Doncças do Glicogênio

of 6
-
Upload
felipe-pereira -
Category
Documents
-
view
67 -
download
0
Transcript of Doncças do Glicogênio
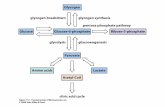
INTRODUO Um nmero de sndromes genticas tem sido identificado e que resultam de algum defeito metablico na sntese ou catabolismo de glicognio. A categoria mais bem compreendida e a mais importante inclui as doenas de armazenamento de glicognio resultantes de deficincia hereditria de uma das enzimas envolvidas na sntese ou degradao seqencial de glicognio. Dependendo da distribuio no tecido ou rgo da enzima especfica no estado normal, o armazenamento de glicognio nesses distrbios pode ser limitado a poucos tecidos, ser mais difundido embora no afete todos os tecidos ou ser sistmico na sua distribuio. O significado de uma deficincia enzimtica especfica mais bem compreendido a partir da perspectiva do metabolismo normal do glicognio. Como bem conhecido, o glicognio uma forma de armazenamento da glicose. A sntese de glicognio comea com a converso de glicose a glicose-6-fosfato pela ao de uma hexoquinase (glicoquinase). Uma fosfoglicomutase em seguida transforma a glicose-6-fosfato em glicose-1-fostato, que por sua vez convertida em uridina-difosfoglicose. Um polmero muito grande e altamente ramificado em seguida construdo contendo acima de 10.000 molculas de glicose ligadas entre si por ligaes de a -1,4-glicosdio. A cadeia e os ramos colaterais do glicognio continuam a ser alongados pela adio de molculas de glicose mediadas por glicognio-sintetases. Durante a degradao, fosforilases distintas no fgado e msculo separam quebrando a glicose-1-fostato do glicognio at que cerca de quatro resduos de glicose permanecem em cada ramo, deixando um oligossacardeo ramificado chamado dextrina limite. Isto pode ser degradado mais apenas pela enzima desramificante. Em adio a essas vias principais o glicognio tambm de degradado nos lisosomos pela maltase cida. Se os lisosomos so deficientes nesta enzima, o glicognio contido dentro deles no acessvel degradao pelas enzimas citoplasmticas como as fosforilases. Com base nas deficincias enzimticas especficas e nos quadros clnicos resultantes, as glicogenoses tm, sido tradicionalmente divididas em uma dzia ou mais de sndromes, designadas por numerais romanos. De uma forma geral, as glicogenoses podem ser divididas em quatro grupos: Formas hepticas. Conforme bem conhecido, o fgado uma pea chave no metabolismo do glicognio. Ele contm enzimas que sintetizam glicognio para armazenamento e que finalmente o quebram em glicose livre, que em seguida liberada no sangue. Uma deficincia herdade das enzimas hepticas que esto envolvidas no metabolismo do glicognio portanto leva no apenas ao armazenamento de glicognio no fgado como tambm a uma reduo no nvel sangneo de glicose (hipoglicemia). A deficincia da enzima glicos-6-fosfatase (doena de von Gierke ou glicogenose tipo I) um exemplo da forma heptica-hipoglicmica da doena de armazenamento de glicognio. Outros exemplos so: deficincia da enzima desramificadora (doena de Cori ou glicogenose tipo III), deficincia de fosforilase heptica (doena de Hers ou glicogenose tipo VI) e outros exemplos como a deficincia de fosfoquinase b heptica (glicogenoses tipos VIII ou IX). Formas miopticas. Nos msculos estriados, ao contrrio do fgado, o glicognio utilizado predominantemente como fonte de energia. Isto derivado pela gliclise, que leva no final formao de lactato. Se as enzimas que servem de combustvel para a via glicoltica estiverem deficientes, o armazenamento de glicognio ocorre nos msculos e est associado com fraqueza muscular em razo da produo prejudicada de energia. Os
