
DISSERTAÇÃO DE MESTRADO CINÉTICA DE … · 1 cinÉtica de cristalizaÇÃo isotÉrmica e nÃo...
96
1 UNIVERSIDADE FEDERAL DE PERNAMBUCO CENTRO DE TECNOLOGIA E GEOCIÊNCIAS PROGRAMA DE PÓS-GRADUAÇÃO EM ENGENHARIA QUÍMICA DISSERTAÇÃO DE MESTRADO CINÉTICA DE CRISTALIZAÇÃO ISOTÉRMICA E NÃO ISOTÉRMICA A FRIO DO PET E DAS BLENDAS PET/PHB EM DIFERENTES CONCENTRAÇÕES Ana Calado Brito Recife / PE Setembro, 2010
Transcript of DISSERTAÇÃO DE MESTRADO CINÉTICA DE … · 1 cinÉtica de cristalizaÇÃo isotÉrmica e nÃo...

1
UNIVERSIDADE FEDERAL DE PERNAMBUCO
CENTRO DE TECNOLOGIA E GEOCIÊNCIAS
PROGRAMA DE PÓS-GRADUAÇÃO EM ENGENHARIA QUÍMICA
DISSERTAÇÃO DE MESTRADO
CINÉTICA DE CRISTALIZAÇÃO ISOTÉRMICA E NÃO
ISOTÉRMICA A FRIO DO PET E DAS BLENDAS PET/PHB EM
DIFERENTES CONCENTRAÇÕES
Ana Calado Brito
Recife / PE
Setembro, 2010

1
CINÉTICA DE CRISTALIZAÇÃO ISOTÉRMICA E NÃO
ISOTÉRMICA A FRIO DO PET E DAS BLENDAS PET/PHB EM
DIFERENTES CONCENTRAÇÕES
ANA CALADO BRITO
RECIFE/PE
Setembro, 2010
Dissertação de Mestrado apresentada ao Programa
de Pós-Graduação em Engenharia Química da
Universidade Federal de Pernambuco.
Orientadora: Prof ª. Drª. Yêda Medeiros Bastos de
Almeida.
Co-orientadora: Drª. Renate Maria Ramos Wellen.

B862c Brito, Ana Calado.
Cinética de cristalização isotérmica e não isotérmica a frio do pet e das blendas PET/PHB em diferentes concentrações / Ana Calado Brito. - Recife: O Autor, 2010.
xii,80f., il : grafs.,tabs. Dissertação (Mestrado) – Universidade Federal de Pernambuco.
CTG. Programa de Pós-Graduação em Engenharia Química, 2010. Orientadora: Profª Dra. Yêda Medeiros Bastos de Almeida Inclui bibliografia e Apêndice. 1. Engenharia Química 2. Cinética 3. Cirstalização isotérmica e não
isotérmica. 4. PET. 5. PHB. 6. Fusão e transição vítrea I.Título. UFPE 660.2 CDD (22. ed.) BCTG/2010-192

1

i
Aos meus pais e irmãos.

ii
Agradecimentos
A professora Drª Yêda Medeiros Bastos de Almeida pela orientação.
A Drª Renate Maria Ramos Wellen pela orientação e pela colaboração na
realização e análise dos resultados.
A Isabel Calado pela colaboração.
Ao Departamento de Engenharia de Materiais de Campina Grande pela obtenção
das amostras e pelas análises de DSC.
A FACEPE pela bolsa no período de março/2008 a agosto/2008.

iii
RESUMO
A cinética de cristalização do PET e Das blendas formadas por PET/PHB foi estudada
por calorimetria exploratória diferencial (DSC). O principal ponto de interesse nesse
estudo foi o pico de cristalização exotérmico observado nas curvas de DSC. Na
cristalização isotérmica do PET e do PET/PHB, foi investigado o efeito de diferentes
temperaturas nas suas taxas de cristalização a frio. Os parâmetros cinéticos da
cristalização isotérmica foram determinados utilizando a teoria de Avrami. Foi
observado que em baixas temperaturas a cristalização ocorre em dois estágios, já para
temperaturas mais elevadas ocorre em apenas um estágio. O expoente de Avrami n, a
constante de velocidade K e o meio tempo de cristalização t0,5 foram analisados para a
cristalização isotérmica a frio do PET e do PET/PHB. A constante K aumentou com a
temperatura de cristalização, t0,5 diminuiu com o aumento da temperatura de
cristalização. Com relação ao expoente n, os valores apresentados ficaram próximos de
2 definindo uma morfologia em forma de disco. Com a adição do PHB ao PET, houve
um aumento na cristalinidade do homopolímero PET. A cinética de cristalização
isotérmica nos possibilita ter um controle sobre as condições de cristalização através do
tempo para uma dada temperatura, já na cristalização não isotérmica, esse controle está
diretamente ligado a taxa de aquecimento durante a formação dos cristais, variando a
temperatura em relação ao tempo. A cinética de cristalização não isotérmica foi
investigada e os parâmetros cinéticos foram determinados através da teoria de Mo para
diferentes taxas de aquecimento e verificou-se que o expoente de Mo a apresentou
valores próximos a 1, a constante de velocidade K’(T) aumentou tanto com o aumento da
temperatura de cristalização quanto com a adição de PHB ao PET, e F(T) apresentou
valores mais baixos para as blendas indicando um aumento da velocidade de
cristalização.
Palavras chave: Cinética, cristalização isotérmica e não isotérmica, PET, PHB, fusão e
transição vítrea.

iv
ABSTRACT
The crystallization kinetics of PET and blends formed by PET/PHB was studied by
differential scanning calorimetry (DSC). The main point of interest in this study were
the crystallization exothermic peak observed in DSC curves. In the study of isothermal
crystallization of PET and PET/PHB, we investigated the effect of temperature on their
rates of cold crystallization. The kinetic parameters of isothermal crystallization were
deteminados using the Avrami theory. It was observed that at low temperatures the
crystallization occurs in two stages, while for higher temperatures in only one stage.
The Avrami exponent n, the rate constant K and the crystallization half-time t0,5 were
analyzed for the isothermal cold crystallization of PET and of PET/PHB. The constant
K increased with crystallization temperature, t0,5 decreased with increasing
crystallization temperature. Regarding the exponent n, the values were close to 2
defining a morphology-shaped disc. With the addition of PHB to PET, there was an
increase in crystallinity of PET homopolymer. The isothermal crystallization kinetics
enables us to have a control over the crystallization conditions through time for a given
temperature, even in non-isothermal crystallization, this control is directly connected to
the heating rate during the formation of crystals by varying the temperature in relation
to time. The kinetics of nonisothermal crystallization was investigated and kinetic
parameters were determined using the theory of Mo for different heating rates and
found that the exponent of a Mo values were close to 1, the rate constant K’(T) both
increased with increasing crystallization temperature and with the addition of PHB to
PET, and F(T) values were more lower for the blends indicating an increase in
crystallization rate.
Keys words: Kinetics, isothermal and non isothermal crystallization, PET, PHB,
fusion and glass transition.

v
SUMÁRIO
Lista de Tabelas vii
Lista de Figuras viii
Lista de Abreviaturas e Símbolos x
1. INTRODUÇÃO 1
2. REVISÃO BIBLIOGRÁFICA 3
2.1. Poli(tereftalato de etileno) (PET) 3
2.2. Poli(hidroxibutirato) (PHB) 4
2.3. Estrutura morfológica 6
2.3.1.Taxa de crescimento dos esferulitos 8
2.4. Blendas poliméricas 9
2.5. Morfologia de blendas poliméricas 11
2.6. Miscibilidade de blendas poliméricas 11
2.6.1.Determinação da Tg 13
2.6.2. Depressão da 14
2.6.3.Blendas formadas por componentes miscíveis no “melt” e na fase amorfa 17
2.6.4.Blendas cujos componentes são parcialmente miscíveis ou imiscíveis 19
2.7. Teoria da taxa de crescimento 20
2.8. Cinética de cristalização a frio 22
2.8.1.Cinética de cristalização isotérmica a frio 23
2.8.1.1. Cinética de cristalização isotérmica a frio – teoria de Avrami 24
2.8.2.Cristalização não isotérmica 30
2.8.2.1.Cinética da cristalização não isotérmica a frio – Teoria de Mo 32
3. PROCEDIMENTO EXPERIMENTAL 35
3.1. Materiais 35
3.2. Obtenção das blendas PET/PHB 35
3.3.Caracterização das amostras 35
3.3.1. Calorimetria exploratória diferencial (DSC) 35
3.3.2. Estudo da cinética de cristalização a frio 36

vi
3.3.3. Cinética de cristalização isotérmica a frio – Teoria de Avrami 36
3.3.3.1. Energia de ativação apara a cristalização isotérmica a frio 37
3.3.3.2. Estudo do comportamento de fusão 37
3.3.4. Cinética de cristalização não isotérmica a frio 38
3.3.4.1. Cinética de cristalização não isotérmica a frio – Teoria de Mo 38
4. RESULTADOS E DISCUSSÃO 39
4.1. Cristalização isotérmica a frio 40
4.1.1. Cristalização isotérmica a frio – Teoria de Avrami 45
4.1.2. Energia de ativação para a cristalização isotérmica a frio 48
4.1.3. Estudo do comportamento de fusão 49
4.2. Cristalização não isotérmica a frio 52
4.2.1. Cristalização não isotérmica a frio – Teoria de Mo 59
5. CONCLUSÕES 65
REFERÊNCIAS 67
APÊNDICE 74

vii
LISTA DE TABELAS
Tabela 1: Valores do expoente e Avrami para vários tipos de nucleação e crescimento.
26
Tabela 2: Porcentagem de cristalização para cada amostra de PET e PETPHB para
cada temperatura correspondente.
45
Tabela 3: Parâmetros de Avrami para a cristalização primária isotérmica a frio do
PET e das blendas PETPHB.
47
Tabela 4: Valores da energia de ativação do PET e das blendas PET/PHB
48
Tabela 5: Valores das temperaturas de fusão do PET e das blendas PET/PHB. Todas as
temperaturas de fusão estão em °C.
50
Tabela 6: Temperaturas de fusão de equilíbrio do PET e das blendas PET/PHB
cristalizadas isotermicamente a frio.
52
Tabela 7: Variação da entropia do sistema.
56
Tabela 8: Parâmetros cinéticos para a cristalização não isotérmica do PET e das
Blendas, determinados pelas equações de Avrami-Ozawa. Parâmetros
determinados para o grau de conversão (Xt’) de 10%.
62
Tabela 9: Parâmetros de Mo em diferentes graus de cristalinidade para cristalização
não isotérmica a frio do PET e das blendas PET/PHB.
64

viii
LISTA DE FIGURAS
Figura 1: Unidade repetitiva do PET 3
Figura 2: Unidade repetitiva do PHB. 5
Figura 3: Estrutura esferulítica do PET. 7
Figura 4: Representação da estrutura de uma blenda binária com uma fase amorfa
homogênea e um componente parcialmente cristalizado.
17
Figura 5: Exemplos de morfologia de acordo com o modo de segregação do
componente amorfo. (1) segregação interlamelar, (2) segregação interesferulítica,
(3) segregação interfibrilar.
18
Figura 6: Curva típica de DSC mostrando um pico de cristalização do PET.
22
Figura 7: Curva de Avrami para a cristalização isotérmica do PET a 115°C.
25
Figura 8: Velocidade de cristalização global, K, em função da temperatura de cristalização
29
Figura 9: Curva de Mo para a cristalização não isotérmica a frio do PET para Xt = 10%. 33
Figura 10: Curva de DSC para a cristalização não isotérmica a frio. 39
Figura 11: Curvas de DSC para a cristalização nas temperaturas e percentuais de PHB
indicadas. 41
Figura 12: Curvas das isotermas de cristalização para o PET e das blendas PET/PHB. 43
Figura 13: Efeito da temperatura no tempo para que seja atingido 50% da cristalinidade máxima.
44
Figura 14: Curvas de Avrami para a cristalização isotérmica com diferentes Tc’s. 46
Figura 15: Efeito das Tc’s na taxa de velocidade de cristalização. 46
Figura 16: Curvas de Arrhenius para a determinação da energia de ativação da cristalização
isotérmica a frio do PET e das blendas PET/PHB.
49
Figura 17: Endotermas de fusão do PET e das blendas PET/PHB, cristalizadas
isotermicamente a frio em diferentes temperaturas.
50
Figura 18: Efeito das condições de cristalização nas temperaturas de fusão do PET e suas blendas com PHB.
51
Figura 19:Determinação da temperatura de fusão de equilíbrio através do método de
Hoffman e Weeks do PET e das blendas PET/PHB em diferentes concentrações.
52
Figura 20: Curvas de DSC para Cristalização não isotérmica. 53

ix
Figura 21: Curvas de DSC para Cristalização não isotérmica para uma taxa de aquecimento
de 30°C/min.
54
Figura 22: Efeito da taxa de aquecimento na temperatura de transição vítrea do PET e das
blendas PET/PHB.
55
Figura 23: Efeito da taxa de aquecimento na temperatura de cristalização do PET e suas
blendas com PHB.
56
Figura 24: Efeito da taxa de aquecimento na temperatura de fusão do PET e suas blendas. 56
Figura 25: Curvas em “S” para a cristalização não isotérmica do PET e das blendas PET/PHB.
58
Figura 26: Efeito das taxas de aquecimento na temperatura para se atingir 50% de cristalinidade do PET e das blendas PET/PHB.
59
Figura 27: Grau de cristalinidade com o tempo (Xt’) para a cristalização não isotérmica do
PET e das blendas PET/PHB
60
Figura 28: Curvas do Log t versus Log [-Ln (1-Xt’)] para a cristalização não isotérmica do
PET e das blendas PET/PHB.
61
Figura 29: Efeito da taxa de aquecimento na constante de velocidade K’(T) para a
cristalização não isotérmica a frio do PET e das blendas PET/PHB
62
Figura 30: Curvas do Log t versus Log da taxa para a cristalização não isotérmica a frio do
PET e das blendas PET/PHB em diferentes faixas de cristalinidade.
63
Figura 31: Variação de F(T) com o grau de conversão para o PET e suas blendas com PHB. 64

x
LISTA DE ABREVIATURA E SÍMBOLOS
ΔE Energia de ativação
ΔGm Energia livre de mistura
ΔH Variação da entalpia
ΔHc Variação do calor de cristalização
ΔHm Variação calor de fusão
ΔS Variação da entropia
ΔTc Intervalo de temperatura do pico de cristalização
ΔTm Intervalo de temperatura do pico de fusão
Δtc Intervalo de tempo do pico de cristalização
δ Local do componente não cristalizável
Energia superficial da face paralela à cadeia molecular (superfície lateral)
a Densidade da fase amorfa
c Densidade da fase cristalina
Taxa de aquecimento/resfriamento
A∞ Área sobre a curva de cristalização do instante zero ao instante t
At Área total sobre a curva de cristalização
a Parâmetro de Mo
B Densidade de energia de interação
BMDPE bimodal-medium-density- polyethylene - (Polietileno de média densidade
grade bimodal)
C0,5 Taxa de cristalização
Cp Calor específico
D Difusão
dd Diâmetro dos domínios de suspensões polidispersas
DSC Calorimetria Exploratória diferencial
F(T) Termo relacionado com a taxa de aquecimento/resfriamento da teoria de Mo
G Taxa de crescimento esferulítico linear
IPN interpenetrating polymer network - (Reticulados Poliméricos Interpenetrantes)
K Constante de velocidade
K(T) Constante de velocidade (equação de Ozawa)
K’(T) Constante de velocidade (equação de Mo)
m Expoente de Ozawa
N0 Número de núcleos heterogêneos por unidade de volume
N’( ) Velocidade de nucleação homogênea

xi
n Expoente de Avrami
P3DDT Poly (3-dodecylthiophene) - (Poli (3-dodecil-tiofeno))
P3ODT poly(3-octadecylthiophene) – (Poli(3-octadecil-tiofeno))
PAr Copolímero poliéster de bifenol A e ácidos tereftálicos e isoftélicos
PBSU Poly(butylenes succinate)- Poli(sucinato de butileno)
PBT Polibutileno Tereftalato - Poli(tereftalato de butireno)
PC Policarbonato
PCL Polycaprolactone - Policaprolactona
PECL Poly(ε-caprolactone) – Poli(ε-caprolactona)
PEEK Poly( ether-ether-ketone) - Poli(éter-éter-cetona)
PEEKK Poly( ether-ether-ketone-ketone)Poli(éter-éter-cetona-cetona)
PEG Poly (ethylene glycol) - Poli(etileno glicol)
PEI Poly (ether imide) – Poli(éter imida)
PEKEKK Poly(ether-ketone-ether-ketone-ketone) - Poli(éter-cetona-éter-cetona-
cetona)
PEN Poly (ethylene naphtalate) - Poli(etileno naftalato)
PEO Poly (ethylene oxide) - Poli(óxido de etileno)
PES Poly (ether sufone) – Poli( éter sulfona)
PET Poly(ethylene terephtalate) - Poli(teraftalato de etileno)
PHA Poly(hydroxyalkanoate) - Poli(hidroxialcanoato)
PHB Poly(hydroxybutyrate) - Poli(hidroxibutirato)
PHB-co-HV Poly(hydroxybutyrate_co_hydroxyvalerate)
Poli(hidroxibutirato_co_hidroxivalerato)
PIP Poly(cis-1, 4-isoprene) - Poli(cis-1,4-isopreno)
PLLA Poly(L-lactic) - Poli(L-láctico)
aPMMA Poly(methyl methacrylate)atactic - Poli(metacrilato de metila)atático
sPMMA Poly(methyl methacrylate)syndiotactic - Poli(metacrilato de metila)sindiotático
PPDO Poly(p-dioxanone) - Poli(p-dioxanona)
PPS Poly(phenylene sulfide) - Poli(sulfeto de fenileno)
PS Polystyrene - Poliestireno
PTT Poli(tereftalato de trimetileno)
PVAc Poly(vinyl acetate) - Poli(acetato de vinila)
PVPh Poly(p-vinyl phenol) - Poli(p-vinil fenol)
R Constante dos gases
r raio dos esferulitos
SAN Styrene acrylonitrile - Copolímero de estireno-acrilonitrila

xii
t∞ Tempo final de cristalização
T0 Temperatura no início da cristalização
t0,5 Tempo necessário para cristalizar 50% do material (isotérmica)
t’0,5 Tempo necessário para cristalizar 50% do material (não isotérmica)
Tc Temperatura de cristalização
Tg Temperatura de transição vítrea
Tm Temperatura de fusão
V Número de cadeias
W Fração em massa molar
Xc Grau de cristalinidade
XT Fração de material cristalizado na temperatura T (não isotérmico)
Xt Fração de material cristalizado no tempo t (isotérmico)
Z Número de coordenação

1
1. INTRODUÇÃO
Com o rápido avanço da tecnologia e a busca constante de alternativas que sejam
capazes de melhorar as condições de vida, muitos cientistas têm trabalhado no
desenvolvimento de materiais com qualidades superiores aos já existentes e com
propriedades específicas. Pelas suas propriedades tais como resistência a tração e ao
impacto, rigidez e principalmente baixa permeabilidade a gases aliadas a um custo
relativamente baixo da matéria-prima, o poli (tereftalato de etileno) - PET tem sido
considerado um dos mais importantes polímeros de engenharia, sendo utilizado em
embalagens, principalmente em recipientes para indústria alimentícia, descartáveis e
utensílios domésticos.
Com toda essa aplicabilidade, vem aumentando consideravelmente a utilização
do PET como matéria-prima para produção de diversos itens de consumo humano. Por
isso uma constante busca pela melhoria das características desse produto vem sendo
estudada em paralelo ao avanço de sua utilização.
Buscando alternativas que contribuam para um melhor desempenho da
produção, foi estudado o efeito que o poli (hidroxibutirato) - PHB causa ao ser
misturado ao PET. Blendas de PET/PHB podem significar uma alternativa viável já que
o PHB é um polímero com características interessantes como o alto grau de
cristalização. A avaliação desta blenda é necessária para verificar o quanto suas
características vão influenciar na formação de um produto que tenha melhor
funcionabilidade.
O termo blenda refere-se à mistura de polímeros que possuem propriedades
físico-químicas diferentes das apresentadas por cada componente separadamente
(Olabisi et al., 1979). Neste trabalho, as blendas utilizadas são misturas de dois
polímeros.
As blendas poliméricas podem ser miscíveis ou parcialmente miscíveis ou
imiscíveis. As blendas são miscíveis quando da mistura dos polímeros, resulta uma
solução homogênea. O termo compatibilidade, neste caso, será utilizado para blendas
poliméricas que atinjam propriedades finais uteis comercialmente, com melhores
características, podendo ou não ter mais de uma fase, do contrário é dita como
incompatível. Para blendas imiscíveis ou parcialmente miscíveis, a mistura apresenta
mais de uma fase, podendo ocorrer incompatibilidade interfacial ou incompatibilidade
total, resultando na separação de fases e na formação de diferentes microestruturas ou

2
morfologia. Neste trabalho o termo miscível será utilizado para definir blendas de dois
componentes que possui uma única fase.
A abordagem utilizada nesta pesquisa foi à adição de baixas concentrações de
PHB ao PET, e elas foram definidas da seguinte maneira, PET/PHB (100/0), (99,5/0,5),
(99/1) e (97/3). Foi investigado também o efeito da adição de PHB nas temperaturas de
transição térmica do PET durante os processos de cristalização isotérmica, não
isotérmica e fusão, representando um estudo inicial do efeito da adição de baixas
concentrações de PHB no comportamento de cristalização a frio e nas propriedades do
PET.
As propriedades finais das blendas PET/PHB dependem da miscibilidade dos
componentes, do grau de cristalinidade e da formação dos cristais. Para isso, é
necessário um controle das propriedades dos polímeros, que é possível através do
estudo da cinética de cristalização. A cristalização a frio do PET e das blendas
PET/PHB foi estudado por calorimetria exploratória diferencial (DSC).
A cristalização isotérmica visa investigar a fase cristalina que ocorre entre a
temperatura de transição vítrea (Tg) e a temperatura de fusão (Tm) do polímero a uma
temperatura constante e os parâmetros cinéticos foram analisados segundo a teoria de
Avrami (Avrami, 1939, 1940, 1941).
Já o estudo da cristalização em meio não isotérmico apresenta grande interesse
prático, pois os processos industriais normalmente ocorrem sob condições não
isotérmicas. Sendo assim, o estudo da cristalização em um ambiente com variação de
temperatura permite não só uma otimização das condições de processamento como
também a obtenção de produtos com melhor desempenho. Esse estudo foi realizado
seguindo a teoria de Mo ( Liu et al., 1997).
É de fundamental importância a realização deste estudo porque através dele
podemos controlar as condições de fabricação de produtos provenientes do PET e
PET/PHB como temperatura, tempo de cristalização e taxa de aquecimento, com a
finalidade de evitar problemas gerados durante o processo de produção como a
cristalização prematura durante a sopragem.

