ERROS INATOS DO METABOLISMO - Marilanda Bellini | … · · 2011-05-25FORMAÇÃO DOS GAMETAS E,...
46
Disciplina: Fundamentos de Genética e Biologia Molecular Turma: Fisioterapia (1 o Ano) E-mail: [email protected] Blog: http://marilandabellini.wordpress.com ERROS INATOS DO METABOLISMO Docente: Profa. Dra. Marilanda Ferreira Bellini
Transcript of ERROS INATOS DO METABOLISMO - Marilanda Bellini | … · · 2011-05-25FORMAÇÃO DOS GAMETAS E,...

Disciplina: Fundamentos de Genética e Biologia Molecular
Turma: Fisioterapia (1o Ano)
E-mail: [email protected]
Blog: http://marilandabellini.wordpress.com
ERROS INATOS DO METABOLISMO Docente: Profa. Dra. Marilanda Ferreira Bellini
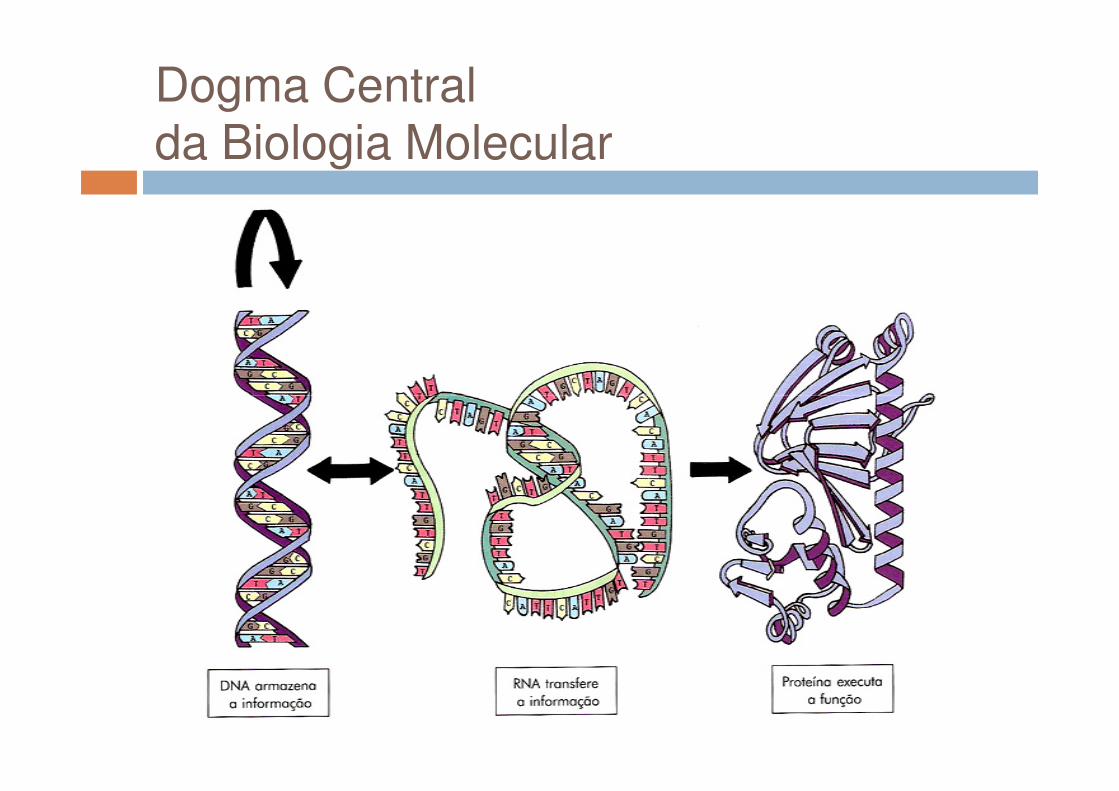
Dogma Central da Biologia Molecular

INTRODUÇÃO
� Gregor Johann Mendel (1822 – 1884)
� 1866: Experimentos emHibridização de Plantas
(Psim sativum)
� 1900
� Hugo de Vries (Holanda)
� Carl Correns (Alemanha)
� Eric von Tschermak-Seysenegg (Áustria)
Aula 03

HERANÇA É DETERMINADA POR APENAS UM
PAR DE FATORES GENÉTICOS (ALELOS - FATORES
MENDELIANOS), QUE SÃO SEPARADOS NA
FORMAÇÃO DOS GAMETAS E, DESTA FORMA, PAI E
MÃE TRANSMITEM APENAS UM PAR PARA SEU
DESCENDENTE. APRESENTANDO GENÓTIPOS E
FENÓTIPOS DISTRIBUÍDOS CONFORME PADRÕES
CARACTERÍSTICOS.
1a. Lei de Mendel
Herança Monogênica
Segregação independente nos gametasAula 03

Gene
Genética – Mendeliana
Porção do cromossomo que determina um único caráter (fenótipo).
Molecular – Beadle e Tatum, 1940
Segmento de material genético que determina ou codifica uma enzima.
1 gene ���� uma enzima (proteína)1 gene ���� uma enzima (proteína)
Um gene codifica pelo menos uma cadeia polipeptídica, um tRNA, ou um
rRNA, como produto final em Eucariotos.
Aula 06

Gene mutante � Bloqueio Metabólico
� 1902:� Sir Archibald E. Garrod
� Reações metabólicas são controladas por genes

