
Dissertação de Mestrado - Sistema de...
129
FACULDADE DE ENGENHARIA QUÍMICA DE LORENA DEPARTAMENTO DE BIOTECNOLOGIA Dissertação de Mestrado CARACTERIZAÇÃO DO BAGAÇO DE CANA PRÉ·TRATADO POR EXPLOSÃO A VAPOR: IDENTIFICAÇÃO DE INIBIDORES POTENCIAIS DE PROCESSOS FERMENTATIVOS E ENZIMÁTICOS Hellen Cristiane Maciel Cunha Lorena - SP - Brasil 1999 Tranferido da Biblioteca do DEBIQ para a Bilblioteca Universitária em Junhoi2004 Proc, n" .202/04 ~---- ..... _ __,
Transcript of Dissertação de Mestrado - Sistema de...

FACULDADE DE ENGENHARIA QUÍMICA DE LORENA DEPARTAMENTO DE BIOTECNOLOGIA
Dissertação de Mestrado
CARACTERIZAÇÃO DO BAGAÇO DE CANA PRÉ·TRATADO POR EXPLOSÃO A VAPOR: IDENTIFICAÇÃO DE INIBIDORES
POTENCIAIS DE PROCESSOS FERMENTATIVOS E ENZIMÁTICOS
Hellen Cristiane Maciel Cunha
Lorena - SP - Brasil 1999 Tranferido da Biblioteca do
DEBIQ para a Bilblioteca Universitária em Junhoi2004
Proc, n" .202/04 ~---- ..... _ __,

FACULDADE DE ENGENHARIA QUÍMICA DE LORENA DEPARTAMENTO DE BIOTECNOLOGIA
PÔS-GRADUAÇÃO EM BIOTECNOLOGIA INDUSTRIAL
CARACTERIZAÇÃO DO BAGAÇO DE CANA PRÉ-TRATADO POR EXPLOSÃO A VAPOR: IDENTIFICAÇÃO DE INIBIDORES POTENCIAIS DE
PROCESSOS FERMENTATIVOS E ENZIMÁTICOS
Dissertação de mestrado apresentada como parte das exigências para a obtenção do título de Mestre em Biotecnologia Industrial
Banca examinadora:
Dr. Flávio Teixeira da Silva (presidente) Dr. José Domingos Fontana Dr. Adilson Roberto Gonçalves
Estudante:
Hellen Cristiane Maciel Cunha
Lorena - SP - Brasil 1999

FACULDADE DE ENGENHARIA QUÍMICA DE LORENA DEPARTAMENTO DE BIOTECNOLOGIA
PÓS-GRADUAÇÃO EM BIOTECNOLOGIA INDUSTRIAL
CARACTERIZAÇÃO DO BAGAÇO DE CANA PRÉ-TRATADO POR EXPLOSÃO A VAPOR: IDENTIFICAÇÃO DE INIBIDORES POTENCIAIS DE
PROCESSOS FERMENTATIVOS E ENZIMÁTICOS
Este exemplar corresponde a versão final da dissertação de mestrado aprovada pela banca examinadora.
Lorena - SP - Brasil 1999

Aos meus pais Paulo e Luiza, que tanto me apóiam
Ao Elias, meu eterno amor

AGRADECIMENTOS
A Deus, que me ilumina e me guia.
Ao Dr. Flávio Teixeira, por sua amizade e por ter me aceito sob sua orientação.
Aos Drs. Adilson Roberto Gonçalves e André Ferraz pelas sugestões e críticas
apresentadas durante a realização deste trabalho.
À Cleyde, que me iniciou na vida científica e ensinou muito do que aprendi no
laboratório.
Aos meus amigos de muitos anos, Andersen, Régis, Márcia, Luane, Luciane,
Sirlene, José Carlos, José Moreira, Jussara, Larissa e Denise.
Aos demais funcionários do DEBIQ e aos outros grupos de pesquisa que me
deram oportunidade de utilizar alguns aparelhos.
Ao CNPq pela bolsa concedida.

LISTA DE ABREVIATURAS:
BSTFA = N, 0-bis(trimetilsilil)fluoroacetamida
CLAE = Cromatografia Líquida de Alta Eficiência
DP = Grau de Polimerização
FA1 = Fase Aquosa 1
F A2 = Fase Aquosa 2
FA3 = Fase Aquosa 3
FIO= Detector de Ionização de Chama
F01 = Fase Orgânica 1
F02 = Fase Orgânica 2
F03 = Fase Orgânica 3
F04 = Fase Orgânica 4
FOS = Fase Orgânica 5
F06 = Fase Orgânica 6
GC/MS = Cromatografia Gasosa acoplada a Espectroscopia de Massas
HPSEC = Cromatografia de Exclusão por Tamanho
TIC = Cromatograma de Íons Reconstituído
TMS = Trimetilsilil

CONTEÚDO
Lista de tabelas i
Lista de figuras iii
Resumo viii
Abstract. ix
1. INTRODUÇÃ0 1
1.1. ASPECTOS GERAIS 1
1.2. CARACTERÍSTICAS ESTRUTURAIS DOS MATERIAIS LIGNOCELULÓSICOS 2
1.2.1. CELULOSE 2
1.2.2. POLIOSES 3
1.2.3. LIGNINA 5
1.2.4. ESTRUTURA DO TECIDO DOS MATERIAIS LIGNOCELULÓSICOS
E A ASSOCIAÇÃO ENTRE CELULOSE, POLIOSES E LIGNINA. 5
1.3. SEPARAÇÃO DOS COMPONENTES DOS MATERIAIS LIGNOCELULÓSICOS 8
1.4. INIBIDORES PRESENTES NO HIDROLISADO HEMICELULÓSIC0 15
2. OBJETIVOS 32
3. MATERIAIS E MÉTODOS 33
3.1. PREPARAÇÃO DO BAGAÇO DE CANA 33
3.2. CARACTERIZAÇÃO QUÍMICA DO BAGAÇO DE CANA. 35
3.2.1. DETERMINAÇÃO DOS EXTRAÍVEIS 35
3.2.2. DETERMINAÇÃO DE LIGNINA KLASON INSOLÚVEL EM MEIO ÁCID0 35
3.2.3. DETERMINAÇÃO DE LIGNINA KLASON SOLÚVEL EM MEIO ÁCID0 36
3.2.4. DETERMINAÇÃO DO TEOR DE CINZAS NO BAGAÇO DE CANA 36
3.2.5. DETERMINAÇÃO DE CARBOIDRATOS E ÁCIDOS ORGÂNICOS POR CLAE 37
3.2.6. DETERMINAÇÃO DE FURFURAL E HIDROXIMETILFURFURAL.. 38
3.3. PRÉ-TRATAMENTO DO BAGAÇO DE CANA POR EXPLOSÃO A VAPOR A
190ºC POR 15 MIN 38
3.4. IDENTIFICAÇÃO DOS OLIGÔMEROS DE GLICOSE E XILOSE POR CLAE 41
3.4.1. HIDRÓLISE DE XILANA E CELULOSE 41
3.5. DISTRIBUIÇÃO DA MASSA MOLAR DOS COMPOSTOS PRESENTES NO
HIDROLISAD0 42
3.5.1. CURVAS DE CALIBRAÇÃO PARA A DISTRIBUIÇÃO DA MASSA
MOLAR DOS COMPOSTOS PRESENTES NO HIDROLISAD0 43
3.6. SEPARAÇÃO DE CARBOIDRATOS E COMPOSTOS AROMÁTICOS
PRESENTES NO HIDROLISAD0 44
3.6.1. SEPARAÇÃO EM CARTUCHOS DE EXTRAÇÃO SÓLIDA (SEP-PAK C18) .44
3.6.2. SEPARAÇÃO POR EXTRAÇÃO LÍQUIDO-LÍQUID0 45

3.7. ANÁLISE POR ESPECTROSCOPIA NO INFRA VERMELH0 46
3.8. IDENTIFICAÇÃO E QUANTIFICAÇÃO DE COMPOSTOS AROMÁTICOS DE
BAIXA MASSA MOLAR PRESENTES NO HIDROLISAD0 48
3.8.1. DERIVATIZAÇÃO DAS AMOSTRAS PARA ANÁLISES DE GC/MS E GC/FID .48
3.8.2. CROMATOGRAFIA GASOSA/ESPECTROMETRIA DE MASSAS .48
3.8.3. CROMATOGRAFIA GASOSA/DETECTOR DE IONIZAÇÃO DE CHAMA 49
4. RESULTADOS E DISCUSSÃ0 51
4.1. PRÉ-TRATAMENTO DO BAGAÇO DE CANA POR EXPLOSÃO A VAPOR. 51
4.2. DETERMINAÇÃO DE OLIGÔMEROS DE XILOSE E GLICOSE POR CLAE 55
4.3. IDENTIFICAÇÃO DE COMPLEXOS LIGNINA-CARBOIDRATO PRESENTES NO
HIDROLISADO DO PRÉ-TRATAMENTO DO BAGAÇO DE CANA POR EXPLOSÃO
AVAPOR 60 4.4. FRACIONAMENTO DOS COMPOSTOS PRESENTES NO HIDROLISADO POR
EXTRAÇÃO LÍQUIDO-LÍQUID0 68
4.4.1. CROMATOGRAFIA GASOSA ACOPLADA A ESPECTROMETRIA
DE MASSAS 69
4.4.2. ESPECTROSCOPIA NO INFRA VERMELH0 85
4.4.3. CONSIDERAÇÕES FINAIS SOBRE OS COMPOSTOS IDENTIFICADOS
POR GC/MS 88
5. CONCLUSÕES 91
6. REFERÊNCIAS BIBLIOGRÁFICAS 92
APÊNDICES

LISTA DE TABELAS
Tabela 1. Concentração de alguns compostos identificados no hidrolisado de
carvalho vermelho e seus efeitos inibitórios na subseqüente fermentação de
xilose a etanol (TRAN e CHAMBERS, 1986) 22
Tabela 2. Compostos derivados de lignina identificados em hidrolisados de
álamo, switchgrass e com stover (FENSKE et ai., 1998) 28
Tabela 3. Proteínas utilizadas na calibração da coluna cromatográfica e suas
respectivas massas molares .43
Tabela 4. Programas de temperatura da coluna para análises das frações
orgânicas nas análises de GC/MS e GC/FID 49
Tabela 5. Balanço de massa do pré-tratamento do bagaço de cana a 190ºC, 15
min. Quantidades expressas em relação a 100 g de bagaço seco 51
Tabela 6: Balanço de massa do pré-tratamento do bagaço de cana a 190ºC,
15 min. Quantidades expressas em relação a 100 g de bagaço seco (SILVA,
1995) 52
Tabela 7. Composição do bagaço de cana pré-tratado a 190ºC por 15 min
comparada aos dados obtidos por SILVA (1995) 53
Tabela 8. Distribuição dos compostos identificados no hidrolisado do pré-
tratamento por explosão a vapor a 190ºC por 15 min. Quantidades expressas
em relação a 100 g de bagaço 54
Tabela 9. Tempos de retenção dos açúcares identificados no hidrolisado
hemicelulósico obtido por explosão a vapor (190ºC/15 min) analisado na coluna
Aminex HPX 42A d t · - d 1· A 56 , para a e ermmaçao e o 1gomeros ..

ii
Tabela 10. Cromatograma de uma amostra padrão contendo xilose, xilobiose e
xilotriose 57
Tabela 11. Tempos de retenção dos xilo-oligômeros obtidos por hidrólise de
xilana (25ºC/7min) e analisados na coluna Aminex HPX 42A 58
Tabela 12. Tempos de retenção dos gluco-oligômeros obtidos por hidrólise de
celulose (45ºC/7min) e analisados na coluna HPX 42A 59
Tabela 13. Rendimento das frações obtidas no procedimento de extração
líquido-líquido 68
Tabela 14. Atribuição dos picos e quantificação dos compostos identificados na
fração F O 1 71
Tabela 15. Atribuição dos picos e quantificação dos compostos identificados na
fração F02 7 4
Tabela 16. Atribuição dos picos e quantificação dos compostos identificados na
fração F03 77
Tabela 17. Atribuição dos picos e quantificação dos compostos identificados na
fração F 04 80
Tabela 18. Atribuição dos picos e quantificação dos compostos identificados na
fração FOS 83
Tabela 19. Quantidade total de cada composto identificado por GC/MS no
hidrolisado obtido do pré-tratamento do bagaço de cana por explosão a
vapor. 88

ili LISTA DE FIGURAS
Figura 1. Estrutura da celulose. Parte central da cadeia molecular (FENGEL e
WEGENER, 1989) 3
Figura 2. Fórmula dos açúcares presentes nas polioses (FENGEL e
WEGENER, 1989) 4
Figura 3. Representação esquemática de uma xilana de gramínea mostrando
alguns grupos substituintes. Xyl = 1,4-0-xilopiranose; Ara = L-arabinofuranose;
(4-Me)-GlcA = ácido (4-0-metil)-D-glucopiranurônico; Ac = acetil; FA = ácido
ferúlico; DDFA = ácido desidroferúlico (McDOUGAL, 1993) .4
Figura 4. Estrutura dos precursores da biossíntese da lignina. (1) álcool p-
cumárico; (li) álcool coniferílico; (Ili) álcool sinápico (FENGEL e WEGENER,
1989) 6
Figura 5. Estrutura da lignina de abeto (Picea abies) proposta por Adler
(FENGEL e WEGENER, 1989) 6
Figura 6: Microscopia eletrônica de transmissão das células de madeira
mostrando as camadas da parede celular: ML= lamela média, P= parede
primária, S1= parede secundária, S2= parede secundária, T= parede terciária e
W= camada de verrugas. (a) (Picea abies) (b) (Fagus sy/vatica) (FENGEL e
WEGENER 1989) 7
Figura 7: Corte ilustrativo do sistema de camadas na parede das células da
madeira (FENGEL e WEGENER, 1989) 7
Figura 8. Representação esquemática do processo de separação dos
componentes do bagaço de cana por explosão a vapor 34

iv
Figura 9. Representação esquemática do sistema de pré-tratamento de
materiais lignocelulósicos por explosão a vapor, em escala de bancada .40
Figura 10. Curva de calibração da coluna cromatográfica Asahipak-320H
eluída com água bidestilada a 1,0 mUmin 44
Figura 11. Representação esquemática do processo de extração do
hidrolisado hemicelulósico do bagaço de cana com diferentes solventes .47
Figura 12. Hidrolisado hemicelulósico obtido por explosão a vapor (190ºC/
15 min) analisado na coluna Aminex HPX 42A, para a determinação de
oligômeros 55
Figura 13. Cromatograma de uma amostra padrão contendo xilose, xilobiose e
xilotriose 56
Figura 14. Xilana hidrolisada com H2SQ4 72% a 25º C e analisada na coluna
Aminex HPX-42A, em um detector de Índice de Refração '. 57
Figura 15. Celulose hidrolisada com H2S04 72% a 45º C e analisada na coluna
Aminex HPX-42A, em um detector de indice de Refração 59
Figura 16. SEC da fração do hidrolisado filtrada somente em membrana, para
retirada de sólidos ;.61
Figura 17. SEC da fração do hidrolisado hemicelulósico filtrada em cartucho de
extração sólida Sep-Pak C18 61
Figura 18. SEC da fração do hidrolisado hemicelulósico retida em cartuchos de
extração sólida Sep-Pak C18 (Waters) e dessorvida com etanol 62

V
Figura 19. Espectro de infravermelho dos compostos presentes no hidrolisado
eluído em uma série de cartuchos de extração sólida Sep-Pak C18. Espectro
obtido a partir de pastilhas de KBr com 0,5% da matéria orgânica presente no
hidrolisado liofilizado 63
Figura 20. Espectros FTIR de transmissão de Euca/yptus regnans e seus
componentes (MICHEL, 1988) 65
Figura 21. Espectro de infravermelho dos compostos presentes no hidrolisado
adsorvido em uma série de cartuchos de extração sólida Sep-Pak C18,
dessorvido com etanol e concentrado a pressão reduzida. Espectro obtido a
partir de pastilhas de KBr com 0,5% da matéria orgânica presente no
hidrolisado adsorvido e seco 66
Figura 22. Espectro FTIR da lignina de bagaço de cana pré-tratado por
explosão a vapor e extraída com NaOH 1 % a 1 OOºC. Espectros tirados de
pastilhas de KBr com 0,5% de lignina (SILVA, 1995) 67
Figura 23. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica F01 após derivatização com BSTFA e detectados por
espectrometria de massas 70
Figura 24. Cromatograma dos compostos presentes na fração orgânica F01
após derivatização com BSTFA e detectados por ionização de chama
(FID) 70
Figura 25. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica F02 após derivatização com BSTFA e detectados por
espectrometria de massas 73

vi
Figura 26. Cromatograma dos compostos presentes na fração orgânica F02
após derivatização com BSTFA e detectados por ionização de chama
(FID) 73
Figura 27. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica F03 após derivatização com BSTFA e detectados por
espectrometria de massas 76
Figura 28. Cromatograma dos compostos presentes na fração orgânica F03
após derivatização com BSTFA e detectados por ionização de chama
(FID) 76
Figura 29. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica F04 após derivatização com BSTFA e detectados por
espectrometria de massas 79
Figura 30. Cromatograma dos compostos presentes na fração orgânica F04
após derivatização com BSTFA e detectados por ionização de chama
(FID) 79
Figura 31. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica FOS após derivatização com BSTFA e detectados por
espectrometria de massas 82
Figura 32. Cromatograma dos compostos presentes na fração orgânica FOS
após derivatização com BSTFA e detectados por ionização de chama
(FID) 82
Figura 33. Cromatograma reconstituído (TIC) da fração orgânica 6 evaporada
a pressão reduzida e derivatizada com BSTFA. 85

vii
Figura 34. Espectro de infravermelho da fração fenólica F01 obtida por
extração com solventes do hidrolisado hemicelulósico de bagaço de cana.
Espectro obtido a partir de pastilhas de KBr com 0,5% da F01 86
Figura 35. Espectro de infravermelho da fração ácida F02 obtida por extração
com solventes do hidrolisado hemicelulósico de bagaço de cana. Espectro
obtido a partir de pastilhas de KBr com 0,5% da F02 86
Figura 36. Espectro de infravermelho da fração total F04 obtida por extração
com solventes do hidrolisado hemicelulósico de bagaço de cana. Espectro
obtido a partir de pastilhas de KBr com 0,5% da F04 87
Figura 37. Espectro de infravermelho da fração neutra F06 obtida por extração
com solventes do hidrolisado hemicelulósico de bagaço de cana. Espectro
obtido a partir de pastilhas de KBr com 0,5% da F06 87

viii Caracterização de bagaço de cana pré-tratado por explosão a vapor: identificação de inibidores potenciais de processos fermentativos e enzimáticos. Hellen Cristiane Maciel Cunha. Dissertação de mestrado. Programa de Pós-graduação em Biotecnologia Industrial, Departamento de Biotecnologia, Faculdade de Engenharia Química de Lorena. Orientador: Flávio Teixeira da Silva (CP 116, 12600000, Lorena, SP, Brasil). Banca examinadora: Dr. José Domingos Fontana e Dr. Adilson Roberto Gonçalves. Dezembro de 1999.
Resumo O pré-tratamento de bagaço de cana por explosão a vapor produz hidrolisados contendo polioses solúveis em H20, além de compostos inibidores de processos fermentativos e enzimáticos, tais como ácido acético e produtos de degradação de açúcares e lignina. Neste trabalho, o bagaço de cana foi pré- tratado a 190ºC por 15 min. O pré-tratamento solubilizou 34% do bagaço de cana. As frações obtidas (bagaço pré-tratado e hidrolisado hemicelulósico) foram caracterizadas quimicamente. Açúcares livres e seus oligômeros foram determinados por Cromatografia Líquida de Alta Eficiência (CLAE), em uma coluna Aminex HPX-87H e HPX-42A, respectivamente. Lignina e cinzas foram determinadas por gravimetria. A análise qualitativa do hidrolisado mostrou a presença de oligômeros de xilose com grau de polimerização variando de 2 a 6, com predominância de xilobiose e xilotriose. Os resultados de HPSEC mostraram que a massa molar média dos carboidratos presentes no hidrolisado foi menor que a dos compostos aromáticos. Tal resultado foi confirmado por CLAE e infravermelho, mostrando que na fração adsorvida em C18 houve a formação de complexo lignina-carboidrato. O uso de C18 foi efetivo para separar os compostos de baixa massa molar derivados da lignina e complexo lignina-carboidrato, mas ineficaz para identificação dos compostos de baixa massa molar por CG/MS. As frações orgânicas obtidas da extração líquido-líquido dos compostos presentes no hidrolisado foram analisadas por CG/MS e quantificadas por CG/FID. Nas frações ácidas (F02 e F03) foram identificados os ácidos trans-cumárico (4,01 % e 2,56%) e ferúlico (0,04% e 0,32%) como principais compostos, além dos ácidos p-hidroxibenzóico (0,05%), vanílico (0,01%), siríngico (0,39%) e cis-cumárico (0,51%), vanilina (0,40%) e siringaldeído (0,36%). Os produtos contidos nas frações fenólicas (F04 e F05} foram similares aos encontrados na fração ácida, sendo o ácido cumárico (0,05% e 2,99%) o principal produto, além dos ácidos ferúlico (O, 14% e 0,45%), p-hidroxibenzóico (0,06%), vanílico (0,67%), cis-cumárico (0,64%), p-hidroxibenzaldeído (1,5%) e siringaldeído (0,51 %). Na fração neutra (F06) nenhum composto foi identificado.

iv Characterization of sugarcane bagasse pretreated by steam explosion: identification of potencial inhibitors for enzimatic and fermentative process. Hellen Cristiane Maciel Cunha. Dissertação de mestrado. Programa de Pós-graduação em Biotecnologia Industrial, Departamento de Biotecnologia, Faculdade de Engenharia Química de Lorena. Orientador: Flávio Teixeira da Silva (CP 116, 12600000, Lorena, SP, Brasil). Banca examinadora: Dr. José Domingos Fontana e Dr. Adilson Roberto Gonçalves. Dezembro de 1999.
Abstract Sugarcane bagasse pretreatment by steam explosion produces hydrolysates containing polyoses soluble in H20, as well as compounds that are inhibitors of the fermentative and enzymatic processes, such as acetic acid and products of sugar and lignin degradation. ln this study, sugarcane bagasse was pretreated at 190ºC for 15 min. The pretreatment solubilized 28% of the sugarcane bagasse. The fractions obtained (pretreated bagasse and hemicellulosic hydrolysate) were chemically characterized. Free sugars and their oligomers were determined by HPLC in two Aminex columns: HPX-87H and HPX-42A, respectively. lignin and ashes were determined by gravimetry. The qualitativa analyses of the hydrolysate showed the presence of xylose oligomers with polimerization degrees ranging from 2 to 6, xylobiose and xylotriose being predominant. HPSEC results showed that the average molar mass of the carbohydrates present in the hydrolysate was smaller than that of the aromatic compounds. This was confirmed by HPLC and infrared, showing that in the fraction in C 18 these was the formation of lignin-carbohydrate complex. The use of C 18 was effective for separating the low molecular weight compounds derived from lignin and lignin-carbohydrate complex, but ineffective for identifying low molecular weight compounds by GC/MS. ln the acid fractions (F02 and F03), the coumaric acid (4.01% and 2.56%) and ferulic acid (0.04% and 0.32%) were identified as the main compounds, besides of them p- hydroxybenzoic acid (0.05%), vanilic acid (0.01%), siring acid (0.39%), eis- coumaric acid (0.51 %), vanilin (0.4%) and syringealdehyde (0.36%). The products contained in the phenolic fractions were similar to those found in the acid fraction, the coumaric acid (0.05% and 2.99%) being the main product, besides of them ferulic acid (0.14% and 0.45%), p-hydroxybenzoic acid (0.06%), vanilic acid (0.67%), cis-coumaric acid (0.64%), p- hydroxybenzaldehyde (1.5%) and syringealdehyde (0.51 %). ln the neutral fraction no compound was identified.

1. INTRODUÇÃO
1.1. ASPECTOS GERAIS
O bagaço de cana é freqüentemente citado na literatura como um
material lignocelulósico promissor para a obtenção de açúcares, que podem
ser transformados em etanol ou outros compostos químicos (SASKA E OZER, 1995).
A cana-de-açúcar, uma gramínea pertencente ao gênero Saccharum, é
originária da Índia. Com o decorrer do tempo, sua cultura expandiu-se por
várias regiões do mundo e foi introduzida no Brasil logo após seu
descobrimento (PAIVA, 1980).
A cana-de-açúcar (Saccharum officinarum) cresce na maioria dos países
tropicais e subtropicais e é usada principalmente para a obtenção de açúcar e
álcool. Após sua moagem, o principal subproduto é o bagaço, que representa
de 12% a 14% da massa seca da cana (ICIDCA, 1987).
Em 1998 foram produzidas no Brasil 287 milhões de toneladas de cana
de açúcar, que foram convertidos em açúcar e álcool, gerando quantidades
superiores a 40 milhões de toneladas de bagaço (MA TIOLI et ai., 1998). Esse
bagaço tem sido utilizado principalmente para geração de vapor e energia, que
são consumidos nas próprias usinas (ARMAS e BIANCHI, 1990). Mesmo
assim, é produzido um grande excedente. Estima-se que é gerado um excesso
de cerca de 1 milhão tonlano, que causa sérios problemas de estocagem e
impacto ao meio ambiente.
Entretanto, o preço do bagaço de cana é baixo e pode ser aproveit_ado
para fins mais nobres, pois é uma fonte abundante de carboidratos com um alto
potencial para ser hidrolisado e fermentado a etanol (MARTIN et ai., 1999). Em
muitos países do mundo, inclusive no Brasil, o bagaço já está sendo usado na
produção de polpa celulósica, papel e papelão (ATCHISON,1993).
MARTIN et ai. (1999) estimaram que as usinas de açúcar e álcool podem
liberar de 30% a 50% do bagaço produzido para usos alternativos, com a
produção de compostos de maior valor econômico, empregando-se a

2
tecnologia já existente (CASTRO et ai., 1995; DESCHAMPS et ai., 1996;
LACORTE et ai .. 1986).
1.2. CARACTERÍSTICAS ESTRUTURAIS DOS MATERIAIS
LIGNOCELULÔSICOS
A dificuldade de se converter o bagaço de cana e outros materiais
lignocelulósicos em insumos químicos é atribuída às suas características
químicas e morfológicas. Esses materiais são um compósito de microfibrilas de
celulose envolvidas em uma matriz amorfa de polioses e lignina. Esta matriz
amorfa age como uma barreira natural ao ataque de microrganismos e/ou
enzimas e torna esses materiais estruturalmente rígidos e poucos reativos
(FENGEL e WEGENER, 1989).
A compreensão da complexidade estrutural e da reatividade dos
materiais lignocelulósicos requer o conhecimento das características e das
propriedades de cada um de seus componentes.
1.2.1. CELULOSE
A celulose é um polímero linear de subunidades de glicose associadas
por ligações J3-(1-A), sendo a celobiose a unidade repetitiva do polímero
(figura 1 ). As cadeias de celulose se encontram agregadas paralelamente para
formar as fibrilas elementares, que são insolúveis em água e apresentam
regiões cristalinas e amorfas (FENGEL e WEGENER, 1989; PARHAM, 1993).
As pontes de hidrogênio inter- e intramolecular são responsáveis pela
manutenção das regiões cristalinas, e tornam a celulose altamente resistente à
hidrólise ácida, alcalina ou enzimática (WOOD e SADDLER, 1988; CONVERSE
e WARE, 1994).

3
H 00 ?°'2~ H 00
'0J,;-~,t-~:'-(º~c1 Hl~o./"''~ H~O.,/\
HOH ~~ Hai ~~
lndade de Celobiose ---
Figura 1. Estrutura da celulose. Parte central da cadeia molecular (FENGEL e
WEGENER, 1989).
1.2.2. POLIOSES
As polioses diferem substancialmente da celulose por serem amorfas,
com estrutura ramificada e compostas pela combinação de vários açúcares
(pentases, hexases, ácidos hexurônicos e deoxiexoses) (figura 2) (PARHAM,
1993; FENGEL e WEGENER, 1989). As polioses são classificadas
basicamente de acordo com os açúcares presentes na cadeia principal do
polímero: xilanas, glucomananas e galactanas.
A cadeia principal pode ser um homopolímero, como no caso das
xilanas, ou um heteropolímero, como no caso das glucomananas e podem
apresentar arabinose, galactose, ácido 4-0-metilglucurônico e grupos acetil
ligados à cadeia principal. As madeiras moles apresentam maior proporção de
galactogluco-mananas do que de xilanas, enquanto as madeiras duras são
mais ricas em xilanas (PARHAM, 1993). A composição de xilanas de
gramíneas foi estudada por vários autores, entre eles por McDOUGALL et ai.
(1993). A representação esquemática de uma xilana típica de gramíneas está representada na figura 3.

4
Pentases Hexases Acides Hexurõnicos
Deoxi-hexoses
~OH
p- D - Xilose
~VOH
Ho"~ HO~OH CH3
H H
p-D -Glicose
HOQOH OH
H
a-L -Arabinopiranose p-D-Manose ácido a-D-4-0-Metilglucurônico a-L-Fucose
"º",º~~ ~q. ~~ a-L -Arabinofuranose a-D -Galactose ácido a-D -Oalacturônico
Figura 2. Fórmula dos açúcares presentes nas polioses (FENGEL e
WEGENER, 1989).
(4-M e )-G lcA (4-M e )-G lcA Ac 1 Ac Ac 1 Ac
I ai 1 1 ai I 2 2 3 3 2 2
-Xyl -p -Xyl- p -Xyl-p -Xyl-p -Xyl-p -Xyl- p -Xyl-p -Xyl-p -Xyl-p -Xyl-p -Xyl- p -Xyl- 3 3 3 3 3
ai Llgnina1 af ai ai ai 1 1 1 1 1
Ara D-Ara Ara D-Ara Llgnlna -FA-Ara
D I D F Ac F
FA -Ara A-Ara A-Ara Ara 1 1 1 1
ai ai ai ai 3 3 3 3
-Xyl-p-Xyl-p -Xyl-p -Xyl-p-Xyl-p-Xyl-p-Xyl- p-Xyl-p-Xyl-p-Xyl-p-Xyl-p -Xyl- 2 3 3 2 2 1 1 1 ai ai
Ac Ac Ac 1 Ac (4-Me)-GlcA
Figura 3. Representação esquemática de uma xilana de gramínea mostrando
alguns grupos substituintes. Xyl = 1,4-D-xilopiranose; Ara = L-arabinofuranose;
(4-Me)-GlcA = ácido (4-0-metil)-D-glucopiranurônico; Ac = acetil; FA = ácido
ferúlico; DDFA = ácido desidroferúlico (McDOUGALL, 1993).

