
DESENVOLVIMENTO E ANÁLISE DAS PROPRIEDADES DE...
195
CECILIA ZORZI BUENO DESENVOLVIMENTO E ANÁLISE DAS PROPRIEDADES DE MEMBRANAS DE QUITOSANA E ALGINATO CONTENDO POLIHEXAMETILENO BIGUANIDA PARA O TRATAMENTO DE LESÕES DE PELE CAMPINAS 2015
Transcript of DESENVOLVIMENTO E ANÁLISE DAS PROPRIEDADES DE...

CECILIA ZORZI BUENO
DESENVOLVIMENTO E ANÁLISE DAS PROPRIEDADES DE
MEMBRANAS DE QUITOSANA E ALGINATO CONTENDO
POLIHEXAMETILENO BIGUANIDA PARA O TRATAMENTO
DE LESÕES DE PELE
CAMPINAS
2015

ii

iii
CECILIA ZORZI BUENO
“DESENVOLVIMENTO E ANÁLISE DAS PROPRIEDADES DE
MEMBRANAS DE QUITOSANA E ALGINATO CONTENDO
POLIHEXAMETILENO BIGUANIDA PARA O TRATAMENTO DE LESÕES
DE PELE”
Tese apresentada à Faculdade de Engenharia Química
da Universidade Estadual de Campinas como parte dos
requisitos exigidos para obtenção do título de Doutora
em Engenharia Química.
Orientadora: Profa. Dra. ÂNGELA MARIA MORAES
Co-orientadora: Profa. Dra. MARA ELGA MEDEIROS BRAGA
ESTE EXEMPLAR CORRESPONDE À VERSÃO
FINAL DA TESE DEFENDIDA PELA ALUNA
CECILIA ZORZI BUENO E ORIENTADA PELA
PROFA. DRA. ÂNGELA MARIA MORAES.
CAMPINAS
2015
UNIVERSIDADE ESTADUAL DE CAMPINAS
Faculdade de Engenharia Química
Departamento de Engenharia de Materiais e de Bioprocessos

iv

v

vi

vii
RESUMO
As lesões de pele representam um grande desafio para os profissionais da saúde, sendo que os
curativos mais modernos disponíveis atualmente são, em sua maioria, importados e de alto custo.
Existe, assim, a necessidade de criar recursos mais acessíveis à população, os quais utilizem
matérias-primas renováveis e sejam produzidos por meio de tecnologias simples e de baixo
custo. A quitosana e o alginato são biopolímeros biocompatíveis, apresentam propriedades
cicatrizantes e são abundantes na natureza, características estas que os tornam atrativos para a
confecção de curativos de lesões de pele. A inclusão de agentes antimicrobianos, como
polihexametileno biguanida (PHMB), aos curativos produzidos a partir destes biopolímeros pode
levar a um melhor desempenho dos mesmos. Neste trabalho de tese, membranas densas e
porosas de quitosana-alginato foram obtidas na presença de diferentes quantidades do tensoativo
Pluronic F68. Os resultados obtidos mostraram que a inclusão de até 10% (m/m) de tensoativo
provoca o aumento da porosidade (de 0,46 para 0,84), da espessura (de 0,08 para 0,50 mm), da
rugosidade (de 1,27 para 21,20 µm), da capacidade de absorção de fluidos (de 14,22 para
21,27 g/g para NaCl a 0,9%) e de vapor d’água (de 15,5 para 36,5%) e a diminuição da
resistência à tração (de 31,1 para 1,1 MPa) e do alongamento na ruptura (de 4,0 para 2,0%) das
membranas. Os materiais obtidos não se mostraram citotóxicos a fibroblastos de camundongos.
O agente antimicrobiano PHMB foi incorporado às membranas por três diferentes métodos:
adição à mistura polimérica, adsorção a partir de solução aquosa e impregnação mediada por
CO2 supercrítico. A incorporação de PHMB por adição à mistura polimérica foi considerada
como a mais satisfatória, pois não provocou alterações no aspecto e morfologia das membranas e
resultou em altas eficiências de incorporação, variando entre 72 e 86 %. Observou-se também
adequado controle da cinética de liberação, sendo que grande parte do antimicrobiano
incorporado não foi liberada em tampão fosfato-salino (PBS), indicando alta afinidade entre o
PHMB e a matriz de quitosana-alginato. As membranas apresentaram atividade antimicrobiana
adequada, o que sugere que possam ser empregadas como barreira contra microrganismos
comumente presentes em lesões de pele, tais como Staphylococcus aureus e Pseudomonas
aeruginosa.
Palavras-chave: quitosana, alginato, polihexametileno biguanida, membranas.

viii

ix
ABSTRACT
Skin lesions represent a great challenge for health professionals, since most of the existing
modern wound dressings are imported and expensive. Therefore, there is a need to create more
accessible products to the public, made from renewable raw materials and processed through
simple and low cost technologies. Chitosan and alginate are biocompatible biopolymers that
have healing properties and are abundant in nature, which are attractive characteristics to the
production of wound dressings. The incorporation of antimicrobial agents, such as
polyhexamethylene biguanide (PHMB), to these dressings can lead to a better performance. In
this work, dense and porous chitosan-alginate membranes were obtained in the presence of
different proportions of the surfactant Pluronic F68. It was noticed that inclusion of up to 10%
(w/w) Pluronic F68 caused an increase in porosity (from 0.46 to 0.84), thickness (from 0.08 to
0.50 mm), roughness (from 1.27 to 21.20 µm), fluids absorption capacity (from 14.22 to
21.27 g/g for 0.9% NaCl) and water vapor absorption capacity (from 15.5 to 36.5%) and a
decrease in tensile strength (from 31.1 to 1.1 MPa) and elongation at break (from 4.0 to 2.0%) of
the membranes. The obtained materials were not toxic to mouse fibroblasts. The antimicrobial
agent PHMB was incorporated to the membranes through three different methods: addition to
the polymeric mixture, adsorption in aqueous solution and via supercritical fluid. The addition to
the polymeric mixture was considered the most promising incorporation method, since it had
high yields (between 72 and 86 %) and did not cause changes in the membranes aspect and
morphology. Release kinetic studies in PBS showed that most of the PHMB loaded remained in
the membranes, indicating high affinity between PHMB and the polymeric matrix. The obtained
membranes showed adequate antimicrobial activity, having potential applicability as barriers
against microorganims commonly found in skin wounds, such as Staphylococcus aureus and
Pseudomonas aeruginosa.
Keywords: chitosan, alginate, polyhehamethylene biguanide, membranes

x

xi
SUMÁRIO
1. INTRODUÇÃO .................................................................................................................... 1
1.1. Objetivos ............................................................................................................................. 4
2. REVISÃO DA LITERATURA ........................................................................................... 6
2.1. Lesões de pele ..................................................................................................................... 6
2.2. Avanços no tratamento de lesões de pele ......................................................................... 8
2.3. Polissacarídeos ................................................................................................................. 11
2.3.1. Quitosana ....................................................................................................................... 12
2.3.2. Alginato .......................................................................................................................... 17
2.3.3. Complexo de polieletrólitos de quitosana-alginato ...................................................... 21
2.4. Membranas de quitosana-alginato ................................................................................. 24
2.5. Surfactantes não-iônicos ................................................................................................. 27
2.5.1. Pluronic F68 .................................................................................................................. 27
2.6. Incorporação de agentes bioativos em curativos .......................................................... 28
2.7. PHMB ............................................................................................................................... 32
2.8. Considerações finais ........................................................................................................ 37
3. MATERIAIS E MÉTODOS .............................................................................................. 38
3.1. Materiais utilizados para o preparo das membranas .................................................. 38
3.2. Método de preparo das membranas .............................................................................. 38
3.2.1. Cinética de evaporação de solventes da membrana durante a secagem ..................... 41
3.3. Incorporação do agente antimicrobiano........................................................................ 41
3.3.1. Incorporação por adição à mistura polimérica ............................................................ 41
3.3.2. Incorporação por adsorção a partir de solução aquosa ............................................... 41
3.3.2.1. Determinação de isotermas de adsorção .................................................................... 42
3.3.2.2. Incorporação por adsorção em duas diferentes condições iniciais .......................... 42
3.3.3. Impregnação de PHMB mediada por CO2 supercrítico ............................................... 42
3.3.3.1. Unidade de processamento e impregnação ................................................................ 43
3.3.3.2. Processamento de membranas sem PHMB em CO2 supercrítico ............................. 43
3.3.3.3. Impregnação de PHMB mediada por CO2 supercrítico ............................................ 44
3.4. Caracterização das membranas ..................................................................................... 44

xii
3.4.1. Morfologia ..................................................................................................................... 44
3.4.2. Espessura ...................................................................................................................... 45
3.4.3. Rugosidade ..................................................................................................................... 45
3.4.4. Coloração ....................................................................................................................... 45
3.4.5. Área superficial e porosidade ........................................................................................ 46
3.4.6. Capacidade de absorção e estabilidade das membranas em soluções aquosas ........... 47
3.4.7. Sorção de vapor d’água ................................................................................................. 48
3.4.8. Transmissão de vapor d’água ....................................................................................... 49
3.4.9. Ângulo de contato .......................................................................................................... 49
3.4.10. Propriedades mecânicas .............................................................................................. 49
3.4.11. Análise térmica simultânea (SDT) (Calorimetria e termogravimetria) ..................... 50
3.4.12. Espectroscopia no infravermelho com transformada de Fourier ............................. 51
3.4.13. Citotoxicidade in vitro indireta .................................................................................... 51
3.4.14. Atividade antimicrobiana ............................................................................................ 52
3.4.15. Determinação da eficiência de incorporação de PHMB nas membranas ................ 53
3.4.16. Cinética de liberação de PHMB incorporado nas membranas ................................. 54
3.4.16.1. Análise da liberação de PHMB incorporado por tecnologia supercrítica ............. 54
3.4.16.2. Análise da liberação de PHMB incorporado por adição direta e por adsorção a
partir de soluções aquosas ....................................................................................................... 55
3.5. Análise dos resultados ..................................................................................................... 55
4. RESULTADOS E DISCUSSÃO ....................................................................................... 56
4.1. Definição das condições mais apropriadas de processamento para a obtenção das
membranas .............................................................................................................................. 56
4.1.1. Efeitos da remoção de acetona da etapa de produção das membranas sobre as
características do material ....................................................................................................... 60
4.1.2. Influência da temperatura em diferentes etapas do processamento nas características
das membranas ........................................................................................................................ 64
4.2. Caracterização detalhada de membranas obtidas na ausência e na presença de
diferentes proporções de Pluronic F68 nas condições mais adequadas de processamento68
4.2.1. Aspecto e morfologia ..................................................................................................... 68
4.2.2. Área superficial e porosidade ........................................................................................ 71

xiii
4.2.3. Espessura e rugosidade ................................................................................................. 72
4.2.4. Coloração ....................................................................................................................... 74
4.2.5. Hidrofilicidade ............................................................................................................... 75
4.2.6. Comportamento das membranas na presença de soluções aquosas ............................ 78
4.2.7. Propriedades mecânicas ................................................................................................ 83
4.2.8. Citotoxicidade in vitro indireta ...................................................................................... 86
4.2.9. Espectroscopia no infravermelho com transformada de Fourier ............................... 87
4.2.10. Análise térmica simultânea (SDT) (Calorimetria e termogravimetria) ..................... 90
4.2.10.1. Análise termogravimétrica (TGA) ............................................................................ 90
4.2.10.2. Calorimetria exploratória diferencial (DSC) ........................................................... 94
4.2.11. Considerações finais acerca da caracterização das membranas produzidas na
ausência e na presença de diferentes proporções de Pluronic F68 ....................................... 95
4.3. Efeitos da incorporação de PHMB por diferentes métodos nas características das
membranas .............................................................................................................................. 97
4.3.1. Incorporação de PHMB por adição à mistura polimérica .......................................... 98
4.3.2. Incorporação de PHMB por adsorção a partir de solução aquosa ........................... 104
4.3.2.1. Estudo do comportamento de adsorção de PHMB às membranas de quitosana-
alginato ................................................................................................................................... 105
4.3.2.2. Caracterização das membranas contendo PHMB incorporado por adsorção a partir
de solução aquosa .................................................................................................................. 113
4.3.3. Incorporação de PHMB mediada por CO2 supercrítico ............................................ 121
4.3.3.1. Análise dos efeitos do processamento com CO2 supercrítico sobre as características
das membranas ...................................................................................................................... 122
4.3.3.2. Caracterização das membranas contendo PHMB incorporado via CO2 supercrítico129
4.3.4. Comparação entre diferentes métodos de incorporação de PHMB em membranas de
diferentes formulações visando à seleção das preparações mais apropriadas .................... 131
4.3.5. Caracterização suplementar das membranas mais promissoras ............................... 132
5. CONCLUSÕES E SUGESTÕES .................................................................................... 138
5.1. Conclusões ...................................................................................................................... 138
5.2. Sugestões para trabalhos futuros ................................................................................. 139
6. REFERÊNCIAS BIBLIOGRÁFICAS ........................................................................... 142

xiv
APÊNDICE I - Determinação da massa molar viscosimétrica média da quitosana e do
alginato .................................................................................................................................. 156
APÊNDICE II – Determinação do grau de desacetilação da quitosan ............................ 159
APÊNDICE III – Determinação da razão M/G do alginato ............................................. 161
APÊNDICE IV – Estabelecimento da estratégia mais apropriada de quantificação do
PHMB incorporado .............................................................................................................. 163
APÊNDICE V – Verificação da estabilidade do PHMB nas condições de preparação das
membranas ............................................................................................................................ 170
APÊNDICE VI – Análise do efeito de potenciais solventes para a solubilização das
membranas ............................................................................................................................ 171
APÊNDICE VII – Ensaios de extração e quantificação de PHMB em soluções básicas 173

xv
AGRADECIMENTOS
A Deus, por tudo.
Aos meus pais, Maria Inês e Gilberto, e à minha irmã Lucia, pelo incentivo e pelo apoio em todos
os momentos.
À professora Dra. Ângela Maria Moraes pela orientação deste trabalho. Agradeço pelo tempo e
atenção dedicados a esta tese e pela forma criteriosa de avaliação.
À Dra. Mara Elga Medeiros Braga pela co-orientação deste trabalho.
Aos membros da banca dos exames de qualificação e da defesa de tese pela contribuição dada a
este trabalho.
Aos professores Dra. Sônia Maria Alves Bueno, Dr. Everson Alves Miranda e Dr. Theo Guenter
Kieckbush pelo uso das dependências dos seus laboratórios.
Ao professor Dr. Hermínio José Cipriano de Sousa, à Dra. Ana Maria Antunes Dias e ao
professor Dr. Paulo de Tarso Vieira e Rosa pela colaboração a este trabalho.
À empresa LM Farma Indústria e Comércio Ltda. pela parceria e por ceder o reagente Cosmocil
CQ e à empresa Arch Chemicals pela doação do reagente Vantocil 100.
À Dra. Eliane Gama Luchesi pela realização dos ensaios de atividade antimicrobiana e à Dra.
Márcia Zilioli Bellini pela realização dos ensaios de citotoxicidade.
Aos funcionários da Faculdade de Engenharia Química que colaboraram para a realização deste
trabalho.
À Fundação de Apoio à Pesquisa do Estado de São Paulo (FAPESP) pela concessão da bolsa de
estudos e da reserva técnica.
À Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) pela concessão da
bolsa de estudos de doutorado sanduíche na Universidade de Coimbra.
Ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pelo apoio
financeiro.

xvi
Aos amigos do LEBC, em especial à Ana Luíza, Andrea, Cecília Westin, Fernanda e Renata, pelo
apoio e pela companhia no laboratório. Agradeço também à Ana Kelly, Priscila e Walter, que
acompanharam as etapas iniciais deste trabalho.
Aos amigos do LEBp e LIMBio, Cecília Mourão, Gabriela, Gisele Atsuko, Gisele Pavan e
Nemailla, pelo companheirismo.
Aos amigos da casa da Martinha, pelos momentos de alegria e descontração.
À Itiara pela amizade, pelo incentivo e pelo auxílio em todos os momentos desde a minha
chegada ao laboratório.
E finalmente a todos que, de alguma forma, fizeram parte desta trajetória.

xvii
“Você nunca sabe que resultados virão da sua ação.
Mas se você não fizer nada, não existirão resultados”
Mahatma Gandhi

xviii

1
1. INTRODUÇÃO
A pele tem diversas funções, atuando na regulação da temperatura corpórea e na detecção
de estímulos, conferindo proteção contra microrganismos, agentes químicos, efeitos nocivos do
sol e perda de líquidos. Assim, alterações da função ou do aspecto da pele podem acarretar
consequências importantes para a saúde, principalmente quando o processo de reparo tecidual é
dificultado. Alguns tipos de lesões, como as feridas crônicas e as queimaduras, representam um
grande desafio para os profissionais da área da saúde.
As feridas crônicas são normalmente decorrentes de tromboses, edemas, diabetes ou
pressão local prolongada e envolvem perdas de tecido progressivas (PAUL e SHARMA, 2004).
Com o envelhecimento da população, o número de pacientes afetados por este tipo de ferida vem
aumentando (STEPHEN-HAYNES et al., 2014). No Brasil, os dados estatísticos sobre feridas
crônicas são escassos e atualizados com pouca frequência. Em 2003, estimava-se que quase 3%
da população brasileira fosse portadora desse tipo de lesão, estatística que se elevava para 10%
quando se considerava apenas a população de diabéticos (MANDELBAUM et al., 2003).
As queimaduras também se mostram como um tipo grave de lesão que acomete um grande
número de pessoas em todo o mundo, no entanto, a compilação de dados de incidência no Brasil
é limitada. Em 2005, as queimaduras eram o terceiro tipo de acidente de maior ocorrência na
população brasileira, representando 2,3% dos atendimentos de emergência em hospitais. Naquele
ano, foram registradas 60451 internações por queimaduras no SIH (Sistema de Informações
Hospitalares), enquanto no SIM (Sistema de Informações sobre Mortalidade) foram registrados
2421 óbitos delas decorrentes (MASCARENHAS et al., 2006).
Apesar da variedade de curativos já comercializados, destinados ao tratamento de feridas
graves e de difícil cicatrização, nenhum produto existente vai ao encontro de todos os
requerimentos de um curativo ideal (MOURA et al., 2013). Assim, existe uma clara demanda
por materiais que possam acelerar a cicatrização e reduzir os riscos para o paciente (WIEGAND
e HIPLER, 2010). Além disso, faz-se necessário um maior investimento em pesquisas neste
campo, com o objetivo de criar recursos e tecnologias mais acessíveis à população
(MANDELBAUM et al., 2003).
O tratamento ideal de lesões de pele deve ser realizado de modo a acelerar a cicatrização,
prevenir infecções, manter um ambiente úmido adequado à formação de tecido de granulação,

2
proteger a ferida, absorver e drenar a secreção produzida e evitar ao máximo a formação de
cicatrizes (MUZZARELLI et al., 2007). Neste contexto, os curativos tradicionais como a gaze
possuem muitas desvantagens. Uma delas é a forte aderência à ferida, levando à sua oclusão e ao
acúmulo de fluido sob o curativo, o que favorece a proliferação de patógenos. Muitas vezes,
estes curativos necessitam ser utilizados em conjunto com géis para reduzir o ressecamento da
ferida e aliviar a dor inerente à troca (YUDANOVA e RESHETOV, 2006).
Os avanços mais recentes no tratamento de lesões de pele estão relacionados ao uso de
materiais com atividade biológica que contribuem para o processo de cicatrização, melhorando a
aparência e funcionalidade do tecido regenerado (MUZZARELLI, 2009). Biopolímeros da
categoria dos polissacarídeos e proteínas são muito utilizados no desenvolvimento destes novos
curativos. Encontram-se neste grupo os polissacarídeos quitosana e alginato. Os biopolímeros
em geral possuem alta biocompatibilidade, biodegradabilidade e frequentemente, atividade
fisiológica (YUDANOVA e RESHETOV, 2006). Além disso, possuem a propriedade de se
associar ao tecido danificado, promovendo a cicatrização, podendo também ser facilmente
misturados com agentes antimicrobianos para melhorar o seu desempenho em feridas crônicas.
Alguns dos curativos já desenvolvidos se mostraram eficazes, contudo, a busca pelo curativo
ideal ainda está em andamento (WIEGAND e HIPLER, 2010).
O complexo de polieletrólitos (PEC) de quitosana e alginato tem sido investigado por
vários grupos de pesquisa ao longo dos últimos anos (MENNINI et al., 2012; DA SILVA et al.,
2012; MIRAFTAB et al., 2011; ANBINDER et al., 2011; YAN et al., 2000; YAN et al., 2001;
WANG et al., 2001; WANG et al., 2002) devido à resistência do complexo a mudanças de pH e
à eficácia na liberação controlada de agentes ativos quando comparado à quitosana ou ao
alginato isolados (KHAN e AHMAD, 2013).
A pesquisa pioneira de Yan et al. (2000) foi seguida por Wang et al. (2001), que
desenvolveram um curativo de quitosana-alginato por meio da mistura de soluções dos
polissacarídeos e da reticulação com íons Ca2+. O material obtido apresentou flexibilidade
adequada e estabilidade em água. Anos depois, Rodrigues et al. (2008) propuseram uma
modificação da metodologia estabelecida por Wang et al. (2001) visando ao escalonamento da
produção. As alterações empregadas por Rodrigues et al. (2008) foram o controle da vazão de
adição da solução de quitosana à solução de alginato, o controle das temperaturas de
complexação entre os polissacarídeos e de secagem do PEC, além de não remover as moléculas

3
de polissacarídeos não complexadas por centrifugação e lavagem do PEC com água. Mais tarde,
Bueno (2010) introduziu mais algumas mudanças na metodologia, objetivando à obtenção de
membranas porosas. Tais mudanças consistiram na adição de tensoativos biocompatíveis, como
o Pluronic F68 e o Tween 80, à suspensão polimérica, sob alta taxa de agitação, permitindo a
incorporação de bolhas de ar, formando uma espuma consistente. As membranas porosas obtidas
por Bueno (2010) por meio de metodologia simples e de baixo custo, foram consideradas
adequadas para aplicações biomédicas, como curativos de lesões de pele ou mesmo como
scaffolds nos quais podem ser cultivadas células previamente à aplicação do material na pele
lesada. Contudo, a proporção de tensoativo utilizada por Bueno (2010) foi considerada muito
alta em relação à quantidade de polissacarídeos utilizada no preparo das membranas, o que
poderia resultar em toxicidade às células da pele, exigindo diversas etapas de lavagem das
membranas a fim de eliminar o excesso de tensoativo.
Desta forma, no presente trabalho, buscou-se melhorar a formulação empregada no
trabalho anterior, a fim de reduzir o seu potencial citotóxico. Para tal, foi dada continuidade aos
estudos anteriores empregando-se menores proporções de tensoativos para a produção das
membranas.
Com o objetivo de facilitar o processo de produção das membranas e reduzir os custos de
material, a obtenção de membranas na ausência de acetona, previamente empregada como co-
solvente da quitosana, foi realizada. Além disso, fez-se um estudo de variação da temperatura de
processo, o que poderia levar à obtenção de membranas mais homogêneas, enquanto que um
estudo de aumento da temperatura de secagem poderia diminuir o tempo de produção das
membranas.
A inclusão de agentes antimicrobianos em curativos mostra-se relevante para o tratamento
de feridas contaminadas, de modo a promover uma ação terapêutica específica e evitar o
crescimento de microrganismos (YUDANOVA e RESHETOV, 2006). Dentre os agentes
antimicrobianos que podem ser utilizados destacam-se o polihexametileno biguanida (PHMB),
os sais de clorexidina, os derivados da furanona, o iodo, o timol, a triclosana, a gentamicina, a
rifampicina ou ainda a prata, na forma de nanopartículas (WEGMANN et al., 2009; MULDER
et al., 2007, STEPHEN-HAYNES et al., 2014). O PHMB é conhecido por sua utilização em
curativos e como ingrediente ativo de elevada estabilidade em soluções de limpeza de lentes de
contato e produtos para assepsia cirúrgica (MULDER et al., 2007; SCHNUCH et al., 2007),

4
entretanto, não foram localizados na literatura trabalhos referentes à incorporação de PHMB em
membranas de quitosana-alginato, abordagem esta explorada neste trabalho pelo uso de três
diferentes estratégias e que lhe confere o caráter de inédito.
Assim, este trabalho enfoca tema de relevância em Engenharia Química, mais
especificamente na área de desenvolvimento de novos materiais, com foco no estabelecimento
de processos de produção de biomateriais aplicáveis na recuperação de lesões de pele e em sua
caracterização.
As atividades foram desenvolvidas em sua maior parte na Faculdade de Engenharia
Química da Unicamp, no Laboratório de Engenharia de Biorreações e Colóides (Departamento
de Engenharia de Materiais e Bioprocessos), incluindo um estágio de sete meses no
Departamento de Engenharia Química da Universidade de Coimbra, em Portugal.
As informações coletadas e os resultados obtidos neste trabalho já geraram um artigo
científico publicado em novembro de 2014 (BUENO et al., 2014). Há ainda dois trabalhos em
fase de elaboração a serem publicados em revistas científicas internacionais ao longo do ano de
2015, além de dois capítulos de livro no prelo.
1.1. Objetivos
Este trabalho de doutorado teve como objetivo geral contribuir na área de desenvolvimento
de novos materiais, mais especificamente no ramo de obtenção de membranas de quitosana e
alginato a serem utilizadas em processos de regeneração de lesões de pele.
Dentre os objetivos específicos deste trabalho, destacam-se:
A melhoria do processo de produção das membranas, de maneira a se ter procedimentos mais
rápidos e de baixo custo. Procurou-se atingir este objetivo pelo uso de adaptações do método
do trabalho anterior desenvolvido por Bueno (2010), tais como a redução da quantidade de
material empregada e a avaliação da influência de diferentes temperaturas de processo e de
secagem.
A análise do efeito de diferentes proporções do tensoativo Pluronic F68 nas características das
membranas, tais como aspecto, morfologia, espessura, porosidade, rugosidade,
comportamento na presença de umidade e fluidos fisiológicos, propriedades mecânicas,

5
citotoxicidade indireta, composição química da superfície e comportamento térmico.
O estudo do efeito da incorporação do agente antimicrobiano PHMB às membranas por três
diferentes técnicas: por adição à mistura polimérica, por adsorção a partir de solução aquosa e
por CO2 supercrítico.

6
2. REVISÃO DA LITERATURA
Os assuntos abordados nesta revisão de literatura visam fornecer informações gerais a
respeito dos principais tipos de lesões de pele e os tratamentos modernos já existentes e em
desenvolvimento, muitos dos quais fazem uso de biopolímeros como a quitosana e o alginato. A
formação do complexo de polieletrólitos de quitosana-alginato e a obtenção de membranas
porosas a partir deste complexo por meio da adição de surfactantes também são abordados nesta
revisão. Outros temas discutidos são a incorporação de fármacos em curativos por diferentes
técnicas e a estrutura e propriedades do agente antimicrobiano PHMB.
2.1. Lesões de pele
Considerada o maior órgão do corpo humano, a pele possui diferentes funções, como
promover proteção contra microrganismos, agentes físicos e químicos; regular a temperatura
corpórea pelo do ajuste do fluxo sanguíneo e da transpiração; prevenir a desidratação e fornecer
suporte a vasos sanguíneos e nervos responsáveis pelas funções sensoriais (calor, frio, pressão,
dor e tato). Este órgão corresponde a 16% do peso corpóreo e possui área superficial de 1,5 a 2,0
m2. Sua espessura pode variar de acordo com a idade e localização no corpo (PAUL e
SHARMA, 2004).
A pele possui três camadas distintas denominadas epiderme, derme e hipoderme. A
primeira é constituída principalmente por células mortas e queratina, o que a torna uma barreira
protetora impermeável. A derme, por sua vez, é constituída por células conjuntivas. Já a
hipoderme promove isolamento e absorve impactos, servindo como suporte às demais camadas.
Na pele ainda podem ser visualizadas diversas estruturas anexas como os pêlos, as unhas e as
glândulas sudoríparas e sebáceas (PAUL e SHARMA, 2004).
Uma lesão de pele é um defeito ou rompimento da sua estrutura, e sua gravidade varia de
acordo com as camadas envolvidas: lesões superficiais atingem a derme, lesões profundas
superficiais atingem o tecido subcutâneo (hipoderme), e lesões profundas totais atingem os
músculos e as suas estruturas adjacentes (BORGES et al., 2007).
A cicatrização é um processo complexo composto por quatro fases: homeostase
(coagulação), inflamação, granulação (proliferação celular, formação de tecido) e re-epitelização

7
(remodelação do tecido), as quais coincidem no decorrer do tempo. O mecanismo envolve vários
tipos celulares, fatores bioquímicos e moléculas extracelulares. Depois que um tecido é
danificado, a coagulação minimiza a perda de fluido corpóreo, além de formar uma barreira que
protege a ferida e serve de suporte ao crescimento celular. Ao mesmo tempo, as plaquetas
presentes no coágulo iniciam a cascata de cicatrização por meio da liberação de compostos
quimioatrativos e fatores de crescimento. Células inflamatórias, como os granulócitos e os
macrófagos, penetram na ferida, protegendo o tecido de partículas estranhas. Os macrófagos
também liberam citocinas pró-inflamatórias que possuem papel importante no processo de
cicatrização, pois induzem a migração celular, assim como a proliferação de fibroflastos e
queratinócitos, além de estimular a síntese de componentes da matriz extracelular. Na fase de
granulação, o tecido da derme se reconstitui e, por fim, na fase de re-epitelização, a epiderme é
restaurada (WIEGAND e HIPLER, 2010).
Dependendo da natureza do processo de reparo da lesão, esta pode ser classificada como
aguda ou crônica. As feridas agudas normalmente cicatrizam completamente, com formação
mínima de cicatrizes, dentro de 8 a 12 semanas. As causas primárias destas feridas são fatores
externos como abrasões, cortes, penetração de objetos, incisões cirúrgicas, queimaduras, choques
e contato com substâncias corrosivas ou radiação (BOATENG et al., 2008).
Dentre as feridas agudas, as queimaduras devem receber atenção especial, uma vez que são
de difícil tratamento, podendo causar danos extensos, perda de fluidos e trauma ao paciente. O
atendimento a vítimas de queimaduras, incluindo os casos complicados por outros tipos de
trauma, exige pessoal com treinamento especializado e acesso a equipamentos e materiais
adequados. O paciente vítima de queimadura é um desafio para todos os profissionais da área da
saúde e exige constante aperfeiçoamento pela equipe (IURK et al., 2010).
As feridas crônicas, por outro lado, cicatrizam lentamente devido a danos constantes ao
tecido, ao mau tratamento da lesão ou a condições fisiológicas como diabetes, infecções
persistentes, tromboses, edemas ou pressão local prolongada. Este tipo de ferida pode conter
níveis elevados de substâncias que perpetuam a fase inflamatória, podendo persistir sem sinais
de cicatrização por meses ou até anos. Exemplos de feridas crônicas são as úlceras de pressão e
as úlceras de perna (BOATENG et al., 2008; PAUL e SHARMA, 2004; WIEGAND e HIPLER,
2010).
A variedade e incidência das lesões de pele existentes e a dificuldade de tratamento de

8
muitas delas são um desafio para os profissionais da saúde. Assim, existe a demanda por
curativos que possam acelerar a cicatrização, facilitando o tratamento e reduzindo os riscos para
o paciente (WIEGAND e HIPLER, 2010), além de diminuir o tempo e os dispêndios com a
internação nos casos mais graves.
2.2. Avanços no tratamento de lesões de pele
No passado, utilizavam-se curativos tradicionais, como bandagens à base de algodão ou lã,
cuja principal função era manter a ferida seca, permitindo a evaporação do exsudato e
prevenindo a entrada de bactérias nocivas. Contudo, mais tarde descobriu-se que para uma
cicatrização rápida e efetiva, deve-se manter um ambiente quente e úmido na ferida, de modo a
mimetizar as condições fisiológicas da matriz extracelular, permitindo trocas gasosas e
impedindo a permeação de microrganismos, de modo que haja migração, proliferação e
organização das células do paciente. Desta forma, a concepção dos curativos modernos se baseia
na criação deste tipo de ambiente (BOATENG et al., 2008; WIEGAND e HIPLER, 2010).
Nos trabalhos de Thomas (2008), Yudanova e Reshetov (2006) e Wiegand e Hipler (2010)
são descritas as características requeridas de uma cobertura ideal para feridas, como é mostrado
na Tabela 2.1. Contudo, sabe-se que o tratamento de uma lesão de pele deve levar em
consideração o tipo da ferida, a fase do processo de cicatrização em que esta se encontra, as
condições de saúde do paciente e até mesmo as condições ambientais e sociais. Deste modo,
existe um curativo adequado a cada situação (BOATENG et al., 2008).
Os curativos modernos podem ser classificados de acordo com o tipo de contato que
estabelecem com a lesão: curativos primários são aqueles usados em contato direto com o tecido
lesado, curativos secundários são aqueles colocados sobre o curativo primário, curativos em ilha
são os que possuem uma região central absorvente cercada por uma porção adesiva. Uma
segunda classificação comum para curativos refere-se à sua forma física: pomadas, filmes,
espumas e géis (BOATENG et al., 2008). Outra classificação divide os curativos de acordo com
a sua atividade: inativos (gaze e outros tecidos), interativos (hidrocolóides, hidrogéis) e ativos
(que contêm em sua composição fatores de crescimento e inibidores de proteases). Apenas os
curativos ativos são capazes de influenciar a cicatrização, enquanto os outros têm a função de

9
promover um ambiente úmido, e prover isolamento térmico e proteção contra microrganismos
(WIEGAND e HIPLER, 2010).
Tabela 2.1: Requerimentos para um curativo ideal (THOMAS, 2008; YUDANOVA e
RESHETOV, 2006; WIEGAND e HIPLER, 2010).
Requerimentos primários Requerimentos secundários
1-Oferecer proteção mecânica
2-Ser livre de substâncias tóxicas e irritantes
3-Proteger contra a entrada de
microrganismos
4-Criar um ambiente úmido para a cicatrização
5-Proteger a pele do exsudato e excesso de
umidade
6-Permitir trocas gasosas com o ambiente
7-Não aderir ao leito da ferida
8-Não incomodar o paciente
9-Não provocar dor quando aplicado ou
removido
10-Moldar-se à região ferida
11-Ser resistente à água e facilmente removível
12-Ser de baixo custo
13-Não liberar partículas ou fibras não
biodegradáveis na ferida
14-Não necessitar de trocas frequentes
15-Funcionar efetivamente sob compressão
16-Manter a ferida a temperatura e pH ótimos
1-Acelerar o processo de reparo
tecidual
2-Possuir atividade antimicrobiana
3-Ser transparente para possibilitar o
monitoramento da cicatrização
4-Possuir atividade hemostática
5-Exibir atividade de desbridamento
(remoção de tecidos necróticos e
corpos estranhos)
6-Ser capaz de combater o mau odor
7-Possuir capacidade de remover ou
inativar enzimas proteolíticas em
feridas crônicas
Boateng et al. (2008) também propõem a divisão dos curativos modernos em:
hidrocolóides, curativos de alginato, hidrogéis, filmes adesivos ou semi-permeáveis, espumas,
matrizes celulares e acelulares, e curativos biológicos.
Os hidrocolóides estão entre os curativos modernos mais utilizados, adequados para feridas

10
com pouca a moderada liberação de exsudato. Estes curativos são obtidos de materiais coloidais
combinados com outros materiais como elastômeros e adesivos. Exemplos de materiais coloidais
são a carboximetilcelulose (CMC), a gelatina e a pectina (BOATENG et al., 2008).
Os curativos de alginato são produzidos a partir dos sais de cálcio e sódio do ácido
algínico. Podem ser comercializados na forma de espumas liofilizadas ou fibras flexíveis.
Formam gel em contato com o exsudato e possuem alta capacidade de absorção da secreção da
ferida, minimizando a contaminação bacteriana (BOATENG et al., 2008).
Os hidrogéis são definidos como redes poliméricas reticuladas que possuem alta
capacidade de reter água, sem se dissolver. A alta absorção de água é devida aos grupos
funcionais hidrofílicos do polímero e a resistência à solubilização se deve aos pontos de
reticulação entre as cadeias poliméricas. Vários tipos de materiais, naturais e sintéticos, podem
ser definidos como hidrogéis (AHMED, 2013).
Os filmes adesivos semi-permeáveis são originalmente feitos de derivados do nylon
suportados por um adesivo de polietileno, o que os tornam oclusivos. Os curativos de nylon,
contudo, não absorvem exsudato de modo suficiente, o que resulta em acúmulo deste abaixo do
curativo, levando à maceração da pele e proliferação de bactérias (BOATENG et al., 2008).
As espumas são normalmente obtidas de poliuretano. Alguns destes curativos possuem
camadas adicionais que evitam aderência a feridas secas ou ainda camadas oclusivas que
previnem perda de fluidos e contaminação bacteriana. Curativos em forma de espuma mantêm
um ambiente úmido na ferida, promovem isolamento térmico e são convenientes de se usar.
Possuem alta capacidade de absorção, sendo esta propriedade controlada pela textura, espessura
e porosidade do material (BOATENG et al., 2008).
As matrizes celulares e acelulares podem ser aplicadas como substitutos temporários da
pele. As matrizes acelulares podem ser produzidas com colágeno combinado com componentes
da matriz extracelular, como ácido hialurônico; também podem ser obtidas de pele natural, sendo
os componentes celulares removidos, preservando-se, entretanto, a arquitetura da derme. As
matrizes celulares incluem filmes poliméricos biodegradáveis que servem como scaffolds para o
cultivo de células da pele (BOATENG et al., 2008). Neste caso, o curativo deve possuir uma
estrutura tridimensional, com grande área superficial, adequada à adesão, migração e
crescimento celular. As propriedades mecânicas também são uma característica importante e
devem ser similares às do tecido danificado. O scaffold também deve se degradar em

11
componentes não-tóxicos, que possam ser eliminados ou reabsorvidos pelo organismo, sendo
que a taxa de degradação deve ser preferencialmente próxima à taxa de formação de tecido novo
(KHAN e AHMAD, 2013; RIBEIRO et al., 2012).
Os curativos biológicos, por sua vez, são feitos de biopolímeros, que são polímeros de
origem natural, possuindo uma variedade de benefícios ambientais, como serem derivados de
fontes renováveis, biodegradáveis e biocompatíveis (BOATENG et al., 2008). Os biopolímeros
são divididos em classes, como os ácidos nucleicos, proteínas, polissacarídeos, polidienos,
ligninas e poli(alfa-hidroxi-alcanoato)s (ELIAS, 2005).
Em alguns casos, os biopolímeros podem ser misturados com componentes ativos como
agentes antimicrobianos e fatores de crescimento passíveis de liberação no ferimento
(BOATENG et al., 2008; YUDANOVA e RESHETOV, 2006; WIEGAND e HIPLER, 2010).
Dentre os biopolímeros, os polissacarídeos recebem especial atenção para aplicações
biomédicas, sendo uma discussão mais detalhada acerca desta classe de biomoléculas
apresentada a seguir.
2.3. Polissacarídeos
Os polissacarídeos são constituídos de monossacarídeos (normalmente hexoses) unidos por
ligações glicosídicas (SILVA et al., 2012), sendo normalmente obtidos na biossíntese em plantas
ou em animais; alguns também podem ser produzidos por microrganismos (RINAUDO, 2008).
Alguns polissacarídeos possuem atividade biológica, como é o caso da quitosana, do ácido
hialurônico (RINAUDO, 2006), da lentinana e da heparina (YANG e ZHANG, 2009). Na Tabela
2.2 são listadas as fontes usuais de alguns polissacarídeos de uso comercial mais intenso.
O uso de polissacarídeos naturais, isolados ou combinados entre si ou com materiais de
origem sintética, como matéria-prima de curativos dérmicos tem sido uma escolha bastante
comum nos últimos anos, sendo que estes materiais apresentam numerosas variações em sua
estrutura, composição e função (BURD e HUANG, 2008). Vários tipos de curativos encontram-
se disponíveis atualmente no mercado, muitos deles contendo polissacarídeos naturais,
destacando-se entre eles a quitosana e o alginato.

12
Tabela 2.2: Fontes usuais de alguns polissacarídeos (adaptada de Da Cunha et al., 2009).
Polissacarídeo Fonte
Alginato algas marrons
Agar algas vermelhas
Carragena algas vermelhas
Goma Arábica exsudato da planta Acacia spp
Goma Tragacanto exsudato da planta Astragalus spp
Goma Guar sementes de Cyamopis tetragonolobus
Goma de Alfarroba sementes de Ceratonia siliqua
Goma de Tamarindo sementes de Tilletia indica
Pectinas maçãs e laranjas
Amido milho, trigo, batatas
Inulina chicória, alcachofra de Jerusalém
Ácido Hialurônico humor vítreo de bovinos, cristas de galináceos
Heparina pulmão de bovinos e intestinos de porcinos
Quitina carapaças de crustáceos
Quitosana carapaças de crustáceos
Glicanas fungos das espécies Pleurotus ostreatus e Agaricus blazei
Xantana bactérias da espécie Xanthomonas spp
Dextrana bactérias da espécie Leuconostoc spp
Gelana bactérias da espécie Sphingmonas elodea
Celulose paredes celulares de plantas
Fucoidina algas
2.3.1. Quitosana
A quitosana é um polissacarídeo catiônico que, juntamente com seu precursor, a quitina,
está se tornando cada vez mais importante no campo da biomedicina, engenharia de alimentos,
biotecnologia, agricultura, cosméticos, entre outros (ARANAZ et al., 2009).

13
A quitina pode ser encontrada na natureza na forma de microfibras cristalinas ordenadas,
presentes na estrutura do exoesqueleto de artrópodes, nas paredes celulares de fungos e leveduras
e também em moluscos. As principais fontes comerciais de quitina são as carapaças de
caranguejos e camarões, na qual a quitina representa 70% dos compostos orgânicos totais,
estando associada a proteínas, pigmentos e carbonato de cálcio (RINAUDO, 2008; HEIN et al.,
2008; GEORGE e ABRAHAM, 2006).
A quitosana é o derivado mais importante da quitina em termos de aplicações (RINAUDO,
2008), sendo obtida por meio da sua desacetilação parcial (mínima de 50%) sob condições
alcalinas (KASAAI, 2009). A fórmula estrutural deste polissacarídeo consiste de um copolímero
de N-acetil glicosamina (β-(1-4)2-acetamido-2-desoxi-D-glicose) (unidade estrutural da quitina)
e D-glicosamina (2-amino-2-desoxi-D-glicose) (unidade estrutural da quitina desacetilada),
representado na Figura 2.1. Destaca-se que este polissacarídeo é um dos poucos de origem
natural que pode apresentar múltiplas cargas positivas em suas moléculas, sendo mais comum na
natureza a ocorrência de poliânions.
Figura 2.1: Fórmula estrutural da quitosana com suas respectivas unidades (Adaptada de
Wiegand e Hipler, 2010).
O
OH
HONH2
O
CH3O
NH
O
OH
O
HO
NH2
O
O
OH
HOO
NH
O
H3C
O
HO
OH
O
NH2
O
OH
NH2
HO
OO
N-acetil glicosamina
D-glicosamina
Quitosana
OH
HO
Quitosana

14
O processo de obtenção da quitosana consiste em separar a quitina de outros componentes
das carapaças de caranguejos ou camarões por um processo químico que envolve etapas de
desmineralização e desproteinização com soluções diluídas de HCl e NaOH, seguidas de
descoloração com KMnO4 e ácido oxálico. A quitina obtida, o biopolímero contendo grupos
acetil (NHCOCH3), é desacetilada com solução concentrada de NaOH, obtendo-se, então, a
quitosana (AZEVEDO et al., 2007). A fração de meros, a sequência e o comprimento de cadeia
da quitosana dependem da sua origem, lote e grau de desacetilação (HEIN et al., 2008).
Geralmente, o grau de desacetilação da quitosana comercial varia de 70 a 95%, e sua massa
molar encontra-se na faixa de 104 a 106 g.mol-1 (CANELLA e GARCIA, 2001; MALAFAYA et
al., 2007).
Considerando-se a quantidade de quitina produzida anualmente em escala global, este
composto é o segundo polímero mais abundante na natureza, depois da celulose (RINAUDO,
2008). Já a quitosana teve produção mundial estimada de 13,7 mil toneladas no ano de 2010,
com previsão de aumento para 21,4 mil toneladas em 2015 (DEMINA et al., 2014).
As propriedades da quitosana variam com a massa molar e sua distribuição, a localização
dos grupos amino livres e acetilados na cadeia polimérica e o grau de acetilação. Dependendo do
valor do grau de acetilação, o pKa da quitosana pode variar entre 6,3 e 7,3 (KARAKEÇILI et al.,
2007; IL’INA e VARLAMOV, 2005). A conformação e tamanho das cadeias em soluções
diluídas também dependem consideravelmente do grau de acetilação e da força iônica.
As principais vantagens da quitosana são a sua biodegradabilidade, biocompatibilidade, o
seu caráter hidrofílico, a presença de grupos polares (-OH e -NH2) capazes de interagir com
outros polímeros e a sua atividade antibacteriana. Além disso, a quitosana é filmogênica,
hidratante, bioadesiva, renovável, antifúngica, não-alergênica, imunoadjuvante,
anticolesterolêmica e antitrombogênica (RINAUDO, 2008). Todas essas propriedades a tornam
interessante para aplicações no campo biomédico.
A quitosana é amplamente utilizada em diferentes formas, como soluções, géis, filmes,
fibras, esponjas e micropartículas (RINAUDO, 2008). Uma de suas principais aplicações ocorre
no campo da liberação controlada de fármacos, uma vez que a sua degradação ocorre
naturalmente no organismo pela ação da enzima lisozima. No entanto, a aplicação mais popular
deste polissacarídeo é como componente em produtos emagrecedores e redutores de colesterol.
Tal aplicação é possível devido à sua natureza catiônica, que dificulta a absorção de lipídeos pelo

15
intestino (GEORGE e ABRAHAM, 2006).
A quitosana também tem sido também estudada visando aplicações na engenharia de
tecidos, na produção de scaffolds biodegradáveis (PAUL e SHARMA, 2004) por ser capaz de ser
processada na forma de matrizes porosas tridimensionais compostas por fibras. Os scaffolds
obtidos a partir de quitosana possuem vantagens sobre outros polímeros comumente utilizados,
como os polilactídeos, os poliglicolídeos, a policaprolactona e seus copolímeros, os quais são
hidrofóbicos e cujos produtos de degradação são ácidos (JAGUR-GRODZINSKI, 2006).
O uso da quitosana na área de terapia de lesões promove a regeneração ordenada da pele,
por meio da formação do tecido de granulação e re-epitelização acompanhada de angiogênese e
deposição regular de fibras de colágeno, limitando a formação de cicatrizes e a retração do tecido
(MUZZARELLI et al., 2007; MUZZARELLI, 2009). De acordo com Muzzarelli (2009), testes
feitos em feridas tratadas com quitosana indicaram que as fibras de colágeno formadas eram
finas e mais maduras em relação às provenientes da cicatrização normal e seu arranjo era mais
próximo ao encontrado na pele. Em sete dias, as feridas estavam completamente re-epitelizadas,
os tecidos de granulação haviam sido quase completamente substituídos por fibroses e os
folículos capilares estavam quase cicatrizados (MUZZARELLI, 2009).
Vários curativos obtidos a partir de quitosana estão disponíveis comercialmente na forma
de não-tecidos, nanofibrilas, compósitos, filmes e esponjas (MUZZARELLI, 2009). Na Tabela
2.3 encontram-se as descrições simplificadas de alguns dos curativos à base de quitosana já
disponíveis comercialmente.
Enquanto a quitina é insolúvel na maioria dos solventes orgânicos, a quitosana é solúvel
em soluções ácidas diluídas com pH menor do que o valor do seu pKa (que pode variar entre 6,3
e 7,3). Isto ocorre devido à presença dos grupos amino, o que faz deste composto uma base forte
de Lewis. Em baixos valores de pH, os grupos amino tornam-se protonados, promovendo a
repulsão das cadeias poliméricas e a consequente solubilização da quitosana (PILLAI et al.,
2009). No entanto, solubilidade é um parâmetro de difícil controle, estando relacionado com o
grau de desacetilação, concentração iônica, pH, natureza do ácido e distribuição dos grupos
acetil ao longo da cadeia (RINAUDO, 2008).
Alguns dos ácidos orgânicos que solubilizam a quitosana são os ácidos fórmico, acético,
lático, málico, cítrico, glioxílico, pirúvico, glicólico e ascórbico (PILLAI et al., 2009). Quanto
aos ácidos inorgânicos, existe uma maior dificuldade para a solubilização do polímero. O ácido

16
nítrico pode solubilizar a quitosana, mas algum tempo após a dissolução é possível observar um
precipitado branco gelatinoso. A solubilização com ácido clorídrico requer aquecimento e
agitação prolongados. O ácido sulfúrico não dissolve a quitosana devido à formação de sulfato
de quitosana, que é um sólido branco cristalino. Já o ácido perclórico pode dissolver a quitosana
facilmente (MUZZARELLI, 1973).
Tabela 2.3: Exemplos de curativos à base de quitosana disponíveis comercialmente
(MUZZARELLI, 2009; STEPHEN-HAYNES et al., 2014; RATNER et al., 2013).
Nome e fabricante do curativo Descrição
Chitodine® - IMS Pó de quitosana com iodo elementar.
Chitoflex® - HemCon Bandagem à base de quitosana.
Chitopack C® - Eisai Quitosana em forma de fibras finas semelhantes ao
algodão.
Chito-Seal® - Abbott Material não-tecido recoberto com gel de quitosana.
HemCon® - HemCon Acetato de quitosana liofilizado.
Tegasorb® - 3M Curativo contendo partículas de quitosana com alta
capacidade de absorção.
TraumaStat® - Ore-Medix Quitosana liofilizada contendo sílica altamente porosa.
Vulnosorb® - Tesla-Pharma Esponja liofilizada de colágeno e quitosana.
KytoCel® - Aspen Medical Fibras de quitosana compactadas em forma de não-
tecido.
Aquanova® - Medtrade Fibras de quitosana e derivados de quitosana não
especificados compactadas em forma de não-tecido.
Celox TM Granules - Celox Medical Grânulos de quitosana.
CeloxTM Gauze - Celox Medical Gaze de alta densidade impregnada com grânulos de
quitosana.
Devido ao grande número de grupos hidroxil e amino, a quitosana pode ser associada a
outros biopolímeros e também a polímeros sintéticos, assim como a fatores de crescimento,
componentes da matriz extracelular e agentes antibacterianos (MUZZARELLI, 2009). Os
compostos de cálcio são os materiais inorgânicos mais estudados em conjunto com este

17
polissacarídeo. Como exemplo desta categoria, tem-se o fosfato de cálcio e o carbonato de
cálcio, os quais podem ser incorporados ao material para promover aumento da sua resistência
mecânica em implantes ortopédicos injetáveis (RATNER et al., 2013).
Já as proteínas mais estudadas em conjunto com a quitosana são o colágeno e a gelatina,
promovendo melhorias nas propriedades dos scaffolds utilizados na engenharia de tecidos para
reparos de pele, ossos, cartilagem e nervos. Alguns glicosaminoglicanos utilizados em conjunto
com a quitosana são a heparina, o sulfato de condroitina e o hialuronato. A heparina, por
exemplo, aumenta a hemocompatibilidade do material (HEIN et al., 2008). Quanto aos
polímeros naturais, tem-se a combinação da quitosana com alginato, pectina, carragena e
xantana, os quais formam materiais atraentes para diversas aplicações em conjunto com a
quitosana, sendo o alginato o biopolímero mais estudado (HEIN et al., 2008; RATNER et al.,
2013).
2.3.2. Alginato
O alginato é um dos polissacarídeos mais estudados nas áreas de engenharia de tecidos e
liberação controlada de agentes bioativos. É obtido principalmente de algas marinhas, nas quais
está presente na parede celular juntamente com outros dois polissacarídeos, o ágar e a carragena,
promovendo flexibilidade e permitindo adaptação do vegetal aos movimentos da água
(RINAUDO, 2008; WIEGAND e HIPLER, 2010; MALAFAYA et al., 2007).
O alginato comercial é comumente obtido de algas marrons, das espécies Laminaria
hyperborea, Ascophyllum nodosum e Macrocystis pyrifera (GEORGE e ABRAHAM, 2006), nas
quais pode estar presente em porcentagens variando de 14 a 40% em relação à massa seca do
vegetal (MUZZARELLI, 1973). Além disso, o alginato pode ser produzido como um polímero
exocelular por bactérias como Azotobacter vinelandii e várias espécies de Pseudomonas
(MAURSTAD et al., 2008).
O termo alginato pode se referir tanto ao ânion do ácido algínico, como também aos seus
sais, ou ao próprio ácido (MCHUGH, 2003). A fórmula estrutural do alginato é normalmente
descrita como sendo um copolímero linear de ânions (1,4) β-D-manuronato (M) e α-L-
guluronato (G). Alguns autores também mencionam a fórmula do ácido algínico para descrever o
alginato: ácido (1,4) β-D-manurônico (M) e α-L-gulurônico (G). Na molécula de alginato, as

18
regiões homopoliméricas, chamadas de blocos M e blocos G, encontram-se interdispersas com
regiões de sequência mista (blocos MG), como mostra a Figura 2.2. A composição química e a
sequência das unidades M e G dependem da origem do alginato, de condições e efeitos
ambientais (WIEGAND e HIPLER, 2010; SÆTHER et al., 2008). A massa molar deste
polímero encontra-se na faixa de 104 a 106 g/mol (KONG et al., 2004; DA SILVA, 2009). O pKa
dos resíduos de manuronato é de 3,38 e o pKa dos resíduos de guluronato é de 3,65 (LI et al.,
2009; SÆTHER et al., 2008).
Figura 2.2: Fórmula estrutural do alginato com suas respectivas unidades: blocos M ((1,4) β-D-
manuronato) e blocos G (α-L-guluronato) (adaptada de Rodrigues, 2008).
Nas algas, o alginato está presente como uma mistura de sais de cátions insolúveis, como o
cálcio e o magnésio (MUZZARELLI, 1973). Para extrair o alginato, as algas marinhas são
moídas e agitadas em uma solução de pH básico aquecida, usualmente com carbonato de sódio.
O processo de extração consiste em converter os sais insolúveis em água (alginato de cálcio e
magnésio) em alginato de sódio, o qual é solúvel em água. O alginato de sódio é então dissolvido
e os resíduos das algas são removidos por filtração. O biopolímero pode então ser recuperado da
solução pela adição de um sal de cálcio. Isso faz com que haja formação de alginato de cálcio
insolúvel. Um ácido é então adicionado para convertê-lo a ácido algínico, o qual é separado
O
OHCOOH
O
OH
O
O
COOH
OHOH
OH
O
COOHOH
O O
OH
OHO
HOOC
O
OH
COOHOH
O
O
OHCOOHOH
O
O
OH
COOH
O
OH
O O
COOH
OH
O
OH
OH
O
COOH
OH
O
OH
O
HOOCO OH
Bloco M Bloco G
Blocos M Blocos M-G Blocos G

19
facilmente da mistura. O ácido algínico passa então por um processo de filtração para retirada de
parte do líquido presente. Adiciona-se então carbonato de sódio até convertê-lo a alginato de
sódio. A pasta de alginato de sódio resultante pode ser extrudada em grãos que são então secos e
moídos (MCHUGH, 2003). Anualmente, a indústria produz cerca de 30 mil toneladas de
alginato, o que não chega a 10% do material biossintetizado anualmente pelas algas marinhas
naturais (KLEINÜBING, 2009). Cerca de 30% do total de alginato produzido é usado na
indústria alimentícia (RINAUDO, 2008).
As principais características dos alginatos são a sua capacidade de absorver e reter água e
suas propriedades gelificantes e estabilizantes. Devido à sua estrutura linear e alta massa molar,
os alginatos formam filmes e fibras fortes no estado sólido (RINAUDO, 2008). A formação de
gel é uma característica muito importante do alginato, ocorrendo na presença de íons divalentes
como Ca2+, Ba2+, Mg2+ e Sr2+, sendo mais comumente empregado o cálcio. A interação dos
cátions com o polímero resulta em uma rede tridimensional de fibras de alginato unidas entre si
por ligações iônicas com os íons cálcio (GEORGE e ABRAHAM, 2006), como pode ser visto na
Figura 2.3.
Figura 2.3: Formação da rede de gel de alginato de cálcio (adaptado de Sacchetin, 2009 e
Kawaguti e Sato, 2008).