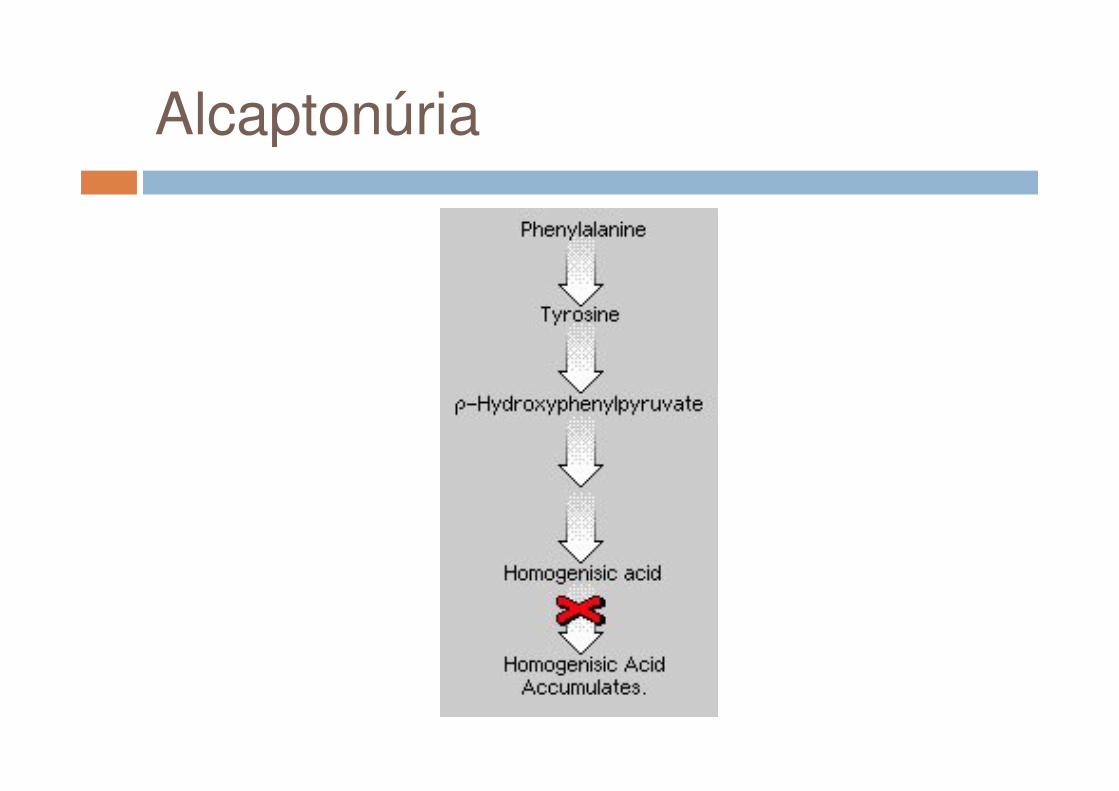
Alcaptonúria
� Doença Autossômica Recessiva;� Mutações (missense, splicesite,
frameshift, nonsense e no-stop) em 3q (3q21 – q23);
↓↓
Deficiência completa da enzima ácido homogentísico oxidase (HGO),
↓
Acúmulo do ácido em diversos órgãos e tecidos e aumento de excreção urinária.
Vilboux et al. Hum Mutat. 2009 December ; 30(12): 1611–1619. doi:10.1002/humu.21120.
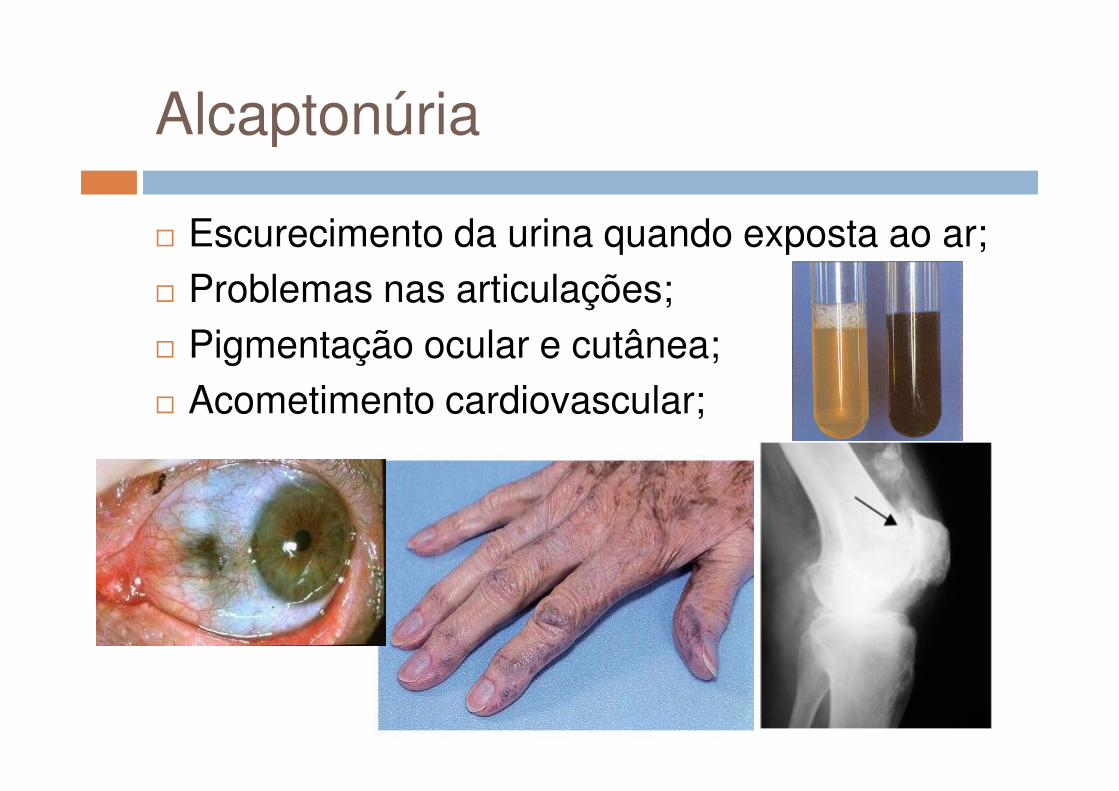
Alcaptonúria
� Escurecimento da urina quando exposta ao ar;� Problemas nas articulações;� Pigmentação ocular e cutânea;� Acometimento cardiovascular;� Acometimento cardiovascular;
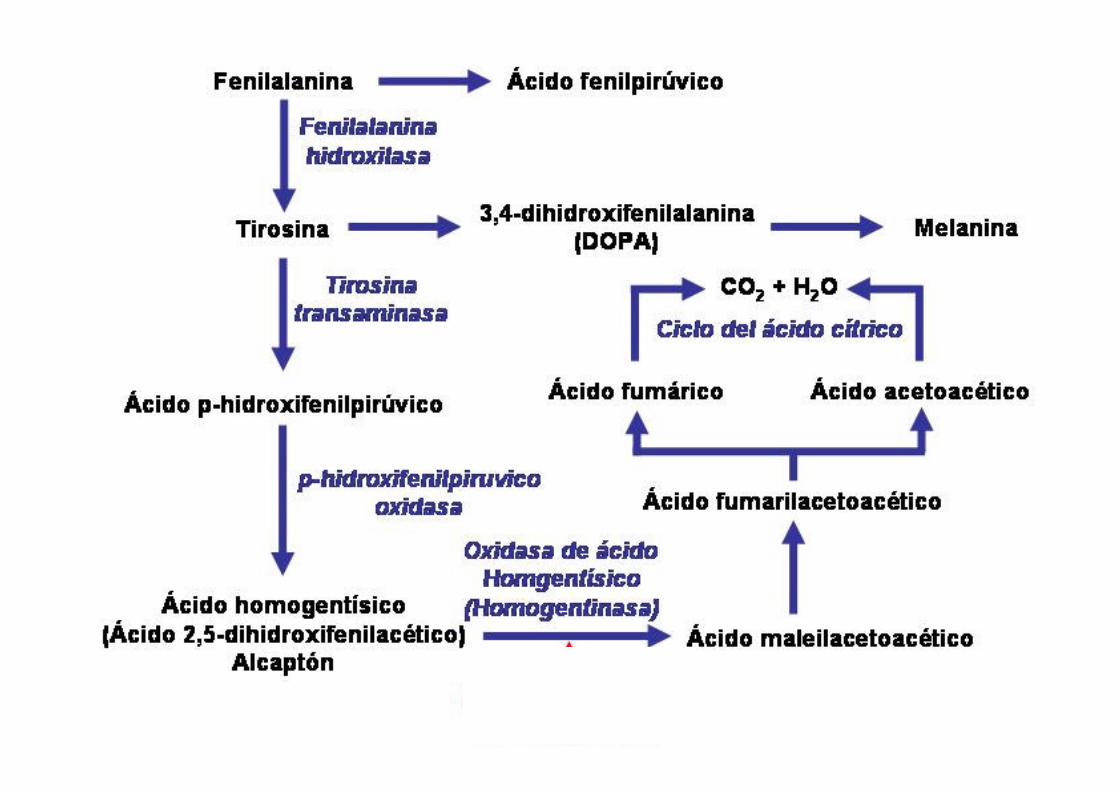