5
1.2.3. LIGNINA
Depois da celulose, a lignina é a macromolécula orgânica mais
importante e abundante do reino vegetal. A lignina aumenta a resistência
mecânica das plantas, de tal forma que árvores de mais de cem metros podem
se manter em pé (FENGEL e WEGENER, 1989).
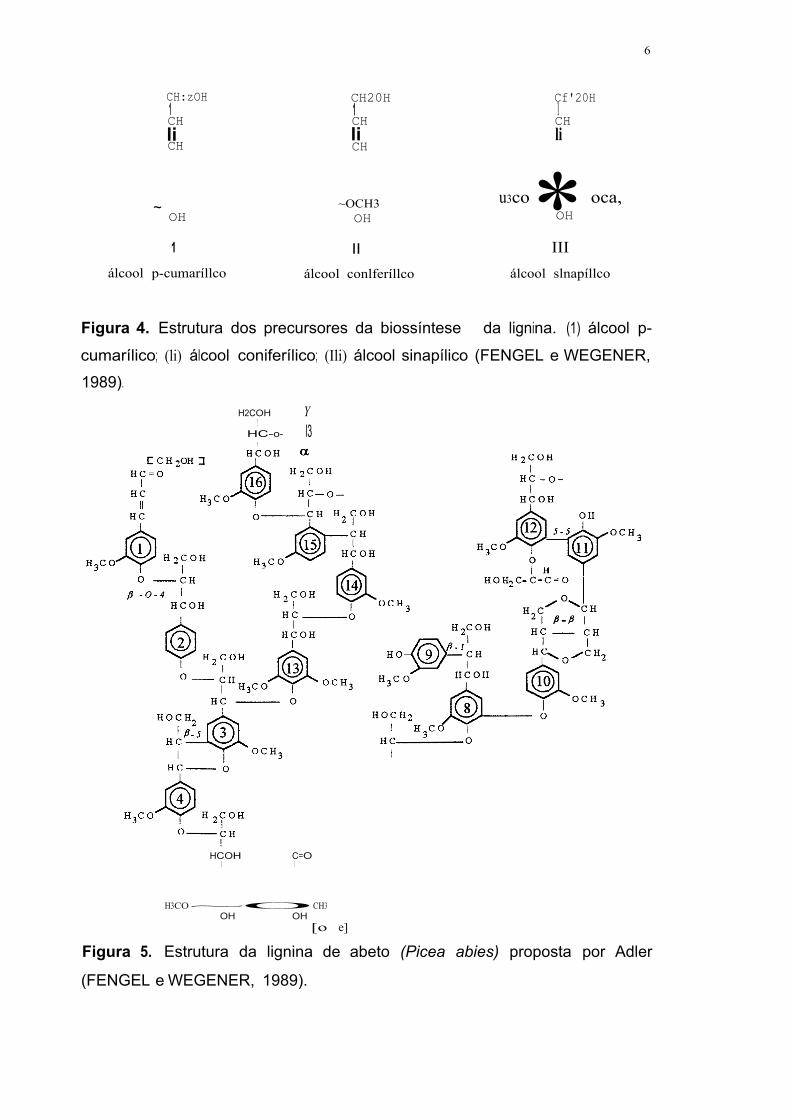
A lignina é uma macromolécula altamente complexa e ramificada,
gerada pela polimerização desidrogenativa dos álcoois hidroxicinamílicos: p-
cumarílico (1), coniferílico (li) e sinapílico (Ili) (figura 4). A lignina é
principalmente constituída de unidades fenilpropano associadas por ligações
estáveis do tipo C-C, aril-éter e aril-aril, sendo as mais abundantes ~-(O-A) e
a-(0--A) (40-60%), ~-5 (10%), ~-1 (5-10%), 5-5 (10%), ~-~ (5%) e 4-0~5
(5%) (HIGUCHI, 1985). Um modelo proposto para a estrutura da lignina de
abeto é mostrado na figura 5.
1.2.4. ESTRUTURA DO TECIDO DOS MATERIAIS LIGNOCELULÓSICOS E
A ASSOCIAÇÃO ENTRE CELULOSE, POLIOSES E LIGNINA
Na parede celular vegetal, celulose, polioses e lignina, organizam-se
formando diferentes camadas (figura 6): parede primária (P), secundária (5) e
terciária (T). As diferentes células encontram-se separadas pela lamela média
(ML), que é uma camada fina que mantém as células coesas e é responsável
pela integridade estrutural do tecido das plantas. A parede primária (P) é a
camada mais fina da parede celular e a primeira a ser depositada nas células.
Do lado de dentro da parede primária é formada a parede secundária, em uma
seqüência de três lamelas, 51, 52 e 53(ou T). Nestas regiões as microfibrilas de
celulose possuem distintas orientações com respeito ao eixo longitudinal da
célula. A parede mais espessa é a 52, na qual as microfibrilas de celulose se
encontram orientadas de forma quase paralela ao eixo axial da célula. As
fibrilas de celulose próximas ao lúmen da célula compreendem a camada
terciária e estão orientadas quase perpendicularmente ao eixo da célula.
6
CH:zOH CH20H Cf'20H 1 1 1 CH CH CH li li li CH CH
* ~ ~OCH3 u3co oca, OH OH OH
1 II III álcool p-cumaríllco álcool conlferíllco álcool slnapíllco
Figura 4. Estrutura dos precursores da biossíntese da lignina. (1) álcool p-
cumarílico; (li) álcool coniferílico; (Ili) álcool sinapílico (FENGEL e WEGENER,
1989).
H2COH Y !
HC-o- l3 1
HCOH
HCOJ@ 3 1
HOCH~ 1 4
HC-----0 1
HCOH C=O 1 1
H3CO~OCH3 OH OH
[o e]
Figura 5. Estrutura da lignina de abeto (Picea abies) proposta por Adler
(FENGEL e WEGENER, 1989).

7
Na parede celular, as fibrilas elementares estão separadas umas das
outras por uma camada de polioses, formando as microfibrilas, que são
envolvidas em uma matriz de lignina, formando a parede celular (FENGEL e
WEGENER, 1989).
A maior quantidade de lignina é encontrada na camada 82, porém se
apresenta em concentração mais elevada na lamela média. A figura 7
apresenta um modelo da construção da parede celular dos materiais
lignocelulósicos, ilustrando a descrição acima.
Figura 6: Microscopia eletrônica de transmissão das células de madeira
mostrando as camadas da parede celular: ML= lamela média, P= parede
primária, 81= parede secundária 1, 82= parede secundária 2, T= parede
terciária e W= camada de verrugas. (a) (Picea abies) (b) (Fagus sy/vatica)
(FENGEL e WEGENER 1989).
Figura 7: Corte ilustrativo do sistema de camadas na parede das células da madeira (FENGEL e WEGENER, 1989).

8
1.3. SEPARAÇÃO DOS COMPONENTES DOS MATERIAIS LIGNOCELULÓSICOS
Para se obter uma conversão simples e efetiva dos três pnnctpars
componentes dos materiais lignocelulósicos (celulose, polioses e lignina) em
produtos de maior econômico, é necessário a separação seletiva dos mesmos.
Há, portanto, a necessidade de se romper o complexo lignina-celulose-poliose
e remover cada fração por técnicas de pré-tratamento e deslignificação (SILVA,
1995).
Várias técnicas de pré-tratamento baseadas na combinação de
processos mecânicos, físicos, químicos e biológicos estão associadas aos
processos de separação e aproveitamento desses componentes (FERRAZ et ai., 1996; McMILLAN, 1994a). Nestas, inclui-se o emprego de álcalí, hidrólise
ácida, explosão a vapor e uso de fluídos supercríticos, todas usadas ou
propostas com o objetivo de produzir combustíveis renováveis e insumos
químicos a partir de biomassa (McMILLAN, 1994a).
A tendência atual está baseada no desenvolvimento e no estudo de
processos para o .aproveitamento integral dos materiais lignocelulósicos
(SILVA, 1995). A produção, prtncipalmente de etanol, através da fermentação
de açúcares obtidos a partir desses materiais, tem sido sistematicamente
estudada (CLARK e MACKIE, 1984; DELGENES, et ai., 1996; HAHN-
HÃGERDAL et ai., 1991; HAHN-HÃGERDAL, 1996; MARTÍN et ai., 1995;
McMILLAN, 1994a;b; PALMQVIST, et ai., 1996a;b; 1997; PFEIFER et ai., 1984;
PRIOR et ai., 1989; RIVARD et ai., 1996; WILSON et ai., 1989). Uma das
vantagens do uso etanol como combustível, por exemplo, é que o dióxido de
carbono produzido não representa um acréscimo de C02 na atmosfera, como
no caso da combustão dos compostos fósseis.
A utilização dos materiais lignocelulósicos requer que o processo de pré-
tratamento proporcione um fracionamento completo ou parcial da biomassa,
como por exemplo, nos processos usados nas indústrias de papel e celulose
(HAHN-HÃGERDAL et ai., 1991 ). O pré-tratamento deve ser eficiente do ponto
de vista energético e químico, para que o processo seja economicamente
vantajoso e deve promover ou proporcionar a conversão efetiva dos
carboidratos a açúcares fermentáveis, para se obter o produto final com alto

9
rendimento. Logo, a degradação ou a perda de carboidratos devem ser
evitadas, bem como a formação de compostos inibidores do metabolismo
celular ou da ação de enzimas usadas nos processos de conversão da
biomassa (McMILLAN, 1994a).
O processo de explosão a vapor tem sido considerado um processo
viável no pré-tratamento de biomassa para a produção econômica de insumos
químicos, combustível, alimentos e polímeros. A explosão a vapor permite a
recuperação de grande parte dos componentes dos materiais lignocelulósicos,
minimizando a sua degradação (AVELLAR e GLASSER, 1998). Além disso, é
uma técnica que tem sido proposta como uma possível alternativa para o
tratamento termo-mecânico dos materiais lignocelulósicos (FOCHER et ai.,
1998).
No processo de explosão a vapor, os materiais lignocelulósicos sofrem
inicialmente reações de hidrólise (EXCOFFIER et ai., 1991; LORA e WAYMAN,
1978). As ligações poliose-lignina são clivadas, solubilizando as polioses e
parte da lignina (AVELLAR e GLASSER, 1998). A ruptura das ligações ~-
(1-+4) entre as unidades de xilose da cadeia principal do polímero com adição
de uma molécula de água (FENGEL E WEGENER, 1989), produzem xilose
livre (equação 1) e seus oligômeros (equação 2) (SADDLER et ai., 1993).
A arabinose é formada pela hidrólise das ligações a-(1~3) (equação 3),
formada entre uma unidade de arabinose e a cadeia principal da xilana (figura
3).
H20,H+
e/ou CALOR ... ~
H
OH OH
+
equação 1

10
CO OH
~
"·: 0~H -<E(º H -Ac
--- o ~ H
equação 2
OH HO",C... r-lH
'Q~A o OH
+ C':OOH
OH OH
OH OH
equação 3
Essas reações de hidrólise são catalisadas pela presença do ácido
acético liberado das reações de hidrólise dos grupos acetil (BOUCHARD et ai.,
1990 e 1991; FOCHER et ai., 1998; SADDLER et ai., 1983; MARTÍN et ai.,
1995), que encontram-se ligados aos átomos de carbono C2 e C3 das unidades
de xilose da cadeia principal da xilana (equação 4) (figura 3).
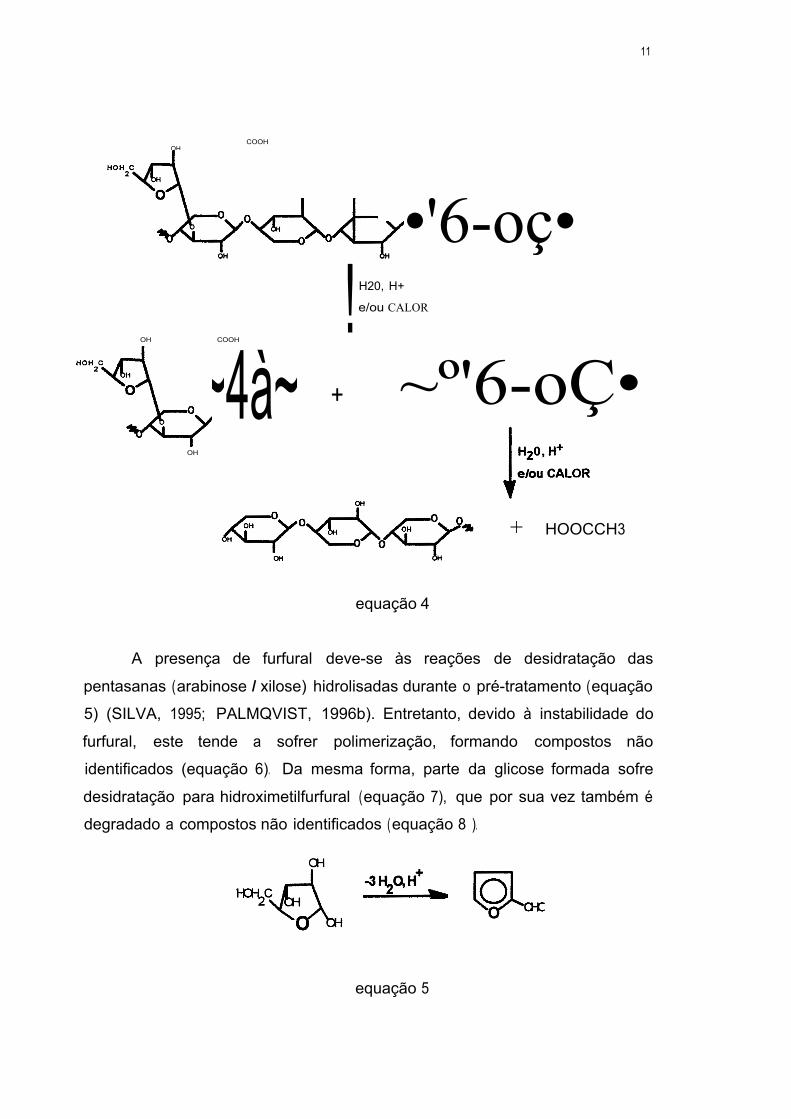
11
OH COOH
",~ OOCCH3
~º •'6-oç• ! H20, H+
e/ou CALOR
+ ~º'6-oÇ• OH COOH
_"·-~4à~ OH
+ HOOCCH3
equação 4
A presença de furfural deve-se às reações de desidratação das
pentasanas ( arabinose I xilose) hidrolisadas durante o pré-tratamento ( equação
5) (SILVA, 1995; PALMQVIST, 1996b). Entretanto, devido à instabilidade do
furfural, este tende a sofrer polimerização, formando compostos não
identificados (equação 6). Da mesma forma, parte da glicose formada sofre
desidratação para hidroximetilfurfural ( equação 7), que por sua vez também é
degradado a compostos não identificados ( equação 8 ).
equação 5
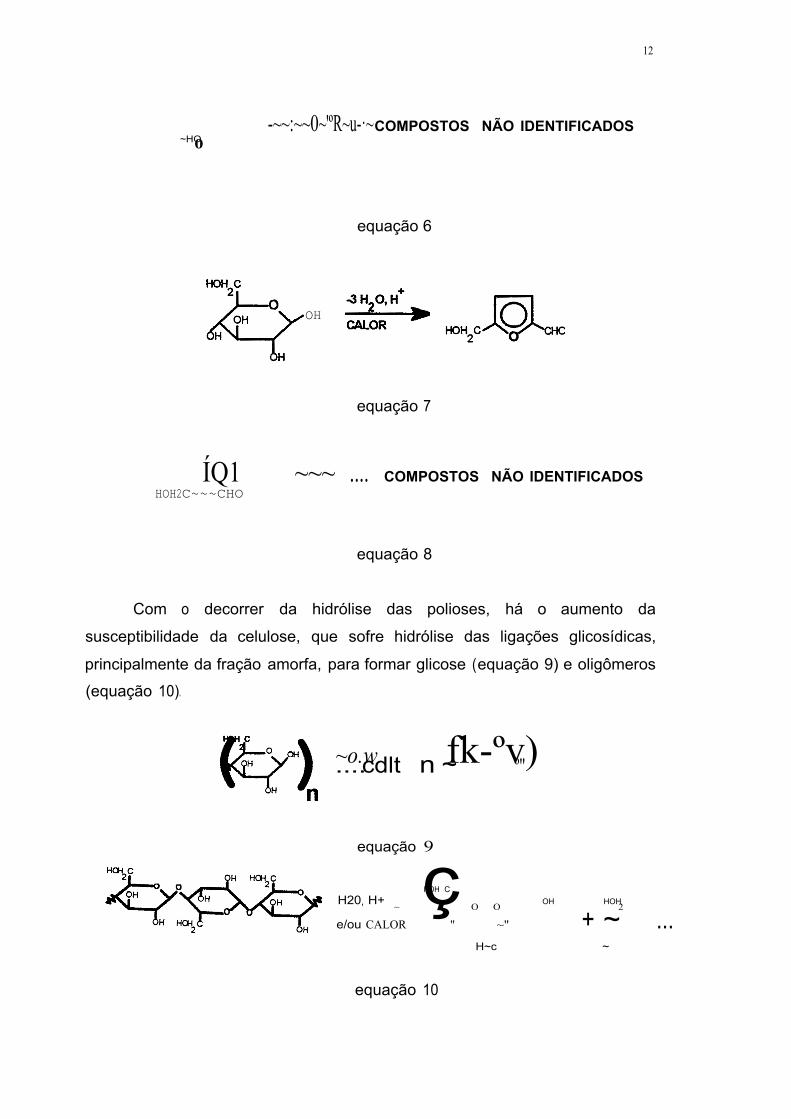
12
~HO o -~~:~~0~'ºR~u-·~ COMPOSTOS NÃO IDENTIFICADOS
equação 6
OH
equação 7
ÍQ1 ~~~ .... COMPOSTOS NÃO IDENTIFICADOS HOH2C~~~CHO
equação 8
Com o decorrer da hidrólise das polioses, há o aumento da
susceptibilidade da celulose, que sofre hidrólise das ligações glicosídicas,
principalmente da fração amorfa, para formar glicose ( equação 9) e oligômeros
(equação 10).
~o.w fk-ºv) .... cdlt n ~ º"
equação 9
HOH C H20 H+ ç OH HOH ' ~ O O 2
e/ou CALOR " ~" + ~ ... H~c ~
equação 10
13
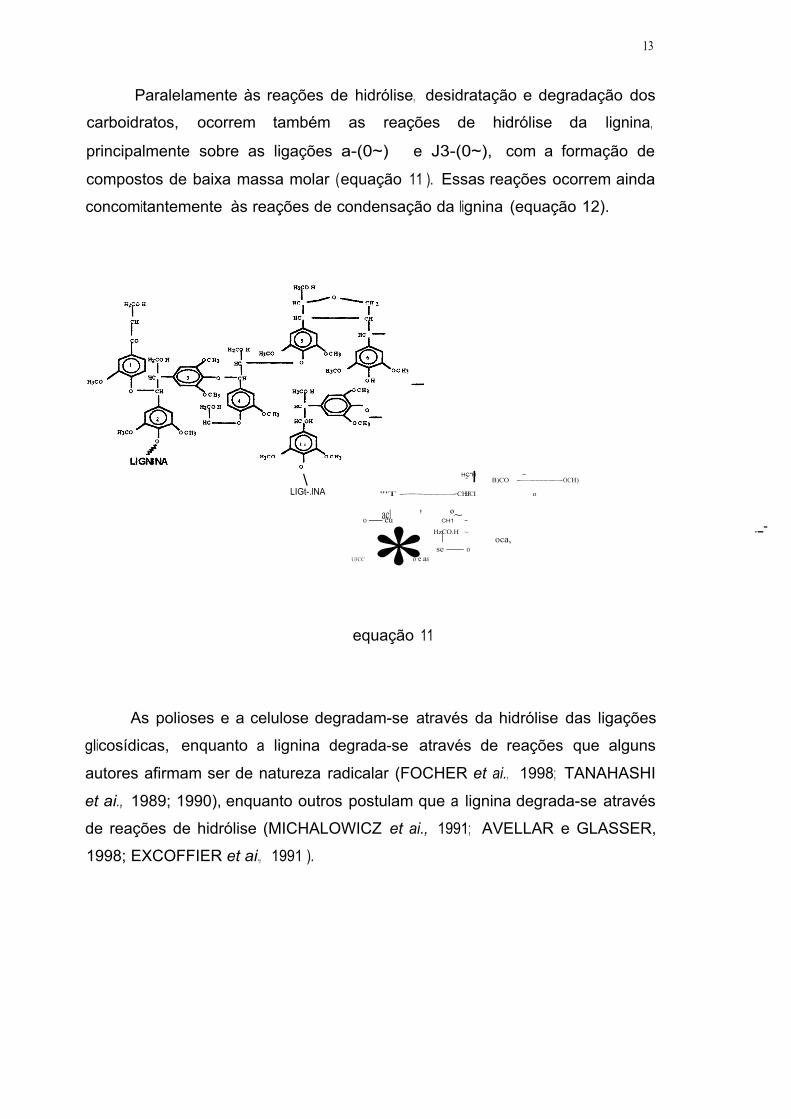
Paralelamente às reações de hidrólise, desidratação e degradação dos
carboidratos, ocorrem também as reações de hidrólise da lignina,
principalmente sobre as ligações a-(0~) e J3-(0~), com a formação de
compostos de baixa massa molar ( equação 11 ). Essas reações ocorrem ainda
concomitantemente às reações de condensação da lignina (equação 12).
\ LIGt-.lNA
HC"H ~ 2 l B)CO ~OCH)
"'T~CHJ HCI o
acl ' º~ o-cu CH1 ~
* HzCO.H ~ I oca, se-o
U3CC o e as
- --
equação 11
As polioses e a celulose degradam-se através da hidrólise das ligações
glicosídicas, enquanto a lignina degrada-se através de reações que alguns
autores afirmam ser de natureza radicalar (FOCHER et ai., 1998; TANAHASHI
et ai., 1989; 1990), enquanto outros postulam que a lignina degrada-se através
de reações de hidrólise (MICHALOWICZ et ai., 1991; AVELLAR e GLASSER,
1998; EXCOFFIER et ai., 1991 ).

14
K,COH - 1 o R: -- -rn, Rz<X>H f f & li:: clt
l * L k l~" r,s º'" 'l •oo .:: ® ca, H <X> li:: .\.:./. O -c!H ~
' -& CH, ~ n,l~oce,
* ff:,COH Q li:: o O
11::1_ ca, s::bt octt,-
11,co H, *11
; H,<X> Clfi LIGNINA O
\ LIGNINA
+ PRODUTOS DA DECOMPOSIÇÃO DOS AÇÚCARES
H+ E/OU CALOR
PRODUTOS DE CONDENSAÇÃO
equação 12
As reações citadas anteriormente são interrompidas pela
descompressão súbita do reator. Neste momento, o material é desfibrado e
reduzido a partículas menores, devido às forças de cisalhamento do processo.
(BARNET et ai., 1989; MICHALOWICZ et ai., 1991; AVELLAR e GLASSER,
1998; EXCOFFIER et ai., 1991; GLASSER e WRIGHT, 1998; MARTÍN et ai., 1995).
Enquanto o efeito térmico da explosão a vapor causa variações
estruturais nos componentes dos materiais lignocelulósicos, o efeito mecânico,
devido à expansão adiabática da água presente nos poros do material, provoca
variações a níveis morfológicos, aumentando a acessibilidade e a área
superficial do material (FOCHER et ai., 1998). Ocorrem mudanças na
macroestrutura (tamanho das partículas) e na microestrutura (distribuição dos
poros) desses materiais. A macroestrutura é modificada em função do
desfibramento, que reduz o tamanho das partículas e a microestrutura em
função do tratamento com vapor, que causa a abertura de microporos (SILVA,

15
1995). Esse fato facilita a ação posterior de reagentes químicos e: de enzimas
para a hidrólise enzimática da celulose (MICHALOWICZ et ai., 1991).
Em geral, tanto a velocidade quanto a extensão da hidrólise enzimática
dos materiais pré-tratados aumentam com o aumento do tempo de duração do ,· .•. ~
pré-tratamento. Isto, porém, só é verdade quando a sacarificação enzimática se
processa sobre o material pré-tratado e lavado. A biomassa pré-tratada bruta
contém produtos de degradação que são inibidores das enzimas e que devem
ser removidos antes da hidrólise enzimática e subseqüente fermentação.
Assim, embora a extensão e a velocidade de hidrólise enzimática dos sólidos
pré-tratados e lavados aumente com a severidade do tratamento, o rendimento
de sacarificação global pode cair devido à remoção dos açúcares solubilizados
na lavagem, como os provenientes da hidrólise das polioses (McMILLAN,
1994a).
O rendimento total de carboidratos é o fator mais importante nos
processos de conversão de biomassa em escala comercial. Estudos para
melhorar os processos de pré-tratamento devem ser direcionados para
minimizar ou eliminar os produtos de degradação dos carboidratos presentes
na biomassa, bem como os derivados da degradação da lignina e dos
extrativos. Os extrativos consistem em um grande número de compostos que
podem ser extraídos da madeira por solventes polares e apoiares. A
concentração desses compostos são maiores em certas regiões das árvores,
como nos galhos, raízes e cascas (FENGEL e WEGENER, 1989).
1.4. INIBIDORES PRESENTES NO HIDROLISADO HEMICELULÔSICO
Hidrolisados hemicelulósicos provenientes do tratamento de biomassa
por explosão a vapor contêm carboidratos como principais produtos.
Entretanto, esses açúcares são contaminados com pequenas quantidades de
uma grande variedade de outros compostos, que inibem a fermentação e o
crescimento de microrganismos. Esses inibidores podem ser divididos em
grupos, em função de sua origem (FENSKE, 1998, HAHN-HAGERDAHL, 1996, McMILLAN, 1994b):

16
( 1) Compostos liberados durante o pré-tratamento. Ex: ácido acético;
(2) Produtos de degradação dos açúcares. Ex: furfural e
hidroximetilfurfural;
(3) Produtos de degradação da lignina. Ex: siringaldeído e vanilina;
(4) Íons metálicos liberados pela corrosão dos equipamentos usados no
processo. Ex: crômio, ferro, etc.;
(5) Produtos de fermentação. Ex: etanol e subprodutos, como ácido
acético, acetaldeído, ácido fórmico e ácido lático.
A identificação e quantificação dos inibidores potenciais dos processos
fermentativos e de sacarificação enzimática podem auxiliar na escolha do
processo ou das condições de pré-tratamento mais apropriadas para um dado
material lignocelulósico (PALMQVIST et ai., 1996a;b). Há uma relação entre a
concentração desses compostos e o efeito inibitório que estes causam no meio
fermentativo.
A presença de compostos inibidores da ação de microrganismos nos
processos de bioconversão dos componentes de madeira pré-tratada com
vapor tem sido relatada por vários autores (ANDO et ai., 1986; MES-HARTREE
e SADDLER, 1983). Geralmente, estes inibidores são compostos de baixa
massa molar, solúveis em água e incluem ácido acético (1), derivados de
açúcares e produtos de degradação da lignina (PULS et ai., 1985; SILVA,
1995). A natureza e a concentração desses inibidores variam com as
condições de pré-tratamento e com a matéria-prima usada. O ácido acético (1)
é liberado durante o pré-tratamento através da hidrólise dos grupos acetil
presentes nas polioses de madeiras duras e gramíneas; o furfural (2) e o
hidroximetilfurfural (3) são formados pela desidratação das pentases e
hexases, produzidas na hidrólise das polioses e da celulose, respectivamente;
e os produtos de degradação da lignina incluem uma grande variedade de
compostos aromáticos, que serão mostrados ao longo deste trabalho.
2
HJC-COOH 1

17
SINEIRO et ai. (1997) reportaram a inibição da atividade de celulases de
Trichoderma reesei por compostos fenólicos, na extração aquosa de óleo de
girassol. O ácido clorogênico (4), testado a concentrações que variaram de
1,20 mM a 4,50 mM, foi o composto que apresentou a menor capacidade
inibitória, seguida pelos ácidos caféico (5) (0,22 mM - 0,70 mM), ferúlico (6)
(0,58 mM- 2,00 mM) e sinápico (7) (0,83 mM - 1,64 mM). Nas concentrações
testadas, a inibição causada pelos ácidos sinápico (7) e ferúlico (6) foi 100 e 1 O vezes maior do que a do ácido clorogênico (4), respectivamente. A inibição
causada por fenóis e polifenóis foi muito maior que a causada pela glicose,
pelo etanol e mesmo pela celobiose, que é considerada um· inibidor forte de
celulases em processos de sacarificação enzimática. O ácido sinápico (7), por
exemplo, apresentou um poder inibitório 1000 vezes maior do que a celobiose
(HOLTZAPPLE et ai., 1990; SINEIRO et ai., 1997).
CHCOOH li H
: ~-- OH
OH
5
COOH
1 ílH CH
~OCTD OH
6
COOH 1 CH li CH
H3CO~OCH3 OH 7
EXCOFFIER et ai. (1991) mostraram que as reações de sacarificação
enzimática de madeira de álamo pré-tratada por explosão a vapor (20-50 bar e
210-260ºC, usando H2S04 0,4% (massa /massa do substrato)) são afetadas
pela inativação das celulases causada por lignina e fenóis solúveis em água. O
rendimento da hidrólise de celulose cristalina por celulases de Trichoderma

18
reesei foi reduzido em até 24% na presença de lignina. Adicionalmente, esses
autores demonstraram a adsorsão e a inativação de endo-1,4-f3-glucosidases
por ligninas. Eles também isolaram e estudaram a ação sobre a atividade das
celulases de ácidos triidroxibutíricos (THBA) (8) e fenóis formados durante o
processo de pré-tratamento. Os resultados mostraram uma diminuição no
rendimento de glicose, o que pode ser explicado pela inibição das f3- glucosidases pelos fenóis e pelos THBA, mesmo em baixas concentrações. Os
THBA podem formar lactonas em soluções aquosas, cuja ação inibitória sobre
as reações de sacarificação enzimática de substratos lignocelulósicos já foi
descrita por DEKKER (1986).
C3H4(0H)3COOH
8
MES-HARTREE e SADDLER (1983) estudaram o efeito de inibidores
produzidos no pré-tratamento por explosão a vapor sobre a hidrólise enzimática
de celulose usando celulases de Trichoderrna harzianum. Como substrato,
usaram palha de trigo e madeira de álamo. A sacarificação enzimática da
celulose presente nos substratos pré-tratados a 250°C por 20 s com e sem a
impregnação dos cavacos com H2S04 só foi eficiente após a remoção dos
compostos de baixa massa molar, através da lavagem dos substratos com
água.
Furfural (2) e hidroximetilfurfural (3) foram durante muito tempo
considerados os principais inibidores produzidos durante o pré-tratamento de
lignocelulósicos. MES-HARTREE e SADDLER (1983) testaram esses
compostos nas concentrações normalmente encontradas nos hidrolisados:
0, 1 g/L - 0,5 g/L em hidrolisado de palha de trigo e 0,01 g/L - 0,05 g/L em
hidrolisado de madeira de álamo e não constataram a inibição das reações de
sacarificação. Testaram também xilana e xilose nos níveis produzidos durante
hidrólise enzimática de substratos lignocelulósicos. Nenhum efeito inibitório foi
constatado. Aparentemente as xilanas foram usadas em parte como substrato
para enzimas hemicelulolíticas presentes nos extratos enzimáticos. Por outro
lado, a adição suplementar do extrato aquoso, isolado do hidrolisado, no
substrato das reações de sacarificação, reduziu drasticamente a produção de

19
glicose, sem afetar significativamente a quantidade de açúcares redutores
totais, o que indica inibição das ~-glucosidases.
PALMQVIST et ai. (1996a) estudaram o efeito inibitório de compostos
solúveis em água sobre a hidrólise enzimática e a fermentação para etanol da
madeira de salgueiro, pré-tratada com vapor, na presença de 802 a 205ºC por
6 min. Os sólidos foram separados do hidrolisado por filtração. O filtrado foi
fracionado por destilação a vácuo para render duas frações, uma de voláteis e
outra de não voláteis, que foram usadas para a determinação do potencial
inibitório dos compostos presentes em cada fração. Os resultados mostraram
que os compostos voláteis não afetaram nem a hidrólise enzimática nem a
fermentação subsequente da glicose para etanol. Por outro lado, a fração do
hidrolisado contendo os compostos não voláteis afetou não só a sacarificação,
mas também a fermentação de glicose usando Saccharomyces cerevisiae.
A susceptibilidade de diferentes microrganismos a um mesmo
hidrolisado contendo inibidores de fermentação foi estudado por BUCHERT et
at. (1988; 1989). Em um desses trabalhos, madeira de bétula foi pré-tratada à 200ºC por 15 min (BUCHERT et at., 1989). Uma parte do extrato aquoso foi
pós-hidrolisado por enzimas de Tricoderma reesei e outra por ácido sulfúrico.
Xilose pura e os pós-hidrolisados foram usados como substrato para a
produção de ácido xilônico (9), etanol e proteína unicelular por Gluconobacter
oxydans, Fusarium oxysporum e Candida utilis, respectivamente. Todos os
microrganismos fermentaram eficientemente xilose pura. Entretanto, quando os
pós-hidrolisados foram usados como fonte de carbono, a fermentescibilidade
foi sofrível. Candida uti/is apresentou tolerância aos inibidores nos
experimentos contendo xilose a concentrações de até 20 g L-1. Por outro lado,
Fusarium oxysporum e Gluconobacter oxydans sofreram forte inibição por parte
dos compostos presentes nos dois hidrolisados. Além disso, a inibição variou
de um microrganismo para outro em função da concentração de inibidores no
hidrolisado. Apesar de terem sido identificados vanilina (10), siringaldeído (11), álcool coniferílico (12), álcool sinapílico (13), furfural (2), hidroximetilfurfural (3)
e alguns ácidos orgânicos nos hidrolisados, nenhum teste de toxicidade foi
efetuado com esses compostos purificados.

