enquanto o gel era formado, de cada uma das 6 amostras de precipitado
de cada solvente (100 μl de amostra por precipitado) foi retirado 20 μL e
transferidos para frasco apropriado. Em cada frasco foi adicionado o volume de
3,30 μL (1/6 da solução da amostra) de corante azul e misturado até atingir
homogeinidade. Para o controle, foi utilizado 20 μL de lisado neutralizado
também com a adição de 3,30 μL de corante. Preparado o gel, o mesmo foi
introduzido no equipamento para realizar a eletroforese e o volume completado
com solução TAE 1x até que o gel ficasse submerso. Em cada espaço do gel,
foi introduzido primeiro a solução controle, isto é, o lisado celular sem passar
pela precipitação com nenhum solvente, e depois as demais soluções de
precipitado em ordem crescente de proporção volumétrica. A eletroforese foi
conduzida a uma voltagem de 80 V pois a 60 V verificou-se que a velocidade de
deslocamento do pDNA era muito baixa. Finalizada a eletroforese, o gel foi
retirado do suporte e colocado em solução de SYBR Safe DNA por 20 min em
local sem iluminação e depois transferido para água destilada e deixado também
por 20 min em lucal sem iluminação. Após esse período, o gelfoi analisado no
equipamento Gel Logic 212 Pro para registrar imagem UV do gel e do
deslocamento do precipitado. As imagens e análises obtidas serão apresentadas
e discutidas na próxima seção.

29
5. RESULTADOS E DISCUSSÃO
5.1. Dados de cultivo celular
Como citado na seção anterior deste relatório, foi realizado o cultivo
celular da Escherichia Coli de modo que fosse possível obter seu lisado e,
consequentemente, realizar as etapas de precipitação. A seguir encontramos, os
dados referentes ao crescimento celular, com sua absorbância e respectivo
intervalo de tempo, até que se atingisse o fim da fase exponencial.
Tabela 3 - Dados de bsorbância em função do tempo para o cultivo de E. Coli.
Hora Tempo Abs(λ=600nm)
08:37 00:00 0,010
08:57 00:20 0,010
09:17 00:40 0,008
09:37 01:00 0,012
09:57 01:20 0,010
10:17 01:40 0,014
10:37 02:00 0,014
11:07 02:30 0,017
11:37 03:00 0,023
12:07 03:30 0,026
12:37 04:00 0,038
14:07 05:30 0,078
14:37 06:00 0,128
15:07 06:30 0,202
15:37 07:00 0,331
16:07 07:30 0,444
16:37 08:00 0,605
17:07 08:30 0,731

30