20
A ligação iônica mais seletiva está estritamente ligada aos blocos GG, sendo que as
unidades G, alinhadas lado a lado, resultam na formação de cavidades onde os íons cálcio
interagem, fazendo a junção entre as cadeias poliméricas. Desta forma, uma maior afinidade por
íons é encontrada em alginatos ricos em ácido gulurônico, formando géis estáveis (SIMPSON et
al., 2004). Muzzarelli (1973) e Martinsen et al. (1991) apontam que alginatos ricos em ácido
gulurônico são normalmente obtidos de Laminaria hyperborea. Por outro lado, em alginatos com
alto teor de ácido manurônico os íons cálcio interagem menos fortemente com a molécula e
podem ser facilmente substituídos por íons sódio, resultando em aumento do intumescimento
quando da absorção de fluidos (WIEGAND e HIPLER, 2010).
O alginato é muito usado como espessante na indústria têxtil e de alimentos (em molhos,
xaropes, coberturas, maioneses e iogurtes). Também é usado como estabilizante, reduzindo a
formação de cristais de gelo e a taxa de derretimento em sorvetes. Além disso, filmes de alginato
de cálcio têm sido usados na preservação de peixes congelados. Na indústria farmacêutica, este
composto tem sido usado na liberação controlada de medicamentos e outros produtos químicos
devido a sua sensibilidade ao pH (MCHUGH, 2003). Além disso, o alginato é um dos
biopolímeros mais estudados na remoção de íons metálicos de soluções diluídas
(KLEINÜBING, 2009).
O alginato pode ser utilizado na produção de curativos para o tratamento de feridas
exsudativas. A absorção do exsudato ocorre por meio de troca iônica entre os íons cálcio do
curativo e os íons sódio do exsudato, resultando na formação de um gel capaz de manter um
ambiente úmido adequado à cicatrização. Lalau et al. (2002) verificaram que pacientes tratados
com curativos de alginato sentiram redução da dor associada com a ferida e com a troca de
curativos devido às propriedades gelificantes do polímero. Além disso, o alginato possui
propriedades hemostáticas, bioadesivas e antibacterianas. A inibição do crescimento de bactérias
ocorre através da imobilização das mesmas na matriz fibrosa do polímero (WIEGAND e
HIPLER, 2010; GEORGE e ABRAHAM, 2006; MA et al., 2007). Na Tabela 2.4 encontram-se
listados alguns dos curativos de alginato disponíveis comercialmente.
A mistura de alginato com outros polímeros pode ter influência na porosidade e
complexidade da rede polimérica, melhorando muitas vezes as características do material.
Podem ser obtidas misturas de alginato com outros polímeros como pectina, quitosana, etil
celulose e Eudragit® (copolímero derivado do ácido metacrílico e metilmetacrílico), que são

21
muito usadas na produção de microcápsulas para liberação controlada (GEORGE e ABRAHAM,
2006). A complexação de alginato com quitosana será mais especificamente discutida a seguir.
Tabela 2.4: Exemplos de curativos de alginato disponíveis comercialmente (RINAUDO, 2008;
MALAFAYA et al., 2007; PAUL e SHARMA, 2004; RODRIGUES, 2008; RATNER et al.,
2013).
Nome e fabricante do curativo Descrição
Algiderm® - Bard Tecido de fibras de alginato de cálcio
Sorbsan® - UDL Laboratories Material não-tecido de fibras de alginato de cálcio
Nu-Derm® - Johnson & Johnson Material não-tecido de fibras de alginato em forma de faixas
Algosteril® - Systagenix Material não-tecido de fibras de alginato de cálcio
Kaltostat® - ConvaTec Material não-tecido de fibras de alginato de cálcio e sódio
Kalginate® - DeRoyal Industries Material não-tecido de fibras espessas de alginato de cálcio
Restore® - Hollister Material não-tecido de fibras de alginato de cálcio e sódio
SeaSorb® - Coloplast Sween Material não-tecido de fibras de alginato de cálcio e
carboximetilcelulose
Maxorb® Extra - Medline Industries Material não-tecido composto por fibras de alginato e
carboximetilcelulose
AlgiSite® - Smith & Nephew Material tecido e não-tecido de fibras de alginato de cálcio
com elevado conteúdo de ácido manurônico
Tegagen HG® - 3M Health Care Fibras de alginato de cálcio em formato de faixas
PhytaCare® - Curafarm Hidrogel de alginate
Silverlon® - Cura Surgical Curativo em duas camadas sendo uma composta por material
não-tecido de fibras de alginato de cálcio de alto conteúdo de
ácido manurônico e outra composta por nylon impregnado
com prata
Hyperion Advanced Film Dressing ®-
Hyperion Medical
Filme transparente de alginate
PolyMem® - Ferris Mfg. Membrana de alginato contendo glicerina e agente surfactante
2.3.3. Complexo de polieletrólitos de quitosana-alginato
Os polieletrólitos são macromoléculas que possuem um número considerável de grupos
funcionais carregados eletrostaticamente, ou que podem se tornar carregados em condições

22
apropriadas de pH. As moléculas podem se apresentar nas formas de policátions, como no caso
da quitosana, ou poliânions, como no caso do alginato, sendo sua carga elétrica dependente da
natureza dos seus grupos funcionais (SIMSEK-EGE et al., 2003).
Os complexos de polieletrólitos (PECs) são formados pela associação de dois ou mais
polieletrólitos complementares, os quais são ligados principalmente devido às forças
eletrostáticas, interações hidrofóbicas, ligações de hidrogênio, forças de van der Waals, ou pela
combinação destas interações. A formação de complexos pode afetar fortemente algumas
propriedades dos polímeros constituintes do sistema, tais como sua solubilidade, reologia,
condutividade elétrica, propriedades mecânicas e permeabilidade, além de modificar a turbidez
das soluções. Um aspecto importante na formação dos PECs está relacionado ao pH do meio, ou
seja, para que ocorra a reação, ambos os polímeros devem estar ionizados e apresentar cargas
opostas. Se as interações iônicas forem muito fortes, pode ocorrer precipitação, impedindo a
formação de hidrogéis homogêneos (LEE et al., 1999; BERGER et al., 2004).
A formação do PEC de quitosana-alginato ocorre por meio da associação dos grupamentos
carboxila –COO- do alginato com os grupamentos amino –NH3+ da quitosana. As propriedades
deste PEC podem diferir daquelas dos polissacarídeos isolados, observando-se, por exemplo,
maior estabilidade a variações de pH e maior eficiência na liberação controlada de princípios
ativos (WANG et al., 2001). Neste sentido, segundo Paul e Sharma (2004), membranas
constituídas por quitosana e alginato simultaneamente podem apresentar desempenho in vivo
mais satisfatório, quando comparadas a filmes constituídos dos mesmos polímeros isolados.
O complexo de polieletrólitos formado pela quitosana e pelo alginato é muito estudado
desde os anos 90, encontrando aplicações na produção de fibras (LI et al., 2009; MIRAFTAB et
al., 2011), órgãos artificiais (HEIN et al., 2008), microcápsulas para liberação de fármacos e de
genes (RINAUDO, 2008; LI et al., 2009; HEIN et al., 2008; SÆTHER et al., 2008; LUO e
WANG, 2014), arcabouços ou scaffolds para regeneração de tecidos (LI et al., 2009; LIN e
YEH, 2010), membranas para cobertura de feridas (RINAUDO, 2008; MENG et al., 2010) e
para uso em processos de separação por pervaporação (LI et al., 2009).
O pH tem grande influência na formação de complexos de polieletrólitos. Na Figura 2.4 é
mostrado o comportamento do PEC de quitosana-alginato de acordo com o pH. Em valores de
pH muito altos, a interação entre o alginato e a quitosana é desfavorecida, assim como em
valores de pH muito baixos, visto que, nestas condições, ocorre preponderância de ionização do

23
alginato e da quitosana isolados, respectivamente. Para promover a formação de um PEC estável,
Cárdenas et al. (2003) propõem o ajuste do pH para 5,28, em um valor intermediário aos valores
de pKa da quitosana (6,3 a 7,3) (KARAKEÇILI et al., 2007) e do alginato (3,38 para os grupos
M e 3,65 para os grupos G) (SIMSEK-EGE et al., 2003; LI et al., 2009; SÆTHER et al., 2008),
garantindo que os grupos amino da quitosana estejam majoritariamente protonados e as
carboxilas do alginato, desprotonadas.
Figura 2.4: Efeito do pH na estrutura e comportamento de intumescimento do complexo de
polieletrólitos formado por quitosana e alginato. Os círculos representam as interações iônicas
entre as cargas negativas (-) do alginato e as cargas positivas da quitosana (+) (Adaptado de
Berger et al., 2004).
Diminuição do pH
Aumento do pH
Meio básico
pH 5,3
Meio ácido
Quitosana
Alginato
H2O
H2O
H2O
H2O
H2O H2O
H2O
H2OH2O
H2O
H2O
H2O
H2O
+-
--
-
-
---
- --
---
--
-
- -
- --
-- -
++
-
-
---
- -
---
--
-
--
-
H2O
H2O
H2O
H2O
H2O
H2O
H2OH2OH2OH2O
H2O
H2O
H2O
+
-+
+- +
-
-+
+
+
+-
-
-
-
H2O H2OH2O
H2O
H2O
H2O
H2O
H2O
H2O
H2O H2OH2O
H2O
H2O
H2O+ +
+ +
+
+ +
+++
+
++
++
++
+ +
++
+ ++
+ +
- -
-
++
+
+
++
+
+++
+ +
+
Diminuição do pH
Aumento do pH

24
Vários fatores afetam as propriedades do PEC de quitosana-alginato. Entre estes estão a
composição do alginato, a massa molar dos polissacarídeos e grau de desacetilação da quitosana
(GEORGE e ABRAHAM, 2006).
Quanto à composição do alginato, a proporção entre os blocos M e G pode afetar
fortemente as características mecânicas do gel formado na presença de cátions divalentes, como
o Ca2+. Alginatos com maior conteúdo de blocos G formam géis quebradiços, enquanto alginatos
com maior conteúdo de blocos M formam géis elásticos (PENMAN e SANDERSON, 1972).
Consequentemente, as propriedades mecânicas de complexos de polieletrólitos de quitosana-
alginato obtidos a partir de alginatos com diferentes razões M/G podem também diferir entre si.
Quanto à massa molar dos polissacarídeos, a literatura mostra que quanto mais próxima a
massa molar da quitosana estiver da do alginato, mais completa é a reação e, consequentemente,
mais cristalino e compacto é o PEC formado, apresentando menor permeabilidade e maior
homogeneidade (YAN et al., 2001). Por outro lado, quando a massa molar da quitosana é muito
maior do que a massa molar do alginato, a conformação da molécula de quitosana muda,
tornando-se menos estendida e diminuindo a acessibilidade dos grupos carboxila do alginato aos
grupos amino da quitosana. Neste caso, o PEC obtido é heterogêneo e menos estável em meios
aquosos (YAN et al., 2001; HONARY et al., 2009).
Quanto ao grau de desacetilação da quitosana, seu aumento favorece as interações entre os
polissacarídeos, devido ao aumento de grupos protonáveis da quitosana (GEORGE e
ABRAHAM, 2006; HSU et al., 2004).
2.4. Membranas de quitosana-alginato
A mistura entre soluções de diferentes polieletrólitos, como a quitosana e o alginato, leva à
formação de coacervados poliméricos. O fenômeno de coacervação, pioneiramente estudado na
década de 1930 por Bungenberg de Jong, é um processo no qual ocorre separação de fases em
uma solução homogênea de macromoléculas carregadas eletricamente, originando uma fase
densa rica em polieletrólitos (BOHIDAR, 2008).
Para obter coacervados adequados visando à sua moldagem na forma de membranas
homogêneas, a taxa de reação entre os dois polímeros deve ser suficientemente lenta, prevenindo
a formação de membranas interfásicas e permitindo, assim, que a reação seja completa (YAN et

25
al., 2000).
As características das membranas obtidas, tais como espessura, conteúdo de umidade,
resistência à tração e alongamento na ruptura, são afetadas pelos parâmetros de produção, como
a razão mássica entre os polímeros, a massa molar, a adição de agente reticulante, o pH, a força
iônica (LUO e WANG, 2014), assim como a taxa de agitação e de adição de uma solução
polimérica a outra (RODRIGUES, 2008).
Uma das técnicas de preparação de membranas densas de quitosana-alginato foi proposta
por Yan et al. (2000) e adaptada por Wang et al. (2001, 2002) e por Rodrigues et al. (2008),
consistindo na mistura de soluções de quitosana e alginato em proporções mássicas iguais, sob
condições pré-estabelecidas de concentração dos polímeros, taxa de agitação e taxa de adição da
solução de quitosana na solução de alginato, dentre outras.
Yan et al. (2001) investigaram a produção de membranas de quitosana-alginato com
quitosanas de diferentes massas molares, variando de 1,3x105 g/mol a 10,0x105 g/mol, sendo a
massa molar do alginato igual a 1,04x105 g/mol. Os autores verificaram que quanto menor a
massa molar da quitosana, maior a sua mobilidade e facilidade de reagir com o alginato,
resultando em membranas mais finas e transparentes e menos permeáveis ao vapor d’água.
Estudos sobre a produção de membranas de quitosana-alginato com alginatos de diferentes
massas molares não foram localizados. No entanto, dados obtidos em experimentos anteriores
não publicados indicaram que é possível produzir membranas com quitosana de massa molar
cerca de 27 vezes maior do que a massa molar do alginato, e que o emprego de outro tipo de
alginato, com massa molar cerca de duas vezes menor, não levou à formação de membranas
(ensaios exploratórios desta tese de doutorado).
O reforço mecânico do PEC de quitosana-alginato pode ser feito pela reticulação com íons
cálcio das cadeias de alginato que não reagiram com a quitosana. O procedimento de preparo
proposto por Rodrigues (2008) contém duas etapas de reticulação com CaCl2. Na primeira etapa
de reticulação, adiciona-se CaCl2 diretamente à mistura polimérica. Na segunda etapa de
reticulação, as membranas secas de quitosana-alginato são imersas em uma solução de CaCl2. A
inclusão de uma segunda etapa de reticulação é necessária para melhorar as características finais
das membranas, visando acentuar a reticulação das regiões das cadeias de alginato ainda não
completamente complexadas com as cadeias de quitosana (RODRIGUES, 2008). Bueno (2010)
observou que o emprego de quantidades de CaCl2 maiores do que a empregada por Rodrigues

26
(2008) na primeira etapa de reticulação causa gelificação excessiva, com consequente
precipitação de agregados poliméricos não-homogêneos, reforçando novamente que há
necessidade do emprego de duas etapas de reticulação.
Membranas lamelares densas de quitosana-alginato produzidas de acordo com Bueno
(2010) apresentaram potencial para utilização como curativos primários em lesões de pele, de
acordo com resultados de ensaios in vivo realizados em ratos Wistar com lesões profundas
provocadas por punching. Nestes ensaios, as membranas de quitosana-alginato foram
previamente hidratadas com solução salina (NaCl a 0,9% m/v) e aplicadas diretamente sobre as
lesões, utilizando-se como curativo secundário gaze fixada com fita adesiva. Os resultados
obtidos indicaram a modulação da fase inflamatória, o aumento da proliferação de fibroblastos e
da formação de colágeno, o que acelerou as fases iniciais do processo de cicatrização e melhorou
a qualidade do tecido formado. As membranas se mostraram não-aderentes e evitaram,
consequentemente, danos secundários ao tecido novo que se formava (CAETANO et al., 2014).
No trabalho de Bueno (2010) também foram produzidas membranas porosas de quitosana-
alginato, por meio da inclusão dos surfactantes não-iônicos Tween 80 ou Pluronic F68 à mistura
polimérica. Tal estrutura porosa permitiria o emprego deste material como matriz tridimensional
ou scaffold para a regeneração de lesões de pele.
A literatura referente ao preparo de membranas e scaffolds porosos menciona, na maioria
das vezes, o uso da liofilização (KUCHARSKA et al., 2008; LAI et al., 2003; YU et al., 2005;
WAN et al., 2007; LI e ZHANG, 2005), contudo esta técnica é demorada e de custo mais
elevado em comparação a outras. O uso de agentes porogênicos, como NaCl ou glicose também
é mencionado frequentemente na literatura, mas o material deve ser lavado sucessivas vezes com
grandes quantidades de água para a remoção destes compostos e dos seus produtos de
degradação (WAN et al., 2007; MOHAN e NAIR, 2005; MA et al., 2001). Além disso, no caso
particular da complexação entre a quitosana e o alginato, o NaCl poderia prejudicar a formação
do PEC devido à competição entre os íons Na+ e Ca2+ pelas carboxilas desprotonadas livres.
Outras formas de se introduzir poros em scaffolds são a inversão de fases (introdução de poros
por meio de um não-solvente do polímero) e a gaseificação por alta pressão. Alternativamente,
os scaffolds porosos podem ser preparados por processos de eletrospinning, que promovem o
depósito de microfibras e nanofibras com formato tridimensional adequado ao crescimento e
diferenciação celular (SHOICHET, 2010). Ainda, os processos assistidos por CO2 supercrítico

27
também são mencionados frequentemente, não requerendo o uso de grandes quantidades de
solventes orgânicos e temperaturas elevadas (DUARTE et al., 2013; REVERCHON e
CARDEA, 2012).
Desta forma, a inclusão de surfactantes não-iônicos se mostra como uma alternativa viável
para obtenção de membranas homogêneas e com estrutura porosa, sendo o processo de produção
mais viável do que outros processos comumente relatados na literatura.
2.5. Surfactantes não-iônicos
A adição de surfactantes a polímeros é muitas vezes realizada para promover
homogeneização, emulsificação, suspensão, controle da reologia e melhora da estabilidade
coloidal (HOLMBERG et al., 2002). Muitos investigadores também exploram o uso dos
tensoativos para melhorar as propriedades de materiais, como tamanho de poro, capacidade de
absorver e reter água, propriedades mecânicas e uniformidade (STOPPEL et al., 2011).
A escolha do surfactante é importante quando se trata de dispositivos biomédicos. Neste
sentido, os surfactantes não-iônicos são mais adequados do que os surfactantes iônicos no caso
de contato direto com a pele saudável, uma vez que não penetram na mesma e não aderem a
regiões eletricamente carregadas (RHEIN et al., 2007). A seguir são discutidas as estruturas e
propriedades do Pluronic F68.
2.5.1. Pluronic F68
Os surfactantes da classe dos Pluronics ou Poloxamers são copolímeros de polioxietileno e
polioxipropileno. Sua fórmula estrutural consiste em um bloco central de polioxipropileno, o
qual é relativamente hidrofóbico, cercado dos dois lados por blocos de polioxietileno, o qual é
relativamente hidrofílico (SIGMA-ALDRICH, 2011; KARMARKAR et al., 2008). A fórmula
estrutural geral dos Pluronics é mostrada na Figura 2.5.
Devido à variação dos índices x e y mostrados na Figura 2.5, existem vários tipos de
Pluronics, os quais se apresentam em diferentes formas físicas. A designação dos Pluronics é
feita do seguinte modo: F para flocos, P para pasta e L para líquido. Particularmente, o Pluronic
F68 apresenta-se na forma de um pó branco, sem cheiro ou sabor, o qual é solúvel em água. Seus

28
índices x e y correspondem a 80 e 27, respectivamente (KARMARKAR et al., 2008), e sua
massa molar é em média 8350 g/mol (SIGMA-ALDRICH, 2011).
Figura 2.5: Fórmula estrutural dos Pluronics (SIGMA-ALDRICH, 2011).
O valor de HLB (equilíbrio hidrófilo-lipófilo) do Pluronic F68, o qual mede a
hidrofilicidade do surfactante com base na porcentagem relativa entre seus grupos hidrofílicos e
hidrofóbicos, é igual a 29 (SIGMA-ALDRICH, 2011), o que indica alta capacidade de formar
soluções aquosas límpidas e espumas. Já o valor de CMC (concentração micelar crítica) em água
do Pluronic F68 é igual a 0,04 mM a 25°C (SIGMA-ALDRICH, 2011).
O Pluronic F68 é muito usado na indústria farmacêutica por ser compatível com diversas
formulações medicamentosas, sendo comumente empregado em sistemas de liberação controlada
de fármacos e proteínas e como gel injetável para administração de medicamentos. Este
tensoativo também é usado como aditivo na engenharia de tecidos, como veículo na terapia
gênica e como agente estabilizante de membranas celulares no caso de cultivo de células animais
em biorreatores agitados (STOPPEL et al., 2011). O Pluronic F68 também foi aprovado pela
FDA para uso na composição de produtos de higienização da pele e, segundo um estudo
realizado com mais de 1000 pacientes que apresentavam lesões dérmicas, a aplicação de
Pluronic F68 sobre as feridas não causou reações adversas, prevenindo a ocorrência de infecções
(RODEHEAVER et al., 1980).
2.6. Incorporação de agentes bioativos em curativos
Curativos contendo agentes bioativos são mais eficazes em diversos aspectos, pois
melhoram o processo de cicatrização da pele e facilitam o tratamento do paciente. Existem
diversos tipos de moléculas que podem ser incorporados a curativos, por exemplo, agentes
antimicrobianos, analgésicos, anti-inflamatórios, anestésicos e anti-alérgicos, entre outros
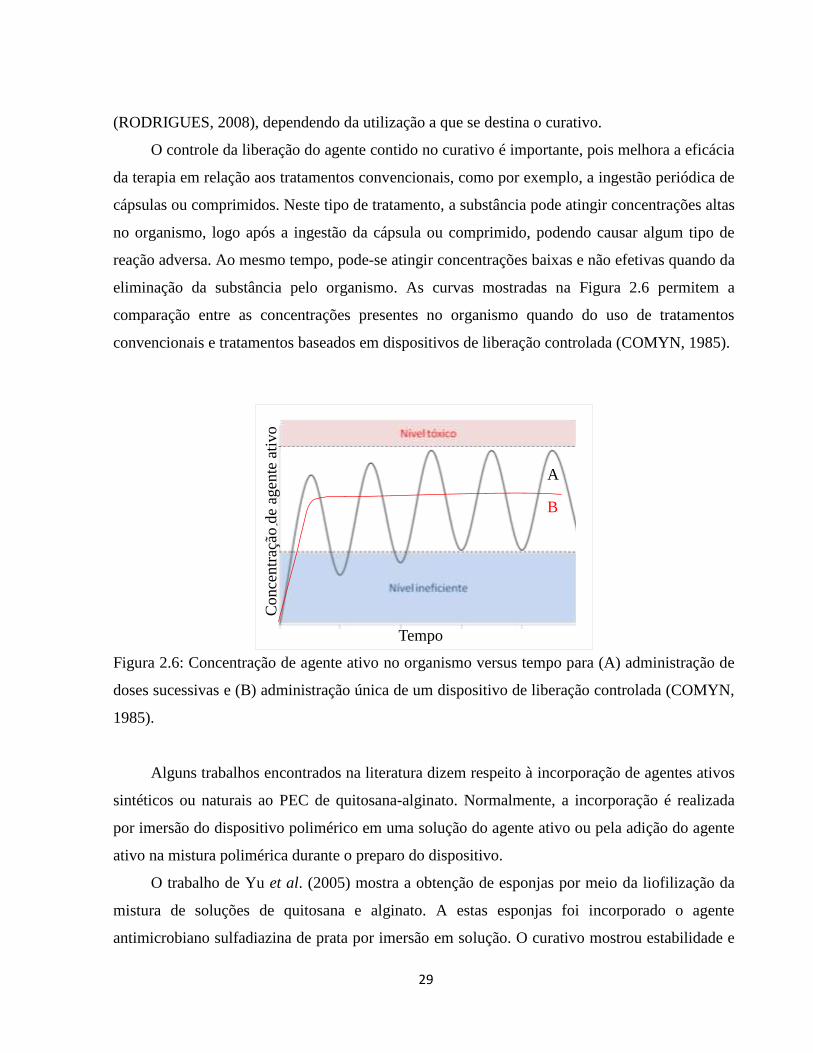
29
(RODRIGUES, 2008), dependendo da utilização a que se destina o curativo.
O controle da liberação do agente contido no curativo é importante, pois melhora a eficácia
da terapia em relação aos tratamentos convencionais, como por exemplo, a ingestão periódica de
cápsulas ou comprimidos. Neste tipo de tratamento, a substância pode atingir concentrações altas
no organismo, logo após a ingestão da cápsula ou comprimido, podendo causar algum tipo de
reação adversa. Ao mesmo tempo, pode-se atingir concentrações baixas e não efetivas quando da
eliminação da substância pelo organismo. As curvas mostradas na Figura 2.6 permitem a
comparação entre as concentrações presentes no organismo quando do uso de tratamentos
convencionais e tratamentos baseados em dispositivos de liberação controlada (COMYN, 1985).
Figura 2.6: Concentração de agente ativo no organismo versus tempo para (A) administração de
doses sucessivas e (B) administração única de um dispositivo de liberação controlada (COMYN,
1985).
Alguns trabalhos encontrados na literatura dizem respeito à incorporação de agentes ativos
sintéticos ou naturais ao PEC de quitosana-alginato. Normalmente, a incorporação é realizada
por imersão do dispositivo polimérico em uma solução do agente ativo ou pela adição do agente
ativo na mistura polimérica durante o preparo do dispositivo.
O trabalho de Yu et al. (2005) mostra a obtenção de esponjas por meio da liofilização da
mistura de soluções de quitosana e alginato. A estas esponjas foi incorporado o agente
antimicrobiano sulfadiazina de prata por imersão em solução. O curativo mostrou estabilidade e
Tempo
Co
nce
ntr
ação
de
agen
te a
tiv
o
A
B

30
capacidade de liberação controlada do agente ativo, suprimindo a proliferação de bactérias das
espécies Pseudomonas aeruginosa e Staphylococcus aureus, frequentemente encontradas na
pele.
Utilizando um método semelhante, Lai et al. (2003) produziram esponjas de alginato de
sódio e quitosana contendo paracetamol a 0,1% m/v adicionado diretamente à mistura
polimérica. Nesse estudo, observou-se que a cinética de liberação de paracetamol é controlada,
havendo uma porcentagem de liberação de mais de 60% do fármaco contido na esponja.
Rodrigues (2008) incorporou o antibiótico bacitracina a membranas de quitosana-alginato
por meio de imersão em solução. O agente ativo apresentou atividade biológica residual
apropriada, não havendo permeação de bactérias através das membranas, apesar da forte
interação do antibiótico com os polissacarídeos.
Girata (2011) incorporou fosfato hidrogenado de zircônio, sódio e prata (Alphasan®
RC2000) a membranas de quitosana-alginato por adição à mistura polimérica, observando
eficiências de incorporação maiores do que 85% e eficácia das membranas contra bactérias
gram-positivas e gram-negativas.
De Souza (2014), por sua vez, incorporou o antibiótico eritromicina a membranas de
quitosana-alginato por adição à mistura polimérica e por adsorção em solução etanólica. A
autora observou que o método de incorporação por adição à mistura polimérica teve maior
eficiência de incorporação (máximo de 54%) e que a liberação do antibiótico em tampão PBS se
estabilizou em torno de 15 horas.
A impregnação de agente ativos também pode ser realizada via fluido supercrítico. No
entanto, não foram localizados estudos referentes à incorporação de moléculas por via
supercrítica em dispositivos à base de quitosana-alginato.
Um fluido supercrítico pode ser definido como uma substância em condições de pressão e
temperatura acima do ponto crítico, apresentando propriedades físicas intermediárias entre
líquido e gás. Como um líquido, o fluido supercrítico apresenta densidade apreciável, com alto
poder de solvatação, enquanto a viscosidade e a difusividade são similares às de um gás,
facilitando a transferência de massa (PASQUALI et al., 2008; DUARTE et al., 2009). O dióxido
de carbono está entre os fluidos supercríticos mais utilizados, pois apresenta a vantagem de não
causar danos ao meio ambiente, ser atóxico, não inflamável, não corrosivo e possuir baixo custo
associado ao alto grau de pureza em comparação a outros fluidos. Além disso, por possuir baixa

31
temperatura crítica e pressão crítica relativamente não muito elevada (Tc de 31 °C e Pc de
73 bar, respectivamente), o CO2 supercrítico é muito estudado para aplicação em sistemas
termossensíveis destinados ao processamento de fármacos e outros compostos bioativos
(DUARTE et al., 2009).
Na impregnação de agentes ativos, o CO2 supercrítico atua reduzindo a temperatura de
transição vítrea de diferentes polímeros, tornando possível a mistura do polímero amolecido com
outras moléculas, preservando a integridade das espécies envolvidas devido principalmente às
baixas temperaturas de trabalho (MCHUGH et al., 2002). As vantagens desta técnica são o baixo
custo operacional, a eliminação do uso de solventes orgânicos e de uma etapa final de secagem
do dispositivo e o fato de não gerar resíduos (DIAS et al., 2012; BHAMIDIPATI et al., 2013). A
impregnação com fluido supercrítico é homogênea e, em geral, não altera negativamente as
propriedades físicas, químicas e mecânicas do material (COOPER, 2000; YAÑEZ et al., 2011).
O uso de CO2 supercrítico na impregnação de agentes ativos apresenta algumas limitações, como
o alto investimento inicial requerido para a implantação de um sistema de impregnação, havendo
a necessidade de aquisição de equipamentos especializados que suportem altas pressões
(COOPER, 2000). Outra limitação é a baixa solubilidade de compostos de alta massa molar em
CO2 supercrítico devido ao seu caráter apolar (DIAS et al., 2012; RINDFLEISCH et al., 1996).
Outros usos do CO2 supercrítico são no processamento de biomateriais, com o objetivo de
modificar a porosidade da matriz, remover solventes ou esterilizar (BHAMIDIPATI et al.,
2013). Por meio do uso da tecnologia supercrítica, é possível alterar a permeabilidade do
material a gases e vapor, assim como o comportamento cinético de liberação de um composto
bioativo. As propriedades desejadas podem ser atingidas pelo controle das condições de
processamento do material, como pressão, temperatura, tempo de residência e taxa de
despressurização (DIAS et al., 2012).
Desta forma, a incorporação de agentes ativos pode ser realizada, dentre outras técnicas,
por simples adição à mistura polimérica durante o preparo do dispositivo, por imersão do
dispositivo em uma solução contendo o agente ativo, ou ainda por impregnação via fluido
supercrítico. Estas três técnicas de incorporação foram utilizadas para impregnar o agente
antimicrobiano polihexametileno biguanida (PHMB) a membranas de quitosana-alginato. As
características e propriedades deste agente antimicrobiano são descritas a seguir.

32
2.7. PHMB
O polihexametileno biguanida (PHMB), também conhecido como poliaminopropil
biguanida e polihexanida é um agente antimicrobiano da família das guanidinas e seu uso é
bastante comum. Pode ser incorporado a uma variedade de produtos, incluindo curativos,
soluções de limpeza de lentes de contato, produtos para assepsia cirúrgica, tecidos e sabonetes
antimicrobianos, produtos para limpeza de piscinas e cosméticos, (MULDER et al., 2007;
SCHNUCH et al., 2007; GAO e CRANSTON, 2008; ARCH CHEMICALS, 2008; ARCH
CHEMICALS, 2005). A variedade de possibilidades de aplicações do PHMB se deve ao fato de
este agente antimicrobiano ser bastante solúvel em água (solubilidade de 41 ± 1 % m/m a 25°C),
estável ao calor, inodoro, compatível com ampla faixa de pH (entre 1,0 e 9,0), além de
apresentar baixa toxicidade e ser de baixo impacto ambiental (ARCH CHEMICALS, 2005;
SUDWORTH, 2002).
Na Figura 2.7 é representada a estrutura do PHMB, o qual consiste de uma mistura
heterodispersa de cadeias de cloreto de polihexametileno biguanida de diferentes tamanhos,
sendo a sua massa molar média igual a aproximadamente 2500 g/mol (GAO e CRANSTON,
2008). O número típico de unidades de hexametileno biguanida na cadeia é normalmente igual a
12 (SCHNUCH et al., 2007; O’MALLEY et al., 2006).
Figura 2.7: Fórmula estrutural do PHMB (n=11 a 15) (adaptado de Gao et al., 2011).
O grupo biguanida tem caráter básico, sendo seu pKa igual a 10,96, o que significa que,
em pH fisiológico, o PHMB possui carga positiva. Consequentemente, o agente antimicrobiano é
adsorvido com facilidade por superfícies aniônicas, como as paredes celulares das bactérias
2+
Cl-

33
(O’MALLEY et al., 2006) e os grupos carboxílicos aniônicos (SIMONCIC e TOMSIC, 2010;
GAO e CRANSTON, 2010) presentes no alginato.
O mecanismo de ação do PHMB foi descrito por vários grupos de pesquisa. Broxton et al.
(1984) demonstraram que a atividade máxima do PHMB ocorre entre pH 5 e 6 e que,
inicialmente, o biocida interage com a superfície da bactéria e então é transferido para seu
citoplasma. Ikeda et al. (1984) mostraram que o PHMB catiônico tem pouco efeito contra os
fosfolipídeos neutros de membranas bacterianas, pois seu efeito é maior em moléculas
negativamente carregadas, nas quais o antimicrobiano adsorve, resultando na liberação de
lipopolissacarídeos e íons potássio através da membrana externa do microrganismo, o qual
eventualmente morre (MULDER et al., 2007).
A literatura descreve a alta eficiência do PHMB contra vários tipos de microrganismos, até
mesmo aqueles resistentes a antibióticos. Até recentemente, a resistência microbiana a este
composto havia sido raramente observada (GAO e CRANSTON, 2008; MOORE e GRAY,
2007).
A concentração mínima inibitória (MIC) é uma medida que indica a menor concentração
de um antimicrobiano necessária para inibir o crescimento visível de um determinado
microorganismo após um período de incubação de 18 a 24 horas, dependendo do tipo de célula,
sendo empregada uma concentração de inóculo padronizada de aproximadamente 105 unidades
formadoras de colônia por mililitro (UFC/mL) (BARRET et al., 2010). Na Tabela 2.5 encontra-
se a concentração mínima inibitória (MIC) do PHMB para diversas bactérias, fungos e
leveduras.
O interesse na produção de curativos com propriedades antimicrobianas é crescente,
principalmente no que se refere ao tratamento de feridas crônicas. Neste tipo de ferida, perpetua
a fase inflamatória, havendo uma população polimicrobiana, composta por quatro ou mais tipos
de bactérias aeróbicas e anaeróbicas, sendo a Staphylococcus aureus uma das mais
problemáticas (WIEGAND e HIPLER, 2010). Como pode ser visto na Tabela 2.5, a
concentração mínima inibitória (MIC) de PHMB para este tipo de bactéria é muito baixa, o que
torna o seu uso muito interessante no tratamento de feridas. Outros microrganismos que podem
estar presentes em feridas crônicas são Pseudomonas aeruginosa, Escherichia coli e Klebsiella
pneumoniae (WIEGAND e HIPLER, 2010).

34
Tabela 2.5: Concentração mínima inibitória de PHMB para diversas bactérias, fungos e
leveduras (adaptado de Arch Chemicals, 2005).
Microrganismo PHMB (µg/mL)
Bactérias
Bacillus subtilis 0,5
Staphylococcus albus 0,2
Staphylococcus aureus 0,2
Staphylococcus epidermidis 1
Enterococcus faecium 5
Streptococcus lactis 5
Enterobacter cloacae 4
Escherichia coli 5
Klebsiella pneumoniae 5
Proteus vulgaris 40
Pseudomonas aeruginosa 20
Pseudomonas putida 5
Serratia marcescens 5
Fungos filamentosos
Aspergillus niger 150
Trichophyton mentagrophytes 5
Leveduras
Candida albicans 60
Saccharomyces cerevisiae (ellipsoideus) 20
Saccharomyces cerevisiae (pastorianus) 20
Saccharomyces cerevisiae (turbidans) 15
As bactérias comumente presentes em hospitais também podem representar um grande
risco ao paciente com lesão de pele. Dados da U.S. National Healthcare Safety Network (2009)
indicam que as bactérias gram-negativas são responsáveis por mais de 30% das infecções
adquiridas em hospitais. Nas unidades de tratamento intensivo (UTI)s dos Estados Unidos, as

35
bactérias gram-negativas são responsáveis por 70% das infecções, e dados similares são
reportados em outras partes do mundo (GAYNES e EDWARDS, 2005). Dentre os
microrganismos gram-negativos, a família Enterobacteriaceae é a mais comumente presente em
infecções adquiridas em hospitais, e outras bactérias resistentes, como Pseudomonas aeruginosa
e Acinetobacter baumannii têm sido reportadas (PELEG e HOOPER, 2010).
A literatura aponta que o PHMB tem maior eficácia contra bactérias gram-positivas, como
Staphylococcus aureus e Bacillus subtilis, do que contra bactérias gram-negativas, como
Pseudomonas aeruginosa e Escherichia coli. Uma possível explicação para esta diferença é que
a parede celular das bactérias gram-negativas é constituída por uma camada a mais do que as
bactérias gram-positivas. Sendo assim, as bactérias gram-negativas seriam mais difíceis de serem
eliminadas pelo antimicrobiano (HÜBNER e KRAMER, 2010; CHEN-YU et al., 2007).
Nota-se também que, na maioria dos casos, a eliminação de fungos e leveduras requer
concentrações ainda maiores do composto ativo, o que pode limitar a indicação deste composto
para o controle destes tipos de microrganismos.
O nível de PHMB considerado ativo em produtos contendo este agente antimicrobiano em
sua formulação é de 0,1% (m/m) (ARCH CHEMICALS, 2005). Dados relatados na literatura
indicam que concentrações acima de 5% (m/m) são irritantes para a pele, de acordo com testes
realizados em ratos (HÜBNER e KRAMER, 2010).
Algumas empresas já comercializam curativos contendo PHMB, os quais protegem a
ferida contra possíveis infecções por meio da diminuição da carga bacteriana, impedindo, ao
mesmo tempo, a penetração de bactérias através do curativo (MOORE e GRAY, 2007). Na
Tabela 2.6 são mostrados exemplos de curativos comercialmente disponíveis contendo PHMB,
assim como a porcentagem mássica de PHMB incorporada nos mesmos.
Algumas patentes referentes a curativos incorporando PHMB também foram localizadas.
Como pode ser visto na Tabela 2.7, as porcentagens de agente antimicrobiano mencionadas
nestas patentes são maiores do que as listadas para os curativos já comercializados.
O PHMB também pode ser usado na forma de soluções aquosas e géis para tratamento e
desinfecção de feridas. Foram localizados três produtos deste tipo disponíveis comercialmente,
todos contendo o agente antimicrobiano na concentração de 0,1% (m/v). São eles o Prontosan
gel (BBraun) e as soluções Aquasept-R (Walkmed) e Lavasept (Fresenius Kabi).
A incorporação de PHMB às membranas de quitosana-alginato mostra-se uma alternativa

36
interessante para se obter curativos de lesões de pele biocompatíveis, biodegradáveis e com
atividade antimicrobiana. Deve-se ressaltar que não foram localizadas na literatura informações a
respeito de incompatibilidade do PHMB com qualquer um dos compostos utilizados para a
produção das membranas de quitosana-alginato.
Tabela 2.6: Exemplos de curativos comercializados contendo PHMB.
Nome e marca Tipo de matriz PHMB (% m/m)
Kerlix®, Covidien gaze 0,2
Curity®, Covidien gaze 0,2
Excilon®, Covidien esponja de rayon-poliéster 0,2
Telfa®, Covidien camada de fibras de algodão
envolta em filme perfurado de
PET
0,2
Xcell®, Medline Inc. filme de microfibras de celulose 0,3
SuprasorbX + PHMB®,
Lohmann & Rauscher
filme de microfibras de celulose 0,3
Tabela 2.7: Registros de patentes encontradas referentes à incorporação de PHMB em curativos.
Patente Tipo de matriz PHMB
(% em massa)
Rohrer et al., 2009 Fibras de alginato 0,1-40
Patel et al., 2007 Múltiplas camadas poliméricas (ex.
poliuretano, celulose, poliéster, entre outros)
0,2-3
Serafica et al., 2004 Película de celulose 0,27-0,37
Feld e Soto, 1987 Adesivo à base de poliuretano 0,45-1,2
Supõe-se que a incorporação de PHMB ao PEC de quitosana-alginato possa envolver
diferentes tipos de interações. Neste caso, como o agente antimicrobiano é positivamente
carregado, poderiam ocorrer interações iônicas entre os grupos biguanida do PHMB e os sítios
negativamente carregados do alginato (-COO-) que não se associaram aos grupos amino da
quitosana (-NH3+) durante o preparo da membrana ou aos íons cálcio (Ca2+) utilizados na etapa

37
de reticulação. Supostamente poderiam ocorrer também ligações de hidrogênio entre os grupos
biguanida e os grupos -OH dos polissacarídeos e interações hidrofóbicas entre as cadeias de
polihexametileno do PHMB e os grupos fracamente hidrofóbicos dos polissacarídeos, como os
grupos -CH da cadeia principal da quitosana e do alginato e os grupos N-acetil da quitosana.
Desta forma, devido às várias possibilidades de interações do PHMB com as membranas de
quitosana-alginato, pode-se supor que a incorporação do agente antimicrobiano tenha alta
eficiência, por outro lado, a liberação do antimicrobiano em meios fisiológicos pode ser limitada.
A incorporação de PHMB pode ser realizada mais facilmente, a princípio, pelos métodos
de adição à mistura polimérica e por adsorção a partir de soluções aquosas, uma vez que o
PHMB possui baixa solubilidade em CO2 supercrítico. Entretanto, para solutos com baixa
solubilidade em CO2 supercrítico, como seria o caso do PHMB, a alta afinidade do soluto pela
matriz polimérica levaria a uma condição de partição favorável (TOMASKO et al., 2003).
2.8. Considerações finais
Tendo em vista os assuntos abordados nesta revisão de literatura, torna-se clara a
necessidade do desenvolvimento de novos curativos com propriedades cicatrizantes e mais
acessíveis à população.
A produção de membranas porosas à base de quitosana-alginato por meio da adição de
tensoativos não-iônicos aparece como uma alternativa pouco onerosa de produção de curativos
biodegradáveis e biocompatíveis, os quais também podem ser empregados como scaffolds para
células da pele. A melhoria das propriedades antimicrobianas destas membranas pode ser
supostamente obtida com a inclusão de polihexametileno biguanida (PHMB), o que pode ser
feito através de diferentes técnicas de incorporação, constituindo o ineditismo do presente
trabalho.

38
3. MATERIAIS E MÉTODOS
3.1. Materiais utilizados para o preparo das membranas
Para o preparo das membranas de quitosana-alginato, utilizou-se quitosana de alta massa
molar obtida de camarão (código C3646, lote 061M0046V) e alginato de sódio de baixa massa
molar (código A2158, lote 090M0092V), ambos da Sigma Chemical Co. A massa molar
viscosimétrica média da quitosana utilizada foi de 1,26x106 g/mol e a massa molar
viscosimétrica do alginato utilizado foi de 4,69x104 g/mol (determinadas por meio de
procedimento descrito no Apêndice I). O grau de desacetilação da quitosana foi de 95%
(procedimento descrito no Apêndice II). A razão entre as unidades de ácido manurônico e
gulurônico (razão M/G) do alginato foi de 1,86 (procedimento descrito no Apêndice III).
Utilizou-se também Pluronic F68 da Sigma Chemical Co., cloreto de cálcio dihidratado e
hidróxido de sódio da Merck, ácido acético glacial e acetona da Synth, solução aquosa de PHMB
a 20% (m/v) (Cosmocil CQ) e PHMB em pó (Vantocil 100) da Arch Chemicals.
Os reagentes utilizados na caracterização das membranas foram tampão fosfato salino
(PBS), tampão Hepes, NaCl, dodecil sulfato de sódio (SDS), dimetil sulfóxido (DMSO) e 3-(4,5-
dimetiltiazol-2-yl)-2,5-difeniltetrazolium brometo (MTT) da Sigma Chemical Co., além de soro
fetal bovino e meio de cultura RPMI-1640 da Nutricell e meio de cultura ágar triptona de soja
(TSA) da Oxoid.
3.2. Método de preparo das membranas
As membranas de quitosana-alginato foram obtidas de acordo com adaptações do método
estabelecido por Bueno (2010). A razão mássica empregada entre a quitosana e o alginato foi de
1:1.
Os parâmetros quantidade de surfactante adicionado, temperatura de processo e de
secagem foram variados a fim de se verificar possíveis modificações nas características das
membranas e promover melhorias no processo produtivo, como explicado anteriormente. A
acetona, previamente utilizada como co-solvente da quitosana, teve seu uso descontinuado neste
trabalho.

39
As soluções empregadas para o preparo das membranas foram: alginato a 0,5% (m/v) em
água deionizada (sistema Milli-Q, Millipore) (pH = 7,7), quitosana a 1% (m/v) em solução
aquosa de ácido acético a 2% (v/v) (pH = 3,5), NaOH a 1 M e CaCl2 a 2% (m/v).
Para o preparo das membranas, adicionou-se o surfactante Pluronic F68 a 180 mL de
solução de alginato. As concentrações empregadas variaram de 0,02 a 0,1% (m/v) em relação ao
volume de solução de alginato. Também foram preparadas membranas sem surfactante.
Em seguida, 90 mL da solução de quitosana, misturados ou não a 90 mL de acetona, foram
adicionados à solução de alginato e surfactante. A adição foi realizada com auxílio de uma
bomba peristáltica Gilson modelo Minipuls 3, a uma vazão baixa de 200 mL/h, de modo a
prevenir a formação de membranas interfásicas, como recomendado por Yan et al. (2000).
A reação ocorreu em um reator de aço inoxidável encamisado com diâmetro interno de
10 cm e altura de 20 cm. Durante a adição da quitosana ao alginato, o sistema foi mantido sob
agitação de 500 rpm com auxílio de um agitador mecânico Q-251 D da Quimis, com hélice do
tipo pás inclinadas de raio de 2,1 cm. A temperatura do sistema foi controlada com auxílio de um
banho ultratermostático (Q-214 M2, Quimis). As temperaturas empregadas foram 25°C, a qual é
uma temperatura muito próxima à temperatura ambiente e atraente do ponto de vista energético;
ou 60°C, a qual, com base no trabalho de Da Silva (2009), é uma temperatura que pode favorecer
a homogeneização dos coacervados. Na Figura 3.1 é mostrado o sistema utilizado para o preparo
das membranas.
Figura 3.1: Sistema utilizado no preparo das membranas.

40
Ao término da adição das soluções, a taxa de agitação foi aumentada para 1000 rpm e a
mistura foi agitada por mais 10 minutos. A seguir, adicionaram-se 16,8 mL de NaOH a 1 M,
visando ajustar o pH de 3,6 para aproximadamente 5,3 (pH no qual a quitosana apresenta-se
protonada e o alginato, desprotonado), mantendo-se a agitação do sistema em 1000 rpm por mais
10 minutos. Por fim, foi feita a reticulação primária da mistura por meio da adição de 3,6 mL de
CaCl2 a 2% (m/v), para que ocorresse a complexação dos grupos carboxila remanescentes do
alginato com os íons Ca2+, mantendo-se a agitação por mais 10 minutos. Deve-se ressaltar que a
as soluções de NaOH e CaCl2 foram gotejadas lentamente por toda a extensão da superfície da
mistura polimérica, o que se mostrou muito importante para garantir a reprodutibilidade do
processo.
Após a etapa de reação, a mistura formada foi dividida igualmente em termos mássicos em
duas placas de Petri de poliestireno de 15 cm de diâmetro e levada à estufa com circulação de ar
(modelo 410D, Nova Ética) a temperatura constante de 37, 50 ou 60°C. O tempo de secagem
dependeu da quantidade de membranas dentro da estufa e da umidade relativa ambiente. Sendo
assim, a secagem pode ser considerada como uma etapa cujo controle se mostra crucial para a
reprodutibilidade do processo.
Após a secagem, cada membrana foi imersa em 150 mL de CaCl2 a 2% (m/v) por 30
minutos para reticular as cadeias de alginato que ainda se encontravam livres (etapa de
reticulação secundária com Ca2+). A reticulação secundária é uma etapa fundamental, visto que,
se as membranas forem expostas diretamente a uma solução aquosa após serem retiradas da
estufa, ocorre o intumescimento dos polímeros e o aparecimento de falhas ao longo de sua
extensão. Tais falhas são decorrentes da solubilização de moléculas de alginato livres que não
foram suficientemente reticuladas (RODRIGUES, 2008). Além disso, o emprego de quantidades
maiores de CaCl2 na etapa de reticulação primária pode levar à formação de coacervados não
homogêneos devido à precipitação de alginato excessivamente reticulado (BUENO, 2010).
Para a retirada do CaCl2 e do surfactante em excesso e visando à neutralização do pH da
membrana, foram feitas mais duas lavagens adicionais de 30 minutos em água deionizada. A
secagem final foi realizada a 37°C por 6 horas, sendo as bordas das membranas presas à placa de
poliestireno para evitar encolhimento, o que, entretanto, não ocasiona o aparecimento de trincas
no material obtido.