20
COOH
1 HC-OH
1 HO-CH
1 HC-oH
1 H2C-OH
9
~ lliC040Clli OH OH 10 11
CH20H
1 CH
li H
CH3 H3CO
12 OH
13
Os processos fermentativos são mais sensíveis aos inibidores do que os
processos de sacarificação enzimática. Em um artigo sobre a fermentação de
D-xilose a etanol por Candida shehatae e Pichia stipitis, PRIOR et ai. (1989)
relacionaram a eficiência das fermentações a fatores nutricionais, temperatura,
pH, concentração de substrato e produto, presença de outros açúcares,
demanda de oxigênio e compostos tóxicos presentes em hidrolisados
hemicelulósicos. Mostraram, por exemplo, que para diferentes cepas de uma
mesma levedura, açúcares outros que a D-xilose, como D-galactose, D-
celobiose, L-arabinose, D-glucose e D-manose, podem funcionar tanto como
co-substratos quanto inibidores. Entretanto, segundo ANDO et ai. (1986) e
McMILLAN (1994b), os maiores problemas dessas fermentações são causados
por outros compostos presentes nos hidrolisados, tais como, ácido acético (1),
furfural (2), hidroximetilfurfural (3) e ácidos, álcoois e cetonas derivados de
lignina, que inibem a fermentação por leveduras.
A forma de ação de alguns inibidores tem sido descrita na literatura.
WEIGERT et ai. (1988), citado em PRIOR et ai. (1989), descobriu que furfural

21
(2) inibiu diretamente a respiração e o crescimento de P. stipitis e foi
imediatamente reduzido a álcool furfurílico (14), o qual também diminuiu a
velocidade da produção de etanol .
14
O ácido acético (1) é, em geral, um inibidor de leveduras e o seu grau
de toxicidade é dependente do pH (HERRERO et ai., 1985), mesmo para
microrganismos modificados geneticamente (LAWFORD e ROUSSEAU,
1993a).
A forma de ação de outros inibidores, diferentes dos compostos
furânicos e ácido acético (1), tem sido pouco descrita na literatura. Entretanto,
alguns autores têm estudado de forma sistemática a ação de alguns compostos
aromáticos derivados da lignin~ (ANDO et ai., 1986; BUCHERT et ai., 1988,
1989; CLARK e MACKIE, 1984; DELGENES et ai., 1996; NISHIKAWA et ai.,
1988;. PFEIFER et ai., 1984; TRAN e CHAMBERS, 1986a,b ).
TRAN e CHAMBERS (1985) investigaram a fermentescibilidade, por
Pichia stipitis, de hidrolisados de xilose produzidos no pré-tratamento de
madeira de carvalho na presença de H2S04. Identificaram e examinaram o
efeito inibitório de compostos modelo derivados das polioses, lignina e
extrativos dessa madeira. Foram identificados e quantificados no hidrolisado,
vanilina (10)-, siringaldeído (11); ácido vanílico (15), álcool coniferílico (12),
ácido siríngico (1&); aldeído sinapíiico (17); álcool diidroconiferílico (18),
siringilmetilcetona (19), álcool diidrosinapHico (20) e álcool f3-oxisinapílico (21),
todos derivados da lignina. Dos extrativos foram identificados os ácidos
capróico (22), pelargônico (23), caprílico (24) e palmítico (24): Além desses
compostos, o hidrolisado continha outros não identificados.
Cada um desses compostos foram misturados a soluções contendo
50 g/L de xilose e nutrientes (como controle) antes da esterilização e
fermentação. Os resultados da inibição por esses compostos podem ser vistos
na tabela 1.
- --

22
Tabela 1. Concentração de alguns compostos identificados no hidrolisado de
carvalho vermelho e seus efeitos inibitórios na subseqüente fermentação de
xilose a etanol (TRAN e CHAMBERS, 1985).
Composto [ ] (gil) Etanol (gil)
Controle 22,3
Furfural 1,32 20,2
Ácido acético 12,14 5,4
Vanilina 0,086 10,2
Siringaldeído 0,213 6,2
Ácido vanílico 0,084 16,7
Ácido siríngico 0,092 19,9
Ácido capróico 0,022 19,4
Ácido caprílico 0,019 18,3
Ácido pelargônico 0,014 17,4
Ácido palmítico 0,016 21,9
Os resultados mostraram que o furfural (2) foi mais inibitório que o ácido
acético (1). Dos modelos de lignina, siringaldeído (11) foi menos tóxico que
vanilina (10) e ácido siríngico (16) menos tóxico que o ácido vanílico (15). O
efeito inibitório em ordem decrescente foi: vanilina > siringaldeído > ácido
vanílico > ácido siríngico. Isto significa que os ácidos aromáticos foram menos
tóxicos que os aldeídos e que um grupo metoxílico adicional reduziu a
toxicidade dos derivados da lignina. Entre os extrativos, os ácidos caproíco
(22), caprílico (24) e pelargônico (23) foram mais inibitórios do que o ácido
palmítico (25). O efeito inibitório em ordem decrescente foi: ácido pelargônico >
ácido caprílico > ácido capróico > ácido palmítico. Esses resultados mostram
que o aumento de tamanho das moléculas é inversamente proporcional ao
efeito inibitório. Esses compostos modelo foram testados a concentrações
similares às encontradas no hidrolisado de carvalho e o estudo sugeriu que os efeitos inibitórios seriam cumulativos.

23
COOH
~ OH
15
COOH
CHO 1 CH li CH
H3CO~OCH3 OH
17
OH
16
H2IOH CH
ijH
":rrb r----Y'ocH3 H O
OH 18
HÍOH
CH
~H
H:r CH20 6 ~OCH3
----o CH3-(CH2)4-COOH
22
20
CH3-(CH2)7-COOH 23
OH
CH3-(CH2 )6-COOH
24
CH3-(CH2)14-COOH 25
Em outro trabalho, TRAN e CHAMBERS (1986b) estudaram a
fermentescibilidade de manose para butanodiol por Klebsiella pneumoniae,
além de identificar e examinar o efeito inibitório dos derivados de lignina e dos
extrativos de madeira de pinho pré-tratada com vapor e ácido sulfúrico. Foram
testados como compostos modelo derivados de lignina: vanilina (10), ácido
vanílico (15), ácido p-hidroxibenzóico (26), ácido protocatecóico (27) e
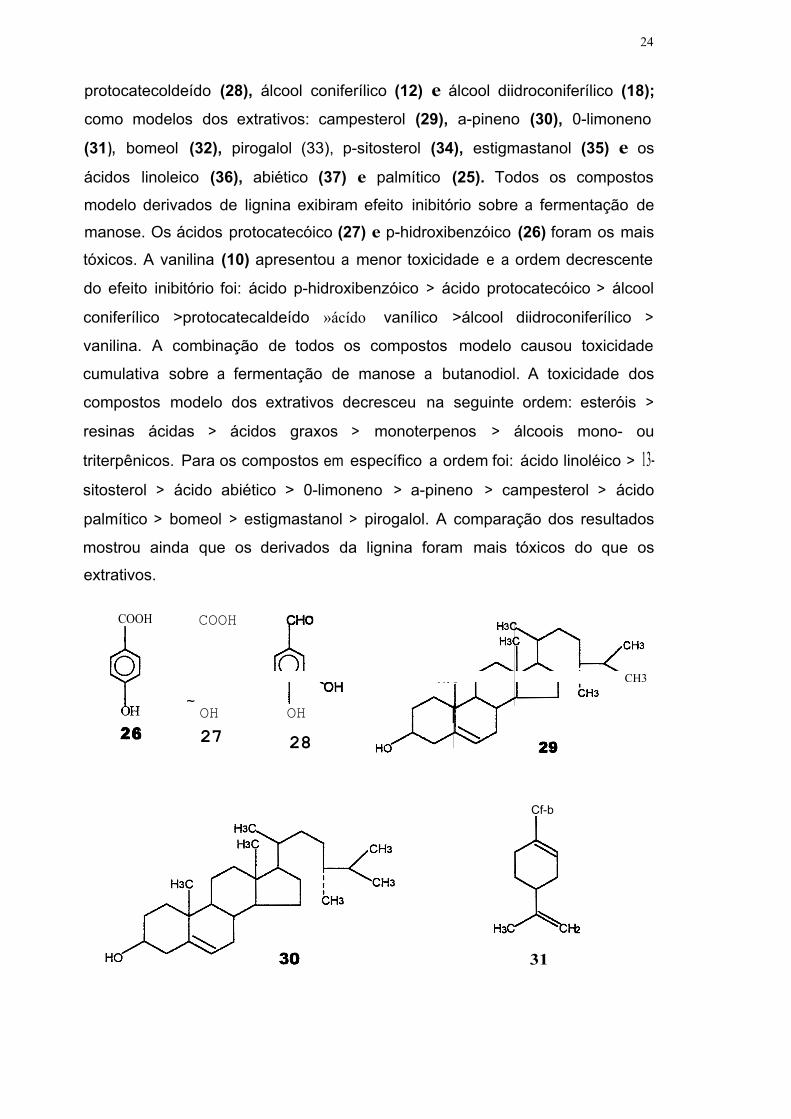
24
protocatecoldeído (28), álcool coniferílico (12) e álcool diidroconiferílico (18);
como modelos dos extrativos: campesterol (29), a-pineno (30), 0-limoneno
(31 ), bomeol (32), pirogalol (33), p-sitosterol (34), estigmastanol (35) e os
ácidos linoleico (36), abiético (37) e palmítico (25). Todos os compostos
modelo derivados de lignina exibiram efeito inibitório sobre a fermentação de
manose. Os ácidos protocatecóico (27) e p-hidroxibenzóico (26) foram os mais
tóxicos. A vanilina (10) apresentou a menor toxicidade e a ordem decrescente
do efeito inibitório foi: ácido p-hidroxibenzóico > ácido protocatecóico > álcool
coniferílico >protocatecaldeído »ácído vanílico >álcool diidroconiferílico >
vanilina. A combinação de todos os compostos modelo causou toxicidade
cumulativa sobre a fermentação de manose a butanodiol. A toxicidade dos
compostos modelo dos extrativos decresceu na seguinte ordem: esteróis >
resinas ácidas > ácidos graxos > monoterpenos > álcoois mono- ou
triterpênicos. Para os compostos em específico a ordem foi: ácido linoléico > 13- sitosterol > ácido abiético > 0-limoneno > a-pineno > campesterol > ácido
palmítico > bomeol > estigmastanol > pirogalol. A comparação dos resultados
mostrou ainda que os derivados da lignina foram mais tóxicos do que os
extrativos.
COOH COOH
~
CH3
OH OH 27 28
Cf-b
31

25 C!-13 LiYOH
32 OH 33
36
H3C-{CH2 )1-CH=CH-CH2-CH=CH-{CH2)4-COOH
37
Esses autores compararam os resultados de fermentação do hidrolisado
sem e com a presença dos compostos citados acima por diferentes métodos. A
remoção dos inibidores proporcionou uma melhora no rendimento das
fermentações.
Além dos compostos modelo testados, TRAN e CHAMBERS (1986b)
identificaram vários derivados da lignina: 4-hidroxi-3-metoxifenilacetaldeído
(38), álcool vanílico (39), aldeído coniferílico (40) e álcool (3-oxiconiferílico (41 ).
Também identificaram os seguintes derivados dos extrativos: a-terpineol (42),
D-verbenona (43), os ácidos nonanóico (= pelargônico) (23), oléico (44), esteárico (45) e desidroabiético (46). Apesar de não terem estudado a
toxicidade desses compostos, postularam, com base nos estudos com
compostos modelo, que os mesmos devem apresentar efeito inibitório sobre a
fermentação de manose para butanodiol.

26
CHO 1
fCHO f fr CH
9'0CHa OCH3 OCH3 OH OH 38 39 OH
40
CHO 1 c=o CH3
lH
9'0CH3 H3
OH ~o CH3
OH 42 43
41
H3C-(CH2)1 s-COOH
45
H3C-(CH2)1-CH=CH-(CH2)1-COOH
44
46
NISHIKAWA et ai. (1988) estudaram a influência de produtos de
degradação da lignina sobre a fermentação de xilose para etanol em meio
sintético, a partir do mesmo microrganismo (K/ebsie/la pneumoniae) estudado
por TRAN e CHAMBERS (1986b). Ácido p-hidroxibenzóico (26); p-
hidroxibenzaldeído (47), ácido vanílico (15), vanilina (10), álcool vanílico (39),
ácido siríngico (16), siringaldeído (11) e dimetoxibenzeno (48) foram testados
em concentrações de O, 1 a 0,6 g/L. O ácido p-hidroxibenzóico (26) e o p-
hidroxibenzaldeído (47) foram mais inibitórios ao crescimento dos
microrganismos e à produção de etanol do que os derivados do tipo guaiacil e
siringil. Logo, o efeito inibitório pode estar associado com a ligação desses
compostos com enzimas, através das posições 3 e 5 do anel aromático.

27
CHO
9 OH 47
lEE e McCASKEY (1983) testaram o efeito de uma mistura de
siringaldeído (11) e vanilina (10) no crescimento da levedura Pachysolen
tannophilus usando como substrato, hidrolisado de Pinus radiata e concluíram
que o crescimento foi completamente inibido a uma concentração de 2,5 gil,
com 50% de inibição a 1,0 g/L. Os autores também testaram a toxicidade da
vanilina (10) e ácido vanílico (15) na fermentação com S. cerevisiae e os
resultados mostraram que ocorreu uma inibição completa pela vanilina (10) a 5,0 g/l, enquanto 50% da inibição correspondeu a 1,3 g/L. Ácido vanílico (15)
foi menos inibitório com 50% de inibição a 3, 7 g/L.
DElGENES et ai. (1996) estudaram o efeito dos produtos de
degradação da lignina na fermentação a etanol de glicose e xilose por S.
cerevisiae, Zymomonas mobilis, Pichia stipitis e Candida shehatae. Para Pichia
stipitis e Candida shehatae, a intensidade da inibição é fortemente relacionada
com a concentração inicial dos compostos testados. Vanilina (10) apresentou o
maior efeito inibitório. Tanto crescimento quanto a produção de etanol foram
totalmente inibidos a uma concentração inicial de vanilina (10) de 1,0 g/L. Uma
comparação dos compostos modelos de lignina indicaram que siringaldeído
(11) foi menos tóxico que p-hidroxibenzaldeído (47) para as duas cepas. S.
cerevisiae foi significativamente inibida pela presença de furfurat (2),
hidroximetilfurfural (3) e vanilina (10), a qual inibiu a ação dessa levedura
quando presente em concentrações muito baixas (0,5 g/l). Zymomonas
mobilis foi fortemente inibida na fermentação de glicose por p-
hidroxibenzaldeído (47) (0,5 gil).
RANATUNGA et ai. (1997) estudaram a fermentação de xilose para
obtenção de etanol pela bactéria Zymomonas mobilis usando como sustrato, o
hidrolisado de uma mistura de madeiras ( carvalho vermelho, carvalho branco e
álamo amarelo). Vários compostos fenólicos foram testados. Vanilina (10) e

28
siringaldeído (11), com concentrações de 43 mg/L e 130 mgll,
respectivamente, apresentaram os mais altos níveis inibitórios. Ácido vanílico
(15) (84 mg/L) e ácido siríngico (16) (93 mg/L) tiveram efeitos inibitórios fracos.
FENSKE et ai. (1998) testaram os efeitos inibitórios de alguns
compostos derivados de lignina em três substratos diferentes: madeira de
álamo, "switchgrass" e "com stover". Os compostos testados foram ácido p- hidroxibenzóico (26), ácido cumárico (49), siringaldeído (11) e vanilina (10) e as
concentrações desses compostos estão mostradas na tabela 2.
IOOH CH
li
~ OH 49
Tabela 2. Compostos derivados de lignina identificados em hidrolisados de
álamo, switchgrass e com stover (FENSKE et ai., 1998).
:s: [ ] (mg/L) Acido p- [ ] (mgll) [ 1 (mgll) [ ] (mgll)
hidroxibenzóico Ácido Siringaldeído Vanilina
cumárico s
AI amo 11, 1 n.d. 29,3 15,1
"Switchgrass" n.d. 7,7 n.d. 16,5
"Com stover" n.d. 10,9 10,0 19,6
n.d. = não detectado.
Os compostos identificados nos três hidrolisados são conhecidos por
inibir fermentação de Pichia stipitis e são: vanilina (10), siringaldeído (11), ácido
acético (1) e furfural (2). Os compostos derivados de lignina, como
siringaldeído (11) e vanilina (10), são particularmente mais inibidores da

29
produção de etanol e do crescimento microbiano, especialmente quando
comparados a ácido acético ou compostos de degradação de açúcares. A
vanilina (10), um composto contendo grupo guaiacil, foi mais inibitório que
compostos contendo grupos siringil e hidroxibenzil.
ANDO et ai. (1986) também relacionaram os diferentes grupos
funcionais dos derivados de lignina à toxicidade. Identificaram e determinaram
quantitativamente vários compostos aromáticos em hidrolisado de madeira de
álamo pré-tratada por explosão a vapor a 205ºC por 1 O min. A influência
desses compostos sobre a fermentação de glicose a etanol por
Saccharomyces cerevisiae foi estudada em meio sintético. Siringaldeído (11 ),
ácido m-hidroxibenzóico (50) e ácido siríngico (16) em concentrações de até
0.1 % (massa /massa seca da madeira pré-tratada) não afetaram a
fermentação. Vanilina (10) apresentou inibição moderada e foi maior do que o
ácido p-hidroxibenzóico (26), que por sua vez foi maior que do p-
hidroxibenzaldeído (47). O ácido cinâmico (51) e o cinamaldeído (52)
interromperam a fermentação quase completamente e promoveram um efeito
tóxico nas leveduras durante a incubação. A vanilina (10) afetou a fermentação
no estágio inicial da incubação, mas após 25 h a fermentação foi recuperada e
o rendimento final de etanol foi o mesmo do controle. Considerando os
resultados de fermentação, os autores estimaram valores emplricos para o
grau de inibição de cada substituinte presente nos compostos aromáticos
examinados: CH=CH = +3,0; CHO = +1,5; p-OH = +1,0; COOH = +0,5; m-OH =
O e OCH3 = -1,0. Segundo esses autores, os valores atribuídos a cada grupo
funcional ligado a um composto podem ser usados para simular o poder
inibitório do mesmo sobre a fermentação. Os resultados obtidos na simulação
mostraram-se concordantes com os resultados das fermentações.
CHO
[ HCCOOH 1 li CH
li HC
~ ~
50 OH
51 52

30
Os trabalhos acima trataram, em sua grande parte, do efeito inibitório
dos compostos formados em diferentes processos de pré-tratamento sobre a
fermentescibilidade de xilose. Resultados semelhantes foram obtidos em
fermentações alcoólicas de hidrolisados contendo glicose como substrato,
usando-se diferentes microrganismos (CLARK e MACKIE, 1984; DELGENES
et ai., 1996; PFEIFER et ai., 1984). Entretanto, não encontramos nenhum
trabalho sistemático envolvendo os possíveis inibidores formados nos
processos de pré-tratamento do bagaço de cana.
O processo de separação dos componentes do bagaço de cana por
explosão a vapor, com ou sem catalisadores, tem sido sistematicamente
estudado em nosso grupo e pesquisa (SILVA, 1995; SILVA et ai. 1997,
MATTOS e SILVA, 1999). Na ausência de catalisador, os melhores resultados
foram alcançados nos pré-tratamentos a 190ºC por 15 min (SILVA, 1995).
Nessas condições, cerca de 36% do bagaço de cana foi solubilizado. 86% da
glucana foi recuperada como celulose no bagaço pré-tratado, 3,2% foi
hidrolisado a glicose, O, 12% foi decomposto a hidroximetilfurfural e 11 % a
compostos desconhecidos. 73% das xilanas foram hidrolisadas a arabinose
(2,5%), a arabinose ligada às xilanas (1%), a xilose (7,4%), a furfural (2,8%), a
oligômeros de xilose (51%) e a compostos não identificados (8,3%). Os grupos
acetil foram hidrolisados a ácido acético (39%) e a grupos acetil ligados às
xilanas (36% ); 12% permaneceram no bagaço pré-tratado e 13% foram
perdidos como compostos não identificados. A quantidade de lignina
hidrolisada a produtos solúveis foi de 20%. Pré-tratamentos sob condições
mais brandas mostraram-se ineficientes quando se buscou altos rendimentos
na recuperação das polioses hidrolisadas e, sob condições mais drásticas,
promoveram reações de condensação entre a lignina, o hidroximetilfurfural e o
furfural. Os resultados obtidos no pré-tratamento de 1 O kg de bagaço (base
seca) a 190°C, 15 min, em um reator de 240 L foram muito próximos daqueles
alcançados em escala de laboratório (SILVA, 1995). Nessas condições, a
quantidade de celulose presente no bagaço pré-tratado foi superior à dos
experimentos de bancada. A quantidade de xilana e dos grupos acetil,
recuperados no licor como compostos conhecidos foi menor, mostrando que as
polioses sofreram maior decomposição em escala piloto. Por outro lado, a

31
composição qualitativa dos hidrolisados foi praticamente a mesma nas duas
escalas.
Nesses estudos, os principais produtos obtidos nos procedimentos de
explosão a vapor (bancada e piloto) foram os oligomêros de xilose, que não
foram determinados quanto ao grau de polimerização. Também não foram
identificados compostos de baixa massa molar derivados da degradação da
lignina.
Nesse trabalho caracterizamos de forma mais completa a fração
solubilizada durante o pré-tratamento, através da identificação e quantificação
de vários compostos. Foram identificados os oligômeros de xilose (principais
produtos obtidos) e foram identificados e quantificados os compostos de baixa
massa molar produzidos pela degradação da lignina. Tais compostos são tidos
como inibidores potenciais de processos fermentativos e enzimáticos.

32
2. OBJETIVOS
O objetivo principal deste trabalho foi identificar e quantificar compostos
que são inibidores potenciais de processos fermentativos e enzimáticos e estão
presentes no hidrolisado hemicelulósico do bagaço de cana pré-tratado por
explosão a vapor.
Para atingirmos esse objetivo, foram propostas as seguintes etapas de
trabalho:
- Caracterização química do bagaço de cana utilizado no pré-tratamento por
explosão a vapor;
Pré-tratamento por explosão a vapor do bagaço de cana, para a obtenção
do hidrolisado hemicelulósico;
- Caracterização da fração solubilizada durante o pré-tratamento;
- Separação das frações do hidrolisado hemicelulósico por diferentes
métodos;
Identificação dos compostos presentes nas diferentes frações obtidas do
pré-tratamento;
- Caracterização espectroscópica por infravermelho das frações contendo
carboidratos e compostos aromáticos.

33
3. MATERIAIS E MÉTODOS
O procedimento experimental foi dividido em 3 tópicos principais:
1 . Preparação (item 3.1 ) e caracterização química do bagaço de cana
(item 3.2);
2. Pré-tratamento do bagaço de cana por explosão a vapor, em escala
de bancada, a 190ºC por 15 min (item 3.3);
3. Separação e caracterização das frações geradas no processo de pré-
tratamento.
O tópico 3 inclui a determinação da composição dos hidrolisados
gerados no pré-tratamento do bagaço de cana por explosão a vapor (itens
3.2.5, 3.4 e 3.5), os procedimentos de separação das frações contendo os
compostos aromáticos e os carboidratos (itens 3.6.1 e 3.6.2) e a identificação e
quantificação de compostos aromáticos de baixa massa molar presentes no
hidrolisado (item 3.8).
A representação esquemática geral do procedimento experimental é
mostrada no diagrama de blocos da figura 8.
3.1. PREPARAÇÃO DO BAGAÇO DE CANA
O bagaço de cana, cedido pela Usina Nova América, foi misturado,
homogeneizado, quarteado, embalado em saco de polietileno contendo
aproximadamente 1 kg, com umidade média de 50% e armazenado em câmara
fria a -20ºC, até sua utilização no experimento de explosão a vapor.
Uma amostra de aproximadamente 200 g foi seca em estufa a 60ºC, por
cerca de 12 h, moída, classificada em partículas de tamanho menor ou igual a
20 mesh e caracterizada quimicamente, de acordo com o procedimento
descrito no item 3.2.

34
BAGAÇO DE CANA DE AÇÚCAR
PREPARAÇÃO E CARACTERIZAÇÃO
' EXPLOSÃO A
VAPOR
1 FILTRAÇÃO 1
'
1 1 HIDROLISADO
HEMICEWLÓSICO BAGAÇO
PRÉ· TRATADO
I CARACTERIZAÇÃO I
SEPARAÇÃO CARBOIDRATOS I
COMPOSTOS BAIXA MASSA MOLAR POR EXTRAÇÃO
LÍQUIDO/LÍQUIDO
SEPARAÇÃO CARBOIDRATOS I
AROMÁTICOS POR C18
•
I CARACTERIZAÇÃO I I CARACTERIZAÇÃO I I CARACTERIZAÇÃO I
Figura 8. Representação esquemática do processo de separação dos
componentes do bagaço de cana por explosão a vapor.

35
3.2. CARACTERIZAÇÃO QUIMICA DO BAGAÇO DE CANA
3.2.1. DETERMINAÇÃO DOS EXTRAiVEIS
O bagaço de cana foi extraído com etanol 95%, em um Soxhlet até o
solvente tomar-se incolor (cerca de 10 sifonações). O bagaço extraído foi seco
ao ar para evaporação do solvente, e em seguida seco em estufa a 105ºC até
massa constante, determinada em balança analítica.
3.2.2. DETERMINAÇÃO DE LIGNINA KLASON INSOLÚVEL EM MEIO
ACIDO
A quantidade de lignina insolúvel em meio ácido foi determinada de
acordo com o método Klason modificado (ASTM, 0.1106-56).
Uma amostra de 2 g de bagaço de cana extraído e seco, pesada com
incerteza de O, 1 mg, foi transferida para um bécher de 100 ml e tratada com
1 O ml de H2S04 72%, sob vigorosa agitação, em um banho termostatizado a
45 ± 0,5°C por 7 min. A reação foi interrompida com a adição de 50 ml de
água destilada. A amostra foi transferida quantitativamente para um
Erlenmeyer de 500 ml, usando-se 275 ml de água.
Para a completa hidrólise dos oligômeros restantes, o Erlenmeyer foi
fechado com papel alumínio e autoclavado por 30 min a 1,05 bar. Após a
descompressão da autoclave, o frasco foi retirado e resfriado à temperatura
ambiente. A mistura reacional foi primeiramente filtrada para um balão
volumétrico de 500 ml, o qual foi posteriormente avolumado com água
destilada. A solução foi armazenada para análises posteriores de lignina
solúvel, cinzas, carboidratos e furfural e hidroximetilfurfural, de acordo com o
procedimento descritos nos itens 3.2.3, 3.2.4, 3.2.5 e 3.2.6, respectivamente.
Os sólidos retidos no papel de filtro foram lavados com
aproximadamente 1,2 L de água destilada e secos em estufa a 105 ± 3°C até
massa constante. O material foi calcinado de acordo com o procedimento
descrito no item 3.2.4. A quantidade de cinzas foi determinada e a massa de
lignina determinada por diferença.

36
3.2.3. DETERMINAÇÃO DE LIGNINA KLASON SOLÚVEL EM MEIO ACIDO
A quantidade de lignina solúvel em meio ácido foi determinada conforme
metodologia descrita por SILVA (1995).
Uma alíquota de 5 mL do hidrolisado obtido no item 3.2.2 foi alcalinizada
com NaOH 6,5 m.L-1 até pH 12,5 (- 2 mL) e diluída com água destilada em um
balão de 100 ml.
A absorbância da solução em 280 nm foi determinada em um
espectrofotômetro UVNisível de duplo feixe, modelo Cintra 20, usando-se água
destilada como referência. A concentração de lignina foi calculada conforme a
equação 13.
Clig = (Ãt.idr2ao - Apct2ao) I &lig
onde:
- Cu9 = concentração de lignina, em gil.
- Atiidr2aa = absorbância do hidrolisado contendo a lignina, em 280
equação 13
nm.
- ~ao = Ctur1 &tur1 + Chmf &hmf = absorbância em 280 nm dos
produtos de decomposição dos açúcares (furfural e hidroximetilfurfural), cujas
concentrações (Ctur1 e Chmt) e as absortividades (&turt e &hmr) foram determinados
previamente por CLAE (Cromatografia Líquida de Alta Eficiência) e por UV,
respectivamente. O valores das absortividades obtidas para furfural e
hidroximetilfurfural foram 146,85 e 114,00 L g-1 cm", respectivamente,
conforme descrito em SILVA (1995).
- &ug = absortividade para lignina (25,20 L g-1 cm") (SILVA,
1995).
3.2.4. DETERMINAÇÃO DO TEOR DE CINZAS NO BAGAÇO DE
CANA
A determinação do teor de cinzas no bagaço de cana ou lignina foi feita
conforme metodologia descrita em SILVA (1995). Cerca de 1,8 a 2,0 g de
bagaço de cana, com umidade conhecida, foram pesados, com incerteza de
O, 1 mg, em um cadinho de porcelana previamente calcinado e tarado. Em

. . 37
seguida, o bagaço de cana foi calcinado inicialmente a 300ºC e depois por
mais 2 h a BOOºC. Após a calcinação, o cadinho foi resfriado em dessecador e
a massa de cinzas determinada. A massa de cinzas da lignina foi usada para .. corrigir o teor de lignina insolúvel, determinado no item 3.2.2.
O teor de cinzas foi calculado pela equação 14:
% czs = (M2 - M1) I M3 x 100
onde:
equação 14
-% czs = porcentagem em massa de cinzas.
= massa do cadinho calcinado vazio, em g.
= massa do cadinho com cinzas, em g.
= massa de lignina seca, em g.
3.2.5. DETERMINAÇÃO DE CARBOIDRATOS E ÁCIDOS
ORGÂNICOS POR CLAE
Os carboidratos e os ácidos orgânicos presentes no hidrolisado foram
identificados em um cromatógrafo ,líquido Shimadzu, modelo CT0-6A (ROCHA
et ai., 1997). Uma alíquota de 40 mL do hidrolisado, obtido no item 3.2.2., teve
o pH ajustado com NaOH 2 moí.L" de 0,6 para a faixa de 1 a 3 e, em seguida,
foi diluída com água destilada em balão volumétrico de 50 mL .
O hidrolisado ácido foi extraído em cartuchos de extração sólida Sep-Pak
C1a (Waters), para a remoção de compostos aromáticos e, então, injetado em
uma coluna Aminex HPX-87H (300 X 7,8 mm, Bio-Rad Laboratories Ltd)
acoplada a uma pré-coluna trocadora de cátions (Bio-Rad Laboratories Ltd),
usando-se H2S04 0,005 mol.L" como fase móvel a uma vazão de 0,6 mUmin
a 45°C, segundo procedimento descrito por SILVA (1995). Os compostos foram
monitorados com um detector de índice de refração (RI) (Shimadzu R10-6A).
As áreas dos picos correspondentes às hexoses e às pentases foram
utilizadas para calcular as massas de glucana e xilana, respectivamente. Essas
massas foram divididas pelo peso seco do material inicial e multiplicadas pelo
fator de hidrólise. Os fatores de hidrólise para conversão de glicose e celobiose

38
em glucana são 0,90 e 0,95, respectivamente. De maneira similar, xilose e
arabinose foram convertidas em xilana e o ácido acético em grupos acetil
usando-se os fatores 0,88 e 0,72, respectivamente.
As concentrações de 'ceíobiose, glicose, xilose, arabinose e ácido acético
foram determinadas a partir de curvas de calibração obtidas com padrões
analíticos.
3.2.6. DETERMINAÇÃO DE FURFURAL E HIDROXIMETILFURFURAL
Furfural e hidroximetilfurfural foram determinados por CLAE em uma
coluna LiChrospher 100 RP-18 (5µm) de 125 x 4mm (Hewlett-Packard),
utilizando-se acetonitrila/água 1 :8 (v/v) com 1 % de ácido acético como fase
móvel a uma vazão de 0,8 mUmin a 25ºC, conforme descrito em SILVA (1995).
O hidrolisado foi previamente diluído com água na razão de 1: 100, filtrado em
membrana com diâmetro de poro de 0,47 µm (Millipore) e injetado com uma
válvula Rheodyne equipada com alça de injeção de 20 µL. Os compostos foram
detectados a 276 nm, em um detetor UVNisível HP 79875. As concentrações
de furfural e hidroximetilfurfural foram determinadas a partir de curvas de
calibração obtidas com os compostos puros.
3.3. PRÉ-TRATAMENTO DO BAGAÇO DE CANA POR EXPLOSÃO A
VAPOR A 190°C POR 15 MIN
O pré-tratamento do bagaço de cana foi realizado em escala de
bancada, usando-se um reator de aço inox 316 com capacidade para 0,65 L,
cuja representação esquemática é mostrada na figura 9 (SILVA, 1995).
No reator [3] pré-aquecido à temperatura de 190ºC foram introduzidos,
com o auxílio de elevador hidráulico (9], 10 g de bagaço seco, pesados com
incerteza de O, 1 mg. O reator foi fechado e o vapor injetado até a temperatura
atingir o equilíbrio, o qual permaneceu por 15 min. A válvula de alimentação de
vapor [2] foi fechada e a válvula de descompressão [4] aberta imediatamente.
Em função da descompressão súbita do reator, o material pré-tratado foi
ejetado para o ciclone (5], do qual foi removido quantitativamente com cerca de
500 mL de água destilada. Os experimentos foram realizados em quatro