Figura 6 - Gráfico da absorbância em função do tempo do cultivo de E. Coli.
Como podemos notar, por volta de 8h30 após o início do crescimento,
atingiu-se o fim da fase exponencial.
5.2. Dados relativos às precipitações
Para interpretação dos resultados, segue a legenda abaixo. Os índices
variam de 1 a 6 indicando as 6 amostras em que foi adicionado diferentes
proporções volumétricas de solvente.
S – Sobrenadante A – Isopropanol
P – Precipitado B – Etanol
C – Acetona
Como discutido anteriormente, após precipitação, seguiu-se análise de
absorbância do precipitado e do sobrenadante pelo espectrofotômetro Nanodrop
2000. Os resultados obtidos para o isopropanol são apresentados abaixo
juntamente com a discussão:
0,000
0,100
0,200
0,300
0,400
0,500
0,600
0,700
0,800
00:00 01:12 02:24 03:36 04:48 06:00 07:12 08:24 09:36
Ab
sorb
ânci
a (n
m)
Tempo (h)
Absorbância em função do tempo do cultivo de E. Coli

31
Tabela 4 - Resultados obtidos pela medição no espectrofotômetro de amostras de isopropanol
Figura 7 - Absorbância do precipitado A medida pelo espectrofotômetro
Figura 8 - Absorbância do sobrenadante A pelo espectrofotômetro
% Solv. Vol. Solv. Conc. (ng/μl) A260 A280 260/280 260/230
SA1 30 0,15 430 8,6 4,253 2,02 1,07
SA2 50 0,25 412 8,239 4,914 1,68 1
SA3 60 0,3 358,7 7,175 4,241 1,69 0,97
SA4 70 0,35 294,6 5,892 3,706 1,59 0,9
SA5 80 0,4 256,3 5,125 3,236 1,58 0,79
SA6 100 0,5 153,3 3,066 1,82 1,68 0,64
% Solv. Vol. Solv. Conc. (ng/μl) A260 A280 260/280 260/230
PA1 30 0,15 95,4 1,908 0,966 1,97 1,29
PA2 50 0,25 181,6 3,633 1,7 2,14 1,92
PA3 60 0,3 129 2,58 1,233 2,09 1,61
PA4 70 0,35 156,1 3,122 1,488 2,1 1,62
PA5 80 0,4 269,3 5,387 2,583 2,09 1,78
PA6 100 0,5 204,1 4,081 2,032 2,01 1,56

32
Figura 9 - Foto da eletroforese realizada com o precipitado de isopropanol
A análise dos resultados do sobrenadante indica que, de acordo com a
tabela 3, conforme aumenta-se a proporção de isopropanol, a concentração de
ácidos nucleicos diminui, o que é verificado pela figura 7, ocorrendo então
precipitação de material genético. A diminuição da absorbância a 260 nm indica
precipitação de RNA e p-DNA. Analisando o precipitado, é verificado que a
concentração de material genético tende a aumentar, mas no geral, não há