ACÚMULO
Alcaptonúria
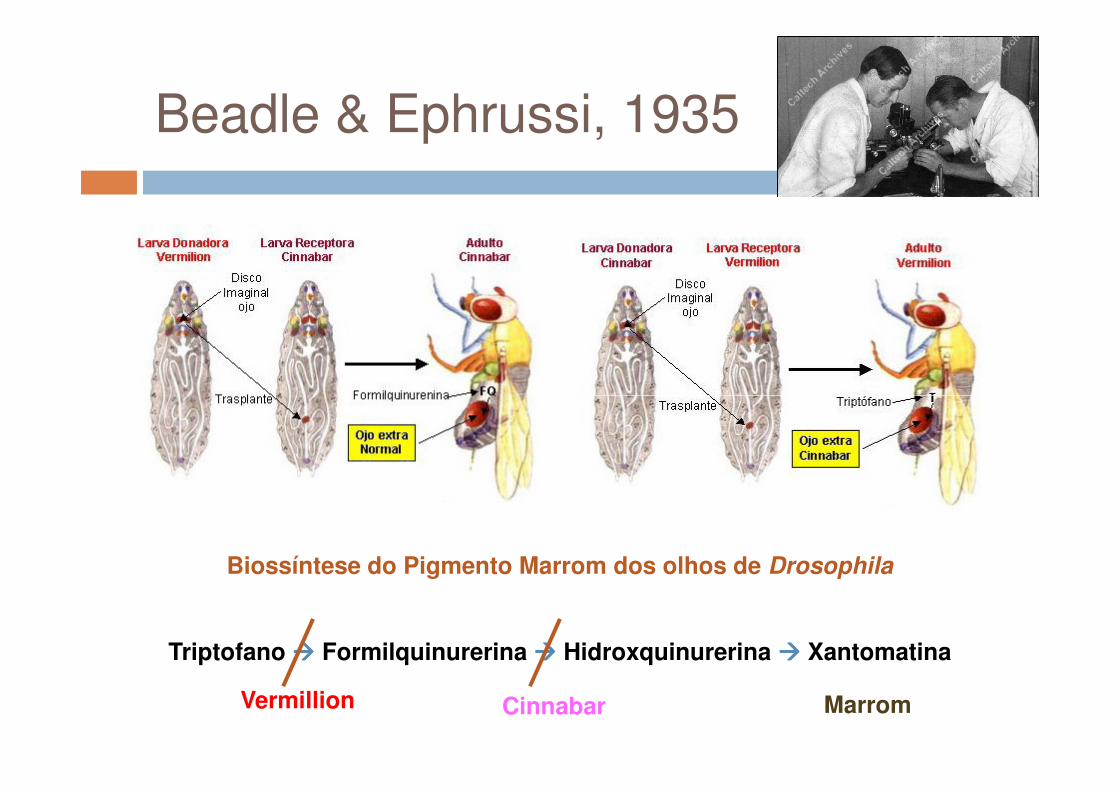
Beadle & Ephrussi, 1935
Biossíntese do Pigmento Marrom dos olhos de Drosophila
Triptofano ���� Formilquinurerina ���� Hidroxquinurerina ���� Xantomatina
Vermillion MarromCinnabar
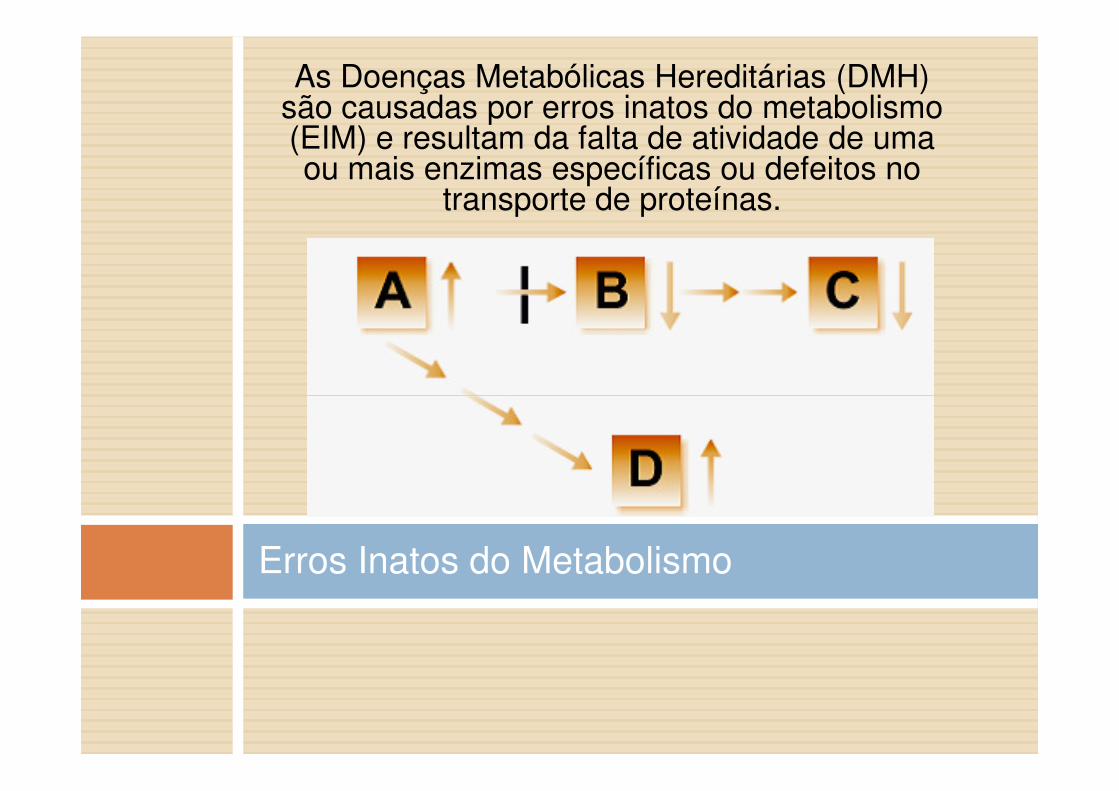
As Doenças Metabólicas Hereditárias (DMH) são causadas por erros inatos do metabolismo (EIM) e resultam da falta de atividade de uma ou mais enzimas específicas ou defeitos no
transporte de proteínas.
Erros Inatos do Metabolismo
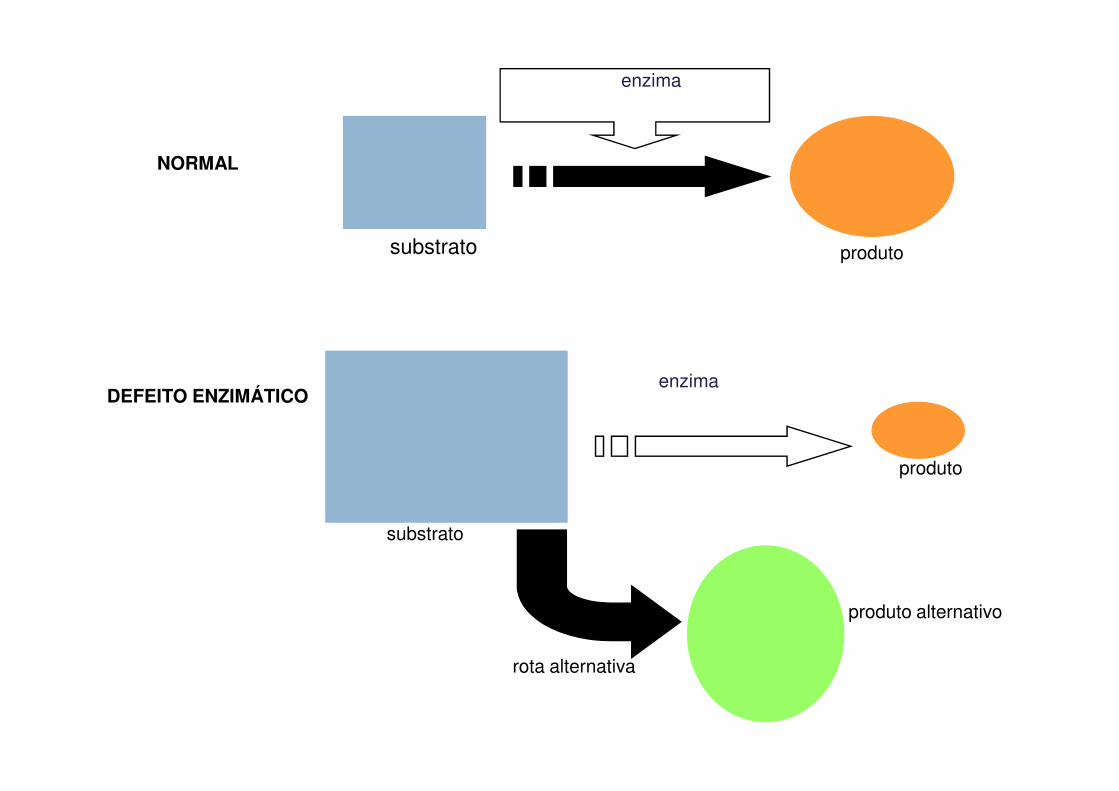
substrato produto
enzima
enzima
NORMAL
DEFEITO ENZIMÁTICO
produto
substrato
rota alternativa
produto alternativo
DEFEITO ENZIMÁTICO
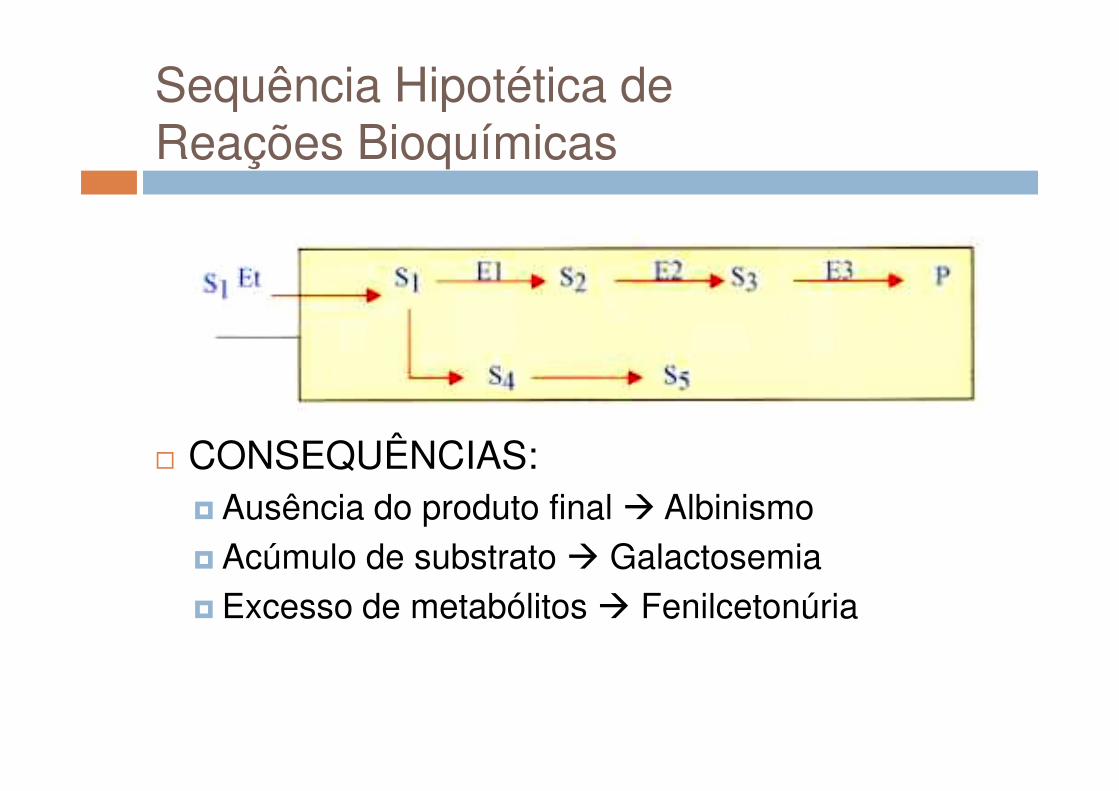
Sequência Hipotética de Reações Bioquímicas
� CONSEQUÊNCIAS:� Ausência do produto final � Albinismo� Acúmulo de substrato � Galactosemia� Excesso de metabólitos � Fenilcetonúria