39
bateladas. As suspensões foram misturadas e a mistura final filtrada em um
funil de Büchner.
O bagaço pré-tratado, retido no funil de Büchner, foi lavado com um
volume adicional de 2000 ml de água destilada a 50ºC. O filtrado foi
transferido quantitativamente do kitassato para provetas de 2000 ml. O volume
final obtido foi de 4200 ml. O bagaço pré-tratado foi lavado com cerca de 2000
ml de água adicionais, até a água de lavagem tomar-se incolor. A água de
lavagem foi desprezada e o bagaço foi levado à estufa a 60ºC até massa
constante, determinada em balança analítica. Em seguida, a amostra foi moída
e classificada em partículas menores ou iguais a 20 mesh, num moinho do tipo
Wiley, e a seguir caracterizada pelo procedimento descrito no item 3.2.
Do filtrado foram retiradas duas alíquotas de 5 ml, sendo uma para
análise da lignina solúvel (item 3.2.3) e a outra para análise do
hidroximetilfurfural e do furfural (item 3.2.6), além de uma alíquota de 40 ml,
para determinação das concentrações dos monômeros de açúcares (item
3.2.5), do ácido acético livre (item 3.2.5) e dos oligômeros de glicose e xilose
(item 3.4).
Outra alíquota de 250 ml foi transferida para um Erlenmeyer de 500 ml
e acidificada com H2S04 a pH 0,6. O frasco foi fechado com papel alumínio e
a amostra foi autoclavada por 30 mina 1,05 bar para a hidrólise completa dos
oligômeros presentes. O hidrolisado foi avolumado para 500 ml, dos quais
foram retiradas duas alíquotas: uma de 5 ml, para determinação de lignina
solúvel (item 3.2.3) e uma de 40 ml para determinação de carboidratos e de
seus produtos de decomposição por CLAE (item 3.2.5).
Outra alíquota de 40 ml do hidrolisado foi extraída em 12 cartuchos de
extração sólida Sep-Pak C 18 (Waters) conectados em série. Os carboidratos e
os oligômeros aluídos nos cartuchos foram determinados por CLAE (itens 3.2.5
e 3.4). Os compostos aromáticos retidos na coluna foram dessorvidos com
etanol (itens 3.2.5 e 3.4). Para as duas frações, foram feitas análises de FTIR
(item 3.7)
Outra alíquota de 580 ml foi extraída com diferentes solventes
sucessivamente, conforme descrito no item 3.6.2. Para cada fração separada,
contendo os compostos aromáticos, foram feitas análises de GC/MS e GC/FID
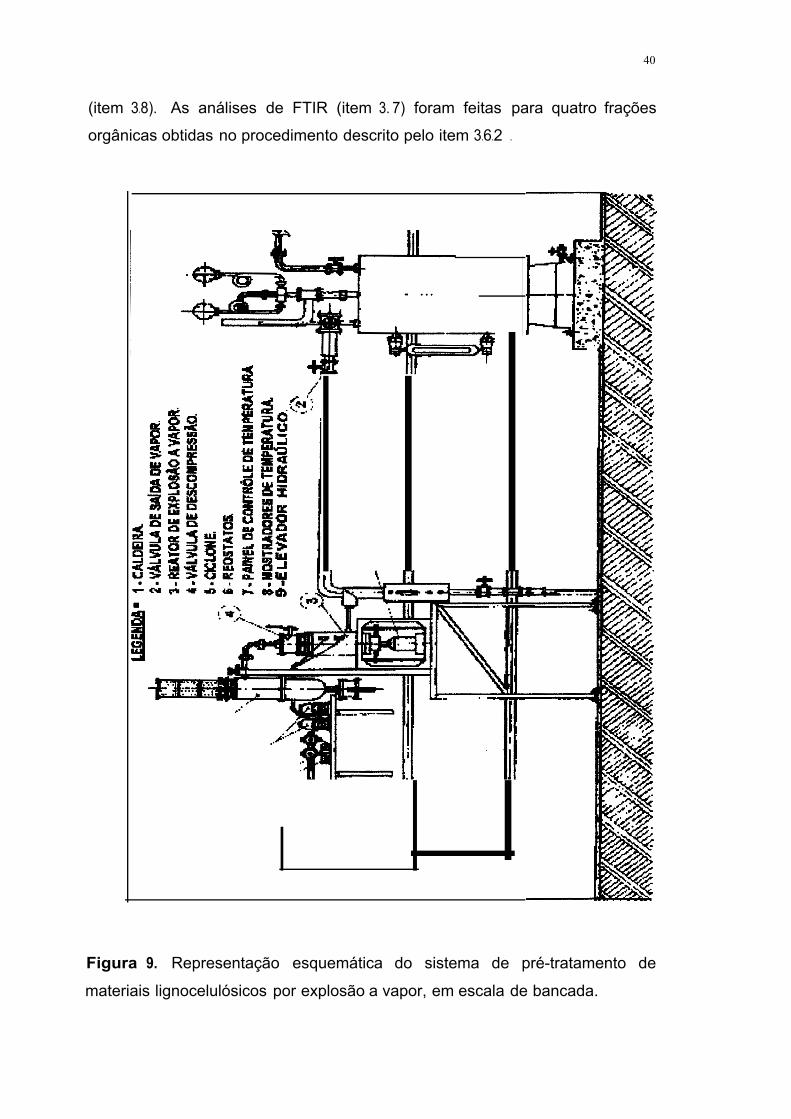
40
(item 3.8). As análises de FTIR (item 3. 7) foram feitas para quatro frações
orgânicas obtidas no procedimento descrito pelo item 3.6.2 .
,
1
/
,· ,,··':'( 46· ... ./
······ ....
c., ,,. 1 1 1 1
1 1
j
(;õJ ... , •· -~.....u.. .q:a,,
(U1'( ...... = .. ;- ~ - - - .... .. •
Figura 9. Representação esquemática do sistema de pré-tratamento de
materiais lignocelulósicos por explosão a vapor, em escala de bancada.

41
3.4. IDENTIFICAÇÃO DOS OLIGÔMEROS DE GLICOSE E XILOSE POR
CLAE
Os oligômeros de glicose e xilose presentes no hidrolisado, obtido no
item 3.3, foram identificados por CLAE em um cromatógrafo líquido Shimadzu,
modelo CT0-6A, de acordo com o procedimento descrito abaixo.
Uma alíquota de 40 ml do hidrolisado teve o pH ajustado para 7 com
NaOH 2 mol. L"1 e em seguida foi diluída com água destilada em balão
volumétrico de 50 ml.
Os compostos aromáticos foram removidos do hidrolisado por extração
em cartuchos Sep-Pak C18 (Waters). O hidrolisado foi injetado diretamente em
uma coluna Aminex HPX-42A (300 X 7,8 mm, Bio-Rad Laboratories ltd)
acoplada a duas pré-colunas: catiônica e aniônica. Como fase móvel foi
empregada água bidestilada com vazão de 0,6 mUmin, a 45°C. Os compostos
foram monitorados com um detector de RI (Shimadzu R10-6A).
A identificação do grau de polimerização dos oligômeros de xilose
presentes no hidrolisado foi feita a partir de uma amostra padrão contendo
xilose, xilobiose e xilotriose, gentilmente cedida pelo Prof. Dr. José Domingos
Fontana, da Universidade Federal do Paraná (UFPR). Os tempos de retenção
da xilotetraose, xilopentaose e xiloexaose foram determinados por
extrapolação, a partir do cromatograma obtido da hidrólise ácida de xilana de
bétula (Sigma).
A identificação dos oligômeros de glicose foi feita de maneira análoga, a
partir da hidrólise de uma amostra de celulose cristalina (Sigmacell - Sigma).
Os procedimentos de hidrólise da xilana e da celulose estão descritos no item
3.4.1.
3.4.1. HIDRÓLISE DE XILANA E CELULOSE
Uma amostra de 1 g de xilana ou de celulose, pesadas com incerteza de
O, 1 mg, foi transferida para um bécher de 100 ml e tratadas com 1 O mL de
H2S04 72%, sob vigorosa agitação, em um banho termostatizado. A xilana foi
hidrolisada à temperatura ambiente (25ºC) e a celulose a 45,0 ± 0,5°C, ambas
por 7 min. A reação foi interrompida com a adição de 50 ml de água destilada.

42
As amostras foram transferidas quantitativamente para erlenmeyers de 500 ml,
usando-se 275 ml de água.
Em seguida, uma alíquota de 25 ml de cada amostra foi retirada e o pH
elevado a 5,5 com auxílio de Ba(OH)2. A solução foi filtrada em papel de filtro e
injetada diretamente na coluna Aminex HPX-42A (300 X 7,8 mm, Bio-rad
Laboratories Ltd) acoplada a duas pré-colunas trocadoras de íons: uma
catiônica e outra aniônica (Bio-Rad Laboratories Ltd). Como fase móvel foi
empregada água bidestilada com vazão de 0,6 mUmin, a 45ºC. Os compostos
foram monitorados em um detector de índice de refração (RI) (Shimadzu
R10-6A).
3.5. DISTRIBUIÇÃO DA MASSA MOLAR DOS COMPOSTOS
PRESENTES HIDROLISADO
A determinação da distribuição da massa molar média dos compostos
presentes no hidrolisado, obtido no item 3.3, foi feita por cromatografia de
exclusão por tamanho (HPSEC) em um cromatógrafo líquido de alta eficiência
Shimadzu modelo CT0-6A, segundo o procedimento descrito por PAIVA et ai.
(1997).
Três frações do hidrolisado foram analisadas: uma fração filtrada em
membrana (Millipore), para remoção de sólidos; uma fração eluída em um
cartucho de extração sólida Sep-Pak C 18 contendo os carboidratos; e a fração
adsorvida em C 18 e dessorvida com etanol, contendo os compostos
aromáticos. As amostras (20 µL) foram aluídas em uma coluna Asahipak GS-
320H com água bidestilada, a uma vazão de 1,0 mUmin. Os compostos foram
monitorados por RI (detector Shimadzu R10-6A).
A distribuição da massa molar dos compostos presentes no hidrolisado
foi determinada a partir de curvas de calibração, usando-se 4 proteínas de
massas molares conhecidas, conforme descrito no item 3.5.1.
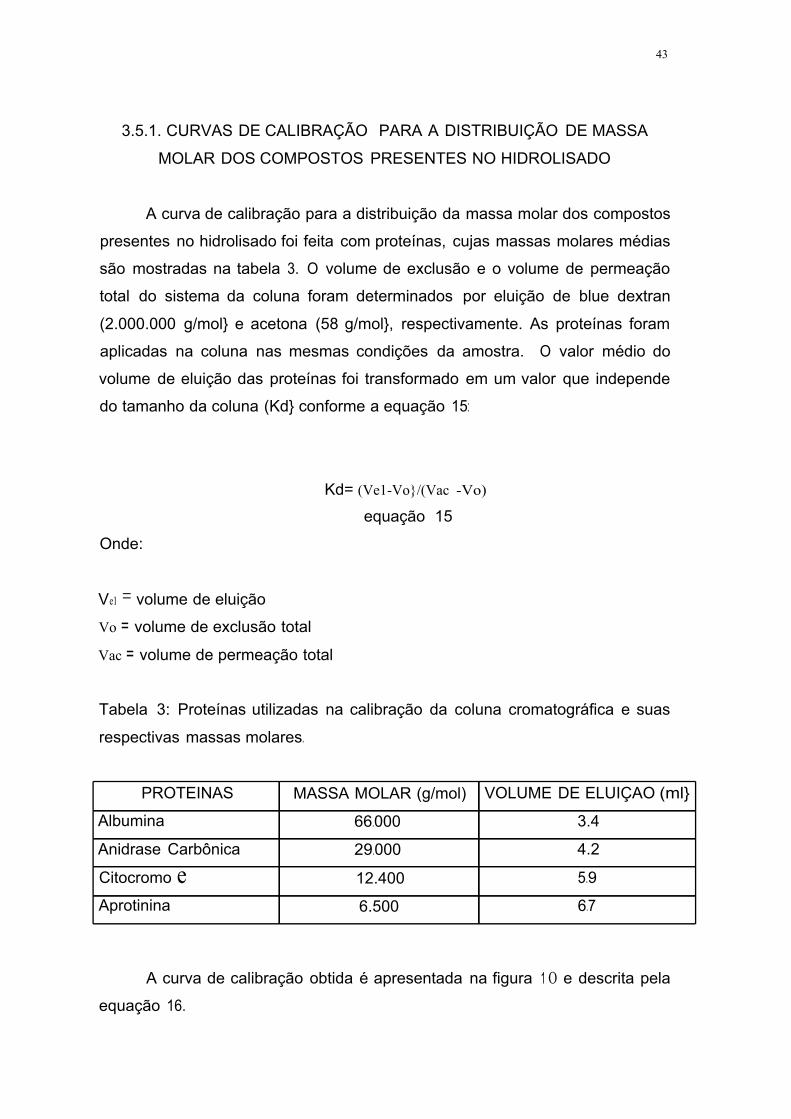
43
3.5.1. CURVAS DE CALIBRAÇÃO PARA A DISTRIBUIÇÃO DE MASSA
MOLAR DOS COMPOSTOS PRESENTES NO HIDROLISADO
A curva de calibração para a distribuição da massa molar dos compostos
presentes no hidrolisado foi feita com proteínas, cujas massas molares médias
são mostradas na tabela 3. O volume de exclusão e o volume de permeação
total do sistema da coluna foram determinados por eluição de blue dextran
(2.000.000 g/mol} e acetona (58 g/mol}, respectivamente. As proteínas foram
aplicadas na coluna nas mesmas condições da amostra. O valor médio do
volume de eluição das proteínas foi transformado em um valor que independe
do tamanho da coluna (Kd} conforme a equação 15:
Kd= (Ve1-Vo}/(Vac -Vo)
equação 15
Onde:
V e1 = volume de eluição
Vo = volume de exclusão total
Vac = volume de permeação total
Tabela 3: Proteínas utilizadas na calibração da coluna cromatográfica e suas
respectivas massas molares.
PROTEINAS MASSA MOLAR (g/mol) VOLUME DE ELUIÇAO (ml}
Albumina 66.000 3.4
Anidrase Carbônica 29.000 4.2
Citocromo e 12.400 5.9
Aprotinina 6.500 6.7
A curva de calibração obtida é apresentada na figura 1 O e descrita pela
equação 16.
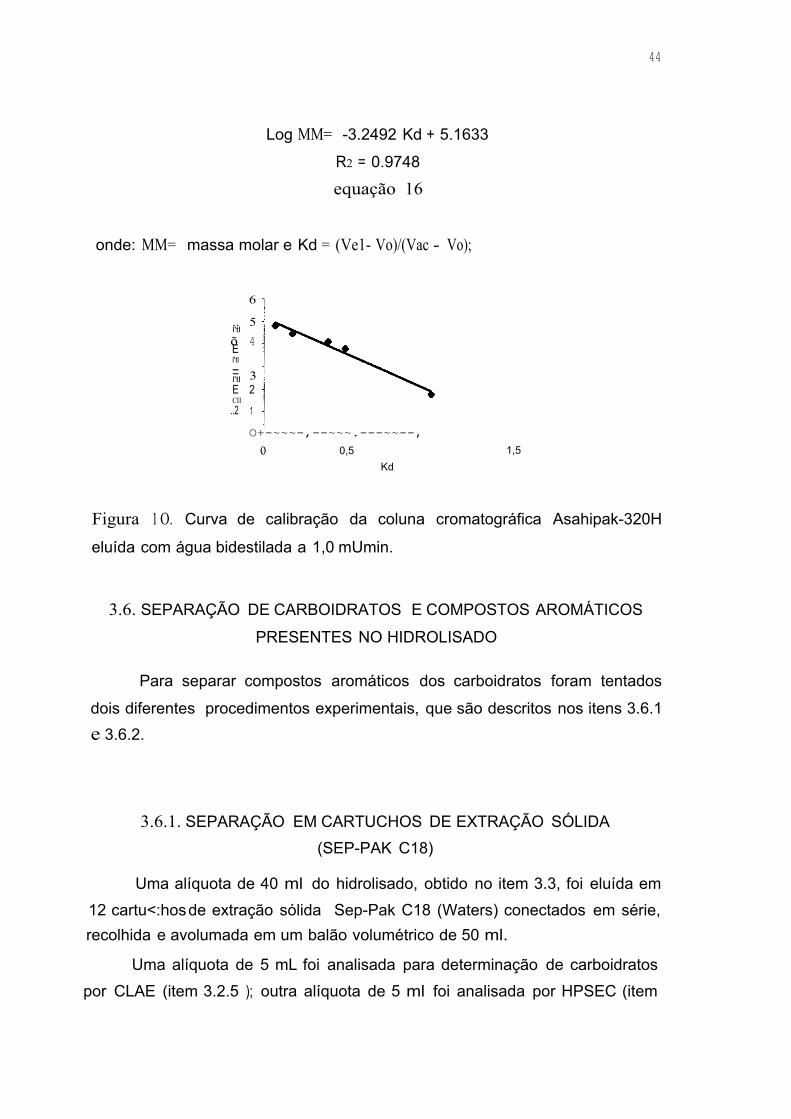
44
Log MM= -3.2492 Kd + 5.1633
R2 = 0.9748
equação 16
onde: MM= massa molar e Kd = (Ve1- Vo)/(Vac - Vo);
6
.. 5 l'tl õ 4 E l'tl = 3 l'tl E 2 CII ..2 1
O+-~~~-,--~~~.---~~--, o 0,5 1,5
Kd
Figura 1 O. Curva de calibração da coluna cromatográfica Asahipak-320H
eluída com água bidestilada a 1,0 mUmin.
3.6. SEPARAÇÃO DE CARBOIDRATOS E COMPOSTOS AROMÁTICOS
PRESENTES NO HIDROLISADO
Para separar compostos aromáticos dos carboidratos foram tentados
dois diferentes procedimentos experimentais, que são descritos nos itens 3.6.1 e 3.6.2.
3.6.1. SEPARAÇÃO EM CARTUCHOS DE EXTRAÇÃO SÓLIDA (SEP-PAK C18)
Uma alíquota de 40 ml do hidrolisado, obtido no item 3.3, foi eluída em 12 cartu<:hos de extração sólida Sep-Pak C18 (Waters) conectados em série, recolhida e avolumada em um balão volumétrico de 50 ml.
Uma alíquota de 5 mL foi analisada para determinação de carboidratos por CLAE (item 3.2.5 ); outra alíquota de 5 ml foi analisada por HPSEC (item

45
3.5), para determinação da distribuição de massa molar dos compostos
presentes; e finalmente, uma alíquota de 40 ml foi liofilizada em um aparelho
Edwards, seca em P20s e analisada por FTIR (item 3. 7).
A fração retida nos cartuchos, contendo os compostos aromáticos, foi
dessorvida com 25 ml de etanol. A partir de uma alíquota de 5 ml dessa
solução, foram feitas análises de açúcar (item 3.2.5) e HPSEC (item 3.5). 20
ml foram evaporados a pressão reduzida, secos em P205 e analisados por
FTIR e GC/MS, conforme descrito nos itens 3.7 e 3.8.2, respectivamente.
3.6.2. SEPARAÇÃO POR EXTRAÇÃO LÍQUIDO-LÍQUIDO
Os carboidratos, os derivados de lignina de massa molar mais alta e os
compostos aromáticos de baixa massa molar solúveis em água, foram
separados por extração líquido-líquido, de acordo com o procedimento
experimental descrito abaixo e ilustrado pelo diagrama de blocos da figura 11. Uma alíquota de 580 ml do hidrolisado, obtido no item 3.3, foi
acidificada com HCI 2 mol.L" até pH 2,0. Em seguida, o hidrolisado foi extraído
com acetato de etila. Após a extração, foram separadas a fração aquosa,
contendo carboidratos (FA1) e a fração orgânica, contendo acetato de etila.
Uma alíquota de 2 ml da fração orgânica foi separada e injetada diretamente
em um cromatógrafo gasoso acoplado a um detector de massas Finnigan,
modelo MAT GCQ™ (item 3.8.2) e analisada por GC/MS (item 3.8). Uma
alíquota de 100 ml foi evaporada, seca em P205, derivatizada e analisada por
GC/MS (F01) (item 3.8.2). A alíquota restante de 500 ml da fração orgânica foi
extraída com uma solução de NaHC03 saturada e NaOH 2 mol.L", três vezes
sucessivamente. As frações solúveis em NaHC03 e NaOH foram acidificadas
com HCI 2 mol.L" e extraídas três vezes com diclorometano e acetato de etila,
sucessivamente.
A fase aquosa obtida da extração com solução saturada de NaHC03 foi
acidificada e extraída com diclorometano, sendo obtidas duas fases: uma
aquosa e outra orgânica. O solvente presente na fase orgânica foi evaporado, a
amostra foi seca em P205 , derivatizada e analisada por GC/MS (F03). A fase
aquosa foi extraída com acetato de etila, três vezes sucessivamente e foram

46
obtidas duas fases: uma aquosa (FA2) e outra orgânica. Dessa fase, o solvente
foi eliminado por rotaevaporação, a amostra seca em P20s, submetida à derivatização e analisada por GC/MS (F02).
O solvente da fase orgânica proveniente da extração com solução
saturada de NaHC03 e extraída com NaOH 2 mol.l," foi evaporado; a amostra
foi seca em P205, submetida à derivatização e analisada por GC/MS (F06). A
fase aquosa foi acidificada e extraída com diclorometano três vezes
sucessivamente. O solvente presente na fase orgânica obtida nessa extração
foi evaporado, a amostra seca em P205 submetida à derivatização e analisada
por GC/MS (FOS). A fase aquosa foi extraída com acetato de etila, três vezes
sucessivamente e separadas duas fasés: uma aquosa (FA3) e outra orgânica,
da qual o solvente foi evaporado, a amostra seca em P20s, submetida à derivatização e analisada por GC/MS (F03). As três frações aquosas (FA1,FA2
e FA3) separadas dos solventes não foram analisadas.
Esse procedimento teve por objetivo separar os compostos aromáticos
presentes no hidrolisado em frações ácida (F02 e F03), fenólica (F04 e FOS)
e neutra (F06) (TANAHASHI et ai, 1989). Para cada uma das frações
orgânicas (F01 - F06), foram feitas análises de GC/MS, conforme descrito no
item 3.8.2. As frações F01, F02, F04 e F06 foram também analisadas por
FTIR, conforme descrito no item 3. 7.
3.7. ANÁLISE POR ESPECTROSCOPIA NO INFRAVERMELHO
As amostras de carboidratos e de compostos de lignina, obtidas no item
3.6.1 e as frações orgânicas F01, F02, F04 e F06, obtidas no 3.6.2, foram
previamente secas em dessecador com pentóxido de fósforo sob vácuo
durante 3 a 4 dias. Foram preparadas pastilhas de KBr contendo 0,5% da
amostra compactadas a 1 O - 12 kgf/cm2 sob vácuo. Os espectros foram
varridos na região de 4.000 a 400 cm", número de scans = 64 e resolução = 4 cm", em um espectrofotômetro FTIR Nicolet 520.
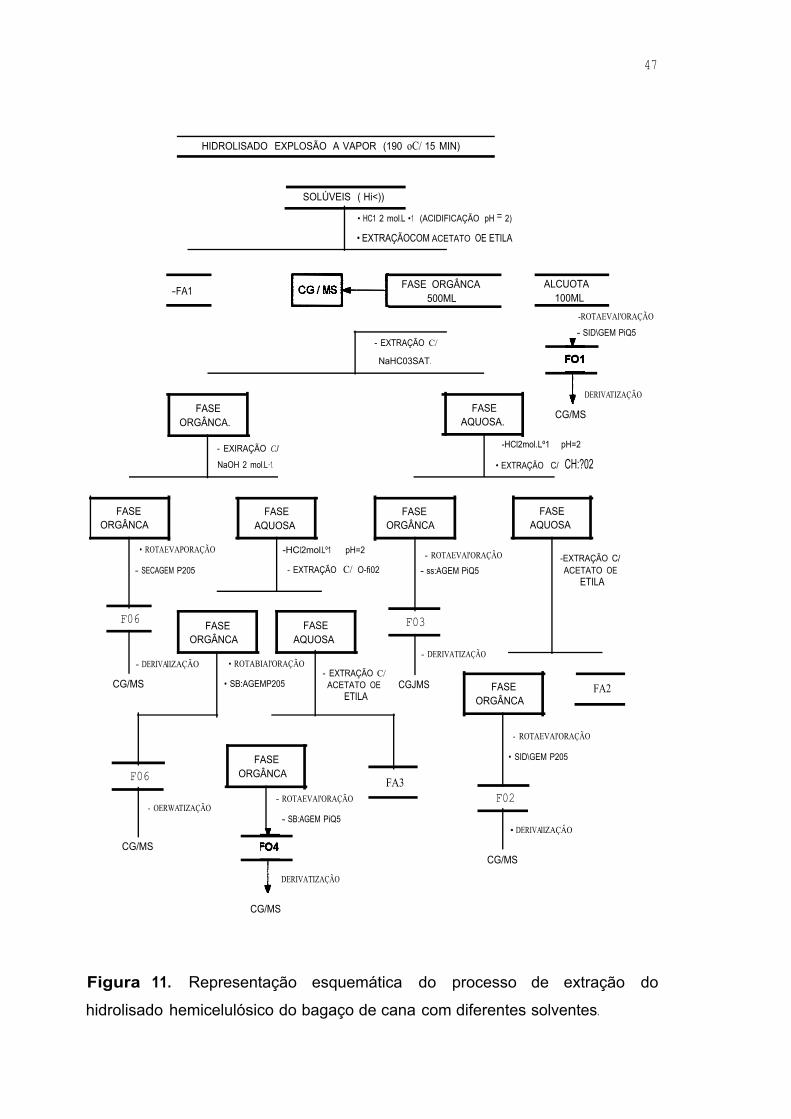
HIDROLISADO EXPLOSÃO A VAPOR (190 oC/ 15 MIN)
SOLÚVEIS ( Hi<))
• HC1 2 mol.L • 1 (ACIDIFICAÇÃO pH = 2)
• EXTRAÇÃO COM ACETATO OE ETILA
-FA1 FASE ORGÂNCA 500ML
- EXTRAÇÃO C/
NaHC03SAT.
FASE ORGÂNCA.
- EXIRAÇÃO CJ
NaOH 2 mol.L·1.
FASE ORGÂNCA
FASE AQUOSA
-HCl2mol.Lº1 pH=2
- EXTRAÇÃO C/ O-fi02
• ROTA EVAPORAÇÃO
- SECAGEM P205
F06 FASE ORGÂNCA
FASE AQUOSA
- EXTRAÇÃO C/ ACETATO OE CGJMS
ETILA FASE
ORGÂNCA
• ROTABIAl'ORAÇÃO
• SB:AGEMP205
- DERIVA llZAÇÃO
CG/MS
FASE AQUOSA.
-HCl2mol.Lº1 pH=2
• EXTRAÇÃO C/ CH:?02
FASE ORGÂNCA
- ROTAEVAl'ORAÇÃO
- ss:AGEM PiQ5
F03
- DERIVATIZAÇÃO
FASE ORGÂNCA F06
- ROTAEVAl'ORAÇÃO
- SB:AGEM PiQ5 - OERWA TIZAÇÃO
CG/MS
- DERIVATIZAÇÃO
CG/MS
FA3
47
ALCUOTA 100ML
-ROTAEVAl'ORAÇÃO
- SID\GEM PiQ5
F02
- DERIVA TIZAÇÃO
CG/MS
FASE AQUOSA
CG/MS
-EXTRAÇÃO C/ ACETATO OE
ETILA
FA2
- ROTAEVAl'ORAÇÃO
• SID\GEM P205
• DERIVA llZAÇÂO
Figura 11. Representação esquemática do processo de extração do
hidrolisado hemicelulósico do bagaço de cana com diferentes solventes.

48
3.8. IDENTIFICAÇÃO E QUANTIFICAÇÃO DE COMPOSTOS
AROMÁTICOS DE BAIXA MASSA MOLAR PRESENTES NO HIDROLISADO
A fração obtida pela dessorção (com etanol) dos compostos retidos na
série de cartuchos Sep-Pak C 18 (item 3.6.1) foi rotoevaporada, a amostra foi
seca em P20s, derivatizada conforme o item 3.8.1 e analisada por GC/MS (item
3.8.2).
Amostras das frações orgânicas F01, F02, F03, F04, F05 e F06,
obtidas no item 3.6.2, foram derivatizadas conforme o item 3.8.1 e analisada
por GC/MS (item 3.8.2). As frações orgânicas F01, F02, F03, F04, FOS e
F06 foram também submetidas a análise por GC/FID, conforme descrito no
item 3.8.3.
3.8.1. DERIVATIZAÇÃO DAS AMOSTRAS PARA ANÁLISES DE
GC/MS E GC/FID
Os compostos de baixa massa molar, presentes na fração dessorvida
da série de cartuchos C18 (item 3.6.1) e nas frações orgânicas (F01 - F06)
(item 3.6.2) foram identificados por GC/MS. Cerca de 2 mg de amostra de cada
fração acima citada foi rotoevaporada, seca em P205, pesada e dissolvida em
100 µL de piridina. A seguir foi adicionado 1 mL de N,0-bis(trimetilsilil)
trifluoroacetamida (BSTFA) às amostras. O frascos foram fechados e mantidos
a 60ºC por 1 h (COTRIM, 1990) e resfriados à temperatura ambiente. Uma
alíquota de 1 µLda mistura reacional foi injetada no cromatógrafo e foi diluída
por um fracionamento mecânico (split) na razão de 1 :100, usando hélio como diluente.
3.8.2. CROMATOGRAFIA GASOSA/ESPECTROMETRIA DE MASSAS
As amostras derivatizadas foram analisadas em um cromatógrafo de
ga~es acoplado a um detector de massas Finnigan, modelo MAT GCQ ™, calibrado com perfluorotributilamina (PFTBA) na faixa de varredura de m/z de

49
20 a 900. Os espectros foram medidos após ionização das moléculas por
impacto de elétrons (70 eV). Foi utilizada uma coluna cromatográfica 08-5 de
30 m x 0,25 mm. O fluxo de hélio usado foi de 33,0 cm/s. A fonte de ionização
foi aquecida a 190ºC, a interfase a 220ºC e a temperatura de transferência de
linha a 275ºC.
Para melhor separação dos compostos presentes nas amostras, foram
utilizadas três diferentes rampas de temperatura da coluna, de acordo com as
condições mostradas na tabela 4.
Tabela 4 . Programas de temperatura da coluna para análises das frações
orgânicas nas análises de GC/MS e GC/FID.
AMOSTRAS INICIAL INTERMEDIARIA FINAL
Temp. Tempo Temp. Tempo Taxa Temp. Tempo Taxa (ºC) (min) (ºC) (min) (ºC/min) (ºC) (min) (ºC/min)
F02, F04, 60 1 140 2 15 280 10 2
F06
F01 80 1 140 4 4 280 25 4
F03, F05 60 1 140 3 30 240 13.33 2
A identificação dos compostos foi feita por comparação dos espectros de
massa obtidos com aqueles de bibliotecas de espectros instalados no
equipamento de GC/MS, bem como por proposições de fragmentações lógicas,
conforme mostrado no apêndice 1.
3.8.3. CROMATOGRAFIA GASOSA/DETECTOR DE IONIZAÇÃO DE CHAMA
As amostras foram analisadas em um cromatógrafo de gases acoplado a
um detector de ionização de chama (FIO), modelo Varian 3400 CX. Foi
utilizada uma coluna cromatográfica DB-5 de 30 m x 0,25 mm. O fluxo de hélio
usado foi de 33,0 cm/se split 1:100. Para quantificar compostos fenólicos, o
fluoranteno foi utilizado como padrão interno e o programa de temperatura da
coluna foi o mesmo usado nas análises de GC/MS (item 3.8.2).