3
2. REVISÃO BIBLIOGRÁFICA
2.1. Poli (tereftalato de etileno) (PET)
O PET é um polímero termoplástico da família dos poliésteres, desenvolvido por
dois químicos britânicos Whinfield e Dickson em 1941, que em sua estrutura química
(Figura 1), apresenta uma unidade repetitiva de grupos de ácido tereftálico (grupo
aromático), representando o segmento rígido, e de etileno glicol (grupo alifático), que é
a estrutura flexível.
Figura 1: Unidade repetitiva do PET. Dados da autora.
Embora a estrutura molecular do PET favoreça a sua forma cristalina a partir do
estado fundido, por ser regular com polaridade média, a presença de grupos aromáticos
na cadeia principal, confere ao polímero baixa mobilidade tornando lento o processo de
cristalização, consequentemente, produtos amorfos são obtidos quando o material é
resfriado rapidamente. Isso ocorre comumente em processos industriais, como na
fabricação de pré-formas injetadas utilizadas na confecção de produtos soprados e em
chapas e filmes extrudados. Em muitos casos (como em injeção-sopro e em
termoformagem) o produto amorfo é submetido a um aquecimento para que adquira a
maleabilidade típica do estado borrachoso, permitindo a conformação na forma final.
Esse fenômeno ocorre a uma temperatura acima da Tg, quando as moléculas do PET
adquirem mobilidade suficiente para se rearranjarem em uma estrutura cristalina
(Rabello, 2008).
O PET tem como características a leveza, boa resistência térmica e química,
bom desempenho mecânico, alto grau de impermeabilidade a gases, transparência e um
baixo custo de produção. Assim, é utilizado na forma de fibras para tecelagem e
embalagens para bebidas (Silva, 1991).
O
C
O
C
O
O CH2
CH2
n

4
Por ser termoplástico, o PET pode ser reprocessado diversas vezes, através do
mesmo tipo de produção ou por diferentes processos de transformação. Quando
aquecido a temperaturas adequadas, esses plásticos amolecem, fundem e podem ser
novamente moldados.
Com a finalidade de aumentar a sua aplicabilidade e obter melhorias
significativas dos produtos derivados desse polímero, muitas pesquisas vêem sendo
realizadas considerando muitos aspectos do PET como, por exemplo, a cinética de
cristalização do PET comercial (Silva, 1991), processos de degradação (Du et al., 2006),
influência do tratamento térmico na estrutura morfológica (Zhao et al., 2002),
propriedades de transporte (Bove et al., 1994), propriedades mecânicas (Viana et al.,
2004) e propriedades de barreira (Hu et al., 2006).
Devido ao problema do acúmulo de produtos na natureza cuja matéria-prima é o
PET e a busca constante de melhorias tecnológicas, tem-se gerado muito interesse ao
desenvolvimento de blendas formadas a partir do PET, como será tratado neste trabalho.
Na literatura, há alguns exemplos dessas blendas: Poli(teraftalato de
etilena)/poliestireno - PET/PS (Wellen, 2007), poli(teraftalato de etileno)/
Poli(tereftalato de trimetileno) - PET/PTT (Mingtao et al., 2009), poli(teraftalato
deetileno)/poli(etileno naftalato) - PET/PEN (Pó et al., 1996).
2.2.Poli (hidroxibutirato) PHB
O PHB foi descoberto em 1925 por Lemoigne, cuja unidade repetitiva está
representada na Figura 2. O PHB geralmente é produzido por fermentação bacteriana, e
tem sido amplamente utilizado na indústria de embalagens como um polímero
biodegradável para minimizar a poluição ambiental. Por se tratar de um polímero com
muitas características interessantes, suas propriedades mecânicas e térmicas foram alvo
de muitos estudos (Ei-Had et al., 2002; Ha et al., 2002).

5
Figura 2: Unidade repetitiva do PHB. Dados da autora.
É um polímero semicristalino, e frágil por natureza, que possui uma Tm
relativamente alta (na faixa de 170-180 °C). A sua Tg é na faixa de 0-5°C. Quando sua
temperatura atinge a Tm há uma instabilidade térmica, pois nessas condições ocorre à
quebra preferencialmente das ligações ésteres entre as unidades repetitivas e a rápida
redução de sua massa molar média (Ha e Cho, 2002).
O PHB de origem natural tem uma estrutura regular perfeita, de elevada pureza e
elevado grau de cristalinidade de modo que ele tem sido considerado como um modelo
para estudar cristalização de polímero e morfologia (Withey et al.,1999). O PHB
apresenta a mesma forma de crescimento dos esferulitos que o PET, mas por outro lado,
possui baixa densidade de nucleação e cristaliza-se formando grandes esferulitos que
podem ser facilmente controlada por suas condições de cristalização (temperatura, taxa
de aquecimento). O estudo do comportamento do PHB na cristalização não isotérmica é
muito importante durante os processos industriais, como a extrusão e moldagem
(Rabello, 2008).
Koning et al (1992) têm mostrado que o PHB também pode sofrer
envelhecimento durante o armazenamento à temperatura ambiente, aumentando a
fragilidade.
Quental et al. (2009) estudaram vários tipos de blendas formadas a partir do PHB e
observaram que blendas de poli (hidroxibutirato)/poli (p-vinil fenol) (PHB/PVPh)
preparadas a partir de solução em epicloridrina mostraram-se miscíveis em todas as suas
composições, apresentando uma única transição vítrea que varia de acordo com a
composição das blendas, já a blenda poli(hidroxibutirato)/poli(acetato de vinila) -
PHB/PVAc é miscível em todas as composições. As blendas de
poli(hidroxibutirato)/poli(cis-1, 4-isopreno) - PHB/PIP mostraram-se imiscíveis e as
propriedades mecânicas indicaram que tais blendas são incompatíveis. Já as blendas de
PHB/PIP-g-PVAc mostraram indícios de existência de interações entre o PHB e o grupo
CH CH2 C
O
CH3 n
O

6
acetato de vinila, com uma pequena diminuição na temperatura de fusão do PHB. Além
disso, a análise morfológica de tais blendas indicou que a mistura com o PIP-g-PVAc
causa uma significativa redução nos tamanhos dos domínios cristalinos quando
comparado ao PIP.
2.3. Estrutura morfológica
O PET e o PHB são polímeros semicristalinos formados por regiões cristalinas e
amorfas como mostrado na Figura 3, e a sua forma, tamanho, fração volumétrica e
orientação durante o processamento pode ser bastante variado (Tan et al., 2000).
A formação dos cristais ocorre quando, no estado amorfo, é aquecido a uma
temperatura superior à Tg, por outro lado, estando no estado fundido, é lentamente
resfriado abaixo do seu ponto de fusão. Enquanto ocorre a cristalização, há a formação
de cristais fibrilares ou lamelares que dependem das condições da cristalização,
podendo dar origem a estruturas do tipo feixe que se transformam em esferulitos
(Billmeyer, 1984).
A formação dos esferulitos é importante na determinação das propriedades
óticas e mecânicas dos polímeros cristalinos. São estruturas compostas por lamelas
cristalinas que crescem radialmente de um núcleo comum em três dimensões e são
conectadas entre si por segmentos moleculares amorfos. O esferulito é composto de
camadas sobrepostas de lamela, em planos paralelos a direção radial, onde as cadeias
são dispostas perpendicularmente ao plano da lamela, como mostra a Figura 3.
O esferulito começa a se formar por apenas uma fibra, cresce formando um
embrião na forma de feixe, e em seguida evolui para o formato de esfera, em que há a
colisão entre vizinhos, tornando-se um poliedro (Basset, 1981).

7
Figura 3: Estrutura esferulítica do PET. (Silva, 1991)
A formação dos esferulitos segue três estágios distintos.
1. Nucleação: é o inicio da cristalização, a formação do núcleo, atuando como
centro de formação no instante zero, e é denominado de nucleação instantânea ou
nucleação heterogênea. Este processo está ligado à presença de heterogeneidades
porosas que asseguram a estabilidade térmica dos embriões cristalinos adsorvidos nas
suas reentrâncias, que continuam em equilíbrio a temperaturas acima de seu ponto de
fusão. Os embriões podem surgir esporadicamente no seio da fase amorfa, neste caso, a
sua formação pode estar ligada à presença de heterogeneidades com afinidade pela fase
cristalina, induzindo à nucleação (nucleação pseudo-homogênea). Outra formação é a
chamada nucleação homogênea, e ocorre quando os núcleos cristalinos se formam por
agregação espontânea de cadeias do polímero (Hoffman et al., 1961).
2. Cristalização primária: ocorre quando há o crescimento do núcleo em todas as
direções, por unidade de tempo. Esse crescimento prossegue a uma taxa acelerada, até
ser atingido um estado de pseudo-equilíbrio, onde o crescimento é retardado à medida
que os núcleos encontram regiões já cristalizadas.

8
3. Cristalização secundária: ocorre no final da cristalização primária; uma
cristalização a taxas muito lentas continua ocorrendo por um período de tempo finito.
Esse comportamento é atribuído à interferência entre cristais, para posterior perfeição
ou reorganização de macromoléculas nas regiões intra ou inter-esferulíticas.
Groeninckx et al. (1980) analisaram o desenvolvimento da estrutura morfológica
do PET em função da temperatura de cristalização a frio (Tc), verificando que em baixas
Tc’s lamelas altamente ramificadas com pequenas dimensões laterais foram formadas
enquanto que em altas Tc’s formaram-se estruturas lamelares comuns. Pingping e Dezhu
(1999) registraram que a cristalização a frio do PET foi facilitada com a presença do
carbonato de cálcio - CaCO3.
Zhao et al (2002) e Lim e Kim (1999) observaram um deslocamento do pico de
cristalização a frio do PET para temperaturas inferiores quando o PET foi reaquecido
em temperaturas próximas a Tg, e quando filamentos de PET foram estirados em altas
velocidades.
Kint et al., (2003) conseguiram diminuir a taxa de cristalização a frio do PET
com a introdução de comonômeros em suas cadeias poliméricas. A utilização de grades
de alta massa molecular como também a adição de diferentes polímeros como o
policarbonato e o PEN reduziram a velocidade de cristalização do PET. Em outros
trabalhos, Kint et al., (2003) avaliaram a cinética de cristalização a frio de chapas
extrudadas de PET, observando que a velocidade de cristalização é fortemente
dependente da temperatura e o comportamento mecânico do material é afetado pela
estrutura cristalina obtida.
2.3.1. Taxa de crescimento dos esferulitos
A taxa de crescimento linear (G) de um cristal é a taxa de avanço macroscópico
da frente do cristal em uma direção. Geralmente essa taxa é conduzida em condições
isotérmicas e depende do monitoramento dos raios dos esferulitos (r) em função do
tempo (t), até que a estrutura cristalize. Essa relação pode ser determinada, pois a uma
temperatura fixa, a curva que caracteriza (r) em função de (t) é linear e a sua inclinação
dá o valor de (G) de acordo com a Equação 1.

9
(1)
Quando a taxa de crescimento ocorre de maneira rápida e a nucleação é lenta,
haverá a formação de poucos esferulitos com tamanhos grandes, já para a rápida
nucleação, os esferulitos terão o seu crescimento reduzido. Ou seja, o controle do
tamanho dos esferulitos está relacionado ao controle da taxa de nucleação.
Keith e Padden (1996) afirmaram que moléculas com vários graus de
cristalizabilidade, como ramificações, finais de cadeia, material atático e unidades de
comonômeros constituem zonas rejeitadas pelo cristal em crescimento, originando a
fase amorfa interlamelar.
Alguns estudos de microscopia eletrônica atribuem para os esferulitos uma
estrutura lamelar para quase todos os polímeros. As lamelas podem apresentar uma
geometria regular assemelhando-se a cristais únicos e dependem do retículo cristalino,
da forma de cristalização e de fatores cinéticos (Woodward, 1995).
Os domínios esferulíticos dependem da competição entre as taxas de nucleação
primária e o crescimento radial e estão na faixa entre 5 e 100µm, tornando-se menores
em baixas temperaturas de cristalização (Baer et al., 1965).
2.4. Blendas poliméricas
Neste texto, o termo blendas poliméricas é definido como misturas de dois
polímeros com a finalidade de se obter um produto final com propriedades físico-
químicas melhores que as apresentadas por cada componente individualmente. Já o
termo miscível será usado para denominar blendas de dois componentes que possuem
uma única fase. O termo compatível será utilizado para blendas que apresentam
propriedades físicas desejáveis, independentemente dos seus componentes serem ou não
miscíveis.
Um sistema de polímeros pode não ser miscível, mas pode ser compatível. Uma
blenda será dita incompatível quando suas propriedades, pelo menos aquelas desejadas,
forem inferiores à de cada componente puro.
Nas blendas poliméricas, a afinidade entre os componentes deve ser satisfatória
para que haja a interação entre as moléculas e consequentemente a troca de

10
propriedades, ou seja, a miscibilidade de uma blenda é um fator importante para existir
a compatibilidade.
A miscibilidade de uma blenda polimérica é um fator muito importante, pois
disso depende a sua morfologia. As forças intermoleculares envolvidas na mistura
contribuem para o decréscimo da energia livre de mistura e, portanto, para a
miscibilidade entre os polímeros. É cada vez mais importante o estudo referente às
blendas poliméricas, pois a indústria de polímeros tem se interessado muito devido à
possibilidade de se obter um produto com propriedades melhores, e que apresenta uma
ótima relação custo/benefício (Paul et al., 1980). Em alguns casos, as propriedades de
uma blenda miscível são intermediárias às dos seus componentes, embora seja de maior
interesse que haja um efeito sinérgico, formando uma blenda de propriedades
superiores.
As blendas podem ser compostas de polímeros amorfos e/ou semicristalinos.
Quando um polímero cristalino é utilizado como integrante de uma blenda polimérica a
obtenção de um sistema totalmente miscível não é possível, já que se parte de um
sistema com duas fases. Geralmente as blendas compatíveis em que pelo menos um
componente é cristalizável, são miscíveis no estado fundido, mas durante o resfriamento
esta blenda se separa em duas ou mais fases. No caso de uma blenda onde um
componente se cristaliza e o outro permanece amorfo, vai existir uma fase
correspondente à parte cristalina do componente cristalizável, uma fase correspondente
à parte amorfas deste polímero que não se mistura com o outro polímero, e uma fase
correspondente à mistura das fases amorfas. O grande problema neste caso é a
existência de inúmeras interfaces dentro do sistema, o que coloca em risco a
compatibilidade deste sistema, já que as concentrações de tensão ocorrem nas interfaces
(Hage Jr., 1989).
As blendas podem ser obtidas por diferentes métodos:
Por solução: método utilizado geralmente em laboratórios e emprega um
solvente comum aos polímeros ou a mistura homogênea das soluções
particulares de ambos os componentes.
Por reticulados poliméricos interpenetrantes (IPN): são caracterizados por
seus constituintes individuais se interpenetrarem e formarem reticulados,
sem que haja reação química entre eles. São obtidos por uma mistura

11
polimérica onde os constituintes estão na forma de reticulados individuais
(Hage Jr., 1989).
Por mistura mecânica: utiliza-se aquecimento para amolecer ou fundir os
componentes da blenda, e o cisalhamento para favorecer a mistura.
2.5. Morfologia de blendas poliméricas
A morfologia das blendas poliméricas cristalizáveis depende da composição, da
interação e da natureza química dos seus componentes, e pode ser descrita avaliando a
sua curva de separação de fases: líquido-líquido e líquido-sólido, sendo a segunda
chamada de curva de cristalização.
As blendas poliméricas cristalizáveis podem ser classificadas como blendas
formadas por componentes miscíveis no estado fundido e na fase amorfa, e blendas
formadas por componentes parcialmente miscíveis ou imiscíveis.
2.6. Miscibilidade de blendas poliméricas
A mistura entre polímeros é considerada uma alternativa econômica para o
desenvolvimento de novos materiais, se comparada à síntese de novos polímeros.
Quando se selecionam polímeros para a produção de uma blenda, dois fatores
importantes devem ser considerados, a miscibilidade e a compatibilidade, os quais
norteiam o desenvolvimento e a aplicação de novos materiais poliméricos.
Segundo Quental et al. (2009), polímeros miscíveis misturam-se em nível
molecular e o processo de mistura deve resultar em uma energia livre de Gibbs próxima
a zero, conforme verificado na Equação 2:
(2)
Sendo
T temperatura absoluta,
p pressão,
ΔGm, ΔHm, ΔSm são as variações da energia livre, entropia e entalpia de
mistura, respectivamente.

12
Uma condição necessária, mas não suficiente para a miscibilidade é que ΔGm =
0. Misturas monofásicas são estáveis termodinamicamente, sendo necessário também
que a Equação 3 seja obedecida.
(3)
Com i sendo a fração volumétrica do polímero “i” na mistura;
ΔGm variações da energia livre.
Se a condição da Equação 3 é satisfeita para toda a faixa de composição da
blenda, então a blenda é miscível, caso contrário ela é imiscível. Alguns autores
consideram que sendo esta condição satisfeita somente para algumas composições, a
blenda é parcialmente miscível (Faves et al., 2000).
Outra forma de se avaliar a miscibilidade em uma blenda é a detecção de uma
única Tg, a qual deve estar situada entre as transições vítreas dos componentes que
constituem a blenda.
O aparecimento de uma única Tg na blenda é indicativo de uma homogeneidade
em nível molecular, na qual seus domínios cristalinos, se existirem, apresentam
diâmetros na faixa 2 ≤ dd ≤ 15 nm. Desta forma sistemas miscíveis binários apresentam
uma única e aguda Tg, cuja temperatura é intermediária entre àquelas dos componentes
puros, e varia com a composição da mistura. Já as blendas parcialmente miscíveis,
apresentam duas Tg’s, uma deslocada na direção da outra. Assim sendo, é necessário
encontrar uma única temperatura de transição vítrea intermediária a dos componentes
isolados (Utracki, 1990).
A taxa de crescimento dos esferulitos (G) depende da temperatura de
cristalização (Tc). O valor de G tende a zero à medida que Tc se aproxima da Tg ou da
temperatura de fusão (Tm) do polímero semicristalino e apresenta um valor máximo
numa temperatura Tmáx intermediária à Tg e Tm. A diluição do polímero semicristalino
por um polímero miscível pode alterar os valores de Tg e Tm, e deslocar Tmáx. O efeito
do componente miscível sobre G irá depender de como a diferença em módulo |Tc -
Tmáx| varia com a composição. Outra característica que pode demonstrar a miscibilidade
de blendas formada por um polímero semicristalino é a depressão da temperatura de
fusão. De acordo com a teoria de Flory-Huggins, uma análise apropriada da depressão

13
da temperatura de fusão pode fornecer informações sobre o parâmetro de interação para
o par polímero-polímero (Quental et al., 2009).
2.6.1. Determinação da Tg
A Tg ocorre quando as cadeias moleculares de um polímero adquirem energia
suficiente (geralmente de fonte térmica) para superar as barreiras de energia necessárias
à rotação de ligações (Paiva et al., 2006). Estruturas de polímeros diferentes necessitam
de diferentes quantidades de energia para “sobrepor” a barreira, isto é, cada estrutura de
polímero tem a sua Tg. Qualquer movimento significativo de uma cadeia é gerado por
rotação em torno das ligações simples que conectam os átomos da cadeia. À medida que
o movimento molecular em um polímero amorfo aumenta, a amostra passa de um
estado vítreo para um borrachoso e até, finalmente, tornar-se fundido, (Olabisi et al.,
1979). Ou seja, abaixo da sua Tg, os polímeros se transformam em sólidos quebradiços,
definidos como frágeis e acima da Tg o polímero passa para o estado flexível definido
como borrachoso. Mudanças em outras propriedades como índice de refração, a
capacidade calorífica, a expansão térmica e o volume específico também são
verificados, quando se observam mudança na temperatura.
A determinação da miscibilidade utilizando a Tg por DSC em uma blenda é
possível quando a diferença das Tg’s dos componentes puros é maior que 20°C.
Para avaliar o grau de dispersão nas blendas algumas equações foram
desenvolvidas utilizando o conceito de Tg. Quando a blenda polimérica é miscível a
correlação entre a Tg da blenda e a sua composição pode ser determinada, através da
correlação de Fox (1956):
(4)
Em que:
Tg,b, Tg1 e Tg2 são as temperaturas de transição vítrea da blenda e dos
componentes 1 e 2, respectivamente.
W1 e W2 são as frações em massa dos respectivos componentes.

