
DISSERTACAO JOEL PRONTA -...
116
SEPARAÇÃO DE HIDROGÊNIO DE MISTURAS GASOSAS ATRAVÉS DE MEMBRANA DE CARBONO DO TIPO FIBRA OCA Joel Santana do Nascimento Dissertação de Mestrado apresentada ao Programa de Pós-graduação em Engenharia Química, COPPE, da Universidade Federal do Rio de Janeiro, como parte dos requisitos necessários à obtenção do título de Mestre em Engenharia Química. Orientador (es): Vera Maria Martins Salim Cristiano Piacsek Borges Rio de Janeiro Março de 2010 COPPE/UFRJ COPPE/UFRJ
Transcript of DISSERTACAO JOEL PRONTA -...

SEPARAÇÃO DE HIDROGÊNIO DE MISTURAS GASOSAS ATRAVÉS DE
MEMBRANA DE CARBONO DO TIPO FIBRA OCA
Joel Santana do Nascimento
Dissertação de Mestrado apresentada ao
Programa de Pós-graduação em Engenharia
Química, COPPE, da Universidade Federal do
Rio de Janeiro, como parte dos requisitos
necessários à obtenção do título de Mestre em
Engenharia Química.
Orientador (es): Vera Maria Martins Salim
Cristiano Piacsek Borges
Rio de Janeiro
Março de 2010
COPPE/UFRJCOPPE/UFRJ

SEPARAÇÃO DE HIDROGÊNIO DE MISTURAS GASOSAS ATRAVÉS DE
MEMBRANA DE CARBONO DO TIPO FIBRA OCA
Joel Santana do Nascimento
DISSERTAÇÃO SUBMETIDA AO CORPO DOCENTE DO INSTITUTO ALBERTO
LUIZ COIMBRA DE PÓS-GRADUAÇÃO E PESQUISA DE ENGENHARIA
(COPPE) DA UNIVERSIDADE FEDERAL DO RIO DE JANEIRO COMO PARTE
DOS REQUISITOS NECESSÁRIOS PARA A OBTENÇÃO DO GRAU DE MESTRE
EM CIÊNCIAS EM ENGENHARIA QUÍMICA.
Examinada por:
______________________________________________________
Profª. Vera Maria Martins Salim, D.Sc. ______________________________________________________
Prof. Cristiano Piacsek Borges, D.Sc. ______________________________________________________
Prof. Fábio Barboza Passos, D.Sc. ______________________________________________________
Profª. Maria Eugênia Ribeiro de Sena Piacsek Borges, D.Sc.
RIO DE JANEIRO, RJ - BRASIL
MARÇO DE 2010

iii
Nascimento, Joel Santana do
Separação de Hidrogênio de Misturas gasosas através
de Membrana de Carbono do Tipo Fibra Oca / Joel Santana
do Nascimento – Rio de Janeiro: UFRJ/ COPPE, 2010.
IX, 107 p: Il.; 29,7 cm.
Orientadores: Vera Maria Martins Salim
Cristiano Piacsek Borges
Dissertação (mestrado) – UFRJ/ COPPE/ Programa
de Engenharia Química, 2010.
Referências Bibliográficas: p. 89-99.
1. Membrana Fibra Oca de Carbono. 2. Separação de
Misturas Gasosas. 3. Hidrogênio. 4. Pirólise. I. Salim, Vera et
al. II. Universidade Federal do Rio de Janeiro, COPPE,
Programa de Engenharia Química. III. Título.

iv
Dedico este trabalho a meus pais, José Santana e
Luzia e a meus irmãos, Luiz, Eliel, Irineu e Míria.

v
AGRADECIMENTOS
- A Deus;
- A minha família pelo amor incondicional;
- A Profª. Vera e Prof. Cristiano pela imprescindível orientação;
- A Profª. Eugênia e Prof. Fábio pela aceitação do convite para avaliação deste
trabalho;
- A todos aqueles que contribuíram direta ou indiretamente para a realização
deste trabalho, em especial a: Amanda, Alan, Felipe e Walter por me ensinarem a
montar extrusora, fazer fiação e utilizar sistema de permeação de gases; Mariana por me
ensinar a fazer análises de MEV; Bob pela ajuda durante a montagem de módulos e
sistemas de permeação; Antônio (Bebezão) pela ajuda durante preparo das misturas;
Anacleto pela imprescindível ajuda durante montagem de sistema e análises em
espectrômetro de massas, Ayr pelas análises cromatográficas das misturas; Beth pelas
análises de TGA.
- A todos os amigos, em especial o pessoal do PAM pelos grandes momentos de
alegria e diversão vividos juntos e os colegas de república: Iran e Danilo pela
companhia e motivação.

vi
Resumo da Dissertação apresentada à COPPE/UFRJ como parte dos requisitos
necessários para a obtenção do grau de Mestre em Ciências (M. Sc.)
SEPARAÇÃO DE HIDROGÊNIO DE MISTURAS GASOSAS ATRAVÉS DE
MEMBRANA DE CARBONO DO TIPO FIBRA OCA
Joel Santana do Nascimento
Março/2010
Orientadores: Vera Maria Martins Salim
Cristiano Piacsek Borges
Programa: Engenharia Química
Reator com membrana para produção de H2 através da reforma do gás natural
demanda membrana com elevada estabilidade térmica. Neste trabalho membrana de
carbono (MC) na forma de fibra oca foi obtida a partir da pirólise de membrana
polimérica (MP) à base de PEI, PVP e sílica. As MP e MC produzidas foram
caracterizadas por microscopia eletrônica de varredura, análise térmica e permeação de
gases. Ambas as membranas mostraram-se bem formadas, isentas de defeitos ou
macrovazios. Enquanto a MP apresentou baixa seletividade a gases (α 1) e baixa
resistência térmica (até 400 °C), a MC resultante apresentou alta estabilidade térmica
(600 °C em atmosfera oxidante e mais que 900 °C em atmosfera inerte) e alta
seletividade (αH2/CO2 > 380 GPU a 30 °C e αH2/CO = 168 a 80 °C). O aumento da
temperatura tem provocado ganho significativo na permeabilidade de H2 na MC (de 0,4
GPU a 27 °C a 2,2 GPU a 180 °C). Estes resultados sugerem que a MC produzida
mostra-se adequada para separação de H2 em processos onde a estabilidade térmica é
necessária.

vii
Abstract of Dissertation presented to COPPE/UFRJ as a partial fulfillment of the
requirements for the degree of Master of Science (M.Sc.).
HYDROGEN SEPARATION FROM GASEOUS MIXTURES THROUGH CARBON
HOLLOW FIBER MEMBRANE
Joel Santana do Nascimento
Março/2010
Advisors: Vera Maria Martins Salim
Cristiano Piacsek Borges
Departament: Chemical Engineering
Membrane reactor for H2 production by natural gas reforming demands
membrane with high thermal stability. In this work, carbon hollow fiber membrane
(CM) was prepared by pyrolysis of polymeric membrane (MP) based on PEI, PVP and
silica. The produced PM and CM were characterized by scanning electron microscopy,
thermal analysis and gas permeation. Both membranes presented good shape and
neither defect nor macrovoid was identified. While the PM showed low selectivity to
gases (α 1) and low thermal resistance (up to 400 °C), the resulting CM showed high
thermal stability (up to 600 °C in oxidizing atmosphere and more than 900 °C in inert
atmosphere) and high selectivity (αH2/CO2 > 380 GPU at 30 °C and αH2/CO = 168 at
80°C). The temperature increase has caused significant gain in the H2 permeability
(from 0.4 GPU at 27 °C to 2.2 GPU at 180 °C). These results suggest that the produced
CM is adequate for H2 separation in cases where thermal stability is required.

viii
Sumário
1. INTRODUÇÃO ....................................................................................................................... 1
2. REVISÃO BIBLIOGRÁFICA ................................................................................................ 6
2.1. FONTE, DEMANDA E PRODUÇÃO INDUSTRIAL DE HIDROGÊNIO ................... 6
2.2. PROCESSOS DE SEPARAÇÃO/PURIFICAÇÃO DE HIDROGÊNIO ........................ 8
2.2.1. Processos clássicos ................................................................................................... 9
2.2.2. Processos com membrana ....................................................................................... 10
2.3. PRODUÇÃO DE MEMBRANAS DE CARBONO PARA SEPARAÇÃO DE H2 ...... 19
2.3.1. Precursores poliméricos .......................................................................................... 20
2.3.2. Pré-tratamento ........................................................................................................ 22
2.3.3. Pirólise .................................................................................................................... 24
2.3.4. Pós-tratamento ........................................................................................................ 25
2.3.5. Envelhecimento da membrana ................................................................................ 26
2.3.6. Adição de carga inorgânica .................................................................................... 27
2.4. CONFIGURAÇÃO DE MÓDULOS ............................................................................. 28
2.5. MECANISMOS DE TRANSPORTE EM MEMBRANAS DE CARBONO ................ 32
3. METODOLOGIA EXPERIMENTAL ................................................................................... 38
3.1. PREPARO DA MEMBRANA POLIMÉRICA ............................................................. 38
3.1.1. Materiais ................................................................................................................. 38
3.1.2. Preparo da solução .................................................................................................. 39
3.1.3. Fiação...................................................................................................................... 39
3.2. PREPARO DA MEMBRANA DE CARBONO ............................................................ 41
3.2.1. Sistema de Pirólise .................................................................................................. 41
3.2.2. Pré-tratamento e Pirólise......................................................................................... 42
3.3. CARACTERIZAÇÃO DAS MEMBRANAS ................................................................ 43
3.3.1. Análise de Resistência Térmica .............................................................................. 43
3.3.2. Análise da Morfologia ............................................................................................ 43
3.3.3. Permeação Gasosa .................................................................................................. 43
3.3.3.1. Sistema de Permeação ............................................................................................ 43
3.3.3.2. Princípios teóricos .................................................................................................. 45
3.4. MONTAGEM DE MÓDULOS COM CONJUNTO DE FIBRAS OCAS .................... 48
3.4.1. Preparo da carcaça .................................................................................................. 48

ix
3.4.2. Preparo das fibras ................................................................................................... 49
3.4.3. Montagem do módulo ............................................................................................. 49
3.5. PERMEAÇÃO EM MÓDULO COM FIBRAS OCAS DE CARBONO ...................... 50
3.5.1. Preparo das Misturas .............................................................................................. 50
3.5.2. Sistema de Permeação ............................................................................................ 52
3.5.3. Curvas de Calibração dos componentes das misturas em EM ................................ 54
4. RESULTADOS E DISCUSSÕES ......................................................................................... 58
4.1. CARACTERIZAÇÃO DAS MEMBRANAS ................................................................ 58
4.1.1. Análise Termogravimétrica .................................................................................... 58
4.1.2. Análise da Morfologia ............................................................................................ 61
4.1.3. Caracterização por Permeação de Gases Puros ...................................................... 65
4.2. PERMEAÇÃO DE MISTURAS .................................................................................... 66
4.2.1. Mistura H2/He/CO2/CH4 ......................................................................................... 67
4.2.2. Permeação de He e H2 puros................................................................................... 74
4.2.4. Permeação da mistura H2/CO ................................................................................. 80
5. CONCLUSÃO ....................................................................................................................... 86
6. SUGESTÕES PARA TRABALHOS FUTUROS ................................................................. 88
7. BIBLIOGRAFIA ................................................................................................................... 89
ANEXO ..................................................................................................................................... 100
TESTES PRELIMINARES DE PRODUÇÃO DE MEMBRANAS, MONTAGEM DE MÓDULOS E PERMEAÇÃO .................................................................................................. 100

1
1. INTRODUÇÃO
O emprego de processos de separação por membrana na indústria química
apresenta uma série de vantagens em relação aos processos tradicionais que vão desde
economia de energia e área construída, simplicidade e flexibilidade operacional e de
projeto, seletividade, qualidade do produto final e redução dos impactos ambientais. A
separação através de membranas em sua grande maioria não envolve mudanças de fases
e isto se traduz em grande economia de energia comparada aos processos tradicionais.
Os processos de separação por membrana são compactos resultando em economia de
espaço e área construída. São sistemas modulares permitindo o aumento de escala sem a
necessidade de novo projeto uma vez que as características observadas em unidades
menores como em escala piloto, por exemplo, se mantêm em grandes escalas. Os
processos de separação por membrana podem substituir ou serem acoplados aos
processos tradicionais de separação, melhorando a eficiência do processo sem, contudo
exigir transformações bruscas e investimentos elevados. A seletividade é uma
característica muito importante dos processos de separação com membrana levando-os a
serem, em alguns casos, a única alternativa técnica de separação, exemplo disso é a
produção de H2 de alta pureza para uso na indústria eletrônica e para aplicações
espaciais. Qualidade do produto e aspectos ambientais são favorecidos por dispensar o
uso de outros produtos químicos para efetuar a separação e pela economia de energia.
Um dos grandes desafios da humanidade atualmente é assegurar o suprimento de
energia concomitante à devida proteção ao meio ambiente. Os combustíveis fósseis têm
sido determinantes nas transformações econômicas, políticas e sociais da população
moderna. No entanto, as incertezas quanto à disponibilidade e preço do petróleo a curto
e longo prazo, bem como, os danos visíveis causados ao meio ambiente pela poluição
gerada devido ao uso desta fonte de energia, têm provocado intensiva busca por fontes
alternativas de energias, mais eficientes e menos poluidoras.
O H2 tem se mostrado bastante promissor na substituição dos combustíveis
fósseis devido a sua alta eficiência energética e principalmente por ser não poluente e
apresentar grande disponibilidade podendo ser obtido de grande quantidade de fontes

2
renováveis. Hidrogênio é o elemento químico mais abundante na natureza, constituindo
75% da massa do universo e está presente em 70% da superfície terrestre fazendo parte
da composição molecular da água e de compostos orgânicos (SOUZA, 2009). H2
apresenta o maior conteúdo energético por unidade de massa: 1,21x108 J/kg (DOE,
2004 citado por BARBOSA-COUTINHO, 2004), sendo sua utilização em células a
combustível muito mais eficiente que a queima de combustíveis fósseis e a fissão
nuclear (BOND, 1987) e tanto em combustão quanto em célula a combustível H2
apresenta liberação zero de poluentes. O hidrogênio é uma substância portadora de
energia possibilitando com isso a exploração de fontes remotas (ondas, hidroelétricas
etc.) ou com distribuição irregular ao longo do tempo (eólica, solar etc.) (SOUZA,
2009).
Embora seja enorme a quantidade de fontes de H2, todas elas ainda apresentam
barreiras que necessitam ser vencidas até a consolidação do uso destas fontes.
Atualmente, a principal fonte de produção de H2 é a reforma com vapor do gás natural.
Este processo gera corrente gasosa contendo principalmente H2 e CO. Mas quando o
interesse não é a produção de gás de síntese, e sim a produção de H2, uma segunda
etapa, reação de deslocamento gás-água, é aplicada ao processo gerando principalmente
H2 e CO2 que necessita de posterior etapa de separação para o H2 atingir a pureza
necessária para a aplicação a que se destina.
Os principais processos de separação de H2 encontrados industrialmente são a
adsorção e a destilação criogênica (OCKWIG E NENOFF, 2007). Processos de
separação de H2 por membranas ainda ocupam uma pequena parcela das aplicações
industriais, sendo representada principalmente por membranas poliméricas, sobretudo
devido à facilidade e o baixo custo de fabricação destas membranas. Além disso, estas
membranas apresentam alta permeabilidade e boas características mecânicas o que torna
fácil a construção de módulos empregando-as. No entanto, estas membranas apresentam
limitações quando se necessita altas seletividades e estão sujeitas à compactação em
altas pressões, bem como, apresentam baixa resistência térmica e química etc. Como
alternativa têm-se as membranas inorgânicas cuja obtenção com propriedades
adequadas, como a uniformidade na distribuição de tamanho de poros e ausência de
defeitos ou rachaduras, é muitas vezes o fator limitante para o emprego deste tipo de
membrana. Dentre as membranas inorgânicas, as membranas de carbono merecem

3
destaque devido apresentar grande flexibilidade de obtenção. Através da escolha
adequada do precursor polimérico, das condições de preparo e pós-tratamento é possível
obter membranas de carbono com uma grande variedade de morfologia e propriedades
de permeação (SAUFI E ISMAIL, 2004). A separação por mecanismo de peneira
molecular tem mostrado superar o limite de permeabilidade e seletividade intrínseca das
membranas poliméricas, representado por diagrama primeiramente proposto por
ROBESON (1991). Contudo, o custo de produção de membrana de carbono ainda é de
uma a três vezes maior que o custo de produção de membranas poliméricas. Então sua
utilização é justificada apenas em condições onde seu desempenho é superior ao
desempenho de membranas poliméricas (OCKWIG E NENOFF, 2007). Exemplo disso
é a aplicação em reator com membrana na reforma do gás natural para produção de H2.
Neste caso a membrana deve apresentar alta seletividade e estabilidade térmica, bem
como, resistência química e resistência a altas pressões.
Neste contexto, este trabalho, desenvolvido no Laboratório de Separação com
Membranas e Polímeros em conjunto com o Núcleo de Catálise, ambos pertencentes ao
Programa de Engenharia Química da COPPE/UFRJ, dá continuidade a uma linha de
pesquisa desenvolvida por MOREIRA (2004), BARBOSA-COUTINHO (2004) e
MOREIRA (2008).
BARBOSA-COUTINHO (2004) desenvolveu estudos iniciais sobre o preparo
de membranas de carbono a partir de membranas poliméricas à base de PEI e PVP,
conseguindo membrana de carbono com permeabilidade a Hélio igual a 5,6.10-8
cm3(CNTP).cm-2.s-1.cmHg-1 a 25 °C e seletividade He/N2 igual a 17. A autora avaliou o
emprego destas membranas de carbono em reatores catalíticos, bem como o emprego de
membrana catalítica, desenvolvidas por MOREIRA (2004) pela deposição de filmes
finos de Pt na superfície destas membranas de carbono, em reatores com membranas
catalíticas para a geração de hidrogênio a partir do gás natural. Não foi observado efeito
significativo do deslocamento de equilíbrio nestes reatores devido à baixa
permeabilidade da membrana.
Em continuidade a estes trabalhos, MOREIRA (2008) fez estudos relacionados à
melhoria das propriedades morfológicas e de transporte das membranas de carbono
conseguindo melhor desempenho em termos de permeabilidade a Hélio (6,0.10-7

4
cm3(CNTP).cm-2.s-1.cmHg-1 a 25 °C) e seletividade He/CO2 (60). A autora investigou
ainda a adição de sílica como aditivo inorgânico gerador de porosidade na membrana de
carbono conseguindo permeabilidade igual a 1,5.10-6 cm3(CNTP).cm-2.s-1.cmHg-1 a 25
°C mantendo a seletividade He/CO2 igual a 60.
Devido a riscos encontrados durante avaliação da permeação de H2 em sistema
elétrico (transdutor de pressão) e à necessidade de quantificação do permeado de
misturas, as propriedades de separação da membrana de carbono nestas condições, bem
como empregando temperatura, ainda não haviam sido averiguadas.
Dentro deste contexto, o objetivo geral deste trabalho de dissertação de mestrado
é a avaliação das propriedades de transporte e separação de H2 de misturas gasosas
através de membrana de carbono em diferentes pressões e temperaturas. Os objetivos
específicos são:
• Obtenção de membrana fibra oca de carbono sintetizada a partir de
membrana polimérica à base de PEI e PVP contendo sílica dispersa em
sua matriz para geração de poros, empregando metodologia previamente
desenvolvida pelo grupo (BARBOSA, 2004 E MOREIRA, 2008);
• Configuração de módulo empregando estas membranas fibras ocas de
carbono e montagem de sistema de permeação acoplado a espectrômetro
de massas para quantificação do permeado;
• Avaliação do desempenho deste módulo, em diferentes pressões e
temperaturas, durante a permeação de gases puros e separação de H2 de
misturas contendo os principais constituintes de corrente resultante de
reforma com vapor do gás natural e de reação de deslocamento gás-
água.
Esta dissertação está estruturada contendo, além desta introdução, uma revisão
bibliográfica, metodologia experimental, resultados e discussões, conclusões, sugestões
para trabalhos futuros e anexo. Na revisão bibliográfica são apresentados de forma geral
assuntos relacionados à produção e purificação industrial de H2, possibilidade de
aplicação de processos com membrana, com ênfase principalmente na produção e
aplicação de membrana de carbono. Na metodologia experimental são descritos os
procedimentos experimentais empregados durante a realização deste trabalho. Nos

5
resultados e discussões, os principais dados experimentais são apresentados e
analisados. Na conclusão são apresentadas as conclusões a que se chegaram a partir da
análise destes resultados. Por fim, na parte de sugestões para trabalhos futuros, são
apresentadas áreas que podem ser exploradas para melhor compreensão dos fenômenos
aqui observados e no anexo são citados testes preliminares envolvendo a produção de
membranas, montagem de módulos e testes de permeação de gases.

6
2. REVISÃO BIBLIOGRÁFICA
2.1. FONTE, DEMANDA E PRODUÇÃO INDUSTRIAL DE HIDROGÊNIO
Hidrogênio é o elemento químico mais abundante na natureza, constituindo 75%
da massa do universo e presente em 70% da superfície terrestre, fazendo parte da
composição molecular da água e de compostos orgânicos (SOUZA, 2009). Na forma
molecular (H2) é altamente inflamável e reage facilmente com outras substâncias sendo
possível encontrá-lo, apenas em quantidades muito pequenas (traços) na atmosfera
(SHREVE E BRINK, 1997). Para aplicação industrial ou como combustível é, então,
necessária a síntese desta substância a partir de hidrocarbonetos ou água (GRAINGER,
2007).
O hidrogênio apresenta crescente demanda nas refinarias para vários processos,
tais como o de hidrotratamentos, hidrocraqueamento, hidrodessulfurização e
hidrodesnitrogenação. Assim como no controle da quantidade de aromáticos e
compostos com enxofre presentes na gasolina, e controle da emissão de NOx. É também
utilizado nas indústrias: químicas (sobretudo para produção de amônia, metanol e
peróxido de hidrogênio), siderúrgicas (produção do aço), indústrias de alimento
(hidrogenação de óleos e gorduras), farmacêuticas (vitaminas), eletrônicas
(semicondutores) e têxteis. Além disso, a crescente preocupação com os efeitos
ambientais causados pela poluição gerada pela queima de combustíveis derivados do
petróleo tem levado a crescente busca por combustíveis alternativos menos poluidores.
Acredita-se que o hidrogênio seja uns dos mais promissores, devido sua alta eficiência
energética e pela não emissão de poluentes durante a sua utilização em células a
combustível (SOUZA, 2009, OCKWIG E NENOFF, 2007).
Os combustíveis fósseis são as principais matérias-primas para a produção de
hidrogênio, sendo o gás natural responsável por 48%, o petróleo por 30% e o carvão por
18% da produção mundial (ARMOR, 1999). Entre estes, o gás natural é o mais
adequado para utilização como matéria-prima devido apresentar maior conteúdo
relativo de hidrogênio, o que resulta em menores quantidades de CO2 liberados durante

7
a geração de hidrogênio. Além disso, as reservas mundiais comprovadas de gás natural
excedem as reservas de petróleo (LUNSFORD, 2000, SOUZA, 2009).
O hidrogênio pode ser obtido do gás natural (cujo principal constituinte é CH4)
através da oxidação parcial, reforma com vapor, reforma com CO2, reforma autotérmica
ou conversão direta em condições não oxidativas. A reforma com vapor é o processo
mais utilizado industrialmente. Neste processo, metano (CH4) e excesso de vapor (H2O)
reagem formando gás de síntese: monóxido de carbono (CO) e hidrogênio (H2) (Reação
1). Este processo é altamente endotérmico, e geralmente ocorre em temperaturas
superiores a 800 °C, o que acaba por consumir elevada quantidade de energia. Excesso
de vapor é normalmente utilizado para redução da formação de coque sobre o
catalisador e aumento da conversão do metano (DYBKJAER, 1995) e, isto provoca
aumento no tamanho dos equipamentos e redução de eficiência energética, com
conseqüente elevação nos custos operacionais (SOUZA, 2009).
3 206 Reação 1
Quando se visa à produção de hidrogênio e não de gás de síntese, hidrogênio
adicional pode ser obtido empregando-se um segundo processo, onde monóxido de
carbono reage com vapor formando H2 e CO2 (Reação 2).
41 Reação 2
Esta reação é conhecida como reação de deslocamento de gás – água (reação de
shift) e é considerada como sendo de alta temperatura quando ocorre na faixa de 340 a
400 °C ou de baixa temperatura, quando ocorre na faixa de 190 a 210 °C. Enquanto a
reação de baixa temperatura favorece a conversão do CO, a de alta temperatura permite

8
a recuperação do calor da reação em um nível de temperatura suficiente para gerar
vapor de alta pressão (SOUZA, 2009, OCKWIG E NENOFF, 2007).
A composição do gás efluente depende da natureza do processo de reação de
deslocamento. Geralmente, reação a alta temperatura (~ 350 °C) e reação a baixa
temperatura (~ 200 °C) geram correntes com as composições próximas àquelas
apresentadas na Tabela 2.1 (MOLBURG, 2003, OCKWIG E NENOFF, 2007).
Tabela 2.1. Composição do gás efluente resultante da reforma com vapor do gás natural de acordo com a temperatura da reação de deslocamento gás-água empregada.
Condição de Reação Temperatura Composição (%)
H2 CO2 CH4 CO Alta Temperatura 350 °C 73,9 17,7 6,9 1
Baixa Temperatura 200 °C 74,1 18,5 6,9 0,1
Para a grande maioria das aplicações a que se destina, o hidrogênio produzido
deste processo necessita de uma posterior etapa de purificação a fim de atingir a
composição desejada.
2.2. PROCESSOS DE SEPARAÇÃO/PURIFICAÇÃO DE HIDROGÊNIO
A indústria de petróleo apresenta crescente necessidade de separação de
hidrogênio de outros gases. Além da separação de H2 durante sua produção, há também
a necessidade de recuperação de H2 de gases de purga em refinarias, tais como corrente
de purga de hidrodessulfurização (separação de H2/CH4) e corrente de purga da amônia

9
(H2/N2), ajuste da composição de gás de síntese (H2/CO) e a recuperação de H2 de
correntes contendo H2 e H2S na gaseificação do carvão, entre outros (NATIONAL
RESEARCH COUNCIL U.S., 1998).
2.2.1. Processos clássicos
A separação de hidrogênio de correntes gasosas em escala industrial é em sua
grande maioria realizada por processos tradicionais: adsorção, processo conhecido como
PSA (sigla originada da expressão inglesa: Pressure Swing Adsorption) ou destilação
criogênica. A destilação criogênica ou destilação fracionada é um processo de separação
baseado na diferença da temperatura de condensação dos componentes da mistura de
alimentação (MILLER E STOECKER, 1989, ADHIKARI E FERNANDO, 2006). H2
tem volatilidade muito elevada quando comparado com hidrocarbonetos, sendo possível
atingir alto grau de recuperação. No entanto só é possível atingir moderado grau de
pureza (até 95 %), não sendo prática a obtenção de hidrogênio de alta pureza através
deste processo. Por ser um processo de separação a baixíssimas temperaturas, há um
considerável consumo de energia (HINCHLIFFE E PORTER, 2000, ADHIKARI E
FERNANDO, 2006). Os primeiros usos da adsorção datam de 1980 e por ser mais
econômico em energia, resultou em ganho de produtividade e se tornou o processo mais
extensivamente utilizado para separação de H2 de misturas gasosas. O princípio de
separação por adsorção se baseia em interações entre os componentes da mistura gasosa
e o adsorvente (peneira molecular, sílica gel ou carvão ativado), submetidos a altas
pressões, já que os adsorventes têm capacidade de adsorver mais impurezas a altas
pressões parciais que em baixas pressões parciais. Ao aproximar das condições de ponto
de orvalho os componentes menos voláteis são adsorvidos. A adsorção é ainda
favorecida por forças eletrostáticas, sendo, portanto, favorecida a adsorção de moléculas
mais polares ou polarizáveis enquanto que o hidrogênio e o hélio, compostos altamente
voláteis e de baixa polaridade, são preferencialmente não adsorvidos (STOCKER,
2005). O processo é dividido em 5 etapas: (i) adsorção, (ii) despressurização, (iii)
despressurização contracorrente, (iv) purga e (v) pressurização. Para ativação do

10
adsorvente, o sistema necessita ser despressurizado e lavado com H2 puro. Além disso,
este é um processo operado em batelada sendo necessária a utilização de vários
adsorventes para a obtenção de fluxo contínuo. A quantidade de H2 recuperada é
dependente da pressão de alimentação, pressão do gás de purga, grau de impurezas e
concentração de H2, sendo, contudo, possível atingir altas seletividades (99,99 %)
(STOCKER, 2005, ADHIKARI E FERNANDO, 2006).
2.2.2. Processos com membrana
Nos últimos anos, os processos com membrana para separação de gases estão
ganhando grande aceitação da indústria e estão competindo no mercado com operações
consolidadas, tais como, a adsorção e a destilação criogênica (BERNARDO et al.,
2009). Isto se deve às melhorias conseguidas nas propriedades das membranas e à série
de vantagens apresentadas pelos processos de separação por membrana em relação aos
processos clássicos. Algumas destas vantagens são apresentadas a seguir (HABERT et
al., 2006).
• Os processos de separação por membrana não necessitam de mudanças de
fases, traduzindo em elevada economia de energia em relação aos
processos clássicos;
• São sistemas compactos o que representa economia em área construída;
• São sistemas modulares o que possibilita aumento de escala aproveitando
as unidades já construídas;
• Apresentam facilidade de operação e possibilidade de funcionamento
contínuo;
• Permitem a integração de processos (sistemas híbridos) possibilitando
ganhos em desempenho. Por exemplo: Combinação de processos de
separação com membranas e processos clássicos para separação gasosa
(ESTEVES E MOTA, 2007) e reator com membrana (ITOH E HARAYA,
2000, BARBOSA-COUTINHO, 2004).