50
A quantificação por GC/FID dos compostos identificados por GC/MS foi
feita por cromatografia gasosa utilizando-se a mesma coluna cromatográfica.
No entanto, a detecção foi feita empregando-se um detector de ionização de
chamas (FIO) para as frações orgânicas derivatizadas com BSTFA. Nesse
detector, as respostas de compostos de diferentes classes são parecidas, o
que não acontece no detector de massas, onde a resposta é mais dependente
da estrutura do composto (GUERRA, 1998).
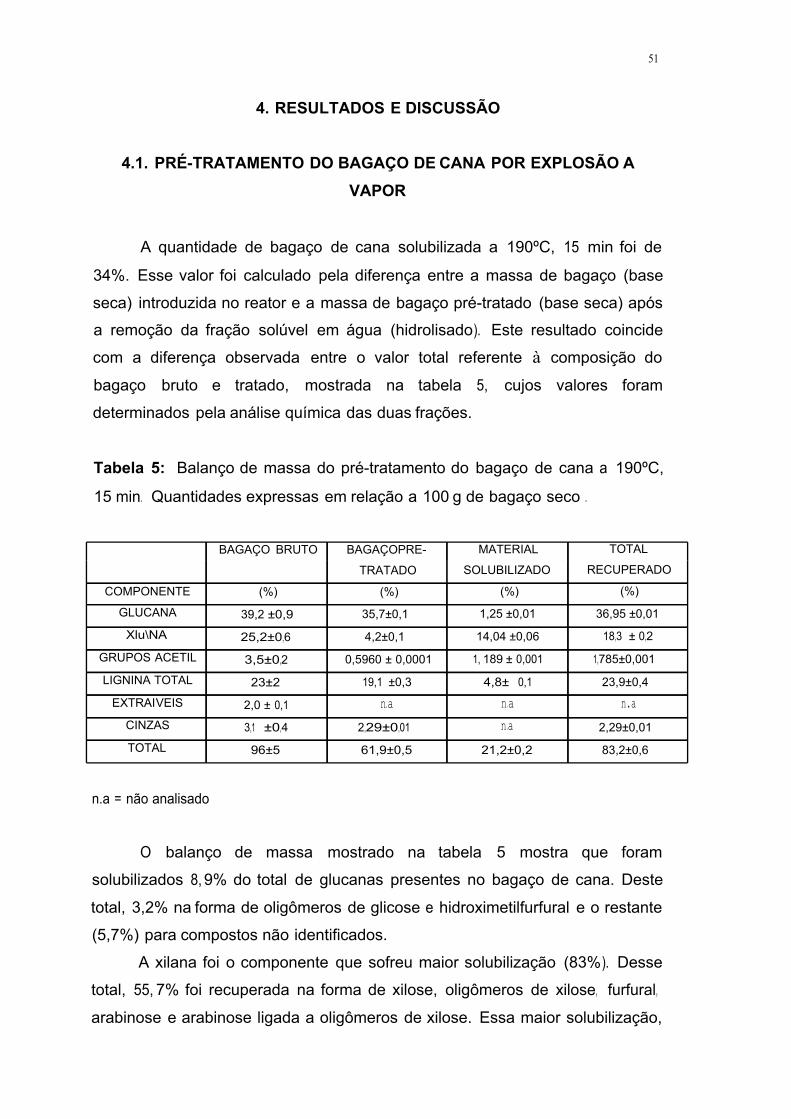
51
4. RESULTADOS E DISCUSSÃO
4.1. PRÉ-TRATAMENTO DO BAGAÇO DE CANA POR EXPLOSÃO A VAPOR
A quantidade de bagaço de cana solubilizada a 190ºC, 15 min foi de
34%. Esse valor foi calculado pela diferença entre a massa de bagaço (base
seca) introduzida no reator e a massa de bagaço pré-tratado (base seca) após
a remoção da fração solúvel em água (hidrolisado ). Este resultado coincide
com a diferença observada entre o valor total referente à composição do
bagaço bruto e tratado, mostrada na tabela 5, cujos valores foram
determinados pela análise química das duas frações.
Tabela 5: Balanço de massa do pré-tratamento do bagaço de cana a 190ºC,
15 min. Quantidades expressas em relação a 100 g de bagaço seco .
BAGAÇO BRUTO BAGAÇOPRE- MATERIAL TOTAL
TRATADO SOLUBILIZADO RECUPERADO
COMPONENTE (%) (%) (%) (%)
GLUCANA 39,2 ±0,9 35,7±0,1 1,25 ±0,01 36,95 ±0,01
Xlu\NA 25,2±0,6 4,2±0,1 14,04 ±0,06 18,3 ± 0,2
GRUPOS ACETIL 3,5±0,2 0,5960 ± 0,0001 1, 189 ± 0,001 1,785±0,001
LIGNINA TOTAL 23±2 19,1 ±0,3 4,8± 0,1 23,9±0,4
EXTRAI VEIS 2,0 ± 0,1 n.a n.a n.a CINZAS 3,1 ±0,4 2,29±0,01 n.a 2,29±0,01
TOTAL 96±5 61,9±0,5 21,2±0,2 83,2±0,6
n.a = não analisado
O balanço de massa mostrado na tabela 5 mostra que foram
solubilizados 8, 9% do total de glucanas presentes no bagaço de cana. Deste
total, 3,2% na forma de oligômeros de glicose e hidroximetilfurfural e o restante
(5,7%) para compostos não identificados.
A xilana foi o componente que sofreu maior solubilização (83% ). Desse
total, 55, 7% foi recuperada na forma de xilose, oligômeros de xilose, furfural,
arabinose e arabinose ligada a oligômeros de xilose. Essa maior solubilização,

52
também foi acompanhada de uma grande quantidade de compostos não
identificados (27,4%). Esses compostos são derivados de furfural, que nas
condições de reação tendem a sofrer reação de polimerização, formando
resinas furânicas e produtos de condensação com a lignina (FENGEL e
WEGENER, 1989).
Os grupos acetil também foram intensamente solubilizados (83,3%),
acompanhando a hidrólise das xilanas. A quantidade de grupos acetil
identificados na forma de ácido acético livre e grupos acetil ligados a
oligômeros de xilana foi de 34%. Uma grande fração dos grupos acetil não foi
identificada (48,9%).
A lignina sofreu uma solubilização apreciável (17%). Por outro lado, a
quantidade de lignina solúvel no hidrolisado foi de 20,9%. Estes resultados
mostram que a quantidade total de lignina recuperada foi de 103,9%, o que
demonstra que houve reações de condensação com os produtos de
degradação dos açúcares.
Esse pré-tratamento foi realizado nas mesmas condições usadas por
SILVA (1995), cujos resultados são apresentados na tabela 6.
Tabela 6: Balanço de massa do pré-tratamento do bagaço de cana a 190ºC, 15
min. Quantidades expressas em relação a 100 g de bagaço seco (SILVA,
1995). BAGAÇO BRUTO BAGAÇOPRE- MATERIAL TOTAL
TRATADO SOLUBILIZADO RECUPERADO
COMPONENTE (%) (%) (%) (%)
GLUCANA 43 ± 1 37,0±0,7 1,5 ± 0,2 38,4±0,9 XILANA 25 ± 1 6,8±0,2 16,2±0,6 23,0±0,8
GRUPOS ACETIL 2,8±0,8 0,33 ±0,05 2,1 ±0,5 2,4±0,6 LIGNINA TOTAL 23±2 18,30 ±0,04 4,6 0,2 22,9 ±0,2
EXTRAIVEIS 3,53 ±0,01 n.a n.a n.a CINZAS 1,5 ± 0,4 1,54 ±0,08 n.a 1,54 ±0,08 TOTAL 99±2 64 ± 1 24±2 88 ±3
n.a = não analisado
A comparação dos dados das tabelas 5 e 6 mostra que os dois
experimentos foram bastante reprodutíveis. A composição química do bagaço

53
de cana foi semelhante, exceto para o conteúdo de glicose, que variou de
39,2% para 43%. As composições do bagaço pré-tratado foram muito
próximas. A principal diferença se deve à quantidade de xilana, que variou de
4,2% para 6,8%. Quanto ao hidrolisado, a maior variação foi observada em
relação ao teor de grupos acetil, que variaram de 1,2% para 2, 1 %. O total de
material recuperado foi de 83,2% contra 88%, sendo a principal diferença sobre
o conteúdo de xilana (18,3% contra 23%). Nestas condições, aproximadamente
21 % da lignina inicial foi solubilizada, contra 20%.
A composição dos componentes do bagaço de cana pré-tratado é
mostrado na tabela 7.
Tabela 7: Composição do bagaço de cana pré-tratado a 190ºC por 15 min
comparada aos dados obtidos por SILVA (1995).
COMPONENTE COMPOSIÇAO (%) COMPOSIÇAO (%)
(SILVA, 1995)
GLUCANA 54,2±0,2 62,0 ±0,6
XILANA 6,4±0,2 7,3 ± 0, 1
GRUPOS ACETIL 0,9057 ± 0,0001 n.d LIGNINA SOLUVEL 0,87 ±0,04 1,19±0,06
LIGNINA INSOLUVEL 28,2±0,4 28,8 ±0,2
LIGNINATOTAL 29,1 ±0,4 30,0±0,2
EXTRAI VEIS n.a n.a CINZAS 3,48 ±0,02 1,51 ± 0,01
TOTAL 94,1 ±0,8 100,8 ±0,9
n.a = não analisado
n.d = não detectado
Os resultados referentes à composição do resíduo celulósico em ambos
os experimentos foram bastante reprodutíveis, com exceção da quantidade de
glucana.
A distribuição dos componentes presentes no hidrolisado do bagaço de
cana, para os pré-tratamentos citados acima, é mostrada na tabela 8.

54
Tabela 8: Distribuição dos compostos identificados no hidrolisado do pré-
tratamento por explosão a vapor a 190ºC por 15 min. Quantidades expressas
em relação a 100 g de bagaço seco.
COMPONENTE COMPOSIÇAO COMPOSIÇAO (%)
(%) (SILVA, 1995)
OLIGOMEROS DE GLICOSE 1,25±0,01 1,4 ± 0,2 HIDROXIMETILFURFURAL 0,077 ± 0,004 0,05 ±0,01 XILOSE 2,70 ±0,01 1,86±0,06 OLIGOMEROS DE XILOSE 10,67 ±0,03 12,8±0,6 ARABINOSE 0,543 ± 0,001 0,63 ±0,08 ARABINOSE LIGADA A OLIGOMEROS 0,13 ±0,01 S0,3 FURFURAL 0,55±0,03 0,71 ±0,01 ACIDO ACETICO 1,063±0,002 1,1 ±0,2 GRUPOS ACETIL LIGADO A OLIGOMEROS 0,2±0,1 1,0±0,7 LIGNINA SOLUVEL NO HIDROLISADO ANTES DA POS- 4,75 ±0,01 4,6±0,2 HIDRÓLISE
LIGNINA SOLUVEL NO HIDROLISADO DEPOIS DA POS- 2,6 ±0,1 1,7 ± 0, 1 HIDRÓLISE
LIGNINA INSOLUVEL NO HIDROLISADO DEPOIS DA n.a 3,0 ± 0,1 PÓS-HIDRÓLISE
TOTAL IDENTIFICADO* 22,0 ±0,3 24±2 TOTAL SOLUBILIZADO** 34,0 36,0 TOTAL SOLUBILIZADO PARA COMPOSTOS NAO 12,0 12,0 IDENTIFICADOS
n.a = não analisado
*valores calculados pela soma de todos os componentes, menos a quantidade de lignina
solúvel no hidrolisado depois da pós-hidrólise e da lignina insolúvel no hidrolisado depois da
pós-hidrólise
**calculado pela diferença, em relação à base seca, entre a massa de bagaço introduzida no
reator e a massa de bagaço pré-tratado após a remoção da fração solúvel em água
(hidrolisado)
Como pode ser visto, os resultados de oligômeros de glicose,
hidroximetilfurfural, furfural, arabinose, grupos acetil e lignina solúvel
apresentam a mesma magnitude dos resultados reportados por SILVA (1995).
A quantidade de oligômeros de xilose foi menor e variou de 10,67% para
12,8%, porém está dentro do erro experimental estimado para o processo.

55
No hidrolisado, 1,25% do bagaço solubilizado provieram da celulose,
14,04% das polioses, 1,189% dos grupos acetil ligados às polioses e 4,8% da
lignina (tabela 5). A celulose foi solubilizada como oligômeros de glicose
(1,25%) e hidroximetilfurfural (0,077%). As polioses foram solubilizadas como
arabinose livre (0,543%), arabinose ligada à oligômeros (O, 13%), xilose
(2,70%}, furfural (0,546%) e oligômeros de xilose (10,67%). Os grupos acetil
foram hidrolisados a ácido acético livre (1,063%) e parte permaneceu ligada a
oligômeros (0,2%) (tabela 8). A quantidade de bagaço hidrolisado a compostos
não identificados foi de 12,0%.
4. 2. DETERMINAÇÃO DE OLIGÔMEROS DE XILOSE E GLICOSE POR CLAE
Os principais produtos identificados no hidrolisado de bagaço de cana
pré-tratado por explosão a vapor foram oligômeros de xilose (10,67%) (tabela
6). A análise cromatográfica do hidrolisado, usando uma coluna apropriada
para separação dos oligômeros de açúcares (HPX-42A), produziu o
cromatograma mostrado na figura 12, cujo tempo de retenção de cada pico é mostrado na tabela 9.
RI
A
o 5 10 15 20 25
t (min)
Figura 12. Hidrolisado hemicelulósico obtido por explosão a vapor ( 190ºC/15
min) analisado na coluna Aminex HPX 42A, para a determinação de
oligômeros.

56
Tabela 9. Tempos de retenção dos açúcares identificados no hidrolisado
hemicelulósico obtido por explosão a vapor (190ºC/15 min) analisado na coluna
Aminex HPX 42A para a determinação de oligômeros.
Açúcar Tempo de Retenção
(min)
H 6,5
F 12,063
E 13, 171
D 14,075
e 15, 171
8 16,464
A 18,023
G 19,952
O cromatograma produzido pela injeção de uma amostra contendo
xilose, xilobiose e xilotriose é mostrado na figura 13, cujo os respectivos
tempos de retenção são mostrados na tabela 1 O.
RI 8
e A
o 5 10 15 20
Figura 13. Cromatograma de uma amostra padrão contendo xilose, xilobiose e xilotriose

57
Tabela 10. Cromatograma de uma amostra padrão contendo xilose, xilobiose e
xilotriose.
Açúcar Tempo de Retenção
(min)
e 15,269
B 16,5
A 17,982
Observou-se que nessa amostra padrão, o oligômero de xilose com grau
de polimerização 2 (xilobiose) apresenta maior concentração, seguido de
xilotriose e por fim, xilose.
O cromatograma produzido pela hidrólise de xilana de bétula é mostrado
na figura 14 e os respectivos tempos de retenção para os picos são mostrados
na tabela 11 .
RI A
8
o 5 10 15 20 25 t(min)
Figura 14. Xilana hidrolisada com H2SQ4 72% a 25º C e analisada na coluna
Aminex HPX-42A, em um detector de Índice de Refração.

58
Tabela 11. Tempos de retenção dos xilo-oligômeros obtidos por hidrólise de
xilana (25ºC/7min) e analisados na coluna Aminex HPX 42A.
Açúcar Tempo de Retenção
(min)
F 12,061
E 13.329
D 14,258
e 15,344
B 16,610
A 18,109
A comparação entre os tempos de retenção dos açúcares presentes na
amostra padrão, com os tempos de retenção dos açúcares presentes nos
hidrolisados (figura 12 e 14), permitiu atribuir os picos A, B e C à xilose,
xilobiose e xilotriose, respectivamente. Por extrapolação, os picos D, E e F
foram atribuídos à xilotetraose, xilopentaose e xitoexaose.
Para se confirmar que no cromatograma do hidrolisado, nenhum dos
picos atribuídos aos oligômeros de xilose estavam sobrepostos aos oligômeros
de glicose, foi preparada uma solução contendo oligômeros de glicose a partir
da hidrólise parcial de uma amostra de celulose cristalina, cujo cromatograma
está mostrado na figura 15.

59
RI
o 5 10 15 20
t (min)
Figura 15. Celulose hidrolisada com H2S04 72% a 45º C e analisada na coluna
Aminex HPX-42A, em um detector de Índice de Refração.
Observou-se a glicose pura em maior quantidade, seguido do seu
dímero e seus oligômeros, com o grau de polimerização variando de 1 a 6. Os
tempos de retenção de cada açúcar pode ser visto na tabela 12.
Tabela 12. Tempos de retenção dos gluco-0ligômeros obtidos por hidrólise de
celulose (45ºC/7min) e analisados na coluna Aminex HPX 42A.
Açúcar Tempo de Retenção
(min)
N 10,344
M 11,229
L 12,334
K 13,736
J 15,524
1 17,680
Os tempos de retenção mostraram-se bastante diferentes dos obtidos
nos cromatogramas contendo oligômeros de xilose. A ausência de oligômeros
de glicose é ainda reforçada pelos resultados mostrados na tabela 6, cuja
quantidade de oligômeros de glicose foi mínima (1,25%). Esses oligômeros

60
provavelmente podem ser atribuídos ao pico de alta massa molar com tempo
de retenção de 6,5 min (pico H).
Desta forma, a análise qualitativa do hidrolisado mostrou a presença de
oligômeros de xilose com grau de polimerização variando de 2 a 6, com
predominância de xilobiose e xilotriose, além de arabinose (figura 12).
A determinação quantitativa dos oligômeros de xilose não pôde ser
efetuada devido à ausência de padrões cromatográficos. Foi feita uma tentativa
usando uma curva padrão de xilose, assumindo-se que o índice de refração
dos oligômeros fosse próximo ao desse carboidrato. O balanço de massa
efetuado a partir do cromatograma obtido da hidrólise parcial de uma
quantidade conhecida de xilana de bétula (Sigma), mostrou que somente
58,5% da massa inicial foi reportada. Isto revela que o índice de refração dos
oligômeros é inferior ao da xilose e não pode ser usado na quantificação.
4.3. IDENTIFICAÇÃO DE COMPLEXOS LIGNINA-CARBOIDRA TO
PRESENTES NO HIDROLISADO DO PRÉ-TRATAMENTO DO BAGAÇO DE
CANA POR EXPLOSÃO A VAPOR
A distribuição da massa molar dos compostos presentes no
hidrolisado é mostrada na figura 16. O cromatograma exibe uma região de alta
massa molar, que equivale a 10% da área total do cromatograma e que foi
eluída entre 3,8 ml e 4,4 ml, correspondendo a uma faixa de 85 kg/mola 33
kg/mol. Uma região de massa molar intermediária, eluída entre 6 ml e 7 ml,
correspondendo a uma variação de 7195 g/mol a 2777 g/mol e outra de massa
molar mais baixa, eluída entre 7,9 e 8,8, correspondendo a massas entre
1297 g/mol e 500 g/mol. Nesta coluna o volume de exclusão (Vo) foi de 2,84 ml
e todas as frações do hidrolisado foram aluídas a volumes maiores que o Vo.

61
RI
3 6 9 12 T(min)
Figura 16. SEC da fração do hidrolisado filtrada somente em membrana, para
retirada de sólidos.
A distribuição da massa molar da fração do contendo os carboidratos e
que foi separada do hidrolisado por filtração em cartucho de extração sólida
Sep-Pak C18, é mostrada na figura 17. Esta fração possui massa molar mais
baixa e foi eluída entre 8 e 1 O ml, correspondendo a uma faixa de massa
molar variando de 1073 a 160 g/mol.
RI
o 2 4 6 8 10 12
Volume de eluição (ml)
Figura 17. SEC da fração do hidrolisado hemicelulósico filtrada em cartucho de
extração sólida Sep-Pak C18.

62
A distribuição de massa molar da fração contendo os compostos
aromáticos retidos no cartucho de extração sólida Sep-Pak C18 e que foi
posteriormente dessorvida com etanol é mostrada na figura 18 . Essa fração foi
eluída entre 4 e 8 ml, correspondendo a uma faixa que variou de 18 kg/mal a
2780 g/mol. A fração eluída entre 8,5 e 12 ml corresponde ao etanol (solvente
usado para dessorção dos compostos de lignina no cartucho C18).
RI
o 2 4 6 8 10 12
Elution Volume (ml)
Figura 18. SEC da fração do hidrolisado hemicelulósico retida em cartuchos de
extração sólida Sep-Pak C18 (Waters) e dessorvida com etanol.
A comparação dos cromatogramas mostrados nas figuras 16, 17 e 18
mostra que a maior parte da fração de massa molar compreendida entre
85 kg/mal a 33 kg/mol (que na figura 16 eluiu entre 3,8 ml e 4,4 ml) ficou
retida nos cartuchos de extração sólida Sep-Pak C 18 e portanto não foi
· detectada nos cromatogramas das figuras 17 e 18.
O fato de uma fração dos compostos presentes no hidrolisado ser
adsorvida em Sep-Pak C 18 indica o caráter aromático desses compostos.
Assim, a fração pode ser composta de derivados de lignina e/ou de complexos
lignina-carboidrato. Como essa fração foi detectada por índice de refração, a
hipótese de que ela seja composta também por complexos lignina-carboidrato
é reforçada.
A presença desses complexos decorre da associação íntima entre os
polissacarídeos e lignina da parede celular (LIN e DENCE, 1992) e esses
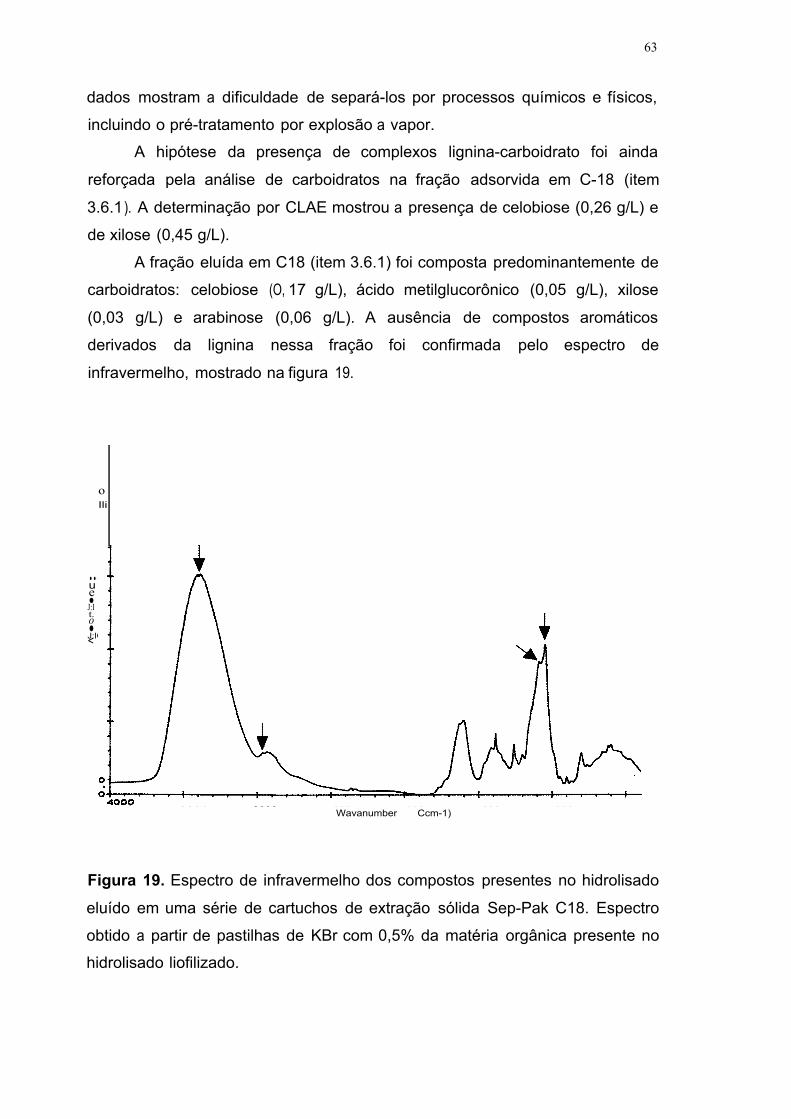
63
dados mostram a dificuldade de separá-los por processos químicos e físicos,
incluindo o pré-tratamento por explosão a vapor.
A hipótese da presença de complexos lignina-carboidrato foi ainda
reforçada pela análise de carboidratos na fração adsorvida em C-18 (item
3.6.1 ). A determinação por CLAE mostrou a presença de celobiose (0,26 g/L) e
de xilose (0,45 g/L).
A fração eluída em C18 (item 3.6.1) foi composta predominantemente de
carboidratos: celobiose (O, 17 g/L), ácido metilglucorônico (0,05 g/L), xilose
(0,03 g/L) e arabinose (0,06 g/L). A ausência de compostos aromáticos
derivados da lignina nessa fração foi confirmada pelo espectro de
infravermelho, mostrado na figura 19.
o Ili
.... u e • J:I t. o • J:IO < • ..
li
o
li
3eoo 3000 2eoo 2000 1eoo Wavanumber Ccm-1)
1000 eoo
Figura 19. Espectro de infravermelho dos compostos presentes no hidrolisado
eluído em uma série de cartuchos de extração sólida Sep-Pak C18. Espectro
obtido a partir de pastilhas de KBr com 0,5% da matéria orgânica presente no
hidrolisado liofilizado.

64
O espectro de infravermelho dessa fração mostrou bandas em
3418 cm", 2964cm-1 e 1094 - 1044 cm", atribuídas a absorção dos grupos
0-H, C-H e C-0, respectivamente. A comparação do espectro dessa fração
com o espectro de infravermelho de celulose e hemicelulose de eucalipto,
apresentados na figura 20 (MICHEL, 1988), mostra as semelhanças entre eles,
apresentando uma banda intensa em aproximadamente 1140 cm", típica
desses compostos. Além disso, no espectro da figura 19, a banda em
151 O cm", característica de compostos aromáticos derivados de lignina, está
ausente.
O espectro de infravermelho da fração do hidrolisado adsorvida em Sep-
Pak C 18 e dessorvida com etanol é apresentada na figura 21. Esse espectro
apresenta uma banda em 1510 cm", indicando a presença de compostos
aromáticos derivados da lignina, cujo espectro é mostrado na figura 22.
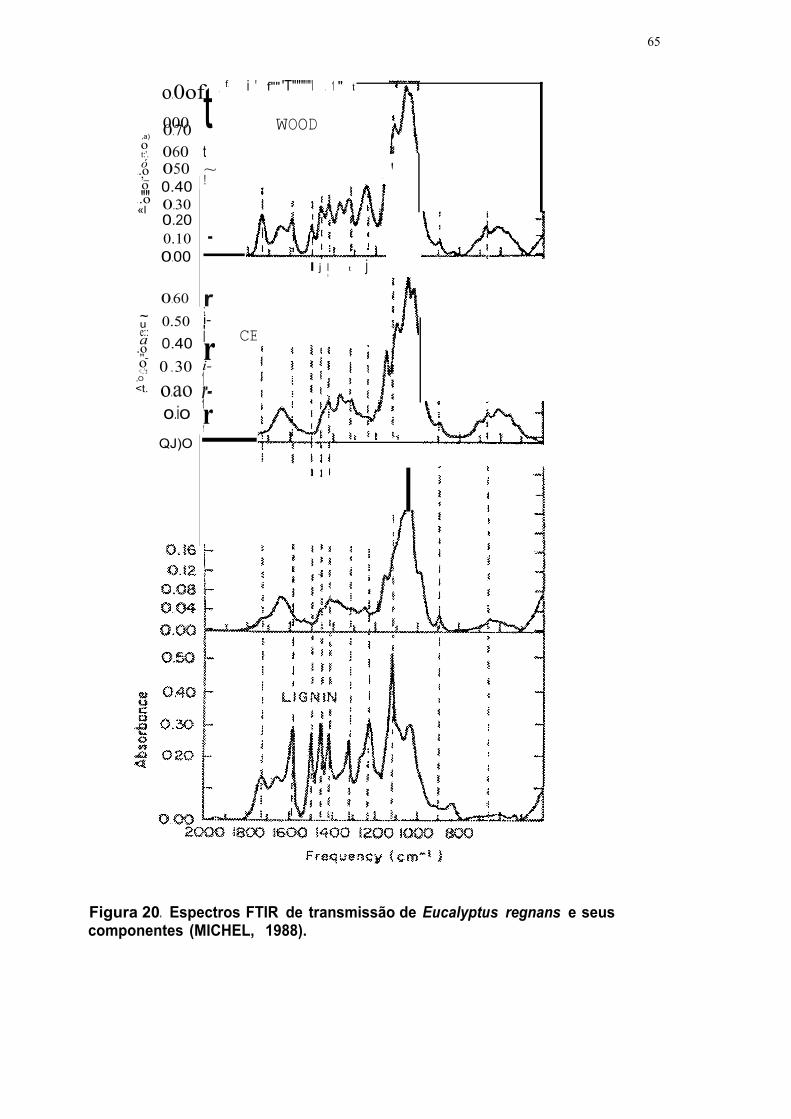
o.0of · f.
000 t ,a) 0.70 o 060 t t:'. ó 050 ~ . .o ,_
0.40 ! o "" . .o 0.30 «l
0.20 0.10 - 0.00
0.60 r ; ~ 0.50 i- u ' e:: l a 0.40 .o r "- o 0 .. 30 i- <..':t 1 ,O
<t. o.ao r- 1 o.io r !
QJ)O
0.32 0.28
tl1 (;} 0.24 e ~ 0.20 ,e, ..... o vi .cs 4
i ' f"" 'T"""""'l . 1 " t
WOOD
. '
l j I t j 1
l l l l j I l l I
I i I l l CELLULOSE
l 1 !
l l a l l I l l í I
HEM 1CELLULOSE 1 ! l
I l
O.iO
600
Figura 20. Espectros FTIR de transmissão de Eucalyptus regnans e seus componentes (MICHEL, 1988).
65
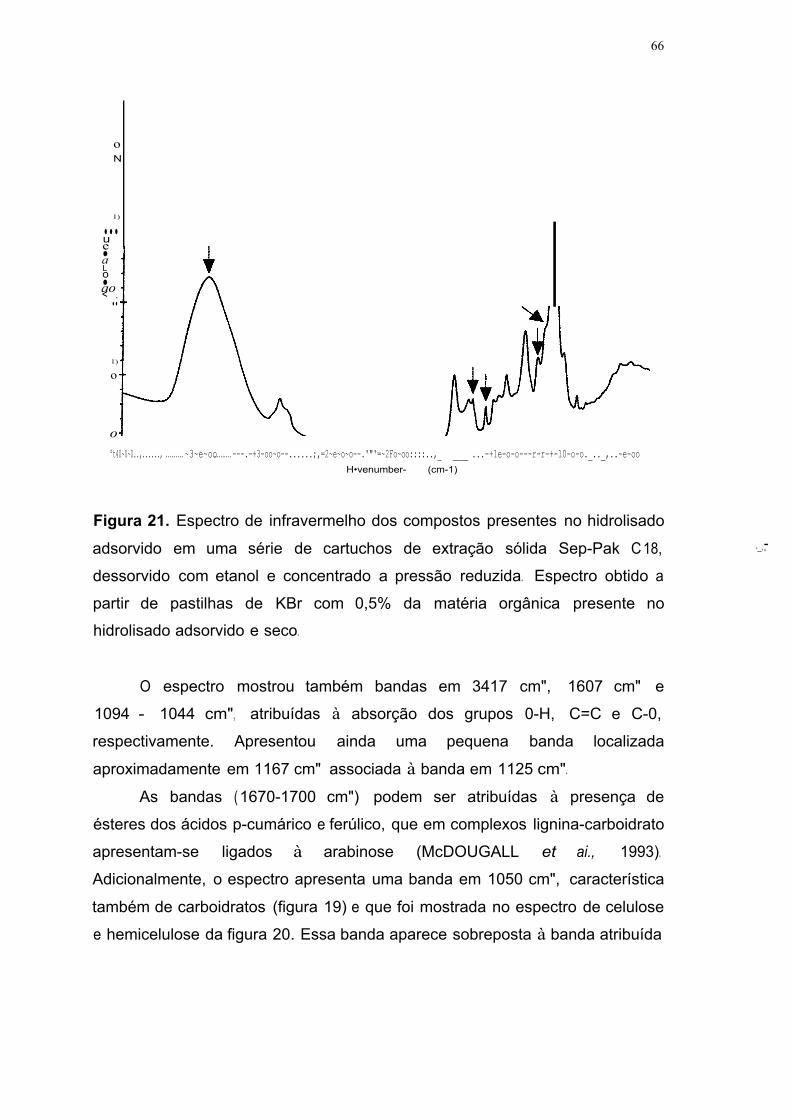
66
o N
1) ... u e • a L o • ao < . ..
1)
o
o ºt40~0~0..,......, ......... ~3~e~oo .......... ---.-+3-oo~o--......;,=2~e~o~o--.'"'=~2Fo~oo::::..,_ __ ...-+1e-o-o---r-r-+-10-o-o._.._,..~e-oo
H•venumber- (cm-1)
Figura 21. Espectro de infravermelho dos compostos presentes no hidrolisado
adsorvido em uma série de cartuchos de extração sólida Sep-Pak C 18,
dessorvido com etanol e concentrado a pressão reduzida. Espectro obtido a
partir de pastilhas de KBr com 0,5% da matéria orgânica presente no
hidrolisado adsorvido e seco.
- -_.,,
O espectro mostrou também bandas em 3417 cm", 1607 cm" e
1094 - 1044 cm", atribuídas à absorção dos grupos 0-H, C=C e C-0,
respectivamente. Apresentou ainda uma pequena banda localizada
aproximadamente em 1167 cm" associada à banda em 1125 cm".
As bandas ( 1670-1700 cm") podem ser atribuídas à presença de
ésteres dos ácidos p-cumárico e ferúlico, que em complexos lignina-carboidrato
apresentam-se ligados à arabinose (McDOUGALL et ai., 1993).
Adicionalmente, o espectro apresenta uma banda em 1050 cm", característica
também de carboidratos (figura 19) e que foi mostrada no espectro de celulose
e hemicelulose da figura 20. Essa banda aparece sobreposta à banda atribuída
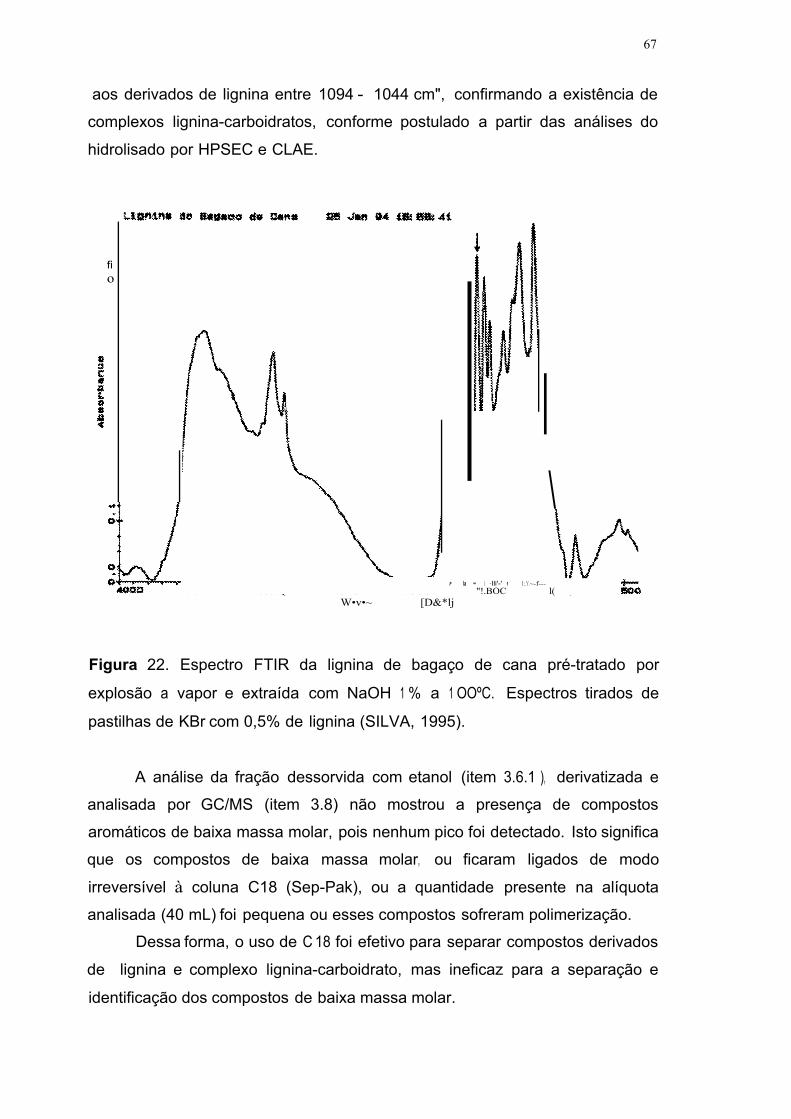
67
aos derivados de lignina entre 1094 - 1044 cm", confirmando a existência de
complexos lignina-carboidratos, conforme postulado a partir das análises do
hidrolisado por HPSEC e CLAE.
fi o
\ . ... •• ,, .• ., .... · ) · .. :-,..~'.'f". · _., P la •• 1 ·Ili'-' 1 1:.Y.~-f--- ............ -+--
3l900 SSOCQ aeno aooo "!.BOC l(:IO(I W•v•~ [D&*lj
Figura 22. Espectro FTIR da lignina de bagaço de cana pré-tratado por
explosão a vapor e extraída com NaOH 1 % a 1 OOºC. Espectros tirados de
pastilhas de KBr com 0,5% de lignina (SILVA, 1995).
A análise da fração dessorvida com etanol (item 3.6.1 ), derivatizada e
analisada por GC/MS (item 3.8) não mostrou a presença de compostos
aromáticos de baixa massa molar, pois nenhum pico foi detectado. Isto significa
que os compostos de baixa massa molar, ou ficaram ligados de modo
irreversível à coluna C18 (Sep-Pak), ou a quantidade presente na alíquota
analisada (40 mL) foi pequena ou esses compostos sofreram polimerização.
Dessa forma, o uso de C 18 foi efetivo para separar compostos derivados
de lignina e complexo lignina-carboidrato, mas ineficaz para a separação e
identificação dos compostos de baixa massa molar.