seletividade entre precipitar RNA e p-DNA de acordo com a eletroforese
realizada, o que é verificado também pelo aumento de absorbância a 260 e 280
nm. A análise da relação 260/280 mostra que quanto mais próximo de 2 a
presença de ácidos nucleicos é verificada, o que ocorre no precipitado.
Para a solução de 30% isopropanol, percebe-se maior precipitação de
RNA e menor precipitação de p-DNA, de acordo com a figura 8 da eletroforese.
Para a solução de 80% isopropanol, a precipitação de p-DNA é ótima, de acordo
com a análise da tabela 3 onde há aumento da concentração e da absorbância
a 260 e 280 nm, o que está de acordo com o encontrado na literatura. Deve
30% 50% 60% 70% 80% 100%% Lisado
33
atentar ao fato de que a análise de absorbância do precipitado pode conter
interferência por conta da diluição.
Abaixo seguem os resultados obtidos da precipitação com etanol:
Tabela 5 - Resultados obtidos pela medição no espectrofotômetro de amostras de etanol
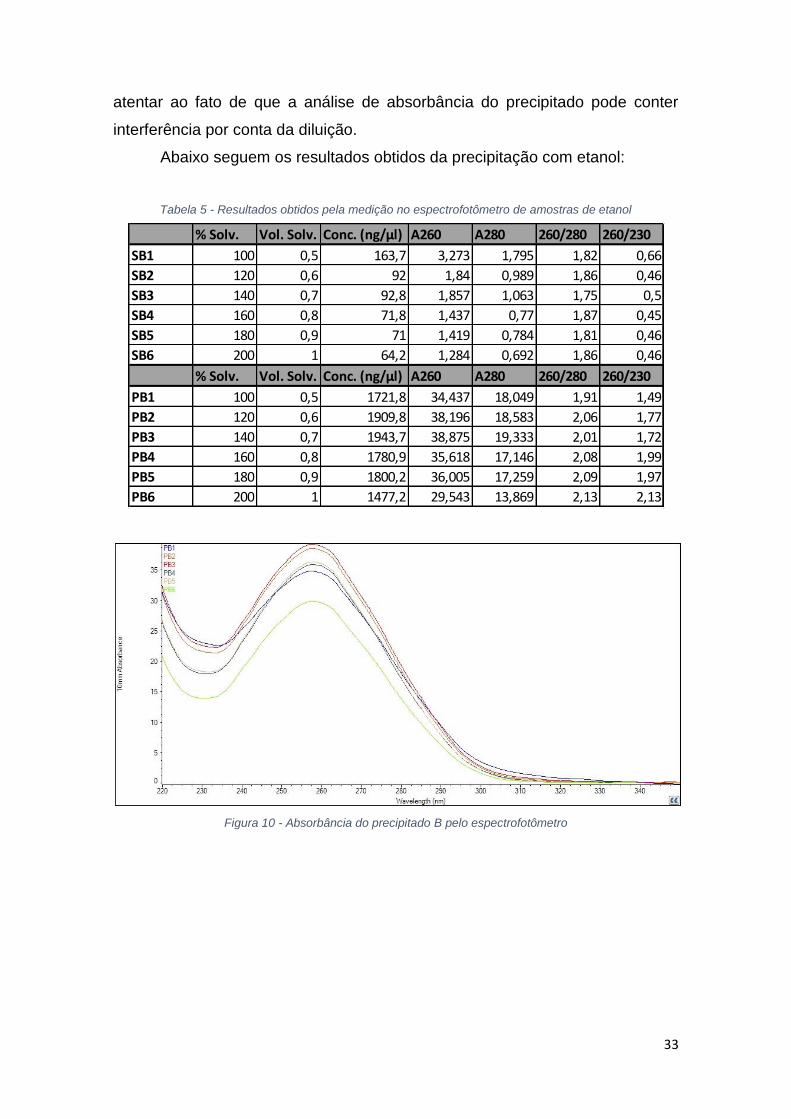
Figura 10 - Absorbância do precipitado B pelo espectrofotômetro
% Solv. Vol. Solv. Conc. (ng/μl) A260 A280 260/280 260/230
SB1 100 0,5 163,7 3,273 1,795 1,82 0,66
SB2 120 0,6 92 1,84 0,989 1,86 0,46
SB3 140 0,7 92,8 1,857 1,063 1,75 0,5
SB4 160 0,8 71,8 1,437 0,77 1,87 0,45
SB5 180 0,9 71 1,419 0,784 1,81 0,46
SB6 200 1 64,2 1,284 0,692 1,86 0,46
% Solv. Vol. Solv. Conc. (ng/μl) A260 A280 260/280 260/230
PB1 100 0,5 1721,8 34,437 18,049 1,91 1,49
PB2 120 0,6 1909,8 38,196 18,583 2,06 1,77
PB3 140 0,7 1943,7 38,875 19,333 2,01 1,72
PB4 160 0,8 1780,9 35,618 17,146 2,08 1,99
PB5 180 0,9 1800,2 36,005 17,259 2,09 1,97
PB6 200 1 1477,2 29,543 13,869 2,13 2,13
34
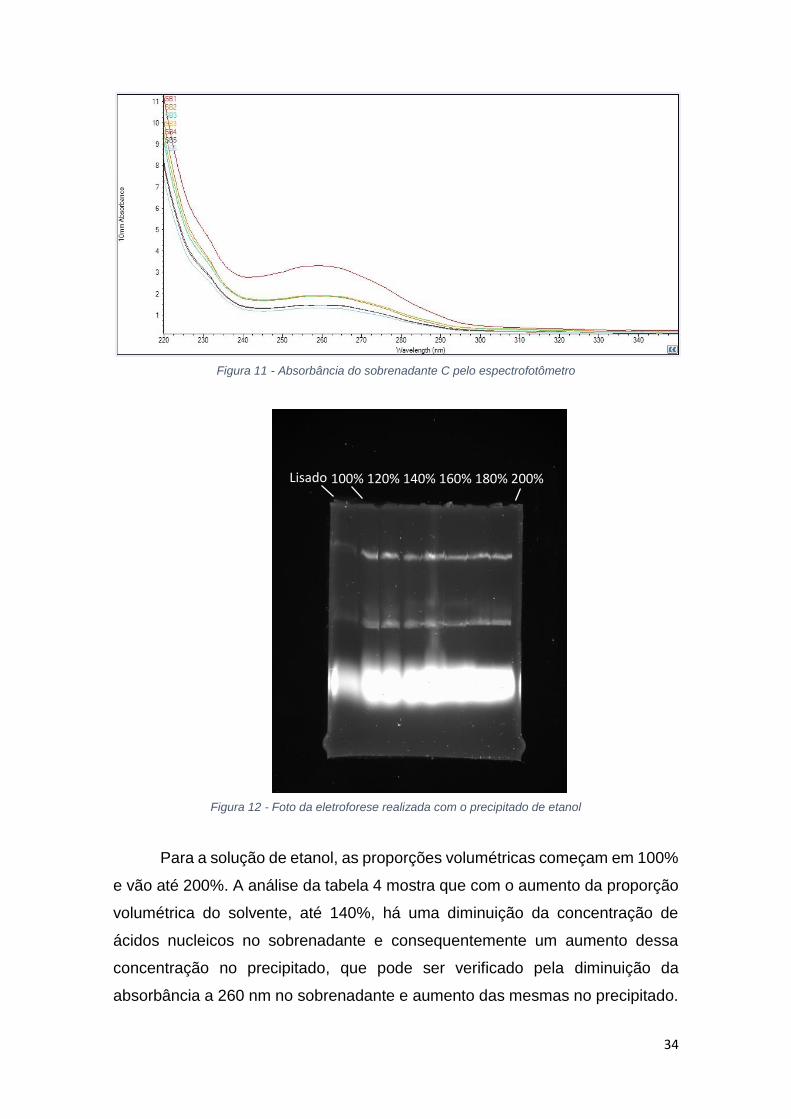
Figura 11 - Absorbância do sobrenadante C pelo espectrofotômetro
Figura 12 - Foto da eletroforese realizada com o precipitado de etanol
Para a solução de etanol, as proporções volumétricas começam em 100%
e vão até 200%. A análise da tabela 4 mostra que com o aumento da proporção
volumétrica do solvente, até 140%, há uma diminuição da concentração de
ácidos nucleicos no sobrenadante e consequentemente um aumento dessa
concentração no precipitado, que pode ser verificado pela diminuição da
absorbância a 260 nm no sobrenadante e aumento das mesmas no precipitado.