11
Os processos de separação por membranas se expandiram na década de 50 com a
descoberta de produção de membranas poliméricas assimétricas através da técnica de
inversão de fase por imersão-precipitação por LOEB E SOURIRAJAN (1963). A
melhoria na seletividade e a redução da resistência ao transporte das espécies
permeantes representavam alterações que poderiam tornar os processos de separação
por membrana mais competitivos do que os processos de separação tradicionais. O
procedimento de preparar membranas compostas, sugerido nos trabalhos de CADOTTE
e FRANCIS (1996), permitiu a exploração comercial de processos de separação por
membrana para separação de misturas gasosas e a empresa americana Monsanto, no
início dos anos 80, foi a pioneira na aplicação industrial de membranas para separação
de gases (HABERT et al., 2006).
Acredita-se que para a hegemonia dos processos de separação por membranas em
separações gasosas, além da conscientização de suas vantagens, serão, ainda,
necessárias melhorias na qualidade das membranas e configuração de processos
(BURGGRAAF E COT, 1996). No entanto, em algumas situações, os processos de
separação por membrana já vêm encontrando seus espaços e se consolidando como a
melhor opção. Exemplo disso é a utilização de membranas poliméricas para recuperação
de H2 em indústrias de amônia e o uso de membranas de paládio para produção de H2
puro para uso em indústrias eletrônicas. Neste caso, o alto custo de produção da
membrana de paládio é justificado pela necessidade de H2 de alta pureza (CLUITERS,
2004). Membrana é, sem dúvida, uma possibilidade na estratégia de intensificação de
processos através de configurações e métodos inovadores de desenvolvimento baseados
na redução dos custos de produção, redução do consumo de energia e rejeitos gerados
(DAUTZEMBERG E MUKHERJEE, 2001, BERNARDO et al., 2009).
Membranas para separação de H2 podem ser dividas em dois grandes grupos de
acordo com sua composição: membranas poliméricas e membranas inorgânicas. Fazem
parte deste último grupo as membranas metálicas e ligas metálicas, membranas
cerâmicas (óxidas, vidro e zeólitas) e membranas de carbono (BURGGRAAF E COT,
1996, CLUITERS, 2004). Quanto à morfologia, elas podem ser densas ou porosas,
isotrópicas (simétricas) ou anisotrópicas (assimétricas), simples ou compostas

12
(HABERT et al., 2006). Como mostrado na Figura 2.1, ao contrário das membranas
anisotrópicas, as membranas isotrópicas possuem morfologia constante ao longo de toda
a seção transversal mantendo-se ou porosa ou densa. No caso de membranas
anisotrópicas a classificação em densa ou porosa é feita com base na superfície
responsável pela separação (superfície menos porosa ou densa, também conhecida como
pele), sendo assim, se a superfície for porosa a membrana é dita porosa, caso contrário,
a membrana é classificada como densa. Ela será integral quando a pele e suporte forem
do mesmo material e, composta quando forem de materiais diferentes. Na Figura 2.1 é
mostrada uma representação esquemática da seção transversal de membranas com as
morfologias previamente discutidas.
Figura 2.1. Representação esquemática da seção transversal de membranas de diferentes tipos de morfologia (HABERT et al., 2006).
As membranas mais empregadas em sistemas de separação de hidrogênio são
poliméricas, as quais são mais estudadas e com maior número de aplicações

13
(CLUITERS, 2004). Isto porque membranas inorgânicas apresentam custo de produção
bastante superior ao custo de produção de membranas poliméricas, sendo, portanto
investigadas para aplicação em condições onde as membranas poliméricas não
apresentam desempenhos desejáveis, como em ambientes com elevadas temperaturas
e/ou em presença de substâncias nocivas às membranas poliméricas. Um exemplo é a
aplicação em reatores catalíticos com membrana (MULDER, 2000, BURGGRAAF E
COT, 1996).
Membranas poliméricas para separação gasosa são membranas densas,
sintetizadas de polímeros elastoméricos ou vítreos. Enquanto membranas de material
elastomérico apresentam altos fluxos de permeação, com baixa seletividade, as
membranas de material vítreo apresentam alta seletividade e baixo fluxo. As membranas
seletivas a H2 são geralmente vítreas, as quais apresentam capacidade de discriminar
moléculas gasosas por tamanho, sendo possível separar, por exemplo, H2 de CO2, cuja
diferença de diâmetro cinético é de apenas 0,41Å (SHAO et al., 2009). Mas poderia ser
empregada, também, membrana seletiva a CO2, mantendo o H2 enriquecido na corrente
de alimentação. Neste caso deveria ser empregada membrana de material elastomérico
onde o transporte de CO2 é favorecido pela sua maior solubilidade no polímero.
As vantagens das membranas poliméricas são o baixo custo de produção,
facilidade de fabricação e montagem de módulos. Encontra-se em estágio avançado de
desenvolvimento sendo encontradas comercialmente (Air Products, Lind, Air Liquid
etc.). Exemplo disso é o módulo Prisma® de membranas poliméricas lançado pela
empresa Monsanto e hoje de propriedade da empresa Air Products para separação de
hidrogênio. Por outro lado, essas membranas possuem limitada estabilidade térmica ( <
100 °C), alta sensibilidade a plasticização e compactação e, baixa resistência química
(CLUITERS, 2004) e limitado desempenho de separação. O desempenho é geralmente
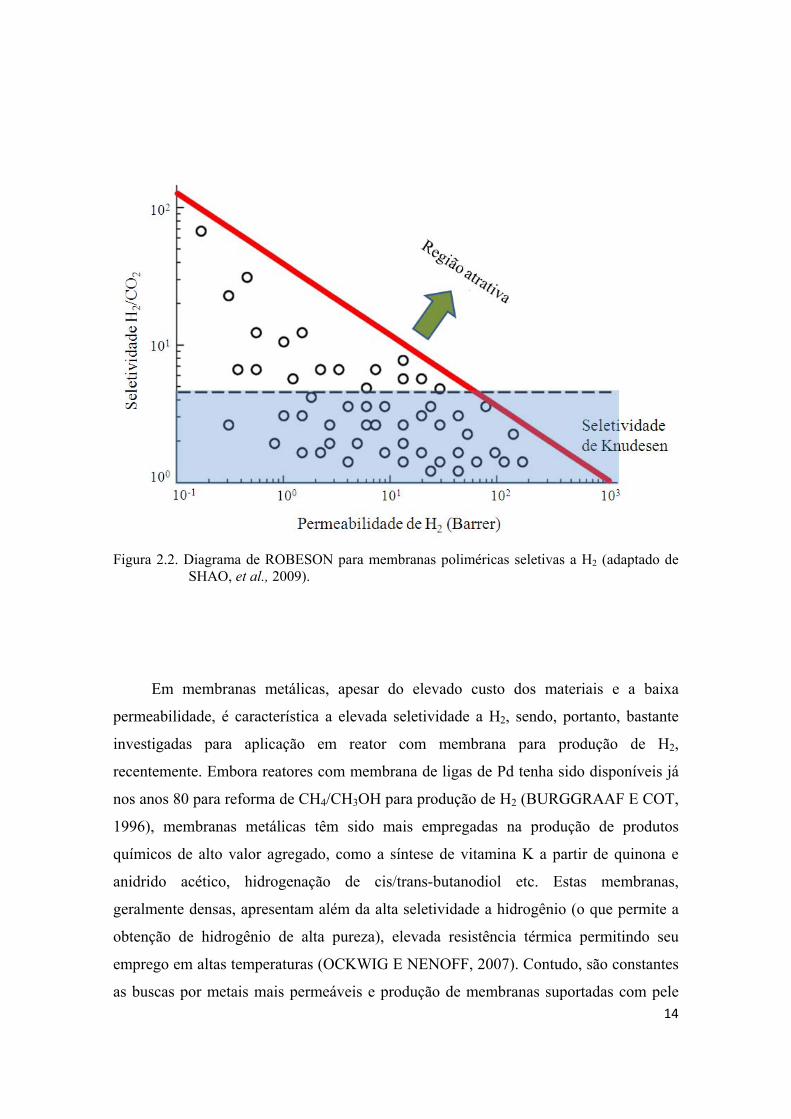
representado por diagrama, primeiramente proposto por ROBESON (1991), de
correlação entre a permeabilidade e seletividade. Na Figura 2.2 é apresentado um
diagrama de permeabilidade de H2 em função da seletividade H2/CO2 para membranas
poliméricas seletivas a H2 a 25 °C (ROBESON et al., 1994).
14
Figura 2.2. Diagrama de ROBESON para membranas poliméricas seletivas a H2 (adaptado de SHAO, et al., 2009).
Em membranas metálicas, apesar do elevado custo dos materiais e a baixa
permeabilidade, é característica a elevada seletividade a H2, sendo, portanto, bastante
investigadas para aplicação em reator com membrana para produção de H2,
recentemente. Embora reatores com membrana de ligas de Pd tenha sido disponíveis já
nos anos 80 para reforma de CH4/CH3OH para produção de H2 (BURGGRAAF E COT,
1996), membranas metálicas têm sido mais empregadas na produção de produtos
químicos de alto valor agregado, como a síntese de vitamina K a partir de quinona e
anidrido acético, hidrogenação de cis/trans-butanodiol etc. Estas membranas,
geralmente densas, apresentam além da alta seletividade a hidrogênio (o que permite a
obtenção de hidrogênio de alta pureza), elevada resistência térmica permitindo seu
emprego em altas temperaturas (OCKWIG E NENOFF, 2007). Contudo, são constantes
as buscas por metais mais permeáveis e produção de membranas suportadas com pele

15
metálica bastante fina (1 a 5 micrômetros), sendo que, neste caso, a diferença de
dilatação térmica e interdifusão de átomos/íons do metal (pele) e suporte entre si podem
afetar a resistência mecânica e propriedades da membrana (BURGGRAAF E COT,
1996).
Pd e Pt são os materiais mais empregados na fabricação de membranas metálicas
devido serem abundantes e apresentarem preços relativamente menores quando
comparados aos outros metais capazes de separar hidrogênio (Nióbio, Vanádio, Tântalo,
Ferro, Níquel e Titânio) (OCKWIG E NENOFF, 2007, ADHIKARI E FERNANDO,
2006). Além disso, BREDSEN et al. (2004) têm conseguido altos fluxos de hidrogênio
em membranas de paládio (3,2 a 160 m3(CNTP)/m2.bar.h).
Para consolidação do uso de membranas metálicas, vários desafios ainda existem
como a produção de pele metálica livre de defeitos, a descoberta de suportes adequados
e solução para o envenenamento por monóxido de carbono (CO), vapor de água e
hidrocarbonetos altamente adsorventes, bem como de substâncias contendo enxofre (S)
e cloro (Cl). Gases contendo impurezas de S (compostos com S como dimetildisulfato,
H2S etc.) ou contendo Cl (CCl4) estão presentes em várias correntes industriais de
hidrocarbonetos e provocam grande perda de permeabilidade. Além disso, membranas
de paládio devem operar a temperaturas altas (300 a 600 °C) caso contrário, o metal em
presença de H2 se torna frágil devido sofrer transições de fases entre diferentes fases
híbridas de Pd-H (fase α e fase β) acarretando perda de resistência mecânica. Para
minimizar estes efeitos, é necessária a formação de ligas de Pd com outros metais (Ag,
Ru, Rh) o que diminui a temperatura de transição de fase e, em alguns casos, melhora a
permeabilidade a H2. Além dos problemas previamente apresentados, carbono
(comumente produzido em reações de reforma) em deposição na superfície da
membrana tem tendência a se dissolver no seio do metal resultando em falha mecânica
da membrana (BURGGRAAF E COT, 1996).
Membranas cerâmicas, porosas ou densas, são construídas pela combinação de
um metal com não-metal na forma de óxido, nitreto ou carbeto. Membranas cerâmicas
porosas são geralmente feitas de duas camadas, a pele responsável pela separação (mais
fina e com tamanho de poros menor) e o suporte (mais espesso e com poros maiores). A
camada de separação é geralmente à base de alumina, zircônia, titânia ou sílica.

16
Estas membranas são mais empregadas em nanofiltração, para separação de gases
estão ainda em estágio inicial, mas apresentam fluxos promissores de hidrogênio
(capazes de superar o desempenho de membranas poliméricas em termos de
permeabilidade e seletividade) e apresentam resistência química e térmica sendo
adequadas para operação em temperaturas entre 200 e 600 °C (KLUITERS, 2004). A
preocupação com este tipo de membranas é a ocorrência de densificação e sinterização
de poros principalmente em presença de vapor. Membranas microporosas, zeólitas
(ZSM-5 e vários outros tipos de zeólitas-A), têm chamado bastante a atenção devido à
alta estabilidade e excelente eficiência de separação (LI et al., 2008). No entanto,
devido à natureza catalítica das zeólitas, elas são passíveis de envenenamentos e
bloqueio dos poros mais estreitos pela formação de coque resultante da quebra catalítica
de hidrocarbonetos (BURGGRAAF E COT, 1996).
BARRER e colaboradores (1973) foram os pioneiros no preparo de membrana de
carbono para separação gasosa. Estes autores prepararam uma membrana microporosa
pela adição sucessiva de pequenas quantidades de pó de grafite em um tubo de aço inox
seguida de alta compressão a cada adição. Em seguida fizeram testes de difusão de
gases puros (He, H2, D2, N2 e NH3) e de misturas binárias (H2/N2, He/NH3, H2/NH3 e
N2/NH3) em temperaturas menores ou iguais a 0 °C. Eles observaram que a
permeabilidade de NH3 (gás condensável) em mistura era maior que a dos outros gases
menos condensáveis. Isto é explicado pelo mecanismo de difusão
superficial/condensação capilar. Neste mecanismo, o gás mais condensável, presente na
alimentação lado de alta pressão, é seletivamente adsorvido na superfície e difunde
através da superfície da membrana até o lado de baixa pressão sendo,
subseqüentemente, dessorvido. A difusão de componentes não condensáveis pode ser
impedida pelo bloqueio de poros (condensação capilar) devido à sorção do componente
desejado, possibilitando atingir altas seletividades (BARRER et al., 1963). Este tipo de
membrana mostra-se atrativa para enriquecimento de H2 em correntes com baixa
quantidade de H2, uma vez que promove o enriquecimento da corrente em H2
mantendo-a pressurizada para posterior purificação por adsorção (PSA) (RAO E
SIRCAR, 1993a, SIRCAR E RAO, 1999). A grande desvantagem deste tipo de
membrana é que devem atuar em temperaturas baixas, uma vez que a adsorção é
desfavorecida pelo aumento da temperatura. Além disso, só é possível separar

17
substâncias com significativa diferença na capacidade de adsorção e, se uma substância
fortemente adsorvente estiver presente na corrente de alimentação, mesmo que em
pequenas quantidades, a capacidade de separação da membrana será governada pela
adsorção-difusão deste componente, impedindo a permeação do gás de interesse (LI et
al., 2008, NUNES E PEINNEMANN, 2006).
Membranas de carbono do tipo peneira molecular são consideradas como muito
promissoras. Estas apresentam poros menores que 5Å e são capazes de desempenhar
separação de gases com pequena diferença de diâmetro cinético. Este tipo de membrana
apresenta, entre os materiais não tradicionais (cerâmicas, zeólitas etc.), um grande
potencial, pois além da alta seletividade, permeabilidade e, estabilidade em operações a
altas temperaturas e em ambientes corrosivos, é de obtenção relativamente fácil.
Membranas de carbono são geralmente obtidas da decomposição térmica (pirólise) de
membranas poliméricas precursoras (LI et al. 2008). Neste processo, a membrana
mantém a forma da membrana precursora o que torna fácil a produção de membranas
em geometrias de elevada área superficial (capilar e fibra oca) e possibilita o controle de
suas propriedades de separação através do ajuste de parâmetros envolvidos durante a
síntese da membrana. Tais parâmetros envolvem a seleção do precursor, preparo da
membrana polimérica, pré-tratamento do precursor, processo de pirólise e pós-
tratamentos (SAUFI E ISMAIL, 2004).
KORESH E SOFFER (1980) foram os pioneiros no preparo de membrana de
carbono do tipo peneira molecular. Estes autores obtiveram membrana plana de carbono
pela pirólise de folha de papel (fonte de celulose). Devido à baixa resistência mecânica
desta configuração para a montagem de módulos eles passaram a produzir membrana
fibra oca de carbono a partir de membrana polimérica fibra oca obtidas de celulose e
polímeros termorresistentes. Estes trabalhos geraram algumas patentes que foram
compradas pela empresa “Carbon Membrane LTDA. (1990-2001)”, primeira empresa a
comercializar membrana peneira molecular de carbono. Em 2001 foi criada na
Alemanha a empresa “Membrana Mundi” que tinha o objetivo de desenvolver
membrana peneira molecular de carbono para separação gasosa com uma nova
tecnologia, membranas planas, em forma de ondas, organizadas em módulos na forma
de favos. Essa nova configuração era proposta com objetivo de melhorar as
propriedades mecânicas da membrana em relação às fibras ocas. Devido à

18
biocompatibilidade do carbono, além do ajuste das propriedades mecânicas e de adesão
a superfícies metálicas de implantes, esta empresa se tornou, mais tarde, especializada
em equipamentos medicinais a base de carbono (“CINVENTION AG”,
http://www.cinvention.com). Atualmente a empresa inglesa MAST Carbon LTDA
produz membrana microporosa de carbono assimétrica (plana, tubular ou monolito)
através da carbonização de um substrato macroporos (Resina NovacarbTM) recoberta
com poliimida (http://www.tech.mastcarbon.co.uk).
Membranas de carbono apresentam custos de produção de um a três vezes
superiores aos custos de produção de membranas poliméricas. Isto faz com que seu uso
seja justificado apenas em condições onde seu desempenho é expressivamente superior
ao das membranas poliméricas (OCKWIG E NENOFF, 2007). Assim as membranas de
carbono são principalmente aplicadas em condições onde resistência química e/ou
resistência térmica é necessária.
Em processos termoquímicos para produção de hidrogênio a partir de
combustíveis fósseis, o emprego de reator com membrana se mostra muito promissor
devido ao deslocamento de equilíbrio pela retirada seletiva de hidrogênio (LU et al.,
2007). ITOH E HARAYA (2000) foram os primeiros a realizarem testes laboratoriais
empregando membrana de carbono em reator com membrana. Eles partiram de fibras
ocas assimétricas de poliimidas, fornecidas pela empresa Japonesa UBE Industry. Estas
fibras foram pré – tratadas em ar a 400 °C por 30 minutos (para evitar o amolecimento e
perda da estrutura assimétrica) e depois pirolisadas a 750 °C sob vácuo por 60 min. Um
módulo foi montado com 20 fibras coladas em uma das extremidades com cola
resistente termicamente e colocadas dentro de um tubo de metal para evitar quebra das
fibras. O módulo foi então colocado no centro do reator de desidrogenação do
ciclohexano, a 195 °C e pressão atmosférica, utilizando argônio como gás diluente. Foi
observado deslocamento no equilíbrio de conversão pela retirada seletiva de H2 (ITOH
E HARAYA, 2000).

19
2.3. PRODUÇÃO DE MEMBRANAS DE CARBONO PARA SEPARAÇÃO
DE H2
É crescente o interesse pelo desenvolvimento de membranas com melhor
desempenho em termos de seletividade, resistência térmica e resistência química do que
as membranas comercialmente disponíveis (membranas poliméricas) para separação de
gases (FUERTES E CENTENO, 1998a). Dentre os materiais não poliméricos, aqueles
com capacidade de separação por peneira molecular, tais como, sílica, zeólitas e
carbono apresentam potencial para superar o limite de permeabilidade e seletividade
para membranas poliméricas representados no diagrama de Robeson (ROBESON, 1991,
BURNS E KOROS, 2003, FREEMAN, 1999, SAUFI E ISMAIL, 2004).
Membranas de carbono têm apresentado bom desempenho na separação de
alcano/alqueno (SUDA E HARAYA, 1997a; HAYASHI et al., 1997; HAYASHI et al.,
1996), separação de isômeros de hidrocarbonetos entre normais e ramificados (SOFFER
et al., 1999), produção de nitrogênio de alta pureza, separação e/ou purificação de
hidrogênio, separação de metano de hidrocarbonetos pesados e separação dos gases
gerados em reações de reforma (SEDIGH E ONSTOT, 1998). Além disso, tem sido
observada alta hidrofobicidade, resistência à corrosão e resistência térmica (KYOTANI,
2000).
Contudo, a aplicação mais promissora para membranas de carbono é a aplicação
em reator com membrana, onde é necessário o uso de membrana resistente térmica e
quimicamente. A retirada seletiva de um dos produtos da reação desloca o equilíbrio da
reação, limitada pelo equilíbrio termodinâmico, como tem sido comprovado pelo uso de
membranas de carbono seletivas a hidrogênio (ITOR E HARAYA, 2000; BARBOSA-
COUTINHO, 2004, SAUFI E ISMAIL, 2004).
A fabricação de membranas de carbono envolve as seguintes etapas: seleção do
precursor, preparo da membrana polimérica, pré-tratamento da membrana polimérica,
pirólise e pós-tratamento.
O precursor deve ser resistente termicamente, capaz de resistir à deformação e
amolecimento durante todas as fases de pirólise (MORTHON-JONES, 1984). Material

20
adequado leva a produção de membranas sem quebra e defeitos (SOFFER et al., 1989).
Membranas de carbono têm sido produzidas através de pirólise (em atmosfera inerte ou
vácuo) de uma variedade de materiais naturais contendo carbono, tais como: resina,
grafite, carvão, piche e plantas e de precursores sintéticos tais como poliamidas e
derivados, poliacrilonitrila, resina fenólica, álcool polifurfúrico, formaldeído fenólico,
celulose entre outros (SAUFI E ISMAIL, 2004). Poliimidas estão entre as classes de
polímeros mais estáveis, sendo possível utilizá-las em temperaturas de até 300 °C ou
superior e geralmente se decompõe antes de atingir o ponto de ebulição. As melhores
membranas de carbono, em termos de separação e propriedades mecânicas são aquelas
produzidas de poliimidas aromáticas (JONES E KOROS, 1994).
Devido à alta resistência, poli (éter imida) (PEI) tem sido muito utilizada na
produção de membranas poliméricas precursoras de membranas de carbono (FUERTES
E CENTENO, 1998b, SEDIGH et al., 1999, BARBOSA-COUTINHO, 2004,
MOREIRA, 2008).
2.3.1. Precursores poliméricos
Membranas de carbono são geralmente sintetizadas a partir de membranas
poliméricas microporosas. Membranas poliméricas deste tipo podem ser obtidas por
sinterização de particulados, estiramento a quente de filmes densos, gravação por
bombardeamento de partículas radioativas e por inversão de fases sendo destas técnicas,
a mais utilizada para preparação de membranas poliméricas isotrópicas ou
anisotrópicas, a inversão de fases (HABERT et al., 2006). Nesta técnica, a solução
polimérica é desestabilizada pela indução do estado de supersaturação, o que pode ser
atingida pela mudança de temperatura da solução (precipitação térmica), pela retirada
do solvente (precipitação por evaporação do solvente em solução contendo um não-
solvente menos volátil que o solvente) ou pela adição de um não-solvente para o
polímero (precipitação por imersão). A solução, então, torna-se termodinamicamente
instável e tende a se separar em pelo menos duas fases: rica e pobre em polímero. A fase
rica se torna a estrutura da membrana enquanto a fase pobre, os poros. A técnica de

21
precipitação por imersão permite uma grande flexibilidade, dependendo da
característica do sistema como: natureza do polímero, do solvente e não-solvente, da
presença de aditivos e das condições em que é realizada a precipitação, pode-se
controlar o processo, obtendo-se diferentes tipos de morfologias (DI LUCCIO, 1997,
HABERT et al., 2006, BARBOSA-COUTINHO, 2004, MORREIRA, 2008).
Estas membranas podem ser produzidas em duas principais configurações:
suportadas ou não-suportadas. As não-suportadas podem ser plana (filme), capilar e
fibra oca enquanto que as suportadas são geralmente planas ou tubulares. Na maioria
dos casos, membranas suportadas são produzidas devido à baixa resistência mecânica (a
fácil quebra) das membranas de carbono não-suportadas. Há uma variedade de métodos
de recobrimento, apropriados à deposição de um filme sobre o suporte para obtenção de
membranas poliméricas suportadas. O método de recobrimento deve ser eficiente no
controle da quantidade e homogeneidade do material depositado além de preservar a
textura original do suporte (ACHARYA, 1999). Devido à redução de volume do
material polimérico durante a pirólise, o procedimento de recobrimento deve ser
repetido até a obtenção de membranas de carbono, do tipo peneira molecular, livre de
defeitos (LINKOV et al., 1994a, ANDERSON et al., 2008). Defeitos presentes no
suporte podem ser transmitidos para o filme de carbono, gerando poros de grandes
dimensões ou orifícios que destroem a propriedade de peneira molecular, necessária
para separação de gases. Para minimizar esses problemas, é necessário recobrimento
prévio da superfície do suporte com uma camada intermediária (FUERTES E
CETENO, 1999). Além disso, ainda é preciso considerar a disponibilidade, custo,
durabilidade, morfologia, características de transferência de calor, reatividade química e
compatibilidade com carbono (SAUFI E SMAIL, 2004). Desta forma, a escolha da
configuração do módulo depende de sua aplicação. Para separação de gases, a maioria
das membranas é do tipo fibras ocas (BAKER, 2002) porque estas chegam a apresentar
densidade de empacotamento (área de permeação por volume do módulo) até 40 vezes
maior que membranas planas (DAVID E SMAIL, 2003), veja Tabela 2.5, resultando em
sistemas muito mais compactos.
Para membrana fibra oca (não-suportada), parâmetros de fiação da solução
polimérica são fatores cruciais que devem ser controlados durante o seu preparo. Estes
parâmetros incluem a quantidade e tipo de polímeros, solventes e aditivos misturados na

22
solução, o fluxo de solução e de líquido interno durante a extrusão, a velocidade de
saída da fibra, distância extrusora – banho de precipitação e a temperatura do banho
(GEISZLER, 1997, CLAUSI E KOROS, 2000, BARBOSA-COUTINHO, 2004,
MOREIRA, 2008). Isto porque há importantes correlações entre as propriedades da
precursora polimérica e a membrana de carbono final.
A utilização de membranas de carbono derivadas de membranas poliméricas à
base de PEI e PVP teve início com o trabalho de BARBOSA-COUTINHO (2004) que
tomou como base a experiência do grupo na obtenção de membranas desses polímeros
com morfologia bem formada (BORGES, 1993). Mais tarde, MOREIRA (2008) avaliou
a influência das propriedades destas nas membranas de carbono resultante. Alterações
nas propriedades da membrana polimérica foram conseguidas através da mudança em
parâmetros durante a síntese, tais como, o teor de aditivo e a distância extrusora-banho.
Segunda a autora, membrana polimérica com macrovazios levaram à formação de
membranas de carbono com defeitos, sugerindo o rompimento destes vazios durante a
pirólise. Enquanto que membranas poliméricas bem formadas, sem macrovazios e com
poros interconectados geraram membranas de carbono bem formadas, com alta
estabilidade térmica e mecânica e, com alta seletividade na separação de gases em
função do diâmetro cinético. Outros aditivos, glicerol e álcool propiônico, foram
testados por BARBOSA-COUTINHO (2004) e embora gerassem membranas
poliméricas bem formadas, estas não se mostraram adequadas para produção de
membranas de carbono devido à fusão incipiente e colapso das fibras durante o
tratamento térmico. Para comprovar a eficiência da PVP como aditivo, a autora
produziu membrana de carbono contendo apenas PEI e observou deformação de sua
estrutura durante a pirólise. Estes resultados evidenciam a influência exercida pela PVP
nas reações durante a pirólise evitando que haja a fusão da fibra.
2.3.2. Pré-tratamento
O pré-tratamento de membranas poliméricas antes do processo de pirólise é
importante para garantir a estabilidade e a preservação da estrutura da membrana.