68
4.4. FRACIONAMENTO DOS COMPOSTOS PRESENTES NO HIDROLISADO POR EXTRAÇÃO LÍQUIDO-LÍQUIDO
O rendimento das diversas frações obtidas no procedimento de extração
líquido-líquido é mostrado na tabela 13.
Tabela 13. Rendimento das frações obtidas no procedimento de extração
líquido-líquido.
FRAÇAO QUANTIDADE (g) RENDIMENTO(%)
Bagaço solubilizado 3,4 34
Fração aquosa (FA1) 2,9463 29,5
Fração orgânica 1 (F01) 0,0907 0,9
Fração orgânica 2 (F02) 0,1372 g 1,4
Fração orgânica 3 (F03) 0,002 0,02
Fração orgânica 4 (F04) 0,087 g 0,87
Fração orgânica 5 (F05) 0,002 0,02
Fração orgânica 6 (F06) 0,2036 2,04
Fração aquosa (FA2) 0,37 3,7
Fração aquosa (FA3) 0,65 6,5
Do bagaço seco submetido à solubilização, cerca de 29,5% dos
compostos ficaram retidos na fração aquosa FA1 e 4,35% foram distribuídos
entre as frações ácidas, fenólicas e neutra (F02 - F06). Esse valor é muito
próximo do valor reportado pela lignina solúvel (4,8%), mostrado na tabela 5.
Portanto, os 29,5% presentes na fração FA1 devem englobar os carboidratos e grupos acetil presentes no hidrolisado (16,5%), além dos 12% de bagaço solubilizado para compostos não identificados (tabela 5), os quais foram
produzidos pelas reações de degradação do furfural e hidroximetilfurfural. O
fato dos compostos não identificados estarem solubilizados na fração aquosa,
indica que são compostos altamente polares, provavelmente ácidos carboxílicos (FENGEL e WEGENER, 1989).

69
O balanço de massa mostrado na tabela 13 indica ainda que a
quantidade de compostos presentes nas frações aquosas F A2. e FA3 devem
ser atribuídas ao NaCI produzido nas etapas de acidificação e neutralização
envolvidas no processo de extração líquido-líquido e não aos compostos
presentes no hidrolisado.
4.4.1. Cromatografia gasosa acoplada a espectroscopia de massas
Com o objetivo de identificar os compostos voláteis de baixa massa
molar presentes na solução de acetato de etila, proveniente da extração do
hidrolisado (item 3.6.2), uma pequena alíquota foi separada e analisada por
GC/MS sem derivatização prévia. Nessa fração, nenhum composto volátil foi
detectado, o que demonstra que os compostos voláteis como o furfural e o
hidroximetílfurfural provavelmente foram eluídos próximos do tempo de
retenção do solvente e portanto foram eliminados no intervalo de corte do
solvente.
Dessa forma, uma outra alíquota de 100 mL foi retirada e seca em P20s (F01 ), derivatizada e analisada por GC/MS e GC/FID. Os cromatogramas TIC
e FIO dessa fração são apresentados nas figuras 23 e 24, respectivamente. A
atribuição dos picos dos compostos identificados, bem como os respectivos
tempos de retenção, massa dos íons moleculares dos derivados sililados,
massa e a quantidade relativa dos compostos em relação à massa total da
fração são mostrados na tabela 14.
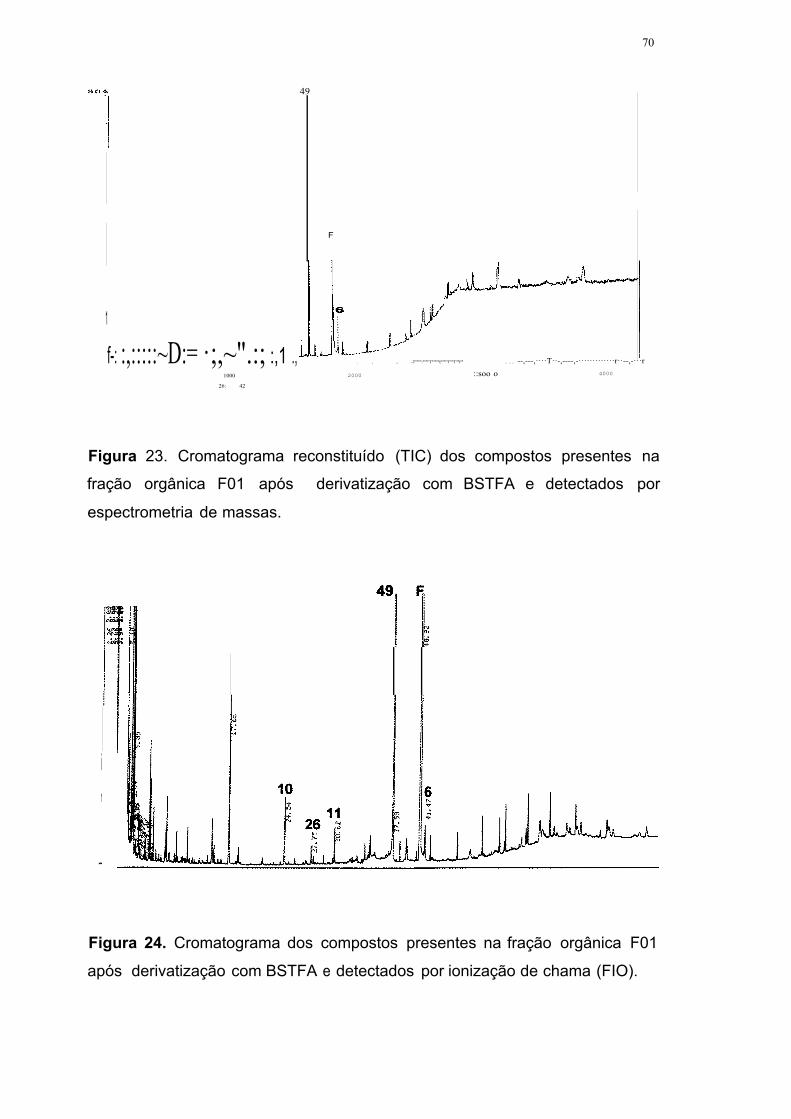
70
49
. , ., -T"""":""""T"""""t""""T'""T""l"" .. , .. --,---,····T··-,----,-·············r··--,-···r
F
1
f-: :,:::::~D:= ·;,~".:; :,1 ., 1000 2000 ::soo o 4000
26: 42
Figura 23. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica F01 após derivatização com BSTFA e detectados por
espectrometria de massas.
47 1
i 1
~
Figura 24. Cromatograma dos compostos presentes na fração orgânica F01
após derivatização com BSTFA e detectados por ionização de chama (FIO).
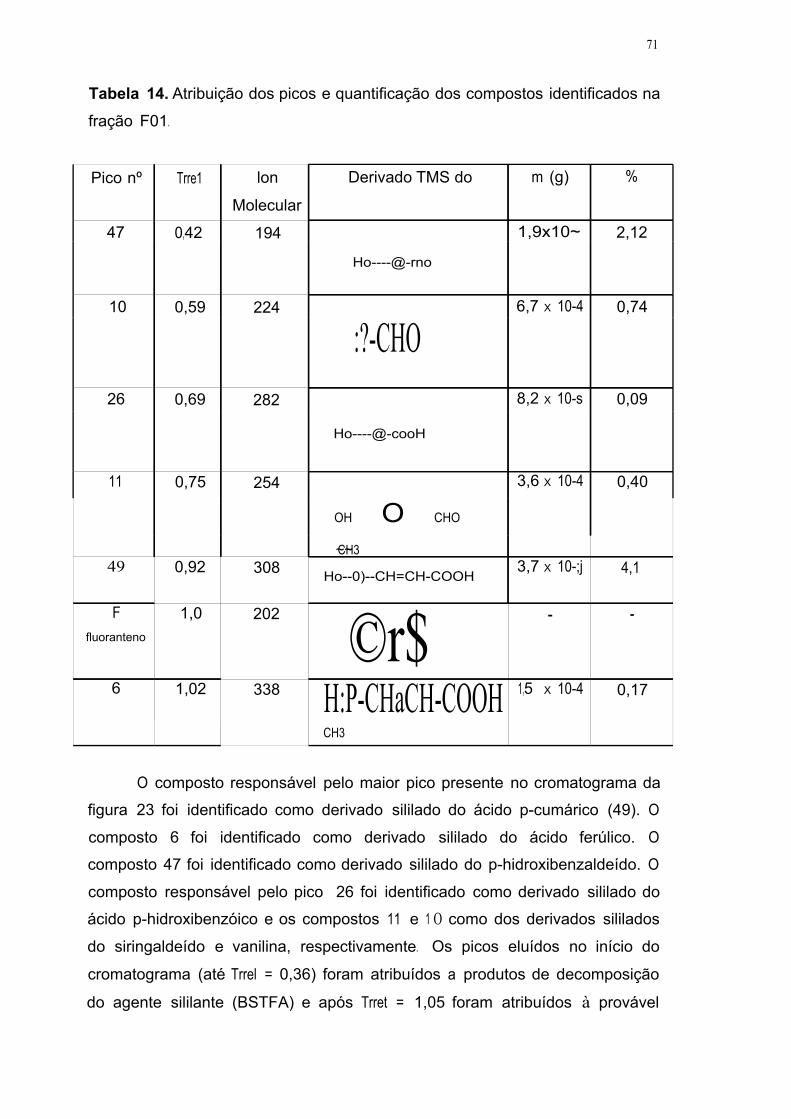
71
Tabela 14. Atribuição dos picos e quantificação dos compostos identificados na
fração F01.
Pico nº Trre1 lon Derivado TMS do m (g) %
Molecular
47 0,42 194 1,9x10~ 2,12
Ho----@-rno
10 0,59 224 6,7 X 10-4 0,74
:?-CHO 26 0,69 282 8,2 X 10-s 0,09
Ho----@-cooH
11 0,75 254
~~
3,6 X 10-4 0,40
OH O CHO
CH3 49 0,92 308 Ho--0)--CH=CH-COOH
3,7 X 10-;j 4,1
F 1,0 202 ©r$ - - fluoranteno
6 1,02 338 H:P-CHaCH-COOH 1,5 X 10-4 0,17
CH3
O composto responsável pelo maior pico presente no cromatograma da
figura 23 foi identificado como derivado sililado do ácido p-cumárico (49). O
composto 6 foi identificado como derivado sililado do ácido ferúlico. O
composto 47 foi identificado como derivado sililado do p-hidroxibenzaldeído. O
composto responsável pelo pico 26 foi identificado como derivado sililado do
ácido p-hidroxibenzóico e os compostos 11 e 1 O como dos derivados sililados
do siringaldeído e vanilina, respectivamente. Os picos eluídos no início do
cromatograma (até Trrel = 0,36) foram atribuídos a produtos de decomposição
do agente sililante (BSTFA) e após Trret = 1,05 foram atribuídos à provável

72
sangria de coluna. Todos os espectros de massas citados anteriormente estão
apresentados no Apêndice 1 .
Para a quantificação dos compostos identificados no GC/MS utilizou-se
uma calibração feita com fluoranteno (GONÇALVES, 1995) . Os resultados
obtidos na quantificação dos compostos dessa fração orgânica são mostrados
na tabela 14. O ácido cumárico foi o composto presente em maior quantidade
(4,1%). Além desse ácido cinamílico também foi detectado na amostra a
presença de pequena quantidade de ácido ferúlico (0, 17%). Esses ácidos
seriam provenientes da hidrólise dos ésteres correspondentes presentes nas
ligninas de gramíneas (FENGEL e WEGENER, 1989). O mesmo ocorre para o
ácido p-hidroxibenzóico (26). O composto 47 identificado como p-
hidroxibenzaldeído foi detectado em quantidade moderada (2, 12%). Esse
aldeído pode ter sido gerado na clivagem das ligações f3-éter das ligninas de
gramíneas (LIN e DENCE, 1992). Os compostos 11 e 1 O identificados como
siringaldeído e vanilina, respectivamente, foram detectados em pequenas
quantidades (0,40% e 0,74%) e foram encontrados como monômeros
proeminentes entre os produtos de degradação da lignina. HISHIYAMA e
SUDO (1989) postulam que esses aldeídos aromáticos são formados pela
clivagem de ligações éter existente entre grupos benzaldeído terminais e a
macromolécula de lignina ou a partir das reações hidrolíticas sobre as ligações
f3-0-4 de grupos terminais contendo grupos hidroxilas livres. TANAHASHI et ai.
( 1989) postulam a quebra homo lítica na posição ex de unidades fenilpropano
terminais.
Somente 7,62% da massa total da fração F01 (tabela 14) corresponde a
compostos identificados. Parte dos compostos volatilizados não puderam ser
identificados e a maior parte dos compostos restantes provavelmente são de
alta massa molar.
Os cromatogramas TIC e FIO da fração ácida (F02) obtida na extração
dos compostos solúveis em acetato de etila após extração com NaHC03,
diclorometano e acetato de etila são apresentados nas figuras 25 e 26,
respectivamente. A atribuição dos picos dos compostos identificados, bem
como os respectivos tempos de retenção, massa dos íons moleculares dos

73
derivados sililados, massa e quantificação relativa dos compostos em relação
à massa total da fração são mostrados na tabela 15.
····r···r•···,·-·,··r···,····r···,--··r-~·-r·--,----,···,····r···, .. -- r- --,--·-i-··r···r··,--------,--··r···,--------1-···r·-·1·'. ,~-r- • ~ººº 3000
~<, s o~
• • 1. ·~
Figura 25. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica F02 após derivatização com BSTFA e detectados por
espectrometria de massas.
,·-, 49 F. - l - . ....; "' ... ,, - ç-,j ,,i fi
6
26
15
Figura 26. Cromatograma dos compostos presentes na fração orgânica F02
após derivatização com BSTFA e detectados por ionização de chama (FIO).
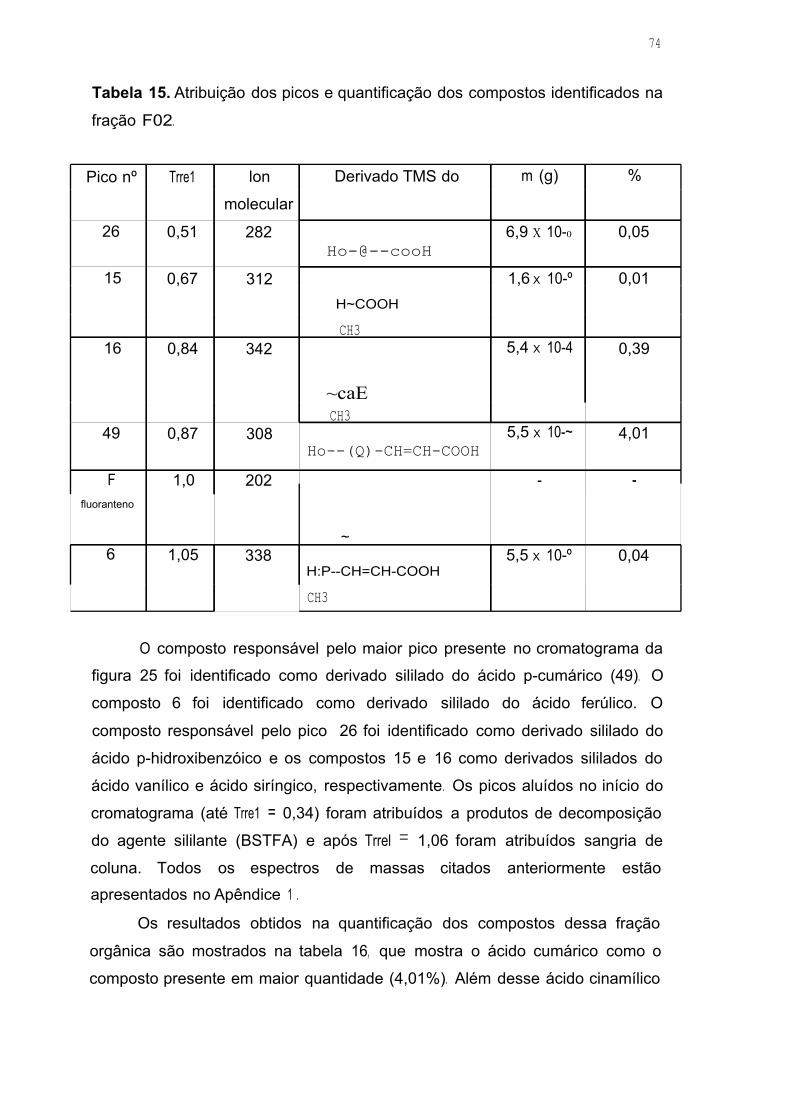
74
Tabela 15. Atribuição dos picos e quantificação dos compostos identificados na
fração F02.
Pico nº Trre1 lon Derivado TMS do m (g) %
molecular
26 0,51 282 Ho-@--cooH
6,9 X 10-0 0,05
15 0,67 312 1,6 X 10-º 0,01 H~COOH
CH3 16 0,84 342
~caE
5,4 X 10-4 0,39
CH3 49 0,87 308
Ho--(Q)-CH=CH-COOH 5,5 X 10-~ 4,01
F 1,0 202 - -
~
fluoranteno
6 1,05 338 H:P--CH=CH-COOH
5,5 X 10-º 0,04
CH3
O composto responsável pelo maior pico presente no cromatograma da
figura 25 foi identificado como derivado sililado do ácido p-cumárico (49). O
composto 6 foi identificado como derivado sililado do ácido ferúlico. O
composto responsável pelo pico 26 foi identificado como derivado sililado do
ácido p-hidroxibenzóico e os compostos 15 e 16 como derivados sililados do
ácido vanílico e ácido siríngico, respectivamente. Os picos aluídos no início do
cromatograma (até Trre1 = 0,34) foram atribuídos a produtos de decomposição
do agente sililante (BSTFA) e após Trrel = 1,06 foram atribuídos sangria de
coluna. Todos os espectros de massas citados anteriormente estão apresentados no Apêndice 1 .
Os resultados obtidos na quantificação dos compostos dessa fração
orgânica são mostrados na tabela 16, que mostra o ácido cumárico como o
composto presente em maior quantidade (4,01%). Além desse ácido cinamílico

75
também foi detectado na amostra a presença de pequenas quantidades do
ácido ferúlico (0,04%). O ácido p-hidroxibenzóico (composto 26) também foi
detectado em quantidades ínfimas (0,05%). Os compostos 15 e 16
identificados como ácido vanílico e ácido siríngico, respectivamente, foram
detectados em pequenas quantidades (0,01% e 0,39%) e podem ser formados
através da hidrólise ácida das ligações éter ou éster das ligninas de gramíneas
(LIN e DENCE, 1992).
Somente 4,5% da massa total da fração F02 (tabela 15) correspondem
a compostos identificados. Parte dos compostos volatilizados não puderam ser
identificados e a maior parte dos compostos restantes provavelmente são de
alta massa molar.
Os cromatogramas TIC e FIO da outra fração ácida (F03), obtida na
extração dos compostos solúveis em diclorometano após extração NaHC03,
são apresentados nas figuras 27 e 28, respectivamente. A atribuição dos picos
dos compostos identificados, bem como os respectivos tempos de retenção,
massa dos íons moleculares dos derivados sililados, massa e a quantidade
relativa dos compostos em relação à massa total da fração são mostrados na
tabela 16.

TOT·
76
49 63
1
F
""'º -re o c 24.f:00 'l.200
.:,o: 02
Figura 27. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica F03 após derivatização com BSTFA e detectados por
espectrometria de massas.
53
t. 1
r-
49
li 6
54 « N .s-
i 11
10 . ·;, ~'. ·~ -e ., ..,
:::i •'< "' :~, ·Õ 1
r, .: ::t •r. z; ,; u iio"> ... "' ... ,,
f 11 ,1 ?. ~· .... :2 -e
i',"" i' e::, (·1 .,
~ j ..: -·
Figura 28. Cromatograma dos compostos presentes na fração orgânica F03
após derivatização com BSTFA e detectados por ionização de chama (FIO).

77
Tabela 16. Atribuição dos picos e quantificação dos compostos identificados na
fração F03.
Pico nº Trrel íon Derivado TMS do m (g) %
molecular
53 0,36 246 Não identificado - -
10 0,40 224 O~CIIO
8,0x 10-4 0,40
CH30 11 0,58 254 :?-rno 7,2 X 10-o 0,36
54 0,68 308 Ho--@-CH=CH-COOH
6,2 X 10-o 0,51
49 0,87 308 Ho--@-CH=CH-COOH
5, 1 X 10-0 2,56
F 1,0 202
©x$ - -
fluoranteno
6 1,05 338 H:P--ffi,Q(.COQH 6,4 X 10-o 0,32
CH3
O composto responsável pelo maior pico presente no cromatograma da
figura 27 foi identificado como derivado sililado do ácido p-cumárico (49). O
tempo de retenção dos compostos 49 e 54 são diferentes. No entanto, o
caminho de fragmentação dos dois compostos são idênticos, sendo atribuído
ao composto 54 o isômero eis do ácido p-cumárico (isômeros eis e trans). O
composto 53 não foi identificado. O composto 6 foi identificado como derivado
sililado do ácido ferúlico. Os compostos 1 O e 11 foram identificados como
derivados sililados da vanilina e siringaldeído, respectivamente. Os picos
aluídos no início do cromatograma (até Trre1 = 0,29) foram atribuídos a produtos
de decomposição do agente sililante (BSTFA) e após Trre1 = 1,06 foram

78
atribuídos à sangria de coluna. Todos os espectros de massas citados
anteriormente estão apresentados no Apêndice 1 .
Os resultados obtidos na quantificação dos compostos dessa fração
orgânica são mostrados na tabela 16, que mostra o ácido cumárico como o
composto presente em maior quantidade (2,56%). Além desse ácido cinamílico
também foi detectado na amostra a presença de uma quantidade muito
pequena de ácido ferúlico (0,32%). Os compostos 10 e 11 identificados como
vanilina e siringaldeído, respectivamente, foram detectados em pequenas
quantidades (0,01% e 0,39%).
Somente 3,95% da massa total da fração F03 (tabela 16) correspondem
a compostos identificados. Parte dos compostos volatilizados não puderam ser
identificados e a maior parte dos compostos restantes provavelmente são de
alta massa molar.
As frações F02 e F03 foram obtidas com o intuito de separar os ácidos
carboxílicos presentes no hidrolisado. No entanto, a quantidade (0,002 g) de
compostos extraídos com diclorometano (F03) foi muito menor do que da
fração (F02) extraída com acetato de etila (O, 1372 g).
Os ácidos trans-cumárico e ferúlico são compostos comuns às duas
frações. Entretanto, na fração F02 o ácido ferúlico e o ácido trans-cumárico
aparecem em quantidades 10 e 100 vezes maiores que na fração F03,
respectivamente.
Esses resultados mostram também que o ácido cis-cumárico não foi
solúvel em acetato de etila, pois aparece apenas na fração F03. O mesmo
acontece com o siringaldeído e com a vanilina. Por outro lado, os resultados
mostram ainda que os três ácidos carboxílicos (ácido p-hidroxibenzóico, ácido
vanílico e ácido siríngico) foram solubilizados apenas na fração extraída com acetato de etila (F02).
Os cromatogramas TIC e FIO da fração fenólica F04, obtida na extração dos compostos solúveis em acetato de etila após extração com NaOH 2N e
diclorometano são apresentados nas figuras 29 e 30, respectivamente. A
atribuição dos picos dos compostos identificados, bem como os respectivos
tempos de retenção, massas íons moleculares dos derivados sililados, massa e
quantidade relativa dos compostos em relação à massa total da fração são mostrados na tabela 17.
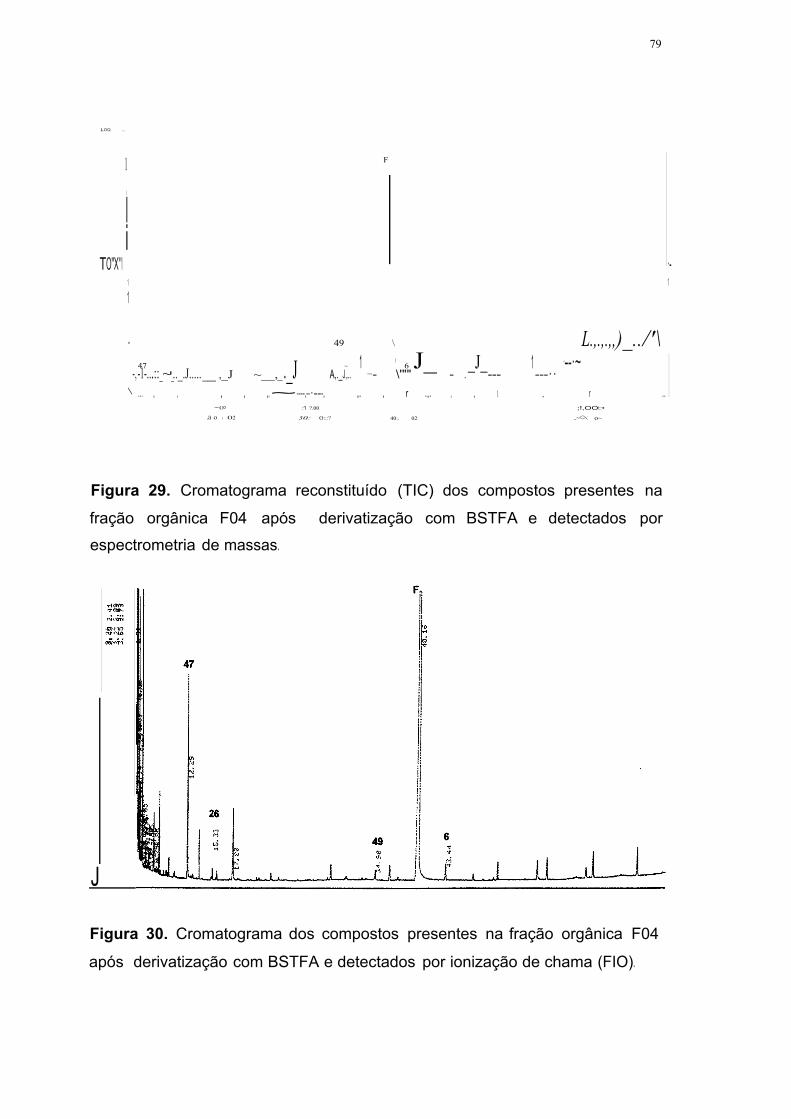
79
LOQ ...
1 1
l i
F
TO"X'''I ·- 1 1 1
\
• 49 \ L.,.,.,,)_../'\ 47 ~ 1 1
6 J_ _J_ 1 ·--·~ -, -1- .... .::_~_ • .._.J..... __ ,_J ~__, __ ._J A,._J,.. ···- \'"" - . --- ---·· ... , , , , ,.~----,--·---, ,. , r .,. , , 1 , r ..
~()0 :'l ?.00 ;!,OOt::•
.. ~<>-: o~ .a o i 02 30: O:::? 40:. 02
Figura 29. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica F04 após derivatização com BSTFA e detectados por
espectrometria de massas.
J
Figura 30. Cromatograma dos compostos presentes na fração orgânica F04
após derivatização com BSTFA e detectados por ionização de chama (FIO).
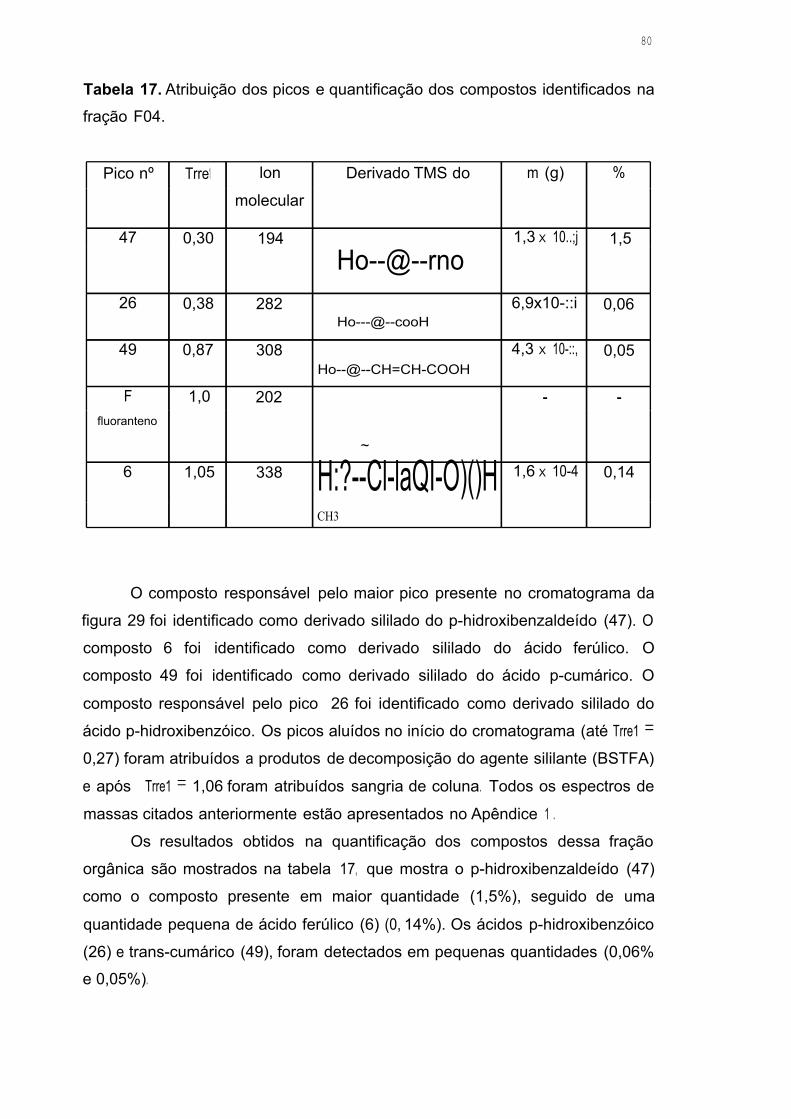
80
Tabela 17. Atribuição dos picos e quantificação dos compostos identificados na
fração F04.
Pico nº Trre1 lon Derivado TMS do m (g) %
molecular
47 0,30 194 1,3 X 10..;j 1,5 Ho--@--rno
26 0,38 282 Ho---@--cooH
6,9x10-::i 0,06
49 0,87 308 Ho--@--CH=CH-COOH
4,3 X 10-::, 0,05
F 1,0 202
~
- - fluoranteno
6 1,05 338 H:?--Cl-laQI-O)()H 1,6 X 10-4 0,14
CH3
O composto responsável pelo maior pico presente no cromatograma da
figura 29 foi identificado como derivado sililado do p-hidroxibenzaldeído (47). O
composto 6 foi identificado como derivado sililado do ácido ferúlico. O
composto 49 foi identificado como derivado sililado do ácido p-cumárico. O
composto responsável pelo pico 26 foi identificado como derivado sililado do
ácido p-hidroxibenzóico. Os picos aluídos no início do cromatograma (até Trre1 = 0,27) foram atribuídos a produtos de decomposição do agente sililante (BSTFA)
e após Trre1 = 1,06 foram atribuídos sangria de coluna. Todos os espectros de
massas citados anteriormente estão apresentados no Apêndice 1 .
Os resultados obtidos na quantificação dos compostos dessa fração
orgânica são mostrados na tabela 17, que mostra o p-hidroxibenzaldeído (47)
como o composto presente em maior quantidade (1,5%), seguido de uma
quantidade pequena de ácido ferúlico (6) (0, 14%). Os ácidos p-hidroxibenzóico
(26) e trans-cumárico (49), foram detectados em pequenas quantidades (0,06%
e 0,05%).