14
Quando se trabalha com blendas cristalizáveis e se relaciona a Tg com a
composição, deve ser levada em consideração a possibilidade de cristalização. Quando
ocorre cristalização, a composição da fase amorfa é diferente da nominal. A composição
real, rica no componente não-cristalizável, pode ser calculada considerando a
quantidade de material cristalizado (Silvestre et al., 1996).
Kim e Burns (1990) adaptaram a equação de Fox, para que esta pudesse fornecer
a fração aparente de cada componente, ou seja, eles desenvolveram uma equação que
possibilita obter a quantidade do componente 1 na fase rica em 1, e 2 na fase rica em 2.
Estas equações apresentam a seguinte forma:
(5)
(6)
Com
Tg 1, Tg 2 sendo as Tg’s dos componentes 1 e 2, respectivamente.
Tg 1,b e Tg 2,b sendo as Tg’s dos componentes 1 e 2 na blenda, respectivamente.
Além da equação de Fox, tem-se também a equação de Gordon-Taylor (1952):
(7)
Sendo
k1 uma constante que está relacionada com a interação dos segmentos de cada
componente.
2.6.2. Depressão da Tm (
Quando uma blenda é formada por um polímero cristalizável e outro amorfo
não-cristalizável, a Tm é usada para verificar a sua miscibilidade. Se os componentes
forem considerados miscíveis, a temperatura de fusão do composto será mais baixa do
que a do componente cristalizável puro. Os efeitos cinéticos e morfológicos para se

15
chegar a esta conclusão, advêm principalmente do fato de que os cristais são formados
em temperaturas abaixo da temperatura de fusão de equilíbrio . Há também um
efeito termodinâmico, onde se conclui que o potencial químico de um polímero diminui
com a adição de diluente miscível, e no caso de um polímero cristalizável tal
diminuição resulta na queda da temperatura de fusão de equilíbrio (Silvestre et al.,
1996; Shiomi et al., 2001).
Vários pesquisadores desenvolveram equações envolvendo as temperaturas de
fusão da blenda (Tm) e do componente puro . A equação mais usada foi
desenvolvida por Nish,Wang e Kwei (1975) e pode ser representada da seguinte forma:
(8)
Considerando
Temperatura de fusão de equilíbrio;
V números de cadeias;
m1 e m2 representam o grau de polimerização do componente 1 e 2
respectivamente;
1 e 2 fração volumétrica do polímero 1 e do polímero 2 respectivamente;
parâmetro de interação polímero-polímero.
Para polímeros m1 e m2 são muito elevados e desta forma a Equação 8 pode ser
simplificada resultando na Equação 9:
(9)
Tem-se que
, em T = Tm, B é a densidade de energia de interação.
ΔH2 é a entalpia de fusão por mol de unidade repetitiva do polímero
cristalizável;
O índice u representa o valor por mol da unidade polimérica.

16
Se no gráfico de versus a curva resultante for uma linha reta,
pode-se concluir que ambos os componentes são miscíveis (Hage Jr., 1989).
Muitos autores têm encontrado dificuldades em correlacionar a equação de Nish,
Wangf e Kwei (1975) com seus dados porque geralmente a curva de
versus apresenta um intercepto com os eixos diferente de 0 (zero), isto ocorre devido
a fatores limitantes da teoria de Flory-Huggins (1914) que considera que é
independente da composição, e é incapaz de predizer a temperatura crítica de solução
superior (LCST) e a dependência da queda do ponto de fusão com fatores morfológicos.
Estes fatores podem ser muito importantes para algumas blendas (Utracki, 1990).
Kwei e Frisch (1978) desenvolveram uma equação para a queda do ponto de
fusão em função da composição, levando em consideração os efeitos morfológicos
sobre a Tm:
(10)
Na qual
C é uma constante que representa os fatores morfológicos.
Nas pesquisas realizadas por Plivelic et al. (2005), em seu estudo sobre blendas
composta por policaprolactona/ poli(ε-caprolactona) - PCL/PECL, observaram que há
uma leve depressão do ponto de fusão com o aumento do teor de PECL . Esse resultado
é esperado considerando que o sistema é miscível, ainda que o parâmetro de interação
polímero/polímero de Flory-Huggins seja próximo a zero.
A determinação do parâmetro de interação em blendas parcialmente miscíveis
é estimada pela queda da Tm, é adimensional e caracteriza a energia de interação para
moléculas diluentes dividido por kT, sendo representada pela equação abaixo:
(11)
Sendo
z o número de moléculas vizinhas;
w a energia requerida para formar os contatos 1-2.

17
A temperatura de fusão pode ser utilizada para calcular o parâmetro de interação
conforme apresentado na Equação 8. Observa-se que para valores negativos de ,
ocorre uma diminuição no ponto de fusão, já para valores positivos de , é observado
um aumento no ponto de fusão.
2.6.3. Blendas formadas por componentes miscíveis no estado fundido e na fase
amorfa
No caso de blendas miscíveis, ambos os componentes da blenda perdem parte de
sua identidade e, geralmente, as propriedades finais representam uma média das
propriedades de ambos os componentes da blenda (Quental et al., 2009). Há a
incorporação da parte não cristalizável durante a cristalização dentro das regiões
interlamelares do esferulito em crescimento. A espessura da fase amorfa e da interface
aumenta com a composição. Para estas blendas a estrutura é constituída de lamelas
cristalinas separadas por regiões amorfas e regiões de transição contendo uma mistura
homogênea dos dois componentes, como ilustrado na Figura 4 (Silvestre et al., 1996).
Figura 4: Representação da estrutura de uma blenda binária com uma fase amorfa
homogênea e um componente parcialmente cristalizado. (Silvestre et al., 1996)
O processo de solidificação tende a separar os componentes, resultando em
segregação do diluente amorfo, e pode ser visto na Figura 5. Existem vários modelos de
segregação. A segregação interlamelar (Figura 5.1), mostra o diluente confinado dentro
das áreas interlamelares; A segregação interesferulítica (Figura 5.2), mostra o diluente
segregado a uma longa distância das regiões dos empacotamentos (bundles) de lamelas.

18
A segregação interfibrilar (Figura 5.3), mostra o componente amorfo segregado a uma
maior distância entre as regiões de esferulitos (Morales, 1994).
Figura 5: Exemplos de morfologia de acordo com o modo de segregação do
componente amorfo. (1) segregação interlamelar, (2) segregação interesferulítica, (3)
segregação interfibrilar. Morales, 1994.
É comum, em polímeros cristalizáveis puros, mantendo uma temperatura fixa,
que haja um crescimento esferulítico constante durante o processo de solidificação, pois
a parte não-cristalizável permanece presa dentro das regiões interlamelares ou
interfibrilares. Já quando é segregada dentro das zonas interesferulíticas, a fase que
representa o estado fundido fica, praticamente, composta pelo componente amorfo,
resultando numa mudança contínua da fase líquida em contato com o cristal (Di
Lorenzo, 2003).
Para a realização destes estudos, foram tomados alguns trabalhos já publicados
que serviram de referência para análises e conclusões, cujos principais resultados são
relatados a seguir:
Silvestre et al. (1987-A) reportaram que nas blendas poli(óxido de
etileno)/poli(metacrilato de metila)atático - PEO/aPMMA e poli(óxido de
etileno)/poli(metacrilato de metila)sindiotático - PEO/sPMMA, a espessura da fase
amorfa e da interfase aumentaram com a adição do PMMA, como resultado da presença
de material não cristalizável nas regiões interlamelares e interfibrilares do PEO.
(1) (2) (3)
15-2
5 n
m
5-25
µm
5-
25µ
m

19
Avella et al. (1991) estudaram o comportamento de cristalização das blendas
PHB/PEO que são miscíveis no “melt”, porém observou-se uma separação de fases com
a cristalização do PHB. Foi verificado que durante o crescimento esferulítico do PHB,
moléculas de PEO foram presas nas regiões interlamelares, formando uma solução
homogênea com o PHB que não cristalizou. Com as moléculas de PEO presas nas
regiões interlamelares a cristalização só foi observada em temperaturas mais baixas,
pois a solução amorfa da blenda de PHB/PEO possui uma Tg mais alta do que a do PEO
puro, o que dificultou a difusão das moléculas de PEO.
Greco e Martuscelli (1989) verificaram nas blendas de PHB/PVAc, miscíveis no
estado fundido, que a estrutura de fases no estado sólido é caracterizada pela presença
de uma fase homogênea amorfa nas regiões interlamelares constituída de moléculas de
PVAc e cadeias não cristalizadas de PHB.
2.6.4. Blendas cujos componentes são parcialmente miscíveis ou imiscíveis
Quando dois polímeros são misturados, seja qual for o método de mistura, o
resultado mais comum é a obtenção de um sistema imiscível. No entanto, a
imiscibilidade é um fator esperado, e muitas vezes desejado (Paul et al., 1988). Em
blendas imiscíveis, tem-se um sistema heterogêneo, onde as propriedades dos
componentes que constituem a blenda permanecem presentes. Algumas propriedades de
um dos componentes, em certa extensão, podem ser camufladas pelas propriedades do
outro componente (Quental et al., 2009).
A blenda polimérica imiscível pode apresentar diferentes tipos de morfologias
de fases, entre elas, uma fase dispersa numa matriz contínua, ou ambas as fases
simultaneamente contínuas, conhecida como morfologia co-contínua. (Utrack e Weiss,
1989).
Nestas blendas o componente não-cristalizável é segregado como uma fase
dispersa. Os domínios separados são compostos por polímero amorfo puro se os
componentes forem completamente imiscíveis, ou podem conter pequenas quantidades
do polímero cristalizável quando existe algum grau de miscibilidade. Os domínios da
fase dispersa podem estar presentes no “melt”, antes da ocorrência da cristalização, ou
podem se desenvolver após a solidificação do componente cristalizável, dependendo do
diagrama de fase do sistema e das condições de solidificação. Em blendas binárias onde
os componentes são imiscíveis ou parcialmente miscíveis, o principal efeito do

20
polímero não-cristalizável depende da perturbação causada pela presença das partículas
dispersas na frente de crescimento esferulítico (Di Lorenzo, 2003).
Na mistura de dois componentes de natureza química diversa, de qualquer
dimensão ou forma, para que ocorra uma interação, é essencial a existência de áreas de
contato entre eles. Quanto maior for essa área, tanto maior será a possibilidade de
ocorrer uma interação de natureza física, química ou físico-química. Assim, em uma
mistura polimérica, os componentes podem interagir somente na interface, ocorrendo
mais de uma fase imiscível com compatibilidade parcial (Feitosa, 2009).
Quando os componentes da blenda são parcialmente miscíveis, a segregação do
polímero amorfo e a mudança na composição da fase líquida podem causar separação
de fases líquido-líquido. O gradiente de concentração na interface fusão-cristal, cuja
espessura depende das taxas relativas de cristalização e difusão, produz uma diminuição
da taxa de crescimento esferulítico com o tempo (Di Lorenzo, 2003).
2.7.Teoria da taxa de crescimento
A taxa de crescimento de um cristal está relacionada à temperatura e se
desenvolve de duas maneiras denominadas de regimes I e II. O regime I é encontrado
em altas temperaturas de cristalização, onde há um alto crescimento cristalino e baixa
nucleação, formando superfícies mais lisas. Já no regime II, ocorre o contrário, é
realizado em baixas temperaturas de cristalização, a nucleação é mais frequente e o
crescimento cristalino é menor, formando superfícies rugosas devido à múltipla
nucleação (Basset, 1981).
A taxa de crescimento referente ao regime I é representada pela equação a
seguir:
(12)
Com:
Sendo:

21
a energia livre superficial da face paralela à cadeia molécula, por unidade de
área superficial lateral;
e a energia livre superficial da face perpendicular à cadeia molécula, por
unidade de área superficial conectada a dobra da cadeia;
U* energia apresentando valor próximo a 6KJmol-1
;
j1 tem a forma exp – (ΔF*/RT);
ΔF energia livre de ativação para o processo de transporte através da interface
líquido superesfriado – cristal.
Para o regime II, sua taxa de crescimento é descrita pela Equação 13:
(13)
Com :
A Equação 14 representa a equação geral para o crescimento esferulítico
(14)
Com:
Para
h calor de fusão por unidade de volume na temperatura de fusão de equilíbrio.
Caso a taxa de nucleação e crescimento primários sejam calculados
separadamente, estes irão aumentar com a redução da temperatura para temperaturas
próximas ao ponto de fusão. Isso se dá devido à diminuição do superesfriamento, que
reduz a energia necessária para a nucleação primária, que limita a taxa de cristalização
(Skoglund e Frasson, 1996).

22
2.8. Cinética de cristalização a frio
O estudo da cinética de cristalização é importante, pois as propriedades finais
dos polímeros e de suas blendas dependem do grau de cristalinidade. Com esse estudo,
pode-se analisar como se desenvolveu o mecanismo de nucleação e crescimento dos
cristais poliméricos.
Um polímero inicia o seu processo de cristalização a frio quando, a partir do seu
estado sólido amorfo, for aquecido em temperaturas acima da sua Tg e com isso, as
moléculas tem energia cinética suficiente para dar início ao movimento molecular.
A temperatura de cristalização situa-se abaixo da Tm e acima da Tg, e o processo
é tanto mais fácil de ocorrer quanto menor for a massa molar do material. Esse
fenômeno, chamado de cristalização a frio pode desenvolver estruturas esferulíticas
normais, e é típico de polímeros como PET, PEN, poli(sulfeto de fenileno) - PPS ou
poli(éter éter cetona) - PEEK (Nogales et al., 2001).
A cristalização a frio pode ser facilmente observada por DSC, como um pico
exotérmico entre Tg e Tm, como mostrado na Figura 6.
Figura 6: Curva típica de DSC mostrando um pico de cristalização do PET. Dados da
autora.
A cristalização a frio resulta na formação de cristais com uma estrutura
imperfeita, mas que pode ser alterada submetendo-se o material a uma temperatura
maior que Tg e menor que Tm.
ENDO
Pico de cristalização

23
Quando um material é bruscamente resfriado e posteriormente cristalizado a frio,
seu grau de cristalinidade pode ser bastante elevado, situando-se relativamente próximo
da amostra resfriada lentamente, mas com lamelas bem mais finas. A cristalização a frio
é altamente recomendável quando se deseja um material com maior resistência
mecânica (Baijal, 1982).
Groeninckx et al. (1980) analisaram a formação de estruturas morfológicas no
PET em função da temperatura de cristalização a frio, observando que em baixas Tcs
(100 – 150°C) ocorreu a formação de lamelas altamente ramificadas com pequenas
dimensões laterais; a temperatura de 200°C formaram-se estruturas lamelares
ramificadas, mas com o desenvolvimento de um feixe de lamelas com orientação radial
e em altas Tc’s (215 – 245°C) estruturas típicas lamelares foram observadas.
Também foi verificado, em seu estudo sobre a cristalização isotérmica do
polihidroxibutirato-copolímero-hidroxivalerato - PHB-co-HV que as condições de
cristalização são tão importantes quanto à adição do componente HV na amostra,
observando-se que a cristalização aumenta com o aumento da concentração de HV. Já o
teor dos cristais diminui com o aumento da temperatura de cristalização (Shuwen et al.,
2002).
Em seu estudo sobre a cristalização do poli(L-láctico) - PLLA quando
adicionado a este uma pequena quantidade de PHB, Yun et al. (2008) verificaram que a
cristalização de um componente afeta a morfologia, a cristalização e as propriedades
mecânicas do outro.
Pingping et al. (2000) avaliaram o efeito da adição CaCO3 ao PET durante a
cristalização não-isotérmica a frio, observando tanto um deslocamento do pico de
cristalização para temperaturas mais baixas, como também um aumento do grau de
cristalinidade do PET após adição do CaCO3, o qual promoveu um aumento da taxa de
nucleação do PET durante o processo de cristalização.
2.8.1.Cinética de cristalização isotérmica a frio
Na cinética de cristalização isotérmica a frio se avalia a formação de cristais do
polímero quando submetido a uma temperatura constante.

24
Essa cinética pode ser estudada através de várias teorias, como é o caso de
Avrami, (1939); Evans, (1945); Flory et al. (1964); entre outros. Neste trabalho foi
utilizada a teoria de Avrami (1939), (1940), (1941).
2.8.1.1. Cinética de cristalização isotérmica a frio – Teoria de Avrami
A cinética de cristalização isotérmica a frio avalia a formação de uma fase
cristalina no interior do material fundido a uma temperatura constante.
Em sua teoria, Avrami considera que a nucleação ocorre ao acaso, a taxa de
crescimento cristalino e a taxa de nucleação são constantes. A primeira equação
aplicada para esse estudo define a variação da cristalinidade com o tempo, sendo
representada a seguir:
(15)
Sendo
X(t) a fração de material cristalizado no tempo t;
K a constante de velocidade, função das velocidades de nucleação e
crescimento;
n o expoente de Avrami e descreve o mecanismo de cristalização, fornecendo
informações qualitativas sobre a natureza dos processos de nucleação e
crescimento.
A fração cristalina é definida como função do tempo de cristalização, e obedece
a seguinte formulação:
(16)
Sendo:
dHc/dt a taxa de evolução de calor;
t0 e t∞ representam o tempo inicial e final da cristalização, respectivamente.
25
A Equação 16 pode ser transformada na Equação 17, já que podemos considerar que as
integrais representam áreas.
(17)
Com
At sendo a área sobre a curva de cristalização do instante zero ao instante t;
A∞ representa a área total sobre a curva de cristalização.
A grandeza X(t) relaciona o grau de cristalinidade desenvolvido em cada instante,
com a cristalinidade total associada a determinadas condições experimentais; com isso o
valor normalizado de X(t) varia de zero a um.
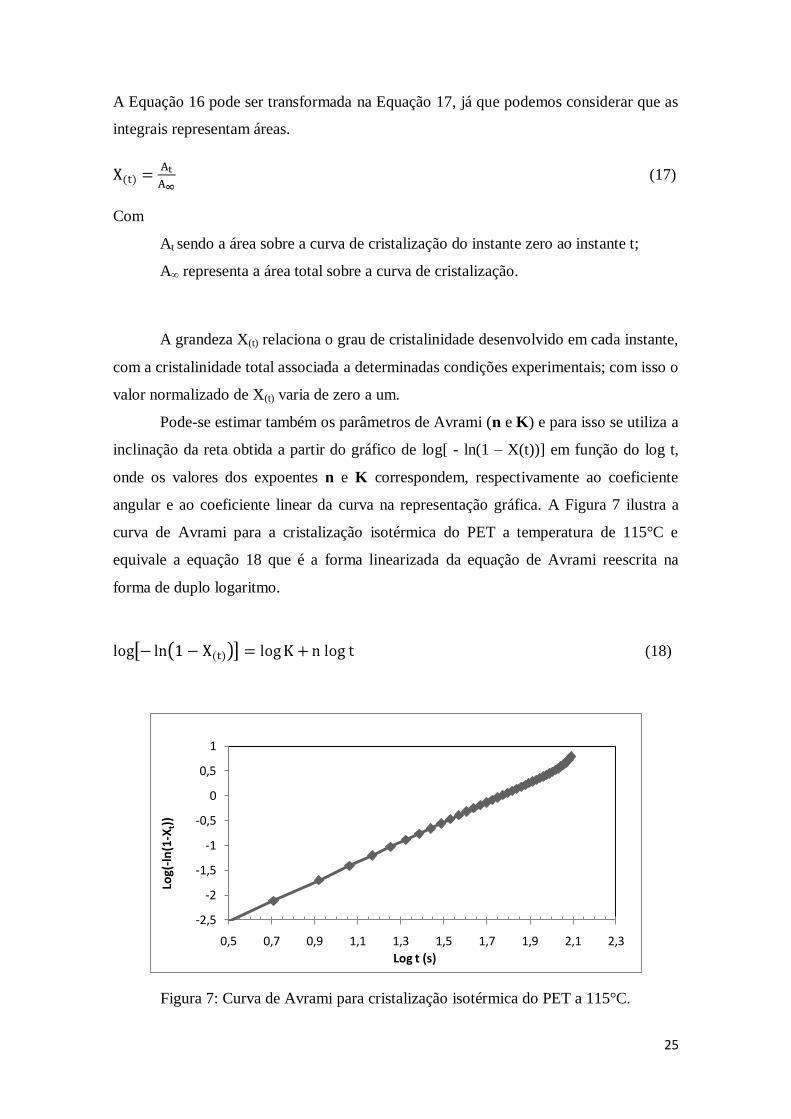
Pode-se estimar também os parâmetros de Avrami (n e K) e para isso se utiliza a
inclinação da reta obtida a partir do gráfico de log[ - ln(1 – X(t))] em função do log t,
onde os valores dos expoentes n e K correspondem, respectivamente ao coeficiente
angular e ao coeficiente linear da curva na representação gráfica. A Figura 7 ilustra a
curva de Avrami para a cristalização isotérmica do PET a temperatura de 115°C e
equivale a equação 18 que é a forma linearizada da equação de Avrami reescrita na
forma de duplo logaritmo.
(18)
Figura 7: Curva de Avrami para cristalização isotérmica do PET a 115°C.
-2,5
-2
-1,5
-1
-0,5
0
0,5
1
0,5 0,7 0,9 1,1 1,3 1,5 1,7 1,9 2,1 2,3
Log(
-ln
(1-X
t))
Log t (s)