100% 120% 140% 160% 180% 200% Lisado

35
As figuras 9 e 10 mostram a absorbância das amostras de precipitado e
sobrenadante, entretanto, para o precipitado há interferência devido a diluição
das amostras. Pela análise dos dados, grande parte do material genético é
precipitado até a solução de 140% de etanol, acima deste valor ocorre
divergência dos dados.
A relação 260/280 na tabela 4 mostra que no sobrenadante ainda há a
presença de proteínas e no precipitado, como está próximo de 2, há a presença
de material genético. Assim como no isopropanol, pela análise da figura 11 da
eletroforese do precipitado de etanol, a precipitação foi eficiente, porém não
seletiva.
Abaixo seguem os dados referentes as precipitações com acetona:
Tabela 6 - Resultados obtidos pela medição no espectrofotômetro de amostras de acetona
% Solv. Vol. Solv. Conc. (ng/μl) A260 A280 260/280 260/230
SC1 40 0,2 1072,1 21,442 16,818 1,27 1,5
SC2 70 0,35 839,7 16,793 13,698 1,23 1,06
SC3 100 0,5 803,2 16,065 12,935 1,24 1,05
SC4 130 0,65 783,9 15,678 12,502 1,25 1,03
SC5 160 0,8 652 13,041 9,696 1,34 0,8
SC6 200 1 628,9 12,578 9,451 1,33 0,77
% Solv. Vol. Solv. Conc. (ng/μl) A260 A280 260/280 260/230