81
Somente 1,75% da massa total da fração F04 (tabela 17) correspondem
a compostos identificados. Parte dos compostos volatilizados não puderam ser
identificados e a maior parte dos compostos restantes provavelmente são de
alta massa molar.
Os cromatogramas TIC e FIO da outra fração fenólica (FOS), obtida na
extração dos compostos solúveis em diclorometano após extração com NaOH
2 mol.L" são mostrados nas figuras 31 e 32, respectivamente. A atribuição dos
picos dos compostos identificados, bem como os respectivos tempos de
retenção, massas íons moleculares dos derivados sililados, massa e
quantidade relativa dos compostos em relação à massa total da fração são
mostrados na tabela 18.

82
'"º-~, i3 1
F
49
1 ~
l t.:«: ,. .•• , •• -·,· ..• y .. ,~K.c, ... 1lJ .... J ... , .... 1\ .. 1 .. r····~·······) ..•. =,····,J=·'i·.·.,cc,~CC ··i r:»; .: ,,... r: , ':.: '. ...... oo 11:100
4 C) ~ ô?.
2400 e o a O:..?
Figura 31. Cromatograma reconstituído (TIC) dos compostos presentes na
fração orgânica FOS após derivatização com BSTFA e detectados por
espectrometria de massas.
Figura 32. Cromatograma dos compostos presentes na fração orgânica FOS
após derivatização com BSTFA e detectados por ionização de chama (FIO).
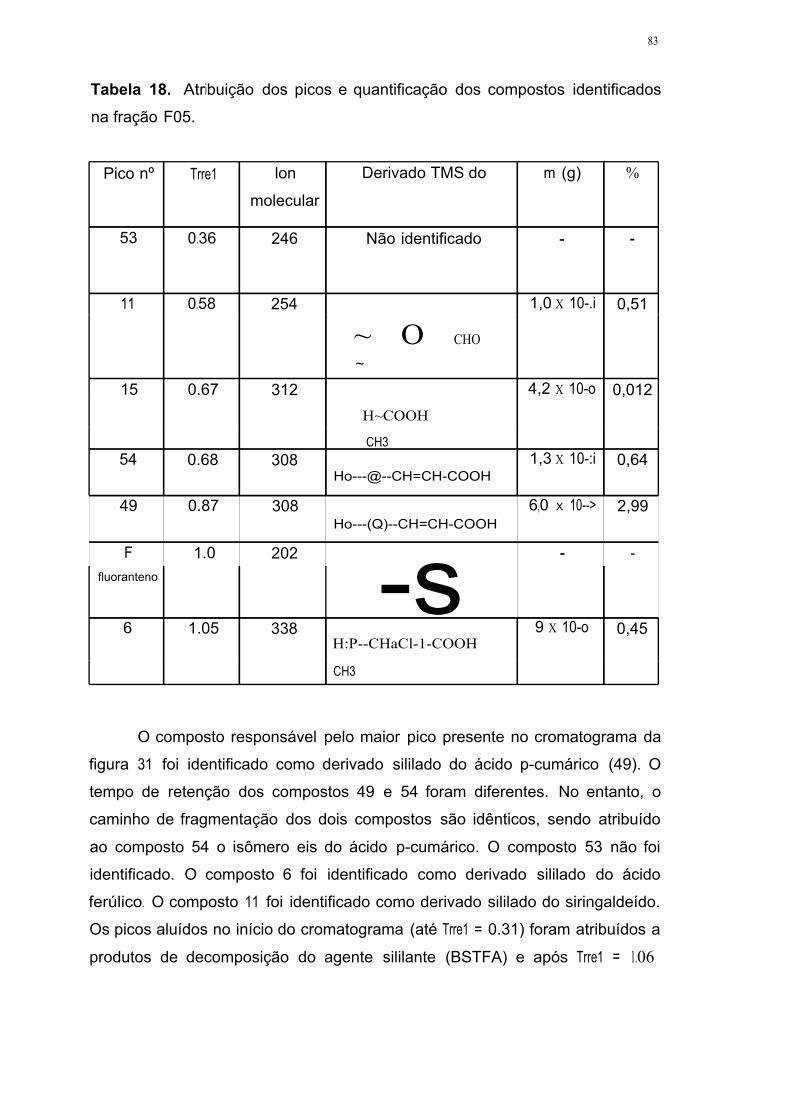
83
Tabela 18. Atribuição dos picos e quantificação dos compostos identificados
na fração F05.
Pico nº Trre1 lon Derivado TMS do m (g) %
molecular
53 0.36 246 Não identificado - -
11 0.58 254
~
1,0 X 10-.i 0,51
~ O CHO
15 0.67 312 4,2 X 10-o 0,012 H~COOH
CH3 54 0.68 308
Ho---@--CH=CH-COOH 1,3 X 10-:i 0,64
49 0.87 308 Ho---(Q)--CH=CH-COOH
6,0 X 10--> 2,99
F 1.0 202 -s - - fluoranteno
6 1.05 338 H:P--CHaCl-1-COOH
9 X 10-o 0,45
CH3
O composto responsável pelo maior pico presente no cromatograma da
figura 31 foi identificado como derivado sililado do ácido p-cumárico (49). O
tempo de retenção dos compostos 49 e 54 foram diferentes. No entanto, o
caminho de fragmentação dos dois compostos são idênticos, sendo atribuído
ao composto 54 o isômero eis do ácido p-cumárico. O composto 53 não foi
identificado. O composto 6 foi identificado como derivado sililado do ácido
ferúlico. O composto 11 foi identificado como derivado sililado do siringaldeído.
Os picos aluídos no início do cromatograma (até Trre1 = 0.31) foram atribuídos a
produtos de decomposição do agente sililante (BSTFA) e após Trre1 = 1.06

84
foram atribuídos sangria de coluna. Todos os espectros de massas citados
anteriormente estão apresentados no Apêndice 1 .
Os resultados obtidos na quantificação dos compostos dessa fração
orgânica são mostrados na tabela 18. O ácido trans-cumárico foi o composto
presente em maior quantidade (2,99%), seguido de seu isômero eis (0,64%).
Além desses ácido cinamílicos, também foi detectada na amostra a presença
de uma quantidade pequena de ácido ferúlico (0,45%). O siringaldeído (11) foi
detectado em pequena quantidade (0,51 %), bem como o ácido vanílico (15)
(0,012%).
Apenas 4,8% da massa total da fração F05 (tabela 18) corresponde a
compostos identificados. Parte dos compostos volatilizados não puderam ser
identificados e a maior parte dos compostos restantes provavelmente são de
alta massa molar.
As frações F04 e F05 foram obtidas com o intuito de separar os fenóis
presentes no hidrolisado. No entanto, a quantidade (0,002 g) de compostos
extraídos com diclorometano (F05) foi bem menor do que a fração F04,
extraída com acetato de etila (0,0866 g).
Os ácidos trans-cumárico e ferúlico foram compostos comuns às duas
frações. Entretanto, na fração F04, o ácido ferúlico aparece em quantidades 18
vezes maiores que na fração F05, enquanto a quantidade de ácido trans-
cumárico foi praticamente a mesma nas duas frações.
Esses resultados mostram também que o ácido cis-cumárico não é solúvel em acetato de etila, aparecendo apenas na fração F05. O mesmo
ocorre com o siringaldeído (11) e com o ácido vanílico (15). Por outro lado, o
ácido p-hidroxibenzóico (26) e o p-hidroxibenzaldeído (47) foram solubilizados
apenas na fração extraída com acetato de etila (F04).
Na fração orgânica F06, considerada fração neutra, nenhum composto
foi identificado. Os picos com Tretre1 = 10,50 min e Tretre1 = 11, 06 min são os
únicos picos que aparecem no cromatograma. Foi feita a injeção de um branco
com os solventes utilizados na análise da fração neutra e foi constatado que os
picos citados são derivados de impurezas contidas nos solventes.

85
F !
>.00 '>.
1 -ro,··1 1
1 ·.x :1
1 1 . J__.... . .1---"' J\· i . /_, ,t __ .L. ·- ...1 L ...l l .. L. .J. _ .. L t . -~ L- __ L. _ _.,,,v- . •···,· · ·· ·--i···•···,---T"": , , , ~ , • , ~- · , , , 1 , ··•r·• • J 1· T ..,.. ••• , ••. ,. ' • r f , • ,
1000 :.2:000 3()0()
~0:02 4000
-4;~ 1 ~2
Figura 33. Cromatograma reconstituído (TIC) da fração orgânica 6 evaporada
a pressão reduzida e derivatizada com BSTFA.
4.4.2. Espectroscopia no Infravermelho
Amostras das frações F01, F02, F04 e F06 foram submetidas a
espectroscopia no infravermelho. Os espectros produzidos são mostrados nas
figuras 34 a 37, respectivamente. As frações F03 e FOS não foram produzidas
em quantidades suficientes para a realização dos espectros e foram usadas
exclusivamente para as análises de GC/MS.
Os espectros das frações F01, F02 e F04 são muito parecidos e
apresentam as características esperadas de compostos aromáticos. Os
espectros mostraram bandas em 3417 cm", 1607 cm" e 1094 - 1044 cm",
atribuídas a absorção dos grupos 0-H, C=C e C-0, respectivamente.
As bandas (1670-1700 cm") podem ser atribuídas à presença de ácidos
carboxílicos aromáticos, tais como, ácidos p-cumárico e ferúlico ou seus
ésteres.
A ausência de uma banda forte na região de 1050 cm", característica de
derivados de celulose e polioses foi observada, o que indica a ausência de
carboidratos nas mesmas frações.

li "'. o O-'- • li; "'. J o.i.
' ... "'.' o.i.
CII• .. ; .ô~ D ' e oJ : --=~ e.o• D j :.; ,e OJ Ô+
J .j Oj ô.;.
l ... OJ ., o~
··i:foo .,,"°º ···············aooo···············eoõ······ . aóoo . S.1100 S.000 "ºº W•venutaDer- (c111-s.J
86
Figura 34. Espectro de infravermelho da fração fenólica F01 obtida por
extração com solventes do hidrolisado hemicelulósico de bagaço de cana.
Espectro obtido a partir de pastilhas de KBr com 0,5% da F01.
o oi '" ó-i.
j
i llj NJ o+
J j
ol 9 NJ ~ ci+ : . '- J ~ 114 .a .. J -e ci-i.
! • gÍ .. J
º:·
i 1
f- ... • ..-- r· r ..... r• •
SIIOO 3000 ... ,- , r· • · • r • ,.. t •·· r· r• ... ,... · •• •• ••·· ..... •
2IIOO aooo S.1100 lOOO 1100 W•v•nulllD•r (ca-ll
Figura 35. Espectro de infravermelho da fração ácida F02 obtida por extração
com solventes do hidrolisado hemicelulósico de bagaço de cana. Espectro
obtido a partir de pastilhas de KBr com 0,5% da F02.

m·
• o . e • . . a,,~ r. •. oo.:. . . ~ ;
o-
87
1•·· .- f·· .- · ,. ••· t· • • ~ · •· , , , r.. f- • r 51500 3000 2!500 ªººº ~1!100
wavenutllltar (cm-~> ... .. ' ~ººº
1-- • 1!100
Figura 36. Espectro de infravermelho da fração total F04 obtida por extração
com solventes do hidrolisado hemicelulósico de bagaço de cana. Espectro
obtido a partir de pastilhas de KBr com 0,5% da F04.
o'
• o . e • • a.,. e, •. ao ... • ~ .
;.. • • ,.. t .,.. r 4000 :91500
1
Figura 37. Espectro de infravermelho da fração neutra F06 obtida por extração
com solventes do hidrolisado hemicelulósico de bagaço de cana. Espectro
obtido a partir de pastilhas de KBr com 0,5% da F06.
'-,.. _ __..,-.,, . . . ' ' ,. ' . 21500 2000
W•venufflbar (c,n- ~)

88
4.4.3. Considerações finais sobre os compostos identificados por GC/MS
A quantidade total de cada composto identificado por GC/MS no
hidrolisado obtido do pré-tratamento do bagaço de cana por explosão a vapor é
mostrada na tabela 19.
Tabela 19. Quantidade total de cada composto identificado por GC/MS no
hidrolisado obtido do pré-tratamento do bagaço de cana por explosão a vapor.
Composto Massa (mg) [composto] (mg/L)
no hidrolisado*
1 - p-Hidroxibenzaldeído 3,2 5,5
2 - Acido p-hidroxibenzóico 0,19 0,33
4 - Ácido trans-cumárico 9,3 16,0
6 - Acido ferúlico 0,38 0,66
7 - Siringaldeído 0,38 0,66
8 - Acido cis-cumárico 1,9 X 10-:.! 3,3x10-;.:
9-Vanilina 1,5 2,60
1 O - Acido vanílico 2,0 X 10-2 3,4 X 10-:.!
11 - Acido siríngico 5,4 X 10-1 0.93
Total 15,5 27.0
* Cálculo efetuado em relação a 580 ml de hidrolisado.
O ácido cumárico (49) foi o principal produto identificado, seguido pelo p-
hidroxibenzaldeído (47), vanilina (10), ácido ferúlico (6), siringaldeído (11),
ácido p-hidroxibenzóico (26) e em quantidades bem menores, ácido siríngico
(16) e ácido vanílico (15), perfazendo um total de 15,5 mg. Esse valor
corresponde a 0,5% de toda a lignina presente no bagaço de cana e 3,2% da
quantidade total de lignina presente no hidrolisado.
A concentração dos compostos contendo grupo p-hidroxifenil foi de
22 mg/L, que foi aproximadamente 6, 7 vezes maior do que a concentração dos
compostos contendo grupo guaiacil (3,3 mg/l) e 14 vezes maior do que a
concentração dos compostos contendo grupo siringil (1,6 mg/l).

89
Dos compostos obtidos da extração líquido-líquido, os considerados
mais tóxicos para vários tipos de fermentação (TRAN e CHAMBERS, 1985;
FENSKE, 1998) foram identificados nesse estudo como derivados sililados dos
ácidos carboxílicos e aldeídos aromáticos. E não foram detectados álcoois e
dímeros.
O hidrolisado obtido no item 3.3 contém 17% de açúcares (3,4 g), que
para ser fermentado teria que ser concentrado por volta de 19 vezes
(McMILLAN, 1994b). Desta forma, os compostos tóxicos também poderiam ser
concentrados 19 vezes, produzindo um hidrolisado contendo esses compostos
em uma faixa de concentração que provavelmente deve contribuir para a
inibição de processos fermentativos, envolvendo o uso de xilose como
substrato.
Por exemplo, TRAN e CHAMBERS (1986a) concluíram que o ácido
siríngico é moderadamente inibitório a P.stipitis e K. pneumoniae quando
presente a níveis de 0,08 mg/l e 0,2-0,5 g/L, respectivamente, mas é
fracamente inibitório a Candida utilis a 4 g/l (MIKULÁSOVÁ et ai., 1990).
Todos esses compostos não são inibitórios a S. cerevisiae a 1.0 g/l (ANDO et
ai., 1986). Similarmente, P. stipitis, K. pneumoniae e C. utilis exibem mais
sensibilidade à vanilina que S. cerevisiae.
O ácido p-hidroxibenzóico mostrou efeito inibitório quando S. cerevisiae
foi utilizada na fermentação de madeira de álamo ou de resíduos agrícolas a
etanol (McMILLAN, 1994b) e p-hidroxibenzaldeído foi inibidor de S. cerevisiae
quando o hidrolisado de álamo foi usado para obter etanol. Ácido siríngico
inibiu a ação de P. sitipis quando os substratos usados foram hidrolisados de
carvalho vermelho, álamo e bétula. Ácido vanílico e vanilina foram inibidores da
mesma levedura quando o substrato usado foi carvalho vermelho na obtenção
de etanol. Siringaldeído foi inibidor de P. stipitis e S.cerevisiae quando
hidrolisados de carvalho vermelho e álamo foram usados como substrato para
fermentação, respectivamente.
Neste trabalho, o efeito inibitório dos compostos aromáticos identificados
no hidrolisado não foi estudado.
A identificação dos compostos aromáticos encontrados no hidrolisado
de bagaço de cana pré-tratado por explosão a vapor abre prespectivas para o
início de um trabalho, que leve em conta o efeito inibitório dos mesmos sobre

90
processos fermentativos de interesse industrial, usando xilose e xilana como substratos.

91
5. CONCLUSÕES
No hidrolisado, os principais componentes obtidos foram oligômeros de
xilose, com grau de polimerização (DP) variando de 2 a 6, com predominância
de xilobiose e xilotriose.
A distribuição de massa molar (HPSEC) dos compostos presentes no
hidrolisado mostrou que os carboidratos compõem a fração de menor massa
molar, enquanto a fração contendo lignina solúvel e complexos lignina-
carboidrato compõe a fração de massa molar média mais elevada.
O uso de C18 foi efetivo para a separação entre os compostos de baixa
massa molar e a fração contendo lignina solúvel e complexos lignina-
carboidrato, mas foi ineficaz para a separação e identificação de compostos de
baixa massa molar.
Os compostos de baixa massa molar foram isolados a partir do
fracionamento do hidrolisado por extração líquido-líquido, permitindo sua
identificação por CG/MS.
O ácido cumárico, principal composto identificado, é proveniente da
lignina e da estrutura de heteroxilana, enquanto p-hidroxibenzaldeído e vanilina
são compostos derivados da lignina.
Todos os compostos aromáticos identificados e quantificados são
considerados inibidores de vários processos fermentativos e enzimáticos.

6. REFERÊNCIAS BIBLIOGRÁFICAS
ARMAS, C.M.; BIANCHI, E. Aporte Energético da Indústria em Usina Sucro-
Alcooleira-Viabilidade Econômica. Stab 8, v. 5/6, p. 41-45, 1990.
ASTM Methods. D 1106-56. Standard test method for lignin in wood.
ATCHISON, J.E. Bagasse. ln: HAMILTON F., LEOPOLD, B. eds. Pulp and
Paper Manufacture: Secundary Fibers and Non-Wood Pulping. 3. ed. Atlanta:
The Joint Text Book Committee of Paper lndustry I Tappi I CPPA, v.3, p. 22-
70, 1993.
AVELLAR, B.K.; GLASSER, W.G. Steam-assisted biomass fractionation. 1. Process considerations and economic evaluation. Biomass and Bioenergy, v.
14, n. 3, p. 205-218, 1998.
BARNET, D.; EXCOFFIER, G.; VIGNON, M .. Valorisation de la biornasse
lignocellulosique: Autohydrolyse rapide de copeaux de bois de peuplier. Bull.
Soe. Chim. Fr.v. 6, p. 836-846, 1989.
BOUCHARD, J.; NGUYEN, T.S.; CHORNET, E.; OVEREND, R.P. Analytical
methodology for biomass pretreatment - Part 1 : Solid residues. Biomass,
Oxford, v. 23, p.243-261, 1990.
BOUCHARD, J.; NGUYEN, T.S.; CHORNET, E.; OVEREND, R.P .. Analytical
methodology for biomass pretreatment. Part 2: Characterization of the
filtrates and cumulativa product distribution as a function of treatment
severity. Bioresource Technol. v. 36, p. 121-131, 1991.
92

93
BUCHERT, J.; PULS, J.; POUTANEN, K. Comparison of Pseudomonas fragi
and Gluconobacter oxydans for production of xylonic acid from hemicellulose
hydrolysates. Applied Microbiologv and Biotechnologv, Berlim, v.28, p.367-
372, 1988.
BUCHERT, J.; PULS, J.; POUTANEN, K. The use of steamed hemicellulose as
substrate in microbial conversions. Applied Biochemistry and Biotechnologv,
New Jersey, v. 20/21, p.309-318, 1989.
CASTRO, F.8.; PAIVA, T.C.B.; ARCARO, I.J. Substitution of sugar cane
bagasse with steam-pretreated eucalyptus (Eucalyptus grandis): effects on
intake and growth rate of dairy heifers. Animal Feed Science and
Technology, Canada, v.52, p.93-100, 1995.
CLARK, T.A.; MACKIE, K.l. Fermentation inhibitors in wood hydrolysates
derived from the softwood Pinus radiata. J.Chem.Tech.Biotechnol. , New
Zealand, v.348, p.101-110, 1984.
CONVERSE, A.O.; WARE, W .. On the reactivity of cellulosic substrates in
enzymatic hydrolysis. ln: IEA/BIOFOR Workshop on applicatons of
Biotechnology in Bioenergy Systems 18-20 de Outubro, Ottawa, Canada,
1994.
COTRIM, A. Separação e caracterização de óleos provenientes da liquefação
direta de ligninas da hidrólise ácida de eucalipto. Campinas: UNICAMP/
Instituto de Química, 1990. 115p. (Tese de Doutorado).
DEKKER, R.F.H. Kinetic, inhibition, and stability properties of a commercial ~-
0-glucosidase ( cellobiase) preparation from Aspergi/Jus niger and its
suitability in the hydrolysis of lignocellulose. Biotechnology and
Bioengineering, New York, v.28, p.1438-1442, 1986.

94
DELGENES, J.P.; MOLETIA, R.; NAVARRO, J.M. Effects of lignocellulose
degradation products on ethanol fermentations of glucose and xylose by
Saccharomyces cerevisiae, Zymomonas mobilis, Pichia stipitis, and Candida
shehatae. Enzyme and Microbial Technology, New York, v.19, p.220-225,
1996.
DESCHAMPS, F.C.; RAMOS, L.P.; FONTANA, J.D. Pretreatment of sugar
cane bagasse for enhanced ruminai digestion. Applied Biochemistry and
Biotechnology, New Jersey, v. 57/58, p.171-182, 1996.
EXCOFFIER, G.;TOUSSAINT, B.; VIGNON, M.R. Saccharification of steam-
exploded poplar wood. Biotechnology and Bioengineering, New York, v.38,
p.1308-1317, 1991.
FENGEL, D.; WEGENER, G. Wood - Chemistry. Ultrastruture and Reactions.
Berlin: Walter de Gruyter, 1989, 613p.
FENSKE, J . .:J.; GRIFFIN, D.A; PENNER, M.H. Comparison of aromatic
monomers in lignocellulosic biomass prehydrolysates. Joumal of Industrial
Microbiology and Biotechnology. n. 20, p. 364-368, 1998.
FERRAZ, A.; MENDONÇA, R.; COTRIM, A.R.; TEIXEIRA DA SILVA, F. The
use of white - rot decay as a pretreatement for organosolv delignification of
Euca/yptus grandis wood. ln: INTERNATIONAL CONFERENCE ON
BIOTECHNOLLOGY IN THE PULP ANO PAPER INDUSTRY. Proceedings.
v.6, p. 1996, 221-224.
FOCHER, B.; MARZETII, A.; BEL TRAME, P.L.; AVELLA, M. Steam exploded
biomass for the preparation of conventional and advanced biopolymer-based
materiais. Biomass and Bioenergy, v. 14, n. 3, p. 187-194, 1998.
GLASSER, W.G.; WRIGHT, R.S. Steam-eassisted biomass fractionation. li.
Fractionation behavior of various biomass resources. Biomass and Bioenergy,
V. 14, n. 3, pp.219-235, 1998

95
GONÇALVES, A R. Oxidacão de ligninas e modelos de lignina com oxigênio
molecular em meio ácido. Campinas: UNICAMP/ Instituto de Química, 1995,
80p. (Tese de Doutorado).
GUERRA, A.A. Polimerizacão de lignina solúvel por polifenoloxidases e
peroxidases de rabanete/H202. Lorena: FAENQUIUDepartamento de
Biotecnologia, 1998. 99p. (Tese de Mestado).
HAHN-HAGERDAL, B. Ethanolic fermentation of lignocellulose hydrolysates.
Applied Biochemistry and Biotechnology, New Jersey, v.57/58, p.195-199,
1996.
HAHN-HAGERDAL, B.; LINDÉN, T.; SENAC, T.; SKOOG, K. Ethanolic
fermentation of pentases in lignocellulose hydrolysates. Applied Biochemistry
and Biotechnology, New Jersey, v.28/29, p.131-144, 1991.
HERRERO, A.A.; GOMEZ, R.F.; SNEDECOR, B.; TOLMAN, C.J.; ROBERTS,
M.F .. Appl. Biachem. Biotechnol.v. 22, n. 53, 1985.
HIGUCHI, T. Biosynthesis and biodegradation of wood components. Florida:
Academic Press, lnc., 1985, 679p.
HISHIYAMA, S.; SUDO, K. Degradation mechaninsm of lignin by steam-
explosion. ln: 6th ISWPC. Proceedings. 1989, p. 405-410.
HOL TZAPPLE, M.; COGNATA, M.; SHU, Y.; HENDRICKSON, e. lnhibition of
Trichoderma reesei cellulase by sugars and solvents. Biotechnology and
Bioengineering, New York, v.36, p.275-287, 1990.
ICIDCA. La industria de los derivados de la cana de azúcar. Editorial Científico-
T écnica. La Habana, 1987.

96
LACORTE, M.C.G.; SURGI, R.; LACORTE, A.J.F. Bagaço de cana hidrolisado
já utilizado em larga escala em confinamento. STAB, v. 5, n. 2, p. 43-52,
1986.
LAWFORD, H.G.; ROUSSEAU, J.D. Effects of pH and acetic acid on glucose
and xylose metabolism by a genetically engineered ethanologenic
Escherichia coli. Applied Biochemistry and Biotechnology, New Jersey,
v.39/40, p.301-322, 1993.
LEE, Y. Y.; McCASKEY, T.A. Hemicellulose hydrolysis and fermentation of
resulting pentases to ethanol. Tappi Journal, Atlanta, v. 66, n.5, p.102-107,
1983.
LIN, S.Y.; DENCE, C.W. Methods in lignin chemistry. Berlin: Springer-Verlag,
1992, 578p.
LORA, J.H.; WAYMAN, M. Delignification of hardwoods by autohydrolysis and
extraction. Tappi, Atlanta, v.61, n.6, p.47-50, 1978.
MARTIN, C.; WAHLBOM, F.; GALBE, M.; JÔNSSON, L.; HAHN-HAGERDAL,
B. Preparation of sugarcane bagasse hydrolysates for alcoholic fermentation
by yeasts. ln: 5TH BRAZ. SYMP. CHEM. LIGNINS ANO OTHER WOOD
COMPON, Guaratinguetá, SP, 25-28 october. Accepted for publication, 1999
MARTÍN, R.S.; AGUILERA, J.M. Steam pretreatment and enzymatic prodution
of D-glucose from Eucalyptus globulus. Biomass, v. 15, p. 281-289, 1988.
MARTÍN, R.S.; PEREZ, C.; BRIONES, R. Simultaneous production of ethanol
and kraft pulp from pine (Pinus radiata) using steam explosion. Bioresource
Technol._v. 53, p. 217-223, 1995.
MATIOLI, C.S.; FRIZZONE, J.; PRES, F.C. Irrigação suplementar de cana-de-
açúcar: modelo de análise de decisão para região norte do Estado de São
Paulo. STAB-Acúcar, álcool e subprodutos, v.17, p.42-45, 1998.

97
MATTOS, L.R.; SILVA, F.T. Steam explosion of sugar cane bagasse catalysed
by H2S04 1. Effect of temperatura, time and acid content on sugar cane
bagasse solubilization. ln: 5TH BRAZ. SYMP. CHEM. LIGNINS AND OTHER
WOOD COMPON, Guaratinguetá, SP, 25-28 october. Accepted for publícetíon, 1999
McDOUGALL, G.J.; MORRISSON, 1.M.; STEWART, D.; WEYERS, J.D.B.;
HILLMAN, J.R. Plant fibres: botany, chemistry and processing for industrial
use. J. Sei. Food Agric., v. 62,. p. 1-20, 1993.
McMILLAN, J.D .. Pretreatment of Lignocellulosic Biomass. ln: HIMMEL, M.E.;
BAKER, J.O. ANO OVEREND, R.P. Enzymatic Conversion of Biomass for
Fuels Production. New York: American Chemical Society, p. 292-324, 1994a.
McMILLAN, J.D .. Conversion of Hemicellulose Hydrolysates to Ethanol. ln:
HIMMEL, M.E.; BAKER, J.O. AND OVEREND, R.P. Enzymatic .Conversion
of Biomass for Fuels Production. New York: American Chemical Society, p.
411-437, 1994b. . :": -
MES-HARTREE, M.; SADDLER, J.N. The nature of inhibitory materiais present
in pretreated lignocellulosic substrates which inhibit the enzymatic hydrolysis
of cellulose. Biotechnology Letters, Andover, v.5, n.8, p.531-536, 1983.
MICHALOWICZ, G.; TOUSSAINT, B.; VIGNON, M.R. Ultrastructural changes in
poplar cell wall during steam explosion treatment. Holzforschung, New York,
v.45, n.3, p. 175-179, 1991.
MICHEL, A .J. lnfrared spectroscopy transformed - new applications in wood
and pulping chemistry. Appita, v. 41, n. 5, p. 375-380, 1988.
MIKULÁSOVÁ, M.; VODNY, S.; PEKAROVICOVÁ, A. Biomass, v. 23, p. 149-
154, 1990.