26
Os valores do expoente n determinados a partir da Equação 18 são apresentados
na Tabela 1:
Tabela 1: Valores do expoente de Avrami para vários tipos de nucleação e
crescimento.(Silva, 1991)
Geometria de
crescimento
Nucleação
instantânea
Nucleação
homogênea
Nucleação
heterogênea
Esferulito 3 4 4 > n < 3
Disco 2 3 3 > n < 2
Bastão 1 2 2 > n < 1
Para o caso mais comum que é o da cristalização esferulítica, a constante de
velocidade K pode ser determinada por:
(19)
Com:
c e a são as densidades das fases cristalina e amorfa, respectivamente;
G é a velocidade de crescimento esferulítico;
N0 é o número de núcleos heterogêneos por unidade de volume;
Xc∞ é o grau de cristalinidade final (considerando tempos longos);
N’( ) é a velocidade de nucleação homogênea.
Os valores para o expoente de Avrami n, geralmente são encontrados fracionados
e podem ser considerados desvios que são atribuídos a processos de cristalização em
que ocorrem os seguintes eventos:
Crescimento simultâneo ou consecutivos de dois tipos diferentes de
estruturas cristalinas;
Formação simultânea de unidades com crescimento similar a partir de
núcleos homogêneos ou heterogêneos;
Para n = 3 (heterogênea)
Para n = 4 (homogênea)

27
Sobreposição dos efeitos de cristalização primária e secundária.
A constante de velocidade K se relaciona com a temperatura de cristalização da
seguinte forma: quanto menor for a Tc maior será o K. Há também uma relação entre K
e n que é dada pela Equação 20:
(20)
Sabe-se que
T0,5 é o tempo necessário para que 50% do material seja cristalizado.
Na cristalização isotérmica, trabalha-se com o tempo necessário para se obter 50%
da cristalização, pois em determinadas temperaturas, o tempo para se chegar próximo a
100% de cristalização pode ser muito longo. Esse tempo é definido como t0,5 e é
determinado a partir de parâmetros cinéticos como mostra a Equação 21 (Skoglund et
al., 1996).
(21)
Portanto a taxa de cristalização será definida por:
(22)
Skoglund et al. (1996) relatou que o tempo necessário para se completar metade
do processo de cristalização é determinado pela competição entre dois processos, a
nucleação e a mobilidade molecular. Em altas temperaturas (baixos superesfriamentos),
a taxa de nucleação é que controla o processo de nucleação, já para baixas temperaturas
a mobilidade molecular é que faz esse controle. Quando o fator dominante é a taxa de
nucleação, o valor de t0,5 aumenta com a temperatura, diminuindo a taxa de
cristalização.

28
A teoria de Avrami foi inicialmente desenvolvida para pequenas moléculas, mas
como os polímeros desenvolvem núcleos cristalinos no seio da fase amorfa formando
macromoléculas, essa teoria precisa de algumas considerações para ser aplicada.
Avrami assumiu que os núcleos podem se desenvolver em uma, duas ou três
dimensões dando origem a formação de bastão, disco ou esfera respectivamente, com a
taxa de crescimento sendo considerada constante durante todo o processo de formação
dos cristais. Pressupõe-se que não há contato entre os núcleos em crescimento, pois o
crescimento cristalino é retardo a medida que os núcleos encontram regiões já
cristalizadas, definida como cristalização primária (Schultz, 1974).
O local de formação dos núcleos é aleatório, a densidade da fase cristalina é a
mesma para todas as estruturas em crescimento e que não ocorre rearranjo cristalino
após a cristalização. Com o procedimento da cristalização os cristalitos vizinhos
começam a colidir (cristalização secundária), então a cristalização se desvia da equação
de Avrami e ocorre uma diminuição na taxa de crescimento. Para cristalização abaixo
desse limite, dados experimentais mostram um comportamento linear de acordo com a
Equação 20 (Skoglund et al., 1996).
Skoglund et al. (1996) calcularam os efeitos de algumas dessas limitações para a
equação de Avrami no parâmetros n e K , concluindo que se trata de uma aproximação
bem razoável para a maioria dos sistemas, exceto quando há mudança na densidade
radial.
Segundo o modelo desenvolvido por Avrami, nas curvas referentes ao percentual
cristalino em função do tempo com diferentes temperaturas, há a sobreposição de uma
curva na outra usando um fator de deslocamento que envolve o meio tempo de
cristalização.
A Figura 8 mostra um exemplo de como se dá a dependência da constante de
velocidade K com a temperatura. A taxa de nucleação máxima ocorre mais próximo da
Tg e a de crescimento máxima, próxima a Tm, já a taxa de cristalização máxima ocorre a
uma temperatura entre Tg e Tm.
No intervalo em que há a cristalização, perto do ponto de fusão, a velocidade de
cristalização é muito lenta. Quando a temperatura é mais baixa, essa velocidade
aumenta. Para muitos polímeros em temperaturas abaixo da Tm, por ser muito elevada,
fica quase impossível de se detectar a temperatura na qual se atinge a velocidade
máxima (Billmeyer, 1984).

29
Figura 8: Velocidade de cristalização global, K, em função da temperatura de
cristalização. (Silva, 1991)
A taxa de cristalização é dependente da temperatura e essa dependência é
determinada pela influência da nucleação e destruição dos núcleos pelo movimento
térmico. Assim, o grau de superesfriamento ΔT (ΔT = - Tc) tem grande importância
para a cristalização. Wellen (2007) descreve que há uma temperatura ótima de
cristalização, na qual acima dela a cristalização é limitada pela baixa nucleação, e
abaixo da mesma é limitada pela alta viscosidade do polímero fundido, dificultando o
movimento molecular. Neste mesmo estudo, foi verificado através da cristalização a frio
do PET e das blendas PET/PS e (poli(taraftalato de etileno)/estireno acrilonitrila)
PET/SAN, que a presença de uma baixa concentração de PS ou SAN, retarda
significativamente a cristalização do PET.
Kong et al. (2002) também em estudo sobre as blendas formadas por
poli(teraftalato de etileno)/policarbonato - PET/PC concluíram que a presença de PC
retarda a cristalização do PET, sendo que o expoente de Avrami n apresentou valores
próximos a 3, indicando uma morfologia esferulítica formada por nucleação
heterogênea.
Yun et al. (2008) estudaram blendas de PHB/PLLA e sugeriram que o PLLA
apresenta miscibilidade limitada com o PHB quando a concentração de PHB na mistura
é de 25%.
Hay et al. (2000) estudaram o efeito que o PVAc provoca na mistura com PHB e
concluíram que a adição de PVAc diminuiu o estado de fragilidade do PHB quando
armazenado por longo períodos de tempo.
Tra
nsi
ção
vít
rea
Po
nto
de
fusã
o
Temperatura de cristalização
K

30
Vários grupos de pesquisa realizaram estudos sobre a cinética de cristalização
isotérmica com a finalidade de estudar o comportamento dos polímeros segundo vários
aspectos como: diferentes modificações estruturais (Li et al., 1999), variação de massa
molar e presença de agentes nucleantes (Van Antwerpen e Van Krevelen, 1972),
diferentes níveis de orientação (Dessai e Abhiraman, 1985), diferentes condições de
deformação (Myung et al., 2001) e diferentes temperaturas de cristalização (Minakov et
al. 2004).
2.8.2.Cristalização não isotérmica a frio
É o estudo realizado da cinética de cristalização em um ambiente em contínua
mudança de temperatura com o tempo.
Estudos realizados por Di Lorenzo et al. (2003) mostraram que a taxa de
cristalização não isotérmica varia com a temperatura. A taxa de
aquecimento/resfriamento também influi na taxa de cristalização.
Para analisar a cinética de cristalização não isotérmica, existem duas teorias que
definem bem esse tipo de cristalização: A teoria de Ozawa (Ozawa, 1971) e a de Mo
(Liu et al., 1997-A e 1997-B).
É importante ressaltar que a teoria de Ozawa apresenta limitações relacionadas
com a determinação da morfologia. Como este modelo se baseia em medidas feitas em
diferentes taxas de aquecimento/resfriamento é difícil determinar a morfologia, pois esta
varia em função deste parâmetro (Wellen, 2007).
Li et al. (1999) pesquisaram o comportamento de cristalização de copolímeros
poli(teraftalato de etileno)-copolímero-poli(éter imida) - PET-co-PEI nas composições
(100/0), (90/10) e (85/15). Para o estudo em condições não-isotérmicas, a teoria de
Ozawa descreveu de forma satisfatória o processo de cristalização. Através da teoria de
Ozawa os autores puderam concluir que o processo de cristalização não-isotérmico dos
copolímeros é dominado pela nucleação heterogênea e observaram também que a
presença de PEI atua retardando a velocidade de cristalização do PET nos copolímeros.
Wellen (2007) relatou que a teoria de Ozawa empregada no estudo da cinética de
cristalização não isotérmica do PET apresentou boa linearidade para algumas taxas de
aquecimento utilizadas. A constante de velocidade K(T) diminuiu com a temperatura no
inicio da cristalização a frio, uma tendência oposta à observada pelas condições
isotérmicas, sendo que o expoente m diminuiu com a temperatura para todas as

31
composições estudadas, concluindo que a teoria de Ozawa pode ser utilizada para o
estudo da cinética de cristalização não isotérmica do PET.
Jabarian (1987) reportou um estudo sobre o comportamento de cristalização do
PET em condições não-isotérmicas em função da massa molecular, do sistema de
catalisador na policondensação e das condições de polimerização. Ele provou que é
possível obter uma taxa de resfriamento mínima requerida para produzir PET sem
cristalinidade detectável através de um estudo sobre cristalização em condições não
isotérmicas. Os requisitos de resfriamento para produzir PET não-cristalino dependem
da massa molecular e, mais importante ainda, do sistema de catalisador usado na etapa
de policondensação. A ausência de cristalinidade é um requerimento necessário para
produzir produtos transparentes.
Em estudo realizado por Li e Lee (1995) sobre a cinética de cristalização não-
isotérmica, foi verificado que o comportamento anisotrópico das fases nemática e
esmética de poliésteres termotrópicos, com espaçadores metilênicos na cadeia principal,
é dependente da velocidade de resfriamento. Foi verificado também que para faixas de
temperatura altas, a cinética de cristalização não satisfaz a teoria de Ozawa, por outro
lado satisfaz, perfeitamente, para faixas de temperaturas mais baixas.
Sajkiewicz et al. (2001) realizaram um estudo sobre a cristalização não
isotérmica do PET a partir do estado fundido e chegaram à conclusão que o modelo de
Ozawa pode ser usado no estudo da cristalização não isotérmica do PET apenas quando
baixas taxas de resfriamento foram empregadas, já que para taxas de resfriamentos
superiores a 20°C.min-1
foram observados grandes desvios de linearidade. Os autores
notaram também desvios do modelo de Ozawa no início e no término do processo de
cristalização não isotérmico, os quais foram atribuídos a constrangimentos espaciais do
crescimento esferulítico.
A validade da teoria de Ozawa foi também pesquisada por Kong e Hay (2002),
estudando a cristalização não isotérmica do copolímero segmentado PET-co-PEO e do
homopolímero PEO. Eles verificaram que a equação de Ozawa descreve com sucesso o
comportamento de cristalização do homopolímero PEO, mas há falhas na descrição da
cristalização do copolímero devido à ocorrência de cristalização secundária nos estágios
finais do processo de cristalização que não pode ser desprezada.
Neste trabalho o estudo aplicado será a teoria de Mo.

32
2.8.2.1. Cinética da cristalização não isotérmica a frio – Teoria do Mo
A teoria desenvolvida por Mo (Liu et al., 1997-A e 1997-B) possibilita o estudo
da cinética de cristalização não isotérmica e foi desenvolvida combinando conceitos
presentes na teoria de Avrami e na teoria de Ozawa.
Durante o processo de cristalização não isotérmico a relação entre o tempo de
cristalização e a temperatura de cristalização é dada pela Equação 23:
(23)
Com:
T é a temperatura no tempo t;
To é a temperatura no início da cristalização (t=0);
é a taxa de aquecimento/resfriamento.
A cristalinidade relativa está correlacionada com a taxa de
aquecimento/resfriamento ( ) e com o tempo (t) (ou temperatura T), e a relação entre o
e t pode ser derivada para um dado grau de cristalinidade. Partindo dos conceitos
presentes na Equação 14, na Equação 22 e na Equação 23, Mo obteve uma nova
equação cinética para cristalização não isotérmica representada pela Equação 24:
(24)
A Equação 24 pode ser reescrita da seguinte forma:
(25)
Assumindo:
F(t) está relacionado com o valor da taxa de aquecimento/ resfriamento e pode
ser obtido através da equação 26:
a é a razão entre o expoente de Avrami (n) e o expoente de Ozawa (m), ou seja,
.

33
(26)
Os parâmetros de Mo (F(T) e a) podem ser estimados para um dado grau de
cristalinidade relativa através da reta obtida a partir do gráfico de Log t versus Log ,
com –a sendo a inclinação e Log F(T) o intercepto da reta. Um gráfico típico está
mostrado na figura 9.
Figura 9: Curva de Mo para a cristalização não isotérmica a frio do PET para Xt=10%.
Dados da autora.
Na teoria de Ozawa analisa-se a cristalinidade em diferentes temperaturas, já
quando se aplica a teoria de Mo pode-se avaliar a cristalização em determinados
intervalos de cristalização. Pela teoria de Ozawa, dependendo das taxas de
aquecimento/resfriamento empregadas, uma amostra pode estar iniciando a cristalização
enquanto a outra pode estar nos últimos estágios da cristalização, o que geralmente
promove desvios de linearidade devido às diferenças morfológicas encontradas em
ambas as amostras (Wellen, 2007).
Gao e Li (2004) estudaram a cinética de cristalização não isotérmica do
polietileno de média densidade grade bimodal - BMDPE utilizando a teoria de Ozawa e
a teoria de Mo. Ao utilizar as curvas de Ozawa, o método foi considerado falho,
conseguindo apenas um método correto ao se trabalhar com a teoria de Mo.
Wellen (2007) estudou também a teoria de Mo para a cinética de cristalização
não isotérmica de blenda de PET/SAN e PET/PS, observando que a cristalização ocorre
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
-0,6 -0,4 -0,2 -1E-15 0,2 0,4 0,6 0,8
Log
Log t (min)
PET
Xc=10%

34
em dois estágios, cristalização primária e secundária e a constante de velocidade K’(T)
aumentou com as taxas de aquecimento.
Qiao et al. (2000) analisaram o comportamento de cristalização não isotérmico
do poli(3-dodecil-tiofeno) - P3DDT e do poli(3-octadecil-tiofeno) - P3ODT pelas
teorias de Ozawa e Mo. A teoria de Ozawa não se mostrou adequada, enquanto que a
teoria de Mo apresentou bons resultados para as duas blendas.
Zheng et al. (2005), analisaram a cinética de cristalização não isotérmica de
blendas poli(p-dioxanona)/poli(etilenoglicol) - PPDO/PEG e mais uma vez a teoria de
Ozawa falhou, enquanto que a teoria de Mo descreveu perfeitamente o processo de
cristalização das blendas.

35
3.PROCEDIMENTO EXPERIMENTAL
3.1. Materiais
O PHB utilizado foi fornecido pela PHB Industrial do Brasil. Já o PET de
procedência da Rhodia foi do tipo grau garrafa (Rhopet S78).
3.2. Obtenção das blendas PET/PHB
O PHB foi seco em estufa com circulação de ar a 80°C por um período de tempo
de 14 horas, e o PET foi seco a 110°C durante 6 horas.As blendas foram obtidas no
equipamento de mistura (misturador interno Rheomix 600) acoplado a um reômetro de
torque System-90 da Haake-Büchler, operando com rotores do tipo roller a uma
temperatura de 260°C com rotação de 60 rpm durante 10 minutos. Ao saírem da câmera
de mistura as amostras foram imediatamente resfriadas em banho de gelo. Para cada
composição foi utilizada 50 gramas de material.
Foram preparadas blendas com as seguintes composições:
PET puro;
PET/PHB (99,5/0,5);
PET/PHB (99/1);
PET/PHB (97/3).
3.3. Caracterização das amostras
3.3.1. Calorimetria exploratória diferencial (DSC)
As transições endotérmicas e exotérmicas ocorridas durante o aquecimento e
resfriamento das amostras foram obtidas por calorimetria exploratória diferencial (DSC)
utilizando-se um equipamento Shimadzu DSC-50, cujas condições de análise e
obtenção de dados foram citadas anteriormente no item obtenção das blendas. Para o
estudo da cinética de cristalização isotérmica a frio foram aplicadas as temperaturas de
115°C, 120°C, 125°C, 130°C, 135°C, 140°C, 145°C e 150°C. Já para o estudo da
cinética de cristalização não isotérmica a frio foram aplicadas taxas de aquecimento de

36
1°C/min; 2,5°C/min; 5°C/min; 7,5°C/min; 10°C/min; 15°C/min; 20°C/min e 30°C/min.
A atmosfera para análise foi o ar.
3.3.2. Estudo da cinética de cristalização a frio
O estudo da cinética de cristalização foi realizado por DSC. O DSC é bastante
utilizado na detecção das transições térmicas que ocorrem durante o aquecimento e
resfriamento de materiais poliméricos. Esta técnica foi empregada neste trabalho com a
finalidade de determinar os parâmetros cinéticos do processo de cristalização do PET e
da blenda PET/PHB.
3.3.3. Cinética de cristalização isotérmica a frio – Teoria de Avrami
Para a obtenção dos dados, houve um aquecimento rápido em torno de
99,9°C/min da temperatura ambiente até a temperatura de cristalização. Como já foi
citada antes, a faixa de temperatura na qual foram realizadas as análises foi de 115°C
até 150°C. As amostras foram embrulhadas em papel alumínio para melhor
transferência de calor. A atmosfera de análise foi o ar, e o peso das amostras ficou entre
5mg e 7mg.
Os parâmetros cinéticos da cristalização isotérmica foram obtidos empregando-
se a teoria de Avrami, representada pela Equação 15 (Avrami 1939, 1940, 1941).
Pela teoria de Avrami, pode-se verificar o grau de cristalinidade em função do
tempo de cristalização. Essa fração cristalina é definida por X(t) e relaciona quanto de
material se cristaliza a cada instante, segundo as Equações 16 e 17.
A partir das informações obtidas com os gráficos gerados por log t em função de
X(t), foram calculados os tempos de início, meio e fim da cristalização. A taxa de
cristalização (C0,5) foi determinada como sendo o inverso de t0,5 como já mencionado, e
representado pela Equação 22.
Os parâmetros de Avrami foram obtidos linearizando a equação de Avrami,
obtendo-se a Equação 18.
Os parâmetros n e K de Avrami foram obtidos a partir do gráfico Log (- Ln( 1 –
Xt )) em função do Log t. Partindo-se da referida figura é possível estimar esses
parâmetros através da inclinação da reta, na qual n é o coeficiente angular e K o
coeficiente linear da referida reta.

37
3.3.3.1. Energia de ativação para a cristalização isotérmica a frio
Para a energia de ativação, a constante de velocidade K é aproximadamente
descrita de acordo com Arrhenius (Cebe et al., 1983), e está representada pela Equação
27.
27
Aplicando a linearização na Equação 27 obtém-se a Equação 28:
28
Sabe-se que:
k0 é um fator pré-exponencial independente da temperatura;
R é a constante dos gases;
ΔE é a energia de ativação da cristalização isotérmica;
Tc é a temperatura de cristalização em Kelvin.
Utilizando-se o gráfico de 1/Tc em função de (1/n) Ln K, e assumindo que ΔE/R
é a inclinação da reta, pode-se chegar a resultados de energia de ativação para a
cristalização isotérmica a frio do PET.
3.3.3.2. Estudo do comportamento de fusão
Para estudar o comportamento de fusão do PET e suas blendas PET/PHB em
todas as composições, partiu-se das endotermas de fusão, a partir de gráficos de Tm (°C)
em função da Tc (°C). Essa análise foi realizada nas amostras com massa entre 5mg e
7mg e aquecidas a aproximadamente 99,9°C/min partindo da temperatura ambiente até
a temperatura de cristalização isotérmica mantendo-se nessa temperatura até não haver
mais nenhuma variação. As amostras foram então aquecidas a 10°C/min a partir da Tc
até a completa fusão da amostra que fica em torno de 253°C. As temperaturas de
cristalização variaram de 115°C até 150°C, com intervalos de 5°C, realizando-se um
ensaio para cada Tc diferente.