exemplos nesta categoria incluem deficincias da fosforilase muscular (doena de McArdle ou glicogenose tipo V) e deficincia da fosfofrutoquinase muscular e outras (glicogenose tipo VII). Formas especficas. Doenas de armazenamento de glicognio associadas com deficincia de a -glicosidades e falta de enzima ramificadora no se adaptam nas categorias descritas acima. Elas esto associadas como o armazenamento de glicognio em muitos rgos. A maltase cida uma enzima lisosmica de glicognio, e dessa maneira, sua deficincia leva ao armazenamento lisosmica de glicognio (doena de Pompe ou glicogenose tipo II). Outra glicogenose de forma especfica a deficincia da enzima desramificadora (doena de Andersen ou glicogenose tipo IV). DESENVOLVIMENTO TIPO I: DOENA DE von GIERKE A deficincia de glicose-6-fosfatase um distrbio gentico autossmico recessivo que incide em uma de cada 100.000 a 400.000 pessoas. Este distrbio costuma manifestar-se j nos primeiros 12 meses de vida atravs da hipoglicemia sintomtica ou pela deteco de hepatomegalia. Pacientes ocasionais experimentam hipoglicemia no perodo neonatal imediato e alguns raros nunca chegam a apresentar hipoglicemia. Os achados caractersticos incluem uma face arredondada e bochechuda, um abdome protuberante devido acentuada hepatomegalia e extremidades finas. A hiperlipidemia pode causar xantomas eruptivos e lipemia retiniana. A esplenomegalia no se manifesta ou descrita, embora um aumento vultoso do lobo heptico esquerdo possa ser confundido com esplenomegalia. O crescimento geralmente normal durante os primeiros meses de vida; posteriormente, sobrevem retardo no crescimento e a adolescncia sofre atrasos. O desenvolvimento metal costuma ser normal, exceto nos casos de leso por hipoglicemia. Como j foi dito uma deficincia da enzima glicose-6-fosfatase, a expresso clnica desta alterao muito diferente da de outras formas de glicogenose. Por exemplo. A hipoglicemia produzida pelo jejum pode ser extrema e, em combinao com acidose lctica, a hiperlipidemia, caracteriza os pacientes do tipo I. o mecanismo responsvel pelas alteraes marcantes do metabolismo dos carbodratos, foi recentemente revisada e resulta sobretudo da produo excessiva de substrato em resposta queda da glicose sangnea. Tratamento: A reverso documentada destas anormalidades atravs do tratamento que mantm os nveis da glicose sangnea entre 80 a 90 mg/dl fortalece o postulado de que estas alteraes resultam de repostas hormonais hipoglicemia. A interveno teraputica tem sido avaliada mais profundamente em pessoas com esses defeitos do que em quaisquer outras. Como resultado, tem sido possvel elaborar um controle diettico razoavelmente eficaz para estes pacientes, o que assegura um desenvolvimento favorvel at a idade adulta. TIPO II: DOENA DE POMPE a deficincia de a -glicosidades, uma enfermidade do armazenamento lisosmico. A incidncia desconhecida, mas parece ser superior a um em 100.000. o distrbio no se associa com hipoglicemia, cetose ou outras anormalidade do metabolismo intermedirio. A forma que se instala na primeira infncia manifesta-se nos primeiros seis meses de vida e pode j existir por ocasio do nascimento. As manifestaes clnicas incluem hipotonia e fraqueza musculares esquelticas, cardiomegalia macia, macrogrossia e graus variveis de hepatomegalia. As enzimas musculares, como a creatinofosquinase e aldolase,