98
NISHIKAWA, N.K.; SUTCLIFFE, R.; SADDLER, J.N. The influence of lignin
degradation products on xylose fermentation by Klebsiella pneumoniae.
Apolied Microbiologv and Biotechnologv, Berlim, v.27, p.549-552, 1988.
PAIVA, L.M.C. Cultivo de Candida utilis em hidrolisados ácidos de bagaço de
cana. Rio de Janeiro: UFRJ, 1980. P.133. (Tese de Mestrado).
PAIVA, T.C.B.; FERRAZ, A.; DURÁN, N. Biotreatment of a pulp mill eop
bleachery effluent with the fungi Penicillium janthinellum and Lentinus
edodes . ln: 5TH BRAZ. SYMP. CHEM. LIGNINS ANO OTHER WOOD
COMPON., Curitiba, PR, Brasil, 31 agosto - 5 setembro 1997. Abstracts,
1997, p. 391.
PALMQVIST, E.; HAHN-HAGERDAL, B.; GALBE, M.; LARSSON, M.;
STENBERG, K.; SZENGYEL, Z.; TENGBORG, C.; ZACCHI, G. Design and
operation of a bench-scale process development unit for the production of
ethanol from lignocellulosics. Bioresource Technologv, London, v.58, p.171-
179, 1996 (a}.
PALMQVIST, E.; HAHN-HAGERDAL, B.; GALBE, M.; ZACCHI, G. The effect of
water-soluble inhibitors from steam-pretreated willow on enzymatic hydrolysis
and ethanol fermentation. Enzvme and Microbial Technologv, New York,
v.19, p.470-476, 1996 (b).
PALMQVIST, E.; HAHN-HAGERDAL, B.; SZENGYEL, Z.; ZACCHI, G.;
RÊCZEY, K. Simultaneous detoxification and enzyme production of
hemicellulose hydrolysates obtained after steam pretreatment. Enzvme and
Microbial Technologv, New York, v.20, p.286-293, 1997.
PARHAM, R.A. Ultra-structure and chemistry. ln: HAMILTON, F; LEOPOLD, B.
Pulp and Paper Manufacture. Atlanta: TAPPI/CPPA, 1993. v. 3, p. 35-45.

99
PFEIFER, P.A.; BONN, G.; BOBLETER, O. lnfluence of biomass degradation
products on the fermentation of glucose to ethanol by Saccharomyces
carlsbergensis W 34. Biotechnology Letters, Andover, v.6, n.8, p.541-546,
1984.
PRIOR, B.A.; KILIAN, S.G.; OU PREEZ, J.C. Fermentation of 0-xylose by the
yeasts Candida shehatae and Pichia stipitis. Process Biochemistry, New
York, v.24, p.21-32, 1989.
PULS, J.; POUTANEN, K.; KÔRNER, H.; VIIKARI, L. Biotechnical utilization of
wood carbohydrates after steaming pretreatment. Applied Microbiology and
Biotechnology, Berlim, v.22, p.416-423, 1985.
RANATUNGA, T.O.; JERVIS, J.; HELM, R.F.; McMILLAN, J.O.; HATZIS, C.
ldentification of inhibitory components toxic toward Zymomonas mobilis
CP4(p285) xylose fermentation. Applied Biochemistry and Biotechnology,
New Jersey, v.67, p.185-198, 1997.
RIVARO, C.J.; ENGEL, R.E.; HAYWARO, T.K.; NAGLE, N.J.; HATZIS, C.;
PHILIPPIOIS, G.P. Measurement of the inhibitory potential and detoxification
of biomass pretreatment hydrolysate for ethanol production. Applied
Biochemistry and Biotechnology, New Jersey, v.57/58, p.183-191, 1996.
ROCHA, G.J.M.; SILVA, F.T.; CURVELO, A.AS.; ARAÚJO, G.T. A fast and
accurate method for determination of cellulose and polyoses by HPLC. ln:
5TH BRAZ. SYMP. CHEM. LIGNINS ANO OTHER WOOO COMPON,
Curitiba, PR, 31 agosto - 5 setembro. Proceedings. 1997, p.3-8.
SAOOLER, J.N.; MES-HARTREE,M.; YU, E.K.C.; BROWNELL, H.H. Enzimatic
hydrolysis of various pretreated lignocellulosic substrates and the
fermentation of the liberated sugars to ethanol and butanediol. Biotechnology
and Bioengineering Symp. Andover, n.13, p.225-238, 1983.

100
SADDLER, J.N.; RAMOS, L.P.; BREUIL, C. Steam pretreatment of
lignocellulosic residues. ln: Bioconversion of Forest and Agricultura! Plant
Residues. Canada: C.A.8. lntemational, 1993. chapter 3, p.73-92.
SASKA, M.; OZER, E. Aqueous extraction of sugarcane bagasse hemicellulose
and production of xylose syrup. Biotechnology and Bioengineering , New
York, v.45, p.517-523, 1995.
SILVA, F.T. Obtenção de insumos químicos a partir do aproveitamento integral
do bagaço de cana. Campinas: UNICAMP/ Instituto de Química, 1995. 106p.
(Tese de Doutorado).
SILVA, F.T.; MATTOS, L.R.; COTRIM, A. R.; FERRAZ, A.; GONÇALVES, AR;
BRUNS, R.E. Acid Steam-Explosion Treatment of Sugar Cane Bagasse
Evaluated by a Using a 23 Factorial Design. ln: 5TH BRAZ. SYMP. CHEM.
LIGNINS AND OTHER WOOD COMPON, Curitiba, PR, 31 agosto - 5
setembro. Abstracts. 1997, p.60.
SINEIRO, J.; DOMINGUEZ, H.; NÚNEZ, M.J.; LEMA, J.M. lnhibition of cellulase
activity by sunflower polyphenols. Biotechnology Letters, Andover, v.19, n.6,
p.521-524, 1997.
TANAHASHI, M.; KARINA, M.; HIGUCHI, T. Degradation mechanism of lignin
accompanying steam explosion li. Mokuzai Gakkaishi, Kyoto, v.36, n.5, p.380-388, 1990.
TANAHASHI, M.; KARINA, M.; TAMABUCHI, K.; HIGUCHI, T. Degradation mechanism of lignin accompanying steam explosion 1. Mokuzai Gakkaishi, Kyoto, v.35, n.2, p.135-143, 1989.
TRAN, A.V.; CHAMBERS, R.P. Ethanol fermentation of red oak acid
prehydrolysate by the yeast Pichia stipitis CBS 5776. Enzvme Microb. Technol., New York, v.8, p.439-444, 1986a.

101
TRAN, A.V.; CHAMBERS, R.P. Lignin and extractives derived inhibitors in the
2,3-butanediol fermentation of mannose-rich prehydrolysates. Applied
Microbiology and Biotechnology , Berlim, v .23, p.191-197, 1986b.
TRAN, A.V.; CHAMBERS, R.P. Red oak wood derived inhibitors in the ethanol
fermentation of xylose by Pichia stipis CBS 5776. Biotechnology Letters,
Andover, v.7, n.11, p.841-846, 1985.
WEIGERT, B.; PRAHL, S.; RIZZI, M.; DELLWEG, H. ln: 8TH INT.
BIOTECHNOL. SYMP., Paris, July 17-22, 1988. Poster 083, 1988.
WILSON, J.J.; DESCHATELETS, L.; NISHIKAWA, N.K. Comparative
fermentability of enzymatic and acid hydrolysates of steam-pretreated
aspenwood hemicellulose by Pichia stipitis CBS 5776. Applied Microbiology
and Biotechnology, Berlim, v.31, p.592-596, 1989.
WOOD, T.M.; SADDLER, J.N. lncreasing the availability of cellulose in biomass
materiais. Methods Enzymol. v. 160, p. 3-11, 1988.

APÊNDICE 1
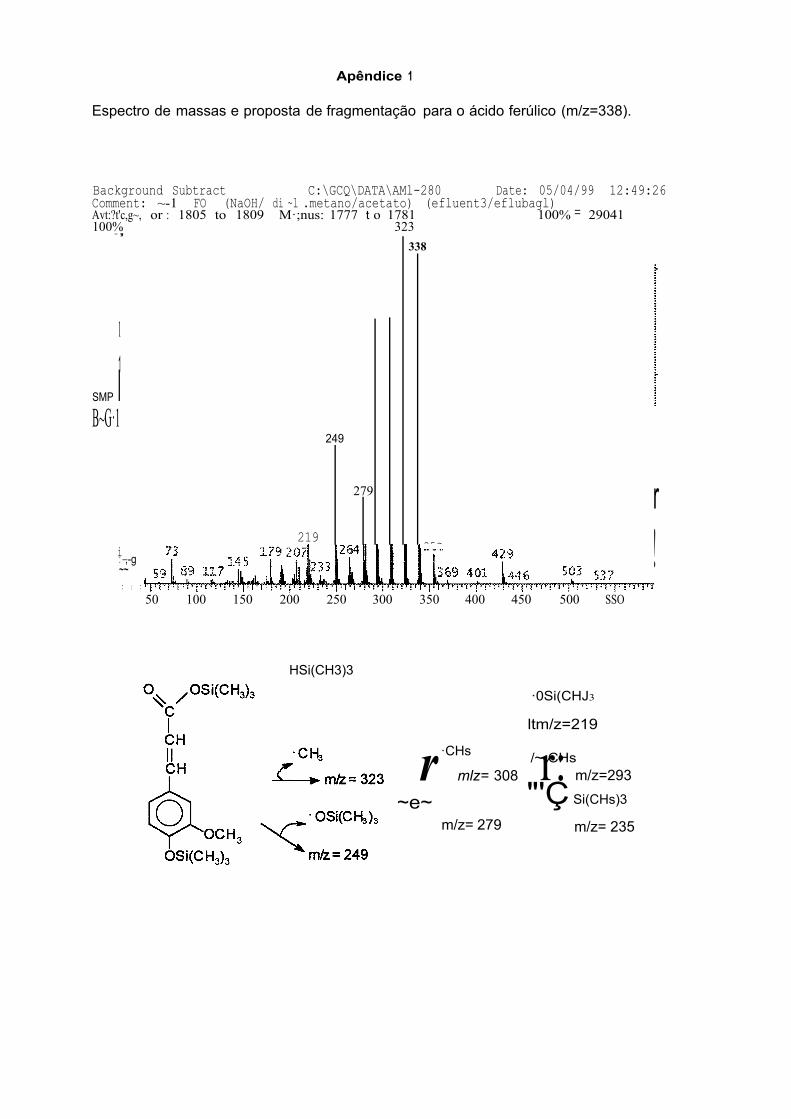
Apêndice 1
Espectro de massas e proposta de fragmentação para o ácido ferúlico (m/z=338).
Background Subtract C:\GCQ\DATA\AMl-280 Date: 05/04/99 12:49:26 Comment: ~-1 FO (NaOH/ di ~l .metano/acetato) (efluent3/eflubagl) Avt:?t'c,g~, or : 1805 to 1809 M·;nus: 1777 t o 1781 100% = 29041 100% 323 .. ,
1 1
SMP l B~G·1
i_ : ,-g ~~
249
219
279
35S
r !
S78j : : :
338
50 100 150 200 250 300 350 400 450 500 SSO
HSi(CH3)3 r: ·CHs r mlz= 308
~e~ m/z= 279
·0Si(CHJ3
ltm/z=219
/~·CHs r: m/z=293 "'Ç Si(CHs)3
m/z= 235
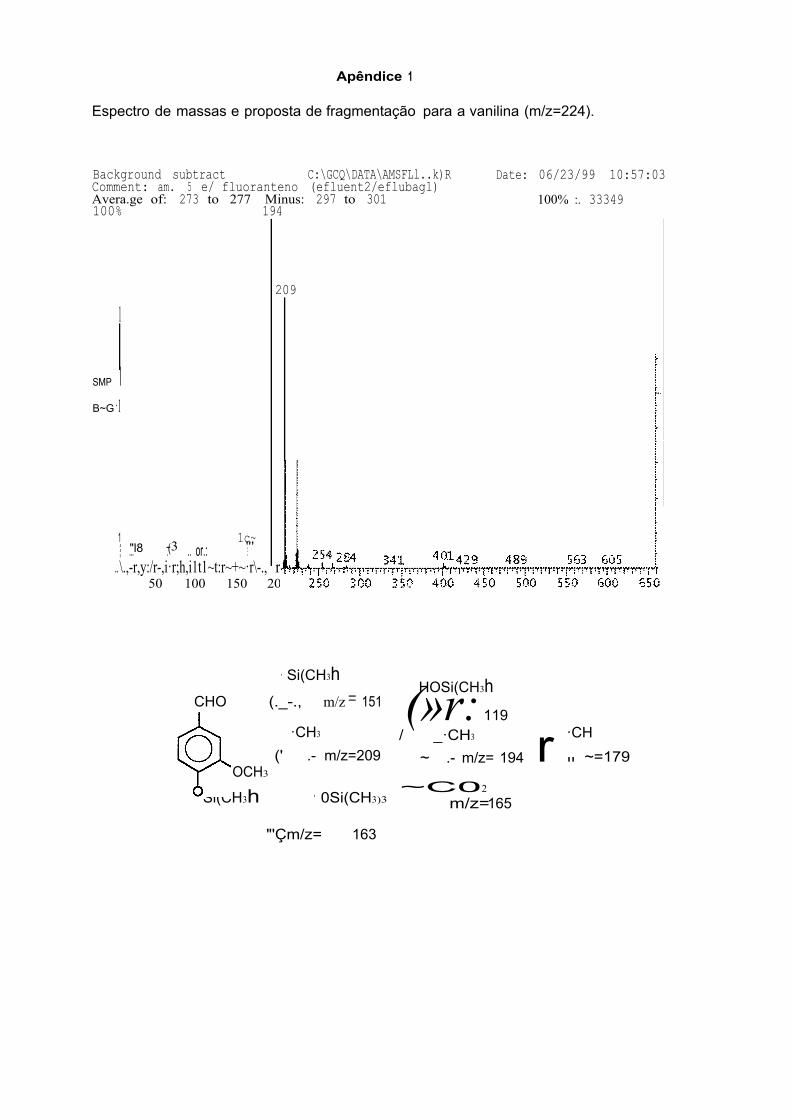
Apêndice 1
Espectro de massas e proposta de fragmentação para a vanilina (m/z=224).
Background subtract C:\GCQ\DATA\AMSFLl..k)R Comment: am. 5 e/ fluoranteno (efluent2/eflubagl) Avera.ge of: 273 to 277 Minus: 297 to 301 100% 194
· Si(CH3h CHO (._-., m/z = 151
·CH3
(' .- m/z=209 OCH3
Si(CH3h · 0Si(CH3)3
"'Çm/z= 163
1
l SMP l B~G ·1
1 1c~ : -3 "' : "l8 ( . , ... . .. or.: : .. \.,-r,y:/r-,i·r;h,i1t1~t:r~+~·r\-., r
50 100 150 200
Date: 06/23/99 10:57:03 100% :. 33349
209
224
HOSi(CH3h ( »r: 119 / _·CH3
~ .- m/z= 194
~C02 m/z= 165
·CH r .. ~=179
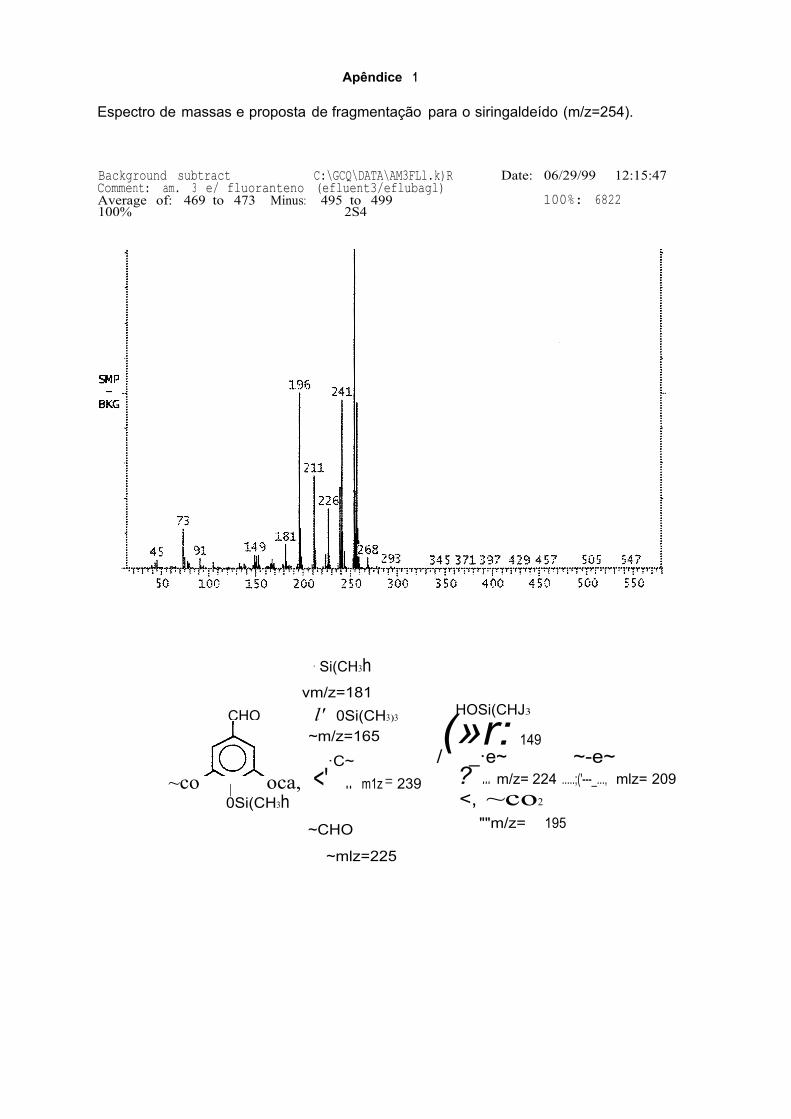
Apêndice 1
Espectro de massas e proposta de fragmentação para o siringaldeído (m/z=254).
Background subtract C:\GCQ\DATA\AM3FLl.k)R Comment: am. 3 e/ fluoranteno (efluent3/eflubagl) Average of: 469 to 473 Minus: 495 to 499 100% 2S4
Date: 06/29/99 12:15:47
100%: 6822
· Si(CH3h vm/z=181
CHO l' 0Si(CH3)3
~m/z=165
·C~ ~co I oca, <' .. m1z = 239
0Si(CH3h ~CHO
~mlz=225
HOSi(CHJ3
( »r: 149 / _·e~ ~-e~
? ... m/z= 224 .....;('---_..., mlz= 209 <, ~co2
""m/z= 195

Apêndice 1
Espectro de massas e proposta de fragmentação para o ácido vanílico (m/z=312)
Background subtract c:\GCQ\DATA\AMl-280 Date: 05/04/99 12:49:26 Ccmment: am.1 FO (NaOH/ dicl.metano/acetato) (efluent3/eflubagl) Average of: 1286 to 1290 M-ir:us: 1268 to 1272 100% : 17319 100% 297
l .:.i:,
~pi :,:::.;._ B~G·1,,·
312
· · 1., 341 385 419447 490 537 607 j .. ,,.,. , ,..!'!-r=·r:·r~t:'h"r"T'i'>"l4t-1.,.1~: · : : : · , "'!'-1 · , • . · , ;'' , ·ryr:·r:·r.,.r:.,.,·,"T'T"l"''l'''fYr:p-rYrfT1":''j''1'"1YFfY;Y'!'l'f'':'''!'''t:"'"r't
50 100 150 20ô 250 300 350 400 450 soo 550 600
267
282
J.81
Ao e~ '-OSi(CHs)3
oc~ ~·OSi(CHs)s
0Si(CHJ3 "'
m/z= 223
HSi(CH3)s r: ·CHs
é ... mlz= 297
HOSi(CH3)3 • 0Si(Ctis}3
( »": 207 {,,m1z= 193
/ _·CHs /(·CHs . ?.., mlz=282---m/z=267
"=C02 ~Si(CHsh m/z = 253 m/z = 209

Apêndice 1
Espectro de massas e proposta de fragmentação para o ácido siríngico (m/z=342)
Background Subtract c:\GCQ\DATA\AM2FLl.OR DaT.e: 06/29/99 16:52:15 Comment: am. 2 e/ fluoranteno (melich 2.0-280-10.0/eflubagl) Aver age of : 1315 te 1319 M1 nus: 1310 to 1314 100% ::. 23 54 100% 327
l .
1,,. 29~12
5>!P J 342 1
r r i ~·1 ·: .., - i
j 4 5 1i3
89 150 .i s < t73 5 5 381 418 446 487 541 1 ··i·r,,1·,\'1-r-r~·r':"'r,·rr11,-r,'rr~1tl'; ... ~·1+r~~~Jr~~';· : ,., T ,·,, fr:·r1..-r'T'T~Fr,.r:·r,-["T'ºí"T'ºPT'T,.:"'T1º:"':'T,.r•·p-;-r,..-;-i-
50 100 150 200 250 300 350 400 450 500 550 600
HSi(CH3)s
Y m/z=268
ho c::,,- 'oSi(CHs)3 . CHs
é • mlz= 327
H3CO/ OC~ . OSi(CHs)s OSi(CHJ,, "'<:
'\J mlz=253 e 0-C-O-ei(CH:i)3
m/z= 221
·CHs
,}' .. mlz= 312 ·CHs r: m/z=297

Apêndice 1
Espectro de massas e proposta de fragmentação para o ácido p-hidroxibenzóico (m/z=282)
Background Subtract C:\GCQ\DATA\AMl-280 D,1te: 05/04/99 12:49:26 Comment: arn.1 FO (NaOH/ dicl .metano/acetato) (efluent3/eflubagl) Average of: 653 to 657 Minus: 672 to 676 J..00% == 24405 100! 267
i
SMP 1 B~G··,
i 1 28
73 193
50 100 150 200
ho e:::-,-- ~ 'osi(CH,),
0Si(C~3
282
2 5 O 3Sú 400 450 500 550
·Si(CH3)3
(___, mlz= 209
(HSi(CH3}s --~·mfz= 238
·CH3
L .. mtz=2a1
"<: · OSi(CHs)3
mfz= 193
HOSi(CH3h ( /mfz= 177 / _·CHs
? m/z=252 ,......._.co2
"'mlz= 223
606 641.
r
1
1 r
f
r •••• ! . ·:
600 650
·CHs
( .. m/z=237

Apêndice 1
Espectro de massas e proposta de fragmentação para o p-hidroxibenzaldeído (m/z=194).
Background subtract C:\GCQ\DATA\AM3FLUOR Date: 06/29/99 12:15:47 C<;mment: ~· 3 e/ fluoran~eno (efluent3/eflubagl) Aver.1ge or : 405 to 409 Minus: 387 to 391 100'% = 17825 100% . 169
r r
1 1:_i,_
SMP i
151 r,.
179
...
.) 28 4 5 65 "l?l:J: ..,/,:, 10~ 123 lt 5 Jli lj5l I l J ll '15 ,_, .. , l .lr . ..l.r'·v··1r1J .r, ~ •• :··",'·!.1-•t· "t'~"'··'rll,.J,., .•. J..; . .1.,...),l ..... ,.J.~ ... r-·,·'J. 'e- t.iJ. , '·v'- , L,..1 . •,····--r·::~'.'r".v···r··-:-,.~; ... , ... ).,
20 40 60 80 100 l20 140 160 180 200 220 240
HOSi(CH3h ( /m/z=89
/ _·CH3
r"' .. m/z= 164
~ ";;i!º: 135
m/z=151
CHO
· Si(CH3h k_mtz=121
('HSi(CH3h --'-----. .... mlz = 12 O
·CH3
~ ., mlz= 179
194
·CH (., ~/z= 149
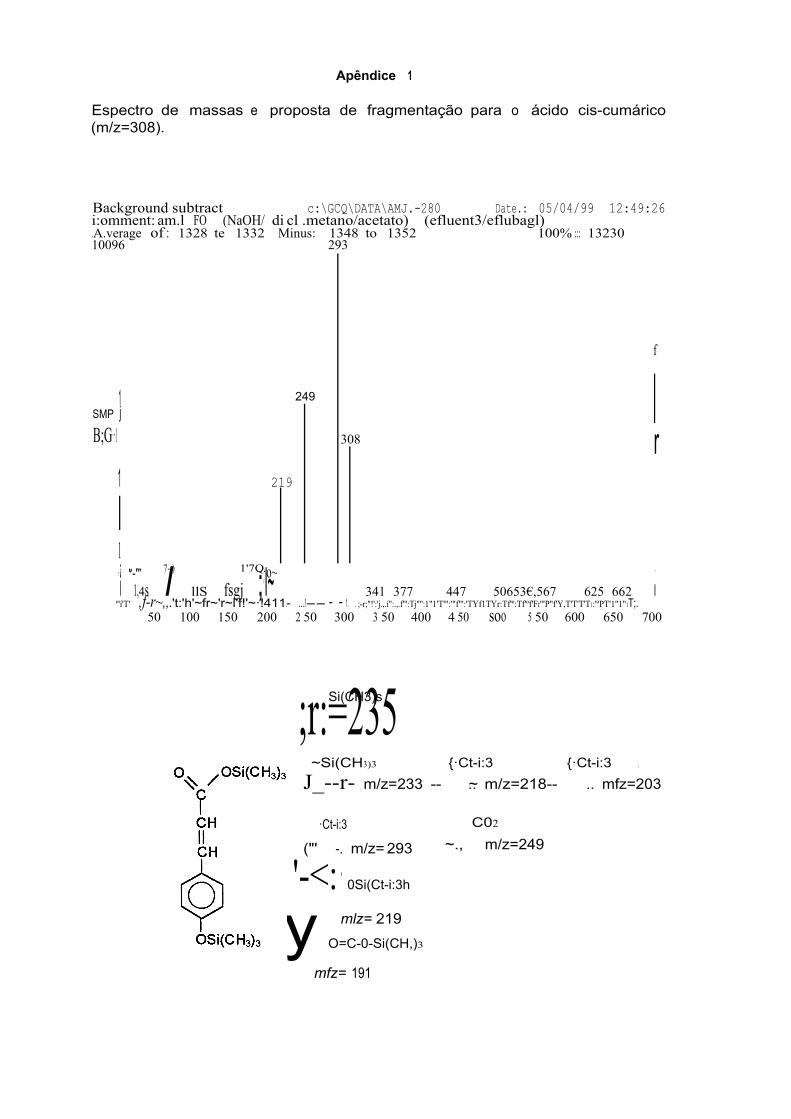
Apêndice 1
Espectro de massas e proposta de fragmentação para o ácido cis-cumárico (m/z=308).
Background subtract c:\GCQ\DATA\AMJ.-280 Date.: 05/04/99 12:49:26 i:omment: am.l FO (NaOH/ di cl .metano/acetato) (efluent3/eflubagl) .A.verage of : 1328 te 1332 Minus: 1348 to 1352 100% ::: 13230 10096 293
1 SMP j B;G··1
1 219
l 1 ' , ~- 7-.) 1'7Q "=,, ·i "-"' · · ···o~ i 1,4s l llS fsgj ;·I~ 341 377 447 50653€,567 625 662 l
º'í'T' ,f-r~,,. 't:'h'~fr~'r~l'f!'~·!411. - .... !--··1. . ;-r;"!':'j.,.í":.,.f"':Tj"":1"1'T'":'"f"':'TYf1TYr:Tf'º:Tf'ºf'Fr'"P"f'Y,T'T'T'T1:'ºPT'1''1"1T;. 50 100 150 200 2 50 300 3 50 400 4 50 soo 5 50 600 650 700
249
308
f
l r
Si(CH3)s ;r:=235 ~Si(CH3)3 {·Ct-i:3 {·Ct-i:3 .
J_--r- m/z=233 -- .. ~ m/z=218-- .. mfz=203
·Ct-i:3
("' -. m/z= 293
C02
~., m/z=249
'-<: · 0Si(Ct-i:3h
mlz= 219 y O=C-0-Si(CH,)3
mfz= 191
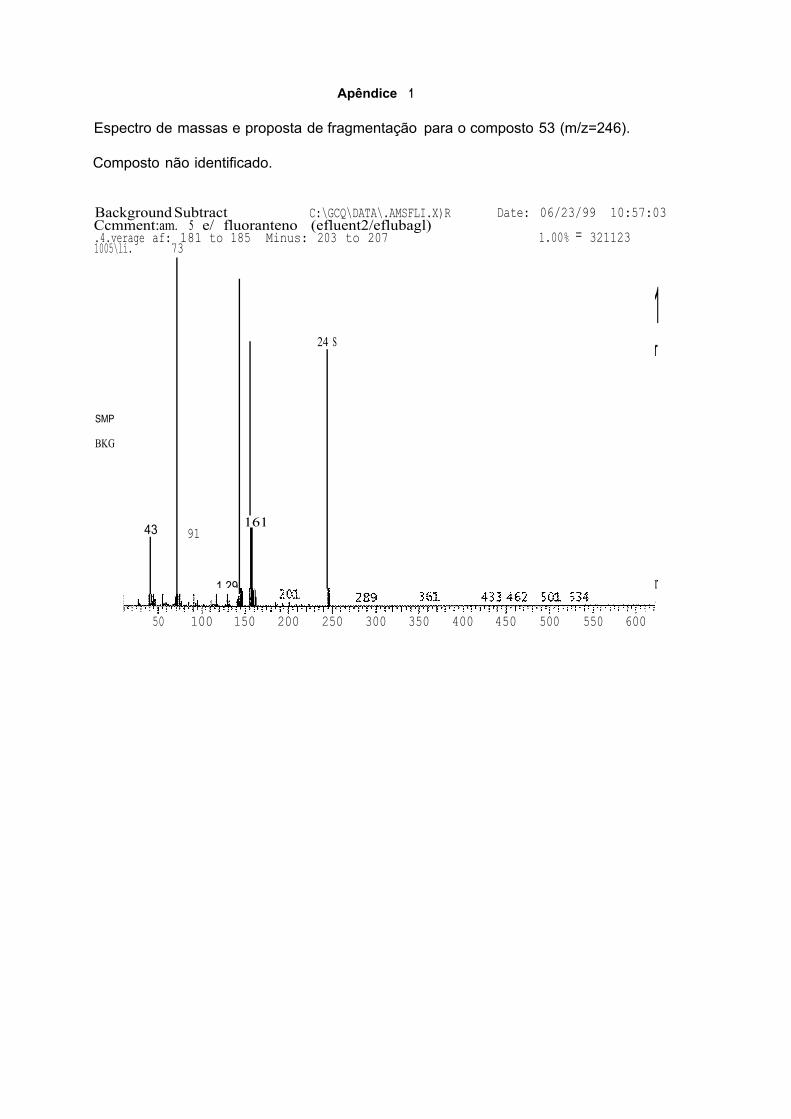
Apêndice 1
Espectro de massas e proposta de fragmentação para o composto 53 (m/z=246).
Composto não identificado.
Background Subtract C:\GCQ\DATA\.AMSFLI.X)R Ccmment: am. 5 e/ fluoranteno (efluent2/eflubagl) .4.verage af: 181 to 185 Minus: 203 to 207 1005\lí. 73
Date: 06/23/99 10:57:03 1.00% = 321123
SMP
BKG
43 91
1 r
r 605\
24 S
161
1.29
50 100 150 200 250 300 350 400 450 500 550 600

Apêndice 1
Espectro de massas e proposta de fragmentação para o ácido trans-cumárico (m/z=308).
Background Subtract c:\GCQ\DATA\AMl-280 D,1te: 05/04/99 12 :49:26 Comment: am.1 FO (NaOH/ dicl .metano/acetato) (efluent3/eflubagl) Average of : 1328 te 1332 M"i nus; 1348 to 13 52 100% ::: 13230 100% 293
267 1 341 371 447 506:i36::67 62:, 662
: ·: ~-~-1 ; .. :'T':''"f•T'T":1·r""·:1·;vrr:,·r,·:"·f""t1·r,·ri-r"·r:·r1·r:·p·r:·r:·:·:·p~-,-:-:T:·f:r:~-c-r:T'1!1-q·:r:r. 250 300 350 400 450 500 550 600 650 700
249
308
Sí(CH3h ;r:=235 1'Si(CH3h
~ m/z=233
·Ct-b (' .., m/z=293
·Ct-b ( m/z=218
-ca, ( mlz= 203
C02 r' • mlz=249
'Ç · OSi(CH,),
m/z= 219 y O=C-0-Si(CH},
mlz= 191


