38
A temperatura de fusão de equilíbrio foi determinada utilizando-se o método
proposto por Hoffman e Weeks (1962), por extrapolação da condição Tm = Tc.
3.3.4. Cinética da cristalização não isotérmica a frio
O estudo da cinética de cristalização não isotérmica é de grande interesse, já que
os processos industriais ocorrem geralmente sob essas condições. As amostras
apresentaram massa variando entre 5mg e 7mg. O aquecimento, como já relatado
anteriormente, seguiu da temperatura ambiente até a fusão completa que gira em torno
dos 253°C. As taxas de aquecimento foram as seguinte: 1°C/min; 2,5°C/min; 5°C/min;
7,5°C/min; 10°C/min; 15°C/min; 20°C/min e 30°C/min.
Foi também estudada a influência das taxas de aquecimento nas Tg’s, Tc’s e
Tm’s.
A partir do desenvolvimento da cristalinidade com a temperatura, pode-se
observar as temperaturas de início, meio e fim da cristalização, e isso é possível através
do gráfico de Ln T (°C) em função de XT.
3.3.4.1. Cristalização não isotérmica a frio - Teoria de Mo
Os parâmetros cinéticos da cristalização não isotérmica a frio foram obtidos
empregando-se a teoria de Mo (Liu et al., 1997-A e 1997-B) que foi desenvolvida
também para verificar as condições de cristalização não isotérmicas, e combinou
conceitos presentes na Teoria de Avrami e na Teoria de Ozawa.
O grau de cristalinidade (Xt’) está relacionado com a taxa de
aquecimento/resfriamento ( ) e com o tempo de cristalização. A relação entre Xt’ e t
pode ser construída para um dado grau de cristalinidade.
Partindo das Equações 25 e 26, foi construído o gráfico Log t versus Log para
determinar os parâmetros de Mo (F(T) e a), sendo estimados para um dado grau de
cristalinidade relativa através da reta obtida, com (–a) sendo a inclinação e Log F(T) o
intercepto da reta.

39
4. RESULTADOS E DISCUSSÃO
O estudo da cristalização isotérmica e não isotérmica foi baseado nas curvas de
DSC ou termogramas do PET e das suas blendas PET/PHB, onde foi possível observar
as suas transições térmicas, como a temperatura de transição vítrea, cristalização e a
fusão. Partindo da temperatura ambiente, houve um aquecimento rápido de
aproximadamente 99,9°C/min até atingir a temperatura de cristalização que, para o
PET, girou em torno dos 127°C, após a cristalização, realizou-se um aquecimento a uma
taxa de 10°C/min até atingir a temperatura de fusão da amostra (∼253°C). A
temperatura de transição vítrea ficou em torno de 68°C.
A Figura 10 apresenta um exemplo de uma curva de DSC para a cristalização
não isotérmica do PET com o detalhamento dos dados obtidos durante o aquecimento.
40.0 80.0 120.0 160.0 200.0 240.0 280.0Temp [C]
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
mWDSC
Figura 10: Curva de DSC para a cristalização a frio. Dados da autora.
Com:
Tc temperatura do pico de cristalização;
Tg temperatura de transição vítrea;
Tmi Tmf
Flu
xo d
e ca
lor
(u.a
.)
40.0 80.0 120.0 160.0 200.0 240.0 280.0 Temperatura (°C)
ΔTm Tg
Pico
exotérmico
Pico
endotérmico
ΔHm
Tc
Tm
ΔTc
ΔHc

40
Tm temperatura de fusão;
ΔHc calor de cristalização;
ΔHm calor de fusão;
ΔTc intervalo de temperatura do pico de cristalização;
ΔTm intervalo de temperatura do pico de fusão.
4.1. Cristalização isotérmica a frio
A Figura 11 apresenta o comportamento das curvas de DSC para o PET e suas
blendas indicando a temperatura de cristalização e através delas, podemos observar a
influência da Tc no processo de transição de fases. À medida que se aumenta a Tc, há
um deslocamento da curva no sentido de diminuir o tempo de transição de fases,
aumentando a sua velocidade de nucleação cristalina, ou seja, o PET cristaliza mais
rapidamente em temperaturas mais altas. Isso ocorre devido à influência que a Tc exerce
sobre a mobilidade molecular, facilitando a ordenação cristalina (Silva, 1991).
Nesta mesma figura, com a adição do PHB, e verificando a mesma temperatura
para diferentes concentrações, observa-se que uma maior concentração de PHB na
mistura acelera o processo de cristalização, isso deve ocorrer porque o PHB possui um
elevado grau de cristalinidade (Duarte et al., 2002).
Para os diferentes percentuais de PHB no PET, observa-se que uma
concentração de 3% de PHB diminui o tempo de cristalização quando comparado a uma
blenda com concentração menor de PHB.
Na literatura, a adição de um polímero ao PET para formar blendas tem
encontrado resultados variados como o PET/SAN e PET/PS, nesses dois casos houve
um retardo na cristalização com relação ao PET puro. Wellen (2007) concluiu que a
redução na velocidade de cristalização do PET na presença de PS ou SAN pode estar
associada com a solubilidade entre os pares de polímeros PET/PS e PET/SAN, embora
essas misturas sejam essencialmente imiscíveis.
Para Bian et al., 2003 sais de ácido carboxílico, como benzoato de sódio e o-
clorobenzoato de sódio podem perfeitamente acelerar a velocidade de cristalização do
PET.

41
Figura 11: Curvas de DSC para a cristalização isotérmica a frio nas temperaturas e
percentuais de PHB indicados.
Na Figura 12 são apresentadas as isotermas de cristalização em diferentes Tc’s,
e se observa formas muito parecidas, indicando que nessas condições, cada amostra de
mesma concentração apresenta o mesmo mecanismo de cristalização sem
descontinuidade, independente da temperatura. Essa característica pode ser comparada
com as observações feitas por Schultz (1974), ao afirmar que as isotermas possuem uma
forma sigmoidal característica da transformação de fases de polímeros. Observa-se que
todas possuem o mesmo crescimento morfológico, havendo variação apenas na taxa de
nucleação e crescimento cristalino, ou seja, no tempo que a amostra levará para atingir o
seu crescimento.
A formação dos cristais segue a uma taxa de aceleração até atingir um estágio
considerado pseudo-equilíbrio, e a partir desse ponto, começa a cristalização secundária,
onde os cristais são formados a uma taxa muito lenta (Lu e Hay, 2001). A fase inicial do
desenvolvimento cristalino é lento e pode ser observada na seguinte faixa de
cristalinidade 0% < Xt < 30% , nessa intervalo a cristalização é controlada pela
-5
-3
-1
1
0 1 2 3 4 5 6 7 8 9 10
110 °C
125°C
150 °C
Tempo (min)
-5
-3
-1
1
0 1 2 3 4 5 6 7 8 9 10
110°C125°C
150°C
Tempo (min)
Flu
xo d
e C
alor
-5
-3
-1
1
0 1 2 3 4 5 6 7 8 9 10
110°C
125°C
150°C
Tempo (min)
Flu
xo
de
Cal
or
(u.a
.)
-5
-3
-1
1
0 1 2 3 4 5 6 7 8 9 10
125°C
150°C
Tempo (min)
PET
(99,5/0,5)
(97/3) (99/1)
110°C
Flu
xo d
e ca
lor
(u.a
.)
Flu
xo d
e C
alor
(u.a
.)

42
nucleação. Para 30% < Xt < 80% é observado um aumento na taxa de cristalização em
toda a porção linear das isotermas, neste caso a cristalização é controlada pelo
crescimento cristalino. Para Xt > 80% é atingido um estado de pseudo-equilíbrio
(cristalização primária), a partir desse ponto, dá-se início a cristalização secundária que
ocorre a taxas muito lentas por um período de tempo finito (Hwang et al., 1997).
Em se tratando de apenas um componente, variando apenas a temperatura,
mostrado no último gráfico da Figura 13, é possível verificar a possibilidade de
superposição das curvas, como já foi mencionado anteriormente, pois não há diferença
na morfologia de formação dos núcleos cristalinos, mas por outro lado há diferenças na
taxa de nucleação e crescimento. Este comportamento foi estudado anteriormente por
alguns autores ao analisarem a cristalização do PET (Reinsch e Rebenfeld, 1996).
Observa-se também que ao se trabalhar com blendas de PET/PHB, há uma
pequena diferença nos tempos de cristalização, pois com o aumento do percentual do
PHB a cristalização é mais rápida.
0
0,2
0,4
0,6
0,8
1
0,1 0,5 0,9 1,3 1,7 2,1 2,5
Xt
(%)
Log t (s)
PET 115 C
0.5% PHB 115 C
1.0% PHB 115 C
3.0% PHB 115 C
0
0,2
0,4
0,6
0,8
1
0,1 0,5 0,9 1,3 1,7 2,1 2,5
Xt
(%)
Log t (s)
PET 120 C
0.5% PHB 120 C
1.0% PHB 120 C
3.0% PHB 120 C
0
0,2
0,4
0,6
0,8
1
0,1 0,5 0,9 1,3 1,7 2,1 2,5
Xt
(%)
Log t (s)
PET 125 C
0.5% PHB 125 C
1.0% PHB 125 C
3.0% PHB 125 C
0
0,2
0,4
0,6
0,8
1
0,1 0,5 0,9 1,3 1,7 2,1 2,5
Xt
(%)
Log t (s)
PET130 C
0.5% PHB130 C
1.0% PHB 130 C
3.0% PHB 130 C

43
Figura 12: Curvas das isotermas de cristalização para o PET e as blendas PET/PHB.
A partir das isotermas de cristalização apresentadas na Figura 12, pode-se
estudar o tempo necessário para que ocorra a cristalização. Como esse tempo é muito
longo, trabalha-se com o tempo em que a amostra atinge 50% da cristalinidade, como
mostra a Figura 13. Pode-se observar que para todos os casos o t0,5 diminui à medida
que se aumenta a Tc, ou seja, uma diminuição no t0,5 indica que há um aumento na
velocidade de cristalização.
Segundo Wellen (2002), esse aumento na velocidade de cristalização pode ser
atribuído a uma diminuição na viscosidade que influencia o movimento segmental. A
0
0,2
0,4
0,6
0,8
1
0,1 0,5 0,9 1,3 1,7 2,1 2,5
Xt
(%)
Log t (s)
PET 135 C
0.5% PHB 135 C
1.0% PHB 135 C
3.0% PHB 135 C
0
0,2
0,4
0,6
0,8
1
0,1 0,5 0,9 1,3 1,7 2,1 2,5
Xt
(%)
Log t (s)
PET 140 C
0.5% PHB 140 C
1.0% PHB 140 C
3.0% PHB 140 C
0
0,2
0,4
0,6
0,8
1
0,1 0,5 0,9 1,3 1,7 2,1 2,5
Xt
(%)
Log t (s)
PET 145 C
0.5% PHB 145 C
1.0% PHB 145 C
3.0% PHB 145 C
0
0,2
0,4
0,6
0,8
1
0,1 0,5 0,9 1,3 1,7 2,1 2,5
Xt
(%)
Log t (s)
PET 150 C
0.5% PHB 150 C
1.0% PHB 150 C
3.0% PHB 150 C
0
0,2
0,4
0,6
0,8
1
0,1 0,5 0,9 1,3 1,7 2,1 2,5
Xt(%
)
Log t (s)
PET
115ºC
120ºC
140ºC
150ºC

44
viscosidade interfere tanto na velocidade de nucleação, quanto na velocidade de
crescimento do cristal.
O mesmo comportamento pode ser verificado nas curvas que correspondem a
adição de PHB ao PET, à medida que se aumenta a concentração de PHB na amostra,
aumenta a sua velocidade de cristalização, confirmando os resultados já obtidos
anteriormente.
Esse comportamento está de acordo com estudos realizados anteriormente por
Gunaratne et al. (2004) e concluiu-se que o PHB cristaliza-se rapidamente e possui uma
alta densidade de nucleação.
Figura 13: Efeito da temperatura no tempo para que seja atingido 50% da
cristalinidade máxima.
De acordo com a Tabela 2, podemos observar melhor o que ocorre em todas as
temperaturas estudadas, e com a adição de PHB na formação de cristais a partir de uma
amostra amorfa de PET. Tomando Δtc como sendo o intervalo de tempo do pico de
cristalização, e sabendo que Δtc = t0,99 - t0,01. Observa-se que o aumento da temperatura
e a adição de PHB contribuem para a cristalização do PET. Isto significa que o
homopolímero PET possui um tempo menor para a ocorrência da nucleação e
crescimento cristalino, o que reduz a velocidade global de cristalização.
0
0,2
0,4
0,6
0,8
1
110 120 130 140 150
t 0,5
(min
)
PET
0
0,2
0,4
0,6
0,8
1
110 120 130 140 150
(99,5/0,5)
0
0,2
0,4
0,6
0,8
1
110 120 130 140 150
Tc (°C)
0
0,2
0,4
0,6
0,8
1
110 120 130 140 150
Tc (°C)
(97/3)
Tc (°C) Tc (°C)
t 0,5 (
min
) t 0
,5 (
min
)
t 0,5 (
min
)
(99/1)

45
Tabela 2: Porcentagem de cristalização para cada amostra de PET e PET/PHB para cada
temperatura correspondente. T=
115oC
PET 0.5%
PHB
1.0%
PHB
xxxx
T=120ºC PET 0.5%
PHB
1.0%
PHB
xxxx
T=125oC PET
0.5%
PHB
1.0%
PHB
Xrel t (s) t (s) t (s) Xrel t (s) t (s) t (s) Xrel t (s) t (s) t (s)
1% 5,6 5,8 6,1 1% 4,1 5,0 5,3 1% 2,9 2,9 3,1
10% 19,0 16,3 18,2 10% 16,3 17,4 18,3 10% 10,6 9,3 10,3
30% 34,7 29,0 35,7 30% 30,6 32,8 35 30% 20,5 13,7 18,7
50% 48,4 39,8 53,7 50% 43,8 47,6 51,5 50% 30,3 25,4 26,2
99% 116,2 101,4 157,9 99% 123,3 142 127,2 99% 81,9 91,3 66,8
T=
130oC
PET 0.5%
PHB
1.0%
PHB xxxx T=135ºC PET
0.5%
PHB
1.0%
PHB xxxx T=140
oC PET
0.5%
PHB
1.0%
PHB
Xrel t (s) t (s) t (s) Xrel t (s) t (s) t (s) Xrel t (s) t (s) t (s)
1% 2,2 1,9 3,0 1% 1,9 2,0 2,5 1% 2,1 1,908 1,95
10% 9,1 6,7 9,4 10% 7,9 6,8 8,1 10% 7,1 6,0 6,1
30% 17,7 12,8 16,6 30% 15 13,2 14,7 30% 13,7 11,5 11,3
50% 25,6 18,7 23,2 50% 21,6 19,5 20,3 50% 19,8 16,7 15,75
99% 77,2 54,4 62,1 99% 58,5 55,0 53,3 99% 53,5 45,8 41,5
T= 145oC PET
0.5%
PHB
1.0%
PHB xxxx T=150ºC PET
0.5%
PHB
1.0%
PHB
Xrel t (s) t (s) t (s) Xrel t (s) t (s) t (s)
1% 1,5 1,7 1,65 1% 1 1,25 1,3
10% 6,1 5,7 5 10% 4 4,5 4,95
30% 11,8 11,1 9,3 30% 7,95 8,65 9,7
50% 17,2 16,1 13,3 50% 11,48 12,6 13,7
99% 45,1 45,7 32,65 99% 27,6 33,6 35,1
4.1.1.Cristalização isotérmica a frio – Teoria de Avrami
As curvas de Avrami em todas as temperaturas estudadas estão representadas
na Figura 14. Gráficos lineares contínuos caracterizam um processo de cristalização em
apenas um estágio, processo este conhecido por cristalização primária. Já, neste caso, ao
final da curva, tem-se o indicativo de cristalização secundária, ou seja, a cristalização

46
ocorre em dois estágios. Este trabalho foi realizado apenas com o primeiro estágio da
cristalização, a chamada cristalização primária.
Em suas pesquisas, Lorenzo et al., (2007) verificaram que quando ocorre a
cristalização em dois estágios, pode resultar em erros na aplicação da teoria de Avrami
para a determinação dos seus parâmetros.
A cristalização em dois estágios ocorre devido à interferência entre os
domínios cristalinos no final da cristalização primária que reduz a velocidade de
cristalização (Groeninckx et al., 1980).
O desvio da linearidade nas curvas de Avrami no final da cristalização
observada para PET é devido à ocorrência da cristalização secundária, onde ocorre o
aperfeiçoamento da cristalinidade com o tempo. Para a blenda, esta cristalização
secundária pode ser decorrente da segregação de moléculas não cristalizáveis de PHB.
Dreezen et al (1999) analisaram a cristalização isotérmica de blendas de poli(óxido de
etileno)/poli(éter sulfona) - PEO/PES, observando que houve a ocorrência de moléculas
não cristalizáveis de PES nas regiões intraesferulíticas e interlamelares do PEO,
diminuindo a taxa de cristalização. As curvas referentes a comparação entre o PET e as
blendas com PHB, em todas as temperaturas, encontram-se no Apêndice.
-3
-2
-1
0
1
0,3 0,6 0,9 1,2 1,5 1,8 2,1 2,4
Log(-
ln(1
-Xt)
)
Log t (s)
PET
115ºC 120ºC
130ºC 135ºC
140°C 145°C
150°C
-3
-2
-1
0
1
2
0,3 0,6 0,9 1,2 1,5 1,8 2,1 2,4
Log(-
ln(1
-Xt)
)
Log t (s)
(99,5/0,5)
110°C 115°C
120°C 125°C
130°C 135°C
140°C 145°C
150°C

47
Figura 14: Curvas de Avrami para a cristalização isotérmica com diferentes Tc’s .
A Figura 15 representa a taxa de cristalização, e é obtida invertendo-se o t0,5 nas
Tc’s analisadas, e se observa que a taxa de cristalização aumenta de forma linear com a
temperatura.
Figura 15: Efeito das Tc’s na taxa de velocidade de cristalização.
Os resultados obtidos para os parâmetros n e K, calculados para a cristalização
primária das curvas mostradas na Figura 16, foram apresentados, para todas as
composições, na Tabela 3. O expoente de Avrami, n, apresentou valores muito próximo
-3
-2
-1
0
1
0,3 0,6 0,9 1,2 1,5 1,8 2,1 2,4
Log(-
ln(1
-Xt)
)
Log t (s)
(99/1)
110°C 115°C
120°C 125°C
130°C 135°C
140°C 145°C
150°C
-3
-2
-1
0
1
0,3 0,6 0,9 1,2 1,5 1,8 2,1 2,4
Log(-
ln(1
-Xt)
)
Log t (s)
(97/3)
100°C 105°C
115°C 120°C
125°C 130°C
135°C 140°C
145°C 150°C
0
1
2
3
4
5
6
105 115 125 135 145 155
0
1
2
3
4
5
6
105 115 125 135 145 155
0
1
2
3
4
5
6
105 115 125 135 145 155
Tc (°C)
0
1
2
3
4
5
6
100 110 120 130 140 150 160
Tc (°C)
PET PET99,5PHB0,5
PET99PHB1 PET97PHB
3
Tax
a de
cris
tali
zaçã
o (
s1)
Tax
a de
cris
tali
zaçã
o (
s-1)
Tax
a de
cris
tali
zaçã
o (
s-1)
Tax
a de
cris
tali
zaçã
o (
s-1)
Tc (°C)
Tc (°C)

48
de 2 para todas as amostras, este resultado é atribuído a um crescimento em forma de
disco gerado por nucleação heterogênea.
Valores muito parecidos foram encontrados na literatura como é o caso de
blendas de PET/PEO (Li et al., 2005), e o PET (Gao et al., 2005).
A constante de velocidade, K, aumenta com o aumento da temperatura, e
também com o aumento da concentração de PHB na amostra. Esta observação confirma
que o aumento no percentual de PHB na amostra de blenda de PET acelera seu ponto de
cristalização.
Tabela 3: Parâmetros de Avrami para a cristalização primária isotérmica a frio do PET e
das blendas PET/PHB.
T (ºC) n n n K (10
-3) K (10
-3) K (10
-3)
PET 0.5% PHB 1.0% PHB PET 0.5% PHB 1.0% PHB
115 2,0 2,10 1,70 0,41 0,49 0,75
120 1,8 1,85 1,88 0,38 1,05 1,42
125 1,8 1,68 2,02 0,68 1,53 1,90
130 1,76 1,89 2,04 1,219 1,55 2,03
135 1,78 1,80 2,06 1,861 2,23 2,30
140 1,84 1,83 1,96 2,782 2,93 2,99
145 1,85 1,83 1,95 2,614 3,05 3,42
150 1,8 1,91 1,89 3,819 4,47 4,96
4.1.2. Energia de ativação para a cristalização isotérmica a frio
Em condições isotérmicas, o parâmetro K pode ser escrito na forma de
Arrhenius para determinar a energia de ativação da cristalização ΔEc, com isso se pode
obter um gráfico de (1/n) Ln K versus 1/Tc, a inclinação da curva determina ΔE/R,
sendo R a constante dos gases (Cebe et al., 1986). A Figura 23 ilustra as curvas da
energia de ativação do PET e suas blendas PET/PHB, e a Tabela 4 mostra os valores de
cada energia de ativação.
Os valores encontrados para ΔEc do PET foi de 48,87KJ/mol. Ao se adicionar
o PHB ao PET, encontrou-se para a blenda de 0,5% um valor de 48,33 KJ/mol e para a
adição de 1% de PHB a energia de ativação foi de 42,92 KJ/mol, observa-se que foram
encontrados valores menores que a energia de ativação obtida para o PET puro, ou seja,