PC1 40 0,2 113,4 2,269 1,252 1,81 1,9
PC2 70 0,35 1251,9 25,038 12,354 2,03 2,32
PC3 100 0,5 1655,2 33,104 16,022 2,07 2,22
PC4 130 0,65 1600,7 32,015 15,959 2,01 2,17
PC5 160 0,8 1680 33,6 17,529 1,92 2
PC6 200 1 997,8 19,955 9,693 2,06 2,28
36
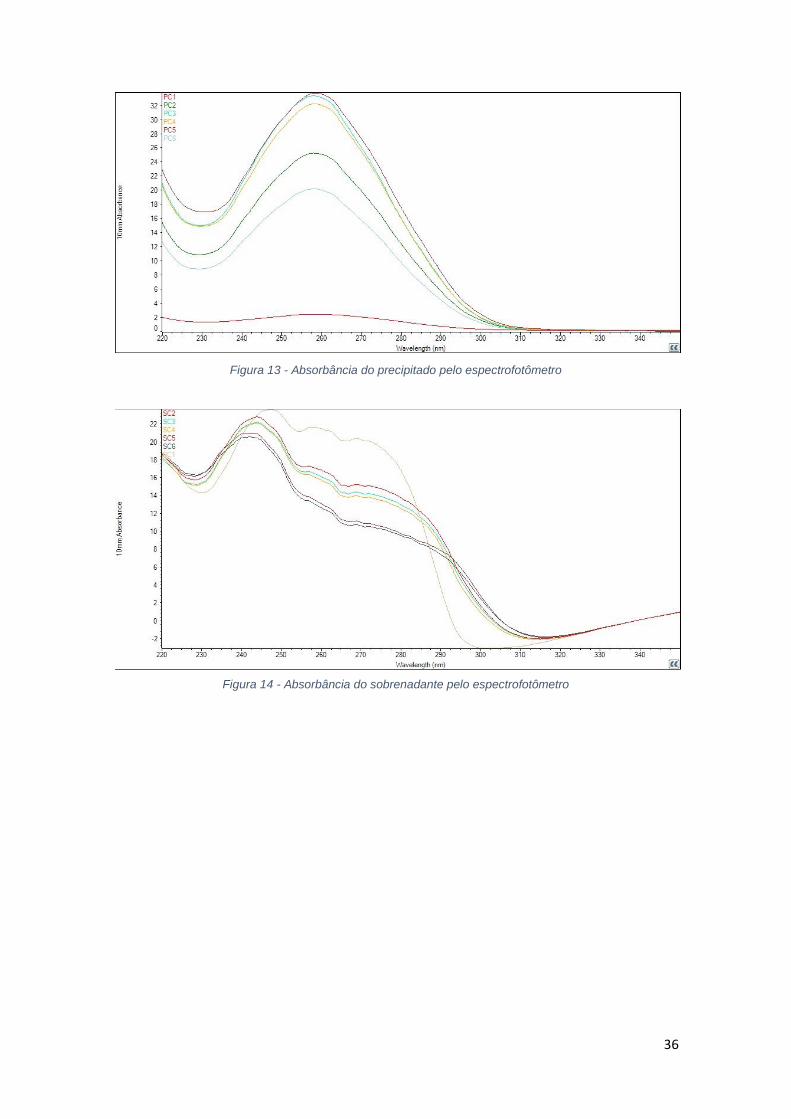
Figura 13 - Absorbância do precipitado pelo espectrofotômetro
Figura 14 - Absorbância do sobrenadante pelo espectrofotômetro
37
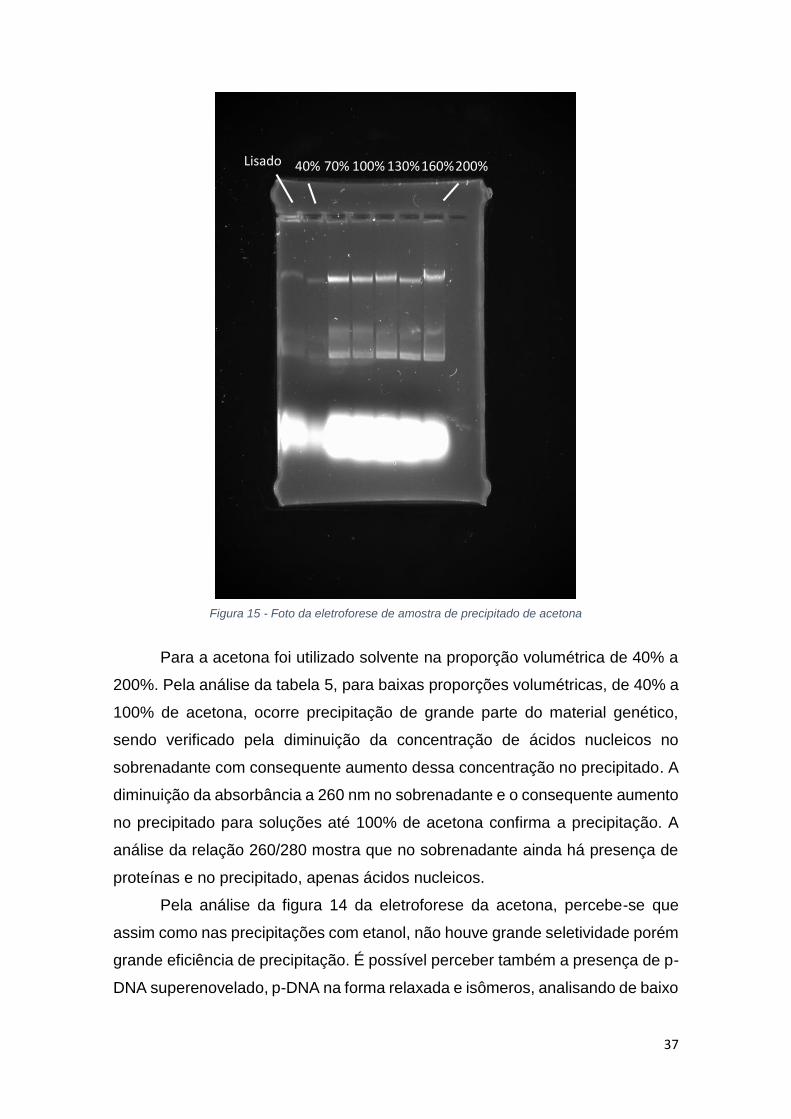
Figura 15 - Foto da eletroforese de amostra de precipitado de acetona
Para a acetona foi utilizado solvente na proporção volumétrica de 40% a
200%. Pela análise da tabela 5, para baixas proporções volumétricas, de 40% a
100% de acetona, ocorre precipitação de grande parte do material genético,
sendo verificado pela diminuição da concentração de ácidos nucleicos no
sobrenadante com consequente aumento dessa concentração no precipitado. A
diminuição da absorbância a 260 nm no sobrenadante e o consequente aumento
no precipitado para soluções até 100% de acetona confirma a precipitação. A
análise da relação 260/280 mostra que no sobrenadante ainda há presença de
proteínas e no precipitado, apenas ácidos nucleicos.