CLASSIFICAÇÃO DOS EIM
CATEGORIA 1 CATEGORIA 2
Saudubray e Charpentier (1995)
Alterações que afetam um único sistema orgânico ou apenas um
órgão. Ex: sistema imunológico e os fatores de coagulação; túbulos
renais; eritrócitos.
Grupo de doenças cujo defeito bioquímico compromete uma via
metabólica comum a diversos órgãos. Ex: doenças lisossomais.
Pode estar restrito a um orgão apenas, porém com manifestações humorais e sistêmicas. Ex: defeitos
no ciclo da uréia.
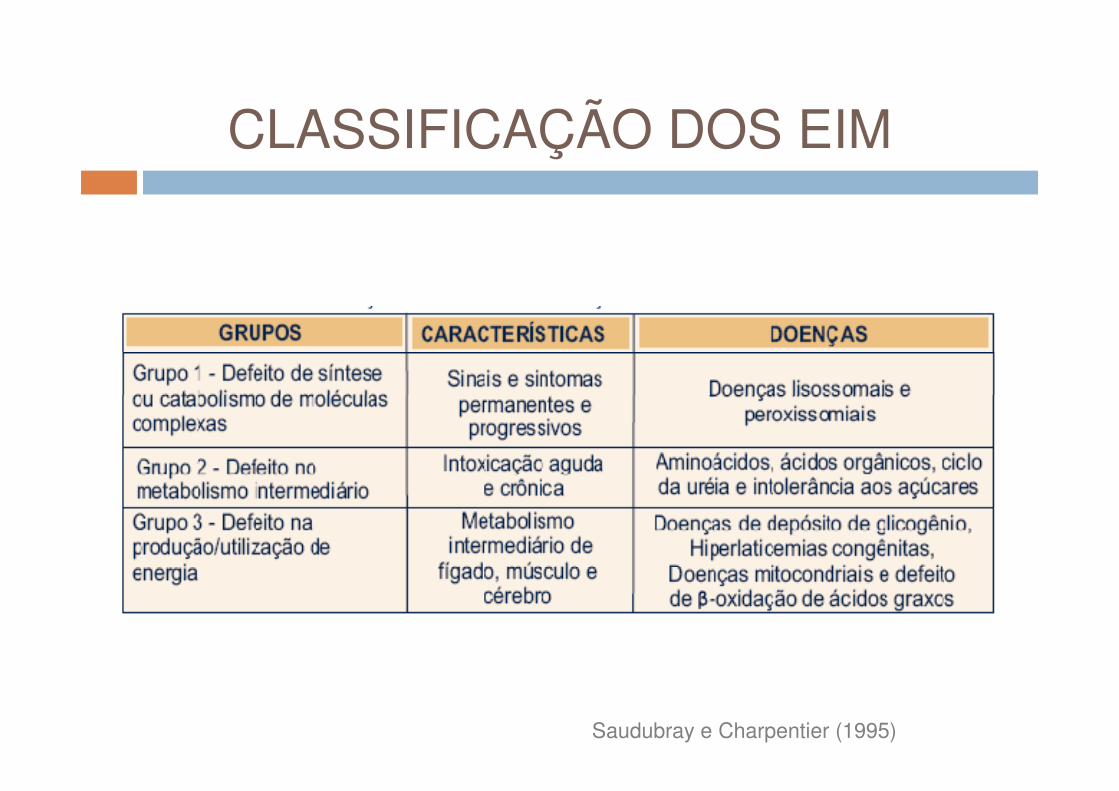
CLASSIFICAÇÃO DOS EIM
Saudubray e Charpentier (1995)
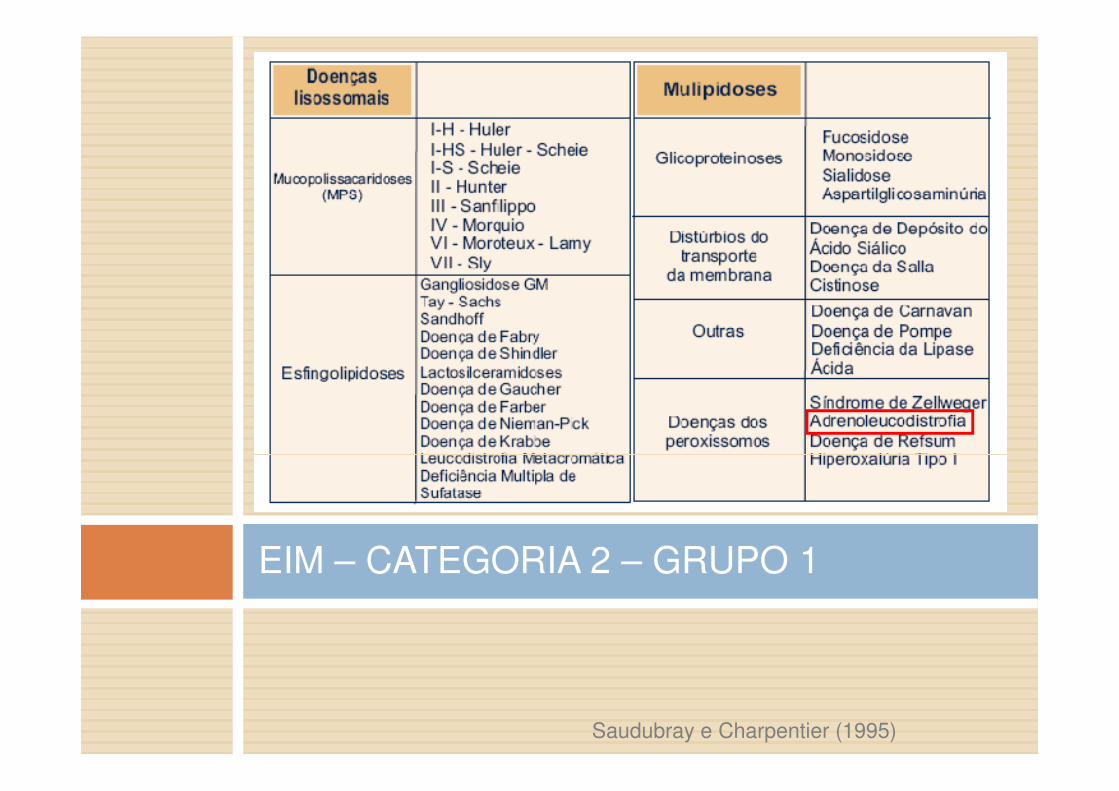
EIM – CATEGORIA 2 – GRUPO 1
Saudubray e Charpentier (1995)
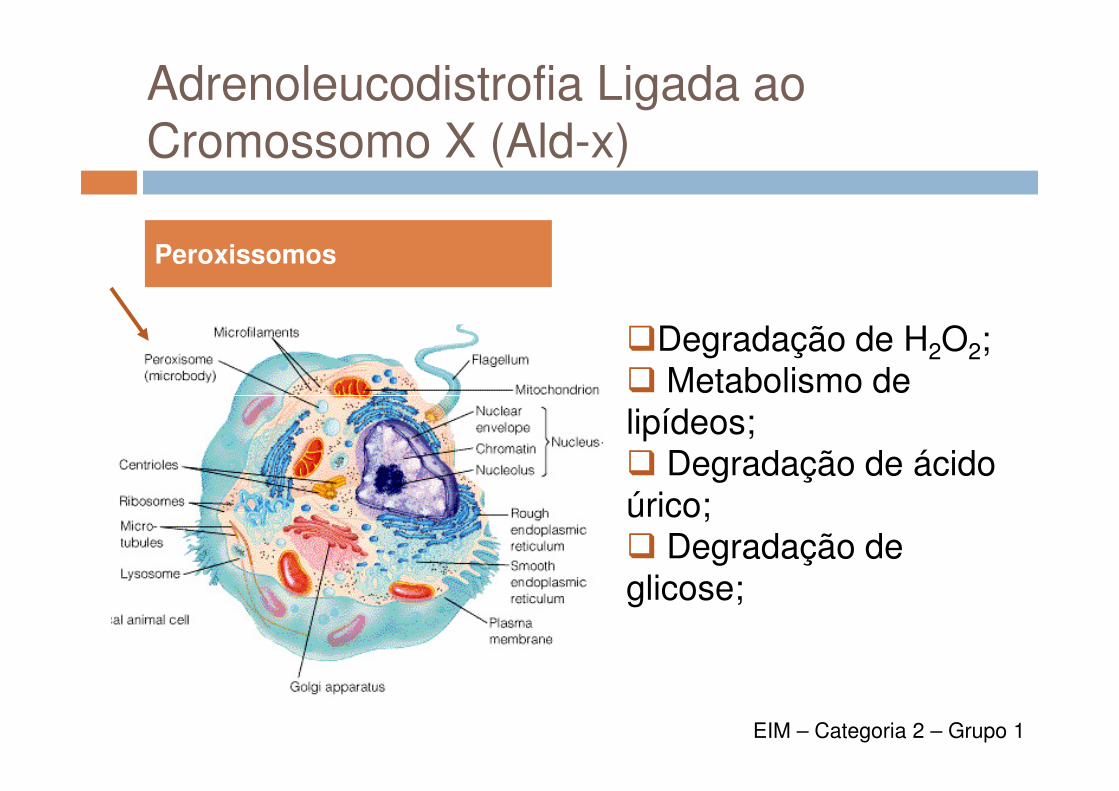