23
KUSUKI et al. (1997) reportaram que certos precursores, quando não pré-oxidados,
sofreram amolecimento durante a pirólise e a membrana de carbono resultante
apresentou baixa permeação. CETENO E FUERTES (2001) observaram que a oxidação
de filmes de resina fenólica em ar, antes da etapa de pirólise, melhorou
significativamente a taxa de permeação gasosa da membrana de carbono resultante.
Essa observação sugere que a oxidação antes da carbonização provoca o aumento de
tamanho dos poros do filme (CETENO e FUERTES, 2001). Estes autores observaram
também perda de permeabilidade e ganho em seletividade em membranas peneira
molecular de carbono obtidas de poli (cloreto co-vinil cloreto) (PVDC-VC) pré-tratadas
em ar a 200 °C por 6 h (CETENO E FUERTES, 2000). Pode-se então concluir que é
necessário aperfeiçoar as condições de pré-tratamento para obtenção do melhor relação
entre permeabilidade e seletividade de membranas de carbono. Além de tudo isso, a
etapa de estabilização ou oxidação tem também o objetivo de evitar a fusão das
membranas, a volatilização de carbono elementar e, com isso, maximizar o rendimento
final de carbono.
Membranas de carbono com diferentes características em termos de estabilidade
e desempenho de separação podem ser obtidas utilizando-se método adequado de pré-
tratamento do precursor. O pré-tratamento pode ser dividido em métodos físicos e
químicos. Os primeiros consistem de estiramento (tensão) da membrana antes da
pirólise (CHEN, 1998, YONEYAMA E NISHIHARA, 1990, SCHINDLER E MAIER,
1990, GRUPTA et al., 1991), enquanto os métodos químicos envolvem a aplicação, no
precursor polimérico, de reagentes químicos (tais como hidrazina, dimetilformamida,
ácido hipoclórico, cloreto de amônia, ácido fosfórico, ácido clorídrico, cloreto de
amônio entre outros (SOFFER et al., 1989, SCHINDLER E MAIER, 1990, LINKOV et
al., 1994b). O método mais empregado na obtenção de membranas de carbono por
pesquisadores tem sido a oxidação, através do aquecimento em presença de oxigênio, ar
ou outra atmosfera oxidativa, no entanto, o precursor pode ser submetido a mais de um
pré-tratamento a fim de atingir a propriedade desejada.

24
2.3.3. Pirólise
Pirólise é o aquecimento do precursor polimérico em atmosfera inerte ou vácuo.
Uma vasta faixa de temperatura pode ser empregada, desde a temperatura de
decomposição à temperatura de grafitização do precursor (geralmente entorno de 3.000
°C). Porém para produção de membrana de carbono, a pirólise é geralmente realizada na
faixa de 500 ºC a cerca de 1.000 ºC (SUDA E HARAYA, 1997b, GEISZLER E
KOROS, 1996). A temperatura, a taxa de aquecimento, tempo de exposição ao patamar
de temperatura, bem como, a atmosfera de pirólise (vácuo ou inerte), vazão do gás em
atmosfera inerte, pressão e concentração são parâmetros que afetam significativamente
o processo de pirólise. Uma pequena mudança nesses parâmetros tem grande impacto
nas propriedades finais da membrana de carbono (JONES E KOROS, 1995, OGAWA E
NAKANO, 1999, SAUFI E ISMAIL, 2004). Foi observado por BARBOSA-
COUTINHO (2008) que quanto maior o tempo de exposição à atmosfera oxidante
durante o pré-tratamento de membranas de PEI/PVP menor é a deformação na seção
transversal da membrana de carbono resultante. Enquanto que para o período de
carbonização, tempos longos levam à formação de estrutura do tipo grafite com
freqüentes fissuras da membrana. A autora avaliou ainda a taxa de aquecimento
concluindo que taxas de aquecimentos muito baixas levam à formação de membranas de
carbono frágeis. A avaliação destes parâmetros permitiu a investigação de condições
adequadas para obtenção de membranas de carbono com características adequadas para
separação de gases por peneira molecular.
Durante a pirólise de polímeros uma grande quantidade de subprodutos de
diferentes volatilidades é liberada causando uma grande perda de massa (STEEL, 2000).
Alguns desses subprodutos são: amônia (NH3), ácido cianídrico (HCN), metano (CH4),
hidrogênio (H2), nitrogênio (N2), monóxido de carbono (CO), dióxido de carbono (CO2)
e outros, dependendo do polímero (GEISZLER E KOROS, 1996). A pirólise de poli
(éter imida), polivinilpirrolidona e da membrana polimérica obtidas destes polímeros
foram acompanhadas por BARBOSA-COUTINHO (2004) e por MOREIRA (2008).
MOREIRA (2008) observou que durante a pirólise da PEI, acompanhada por
espectrômetro de massas, não houve consumo de oxigênio. No entanto, durante a
pirólise de PVP e da membrana polimérica, oxigênio foi consumido acompanhado da

25
liberação de H2O, CO, CO2 e H2. Isto caracteriza a susceptibilidade de degradação da
PVP, tanto pura, como na matriz da membrana polimérica onde as reações (durante a
degradação) são importantes para estabilização e formação de poros na membrana de
carbono. A liberação desses gases resulta na formação de ligações de reticulação entre
os segmentos das cadeias poliméricas do precursor, impedindo a formação de estruturas
ordenadas e favorecendo a obtenção de um material amorfo com estreita distribuição de
tamanho de poros (SAUFI E SMAIL, 2004). Os poros variam em tamanho, forma e
grau de conectividade dependendo da morfologia do precursor orgânico e das condições
de pirólise. Uma estrutura idealizada foi proposta por STEEL (2000) que considerou os
poros de membrana de carbono como sendo não homogêneos, apresentando regiões
com diâmetro relativamente grande acompanhados por constrições, Figuras 2.3.
Figura 2.3. Estrutura idealizada de poros em material de carbono apresentando regiões com diâmetro relativamente grande acompanhado por constrições (STEEL, 2000 citado por LI et al., 2008).
2.3.4. Pós-tratamento
Vários métodos de pós-tratamentos podem ser aplicados à membrana de carbono
para minimizar defeitos, aperfeiçoar a estrutura de poros e propriedades de separação.
Dentre estes métodos estão a oxidação ou ativação, deposição química de vapor pós-
pirólise e recobrimento. A oxidação é o pós-tratamento preferido pelos pesquisadores
para alteração da estrutura dos poros de membranas de carbono, geralmente aumentando
o tamanho dos poros (SOFFER et al., 1989, SCHINDLER E MAIER, 1990, SOFFER et
al., 1997, SOFFER et al., 1987, FUERTES, 2001). Esta etapa pode ser feita utilizando
oxigênio puro, mistura de oxigênio com outros gases, ar, vapor, dióxido de carbono,
óxidos de nitrogênio, óxidos de cloro, soluções de ácido nítrico, misturas de ácidos

26
nítricos e sulfúricos, soluções de ácido crômico e peróxido a elevadas temperaturas
(SOFFER et al., 1987). KUSAKABE et al. (1998) mostraram que a oxidação de
membranas de carbono de poliimidas em uma mistura de O2-N2 aumentou a
permeabilidade de CO2 e O2 sem alterar significativamente a seletividade. O volume
microporoso aumentou significativamente devido ao processo de oxidação, contudo a
distribuição de tamanho de poros não sofreu alteração (SAUFI E ISMAIL, 2004). Já
FUERTES (2001) observou que o aumento da temperatura de oxidação aumentou a
permeação de todos os gases (permanentes e hidrocarbonetos) e diminuiu a seletividade
de pares de gases permanentes (O2/N2 ou CO2/N2). No entanto, hidrocarbonetos (n-
C4H10, C2H4, C3H6) exibiram permeação maior que gases permanentes (He, N2) o que é
conseqüência do transporte através da adsorção em microporos. A variação observada
na seletividade sugere que para gases permanentes o mecanismo de transporte através
da membrana altera com o progresso da oxidação de mecanismo de peneira molecular
para difusão superficial para amostras altamente oxidadas (FUERTES, 2001, SAUFI E
ISMAIL, 2004). Contudo, foi observado por ANDERSON et al. (2008) que quanto
mais porosa as membranas de carbono (obtidas a maiores temperaturas de pirólise) mais
quebradiças são estas membranas.
2.3.5. Envelhecimento da membrana
Membranas peneira molecular de carbono sofrem queda de permeabilidade com
o tempo de vida (envelhecimento). Isto ocorre porque o carbono apresenta, na
superfície, sítios ativos com alta capacidade de fisissorção e mesmo de quimissorção de
substâncias presentes na atmosfera, principalmente oxigênio resultando em redução do
diâmetro efetivo dos poros (LAGORSSE et al., 2008, JONES E KOROS, 1994,
DUBININ, 1980 citado por GRAINGER, 2007).
ANDERSON, et al. (2008) destacaram o fato de membranas nanoporosas de
carbono ser muito afetadas pela presença combinada de água e oxigênio, quimissorção
de oxigênio e fisissorção de água da atmosfera. Para evitar este comportamento, estes
autores mantinham as membranas, quando não utilizadas, em dessecador de umidade
sobre purga de argônio de alta pureza.

27
Vários métodos de regeneração têm sido testados por outros pesquisadores. Para
remover a água, a membrana pode ser evacuada (<1mbar) ou aquecida a 120 °C sob
purga de N2. Para remoção do O2 quimissorvido, temperaturas cerca de 600-800 °C são
necessárias. Como não é possível remover o oxigênio quimissorvido sem alterar a
estrutura de carbono, apenas recuperação parcial do desempenho original pode ser
conseguida removendo água fisissorvida através de regeneração (SAUFI E ISMAIL,
2004).
2.3.6. Adição de carga inorgânica
Com o objetivo de funcionalizar membranas de carbono obtidas a partir de
poliimida, BARSEMA e colaboradores (2003) adicionaram nitrato de prata ou acetato
de prata à membrana precursora. Foi observado que a maior parte da prata migra para a
superfície da membrana, formando uma camada que provoca a redução da
permeabilidade, sendo maior, quanto maior a temperatura de pirólise e o tempo na
temperatura máxima. No entanto, para temperatura abaixo de 600 °C foi constatado
aumento de permeabilidade e redução da seletividade devido à prata agir como
espaçador de poros, sendo que altas concentrações de prata resultaram em membranas
muito frágeis.
LIE E HÄGG (2005) investigaram a adição de óxidos de cálcio, magnésio, ferro
(III) e silício e, nitratos de prata, cobre e ferro às membranas de carbono obtidas de
celulose. Nitrato de cobre e nitrato de prata melhorou a seletividade de separação dos
pares O2/N2 e CO2/CH4 e a permeabilidade de H2 chegando a atingir 1100 e 1500 Barrer
(ou 1,1.10-7 e 1,5.10-7 cm3(CNTP).cm.cm-2.s-1.cmHg-1) para o nitrato de cobre e de
prata, respectivamente (GRAINGER, 2007).
MOREIRA (2008) estudou a adição de sílica à membrana polimérica como
carga inorgânica, a fim de aumentar a porosidade da membrana de carbono. A
membrana de carbono obtida era submetida à lixiviação ácida para retirada da sílica
resultando em membrana de carbono com alta porosidade. No entanto, a membrana
resultante mostrou-se altamente frágil, sendo impossível o seu manuseio. A

28
permeabilidade da membrana de carbono com sílica, antes da lixiviação ( He =
1,5 GPU), foi superior à permeabilidade obtida para membrana de carbono (obtida do
mesmo precursor) sem o aditivo inorgânico ( He = 0,6 GPU) enquanto que a
seletividade foi mantida ( / 60). Desta forma, nesta dissertação foi preparada
membrana de carbono com sílica, sem, contudo, aplicar o processo de lixiviação.
2.4. CONFIGURAÇÃO DE MÓDULOS
A eficiência do processo com membrana em uma dada aplicação depende da
escolha adequada da geometria da membrana bem como da forma de confecção do
módulo (STRATHMANN, 1999, YONEYAMA et al., 1992). A seleção do módulo é
principalmente determinada por razões econômicas, o que deve levar em conta todos os
fatores de custo e não apenas o custo do módulo. A configuração mais barata não é
sempre a melhor escolha dependendo do tipo de aplicação e com isso a funcionalidade
do módulo, é também um fator importante (YONEYAMA et al., 1992).
Para aplicações industriais é preferível a fabricação de módulos com estrutura
assimétrica e configurações capilares ou fibra oca para aumentar a taxa de permeação
dos produtos (PETERSEN et al., 1997). Em geral, as características de módulos que
devem ser consideradas para sistemas de todas as configurações são resumidas na
Tabela 2.2.
Várias configurações de módulos têm sido propostas para membranas de carbono, mas
o maior desafio é melhorar a resistência mecânica da membrana principalmente para o
caso de fibra oca que são membranas auto-suportadas (SAUFI E ISMAIL, 2004).
Módulos são os elementos centrais em sistemas de separação e purificação por
membranas, suas configurações podem afetar drasticamente o desempenho do sistema,
devendo, portanto, ser adotadas configurações apropriadas durante suas fabricações (LI
et al., 2004).
29
Tabela 2.2. Considerações importantes para a construção de módulos com membrana (SMITH et al., 1995, YONEYAMA et al., 1992, BAKER, 2004).
Categorias Características
Produção Alta densidade de empacotamento Facilidade de obtenção em uma dada configuração e estrutura Natureza do material da membrana
Custo Baixo custo de operação Baixo custo de investimento
Durabilidade Baixa tendência à queda de permeação (“fouling”) Facilidade de limpeza Possibilidade de troca de membrana
Eficiência Extensão do uso Natureza da separação desejada Resistência estrutural
Em geral, módulos de membrana podem ser classificados nos seguintes tipos:
plana (placa e quadro, espiral), tubular, capilar e fibra oca (outras geometrias possíveis,
mas menos comum, são monolito e na forma de favos). Mais de 80% dos sistemas com
membrana em aplicação industrial para separação de gases utilizam módulos do tipo
fibra oca (SOFFER et al., 1999, LI et al., 2004) por ser auto-suportadas, apresentarem
alta densidade de empacotamento e alta relação de área superficial por volume.
Módulos do tipo fibra oca chegam a apresentar densidade de empacotamento de 8 a 40
vezes maior que módulos em geometria plana (LI et al., 2004). A tabela 2.3 mostra uma
classificação geral de módulos de geometria cilíndrica.
Tabela 2.3. Classificação geral de membranas de geometria cilíndrica de acordo com o diâmetro das mesmas.
Geometria cilíndrica Diâmetro Tubular > 5mm Capilar 0,5 < diâmetro < 5mm
Fibra oca < 0, 5 mm

30
As principais características desejadas na construção de módulos com fibra oca
para separação de gases são:
• Resistência mecânica: membrana e carcaça devem resistir às condições como
pressão e temperatura de operação, ataques químicos e, além disso, apresentar
longa vida útil.
• Fibras e arranjo das fibras: a qualidade das fibras deve ser controlada e elas
devem apresentar uma alta densidade de empacotamento no módulo.
• Padrão de escoamento: o escoamento dos gases pode ser no mesmo sentido,
contracorrente ou tangencial de acordo com as necessidades e especificações do
produto. O gás pode ser alimentado tanto internamente como externamente as
fibras.
• Economia: Os custos da membrana e da carcaça do módulo, juntamente com o
período de vida útil devem ser considerados bem como conveniência para
manutenção e troca de membranas se necessário.
Módulos com fibras ocas podem apresentar diferentes configurações para
atender as necessidades de diferentes aplicações. A configuração mais comum é aquela
onde a alimentação é feita pela carcaça do módulo, parte externa das fibras como no
módulo Prisma® de membranas poliméricas fabricado pela empresa Monsanto para
recuperação de hidrogênio, Tabela 2.4. Isto possibilita o uso de pressões maiores que as
que seriam possíveis se a alimentação fosse feita pelo orifício interno das fibras, como é
o caso do módulo Generon® lançado em 1985 pela empresa Dow Chemical para
separação de ar (SHIFLETT E FOLEY, 2000). A aplicação de gás de arraste durante a
separação pode melhorar a eficiência do processo, embora seja necessária uma etapa
posterior de separação para recuperação do gás de arraste.
O tamanho do módulo, também, afeta o desempenho de separação.
Normalmente módulos feitos com uma pequena quantidade de fibras, para avaliação do
desempenho em escala de laboratório, podem não promover suficiente informação sobre
aplicações industriais, pois negligenciam fatores como variação de temperatura, padrão
de escoamento e variação da fibra. LI et al. (2004) propuseram um método simples de
obtenção de módulos de fibras ocas em escala laboratorial, com densidade de

31
empacotamento próxima à dos módulos comerciais e usando conexões disponíveis
comercialmente. Basicamente o procedimento de fabricação de módulos envolve cinco
etapas: preparo da carcaça (que deve ser de montagem simples, confiável em termo de
desempenho do módulo e possibilidade de reutilização dos materiais), preparo do
conjunto de fibras, montagem do módulo e colagem (para isolamento da corrente de
permeado do concentrado).
Tabela 2.4. Características de módulo comercial (empresa Monsanto) de membrana polimérica para recuperação de H2 (KING et al., 1978, LI et al., 2004).
Material da membrana Polisulfona (P3500) Diâmetro interno (µm) Diâmetro externo (µm)
120-260 450-540
Permeabilidade de H2 (GPU) 50 Seletividade do módulo (H2/CH4) 30 Diâmetro interno do módulo (cm) 10 Comprimento (m) 3 Número de fibras ~ 20.000 Área efetiva de membranas (m2) ~ 93 Densidade de empacotamento (%) 43-56 Padrão de escoamento Contracorrente Gradiente de pressão através da membrana (MPa) ~ 6,6 Temperatura (°C) ~ 30
A Tabela 2.5 apresentada a seguir, compara os diferentes tipos de módulos em
termos de densidade de empacotamento (área de permeação/volume do módulo)
(HABERT et al., 2006).
Tabela 2.5. Densidade de empacotamento para diferentes tipos de módulos.
Tipo de módulo Densidade de empacotamento (m2/m3) Tubular 30 Plana (arranjada na forma de placa e quadro) 500 Capilar 1.000 Fibra oca 10.000

32
2.5. MECANISMOS DE TRANSPORTE EM MEMBRANAS DE CARBONO
Membrana pode ser definida como uma barreira seletiva, entre duas fases, capaz
de restringir total ou parcialmente o transporte de uma ou mais espécies químicas em
detrimento da passagem de outra(s) espécie(s) através de sua matriz (MULDER, 2000).
Independente da estrutura da membrana, a força motriz para a separação é o gradiente
de potencial químico devido à diferença de concentração, pressão, temperatura ou
potencial elétrico. No caso de gases, a diferença de potencial químico é devida à
diferença na fugacidade (ou pressão parcial se o gás se comporta como ideal) dos
componentes entre os lados da alimentação e do permeado. Em membranas densas, o
transporte ocorre pelo mecanismo de sorção e difusão originalmente proposto por
GRAHAM (1866). As principais variáveis envolvidas na separação são a temperatura,
pressão, concentração, massa molar, tamanho e forma da molécula penetrante etc. A
sorção está associada a aspectos termodinâmicos (incorporação da molécula na matriz
da membrana), enquanto a difusão está associada a fatores cinéticos (mobilidade da
molécula/íon na membrana) (HABERT et al., 2006). Uma equação descrevendo a
difusão em estado estacionário foi proposta por Fick em 1855 considerando a força
motriz como o gradiente de concentração (Equação 2.1):
Equação 2.1
Onde é o fluxo da espécie i (cm3/s), é o coeficiente de difusão da espécie
permeante através da membrana, a concentração e x a espessura da membrana onde a
difusão é considerada. Se a concentração da espécie permeante na interface da
membrana ( ) está em equilíbrio com a pressão parcial desta mesma espécie na fase
vapor em cmHg) e se essas variáveis seguem uma relação linear (Lei de Henry)
(Equação 2.2):
Equação 2.2

33
Onde é o coeficiente de solubilidade do permeante na matriz da membrana. Então
Equação 2.3
Onde é o gradiente de pressão parcial do permeante através da membrana e l
corresponde à espessura da membrana (em cm). Desta forma o fluxo está relacionado à
variação da pressão da seguinte forma (Equação 2.4).
Equação 2.4
Onde é a permeabilidade (cm3cm-2s-1cmHg-1) do permeante através da membrana
normalizado pela espessura da membrana.
Para avaliar a eficiência de separação de dois gases (i e j) pela membrana é
calculada a seletividade ( / ), que é a razão entre a permeabilidade dos dois gases
(Equação 2.5).
/ Equação 2.5
Em membranas porosas, o transporte é do tipo convectivo ou difusivo descrito
por um ou combinação dos seguintes mecanismos, de acordo com o tamanho dos poros:
Fluxo viscoso, difusão de Knudsen e peneira molecular (Figura 2.4).

34
a) Fluxo viscoso, não há separação. O diâmetro dos poros ( ) é muito grande para
efetuar separação.
b) Difusão de Knudsen é observada quando colisões entre moléculas e as paredes
dos poros se tornam significativas (poros de 2 a 100 nm) (BURGRAAF E COT,
1996). A seletividade entre dois gases i e j é dada pela raiz quadrada do inverso
da razão entre as massas moleculares destes gases (Equação 2.6).
/ Equação 2.6
c) Peneira molecular (ou difusão ativada) é dominante quando o diâmetro efetivo
dos poros é da ordem do diâmetro cinético dos gases (3 a 5 Å). A separação é
função da maior taxa de difusão quanto menor a molécula (LU et al., 2007).
Figura 2.4. Mecanismos de separação em membranas porosas: a) fluxo viscoso, b) difusão de Knudsen e c) peneira molecular (LU et al., 2007).
KORESH E SOFFER (1980) postularam que o mecanismo cinético-estatístico
deve ser responsável pela habilidade específica da membrana peneira molecular de
discriminar diferentes moléculas. Nesse modelo, cada constrição contribui para a

35
probabilidade total da molecular de passar através da camada, contendo N constrições.
Em termos da teoria cinética, a probabilidade de a molécula atravessar a constrição é
dada por
Equação 2.7
Onde é o fator pré-exponencial, é a energia de ativação da difusão (relacionada à
parte repulsiva do potencial de adsorção durante a aproximação da molécula à
constrição), onde quanto maior a molécula maior é a (BURGRAAF E COT, 1996), k
é a constante dos gases ideais e T a temperatura. A probabilidade para N constrições é:
Equação 2.8
A seletividade entre dois gases i e j é dada pela razão entre as permeabilidades
/ Equação 2.9
Assim, mesmo que pequena a diferença entre as energias de ativação, depois de
multiplicada N vezes, faz grande diferença. Isto explica porque membrana peneira
molecular com ampla distribuição de tamanho de poros é capaz de separar moléculas
com tamanho similar tão efetivamente. Das equações 2.8 e 2.9 é esperado que a
permeabilidade aumente com o aumento da temperatura e se j, a maior molécula,
apresenta maior espera-se que a seletividade diminua com o aumento da temperatura
(GRAINGER, 2007).

36
Exemplo de separação por este tipo de mecanismo é a separação de gases com
tamanho muito próximo entre si, H2/CO2, H2/CO, H2/N2, CO2/N2, CO2/CH4 etc., em
membranas porosas de carbono, cerâmicas, zeólitas etc. Membranas com essas
características apresentam vantagens em relação às membranas poliméricas por
apresentarem um desempenho melhor na separação de gases semelhantes (pontos acima
da curva limite do diagrama de Robesson (ROBESON, 1991) para membranas
poliméricas). Membranas de carbono para separação de H2 a partir de misturas gasosas
contendo CO2, N2, CO, CH4 etc. apresentam poros com diâmetros menores que 4Å,
sendo, portanto da ordem do diâmetro cinéticos dos gases presentes em corrente de
reforma do gás natural (Tabela 2.6).
Tabela 2.6. Diâmetro cinético de Lennard – Jones (Øcinético) dos principais gases encontrados em correntes de reforma do gás natural, valores determinados por adsorção em zeólita (Breck, 1975).
Gás He H2 CO2 Ar O2 H2S N2 CO CH4 Øcinético (Ǻ) 2,60 2,89 3,30 3,4 3,46 3,6 3,64 3,7 3,80
Quando gases condensáveis estão presentes o mecanismo de transporte é
significativamente alterado devido à condensação destes gases nos poros da membrana
(RAO E SIRCAR, 1993a). O mecanismo passa a ser então do tipo difusão superficial
(Selective Surface Flow), onde o gás mais condensável na corrente de alimentação se
adsorve seletivamente na superfície da membrana, difunde até a região de baixa pressão
(lado do permeado) e subseqüentemente se desorve (LI et al., 2008). Este mecanismo é
comum em membranas de carbono com diâmetro de poros entre 5 e 7Å, para
enriquecimento de H2 através da permeação de hidrocarbonetos (ARNAND et al., 1995,
1997, PARANJABE et al., 1998, RAO E SIRCAR, 1993 a e b, 1996, SIRCAR et al.,
1999, ZHOU et al., 2003, FUERTES, 2001). Dependendo do diâmetro do poro e da
temperatura pode ocorrer a condensação capilar do gás mais condensável, bloqueando a
passagem dos gases não condensáveis, aumentando, com isso, a seletividade da
membrana (Figura 2.5). Desvantagem deste tipo de membrana, como já mencionado
anteriormente, é que hidrocarbonetos grandes, mesmo que presentes em pequenas

37
quantidades podem sofrer adsorção permanente levando ao bloqueio dos poros com
conseqüente redução na permeabilidade (BAKER, 2004).
Figura 2.5. Representação esquemática dos mecanismos de separação em membranas porosas: a) Adsorção superficial, b) Condensação capilar (adaptado de BAKER, 2004).
Além disso, por possuírem custo de produção mais elevado que membranas
poliméricas, membranas de carbono são mais adequadas para condições onde as
membranas poliméricas não são adequadas como, por exemplo, em altas temperaturas.
Uma vez que o mecanismo de difusão superficial é desfavorecido pelo aumento de
temperatura, as membranas mais adequadas são as do tipo peneira molecular. O
aumento da temperatura favorece o desbloqueio dos poros por gases condensáveis,
melhorando a permeabilidade em membrana deste tipo.

3. M
3
(éter
trans
Com
360.0
solve
2008
consi
acord
utiliz
afini
(BAR
vidra
soluç
Figur
METOD
3.1. PREPA
3.1.1. M
No prepa
imida) – P
sição vítrea
mo aditivo
000 Da e T
entes orgân
8). Aerosil 2
iste de partí
do com o fa
zado N-me
dade com
RBOSA-CO
arias foram
ções.
ra 3.1 Fórmempr
DOLOG
ARO DA M
Materiais
aro de mem
PEI (Ultem
(Tg) de 21
orgânico u
Tg de 177 °
nicos (BOR
200 (Degus
ículas nanom
abricante), a
etil-2-pirroli
a água e
OUTINHO,
secos em
mulas estruregados no p
GIA EX
MEMBRAN
mbranas pol
m® 1000, G
5 °C e mas
utilizou-se
C, polímero
RGES, 1993
ssa-Hüls) fo
métricas de
alta estabilid
idona – NM
elevada tem
2004, MO
estufa a 60
turais das preparo da so
XPERIM
NA POLIM
liméricas fo
G.E.), polím
ssa molar n
a poli (vin
o hidrossolú
3, BARBOS
oi utilizado
e sílica – SiO
dade térmic
MP (Aldri
mperatura
OREIRA, 2
0 °C por n
unidades molução polim
MENTA
MÉRICA
oi utilizado
mero termoe
numérica mé
nil pirrolid
úvel, miscív
SA–COUTI
como aditi
O2 – com ta
ca e inércia
ch) – subs
de ebuliçã
2008). Tod
no mínimo
monoméricasmérica (MORE
AL
como polím
estável com
édia (Mn) d
dona) – PV
vel com vá
INHO, 200
ivo inorgân
amanho méd
química. Co
stância que
o (202 °C
dos os mat
12 h antes
s dos polímEIRA, 2008)
mero base a
m temperatu
de 32.800 g
VP-K90 (Fl
ários políme
4 e MORE
nico. Aerosi
dio de 12 nm
omo solven
e apresenta
a 760 mm
teriais sólid
do preparo
meros e sol).
38
a poli
ura de
g/mol.
luka),
eros e
EIRA,
il 200
m (de
nte foi
a alta
mHg)
dos e
o das
lvente

39
3.1.2. Preparo da solução
Prepararam-se 1000 g de solução PEI/PVP/NMP/Aerosil 200 na proporção de
17/10/73 acrescido de 1% de Aerosil (% m/m). A fim de evitar a aglomeração de
partículas de sílica durante o preparo da mistura, fez-se inicialmente a dispersão do
Aerosil 200 em solução de PVP/NMP. Para isso, foram preparadas duas soluções, uma
contendo PVP/NMP (3% m/m) preparada com 30% da NMP que seria utilizada para o
preparo de toda a solução e outra contendo PEI/PVP/NMP preparada com o restante dos
materiais. Ambas as soluções foram deixadas sob agitação mecânica e aquecimento (60
– 80 °C) por 12 h. Aerosil 200 foi, então, adicionado à solução PVP/NMP e agitada
mecanicamente a uma rotação de 16000 rpm por 15 a 20 minutos utilizando um
agitador Ultra Turrax (Ika Labortechnik). Em seguida, a suspensão resultante foi
adicionada à solução de PEI/PVP/NMP, mantendo-se a agitação mecânica (MOREIRA,
2008). Após homogeneização a mistura resultante foi vertida em tanque de fiação e
deixada em repouso por 12 h para evitar o aparecimento de defeitos na fibra devido à
presença de bolhas de ar na mistura. É importante manter o tanque bem vedado para
evitar a absorção de água do ambiente e a precipitação da mistura antes da extrusão.
3.1.3. Fiação
Membranas na forma de fibras ocas foram obtidas a partir da solução contendo
sílica por inversão de fase, utilizando-se o método de precipitação por imersão. As
fibras foram obtidas por extrusão simples da solução seguido de imersão em um banho
de água (tanque de 100L). Água foi utilizada também como líquido interno. A extrusão
foi realizada pressurizando a solução a 7,0 bar com N2. As condições de fiação são
apresentadas na Tabela 3.1.