41
3.2.1. Cinética de evaporação de solventes da membrana durante a secagem
A cinética de evaporação de solventes durante a secagem em estufa foi avaliada
gravimetricamente, com auxílio de uma balança analítica, por meio da pesagem periódica das
placas de poliestireno contendo a mistura polimérica. Com os dados obtidos, construiu-se o
gráfico da cinética de evaporação de solventes, descontando-se a massa da placa de poliestireno
vazia.
3.3. Incorporação do agente antimicrobiano
A incorporação de PHMB foi realizada de três formas: por simples adição do composto à
mistura polimérica durante o preparo do dispositivo, pela adsorção do composto contido em
solução e mediada por CO2 supercrítico. Estes três métodos de incorporação são descritos a
seguir.
3.3.1. Incorporação por adição à mistura polimérica
A incorporação por adição à mistura polimérica foi realizada após a etapa de reação e
reticulação primária do PEC, por meio da adição de 1 mL de solução aquosa de PHMB em
diferentes concentrações (18 mg/mL e 180 mg/mL) sob agitação constante por 5 minutos, de
forma a resultar nas porcentagens almejadas em relação à massa total de polissacarídeos, de
1% m/m (10 mg/g) e 10% m/m (100 mg/g). As etapas remanescentes do procedimento de
produção de membranas seguiram normalmente.
3.3.2. Incorporação por adsorção a partir de solução aquosa
A incorporação por adsorção foi realizada em membranas prontas, através da imersão em
soluções aquosas de PHMB. Primeiramente, obteve-se a isoterma de adsorção em uma ampla
faixa de concentrações, analisando-se detalhadamente os mecanismos de adsorção do
antimicrobiano às membranas. Em seguida, selecionaram-se duas diferentes condições de
incorporação para caracterização detalhada. Tais procedimentos são descritos a seguir.

42
3.3.2.1. Determinação de isotermas de adsorção
A isoterma de adsorção de PHMB foi determinada por meio da imersão de corpos de prova
medindo 4 cm x 1 cm em 15 mL de soluções aquosas de PHMB de diversas concentrações.
Trabalhou-se com uma ampla faixa de concentrações, variando entre um valor muito baixo, igual
a 1 µg/mL (4,0x10-7 mol/L), até uma concentração muito próxima à do Cosmocil CQ puro, igual
a 190000 µg/mL (7,6x10-2 mol/L). Os ensaios foram realizados em duplicata a 37°C e 100 rpm
por 24 horas. O tempo necessário para que ocorresse o equilíbrio de adsorção foi avaliado
inicialmente à concentração inicial de 2x10-2 mol/L.
Optou-se por trabalhar com uma temperatura maior do que a ambiente para proporcionar o
aumento da solubilidade do PHMB em água e, principalmente, maior intumescimento das
membranas nas soluções do antimicrobiano, permitindo uma incorporação eficaz.
3.3.2.2. Incorporação por adsorção em duas diferentes condições iniciais
Nestes ensaios, foi necessário utilizar corpos de prova de tamanhos maiores do que os
utilizados na determinação das isotermas de adsorção de forma a possibilitar as caracterizações
das membranas.
Utilizaram-se corpos de prova medindo 4 cm x 4 cm, os quais foram imersos em 60 mL de
soluções aquosas de PHMB de 4,0x10-5 mol/L e 2,0x10-2 mol/L. Estes ensaios também foram
realizados em duplicata.
3.3.3. Impregnação de PHMB mediada por CO2 supercrítico
O PHMB foi impregnado nas membranas na presença de CO2 em diferentes condições de
temperatura e pressão, em um sistema experimental específico para este fim. Anteriormente aos
ensaios, a estabilidade e o efeito do processamento das membranas na presença de CO2
supercrítico foram avaliados.
Os ensaios descritos neste item foram realizados no Departamento de Engenharia Química
da Universidade de Coimbra, Portugal.

43
3.3.3.1. Unidade de processamento e impregnação
Para a realização dos processamentos e impregnações das membranas via CO2 supercrítico
utilizou-se CO2 a 99,998% (Praxair). A unidade experimental consistiu de uma bomba de CO2
líquido operada com ar comprimido, uma célula de impregnação de aço inoxidável com
aproximadamente 10 cm3 de volume interno, um banho termostático (Haake DC30, Thermo
Electron Corporation), uma placa de agitação magnética (Agimatic-Rev-W, P Selecta) para
homogeneizar a mistura a alta pressão (fármaco + CO2) e um manômetro (Datum 2000TM, Setra).
O diagrama da unidade de impregnação é mostrado na Figura 3.2.
Figura 3.2: Sistema utilizado para o processamento das membranas com CO2 supercrítico (C:
compressor, MS: placa de agitação magnética, TC: controlador de temperatura, M: manômetro,
v: válvulas, IC: célula de impregnação, tr: frasco para recolher resíduos durante a
despressurização).
3.3.3.2. Processamento de membranas sem PHMB em CO2 supercrítico
Nestes ensaios, analisou-se o efeito do processamento com CO2 supercrítico nas
características das membranas. As condições empregadas foram: tempo de processamento de
2 horas, temperatura de 45°C, taxa de despressurização de 5 bar/min e pressão variando de 100 a
300 bar. As membranas foram cortadas previamente em corpos de prova de dimensões
1 cm x 1 cm, sendo posicionados em 2 compartimentos dispostos verticalmente dentro da célula
de impregnação, cada um contendo 4 corpos de prova. A homogeneização do CO2 supercrítico
V-1 V-2C
V-3
V-4
V-5
V-6
V-7 V-8 V-9
tr
IC
MS
M
TC

44
dentro da célula foi realizada com auxílio de agitação magnética a 900 rpm. As formulações
obtidas foram caracterizadas quanto à morfologia, espessura, sorção e transmissão de vapor
d’água e comportamento térmico (calorimetria e termogravimetria).
3.3.3.3. Impregnação de PHMB mediada por CO2 supercrítico
As condições empregadas nos ensaios de incorporação foram tempo de 2 horas,
temperaturas de 45 e 55°C, pressões de 100, 150, 200 e 300 bar e taxa de despressurização de
5 bar/min. As membranas foram cortadas previamente em quatro corpos de prova de dimensões
1 cm x 1 cm, os quais foram dispostos lado a lado dentro da célula de impregnação. A
quantidade de 2 mg de PHMB na forma sólida foi colocada no fundo da célula, e a
homogeneização da mistura contendo CO2 e fármaco foi realizada com auxílio de agitação
magnética a 900 rpm.
3.4. Caracterização das membranas
A seguir são apresentadas as técnicas empregadas para a caracterização das membranas
obtidas. Parte dos ensaios foi realizada na Faculdade de Engenharia Química da Unicamp e parte
no Departamento de Engenharia Química da Universidade de Coimbra, sendo estes últimos a
caracterização das membranas quanto à coloração, área superficial e porosidade, sorção e
transmissão de vapor d’água, ângulo de contato, análise térmica simultânea (SDT) e
espectroscopia no infravermelho (FTIR-ATR).
A maior parte dos métodos utilizados foi baseada nos trabalhos de Rodrigues (2008) e
Bueno (2010) e os restantes foram baseados nos trabalhos de da Silva (2009) e Dias et al.
(2013). Outras referências específicas são explicitadas ao longo do texto.
3.4.1. Morfologia
A morfologia das membranas foi avaliada macroscopicamente (utilizando uma câmera
fotográfica modelo DMC-F2, Panasonic) e microscopicamente (utilizando um microscópio ótico
Olympus BH-2 e um microscópio eletrônico de varredura modelo LEO 440 da marca Leica

45
operando a 10kV e 50pA).
Previamente às análises de microscopia óptica, as membranas foram cortadas em corpos de
prova de 2 cm x 2 cm e mantidas em dessecador com sílica por no mínimo 24 horas.
Para as análises de microscopia eletrônica de varredura, as membranas foram cortadas com
tesoura em corpos de prova de 1 cm x 1 cm e mantidas em dessecador com sílica por no mínimo
24 horas. Cortes feitos por criofratura foram também avaliados no início dos ensaios, mas
resultavam em amostras com certo grau de deformação e, portanto, tal abordagem foi
abandonada. Em seguida, os corpos de prova foram recobertos com uma camada ultrafina de
ouro (92Å) em um mini sputter coater (SC 7620, VG Microtech).
3.4.2. Espessura
A espessura das membranas foi medida com auxílio de micrômetro digital (Mitutoya,
modelo MDC-25S, Japão), sendo realizadas 10 medidas em cada formulação. Antes das
medidas, as membranas foram previamente acondicionadas em umidade relativa de 20% por, no
mínimo, 48 horas. A armazenagem prévia em umidade controlada é importante para evitar
variações nos resultados, tendo sido escolhido um baixo valor de umidade relativa (20%) de
modo a evitar que as amostras ficassem excessivamente ressecadas.
3.4.3. Rugosidade
A análise de rugosidade foi realizada utilizando-se um rugosímetro digital Mitutoyo SJ-
201, executando-se 10 leituras em cada filme. Os comprimentos de amostragem (cutoff) e
comprimento de medição total foram programados no equipamento antes da realização das
medidas, sendo tais valores escolhidos de acordo com a recomendação das normas ISO 4288
(1996) e ASME B46.1 (2002) em relação ao valor de rugosidade média Ra (µm). As membranas
foram previamente acondicionadas em umidade relativa de 20% por 48 horas.
3.4.4. Coloração
A avaliação de cor foi realizada a fim de verificar quantitativamente a influência da

46
presença do tensoativo Pluronic F68 sobre o aspecto visual das membranas, uma característica
que poderia ser relevante do ponto de vista de atratividade para potenciais usuários.
As membranas foram previamente acondicionadas em umidade relativa de 20% por
48 horas e a análise foi realizada em triplicata com auxílio de colorímetro portátil Minolta CT-
310, o qual utiliza o padrão CIELAB. Neste sistema, a luminosidade e a cor são determinadas
pelos parâmetros L, a e b. O parâmetro L varia de 0 (preto) a 100 (branco), o parâmetro a varia
de -60 (verde) a +60 (vermelho) e o parâmetro b varia de -60 (azul) a +60 (amarelo). A obtenção
destes parâmetros permitiu calcular a tonalidade métrica (Hue), que indica a cor do material, o
croma métrica (Cr), relacionado à intensidade de cor em uma escala de 0 a 16, e a diferença de
cor em relação a um padrão (ΔE), que no caso do presente trabalho foi definido como a
membrana sem surfactante. Os cálculos foram realizados de acordo com as Equações 1, 2 e 3:
)a/b(arctanHue (1)
22 baCr (2)
222 baLE (3)
3.4.5. Área superficial e porosidade
A área superficial das membranas foi medida por adsorção de N2 (Micrometrics ASAP
2000 V2.04), sendo calculada por meio da equação BET.
A porosidade das membranas foi calculada de acordo com a Equação 4:
sólidoaparente1P (4)
onde aparente (g/cm3) é a densidade aparente (calculada como sendo a razão entre a massa e o
volume de cada amostra) e sólido (g/cm3) é a densidade real medida por picnometria de hélio
(Accupyc 1330 Micromeritics, USA).
Previamente aos ensaios de adsorção de N2 e picnometria de hélio, as membranas foram

47
cortadas em pequenos quadrados de 1 cm x 1 cm e acondicionadas em umidade relativa de 20%
por 48 horas.
3.4.6. Capacidade de absorção e estabilidade das membranas em soluções aquosas
A capacidade de absorção e a estabilidade das membranas foi avaliada em diferentes
soluções aquosas de modo a simular as suas condições de aplicação. Nestes ensaios utilizou-se
água deionizada; solução salina (NaCl a 0,9% m/v, SS), a qual seria potencialmente empregada
para a hidratação das membranas antes de seu uso in vivo; fluido corpóreo simulado (FCS)
(preparado de acordo com Kokubo et al., 1990) e tampão fosfato salino (PBS), os quais são
soluções isotônicas com concentração de sais próxima à do corpo humano; soro fetal bovino
(SFB), o qual simula um ambiente mais complexo e mais próximo ao sangue animal; e meio
RPMI-1640 (0,3 g/L de L-glutamina, 2,0 g/L de D-glicose, 2,0 g/L de NaHCO3, 10000 UI/L de
penicilina e 0,05 g/L de estreptomicina) suplementado com Hepes (5,958 g/L) e soro fetal
bovino (10% v/v), o qual seria potencialmente empregado como meio de cultivo de células da
pele nas membranas.
Os dois ensaios, de análise da capacidade de absorção e da estabilidade, foram realizados
em triplicata, utilizando três corpos de prova para cada solução aquosa. As membranas foram
previamente cortadas em corpos de prova de 6 cm x 1 cm, os quais foram armazenados em
dessecador com sílica por 24 horas para remoção de umidade e, em seguida, sua massa inicial foi
determinada em balança analítica.
Para a determinação da capacidade de absorção, os corpos de prova foram imersos em
10 mL das referidas soluções a 37°C por 24 horas, de forma a atingir o equilíbrio de absorção.
Após este período o excesso de solvente foi removido levemente com papel de filtro por cerca de
5 segundos e a massa final foi imediatamente determinada em balança analítica. A capacidade
máxima de absorção (Ab) foi calculada de acordo com a Equação 5:
s
su
M
)MM(Ab
(5)
onde Mu refere-se à massa da amostra úmida e Ms refere-se à massa da amostra seca.

48
Para a determinação da estabilidade das membranas, expressa como perda de massa de
materiais de formação e estabilização da matriz, os corpos de prova foram imersos nas diferentes
soluções a 37°C por um período prolongado, de uma semana. Após o período de exposição, cada
corpo de prova foi imerso em 20 mL de água deionizada por 5 minutos, por 5 vezes, para
remoção de compostos retidos, mas não ligados, como íons, proteínas e polissacarídeos,
dependendo da solução utilizada. Em seguida, realizou-se a secagem em estufa a 37°C por no
mínimo 72 horas até se atingir massa constante. A perda de massa (Pm) foi determinada por
meio da Equação 6:
100M
)MM(Pm
i
fi
(6)
onde Mi refere-se à massa inicial e Mf refere-se à massa final das amostras após o período de
incubação nas soluções.
3.4.7. Sorção de vapor d’água
A sorção de vapor d’água foi avaliada gravimetricamente, em triplicatas, segundo o
método proposto por Dias et al. (2011). As membranas foram cortadas em corpos de prova de
1 cm x 1 cm e armazenadas por 48 horas em um recipiente contendo solução saturada de LiCl
para manter a umidade relativa em 20% à temperatura ambiente. Em seguida, mediram-se as
massas iniciais dos corpos de prova, os quais foram então transferidos para um recipiente
hermético contendo solução saturada de K2SO4 para manter a umidade relativa em 95% a 32oC.
As massas dos corpos de prova foram medidas periodicamente até completar 9 horas de ensaio.
A capacidade de sorção de vapor (SV) foi calculada de acordo com a Equação 7, onde Mt é
a massa da amostra no tempo t e M0 é a massa inicial da amostra:
100M
)MM(SV
0
0t
(7)

49
3.4.8. Transmissão de vapor d’água
A transmissão de vapor d’água foi avaliada por um método gravimétrico adaptado da
norma ASTM E96-95 (1995) (DIAS et al., 2011). As membranas foram cortadas em corpos de
prova de 1,2 cm x 1,2 cm e acondicionadas em um recipiente hermético contendo solução
saturada de Mg(NO3)2, o qual proporcionou a manutenção da umidade relativa em 50%, à
temperatura ambiente por 48 horas. Em seguida, os corpos de prova foram fixados no topo de
frascos contendo sílica gel, com área disponível para a entrada de vapor d’água igual a
0,6362 cm2, os quais foram armazenados em umidade relativa de 50% a 32oC por 24 horas. As
massas dos frascos foram medidas periodicamente, sendo os ensaios realizados em triplicata.
A transmissão de vapor d’água (TVA) foi calculada de acordo com a Equação 8 onde MVt é
a massa de vapor d’água que permeou através da amostra no tempo t e A é a área da amostra
exposta ao ambiente:
A.t
MVTVA t (8)
3.4.9. Ângulo de contato
As medidas de ângulo de contato foram feitas no equipamento OCA20 (Dataphysics).
Devido à estrutura porosa e irregular da superfície das membranas mais porosas, as mesmas
foram previamente compactadas com auxílio de uma prensa manual e acondicionadas em
umidade relativa de 20% por 48 horas. Os resultados foram obtidos a partir de 20 medidas
independentes.
3.4.10. Propriedades mecânicas
A resistência à tração e o alongamento na ruptura das membranas foram determinados
segundo o método D882 (ASTM, 1995), utilizando-se um texturômetro TA.XT2 (Stable
Microsystems SMD) com célula de carga de 50 N.
As membranas foram cortadas em corpos de prova de 10 cm x 2,54 cm e armazenadas em

50
dessecador com sílica por 24 horas para o controle da umidade.
Os corpos de prova foram então fixados por duas garras corrugadas do equipamento
distantes 5,0 cm entre si, uma das quais foi afastada a uma velocidade de 0,1 cm/s.
A resistência à tração foi calculada dividindo-se a força máxima no rompimento pela área
da seção transversal da amostra, de acordo com a Equação 9:
s
m
A
FRT (9)
onde RT é a resistência à tração (MPa), Fm é a força máxima no rompimento (N) e As é a área da
seção transversal inicial da amostra (m2).
O alongamento na ruptura foi determinado pela Equação 10:
100d
dAR
i
r (10)
onde AR é o alongamento na ruptura, di é o afastamento inicial das garras do equipamento (5 cm)
e dr é o afastamento das garras no momento da ruptura.
3.4.11. Análise térmica simultânea (SDT) (Calorimetria e termogravimetria)
O comportamento térmico das membranas foi determinado no equipamento Simultaneous
Differential Thermal Equipment (TA Q600) utilizando-se panelas de alumínio. As medidas
foram realizadas com amostras com massa de cerca de 7 a 10 mg na faixa de temperatura entre
25 e 700ºC a uma taxa de aquecimento de 10 ºC/min. O equipamento foi calibrado com índio. A
perda de massa e as temperaturas características foram calculadas utilizando-se o software TA
Universal Analysis. Os experimentos foram realizados em duplicata, sendo as membranas
previamente armazenadas em dessecador com sílica por 24 horas para reduzir a umidade
residual.

51
3.4.12. Espectroscopia no infravermelho com transformada de Fourier
A caracterização por espectroscopia no infravermelho (FTIR-ATR) foi realizada em
espectrofotômetro FTIR 4200 (Jasco). As membranas foram previamente cortadas em dimensões
de 1 cm x 1 cm e mantidas em dessecador com sílica por 24 horas. A varredura de absorção foi
realizada de 4000 a 500 cm-1, com resolução de 4 cm-1 e 128 varreduras acumuladas, sendo os
ensaios realizados em triplicata.
3.4.13. Citotoxicidade in vitro indireta
O princípio dos ensaios de citotoxicidade in vitro indireta, de acordo com a norma ISO
10993-5e, consiste na obtenção do extrato das membranas, sendo uma alíquota do extrato
adicionada a uma cultura de células, avaliando-se a proliferação das mesmas na presença dos
componentes extraídos das membranas.
Nestes ensaios, foram utilizadas células L929 (fibroblastos de camundongos) e meio de
cultura RPMI-1640 suplementado com L-glutamina (0,3 g/L), D-glicose (2,0 g/L), NaHCO3
(2,0 g/L), penicilina (10.000 UI/L), estreptomicina (0,05 g/L), Hepes (5,958 g/L) e soro fetal
bovino (SFB) 10%. Para avaliar a proliferação celular, utilizou-se o reagente MTT (3-(4,5-
dimeteiltiazol-2-il)-2,5-difenil tetrazolium brometo), que é clivado somente por células vivas e
metabolicamente ativas, formando um precipitado azul denominado formazan, detectável
espectrofotometricamente.
Primeiramente, obteve-se o extrato das membranas. Para isso, amostras de 0,05 g foram
mantidas em 1 mL de meio de cultura em estufa (Tecnal, TE-399) por 48 horas a 37°C e 5% de
CO2. Em seguida, alíquotas de 100 µL de uma suspensão de 1x105 células/mL em meio de
cultura foram transferidas para uma placa de poliestireno de 96 poços de fundo chato (TPP), que
foi mantida por 24 horas em estufa nas mesmas condições. Após este período, os sobrenadantes
foram retirados, adicionando-se 100 µL do extrato em cada poço, e a placa permaneceu em
estufa por mais 24 horas. Os extratos foram então removidos, lavando-se cada poço duas vezes
com 100 µL de tampão PBS/EDTA. Em seguida, adicionaram-se em cada poço 100 µL de meio
de cultura, etapa seguida pela adição de 10 µL de MTT a 5 mg/mL em tampão PBS/EDTA. A
placa foi então mantida em estufa a 37°C e 5% de CO2 por quatro horas e, após este período,

52
adicionaram-se 100 µL de isopropanol ácido (30 µL de HCl em 10 mL de isopropanol) em cada
poço, visando solubilizar os cristais formazan. A leitura da absorbância de cada poço foi feita em
espectrofotômetro de placas ELISA (Thermo Scientific, Multiskan FC) a 620 nm, após
incubação em estufa a 37°C e 5% de CO2 por uma hora. Utilizou-se uma amostra de látex como
controle positivo e extrato de fragmentos da placa de cultura como controle negativo. O cálculo
da citotoxicidade indireta foi feito de acordo com a Equação 11:
100AbsAbs
AbsAbs1edadicixototiC
branconegativocontrole
brancoamostra
(11)
onde Absamostra é a absorbância da amostra em análise, Abscontrole negativo é a absorbância do
controle negativo e Absbranco é a absorbância do branco (meio de cultura sem células).
Previamente aos ensaios de citotoxicidade, as membranas foram lavadas uma vez em
250 mL de tampão Hepes 10 mM e outra em 500 mL de água deionizada para remover o
surfactante residual.
3.4.14. Atividade antimicrobiana
A atividade antimicrobiana das membranas contra dois tipos de bactérias comumente
presentes em lesões de pele, Staphylococcus aureus e Pseudomonas aeruginosa, foi avaliada por
meio de formação de halo de inibição e da permeação através do material, a fim de verificar a
eficácia dos curativos produzidos. Os ensaios foram realizados na empresa IPEL Itibanyl
Produtos Especiais Ltda (Jarinú, SP).
Para avaliar a formação de halo de inibição, as membranas foram cortadas em corpos de
prova de 1,5 cm x 1,5 cm e hidratadas por 1 minuto em água esterilizada em uma câmara de fluxo
laminar (VLFS 12, Veco). Em seguida, foram colocadas na superfície de placas de Petri de 9 cm
de diâmetro contendo 20 a 25 mL de meio de cultura Ágar Triptona de Soja (Tryptic Soy Agar -
TSA, Oxoid) a 40 g/L em água e previamente inoculado com 1% (v/v) de suspensão de
Staphylococcus aureus contendo 4,2x108 unidades formadoras de colônia (UFC) por mililitro ou
de Pseudomonas aeruginosa contendo 3,9x108 UFC/mL. As placas foram incubadas a 35 ± 2°C

53
em estufa de cultura (BOD, Quimis) por 48 h. Após este período, a formação de uma zona sem
crescimento de células ao redor das membranas, chamada de halo de inibição, foi medida.
Para avaliar a permeação de bactérias através das membranas, cortaram-se corpos de prova
de 2,5 cm x 2,5 cm, os quais foram hidratados em água estéril por 30 segundos no interior da
câmara de fluxo laminar. Em seguida, os corpos de prova foram colocados na superfície de placas
de Petri contendo 20 a 25 mL de meio de cultura TSA solidificado. Adicionaram-se 100 μL de
uma suspensão aquosa de Staphylococcus aureus (4,2x108 UFC/mL) ou Pseudomonas
aeruginosa (3,9x108 UFC/mL) na superfície da amostra e as placas foram incubadas a 35 ± 2 °C
por 48 h. Avaliou-se, em seguida, a presença de colônias que indicassem crescimento bacteriano
no meio de cultura.
3.4.15. Determinação da eficiência de incorporação de PHMB nas membranas
Diferentes métodos de quantificação do PHMB incorporado nas membranas foram
testados visando determinar qual seria mais apropriado para a finalidade proposta, tendo em vista
que os componentes das membranas poderiam atuar como importantes interferentes na análise.
Os métodos testados foram a espectrofotometria no UV-VIS, o método colorimétrico
envolvendo a reação entre níquel e nioxima proposto por Rowhani e Lagalante (2007) e o
método de cromatografia líquida de alta eficiência (HPLC) proposto por Arora et al. (2009). Tais
ensaios estão detalhadamente descritos no Apêndice IV, observando-se que, de forma geral, a
abordagem mais apropriada foi baseada na análise direta da absorbância do antimicrobiano por
espectrofotometria no UV-VIS.
No caso do método de adição direta do agente bioativo à mistura polimérica, computaram-
se as perdas de antimicrobiano nas etapas de reticulação secundária em solução de CaCl2 e nas
etapas de lavagem com água por meio de medidas de absorbância por espectrofotometria no UV-
VIS a 235 nm. Já no caso do método de adsorção às membranas em solução aquosa, realizou-se
a leitura de absorbância a 235 nm da solução de PHMB antes e depois do procedimento de
impregnação.
A eficiência de incorporação (Ef) foi calculada segundo a Equação 12, em que
PHMBadicionado ou inicial é a massa de PHMB adicionado à mistura ou presente inicialmente em
solução, dependendo do método de incorporação, e PHMBincorporado refere-se à massa de PHMB

54
efetivamente incorporado à membrana:
100PHMB
PHMBEf
inicialouadicionado
oincorporad (12)
No caso da incorporação de PHMB mediada por CO2 supercrítico, devido à não
solubilização das membranas em várias soluções com valores de pH extremos e à ineficiência
dos testes de extração do PHMB incorporado, como mencionado nos Apêndices VI e VII, não
foi possível estabelecer um método confiável de quantificação.
3.4.16. Cinética de liberação de PHMB incorporado nas membranas
A cinética de liberação de agentes ativos contidos em dispositivos biomédicos pode ser
analisada através da determinação de sua concentração em um meio líquido extrator de
composição apropriada. O tampão PBS é bastante utilizado para esta finalidade por possuir pH
igual a 7,4, simulando condições fisiológicas, como por exemplo, no caso de uma ruptura da
pele.
Para a realização deste ensaio, corpos de prova de massas conhecidas, contendo ou não
PHMB, foram colocados em um volume pré-estabelecido de PBS conforme descritos nos
subitens que se seguem. Os sistemas contendo as membranas em PBS foram mantidos em estufa
a 37oC sob agitação de 100 rpm por até 48 horas, sendo a leitura de absorbância realizada em
seguida por meio de um espectrofotômetro DU640 (Beckman).
3.4.16.1. Análise da liberação de PHMB incorporado por tecnologia supercrítica
Este método foi empregado para corpos de prova de pequenas dimensões, de 1 cm x 1 cm,
obtidos após as impregnações de PHMB via CO2 supercrítico, e consistiu na montagem de
sistemas de liberação em cubetas de quartzo de 4 mL.
Os corpos de prova foram previamente pesados e colocados em cestos de nylon, os quais
foram colocados na parte superior de cubetas contendo 3,5 mL de PBS. As cubetas foram
mantidas por 48 horas em estufa a 37oC com agitação orbital a 100 rpm. A absorbância do meio

55
de liberação a 235 nm foi medida periodicamente. Utilizou-se como referência membranas sem
PHMB. Os ensaios foram realizados em triplicata.
Destaca-se que este método é recomendado somente para membranas contendo pequenas
quantidades de PHMB incorporado, para as quais não é necessário diluir as soluções antes das
medidas diretas de absorbância.
3.4.16.2. Análise da liberação de PHMB incorporado por adição direta e por adsorção a partir
de soluções aquosas
Neste ensaio, realizado em triplicata, corpos de prova de 4 cm x 4 cm foram colocados em
frascos de vidro contendo 60 mL de PBS, de forma a se ter uma relação entre a massa da
membrana e o volume de PBS próxima da utilizada nos ensaios realizados para as membranas
processadas em condições supercríticas. Os frascos foram mantidos em estufa a 37°C sob
agitação de 100 rpm por até 48 horas. A quantidade de PHMB liberada foi avaliada
periodicamente, pela coleta de 1 mL do meio de liberação seguida de análise de absorbância a
235 nm. Adicionou-se, em seguida, 1 mL de PBS fresco ao frasco. Quando necessário, o volume
de 1 mL retirado da amostra foi diluído apropriadamente em PBS para possibilitar a leitura de
absorbância.
3.5. Análise dos resultados
O software Statistica 7® foi utilizado para analisar os resultados numéricos referentes às
propriedades das membranas, empregando-se o teste de comparação de médias de Tukey com
nível de significância de 5%.
Todos os resultados são expressos como a média das medidas experimentais. Cada valor
médio é acompanhado de seu respectivo erro padrão, o que é indicado para realizar comparações
entre médias de diferentes populações (PAES, 2008).

56
4. RESULTADOS E DISCUSSÃO
Neste capítulo são apresentados os resultados referentes à produção de membranas de
quitosana-alginato na presença ou ausência de acetona. São também mostrados os estudos dos
efeitos das temperaturas de processo e de secagem sobre as características das membranas. Em
seguida, são apresentados os resultados de caracterização das membranas produzidas na ausência
e na presença de diferentes proporções do tensoativo Pluronic F68. Por fim, são apresentados os
resultados dos ensaios de incorporação de PHMB por adição à mistura polimérica, por adsorção
a partir de solução aquosa e mediado por CO2 supercrítico.
Nas Figuras 4.1 e 4.2 são apresentadas as atividades desenvolvidas neste trabalho na forma
de diagramas. O trabalho foi dividido em duas partes principais, sendo que a primeira consiste na
definição das condições de processo e da massa de material empregado na produção das
membranas e a segunda consiste na caracterização detalhada das formulações obtidas e nos
ensaios de impregnação de PHMB por diferentes técnicas.
4.1. Definição das condições mais apropriadas de processamento para a obtenção das
membranas
No trabalho anterior de Bueno (2010), empregou-se um método de produção de
membranas que utilizou os mesmos equipamentos empregados no presente trabalho. O
procedimento consistia na adição controlada de uma solução de quitosana (90 mL de quitosana a
1% m/v em ácido acético 2% v/v misturada a 90 mL de acetona) a 180 mL de uma solução
aquosa de alginato a 0,5% m/v contendo ou não 1% (m/v) dos tensoativos Tween 80 ou Pluronic
F68. A adição foi realizada sob taxa de agitação de 500 rpm, sendo a suspensão obtida
homogeneizada a 1000 rpm por 10 minutos. Realizou-se em seguida a adição de solução de
NaOH a 1 M para ajuste do pH para aproximadamente 5,3, sendo a taxa de agitação mantida por
mais 10 minutos. Adicionou-se então 3,6 mL de CaCl2 a 2% (m/v), mantendo-se a taxa de
agitação por 10 minutos. Durante o processo, a temperatura foi mantida em 25°C. Após a divisão
da mistura em duas placas de poliestireno circulares de 15 cm de diâmetro, a secagem foi
realizada em estufa com circulação de ar a 37°C, sendo o tempo total de secagem de
aproximadamente 24 horas. As etapas posteriores de reticulação e secagem foram realizadas por
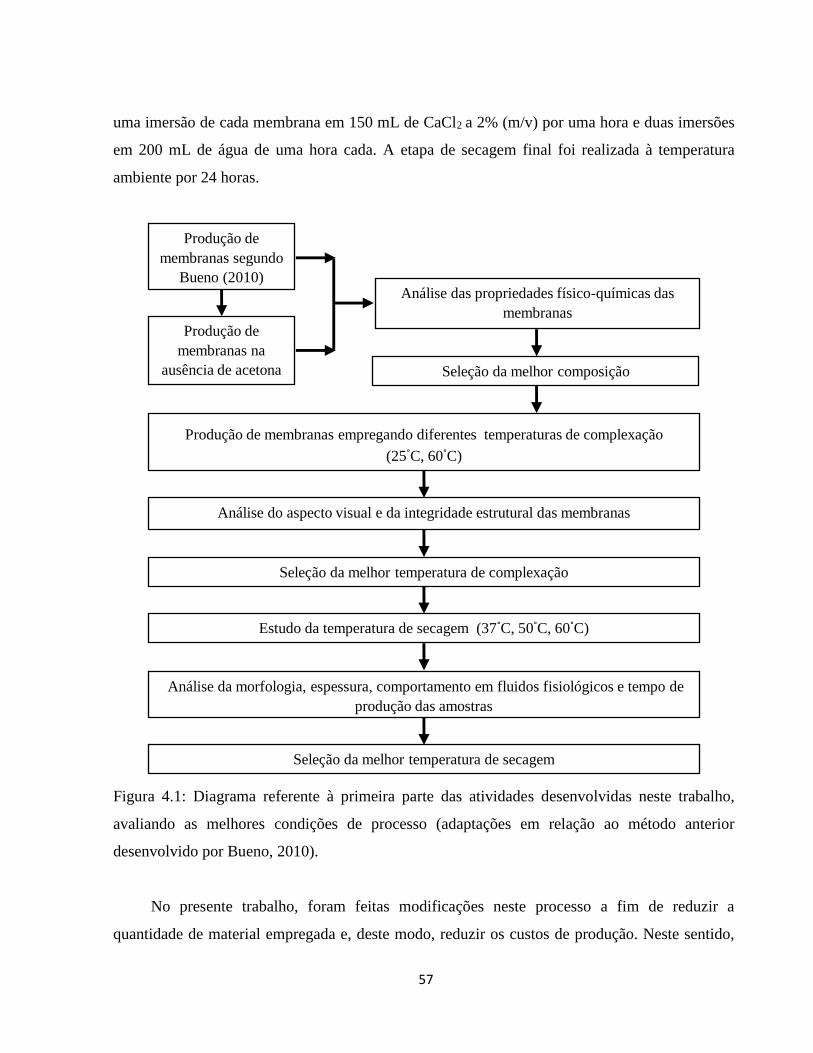
57
uma imersão de cada membrana em 150 mL de CaCl2 a 2% (m/v) por uma hora e duas imersões
em 200 mL de água de uma hora cada. A etapa de secagem final foi realizada à temperatura
ambiente por 24 horas.
Figura 4.1: Diagrama referente à primeira parte das atividades desenvolvidas neste trabalho,
avaliando as melhores condições de processo (adaptações em relação ao método anterior
desenvolvido por Bueno, 2010).
No presente trabalho, foram feitas modificações neste processo a fim de reduzir a
quantidade de material empregada e, deste modo, reduzir os custos de produção. Neste sentido,
Seleção da melhor composição
Produção de
membranas na
ausência de acetona
Análise das propriedades físico-químicas das
membranas
Produção de membranas empregando diferentes temperaturas de complexação
(25°C, 60°C)
Estudo da temperatura de secagem (37°C, 50°C, 60°C)
Produção de
membranas segundo
Bueno (2010)
Seleção da melhor temperatura de complexação
Seleção da melhor temperatura de secagem
Análise do aspecto visual e da integridade estrutural das membranas
Análise da morfologia, espessura, comportamento em fluidos fisiológicos e tempo de
produção das amostras

58
tentou-se produzir membranas na ausência de acetona e a quantidade de tensoativo foi
significativamente reduzida, mas de modo a não prejudicar a formação de poros. A redução da
proporção de tensoativo também teve como meta a redução do potencial citotóxico das
membranas.
Figura 4.2: Diagrama referente à segunda parte das atividades desenvolvidas neste trabalho,
visando à seleção da melhor formulação de membrana em relação às propriedades físico-
químicas e biológicas.
Outra modificação realizada em relação ao trabalho anterior foi a suspensão dos estudos
com o tensoativo Tween 80, que, assim como o Pluronic F68, também era empregado para a
obtenção de membranas porosas. Verificou-se que o Tween 80 possui maior potencial citotóxico
em comparação ao Pluronic F68 (BELLINI et al., 2012; SAKAI et al., 2001; BOGMAN et al.,
2003). Além disso, a maior facilidade de manipulação do Pluronic F68, o qual é um sólido em
Formulações contendo diferentes proporções de Pluronic F68
Caracterização quanto a propriedades físico-químicas e biológicas
Incorporação de PHMB por diferentes métodos:
- Adição direta
- Adsorção
- Com CO2 supercrítico
-Caracterização quanto a propriedades físico-químicas e biológicas
- Análise da eficiência de incorporação e cinética de liberação
Seleção dos dispositivos mais promissores
Caracterização suplementar e seleção dos dispositivos mais
apropriados

59
pó, em comparação ao Tween 80, o qual é viscoso, também foi um fator que contribuiu para a
continuação dos estudos apenas com o surfactante Pluronic F68.
Outras modificações realizadas foram o aumento das temperaturas de processo e de
secagem das membranas. Acredita-se que o aumento da temperatura de processo poderia
contribuir para melhorar a homogeneidade dos coacervados obtidos e evitar a gelificação
localizada, como observado por Da Silva (2009). O aumento da temperatura de secagem, por sua
vez, poderia acelerar o processo de produção. Além disso, com base na análise de dados de
solubilidade do ar em água (HIMMELBLAU, 1960), pode-se supor que o aumento das
temperaturas de processo e secagem contribuiria para a diminuição da solubilidade do ar na
mistura polimérica, reduzindo a quantidade de bolhas, o que é vantajoso na produção de
membranas densas.
Na Tabela 4.1, encontram-se sumarizadas as modificações de processo realizadas neste
trabalho em relação ao trabalho anterior desenvolvido por Bueno (2010).
Tabela 4.1: Modificações no processo de produção das membranas realizadas no presente
trabalho em relação ao trabalho anterior desenvolvido por Bueno (2010).
Alteração Bueno (2010) Presente trabalho
Uso de acetona Sim Não
Proporção de tensoativo
(massa de tensoativo/massa total de polissacarídeos) 1:1 0,02:1 a 0,1:1
Temperatura de processo (°C) 25 25 ou 60
Temperatura de secagem (°C) 37 37, 50, 60
Duração da reticulação secundária em CaCl2 (min) 60 30
Duração de cada lavagem em água (min) 60 30
A seguir são mostrados os resultados obtidos nesta primeira etapa do trabalho referente às
alterações do método de produção das membranas. Os efeitos destas alterações foram analisados
sobre o aspecto visual, espessura, propriedades mecânicas e capacidade de absorção e perda de
massa em fluidos fisiológicos.

60
4.1.1. Efeitos da remoção de acetona da etapa de produção das membranas sobre as
características do material
Uma adaptação importante realizada no método de produção de membranas utilizado por
Bueno (2010) foi a suspensão do uso de acetona como cossolvente da quitosana. Este solvente
orgânico de baixa polaridade atua de forma que a quitosana assuma uma conformação menos
estendida, reduzindo a acessibilidade aos seus grupos reativos e, consequentemente, diminuindo
a taxa de reação entre a quitosana e alginato (YAN et al., 2000). Sabe-se também que a adição
de acetona a uma solução de alginato provoca inicialmente o aumento de viscosidade, seguido de
precipitação (WHISTLER e BEMILLER, 1993).
Na Figura 4.3 são mostrados os aspectos das membranas de quitosana-alginato obtidas na
presença e na ausência de acetona sem adição de tensoativo e à temperatura de secagem de 37°C.
Nota-se que a retirada do cossolvente não afetou significativamente a aparência das membranas.
Figura 4.3: Aspecto visual das membranas sem tensoativo: A) membrana preparada na presença
de acetona, B) membrana preparada na ausência de acetona.
Contudo, as imagens de microscopia eletrônica de varredura (MEV), mostradas na Figura
4.4, mostram que existem diferenças entre as morfologias das membranas preparadas na
presença e na ausência de acetona. Nota-se que, apesar de ambas as formulações apresentarem
seção transversal lamelar, nas obtidas na ausência de acetona, o número de lamelas é reduzido,
aumentando a compactação do material. Observa-se também maior número de agregados de
fibras nas membranas preparadas com acetona.
Verificou-se também que, ao contrário do que foi relatado por Yan et al. (2000), os
coacervados obtidos na presença de acetona são heterogêneos, possuindo aglomerados
A B
3 cm 3 cm

61
poliméricos visíveis a olho nu. Já os coacervados obtidos na ausência de acetona são mais
homogêneos, sem aglomerados visíveis.
Figura 4.4: Microscopia eletrônica de varredura das membranas sem tensoativo: A) membrana
preparada na presença de acetona, B) membrana preparada na ausência de acetona.
Estes resultados seriam consequências diretas da conformação das moléculas dos
polissacarídeos. Notou-se também que várias das propriedades das membranas foram afetadas
pela retirada da acetona, como indicado nas Tabelas 4.2 e 4.3.
Tabela 4.2: Espessura e propriedades mecânicas das membranas sem tensoativo preparadas na
presença e na ausência de acetona.
Propriedade Com acetona Sem acetona
Espessura (µm) 87,0 ± 2,0 a 72,2 ± 7,6 b
Resistência à tração (MPa) 9,8 ± 1,5 a 22,2 ± 4,3 b
Alongamento na ruptura (%) 3,3 ± 1,4 a 3,2 ± 0,4 a
Mesma letra na mesma linha indica que não há diferença significativa entre os valores médios (Teste
de Tukey, p<0,05).
Observou-se que a espessura das membranas produzidas na ausência de acetona é menor,
indicando maior compactação do material, corroborando os resultados observados na Figura 4.4.
Quanto às propriedades mecânicas, as membranas produzidas na ausência de acetona
apresentaram resistência à tração cerca de 2 vezes maior e alongamento na ruptura semelhante às
membranas produzidas na presença de acetona. Estes dados indicam que as membranas sem
acetona possuem vantagens em relação às propriedades mecânicas, apresentando mais
possibilidades de aplicação in vivo.
30 µm 30 µmA B
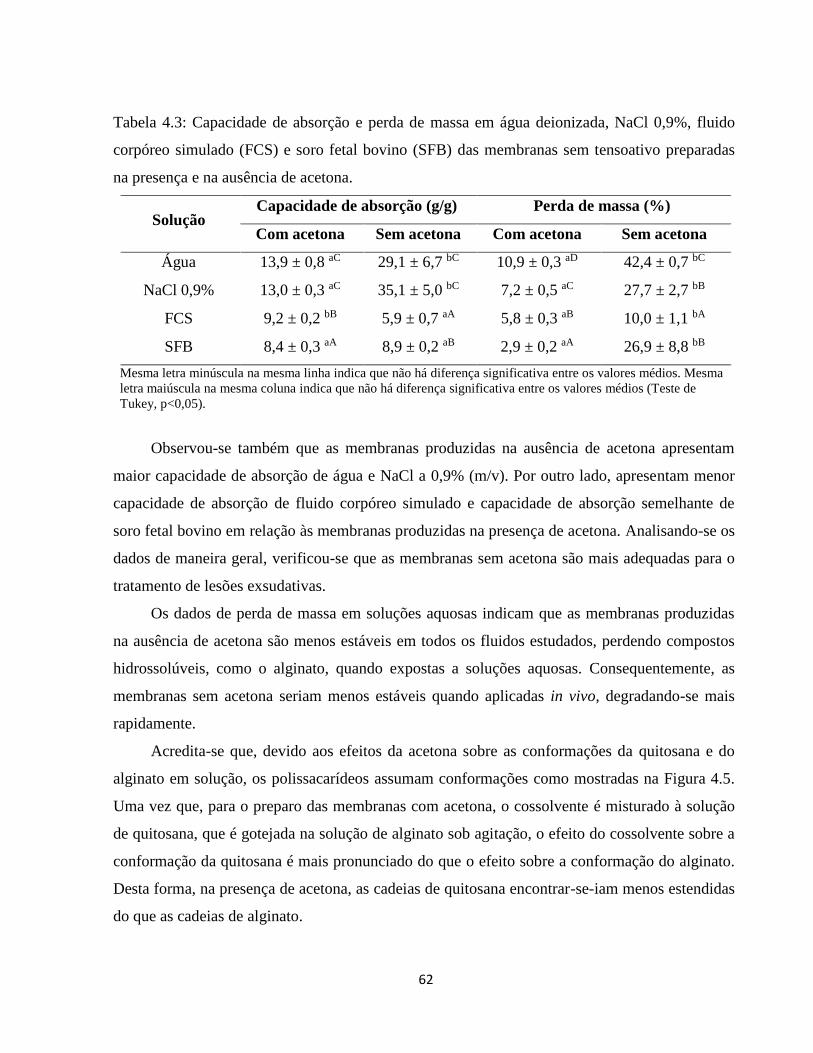
62
Tabela 4.3: Capacidade de absorção e perda de massa em água deionizada, NaCl 0,9%, fluido
corpóreo simulado (FCS) e soro fetal bovino (SFB) das membranas sem tensoativo preparadas
na presença e na ausência de acetona.
Solução Capacidade de absorção (g/g) Perda de massa (%)
Com acetona Sem acetona Com acetona Sem acetona
Água 13,9 ± 0,8 aC 29,1 ± 6,7 bC 10,9 ± 0,3 aD 42,4 ± 0,7 bC
NaCl 0,9% 13,0 ± 0,3 aC 35,1 ± 5,0 bC 7,2 ± 0,5 aC 27,7 ± 2,7 bB
FCS 9,2 ± 0,2 bB 5,9 ± 0,7 aA 5,8 ± 0,3 aB 10,0 ± 1,1 bA
SFB 8,4 ± 0,3 aA 8,9 ± 0,2 aB 2,9 ± 0,2 aA 26,9 ± 8,8 bB
Mesma letra minúscula na mesma linha indica que não há diferença significativa entre os valores médios. Mesma
letra maiúscula na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de
Tukey, p<0,05).
Observou-se também que as membranas produzidas na ausência de acetona apresentam
maior capacidade de absorção de água e NaCl a 0,9% (m/v). Por outro lado, apresentam menor
capacidade de absorção de fluido corpóreo simulado e capacidade de absorção semelhante de
soro fetal bovino em relação às membranas produzidas na presença de acetona. Analisando-se os
dados de maneira geral, verificou-se que as membranas sem acetona são mais adequadas para o
tratamento de lesões exsudativas.
Os dados de perda de massa em soluções aquosas indicam que as membranas produzidas
na ausência de acetona são menos estáveis em todos os fluidos estudados, perdendo compostos
hidrossolúveis, como o alginato, quando expostas a soluções aquosas. Consequentemente, as
membranas sem acetona seriam menos estáveis quando aplicadas in vivo, degradando-se mais
rapidamente.
Acredita-se que, devido aos efeitos da acetona sobre as conformações da quitosana e do
alginato em solução, os polissacarídeos assumam conformações como mostradas na Figura 4.5.
Uma vez que, para o preparo das membranas com acetona, o cossolvente é misturado à solução
de quitosana, que é gotejada na solução de alginato sob agitação, o efeito do cossolvente sobre a
conformação da quitosana é mais pronunciado do que o efeito sobre a conformação do alginato.
Desta forma, na presença de acetona, as cadeias de quitosana encontrar-se-iam menos estendidas
do que as cadeias de alginato.