49
a presença de PHB facilita o processo de cristalização, pois uma menor energia deve ser
fornecida para que se desenvolva o processo de cristalização, o que confirma os
resultados mostrados anteriormente.
Figura 16: Curvas de Arrhenius para a determinação da energia de ativação da
cristalização isotérmica a frio do PET e das blendas PET/PHB.
Tabela 4: Valores da energia de ativação do PET e das blendas PET/PHB.
Composição Energ. (kJ/mol)
PET 48,87
0.5% PHB 48,33
1.0% PHB 42,92
4.1.3. Estudo do comportamento de fusão
A Figura 17 representa a endoterma de fusão do PET e de suas blendas com
PHB e pode-se observar que, aparentemente, o comportamento de fusão do PET não
y = -5,8734x + 11,048
R² = 0,9685
-6
-5
-4
-3
-2
2,2 2,3 2,4 2,5 2,6 2,7 2,8
1/n
Ln
(K
)
1/Tc (10-3 K-1)
PET
y = -6,0146x + 11,467
R² = 0,8778
-6
-5
-4
-3
-2
2,2 2,3 2,4 2,5 2,6 2,7 2,8
1/n
Ln
(K
)1/Tc (10-3 K-1)
0.5% PHB
y = -8,3508x + 17,235
R² = 0,9821
-6
-5
-4
-3
-2
2,2 2,3 2,4 2,5 2,6 2,7 2,8
1/n
Ln
(K
)
1/Tc (10-3 K-1)
1.0% PHB

50
alterou de forma significativa com relação às temperaturas de cristalização e nem com a
adição de PHB. Isso ocorre devido à fusão acontecer em uma ampla faixa de
temperaturas em virtude de diferentes tamanhos e graus de perfeição dos cristais
presentes. A Tm é determinada através do pico da curva. Como definido na Figura 14, a
Tabela 5 mostra os valores para o inicio da temperatura de fusão (Tmi), o pico no qual
ela ocorre (Tm) e o valor do final dessa temperatura (Tmf).
220 240 260 280Temp [C]
m W
DSC
120ºC
130ºC
140ºC
150ºC
PET
220 240 260 280Temp [C]
m W
DSC
120120ºC
130130ºC
140140ºC
150150ºC
0.5% PHB
220 240 260 280Temp [C]
m W
DSC
120ºC
130ºC
140ºC
150ºC
1% PHB
220 240 260 280Temp [C]
m W
DSC
120ºC
130ºC
140ºC
150ºC
3% PHB
Figura 17: Endotermas de fusão do PET e das blendas PET/PHB, cristalizadas
isotermicamente a frio em diferentes temperaturas.
PET 0,5% PHB
1% PHB 3% PHB

51
Tabela 5: Valores das temperaturas de fusão do PET e das blendas PET/PHB. Todas as
temperaturas de fusão estão em °C.
T(°C) PET 0,5% PHB 1% PHB 3% PHB
Tmi Tm Tmf Tmi Tm Tmf Tmi Tm Tmf Tmi Tm Tmf
120 242 254 260 244 252 262 241 253 265 240 253 262
130 241 251 262 244 254 263 241 252 264 236 252 263
140 243 252 264 243 253 263 241 252 265 248 254 263
150 240 251 260 242 252 263 239 252 263 240 253 264
Partindo dos gráficos expostos na Figura 24 foi determinada a influência da
temperatura de cristalização na temperatura de fusão, os resultados encontram-se na
Figura 18, e se observa que houve uma pequena variação na Tm. Para uma melhor
visualização, determina-se a temperatura de fusão de equilíbrio que representa a
estabilidade de cristais de tamanho infinito, e assim avaliar as diferenças de estabilidade
térmica em diferentes composições (Utracki, 1990).
Figura 18:Efeito das condições de cristalização nas temperaturas de fusão do PET e suas
blendas com PHB.
A partir das endotermas da Figura 24, pode-se calcular a , através do
método proposto por Hoffman e Weeks (Hoffman et al., 1962) por extrapolação da
condição Tm = Tc utilizando-se amostras cristalizadas em várias temperaturas, ver
Figura 19. Estes valores estão dispostos na Tabela 6.
O PET apresentou uma ≈ 251°C, o valor encontrado está de acordo com a
literatura que apresenta uma faixa de temperatura de 245°C a 260°C (Reinsch et
al.,1996; Groeninckx et al.,1980; Kong et al., 2002).
245
250
255
260
110 115 120 125 130 135 140 145 150 155
Tm
(°C
)
Tc(°C)
PET
0.5% PHB
1% PHB
3% PHB

52
Com a adição de 0,5% de PHB ao PET, a ≈ 255°C e com a adição de 1%
de PHB, a ≈ 255,5°C. Um dos métodos utilizados para se avaliar a miscibilidade de
blendas é a depressão da do componente cristalizável. Em sistemas miscíveis a
temperatura de fusão da blenda será mais baixa do que a do componente cristalizável
puro (Sivestre et al., 1996). Portanto, como esse fato não foi observado, sendo
verificado o contrário, é provável que as blendas de PET e PHB não sejam miscíveis.
Figura 19: Determinação da temperatura de equilíbrio através do método de Hoffman e
Weeks do PET e das blendas PET/PHB em diferentes concentrações. Dados da autora
Tabela 6: Temperatura de fusão de equilíbrio do PET e das blendas PET/PHB
cristalizadas isotermicamente a frio.
Composição (oC)
PET 251,0
0.5% PHB 255,0
1.0% PHB 255,5
4.2. Cristalização não isotérmica a frio
De acordo com a Figura 10 que representa uma curva para a cristalização,
pode-se encontrar os valores para a temperatura vítrea, a temperatura de cristalização e a
temperatura de fusão. De acordo com as curvas para cristalização não isotérmica,
observando o primeiro pico, denominado de cristalização, à medida que aumenta a
porcentagem de PHB na mistura, esse pico aparece a uma temperatura mais baixa. Com
100
150
200
250
300
100 150 200 250 300
Tc (°C)
Tm = Tc
Temperatura de
fusão de equilíbrio

53
essas curvas, partiu-se para um estudo mais aprofundado da cinética de cristalização não
isotérmica. Todas as temperaturas de transição vítrea, temperaturas de cristalização e
temperaturas de fusão e em todas as taxas de aquecimento, foram listadas através de
tabelas no Apêndice.
Na Figura 20 está ilustrada a influência da taxa de aquecimento na cristalização
não isotérmica do PET e das blendas PET/PHB e verificou-se que há um deslocamento
das exotermas de cristalização para temperaturas mais elevadas à medida que se
aumenta a taxa de aquecimento. Isso ocorre porque durante o aquecimento, a
cristalização depende da velocidade de nucleação e do crescimento cristalino. Segundo
Wellen (2002), esse fenômeno pode ser explicado porque em baixas velocidades de
aquecimento tem-se um maior tempo disponível para a cristalização.
As endotermas de fusão aparentemente não apresentaram alterações, efeito
causado, provavelmente, porque há uma resposta mais lenta da fusão dos cristais
quando comparado com a taxa de aquecimento (Miyagi et al., 1972).
Na Figura 21, podemos comparar os resultados obtidos para adição de PHB ao
PET. Através das exotermas de cristalização das blendas, observa-se uma cristalização
em temperaturas mais baixas que as do PET à medida que se aumenta o percentual de
PHB.
Na Figura 21, há uma melhor comparação quanto à adição do PHB ao PET, e se
verifica também que as endotermas de fusão não apresentaram alterações significativas
em função das diferentes taxas de aquecimento e concentrações de PHB na mistura.

54
.
50 100 150 200 250Temp [C]
- 10 . 0
0 . 0
m W
DSC
1
2.5
5
7.5
10
15
20
30
PET
50 100 150 200 250Temp [C]
- 30 . 0 0
0 . 0 0
m W
DSC
1
2.5
57.5
10
15
20
30
0.5% PHB
50 100 150 200 250Temp [C]
- 25 . 0 0
0 . 0 0
m W
DSC
1
2.5
5
7.5
10
15
20
30
1% PHB
50 100 150 200 250Temp [C]
- 30 . 0 0
0 . 0 0
m W
DSC
1
2.5
7.5
10
15
20
30
3% PHB
Figura 20: Curvas de DSC para Cristalização não isotérmica.
50 100 150 200 250Temp [C]
- 0. 00
m W
DSC
3% PHB
1% PHB
PET
0.5% PHB
Figura 21: Curvas de DSC para cristalização não isotérmica para uma taxa de
aquecimento de 30°C/min.
Partindo das curvas mostradas na Figura 27, estudou-se a influência das taxas de
aquecimento na Tg (temperatura de transição vítrea), na Tc (temperatura do pico de
cristalização a frio) e na Tm (temperatura do pico de fusão) do PET e das blendas
PET/PHB.
A Figura 22 é a representação gráfica da influência da taxa de aquecimento
sobre a Tg e observa-se que tanto para o PET quanto para suas blendas, a Tg aumenta a
medida que aumenta a taxa de aquecimento.

55
A transição vítrea é um fenômeno de relaxação molecular. Quanto mais rápido
for o aquecimento menos tempo tem-se para a relaxação, que passa então a ser
observada em temperaturas mais elevadas (Wellen, 2002).
Quanto à adição de PHB ao PET, as Tg’s apresentadas pelas blendas são
inferiores as do PET, e na medida em que se aumenta a concentração de PHB à mistura,
a Tg decai, isso pode ocorrer devido ao fato de que a Tg do PHB é mais baixa que a do
PET, influenciando assim quando formam as blendas.
Essas variações também foram observadas na literatura com blendas de
poli(teraftalato de etileno)/poli(teraftalato de butireno) - PET/PBT (Avramova, 1995),
poli(teraftalato de etileno)/poli(ADP-ribose) - PET/PAr (Porter et al., 1992), PET/OS
(Jang et al., 2000), poli (hidroxibutirato)/poli (sicinato de butileno) - PHB/PBSU (Gao
et al.,2005).
Figura 22: Efeito da taxa de aquecimento na temperatura de transição vítrea do PET e
das blendas PET/PHB.
Ainda partindo das curvas de DSC da Figura 20, foi determinada a temperatura
do pico de cristalização a frio (Tc) e ilustrada na Figura 23, que representa o efeito da
taxa de aquecimento na temperatura de cristalização. Verifica-se que as Tc’s tanto do
PET quanto das blendas aumentaram com o aumento da taxa de aquecimento. Isso é
explicado porque quanto maior a taxa de aquecimento, menor é o tempo para que haja o
crescimento cristalino, reduzindo assim a velocidade de cristalização. Ou seja, à medida
que se aumenta a taxa de aquecimento, torna-se necessária uma temperatura mais alta
para formação dos cristais. O aumento na temperatura de cristalização a frio com as
50
55
60
65
70
75
80
0 5 10 15 20 25 30
Tem
per
atu
ra d
e tr
ansi
ção v
ítre
a (°
C)
Taxa de aquecimento (°C/min)
PET
0.5% PHB

56
taxas de aquecimento foi também observado por vários autores em diferentes sistemas
poliméricos como encontrado durante a cristalização a frio do PET (Lu e Hay., 2001),
poli(éter-cetona-éter-cetona-cetona) - PEKEKK (Qiu et al., 2000), PPS (Martinelli et
al., 2005).
Neste trabalho verificou-se que as blendas apresentaram Tc’s inferiores ao
homopolímero PET. Quental et al. (2009) estudaram o efeito da Tc sobre a energia para
a relaxação das blendas e chegaram à conclusão de que uma menor Tc requer uma
quantidade menor de energia para a relaxação, por isso há uma maior facilidade na
obtenção dos cristais.
Figura 23: Efeito da taxa de aquecimento na temperatura de cristalização do PET e suas
blendas com PHB.
A Figura 24 apresenta o efeito das taxas de aquecimento na temperatura de fusão
do PET e suas blendas PET/PHB. Observa-se que há uma pequena variação na Tm com
as taxas de aquecimento analisadas, e que as Tm das blendas possuem valores
ligeiramente superiores ao PET.
Utilizando a Equação 2 e considerando ΔG=0 (pois ao final da cristalização
primária, atinge-se um estado de pseudo-equilíbrio), foi montada uma tabela para os
valores da variação da entropia do sistema. A partir da Tabela 7, observa-se que com o
aumento da taxa de aquecimento, diminui a variação da entropia do sistema tanto para o
PET quanto para a blenda, porque com o aumento da taxa de aquecimento dificulta o
processo de cristalização, pois o sistema precisa de mais energia para cristalizar. Os
valores correspondentes a 0,5 e 1% de PHB encontram-se no apêndice.
95
100
105
110
115
120
125
130
135
140
145
0 5 10 15 20 25 30
Tem
per
atu
ra d
e cr
ista
liza
ção (
°C)
Taxa de aquecimento (°C/min)
PET
0.5% PHB
1% PHB
3% PHB

57
Tabela 7: Variação da entropia do sistema em todas as taxas de aquecimento. Φ
(°C/min)
PET 3% PHB
Tm (°C) ΔHm (J/g) ΔS(J/g°C) Tm (°C) ΔHm (J/g) ΔS(J/g°C)
5 250,4 38,6 0,16 251,7 49,1 0,19
7,5 249,3 36,7 0,15 251,5 46,8 0,18
10 250,6 36,3 0,14 252,1 42,6 0,17
15 251,2 33,6 0,13 254,1 38,3 0,15
20 251,4 31,5 0,12 252,3 34,4 0,14
30 255,1 22,9 0,09 256,1 33,4 0,13
Fann et al. (1998) estudaram o efeito da adição de PET reciclado ao PET (R-
PET), através das curvas de DSC, e observaram que houve uma ligeira diminuição da
Tm do PET, e sua conclusão foi que com a adição do PET reciclado houve um aumento
da variação da entropia (ΔS) do sistema.
Figura 24: Efeito da taxa de aquecimento na temperatura de fusão do PET e suas
blendas.
Trabalhando a partir das exotermas de DSC da Figura 20, foram obtidas curvas
que mostram o desenvolvimento da cristalinidade com a temperatura, XT, e encontram-
se ilustradas na Figura 25. Estas curvas apresentaram um processo de transformação de
fases sem descontinuidades, característico da transformação de fases em polímeros.
Na Figura 25 observa-se que em todos os casos as curvas de XT são deslocadas
para temperaturas mais elevadas com o aumento da taxa de aquecimento, estando de
245
250
255
260
0 5 10 15 20 25 30
Tem
per
atu
ra d
e fu
são (
°C)
Taxa de aquecimento (°C/min)
PET
0.5% PHB
1% PHB
3% PHB

58
acordo com as curvas da Figura 20. É possível observar que as curvas apresentam
formas similares, o que indica o mesmo mecanismo de cristalização, ou seja, o mesmo
crescimento morfológico, diferindo apenas na taxa de nucleação e crescimento
cristalino.
Ainda na Figura 25, pode-se verificar que o PET apresentou o inicio da
cristalização mais lenta, enquanto que nas blendas foi mais rápido, sugerindo que a
presença de PHB acelerou o processo de cristalização, o que é um indicativo de que o
PHB pode estar atuando como agente nucleante.
Kong et al., (2001) observaram curvas sigmoidais reversas durante a
cristalização não isotérmica do copolímero PET-PEO. Tankhiwale et al., (2002)
observaram que há um retardo inicial da cristalização do PET nas blendas de PET/PS, o
que foi explicado devido a diminuição da taxa de nucleação. As curvas referentes a
comparação do PET com as blendas PET/PHB, em todas as taxas de aquecimento,
encontram-se no Apêndice.
0
0,2
0,4
0,6
0,8
1
4,5 4,6 4,7 4,8 4,9 5 5,1Ln T (
oC)
XT (
%)
1 2.5
5 7.5
10 15
20 30
PET
0
0,2
0,4
0,6
0,8
1
4,5 4,6 4,7 4,8 4,9 5 5,1
Ln T (oC)
XT (
%)
1 2.5
5 7.5
10 15
20 30
0.5% PHB
0
0,2
0,4
0,6
0,8
1
4,5 4,6 4,7 4,8 4,9 5 5,1
Ln T (oC)
XT (
%)
1 2.55 7.510 1520 30
1% PHB
0
0,2
0,4
0,6
0,8
1
4,5 4,6 4,7 4,8 4,9 5 5,1
Ln T (oC)
XT (
%)
1 2.55 7.510 1520 30
3% PHB

59
Figura 25: Curvas em “S” para a cristalização não isotérmica do PET e das blendas
PET/PHB.
A Figura 26 mostra o efeito da taxa de aquecimento no parâmetro T0,5,
ilustrando o efeito que a taxa de aquecimento exerce para atingir 50% de cristalização.
Pode-se verificar que T0,5 aumentou com a taxa de aquecimento, e pode ser atribuído a
uma diminuição na viscosidade (Hage et al., 1999). Ainda na Figura 26 é possível
verificar que o desenvolvimento da cristalinidade do PET é mais lento quando
comparado com as blendas PET/PHB, o que está de acodo com as exotermas de
cristalização da Figura 25.
Figura 26: Efeito da taxa de aquecimento para se atingir 50% de cristalinidade do PET e
suas blendas PET/PHB.
4.2.1. Cristalização não isotérmica a frio – Teoria de Mo
Para um melhor entendimento da cristalização não isotérmica do PET e suas
blendas PET/PHB em diferentes concentrações foi empregada a teoria de Mo (Liu et al.,
0
0,2
0,4
0,6
0,8
1
4,75 4,8 4,85 4,9 4,95
Ln T (oC)
XT (
%)
PET
0.5% PHB
1% PHB
3% PHB
10 oC.min
-1
95
105
115
125
135
145
0 5 10 15 20 25 30
Taxa de aquacimento (ºC.min-1
)
T0.
5 (o C
)
PET
0.5% PHB
1% PHB
3% PHB

60
1997), que é uma combinação da teoria de Avrami (Avrami, 1939, 1940, 1941) e Ozawa
(Ozawa, 1971). A partir das exotermas de DSC mostradas na Figura 20, foi obtido o
gráfico referente a cristalinidade em função da temperatura mostrado na Figura 22.
Assim como na observação feita em relação à cristalização isotérmica; no caso da não
isotérmica, as curvas de cristalização apresentam uma forma sigmoidal caracterizando
um processo de transformação de fases sem descontinuidades.
Já a Figura 27 apresenta o comportamento do desenvolvimento da cristalinidade
com o tempo em diferentes taxas de aquecimento. Como já foi mencionado, o grau de
cristalinidade está relacionado com a taxa de aquecimento ( ) e com o tempo de
cristalização (ou temperatura). A relação entre a taxa de aquecimento e o tempo t pode
ser construída para um dado grau de cristalinidade (Xt' ).
Observa-se na Figura 27 que quanto mais alta a taxa de aquecimento menos
tempo se tem para que ocorra o processo de cristalização, ou seja, há uma diminuição da
cristalinidade com a taxa de aquecimento.
Figura 27: Grau de cristalinidade com o tempo (Xt’) para a cristalização não
isotérmica do PET e das blendas PET/PHB.
.
PET
0
0,2
0,4
0,6
0,8
1
0 2 4 6 8 10 12
t (min)
Xt'(%
)
1 2.5
5 7.5
10 15
20 30
0.5% PHB
0
0,2
0,4
0,6
0,8
1
0 2 4 6 8 10 12
t (min)
Xt'(%
)
1 2.5
5 7.5
10 15
20 30
1% PHB
0
0,2
0,4
0,6
0,8
1
0 2 4 6 8 10 12
t (min)
Xt'(%
)
1 2.5
5 7.5
10 15
20 30

61
A Figura 28 apresenta as curvas do Log t versus Log [-Ln (1-Xt’)] para a
cristalização não isotérmica do PET e das blendas nas diferentes taxas de aquecimento.
Na referida curva, observa-se que ao final da curva, obtemos um desvio de linearidade,
o que corresponde ao final da cristalização primária. Ou seja, nestes casos, a
cristalização ocorreu em dois estágios, o primário e o secundário. E conclui-se que
quanto maior é a taxa de aquecimento menor é a faixa em que ocorre a cristalização
secundária. Este comportamento foi estudado anteriormente para o PEEK (Cebe, 1988),
compósitos de PEN com nano-partículas de sílica (Kim et al., 2003), Nylon 1212 (Lui et
al., 2003).
Neste trabalho, a teoria de Mo foi aplicada apenas para o primeiro estágio da
cristalização não isotérmica a frio do PET e suas blendas PET/PHB.
Figura 28: Curvas do Log t versus Log [-Ln (1-Xc)] para a cristalização não isotérmica
do PET e das blendas PET/PHB.
A partir da Figura 28 os parâmetros cinéticos da cristalização não isotérmica a
frio do PET e das blendas PET/PHB foram determinados utilizando-se a teoria de Mo
apenas no primeiro estágio da cristalização não isotérmica. Os parâmetros, n’ e K’(T) da
cristalização não isotérmica e os resultados obtidos estão dispostos na Tabela 8. As
PET
-2,2
-1,7
-1,2
-0,7
-0,2
0,3
0,8
-1,5 -1 -0,5 0 0,5 1 1,5
Log t (min)
Lo
g [
-Ln
(1
-Xt’)]
1 2.5
5 7.510 15
20 30
0.5% PHB
-2,2
-1,7
-1,2
-0,7
-0,2
0,3
0,8
-1,5 -1 -0,5 0 0,5 1 1,5
Log t (min)
Lo
g [
-Ln
(1
-Xt’)]
1 2.5
5 7.5
10 15
20 30
1% PHB
-2,2
-1,7
-1,2
-0,7
-0,2
0,3
0,8
-1,5 -1 -0,5 0 0,5 1 1,5
Log t (min)
Lo
g [
-Ln
(1
-Xt’)]
1 2.5
5 7.5
10 15
20 30