40
Após o preparo, a membrana foi mantida em banho com água a 60 °C por 48 h
para retirada do solvente residual. Trocou-se a água e em seguida fez-se troca de
solventes para evitar o colapso dos poros da membrana durante a secagem devido à alta
tensão superficial da água (HABERT et al., 2006). A troca de solventes foi feita
deixando as fibras por 2 h imersas em etanol, seguido de 2 h imersas em hexano.
Tabela 3.1 Condições de fiação.
Distância Extrusora – Banho (DEB) 1,0 cm
Temperatura Ambiente 25 °C
Umidade Relativa do ar 53%
Vazão mássica da solução 2 g/min
Vazão do Líquido Interno 2,2 mL/min
Velocidade de Fiação (comprimento de fibra por minuto) 80 cm/min
Legenda: 1 – Válvula 2 – Tanque de Líquido Interno 3 – Tanque de Solução 4 – Bomba 5 – Extrusora 6 – Banho
Figura 3.2 Diagrama esquemático do sistema de fiação de membrana polimérica (BARBOSA-COUTINHO, 2004).

41
3.2. PREPARO DA MEMBRANA DE CARBONO
Membranas de carbono na forma de fibras ocas foram obtidas da pirólise de
fibras poliméricas previamente preparadas.
3.2.1. Sistema de Pirólise
Fibras ocas poliméricas eram colocadas em um suporte de aço inoxidável e
então inseridas em um reator tubular de quartzo de 50 cm de comprimento e 3 cm de
diâmetro. O reator, por sua vez, era introduzido em forno tubular vertical (30 cm de
comprimento) acoplado com controlador de temperatura (controlador/programador,
therma – TH2031, termopar tipo K – Cromel-Alumel) (Figura 3.3).
Figura 3.3. Figura do sistema de pirólise (BARBOSA-COUTINHO, 2004, MOREIRA, 2008).

42
Em uma das extremidades do reator tinha-se a alimentação de ar sintético (para
pré-tratamento) e nitrogênio (pirólise). A alimentação era com vazão de 1500 mL/min
ajustada por um rotâmetro (AALBORG Instruments, 102 – 05ST). Na outra
extremidade tinha-se o sistema de exaustão. As conexões de alimentação e exaustão
com o tubo de quartzo eram de alumínio e eram mantidas cerca de 10 cm da
extremidade do forno para evitar superaquecimento.
3.2.2. Pré-tratamento e Pirólise
O pré-tratamento químico era feito com fluxo de ar, aquecendo-se o reator a uma
taxa de 3 °C/min até atingir 400 °C e mantinha-se a esta temperatura por mais uma hora.
Terminado o pré-tratamento trocava-se a condição de oxidante para inerte (fluxo de N2)
aquecendo-se a 3 °C até atingir 800 °C e era mantida a esta temperatura por mais 1 h. O
forno era então resfriado lentamente sob atmosfera inerte até temperaturas inferiores a
200 °C. As condições de pré-tratamento e pirólise são mostradas na Figura 3.4.
Figura 3.4 Condições de pré-tratamento (Fluxo de ar) e pirólise (Fluxo de N2) (BARBOSA-COUTINHO, 2004, MOREIRA, 2008).

43
3.3. CARACTERIZAÇÃO DAS MEMBRANAS
3.3.1. Análise de Resistência Térmica
Membrana polimérica e de carbono foram avaliadas quanto à estabilidade
térmica via método termogravimétrico (TGA) em atmosfera oxidante e atmosfera inerte
utilizando o equipamento Pyris1 TGA PERKELMER. Para atmosfera oxidante foi
utilizada uma mistura O2 em N2 (14% v/v) e vazão de 60 mL/min e para atmosfera
inerte N2 a 50 mL/min. O aquecimento foi até 900 °C com taxa de 10 °C/min.
3.3.2. Análise da Morfologia
A investigação da morfologia das membranas foi feita por meio de Microscopia
Eletrônica de Varredura (MEV), utilizando o microscópio eletrônico JEOL JSM – 5300
e o LEICA® (modelo S440).
A fim de reduzir as deformações, as seções transversais das membranas
poliméricas foram fraturadas depois de congeladas em nitrogênio líquido durante
amostragem. Uma fina camada de ouro foi aplicada sobre as amostras de membranas
poliméricas utilizando metalizador JFC-1500, JEOL a fim de reduzir os danos às
amostras durante as observações no MEV (MULDER, 2000).
3.3.3. Permeação Gasosa
3.3.3.1.Sistema de Permeação
Para avaliação da permeabilidade gasosa de membranas poliméricas e de
carbono, primeiramente foram montados módulos com uma fibra, como mostrado na

Figur
com
inser
form
mem
quen
ser r
tubo
então
fibra
para
2004
Figur
most
CO2
a pre
press
da m
ra 3.5. Este
cola quente
rida em tub
ma que o ori
mbranas poli
nte, enquant
rígido, evita
tinha o me
o cortada na
a livre para
fixação e v
, MOREIRA
ra 3.5. Módu
Pronto o
trado na Fig
e N2. A lim
esença de a
surizado com
membrana e
es módulos
e (à base de
bo de 1/4 d
ifício intern
iméricas o t
to que para
ando assim
esmo diâme
a extremida
passagem
vedação do
A, 2008).
ulo com uma
o módulo, el
gura 3.6. O
mpeza do sis
apenas o gá
m o gás de
a permeaçã
s foram feit
e resina sinté
de polegada
no da memb
tubo utilizad
membrana
a quebra d
etro e era p
ade do tubo
do permea
o módulo ao
fibra de carb
le era introd
sistema pod
stema era fe
ás de intere
interesse, d
ão era avalia
tos obstruin
ética e cera)
preenchido
brana ficas
do era de pl
as de carbon
da membran
preenchido
o de cobre d
ado. Estes t
o sistema d
bono.
duzido em u
de ser alime
eita por purg
esse) e/ou p
de forma a s
ada por bolh
ndo-se uma
) enquanto
o com cola
se livre par
lástico (poly
no era utiliz
na por torç
com cola A
de forma a
tubos receb
de permeaçã
um sistema
entado pelos
ga do gás de
por vácuo.
se ter uma d
hômetro ou
a das extrem
que a outra
e, após a
ra saída do
y-flux) pree
zado um tub
ão durante
Araldite®. A
deixar o or
iam posteri
ão (BARBO
de permeaç
s seguintes
e alimentaç
Em seguid
diferença de
por transdu
midades da
a extremidad
cura, cortad
permeado.
nchido com
bo de cobre
o manuseio
Após a cur
rifício intern
iormente an
OSA-COUTIN
ção gasosa,
gases puros
ção (para ga
da o sistem
e pressão at
utor de press44
fibra
de era
da de
Para
m cola
e, por
o. O
ra era
no da
nilhas
NHO,
como
s: He,
arantir
ma era
través
são.

45
Figura 3.6. Sistema de permeação gasosa.
3.3.3.2.Princípios teóricos
No caso de bolhômetro, a vazão (Q, em cm3/s) do gás permeado, na temperatura
do experimento (T, em K), era calculada pela Equação 3.1.
é =
∑ Equação 3.1
Em que Vm é o volume (cm3) percorrido pela bolha no bolhômetro, tmédio é a
média dos tempos experimentais (ti, em s) obtidos durante o experimento e m é o
número de tomadas de tempo durante o experimento. Para o cálculo de valores médios,
realizaram-se no mínimo três medidas de tempo (m = 3).
O coeficiente de permeabilidade da membrana (P/l, em cm3.cm-2.s-1.cmHg-1 ou
em GPU – Unidade de Permeação de Gás: 1GPU = 1.10-6 cm3.cm-2.s-1.cmHg-1) foi

46
obtido admitindo-se o modelo de sorção e difusão (MULDER, 2000), conforme a
Equação 3.2.
∆ Equação 3.2
Em que JCNTP é o fluxo volumétrico de gás permeado (cm3.cm-2.s-1) nas
Condições Normais de Temperatura e Pressão (CNTP) e ∆p (cmHg) é a diferença de
pressão através da membrana. A pressão do permeado foi sempre mantida nas
condições atmosféricas. A área de permeação (A, em cm2) foi calculada a partir do
comprimento útil (L, em cm) da fibra e do perímetro externo (π.Dext, em cm), obtido a
partir de micrografias, em que Dext é o diâmetro externo da fibra. O fluxo volumétrico
permeado (J) foi obtido pela Equação 3.3.
· ·
Equação 3.3
Para normalizar a medida de fluxo de gás permeado para as CNTP, considera-se
a equação dos gases ideais, Equação 3.4.
Equação 3.4
Obtendo-se a permeabilidade das fibras ocas com a Equação 3.5.
·
Equação 3.5

47
Quando o fluxo permeado é baixo para ser medido pelo deslocamento de bolhas,
o gás permeado era coletado em um volume calibrado (Vsist, em cm3), fechando-se a
válvula de saída do lado do permeado. Este recipiente está conectado a um transdutor de
pressão, cujo sinal (S, em mA) é convertido em pressão através de uma calibração
prévia. Desta forma, tem-se o aumento de pressão no lado do permeado (p, em cmHg)
em função do tempo. A partir do coeficiente angular da variação da pressão do sistema
com o tempo (dpsist/dt), pode-se calcular a variação do número de moles permeados
(dn/dt) com o tempo. Este valor é convertido, pela equação dos gases ideais, em
variação do volume de gás permeado (dV/dt) com o tempo nas CNTP, determinando-se
a vazão de permeado (QCNTP) conforme indicado pela Equação 3.6.
Equação 3.6
O coeficiente de permeabilidade da membrana é obtido, então, pela Equação 3.7
(BARBOSA-COUTINHO, 2004, MOREIRA, 2008).
· ∆ · · Equação 3.7
A seletividade ideal (α) entre dois gases puros (A e B) é calculada em termos da
razão entre os coeficientes de permeabilidade destes gases, conforme Equação 3.8.
/ Equação 3.8

48
Mantendo a pressão de alimentação constante, a permeabilidade dos gases puros
é obtida através do aumento da pressão no lado do permeado. A pressão nesse lado é
convertida em sinal elétrico (milivoltímetro) com o auxílio de um transdutor de pressão
(Cole-Parmer, modelo 07356-12). O transdutor foi previamente calibrado aplicando-se
pressões conhecidas e avaliando-se o sinal (mA). Durante a permeação os dados da
corrente elétrica relativos à variação de pressão do permeado foram armazenados em
um sistema de aquisição de dados (IQ Logger VmA-40).
3.4. MONTAGEM DE MÓDULOS COM CONJUNTO DE FIBRAS OCAS
3.4.1. Preparo da carcaça
Módulos com várias fibras foram montados utilizando conexões (DETROIT) e
tubo de aço inoxidável de ½ polegadas para montagem da carcaça (Figura 3.7a). Estas
conexões foram escolhidas porque o metal apresentar baixa permeação a hidrogênio e é
resistente a altas temperaturas, características necessárias para posterior aplicação do
módulo em reator com membrana.
Após montagem da carcaça, colou-se com cola Araldite® as duas extremidades
e alimentou-se o módulo com He a 5 bar de pressão e adicionou-se um líquido detector
de vazamentos (Snoop®, Swagelok) em todas as conexões do módulo a fim de garantir a
ausência de vazamentos. Em seguido, retirou-se a cola e lavou-se a carcaça com ácido
sulfúrico diluída (2,0 Normal) para garantir boa aderência da cola à carcaça durante a
montagem do módulo.

49
3.4.2. Preparo das fibras
As fibras de carbono foram previamente testadas quanto à obstrução do canal
interno aplicando-se ar em uma das extremidades com uma seringa e tendo a outra
extremidade imersa em meio liquido (água), visto que algumas fibras eram obtidas
obstruídas. As fibras eram então secas, em estufa a vácuo, e sob aquecimento (60 °C).
Em seguida as fibras tinham as duas extremidades coladas com uma pequena
quantidade de cola Araldite® e deixadas secar. Este procedimento foi adotado a fim de
evitar fibras obstruídas no módulo devido à entrada de cola no interior das fibras
durante a montagem do módulo.
3.4.3. Montagem do módulo
Após montagem e limpeza da carcaça e preparo das fibras, o passo seguinte foi a
montagem do módulo. As fibras foram juntadas em um feixe por uma de suas
extremidades e amarrada com um pedaço de Teflon (material macio) sem apertar muito
para que as fibras pudessem se acomodar durante a inserção na carcaça. A parte
amarrada foi, então, cuidadosamente introduzida na carcaça. A outra extremidade
(aquela com as fibras sem amarração) foi então preparada para colagem. A extremidade
da carcaça foi enrolada com Teflon (para evitar contato da cola com a parte externa do
tubo) e em seguida com um filme plástico formando um canal por onde a cola era
inserida. A cola foi preparada, na proporção de 1:1 (v/v) de resina e endurecedor e
inserida no módulo através de uma seringa. Deve-se ter o cuidado de não deixar as
fibras se deslocarem durante a inserção da cola.
A cola foi curada em estufa a 60 °C por 2 h ou até endurecimento. Após a
colagem de uma das extremidades, desamarraram-se as fibras e repetiu-se o processo de
colagem para a outra extremidade. A cola pode ser cortada, para isso não se deve deixar
curar completamente antes do corte, mas também não se deve cortar com a cola ainda
muito mole o que pode levar a obstrução das fibras. Outra opção é deixar a cola ficar
bastante rígida, bem curada, e fazer um pequeno corte superficial e em seguida quebrar

a col
prov
Figur
3
apres
30:50
la neste loc
eniente do c
ra 3.7. Montadas fib(orifícsaída d
3.5. PERME
3.5.1. P
Duas m
sentado na
0:10:10 e ou
cal. Neste ú
corte.
agem do módbras de carbcio interno ddo permeado
EAÇÃO EM
Preparo das
isturas fora
Figura 3.
utra conten
último caso
dulo: a) conebono na carcdas fibras poo (membrana
M MÓDUL
s Misturas
am prepara
8, uma co
do H2 e CO
as fibras fi
exões utilizadaça, c) visão
or onde sai oa).
LO COM F
adas no lab
ontendo H2
O na proporç
ficam comp
das para mono lateral do o permeado)
FIBRAS OC
boratório u
, He, CO2
ção de 1:1.
letamente l
ntagem da camódulo apó
) e d) amplia
CAS DE C
utilizando u
2 e CH4 n
Ao conecta
limpas, sem
arcaça, b) inss colagem eação da regi
CARBONO
um sistema
na proporçã
ar os cilindr
50
m cola
serção e corte ião de
com
ão de
ros no

51
sistema de misturas uma válvula de escape foi utilizada para fazer vácuo e purga,
garantindo assim, a limpeza das tubulações. Antes da mistura fez-se limpeza no cilindro
da mistura fazendo-se vácuo, seguido de lavagem com o gás de maior proporção (He)
na primeira mistura e por H2 na segunda mistura. O procedimento de limpeza foi
repetido por no mínimo três vezes para garantir a limpeza do cilindro. Na primeira
mistura adicionou-se em cilindro de 10 L, pressão de 90 psi de CH4, seguido de 180 psi
de CO2, 400 psi de H2 e finalmente 900 psi de He. A segunda mistura foi preparada em
cilindro de 5 L adicionando-se 450 psi de CO seguido de 900 psi de H2. Para garantir a
homogeneização da mistura elas só foram utilizadas 48 h após o preparo. A primeira
mistura foi quantificada por cromatografia gasosa em cromatógrafo a gás Micro-GC -
VARIAN, modelo CP-380, dotado de detector de ionização de chamas e detector de
condutividade térmica com metanador e colunas cromatográficas Peneiro Molecular 5m
e Poraplot Q. A composição determinada para esta mistura foi de 30:50:9,91:10,09 para
H2, He, CO2 e CH4 respectivamente.
Figura 3.8. Diagrama esquemático do sistema de preparo de misturas.
CO
H2
Ar
CO2
CH4
P I
He
Painel de Gases
Cilindro
Manômetro
Válvula
Bomba de Vácuo

de qu
press
ocas
mem
cone
confo
Figur
este
por a
2,89Å
tamb
3.5.2. S
Devido à
uantificar o
são. Com is
de carbon
mbrana, já p
ctada a um
forme mostr
ra 3.9. Fotogperm
Argônio
gás ser iner
apresentar u
Å, respectiv
bém era ut
Sistema de P
à mistura se
o permeado
sso, para os
no foi utili
para os te
m Espectrôm
rado na Figu
grafia do sismeado de mis
(Ar) foi ut
rte, não est
um diâmetro
vamente). O
tilizado par
Permeação
er multicom
o, o que nã
s testes de p
izado o m
stes de pe
metro de Ma
ura 3.9 e Fig
stema experisturas.
tilizado com
ar presente
o cinético gr
O Ar era co
ra promove
o
mponente, h
ão é possíve
permeação
mesmo siste
rmeação d
assas (EM)
gura 3.10.
imental emp
mo gás de a
em nenhum
rande (3,4Å
onectado a
er ajuste d
há a necessi
el utilizand
de gases pu
ema utiliza
e misturas
Quadrupol
pregado na p
arraste nos t
ma das mist
Å) quando c
um contro
da composi
idade de um
do apenas u
uros em mó
do para ca
a linha d
ar Balzers/P
permeação e
testes de pe
turas, não s
omparado a
lador de va
ição da mi
m detector c
um transdut
ódulo com f
aracterizaçã
de permead
Prisma-QM
e quantificaç
ermeação d
ser interfere
a He e H2 (2
azão (MKS
istura duran
52
capaz
tor de
fibras
ão da
do foi
MS200
ção do
evido
ente e
2,65 e
) que
nte a

53
determinação da curva de calibração. O MKS era conectado diretamente no EM. Uma
válvula de 4 vias localizada entre o MKS e o EM permitia a escolha de injeção direta do
gás de arraste (limpeza do EM) ou mistura de calibração sem passagem pelo módulo ou
a injeção do gás de arraste depois de passagem pelo módulo (durante limpeza do
módulo ou durante análise do permeado). Após limpeza do sistema, o módulo era
alimentado com a mistura e o permeado era injetado diretamente no EM.
Figura 3.10. Diagrama esquemático do sistema experimental empregado na permeação e quantificação do permeado de misturas.
Depois da montagem do sistema, antes da primeira análise, fez-se vácuo no
módulo por uma hora. Depois disso teve-se o cuidado de deixar o sistema fechado, sem
contato com ar, entre as análises. Antes de cada análise o sistema (lado do permeado e
EM) era lavado com gás de arraste até estabilização dos sinais. Ao alimentar o módulo
era também aliviado um pouco do gás de alimentação para limpeza do módulo. Os
testes de permeação foram feitos alimentando-se o módulo com a mistura e controlando
a pressão e temperatura. A válvula do concentrado era mantida fechada durante as
análises em um sistema estático a fim de economizar a mistura do cilindro.

54
3.5.3. Curvas de Calibração dos componentes das misturas em EM
Uma vez que os sinais gerados em espectrômetro de massas por cada
componente das misturas não possuem interferências entre si, as curvas de calibração
dos componentes destas misturas foram obtidas variando-se a composição das mesmas
em argônio. Argônio foi utilizado por ser o gás de arraste empregado em todas as
análises de permeação através do espectrômetro de massas. A intensidade do sinal de
cada uma das espécies presentes nas misturas eram coletada e utilizada na construção de
uma curva do sinal de corrente iônica (mA), resposta do EM, em função da
concentração (% V/V) da espécie desejada na corrente injetada no EM.
As curvas de calibração de H2 e He foram obtidas variando-se de 0 a 100% a
composição da mistura H2/He/CO2/CH4 em Argônio. A variação da composição da
mistura em argônio foi efetuada em um misturador de gases (MKS) em corrente
mantida com vazão total constante e igual a 1,0 cm3/s. Estas curvas de calibração são
apresentadas na Figura 3.11a e b.
A resposta do espectrômetro de massas à variação de concentração dos gases
presentes nas misturas não é linear. Então para que as curvas de calibração dos gases
mais permeáveis (He e H2) cobrissem toda a região de concentrações possíveis partido
desta mistura, os pontos experimentais foram ajustados a curvas polinomiais
apresentadas nas Equações 3.9 e 3.10.
2,45 · 10 3,90 · 10 · 4,77 · 10 · 3,63 · 10 ·
0,99992 Equação 3.9
8,99 · 10 3,63 · 10 · 2,31 · 10 · 1,35 · 10 ·
0,99994 Equação 3.10
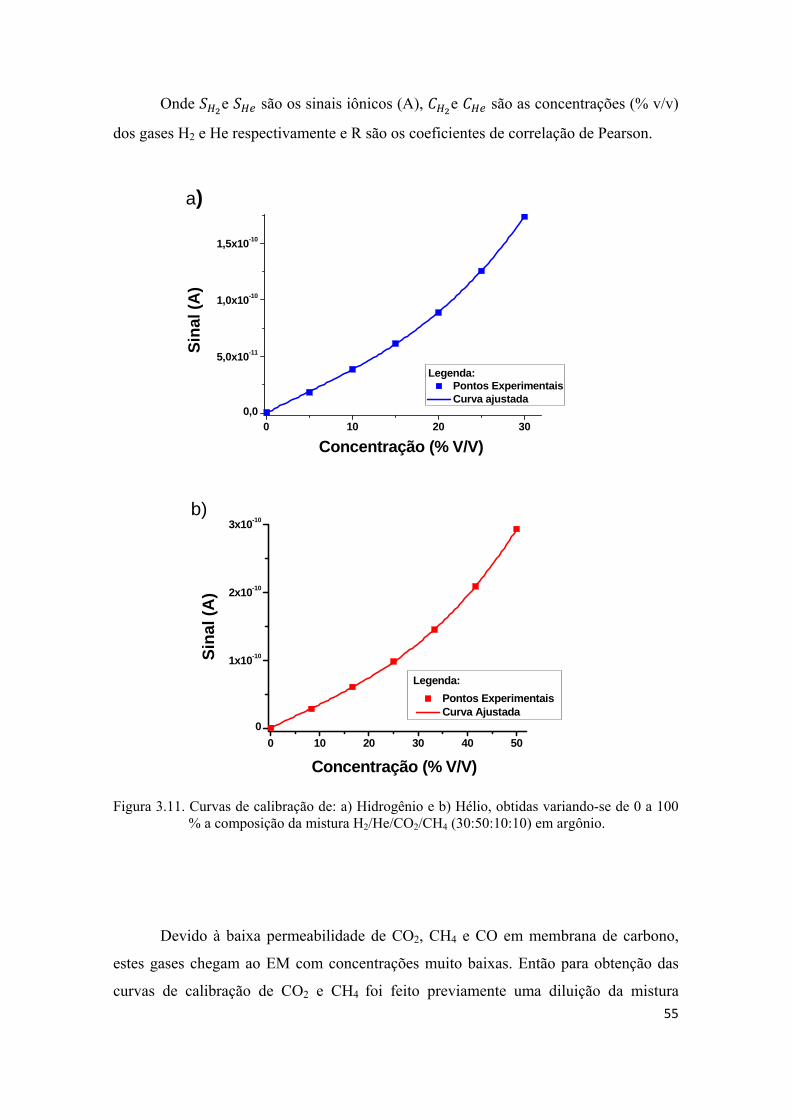
55
Onde e são os sinais iônicos (A), e são as concentrações (% v/v)
dos gases H2 e He respectivamente e R são os coeficientes de correlação de Pearson.
Figura 3.11. Curvas de calibração de: a) Hidrogênio e b) Hélio, obtidas variando-se de 0 a 100 % a composição da mistura H2/He/CO2/CH4 (30:50:10:10) em argônio.
Devido à baixa permeabilidade de CO2, CH4 e CO em membrana de carbono,
estes gases chegam ao EM com concentrações muito baixas. Então para obtenção das
curvas de calibração de CO2 e CH4 foi feito previamente uma diluição da mistura
0 10 20 300,0
5,0x10-11
1,0x10-10
1,5x10-10
Concentração (% V/V)
Sina
l (A
)
Pontos Experimentais Curva ajustada
Legenda:
a)
0 10 20 30 40 500
1x10-10
2x10-10
3x10-10
Concentração (% V/V)
Sina
l (A
)
Pontos Experimentais Curva Ajustada
Legenda:
b)

56
H2/He/CO2/CH4 colocando 2,6 bar da mistura e 20 bar de Ar em cilindro de 1,0 L,
resultando assim, em uma mistura de calibração com proporções nominais de
3,9:6,5:1,3:1,3 de H2, He, CO2 e CH4, respectivamente. Já para a curva de calibração de
CO foi aplicado, também em cilindro de 1,0L, uma pressão de 120 psi de Ar seguida de
140 psi da mistura de H2/CO. Com isso espera-se uma mistura de calibração contendo
H2, CO e Ar com proporção nominal de 7,15:7,15:85,7. Estas misturas foram
conectadas em MKS e tiveram suas composições variadas de 0 a 100% em Ar do
misturador, resultando nas curvas de calibrações apresentadas nas Figuras 3.12 a, b e c.
Os pontos experimentais para estas concentrações ajustam-se bem a equações
lineares dadas pelas Equações 3.11, 3.12 e 3.13.
8,8431 · 10 · 0,0299. 10 0,9999 Equação 3.11
1,2921 · 10 · 0,0052. 10 0,9995 Equação 3.12
8,8855 · 10 · 0,0006. 10 0,9998 Equação 3.13
Onde , e são os sinais iônicos (A), , e as
concentrações (% v/v) dos gases CH4, CO2 e CO respectivamente e R são os
coeficientes de correlação de Pearson.
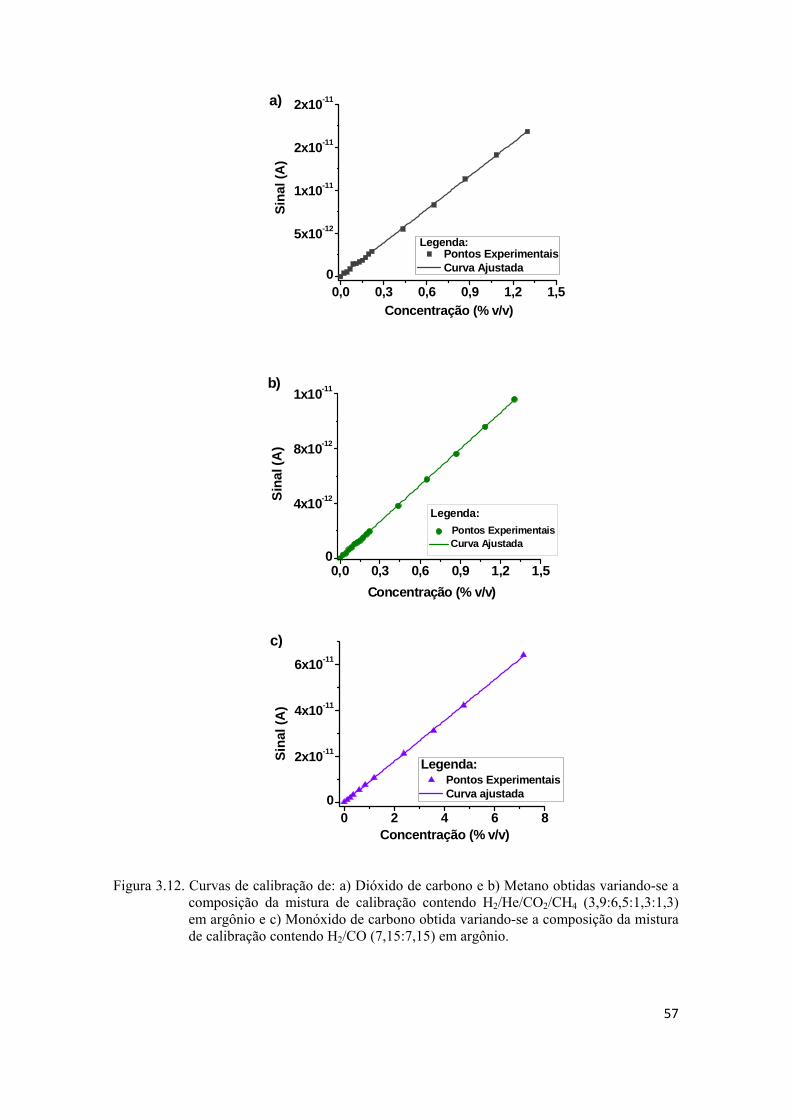
57
Figura 3.12. Curvas de calibração de: a) Dióxido de carbono e b) Metano obtidas variando-se a composição da mistura de calibração contendo H2/He/CO2/CH4 (3,9:6,5:1,3:1,3) em argônio e c) Monóxido de carbono obtida variando-se a composição da mistura de calibração contendo H2/CO (7,15:7,15) em argônio.
0,0 0,3 0,6 0,9 1,2 1,50
5x10-12
1x10-11
2x10-11
2x10-11
Pontos Experimentais Curva Ajustada
Concentração (% v/v)
Sina
l (A
)
a)
Legenda:
0 2 4 6 80
2x10-11
4x10-11
6x10-11
Pontos Experimentais Curva ajustada
Sina
l (A
)
Concentração (% v/v)
Legenda:
c)
0,0 0,3 0,6 0,9 1,2 1,50
4x10-12
8x10-12
1x10-11
Pontos Experimentais Curva Ajustada
Sina
l (A
)
Concentração (% v/v)
b)
Legenda:

58
4. RESULTADOS E DISCUSSÕES
Membranas na forma de fibras ocas foram preparadas pela técnica de inversão
de fases através de fiação úmida, sendo utilizadas como precursoras para fibras ocas de
carbono sintetizadas através do processo de pirólise. As fibras ocas de carbono foram
acondicionadas em módulos de permeação e utilizadas para investigar a permeação
seletiva de hidrogênio. Os principais resultados obtidos e a discussão dos efeitos
observados são apresentados a seguir.
4.1. CARACTERIZAÇÃO DAS MEMBRANAS
As membranas poliméricas e de carbono foram caracterizadas quanto à
estabilidade térmica através de análise termogravimétrica (TGA), quanto à morfologia
através de microscopia eletrônica de varredura (MEV) e quanto à permeabilidade
gasosa (PG). Os resultados destas análises são apresentados e discutidos a seguir.
4.1.1. Análise Termogravimétrica
A análise de estabilidade térmica das membranas polimérica e de carbono foi
realizada em atmosfera oxidante com a presença de oxigênio e em atmosfera inerte
contendo apenas nitrogênio. Em ambas as atmosferas o aquecimento foi realizado a taxa
constante (veja seção 3.3.1). As perdas percentuais de massa em função da temperatura
para cada condição experimental são apresentadas na Figura 4.1.
A membrana polimérica, quando submetida ao aquecimento em atmosfera
oxidante (curva MPO), apresenta três faixas de temperatura onde ocorrem expressivas
perdas de massa. A primeira perda de massa ocorre na faixa de 300 °C a 450 °C, a

59
segunda de 500 °C a 550 °C e a terceira acima de 600 °C, na qual ocorre a degradação
de todo material orgânico. Dentre estas três faixas de temperatura, para o processo de
pirólise, a primeira perda de massa é bastante importante porque está relacionada com
as reações que possibilitam a formação de grupos funcionais (carbonilas, hidroxilas,
etc.) que estabilizam a membrana durante o processo de pirólise. Até atingir 400 °C
(temperatura máxima da etapa de pré-tratamento no processo de pirólise) a membrana
perdeu cerca de 4% (3,39%) de sua massa. Ao final dessa etapa de degradação (450 °C)
a perda de massa foi de 10%, valor próximo àquele observado por MOREIRA
(MOREIRA, 2008). O procedimento utilizado por esse autor para a pirólise das fibras
ocas é empregado neste trabalho, o qual envolve uma etapa de pré-tratamento,
mantendo o precursor polimérico na temperatura igual a 400 °C por uma hora. Segundo
MOREIRA, nesta etapa ocorre a degradação preferencial da poli(vinil pirrolidona),
PVP, enquanto que a degradação da poli(éter imida), PEI, ocorre em temperaturas mais
elevadas.
0 200 400 600 800 10000
25
50
75
100
MPO MPI MCO MCId
bc
a
Mas
sa P
erce
ntua
l (%
)
Temperatura (°C)
Legenda:a
c
d
b
Figura 4.1. Curvas termogravimétricas da membrana polimérica em atmosfera oxidante (MPO) e em atmosfera inerte (MPI), e da membrana de carbono em atmosfera oxidante (MCO) e em atmosfera inerte (MCI).