costumam estar elevadas e o ECG pode evidenciar complexos QRS amplos e um intervalos PR encurtado. Podem estar presentes fraqueza motora e retardo do desenvolvimento. O bito ocorre nos primeiros dois a trs meses de vida e, na maioria das vezes, secundrio ao comprometimento cardaco. A forma juvenil apresenta caractersticas sugestiva de uma forma progressiva de distrofia muscular. Estes pacientes apresentam anormalidade de marcha, sem sinais e sintomas cardacos. Uma forma adulta ainda mais branda manifesta-se como fraqueza muscular esqueltica entre a terceira e Quinta dcada de vida. O diagnstico especfico firmado pelo ensaio enzimtico em material de biopsia oriundo do msculo e do fgado. O diagnstico pr-natal confivel e tem sido muito empregado para a forma infantil. Tratamento: Foram tentados vrios processos de terapia com infuso enzimtica, sem muita eficcia. TIPO III: DOENA DE CORI A doena de Cori, tambm chamada deficincia da enzima desramificadora, um distrbio autossmico recessivo e pode ser considerada com uma das mais comuns deficincias do armazenamento do glicognio. Os pacientes costumam apresentar hipoglicemia e hepatomegalia durante o primeiro ano de vida. Essa doena causa miopatia esqueltica e, em alguns casos, cardaca que pode ser agravada em adultos, podendo at incapacit-lo. A doena de Cori pode ser dividida como tipo IIIb, vista em pessoas que no apresentam comprometimento muscular e tipo IIIa, quando o paciente apresenta deficincia enzimtica tanto no fgado como no msculo. Cerca de 80% dos pacientes apresentam hipoglicemia de jejum, j que a resposta da glicose administrao do glucagon ou da adrenalina anormal nesta situao. Verifica-se, tambm, que o teste de tolerncia galactose costuma ser bem satisfatrio, assim como o lactato sangneo, porm o nvel de cetose chama a ateno. Para se definir o diagnstico faz-se a anlise do glicognio e a dosagem da enzima desramificadora atravs de amostras de decido do paciente. Logo v-se que o teor de glicognio nas hemcias e no fgado aumenta na maioria dos indivduos afetados, no entanto o teor de glicognio no msculo aumenta em apenas alguns dos pacientes. As experincias mostram que a definio do diagnstico por ensaio enzimtico muito mais complicada, tanto por problemas metablicos quanto pela heterogeneidade gentica. Portanto a documentao da estrutura anormal de glicognio um achado muito mais consistente, mediante o emprego de tcnicas espectrofotomtricas. Assim, embora seja possvel fazer o diagnstico de alguns pacientes atravs das hemcias, leuccitos ou fibroblastos. prefervel documentar a estrutura anormal do glicognio e a deficincia enzimtica diretamente pelo material extrado do fgado ou do msculo. Tratamento: importante que o paciente se alimente muito bem. A glicognese normal, sendo que o paciente deve ingerir galactose, frutose ou protenas para ajudar a manter a glicemia. Por isso, a dieta deve incluir uma quantidade alta de calorias sob a forma de protenas, mas importante ingerir carbodratos, correspondente a 40 a 50% do total. s vezes uma simples refeio suficiente para evitar a hipoglicemia, no entanto pode ser necessria uma alimentao por cateter nasogstrico ou terapia com amido de milho para as crianas gravemente comprometidas. A enzima desramificadora, tambm chamada de amido-1,6-glicosidase, faz uma ciso hidroltica dos laos a -1,6 (ligaes glicosdicas que unem resduos de glicose ramificando
a cadeia de glicognio) na glicogenlise. A falta dessa enzima causa o acmulo de um polissacardeo do tipo dextrina limite (C6H10O5)n.xH2O. TIPO IV: DOENA DE ANDERSEN A deficincia da enzima ramificadora, tambm conhecida como doena de Andersen ou glicogenose do tipo IV tem origem em uma anomalia gentica, ou seja, causada por um gene autossmico recessivo e considerada como uma molstia rara, esse gene afeta a produo da enzima amilo(1,4-1,6)trans-glicosidase, causando uma amilo-pectinose no indivduo afetado. Neste caso o glicognio no ramificado, assim, adquire uma forma muito longa, ou seja, no apresenta as ligaes ?