62
taxas de aquecimento de 1°C/min e 2,5°C/min não foram utilizadas pois , como mostra
a Figura 27, essas taxas apresentam grandes desvios de linearidade no início da
cristalização. Os valores apresentados para o expoente n’ ficaram entre 2 e 3 tanto para
o PET quanto para as blendas, o que significa dizer que há um crescimento em forma de
disco gerado por nucleação heterogênea.
A Figura 29 apresenta a influência da taxa de aquecimento na constante de
velocidade K’(T). Verifica-se um aumento de K’(T) em todas as composições no sentido
da taxa crescente. Verifica-se também que as blendas apresentaram valores mais
elevados para a constante K’(T) sugerindo que o PHB facilita a cristalização do PET,
resultados esses que concordam com as curvas de Xt’ e segue a mesma tendência da
cristalização isotérmica a frio do PET e suas blendas PET/PHB.
De acordo com a literatura, alguns casos foram estudados anteriormente como
Xiao et al. (2002) verificaram que aplicando a mesma teoria, o PET apresentou valores
de n’ entre 2 e 3. Já Cebe et al. (1986), com relação ao PEEK, verificaram que o K’(T)
aumentou com as taxas de aquecimento/resfriamento.
Tabela 8: Parâmetros cinéticos para a cristalização não isotérmica a frio do PET e das
Blendas determinados pelas equações de Avrami-Ozawa. Parâmetros determinados para
o grau de conversão (Xt’) de 10%.
Taxa PET 0.5% PHB 1%PHB
n’ n’ n’
5 2.5 177.70 0.9967 2.5 182.47 0.9986 2.0 364.33 0.9908
7.5 2.7 321.00 0.9968 2.2 384.33 0.9908 3.0 519.40 0.9971
10 2.5 705.67 0.9962 2.5 978.14 0.9993 2.6 1164.93 0.9993
15 2.3 1538.86 0.9979 2.2 2186.25 0.9976 2.5 2457.14 0.9974
20 2.6 2189.78 0.9940 2.5 3833.54 0.9996 2.5 4086.02 0.9998
T'K 2r T'K 2r T'K 2r

63
Figura 29: Efeito da taxa de aquecimento na constante de velocidade K’(T) para a
cristalização não isotérmica a frio do PET e das suas blendas PET/PHB.
A Figura 30 apresenta as curvas do Log t (min) em função de Log (°C/min)
em diferentes graus de conversão para o PET e suas blendas PET/PHB, a partir dela, os
parâmetros de R2, a e F(T), foram determinados e os resultados obtidos foram
apresentados na Tabela 9.
O parâmetro de Mo a apresentou valores próximo a 1 para todas as composições
analisadas. Verificou-se também, que houve um aumento do a com o aumento do grau
de conversão. A constante de velocidade F(T) aumentou com o grau de conversão em
todas as composições analisadas, Figura 31. Isso significa que para uma dada unidade
de tempo da cristalização uma maior velocidade de aquecimento deve ser usada para
que consiga um maior grau de cristalinidade.
A constante de velocidade F(T) também pode ser considerada como um
parâmetro da taxa de cristalização não isotérmica dos sistemas poliméricos em estudo.
Valores mais baixos para F(T) indicam que a cristalização não isotérmica procedeu de
forma mais acelerada (Huang et al., 2006). As blendas PET/PHB apresentaram valores
mais baixos para F(T) do que o PET, isto significa dizer que as blendas apresentaram
uma velocidade de cristalização mais rápida do que o PET.
A teoria de Mo vem sendo bastante empregada no estudo da cristalização não
isotérmica como PET e o copolímero PETI (Xiao et al., 2002), P3DDT e P3ODT (Qiao
et al., 2000), Nylon (Liu et al., 2003), PPDO e PEG (Zheng et al., 2005), observando,
em todos os casos, que houve um aumento do parâmetro F(T) com o grau de conversão.
0
2000
4000
6000
8000
10000
12000
1 6 11 16 21 26
K' (
T)(m
in-1
10
-3)
Taxa de aquecimento (oC/min)
PET
0.5% PHB
1% PHB

64
Figura 30: Curvas do Log t versus Log da taxa para a cristalização não isotérmica a frio
do PET e das blendas PET/PHB em diferentes faixas de cristalinidade.
Tabela 9: Parâmetros de Mo em diferentes graus de cristalinidade para cristalização
não isotérmica a frio do PET e das blendas PET/PHB.
Xt’
(%)
PET 0.5% PHB 1%PHB
a F(T) a F(T) a F(T)
10 1.27 0.59 0.9885 1.18 0.52 0.9951 1.06 0.60 0.9864
30 1.26 0.81 0.9902 1.19 0.75 0.9968 1.08 0.79 0.9857
50 1.25 0.93 0.9914 1.19 0.87 0.9977 1.22 0.86 0.9828
70 1.27 1.04 0.9917 1.20 0.98 0.9981 1.21 0.97 0.9885
90 1.30 1.17 0.9911 1.23 1.12 0.9985 1.20 1.10 0.9915
PET
0
0,5
1
1,5
2
-0,8 -0,5 -0,2 0,1 0,4 0,7 1
Log t (min)
Lo
g T
ax
aXc=10%
Xc=30%
Xc=50%
Xc=70%
Xc=90%
0.5% PHB
0
0,5
1
1,5
2
-0,8 -0,5 -0,2 0,1 0,4 0,7 1
Log t (min)
Lo
g T
ax
a
Xc=10%
Xc=30%
Xc=50%
Xc=70%
Xc=90%
1.0% PHB
0
0,5
1
1,5
2
-0,8 -0,5 -0,2 0,1 0,4 0,7 1
Log t (min)
Lo
g T
ax
a
Xc=10%
Xc=30%
Xc=50%
Xc=70%
Xc=90%
2r 2r 2r

65
Figura 31: Variação de F (T) com o grau de conversão para o PET e suas blendas com
PHB
0,5
0,7
0,9
1,1
10 30 50 70 90
F(T
)
Xt'(%)
PET
0.5% PHB
1% PHB

66
5. CONCLUSÕES
O estudo da cinética de cristalização isotérmica e não isotérmica a frio do PET e
das blendas PET/PHB foi realizado por DSC. As conclusões de maior importância estão
descritas abaixo.
A velocidade com que houve a cristalização nos polímeros analisados é
fortemente influenciada pela temperatura de cristalização. Isso porque quando o
polímero é cristalizado a uma temperatura de cristalização mais elevada, diminui o
tempo de transição de fases, diminuindo a sua velocidade de nucleação cristalina.
Na cristalização isotérmica, a investigação foi realizada na fase primária e o
expoente n de Avrami para esse estágio ficou próximo do valor 2 tanto para o PET
quanto para suas blendas, o que corresponde a um crescimento na forma de discos
formado por nucleação heterogênea. Já para a constante de velocidade, K, observa-se
um aumento com o aumento da temperatura, e também com o aumento da concentração
de PHB na amostra.
Na cristalização não isotérmica, o aumento da taxa de aquecimento favoreceu o
deslocamento das isotermas para temperaturas mais elevadas.
Com a adição do PHB, as temperaturas de transição vítrea e cristalização a frio
do PET diminuíram, isso pode ter ocorrido devido a uma diminuição da barreira de
energia térmica de sobreposição da região de transição vítrea e uma maior facilidade de
cristalização.
A temperatura para se atingir 50% da cristalização aumentou com as taxas de
aquecimento e diminuiu com a adição de PHB.
A taxa de cristalização não isotérmica a frio do PET apresentou valores mais
baixos nas blendas devido, provavelmente, a uma maior facilidade de cristalização das
blendas.
A adição de PHB (0,5%, 1% e 3%) acelerou a taxa de cristalização a frio do
PET.

67
Na cristalização não isotérmica, foi utilizada a teoria de Mo para determinar os
parâmetros cinéticos. O parâmetro de Mo a apresentou valores próximo a 1 e a
constante de velocidade F(T) aumentou com o grau de conversão para as amostras. A
teoria de Mo foi capaz de descrever com sucesso a cinética de cristalização não
isotérmica a frio do PET e das blendas PET/PHB.
Os resultados obtidos tanto para a cristalização isotérmica quanto para a não
isotérmica a frio do PET e das blendas PET/PHB permitiram conclusões semelhantes,
ou seja, a teoria de Mo aplicada nas condições de cristalização não isotérmica confirma
a teoria de Avrami aplicada a cristalização isotérmica a frio do PET e das blendas
PET/PHB.

68
REFERÊNCIAS
Avella, M.; Martuscelli, E.; Greco, P. Crystallization behaviuor of poli (ethylene oxide) from poli (3-hydroxybutyrate)/poly (ethylene oxide) blends: phase structuring, morphology and
thermal behavior. Polymer 32, 1647-1653, 1991.
Avrami, M. Kinetics of Phase Change. I. General Theory.J. Chen. Phys. 7, 1103-1112, 1939.
Avrami, M. Kinetics of Phase Change. II. Transformation-time relations for random distribution of nuclei. J. Chen. Phys. 8, 212-224, 1940.
Avrami, M. Kinetics of Phase Change. II. Granulation, phase change, andmicrostructure.J.
Chen. Phys. 9, 177-184, 1941.
Avramova, N. Amorphous poly (ethylene terephthalate/poly(butylenes terephthalate) blends:
miscibility and properties. Polymer 36, 801-808, 1995.
Baer, E.; Collier, J.R.; Carter, D.R. Kinetics of spherulitic crystallization. Polym. Eng. Sci. 5,
22-28, 1965.
Basset, D. C. Principles of polymer morphology. Cambridge University Press, Cambridge,
1981.
Baijal, M. Plastics polymer science and technology. Jonh Wiley & Sons, New York, 1982.
Bian J., Ye S., Feng L. Heterogeneous nucleation on the crystallization poly(ethylene terephthalate). Wiley interscience (www.interscience.wiley.com), pesquisado dia 30 de julho de
2009 as 20:30. Publicado em 2003.
Billmeyer Jr, F.W. Textbook of science. Wiley Interscience Publication, New York. 1984.
Bove, L., D’Aniello, C., Gorrasi, G., Guadagno, L., Vittoria, V. Transport properties of poly
(ethylene terephthalate) cryatalizedfrom the glassy state. Poly. Adv. Tech. 7, 858-862, 1994.
Cebe, P. Non-isothermal crystallization of poly (ether ether ketone). Aromatic polymer
composite. Polym. Comp. 9, 271-279, 1988.
Dessai, P.; Abhiraman, A.S. Crystalization in oriented poly (ethylene terephthalate) fibers. I.
Fundamental aspects. J. Polym. Sci. Polym. Phys. Edn. 23, 653-674, 1985.
Derenzo, S.; Saito, R. M.; Garófalo, G. M. C.; Ribeiro, A. M. M.; Bueno Netto, C. L.;
Mantelatto, P. E.; Rossel, C. E. V.; 2003.
Di Lorenzo, M.L.; Spherulite growth rates in binary polymer blends. Prog. Polym. Sci. 28, 663-
689, 2003.
Dreezen, G.; Mischenko, N.; Koch, M. H. J.; Reynaers, H. and Groeninckx, G. Miscible Binary
Blends of Poly(ethylene oxide) and Poly(ether sulfone). 2. Real-Time Small-Angle X-ray
Scattering Investigation of the Semicrystalline Morphology. ACS Publications. Publication Date
(Web): May 29, 1999. Copyright © 1999 American Chemical Society

69
Du, X.H., Zhao, C.S., Wang, Y.Z., Zhou, Q., Deng, Y., Qu, M.H., Yang, B. Thhhermal
Oxidative degradation behaviours of flame-retardant thermotropic liquid crystal
copolllyester/PET blends. PET/Polymer 48, 172-177, 2006.
Duarte, M. A.; Pezzin, A. P. T.; Pezzin, S. H.; Avaliação da cristalinidade de poli(3-
hidroxibutirato) produzido a partir da cana de açúcar, Anais X Encontro de Química da Região Sul, Joinville, p. 242, 2002.
El-Hadi, A., Schnabel, R., Straube, E., Müller, G., Henning, S. (2002). Correlation between
degree of crystallinity, morphology, glass temperature, mechanical properties and biodegradation of poly(3-hydroxyalkanoate) PHAs and their blends. Polymer Testing, 21(6),
665-674, 2002.
Fann, D.M.; Huang, S.K.; Lee, J.Y. DSC studies on the crystallization characteristics of poly
(ethylene terephthalate) for blow applications. Polym. Eng. Sci. 38, 265-272, 1998.
Favis, B. D. Em Polymer Blends in Factors Influencing the Morphology of Immiscible
Polymer Blends in Melt Processing; Paul, D. R.; Bucknall, C. B., eds.; John Wiley &
Sons: New York, 2000, vol. 1.
Feitosa M.A.F. Compatibilização de blenda polimérica de poliamida 6,6/ polietileno de baixa densidade utilizando radiação ionizante de feixe de elétrons. São Paulo, Universidade de são
Paulo, Instituto de pesquisas energéticas e nucleares (IPEN). 2009. Dissertação de mestrado.
Fox, T.G. Influence of diluent ando f copolymer composition on the glass temperature of a
polymer system. Bull. Am. Phys. Sco. 1, 123, 1956.
Gao, J.; Yu, M.; Li, Z. Nonisothermal crystallization kinetics and melting behaviour of bimodal medium densitypolyethlene/low density polyethylene blends. Eur. Polym. J. 44, 1533-
1539, 2004.
Gao, G.; Zhang, L.; Sun, H. Chen, G.; Zhang, M.; Ma, R.; Liu, F. Crystalline end thermal
behaviourof poly (ethylene terephthalate)/polyphenox blends. J. Appl. Polym. Sci. 97, 878-885,
2005.
Greco, P.; Martuscelli, E. Crysatallization and thermal behaviour of poly (D(-) -3-
hydroxybutyrate)-based blends.
Groeninckx, G.; Reynaeres, H.; Berghmans, H.; Smerts, G. Morphology and melting behavior
of semicrystaline poly (ethylene terephthalate). I. Isothermally crystallized PET. J. Polym. Sci.; Polym. Phys. Edn. 18, 1311-1324, 1980.
Gunaratne L.M.W.K., Shanks R.A., Amarasinghe G. Thermal history effects on crystallisation and melting of poly(3-hydroxybutyrate). School of Applied Science, RMIT University, GPO
Box 2476V, Melbourne, Vic. 3001, Australia , 2004.
Ha, C. S.; Cho, W. J. Miscibility, properties, and biodegradability of microbial polyester containing blends. Prog. Polym. Sci. 2002, 27, 759.
Hage Jr, E. Compósitos e blendas poliméricas: Curso de blendas poliméricas. São Carlos. Departamento de Engenharia de Materiais, Universidade Federal de São Carlos, 1989. Apostila.
Hay, J.N. Crystallisation of poly(3-hydroxybutyrate)/polyvinyl acetate blends. The School of Metallurgy and Materials, The University of Birmingham, Edgbaston, 2000.

70
Hoffman, J.D.; Weeks, J.J. melting process and the equilibrium melting temperature of
polyclorotrifluoroethylene. J. Research Natl. Bur. Standards, 66-A, 13-28, 1962.
Hu, Y.S.; Liu, R.Y.F.; Rogunova, M.; Schiraldi, D.A.; Nazarenko, S.; Hiltner, A.; Baer, E.
Oxygen-barrier properties of cold-crystallized and melt-crystallized poly (etfilene terephthalate-co-4,4-bibenzoate). J. Appl. Polym. Sci. Part B: Polym. Phys. 40, 2685-2698, 2005.
Huang, H.; Ozaki, Y. Non-isothermal crystallization and thermal transitions of a biodegradable,
partially hydrolyzed poly(vinyl alcohol). Polymer, 2006, 47, 3935.
Hwang, S.H.; Chen, C.C.; Chen, H.L.; Yang, W.C.O. Analysis of two-stages crystallization
kinetics for poly (ethylene therephthalate)/poly (ether imide) blends. Polymer 38, 4097-4101, 1997.
Jabarian, S.A. Crystallizationkinetcis of polyetilene teraphtalate. J. Appl. Polym, 1987.
Jang, L.W.; Lee, K.H.; Lee, D.C.; Yoon, J.S.; Chin,I.; Choi, H.J. Alternating copolymer as
compatibilizerfor blends of poly (ethylene terephthalate) and polystyrene. J. Appl. Polym. Sci.
78, 1998-2007, 2000.
Keith, H.D.; Padden, F.J.– Banding in polyethylene and other spherulites. Mcromolecules 29,
7776-7786, 1996.
Keller, A. The spherulitic structure of crystalline polymers. Part. I. Investigations with the
polarizingmicroscoope. J. polym. Sci. 17, 291-308, 1955.
Kim, W.N; Bunrs, C.M. phase beaviour of blends of PC with partially miscible polymer. J.
Appl. Polym. Sci. 41, 1575-1593, 1990.
Kint, D.P.R.; Ilarduya, A.M.; Sansalvadó, A.; Ferrer, J.;Muñoz-Guerra, S. Microstucture and
crystallization of melt-mixed poly (ethilene terephthalate)/poly(ethylene isophitalate) blends. J.
Appl. Polym. Sci. 90, 3076-3086, 2003.
Kint, D.P.R.; Rudé, E.; Llorens, J.; Muñoz-Guerra, S. Crystalization and proerties of poly
(ethilene terephthalate copolymers contain 5-tert-butyl isophitalic units. Polymer 43. 7529-
7537, 2002.Lim, J.Y. Properties of high-speed spun high molecular weight poly (ethilene terephthalate) filaments. J. Appl. Polym. Sci. 71, 1283-1291, 1999.
Kong, Y.; Hay, J.N. Miscibility and crystallization beaviour of poly (ethylene theraphytalate)/ polycarbonate blends. Polymer 43, 1805-1811, 2002.
Koning G.J.M.; Lemstra, P.J.; Hill, D.J.T.; Carswell ,T.G.; O’Donnell ,J.H.; Ageing phenomena in bacterial poly[(R)-3-hydroxybutyrate]: 1. A study on the mobility in poly[(R)-3-
hydroxybutyrate] powders by monitoring the radical decay with temperature after γ-radiolysis at
77 K Polymer 33 (1992) 3295–3297.
Kwei, K; Frisch, H.L. Interaction parameter in polimer mixtures. Macromolecules 11, 1267-
1271, 1978.
Li, B.; Yu, J.; Lee, S.; Ree, M. Crystallization of poly (ethylene terephtalate co ethylene
isophthalate). Polymer 40, 5371-5375, 1999.
Lim, J.Y.; Kim, S.Y. Properties of high-speed spun high molecular weight poly (ethylene terephthalate) filaments. J. Appl. Polym. Sci. 71, 1283-1291, 1999.