60
A massa residual, observada em atmosfera oxidante, para a membrana
polimérica ou de carbono pode ser utilizada para determinação do teor de sílica
originalmente presente na membrana. Uma vez que a sílica é um material com alta
estabilidade térmica, seu teor pode ser determinado através da massa resultante após a
degradação completa do material da membrana. Os valores obtidos ao final da
degradação da membrana polimérica e de carbono foram cerca de 4,48% m/m e 7,85%
m/m, respectivamente. MOREIRA (2008) relata para membranas de carbono valores
próximos a 8% m/m. O teor de sílica em membrana polimérica é inferior ao observado
para membranas de carbono, pois durante a pirólise da membrana polimérica há perda
de cerca de 50% de massa, resultando em aumento no teor de material inorgânico
(sílica).
A membrana polimérica, quando submetida ao aquecimento em atmosfera inerte
(Curva MPI), apresentou duas regiões de perda de massa bastante semelhantes às
primeiras etapas ocorridas em atmosfera oxidante. Estas regiões correspondem a
reações de degradação dos polímeros utilizados (MOREIRA, 2008). Na primeira região
a perda de massa (12%) e a temperatura de decomposição (300 - 450 ºC) correspondem,
assim como observado em atmosfera oxidante, à decomposição da PVP. Na segunda
região a perda de massa se inicia em cerca de 500 ºC, superior à temperatura de
degradação da PEI, no entanto semelhante à temperatura encontrada por MOREIRA
(2008) para membrana polimérica contendo sílica. A presença de sílica aumenta a
temperatura em que se inicia a segunda região de degradação, que tem sido atribuído
aos grupos OH superficiais presentes na sílica, cuja interação com os grupos funcionais
das cadeias poliméricas aumentam sua estabilidade térmica (BARBOSA-COUTINHO,
2004). Observa-se que a perda de massa é similar em atmosfera inerte e em atmosfera
oxidante até a temperatura de 690 °C. Acima desta temperatura a membrana se degrada
rapidamente em atmosfera oxidante restando apenas material inorgânico, enquanto que
em atmosfera inerte ocorre o processo conhecido como pirólise, dando origem a
carbono poroso, restando ao final do processo 49% da massa inicial. Cabe salientar que
a degradação da PEI somente ocorre em temperatura elevada, tanto em atmosfera
oxidante quanto inerte, evidenciando a grande estabilidade térmica deste polímero, o
que o torna adequado para a fabricação de membranas de carbono.

61
O aquecimento de membrana de carbono em atmosfera inerte (Curva MCI)
mostra uma variação percentual de massa muito pequena (5,52%), o que representa um
ganho de estabilidade em relação à sua precursora. Uma vez que a membrana já foi
submetida a estas condições durante a pirólise, a pequena perda de massa observada
pode estar associada, em grande parte, à liberação de gases fisissorvidos e
quimissorvidos na membrana de carbono. Este resultado corrobora com outros autores
(ADHIKARI E FERNANDO, 2006) que mencionam a utilização de membranas de
carbono em atmosferas não oxidantes na faixa de 500 a 900 °C.
O aquecimento da membrana de carbono em atmosfera oxidante (Curva MCO)
mostra, também, o ganho de resistência térmica da membrana de carbono em atmosfera
oxidante quando comparada à membrana polimérica precursora. Apenas a partir de 570
°C a perda de massa se torna expressiva, até então a perda de massa não havia
ultrapassado 3,55%.
4.1.2. Análise da Morfologia
As morfologias das membranas poliméricas e de carbono foram avaliadas
através de microscopia eletrônica de varredura (MEV). As fotomicrografias da
membrana polimérica e da membrana de carbono são apresentadas na Figura 4.2 e na
Figura 4.3, respectivamente.
Através destas imagens é possível verificar que a membrana polimérica não
possui macrovazios em sua seção transversal, apresentando poros interconectados, cujo
tamanho aumenta em direção à região central ao longo da espessura da membrana
(fotomicrografias A, B e C). A superfície externa mostrou poros de maior tamanho
(fotomicrografia E e F), não sendo possível observar a presença de poros na superfície
interna na resolução observada (fotomicrografia D).
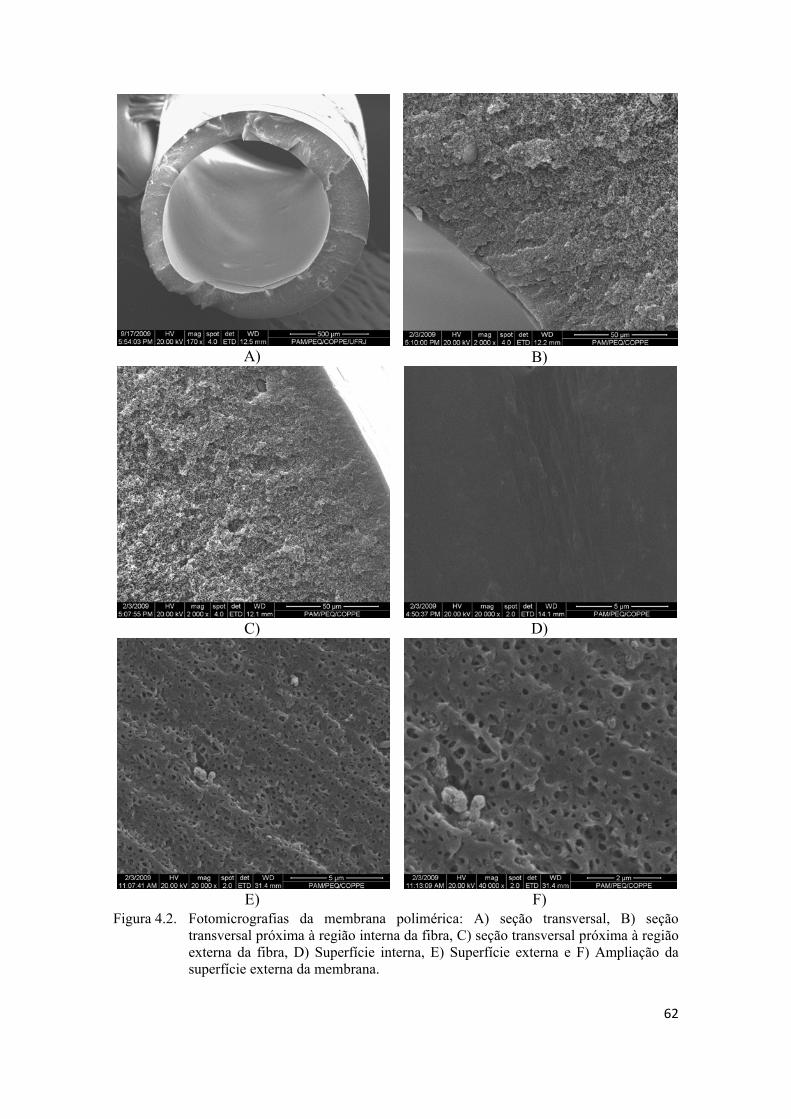
62
A) B)
C) D)
E) F) Figura 4.2. Fotomicrografias da membrana polimérica: A) seção transversal, B) seção
transversal próxima à região interna da fibra, C) seção transversal próxima à região externa da fibra, D) Superfície interna, E) Superfície externa e F) Ampliação da superfície externa da membrana.

63
A morfologia observada para a membrana polimérica era esperada, pois
durante o preparo da membrana utiliza-se água (não solvente) como líquido interno. A
precipitação rápida da solução polimérica na região próxima à superfície interna
favorece a formação de uma superfície de baixa porosidade. Já na parte externa da
membrana o DEB de 1,5 cm permite o contato da solução com vapores de água,
presentes no ambiente, antes do contato com o banho. Isto provoca uma precipitação
mais lenta, favorecendo o aumento do tamanho dos poros. A precipitação da solução
nas regiões próximas às superfícies interna e externa, resulta em transferência de massa
mais dificultada na região central da fibra, favorecendo a etapa de crescimento dos
núcleos da fase diluída e o aumento do tamanho dos poros resultantes (DI LUCCIO,
1997, BARBOSA-COUTINHO, 2004).
A membrana de carbono obtida, também se mostrou bem formada, sem defeitos
e com ausência de macrovazios. A ausência de macrovazios é importante porque
aumenta a resistência mecânica da membrana possibilitando sua utilização no processo
de separação de gases em pressões elevadas (LI, 1993). A redução dos diâmetros
interno e externo da fibra de carbono em relação à fibra polimérica é devido à perda de
massa durante a pirólise. Com as resoluções empregadas para obtenção das
fotomicrografias (ampliações de até 150.000 vezes) não foi possível observar poros nas
superfícies ou na seção transversal da membrana de carbono. Desta forma, não é
possível verificar se a membrana de carbono mantém a variação no tamanho de poros ao
longo da seção transversal, conforme observado na morfologia da membrana polimérica
precursora. No entanto, a morfologia observada para a membrana de carbono sugere que
os poros da membrana polimérica sofreram sinterização durante o processo de pirólise
com conseqüente redução expressiva no diâmetro destes poros.

Figurra 4.3. FototransexterSupe
A)
C)
E) omicrografiassversal próxirna da fibra,erfície extern
s da membima à região , D) Superfína.
brana de cainterna da ficie interna,
rbono: A) ibra, C) seçãE) Superfíci
B)
D)
F) Seção transo transversalie externa e
sversal, B) l próxima à rF) Ampliaç
64
seção região ção da
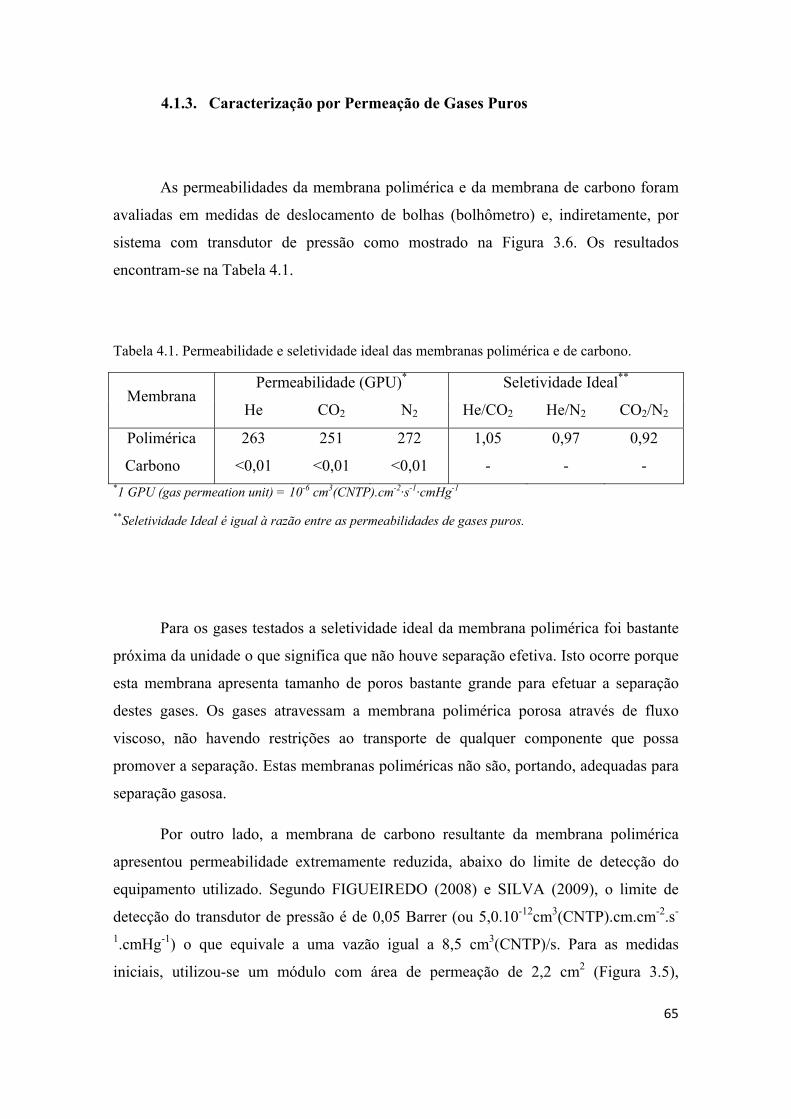
65
4.1.3. Caracterização por Permeação de Gases Puros
As permeabilidades da membrana polimérica e da membrana de carbono foram
avaliadas em medidas de deslocamento de bolhas (bolhômetro) e, indiretamente, por
sistema com transdutor de pressão como mostrado na Figura 3.6. Os resultados
encontram-se na Tabela 4.1.
Tabela 4.1. Permeabilidade e seletividade ideal das membranas polimérica e de carbono.
Membrana Permeabilidade (GPU)* Seletividade Ideal**
He CO2 N2 He/CO2 He/N2 CO2/N2
Polimérica 263 251 272 1,05 0,97 0,92
Carbono <0,01 <0,01 <0,01 - - - *1 GPU (gas permeation unit) = 10-6 cm3(CNTP).cm-2·s-1·cmHg-1
**Seletividade Ideal é igual à razão entre as permeabilidades de gases puros.
Para os gases testados a seletividade ideal da membrana polimérica foi bastante
próxima da unidade o que significa que não houve separação efetiva. Isto ocorre porque
esta membrana apresenta tamanho de poros bastante grande para efetuar a separação
destes gases. Os gases atravessam a membrana polimérica porosa através de fluxo
viscoso, não havendo restrições ao transporte de qualquer componente que possa
promover a separação. Estas membranas poliméricas não são, portando, adequadas para
separação gasosa.
Por outro lado, a membrana de carbono resultante da membrana polimérica
apresentou permeabilidade extremamente reduzida, abaixo do limite de detecção do
equipamento utilizado. Segundo FIGUEIREDO (2008) e SILVA (2009), o limite de
detecção do transdutor de pressão é de 0,05 Barrer (ou 5,0.10-12cm3(CNTP).cm.cm-2.s-
1.cmHg-1) o que equivale a uma vazão igual a 8,5 cm3(CNTP)/s. Para as medidas
iniciais, utilizou-se um módulo com área de permeação de 2,2 cm2 (Figura 3.5),

66
correspondente a uma única fibra oca de carbono com comprimento de 10 cm e a um
limite de detecção de 0,01 GPU. Para aumentar a vazão de permeado e atingir o limite
de detecção do transdutor foi construído um módulo com área de permeação igual a 80
cm2 (Figura 3.7), com 40 fibras ocas de carbono, ampliando o limite de detecção para
0,0003 GPU. Os resultados dos testes de permeação de gases puros para este módulo
são apresentados na Tabela 4.2.
Tabela 4.2. Permeabilidade de gases puros em membranas de carbono utilizando um módulo com 80 cm² de área de permeação.
Permeabilidade (GPU) Seletividade Ideal He CO2 N2 He/CO2 He/N2
0,17 < 0,0003 < 0,0003 > 570 > 570
1 GPU = 10-6cm3(CNTP)·cm-2·s-1·cmHg-1
Os resultados na Tabela 4.2 mostram que mesmo utilizando um módulo com
área de permeação de 80 cm2, a vazão de CO2 e CH4 é ainda inferior ao limite de
detecção do aparelho, permeando apenas o gás com o menor diâmetro cinético (He).
Isto comprova que a separação destes gases pela membrana de carbono ocorre por
exclusão por tamanho, ou seja, por mecanismo de separação do tipo peneira molecular.
4.2. PERMEAÇÃO DE MISTURAS
Para investigar a permeação de misturas de gases através de membrana de
carbono foi necessário aumentar a sensibilidade do método de detecção, escolhendo-se
utilizar espectrômetro de massas (EM). Os resultados são apresentados e discutidos a
seguir.
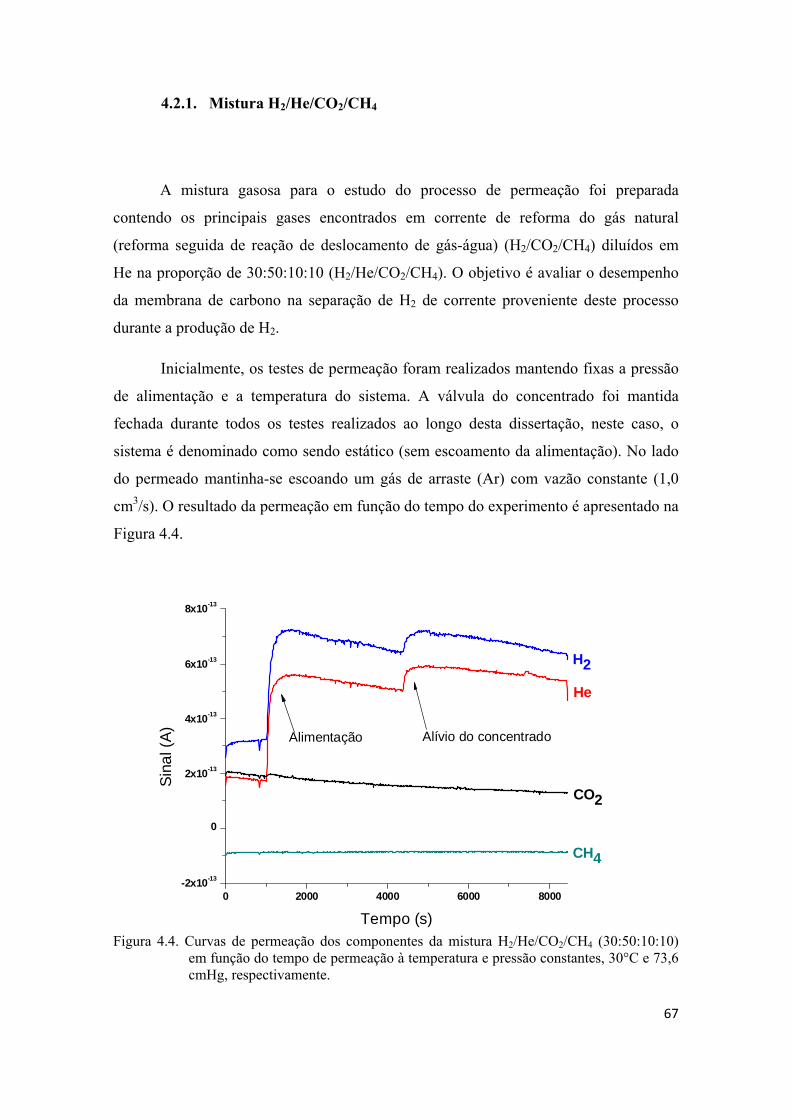
67
4.2.1. Mistura H2/He/CO2/CH4
A mistura gasosa para o estudo do processo de permeação foi preparada
contendo os principais gases encontrados em corrente de reforma do gás natural
(reforma seguida de reação de deslocamento de gás-água) (H2/CO2/CH4) diluídos em
He na proporção de 30:50:10:10 (H2/He/CO2/CH4). O objetivo é avaliar o desempenho
da membrana de carbono na separação de H2 de corrente proveniente deste processo
durante a produção de H2.
Inicialmente, os testes de permeação foram realizados mantendo fixas a pressão
de alimentação e a temperatura do sistema. A válvula do concentrado foi mantida
fechada durante todos os testes realizados ao longo desta dissertação, neste caso, o
sistema é denominado como sendo estático (sem escoamento da alimentação). No lado
do permeado mantinha-se escoando um gás de arraste (Ar) com vazão constante (1,0
cm3/s). O resultado da permeação em função do tempo do experimento é apresentado na
Figura 4.4.
Figura 4.4. Curvas de permeação dos componentes da mistura H2/He/CO2/CH4 (30:50:10:10) em função do tempo de permeação à temperatura e pressão constantes, 30°C e 73,6 cmHg, respectivamente.
0 2000 4000 6000 8000-2x10-13
0
2x10-13
4x10-13
6x10-13
8x10-13
Sina
l (A)
Tempo (s)
Alimentação Alívio do concentrado
H2
He
CO2
CH4

68
Como pode ser observado na Figura 4.4, pressurizando-se o módulo com a
mistura de alimentação há detecção imediata dos sinais relativos ao He e H2,
demonstrando a permeação preferencial destes componentes e a seletividade da
membrana. Como a alimentação é mantida confinada, observa-se que com o tempo o
sinal relativo aos componentes permeáveis pela membrana vai sofrendo redução. A
abertura da válvula de concentrado, promovendo uma purga da alimentação, levou a
recuperação imediata do sinal dos componentes permeáveis. O comportamento
observado com o tempo pode ser atribuído a uma redução da pressão parcial dos gases
permeáveis devido a sua transferência preferencial para o permeado. Com a redução da
pressão parcial do gás na alimentação, há a redução do fluxo permeado. Desta maneira,
para os testes seguintes foi estipulado um tempo de permeação de 15 minutos, suficiente
para atingir a máxima intensidade do sinal de massas. Esses valores foram utilizados
para os cálculos da quantidade das substâncias permeadas.
Na Figura 4.4, pode-se observar também que não ocorrem aumentos
significativos nos sinais de CO2 ou CH4. Este resultado indica que não houve
permeação destes gases ou que a quantidade permeada foi menor que o limite de
detecção do espectrômetro de massas. Este resultado evidencia a alta seletividade da
membrana, uma vez que a elevação na pressão parcial destes componentes na mistura
não resultou em aumento significativo na permeação dos mesmos. Desta forma, nas
próximas figuras apenas os resultados de permeação de He e H2 são apresentados.
Para o cálculo da permeabilidade, considerou-se que a vazão do gás de arraste
(1,0 cm3/s) era muito maior que a vazão do permeado. Desta forma, através da
concentração determinada por espectrômetro de massas chega-se facilmente à vazão
permeada de cada gás ( em cm3/s), que é diretamente proporcional a sua concentração
( em % V/V), conforme representado pela Equação 4.1.
· Equação 4.1
Onde é a vazão do gás de arraste em cm3/s.

69
O fluxo permeado volumétrico nas CNTP da espécie i (i = H2, He, CO2 ou CH4)
é, então, obtido pela Equação 4.2.
· Equação 4.2
Onde é o fluxo permeado volumétrico nas CNTP, A é a área de permeação
do módulo que é de 80 cm2, T é a temperatura do experimento e é a temperatura
nas CNTP (273 K).
A permeabilidade nas CNTP é o coeficiente angular da curva entre o fluxo
permeado volumétrico e a diferença de pressão parcial através da membrana, conforme
representado pela Equação 4.3.
· ∆ Equação 4.3
Onde é a permeabilidade nas CNTP, é a espessura da membrana e ∆pi é
a diferença de pressão parcial do componente i através da membrana.
As Figuras 4.5 e 4.6, a seguir, apresentam para o He e H2, respectivamente, a
variação do sinal do espectrômetro de massas e do fluxo permeado em função da
diferença de pressão parcial através da membrana. Na Tabela 4.3 são apresentados os
diâmetros cinéticos dos gases componentes da mistura e as suas permeabilidades na
membrana de carbono. O limite de detecção do espectrômetro de massas foi estipulado
como sendo a quantidade mínima de gás que deve estar presente no permeado para ser
observada uma variação significativa no sinal do equipamento (2 x 10-14 unidades
arbitrárias), levando em conta a máxima pressão parcial aplicada durante a permeação.
Com a vazão de gás de arraste mantida em 1cm3/s, este sinal corresponde a uma vazão
permeada de 4,3.10-5 cm3/s, ou para o módulo em questão (80 cm2) equivalente a uma
permeabilidade de 0,001 GPU.
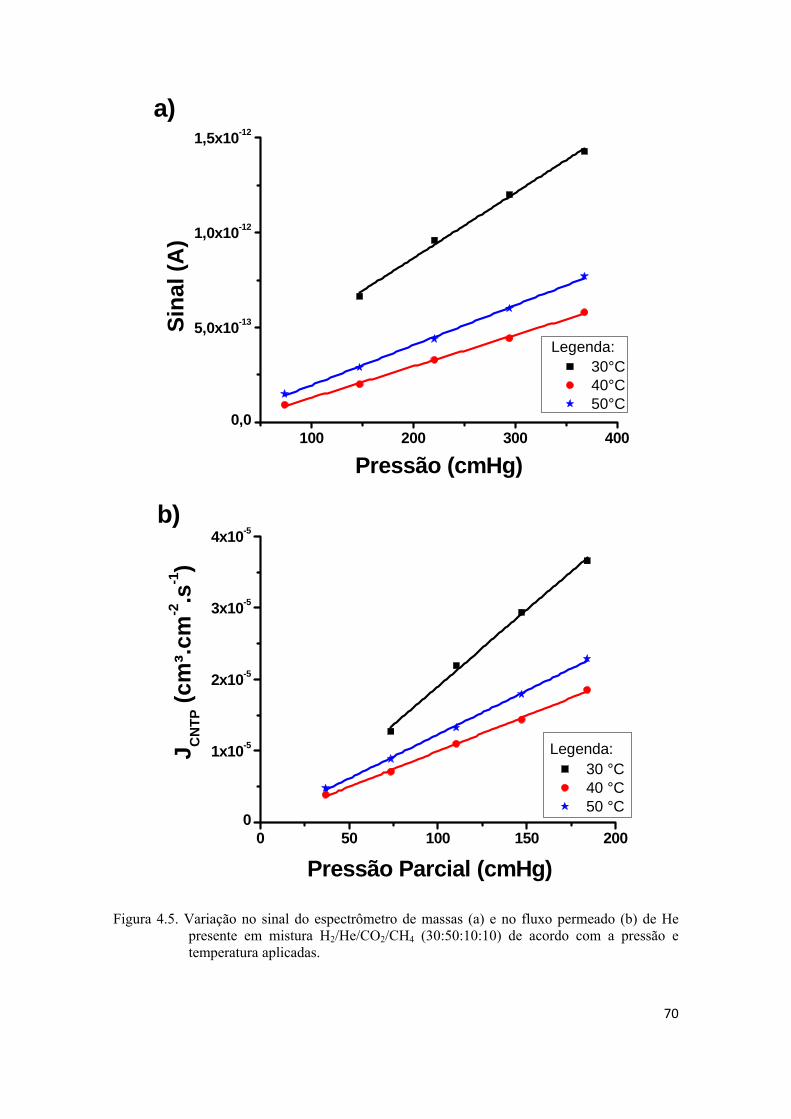
70
Figura 4.5. Variação no sinal do espectrômetro de massas (a) e no fluxo permeado (b) de He presente em mistura H2/He/CO2/CH4 (30:50:10:10) de acordo com a pressão e temperatura aplicadas.
100 200 300 4000,0
5,0x10-13
1,0x10-12
1,5x10-12
Sina
l (A
)
Pressão (cmHg)
30°C 40°C 50°C
Legenda:
a)
0 50 100 150 2000
1x10-5
2x10-5
3x10-5
4x10-5b)
Pressão Parcial (cmHg)
J CN
TP (c
m³.c
m-2.s
-1)
30 °C 40 °C 50 °C
Legenda:
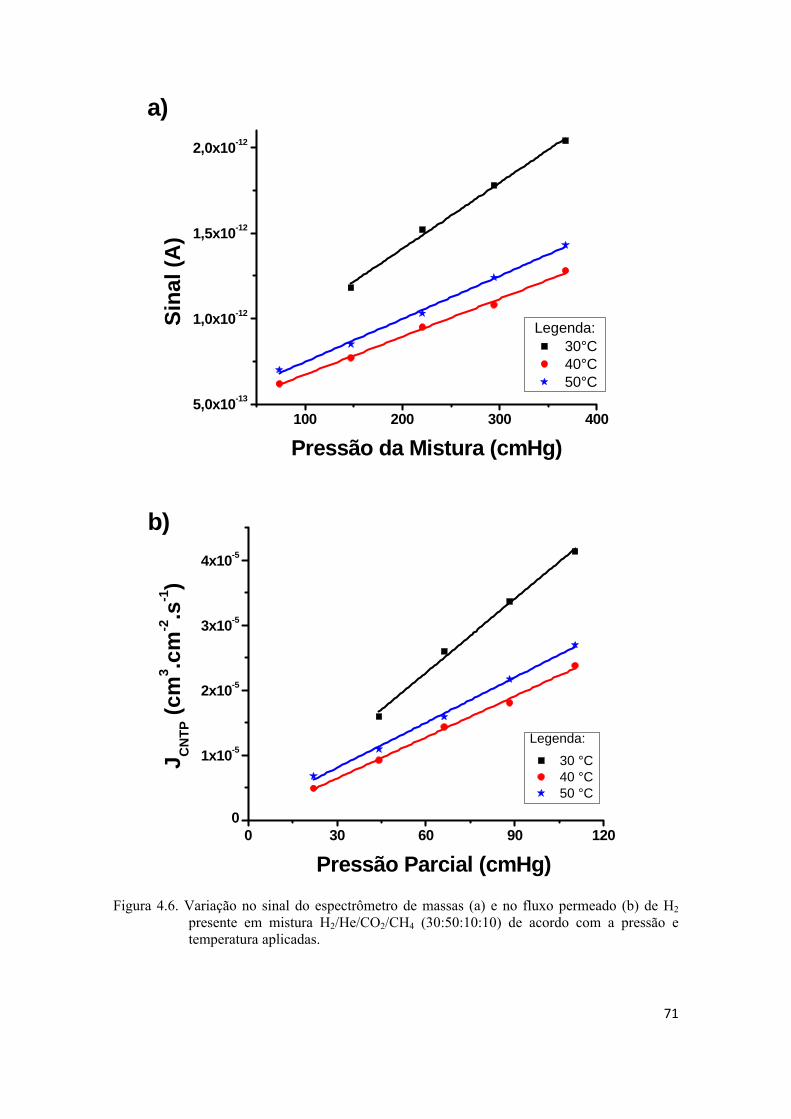
71
Figura 4.6. Variação no sinal do espectrômetro de massas (a) e no fluxo permeado (b) de H2
presente em mistura H2/He/CO2/CH4 (30:50:10:10) de acordo com a pressão e temperatura aplicadas.
100 200 300 4005,0x10-13
1,0x10-12
1,5x10-12
2,0x10-12
30°C 40°C 50°C
Sina
l (A
)
Pressão da Mistura (cmHg)
Legenda:
a)
0 30 60 90 1200
1x10-5
2x10-5
3x10-5
4x10-5
Pressão Parcial (cmHg)
J CN
TP (c
m3 .c
m-2.s
-1)
30 °C 40 °C 50 °C
Legenda:
b)