-1,6 glicosdicas, que permite a condensao do glicognio que, dessa forma, torna-se menos eficiente na sua funo de armazenar a glicose e, ao ser "lisada", a liberar glicoses, como pode ser visto no esquema. Com tais deficincias surge um problema para o indivduo afetado: os rgo que devem armazenar o glicognio (principalmente o fgado e os msculos) comeam a sofrer as conseqncias desta doena, ou seja, o indivduo passa a sofrer de hepatoesplenomegalia (aumento do fgado e do bao), e a partir da uma insuficincia heptica; alm de uma miopatia (hipotonia e atrofia muscular) e retardo do crescimento. A doena de Andersen pode ser identificada atravs de alteraes laboratoriais como por exemplo as verificadas em ensaio enzimtico no fgado, msculos, leuccitos ou fibroblastos e pela anlise de glicognio, que no chama ateno pelo seu teor mas sim pela sua estrutura anormal. H uma ausncia de hipoglicemia, porm com alteraes tpicas de patologias hepticas graves. O diagnstico pode ser feito tambm por meio de cultura de clulas amniticas. Tratamento: uma patologia letal, geralmente o indivduo morre entre 3 e 4 anos de vida, embora possa haver uma variao mais amena podendo evoluir de um modo mais benigno. TIPO V: DOENA DE McARDLE A sndrome de McArdle ou glicogenose do tipo V causada pela deficincia de miofosforilase. Os portadores dessa doena apresentam dificuldades em realizar exerccios fsicos. Seus msculos apresentam um elevado teor de glicognio (entre 2,5 e 4,1%), no entanto, seu sangue possui pouco lactato sangneo aps a liberao de glucagon ou adrenalina, indicando que a fosforilase heptica est em seu nvel normal. Existem casos relatados em que a mioglobinria apresenta um achado associado. A fosforilase atua nas ligaes entre as glicoses no ramificadas (ligaes a -1,4 glicosdicas), desligando-as e liberando uma molcula de glicose-1-P, como pode ser visto no esquema. O paciente com tal deficincia apresenta sintomas de dor e cibras aps os exerccios. Eles so, sob outros aspectos, normais, no apresentando distrbios hepticos, musculares ou cardacos. Para diagnosticar a doena feito, com grande eficincia, um teste isqumico de exerccios. Assim, o diagnstico definido por uma documentao do teor elevado de glicognio e de uma reduo na atividade da fosforilase no tecido muscular biopsiado, j que sabida a sua regio de depsito (regies subsarcolmicas do msculo). Hoje, sabe-se que o nmero de homens afetados superior ao de mulheres e o gene para a miofosforilase localiza-se no cromossoma 11, em consonncia com a natureza autossmica recessiva dessa doena.
Tratamento: Para controlar a deficincia de miofosforilase, deve-se, em primeiro lugar, ter muito cuidado, ou seja, no se deve praticar exerccios fsicos vigorosos e deve-se ingerir glicose ou frutose antes de qualquer exerccio, mesmo sendo este de pequena intensidade ou pouco vigoroso. VI: DOENA DE HERS O diagnstico de deficincia de fosforilase heptica, foi previamente aplicado a um grupo variado de pacientes que apresentava redues dos nveis de fosforilase heptica secundrias a diversas causas, mas atualmente limitado a pacientes nos quais a deficincia de fosforilase heptica corresponde a um defeito primrio. Esta dificuldade nosolgica uma conseqncia do fato de que a fosforilase existe tanto na forma ativa quanto na forma inativa e que muitos fatores so capazes de inibir a ativao secundria da enzima. Consequentemente, este diagnstico exige a documentao do fato de que a fosforilase heptica deficiente e de que fosforilase b-quinase responsvel por uma ativao normal. O distrbio provavelmente deve-se a uma mutao autossmica recessiva. Tratamento: A maioria dos