71
Liu,T.; Mo, Z.; Wang, S.; Zhang, H. Noisothermal Melt and cold crystallization kinects of poly
(aryl ether ether ketone ketone). Polym. Eng. Sci. 37, 568-575, 1997. A
Liu,T.; Mo, Z.; Wang, S.; Zhang, H. isothermal Melt and cold crystallization kinects of poly
(aryl ether ether ketone ketone) (PEEKK). Eur. Polym. J. 33, 1405-1414, 1997. B
Liu, M.; Zhao, Q.; Wang, Y.; Zhang, C.; Mo, Z.; Cao, S. Melting behaviors, isothermal and
non-isothermal crystallization kinetics of nylon 1212. Polymer, 2003, 44, 2537.
Lorenzo, A.T.; Arnal, M.L.; Albuerne, J.; Müller, A.J. DSC isothermal polymer crystallization
kinetics measurements and the use of the Avrami equation to fit the data: Guidelines to avoid
common problems. Polym. Test., 2007, 26, 222.
Lu,X.F.; Hay, J.N. Isothermal crystallization kinects and melting behavior of poly (ethylene
terephthalate). Polymer. 42, 9423-9432, 2001.
Martinelli, A.; D’llario, L.; Caminiti, R. Poly (p-phenylene sulfide) nonisothermal cold-
crystallization. J. Polym. Sci., Part B: Polym. Phys. 43, 2725-2736, 2005.
Minakov, A.A.; Mordvintsev, D.A.; Schick, C. Melting and reorganization of poly (ethylene
terephthalate) on fast heating (1000 K/s). Polymer 45, 3755-3763, 2004.
Mingtao R.; Yanping H.; Chenguang Y. Melt-crystallization behavior and isothermal
crystallization kinetics of crystalline/crystalline blends of poly(ethylene
terephthalate)/poly(trimethylene terephthalate). Thermochimica Acta 495 (2009) 51–56.
Miyagi, A.; Wunderlich, B. Superheating and reorganization on melting of poly (ethylene
terephthalate). J. Polym. Sci., Part A-2: Polym. Phys. 10, 1401-1405, 1972.
Myung, H.S.; Yoon. W.J.; Yoo, E.S.; Kim, B.C.; Im, S.S. Effect of shearing on crystallization
beaviour of poly (ethylene terephthalate). J. Appl. Polym. Sci. 80, 2640-2646, 2001.
Nishi, T.; Wang, T.T.; Kwei, T.K. Thermally Induced phase separation beaviour of compatible
polymer mixtures. Macromolecules 8, 227-234, 1975.
Nogales, A.; Esquerra, T.A.; Denchev, Z.; Calleja, F.J.B. Introduction time for cold crystallization in semi-rigid polymers: PEN and PEEK. Polymer 42, 5711-5715, 2001.
Olabisi,O; Robeson, L.M.; Shaw, M.T. Polymer-Polymer Miscibility, Academic Press, New York, 1979.
Ozawa, T. Kinetics of non-isothermal crystallization. Polymer12, 150-158,01971.
Paiva, J.M.F.; Mayer, S.; Cândido, G.M.; Rezende, M.C. Avaliação da temperatura de transição
vítrea de compósitos poliméricos reparados de uso aeronáutico. Polímeros:ciência e tecnologia,
janeiro-março, vol.16, número 001. Associação brasileira de polímeros. São Carlos, Basil. PP.79-87. 2006.
Paul, D. R.; Barlow J. W. Polymer blends (or alloys). J. Macromol. Sci. Rev.
Macromol. Chem., 109-168, 1980.

72
Paul, D. R.; Barlow J. W.; Keskkula, H. Em Encyclopedia of Polymer Science and
Engineering; Mark, H. F.; Bikales, N. M.; Overberger, C. G.; Menges, G.; Kroschwits,
J. I., eds.; J. Wiley & Sons: New York, 1988, vol. 12. Pingping, Z.; Dezhu, M. Double cold crystallization peaks of poly (ethylene terephthalate) – 1.
Samples isothermally crystallized at low temperature. Eur. Polym. J.. 33, 1817-1818, 1990.
Plivelic, T.S.; Cassu, S.N.; Gonçalves, M.C.; Torriani, I.L. Experiências simultâneas de espalhamento de raios X e DSC com resolução temporal utilizando radiação síncrotron. São
Carlos. Artigo Técnico científico. Polímeros v.15 n°3, 2005.
Pó, R.; Occhiello, E.; Giannotta, G; Pelosini, L; Abis, L. New polymeric materiles for
containers manufacture based on PET/PEN compolyesteres and blends. Polym. Adv. Tech. 7,
365-373, 1996.
Porter, R.S.; Wang,L. Compatibility and transesterification in binary polymer blends. Polymer
33, 2019-2030, 1992.
Quental, A. C.; Carvalho, F.P.; Tada, E.S. e Felisberti, M. I. Blendas de PHB e seus
copolímeros: miscibilidade e compatibilidade – Quim. Nova, Vol. XY, No. 00, 1-9, 2009.
Instituto de Química, Universidade Estadual de Campinas, Campinas - SP, Brasil.
Qiao, X.; Wang, X.; Zhao, X.; Mo, Z.; Zhang, H. Nonisothermal crystallization of poly (3-
dodecylthiophene) and poly (octadodecylthiophene). Synthetic Metals 113, 1-6, 2000.
Qiu,Z., Mo, Z.; Zhang, H.; Sheng, S.; Song, C. Nonisothermal melt and cold crystallization
kinetics of poly (aryl ether ketone ether ketone ketone). J. Appl. Polym. Sci. 77, 2865-2871,
2000.
Rabello, M.S. Estudo da cristalização a frio do PET para produção de embalagens. Revista
eletrônica de materiais e processos. V.3.2, 2008.
Reinsch, V.E.Rebenfeld, L. Crystallization of poly (ethilene terephthalate)/ polycarbonate
blends. J. Appl. Polym. 1996.
Rodrigues F.J.M.; Guuillen, R.L.; Mendoza, M.A.W. solid-state polymerization and bulk
crystallization behavior of poly (ethilene terephthalate). J. Appl. Polym. Sci. 75, 78-86, 2000.
Sajkiewicz, P.; Carpanet, L.; Wasiak, A. Apliccation of the Ozawa model to non-isothermal
cryztallization of poly (ethilene terephthalate). Polymer 42, 5365-5370, 2001.
Schultz, J.M. Polymer Materials Science. Prentice-Hall, New Jersey, 1974.
Shiomi, T.; Tsukada, H.; Takeshita, H.; Takenaka, K.; Tekuza, Y. Crystallization of
semicrystalline block copolymers containing a glassy amorphous component. Polymer 42, 4997-5004, 2001.
Silva, A.M.L.M.V. Cinética de cristalização do Poli (tereftalato de etileno) comercial. Rio de Janeiro, Universidade Federal do Rio de Janeiro, IMA, 1991. Dissertação de mestrado.
Silva, L. F.; Gomez, J. G. C.; Oliveira, M. S.; Torres, B. B.; J. Biotechnol. 2000, 76, 165.
Silvestre, C.; Cimino, S.; Di Pace, E. Crystalizable polymer blends. 1595-1609, 1996.

73
Silvestre, C.; Cimino, S.; Martuscelli, E.; Karasz, F.E.; Macknight, W.J. Poly (ethylene
oxide)/poly (methacrylate) blends: Influence of tacticity of poly (methyl methacrylate) on blend
structure and miscibility. Polymer 28, 1190-1199, 1987. A.
Skoglund, P.; Frasson, A. Continuous cooling and isothermal crystallization of
polycaproprolactone. J. Appl. Polym. Sci. 61,2455-2465, 1996.
Shuwen P., Yuxian A., Cheng C., Bin F., Yugang Z., Lisong D. Isothermal crystallization of poly(3-hydroxybutyrate-co-3-hydroxyvalerate). www.elsevier.com/locate/polymer, pesquisado
dia 2 de agosto de 2009 às 23:15, publicado em 2002.
Tan, S.; Su, A.; Li, W.; Zhou, E. New insight into melting and crystallization behaviour in semicrystalline poly (ethylene terephthalate). J. Appl. Polym. Sci. 38, 53-60, 2000.
Tankhiwale, S.; Gupta, M. C.; Viswanath, S. G. J. Crystallization studies of crystalline-
amorphous blends: Polyethylene terephthalate (PET)-polystyrene (PS). Polym Plast Technol
Eng 2002, 41, 171–181.
Utracki and Weiss. Multiphase Polymers Blend and Ionomers Acs Symposium. Editors, 1989,
pag. 101, chapter 16.
Van Antwerpen, F.; Van Krevelen, D.W. Light-Scattering Method for investigation of the
kinetics of crystallization of spherulites. J. Polym. Sci., Polym. Phys. Edn. 10, 2409-2421, 1972.
Viana, J.C.; Alves, N.M.; Mano, J.F. Morfology andmechanical properties of injection molded
poly(ethilene terephthalate). Polym. Eng. Sci. 44, 2174-2184, 2004.
Xiao, J.; Zhang, H.; Wan, X.; Zhang, D.; Zhou, Q.; Woo, E.M. and Turner, S.R. Crystallization
kinetics of new copoly(ethylene terephthalate-imide)s. Polymer, 2002, 43, 3683.
Wellen, R.M.R. ; Rabelo, M.S. Cristalização a frio do PET: como ela ocorre e quais são as suas conseqüências. Plástico industrial 96, 96-11, 2005.
Wellen, R.M.R. ; Rabelo, M.S. The kinetics of isothermal Cryztallization and tensile properties of poly (ethylene) terephthalate. J. Mater. Sci. 40, 6099-6104, 2005.
Wellen, R.M.R. ; Rabelo, M.S. A cristalização a frio de pre-formas de PET: suas causas e como
retardá-la.2006.
Wellen, R.M.R. Cristalização a frio do PET e das blendas PET/SAN e PET/PS. Tese de
doutorado, 2007.
Withey, R.E.; Hay, J.N.; The effect of seeding on the crystallisation of
poly(hydroxybutyrate), and co-poly(hydroxybutyrate-co-valerate). Polymer 40: 5147-
5152, 1999. Woodward, A.E. Introduction to the series. Hanser/Gardner Publishers, 1995.
Wunderlich B., Okazaki I., Ishikiriyama K., Boller A., Melting by temperature-modulated calorimetry. Thermochim.Acta 324 (1998) 77.
Yun H.; Harumi S.; Jianming Z.; Isao N.; Yukihiro O. Crystallization behavior of poly(L-lactic acid) affected by the addition of a small amount of poly(3-hydroxybutyrate).
www.elsevier.com/locate/polymer.

74
Zhao, J.; Yang, J.; Song, R.; Linghu, X.; Fan, Q. The affect of annealing on the subsequent cold
crystallization of amourfous poly (ethilene terephthalate). Eur. Polym. J. 38, 645-648, 2002.
Zheng, L.; Wang, Y.Z.; Yang, K.K.; Wang, X.L.; Chen, S.C.; Li, J. Effect of PEG on the
crystallization of PPDO/PEG blends. Eur. Polym.. J. 41, 1243-1250, 2005.

75
Apêndice
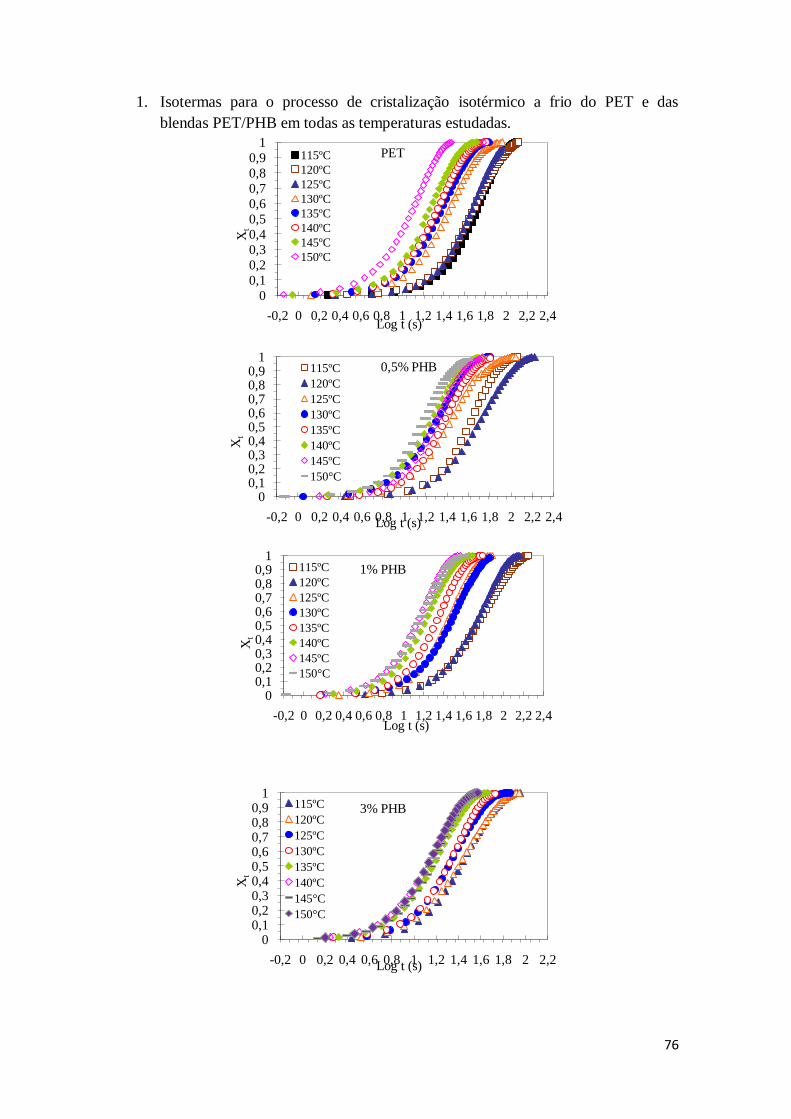
76
1. Isotermas para o processo de cristalização isotérmico a frio do PET e das
blendas PET/PHB em todas as temperaturas estudadas.
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
-0,2 0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2 2,2 2,4
Xt
Log t (s)
PET115ºC
120ºC
125ºC
130ºC
135ºC
140ºC
145ºC
150ºC
00,10,20,30,40,50,60,70,80,9
1
-0,2 0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2 2,2 2,4
Xt
Log t (s)
0,5% PHB115ºC
120ºC
125ºC
130ºC
135ºC
140ºC
145ºC
150°C
00,10,20,30,40,50,60,70,80,9
1
-0,2 0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2 2,2 2,4
Xt
Log t (s)
1% PHB115ºC
120ºC
125ºC
130ºC
135ºC
140ºC
145ºC
150°C
00,10,20,30,40,50,60,70,80,9
1
-0,2 0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2 2,2
Xt
Log t (s)
3% PHB115ºC
120ºC
125ºC
130ºC
135ºC
140ºC
145°C
150°C

77
2. Curvas de Avrami para a cristalização isotérmica a frio do PET e das blendas
PET/PHB em todas as temperaturas estudadas.
-3
-2,5
-2
-1,5
-1
-0,5
0
0,5
1
0,4 0,9 1,4 1,9 2,4
Log(-
ln(1
-Xt)
)
Log t (s)
115ºC
PET
0.5% PHB
1% PHB
3% PHB
-3
-2,5
-2
-1,5
-1
-0,5
0
0,5
1
0,4 0,9 1,4 1,9 2,4
Log(-
ln(1
-Xt)
)
Log t (s)
120ºC
PET
0.5% PHB
1% PHB
3% PHB
-3
-2,5
-2
-1,5
-1
-0,5
0
0,5
1
0,4 0,9 1,4 1,9 2,4
Log
(-ln
(1-X
t))
Log t (s)
125ºC
PET
0.5% PHB
1% PHB
3% PHB
-3
-2,5
-2
-1,5
-1
-0,5
0
0,5
1
0,4 0,9 1,4 1,9 2,4
Log
(-ln
(1-X
t))
Log t (s)
130ºC
PET
0.5% PHB
1% PHB
3% PHB
-3
-2,5
-2
-1,5
-1
-0,5
0
0,5
1
0,4 0,9 1,4 1,9 2,4
Log
(-ln
(1-X
t))
Log t (s)
135ºC
PET
0.5% PHB
1% PHB
3% PHB
-3
-2,5
-2
-1,5
-1
-0,5
0
0,5
1
0,4 0,9 1,4 1,9 2,4
Log
(-ln
(1-X
t))
Log t (s)
140ºC
PET
0.5% PHB
1% PHB
3% PHB
-3
-2,5
-2
-1,5
-1
-0,5
0
0,5
1
0,4 0,9 1,4 1,9 2,4
Log
(-ln
(1-X
t))
Log t (s)
145ºC
PET
0.5% PHB
1% PHB
3% PHB
-3
-2,5
-2
-1,5
-1
-0,5
0
0,5
1
0,4 0,9 1,4 1,9 2,4
Log
(-ln
(1-X
t))
Log t (s)
150ºC
PET
0.5% PHB
1% PHB
3% PHB

78
3. Curvas em “S” para a cristalização não isotérmica a frio do PET e das blendas
PET/PHB, em todas as taxas de aquecimento estudadas.
0
0,2
0,4
0,6
0,8
1
3,6 4,1 4,6 5,1 5,6 6,1 6,6 7,1
XT
(%)
Ln T (°C)
1 °C/min
PET
0,5% PHB
1% PHB
3% PHB
0
0,2
0,4
0,6
0,8
1
3,6 4,1 4,6 5,1 5,6 6,1 6,6 7,1
XT
(%)
Ln T (°C)
2,5 °C/min
PET
0,5% PHB
1% PHB
3% PHB
0
0,2
0,4
0,6
0,8
1
3,6 4,1 4,6 5,1 5,6 6,1 6,6 7,1
XT
(%)
Ln T (°C)
5 °C/min
PET
0,5% PHB
1% PHB
3% PHB
0
0,2
0,4
0,6
0,8
1
3,6 4,1 4,6 5,1 5,6 6,1 6,6 7,1
XT
(%)
Ln T (°C)
7,5 °C/min
PET
0,5% PHB
1% PHB
3% PHB
0
0,2
0,4
0,6
0,8
1
3,6 4,1 4,6 5,1 5,6 6,1 6,6 7,1
XT
(%)
Ln T (°C)
10 °C/min
PET
0,5% PHB
1% PHB
3% PHB
0
0,2
0,4
0,6
0,8
1
3,6 4,1 4,6 5,1 5,6 6,1 6,6 7,1
XT
(%)
Ln T (°C)
15 °C/min
PET
0,5% PHB
1% PHB
3% PHB
0
0,2
0,4
0,6
0,8
1
3,6 4,1 4,6 5,1 5,6 6,1 6,6 7,1
XT
(%)
Ln T (°C)
20 °C/min
PET
0,5% PHB
1% PHB
3% PHB
0
0,2
0,4
0,6
0,8
1
3,6 4,1 4,6 5,1 5,6 6,1 6,6 7,1
XT
(%)
Ln T (°C)
30 °C/min
PET
0,5% PHB
1% PHB
3% PHB

79
4. Tabelas com as temperaturas vítreas, de fusão e de cristalização para todas as
taxas de aquecimento e todas as amostras analisadas.
(°C/min)
PET
Tg (°C)
0,5 % PHB
Tg (°C)
1% PHB
Tg (°C)
3%PHB
Tg (°C)
1 62,5 62,4 57,3 53,9
2,5 61,3 61,3 65,7 56,4
5 65,3 63,5 63,0 63,7
7,5 64,2 64,3 65,4 59,7
10 76,3 66,5 66,7 60,8
15 67,6 71,0 70,2 63,8
20 72,5 71,1 71,7 63,7
30 73,5 74,0 74,6 67,3
(°C/min)
PET
T (°C)
0,5 % PHB
T (°C)
1% PHB
T (°C)
3%PHB
T (°C)
Tci Tc Tcf Tci Tc Tcf Tci Tc Tcf Tci Tc Tcf
1 103,1 112,7 123,4 100,2 108,5 118,5 97,9 108,7 118,5 87,2 98,3 108,2
2,5 108,6 115,8 126,5 103,4 115,5 127,6 100,8 110,9 121,3 94,0 106,2 116,8
5 113,1 122,1 135,5 107,6 119,5 132,8 106,0 116,5 129,5 97,8 108,9 125,2
7,5 112,5 124,2 140,0 109,2 122,6 139,2 109,6 120,9 135,3 99,7 112,2 127,5
10 115,5 127,7 144,6 115,1 125,7 143,0 110,9 123,0 139,5 99,9 114,6 133,7
15 119,8 132,5 148,1 118,4 131,9 153,0 116,1 129,4 148,1 104,7 121,5 137,9
20 123,3 136,6 155,5 119,2 134,8 155,4 119,1 132,8 151,3 106,8 121,8 145,0
30 130,6 144,3 162,58 125,7 140,4 163,9 126,7 141,1 161,3 112,2 129,7 151,5

80
(°C/min)
PET
T (°C)
0,5 % PHB
T (°C)
1% PHB
T (°C)
3%PHB
T (°C)
Tmi Tm Tmf Tmi Tm Tmf Tmi Tm Tmf Tmi Tm Tmf
1 237,7 259,4 265,9 218,3 253,4 263,4 219,2 257,4 261,3 222,3 252,3 262,5
2,5 229,2 251,6 260,9 220,2 251,6 263,1 215,5 251,3 264,4 230,4 253,5 261,0
5 213,8 250,6 267,2 216,0 251,1 267,0 216,7 251,4 264,2 235,7 251,7 265,2
7,5 213,6 249,3 264,1 213,2 152,7 271,5 213,6 250,9 266,8 228,5 251,5 263,7
10 215,7 250,6 264,9 216,6 253,2 273,8 214,1 251,1 265,9 232,4 252,7 267,2
15 218,5 251,2 268,7 217,8 254,2 272,3 216,8 252,8 268,6 214,4 254,1 273,1
20 218,9 251,4 269,5 219,1 253,8 275,8 219,5 255,3 276,6 228,4 275,3 252,3
30 226,9 255,0 277,4 219,9 254,2 279,4 220,8 254,1 286,4 214,4 256,0 282,9
5. Tabela Variação da entropia do sistema em todas as taxas de aquecimento e
concentração de PHB indicada.
Φ
(°C/min)
0,5% PHB 1% PHB
Tm (°C) ΔHm (J/g) ΔS(J/g°C) Tm (°C) ΔHm (J/g) ΔS(J/g°C)
5 251,1 44,9 0,18 251,4 45,1 0,18
7,5 252,7 44,2 0,17 250,9 44,6 0,18
10 253,2 42,3 0,17 251,1 42,5 0,17
15 254,2 34,6 0,14 252,8 34,9 0,14
20 253,8 33,5 0,13 255,3 33,8 0,13
30 254,2 32,1 0,12 254,1 32,3 0,13


