63
Figura 4.5: Prováveis conformações das moléculas de quitosana e alginato na mistura polimérica
durante o preparo das membranas na presença e na ausência de acetona.
Na presença de acetona, a conformação menos estendida de ambos os polissacarídeos,
principalmente da quitosana, resultaria, devido ao impedimento estérico, em um baixo número
de ligações entre a quitosana e o alginato e numa distribuição polimérica irregular. Desta forma,
a reticulação do complexo com CaCl2 ocorreria de maneira não uniforme, sendo possível
observar vários pontos de gelificação, o que pode ter sido a causa de quebras precoces nos
ensaios de tração. Por outro lado, devido ao baixo número de ligações entre a quitosana e o
alginato, a intensidade de reticulação do alginato com CaCl2 seria alta, o que pode ter aumentado
a estabilidade do complexo em soluções aquosas.
Na ausência de acetona, ter-se-ia um aumento da densidade de interações entre os
polissacarídeos devido à conformação estendida das moléculas, formando coacervados mais
organizados. Desta forma, as membranas obtidas nesta situação seriam mais homogêneas e mais
resistentes mecanicamente. A reticulação com CaCl2, neste caso, ocorreria em menor proporção
em comparação à outra situação.
Considerou-se que as membranas produzidas na ausência de acetona são mais adequadas
devido à sua maior capacidade de absorção de água e solução salina, melhores propriedades
mecânicas, menor potencial citotóxico e menor custo em termos de matéria-prima. Além disso,
uma instalação industrial para a produção deste tipo de material não requer o uso de tanques à
base de materiais especiais e à prova de explosão, e é mais segura ao meio ambiente.
Sendo assim, as membranas de quitosana-alginato deste trabalho passaram a ser
produzidas sem acetona, sendo o procedimento testado também com sucesso para formulações
porosas obtidas na presença de Pluronic F68 nas proporções de 0,02:1 e 0,1:1 (m/m) em relação
Quitosana Alginato Quitosana
Alginato
Com acetona Sem acetona

64
aos polissacarídeos. Notou-se adequada homogeneidade quanto ao aspecto da mistura polimérica
antes da secagem, a qual se apresentou na forma de uma espuma homogênea, e adequada
homogeneidade quanto à aparência final das membranas, como mostrado na Figura 4.6.
Figura 4.6: Aspecto visual das membranas produzidas na presença de Pluronic F68 nas
proporções de 0,02:1 e 0,1:1 (m/m) na ausência de acetona.
4.1.2. Influência da temperatura em diferentes etapas do processamento nas características
das membranas
Nesta etapa do trabalho, estudou-se o efeito das temperaturas de obtenção do PEC e de
secagem sobre algumas propriedades importantes das membranas. Neste estudo, a temperatura
de complexação variou de 25 a 60°C, enquanto a temperatura de secagem variou de 37 a 60°C.
As membranas utilizadas neste estudo foram produzidas na presença de 0,02:1 de Pluronic F68
em relação à massa de polímeros devido à sua condição intermediária em relação à porosidade e
à quantidade de Pluronic F68 em relação às demais formulações.
Na Tabela 4.4 são mostradas as temperaturas de degradação térmica dos compostos de
maior relevância das membranas, o que foi levado em consideração para a escolha das condições
de trabalho.
Da Silva (2009) relatou que o emprego de uma alta temperatura de complexação entre a
quitosana e o alginato, de 70°C, favorece a homogeneização de coacervados. No entanto, no caso
do presente trabalho, no qual a formação de bolhas estáveis é um aspecto importante, o aumento
da temperatura de complexação para 60°C não favoreceu a produção de membranas porosas. Os
aspectos típicos das membranas preparadas a diferentes temperaturas são mostrados na Figura
4.7.
0,02:1 0,1:1
3 cm 3 cm
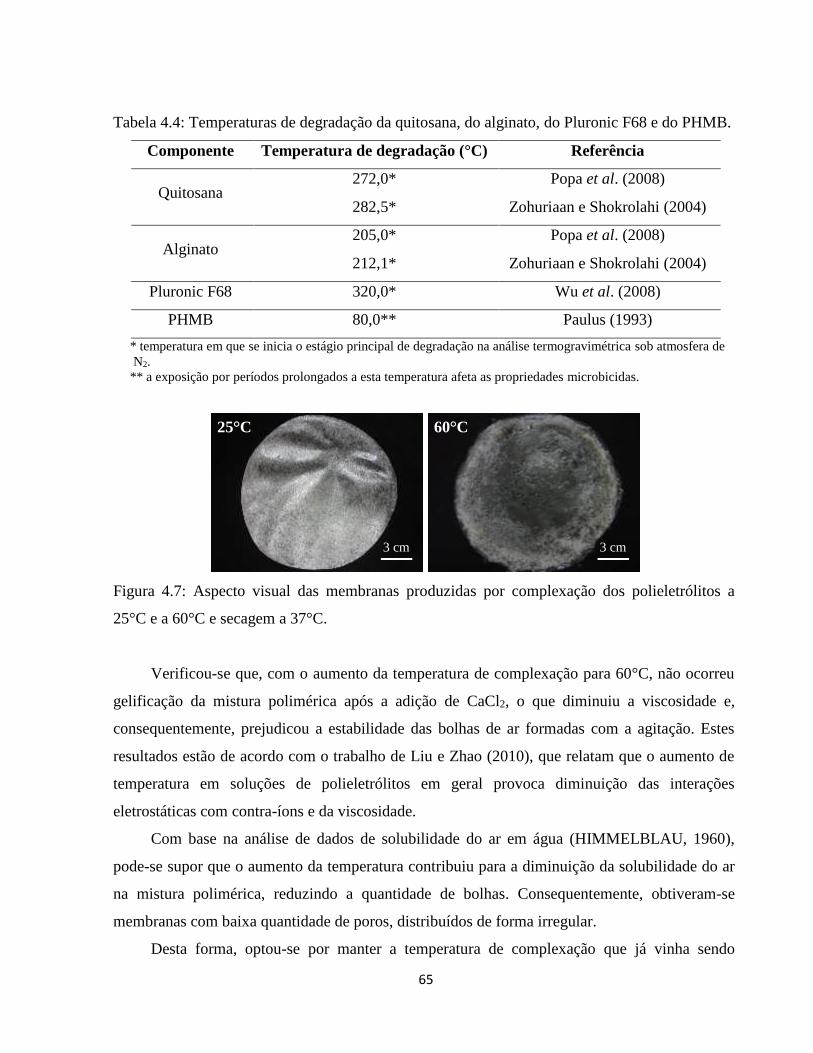
65
Tabela 4.4: Temperaturas de degradação da quitosana, do alginato, do Pluronic F68 e do PHMB.
Componente Temperatura de degradação (°C) Referência
Quitosana 272,0* Popa et al. (2008)
282,5* Zohuriaan e Shokrolahi (2004)
Alginato 205,0* Popa et al. (2008)
212,1* Zohuriaan e Shokrolahi (2004)
Pluronic F68 320,0* Wu et al. (2008)
PHMB 80,0** Paulus (1993)
* temperatura em que se inicia o estágio principal de degradação na análise termogravimétrica sob atmosfera de
N2.
** a exposição por períodos prolongados a esta temperatura afeta as propriedades microbicidas.
Figura 4.7: Aspecto visual das membranas produzidas por complexação dos polieletrólitos a
25°C e a 60°C e secagem a 37°C.
Verificou-se que, com o aumento da temperatura de complexação para 60°C, não ocorreu
gelificação da mistura polimérica após a adição de CaCl2, o que diminuiu a viscosidade e,
consequentemente, prejudicou a estabilidade das bolhas de ar formadas com a agitação. Estes
resultados estão de acordo com o trabalho de Liu e Zhao (2010), que relatam que o aumento de
temperatura em soluções de polieletrólitos em geral provoca diminuição das interações
eletrostáticas com contra-íons e da viscosidade.
Com base na análise de dados de solubilidade do ar em água (HIMMELBLAU, 1960),
pode-se supor que o aumento da temperatura contribuiu para a diminuição da solubilidade do ar
na mistura polimérica, reduzindo a quantidade de bolhas. Consequentemente, obtiveram-se
membranas com baixa quantidade de poros, distribuídos de forma irregular.
Desta forma, optou-se por manter a temperatura de complexação que já vinha sendo
25 C 60 C
3 cm 3 cm

66
utilizada, 25°C, o que além de levar à obtenção de membranas de características mais adequadas,
também é vantajoso do ponto de vista energético e, consequentemente, econômico, uma vez que
os gastos com utilidades para aquecer ou resfriar o reator são menores, por se trabalhar com uma
temperatura mais próxima do ambiente.
Dando continuidade aos estudos, procederam-se os ensaios de análise dos efeitos da
temperatura de secagem das membranas. Verificaram-se reduções significativas dos tempos de
produção em função da temperatura, sendo os tempos de secagem iguais 24 horas para 37°C,
19 horas para 50°C e 13 horas para 60°C.
Na Tabela 4.5 são mostrados os resultados referentes à espessura, absorção e perda de
massa em água e solução salina das membranas obtidas a diferentes temperaturas de secagem.
Na Figura 4.8 são mostrados os resultados da análise de morfologia por microscopia eletrônica
de varredura (MEV).
Tabela 4.5: Espessura, capacidade de absorção e perda de massa das membranas preparadas na
presença de 0,02:1 (m/m) de Pluronic F68:polissacarídeos à temperatura de complexação de
25°C e a diferentes temperaturas de secagem.
T
(˚C)
Espessura
(mm)
Absorção (g/g) Perda de massa (%)
Água NaCl 0,9% Água NaCl 0,9%
37 0,58±0,04 b 36,1±1,5 cB 27,4±0,7 bA 32,3±1,0 cB 17,2±0,6 bA
50 0,49±0,03 b 29,5±0,7 bB 23,4±1,3 bA 27,8±0,9 bB 15,2±1,1 abA
60 0,36±0,03 a 21,5±1,0 aB 17,5±0,9 aA 21,1±0,3 aB 13,5±0,2 aA
Mesma letra minúscula na mesma coluna indica que não há diferença significativa entre os valores médios. Mesma
letra maiúscula na mesma linha indica que não há diferença significativa entre os valores médios. Obs: os valores de
absorção e perda de massa não foram comparados entre si (Teste de Tukey, p<0,05).
Figura 4.8: Microscopia eletrônica de varredura das membranas preparadas na presença de
0,02:1 (m/m) de Pluronic F68 secas a 37, 50 e 60°C.
300 µm37˚C 50˚C 60˚C300 µm 300 µm

67
Os resultados da Tabela 4.5 mostram que, com o aumento da temperatura de secagem, a
espessura, a capacidade de absorção e a perda de massa em água e NaCl a 0,9% (m/v)
diminuíram. Já os resultados da Figura 4.8 mostram que houve diminuição da quantidade de
bolhas retidas no material, obtendo-se membranas mais compactas. É possível supor que estes
resultados se devam à diminuição da solubilidade do ar na mistura polimérica com o aumento da
temperatura de secagem.
A obtenção de um material mais compacto e consequentemente mais estável frente a
soluções aquosas pode ser diretamente relacionada a um maior tempo de aplicação das
membranas como curativos. Além disso, a redução significativa do tempo de produção foi
considerada um resultado bastante positivo do ponto de vista de produção das membranas em
escala industrial. Desta forma, optou-se por continuar os estudos utilizando-se a temperatura de
secagem de 60°C.
Na Figura 4.9 é mostrado o comportamento cinético na condição de secagem a 60°C, no
que se refere à variação de massa com o tempo devido à evaporação dos solventes da mistura
polimérica, constituídos principalmente por água. As maiores taxas de secagem foram
observadas no início do processo.
Figura 4.9: Cinética de evaporação de solventes da membrana produzida na presença de
0,02:1 (m/m) de Pluronic F68 com a secagem a 60°C.
0
20
40
60
80
100
120
140
0 3 6 9 12 15
Mass
a (
g)
Tempo de secagem (h)
Massa final
0,9 g

68
4.2. Caracterização detalhada de membranas obtidas na ausência e na presença de
diferentes proporções de Pluronic F68 nas condições mais adequadas de processamento
Após definir o procedimento de produção das membranas de quitosana-alginato em relação
à ausência de acetona e às temperaturas de complexação (25°C) e de secagem (60°C), foram
produzidas membranas na ausência e na presença de diferentes proporções de Pluronic F68
(0,02:1 e 0,1:1) em relação à massa de polímeros, sendo as proporções selecionadas menores do
que a empregada por Bueno (2010), de 1:1 (m/m), para reduzir seu potencial citotóxico e a
quantidade de material empregada, como explicado anteriormente.
As membranas produzidas nestas condições foram nomeadas P0% (para formulações
produzidas na ausência de tensoativo), P2% (para a razão 0,02:1 m/m de Pluronic F68 em
relação aos polissacarídeos) e P10% (para a razão 0,1:1 m/m). As formulações obtidas foram
caracterizadas detalhadamente.
4.2.1. Aspecto e morfologia
Na Figura 4.10 são mostradas as imagens do aspecto visual das membranas e as imagens
obtidas por microscopia óptica e por microscopia eletrônica de varredura das membranas
preparadas na presença de diferentes quantidades de Pluronic F68. É possível notar o aumento da
porosidade das membranas, assim como de sua espessura, com o aumento da quantidade de
Pluronic F68 empregada.
As imagens obtidas por microscopia óptica mostram a presença de muitas cavidades dentro
das membranas preparadas na presença de Pluronic F68. Aparentemente, as cavidades da
formulação P10% são maiores, distribuídas de forma irregular, e algumas delas apresentam
rupturas nas suas paredes, o que indica que elas podem ter se rompido durante o processo de
secagem na estufa com circulação de ar.
As imagens de MEV indicam que as membranas possuem estrutura lamelar e que os poros
não atravessam integralmente a sua espessura, característica que favorece a aplicação do
biomaterial no tratamento de lesões de pele, uma vez que a penetração de microrganismos na
ferida seria evitada (BUENO e MORAES, 2011; HINRICHS et al., 1992). Verificou-se que a
distribuição de tamanho de poros é irregular nas membranas porosas, observando-se poros de

69
cerca de 300 µm na formulação P2% e de cerca de 900 µm na formulação P10%.
Figura 4.10: Aspecto visual e morfologia das membranas preparadas na ausência e na presença
de diferentes proporções de Pluronic F68: A) aspecto visual, B) microscopia óptica, C)
microscopia eletrônica de varredura (MEV).
A mudança na morfologia das membranas com a inclusão de Pluronic F68 pode ser
explicada pelo alto valor do equilíbrio hidrófilo-lipófilo (HLB) deste surfactante, igual a 29, o
que implica em uma boa capacidade de formação de espuma em meios aquosos. Observou-se
que quanto maior a quantidade de surfactante, maior a retenção de ar durante a agitação
mecânica da mistura polimérica, levando consequentemente ao aumento do tamanho dos poros
do material e ao aumento do espaçamento entre as camadas observadas na estrutura transversal
da membrana. A espuma formada pela mistura polimérica durante o preparo da membrana
tornou-se ainda mais estável após a reticulação de grupos carboxila do alginato, que não
interagiram com os grupos amino da quitosana, com íons Ca2+. Esta espuma apresentou
consistência suficiente para manter a sua estrutura mesmo durante a secagem do material em
estufa.
P0%
P2%
P10%
1 cm
200 m
200 m
200 m
1 cm
1 cm 300 µm
300 µm
300 µm
(A) (B) (C)

70
As concentrações de Pluronic F68 na mistura polimérica durante o preparo das membranas
foram de 0,015 mM para a formulação P2% e de 0,074 mM para a formulação P10%. Levando-se
em consideração a concentração micelar crítica (CMC) do Pluronic F68 em água, de 0,04 mM a
25°C (SIGMA-ALDRICH, 2011), tem-se que, durante o preparo da formulação P2%, trabalhou-
se com uma concentração de surfactante provavelmente abaixo da CMC, enquanto que para a
formulação P10%, trabalhou-se com uma concentração possivelmente acima da CMC. De acordo
com Lee et al. (2014), espumas formadas na presença de surfactantes acima da CMC são mais
estáveis cineticamente do que as espumas formadas abaixo da CMC. No entanto, o valor de CMC
localizado na literatura é válido apenas para água, não tendo sido localizados dados a respeito da
CMC do Pluronic F68 em meios complexos como no caso do presente trabalho.
Acredita-se que o Pluronic F68 interage com a quitosana e o alginato principalmente por
meio de interações hidrofílicas, devido ao alto conteúdo de grupos –OH dos polissacarídeos, o
que faz com que os polímeros assumam uma conformação estendida na solução (HOLMBERG
et al., 2002). De acordo com o seu caráter anfifílico, as moléculas de Pluronic F68 devem se
orientar com as suas cadeias polares (duas cadeias de polioxietileno) voltadas para o interior do
filme aquoso, interagindo com a água ou com os polissacarídeos, e com a sua cadeia apolar (uma
cadeia de polioxipropileno) voltada para o ar contido na bolha formada durante a agitação da
mistura. Enquanto a água é drenada ou evapora do filme líquido da espuma, a integridade
estrutural da mesma é mantida, provavelmente devido à forte associação entre as
macromoléculas e à alta viscosidade do sistema. A estabilidade da espuma também pode ser
atribuída ao aumento da concentração de Pluronic F68 durante a evaporação de água, levando à
diminuição da tensão interfacial (HOLMBERG et al., 2002). Além disso, a drenagem rápida da
água contida nas bolhas pode ter sido dificultada pela presença de Pluronic F68 (CHISTI, 2000),
favorecendo a manutenção de estruturas porosas estáveis durante a secagem.
A forte associação entre o Pluronic F68 e o alginato foi estudada por Stoppel et al. (2011).
Os autores reportaram que as cadeias de Pluronic F68 e alginato podem formar um complexo por
meio de interações majoritariamente do tipo ligações de hidrogênio, o que aumentaria o volume
do polímero em água, levando a um aumento na viscosidade dinâmica da solução. A adição de
5% (m/v) de Pluronic F68 a uma solução de alginato a 1% (m/v) aumenta a viscosidade
dinâmica em 59% e aumenta o tamanho do complexo polimérico em 110% (STOPPEL et al.,
2011).
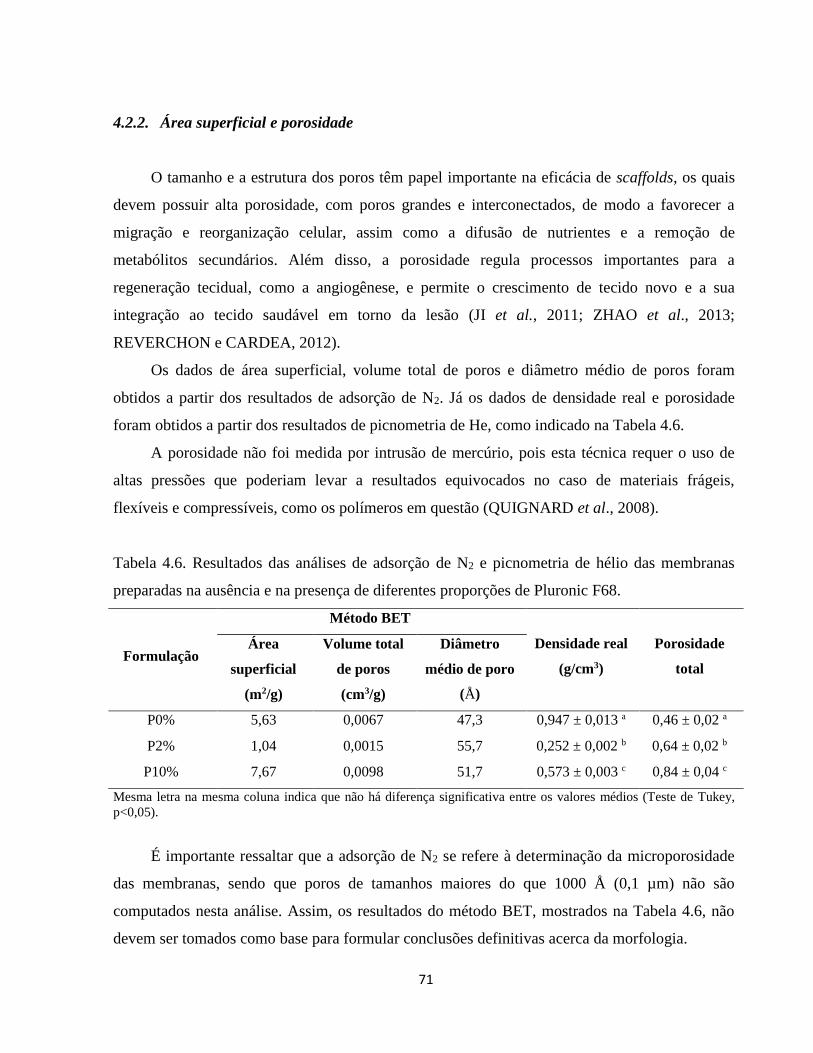
71
4.2.2. Área superficial e porosidade
O tamanho e a estrutura dos poros têm papel importante na eficácia de scaffolds, os quais
devem possuir alta porosidade, com poros grandes e interconectados, de modo a favorecer a
migração e reorganização celular, assim como a difusão de nutrientes e a remoção de
metabólitos secundários. Além disso, a porosidade regula processos importantes para a
regeneração tecidual, como a angiogênese, e permite o crescimento de tecido novo e a sua
integração ao tecido saudável em torno da lesão (JI et al., 2011; ZHAO et al., 2013;
REVERCHON e CARDEA, 2012).
Os dados de área superficial, volume total de poros e diâmetro médio de poros foram
obtidos a partir dos resultados de adsorção de N2. Já os dados de densidade real e porosidade
foram obtidos a partir dos resultados de picnometria de He, como indicado na Tabela 4.6.
A porosidade não foi medida por intrusão de mercúrio, pois esta técnica requer o uso de
altas pressões que poderiam levar a resultados equivocados no caso de materiais frágeis,
flexíveis e compressíveis, como os polímeros em questão (QUIGNARD et al., 2008).
Tabela 4.6. Resultados das análises de adsorção de N2 e picnometria de hélio das membranas
preparadas na ausência e na presença de diferentes proporções de Pluronic F68.
Formulação
Método BET
Densidade real
(g/cm3)
Porosidade
total
Área
superficial
(m2/g)
Volume total
de poros
(cm3/g)
Diâmetro
médio de poro
(Å)
P0% 5,63 0,0067 47,3 0,947 ± 0,013 a 0,46 ± 0,02 a
P2% 1,04 0,0015 55,7 0,252 ± 0,002 b 0,64 ± 0,02 b
P10% 7,67 0,0098 51,7 0,573 ± 0,003 c 0,84 ± 0,04 c
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de Tukey,
p<0,05).
É importante ressaltar que a adsorção de N2 se refere à determinação da microporosidade
das membranas, sendo que poros de tamanhos maiores do que 1000 Å (0,1 µm) não são
computados nesta análise. Assim, os resultados do método BET, mostrados na Tabela 4.6, não
devem ser tomados como base para formular conclusões definitivas acerca da morfologia.

72
Os resultados obtidos indicaram a presença de maiores quantidades de microporos na
formulação P10%, seguida pelas formulações P0% e P2%. Os dados também indicam que, com
o aumento da concentração de Pluronic F68, a área superficial das membranas não seguiu uma
tendência de variação, sendo observado um aumento da área superficial apenas para a
formulação P10% (preparada na presença da maior quantidade de surfactante).
Notou-se que o volume total de poros e o diâmetro médio dos poros também não seguiram
uma tendência de aumento ou diminuição com a proporção de Pluronic F68 empregada no
preparo das membranas. Verificou-se que a formulação P2%, cuja proporção de Pluronic F68 é
intermediária às das demais formulações, apresentou o menor volume total de poros e o maior
diâmetro médio de poros.
Os resultados obtidos por picnometria de He indicam que a formulação P0% é a mais densa,
seguida pelas formulações P10% e P2%, nesta ordem. De acordo com Stoppel et al. (2011), géis
de alginato e Pluronic contraem-se com a gelificação por íons divalentes. A diminuição de
tamanho das partículas de gel aumenta com a concentração de Pluronic F68, resultando no
aumento da densidade de reticulação (STOPPEL et al., 2011). Portanto, o Pluronic F68 poderia
influenciar a densidade real das membranas de quitosana-alginato, sendo este efeito observado
apenas para a formulação preparada na presença da maior quantidade de Pluronic F68 (P10%).
Observou-se uma tendência clara de aumento da porosidade total das membranas com o
aumento da proporção de Pluronic F68, o que está de acordo com os resultados de morfologia
discutidos anteriormente. A literatura referente a substitutos temporários da pele recomenda que
a porosidade dos scaffolds seja maior do que 0,9 (KHAN e AHMAD, 2013; GREAVES et al.,
2013). Neste sentido, a formulação P10% seria a mais indicada para uso como scaffold, uma vez
que seu valor de porosidade se aproxima do valor mínimo recomendado na literatura.
4.2.3. Espessura e rugosidade
Na Tabela 4.7 são mostrados os dados de espessura e rugosidade média (Ra) das
membranas preparadas na presença de diferentes quantidades de Pluronic F68. A análise destes
dados mostra que o aumento da proporção de Pluronic F68 levou a aumentos significativos em
ambas as propriedades, de até 6 e 17 vezes, respectivamente. O aumento da espessura era
esperado, uma vez que maiores quantidades de surfactante levam a maior incorporação de ar no

73
material, como discutido anteriormente. O aumento da rugosidade, por sua vez, seria também
consequência da incorporação de ar, na forma de bolhas, as quais podem ser observadas nas
imagens de MEV (Figura 4.10), como discutido anteriormente.
Tabela 4.7: Espessura e rugosidade média (Ra) das membranas preparadas na ausência e na
presença de diferentes proporções de Pluronic F68.
Formulação Espessura (mm) Ra (µm)
P0% 0,08 ± 0,01 a 1,27 ± 0,08 a
P2% 0,36 ± 0,03 b 17,57 ± 0,88 b
P10% 0,50 ± 0,04 c 21,20 ± 0,74 c
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de Tukey,
p<0,05).
De acordo com Ma et al. (2001), os substitutos poliméricos da derme são geralmente mais
finos do que a pele humana, cuja espessura varia de 0,5 a 2 mm, dependendo da idade, sexo e
região do corpo. Considerando esta informação, pode-se concluir que as membranas produzidas
têm potencial de serem utilizadas como curativos para a pele. A literatura não aponta uma
espessura ideal para scaffolds porosos, uma vez que esta propriedade depende da região do corpo
a ser tratada. Contudo, o uso de scaffolds de espessura menor do que 1 mm é relatado (WALLES
et al., 2003), o que mostra que as membranas produzidas neste trabalho também possuem
potencial para este tipo de aplicação.
A rugosidade de curativos e scaffolds dérmicos influencia a adesão e proliferação celular,
assim como o formato assumido pelas células quando cultivadas sobre uma determinada
superfície. Sendo assim, a rugosidade é um fator importante que determina a biocompatibilidade
de um material (VON RECUM et al., 1996). De acordo com Milleret et al. (2012), quanto mais
rugosa a superfície, maior a adesão de plaquetas e a formação de trombina, que são condições
favoráveis para acelerar a cicatrização de lesões de pele. Também foi reportado que quanto
maior a rugosidade de um curativo de pele, maior o ancoramento entre a superfície e o tecido
necrótico da ferida (BOEHRINGER et al. 2011). Desta forma, quando o curativo é trocado, a
ferida sofre desbridamento, ou seja, a remoção de tecidos desvitalizados, necróticos e de corpos
estranhos, estimulando a formação de tecido novo, apesar de tal processo ser geralmente
associado a dor ou desconforto.

74
Neste trabalho, foram observados valores de rugosidade de 1,3 a 21 µm. O aumento da
rugosidade com a concentração de Pluronic F68 pode ser considerado positivo para as aplicações
propostas, uma vez que valores de rugosidade de até 200 µm são reportados na literatura e
indicados para o tratamento de lesões nos estágios iniciais de cicatrização (BOEHRINGER et
al., 2011).
4.2.4. Coloração
As colorações das membranas preparadas na ausência e na presença de diferentes
proporções de Pluronic F68 foram avaliadas com o objetivo de verificar quantitativamente a
influência do tensoativo sobre o aspecto do material. Os parâmetros de cor obtidos são
mostrados na Tabela 4.8, indicando que a luminosidade das membranas (L) diminui com o
aumento da quantidade de surfactante, o que se reflete no aumento de opacidade.
Tabela 4.8: Parâmetros de cor das membranas preparadas na ausência e na presença de diferentes
proporções de Pluronic F68.
Formulação Parâmetro
L a b Hue (º) Croma ΔE*
P0% 96,1 ± 0,1 a -0,4 ± 0,1 a 1,6 ± 0,1 a 103,8 ± 1,6 a 1,7 ± 0,1 a 0
P2% 67,7 ± 2,1 b -0,6 ± 0,1 a 3,5 ± 0,2 b 100,2 ± 0,9 a 3,6 ± 0,2 b 28,5 ± 2,1 a
P10% 59,9 ± 1,2 c -0,6 ± 0,1 a 3,4 ± 0,1 b 99,6 ±1,3 a 3,5 ± 0,1 b 36,3 ± 1,2 b
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de Tukey,
p<0,05).
*ΔE: em relação à formulação P0%.
Os parâmetros a e b foram utilizados para calcular a tonalidade métrica das membranas
(Hue), as quais não diferiram estatisticamente entre si, ou seja, todas as formulações apresentam
tonalidades semelhantes, tendendo ao amarelo.
A intensidade da cor das membranas, avaliada por meio do parâmetro Croma, cujo valor
máximo pode chegar a 16 (KONICA MINOLTA INC., 2007) aumentou com a inclusão de
surfactante. De maneira geral, observaram-se valores baixos de intensidade de cor para todas as
formulações.
O parâmetro ΔE, que mede a diferença de cor, foi calculado utilizando-se a formulação

75
P0% como padrão de referência. Como era esperado, notaram-se grandes diferenças de cor entre
as membranas porosas e a membrana P0%, a qual é preparada na ausência de Pluronic F68 e
apresenta estrutura mais densa e sem poros.
A princípio, a literatura não recomenda uma cor característica ideal para curativos,
existindo curativos transparentes e opacos. Alguns usuários preferem curativos transparentes, por
possibilitar um melhor monitoramento da cicatrização, o que não indica melhor funcionalidade,
apenas melhor controle. O curativo Hydrofera Blue®, por outro lado, apresenta funcionalidade de
cor, sendo constituído de PVA e dois tipos de pigmento orgânico (azul de metileno e cristal
violeta). Tal biomaterial sofre mudança de cor de azul para branco quando necessita ser trocado
e, em lesões com alto grau de infecção, a mudança de cor ocorre mais rapidamente, devido à
liberação dos pigmentos que funcionam como agentes antimicrobianos.
4.2.5. Hidrofilicidade
A hidrofilicidade de materiais utilizados em contato com a pele é uma característica
importante a ser avaliada, pois os mesmos devem ser capazes de reter e transportar a umidade
em níveis apropriados, de modo a evitar o ressecamento ou a maceração da ferida devido ao
acúmulo de líquido. Além disso, a hidrofilicidade influencia os mecanismos de adesão,
crescimento e migração celular (TANODEKAEW et al., 2004). Tendo em vista a importância
desta propriedade, o grau de hidrofilicidade do material obtido foi analisado avaliando-se seu
desempenho em presença de umidade e quanto ao ângulo de contato.
Na Figura 4.11 são mostradas as cinéticas de sorção de vapor d’água das membranas
produzidas na presença de diferentes quantidades de Pluronic F68. Foram observadas taxas de
sorção maiores nas primeiras horas de ensaio, sendo o equilíbrio atingido em torno de 5 horas
para as formulações P0% e P2% e 7 horas para a formulação P10%. A cinética de sorção da
formulação P0% aparenta ter uma pequena queda em torno de 8 horas, no entanto, devido às
dimensões das barras de erro, considerou-se que não houve diminuição da quantidade de vapor
sorvido pela membrana.
As capacidades de sorção de vapor das membranas produzidas foram de 15,5 ± 1,3% para
a formulação P0%, 22,5 ± 1,7% para a formulação P2% e 36,5 ± 0,3% para a formulação P10%.
Assim, observou-se que as membranas mais porosas apresentam maior capacidade de sorção de

76
vapor, o que está de acordo com seu maior espaço disponível para reter moléculas de água.
Figura 4.11: Cinética de sorção de vapor em UR igual a 95% e 32°C das membranas preparadas
na ausência e na presença de diferentes proporções de Pluronic F68 (amostras previamente
acondicionadas em UR igual a 20% a temperatura ambiente).
Em comparação com a literatura, Dias et al. (2011) observaram capacidades de sorção de
vapor que variaram de cerca de 30 a 50% para curativos à base de N-carboxibutilquitosana e
agarose, valores considerados desejáveis para curativos comerciais. Já a capacidade de sorção de
vapor de curativos à base de colágeno e celulose, N-carboxibutilquitosana e ácido hialurônico
foram de 57%, 29% e 36%, respectivamente, e estes valores também foram considerados
convenientes (DIAS et al., 2013).
Dado que capacidades de sorção de vapor entre 29 e 57% são consideradas como
adequadas para o tratamento de lesões de pele, a formulação P10% produzida neste trabalho
seria a mais adequada quanto a esta propriedade.
Na Tabela 4.9 são mostrados os valores de taxa de transmissão de vapor d’água (TVA) e
ângulo de contato das membranas com água. Nota-se que, assim como observado para a sorção
de vapor, a adição do tensoativo resultou no aumento da TVA das membranas, devido à sua
estrutura mais porosa. Em comparação com a literatura, filmes de quitina apresentam taxa de
transmissão de vapor de 600 g/m2.dia, similar a filmes comerciais de poliuretano
(MUZZARELLI, 2009), valor comparável à formulação P10%. Por outro lado, nanofibras
0
10
20
30
40
0 2 4 6 8 10
So
rçã
o (
%)
Tempo (h)
P0% P2% P10%

77
antimicrobianas de compósito de acetato de celulose e poliéster uretano apresentam taxa de
transmissão de vapor de 204 g/m2.dia, valor próximo ao da pele normal (LIU et al., 2012).
Tabela 4.9: Taxa de transmissão de vapor em UR igual a 50% e 32°C e ângulo de contato das
membranas preparadas na ausência e na presença de diferentes proporções de Pluronic F68.
Formulação TVA (g/m2.dia) Ângulo de contato (°)
P0% 237,0 ± 7,8 a 62,5 ± 2,9 a
P2% 481,5 ± 31,3 b 63,2 ± 2,7 a
P10% 576,2 ± 29,0 b 67,0 ± 3,2 a
Mesma letra na mesma coluna indica que não há diferença significativaentre os valores médios (Teste de
Tukey, p<0,05).
Shah et al. (2010) recomendam que a TVA de curativos esteja na faixa de 300 a
3000 g/m2.dia, dependendo do tipo da lesão e da fase de cicatrização em que a mesma se
encontra. De acordo com Lee et al. (2012), queimaduras de primeiro grau e feridas em estágio de
granulação apresentam taxas de transmissão de vapor de 279 g/m2.dia a 5138 g/m2.dia,
respectivamente, o que é uma variação considerável e que deve ser levada em consideração na
seleção do curativo.
Desta forma, tem-se que as membranas produzidas neste trabalho apresentam valores de
TVA adequadas à finalidade proposta, sendo mais indicadas para o tratamento de lesões com
baixa liberação de exsudato.
Os ângulos de contato obtidos indicam que as membranas apresentam caráter hidrofílico,
uma vez que são menores do que 90º (FÖRCH et al., 2009). Verificou-se que a presença de
tensoativo não alterou significativamente a hidrofilicidade do material em si, o que indica que as
composições químicas das membranas são semelhantes devido à remoção total ou parcial do
tensoativo durante as etapas de reticulação secundária e lavagens em água. Desta forma, a
inclusão de Pluronic F68 afetaria fundamentalmente a morfologia, de modo a aumentar o
distanciamento das lamelas das membranas, como observado nas análises de porosidade e
microscopia. Os efeitos da morfologia sobre os resultados do ângulo de contato foram
minimizados com a compactação das membranas previamente às medidas.
Um valor de ângulo de contato similar ao obtido neste trabalho, de 65,2º, foi reportado
para curativos à base de quitosana e PVA (KANG et al., 2010), enquanto valores maiores,
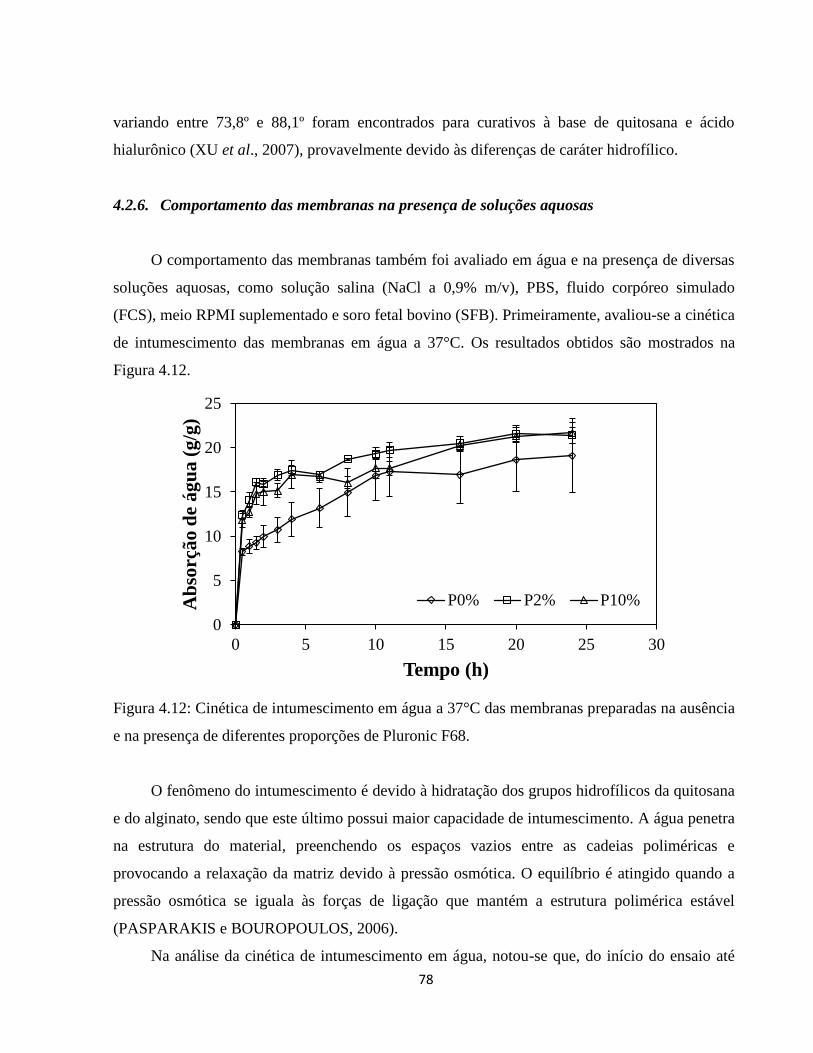
78
variando entre 73,8º e 88,1º foram encontrados para curativos à base de quitosana e ácido
hialurônico (XU et al., 2007), provavelmente devido às diferenças de caráter hidrofílico.
4.2.6. Comportamento das membranas na presença de soluções aquosas
O comportamento das membranas também foi avaliado em água e na presença de diversas
soluções aquosas, como solução salina (NaCl a 0,9% m/v), PBS, fluido corpóreo simulado
(FCS), meio RPMI suplementado e soro fetal bovino (SFB). Primeiramente, avaliou-se a cinética
de intumescimento das membranas em água a 37°C. Os resultados obtidos são mostrados na
Figura 4.12.
Figura 4.12: Cinética de intumescimento em água a 37°C das membranas preparadas na ausência
e na presença de diferentes proporções de Pluronic F68.
O fenômeno do intumescimento é devido à hidratação dos grupos hidrofílicos da quitosana
e do alginato, sendo que este último possui maior capacidade de intumescimento. A água penetra
na estrutura do material, preenchendo os espaços vazios entre as cadeias poliméricas e
provocando a relaxação da matriz devido à pressão osmótica. O equilíbrio é atingido quando a
pressão osmótica se iguala às forças de ligação que mantém a estrutura polimérica estável
(PASPARAKIS e BOUROPOULOS, 2006).
Na análise da cinética de intumescimento em água, notou-se que, do início do ensaio até
0
5
10
15
20
25
0 5 10 15 20 25 30
Ab
sorç
ão
de
ág
ua
(g
/g)
Tempo (h)
P0% P2% P10%

79
cerca de 6 horas, as formulações preparadas na presença de Pluronic F68 apresentaram maiores
capacidades de absorção de água (de 13,2 g/g para a formulação P0%, 16,9 g/g para a P2% e
16,7 g/g para a P10%). A partir de cerca de 10 horas, as capacidades de absorção tornaram-se
mais próximas e, ao final de 24 horas, não foram mais observadas diferenças estatísticas entre as
capacidades de absorção (de 19,1 g/g para a formulação P0%, 21,4 g/g para a P2% e 21,7 g/g
para a P10%). Para todas as membranas, ocorreu a formação de um platô em torno de 20 horas, o
que indica que é recomendável que a troca do curativo aplicado sobre uma lesão exsudativa seja
realizada neste intervalo.
Na Figura 4.13 são mostrados os resultados referentes à capacidade de absorção destas
membranas em diferentes meios aquosos após 24 horas de imersão, o qual foi considerado como
tempo suficiente para se atingir o estado de equilíbrio.
Figura 4.13: Capacidade de absorção após 24 horas a 37°C em água deionizada, solução salina
de NaCl a 0,9% (SS), tampão fosfato salino (PBS), meio RPMI-1640 suplementado (RPMI),
fluido corpóreo simulado (FCS) e soro fetal bovino (SFB) das membranas preparadas na
ausência e na presença de diferentes proporções de Pluronic F68 (os sinais * foram utilizados
para indicar valores estatisticamente semelhantes, entre grupos específicos, de acordo com o
teste de Tukey, p<0,05).
0
5
10
15
20
25
30
**********
****
******
****
****
**
*
**
*
**
**
**
Abs
orçã
o (g
/g)
Água SS PBS RPMI FCS SFB
P0%
P2%
P10%
*

80
Em geral, as membranas produzidas neste trabalho apresentaram maior capacidade de
absorção em água, seguido por solução salina (NaCl a 0,9% m/v) e tampão PBS, os quais não
apresentaram comportamento significativamente diferentes na maioria dos casos. A capacidade
de absorção em meio RPMI suplementado foi intermediária em comparação aos demais fluidos.
As menores capacidades de absorção foram observadas para fluido corpóreo simulado (FCS) e
soro fetal bovino (SFB).
Sabe-se que a presença de íons em todos os fluidos (com exceção da água) pode induzir à
blindagem de cargas dos polissacarídeos, reduzindo a repulsão eletrostática entre as cadeias
poliméricas, originando uma estrutura mais empacotada e menos hidrofílica, com menor
capacidade de absorção de líquidos (MAURSTAD et al., 2008; BARTKOWIAK, 2002;
STRAND et al., 2001). Além disso, o gradiente de força iônica no caso das soluções salinas é
maior, o que reduz o fluxo de água para dentro da matriz polimérica. Não se observou correlação
direta entre os dados capacidade de absorção e o pH das soluções.
Notou-se que as capacidades de absorção de água e meio RPMI suplementado não
diferiram estatisticamente entre as diferentes formulações, indicando que, independentemente da
porosidade, as membranas absorvem quantidades semelhantes, o que não era esperado. Já para
os demais fluidos, notou-se que as membranas mais porosas (P2% e P10%) apresentaram
maiores capacidades de absorção em comparação à formulação P0%.
As membranas estudadas apresentaram, em geral, maiores valores de capacidade de
absorção em comparação aos dados anteriores reportados por Bueno (2010), que foram de
13,83 g/g em água, 11,96 g/g em NaCl a 0,9%, 7,74 g/g em FCS e 7,95 g/g em SFB, para
membranas preparadas com Pluronic F68 a 1:1 (m/m) em relação à massa de polissacarídeos.
Os valores de capacidade máxima de absorção de água obtidos nestes ensaios são
comparáveis aos curativos de quitosana e alginato produzidos por Rodrigues et al. (2008), que
absorveram de 11 a 19 g/g. Membranas de quitosana e xantana na proporção mássica de 1:1, por
outro lado, podem apresentar cerca do dobro da capacidade de absorção de água observada neste
trabalho, variando de 16 a 40 g/g. Além disso, tais membranas apresentam também diferenças
em relação ao comportamento de hidratação, sendo que a absorção de água ocorre ao longo de
todo o ensaio (24 horas), não se atingindo um platô (VEIGA e MORAES, 2012). Este
comportamento pode ser explicado pelas diferentes composições das membranas e pelo fato de
que as membranas de quitosana-xantana não foram reticuladas com cálcio. Altos valores de

81
capacidade de absorção de água também foram reportados por Tanodekaew et al. (2004) para
um hidrogel de β-quitina e poliácido acrílico, que variaram de 30 a 60 g/g, dependendo do
conteúdo de ácido acrílico. A cinética de intumescimento destes curativos foi estudada ao longo
de 7 dias, atingindo-se o equilíbrio de absorção de água após 3 a 4 dias.
Membranas de quitosana-xantana apresentaram menor capacidade de absorção em meio
RPMI suplementado após um período de incubação de 72 horas (8,6 g/g) (BELLINI et al.,
2012), o que mostra que as membranas obtidas no presente trabalho têm maior possibilidade de
aplicação para cultivo celular em comparação às membranas de quitosana-xantana, permitindo
maior fornecimento de nutrientes às células no interior do scaffold. As membranas de quitosana-
xantana apresentaram diâmetro de poro variando entre 90 e 300 um, enquanto que as membranas
de quitosana-alginato obtidas neste trabalho apresentaram diâmetro de poro muitas vezes
maiores do que 300 µm, como observado nas análises de MEV. Assim, as diferenças entre as
capacidades de absorção destas diferentes membranas podem ser devidas às suas diferentes
arquiteturas.
Na Figura 4.14 são mostrados os resultados referentes à perda de massa das membranas em
diferentes meios aquosos após 7 dias a 37ºC. Notou-se que, de modo geral, ocorreu maior
desestabilização das membranas quando imersas em água em comparação às outras soluções
contendo sais e outros componentes eletricamente carregados (como proteínas, no caso do SFB).
Como discutido anteriormente, a blindagem de cargas dos polissacarídeos promovida pelos íons
em solução faz com que as membranas assumam uma conformação mais empacotada, que
dificulta a penetração de água na estrutura do material (MAURSTAD et al., 2008;
BARTKOWIAK, 2002; STRAND et al., 2001), melhorando, consequentemente, a estabilidade
das membranas. Assim, apesar da possibilidade de ocorrência de troca iônica entre os íons Na+
da solução e Ca2+ provenientes das etapas de reticulação, as membranas não apresentaram maior
degradação nas soluções salinas contendo Na+. Também não se observaram grandes perdas de
massa em PBS, o qual poderia quelatar os íons Ca2+, supondo-se, com isso, que a intensidade da
interação das carboxilas do alginato com as aminas da quitosana e os íons cálcio introduzidos
nas etapas de reticulação era elevada o suficiente para estabilizar a estrutura da matriz quando de
sua exposição às soluções aquosas.
As análises de estabilidade das membranas indicaram que a formulação P10%, que
possui o maior valor de porosidade (0,84) apresentou maiores perdas de massa em quase todas as
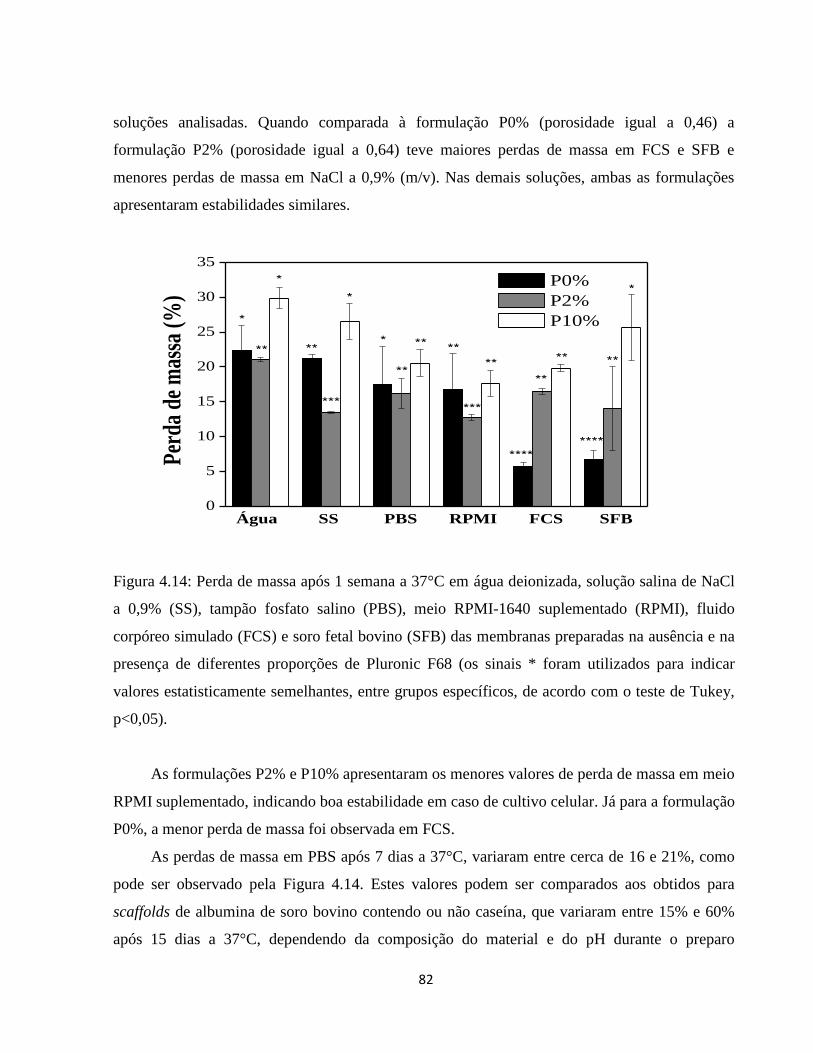
82
soluções analisadas. Quando comparada à formulação P0% (porosidade igual a 0,46) a
formulação P2% (porosidade igual a 0,64) teve maiores perdas de massa em FCS e SFB e
menores perdas de massa em NaCl a 0,9% (m/v). Nas demais soluções, ambas as formulações
apresentaram estabilidades similares.
Figura 4.14: Perda de massa após 1 semana a 37°C em água deionizada, solução salina de NaCl
a 0,9% (SS), tampão fosfato salino (PBS), meio RPMI-1640 suplementado (RPMI), fluido
corpóreo simulado (FCS) e soro fetal bovino (SFB) das membranas preparadas na ausência e na
presença de diferentes proporções de Pluronic F68 (os sinais * foram utilizados para indicar
valores estatisticamente semelhantes, entre grupos específicos, de acordo com o teste de Tukey,
p<0,05).
As formulações P2% e P10% apresentaram os menores valores de perda de massa em meio
RPMI suplementado, indicando boa estabilidade em caso de cultivo celular. Já para a formulação
P0%, a menor perda de massa foi observada em FCS.
As perdas de massa em PBS após 7 dias a 37°C, variaram entre cerca de 16 e 21%, como
pode ser observado pela Figura 4.14. Estes valores podem ser comparados aos obtidos para
scaffolds de albumina de soro bovino contendo ou não caseína, que variaram entre 15% e 60%
após 15 dias a 37°C, dependendo da composição do material e do pH durante o preparo
0
5
10
15
20
25
30
35
********
******
****
**
**
****
**
****
*
*
*
*
Água SS PBS RPMI FCS SFB
Per
da d
e m
assa
(%
)
P0%
P2%
P10%*

83
(RIBEIRO et al., 2012). Já os valores de capacidade de absorção observados para os mesmos
scaffolds após 24 h a 37ºC foram bem menores (variando entre 0,075 a 0,16 g/g) do que os
obtidos no presente trabalho (variando de 13,39 a 19,79 g/g).
Verma et al. (2012) prepararam filmes de quitosana-alginato destinados a serem usados
como barreiras anti-adesivas pós neurocirurgia. Os filmes foram obtidos pela adição controlada
de uma solução de quitosana a 1% (m/v) a uma solução de alginato a 1% (m/v), seguida por uma
etapa de sonicação da mistura e secagem com ar. As amostras contendo quitosana e alginato na
proporção de 1:1 absorveram uma quantidade de água significativamente menor, de 1,1 g/g, do
que as membranas obtidas neste trabalho na ausência ou na presença do tensoativo Pluronic F68,
o que pode ser atribuído às várias diferenças do método de preparo utilizado pela equipe de
Verma et al., por exemplo, a etapa de sonicação, a qual pode ter modificado a geometria do PEC.
A pequena capacidade de absorção de água desse material está diretamente relacionada com a
sua alta estabilidade em PBS a 37°C por até um mês, sendo que após este período não se
notaram perdas de massa.
Como conclusão das análises de capacidade de absorção e perda de massa das membranas
em diferentes soluções aquosas, pode se assumir que a formulação P0% é adequada para
aplicações que requeiram estabilidade do biomaterial, por exemplo, no caso de um curativo que
deve permanecer em contato com a pele por um longo período, enquanto as formulações P2% e
P10% seriam mais adequadas para aplicações que requeiram alta capacidade de absorção de
fluidos, por exemplo, no caso de feridas exsudativas, ou ainda, para aplicações em que o aspecto
poroso seja desejável para permitir crescimento celular, como no caso dos scaffolds, sendo
desejável também que o material apresente taxa de degradação adequada quando em contato
com fluidos fisiológicos.
4.2.7. Propriedades mecânicas
Na Tabela 4.10 são mostrados os resultados referentes à resistência à tração (RT) e ao
alongamento na ruptura (AR) das membranas preparadas na presença de diferentes quantidades
de Pluronic F68. As análises foram realizadas com membranas secas.
Apesar da condição de uso das membranas como curativos ser no estado úmido, não foi
possível realizar os ensaios mecânicos com amostras intumescidas, pois ocorre perda da solução

84
de hidratação no momento de compressão dos extremos das membranas pelas garras de fixação
do equipamento, o que também foi observado por Rodrigues (2008). Vale ressaltar que os
valores obtidos para as propriedades mecânicas das membranas no estado seco não diferem
significativamente dos valores obtidos em umidades relativas maiores, variando até 75% (DE
SOUZA, 2014).
Tabela 4.10: Resistência à tração e alongamento na ruptura das membranas preparadas na
ausência e na presença de diferentes proporções de Pluronic F68.
Formulação RT (MPa) AR (%)
P0% 31,1 ± 1,7 b 4,0 ± 0,2 b
P2% 3,1 ± 0,3 a 3,6 ± 0,2 b
P10% 1,1 ± 0,1 a 2,0 ± 0,1 a
Mesma letra na mesma coluna indica que não há diferença significativaentre os valores médios
(Teste de Tukey, p<0,05).
Nota-se que as membranas preparadas na ausência de Pluronic F68 apresentaram um valor
significativamente maior de resistência à tração. Já a resistência à tração das demais formulações
não diferiu significativamente entre si. Esta diminuição do valor da RT já era esperada, uma vez
que as membranas preparadas com Pluronic F68 são porosas e apresentam estrutura mais frágil.
Quanto ao alongamento na ruptura, observou-se uma diminuição gradual proporcional ao
aumento da quantidade de Pluronic F68. No entanto, esta diminuição não foi tão significativa
quanto a diminuição da resistência à tração.
Nota-se também que as membranas P0% secas a 60°C apresentam propriedades mecânicas
superiores às membranas preparadas na ausência de acetona secas a 37°C (resistência à tração de
22,2 MPa e alongamento na ruptura de 3,2%), como mostrado na Tabela 4.2. Esta diferença pode
ser explicada pela maior compactação das membranas secas a 60°C, como mencionado no item
4.1.1.
As propriedades mecânicas encontradas no presente trabalho são superiores às
propriedades mecânicas das membranas do trabalho anterior (BUENO, 2010; BUENO e
MORAES, 2011), as quais foram preparadas com os mesmos polissacarídeos e surfactante, mas
de acordo com outro protocolo experimental, apresentando resistência à tração e alongamento na
ruptura de 0,98 MPa e 1,96%, respectivamente. Desta forma, as modificações realizadas no

85
processo de produção em relação ao trabalho anterior não apenas facilitaram a produção das
membranas e reduziram o custo, como também melhoraram significativamente as propriedades
mecânicas.
As propriedades mecânicas desejáveis para curativos de pele dependem da região do corpo
a ser tratada, não havendo um consenso geral a respeito dos valores ideais. Além disso, deve-se
considerar a provável necessidade de aplicação das membranas como curativos primários em
conjunto com curativos secundários, como um adesivo ou uma gaze, por exemplo, o que afetaria
significativamente as propriedades mecânicas. Em adição, faz-se necessária a hidratação das
membranas antes da sua aplicação in vivo. De acordo com Rodrigues (2008), quando hidratadas
por 30 minutos, as membranas de quitosana-alginato apresentam alongamento na ruptura cerca
de 5 vezes maior, porém sua resistência à tração diminui em cerca de 28 vezes.
Caso as membranas sejam aplicadas como substitutos dérmicos, como no caso dos
scaffolds, é necessário fazer uma comparação entre as suas propriedades mecânicas com as da
pele humana. Tem-se assim que, com exceção da formulação P10%, os valores de resistência à
tração das membranas produzidas no presente trabalho, quando no estado seco, são comparáveis
ao da pele normal, que varia de 2,5 a 16 MPa (WANG et al., 2002). Contudo, os valores de
alongamento na ruptura das membranas são bastante inferiores ao da pele normal, que é de cerca
de 70% (HANSEN e JEMEC, 2002).
A melhoria das propriedades mecânicas poderia ser conseguida por meio da adição de
agentes plastificantes, como por exemplo, o glicerol. Contudo, no trabalho de Rodrigues (2008),
a adição deste plastificante à mistura polimérica de quitosana e alginato não gerou resultados
satisfatórios, uma vez que o glicerol foi removido das membranas durante as etapas de
reticulação secundária e lavagens com água. Sendo assim, este plastificante seria melhor
empregado como meio de estocagem das membranas, prevenindo quebras e encolhimento do
material (RODRIGUES, 2008).
Outro plastificante já estudado pelo grupo de pesquisa é o agente siliconado Silpuran®
A/B, cuja adição à mistura polimérica de quitosana e alginato na proporção de 10% (m/m)
resultou em membranas com boa resistência à tração, de até 60 MPa, contudo, este não
aumentou o alongamento na ruptura, que se manteve entre 3 e 5% (PIRES, 2013).
Uma estratégia ainda não testada para se obter membranas de quitosana-alginato com
melhores propriedades mecânicas é o uso de ácidos alternativos ao ácido acético para o preparo

86
da solução de quitosana. Na literatura existem relatos sobre a obtenção de membranas de
quitosana com diferentes propriedades de acordo com o ácido utilizado na solução de partida.
Chen et al. (2007) testaram a solubilização de quitosana a 3% (m/m) em diferentes ácidos
(0,2 M), obtendo membranas porosas por liofilização das soluções. O uso de ácido ascórbico
(C6H8O6) aumentou a resistência à tração em aproximadamente 100% e a capacidade de
absorção de água em 50% em comparação à da membrana preparada na presença de ácido
acético. Já o ácido glicólico (C2H4O3) aumentou em 15% a resistência à tração e em 90% a
absorção de água das membranas. A melhora das propriedades mecânicas obtidas com o uso de
ácido ascórbico pode ter ocorrido devido à sua oxidação, que ocasionou o surgimento de três
grupos carbonila na molécula de ácido. Estes grupos carbonila podem ter reagido com os grupos
amino ou hidroxila das cadeias de quitosana, reticulando a membrana (CHEN et al., 2007).
Kucharska et al. (2008) prepararam esponjas de quitosana-alginato por liofilização, as
quais apresentaram menor resistência à tração do que as membranas obtidas neste trabalho, ao
redor de 0,6MPa (valor estimado a partir da tenacidade fornecida pelos autores). Por outro lado,
o alongamento na ruptura encontrado por estes autores foi maior, de 10%. Lai et al. (2003)
também produziram esponjas de quitosana-alginato que apresentaram alongamento na ruptura
comparável aos valores encontrados neste trabalho, sendo este de 3% para uma proporção de
1:1 (m/m) entre os polissacarídeos. A força máxima suportada por essas esponjas antes de sua
ruptura foi de 5 N, a qual não pode ser comparada aos dados deste trabalho, uma vez que os
autores não forneceram detalhes a respeito das dimensões dos corpos de prova utilizados nos
ensaios. Contudo, uma vez que não existe um consenso a respeito das condições em que os
ensaios são realizados (dimensões dos corpos de prova, velocidade de distensão e força aplicada
à amostra, distância entre as garras do equipamento, umidade relativa, etc.), comparações diretas
entre os dados da literatura podem eventualmente levar a conclusões equivocadas.
4.2.8. Citotoxicidade in vitro indireta
A citotoxicidade das membranas a células L929 foi analisada por via indireta e os
resultados obtidos são mostrados na Figura 4.15. Os resultados obtidos mostram que nenhuma
das formulações afeta negativamente o crescimento celular. Bellini et al. (2012) também
observaram baixos valores de citotoxicidade a células L929 para membranas de quitosana-

87
xantana (1,6% para membranas preparadas na ausência de Pluronic F68 e 2,6% para membranas
preparadas na presença de 0,75% m/m de Pluronic F68).
Figura 4.15: Citotoxicidade in vitro indireta a células L929 das membranas preparadas na
ausência e na presença de diferentes proporções de Pluronic F68, comparadas aos controles
negativo (C.N) e positivo (C.P.).
Os baixos valores de citotoxicidade encontrados já eram esperados uma vez que
surfactantes macromoleculares, como o Pluronic F68, apresentam baixa toxicidade e efeitos de
irritação (TADROS, 2005). O Pluronic F68 não é tóxico a células P388 (células leucêmicas de
camundongos) em concentrações variando de 2×10-5 a 0,5% (m/v) (BOGMAN et al., 2003).
Mesmo considerando que todo o Pluronic F68 usado na preparação das membranas
permanecesse no material após as lavagens, e que todo o surfactante fosse extraído pelo meio de
cultura RPMI-1640 utilizado nos ensaios de citotoxicidade, a concentração final de Pluronic F68
no meio de cultura seria de 0,1% (m/v) e 0,5% (m/v) para as formulações P2% e P10%,
respectivamente, não superando o limite superior apontado por Bogman et al. (2003).
4.2.9. Espectroscopia no infravermelho com transformada de Fourier
A técnica de FTIR-ATR foi utilizada para avaliar a presença de Pluronic F68 residual nas
membranas preparadas com o surfactante e a formação do complexo de polieletrólitos entre a
quitosana e o alginato.
-20
0
20
40
60
80
100
120
Cit
oto
xic
idad
e (%
)
P0% P2% P10% C.N. C.P.