Adrenoleucodistrofia Ligada aoCromossomo X (Ald-x)
�Degradação de H2O2;� Metabolismo de
Peroxissomos
� Metabolismo de lipídeos;� Degradação de ácidoúrico;� Degradação de glicose;
EIM – Categoria 2 – Grupo 1
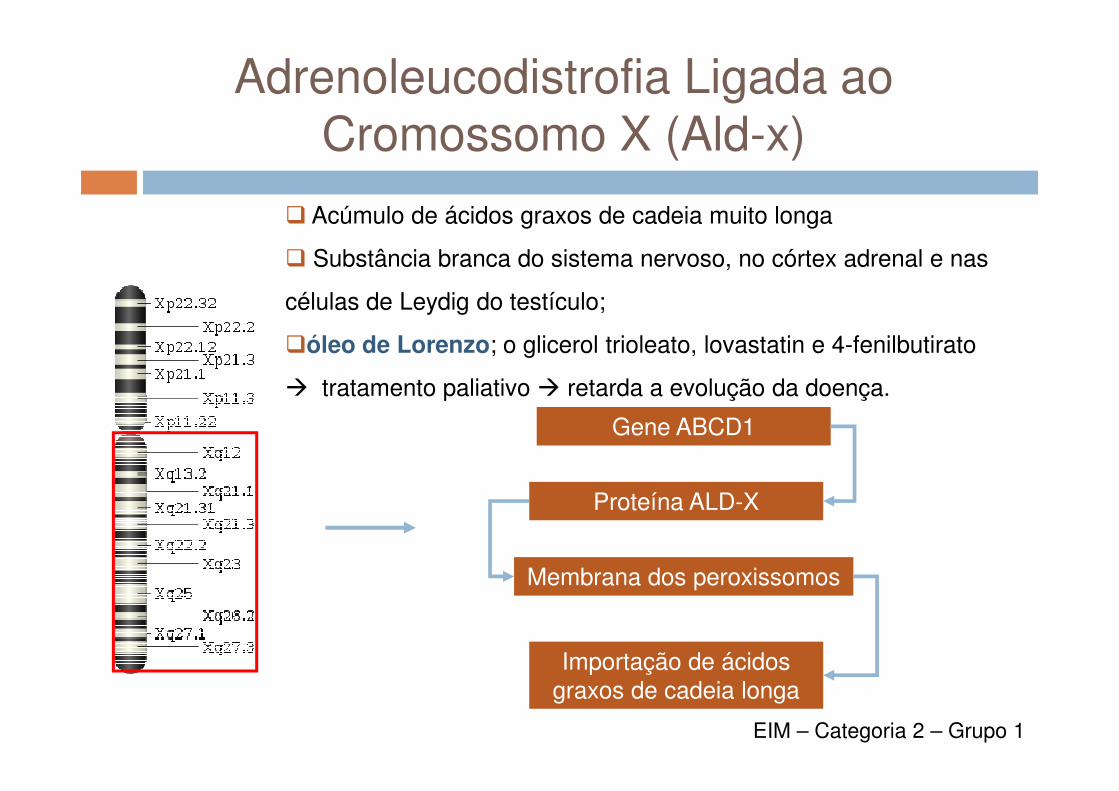
Adrenoleucodistrofia Ligada aoCromossomo X (Ald-x)
� Acúmulo de ácidos graxos de cadeia muito longa
� Substância branca do sistema nervoso, no córtex adrenal e nas
células de Leydig do testículo;
�óleo de Lorenzo; o glicerol trioleato, lovastatin e 4-fenilbutirato
� tratamento paliativo � retarda a evolução da doença.
EIM – Categoria 2 – Grupo 1
� tratamento paliativo � retarda a evolução da doença.
Gene ABCD1
Proteína ALD-X
Membrana dos peroxissomos
Importação de ácidos graxos de cadeia longa