Pela análise da figura 14 da eletroforese da acetona, percebe-se que
assim como nas precipitações com etanol, não houve grande seletividade porém
grande eficiência de precipitação. É possível perceber também a presença de p-
DNA superenovelado, p-DNA na forma relaxada e isômeros, analisando de baixo
40% 70% 100% 130% 160% 200% Lisado

38
para cima a figura, respectivamente. Mais uma vez, a figura 12 que mostra a
absorbância do precipitado nas amostras de acetona apresenta interferência
devido a diluição das amostras.

39
6. CONCLUSÃO
Para os ensaios realizados com isopropanol, para a solução com 30% em
volume ocorre maior precipitação de RNA do p-DNA. Para solução de 80%,
ocorre boa precipitação de p-DNA, como de acordo com a literatura. Não houve
boa seletividade nem boa eficiência de precipitação. A esse problema podemos
associar erros de operação do experimento, contaminação cruzada de amostras
e interferências de diluição.
Para os ensaios com etanol, precipitações com soluções de até 140% em
volume mostram que ocorre precipitação da maior parte dos ácidos nucleicos
presentes. A precipitação entretanto não mostra seletividade, porém apresenta
alta eficiência. Os erros apresentados podem ser associados a erro de operação,
contaminação cruzada entre as amostras e interferências de diluição. Portanto,
a princípio a precipitação em etanol é mais eficiente que em isopropanol.
Para os ensaios com acetona, para soluções de até 100% em volume,
ocorre grande parte da precipitação de ácidos nucleicos presentes. As
precipitações são eficientes, porém pouco seletivas.
Não foram identificadas condições ótimas onde há precipitação seletiva
em nenhum dos solventes. Pelo trabalho realizado, o isopropanol não é tão
eficiente como etanol e acetona. Além disso, a utilização do etanol é mais
vantajosa em relação aos custos associados, além das questões de proteção
ambiental.

40
7. SUGESTÕES PARA TRABALHOS FUTUROS
Buscando um novo estudo das seletividades, a recomendação seria
refazer as precipitações, buscando melhores resultados. Deve-se, no entanto,
desta vez, compreender previamente e evitar possíveis erros que tenham
interferido na primeira realização dos experimentos, tais como problemas de
dilução. Tendo resultados que satisfizessem o propósito inicial do trabalho, seria
possível decidir de fato qual o mais eficiente e seletivo.
A partir de resultados melhores definidos, duas novas possibilidades
surgiriam: o uso de outros solventes ou um estudo da influência da mistura
daqueles estudados, verificando se há uma melhoria do processo usando, ao
mesmo tempo, os melhores resultados de diferentes pares desses solventes, por
exemplo.

41
8. REFERÊNCIAS BIBLIOGRÁFICAS
ADLER, A. F.; LEONG, K. W. Emerging links between surface
nanotechnology and endocytosis: impact on nonviral gene delivery. Nano Today, p.
553-569, 2010.
BIRBOIM, H.C.; DOLY, J. A. rapid alkaline extraction procedure for screening
recombinant plasmid DNA. Nucl. Acids Res., v. 7, p. 1513-1523, 1979.
BONTURI, N. Purificação em etapa cromatográfica única de DNA plasmidial
a partir de lisado neutralizado visando a sua aplicação em estudos de terapia e
vacinação gênica. 2011. 175 f. Dissertação (Mestrado em Engenharia Química) -
Faculdade de Engenharia Química, Universidade Estadual de Campinas, São
Paulo. 2011.