88
Na Figura 4.16 são mostrados os espectros de absorção no infravermelho (FTIR-ATR) dos
polímeros quitosana e alginato e do surfactante Pluronic F68. Na Tabela 4.11 encontram-se os
picos de absorção característicos, que podem ser utilizados para identificar os compostos
presentes nas membranas.
Figura 4.16: Espectros de absorção obtidos por FTIR-ATR dos componentes puros das
membranas (quitosana, alginato e Pluronic F68).
Tabela 4.11: Picos característicos observados nos espectros de absorção FTIR-ATR da
quitosana, do alginato e do Pluronic F68 e suas respectivas atribuições.
Pico (cm-1) Grupo Composto Referência
1098 Éter alifático Pluronic F68 Wang et al. (2010)
1150 Éter alifático Pluronic F68 Maghraby e Alomrani (2009)
1405 -COOH Alginato Sankalia et al. (2007)
1560 -NH Quitosana Sankalia et al. (2007)
1580 -NH2 Quitosana Sankalia et al. (2007)
1600 -COOH Alginato Sankalia et al. (2007)
1650 -C=O do grupo amida Quitosana Sankalia et al. (2007)
Como pode ser observado, ambos os espectros de absorção dos biopolímeros quitosana e
4000 3500 3000 2500 2000 1500 1000 500
Pluronic F68
Alginato
Quitosana
Ab
sorç
ão
Número de onda (cm-1)

89
alginato mostram uma banda larga na região de 3600-3000 cm-1, aproximadamente. De acordo
com Lawrie et al. (2007), esta banda pode ser atribuída ao estiramento dos grupos -OH,
presentes tanto na quitosana como no alginato. O pico a 2900 cm-1 representa as vibrações dos
grupos metil e metileno, presentes também em ambos os compostos (SANKALIA et al., 2007;
LAWRIE et al,. 2007). Deve-se ressaltar que, no espectro da quitosana, o pico a 1560 cm-1, que
indica a vibração do grupo -NH dos resíduos acetilados, não foi observado em intensidade
significativa, sendo compatível com o alto grau de desacetilação do biopolímero utilizado. O
espectro de absorção do Pluronic F68 possui picos de absorção a 2900 e 1470 cm-1, os quais
podem ser atribuídos aos grupos metil e metileno, comuns a vários dos compostos utilizados
para o preparo das membranas.
Na Figura 4.17 são mostrados os espectros de absorção da mistura física entre os polímeros
quitosana e alginato em pó e da membrana formada pela complexação de quitosana e alginato,
ambos obtidos na ausência do tensoativo Pluronic F68.
Figura 4.17: Espectros de absorção obtidos por FTIR-ATR da mistura física dos polímeros em
pó e da membrana sem surfactante (destaque para o alargamento da banda a 1590 cm-1).
Não foram notadas diferenças significativas entre os espectros de absorção da mistura
física entre a quitosana e o alginato e da membrana em si, como mostrado na Figura 4.17. De
acordo com Lawrie et al. (2007), Sankalia et al. (2007) e Li et al. (2009), os grupos carboxila e
amina absorvem a radiação em comprimentos de onda semelhantes, na região de 1560 a
4000 3500 3000 2500 2000 1500 1000 500
Membrana
Abso
rção
Número de onda (cm-1)
Mistura física
1800 1600 1400 1200
Membrana
Abso
rção
Número de onda (cm-1)
Mistura física
1590 cm-1

90
1640 cm-1. Desta forma, é difícil identificar a presença de um ou outro composto livre, assim
como seu estado de protonação, o que seria importante para detectar a formação do complexo de
polieletrólitos (PEC).
Devido a esta sobreposição de comprimentos de onda característicos da quitosana e do
alginato, quando o espectro da mistura física entre os polímeros é comparado ao espectro da
membrana, observa-se um alargamento da banda em torno de 1590 cm-1, como pode ser
observado pela Figura 4.17.
Lawrie et al. (2007) observaram que os espectros de FTIR de vários tipos de mistura de
quitosana e alginato, como precipitação conjunta, formação de filmes densos e de filmes em
camadas alternadas, são muito similares, apesar do variado grau de interação entre os grupos
funcionais dos polissacarídeos. Isso faz com que a técnica de FTIR seja limitada para confirmar
a formação de PEC.
Na Figura 4.18 são mostrados os espectros de absorção das membranas obtidas na
presença de diferentes quantidades de Pluronic F68, podendo-se concluir que os mesmos não
apresentam diferenças. Pode-se supor que houve sobreposição entre os picos característicos do
Pluronic F68 e dos polissacarídeos, devido à presença de uma baixa quantidade do surfactante.
4.2.10. Análise térmica simultânea (SDT) (Calorimetria e termogravimetria)
O comportamento térmico dos polímeros isolados e das membranas obtidas na ausência e
na presença de diferentes quantidades de Pluronic F68 foi analisado por análise
termogravimétrica (TGA) e calorimetria exploratória diferencial (DSC). Os resultados obtidos
são mostrados na Tabela 4.12 e na Figura 4.19.
4.2.10.1. Análise termogravimétrica (TGA)
A análise de TGA da quitosana e do alginato resultou em dois eventos térmicos. O
primeiro evento ocorreu em temperaturas menores do que 100°C, podendo ser atribuído à perda
de água residual contida nas amostras, mesmo após seu armazenamento em dessecador com
sílica por 24 horas. Destaca-se que a umidade relativa no interior do dessecador era de cerca de
11%. Os eventos térmicos subsequentes foram associados à degradação dos polímeros.
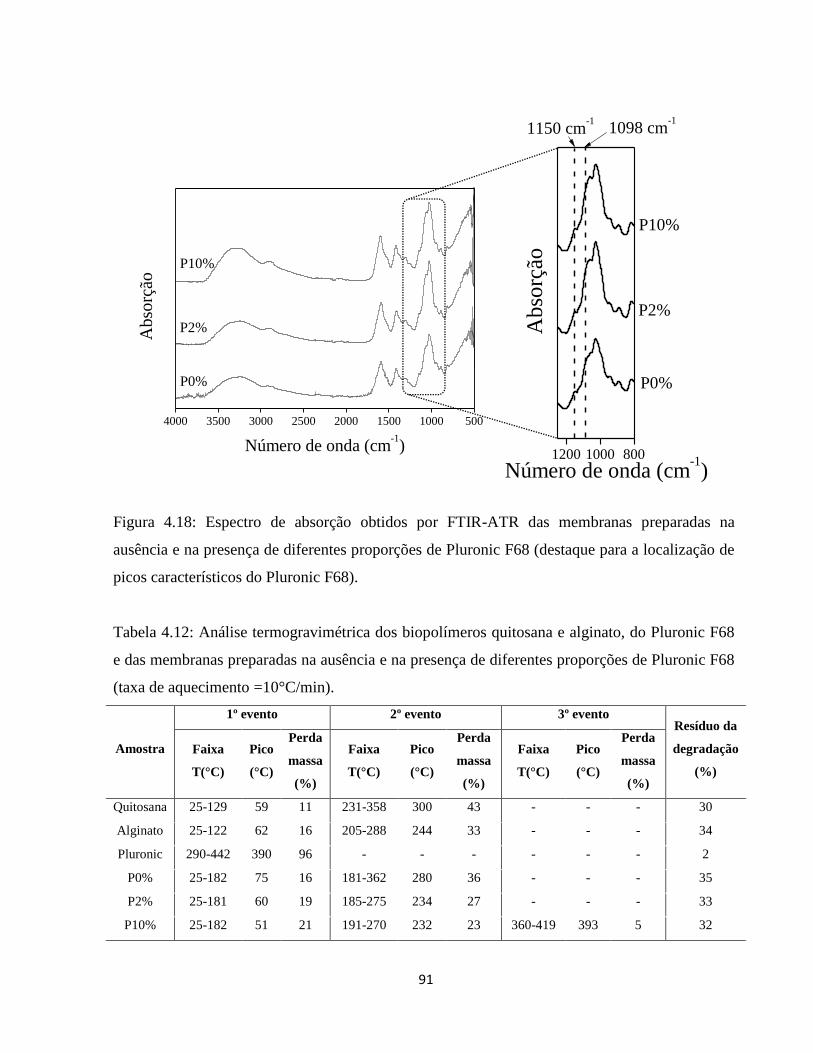
91
Figura 4.18: Espectro de absorção obtidos por FTIR-ATR das membranas preparadas na
ausência e na presença de diferentes proporções de Pluronic F68 (destaque para a localização de
picos característicos do Pluronic F68).
Tabela 4.12: Análise termogravimétrica dos biopolímeros quitosana e alginato, do Pluronic F68
e das membranas preparadas na ausência e na presença de diferentes proporções de Pluronic F68
(taxa de aquecimento =10°C/min).
Amostra
1º evento 2º evento 3º evento Resíduo da
degradação
(%)
Faixa
T(°C)
Pico
(°C)
Perda
massa
(%)
Faixa
T(°C)
Pico
(°C)
Perda
massa
(%)
Faixa
T(°C)
Pico
(°C)
Perda
massa
(%)
Quitosana 25-129 59 11 231-358 300 43 - - - 30
Alginato 25-122 62 16 205-288 244 33 - - - 34
Pluronic 290-442 390 96 - - - - - - 2
P0% 25-182 75 16 181-362 280 36 - - - 35
P2% 25-181 60 19 185-275 234 27 - - - 33
P10% 25-182 51 21 191-270 232 23 360-419 393 5 32
4000 3500 3000 2500 2000 1500 1000 500
Abso
rção
Número de onda (cm-1)
P0%
P2%
P10%
1200 1000 800
1150 cm-1
Ab
sorç
ão
Número de onda (cm-1)
P0%
P2%
P10%
1098 cm-1
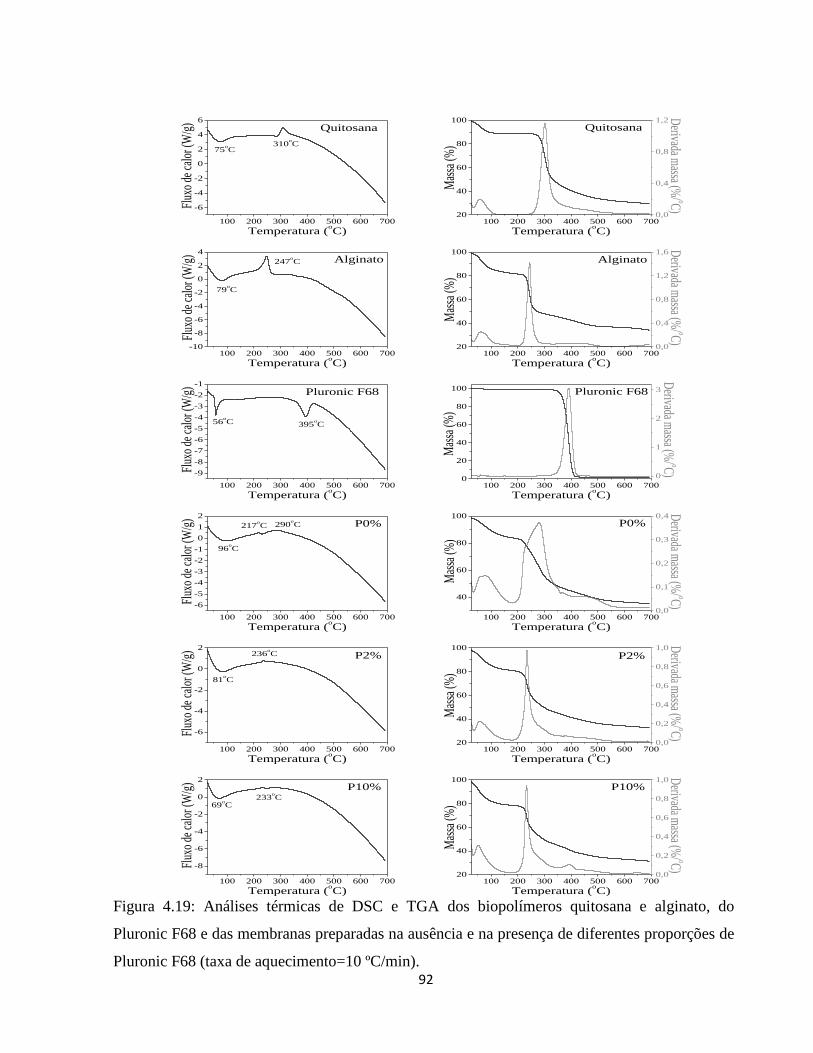
92
Figura 4.19: Análises térmicas de DSC e TGA dos biopolímeros quitosana e alginato, do
Pluronic F68 e das membranas preparadas na ausência e na presença de diferentes proporções de
Pluronic F68 (taxa de aquecimento=10 ºC/min).
100 200 300 400 500 600 700
-6
-4
-2
0
2
4
6
Flux
o de
cal
or (W
/g)
Temperatura (oC)
Quitosana
310oC
75oC
100 200 300 400 500 600 70020
40
60
80
100
Quitosana
Temperatura (oC)
Mas
sa (%
)
0,0
0,4
0,8
1,2
Derivada m
assa (%/ oC
)
100 200 300 400 500 600 700-10
-8
-6
-4
-2
0
2
4
247oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
Alginato
79oC
100 200 300 400 500 600 70020
40
60
80
100
Alginato
Temperatura (oC)
Mas
sa (%
)
0,0
0,4
0,8
1,2
1,6
Derivada m
assa (%/ oC
)
100 200 300 400 500 600 700
-9
-8
-7
-6
-5
-4
-3
-2
-1
395oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
Pluronic F68
56oC
100 200 300 400 500 600 7000
20
40
60
80
100
Pluronic F68
Temperatura (oC)
Mas
sa (%
)
0
1
2
3
Derivada m
assa (%/ oC
)
100 200 300 400 500 600 700
-6
-5
-4
-3
-2
-1
0
1
2
290oC217
oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P0%
96oC
100 200 300 400 500 600 700
40
60
80
100
P0%
Temperatura (oC)
Mas
sa (%
)
0,0
0,1
0,2
0,3
0,4
Derivada m
assa (%/ oC
)
100 200 300 400 500 600 700
-6
-4
-2
0
2236
oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P2%
81oC
100 200 300 400 500 600 70020
40
60
80
100
P2%
Temperatura (oC)
Mas
sa (%
)
0,0
0,2
0,4
0,6
0,8
1,0
Derivada m
assa (%/ oC
)
100 200 300 400 500 600 700
-8
-6
-4
-2
0
2
233oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P10%
69oC
100 200 300 400 500 600 70020
40
60
80
100
P10%
Temperatura (oC)
Mas
sa (%
)
0,0
0,2
0,4
0,6
0,8
1,0
Derivada m
assa (%/ oC
)

93
Na análise da quitosana, verificou-se perda de massa de 43% no segundo evento térmico,
com pico a 300°C. Resultados similares foram encontrados por Martins et al. (2011), que
observaram um pico de degradação a 312°C com perda de massa de 59,8%. Esta etapa de
degradação pode ser atribuída à despolimerização e decomposição pirolítica do polissacarídeo
(MARTINS et al., 2012).
Verificou-se uma perda de massa de 33% para a degradação térmica do alginato no
segundo evento, com pico a 244°C. Encontraram-se na literatura dados um pouco diferentes,
sendo a perda de massa de 40%, atribuída à formação de Na2CO3 e material carbonizado, e o
pico a 226°C (POPA et al., 2008). As diferenças entre os resultados podem estar relacionadas às
razões entre os blocos M/G ou às massas molares dos diferentes tipos de alginato analisados.
Diferentemente da quitosana e do alginato, o Pluronic F68 apresentou um único evento
térmico, com pico de degradação a 390°C e perda de massa de 96%. Wu et al. (2008) também
observaram que o Pluronic F68 se degrada em uma única etapa, mas a uma temperatura menor,
de 320°C.
Na análise das membranas, o primeiro evento térmico ocorreu em temperaturas variando
de 51 a 75°C, com perdas de massa variando de 16 a 21%. Este evento está relacionado à perda
de água residual presente nas amostras, mesmo após à manutenção das mesmas em dessecador
com sílica (umidade relativa igual a 11%). Observou-se que as membranas mais porosas tiveram
maiores perdas de massa nesta etapa, o que pode ser explicado pela maior capacidade de
absorção de água destas, o que faz com que tenham maior conteúdo de umidade.
As temperaturas dos picos de degradação das membranas P2% e P10% foram de 234 e
232°C, respectivamente, as quais são temperaturas menores do que a temperatura de degradação
do alginato puro. A membrana P0%, por outro lado, apresentou um comportamento diferente,
com pico largo de degradação em 280°C, temperatura menor do que a temperatura de degradação
da quitosana pura, possuindo também um ombro em cerca de 230°C, temperatura menor do que a
temperatura de degradação do alginato puro. As diferenças no comportamento térmico das
formulações preparadas na presença e na ausência do tensoativo Pluronic F68 podem ser devidas
a distintas formas de interação na estabilização do PEC.
A presença de uma única etapa de degradação pode ser considerada um indicativo da
formação de um complexo de polieletrólitos (PEC) de acordo com Smitha et al. (2005), que
observaram um único pico de degradação em torno de 260°C para o complexo quitosana-

94
alginato. Anbinder et al. (2011) também observaram que cápsulas de alginato cobertas com
quitosana e reticuladas com íons cálcio apresentaram uma única etapa de degradação a 250°C,
com perda de massa de 40%. Assim, pode-se inferir que, na presença de Pluronic F68, a
interação entre a quitosana e o alginato seria favorecida.
A formulação P10% também apresentou um pequeno pico de degradação a 393°C, que
pode indicar que uma dada fração de Pluronic F68 permanece na estrutura das membranas
mesmo após sucessivas etapas de lavagem. Contudo, a presença de Pluronic F68 residual não
afeta negativamente a biocompatibilidade, como indicado nos ensaios de citotoxicidade indireta
a células L929.
4.2.10.2. Calorimetria exploratória diferencial (DSC)
As análises de DSC dos polissacarídeos quitosana e alginato isolados, mostradas na Figura
4.19 (coluna à esquerda), resultaram em dois eventos térmicos, assim como observado nas
análises de TGA. Os picos endotérmicos a 75 e 79°C podem ser atribuídos à vaporização da
água adsorvida, corroborando com os resultados de TGA.
Os picos exotérmicos observados para os polímeros também corroboram com os resultados
de TGA e com a literatura (ANBINDER et al., 2011). O alginato apresentou um pico de
degradação a 247°C e a quitosana apresentou um pico de degradação a 310°C.
A análise de DSC do Pluronic F68 resultou em dois picos endotérmicos, um a 56°C e outro
a 395°C. O primeiro pode ser atribuído à fusão, de acordo com dados similares obtidos por
Verger et al. (1998). O segundo pode estar relacionado à oxidação ou decomposição, uma vez
que observou-se também um pico de degradação a 390°C na análise de TGA, como comentado
anteriormente.
Nas análises de DSC das membranas, o primeiro pico endotérmico foi atribuído à perda de
água. A membrana P0% apresentou também dois picos exotérmicos, o primeiro a 217°C e o
segundo a 290°C. Já as formulações P2% e P10% apresentaram picos exotérmicos em 236°C e
233°C, respectivamente.
Acredita-se que, na formulação P0%, as temperaturas dos picos característicos da
quitosana e do alginato isolados foram deslocadas em -20°C e -30°C, respectivamente. Já nas
formulações P2% e P10%, verificou-se a presença de apenas um pico, que pode ser atribuído ao

95
deslocamento da temperatura do pico característico do alginato. Observou-se que, assim como
nas análises de TGA, que a presença de Pluronic F68 alterou o comportamento térmico das
membranas.
De acordo com Anbinder et al. (2011), o deslocamento da temperatura de pico
característico do alginato na análise térmica de cápsulas de quitosana-alginato poderia indicar a
presença de um complexo de polieletrólitos. Por outro lado, a análise térmica por DSC do PEC
de quitosana-alginato realizada por Li et al. (2009) resultou em um pico exotérmico em uma
temperatura intermediária às temperaturas de pico referentes à degradação da quitosana e do
alginato isolados, o que foi interpretado por estes autores como um indicativo da interação entre
os polissacarídeos. A diferença entre os resultados obtidos por Li et al. (2009) e os resultados
obtidos no presente trabalho podem ser atribuídas aos diferentes tipos de quitosana e alginato
empregados com relação às suas origens e massas molares, as quais não foram informadas em
detalhes por aqueles autores, e também aos diferentes procedimentos de preparo do PEC, que, no
primeiro caso, envolveu a liofilização de uma mistura de quitosana e alginato na proporção de
1:0,5, seguida de reticulação por imersão em solução de CaCl2 e por mais uma etapa de
liofilização.
4.2.11. Considerações finais acerca da caracterização das membranas produzidas na ausência
e na presença de diferentes proporções de Pluronic F68
Na Tabela 4.13 é mostrada uma comparação geral entre as propriedades das membranas
obtidas no presente trabalho com dados relatados anteriormente por Bueno (2010) e com valores
recomendados ou comumente verificados na literatura para materiais destinados a aplicações na
pele. Entretanto, destaca-se que no presente trabalho, realizou-se uma caracterização mais
detalhada em relação ao trabalho anterior de Bueno (2010), por isso não é possível a comparação
de alguns dos dados.
De forma geral, observou-se que todas as formulações obtidas são superiores às de Bueno
(2010) em termos das propriedades mecânicas, da capacidade de absorção e da estabilidade em
água.
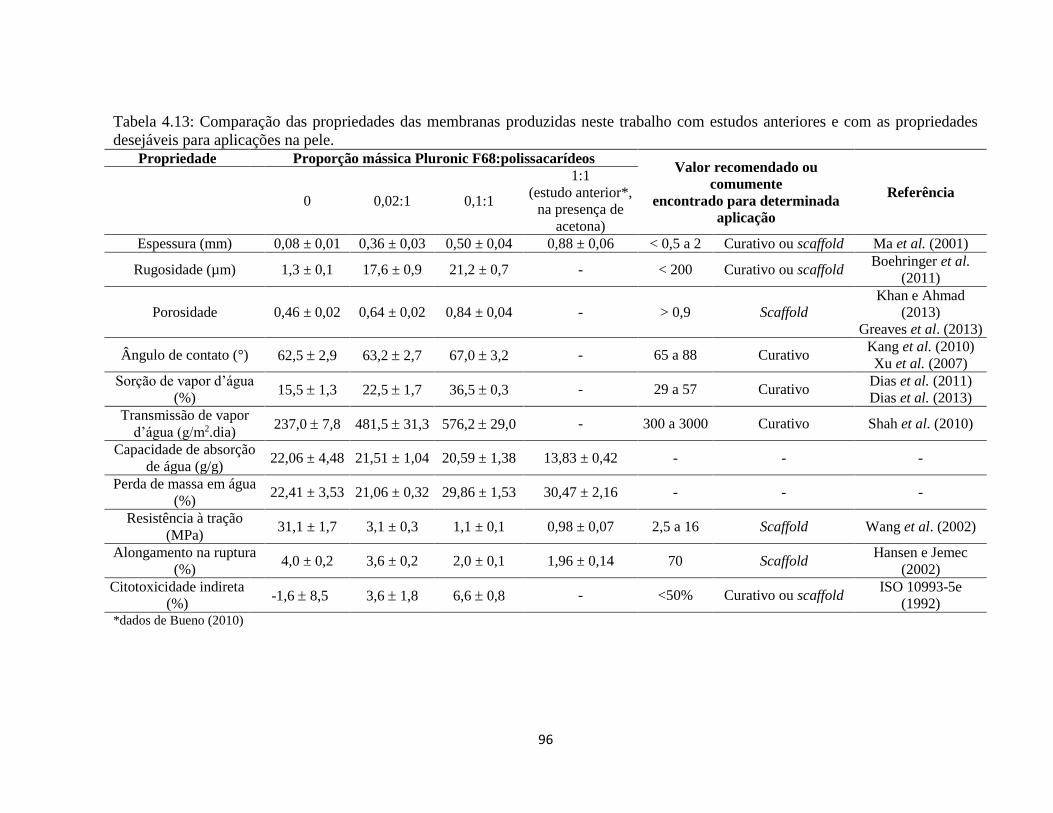
96
Tabela 4.13: Comparação das propriedades das membranas produzidas neste trabalho com estudos anteriores e com as propriedades
desejáveis para aplicações na pele.
Propriedade Proporção mássica Pluronic F68:polissacarídeos Valor recomendado ou
comumente
encontrado para determinada
aplicação
Referência 0 0,02:1 0,1:1
1:1
(estudo anterior*,
na presença de
acetona)
Espessura (mm) 0,08 ± 0,01 0,36 ± 0,03 0,50 ± 0,04 0,88 ± 0,06 < 0,5 a 2 Curativo ou scaffold Ma et al. (2001)
Rugosidade (µm) 1,3 ± 0,1 17,6 ± 0,9 21,2 ± 0,7 - < 200 Curativo ou scaffold Boehringer et al.
(2011)
Porosidade 0,46 ± 0,02 0,64 ± 0,02 0,84 ± 0,04 - > 0,9 Scaffold
Khan e Ahmad
(2013)
Greaves et al. (2013)
Ângulo de contato (°) 62,5 2,9 63,2 2,7 67,0 3,2 - 65 a 88 Curativo Kang et al. (2010)
Xu et al. (2007)
Sorção de vapor d’água
(%) 15,5 1,3 22,5 1,7 36,5 0,3 - 29 a 57 Curativo
Dias et al. (2011)
Dias et al. (2013)
Transmissão de vapor
d’água (g/m2.dia) 237,0 7,8 481,5 31,3 576,2 29,0 - 300 a 3000 Curativo Shah et al. (2010)
Capacidade de absorção
de água (g/g) 22,06 ± 4,48 21,51 ± 1,04 20,59 ± 1,38 13,83 ± 0,42 - - -
Perda de massa em água
(%) 22,41 ± 3,53 21,06 ± 0,32 29,86 ± 1,53 30,47 ± 2,16 - - -
Resistência à tração
(MPa) 31,1 ± 1,7 3,1 ± 0,3 1,1 ± 0,1 0,98 ± 0,07 2,5 a 16 Scaffold Wang et al. (2002)
Alongamento na ruptura
(%) 4,0 ± 0,2 3,6 ± 0,2 2,0 ± 0,1 1,96 ± 0,14 70 Scaffold
Hansen e Jemec
(2002)
Citotoxicidade indireta
(%) -1,6 8,5 3,6 1,8 6,6 0,8 - <50% Curativo ou scaffold
ISO 10993-5e
(1992) *dados de Bueno (2010)

97
Considerando-se a aplicação como curativos, pode-se afirmar que todas as formulações
preparadas na presença de diferentes proporções de Pluronic F68 são adequadas em termos da
sua espessura, rugosidade, ângulo de contato, transmissão de vapor d’água, propriedades
mecânicas e citotoxicidade. Destaca-se novamente que as membranas seriam mais adequadas
para o tratamento de lesões com baixa liberação de exsudato. A formulação P10% destaca-se
quanto à sua capacidade de sorção de vapor e de soluções fisiológicas. Já a formulação P0%
destaca-se por sua estabilidade e resistência à tração. Assim, dentre os materiais obtidos, as
formulações P0% e P10% seriam as mais recomendáveis para o tratamento de feridas. Contudo,
caso as membranas necessitem ser associadas a curativos secundários, uma análise mais
detalhada das suas propriedades na presença dos mesmos de se faz necessária.
Se o foco de utilização das membranas é a área de Engenharia de Tecidos, com aplicação
como scaffolds para o cultivo de células destinadas ao tratamento de lesões de pele, tem-se que
todas as formulações são adequadas quanto à espessura e citotoxicidade. A formulação P10%
destaca-se no que se refere à sua rugosidade e porosidade.
4.3. Efeitos da incorporação de PHMB por diferentes métodos nas características das
membranas
Neste item são apresentados os resultados referentes à caracterização das membranas P0%,
P2% e P10% contendo PHMB impregnado por adição à mistura polimérica, por adsorção a partir
de solução aquosa e por CO2 supercrítico. Assumiu-se que, devido às fortes interações que
podem se estabelecer entre o PHMB e os polissacarídeos, a incorporação de proporções maiores
do que aquelas encontradas em curativos já disponíveis no mercado (0,2 a 0,3% m/m) seria
necessária para atingir valores adequados de biodisponibilidade. Assim, as porcentagens de
PHMB incorporado às membranas por adição à mistura polimérica variaram de 1% m/m
(10 mg/g) a 10% m/m (100 mg/g). Para a impregnação via CO2 supercrítico, trabalhou-se apenas
com uma condição, adicionando-se na célula de impregnação uma quantidade de PHMB
equivalente a 7% (m/m) da massa de polissacarídeos, que seria uma condição aproximadamente
intermediária à utilizada na adição à mistura polimérica. Não foi escolhida uma concentração
menor do que 7% (m/m) devido à baixa solubilidade do PHMB em CO2 supercrítico, o que
poderia resultar na incorporação de quantidades muito baixas, ou mesmo na ineficiência de

98
incorporação. Concentrações maiores do que 7% (m/m) também não foram utilizadas devido à
baixa solubilidade do PHMB em CO2 supercrítico, o que poderia resultar em baixas eficiências
de incorporação. Para a incorporação por adsorção a partir de solução aquosa, trabalhou-se com
uma faixa ampla de concentrações, variando de 4,0x10-7 mol/L a 7,6x10-2 mol/L
As membranas foram analisadas com relação à sua integridade estrutural e homogeneidade
no aspecto por avaliação visual. Também foi realizada a análise da morfologia por microscopia,
avaliando-se a ocorrência de material depositado irregularmente, quebras, ou mesmo a formação
de orifícios. Analisou-se também a variação de espessura das membranas após os procedimentos
de impregnação de PHMB, assim como a eficiência de incorporação e a cinética de liberação do
PHMB.
4.3.1. Incorporação de PHMB por adição à mistura polimérica
Neste item são mostrados os resultados referentes à incorporação de PHMB por adição à
mistura polimérica nas proporções de 10 e 100 mg/g. O aspecto visual e morfologia das
membranas são mostrados nas Figuras 4.20 e 4.21.
Figura 4.20: Aspecto visual das membranas preparadas na ausência e na presença de diferentes
proporções mássicas de PHMB: A) sem PHMB, B) PHMB =10 mg/g, C) PHMB =100 mg/g.
P0%
P2%
P10%
(A)
(A)
(A)
(B)
(B)
(B)
(C)
(C)
(C)
3 cm 3 cm 3 cm
3 cm 3 cm 3 cm
3 cm 3 cm 3 cm

99
Figura 4.21: Microscopia eletrônica de varredura (MEV) das membranas preparadas na ausência
e na presença de diferentes proporções mássicas de PHMB: A) sem PHMB, B) PHMB=10 mg/g,
C) PHMB=100 mg/g.
Não foram notadas alterações significativas no aspecto visual e morfologia das membranas
contendo ou não PHMB. Pode-se supor que o PHMB esteja homogeneamente disperso entre as
lamelas das membranas, o que dificulta a sua visualização. Destaca-se que análises de
espectrofotometria no UV-VIS indicaram que o antimicrobiano é estável nas condições de
incorporação, como mostrado no Apêndice V.
Por sua vez, os dados de espessura mostrados na Tabela 4.14 não indicaram uma tendência
de variação com a ausência ou presença de diferentes quantidades de PHMB.
A eficiência de incorporação do PHMB foi calculada por meio das medidas de absorbância
a 235 nm das soluções de lavagem das membranas como explicado no item 3.4.15. Verificaram-
se altos valores de eficiência de incorporação por adição à mistura polimérica, variando de cerca
de 72 a 86%, como mostrado na Tabela 4.15.
A adição de PHMB a 100 mg/g resultou em maiores massas de PHMB incorporado, mas
em menores eficiências de incorporação em comparação com a adição de 10 mg/g. Este
resultado pode ter ocorrido porque a adição de uma maior quantidade de PHMB faz com que
300µm
P2%
300µm
P10%
300µm 300µm
300µm
P0%
300µm 300µm
300µm
300µm
(A) (B) (C)
(A)
(A)
(B)
(B)
(C)
(C)
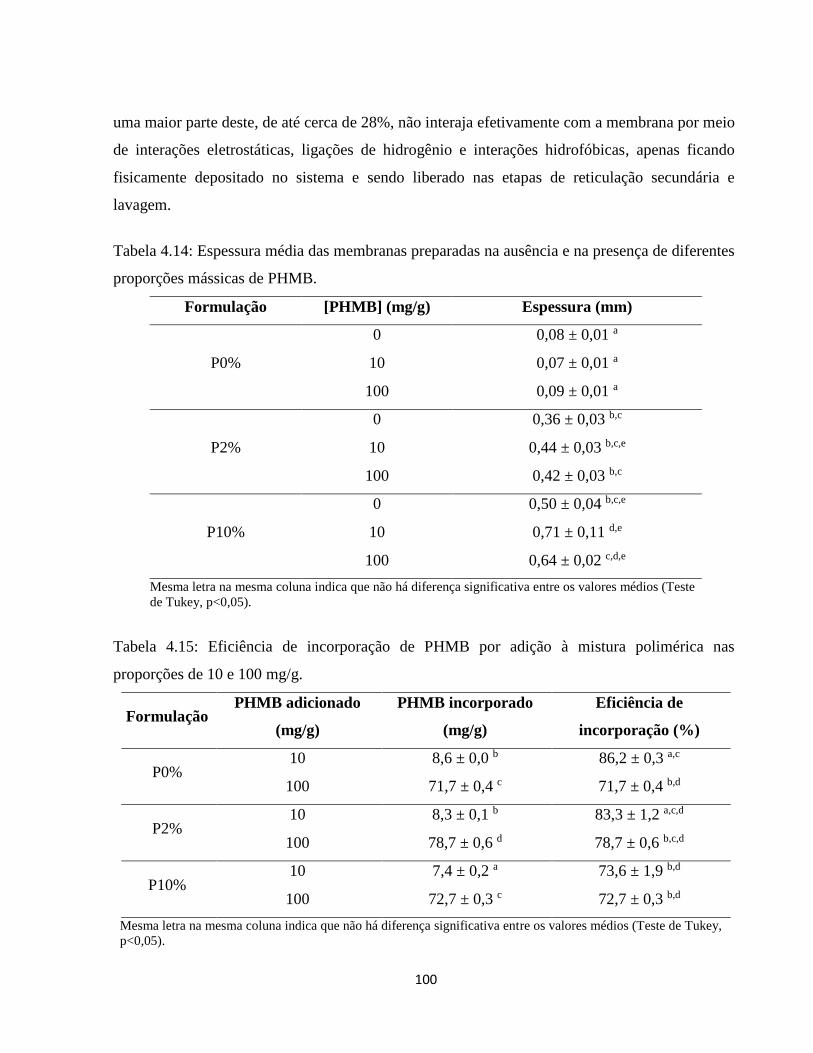
100
uma maior parte deste, de até cerca de 28%, não interaja efetivamente com a membrana por meio
de interações eletrostáticas, ligações de hidrogênio e interações hidrofóbicas, apenas ficando
fisicamente depositado no sistema e sendo liberado nas etapas de reticulação secundária e
lavagem.
Tabela 4.14: Espessura média das membranas preparadas na ausência e na presença de diferentes
proporções mássicas de PHMB.
Formulação [PHMB] (mg/g) Espessura (mm)
P0%
0 0,08 ± 0,01 a
10 0,07 ± 0,01 a
100 0,09 ± 0,01 a
P2%
0 0,36 ± 0,03 b,c
10 0,44 ± 0,03 b,c,e
100 0,42 ± 0,03 b,c
P10%
0 0,50 ± 0,04 b,c,e
10 0,71 ± 0,11 d,e
100 0,64 ± 0,02 c,d,e
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste
de Tukey, p<0,05).
Tabela 4.15: Eficiência de incorporação de PHMB por adição à mistura polimérica nas
proporções de 10 e 100 mg/g.
Formulação PHMB adicionado
(mg/g)
PHMB incorporado
(mg/g)
Eficiência de
incorporação (%)
P0% 10 8,6 ± 0,0 b 86,2 ± 0,3 a,c
100 71,7 ± 0,4 c 71,7 ± 0,4 b,d
P2% 10 8,3 ± 0,1 b 83,3 ± 1,2 a,c,d
100 78,7 ± 0,6 d 78,7 ± 0,6 b,c,d
P10% 10 7,4 ± 0,2 a 73,6 ± 1,9 b,d
100 72,7 ± 0,3 c 72,7 ± 0,3 b,d
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de Tukey,
p<0,05).
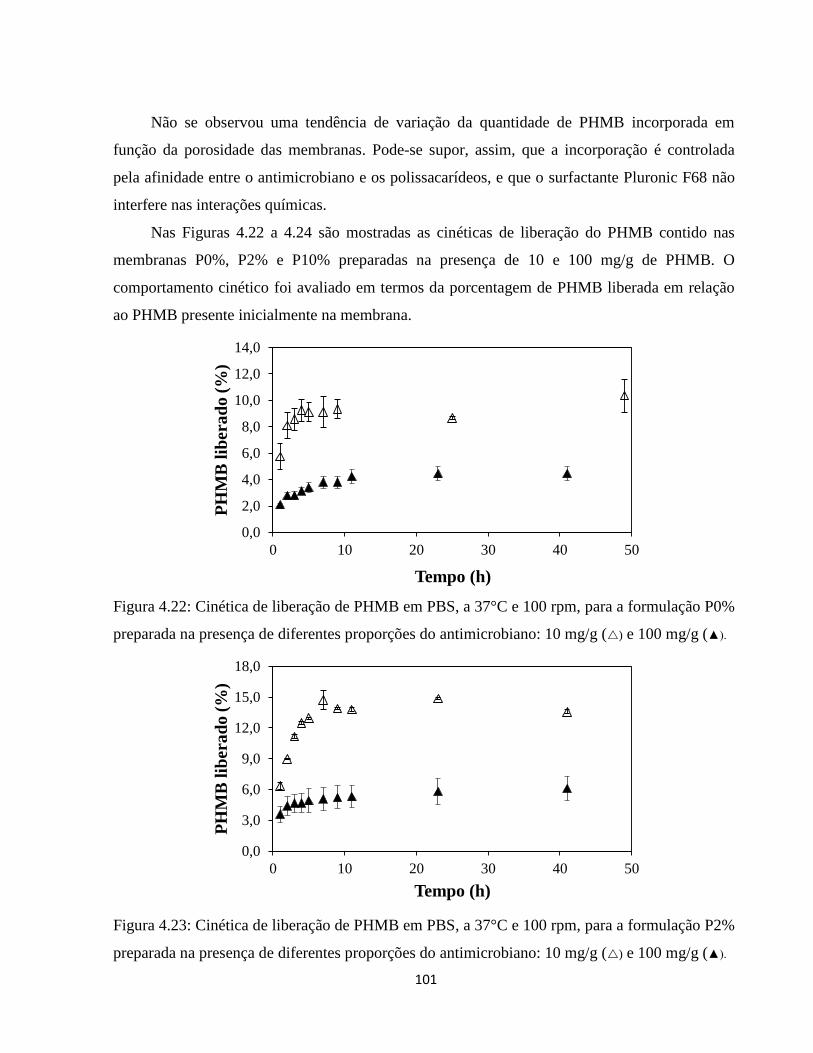
101
Não se observou uma tendência de variação da quantidade de PHMB incorporada em
função da porosidade das membranas. Pode-se supor, assim, que a incorporação é controlada
pela afinidade entre o antimicrobiano e os polissacarídeos, e que o surfactante Pluronic F68 não
interfere nas interações químicas.
Nas Figuras 4.22 a 4.24 são mostradas as cinéticas de liberação do PHMB contido nas
membranas P0%, P2% e P10% preparadas na presença de 10 e 100 mg/g de PHMB. O
comportamento cinético foi avaliado em termos da porcentagem de PHMB liberada em relação
ao PHMB presente inicialmente na membrana.
Figura 4.22: Cinética de liberação de PHMB em PBS, a 37°C e 100 rpm, para a formulação P0%
preparada na presença de diferentes proporções do antimicrobiano: 10 mg/g () e 100 mg/g (▲).
Figura 4.23: Cinética de liberação de PHMB em PBS, a 37°C e 100 rpm, para a formulação P2%
preparada na presença de diferentes proporções do antimicrobiano: 10 mg/g () e 100 mg/g (▲).
0,0
2,0
4,0
6,0
8,0
10,0
12,0
14,0
0 10 20 30 40 50
PH
MB
lib
era
do
(%
)
Tempo (h)
0,0
3,0
6,0
9,0
12,0
15,0
18,0
0 10 20 30 40 50
PH
MB
lib
era
do
(%
)
Tempo (h)
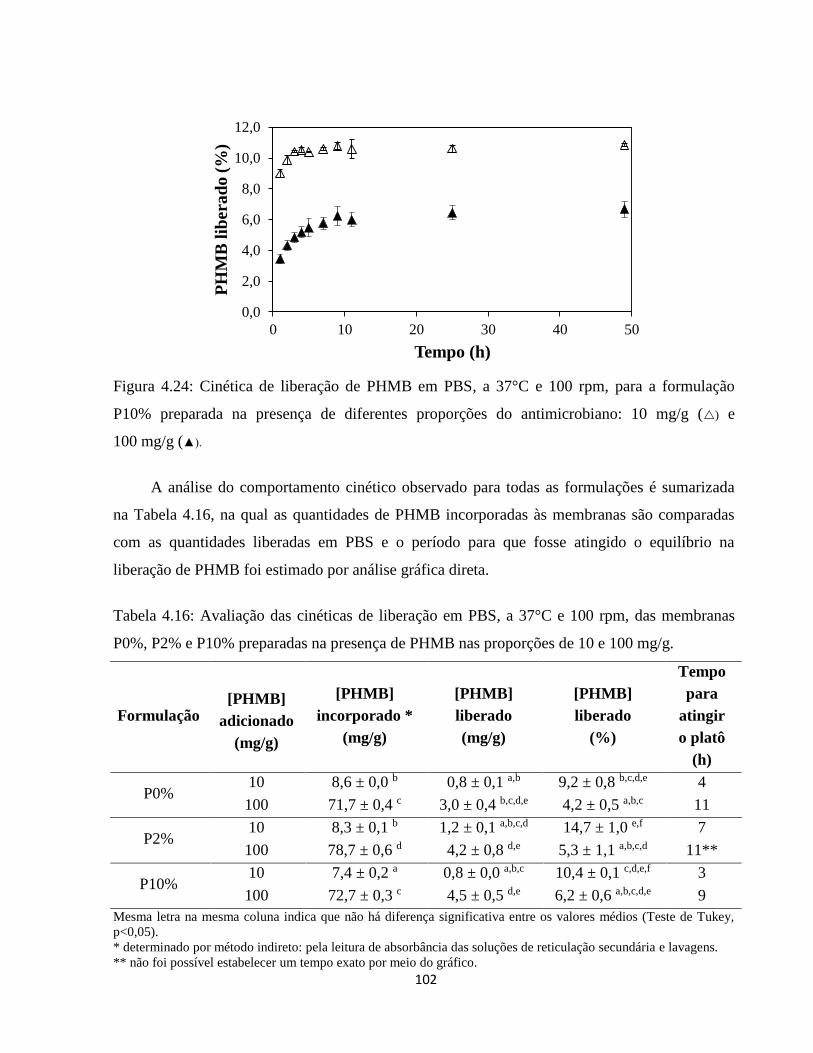
102
Figura 4.24: Cinética de liberação de PHMB em PBS, a 37°C e 100 rpm, para a formulação
P10% preparada na presença de diferentes proporções do antimicrobiano: 10 mg/g () e
100 mg/g (▲).
A análise do comportamento cinético observado para todas as formulações é sumarizada
na Tabela 4.16, na qual as quantidades de PHMB incorporadas às membranas são comparadas
com as quantidades liberadas em PBS e o período para que fosse atingido o equilíbrio na
liberação de PHMB foi estimado por análise gráfica direta.
Tabela 4.16: Avaliação das cinéticas de liberação em PBS, a 37°C e 100 rpm, das membranas
P0%, P2% e P10% preparadas na presença de PHMB nas proporções de 10 e 100 mg/g.
Formulação [PHMB]
adicionado
(mg/g)
[PHMB]
incorporado *
(mg/g)
[PHMB]
liberado
(mg/g)
[PHMB]
liberado
(%)
Tempo
para
atingir
o platô
(h)
P0% 10 8,6 ± 0,0 b 0,8 ± 0,1 a,b 9,2 ± 0,8 b,c,d,e 4
100 71,7 ± 0,4 c 3,0 ± 0,4 b,c,d,e 4,2 ± 0,5 a,b,c 11
P2% 10 8,3 ± 0,1 b 1,2 ± 0,1 a,b,c,d 14,7 ± 1,0 e,f 7
100 78,7 ± 0,6 d 4,2 ± 0,8 d,e 5,3 ± 1,1 a,b,c,d 11**
P10% 10 7,4 ± 0,2 a 0,8 ± 0,0 a,b,c 10,4 ± 0,1 c,d,e,f 3
100 72,7 ± 0,3 c 4,5 ± 0,5 d,e 6,2 ± 0,6 a,b,c,d,e 9
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de Tukey,
p<0,05).
* determinado por método indireto: pela leitura de absorbância das soluções de reticulação secundária e lavagens.
** não foi possível estabelecer um tempo exato por meio do gráfico.
0,0
2,0
4,0
6,0
8,0
10,0
12,0
0 10 20 30 40 50
PH
MB
lib
erad
o (
%)
Tempo (h)

103
As quantidades de PHMB liberadas das membranas foram baixas em relação à quantidade
de PHMB incorporada, e variaram entre cerca de 0,8 e 4,5 mg/g. Estas concentrações são
menores do que a concentração mínima considerada como irritante para a pele de 5% (m/m),
equivalente a 50 mg/g (HÜBNER e KRAMER, 2010). As formulações P0% e P10% preparadas
na presença de 10 mg/g de PHMB liberaram cerca de 0,8 mg/g, ou seja, quantidades abaixo do
nível mínimo considerado ativo, de 1 mg/g (ARCH CHEMICALS, 2005).
Verificou-se que a quantidade de PHMB liberado é diretamente proporcional à quantidade
de PHMB incorporado. No entanto, em termos de porcentagem, a quantidade de PHMB liberado
é inversamente proporcional à quantidade de PHMB incorporado. Resultados similares foram
encontrados por Gupta e Kumar (2000), na análise de partículas de quitosana incorporando
diclofenaco de sódio, por Meng et al. (2010), na análise de membranas de quitosana-alginato
incorporando sulfadiazina de prata e por Girata (2011), na análise de membranas de quitosana-
alginato incorporando fosfato hidrogenado de zircônio, sódio e prata (Alphasan® RC2000).
Uma hipótese para explicar esse comportamento é que a difusão do PHMB foi facilitada à
medida em que a sua concentração inicial diminuía, comportamento típico de sistemas de
liberação do tipo monolíticos, cuja liberação é majoritariamente controlada pela difusão.
A saber, os sistemas de liberação controlada por difusão podem ser classificados em duas
categorias: sistemas monolíticos, nos quais o agente terapêutico está disperso numa matriz
polimérica e sua liberação é controlada pela difusão através da matriz; e sistemas tipo
reservatório, nos quais o agente ativo está contido num núcleo cercado por uma membrana
polimérica e a liberação ocorre por difusão através desta membrana (RAVAL et al., 2010).
Nos sistemas monolíticos, a taxa de liberação depende da concentração inicial do agente
ativo na matriz polimérica. Se a concentração do ativo é menor do que o limite de solubilidade
na matriz, a difusão através da matriz limita a taxa de liberação. Por outro lado, se a
concentração do ativo é maior do que o limite de solubilidade na matriz, a dissolução deste na
matriz polimérica limita a taxa de liberação (RAVAL et al., 2010).
Observou-se também que o PHMB incorporado por adição à mistura polimérica foi
liberado das membranas de maneira não totalmente controlada, sendo que as cinéticas se
estabilizaram em 11 horas ou menos. Verificou-se também que o tempo de estabilização da
concentração de PHMB não parece ser diretamente influenciado pela porosidade, o que sugere
que o PHMB está associado às moléculas que compõem a membrana e não somente retido