EIM – CATEGORIA 2 – GRUPO 2

� Incidência: 1:2.900 neonatos� Heterogeneidade alélica: +400 alelos diferentes
� Excesso de metabólitos
Fenilcetonúria (PKU)
� Excesso de metabólitos� Hiperfenilalaninemia → Fenilcetonúria (PKU)� Defeito enzimático: Fenialanina Hidroxilase
Fenilalanina Tirosina
metabólitos tóxicos
(excretados na urina)

Fenilalanina hidroxilase (PAH) Fenilalanina � tirosina
Fenilcetonúria (PKU)
EIM – Categoria 2 – Grupo 2
Tetraidrobiopterina

� Incapacidade de metabolizar a fenilalanina� ↑ líquidos corporais� Prejuízo no desenvolvimento do cérebro
Retardo mental Microcefalia
Fenilcetonúria (PKU)
EIM – Categoria 2 – Grupo 2
MicrocefaliaHipopigmentação da pele, cabelos e olhosIrritabilidadeEpilepsiaDéficit do crescimento somático pós-natalDistúrbios do comportamentoOutras alterações neurológicas (tremores, hipertonia, hiper-refelixa tendinosa profunda
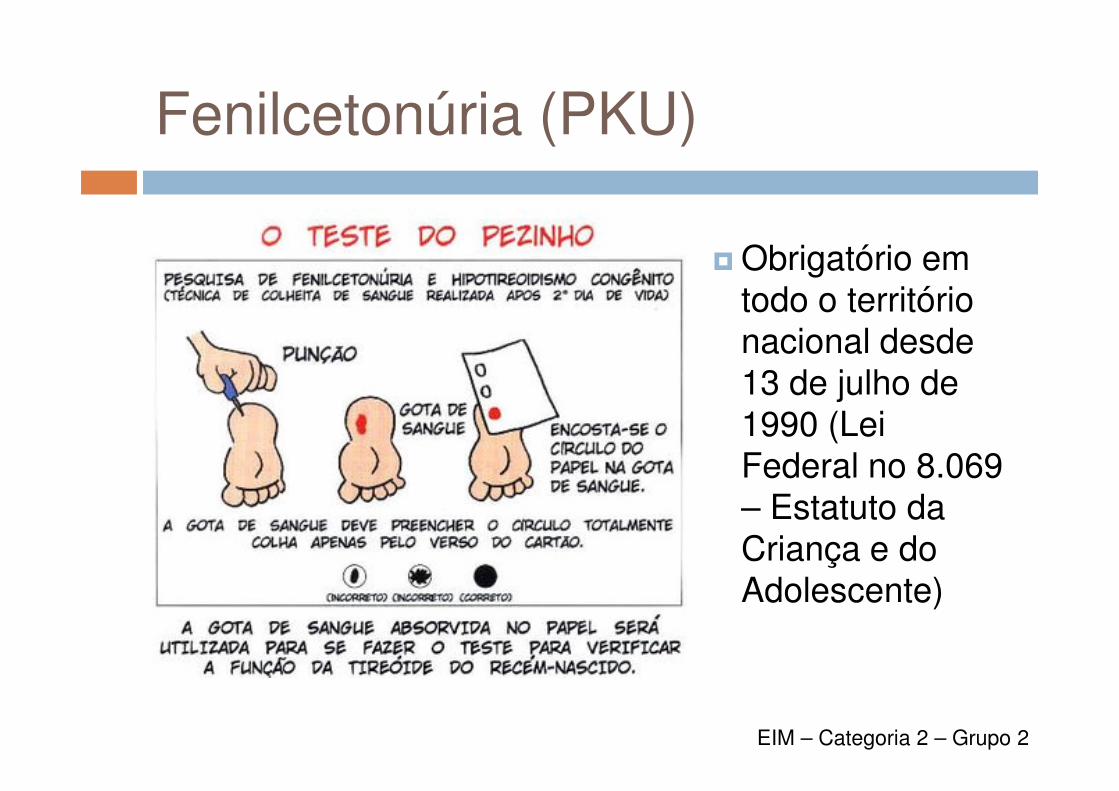
Fenilcetonúria (PKU)
� Obrigatório em todo o território nacional desde 13 de julho de 13 de julho de 1990 (Lei Federal no 8.069 – Estatuto da Criança e do Adolescente)
EIM – Categoria 2 – Grupo 2