pacientes tem manifestaes semelhantes quelas verificadas no tipo III, mas numa forma mais branda. O diagnstico suspeito a partir de uma hepatomegalia ou de uma hipoglicemia e os pacientes costumam responder a condutas dietticas semelhantes quelas empregadas na doena do tipo III. TIPO VII: DEFICINCIA DE FOSFOFRUTOQUINASE Essa deficincia manifestas-se com mialgias e fadiga que ocorrem depois de exerccios e representam as manifestaes clnicas predominantes. Ao contrrio da doena de McArdle, os pacientes com deficincia de fosfofrutoquinase pioram com a ingesto de carbodratos. Os pacientes se apresentam com rabdomilise e mioglobinria. A paresia permanente dos membros menos comum do que na doena de McArdle. Em virtude de deficincia eritrocitria, uma leve anemia hemoltica muitas vezes ocorre e pode ser til no diagnstico. A deficincia de fosfofrutoquinase transmitida como trao autossmico recessivo. O distrbio tem um predomnio masculino incomumente acentuado e representao exagerada entre judeus asquenazi. Tratamento: A anemia da deficincia no costuma exigir tratamento. A manipulao da dieta no altera os sintomas musculares. OUTRAS DEFICINCIAS: TIPO VIII: DEFICINCIA DE FOSFORILASE-QUINASE HEPTICA Nessa deficincia temos a principal estrutura afetado o fgado, a estrutura do glicognio no sofre alteraes. TIPO IX: DEFICINCIA DE SINTETASE HEPTICA Neste caso, na verdade, no temos uma glicogenose e sim uma hipoglicogenose da enzima sintetase heptica. O principal rgo afetado o fgado e a estrutura do glicognio no sofre alteraes. TIPO X: DEFICINCIA DE FOSFOGLICERATO MUTASE Nesse raro distrbio, os pacientes se apresentam com mialgia, cibras musculares e mioglobinria depois de exerccio intenso. Nenhum outro rgo do sistema comprometido. O padro da herana provavelmente autossmico recessivo, embora no tenham sido descritas famlias com vrias geraes ou parentes acometidos.
Tratamento: No se dispe de tratamento especfico. TIPO XI: DEFICINCIA DE DESIDROGENASE LTICA A desidrogenase ltica (LDH) uma enzima tetramrica composta por duas subunidades, M e H. as isoenzimas da subunidade M predominam no msculo esqueltico, enquanto as isoenzimas que contm a subunidade H so os componentes principais da LDH no corao Os pacientes com deficincia de LDH-M apresentam intolerncia ao esforo, dor muscular e mioglobinria em adolescentes. Nenhum outro sistema orgnico est envolvido. A doena pode ser herdada como trao autossmico recessivo. A subunidade M da LDH se localiza no cromosssomo 11. Uma deficincia da subunidade da LDH, a isoforma cardaca, foi descrita mas no se associa a sintomas do msculo esqueltico. Tratamento: Atualmente no h tratamento para esse distrbio. DIAGNSTICO E DIAGNSTICO PR-NATAL DAS GLICOGENOSES A anlise enzimtica, para fins diagnsticos, em tecidos hepticos ou musculares de todos os tipos de doenas do armazenamento do glicognio, est atualmente concentrada no Duke medical Center, Diviso de Gentica. O diagnstico pr-natal de trs tipos de doenas de armazenamento do glicognio (tipos II, III e IV) tambm possvel, sendo realizado atravs de culturas de clulas amniticas neste laboratrio. CONCLUSO As Doenas de de Armazenamento de Glicognio, ou Glicogenoses, so de fundo gentico, isto , esto relacionadas a falhas geradas durante a transcrio e traduo de protenas ou durante a fecundao, ou ainda durante o desenvolvimento embriolgico. So patologias raras, de difcil diagnstico, geralmente letais quando no diagnosticadas j ao nascimento e que se percebidas, com uma dieta especfica e exerccios nos casos de distrbios com fisiopatologia energtica muscular, podem at serem controladas Fonte: http://members.tripod.com/medworks/Bioquimica/glicogenoses.htm