104
fisicamente, como consequência provável das fortes ligações iônicas e de hidrogênio entre o
antimicrobiano e a matriz polimérica de quitosana-alginato.
Uma alternativa para diminuir as interações iônicas entre o PHMB e a matriz polimérica
seria aumentar a proporção mássica entre a quitosana e o alginato, diminuindo assim a
quantidade de cargas negativas do PEC. Neste sentido, foram produzidas membranas contendo
uma proporção mássica entre a quitosana e o alginato de 0,75:0,25. No entanto, tais membranas
não apresentaram aspecto e maleabilidade adequados.
Contudo, o fato de não haver liberação total do PHMB incorporado pode ser interessante
do ponto de vista de barreira física e bioativa promovida pelo curativo, prevenindo a entrada e
proliferação nas lesões de microrganismos provenientes do ambiente externo mesmo após o
término da liberação.
Rodrigues (2008) obteve resultados semelhantes aos do presente trabalho ao analisar a
liberação do antibiótico bacitracina, o qual é constituído por um complexo polipeptídico,
incorporado em membranas densas de quitosana e alginato. A autora observou que uma alta
porcentagem do fármaco incorporado, igual a 69,8%, permaneceu retido na membrana, não
sendo liberado em PBS após 96 horas de ensaio. Estes resultados se devem provavelmente às
interações iônicas entre o fármaco e os polissacarídeos.
Por outro lado, Wilhelms et al. (2007) verificaram que mais de 75% do PHMB contido em
curativos à base de celulose é liberado durante 24 horas em solução fisiológica de NaCl, o que se
deve provavelmente à baixa interação que se estabelece entre a celulose e o PHMB. A celulose é
um polissacarídeo cuja unidade monomérica é a glicose, que não possui cargas, já o PHMB
possui cargas positivas. Desta forma, não ocorrem interações eletrostáticas entre estes dois
compostos, diferentemente do que se esperaria para o PHMB e as carboxilas livres do alginato
usado nas formulações testadas no presente trabalho.
4.3.2. Incorporação de PHMB por adsorção a partir de solução aquosa
A incorporação de PHMB por adsorção a partir de solução aquosa foi realizada como
descrito no item 3.3.2. Inicialmente foram obtidas as isotermas de adsorção em uma ampla faixa
de concentração de soluções aquosas de PHMB, visando elucidar o mecanismo de incorporação
do composto no dispositivo, o que traria benefícios também na interpretação dos dados obtidos
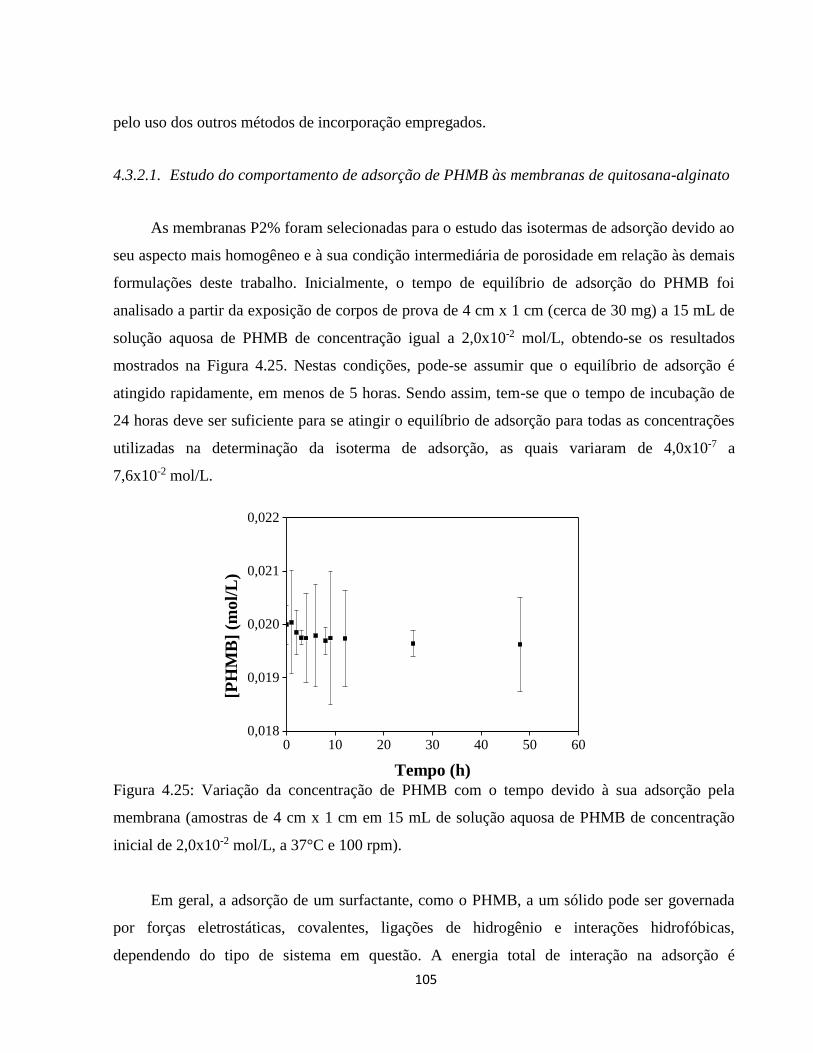
105
pelo uso dos outros métodos de incorporação empregados.
4.3.2.1. Estudo do comportamento de adsorção de PHMB às membranas de quitosana-alginato
As membranas P2% foram selecionadas para o estudo das isotermas de adsorção devido ao
seu aspecto mais homogêneo e à sua condição intermediária de porosidade em relação às demais
formulações deste trabalho. Inicialmente, o tempo de equilíbrio de adsorção do PHMB foi
analisado a partir da exposição de corpos de prova de 4 cm x 1 cm (cerca de 30 mg) a 15 mL de
solução aquosa de PHMB de concentração igual a 2,0x10-2 mol/L, obtendo-se os resultados
mostrados na Figura 4.25. Nestas condições, pode-se assumir que o equilíbrio de adsorção é
atingido rapidamente, em menos de 5 horas. Sendo assim, tem-se que o tempo de incubação de
24 horas deve ser suficiente para se atingir o equilíbrio de adsorção para todas as concentrações
utilizadas na determinação da isoterma de adsorção, as quais variaram de 4,0x10-7 a
7,6x10-2 mol/L.
Figura 4.25: Variação da concentração de PHMB com o tempo devido à sua adsorção pela
membrana (amostras de 4 cm x 1 cm em 15 mL de solução aquosa de PHMB de concentração
inicial de 2,0x10-2 mol/L, a 37°C e 100 rpm).
Em geral, a adsorção de um surfactante, como o PHMB, a um sólido pode ser governada
por forças eletrostáticas, covalentes, ligações de hidrogênio e interações hidrofóbicas,
dependendo do tipo de sistema em questão. A energia total de interação na adsorção é
0 10 20 30 40 50 600,018
0,019
0,020
0,021
0,022
[PH
MB
] (m
ol/
L)
Tempo (h)

106
normalmente o resultado cumulativo de algumas ou de todas estas forças. A isoterma de
adsorção de um surfactante pode ser classificada basicamente em três tipos, de acordo com o seu
formato: tipo Langmuir (L), tipo S (S), ou tipo dois platôs (L-S), com características de ambas as
isotermas anteriores (ZHANG e SOMASUNDARAN, 2006). A equação proposta por Zhu e Gu
(1989), mostrada na Equação 13, é um modelo geral para a adsorção de surfactantes em
interfaces sólido-líquido e engloba esses três tipos de isotermas:
1n
21
1n
21max
Ck1Ck1
Ckn
1Ckq
q
(13)
onde q é a quantidade de surfactante adsorvida por unidade de massa do adsorvente; qmax é a
capacidade máxima de adsorção do adsorvente; C é a concentração de surfactante no equilíbrio;
k1 é a constante de equilíbrio da primeira etapa da adsorção, quando há a interação direta entre o
surfactante e a superfície adsorvente; k2 é a constante de equilíbrio da segunda etapa da adsorção,
na qual ocorre a formação de hemimicelas (agregados de moléculas que se estruturam formando
uma monocamada sobre a superfície sólida em decorrência da interação entre as moléculas de
surfactante adsorvidas e as moléculas de surfactante em solução) a partir da concentração
hemimicelar crítica (CHC), etapa esta na qual ocorre um aumento significativo da quantidade de
moléculas adsorvidas; e n é um parâmetro que se refere ao número de agregação nas
hemimicelas, ou seja, o número de moléculas por hemimicela.
Os parâmetros da Equação de Zhu e Gu (1989) podem assumir diversos valores. Um dos
casos que simplificam esta equação é quando k2 tende a zero e n tende a um, o que faz com que a
mesma se reduza a uma isoterma tipo Langmuir, como mostrado na Equação 14:
Ck1
Ckqq
1
1max
(14)
A adsorção de surfactantes em interfaces sólido-líquido, em particular do PHMB em
celulose (BLACKBURN et al., 2006), pode ser também descrita pelo modelo de Freundlich, que
prevê a formação de múltiplas camadas em decorrência de interações entre as moléculas
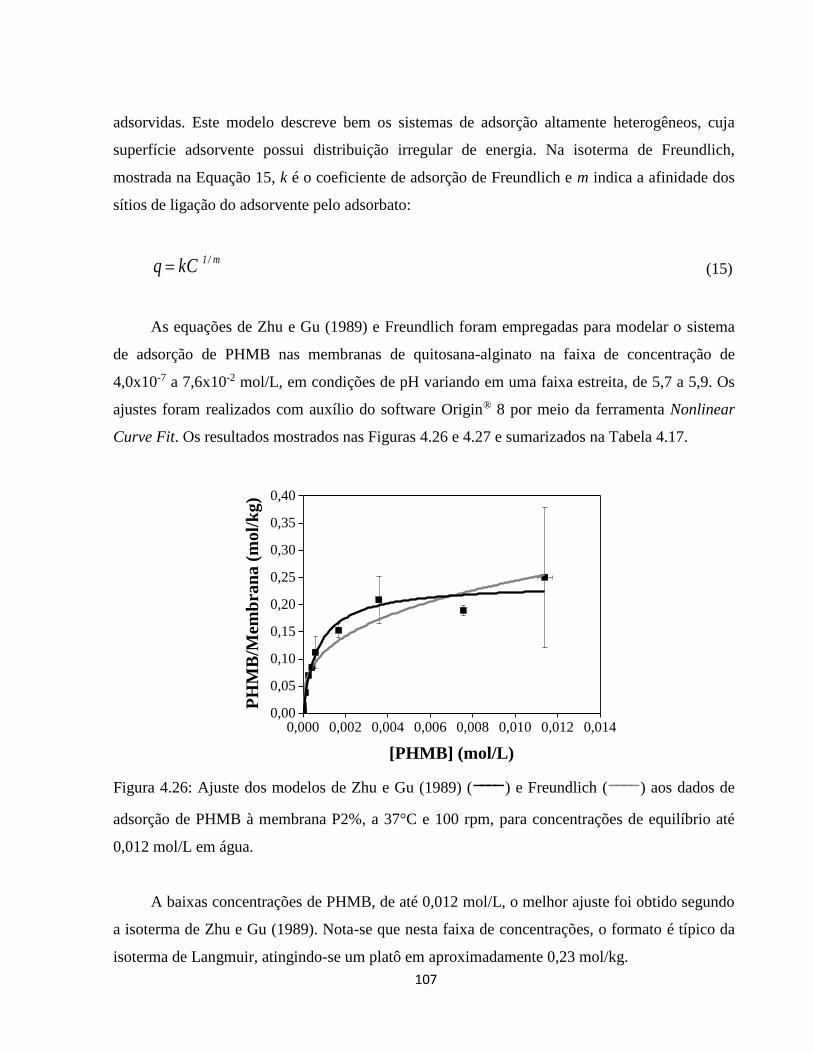
107
adsorvidas. Este modelo descreve bem os sistemas de adsorção altamente heterogêneos, cuja
superfície adsorvente possui distribuição irregular de energia. Na isoterma de Freundlich,
mostrada na Equação 15, k é o coeficiente de adsorção de Freundlich e m indica a afinidade dos
sítios de ligação do adsorvente pelo adsorbato:
m/1kCq (15)
As equações de Zhu e Gu (1989) e Freundlich foram empregadas para modelar o sistema
de adsorção de PHMB nas membranas de quitosana-alginato na faixa de concentração de
4,0x10-7 a 7,6x10-2 mol/L, em condições de pH variando em uma faixa estreita, de 5,7 a 5,9. Os
ajustes foram realizados com auxílio do software Origin® 8 por meio da ferramenta Nonlinear
Curve Fit. Os resultados mostrados nas Figuras 4.26 e 4.27 e sumarizados na Tabela 4.17.
Figura 4.26: Ajuste dos modelos de Zhu e Gu (1989) ( ̶ ̶ ̶ ̶ ) e Freundlich ( ̶ ̶ ̶ ̶ ) aos dados de
adsorção de PHMB à membrana P2%, a 37°C e 100 rpm, para concentrações de equilíbrio até
0,012 mol/L em água.
A baixas concentrações de PHMB, de até 0,012 mol/L, o melhor ajuste foi obtido segundo
a isoterma de Zhu e Gu (1989). Nota-se que nesta faixa de concentrações, o formato é típico da
isoterma de Langmuir, atingindo-se um platô em aproximadamente 0,23 mol/kg.
0,000 0,002 0,004 0,006 0,008 0,010 0,012 0,0140,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
PH
MB
/Mem
bra
na
(m
ol/
kg
)
[PHMB] (mol/L)
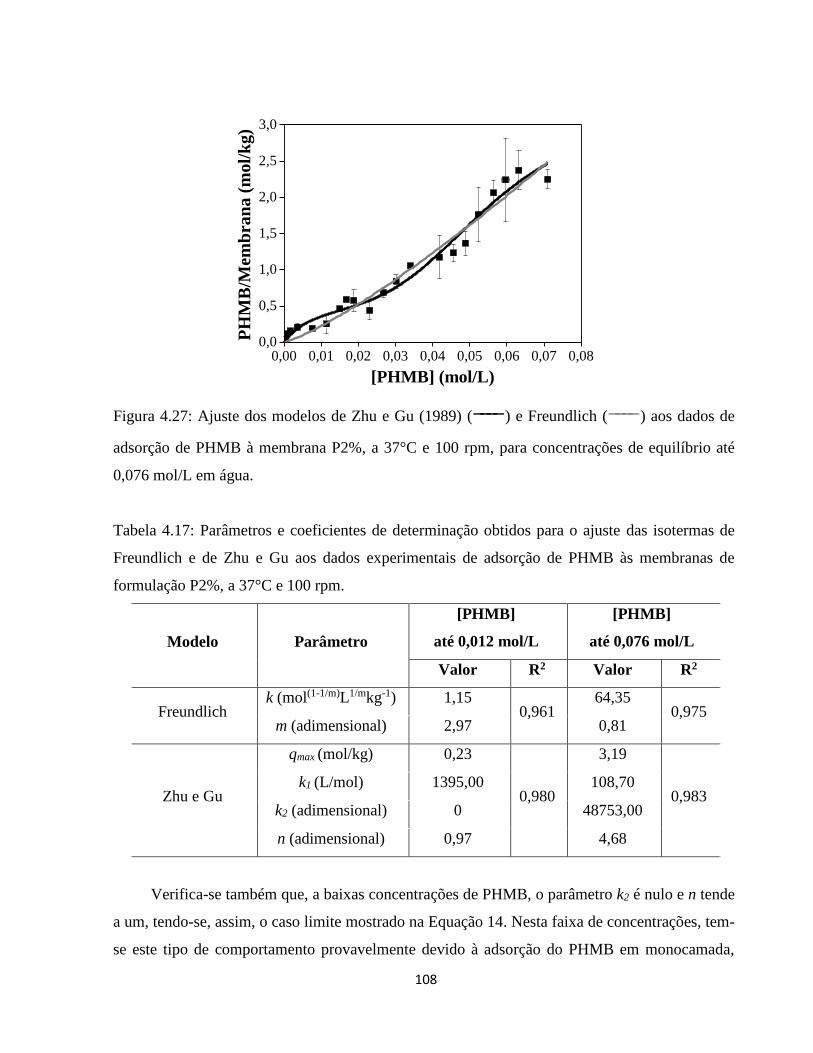
108
Figura 4.27: Ajuste dos modelos de Zhu e Gu (1989) ( ̶ ̶ ̶ ̶ ) e Freundlich ( ̶ ̶ ̶ ̶ ) aos dados de
adsorção de PHMB à membrana P2%, a 37°C e 100 rpm, para concentrações de equilíbrio até
0,076 mol/L em água.
Tabela 4.17: Parâmetros e coeficientes de determinação obtidos para o ajuste das isotermas de
Freundlich e de Zhu e Gu aos dados experimentais de adsorção de PHMB às membranas de
formulação P2%, a 37°C e 100 rpm.
Modelo Parâmetro
[PHMB]
até 0,012 mol/L
[PHMB]
até 0,076 mol/L
Valor R2 Valor R2
Freundlich k (mol(1-1/m)L1/mkg-1) 1,15
0,961 64,35
0,975 m (adimensional) 2,97 0,81
Zhu e Gu
qmax (mol/kg) 0,23
0,980
3,19
0,983 k1 (L/mol) 1395,00 108,70
k2 (adimensional) 0 48753,00
n (adimensional) 0,97 4,68
Verifica-se também que, a baixas concentrações de PHMB, o parâmetro k2 é nulo e n tende
a um, tendo-se, assim, o caso limite mostrado na Equação 14. Nesta faixa de concentrações, tem-
se este tipo de comportamento provavelmente devido à adsorção do PHMB em monocamada,
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,07 0,080,0
0,5
1,0
1,5
2,0
2,5
3,0
PH
MB
/Mem
bra
na (
mo
l/k
g)
[PHMB] (mol/L)

109
havendo interações somente entre as moléculas de PHMB adsorvidas e a superfície da
membrana. Tais interações seriam provavelmente de ordem eletrostática entre os grupos
biguanida do PHMB e as carboxilas do alginato, podendo também ocorrer ligações de
hidrogênio entre os grupos biguanida e os grupos –OH dos polissacarídeos, além de interações
hidrofóbicas entre as cadeias de polihexametileno do PHMB e os grupos fracamente
hidrofóbicos dos polissacarídeos, como os grupos –CH da cadeia principal da quitosana e do
alginato e os grupos N-acetil da quitosana. Destaca-se que, uma vez que o pKa do PHMB é igual
a 10,96 (O’MALLEY et al., 2006), é possível afirmar que o mesmo se encontra positivamente
carregado nas condições de incorporação testadas. Na Figura 4.28 é mostrada uma representação
das interações entre as moléculas de PHMB e a superfície das membranas.
Figura 4.28: Grupos envolvidos em diferentes tipos de interações entre as moléculas de PHMB e
a superfície das membranas (grupos envolvidos em interações eletrostáticas marcados em verde,
ligações de hidrogênio em vermelho e interações hidrofóbicas em azul).
A concentrações maiores de PHMB, de até 0,076 mol/L, o melhor ajuste foi novamente
obtido para a isoterma de Zhu e Gu (1989). O formato da isoterma da Figura 4.27 é complexo
(classificação do tipo L-S, com mais de um platô), devendo-se provavelmente à crescente
reorganização das moléculas de PHMB na superfície e à adsorção de moléculas subsequentes,
por meio da associação entre moléculas de PHMB, possibilitando atingir a capacidade máxima
teórica de adsorção das membranas (qmax) de cerca de 3,2 mol/kg, equivalente a 8,0 g/g.
Com base nos trabalhos de Blackburn et al. (2006), Zhu e Gu (1989) e De Paula et al.
(2011), a associação entre as moléculas de PHMB ocorreria por meio de interações hidrofóbicas,
ligações de hidrogênio e interações eletrostáticas com os contra-íons cloreto (Cl-). Na Figura
4.29 são representados os tipos de interações entre as moléculas de PHMB, as quais possibilitam
a sua adsorção às membranas em mais de uma camada.
......
-COO- -OH -OH -CH -CH
Cadeia de PHMB
Membrana

110
Figura 4.29: Grupos envolvidos em diferentes tipos de interações entre as moléculas de PHMB
(grupos envolvidos em ligações de hidrogênio marcados em vermelho, interações hidrofóbicas
em azul e interações eletrostáticas com contra-íons cloreto em verde).
Sabe-se ainda que o PHMB comporta-se como um surfactante, possuindo concentração
micelar crítica (CMC) em torno de 0,016 a 0,04 mol/L quando água é usada como solvente. As
micelas de PHMB seriam formadas por uma única molécula, sendo que os segmentos
hidrofóbicos de hexametileno se direcionam para o centro da micela supostamente esférica, e os
segmentos hidrofílicos protonados de biguanida se direcionam para fora da micela (DE PAULA
et al., 2011).
A literatura referente à adsorção de surfactantes sobre superfícies sólidas em geral também
menciona a formação de hemimicelas, admicelas e multicamadas adsorvidas. As hemimicelas
são agregados de moléculas de surfactante que se estruturam formando uma monocamada sobre
uma superfície sólida a partir da concentração hemimicelar crítica (CHC). Já as admicelas são
agregados do tipo bicamadas ordenadas (ZHU et al., 1989, BLACKBURN et al., 2006).
No sistema em questão, a concentração hemimicelar crítica (CHC), a partir da qual se
formam as hemimicelas, pode ser calculada por meio da Equação 16, proposta por Zhu et al.
(1989):
1n
1
2
1n
n
kn
2nCHC
(16)
onde n e k2 são parâmetros obtidos do ajuste segundo a isoterma de Zhu e Gu (1989).
......
......
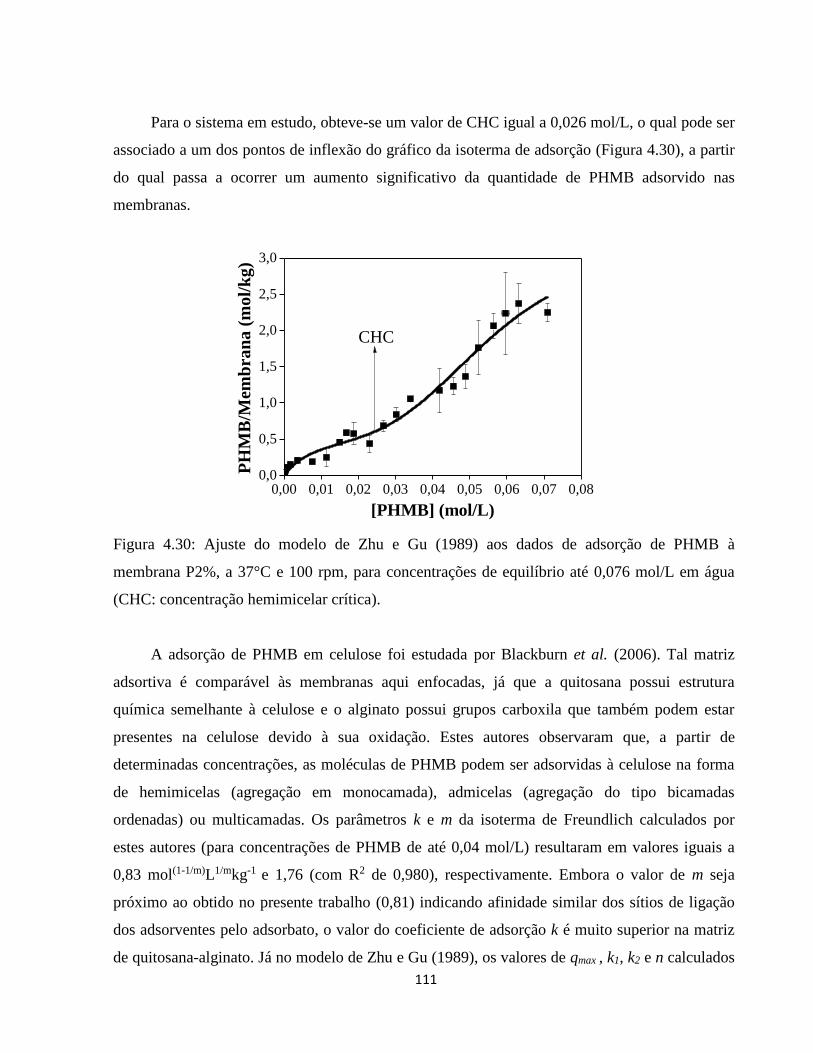
111
Para o sistema em estudo, obteve-se um valor de CHC igual a 0,026 mol/L, o qual pode ser
associado a um dos pontos de inflexão do gráfico da isoterma de adsorção (Figura 4.30), a partir
do qual passa a ocorrer um aumento significativo da quantidade de PHMB adsorvido nas
membranas.
Figura 4.30: Ajuste do modelo de Zhu e Gu (1989) aos dados de adsorção de PHMB à
membrana P2%, a 37°C e 100 rpm, para concentrações de equilíbrio até 0,076 mol/L em água
(CHC: concentração hemimicelar crítica).
A adsorção de PHMB em celulose foi estudada por Blackburn et al. (2006). Tal matriz
adsortiva é comparável às membranas aqui enfocadas, já que a quitosana possui estrutura
química semelhante à celulose e o alginato possui grupos carboxila que também podem estar
presentes na celulose devido à sua oxidação. Estes autores observaram que, a partir de
determinadas concentrações, as moléculas de PHMB podem ser adsorvidas à celulose na forma
de hemimicelas (agregação em monocamada), admicelas (agregação do tipo bicamadas
ordenadas) ou multicamadas. Os parâmetros k e m da isoterma de Freundlich calculados por
estes autores (para concentrações de PHMB de até 0,04 mol/L) resultaram em valores iguais a
0,83 mol(1-1/m)L1/mkg-1 e 1,76 (com R2 de 0,980), respectivamente. Embora o valor de m seja
próximo ao obtido no presente trabalho (0,81) indicando afinidade similar dos sítios de ligação
dos adsorventes pelo adsorbato, o valor do coeficiente de adsorção k é muito superior na matriz
de quitosana-alginato. Já no modelo de Zhu e Gu (1989), os valores de qmax , k1, k2 e n calculados
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,07 0,080,0
0,5
1,0
1,5
2,0
2,5
3,0P
HM
B/M
emb
ran
a (
mo
l/k
g)
[PHMB] (mol/L)
CHC
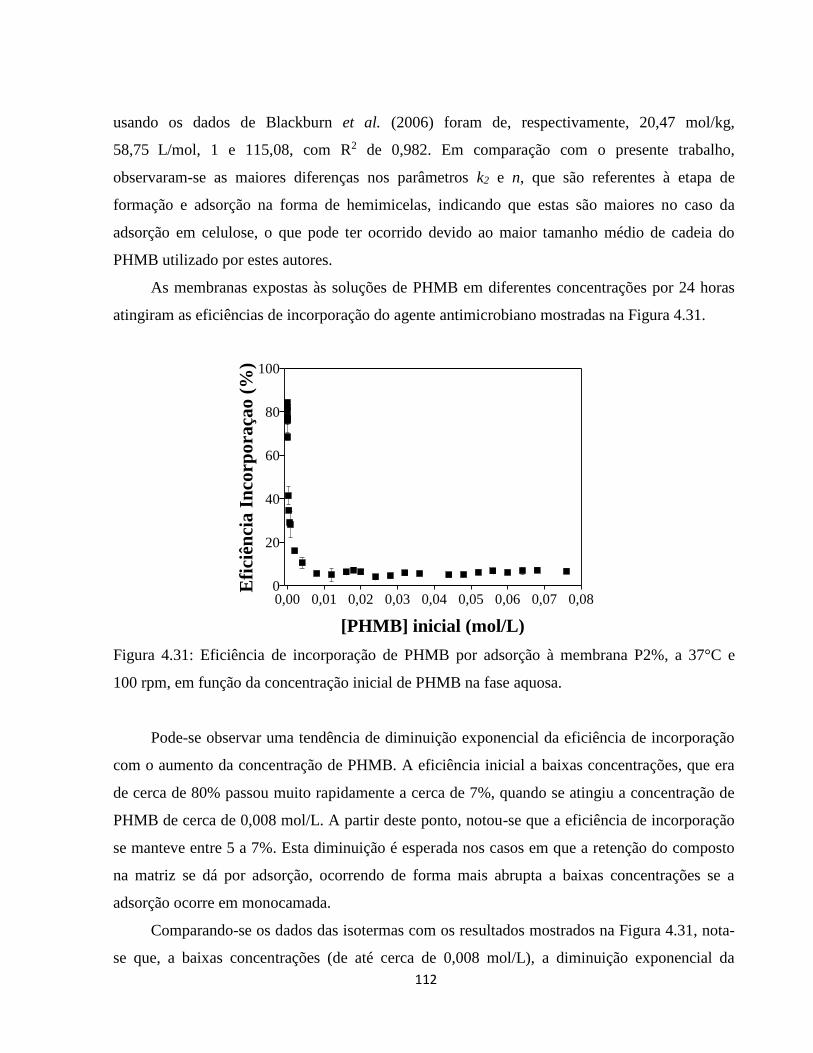
112
usando os dados de Blackburn et al. (2006) foram de, respectivamente, 20,47 mol/kg,
58,75 L/mol, 1 e 115,08, com R2 de 0,982. Em comparação com o presente trabalho,
observaram-se as maiores diferenças nos parâmetros k2 e n, que são referentes à etapa de
formação e adsorção na forma de hemimicelas, indicando que estas são maiores no caso da
adsorção em celulose, o que pode ter ocorrido devido ao maior tamanho médio de cadeia do
PHMB utilizado por estes autores.
As membranas expostas às soluções de PHMB em diferentes concentrações por 24 horas
atingiram as eficiências de incorporação do agente antimicrobiano mostradas na Figura 4.31.
Figura 4.31: Eficiência de incorporação de PHMB por adsorção à membrana P2%, a 37°C e
100 rpm, em função da concentração inicial de PHMB na fase aquosa.
Pode-se observar uma tendência de diminuição exponencial da eficiência de incorporação
com o aumento da concentração de PHMB. A eficiência inicial a baixas concentrações, que era
de cerca de 80% passou muito rapidamente a cerca de 7%, quando se atingiu a concentração de
PHMB de cerca de 0,008 mol/L. A partir deste ponto, notou-se que a eficiência de incorporação
se manteve entre 5 a 7%. Esta diminuição é esperada nos casos em que a retenção do composto
na matriz se dá por adsorção, ocorrendo de forma mais abrupta a baixas concentrações se a
adsorção ocorre em monocamada.
Comparando-se os dados das isotermas com os resultados mostrados na Figura 4.31, nota-
se que, a baixas concentrações (de até cerca de 0,008 mol/L), a diminuição exponencial da
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,07 0,080
20
40
60
80
100
Efi
ciên
cia I
nco
rp
ora
çao (
%)
[PHMB] inicial (mol/L)

113
eficiência de incorporação deve ocorrer devido à adsorção do PHMB em monocamada,
exaurindo progressivamente os sítios disponíveis para adsorção na superfície da matriz. A
maiores concentrações, as moléculas de PHMB passam a adsorver em multicamadas sobre as
moléculas de PHMB já adsorvidas. A partir deste ponto, a concentração de PHMB torna-se alta
em relação aos sítios disponíveis para adsorção, diminuindo a eficiência de incorporação, mas
não a fazendo tender a zero por não se atingir na faixa estudada o platô de máxima adsorção,
estimado em 3,19 mol/kg (equivalente a 7,97 g/g), conforme indicado pelo parâmetro qmax da
Tabela 4.17.
Tendo em vista os diferentes resultados obtidos na faixa de concentrações de PHMB
estudada, selecionaram-se duas condições bastante distintas entre si para comparação em termos
do aspecto, morfologia, espessura, comportamento nas soluções de PHMB, eficiência de
incorporação e cinética de liberação. As formulações selecionadas foram as de concentrações
iniciais de PHMB de 4x10-5 e 2x10-2 mol/L. A primeira delas apresentou alta eficiência de
incorporação, de cerca de 70%. Já a segunda formulação apresentou eficiência de incorporação
de apenas cerca de 6%. Não foram selecionadas formulações das extremidades da faixa de
concentrações testada pois, a concentrações muito altas de PHMB, as membranas se tornaram
muito quebradiças e de difícil manipulação. Já a concentrações muito baixas de PHMB, a
pequena quantidade impregnada possivelmente dificultaria a medida precisa da cinética de
liberação em razão da limitada sensibilidade do método de detecção, como observado no
Apêndice IV.
4.3.2.2. Caracterização das membranas contendo PHMB incorporado por adsorção a partir de
solução aquosa
Nas Figuras 4.32 e 4.33 são mostrados os aspectos visuais e as imagens obtidas por
microscopia eletrônica de varredura (MEV) das membranas de tamanho inicial de 4 cm x 4 cm
contendo PHMB incorporado por adsorção a partir de solução aquosa a concentrações iniciais de
4x10-5 e 2x10-2 mol/L.
É possível observar pela Figura 4.32 que as membranas contendo PHMB incorporado por
adsorção na solução de menor concentração inicial, de 4x10-5 mol/L, mostraram-se mais

114
enrugadas do que as membranas expostas à solução de maior concentração inicial, de
2x10-2 mol/L.
Figura 4.32: Aspecto visual das membranas contendo PHMB incorporado por adsorção a 37°C e
100 rpm a partir de soluções aquosas de diferentes concentrações: A) 4x10-5 mol/L,
B) 2x10-2 mol/L.
Figura 4.33: Microscopia eletrônica de varredura (MEV) das membranas contendo ou não
PHMB incorporado por adsorção a 37°C e 100 rpm a partir de soluções aquosas de diferentes
concentrações: A) sem PHMB, B) 4x10-5 mol/L, C) 2x10-2 mol/L.
P0% P2% P10%(A)
(B)
1 cm 1 cm 1 cm
1 cm 1 cm 1 cm
(A) (A)
(B) (B)P0% P2% P10%
300µm
P2%
P10%
300µm
300µm
P0%
300µm
300µm
300µm
300µm
300µm
300µm
(A) (B) (C)
(A)
(A)
(B)
(B)
(C)
(C)

115
As imagens obtidas por microscopia eletrônica de varredura (Figura 4.33) também indicam
que houve deformação da estrutura das membranas na solução de 4x10-5 mol/L, podendo-se
observar superfícies menos lisas e lamelas transversais menos perceptíveis. Por outro lado, na
solução de 2x10-2 mol/L, as membranas apresentaram superfície mais lisa e estrutura da seção
transversal próxima à observada para membranas sem PHMB.
Pode-se supor que as diferenças de aspecto e morfologia entre as membranas expostas às
diferentes soluções são devidas à forma com que o PHMB se depositou sobre a superfície.
Enquanto na solução de 2x10-2 mol/L houve o depósito de múltiplas camadas, tornando a
superfície mais lisa; na solução de 4x10-5 mol/L, o depósito de PHMB sobre a superfície ocorreu
de forma irregular.
As variações de espessura e de massa após a impregnação por adsorção também foram
medidas, como mostrado nas Tabelas 4.18 e 4.19.
Tabela 4.18: Espessura média das membranas secas contendo ou não PHMB incorporado por
adsorção a partir de solução aquosa a 4x10-5 mol/L e 2x10-2 mol/L.
[PHMB] inicial
(mol/L) Formulação
Espessura inicial
(mm)
Espessura final
(mm)
4x10-5
P0% 0,08 ± 0,01 aA 0,11 ± 0,01 bA
P2% 0,36 ± 0,03 aB 0,12 ± 0,01 bA
P10% 0,50 ± 0,04 aC 0,41 ± 0,02 bC
2x10-2
P0% 0,08 ± 0,01 aA 0,08 ± 0,00 aA
P2% 0,36 ± 0,03 aB 0,29 ± 0,01 bB
P10% 0,50 ± 0,04 aC 0,51 ± 0,04 aD
Mesma letra minúscula na mesma linha indica que não há diferença significativa entre os valores médios.
Mesma letra maiúscula na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de
Tukey, p<0,05).
Não foi verificada influência significativa da porosidade das membranas sobre a variação
de massa das mesmas nas soluções de PHMB. Observou-se que, na solução de menor
concentração (4x10-5 mol/L), as membranas P0%, P2% e P10% apresentaram perdas de massa de
cerca de 9%, 11% e 13%, respectivamente. Já na solução de maior concentração (2x10-2 mol/L),
foram observados ganhos de massa de cerca de 67%, 70% e 65%, respectivamente. As

116
formulações P2% e P10% tiveram suas espessuras reduzidas nas soluções de 4x10-5 mol/L,
corroborando os dados de perda de massa. Já na solução de 2x10-2 mol/L, a formulação P2%
teve uma pequena redução da espessura, o que poderia ser relacionado à compactação das
lamelas, e não à perda de massa como no caso anterior.
Tabela 4.19: Capacidade de absorção e variação de massa das membranas em soluções aquosas
de PHMB a 4x10-5 mol/L e 2x10-2 mol/L.
Concentração inicial
de PHMB (mol/L) Formulação Absorção (g/g) Variação de massa (%)
4x10-5
P0% 29,15 ± 10,67 a,b,c -9,04 ± 3,80 a
P2% 37,93 ± 0,11 a,b -11,32 ± 2,89 a
P10% 22,17 ± 2,19 a,b,c -13,23 ± 1,75 a
2x10-2
P0% 8,24 ± 0,58 a,c 67,42 ± 6,24 b
P2% 7,42 ± 0,43 a,c 70,09 ± 1,27 b
P10% 17,01 ± 0,06 a,b,c 64,89 ± 7,61 b
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de Tukey,
p<0,05).
A capacidade de absorção das membranas variou de 22 a 38 g/g na solução de
4x10-5 mol/L e de 7 a 17 g/g na solução de 2x10-2 mol/L, não sendo verificada influência
significativa da porosidade das membranas sobre os valores obtidos. Estes resultados corroboram
os dados de variação de massa e espessura, uma vez que, na solução de menor concentração de
PHMB foi observada maior desestabilização das membranas, provavelmente devido à maior
entrada de água na estrutura. Já na solução de maior concentração de PHMB, a entrada de água
na estrutura seria desfavorecida pela presença do antimicrobiano, o qual blindaria as cargas dos
polissacarídeos, como observado para as diversas soluções salinas mencionadas no item 4.2.6.
Verifica-se que os resultados obtidos nas análises de aspecto visual, microscopia,
espessura, capacidade de absorção e variação de massa foram bem diferentes nas duas
concentrações iniciais de PHMB estudadas. Acredita-se que, na solução de maior concentração
(2x10-2 mol/L), a ação reticulante do PHMB prevaleceu, tendo ocorrido interação entre os
grupos biguanida do antimicrobiano e os grupos carboxila livres do alginato, o que manteve a
estrutura das membranas íntegra e fez com que as mesmas ganhassem massa devido ao PHMB

117
incorporado, contudo, verificou-se que o material tornou-se quebradiço nestas condições. Era
esperado que as membranas apresentassem também aumento de espessura, devido ao ganho de
massa. Observou-se, no entanto, que a ação do PHMB ocorreu no sentido de manter a espessura
ou mesmo compactar as membranas, como foi observado para a formulação P2%.
Por outro lado, nas soluções de menor concentração (4x10-5 mol/L), o efeito reticulante do
PHMB não foi percebido, havendo alteração do aspecto visual e microscópico, perda de massa e,
na maioria dos casos, redução significativa da espessura decorrente da perda de massa. Desta
forma, a baixas concentrações, prevaleceria o efeito da água presente no meio.
Os dados de quantidade de PHMB incorporado às membranas e eficiência de incorporação
por adsorção a partir de soluções aquosas são mostrados na Tabela 4.20.
Tabela 4.20: Eficiência de incorporação de PHMB por adsorção a partir de soluções aquosas
com concentração de 4x10-5 mol/L e 2x10-2 mol/L.
Concentração inicial
de PHMB (mol/L) Formulação
PHMB incorporado
(mg/g)
Eficiência de
incorporação (%)
4x10-5
P0% 49,9 ± 0,3 b 58,4 ± 7,6 a
P2% 45,5 ± 0,1 b 68,4 ± 1,8 a
P10% 33,8 ± 1,8 a 77,2 ± 2,9 a
2x10-2
P0% 2002,9 ± 497,3 c 5,0 ± 1,0 b
P2% 1412,9 ± 330,6 c 6,3 ± 1,2 b
P10% 1651,5 ± 14,2 c 7,5 ± 0,4 b
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de Tukey,
p<0,05).
As quantidades de PHMB incorporado e os dados de eficiência de incorporação mostrados
na Tabela 4.20 indicam que na menor concentração inicial de PHMB (4x10-5 mol/L) foram
alcançadas maiores eficiências de incorporação do antimicrobiano. Este resultado se deve
provavelmente à grande quantidade de sítios disponíveis para adsorção em comparação à
quantidade de PHMB em solução. Já na maior concentração inicial (2x10-2 mol/L), a quantidade
de PHMB em solução seria muito alta em relação aos sítios disponíveis para adsorção, mesmo
supondo que o antimicrobiano adsorve em multicamadas.
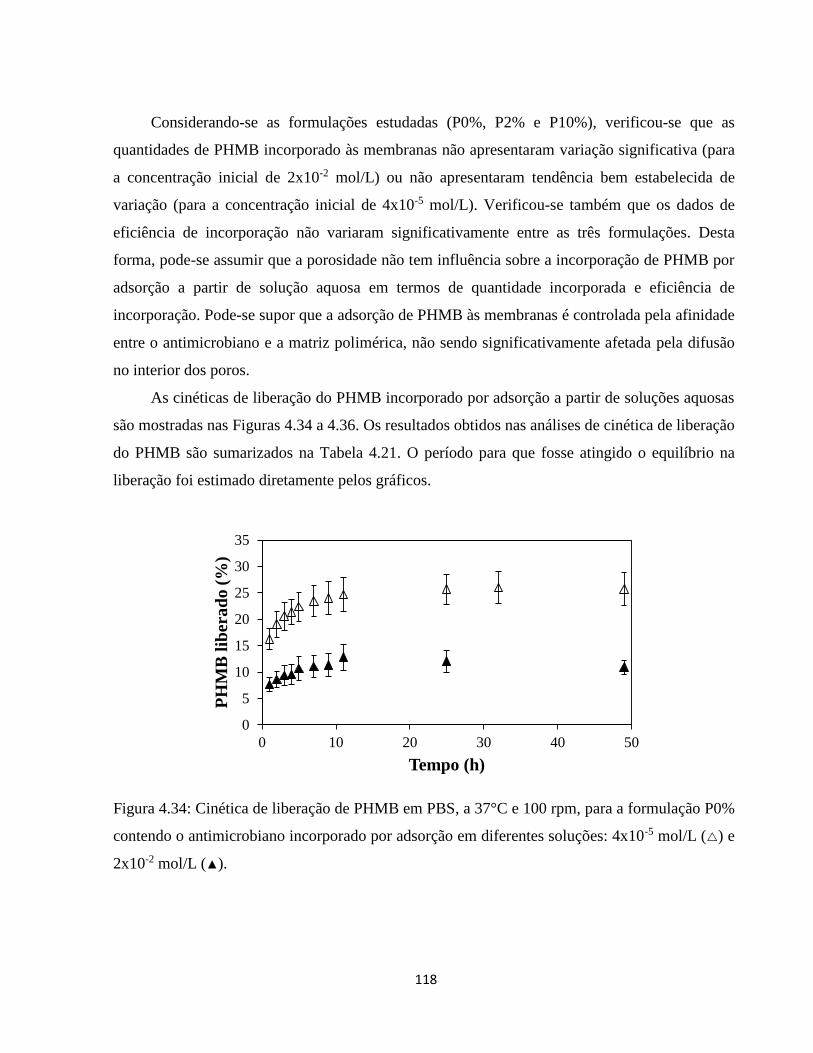
118
Considerando-se as formulações estudadas (P0%, P2% e P10%), verificou-se que as
quantidades de PHMB incorporado às membranas não apresentaram variação significativa (para
a concentração inicial de 2x10-2 mol/L) ou não apresentaram tendência bem estabelecida de
variação (para a concentração inicial de 4x10-5 mol/L). Verificou-se também que os dados de
eficiência de incorporação não variaram significativamente entre as três formulações. Desta
forma, pode-se assumir que a porosidade não tem influência sobre a incorporação de PHMB por
adsorção a partir de solução aquosa em termos de quantidade incorporada e eficiência de
incorporação. Pode-se supor que a adsorção de PHMB às membranas é controlada pela afinidade
entre o antimicrobiano e a matriz polimérica, não sendo significativamente afetada pela difusão
no interior dos poros.
As cinéticas de liberação do PHMB incorporado por adsorção a partir de soluções aquosas
são mostradas nas Figuras 4.34 a 4.36. Os resultados obtidos nas análises de cinética de liberação
do PHMB são sumarizados na Tabela 4.21. O período para que fosse atingido o equilíbrio na
liberação foi estimado diretamente pelos gráficos.
Figura 4.34: Cinética de liberação de PHMB em PBS, a 37°C e 100 rpm, para a formulação P0%
contendo o antimicrobiano incorporado por adsorção em diferentes soluções: 4x10-5 mol/L () e
2x10-2 mol/L (▲).
0
5
10
15
20
25
30
35
0 10 20 30 40 50
PH
MB
lib
era
do
(%
)
Tempo (h)
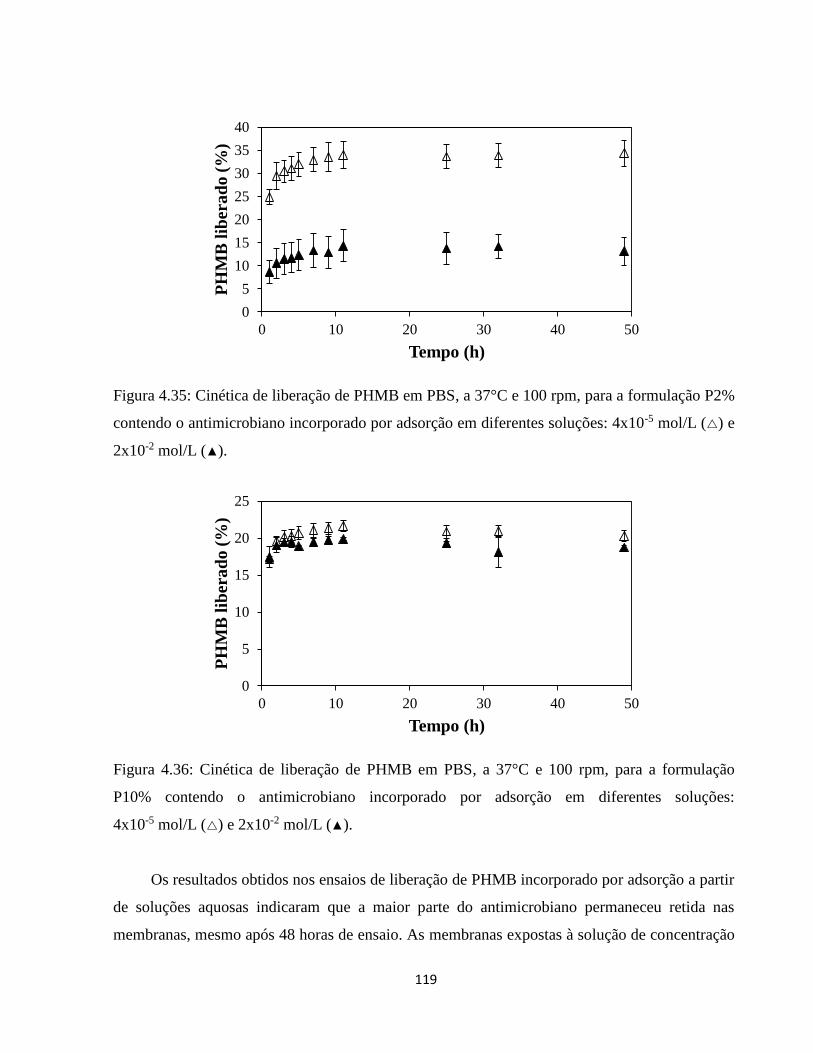
119
Figura 4.35: Cinética de liberação de PHMB em PBS, a 37°C e 100 rpm, para a formulação P2%
contendo o antimicrobiano incorporado por adsorção em diferentes soluções: 4x10-5 mol/L () e
2x10-2 mol/L (▲).
Figura 4.36: Cinética de liberação de PHMB em PBS, a 37°C e 100 rpm, para a formulação
P10% contendo o antimicrobiano incorporado por adsorção em diferentes soluções:
4x10-5 mol/L () e 2x10-2 mol/L (▲).
Os resultados obtidos nos ensaios de liberação de PHMB incorporado por adsorção a partir
de soluções aquosas indicaram que a maior parte do antimicrobiano permaneceu retida nas
membranas, mesmo após 48 horas de ensaio. As membranas expostas à solução de concentração
0
5
10
15
20
25
30
35
40
0 10 20 30 40 50
PH
MB
lib
era
do
(%
)
Tempo (h)
0
5
10
15
20
25
0 10 20 30 40 50
PH
MB
lib
era
do
(%
)
Tempo (h)
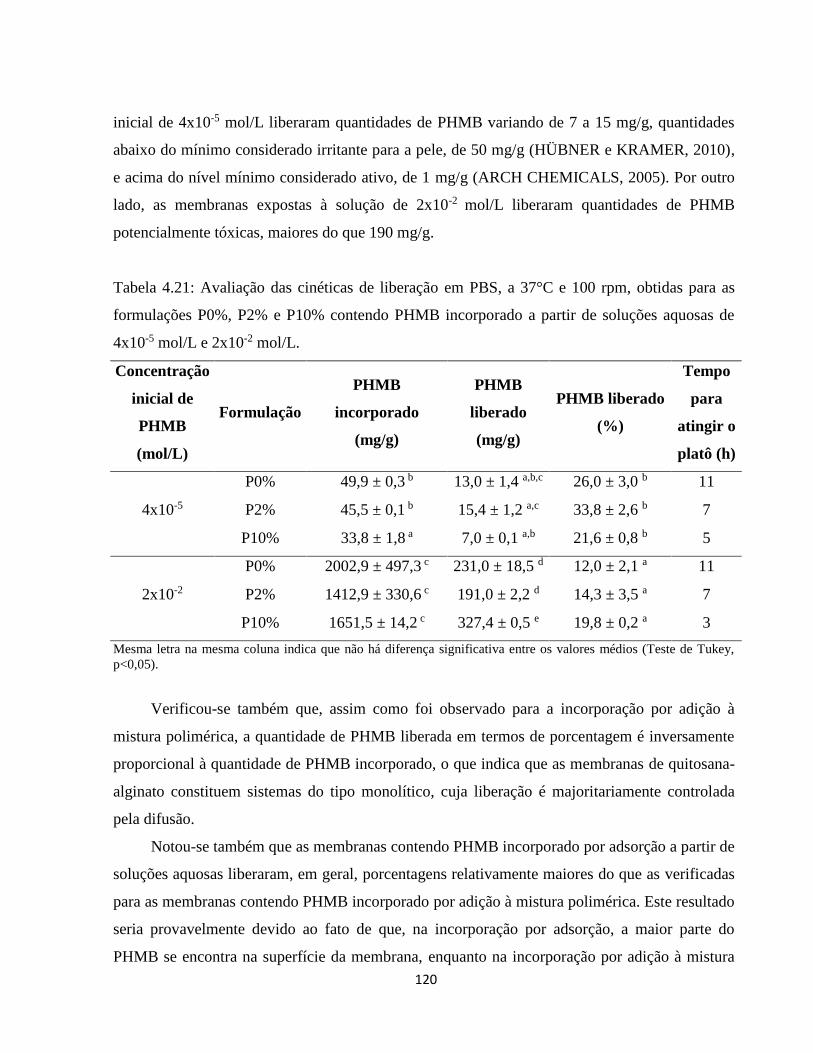
120
inicial de 4x10-5 mol/L liberaram quantidades de PHMB variando de 7 a 15 mg/g, quantidades
abaixo do mínimo considerado irritante para a pele, de 50 mg/g (HÜBNER e KRAMER, 2010),
e acima do nível mínimo considerado ativo, de 1 mg/g (ARCH CHEMICALS, 2005). Por outro
lado, as membranas expostas à solução de 2x10-2 mol/L liberaram quantidades de PHMB
potencialmente tóxicas, maiores do que 190 mg/g.
Tabela 4.21: Avaliação das cinéticas de liberação em PBS, a 37°C e 100 rpm, obtidas para as
formulações P0%, P2% e P10% contendo PHMB incorporado a partir de soluções aquosas de
4x10-5 mol/L e 2x10-2 mol/L.
Concentração
inicial de
PHMB
(mol/L)
Formulação
PHMB
incorporado
(mg/g)
PHMB
liberado
(mg/g)
PHMB liberado
(%)
Tempo
para
atingir o
platô (h)
4x10-5
P0% 49,9 ± 0,3 b 13,0 ± 1,4 a,b,c 26,0 ± 3,0 b 11
P2% 45,5 ± 0,1 b 15,4 ± 1,2 a,c 33,8 ± 2,6 b 7
P10% 33,8 ± 1,8 a 7,0 ± 0,1 a,b 21,6 ± 0,8 b 5
2x10-2
P0% 2002,9 ± 497,3 c 231,0 ± 18,5 d 12,0 ± 2,1 a 11
P2% 1412,9 ± 330,6 c 191,0 ± 2,2 d 14,3 ± 3,5 a 7
P10% 1651,5 ± 14,2 c 327,4 ± 0,5 e 19,8 ± 0,2 a 3
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de Tukey,
p<0,05).
Verificou-se também que, assim como foi observado para a incorporação por adição à
mistura polimérica, a quantidade de PHMB liberada em termos de porcentagem é inversamente
proporcional à quantidade de PHMB incorporado, o que indica que as membranas de quitosana-
alginato constituem sistemas do tipo monolítico, cuja liberação é majoritariamente controlada
pela difusão.
Notou-se também que as membranas contendo PHMB incorporado por adsorção a partir de
soluções aquosas liberaram, em geral, porcentagens relativamente maiores do que as verificadas
para as membranas contendo PHMB incorporado por adição à mistura polimérica. Este resultado
seria provavelmente devido ao fato de que, na incorporação por adsorção, a maior parte do
PHMB se encontra na superfície da membrana, enquanto na incorporação por adição à mistura