Fenilcetonúria (PKU)
� Teste da fralda: � adicionar na urina cloreto férrico → verde escuro
(ácico fenilpirúvico)
Diagnóstico precoce até 6 meses e dieta pobre em fenilalanina (300-500 mg de fenilalanina/dia) até 7 anos de idade ou tempo indeterminado.
EIM – Categoria 2 – Grupo 2

CLASSIFICAÇÃO
Fenilcetonúria (PKU)
EIM – Categoria 2 – Grupo 2
Valores normais: 2-4mg/dl
Controle da dieta

EIM – CATEGORIA 2 – GRUPO 2

Galactosemia Clássica
� Incidência: 1:55.000 neonatos� Ausência: galactose -1- fosfato uridil-
transferase
galactose � glicose
Deficiência da enzima↓
ACÚMULO DE SUBSTRATO
EIM – Categoria 2 – Grupo 2

Galactosemia
Metabolização alternativa:
galactitol GALACTOSE galactonato

Galactossemia Clássica
Manifestação clínica:problemas gastrointestinais(ingestão de leite) -hepatoesplenomegalia
-cirrose hepática-catarata-lesões do SNC e RM
http://www.nytimes.com/imagepages/2007/08/01/health/adam/17187Galactosemia.html
-lesões do SNC e RM
Triagem neonatal: dosagem de GAL-1-P uridil transferase
Tratamento: até os 3 anos-suspensão do leite e seus derivados (leite desprovido de galactose, leite de soja, hidrolisados de proteínas)-evitar lactose durante toda a vida

EIM – CATEGORIA 2 – GRUPO 3

� Herança Mitocondrial Materna
Doença de Leigh
EIM – Categoria 2 – Grupo 3
Doença de Leigh
EIM – Categoria 2 – Grupo 3

Doença de Leigh
� Neuropatia
EIM – Categoria 2 – Grupo 3
� Neuropatia� Ataxia� Retinite pigmentosa� Retardo de desenvolvimento� Retardo mental� Acidemia lática
Incidência de EIM�Incidência acumulativa 1: 5000 nascidosvivosvivos
� Importância do diagnóstico precoce
� Quadros clínicos de difícil explicação pelafisiopatologia da doença - EIM

Estratégias de Tratamento
� Restrição alimentar
� Reposição
� Desvio� Desvio

Restrição A
limentar
� não ingerir o substrato� alteração do estilo de vida� custo financeiro elevado
distúrbios emocionais
Estratégias de Tratamento
Restrição A
limentar
� distúrbios emocionais� altamente eficaz� Requer o cumprimento vitalício de
uma dieta restrita, artificial e cara
Reposição
� Administrar a enzima ausente;� É a forma que tem mais êxito no
tratamento;Poucas doenças tratáveis.
Estratégias de Tratamento
Reposição
� Poucas doenças tratáveis.

Desvio
� Consiste em desviar a rotametabólica alterada para umarota alternativa desde que oproduto da rota alternativa não
Estratégias de Tratamento
Desvio
produto da rota alternativa nãoseja tóxico

O diagnóstico definitivo de uma DMH é feito através de determinação da atividade enzimática ou identificação do defeito
DIAGNÓSTICO
enzimática ou identificação do defeito molecular, métodos estes pouco
disponíveis, principalmente no nosso meio.
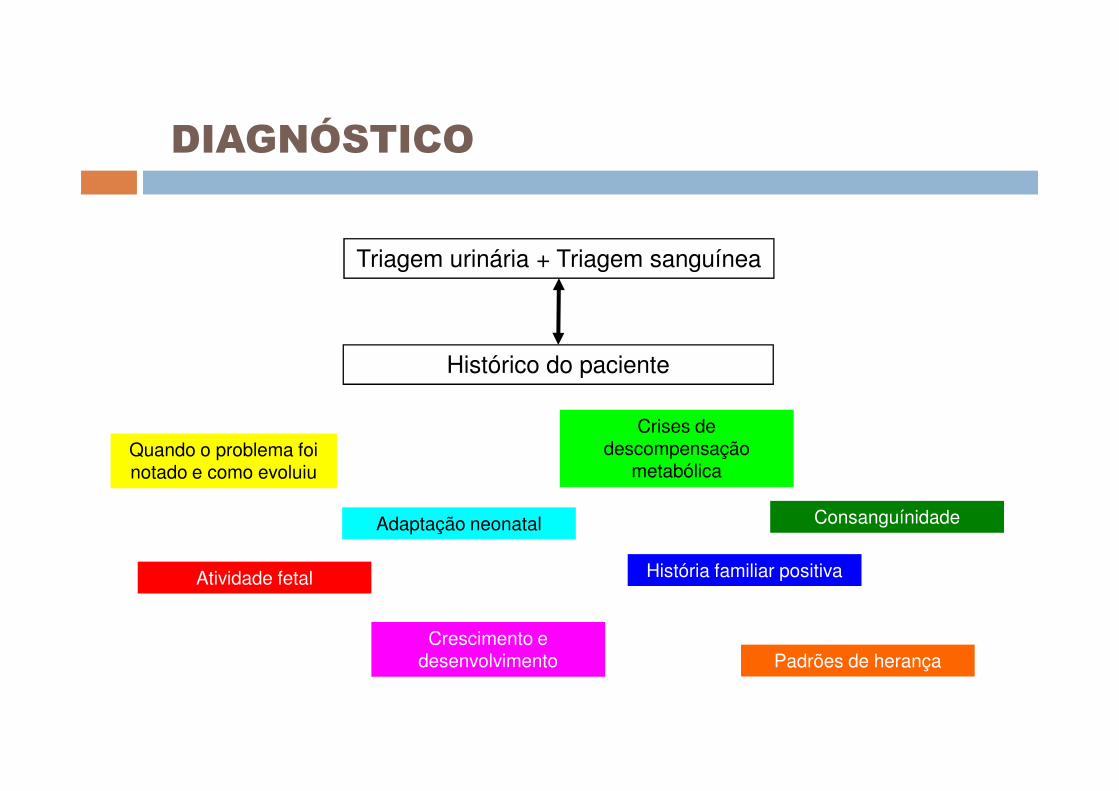
DIAGNÓSTICO
Triagem urinária + Triagem sanguínea
Histórico do paciente
Quando o problema foi notado e como evoluiu
Atividade fetal
Adaptação neonatal
Crescimento e desenvolvimento
Crises de descompensação
metabólica
História familiar positiva
Padrões de herança
Consanguínidade
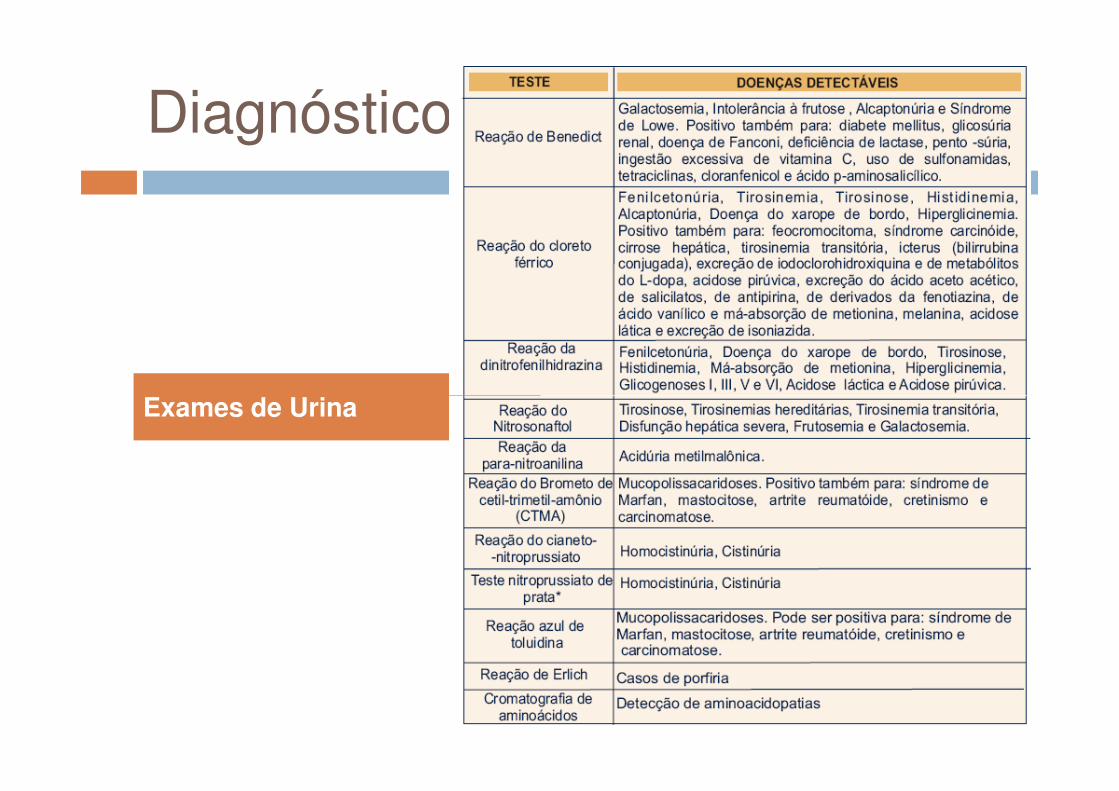
Diagnóstico
Exames de UrinaExames de Urina