72
Tabela 4.3. Diâmetro cinético e permeabilidade através da membrana de carbono dos gases constituintes da mistura H2/He/CO2/CH4 (30:50:10:10) em diferentes temperaturas.
Gás Diâmetro Cinético (Å)
Permeabilidade (GPU)* Seletividade H2/i 30 °C 40 °C 50 °C 30 °C 40 °C 50 °C
H2 2,89 0,380 0,211 0,232
He 2,65 0,215 0,100 0,123 1,8 2,1 1,9
CO2 3,46 < 0,001 < 0,001 < 0,001 >380 >211 >232
CH4 3,80 < 0,001 < 0,001 < 0,001 >380 >211 >232
*1GPU = 1·10-6cm3·cm-2·s-1·cmHg-1
Como pode ser observado nas Figuras 4.5 e 4.6, o fluxo permeado variou
linearmente com a diferença de pressão através da membrana, validando o uso do
modelo linear para correlacionar a força motriz e o fluxo permeado. Em todas as
condições não foram observadas variações nos sinais relativos ao CO2 e CH4, indicando
uma seletividade extremamente elevada para a membrana de carbono, ou seja, é
praticamente impermeável a estes compostos.
A Figura 4.7 apresenta a variação da permeabilidade do He e H2 com a
temperatura do sistema.
30 40 50
0,0
0,1
0,2
0,3
0,4
He H2
Perm
eabi
lidad
e (G
PU)
Temperatura (°C)
Legenda:
Figura 4.7. Permeabilidade dos componentes presentes em mistura H2/He/CO2/CH4 (30:50:10:10) de acordo com a temperatura.

73
Pode ser observado no gráfico da Figuras 4.7 que a permeabilidade do He e H2 a
30 °C é superior às suas respectivas permeabilidades em temperaturas mais altas (40 e
50 °C). Este comportamento pode ser atribuído à competição entre mecanismos de
permeação: escoamento por difusão de Knudsen e difusão superficial. O aumento da
temperatura eleva a viscosidade e o livre percurso médio dos gases, aumentando com
isso a resistência ao transporte através da difusão de Knudsen. No entanto, o aumento
da temperatura facilita o salto difusional das moléculas adsorvidas entre os sítios de
adsorção favorecendo com isso o transporte por difusão superficial. Desta forma, o
comportamento observado na Figura 4.7 sugere que inicialmente o transporte era
principalmente através de difusão de Knudsen o que foi desfavorecido com o aumento
da temperatura de 30 para 40 °C. A partir de então a permeabilidade apresentou
elevação com o aumento da temperatura indicando que o mecanismo passou a ser
preponderantemente do tipo difusão superficial.
A ausência da permeação dos gases CO2 e CH4 deixa evidente que além dos
mecanismos previamente discutidos, ocorre também na membrana de carbono a
separação pelo mecanismo de peneira molecular, onde apenas os gases com diâmetro
cinético menor que o diâmetro efetivo dos poros da membrana (He e H2) são capazes de
atravessá-la. Diferente do que foi observado por SHELERKHIN et al. (1992) em
membrana de sílica com tamanho de poros menor que 20Å, na membrana de carbono
utilizada nestes experimentos a permeabilidade do H2, que possui diâmetro cinético
maior que o He, foi ligeiramente superior à permeabilidade de He. Este resultado sugere
que há adsorção preferencial das moléculas de H2 na superfície da membrana de
carbono, resultando em maior permeabilidade deste gás em relação a He. Este
comportamento também foi observado por outros autores (SUDA E HARAYA, 1997b,
HARAYA et al., 1981, RAO e SIRCAR, 1993b).
A membrana mostrou-se bastante eficiente nas temperaturas testadas para
separação de gases proveniente da reação de deslocamento gás-água. Não havendo
Hélio na corrente, será obtido H2 com alta pureza, já que a seletividade H2/CO2 e
H2/CH4 é bastante elevada.
Vale lembrar que a permeabilidade da membrana de carbono é bastante afetada
devido à adsorção de gases presentes na atmosfera e pode haver dificuldade de se evitar

74
o contato da membrana com esses gases durante a montagem dos equipamentos. Neste
sentido, a permeação em temperatura próxima à temperatura ambiente não se mostra
adequada para avaliação do desempenho de membranas de carbono. Com o aumento da
temperatura a maioria dos gases é dessorvida da superfície da membrana, esperando-se
com isso um aumento da permeabilidade dos gases com baixo diâmetro cinético.
4.2.2. Permeação de He e H2 puros
Nos experimentos anteriores não se observou sinal no espectrômetro de massas
para os gases com diâmetros cinéticos maiores. Desta forma, para os experimentos
posteriores foi utilizada a menor vazão possível para o gás de arraste, apenas a
quantidade necessária para fazer a limpeza do equipamento. Esta quantidade de gás de
arraste foi incapaz de ser quantificada através do medidor de vazão disponível
(bolhômetro) e foi determina de forma indireta como explicado a seguir.
Inicialmente a permeabilidade de He puro foi determinada com o auxílio de um
medidor de deslocamento de bolhas (bolhômetro) a 30 °C, cujo valor obtido nas CNTP
foi de 0,17 GPU. Em seguida, foram realizados testes de permeação do mesmo gás (He
puro) no sistema acoplado ao espectrômetro de massas, cujos resultados são
apresentados na Figura 4.8a e b. Com o valor da permeabilidade obtida em bolhômetro
é possível determinar indiretamente a vazão do gás de arraste utilizando as
concentrações do He determinadas a partir do sinal do espectrômetro de massas (Figura
4.8a). A Equação 4.4 relaciona a concentração de He com sua vazão no permeado e a
vazão do gás de arraste (Argônio).
100xQQ
QCgaHe
HeHe += Equação 4.4
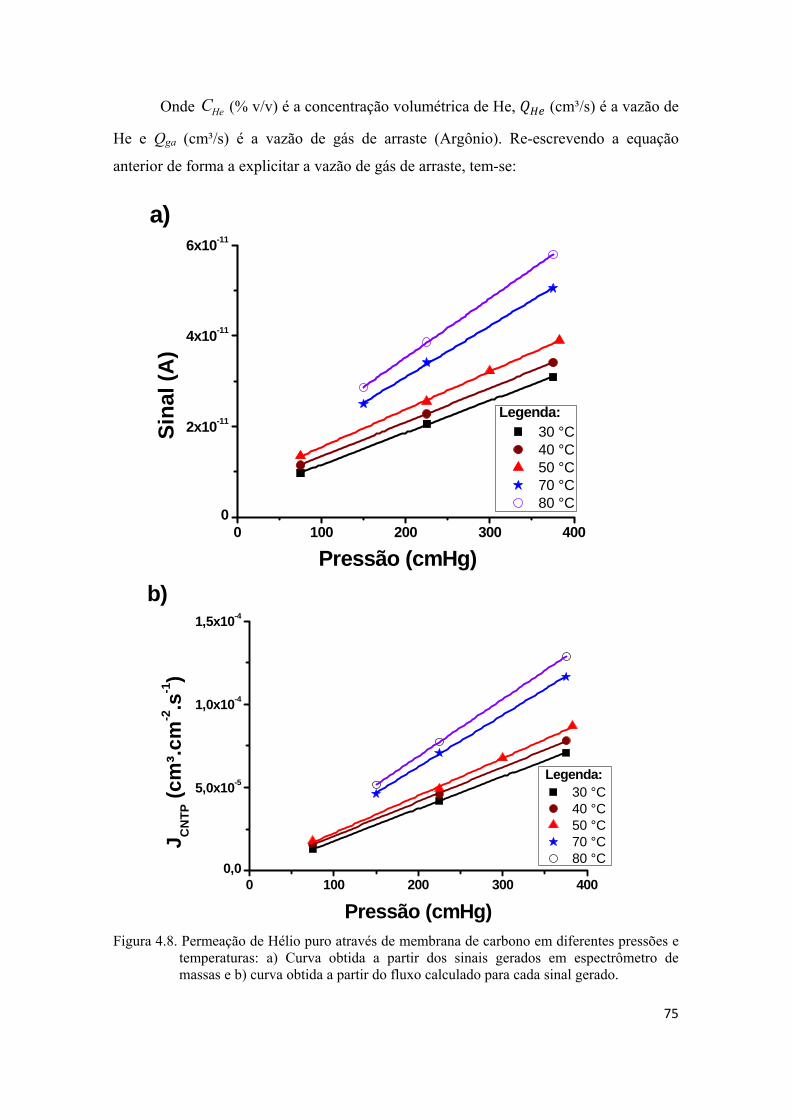
75
Onde HeC (% v/v) é a concentração volumétrica de He, (cm³/s) é a vazão de
He e Qga (cm³/s) é a vazão de gás de arraste (Argônio). Re-escrevendo a equação
anterior de forma a explicitar a vazão de gás de arraste, tem-se:
Figura 4.8. Permeação de Hélio puro através de membrana de carbono em diferentes pressões e temperaturas: a) Curva obtida a partir dos sinais gerados em espectrômetro de massas e b) curva obtida a partir do fluxo calculado para cada sinal gerado.
0 100 200 300 4000
2x10-11
4x10-11
6x10-11
Sina
l (A
)
Pressão (cmHg)
30 °C 40 °C 50 °C 70 °C 80 °C
Legenda:
a)
0 100 200 300 4000,0
5,0x10-5
1,0x10-4
1,5x10-4
J CN
TP (c
m³.c
m-2.s
-1)
Pressão (cmHg)
30 °C 40 °C 50 °C 70 °C 80 °C
Legenda:
b)

76
⎟⎟⎠
⎞⎜⎜⎝
⎛−= 1100
HeHega C
QQ Equação 4.5
A partir das equações 4.2 e 4.3, tem-se:
∆ Equação 4.6
Então, combinando a Equação 4.5 e 4.6, tem-se:
⎟⎟⎠
⎞⎜⎜⎝
⎛−⎥
⎦
⎤⎢⎣
⎡∆⎟
⎠⎞
⎜⎝⎛= 1100...
HeHe
Hega C
pAl
PQ Equação 4.7
Utilizando os dados de permeação do He, a 30 °C, com detecção pelo
espectrômetro de massas em combinação com o valor da permeabilidade do Hélio nesta
temperatura °
0,19 GPU obtida por deslocamento de bolhas, determinou-
se que a vazão de gás de arraste era em torno de 0,078 cm3/s.
A vazão de gás de arraste foi mantida constante nos demais experimentos,
possibilitando determinar a vazão permeada de cada componente. A vazão volumétrica
de permeado de cada componente ( em cm3/s) pode ser calculada através da Equação
4.8.
100 Equação 4.8
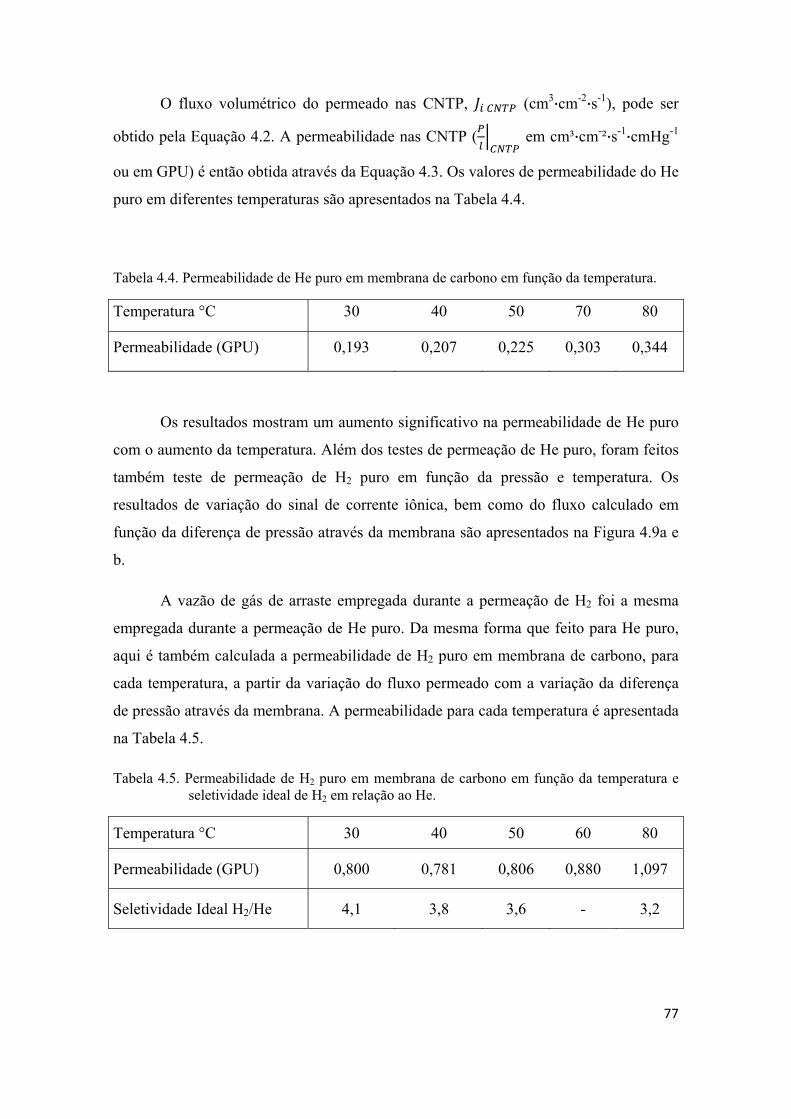
77
O fluxo volumétrico do permeado nas CNTP, (cm3·cm-2·s-1), pode ser
obtido pela Equação 4.2. A permeabilidade nas CNTP ( em cm³·cm-²·s-1·cmHg-1
ou em GPU) é então obtida através da Equação 4.3. Os valores de permeabilidade do He
puro em diferentes temperaturas são apresentados na Tabela 4.4.
Tabela 4.4. Permeabilidade de He puro em membrana de carbono em função da temperatura.
Temperatura °C 30 40 50 70 80
Permeabilidade (GPU) 0,193 0,207 0,225 0,303 0,344
Os resultados mostram um aumento significativo na permeabilidade de He puro
com o aumento da temperatura. Além dos testes de permeação de He puro, foram feitos
também teste de permeação de H2 puro em função da pressão e temperatura. Os
resultados de variação do sinal de corrente iônica, bem como do fluxo calculado em
função da diferença de pressão através da membrana são apresentados na Figura 4.9a e
b.
A vazão de gás de arraste empregada durante a permeação de H2 foi a mesma
empregada durante a permeação de He puro. Da mesma forma que feito para He puro,
aqui é também calculada a permeabilidade de H2 puro em membrana de carbono, para
cada temperatura, a partir da variação do fluxo permeado com a variação da diferença
de pressão através da membrana. A permeabilidade para cada temperatura é apresentada
na Tabela 4.5.
Tabela 4.5. Permeabilidade de H2 puro em membrana de carbono em função da temperatura e seletividade ideal de H2 em relação ao He.
Temperatura °C 30 40 50 60 80
Permeabilidade (GPU) 0,800 0,781 0,806 0,880 1,097
Seletividade Ideal H2/He 4,1 3,8 3,6 - 3,2
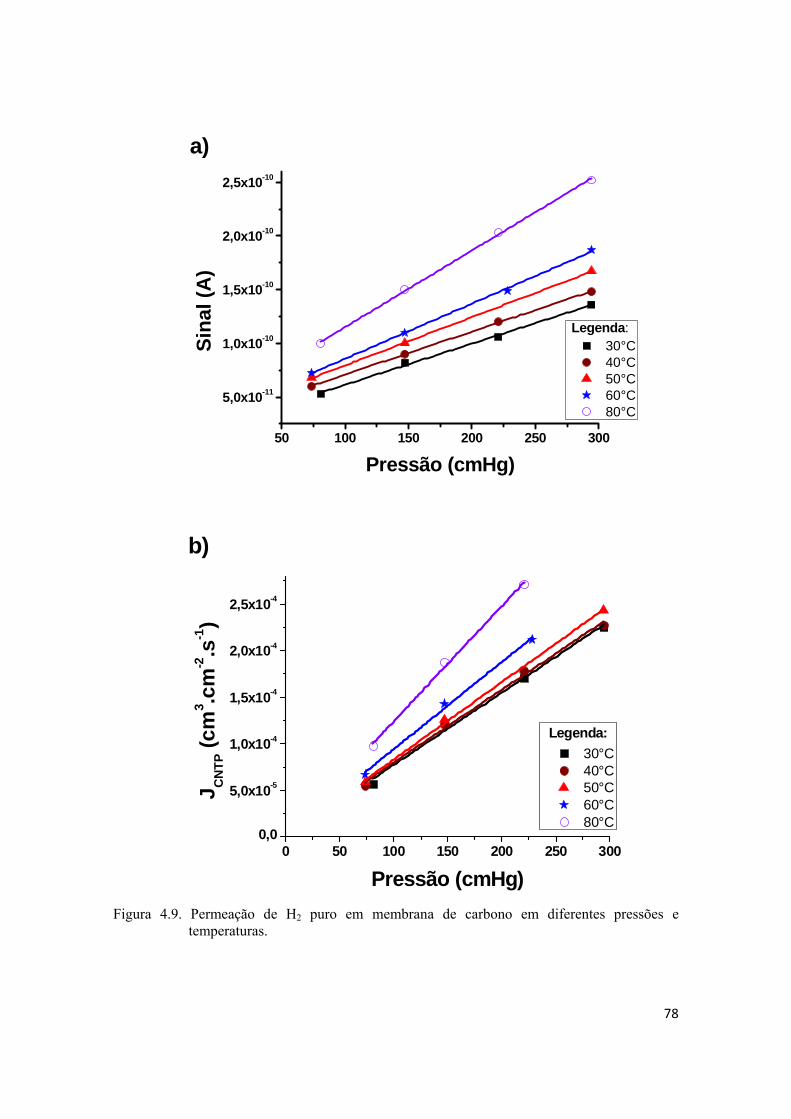
78
Figura 4.9. Permeação de H2 puro em membrana de carbono em diferentes pressões e temperaturas.
50 100 150 200 250 300
5,0x10-11
1,0x10-10
1,5x10-10
2,0x10-10
2,5x10-10
Sina
l (A
)
Pressão (cmHg)
30°C 40°C 50°C 60°C 80°C
Legenda:
a)
0 50 100 150 200 250 3000,0
5,0x10-5
1,0x10-4
1,5x10-4
2,0x10-4
2,5x10-4
J CN
TP (c
m3 .c
m-2.s
-1)
Pressão (cmHg)
30°C 40°C 50°C 60°C 80°C
Legenda:
b)
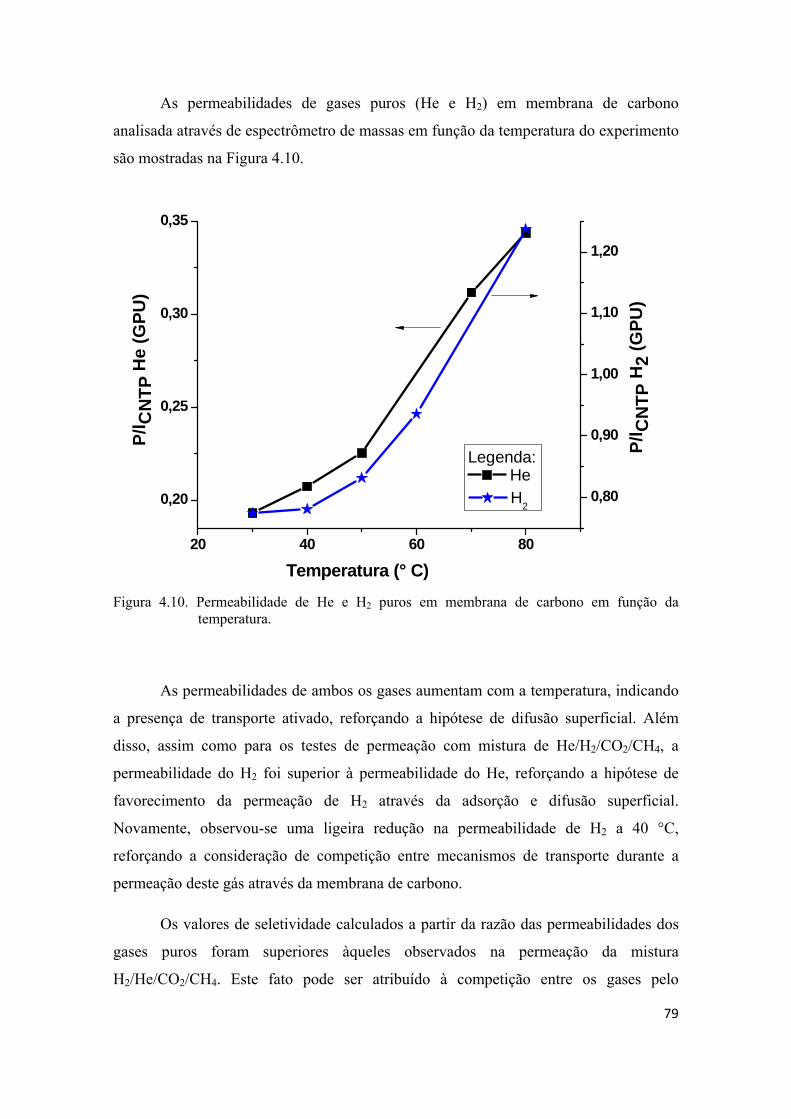
79
As permeabilidades de gases puros (He e H2) em membrana de carbono
analisada através de espectrômetro de massas em função da temperatura do experimento
são mostradas na Figura 4.10.
Figura 4.10. Permeabilidade de He e H2 puros em membrana de carbono em função da temperatura.
As permeabilidades de ambos os gases aumentam com a temperatura, indicando
a presença de transporte ativado, reforçando a hipótese de difusão superficial. Além
disso, assim como para os testes de permeação com mistura de He/H2/CO2/CH4, a
permeabilidade do H2 foi superior à permeabilidade do He, reforçando a hipótese de
favorecimento da permeação de H2 através da adsorção e difusão superficial.
Novamente, observou-se uma ligeira redução na permeabilidade de H2 a 40 °C,
reforçando a consideração de competição entre mecanismos de transporte durante a
permeação deste gás através da membrana de carbono.
Os valores de seletividade calculados a partir da razão das permeabilidades dos
gases puros foram superiores àqueles observados na permeação da mistura
H2/He/CO2/CH4. Este fato pode ser atribuído à competição entre os gases pelo
20 40 60 80
0,20
0,25
0,30
0,35
P/l C
NTP
H2
(GPU
)
P/l C
NTP
He
(GPU
)
He
Temperatura (° C)
0,80
0,90
1,00
1,10
1,20
H2
Legenda:

80
transporte através da membrana, reduzindo assim a área disponível para a permeação de
um dado componente da mistura. Cabe mencionar que o H2 continua apresentando
permeabilidade superior à permeabilidade do He. O favorecimento da permeação do H2
foi atribuída à adsorção seletiva seguida de difusão superficial. Contudo, a adsorção é
desfavorecida com o aumento da temperatura, o que por sua vez gera um decréscimo na
seletividade ideal de H2/He com o aumento na temperatura.
4.2.4. Permeação da mistura H2/CO
A separação de H2 de mistura H2/CO foi realizada com o objetivo de avaliar o
desempenho da membrana de carbono na separação deste gás em corrente resultante da
reforma com vapor do gás natural para produção de H2, bem como para aplicações em
que é necessário o ajuste de composição do gás de síntese.
Foram feitos testes de permeação com a mistura contendo H2 e CO na proporção
de 1:1. Os resultados destes testes são apresentados nas Figuras 4.11a e b, 4.12a e b e,
4.13 e Tabela 4.6.
São observadas variações mais intensas nos sinais dos gases a temperaturas mais
elevadas (Figura 4.10a), o que é resultado do aumento no fluxo permeado. Como a
vazão de gás de arraste é baixa, é necessário levar em conta que a quantidade de H2
permeada tem influência na concentração de CO e vice-versa. Então as concentrações
de H2 e CO no permeado são dadas pelas Equações 4.10 e 4.11, respectivamente.
100 Equação 4.10
100 Equação 4.11
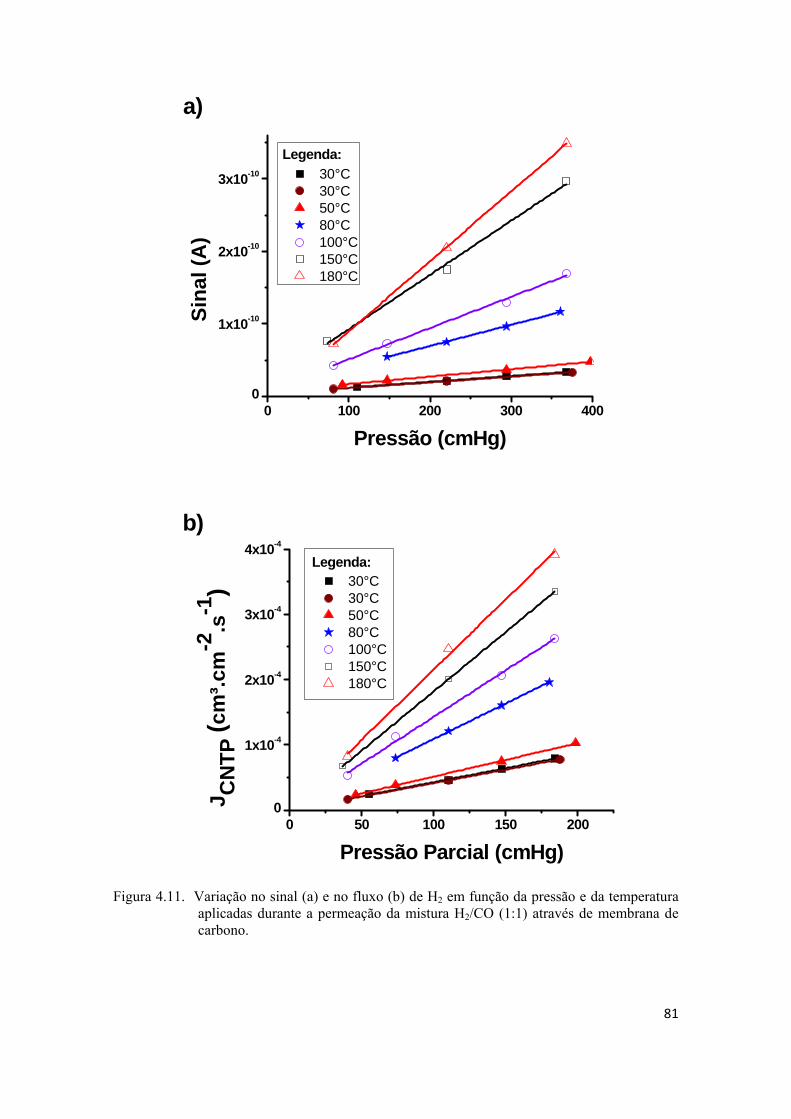
81
Figura 4.11. Variação no sinal (a) e no fluxo (b) de H2 em função da pressão e da temperatura aplicadas durante a permeação da mistura H2/CO (1:1) através de membrana de carbono.
0 100 200 300 4000
1x10-10
2x10-10
3x10-10
Sina
l (A
)
Pressão (cmHg)
30°C 30°C 50°C 80°C 100°C 150°C 180°C
Legenda:
a)
0 50 100 150 2000
1x10-4
2x10-4
3x10-4
4x10-4
30°C 30°C 50°C 80°C 100°C 150°C 180°C
J CN
TP (c
m³.c
m-2
.s-1
)
Pressão Parcial (cmHg)
Legenda:
b)
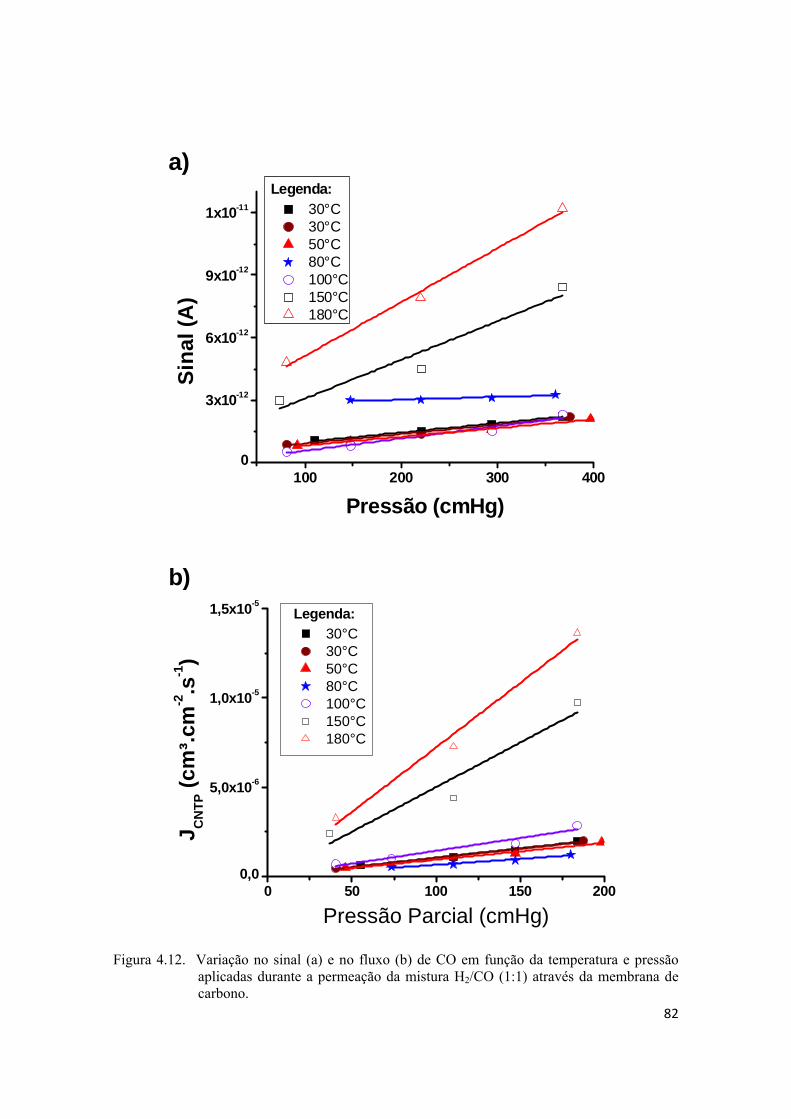
82
Figura 4.12. Variação no sinal (a) e no fluxo (b) de CO em função da temperatura e pressão aplicadas durante a permeação da mistura H2/CO (1:1) através da membrana de carbono.
100 200 300 4000
3x10-12
6x10-12
9x10-12
1x10-11
Sina
l (A
)
Pressão (cmHg)
30°C 30°C 50°C 80°C 100°C 150°C 180°C
Legenda:a)
0 50 100 150 2000,0
5,0x10-6
1,0x10-5
1,5x10-5
30°C 30°C 50°C 80°C 100°C 150°C 180°C
J CN
TP (c
m³.c
m-2.s
-1)
Pressão Parcial (cmHg)
Legenda:
b)

83
Onde , , e são as vazões e concentrações volumétricas de H2 e
CO, respectivamente.
Como a vazão do gás de arraste é conhecida e as concentrações dos
componentes determinadas através do espectrômetro de massas, empregando as
Equações 4.10 e 4.11 podem-se determinar as vazões de H2 e CO, como apresentado
nas equações 4.12 e 4.13.
Equação 4.12
Equação 4.13
A partir dos valores das vazões permeadas, foram calculados os fluxos
volumétricos permeados para cada componente e representados em função da diferença
de pressão parcial do componente através da membrana. A permeabilidade é obtida a
partir do coeficiente angular da reta que relaciona o fluxo à diferença de pressão através
da membrana. Os resultados são apresentados na Tabela 4.6 e Figura 4.13.
Tabela 4.6. Permeabilidade de H2 e CO determinadas a partir da permeação da mistura H2/CO (1:1) em membrana de carbono em função da temperatura.
Temperatura (°C) H2 (GPU) CO (GPU) Seletividade H2/CO
27* 0,420 0,008 0,0104 0,0006 40 1 50 0,514 0,0093 55 80 1,090 0,0065 168 100 1,429 0,0143 100 150 1,820 0,0500 36 180 2,154 0,0722 30
*Experimento em duplicata.
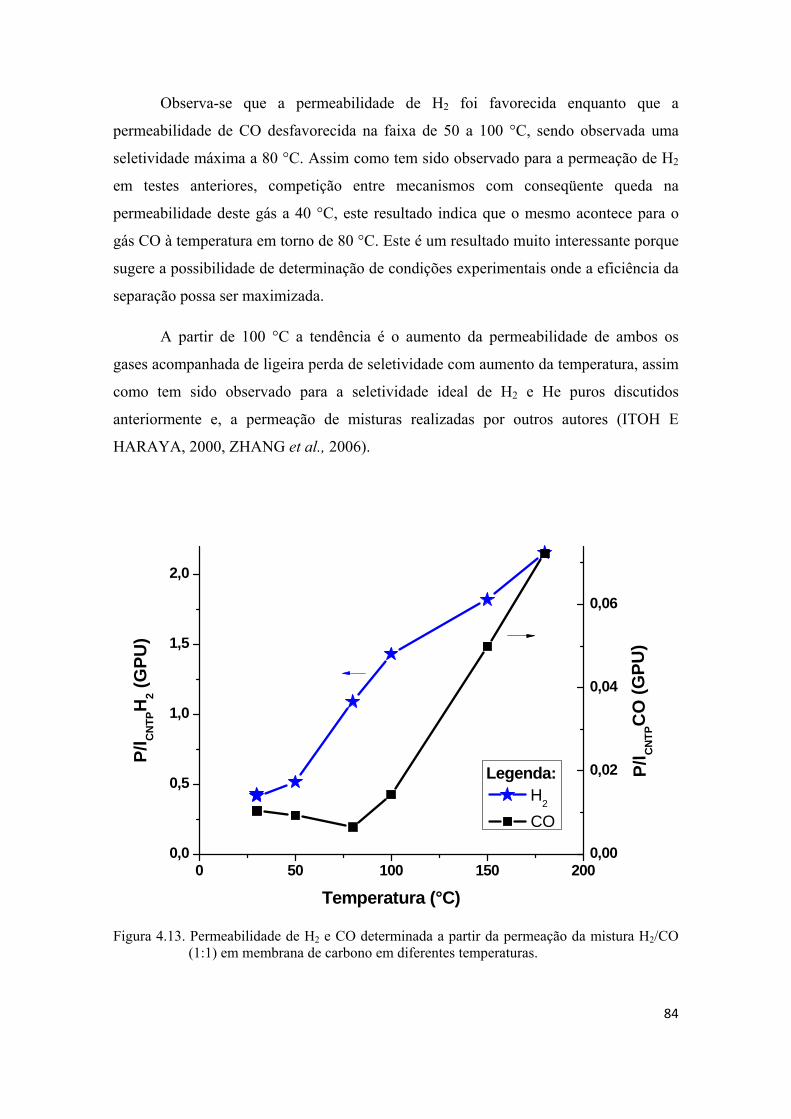
84
Observa-se que a permeabilidade de H2 foi favorecida enquanto que a
permeabilidade de CO desfavorecida na faixa de 50 a 100 °C, sendo observada uma
seletividade máxima a 80 °C. Assim como tem sido observado para a permeação de H2
em testes anteriores, competição entre mecanismos com conseqüente queda na
permeabilidade deste gás a 40 °C, este resultado indica que o mesmo acontece para o
gás CO à temperatura em torno de 80 °C. Este é um resultado muito interessante porque
sugere a possibilidade de determinação de condições experimentais onde a eficiência da
separação possa ser maximizada.
A partir de 100 °C a tendência é o aumento da permeabilidade de ambos os
gases acompanhada de ligeira perda de seletividade com aumento da temperatura, assim
como tem sido observado para a seletividade ideal de H2 e He puros discutidos
anteriormente e, a permeação de misturas realizadas por outros autores (ITOH E
HARAYA, 2000, ZHANG et al., 2006).
Figura 4.13. Permeabilidade de H2 e CO determinada a partir da permeação da mistura H2/CO (1:1) em membrana de carbono em diferentes temperaturas.
0 50 100 150 2000,0
0,5
1,0
1,5
2,0
P/l C
NTP
CO
(GPU
)
P/l C
NTP
H2 (G
PU)
Temperatura (°C)
H2
Legenda:
0,00
0,02
0,04
0,06
CO

85
Após os experimentos com a mistura, que envolveram temperaturas elevadas,
repetiu-se o teste de permeação com He puro, medindo-se o fluxo permeado por
deslocamento de bolhas (bolhômetro). O valor da permeabilidade nas CNTP foi de 0,44
GPU. Este valor é 2,6 vezes maior que a permeabilidade obtida para o mesmo gás antes
dos experimentos com as misturas. Resultado que evidencia a ocorrência de
desbloqueio dos poros da membrana por dessorção, promovido pela utilização de gás de
arraste e temperatura elevada.

86
5. CONCLUSÃO
Este trabalho representa a continuidade de uma linha de pesquisa envolvendo o
desenvolvimento de membranas de carbono seletivas a H2 para aplicação em reatores de
reforma do gás natural.
As conclusões chegadas com a análise dos dados experimentais são apresentadas
a seguir:
• A membrana de carbono tem sua morfologia (diâmetro de poros) alterada
durante a pirólise acompanhada de perda de massa, perda de
permeabilidade e aumento da seletividade e resistência térmica.
• A membrana de carbono obtida com metodologia empregada em
trabalhos anteriores, embora tenha apresentado baixa permeabilidade à
temperatura ambiente, mostrou-se extremamente seletiva a H2.
• Membrana de carbono apresentou muito alta seletividade a H2 em relação
a CO2 e CH4 em mistura contendo H2/He/CO2/CH4.
• Membrana de carbono teve sua permeabilidade aumentada com o
aumento da temperatura durante a permeação de gases puros.
• A permeabilidade de componentes de mistura tem influência positiva
com o aumento da temperatura, contudo há perda de seletividade (de H2
em relação a CO), o que evidencia a necessidade de preparo de
membrana de carbono com alta seletividade a temperatura ambiente.
Aparentes desvios destas tendências merecem investigação mais
criteriosa a fim de comprovar a existência de competição entre
mecanismos de transporte dos gases em membranas de carbono e,
conseqüentemente a possibilidade de existência de regiões onde a
separação de misturas possa ser maximizada.
• O módulo montado mostrou-se eficiente devido ao aumento de área de
permeação, possibilitando o aumento da quantidade permeada e
conseqüentemente possibilitando a análise do permeado com menor erro
de quantificação.

87
• A configuração do módulo permitiu a análise direta do permeado que por
sua vez possibilita a análise das variações com o tempo em sistemas
estáticos.
• O emprego de sistema com capacidade de quantificação dos
componentes presentes em permeado de mistura, através do emprego de
espectrômetro de massas mostrou-se adequada, devido à simplicidade e
continuidade da coleta de dados permitindo a avaliação de todas as
interferências ocasionadas durante o tempo de permeação.

88
6. SUGESTÕES PARA TRABALHOS FUTUROS
• Avaliação mais detalhada dos desvios observados para a permeação de gases
puros e misturas, a fim de averiguar a existência de competição por mecanismo e
a possibilidade de melhorias das condições de separação de misturas.
• Testes em sistema dinâmico.
• Emprego da membrana de carbono em reator de reforma do gás natural e/ou
etanol para produção de H2 na configuração de reator assistido por membrana e
reator com membrana.
• Emprego de temperaturas e pressões mais elevadas em permeação de misturas.
• Aplicação de pós-tratamentos em membranas de carbono, se necessário ajuste
das propriedades de separação em temperaturas mais elevadas.

89
7. BIBLIOGRAFIA
ACHARYA, M., 1999, Engineering design and theoretical analysis of nanoporous
carbon membranes for gas separation, PhD thesis, University of Delaware,
Delaware, USA.
ADHIKARI, S., FERNANDO, S., 2006, Hydrogen separation techniques - reviews,
Ind. Eng. Chem. Res., n. 45, pp. 875-881.
ANDERSON, C.J., PAS, S. J., ARORA, G., et al., 2008, Effect of pyroslysis
temperature and operating temperature on the performance of nanoporous
carbon membranes, Journal of Membrane Science, n. 322, pp. 19-27.
ARMOR, J. N., 1999, The multiple roles for catalysis in the production for H2,
Applied Catalysis A: General, v. 176, pp. 159-176.
ARNAND, M., LANGSAM, M., RAO, M. B., SIRCAR, S., 1997, Multicomponent gas
separation by selective surface flow (SSF) and poly-trimethylpropyne (PTMSP)
membranes. J. Membr. Sci. 123, 17–25.
ARNAND, M., RAO, M. B., SIRCAR, S., 1995, Hydrogen recovery by adsorbent
membranes, U.S. Patent 5435836.
BAKER, R. W., 2002, Future directions of membrane gas separation technology, Ind.
Eng. Chem. Res., v. 41, n. 6, pp. 1393-1411.
BAKER, R. W., 2004, Membrane Technology and Applications, 4 edn., New York,
John Wiley & Sons, Ltd.
BARBOSA-COUTINHO, E., 2004, Membranas catalíticas para a geração de
hidrogênio, COPPE/UFRJ, Tese de D.Sc., COPPE/UFRJ, Rio de Janeiro, RJ,
Brasil.

90
BARRER, R. M., ASH, R., 1973, Transport of Single Gas and of Binary Gas
Mixtures in a Microporous Carbon Membrane. Phisical Chem. Lab., Chem.
Department, Imperial College, Londom SW7 2AY.
BARRER, R. M., ASH, R., POPE, C. G., 1963, Flow of adsorbable gases and vapors
in a microporous medium. Part II. Binary Mixtures. Proc. Roy. Soc. A271, pp.
19.
BARSEMA, J. N., BALSTER, J., JORDAN, V., et al., 2003, Functionalized carbon
molecular sieve membranes containing Ag-nanoclusters. Journal of Membrane
Science n. 219, pp. 47-57.
BERNARDO, P.,DRIOLI, E., GOLEMME, G., 2009, Membrane Gas Separation: A
Review/State of the Art. Ind. Eng. Chem. Res., n. 48, pp. 4638-4663.
BOND, G.C., 1987, Heterogeneous Catalysis: Principles and Applications, 2 ed.,
Oxford, Oxford University Press.
BORGES, C.P., 1993, Fibras Ocas Compostas para a Remoção de Poluentes
Orgânicos de Soluções Aquosas pelo Processo de Pervaporação, Tese de D.Sc.,
COPPE/UFRJ, Rio de Janeiro, RJ, Brasil.
BREDSEN, R., JORDAL, K., BOLLARD, O., 2004, Chem. Eng. Process, n. 43, pp.
1129.
BURGGRAAF, A. J., COT, L., 1996, Fundamentals of inorganic membrane science
and technology, Membrane Science and Technology Series, 4, Elsiever:
Amsterdam, The Nerthlands.
BURNS, R. L., AND KOROS, W. J., 2003, Defining the challenges for C3H6/C3H8
separation using polymeric membranes, J. Membr. Sci., n. 211, v. 2, pp. 299-
309.
CADOTTE E FRANCIS, 1996, Highly anisotropic membranes, United States Patent
4333972 .

91
CETENO, T. A., FUERTES, A. B., 2000, Carbon molecular sieve gas separation
membranes based on poly(vinylidene chloride-co-vinyl chloride). Carbon, n. 38,
pp. 1067–1073.
CETENO, T. A., FUERTES, A. B., 2001, Carbon molecular sieve membranes derived
from a phenolic resin supported on porous ceramic tubes. Sep. Purif. Tech., n.
25, pp. 379–84.
CETENO, T. A., FUERTES, A. et al., 1997, Gas permeation properties and
characterization of asymmetric carbon membranes prepared by pyrolyzing
asymmetric polyimide hollow fiber membrane. J. Membr. Sci., n. 134, pp. 245–
53.
CHEN, J. C., 1998, Modification of polyacrylonitrile (PAN) precursor fiber via post-
spinning plasticization and stretching. PhD. Thesis, The Pennsylvania State
University, Pennsylvania, USA.
CLAUSI, D. T., KOROS, W. J., 2000, Formation of defect-free polyimide hollow fiber
membranes for gas separations, J. Membran. Sci., n. 167, pp. 79-89.
CLUITERS, S. C. A., 2004, Status review on membrane systems for Hydrogen
separation, Energy Center of The Netherlands: Petten, The Netherlands.
CONTE, M., LACOBAZZI, A., ROCHETTI, M., VELLONE, R., 2001, Hydrogen
economy for a sustainable development: state-of-the-art and technological
perspectives. Journal of Power Sources, v. 100, pp. 171-187.
COSTAMAGNA, P., SRINIVASAN, S., 2001, Quantum jumps in the PEMFC
science and technology from the 1960s to the year 2000, Part I Fundamental
scientific aspects. Journal of Power Sources, v. 102, pp. 242-252.
COURTY, P. R., CHAUVEL, A., 1996, Catalysis, the turntable for a clean future.
Catalysis Today, v. 29, pp. 3-15.
DAUTZEMBERG, F. M., MUKHERJEE, M., 2001, Process intensification using
multifunctional reators. Chem. Eng. Sci., n. 56, pp. 251.

92
DAVID, L. I. B., ISMAIL, A. F., 2003, Influence of the thermostabilization process
and soak time during pyrolisis process on the polyacrylonitrile carbon
membranes for O2/N2 separation, J. Membran. Sci., n. 213, pp. 285-291.
DI LUCCIO, M., 1997, Membranas Microporosas Planas e do Tipo Fibra-oca a partir
de Sistemas contendo policarbonato como polímero base, Tese de M.Sc.,
COPPE/UFRJ, Rio de Janeiro, RJ, Brasil.
DYBKJAER, I., 1995, Tubular reforming and autothermal reforming of natural gas –
an overview of available processes, Fuel Processing Technology, v. 42, p. 85-
107.
ESTEVES, I. A. A. C., MOTA, J. P. B., 2007, Hybrid Membrane/PSA Processes for
CO2/N2 Separation. Adsorp. Sci. Technol., n. 29, pp. 693-715.
FIGUEIREDO, K. C. S, 2008, Desenvolvimento de membrana com mioglobina para a
permeação seletiva de oxigênio, Tese de D.Sc., COPPE/UFRJ, Rio de Janeiro,
RJ, Brasil.
FREEMAN, B. D., 1999, Basis of permeability/selectivity tradeoff relations in
polymeric gas separation membranes, Macromolecules, 32(2), 375-380.
FUERTES, A. B., 2001, Effect of air oxidation on gas separation properties of
adsorption-selective carbon membranes. Carbon, n. 39, pp. 697–706.
FUERTES, A. B., CENTENO, T. A., 1999, Preparation of supported carbon
molecular sieve membrane, Carbon, n. 37, pp. 679-684.
FUERTS, A. B., CENTENO, T. A., 1998a, Preparation of supported asymmetric
carbon molecular sieve membranes, J. Membr. Sci., n. 144, pp. 105-111.
FUERTS, A. B., CENTENO, T. A., 1998b, Carbon molecular sieve membranes from
polyetherimide, Microporous, Mesoporous Mater, n. 26, pp. 23-26.
GEISZLER, V. C., (1997) Polyimide precursor for carbon membranes, PhD thesis,
University of Texas, Texas, USA.

93
GEISZLER, V. C., KOROS, W. J., 1996, Effect of polyimide pyrolysis conditions on
carbon molecular sieve membrane properties, Ind. Eng. Chem. Res., n. 35, pp.
2999–3003.
GRAHAM, T., 1866, On the absorption and dialytic separation of gases by colloid
septa, Phil. Magazine, n. 32, pp. 401–420.
GRAINGER, D., 2007, Development of carbon membranes for hydrogen recovery,
PhD thesis, Norwegian University of Science and Technology, Trondheim,
Norway.
GRUPTA, A. K., PALIWAL D. K., BAJA P., 1991, JMS-Rev. Macromol. Chem.
Phys., C31:1.
HABERT, A. C., BORGES, C. P., NÓBREGA, R., 2006, Processos e Separação por
Membranas, Rio de Janeiro, e-papers.
HARAYA, K., SUDA, H., YANAGISHITA, H., MATSUDA, S., 1995, Asymmetric
Capillary Membrane of a Carbon Molecular Sieve. J. Chem. Soc., Chem.
Commun., n. 17, pp. 1781–1782.
HAYASHI, J., MIZUTA, H., YAMAMOTO, M. et al., 1996, Separation of
ethane/ethylene and propane/propylene system with a carbonized BPDA-
pp0ODA polyimide. Ind. Eng. Chem. Res., n. 35, pp. 4176–4181.
HAYASHI, J., YAMAMOTO, M., KUSAKABE K., MOROOKA, S., 1997, Effect of
oxidation on gas permeation of carbon molecular sieve membranes based on
BPDA-pp’ODA polyimide, Ind. Eng. Chem. Res. n. 36, pp. 2134-2140.
HINCHLIFFE, A. B., PORTER, K. E., 2000, A comparison of membrane separation
and distillation. Trans. Inst. Chem. Eng., n. 78, pp. 255 – 268.
http://www.cinvention.com, acesso em 01/02/2010.
http://www.tech.mastcarbon.co.uk, acesso em 04/02/2008.
ITOH, N., HARAYA, K., 2000, A carbon membrane reactor, Cat Today, n. 56, pp.
103-111.

94
JONES, C. W., KOROS, W. J., 1994, Carbon molecular sieve gas separation
membranes I – preparation and characterization base on polyimide precursors,
Carbon, n. 32, pp. 1419-1425.
JONES, C. W., KOROS, W.J., 1995, Characterization of ultramicroporous carbon
membranes with humidified feeds. Ind. Eng. Chem. Res., n. 34, pp. 158–163.
KING, D. E., HARRIS, B. J., MACLEAN, D. L., GRAHAM, T. E., 1978, Hollow
fiber permeator apparatus, US Patent 4315819
KORESH, J. E., SOFFER, A., 1980, Study of molecular sieve carbons. Part 1- Pore
structure, gradual pore opening and mechanism of molecular sieving. J. Chem.
Soc. Faraday Trans, n. 76, pp. 2457-2571.
KOROS, W. J., MAHAJAN, R., 2000, Pushing the Limits in Possibilities for Large
Scale Gas Separation: Which Strategies, Journal of Membrane Science, v. 175,
pp. 181-196.
KUSAKABE, K., YAMAMOTO, M., MOROOKA, S., 1998, Gas permeation and
micropore structure of carbon molecular sieving membranes modified by
oxidation. J. Membr. Sci., n.149, pp. 59–67.
KUSUKI, Y., SHIMAZAKI, H., TANIHARA, N. et al., 1997, Gas permeation
properties and characterization of asymmetric carbon membranes prepared by
pyrolyzing asymmetric polyimide hollow fiber membrane. J. Membr. Sci., n. 134,
pp. 245–53.
KYOTANI, T., 2000, Control pore in carbon, Carbon, n. 38, pp. 269-286.
LAGORSSE, S., MAGALHÃES, F. D., MENDES, A., 2008, Aging study of carbon
molecular sieve membranes, Journal of Membrane Science, n. 310, pp. 494–502.
LI, D., WANG, R., CHUNG, T., 2004, Fabrication of lab-scale hollow fiber
membrane modules with high packing density, Separation and Purification
Technology, n. 40, pp. 15-30.

95
LI, N, N., FANE, A. G., WINSTON HO, W. S., MATSUURA, T., 2008, Advanced
Membrane Technology and Applications, Hoboken, New Jersey, A John Wiley
& Sons Inc., Publication.
LI, S-G., 1993, Preparation of Hollow Fiber Membranes for Gas Separation, Ph.D.
Thesis, University of Twente, Holanda.
LIE, J. A., HÄGG, M.-B., 2005, Carbon membranes from cellulose and metal loaded
cellulose. Carbon v. 43, pp. 2600–2607.
LINKOV, V. M., SANDERSON, R. D. LAPIDUS, A. L., KRYLOVA, A. J., 1994a,
Carbon membrane-based catalyst for hydrogenation of CO. Catal. Lett., n. 27,
pp. 97-101.
LINKOV, V. M., SANDERSON, R. D., JACOBS, E. P., 1994b, Carbon membranes
from precursors containing low-carbon residual polymers. Polym. Inter., n. 35,
pp. 239–242.
LOEB, S., SOURIRAJAN, S., 1963, Sea Water Demineralization by Means of an
Osmotic Membrane, in Saline Water Conversion–II, Advances in Chemistry
Series Number 28, American Chemical Society, Washington, DC, pp. 117–132.
LU, G. Q., DINIZ DA COSTA, J. C., DUKE, M. et al., 2007, Inorganic membranes
for hydrogen production and purification: A critical review and perspective,
Journal of Colloid and Interface Science, n. 314, pp. 589-603.
LUNSFORD, J. H., 2000, Catalytic Conversion of methane to more useful chemicals
and fuels: a challenge for the 21st century. Catalysis Today, v. 63, p. 165-174.
MENENDEZ, I., FUERTES, A.B., 2001, Aging of carbon membranes under different
environments, Carbon, n. 39, v. 5, pp. 733–740.
MILLER, G. Q., STOECKER, J., 1989, Selection of a hydrogen separation process;
National Petrochemical and Refiners Association: San Francisco, CA.
MOLBURG, J. C.; DOCTOR, R. D., 2003, 20th Annual International Pittsburg Coal
Conference, Pittsburg, PA, USA.

96
MORTHON-JONES D. H., 1984, Polymer processing, London: Chapman and Hall;
[Chapter 2].
MULDER, M., 2000, Basic principles of membrane technology, 2nd Ed, Holanda:
Kluwer Academic Publisher.
NATIONAL RESEARCH COUNCIL (U.S.). PANEL ON SEPARATION
TECHNOLOGY FOR INDUSTRIAL REUSE AND RECYCLING, 1998,
Separation technologies for the industries of the future, National Academies
Press, ISBN 0309063779, 9780309063777.
NUNES, S. P., PEINEMANN, K.-V., 2006, Membrane Technology in the Chemistry
Industry. 2nd edn., Weinheim, Germany, Wiley-VCH Verlag GmbH & Co.
KGaA,
OCKWIG, N. W. and NENOFF, T. M., 2007, Membranes for Hydrogen Separation.
Chem. Rev., n. 107, pp. 4078-4110.
OGAWA, M., NAKANO, Y., 1999, Gas permeation through carbonized hollow fiber
Membranes prepared by gel modification of polyamic acid. J. Membr. Sci., n.
162, pp. 189–198.
PARANJAPE, M., CLARKE, P. F., PRUDEN, B. B. et al., 1998, Separation of bulk
carbon dioxide-hydrogen mixtures by selective surface flow membranes.
Adsorption, n. 4, pp. 355–360.
PETERSEN, J., MATSUDA, M., HARAYA, K., 1997, Capillary carbon molecular
sieve membranes derived from kapton for high temperature gas separation. J.
Membr. Sci., n. 131, pp. 85–94.
RAO, M. B., SIRCAR, S., 1993a, Nanoporous carbon membrane for gas separation.
Gas Sep. Purif., n. 7, v. 4, pp. 279–284.
RAO, M. B., SIRCAR, S., 1993b, Nanoporous carbon membranes for separation of
gas mixtures by selective surface flow. Journal of Membrane Science, n. 85, pp.
253-264.