121
polimérica, ter-se-ia boa parte do antimicrobiano distribuída no interior das lamelas. Sendo
assim, a liberação do PHMB incorporado por adsorção a partir de solução aquosa ocorreria mais
facilmente.
Diferentemente do que foi verificado para a incorporação de PHMB por adição à mistura
polimérica, a porosidade das membranas pareceu influenciar os tempos de estabilização da
cinética de liberação. As membranas densas (P0%) apresentaram, em ambos os casos, tempo de
estabilização igual a 11 horas. As membranas de porosidade intermediária (P2%) apresentaram
tempo de estabilização igual a 7 horas em ambos os casos. Já as membranas mais porosas
(P10%) apresentaram tempos de estabilização de 5 horas para a solução de concentração inicial
de 4x10-5 mol/L e de 3 horas para a solução de concentração inicial de 2x10-2 mol/L. Estes
resultados indicam que as membranas mais porosas liberam o PHMB incorporado mais
rapidamente.
Como conclusão desta etapa, verificou-se que, em termos da eficiência de incorporação,
seria mais vantajoso empregar soluções de PHMB com concentração inicial abaixo de
0,8x10-2 mol/L, pois, como observado no item 4.3.2.1, a eficiência de incorporação a
concentrações maiores do que 0,8x10-2 mol/L é muito baixa, menor do que 7%. Além disso, com
o aumento da concentração inicial de PHMB, pode-se atingir níveis potencialmente tóxicos do
antimicrobiano, como observado para a solução de 2x10-2 mol/L.
4.3.3. Incorporação de PHMB mediada por CO2 supercrítico
O CO2 supercrítico tem sido muito estudado para o processamento de polímeros. As
principais razões para a sua utilização são o efeito plastificante e as temperaturas amenas de
trabalho, com aplicação no processamento de materiais termossensíveis. Existe também o
potencial uso do CO2 supercrítico para a impregnação de fármacos em matrizes poliméricas sem
a necessidade do uso de solventes cujos resíduos possam ser deletérios ao paciente, um processo
que, com frequência, não requer a posterior secagem do material.
Primeiramente, as membranas foram submetidas ao processamento com CO2 supercrítico
na ausência de PHMB em condições controladas de pressão (100 e 300 bar), temperatura (45°C)
e taxa de despressurização (5 bar/min), de forma a verificar se o material suportaria possíveis
condições de incorporação de agentes ativos por via supercrítica. Os efeitos do processamento
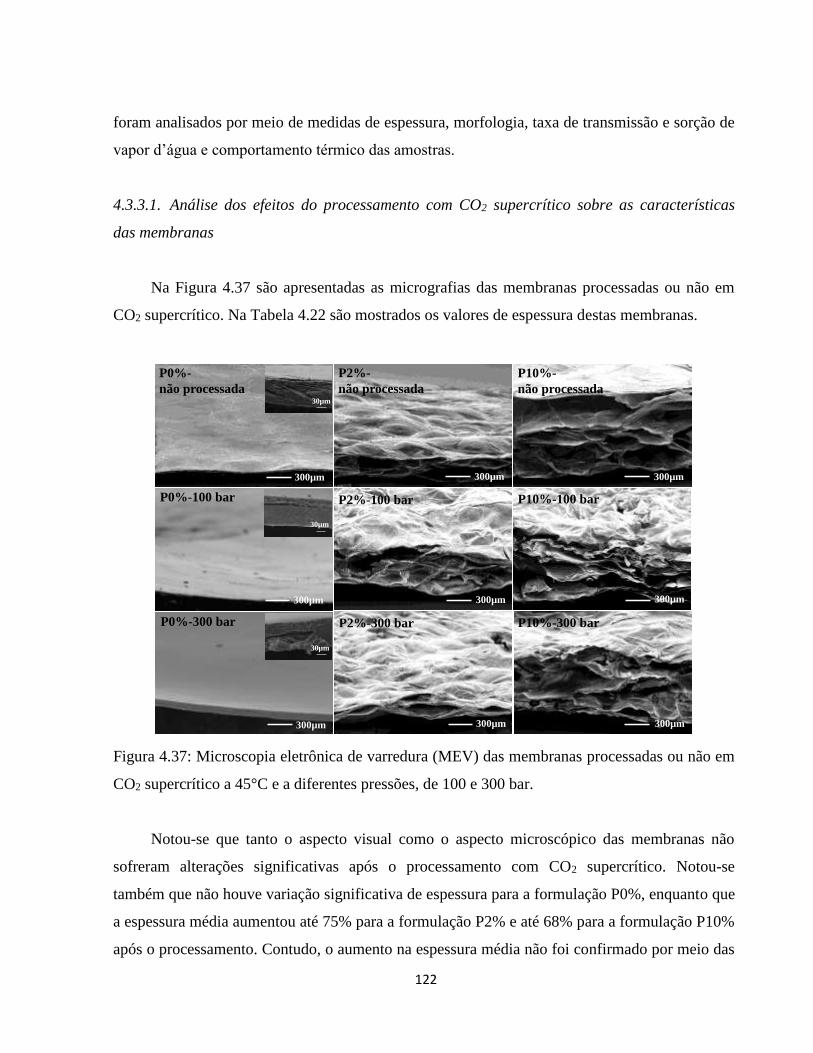
122
foram analisados por meio de medidas de espessura, morfologia, taxa de transmissão e sorção de
vapor d’água e comportamento térmico das amostras.
4.3.3.1. Análise dos efeitos do processamento com CO2 supercrítico sobre as características
das membranas
Na Figura 4.37 são apresentadas as micrografias das membranas processadas ou não em
CO2 supercrítico. Na Tabela 4.22 são mostrados os valores de espessura destas membranas.
Figura 4.37: Microscopia eletrônica de varredura (MEV) das membranas processadas ou não em
CO2 supercrítico a 45°C e a diferentes pressões, de 100 e 300 bar.
Notou-se que tanto o aspecto visual como o aspecto microscópico das membranas não
sofreram alterações significativas após o processamento com CO2 supercrítico. Notou-se
também que não houve variação significativa de espessura para a formulação P0%, enquanto que
a espessura média aumentou até 75% para a formulação P2% e até 68% para a formulação P10%
após o processamento. Contudo, o aumento na espessura média não foi confirmado por meio das
P10%-
não processada
P10%-100 bar
P10%-300 bar
P2%-100 bar
P2%-300 bar
300µm
P2%-
não processada
P0%-
não processada
P0%-100 bar
P0%-300 bar
300µm
300µm
300µm
300µm
300µm 300µm
300µm 300µm
30µm
30µm
30µm
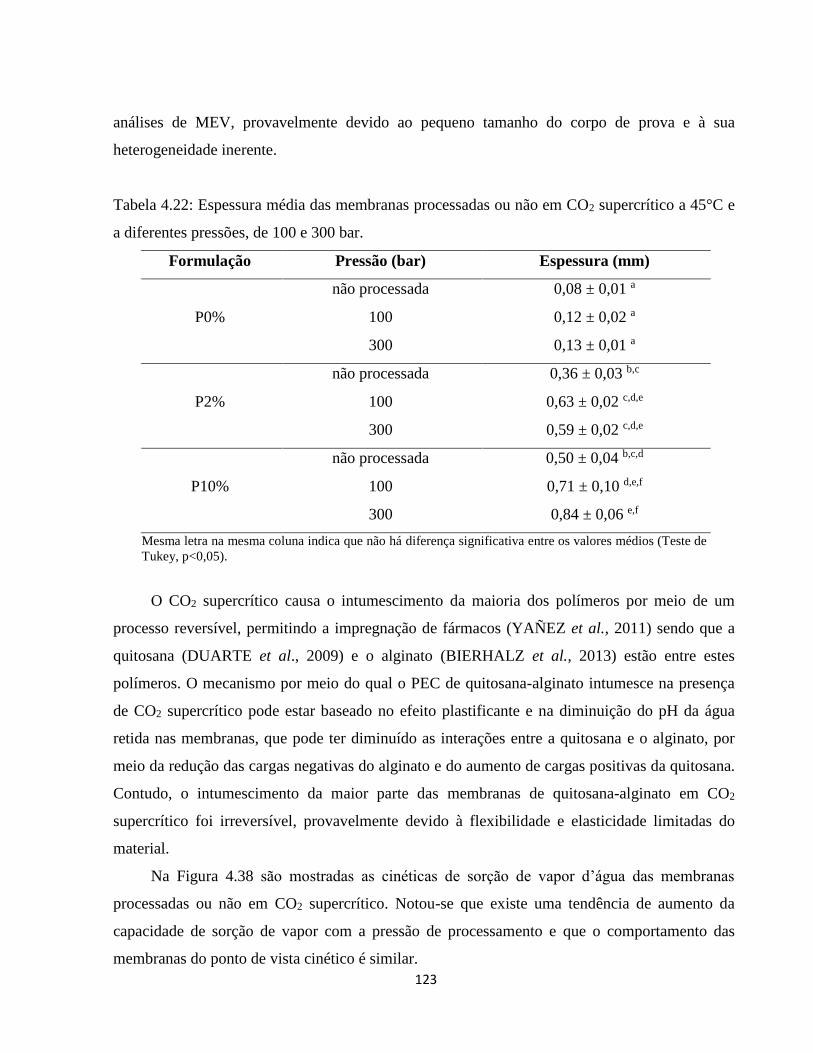
123
análises de MEV, provavelmente devido ao pequeno tamanho do corpo de prova e à sua
heterogeneidade inerente.
Tabela 4.22: Espessura média das membranas processadas ou não em CO2 supercrítico a 45°C e
a diferentes pressões, de 100 e 300 bar.
Formulação Pressão (bar) Espessura (mm)
P0%
não processada 0,08 ± 0,01 a
100 0,12 ± 0,02 a
300 0,13 ± 0,01 a
P2%
não processada 0,36 ± 0,03 b,c
100 0,63 ± 0,02 c,d,e
300 0,59 ± 0,02 c,d,e
P10%
não processada 0,50 ± 0,04 b,c,d
100 0,71 ± 0,10 d,e,f
300 0,84 ± 0,06 e,f
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de
Tukey, p<0,05).
O CO2 supercrítico causa o intumescimento da maioria dos polímeros por meio de um
processo reversível, permitindo a impregnação de fármacos (YAÑEZ et al., 2011) sendo que a
quitosana (DUARTE et al., 2009) e o alginato (BIERHALZ et al., 2013) estão entre estes
polímeros. O mecanismo por meio do qual o PEC de quitosana-alginato intumesce na presença
de CO2 supercrítico pode estar baseado no efeito plastificante e na diminuição do pH da água
retida nas membranas, que pode ter diminuído as interações entre a quitosana e o alginato, por
meio da redução das cargas negativas do alginato e do aumento de cargas positivas da quitosana.
Contudo, o intumescimento da maior parte das membranas de quitosana-alginato em CO2
supercrítico foi irreversível, provavelmente devido à flexibilidade e elasticidade limitadas do
material.
Na Figura 4.38 são mostradas as cinéticas de sorção de vapor d’água das membranas
processadas ou não em CO2 supercrítico. Notou-se que existe uma tendência de aumento da
capacidade de sorção de vapor com a pressão de processamento e que o comportamento das
membranas do ponto de vista cinético é similar.
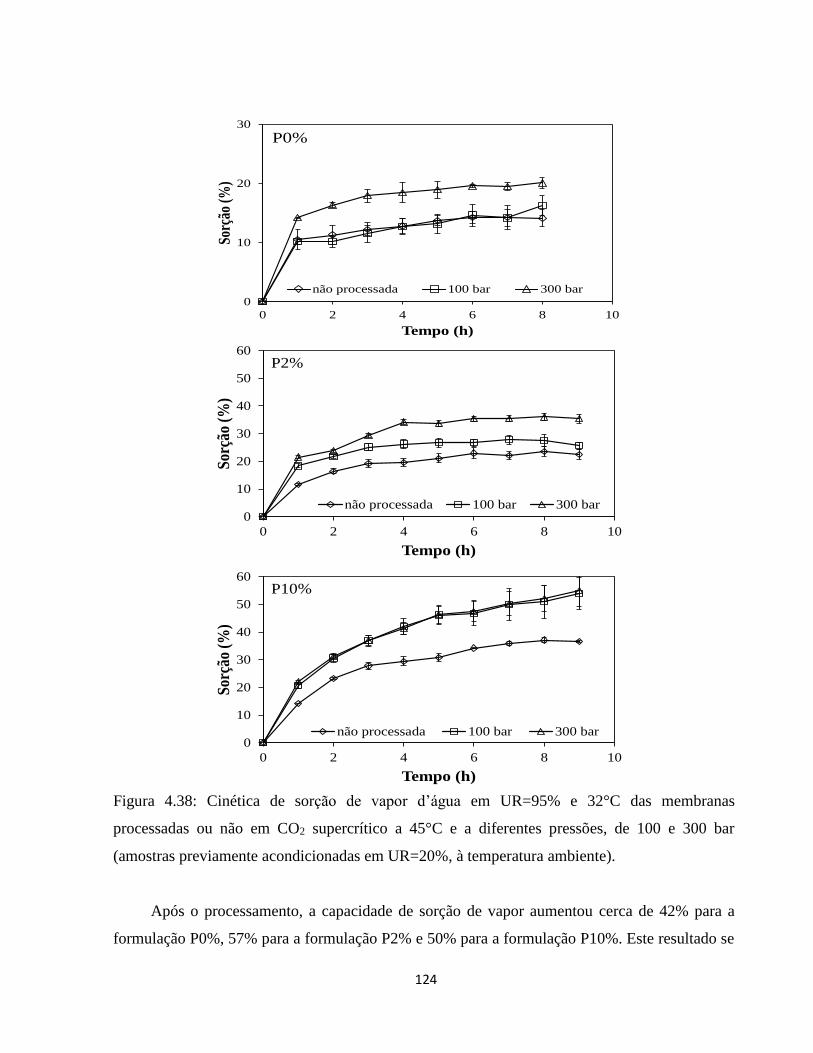
124
Figura 4.38: Cinética de sorção de vapor d’água em UR=95% e 32°C das membranas
processadas ou não em CO2 supercrítico a 45°C e a diferentes pressões, de 100 e 300 bar
(amostras previamente acondicionadas em UR=20%, à temperatura ambiente).
Após o processamento, a capacidade de sorção de vapor aumentou cerca de 42% para a
formulação P0%, 57% para a formulação P2% e 50% para a formulação P10%. Este resultado se
0
10
20
30
0 2 4 6 8 10
Sorç
ão (
%)
Tempo (h)
não processada 100 bar 300 bar
P0%
0
10
20
30
40
50
60
0 2 4 6 8 10
Sor
ção
(%)
Tempo (h)
não processada 100 bar 300 bar
P2%
0
10
20
30
40
50
60
0 2 4 6 8 10
Sor
ção
(%)
Tempo (h)
não processada 100 bar 300 bar
P10%

125
deve provavelmente à acidificação do PEC pelo CO2 supercrítico, o que pode ter diminuído as
interações entre a quitosana e o alginato, aumentando o espaçamento entre as cadeias
poliméricas. Este resultado foi considerado positivo para a finalidade proposta, indicando que as
membranas processadas também poderiam apresentar uma absorção mais eficiente de fármacos e
do exsudato liberado por lesões cutâneas.
Como comentado anteriormente, capacidades de sorção de vapor entre 29% e 57% são
consideradas adequadas para o tratamento de lesões de pele (DIAS et al., 2011; DIAS et al.,
2013). Observou-se que a formulação P2% processada a 300 bar e que a formulação P10%
processada e não-processada apresentaram capacidades de sorção de vapor dentro da faixa
desejada. Assim, em relação à capacidade de sorção de vapor, essas formulações podem ser
consideradas como as mais adequadas para o tratamento de lesões de pele.
Na Tabela 4.23 são mostrados os valores de ângulo de contato e taxa de transmissão de
vapor d’água (TVA) através das membranas.
Tabela 4.23: Taxa de transmissão de vapor em UR igual a 50% e 32°C através das membranas
processadas ou não em CO2 supercrítico a 45°C e a diferentes pressões, de 100 e 300 bar.
Formulação Pressão (bar) TVA (g/m2.dia) Ângulo de contato (°)
P0%
não processada 237,0 ± 7,8 a 62,5 ± 2,9 a,b,d,e
100 591,0 ± 22,1 b,d,e,f 72,2 ± 2,2 a,b,c,d,e
300 320,1 ± 38,6 a 71,9 ± 3,4 a,b,c,d,e
P2%
não processada 481,5 ± 31,3 b,c,d,e 63,2 ± 2,7 a,b,d,e
100 488,1 ± 20,1 b,c,d,e 68,1 ± 2,2 a,b,c,d,e
300 461,7 ± 36,7 b,c,d 78,2 ± 2,8 b,c,e
P10%
não processada 576,2 ± 29,0 b,c,d,e,f 67,0 ± 3,2 a,b,c,d,e
100 768,9 ± 0,6 f,g 62,7 ± 2,5 a,b,d
300 710,3 ± 8,1 d,e,f,g 74,0 ± 2,4 a,b,c,e
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de Tukey,
p<0,05).
Verificou-se que a transmissão de vapor (TVA) não apresentou uma tendência de mudança
no comportamento com a variação da pressão de processamento, sendo que os valores medidos
são adequados para o tratamento de feridas pouco exudativas, de acordo com recomendações da
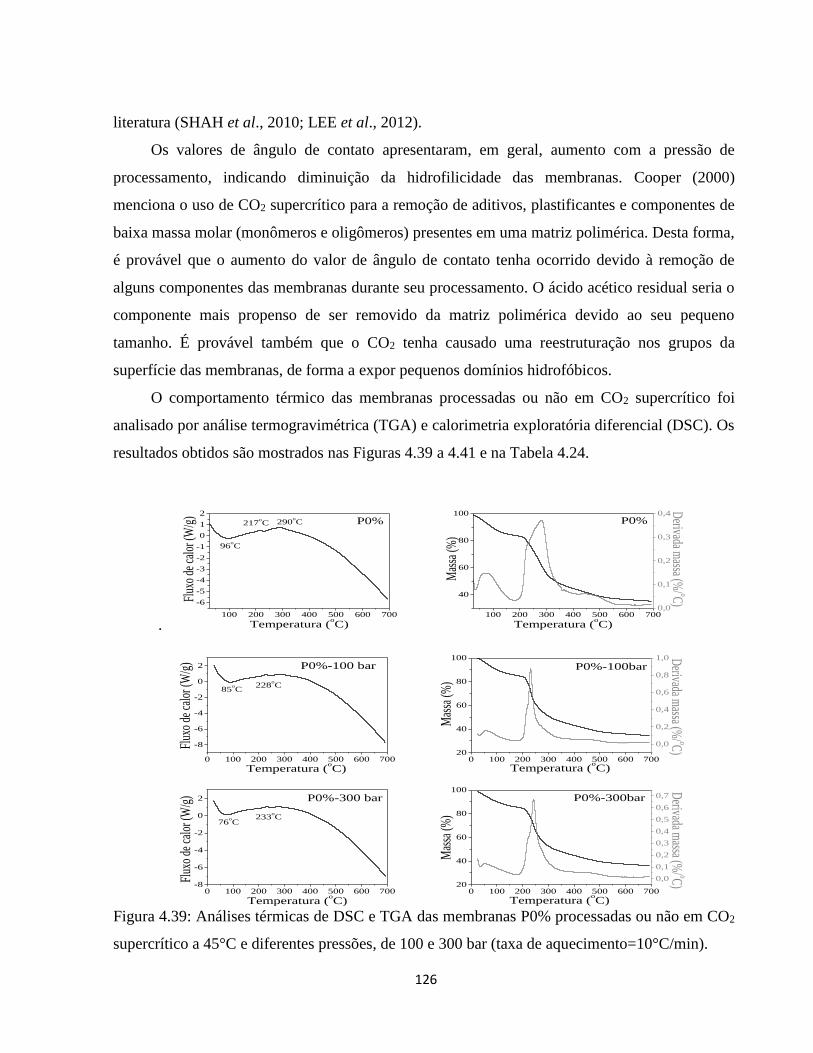
126
literatura (SHAH et al., 2010; LEE et al., 2012).
Os valores de ângulo de contato apresentaram, em geral, aumento com a pressão de
processamento, indicando diminuição da hidrofilicidade das membranas. Cooper (2000)
menciona o uso de CO2 supercrítico para a remoção de aditivos, plastificantes e componentes de
baixa massa molar (monômeros e oligômeros) presentes em uma matriz polimérica. Desta forma,
é provável que o aumento do valor de ângulo de contato tenha ocorrido devido à remoção de
alguns componentes das membranas durante seu processamento. O ácido acético residual seria o
componente mais propenso de ser removido da matriz polimérica devido ao seu pequeno
tamanho. É provável também que o CO2 tenha causado uma reestruturação nos grupos da
superfície das membranas, de forma a expor pequenos domínios hidrofóbicos.
O comportamento térmico das membranas processadas ou não em CO2 supercrítico foi
analisado por análise termogravimétrica (TGA) e calorimetria exploratória diferencial (DSC). Os
resultados obtidos são mostrados nas Figuras 4.39 a 4.41 e na Tabela 4.24.
.
Figura 4.39: Análises térmicas de DSC e TGA das membranas P0% processadas ou não em CO2
supercrítico a 45°C e diferentes pressões, de 100 e 300 bar (taxa de aquecimento=10°C/min).
100 200 300 400 500 600 700
-6
-5
-4
-3
-2
-1
0
1
2
290oC217
oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P0%
96oC
100 200 300 400 500 600 700
40
60
80
100
P0%
Temperatura (oC)
Mas
sa (%
)
0,0
0,1
0,2
0,3
0,4
Derivada m
assa (%/ oC
)
0 100 200 300 400 500 600 700
-8
-6
-4
-2
0
2
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P0%-100 bar
85oC
228oC
0 100 200 300 400 500 600 70020
40
60
80
100
P0%-100bar
Temperatura (oC)
Mas
sa (%
)
0,0
0,2
0,4
0,6
0,8
1,0 Derivada m
assa (%/ oC
)
0 100 200 300 400 500 600 700-8
-6
-4
-2
0
2
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P0%-300 bar
76oC
233oC
0 100 200 300 400 500 600 70020
40
60
80
100
P0%-300bar
Temperatura (oC)
Mas
sa (%
)
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
Derivada m
assa (%/ oC
)
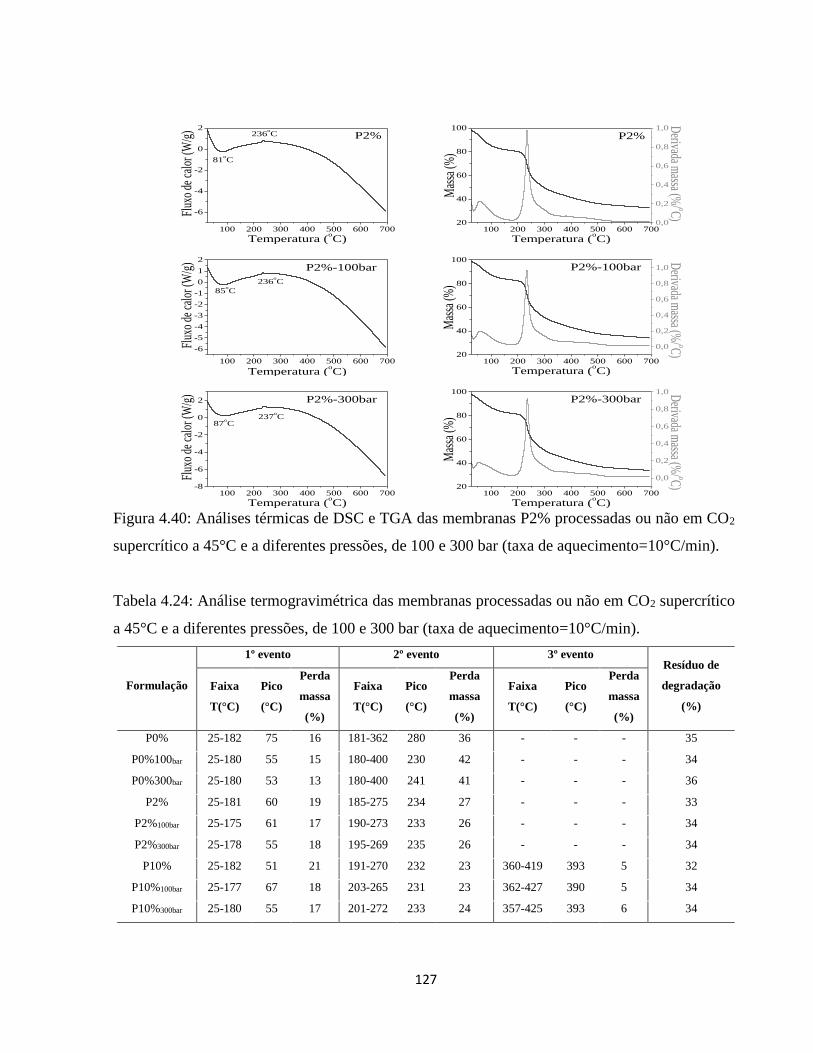
127
Figura 4.40: Análises térmicas de DSC e TGA das membranas P2% processadas ou não em CO2
supercrítico a 45°C e a diferentes pressões, de 100 e 300 bar (taxa de aquecimento=10°C/min).
Tabela 4.24: Análise termogravimétrica das membranas processadas ou não em CO2 supercrítico
a 45°C e a diferentes pressões, de 100 e 300 bar (taxa de aquecimento=10°C/min).
Formulação
1º evento 2º evento 3º evento Resíduo de
degradação
(%)
Faixa
T(°C)
Pico
(°C)
Perda
massa
(%)
Faixa
T(°C)
Pico
(°C)
Perda
massa
(%)
Faixa
T(°C)
Pico
(°C)
Perda
massa
(%)
P0% 25-182 75 16 181-362 280 36 - - - 35
P0%100bar 25-180 55 15 180-400 230 42 - - - 34
P0%300bar 25-180 53 13 180-400 241 41 - - - 36
P2% 25-181 60 19 185-275 234 27 - - - 33
P2%100bar 25-175 61 17 190-273 233 26 - - - 34
P2%300bar 25-178 55 18 195-269 235 26 - - - 34
P10% 25-182 51 21 191-270 232 23 360-419 393 5 32
P10%100bar 25-177 67 18 203-265 231 23 362-427 390 5 34
P10%300bar 25-180 55 17 201-272 233 24 357-425 393 6 34
100 200 300 400 500 600 700
-6
-4
-2
0
2236
oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P2%
81oC
100 200 300 400 500 600 70020
40
60
80
100
P2%
Temperatura (oC)
Mas
sa (%
)
0,0
0,2
0,4
0,6
0,8
1,0
Derivada m
assa (%/ oC
)
100 200 300 400 500 600 700
-6
-5
-4
-3
-2
-1
0
1
2
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P2%-100bar
236oC
85oC
100 200 300 400 500 600 70020
40
60
80
100
P2%-100bar
Temperatura (oC)
Mas
sa (%
)
0,0
0,2
0,4
0,6
0,8
1,0
Derivada m
assa (%/ oC
)
100 200 300 400 500 600 700-8
-6
-4
-2
0
2
237oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P2%-300bar
87oC
100 200 300 400 500 600 70020
40
60
80
100
P2%-300bar
Temperatura (oC)
Mas
sa (%
)
0,0
0,2
0,4
0,6
0,8
1,0 Derivada m
assa (%/ oC
)
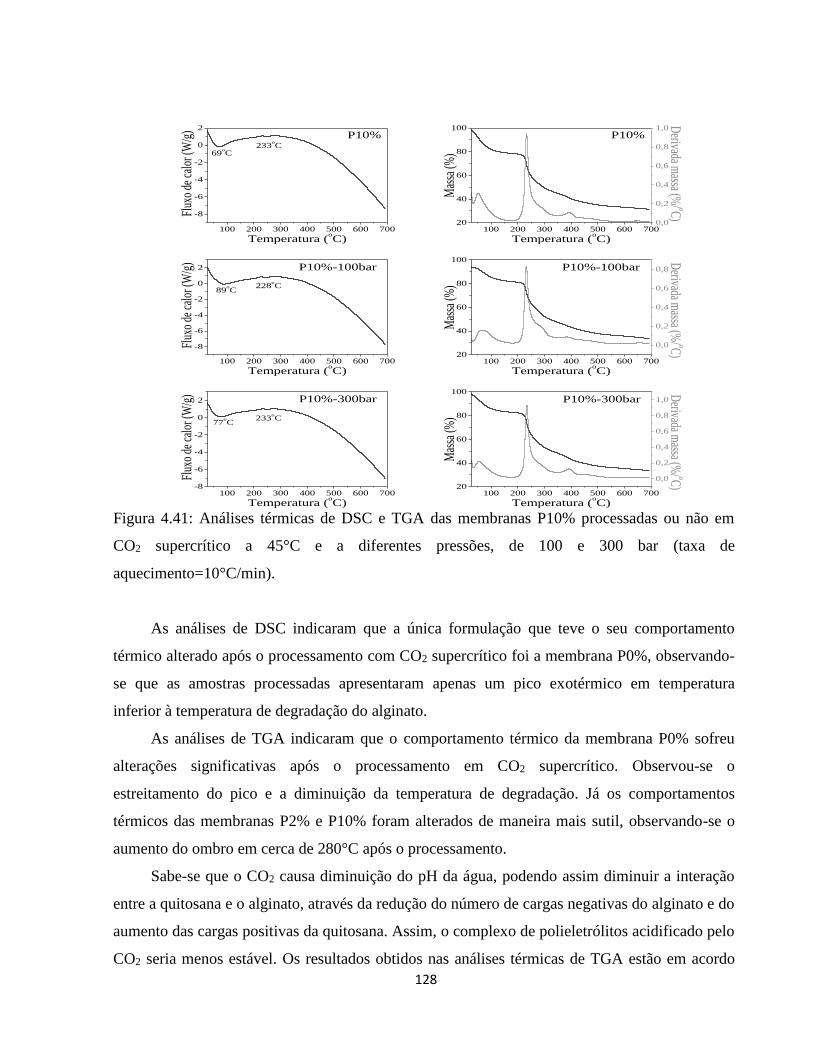
128
Figura 4.41: Análises térmicas de DSC e TGA das membranas P10% processadas ou não em
CO2 supercrítico a 45°C e a diferentes pressões, de 100 e 300 bar (taxa de
aquecimento=10°C/min).
As análises de DSC indicaram que a única formulação que teve o seu comportamento
térmico alterado após o processamento com CO2 supercrítico foi a membrana P0%, observando-
se que as amostras processadas apresentaram apenas um pico exotérmico em temperatura
inferior à temperatura de degradação do alginato.
As análises de TGA indicaram que o comportamento térmico da membrana P0% sofreu
alterações significativas após o processamento em CO2 supercrítico. Observou-se o
estreitamento do pico e a diminuição da temperatura de degradação. Já os comportamentos
térmicos das membranas P2% e P10% foram alterados de maneira mais sutil, observando-se o
aumento do ombro em cerca de 280°C após o processamento.
Sabe-se que o CO2 causa diminuição do pH da água, podendo assim diminuir a interação
entre a quitosana e o alginato, através da redução do número de cargas negativas do alginato e do
aumento das cargas positivas da quitosana. Assim, o complexo de polieletrólitos acidificado pelo
CO2 seria menos estável. Os resultados obtidos nas análises térmicas de TGA estão em acordo
100 200 300 400 500 600 700
-8
-6
-4
-2
0
2
233oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P10%
69oC
100 200 300 400 500 600 70020
40
60
80
100
P10%
Temperatura (oC)
Mas
sa (%
)
0,0
0,2
0,4
0,6
0,8
1,0
Derivada m
assa (%/ oC
)
100 200 300 400 500 600 700
-8
-6
-4
-2
0
2
228oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P10%-100bar
89oC
100 200 300 400 500 600 70020
40
60
80
100
P10%-100bar
Temperatura (oC)
Mas
sa (%
)
0,0
0,2
0,4
0,6
0,8
Derivada m
assa (%/ oC
)
100 200 300 400 500 600 700-8
-6
-4
-2
0
2
233oC
Flux
o de
cal
or (W
/g)
Temperatura (oC)
P10%-300bar
77oC
100 200 300 400 500 600 70020
40
60
80
100
P10%-300bar
Temperatura (oC)
Mas
sa (%
)
0,0
0,2
0,4
0,6
0,8
1,0
Derivada m
assa (%/ oC
)

129
com esta suposição, uma vez que, para a membrana P0%, a diminuição da temperatura de
degradação indicaria o enfraquecimento das interações entre os polissacarídeos. Para as
membranas P2% e P10%, por sua vez, o aumento do ombro a 280°C estaria indicando a
tendência ao aparecimento de um novo pico de degradação, referente à presença de quitosana
isolada.
Verificou-se, assim, que o processamento com CO2 supercrítico alterou algumas
propriedades das membranas de quitosana-alginato, observando-se, na maioria dos casos,
aumento da espessura, da capacidade de sorção de vapor d’água, da taxa de transmissão de vapor
d’água e do ângulo de contato. Apesar de os resultados das análises térmicas indicarem a
desestabilização do complexo quitosana-alginato após o processamento, a microestrutura das
membranas não foi alterada, mostrando que a integridade do material foi mantida. Desta forma,
os resultados foram considerados positivos e mostram que a impregnação de fármacos via CO2
supercrítico em membranas de quitosana-alginato pode ser encarada como uma rota promissora.
4.3.3.2. Caracterização das membranas contendo PHMB incorporado via CO2 supercrítico
Na Figura 4.42 são mostradas as imagens do aspecto visual das membranas submetidas à
impregnação de PHMB mediada por CO2 supercrítico nas diferentes condições testadas. Não
foram notadas alterações significativas na massa e no aspecto visual do material.
Embora se tenha verificado que a incorporação de fármacos em membranas de quitosana-
alginato via CO2 supercrítico é promissora, não se detectou quantidades mensuráveis de PHMB
liberado de membranas expostas a PBS nas condições testadas. Observou-se que as leituras de
absorbância dos meios de liberação resultaram em valores muito baixos e com desvio padrão
elevado.
Os valores de eficiência de incorporação nesta parte do estudo também não puderam ser
determinados. Neste caso, duas estratégias poderiam ter sido empregadas, mas não tiveram
resultados satisfatórios. Uma delas seria a medição do PHMB remanescente e eluído do sistema
supercrítico. No entanto, notou-se que, em todos os ensaios, o antimicrobiano presente
inicialmente na célula de impregnação foi arrastado pelo CO2. Além disso, não foram detectadas
quantidades mensuráveis de PHMB eluído do sistema por absorbância no UV-VIS. A segunda
estratégia seria quantificar a massa de PHMB retida nas membranas. Neste último caso, seria

130
necessário realizar a extração do PHMB total incorporado ou solubilizar as membranas com um
solvente apropriado e, dentre as alternativas testadas, nenhuma se mostrou efetiva, como
sumarizado nos Apêndices VI e VII.
Figura 4.42: Aspecto visual das membranas submetidas à incorporação de PHMB mediada por
CO2 supercrítico na condição inicial de 0,07 mg de agente antimicrobiano por miligrama de
membrana.
Supõe-se que, caso tenha ocorrido de fato impregnação do agente bioativo nas membranas,
este deva ter ficado firmemente ligado à matriz, não sendo dela deslocado quando da incubação
em solução salina tamponada. No entanto, acredita-se que, devido aos resultados obtidos
referentes às tentativas de quantificação, não houve impregnação detectável de PHMB via CO2
supercrítico.
P2%
P10%
45 C
55 C
45 C
55 C
100 bar 150 bar 200 bar 300 bar

131
4.3.4. Comparação entre diferentes métodos de incorporação de PHMB em membranas de
diferentes formulações visando à seleção das preparações mais apropriadas
Com base nos resultados obtidos acerca das membranas de quitosana-alginato contendo
PHMB, é possível estabelecer uma comparação apenas entre a incorporação por adição à mistura
polimérica e a incorporação por adsorção a partir de solução aquosa, uma vez que os resultados
obtidos na incorporação por tecnologia supercrítica foram inconclusivos. A comparação entre os
métodos encontra-se sumarizada na Tabela 4.25.
Tabela 4.25: Comparação entre os métodos de incorporação de PHMB por adição à mistura
polimérica e por adsorção a partir de solução aquosa.
Adição à mistura polimérica Adsorção a partir de solução aquosa
Eficiência de incorporação alta nas duas
condições testadas (72 a 86%)
Eficiência de incorporação alta (>70%) apenas
para baixas quantidades iniciais de PHMB (até
4x10-5 mol/L)
Liberação de baixas porcentagens do PHMB
incorporado (4 a 15%)
Liberação de baixas porcentagens do PHMB
incorporado (12 a 34%)
Não houve alteração de aspecto ou
desestabilização das membranas
Perda de massa e alteração de aspecto para as
maiores eficiências de incorporação
Em geral, a adição à mistura polimérica e a adsorção a partir de solução aquosa resultaram
em altas eficiências de incorporação quando foram empregadas baixas quantidades de PHMB.
No entanto, no caso da adsorção, a diminuição exponencial da eficiência de incorporação com o
aumento da concentração de PHMB faz com que este método não seja vantajoso para a
impregnação de maiores quantidades do antimicrobiano.
Outras desvantagens da incorporação a partir de solução aquosa são o fato de as
membranas apresentarem perda de massa e de haver alteração de aspecto das mesmas, como
observado no item 4.3.2.2. Além disso, este tipo de incorporação requer uma etapa a mais no
preparo das membranas, havendo gastos de energia para o controle de temperatura e agitação das
soluções durante a impregnação, como também para a etapa final de secagem após a
incorporação.

132
Com base nestes dados e nestas observações, é possível concluir que a incorporação de
PHMB por adição à mistura polimérica é mais vantajosa do que a incorporação a partir de
solução de aquosa.
4.3.5. Caracterização suplementar das membranas mais promissoras
Como mostrado nos itens 4.3.1 e 4.3.2, os ensaios de avaliação da cinética de liberação de
PHMB indicaram que a maior parte do antimicrobiano incorporado ficou retida na matriz
polimérica, independentemente do método de incorporação. Verificou-se, assim, a alta afinidade
do PHMB pelos polissacarídeos quitosana e alginato.
De acordo com Powell et al. (2010), o caráter hidrofílico e a alta densidade de cargas
positivas do PHMB favorecem sua interação com hidrogéis, especialmente aqueles que possuem
carga negativa, impedindo que a sua liberação seja muito rápida. Além disso, de acordo com Gao
e Cranston (2008), se a interação iônica for muito forte, pode haver diminuição da quantidade de
PHMB livre e, consequentemente, da atividade antimicrobiana.
Sendo assim, os testes de halo de inibição e permeação das bactérias Staphylococcus
aureus (gram positiva) e Pseudomonas aeruginosa (gram negativa) foram realizados a fim de
verificar a atividade antimicrobiana das membranas. Selecionaram-se para estes ensaios as
membranas contendo ou não PHMB incorporado por adição à mistura polimérica, que foi o
método de incorporação mais vantajoso dentre os empregados neste trabalho. Os resultados são
mostrados na Tabela 4.26 e nas Figuras 4.43 a 4.46.
Verificou-se que todas as membranas preparadas na ausência ou na presença de PHMB
apresentaram atividade antimicrobiana, sendo capazes de impedir o crescimento de
Staphylococcus aureus e Pseudomonas aeruginosa nas suas superfícies, o que indica potencial
uso no tratamento de lesões de pele.
As membranas preparadas na ausência de PHMB não apresentaram formação de halo de
inibição, representado por uma zona mais clara ao redor da amostra. Ressalta-se que a ausência
de halo de inibição não implica, necessariamente, na ausência total de atividade antimicrobiana,
uma vez que as membranas obtidas foram capazes de impedir o crescimento dos microrganismos
testados nas suas superfícies.

133
Tabela 4.26: Propriedades antimicrobianas das membranas preparadas na ausência e na presença
de diferentes proporções mássicas de PHMB.
Formulação [PHMB] mg/g
Staphylococcus aureus Pseudomonas aeruginosa
C.M. H.I. P. C.M. H.I. P.
P0%
0 - - - - - -
10 - - - - - -
100 - + - - - -
P2%
0 - - - - - -
10 - - - - - -
100 - + - - - +
P10%
0 - - + - - -
10 - - + - - +
100 - + - - - +
C.M.: Crescimento microbiano na superfície da membrana; H.I.: Halo de inibição, P.: Permeação
Resultados similares foram encontrados por Rodrigues (2008) para membranas lamelares
de quitosana-alginato e por Bueno e Moraes (2011) para membranas porosas de quitosana-
alginato na ausência de agentes ativos. Veiga (2012) observou que membranas densas de
quitosana-xantana também não apresentaram formação de halo de inibição. Verifica-se, assim,
que membranas de quitosana complexada com outros polímeros na ausência de agentes ativos
não liberam substâncias antimicrobianas no meio de cultura. Por outro lado, segundo Yu et al.
(2005), a dissociação gradual do complexo quitosana-alginato, quando in vivo, ocasionaria a
liberação de macromoléculas livres dos polissacarídeos, possibilitando a interação dos grupos
amino positivos da quitosana com os componentes negativos da parede celular das bactérias,
havendo, deste modo, a atuação da quitosana como agente antimicrobiano.

134
Figura 4.43: Formação de halo de inibição em culturas de Staphylococccus aureus por exposição
a membranas preparadas na ausência e na presença de diferentes proporções de PHMB.
A formação de halo de inibição a bactérias Staphylococcus aureus ocorreu somente para as
membranas preparadas na presença da maior proporção de PHMB (100 mg/g), indicando que o
antimicrobiano liberado no meio de cultura foi suficiente para impedir a proliferação dos
microrganismos. Deve-se destacar, porém, que os halos de inibição formados continham traços
de crescimento microbiano. As membranas preparadas na presença de 10 mg/g de PHMB não
formaram halo de inibição provavelmente porque a quantidade de antimicrobiano liberada pela
membrana no meio de cultura não atingiu a concentração mínima inibitória (MIC) para as
bactérias Staphylococcus aureus (MIC=0,2 µg/mL). Além disso, verificou-se na análise da
cinética de liberação de PHMB em PBS, que a maioria das membranas preparadas na presença de
10 mg/g liberaram quantidades de antimicrobiano abaixo do nível mínimo considerado ativo, de
1 mg/g (ARCH CHEMICALS, 2005).
P0% P0%
PHMB 10 mg/g
P0%
PHMB 100 mg/g
P2% P2%
PHMB 10 mg/g
P2%
PHMB 100 mg/g
P10% P10%
PHMB 10 mg/g
P10%
PHMB 100 mg/g

135
Figura 4.44: Permeação de Staphylococccus aureus através de membranas preparadas na
ausência e na presença de diferentes proporções de PHMB.
Os testes com Pseudomonas aeruginosa não resultaram na formação de halo de inibição
para nenhuma das membranas, o que ocorreu provavelmente porque a eficácia do PHMB contra
bactérias gram positivas, como a Staphylococcus aureus (MIC=0,2 µg/mL), é maior do que a
eficácia contra bactérias gram-negativas, como a Pseudomonas aeruginosa (MIC=20 µg/mL).
Como já mencionado no item 2.7, as bactérias gram-positivas têm uma parede celular mais
simples do que as bactérias gram-negativas, as quais possuem uma membrana externa. Desta
forma, o PHMB teria maior facilidade de permear a parede celular das bactérias gram-positivas
(HÜBNER e KRAMER, 2010; CHEN-YU et al., 2007).
A permeação de Staphylococcus aureus através das membranas foi observada somente para
a formulação P10% preparada na ausência de PHMB e na presença de 10 mg/g de PHMB. Foi
possível concluir que a permeação das bactérias ocorreu devido à maior porosidade dessas
P0% P0%
PHMB 10 mg/g
P0%
PHMB 100 mg/g
P2% P2%
PHMB 10 mg/g
P2%
PHMB 100 mg/g
P10% P10%
PHMB 10 mg/g
P10%
PHMB 100 mg/g

136
membranas. A presença de uma maior quantidade de PHMB, de 100 mg/g, impediu a permeação
desses microrganismos através da formulação P10%. Destaca-se que não é possível identificar a
ocorrência de permeação por meio das fotos, uma vez que neste teste analisa-se somente o lado
da membrana em contato com o meio de cultura, sendo os pontos amarelados inerentes ao
inóculo de bactérias.
Figura 4.45: Formação de halo de inibição em culturas de Pseudomonas aeruginosa por
exposição a membranas preparadas na ausência e na presença de diferentes proporções de
PHMB.
A permeação de Pseudomonas aeruginosa foi observada para um maior número de
membranas em comparação à permeação de Staphylococcus aureus, provavelmente porque a
eficácia do PHMB contra esta última é maior. Observou-se que a permeação ocorreu apenas
P0% P0%
PHMB 10 mg/g
P0%
PHMB 100 mg/g
P2% P2%
PHMB 10 mg/g
P2%
PHMB 100 mg/g
P10% P10%
PHMB 10 mg/g
P10%
PHMB 100 mg/g

137
através de membranas porosas. Contraditoriamente, não foi observada uma relação direta entre a
permeação de Pseudomonas aeruginosa e a quantidade de PHMB incorporado.
Figura 4.46: Permeação de bactérias Pseudomonas aeruginosa através de membranas preparadas
na ausência e na presença de diferentes proporções de PHMB.
Os resultados obtidos nestes experimentos sugerem que as membranas de quitosana-alginato
densas e porosas têm potencial para serem utilizadas no tratamento de lesões de pele. As
formulações obtidas poderiam atuar como barreiras contra microrganismos, visto que não houve
crescimento bacteriano sobre as suas superfícies. Assim, apesar da alta afinidade entre o PHMB e
a matriz polimérica de quitosana-alginato, verificou-se que a atividade antimicrobiana foi
preservada. Por outro lado, a formulação P10% requer cautela no seu uso, uma vez que, mesmo na
presença do antimicrobiano, as bactérias testadas permearam através da mesma devido à sua
porosidade.
P0% P0%
PHMB 10 mg/g
P0%
PHMB 100 mg/g
P2% P2%
PHMB 10 mg/g
P2%
PHMB 100 mg/g
P10% P10%
PHMB 10 mg/g
P10%
PHMB 100 mg/g

138
5. CONCLUSÕES E SUGESTÕES
5.1. Conclusões
Neste trabalho, membranas de quitosana-alginato densas e porosas, com ótimo potencial
para serem usadas como curativos ou scaffolds dérmicos, foram produzidas através de uma
metodologia simples e escalonável.
As adaptações do método de produção utilizado por Bueno (2010), que consistiram na
diminuição da quantidade de material empregada na produção das membranas e no aumento da
temperatura de secagem, permitiram obter membranas com propriedades mecânicas melhores e
reduzir o tempo de produção em cerca de 11 horas.
As membranas produzidas com variadas proporções de Pluronic F68 apresentaram
diferenças com relação ao aspecto e propriedades físico-químicas. Verificou-se que o aumento
da proporção do tensoativo levou ao aumento da porosidade, rugosidade, opacidade, espessura,
capacidade de absorção e perda de massa em soluções aquosas, sorção e transmissão de vapor
d’água. Por outro lado, observou-se diminuição da resistência à tração e do alongamento na
ruptura devido à incorporação de ar.
Levando-se em conta a aplicação das membranas como curativos de pele, acredita-se que a
formulação preparada na ausência de tensoativo é bastante promissora, uma vez que a sua
transparência permite o monitoramento do processo de cicatrização sem a necessidade de
remover o curativo. Além disso, esta formulação mostrou boas estabilidade em soluções aquosas
e resistência mecânica. A membrana preparada com 10% (m/m) de Pluronic F68 também pode
ser considerada promissora para esta aplicação, possuindo boa capacidade de absorção de
soluções fisiológicas e de vapor d’água.
Por outro lado, quando se considera a aplicação das membranas como scaffolds porosos
para o cultivo de células da pele, tem-se que a formulação preparada com 10% (m/m) de
Pluronic F68 seria a mais promissora, possuindo rugosidade e porosidade adequadas.
A incorporação de PHMB por adição à mistura polimérica nas proporções de 10 e
100 mg/g teve alta eficiência, variando de 72 a 86%. Já a avaliação da cinética de liberação do
PHMB mostrou que apenas de 4 a 15% do antimicrobiano incorporado é efetivamente liberado
em PBS, o que ocorreu devido à alta afinidade do PHMB pelos principais constituintes das

139
membranas. A incorporação por este método não resultou em alterações no aspecto ou
morfologia das membranas.
A incorporação de PHMB por adsorção a partir de solução aquosa resultou em um bom
ajuste segundo a isoterma proposta por Zhu e Gu (1989). Observou-se que é possível impregnar
quantidades muito altas do antimicrobiano às membranas, de cerca de 6 g/g, sendo o máximo
teórico previsto pelo modelo de cerca de 8 g/g. No entanto, este método de incorporação
apresentou desvantagens como a alteração do aspecto do material e perda de massa em soluções
de baixas concentrações do antimicrobiano, para as quais foram obtidas as maiores eficiências de
incorporação. Os ensaios de cinética de liberação em PBS indicaram que a maior parte do
PHMB incorporado permanece retida nas membranas mesmo após 48 horas de ensaio, assim
como foi observado para a incorporação por adição à mistura polimérica. Notou-se ainda que
para soluções de impregnação de 2x10-2 mol/L, as membranas liberaram quantidades
potencialmente tóxicas do antimicrobiano, indicando a necessidade de se trabalhar com
concentrações menores.
O processamento das membranas em CO2 supercrítico na ausência de PHMB indicou que a
impregnação de fármacos por esta técnica é promissora, sendo observados resultados positivos
como a manutenção do aspecto e integridade estrutural das membranas, além do aumento da
capacidade de sorção de vapor d’água devido ao intumescimento dos polímeros na presença do
CO2 supercrítico. Contudo, não foi detectada a liberação do antimicrobiano em PBS por medidas
de absorbância no UV-VIS.
Com base nos resultados referentes à eficiência de incorporação, cinética de liberação,
aspecto e morfologia, é possível concluir que o método de adição à mistura polimérica é o mais
vantajoso dentre os métodos de incorporação de PHMB empregados neste trabalho, resultando
em membranas com grande potencial de uso como curativos com propriedades antimicrobianas.
5.2. Sugestões para trabalhos futuros
a) Produção de membranas utilizando ácidos alternativos ao ácido acético para a
solubilização da quitosana, como os ácidos glicólico e ascórbico. O ácido ascórbico é capaz de
aumentar a resistência à tração de membranas de quitosana em aproximadamente 100% em
comparação à da membrana preparada com ácido acético, enquanto que o ácido glicólico pode

140
aumentar em 100% a capacidade de absorção de água das membranas (CHEN et al., 2007).
b) Produção de membranas a partir da mistura dos polissacarídeos a alta temperatura sob
agitação para obter uma boa dispersão, seguida da aeração do sistema por agitação ou por meio
de um dispersor de ar.
c) Incorporação de PHMB às membranas por adsorção em fluxo, com reciclo, para facilitar
a entrada do antimicrobiano na matriz polimérica e reduzir os gastos de solução de agente
antimicrobiano.
d) Medida do módulo de Young, ou módulo de elasticidade das membranas, definido como
a razão entre a tensão aplicada e a deformação elástica longitudinal. Esta medida permitiria
analisar mais detalhadamente o comportamento do material durante o estiramento, podendo ser
relacionada às interações entre as moléculas dos polissacarídeos.
e) Produção de membranas com diferentes composições de forma a diminuir as interações
entre o PHMB e o alginato e, desta forma, aumentar a liberação do antimicrobiano. Neste
sentido, as sugestões seriam aumentar a proporção mássica entre a quitosana e o alginato, não
excedendo a maior proporção já testada, de 0,75:0,25, a qual não se mostrou adequada. Outra
sugestão seria aumentar a proporção de cálcio nas etapas de reticulação, mas de forma a não
tornar as membranas quebradiças.
f) Estudo de escalonamento e de viabilidade econômica do processo, visando à produção
industrial das membranas. Neste sentido, o maior obstáculo seria a etapa de secagem da mistura
polimérica em estufa com circulação de ar, sendo necessário buscar alternativas a este método de
secagem.
g) Seleção de curativos secundários adequados para serem utilizados em conjunto com as
membranas obtidas neste trabalho e análise das propriedades físico-químicas das membranas na
presença destes curativos secundários.
h) Análise da distribuição de cargas superficiais das membranas, por meio da medida de
potencial zeta, para obter um resultado mais confiável acerca das interações entre a quitosana e o
alginato e da melhor proporção entre os polissacarídeos.
i) Análise da cinética de liberação de PHMB utilizando célula de Franz, de modo a simular
as condições reais de aplicação dos curativos.
j) Produção de membranas densas e porosas na forma de compósitos em camadas,
possibilitando a obtenção de um material com propriedades mecânicas melhores em relação às

141
membranas porosas e com uma barreira protetora à permeação de microrganismos.
k) Realização de testes de capacidade de absorção e perda de massa in vivo, obtendo-se
resultados mais próximos da realidade acerca do intumescimento e estabilidade das membranas.
l) Avaliação do desempenho membranas como scaffolds, por meio da realização de cultivo
celular in vitro e testes in vivo.