• hemograma• gasometria venosa• determinação de sódio (Na), potássio (K), cloro (Cl)
Diagnóstico
Exames Sanguíneospotássio (K), cloro (Cl)• glicemia• transaminases hepáticas• colesterol total e frações, triglicérides• ácido úrico, lactato, piruvato eamônia.
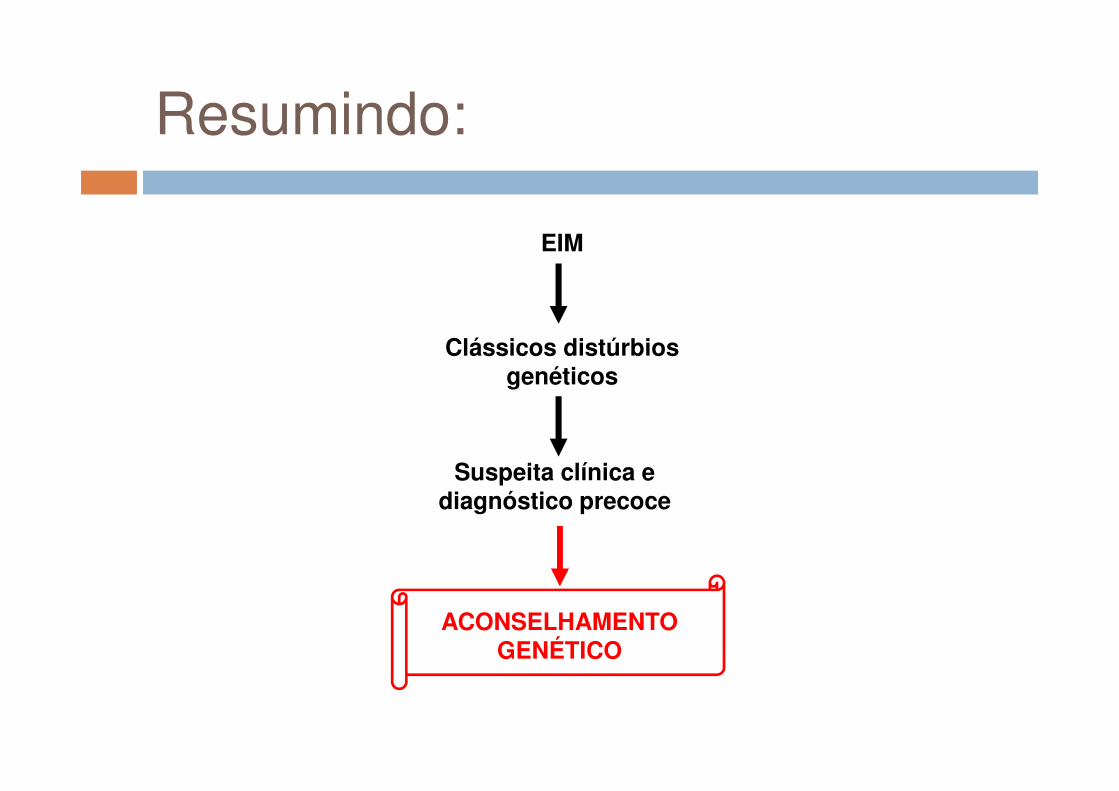
EIM
Clássicos distúrbios genéticos
Resumindo:
Suspeita clínica e diagnóstico precoce
ACONSELHAMENTO GENÉTICO

Referências
� Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Biologia Molecular da Célula. 4 ed, Porto Alegre: Artmed, 2004.
� Lourenço LB, Felisbino SL, Carvalho HF. Peroxissomos. In: A Célula, Ed. Carvalho HF, Recco-Pimentel S. 2 ed, Barueri: Manole, 2007. Mir L. Genômica. São Paulo: Atheneu, 2004.
� Mir L. Genômica. São Paulo: Atheneu, 2004.� Nussbaum RL, Mcinnes RR, Willard, HF. Thompson & Thompson -
Genética médica. 7 ed, Rio de Janeiro: Guanabara Koogan, 2008.� Passarge E. Color Atlas of Genetics. Thieme, 1995.� www.supportnet.com.br/artigos/pdf/monografia.pdf� El Husny AS, Fernandes-Caldato MC. Erros inatos do metabolismo:
Revisão de Literatura. Revista Paraense de Medicina, 20(2), 2006.