97
RAO, M. B., SIRCAR, S., 1996, Performance and pore characterization of
nanoporous carbon membranes for gas separation. J. Membr. Sci., n. 110, pp.
109–118.
ROBESON, L. M., 1991, Correlation of separation factor versus permeability for
polymeric membranes. J. Membran. Sci., n. 62, vol. 2, pp. 165-185.
ROBESON, L. M., BURGOYNE, W. F., LANGSAM, M. et al., 1994, High
performance polymers for membrane separation. Polymer, n. 35, pp. 4970-4978.
ROSTRUP-NIELSON, J. R.; SÉESTED, J.; NORSKOV, J. K., 2002, Hydrogen and
Synthesis gas by steam and CO2 reforming. Advances in Catalysis, v. 47, p. 65-
139.
SAUFI, S. M., ISMAIL, A. F., 2004, Fabrication of carbon membranes for gas
separation – a review. Carbon, n. 42, pp. 241-259.
SCHINDLER, E., MAIER, F., 1990, Manufacture of porous carbon membranes. US
patent 4919860.
SEDIGH, M. G., ONSTOT, W. J., XU, L., 1998, Experiments and a simulation of
transport and separation of gas mixtures in carbon molecular sieve membranes.
Journal of Physical Chemistry, n. 102, pp. 8580-8589.
SEDIGH, M.G., XU, L., TSOTSIS, T.T., SAHIMI, M., 1999, Transport and
Morphological Characteristics of Polyetherimide-Based Carbon Molecular
Sieve Membranes. Ind. Eng. Chem. Res., n. 38, pp. 3367-3380.
SHAO, L., LOW, B. T., CHUNG, T-S., et al., 2009, Polymeric membranes for the
hydrogen economy: Contemporary approaches and prospects for the future –
review. J. Membr. Sci., n. 327, pp. 18-31.
SHELEKHIN, A. B., DIXON, A. G., MA, Y. H., 1992, J. Membr. Sci., n. 75, p. 233.
SHIFLETT, M. B., FOLEY, H. C., 2000, On the preparation of supported nanoporous
carbon membranes. J. Membr. Sci., n. 179, pp. 275–82.

98
SHREVE, R. N., BRINK JR., J. A., 1997, Indústrias de processos químicos. 4 ed., Rio
de Janeiro, Editora Guanabara Koogan S. A.
SILVA, S. E., 2009, Caracterização do Transporte Facilitado em Membranas
contendo Biotransportadores para Separação do Oxigênio, Tese de D.Sc.,
COPPE/UFRJ, Rio de Janeiro, RJ, Brasil.
SIMONSEM, K. A., O’KEEFE, L., FONG, W. F. Oil & gas Journal 22 (March-1993)
45.
SIRCAR, S., RAO, M. B., THAEROM, M. A., 1999, Selective surface flow membrane
for gas separation. Separation Science Technology, n. 34, pp. 2081.
SMITH, S. P. J., LINKOV V. M., SANDERSON R. D. et al., 1995, Preparation of
hollow-fibre composite carbon–zeolite membranes. Micro. Mat., n. 4, pp. 385–
90.
SOFFER, A., AZARIAH, A., AMAR, A. et al., 1997, Method of improving the
selectivity of carbon membranes by chemical vapor deposition. US patent, n.
5695818.
SOFFER, A., GILRON, J., COHEN, H., 1999, Separation of linear from branched
hydrocarbons using a carbon membrane. US patent 5914434.
SOFFER, A., KORESH, J. SAGGY, S., 1987, Separation device. US patent, n.
4685940.
SOFFER, A., ROSEM, D., SAGUEE. S., KORESH, J., 1989, Carbon membranes. GB
patent, n. 2207666.
SOUZA, M. M. V. M., 2009, Tecnologia do Hidrogênio. Sinergia: FAPERJ.
STEEL, K. M., 2000, Carbon membranes for challenging gas separations. PhD thesis,
University of Texas, Texas, USA.
STOCKER, J.; WHYSALL, M.; MILLER, G. Q., 2005, 30 years of PSA technology
for hydrogen purification. UOP LLC: Des Plaines, IL, 1998.

99
STRATHMANN, H., 1999, Membrane processes for sustainable industrial growth.
Membr. Tech., n. 113, pp. 9–11.
SUDA, H., HARAYA, K., 1997a, Alkene/alkanepermeselectivities of a carbon
molecular sieve membrane. J. Chem. Soc. Chem. Commun., n. 1, pp. 93-94.
SUDA, H., HARAYA, K., 1997b, Gas permeation through micropores of carbon
molecular sieve membranes derived from kapton polyimide. J. Phys. Chem. B, n.
101, pp. 3988-3994.
Vu, D.Q., Koros, W.J., Miller, S.J., 2003, Fouling of carbon molecular sieve hollow-
fiber membranes by condensable impurities in carbon dioxide-methane
separations, Industrial & Engineering Chemistry Research, n. 42, v. 5, pp. 1064–
1075.
YONEYAMA, H., NISHIHARA, Y., 1990, Porous hollow carbon fiber film and
method of manufacturing the same. EP patent 0394449.
YONEYAMA, H., NISHIHARA, Y., 1992, Carbon based porous hollow fiber
membrane and method for producing same. US patent, n. 5089135.
ZHANG, X., HU, H., ZHU, Y. et al., 2006, Methanol Steam Reforming to Hydrogen
in a Carbon Membrane Reactor System. Ind. Eng. Chem. Res., n.45, pp. 7997-
8001.
ZHOU, W., YOSHINO, M., KITA, H., OKAMOTO, K.-I., 2003, Preparation and gas
permeation properties of carbon molecular sieve membranes based on
sulfonated phenolic resin. J. Membr. Sci., n. 217, pp. 55–67.
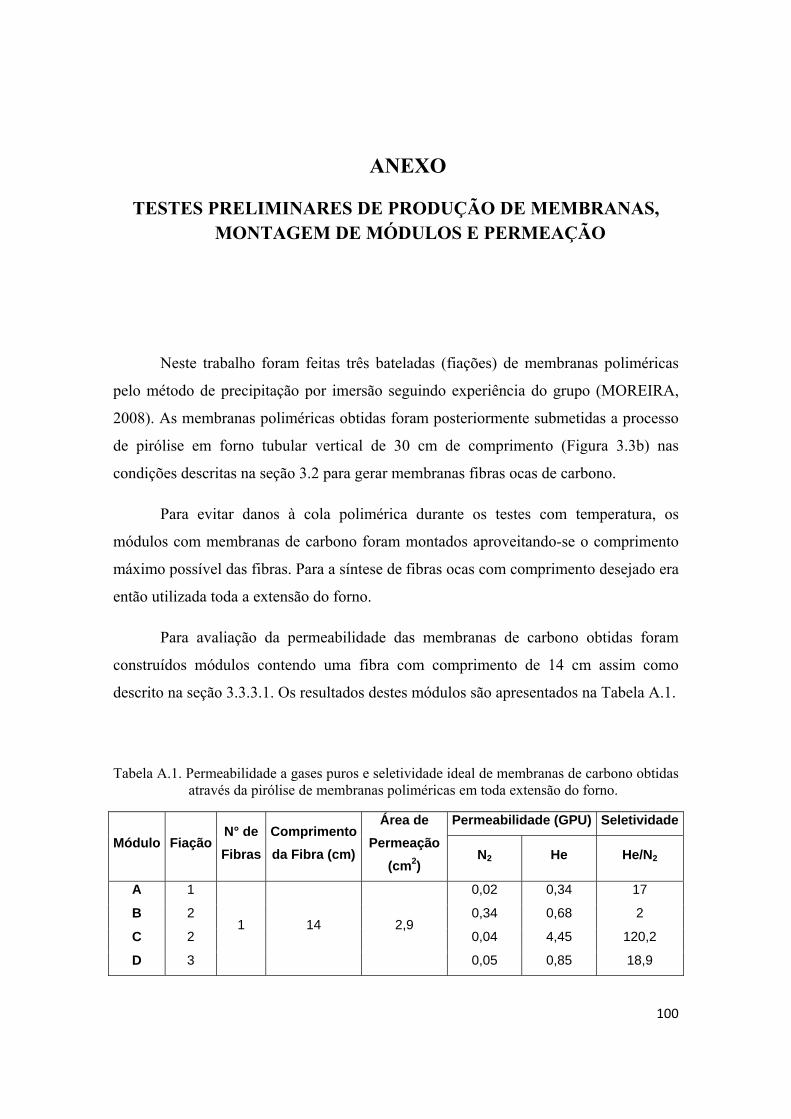
100
ANEXO
TESTES PRELIMINARES DE PRODUÇÃO DE MEMBRANAS, MONTAGEM DE MÓDULOS E PERMEAÇÃO
Neste trabalho foram feitas três bateladas (fiações) de membranas poliméricas
pelo método de precipitação por imersão seguindo experiência do grupo (MOREIRA,
2008). As membranas poliméricas obtidas foram posteriormente submetidas a processo
de pirólise em forno tubular vertical de 30 cm de comprimento (Figura 3.3b) nas
condições descritas na seção 3.2 para gerar membranas fibras ocas de carbono.
Para evitar danos à cola polimérica durante os testes com temperatura, os
módulos com membranas de carbono foram montados aproveitando-se o comprimento
máximo possível das fibras. Para a síntese de fibras ocas com comprimento desejado era
então utilizada toda a extensão do forno.
Para avaliação da permeabilidade das membranas de carbono obtidas foram
construídos módulos contendo uma fibra com comprimento de 14 cm assim como
descrito na seção 3.3.3.1. Os resultados destes módulos são apresentados na Tabela A.1.
Tabela A.1. Permeabilidade a gases puros e seletividade ideal de membranas de carbono obtidas através da pirólise de membranas poliméricas em toda extensão do forno.
Módulo Fiação N° de Fibras
Comprimento da Fibra (cm)
Área de Permeação
(cm2)
Permeabilidade (GPU) Seletividade
N2 He He/N2
A 1
1 14 2,9
0,02 0,34 17
B 2 0,34 0,68 2
C 2 0,04 4,45 120,2
D 3 0,05 0,85 18,9
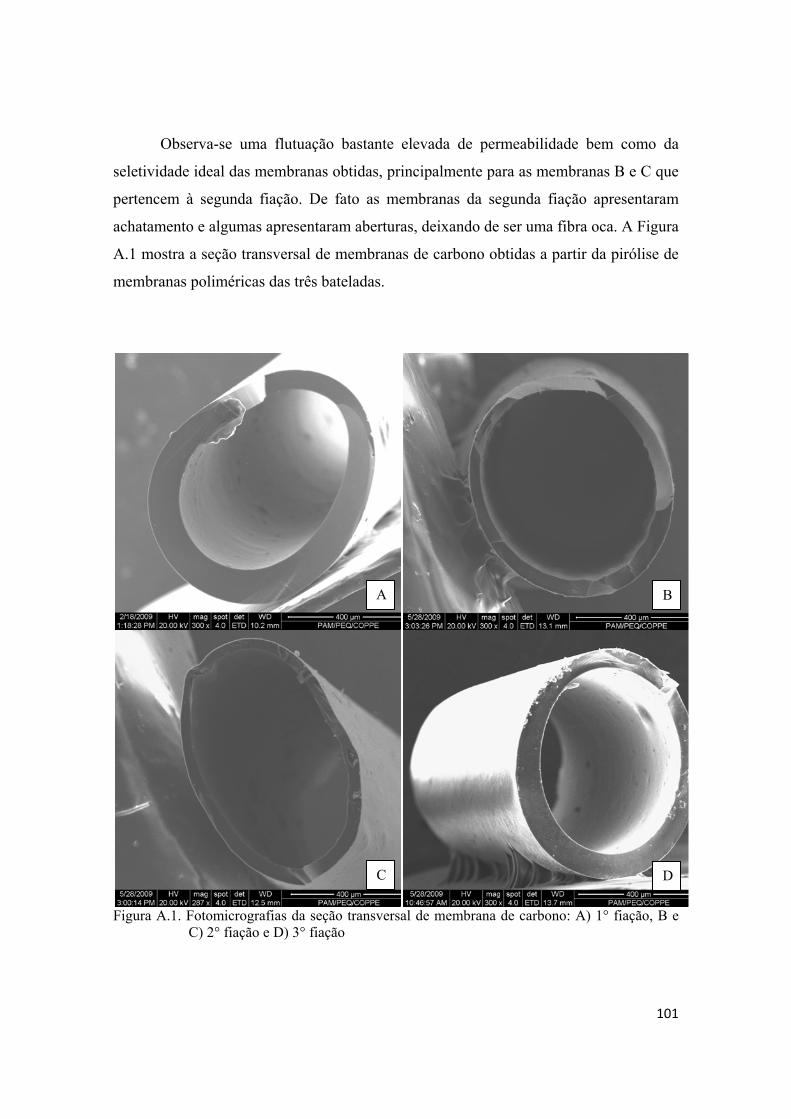
101
Observa-se uma flutuação bastante elevada de permeabilidade bem como da
seletividade ideal das membranas obtidas, principalmente para as membranas B e C que
pertencem à segunda fiação. De fato as membranas da segunda fiação apresentaram
achatamento e algumas apresentaram aberturas, deixando de ser uma fibra oca. A Figura
A.1 mostra a seção transversal de membranas de carbono obtidas a partir da pirólise de
membranas poliméricas das três bateladas.
Figura A.1. Fotomicrografias da seção transversal de membrana de carbono: A) 1° fiação, B e C) 2° fiação e D) 3° fiação
A B
C D

apres
resist
duran
na m
resul
(foto
trans
não e
mem
morf
Abai
polim
mem
Figur
Observa-
sentam pare
tência da m
nte a pirólis
membrana o
ltante bem
omicrografia
sversal. Um
era visualm
mbrana de c
fologia bem
ixo (Figura
méricas da
mbranas de c
ra A.2. FotomB) 3
-se que as
ede com es
membrana g
se causam o
que por su
como na re
a A) apresen
m pequeno ac
mente percep
carbono da
m formada o
a A.2) são
primeira
carbono com
micrografias° fiação
membrana
spessura ba
gerando em
o achatamen
ua vez refle
esistência a
nta parede m
chatamento
ptível como
a terceira f
o que com
apresentad
e terceira
m relação à
s da seção tra
as da segu
stante redu
m alguns ca
nto e podem
te nas carac
a altas pres
mais espess
o também fo
o ocorria com
fiação (foto
certeza fav
das também
fiação a
morfologia
ansversal de
A
nda fiação
uzida, o que
sos membr
m também p
cterísticas d
sões. A me
sa, mas com
oi observado
m as memb
omicrografi
vorecerá su
m as fotomi
fim de co
de suas pre
membranas
(fotomicro
e conseqüen
anas achata
provocar fra
de permeaçã
embrana da
m variação a
o para esta m
branas da se
ia D) most
as propried
icrografias
omparar a
ecursoras.
poliméricas
ografias B
ntemente af
adas. As ten
aturas ou de
ão da memb
a primeira f
o longo da
membrana
egunda fiaç
tra-se com
dades mecân
das memb
morfologia
s da: A) 1° f
102
e C)
feta a
nsões
feitos
brana
fiação
seção
o que
ão. A
uma
nicas.
branas
a das
fiação,
B
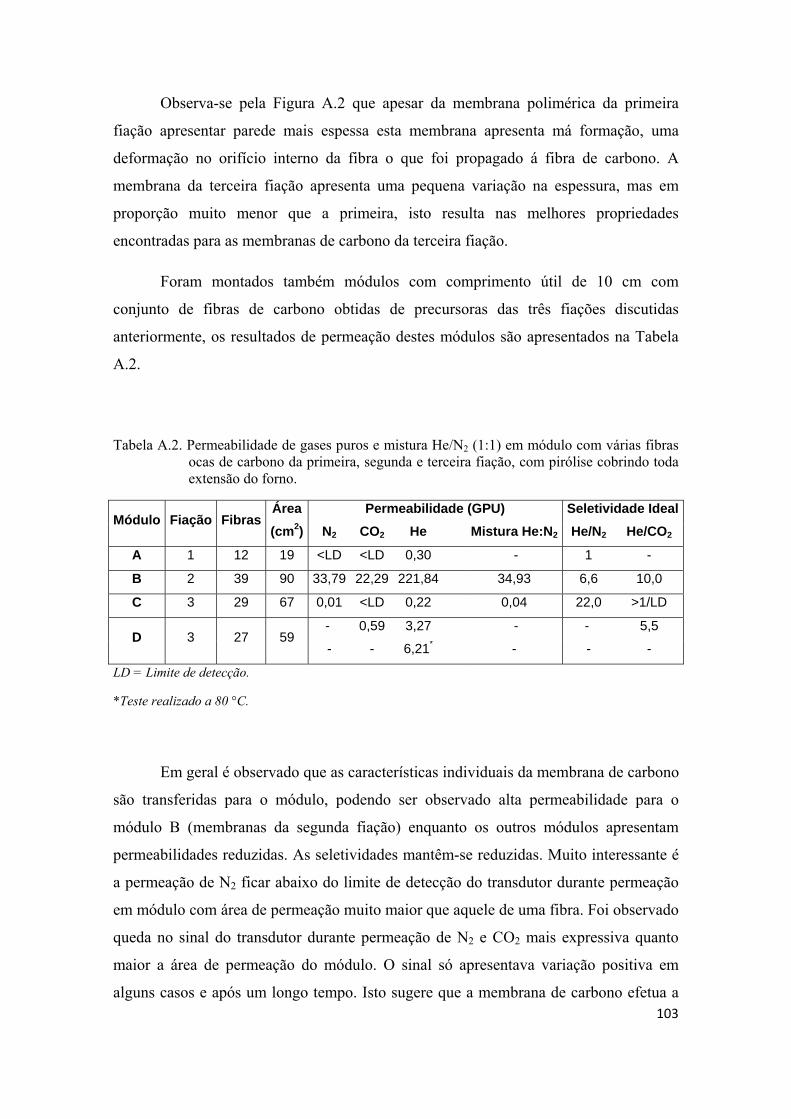
103
Observa-se pela Figura A.2 que apesar da membrana polimérica da primeira
fiação apresentar parede mais espessa esta membrana apresenta má formação, uma
deformação no orifício interno da fibra o que foi propagado á fibra de carbono. A
membrana da terceira fiação apresenta uma pequena variação na espessura, mas em
proporção muito menor que a primeira, isto resulta nas melhores propriedades
encontradas para as membranas de carbono da terceira fiação.
Foram montados também módulos com comprimento útil de 10 cm com
conjunto de fibras de carbono obtidas de precursoras das três fiações discutidas
anteriormente, os resultados de permeação destes módulos são apresentados na Tabela
A.2.
Tabela A.2. Permeabilidade de gases puros e mistura He/N2 (1:1) em módulo com várias fibras ocas de carbono da primeira, segunda e terceira fiação, com pirólise cobrindo toda extensão do forno.
Módulo Fiação Fibras Área (cm2)
Permeabilidade (GPU) Seletividade IdealN2 CO2 He Mistura He:N2 He/N2 He/CO2
A 1 12 19 <LD <LD 0,30 - 1 -
B 2 39 90 33,79 22,29 221,84 34,93 6,6 10,0
C 3 29 67 0,01 <LD 0,22 0,04 22,0 >1/LD
D 3 27 59 - 0,59 3,27 - - 5,5
- - 6,21* - - -
LD = Limite de detecção.
*Teste realizado a 80 °C.
Em geral é observado que as características individuais da membrana de carbono
são transferidas para o módulo, podendo ser observado alta permeabilidade para o
módulo B (membranas da segunda fiação) enquanto os outros módulos apresentam
permeabilidades reduzidas. As seletividades mantêm-se reduzidas. Muito interessante é
a permeação de N2 ficar abaixo do limite de detecção do transdutor durante permeação
em módulo com área de permeação muito maior que aquele de uma fibra. Foi observado
queda no sinal do transdutor durante permeação de N2 e CO2 mais expressiva quanto
maior a área de permeação do módulo. O sinal só apresentava variação positiva em
alguns casos e após um longo tempo. Isto sugere que a membrana de carbono efetua a

104
adsorção destes gases inseridos no permeado durante a etapa de limpeza do sistema.
Como a permeabilidade desses gases é muito baixa, a membrana pode às vezes
apresentar apenas o efeito da adsorção o que é mais expressivo quanto maior a área de
membrana.
A permeabilidade de mistura apresenta valores intermediários, entre a
permeabilidade do gás mais permeável e a permeabilidade do gás menos permeável.
Surge, aqui, a necessário de um método de análise capaz de quantificar o permeado a
fim de avaliar a permeabilidade individual de cada um dos componentes da mistura
levando em conta sua pressão parcial nos cálculos de permeabilidade.
O primeiro módulo apresentou aumento na permeabilidade de todos os gases
puros testados (He, H2, CO2 e N2 todos com permeabilidade igual a 0,45 GPU)
acompanhada de perda total de seletividade após testes de permeação de He a 128 °C
cujo valor de permeação a esta temperatura foi de 0,73 GPU. Isto foi resultado do
aquecimento excessivo da cola polimérica resultando em danos ao módulo. Os outros
módulos também apresentaram o mesmo problema depois de submetidos a vários ciclos
de aquecimento e resfriamento devido à armazenagem destes módulos em estufa
aquecida a 60 °C a qual pode apresentar regiões com temperaturas mais elevadas. Para
recuperação dos módulos foi feito aplicação de uma nova cola, cola Hard® (resina
GM958A e endurecedor GM958B), mais fluida que a Araldite® utilizada na confecção
dos módulos. No entanto a vedação dos módulos resultou em uma elevada perda de
permeabilidade, chegando a não ser possível quantificar a permeabilidade de gases a
não ser do hélio e hidrogênio, cujos diâmetros cinéticos são bastante pequenos. Mais
tarde foi observado que a cola Hard® apresenta migração nas fibras por capilaridade
devido à alta proximidade destas entre si no módulo, resultando em redução na área
efetiva de permeação e conseqüentemente reduzido a permeabilidade do módulo.
Além da grande variação de permeabilidade e seletividade das membranas de
carbono obtidas da pirólise de membranas poliméricas, foi observada visualmente uma
diminuição no diâmetro externo das fibras em suas extremidades, além de rompimentos
freqüentes próximo às extremidades destas fibras principalmente na parte inferior do
reator de pirólise. Uma análise da seção transversal destas regiões de uma mesma fibra
(terceira batelada) através de MEV é apresentada na Figura A.3

105
Figura A.3. Fotomicrografias da seção transversal de membrana de carbono: A) Visão da seção transversal de amostra da extremidade da fibra (à esquerda) e da região intermediária da mesma fibra (à direita) B) Visão ampliada da região intermediária da fibra, C) Visão ampliada da extremidade da fibra.
A parte da fibra, pirolisada na região central do forno, se mostrou bem formada,
isenta de defeitos e de macrovazios enquanto que a parte pirolisada em região próxima
da extremidade apresentou morfologia completamente diferente sendo observadas
deformações e macrovazios. Estas características podem levar à formação de defeitos ou
poros não uniformemente distribuídos, resultando em alteração nas propriedades de
separação das fibras. Este efeito nas extremidades pode ser causado pela irregularidade
nas condições de pirólise ao longo do comprimento do forno.
Foi, então, levantado o perfil de distribuição de temperatura no forno de pirólise
mantendo-se fixo o termopar utilizado para controle de temperatura no forno durante a
pirólise e com um novo termopar foi feita a varredura dentro do reator onde as fibras
são localizadas durante o processo de pirólise. As curvas de temperatura do forno
medidas durante o patamar de temperatura da etapa de estabilização ou pré-tratamento
(400 °C) e durante o patamar da etapa de pirólise (800 °C) são apresentadas na Figura
A.4.
A B C
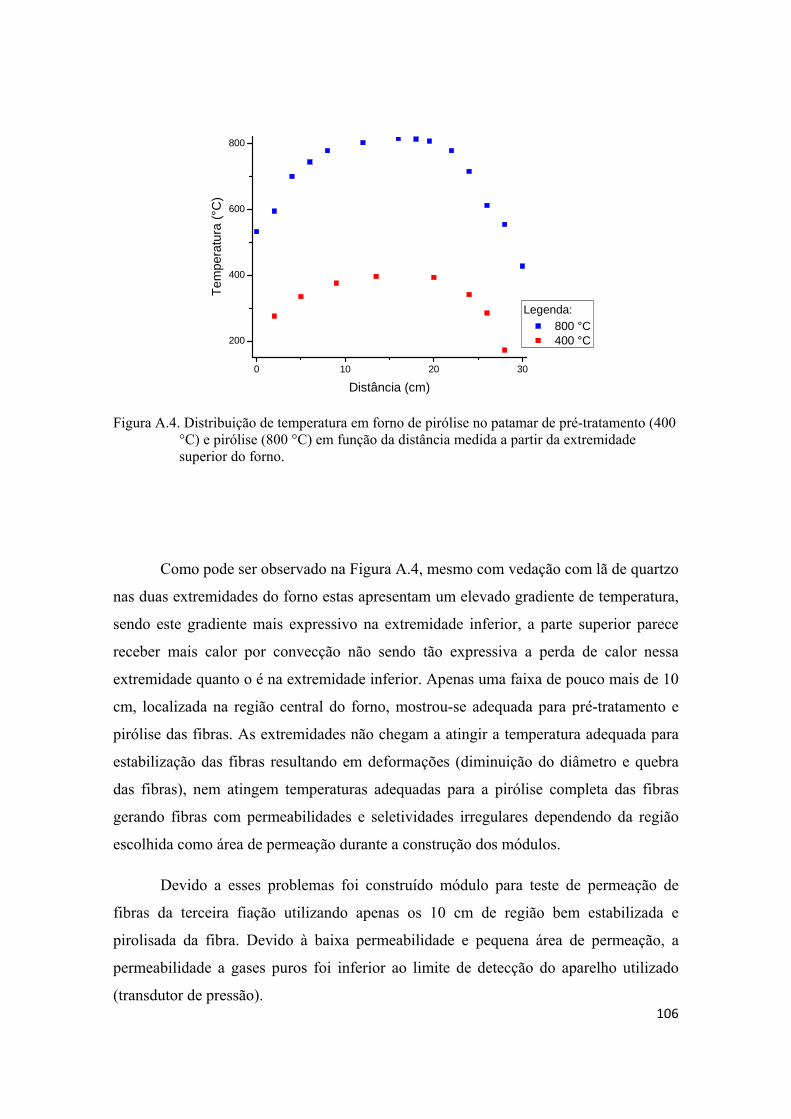
106
0 10 20 30
200
400
600
800
Tem
pera
tura
(°C
)
800 °C 400 °C
Distância (cm)
Legenda:
Figura A.4. Distribuição de temperatura em forno de pirólise no patamar de pré-tratamento (400 °C) e pirólise (800 °C) em função da distância medida a partir da extremidade superior do forno.
Como pode ser observado na Figura A.4, mesmo com vedação com lã de quartzo
nas duas extremidades do forno estas apresentam um elevado gradiente de temperatura,
sendo este gradiente mais expressivo na extremidade inferior, a parte superior parece
receber mais calor por convecção não sendo tão expressiva a perda de calor nessa
extremidade quanto o é na extremidade inferior. Apenas uma faixa de pouco mais de 10
cm, localizada na região central do forno, mostrou-se adequada para pré-tratamento e
pirólise das fibras. As extremidades não chegam a atingir a temperatura adequada para
estabilização das fibras resultando em deformações (diminuição do diâmetro e quebra
das fibras), nem atingem temperaturas adequadas para a pirólise completa das fibras
gerando fibras com permeabilidades e seletividades irregulares dependendo da região
escolhida como área de permeação durante a construção dos módulos.
Devido a esses problemas foi construído módulo para teste de permeação de
fibras da terceira fiação utilizando apenas os 10 cm de região bem estabilizada e
pirolisada da fibra. Devido à baixa permeabilidade e pequena área de permeação, a
permeabilidade a gases puros foi inferior ao limite de detecção do aparelho utilizado
(transdutor de pressão).

107
Em seguida foi montado um módulo com 21 cm de comprimento, sendo 10 cm o
comprimento efetivo, o restante preenchido com cola. Este módulo continha 40 fibras
ocas de carbono, tendo-se o cuidado de manter a área de permeação na região da fibra
adequadamente estabilizada e pirolisada enquanto que as extremidades foram utilizadas
apenas para colagem das fibras no módulo.
Conclui-se que a obtenção de membrana polimérica bem formada e com
morfologia adequada é necessária para obtenção de membranas de carbono com
morfologia e propriedades adequadas para aplicações em separações gasosas a altas
temperaturas e pressões. Além das características da membrana polimérica precursora
as condições de pirólise também exercem grandes efeitos sobre a membrana de carbono
como pode ser observado pela influência nas propriedades da membrana de carbono
exercida pela variação de temperatura a que foi aplicada esta membrana durante o
processo de pirólise.


