DIOGO, M. M.; QUEIROZ, J. A.; PRAZERES, D. M. F. Chromatography of
plasmid DNA. J. Chromatogr. A, v. 1069, p. 3-22, 1999.
DIOGO, M. M.; QUEIROZ, J.A.; MONTEIRO, G. A.; MARTINS, S. A.;
FERREIRA, G. N. M.; PRAZERES, D.M.F. Purification of a Cystic Fibrosis Plasmid
Vector for Gene Therapy Using Hydrophobic Interaction Chromatography.
Biotechnology and Bioengineering, v. 68, p. 576-583, 2005.
FERREIRA, G. N. M.; MONTEIRO, G.A.; PRAZERES, D.M.F.; CABRAL,
J.M.S. Downstream processing of plasmid DNA for gene therapy and DNA vaccine
applications. Trends Biotechnol., v. 18, p. 380-388, 2000.
FERREIRA, G. N. M.; PRAZERES, D. M. F.; CABRAL, J. M. S., SCHLEEF,
M. Plasmids for Therapy and Vaccination, in: Schleef, M. (Ed.), Plasmids for Therapy
and Vaccination, Wiley-VCH Verlag Gmbh & Co., Weinhein, p. 193-236, 2001.
FREITAS, S. Development and optimization of a scalable plasmid DNA
production process based on hydrophobic interaction chromatography and aiming at
gene therapy and DNA vaccination. Lisboa: Universidade Técnica de Lisboa, 2007,
176 p. Tese (Doutorado)- Programa de Doutorado em Biotecnologia, Instituto
Superior Técnico, Universidade Técnica de Lisboa, Lisboa. 2007.
GONÇALVES, G. A. L.; PRATHER, K. L. J.; MONTEIRO, G. A. M.; CARNES,
A. E.; PRAZERES, D. M. F. Plasmid DNA production with Escherichia coli GALG20,
a pgi-geneknockout strain: Fermentation strategies and impact on
downstreamprocessing. Journal of Biotechnology, v. 186, p. 119-127, 2014.

42
GONÇALVES, G. A. L.; PRATHER, K. L. J.; MONTEIRO, G. A.; PRAZERES,
D. M. F. Engineering of Escherichia coli strains for plasmid biopharmaceutical
production: Scale-up challenges. Vaccine, v. 32, p. 2847-2850. 2014.
HORN, N.A.; MEEK, J. A.; BUDAHAZI, G.; MARQUET, M. Cancer gene
therapy using plasmid DNA: purification of DNA for human clinical trials. Hum. Gene
Ther., v. 6, p. 565-573, 1995.
OLIVEIRA, P. H.; PRATHER, K. J.; PRAZERES, D. M. F.; MONTEIRO, G. A.
Structural instability of plasmid biopharmaceuticals: challenges and implications.
Trends in Biotechnology, v. 27, p. 503-511, 2009.
PRAZERES, D. M. F.; FERREIRA, G. N. M., MONTEIRO, G. A., COONEY,
C. L.; CABRAL, J. M. S. Large-scale production of pharmaceutical-grade plasmid
DNA for gene therapy: problems and bottlenecks. Tibtech, v. 17, p. 169-174, 2001.
PRAZERES, D. M. F.; SCHLUEP, T.; COONEY, C. Preparative purification of
supercoiled plasmid DNA using anion-exchange chromatography. J. Chromatogr. A,
v. 806, p. 31-45, 1998.
REIMANN, J.; KWISSA, M.; SCHIMBERK, R. Genetic Vaccination With
Plasmid Vectors, in: Schleef, M. (Ed.), Plasmids for Therapy and Vaccination, Wiley-
VCH Verlag Gmbh & Co., Weinhein, p. 45-73, 2001.
SCHMIDT, T.; FRIEHS, K.; FLASCHEL, E. Structures of plasmid, in: Schleef,
M. (Ed.), Plasmids for Therapy and Vaccination, Wiley-VCH Verlag Gmbh & Co.,
Weinhein, p. 29-32, 2001.
SOUSA, A.; SOUSA, F.; QUEIROZ, J. A. Biorecognition of supercoiled
plasmid DNA isoform in lysine-affinity chromatography. J. of Chromatography, v.
877, p. 3257-3260, 2009.
STADLER, J.; LEMMENS, R.; NYHAMMAR, T. Plasmid DNA purification. J.
Gene Med., v. 6, p. S54-S66, 2004.


