142
6. REFERÊNCIAS BIBLIOGRÁFICAS
AHMED, E. M. Hydrogel: Preparation, characterization, and applications, Journal of
Advanced Research, 2013. Disponível em <http://dx.doi.org/10.1016/j.jare.2013.07.006>. Acesso
em Novembro/2014.
AMERICAN SOCIETY FOR TESTING AND MATERIALS. ASTM D882; Standard test
method for tensile properties of thin plastic sheeting, 1995.
AMERICAN SOCIETY FOR TESTING AND MATERIALS. ASTM E 96-95; Standard
test methods of water vapor transmission of material, 1995.
ANBINDER, P. S.; DELADINO, L.; NAVARRO, A. S.; AMALVY, J. I.; MARTINO, M.
N. Yerba mate extract encapsulation with alginate and chitosan systems: interactions between
active compound encapsulation polymers. Journal of Encapsulation and Adsorption Sciences, v.
1, p. 80-87, 2011.
ARANAZ, I.; MENGÍBAR, M.; HARRIS, R.; PAÑOS, I.; MIRALLES, B.; ACOSTA, N.;
GALED, G.; HERAS, A. Functional characterization of chitin and chitosan. Current Chemical
Biology, v. 3, p. 203-230, 2009.
ARCH CHEMICALS. Polyhexamethylene Biguanide (PHMB). Product stewardship
summary. 2008. Disponível em <http://www.archchemicals.com/Fed/Corporate/Docs/ACC/
ACC_PHMB. pdf>. Acesso em Abril/2014.
ARCH CHEMICALS. Product Data - Cosmocil® CQ. 2005. Disponível em
<http://www.in-cosmetics.com/ExhibitorLibrary/3/CosmocilCQv1.6.pdf>. Acesso em Abril/
2011.
AZEVEDO, V. V. C.; CHAVES, S. A.; BEZERRA, D. C.; LIA FOOK, M. V.; COSTA,
A. C. F. M. Quitina e quitosana, aplicações como biomateriais. Revista Eletrônica de Materiais e
Processos, v. 2.3, p. 27-34, 2007.
BARRETT, S.; BATTACHARYYA, M.; BUTCHER, M.; ENOCH, S.; FUMAROLA, S.;
GRAY, D.; HAYNES, J. S.; EDWARDS-JONES, V.; LEAPER, D.; STROHAL, R.; WHITE,
R.; WICKS, G.; YOUNG, T. PHMB and its potential contribution to wound management.
Consensus Document. Wounds UK, p. 1-16, 2010.
BARTKOWIAK, A. Effect of the ionic strength on properties of binary
alginate/oligochitosan microcapsules. Colloids and Surfaces A: Physicochemical and
Engineering Aspects, v. 204, p. 117-124, 2002.
BELLINI, M. Z.; PIRES, A. L. R.; VASCONCELOS, M. O.; MORAES, A. M.
Comparison of the properties of compacted and porous lamellar chitosan–xanthan membranes as
dressings and scaffolds for the treatment of skin lesions. Journal of Applied Polymer Science, v.
125, n. S2, p. E421-E431, 2012.
BERGER, J.; REIST, M.; MAYER, J. M.; FELT, O.; GURNY, R. Structure and
interactions in chitosan hydrogels formed by complexation or aggregation for biomedical
applications. European Journal of Pharmaceutics and Biopharmaceutics, v. 57, p. 35-52, 2004.
BHAMIDIPATI, M.; SCURTO, A. M.; DETAMORE, M. S. The future of carbon dioxide

143
for polymer processing in tissue engineering. Tissue Engineering: Part B, v. 19, n. 3, p. 221-232,
2013.
BIERHALZ, A. C. K.; DA SILVA, M. A.; DE SOUSA, H. C.; BRAGA, M. E. M.;
KIECKBUSCH, T. G. Influence of natamycin loading methods on the physical characteristics of
alginate active films. Journal of Supercritical Fluids, v. 76, p. 74– 82, 2013.
BLACKBURN, R. S.; HARVEY, A.; KETTLE, L. L.; PAYNE, J. D.; RUSSELL, S. J.
Sorption of Poly(hexamethylenebiguanide) on cellulose: mechanism of binding and molecular
recognition. Langmuir, v. 22, p. 5636-5644, 2006.
BOATENG, J. S.; MATTHEWS, K. H.; STEVENS, H. N. E.; ECCLESTON, G. M.
Wound healing dressings and drug delivery systems: A review. Journal of Pharmaceutical
Sciences, v. 97, p. 2892-2923, 2008.
BOEHRINGER, J. R.; KARPOWICZ, J.; MITRA, A.; RADL, C. L. Growth stimulating
wound dressing with improved contact surfaces. United States Patent US7951124 B2,
31.Maio.2011.
BOGMAN, K.; ERNE-BRAND, F.; ALSENZ, J.; DREWE, J. The role of surfactants in
the reversal of active transport mediated by multidrug resistance proteins. Journal of
Pharmaceutical Sciences, v. 92, n. 6, p. 1250-1261, 2003.
BOHIDAR, H. B. Coacervates: a novel state of soft matter - an overview. Journal of
Surface Science and Technology, v. 24, n. 3-4, p. 105-124, 2008.
BORGES, E. L.; SAAR, S. R. C.; LIMA, V. L. A. N.; GOMES, F. S. L.; MAGALHÃES,
M. B. B. Feridas: Como tratar. 2.ed. Belo Horizonte: Editora Coopmed, 2007.
BROXTON, P.; WOODCOCK, P. M.; GILBERT, P. Binding of some polyhexamethylene
biguanides to the cell envelope of Escherichia coli ATCC 8739. Microbios, v. 41, n. 163, p. 15–
22, 1984.
BUENO, C. Z. Influência do tipo de estratégia de reforço das propriedades mecânicas nas
características de membranas de quitosana e alginato projetadas para o recobrimento de lesões
de pele. Campinas: Faculdade de Engenharia Química, Universidade Estadual de Campinas,
2010. Dissertação (Mestrado).
BUENO, C. Z.; DIAS, A. M. A.; DE SOUSA, H. J. C.; BRAGA, M. E. M.; MORAES, A.
M. Control of the properties of porous chitosan–alginate membranes through the addition of
different proportions of Pluronic F68. Materials Science and Engineering C, v. 44, p. 117–125,
2014.
BUENO, C. Z.; MORAES, A. M. Development of porous lamellar chitosan-alginate
membranes: Effect of different surfactants on biomaterial properties. Journal of Applied Polymer
Science, v. 122, p. 624–631, 2011.
BURD, A.; HUANG, L. Carbohydrates and cutaneous wound healing. In: GARG, H. G.;
COWMAN, M. K.; HALES, C. A. Carbohydrate chemistry, biology and medical applications.
Amsterdam: Elsevier Ltd., 2008.
CAETANO, G. F.; FRADE, M. A. C.; ANDRADE, T. A. M.; LEITE, M. N.; BUENO, C.
Z.; MORAES, A. M.; RIBEIRO-PAES, J. T. Chitosan-alginate membranes accelerate wound
healing. Journal of Biomedical Materials Research Part B: Applied Biomaterials, 2014.

144
DOI: 10.1002/jbm.b.33277.
CANELLA, A. M. N. C.; GARCIA, R. B. Caracterização de quitosana por cromatografia
de permeação em gel – influência do método de preparação e do solvente. Química Nova, v. 24,
n. 1, p. 13-17, 2001.
CÁRDENAS, A.; ARGUELLES-MONAL, W.; GOYCOOLEA, F. M.; HIGUERA-
CIAPARA, I.; PENICHE, C. Diffusion through membranes of the polyelectrolyte complex of
chitosan and alginate. Macromolecular Bioscience, v. 3(10), p. 535-539, 2003.
CHEN, P. H.; HWANG, Y. H.; KUO, T. Y.; LIU, F. H.; LAI, J. Y.; HSIEH, H. J.
Improvement in the properties of chitosan membranes using natural organic acid solutions as
solvents for chitosan dissolution. Journal of Medical and Biological Engineering, v. 27, n. 1, p.
23-28, 2007.
CHEN-YU, J. H.; EBERHARDT, D. M.; KINCADE, D. H. Antibacterial and laundering
properties of AMS and PHMB as finishing agents on fabric for health care workers’ uniforms.
Clothing & Textiles Research Journal, v. 25, n. 3, p. 258-272, 2007.
CHISTI, Y. Animal-cell damage in sparged bioreactors. Trends in Biotechnology, v. 18, n.
10, p. 420–432, 2000.
COMYN, J. Polymer Permeability. 1a ed. Londres: Elsevier Applied Science, 1985
COOPER, A.I. Polymer synthesis and processing using supercritical carbon dioxide.
Journal of Materials Chemistry, v. 10, p. 207-234, 2000.
DA CUNHA, P. L. R.; DE PAULA, R. C. M.; FEITOSA, J. P. A. Polissacarídeos da
biodiversidade brasileira: uma oportunidade de transformar conhecimento em valor econômico.
Química Nova, v. 32, n. 3, p. 649-660, 2009.
DA SILVA, M. A. Desenvolvimento e caracterização de filmes compostos de alginato e
quitosana contendo natamicina como agente antimicótico. Campinas: Faculdade de Engenharia
Química, Universidade Estadual de Campinas, 2009. Tese (Doutorado).
DA SILVA, M. A.; BIERHALZ, A. C. K.; KIECKBUSCH, T. G. Modelling natamycin
release from alginate/chitosan active films. International Journal of Food Science and
Technology, v. 47, p. 740–746, 2012.
DEMINA, T. S.; GILMAN, A. B.; AKOPOVA, T. A.; ZELENETSKII, A. N.
Modification of the chitosan structure and properties using high-energy chemistry methods. High
Energy Chemistry, v. 48, n. 5, p. 293–302, 2014.
DE PAULA, G. F.; NETTO, G. I.; MATTOSO, L. H. C. Physical and chemical
characterization of Poly(hexamethylene biguanide) hydrochloride. Polymers, v. 3, p. 928-941,
2011.
DE SOUZA, R. F. B. Membranas de quitosana complexada com alginato e xantana:
Comportamento na presença de diferentes proporções de água e incorporação de eritromicina.
Campinas: Faculdade de Engenharia Química, Universidade Estadual de Campinas, 2014.
Dissertação (Mestrado).
DIAS, A. M. A.; BRAGA, M. E. M.; SEABRA, I. J.; FERREIRA, P.; GIL, M. H.; DE
SOUSA, H. C. Development of natural-based wound dressings impregnated with bioactive

145
compounds and using supercritical carbon dioxide. International Journal of Pharmaceutics, v.
408, p. 9-19, 2011.
DIAS, A. M. A.; BRAGA, M. E. M.; SEABRA, I. J.; DE SOUSA. H. C. Supercritical
solvent impregnation of natural bioactive compounds in N-carboxybutylchitosan and agarose
membranes for the development of topical wound healing applications. In: JORGE, R. M. N.;
TAVARES, J. M. R. S.; BARBOSA, M. P.; SLADE. A. P. Lecture notes in computational
vision and biomechanics. Technologies for medical sciences. Volume 1. Springer, 2012.
DIAS, A. M. A.; REY-RICO, A.; OLIVEIRA, R. A.; MARCENEIRO, S.; ALVAREZ-
LORENZO, C.; CONCHEIRO, A.; JUNIOR, R. N. C.; BRAGA, M. E. M.; DE SOUSA, H. C.
Wound dressings loaded with an anti-inflammatory juca (Libidibia ferrea) extract using
supercritical carbon dioxide technology. Journal of Supercritical Fluids, v. 74, p. 34-45, 2013.
DUARTE, A. R. C.; MANO, J. F.; REIS, R. L. Supercritical fluids in biomedical and
tissue engineering applications: a review. International Materials Reviews, v. 54, n. 4, p. 214-
222, 2009.
DUARTE, A. R. C.; SANTO, V. E.; ALVER, A.; SILVA, S. S.; MIREIRA-SILVA, J.;
SILVA, T. H.; MARQUES, A. P.; SOUZA, R. A.; GOMES, M. E.; MANO, J. F.; REIS, R. L.
Unleashing the potential of supercritical fluids for polymer processing in tissue engineering and
regenerative medicine. Journal of Supercritical Fluids, v. 79, p. 177–185, 2013.
ELIAS, H. G. Biological polymerization. In: ELIAS, H. G. Macromolecules. Weinheim:
Wiley-VCH, 2005.
FELD, D.; SOTO, T. A. Antimicrobial dressing. US Patent 4643180. 17.Fevereiro.1987.
FÖRCH, R.; SCHÖNHERR, H.; JENKINS, T. A. Surface design: Applications in
bioscience and nanotechnology. 1a ed. Weinheim: Wiley-VCH, 2009.
GAO, Y.; CRANSTON, R. An effective antimicrobial treatment for wool using
polyhexamethylene biguanide as the biocide, Part 1: Biocide uptake and antimicrobial activity.
Journal of Applied Polymer Science, v. 117, p. 3075-3082, 2010.
GAO, Y.; CRANSTON, R. Recent advances in antimicrobial treatments of textiles. Textile
Research Journal, v. 78, n. 1, p. 60–72, 2008.
GAO, Y.; YU, X.; PIERLOT, A. P.; DENNING, R. J.; R. CRANSTON. A simultaneous
antimicrobial and shrink resistance treatment of wool woven fabrics using the polymeric biocide
polyhexamethylene biguanide. Journal of Materials Science, v. 46, p. 3020–3026, 2011.
GAYNES, R.; EDWARDS, J. R. Overview of nosocomial infections caused by gram-
negative bacilli. Clinical Infectious Diseases, v. 41, p. 848-854, 2005.
GEORGE, M.; ABRAHAM T. E. Polyionic hydrocolloids for the intestinal delivery of
protein drugs: Alginate and chitosan — a review. Journal of Controlled Release, v. 114, p. 1–14,
2006.
GIRATA, A. K. Desenvolvimento de curativos de quitosana e alginato contendo fosfato
hidrogenado de zircônio, sódio e prata. Campinas: Faculdade de Engenharia Química,
Universidade Estadual de Campinas, 2011. Dissertação (Mestrado).
GREAVES, N. S.; IQBAL, S. A.; BAGUNEID, M.; BAYAT, A. The role of skin

146
substitutes in the management of chronic cutaneous wounds. Wound Repair and Regeneration,
v. 21, p. 194–210, 2013.
GUNASEKARAN, S.; NATARAJAN, R. K.; RENGANAYAKI, V.; NATARAJAN, S.
Vibrational spectra and thermodynamic analysis of metformin. Indian Journal of Pure and
Applied Physics, v. 44, p. 495-500, 2006.
GUPTA, K. C.; KUMAR, M. N. V. R. Drug release behavior of beads and microgranules
of chitosan. Biomaterials, v. 21, p. 1115-1119, 2000.
HANSEN, B.; JEMEC, G. B. E. The mechanical properties of skin in osteogenesis
imperfecta. Archives of Dermatology, v. 138(7), p. 909-911, 2002.
HEIN, S.; WANG, K.; STEVENS, W. F.; KJEMS, J. Chitosan composites for biomedical
applications: status, challenges and perspectives. Materials Science and Technology, v. 24, p.
1053-1061, 2008.
HIMMELBLAU, D. M. Solubilities of inert gases in water. Journal of Chemical and
Engineering Data, v. 5, n. 1, p. 10-15, 1960.
HINRICHS, W. L. J.; LOMMEN, E. J. C. M. P.; WILDEVUUR, R. H.; FEIJEN, J.
Fabrication and characterization of an asymmetric polyurethane membrane for use as a wound
dressing. Journal of Applied Biomaterials, v. 3, n. 4, p. 287-303, 1992.
HOLMBERG, K.; JÖNSSON, B.; KRONBERG, B.; LINDMAN, B. Surfactants and
polymers in aqueous solutions. 2a ed. Chichester: John Wiley & Sons, 2002.
HONARY, S.; MALEKI, M.; KARAMI, M. The effect of chitosan molecular weight on
the properties of alginate/chitosan microparticles containing prednisolone. Tropical Journal of
Pharmaceutical Research, v. 8, n. 1, p. 53-61, 2009.
HSU, S. H.; WHU, S. W.; TSAI, C. L.; WU, Y. H.; CHEN, H. W.; HSIEH, K. H. Chitosan
as scaffold materials: Effects of molecular weight and degree of deacetylation. Journal of
Polymer Research, v. 11, p. 141-147, 2004.
HÜBNER, N. O.; KRAMER, A. Review on the efficacy, safety and clinical applications of
polihexanide, a modern wound antiseptic. Skin Pharmacology and Physiology, v. 23, n. 1, p. 17-
27, 2010.
IKEDA, T.; LEDWITH, A.; BAMFORD, C. H.; HANN, R. A. Interaction of a polymeric
biguanide biocide with phospholipids membranes. Biochimica et Biophysica Acta, v. 769, n. 1, p.
57–66, 1984.
IL’INA, A. V.; VARLAMOV, V. P. Chitosan-based polyelectrolyte complexes: a review.
Applied Biochemistry and Microbiology, v. 41, n. 1, p. 5-11, 2005.
INTERNATIONAL ORGANIZATION FOR STANDARDIZATION. ISO 10993-5e;
Biological evaluation of medical devices. Parte 5: Tests for cytotoxicity: In vitro methods, 1992.
INTERNATIONAL ORGANIZATION FOR STANDARDIZATION. ISO 4288;
Geometrical product specifications (GPS) - Surface texture: Profile method-rules and procedures
for the assessment of surface texture, 1996.
IURK, L. K.; OLIVEIRA, A. F.; GRAGNANI, A.; FERREIRA, L. M. Evidências no
tratamento de queimaduras. Revista Brasileira de Queimaduras, v. 9, n. 3, p. 95-99, 2010.

147
JAGUR-GRODZINSKI, J. Polymers for tissue engineering, medical devices, and
regenerative medicine. Concise general review of recent studies. Polymers for Advanced
Technologies, v. 17, p. 395-418, 2006.
JI, C.; ANNABI, N.; KHADEMHOSSEINI, A.; DEHGHANI, F. Fabrication of porous
chitosan scaffolds for soft tissue engineering using dense gas CO2. Acta Biomaterialia, v. 7, p.
1653–1664, 2011.
KANG, Y. O.; YOON, I. S.; LEE, S. Y.; KIM, D. D.; LEE, S. J.; PARK, W. H.;
HUDSON, S. M. Chitosan-coated poly(vinyl alcohol) nanofibers for wound dressings. Journal
of Biomedical Materials Research Part B: Applied Biomaterials, v. 92B, p. 568-576, 2010.
KARAKEÇILI, A. G.; SATRIANO, C.; GÜMÜŞDERELIOĞLU, M.; MARLETTA, G.
Surface characteristics of ionically crosslinked chitosan membranes. Journal of Applied Polymer
Science, v. 106, p. 3884-3888, 2007.
KARMARKAR, A. B.; GONJARI, I. D.; HOSMANI, A. H. Poloxamers and their
applications. Disponível em: <http://www.pharmainfo.net/pharma-student-
magazine/poloxamers-and-their-applications-0>. 2008. Acesso em Fevereiro/2011.
KASAAI, M. R. Various methods for determination of the degree of n-acetylation of chitin
and chitosan: a review. Journal of Agricultural and Food Chemistry, v. 57, p. 1667–1676, 2009.
KAWAGUTI, H. Y.; SATO, H. H. Produção de isomaltulose, um substituto da sacarose,
utilizando glicosiltransferase microbiana. Química Nova, v. 31, n. 1, p. 134-143, 2008.
KAŹMIERSKA, K. A.; KUC, K.; CIACH, T. Polyninylpyrrolidone-polyurethane
interpolymer hydrogel coating as a local drug delivery system. Acta Poloniae Pharmaceutica -
Drug Research, v. 65, n. 6, p. 763-766, 2008.
KHAN, F.; AHMAD, S. R. Polysaccharides and their derivatives for versatile tissue
engineering application. Macromolecular Bioscience, v. 13, n. 4, p. 395–421, 2013.
KLEINÜBING, S. J. Bioadsorção competitiva dos íons níquel e cobre em alginato e alga
marinha Sargassum Filipendula. Campinas: Faculdade de Engenharia Química, Universidade
Estadual de Campinas, 2009. Tese (Doutorado).
KOKUBO, T.; KUSHITANI, H.; SAKKA, S.; KITSUGI, T.; YAMAMURO, T. Solutions
able to reproduce in vivo surface-structure changes in bioactive glass-ceramic A-W. Journal of
Biomedical Materials Research, v. 24, p. 721-734, 1990.
KONG, H. J.; KAIGLER, D.; KIM, K.; MOONEY, D. J. Controlling rigidity and
degradation of alginate hydrogels via molecular weight distribution. Biomacromolecules, v. 5, p.
1720-1727, 2004.
KONICA MINOLTA INC. Precise color communication – Color control from perception
to instrumentation, Japan 2007.
KUCHARSKA, M.; NIEKRASZEWICZ, A.; WIŠNIEWSKA-WRONA, M.; BRZOZA-
MALCZEWSKA, K. Dressing sponges made of chitosan and chitosan-alginate fibrids. Fibres &
Textiles in Eastern Europe, v. 16, n. 3, p. 109-113, 2008.
LAI, H. L.; ABU’KHALIL, A.; CRAIG, D. Q. M. The preparation and characterization of
drug-loaded alginate and chitosan sponges. International Journal of Pharmaceutics, v. 251, p.

148
175-181, 2003.
LALAU, J. D.; BRESSON, R.; CHARPENTIER, P.; COLICHE, V.; ERLHER, S.;
HAVAN, G.; MAGALON,G.; MARTINI, J.; MOREAU, Y.; PRADINES, S.; RIGAL, F.;
WEMEAU, J. L.; RICHARD, J. L. Efficacy and tolerance of calcium alginate versus vaseline
gauze dressings in the treatment of diabetic foot lesions. Diabetes & Metabolism, v. 28, n. 3, p.
223-229, 2002.
LAWRIE, G.; KEEN, I.; DREW, B.; CHANDLER-TEMPLE, A.; RINTOUL, L.;
FREDERICKS, P.; GRØNDAHL, L. Interactions between alginate and chitosan biopolymers
characterized using FTIR and XPS. Biomacromolecules, v. 8, p. 2533-2541, 2007.
LEE, J.; NIKOLOV, A.; WASAN, D. Foam stability-The importance of film size and the
micellar structuring phenomenon. The Canadian Journal of Chemical Engineering,
DOI: 10.1002/cjce.22071, 2014.
LEE, J. W.; KIM, S. Y.; KIM, S. S.; LEE, Y. M.; LEE, K. H.; KIM S. J. Synthesis and
characteristics of interpenetrating polymer network hydrogel composed of chitosan and poly
(acrylic acid). Journal of Applied Polymer Science, v. 73, p. 113-120, 1999.
LEE, Y. H.; CHANG, J. J.; YANG, M. C.; CHIEN, C. T.; LAI, W. F. Acceleration of
wound healing in diabetic rats by layered hydrogel dressing. Carbohydrate Polymers, v. 88, p.
809–819, 2012.
LI, X.; XIE, H.; LIN, J.; XIE, W.; MAA, X. Characterization and biodegradation of
chitosan–alginate polyelectrolyte complexes. Polymer Degradation and Stability, v. 94, p. 1–6,
2009.
LI, Z.; ZHANG, M. Chitosan-alginate as scaffolding material for cartilage tissue
engineering. Journal of Biomedical Materials Research Part A, v. 75, n. 2, p. 485-493, 2005.
LIN, H. Y.; YEH, C. T. Alginate-crosslinked chitosan scaffolds as pentoxifylline delivery
carriers. Journal of Materials Science: Materials in Medicine, v. 21, p. 1611-1620, 2010.
LIU, C. Y.; ZHAO, K. S. Dielectric relaxations in chitosan solution with varying
concentration and temperature: analysis coupled with a scaling approach and thermodynamical
functions. Soft Matter, v. 6, p. 2742-2750, 2010.
LIU, X.; LIN, T.; GAO, Y.; XU, Z.; HUANG, C.; YAO, G.; JIANG, L.; TANG, Y.;
WANG, X. Antimicrobial electrospun nanofibers of cellulose acetate and polyester urethane
composite for wound dressing. Journal of Biomedical Materials Research Part B, v. 100B, p.
1556–1565, 2012.
LUO, Y.; WANG, Q. Recent development of chitosan-based polyelectrolyte complexes
with natural polysaccharides for drug delivery. International Journal of Biological
Macromolecules, v. 64, p. 353-367, 2014.
MA, J.; WANG, H.; HE, B.; CHEN, J. A preliminary in vitro study on the fabrication and
tissue engineering applications of a novel chitosan bilayer material as a scaffold of humam
neofetal dermal fibroblasts. Biomaterials, v. 22, p. 331-336, 2001.
MA, L.; YU, W.; MA, X. Preparation and characterization of novel sodium
alginate/chitosan two ply composite membranes. Journal of Applied Polymer Science, v. 106, p.
394–399, 2007.

149
MAGHRABY, G. M. E.; ALOMRANI, A. H. Synergistic enhancement of itraconazole
dissolution by ternary system formation with Pluronic F68 and hydroxypropylmethylcellulose.
Scientia Pharmaceutica, v. 77, p. 401-417, 2009.
MALAFAYA, P. B.; SILVA, G. A.; REIS, R. L. Natural–origin polymers as carriers and
scaffolds for biomolecules and cell delivery in tissue engineering applications. Advanced Drug
Delivery Reviews, v. 59, p. 207–233, 2007.
MANDELBAUM, S. H.; DI SANTIS, E. P.; MANDELBAUM, M. H. S. Cicatrização:
conceitos atuais e recursos auxiliares – Parte II. Anais Brasileiros de Dermatologia, v. 78(5), p.
525-542, 2003.
MARTINS, A. F.; PEREIRA, A. G. B.; FAJARDO, A. R.; RUBIRA, A. F.; MUNIZ, E. C.
Characterization of polyelectrolytes complexes based on N,N,N-trimethyl chitosan/heparin
prepared at different pH conditions. Carbohydrate Polymers, v. 86, p. 1266-1272, 2011.
MARTINS, J. T.; CERQUEIRA, M. A.; VICENTE, A. A. Influence of α-tocopherol on
physicochemical properties of chitosan-based films. Food Hydrocolloids, v. 27, p. 220-227,
2012.
MARTINSEN, A.; SKJÅK-BRÆK, G.; SMIDSRØD, O.; ZANETTI, F.; PAOLETTI, S.
Comparison of different methods for determination of molecular weight and molecular weight
distribution of alginates. Carbohydrate Polymers, v. 15, p. 171-193, 1991.
MASCARENHAS, M. D. M.; DA SILVA, M. M. A.; MALTA, D. C.; DE MOURA, L.;
GAWRYSZEWSKI, V. P.; COSTA, V. C.; SOUZA, M. F. M.; DE MORAIS NETO, O. L.
Atendimentos de emergência por acidentes na rede de vigilância de violências e acidentes –
Brasil, 2006. Disponível em <http://portal.saude.gov.br/portal/arquivos/pdf/VIVA 2006
_Rev_Cienc_Saude_Col_ 2008.pdf>. Acesso em Abril/2011.
MAURSTAD, G.; MØRCH, Y. A.; BAUSCH, A. R.; STOKKE, B. T. Polyelectrolyte
layer interpenetration and swelling of alginate–chitosan multilayers studied by dual wavelength
reflection interference contrast microscopy. Carbohydrate Polymers, v. 71, p. 672–681, 2008.
MCHUGH, D. J. A guide to the seaweed industry. FAO Fisheries Technical Paper, no 441,
2003.
MCHUGH, M. A.; MANDEL, F. S.; WANG, J. D. Supercritical fluid processing of
polymeric materials. In: SUN, Y. P. Supercritical fluid technology in materials science and
engineering – Syntheses, properties and applications. New York: Marcel Dekker Inc, 2002.
MENG, X.; TIAN, F.; YANG, J.; HE, C. N.; XING, N.; LI, F. Chitosan and alginate
polyelectrolyte complex membranes and their properties for wound dressing application. Journal
of Materials Science: Materials in Medicine, v. 21, p. 1751-1759, 2010.
MENNINI, N.; FURLANETTO, S.; CIRRI, M.; MURA, P. Quality by design approach for
developing chitosan-Ca-alginate microspheres for colon delivery of celecoxib-hydroxypropyl-b-
cyclodextrin-PVP complex. European Journal of Pharmaceutics and Biopharmaceutics, v. 80,
p. 67–75, 2012.
MILLERET, V.; HEFTI, T.; HALL, H.; VOGEL, V.; EBERLI, D. Influence of the fiber
diameter and surface roughness of electrospun vascular grafts on blood activation. Acta
Biomaterialia, v. 8, p. 4349–4356, 2012.

150
MIRAFTAB, M.; BARNABAS, J.; KENNEDY, J. F.; MASOOD, R. Antimicrobial
properties of alginate-chitosan (alchite) fibers developed for wound care applications. Journal of
Industrial Textiles, v. 40, n. 4, p. 345-360, 2011.
MOHAN, N.; NAIR, P. D. Novel porous, polysaccharide scaffolds for tissue engineering
applications. Trends in Biomaterial and Artificial Organs, v. 18, p. 219-224, 2005.
MOORE, K.; GRAY, D. Using PHMB antimicrobial to prevent wound infection. Wounds
UK, v. 3, n. 2, p. 96-102, 2007.
MOURA, L. I. F.; DIAS, A. M. A.; CARVALHO, E.; DE SOUSA, H. C. Recent advances
on the development of wound dressings for diabetic foot ulcer treatment – A review. Acta
Biomaterialia, v. 9, p. 7093-7114, 2013.
MULDER, G., CAVORSI, J., LEE, D. Polyhexamethylene biguanide (PHMB): an
addendum to current topical antimicrobials. Wounds, v. 19, n. 7, p. 173-182, 2007.
MUZZARELLI, R. A. A. Chitins and chitosans for the repair of wounded skin, nerve,
cartilage and bone. Carbohydrate Polymers, v. 76, p. 167–182, 2009.
MUZZARELLI, R. A. A. Natural chelating polymers. Alginic acid, chitin and chitosan.
Nova Iorque: Pergamon Press, 1973.
MUZZARELLI, R. A. A.; MORGANTI, P.; MORGANTI, G.; PALOMBO, P.;
PALOMBO M.; BIAGINI, G.; BELMONTE, M. M.; GIANTOMASSI, F.; ORLANDI, F.;
MUZZARELLI, C. Chitin nanofibrils/chitosan glycolate composites as wound medicaments.
Carbohydrate Polymers, v. 70, p. 274–284, 2007.
O’MALLEY, L. P.; COLLINS, A. N.; WHITE, G. F. Biodegradability of end-groups of
the biocide polyhexamethylene biguanide (PHMB) assessed using model compounds Journal of
Industrial Microbiology & Biotechnology, v. 33, p. 677–684, 2006.
PAES, A. T. Desvio padrão ou erro padrão: qual utilizar? Revista Educação Continuada
em Saúde, v. 6, p. 107-108, 2008.
PASPARAKIS, G.; BOUROPOULOS, N.. Swelling studies and in vitro release of
verapamil from calcium alginate and calcium alginate chitosan beads. International Journal of
Pharmaceutics, v. 323, p. 34–42, 2006.
PASQUALI, I.; BETTINI, R.; GIORDANO, F. Supercritical fluid technologies: An
innovative approach for manipulating the solid-state of pharmaceuticals. Advanced. Drug
Delivery Reviews, v. 60, p. 399-410, 2008.
PATEL, H. A.; SWANIKER, H. P.; HEAGLE, D. G.; WARD, K.;
TRANCHEMOTAGNE, A. Multi-layer wound dressing. US Patent 2007/0237812 A1.
11.Outubro.2007.
PAUL, W.; SHARMA, C. P. Chitosan and alginate wound dressings: a short review.
Trends in Biomaterials & Artificial Organs, v. 18, p. 18-23, 2004.
PAULUS, W. Microbicides for the protection of materials – a Handbook. 1a ed. Londres:
Chapman & Hall, 1993.
PELEG, A. Y.; HOOPER, D. C. Hospital-acquired infections due to gram-negative
bacteria. The New England Journal of Medicine, v. 362, p. 1804-1813, 2010.

151
PENMAN, A.; SANDERSON, G. R. A method for the determination of uronic acid
sequence in alginates. Carbohydrate Research, v. 25, p. 273-282, 1972.
PILLAI, C. K. S.; PAUL, W.; SHARMA, C. P. Chitin and chitosan polymers: Chemistry,
solubility and fiber formation. Progress in Polymer Science, v. 34, p. 641–678, 2009.
PIRES, A. L. R. Desenvolvimento de curativos flexíveis e neutralizados de quitosana e
alginato contendo Alphasan® RC2000. Campinas: Faculdade de Engenharia Química,
Universidade Estadual de Campinas, 2013. Dissertação (Mestrado).
POPA, M. I.; LISA, G.; AELENEI, N. Thermogravimetric characterization of
chitosan/alginate microparticles loaded with different drugs. Polymer Bulletin, v. 61, p. 481-490,
2008.
POWELL, C. H.; LALLY, J. M.; HOONG, L. D.; HUTH, S. W. Lipophilic versus
hydrodynamic modes of uptake and release by contact lenses of active entities used in
multipurpose solutions. Contact Lens & Anterior Eye, v. 33, p. 9–18, 2010.
QUIGNARD, F.; VALENTIN, R.; DI RENZO, F. Aerogel materials from marine
polysaccharides. New Journal of Chemistry, v. 32, n. 8, p. 1300-1310, 2008.
RATNER, B. D.; HOFFMAN, A. S.; SCHOEN, F. J.,; LEMONS, J. E. Biomaterials
science – An introduction to materials in medicine. 3a ed. Oxford: Elsevier, 2013.
RAVAL, A.; PARIKH, J.; ENGINEER, C. Mechanism of controlled release kinetics from
medical devices. Brazilian Journal of Chemical Engineering, v. 27, n.2, p. 211-225, 2010.
REVERCHON, E.; CARDEA, S. Supercritical fluids in 3-D tissue engineering. Journal of
Supercritical Fluids, v. 69, p. 97-107, 2012.
RHEIN, L. D.; SCHLOSSMAN, M.; O’LENICK, A.; SOMASUNDARAN, P. Surfactants
in personal care products and decorative cosmetics. 3a ed. Surfactant series v. 135. New York:
CRC Press, Taylor and Francis, 2007.
RIBEIRO, A. J. A. M.; GOMES, A. C.; CAVACO-PAULO, A. M. Developing scaffolds
for tissue engineering using the Ca2+-induced cold gelation by an experimental design approach.
Journal of Biomedical Materials Research B: Applied Biomaterials, v. 100B, p. 2269–2278,
2012.
RINAUDO, M. Characterization and properties of some polysaccharides used as
biomaterials. Macromolecular Symposia, v. 245–246, p. 549–557, 2006.
RINAUDO, M. Main properties and current applications of some polysaccharides as
biomaterials. Polymer International, v. 57, p. 397–430, 2008.
RINDFLEISCH, F.; DINOIA, T. P.; MCHUGH, M. A. Solubility of polymers and
copolymers in supercritical CO2. The Journal of Physical Chemistry, v. 100, n. 38, p.15581-
15587, 1996.
RODEHEAVER, G. T.; KURTZ, L.; KIRCHER, B. J.; EDLICH, R. F. Pluronic F68: A
promising new skin wound cleanser. Annals of Emergency Medicine, v. 9, n. 11, p. 572-576,
1980.
RODRIGUES, A. P. Preparação e caracterização de membranas de quitosana e alginato

152
para aplicação na terapia de lesões. Campinas: Faculdade de Engenharia Química,
Universidade Estadual de Campinas, 2008. Tese (Doutorado).
RODRIGUES, A. P.; SANCHEZ, E. M. S.; COSTA, A. C.; MORAES, A. M. The
influence of preparation conditions on the characteristics of chitosan-alginate dressings for skin
lesions. Journal of Applied Polymer Science, v. 109, p. 2703-2710, 2008.
ROHRER, C.; LEUPRECHT, H.; ALUPEI, I. C. Antiseptic alginate preparation. US
Patent 2009/0306157 A1. 10. Dezembro.2009.
SACCHETIN, P. S. C. Incorporação de Flavobacterium columnare inativado em
micropartículas de alginato e quitosana para a imunização de tilápia do nilo (oreochromis
niloticus) por via oral. Campinas: Faculdade de Engenharia Química, Universidade Estadual de
Campinas, 2009. Dissertação (Mestrado).
SÆTHER, H. V.; HOLME, H. K.; MAURSTAD, G.; SMIDSRØD, O.; STOKKE, B. T.
Polyelectrolyte complex formation using alginate and chitosan. Carbohydrate Polymers, v. 74, p.
813–821, 2008.
SAKAI, K.; HAYASHI, C.; YAMAJI, H.; FUKUDA, H. Use of nonionic surfactants for
effective supply of phosphatidic acid in serum-free culture of chinese hamster ovary cells.
Journal of Bioscience and Bioengineering, v. 92, n. 3, p. 256-261, 2001.
SANKALIA, M. G.; MASHRU, R. C.; SANKALIA, J. M.; SUTARIYA, V. B. Reversed
chitosan–alginate polyelectrolyte complex for stability improvement of alpha-amylase:
Optimization and physicochemical characterization. European Journal of Pharmaceutics and
Biopharmaceutics, v. 65, p. 215–232, 2007.
SCHNUCH, A.; GEIER, J.; UTER, W.; BASKETTER, D. A.; JOWSEY, I. R. The biocide
polyhexamethylene biguanide remains an uncommon contact allergen. Contact Dermatitis, v. 56,
p. 235–239, 2007.
SERAFICA, G.; MORMINO, R.; OSTER, G. A.; LENTZ, K. E.; KOEHLER, K. P.
Microbial cellulose wound dressing for treating chronic wounds. US Patent 2004/0028722 A1.
12.Fevereiro.2004.
SHAH, C. B.; VITARIS, R. F.; FINK, E. D.; ORR, S.; DOWD, B.;
TRANCHEMONTAGNE, A.; WARD, K.; SWANIKER, H.; PATEL, H. A. Antimicrobial foam
compositions, articles and methods. United States Patent Application Publication
US2010/0260824 A1, 14.Outubro.2010.
SHOICHET, M. S. Polymer scaffolds for biomaterials applications. Macromolecules, v.
43, p. 581-591, 2010.
SIGMA-ALDRICH. Pluronic®F68 P1300. Disponível em: <http://www.sigmaaldrich.
com/catalog/ProductDetail.do?lang=pt&N4=P1300|SIGMA&N5=SEARCH_CONCAT_PNO|B
RAND_KEY&F=SPEC>. Acesso em Fevereiro/2011.
SILVA, T. H.; ALVES, A.; FERREIRA, B. M.; OLIVEIRA, J. M.; REYS, L. L.;
FERREIRA, R. J. F.; SOUSA, R. A.; SILVA, S. S.; MANO, J. F.; REIS, R. L. Materials of
marine origin: a review on polymers and ceramics of biomedical interest. International
Materials Reviews, v. 57, n. 5, p. 276-307, 2012.
SIMONCIC, B.; TOMSIC, B. Structures of novel antimicrobial agents for textiles – A

153
review. Textile Research Journal, v. 80, n. 16, p. 1721-1737, 2010.
SIMPSON, N. E.; STABLER, C. L.; SIMPSON, C. P.; SAMBANIS, A.;
CONSTANTINIDIS, I. The role of the CaCl2-guluronic acid interaction on alginate encapsulated
βTC3 cells. Biomaterials, v. 25, p. 2603-2610, 2004.
SIMSEK-EGE, F. A.; BOND, G. M.; STRINGER, J. Polyelectrolye complex formation
between alginate and chitosan as a function of pH. Journal of Applied Polymer Science, v. 88, p.
346-351, 2003.
SMITHA, B.; SRIDHAR, S.; KHAN, A. A. Chitosan–sodium alginate polyion complexes
as fuel cell membranes. European Polymer Journal, v. 41, p. 1859–1866, 2005.
STEPHEN-HAYNES, J.; GIBSON, E.; GREENWOOD, M. Chitosan: a natural solution
for wound healing. Journal of Community Nursing, v. 28, n. 1, p. 48-53, 2014.
STOPPEL, W. L.; WHITE, J. C.; HORAVA, S. D.; BHATIA, S. R.; ROBERTS, S. C.
Transport of biological molecules in surfactant-alginate composite hydrogels. Acta
Biomaterialia, v. 7, p. 3988–3998, 2011.
STRAND, S. P.; VANDVIK, M. S.; VÅRUM, K. M.; ØSTGAARD, K. Screening of
chitosans and conditions for bacterial flocculation. Biomacromolecules, v. 2, p. 126-133, 2001.
SUDWORTH, J. Physico-chemical data. Project 1270585. Analytical Science Group,
Manchester, 2002.
TADROS, T. F. Applied surfactants – Principles and Applications. 1a ed. Berkshire:
Wiley-VCH, 2005.
TANODEKAEW, S.; PRASITSILP, M.; SWASDISON, S.; THAVORNYUTIKARN, B.;
POTHSREE, T.; PATEEPASEN, R. Preparation of acrylic grafted chitin for wound dressing
application. Biomaterials, v. 25, p. 1453–1460, 2004.
THE AMERICAN SOCIETY OF MECHANICAL ENGINEERS. ASME B46.1; Surface
texture (surface roughness, waviness, and lay). 2002.
THOMAS, S. The role of dressings in the treatment of moisture-related skin damage.
Disponível em: <http://www.worldwidewounds.org/2008/march /Thomas/Maceration-and-the-
role-of-dressings.html> 2008. Acesso em Fevereiro/2011.
TOMASKO, D. L.; LI, H.; LIU, D.; HAN, X.; WINGERT, M. J.; LEE, L. J.; KOELLING,
K. W. Review of CO2 applications in the processing of polymers. Industrial & Engineering
Chemistry Research, v. 42, p. 6431-6456, 2003.
VEIGA, I. G. Produção e caracterização de membranas de quitosana associada com
outros biopolímeros para liberação controlada de anti-inflamatórios. Campinas: Faculdade de
Engenharia Química, Universidade Estadual de Campinas, 2012. Tese (Doutorado).
VEIGA, I. G.; MORAES, A. M. Study of the swelling and stability properties of chitosan–
xanthan membranes. Journal of Applied Polymer Science, v. 124, p. E154-E160, 2012.
VERGER, M. L. L.; FLUCKIGER, L.; KIM, Y. I.; HOFFMAN, M.; MAINCENT, P.
Preparation and characterization of nanoparticles containing an antihypertensive agent.
European Journal of Pharmaceutics and Biopharmaceutics, v. 46, p. 137–143, 1998.

154
VERMA, D.; PREVITERA, M. L.; SCHLOSS, R.; LANGRANA, N. Polyelectrolyte
complex membranes for prevention of post-surgical adhesions in neurosurgery. Annals of
Biomedical Engineering, v. 40, n. 9, p. 1949–1960, 2012.
VON RECUM, A. F.; SHANNON, C. E.; CANNON, C. E.; LONG, K. J.; VAN
KOOTEN, T. G.; MEYLE, J. Surface roughness, porosity, and texture as modifiers of cellular
adhesion. Tissue Engineering, v. 2, n. 4, p. 241-253, 1996.
WALLES, T.; HERDEN, T.; HAVERICH, A. E MERTSCHING, H. Influence of scaffold
thickness and scaffold composition on bioartificial graft survival. Biomaterials, v. 24 p. 1233-
1239, 2003.
WAN, Y.; WU, Q.; WANG, S.; ZHANG, S.; HU, Z. Mechanical properties of porous
polylactide/chitosan blend membranes. Macromolecular Materials and Engineering, v. 292, p.
598-607, 2007.
WANG, L.; KHOR, E.; LIM, L. Y. Chitosan-alginate-CaCl2 system for membrane coat
application. Journal of Pharmaceutical Sciences, v. 90, p. 1134-1142, 2001.
WANG, L.; KHOR, E.; WEE, A.; LIM, L. Y. Chitosan-alginate PEC membrane as wound
dressing: Assessment of incisional wound healing. Journal of Biomedical Materials Research, v.
63, p. 610-618, 2002.
WANG, T.; WU, Y.; ZENG, A. J. Synthesis and characterization of amphiphilic Pluronic
(F68)-1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine copolymers and their micelles as a
drug carrier. Journal of Applied Polymer Science, v. 117, p. 604–613, 2010.
WEGMANN, J.; ODERMATT, E.; BLENDER, B. Layered wound dressing. US Patent
2009/0280162 A1. 12.novembro.2009.
WHISTLER, R. L.; BEMILLER, J. N. Industrial gums – Polysaccharides and their
derivatives. 3a ed. San Diego: Academic Press, 1993.
WIEGAND, C.; HIPLER, U. C. Polymer-based biomaterials as dressings for chronic
stagnating wounds. Macromolecular Symposia, v. 294-II, p. 1-13, 2010.
WILHELMS, T.; SCHULZE, D.; ALUPEI, I. C.; ROHRER, C.; ABEL, M.; WIEGAND,
C.; HIPLER, U. C. Release of polyhexamethylene biguanide hydrochloride (PHMB) from a
hydrobalanced cellulose wound dressing with PHMB. In 17TH CONFERENCE OF THE
EUROPEAN WOUND MANAGEMENT ASSOCIATION, 2007.
WU, Y.; CHE, F. B.; CHEN, J. H. Synthesis and characterization of a amphiphilic
Pluronic-poly(D,L-lactide-co-glycolide) copolymer and their nanoparticles as protein delivery
systems. Journal of Applied Polymer Science, v. 110, p. 1118-1128, 2008.
XU, H.; MA, L.; SHI, H.; GAO, C.; HAN, C. Chitosan–hyaluronic acid hybrid film as a
novel wound dressing: in vitro and in vivo studies. Polymers for advanced technologies, v. 18, p.
869–875, 2007.
YAN, X. L.; KHOR, E.; LIM, L. Y. Chitosan-alginate films prepared with chitosans of
different molecular weights. Journal of Biomedical Materials Research Part B: Applied
Biomaterials, v. 58, p. 358-365, 2001.
YAN, X.; KHOR, E.; LIM, L. Y. PEC films prepared from chitosan-alginate coacervates.

155
Chemical and Pharmaceutical Bulletin, v. 48(7), p. 941-946, 2000.
YAÑEZ, F.; MARTIKAINEN, L.; BRAGA, M. E. M.; ALVAREZ-LORENZO, C.;
CONCHEIRO, A.; DUARTE, C. M. M.; GIL, M. H.; DE SOUSA, H. C. Supercritical fluid-
assisted preparation of imprinted contact lenses for drug delivery. Acta Biomaterialia, v. 7, p.
1019-1030, 2011.
YANG, L.; ZHANG, L. M. Chemical structural and chain conformational characterization
of some bioactive polysaccharides isolated from natural sources. Carbohydrate Polymers, v. 76,
p. 349-361, 2009.
YU, S. H.; MI, F. L.; WU, Y. B.; PENG, C. K.; SHYU, S. S.; HUANG, R. N.
Antibacterial activity of chitosan-alginate sponges incorporating silver sulfadiazine: effect of
ladder-loop transition of interpolyelectrolyte complex and ionic crosslinking on the antibiotic
release. Journal of Applied Polymer Science, v. 98, p. 538-549, 2005.
YUDANOVA, T. N.; RESHETOV, I. V. Modern wound dressings: manufacturing and
properties. Pharmaceutical Chemistry Journal, v. 40, n. 2, p. 85-92, 2006.
ZHANG, R.; SOMASUNDARAN, P. Advances in adsorption of surfactants and their
mixtures at solid/solution interfaces. Advances in Colloid and Interface Science, v. 123-126, p.
213–229, 2006.
ZHAO, W.; JIN, X.; CONG, Y.; LIU, Y.; FU, J. Degradable natural polymer hydrogels for
articular cartilage tissue engineering. Journal of Chemical Technology and Biotechnology, v. 88,
p. 327–339, 2013.
ZHU, B. Y.; GU, T. General isotherm equation for adsorption of surfactants at solid/liquid
interfaces. Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in
Condensed Phases, v. 85, n. 11, p. 3813-3817, 1989.
ZHU, B. Y.; GU, T.; ZHAO, X. General isotherm equation for adsorption of surfactants at
solid/liquid interfaces. Part 2. Applications. Journal of the Chemical Society, Faraday
Transactions 1, v. 85, p. 3819-3824, 1989.
ZOHURIAAN, M. J.; SHOKROLAHI, F. Thermal studies on natural and modified gums.
Polymer Testing, v. 23, p. 575–579, 2004.

156
APÊNDICE I - Determinação da massa molar viscosimétrica média da quitosana e do
alginato
As massas molares viscosimétricas dos biopolímeros quitosana e alginato foram
determinadas segundo da Silva (2009). Primeiramente, foram preparadas soluções diluídas dos
biopolímeros a diferentes concentrações. As soluções de alginato foram preparadas nas
concentrações de 0,002; 0,005; 0,007; 0,010; 0,015 e 0,020 g/mL utilizando como solvente uma
solução de NaCl a 0,1M. Já as soluções de quitosana foram preparadas nas concentrações de
0,001; 0,003; 0,006; 0,009 e 0,012 g/mL utilizando como solvente uma solução de ácido acético
a 0,1M e NaCl a 0,2M, sendo previamente filtradas em funil de placa porosa antes das medidas.
Os tempos de escoamento das soluções foram medidos em um viscosímetro capilar tipo
Ostwald-Cannon-Fenske (núm. 200) imerso em um banho termostático com circulação a 25°C,
sendo o volume utilizado igual 10 mL. Para cada solução, foram realizadas 5 medidas.
A viscosidade intrínseca [ ] foi calculada de acordo com as Equações A.1 a A.5:
0
relt
t (A.1)
0
0relesp
t
tt1
(A.2)
c
esp
red.esp
(A.3)
c
)ln( reline
(A.4)
)(lim)(lim espred0cine0c (A.5)
onde rel é a viscosidade relativa, esp é a viscosidade específica, red.esp é a viscosidade
específica reduzida, ine é a viscosidade inerente, t é o tempo de escoamento da solução no
viscosímetro, 0t é o tempo de escoamento do solvente puro no viscosímetro, c é a concentração
do polímero na solução.

157
A massa molar viscosimétrica média vM foi calculada de acordo com a Equação de Mark-
Houwink-Sakurada (Equação A.6):
a
vMK][ (A.6)
As constantes K e a foram obtidas da literatura. Segundo Canella e Garcia (2001), as
constantes para a quitosana no solvente utilizado são gmLK /108,1 3 e 93,0a . Por sua
vez, segundo Mancini et al. (1996), as constantes para o alginato no solvente utilizado são
gmLK /102,1 2 e 96,0a .
Na Figura A.1 é mostrado o gráfico da viscosidade inerente versus concentração para as
soluções de quitosana. A extrapolação da curva para concentração zero permitiu a obtenção da
viscosidade intrínseca ([η]= 848,47 mL/g), a partir da qual se calculou que a massa molar
viscosimétrica média da quitosana é de 1,26x106 g/mol, de acordo com a equação de Mark-
Houwink-Sakurada. De acordo com Yan et al. (2001), a quitosana utilizada neste trabalho seria
classificada como sendo de alta massa molar.
Figura A.1: Viscosidade inerente versus concentração para soluções diluídas de quitosana.
Na Figura A.2 é mostrado o gráfico da viscosidade inerente versus concentração para
soluções diluídas de alginato, que permitiu calcular a viscosidade intrínseca ([η]=366,39 mL/g) e
y = -32153x + 848,47
R² = 0,9174
0
200
400
600
800
1000
0 0,002 0,004 0,006 0,008 0,01 0,012 0,014
Vis
co
sid
ad
e i
nere
nte
(m
L/g
)
Concentração (g/mL)
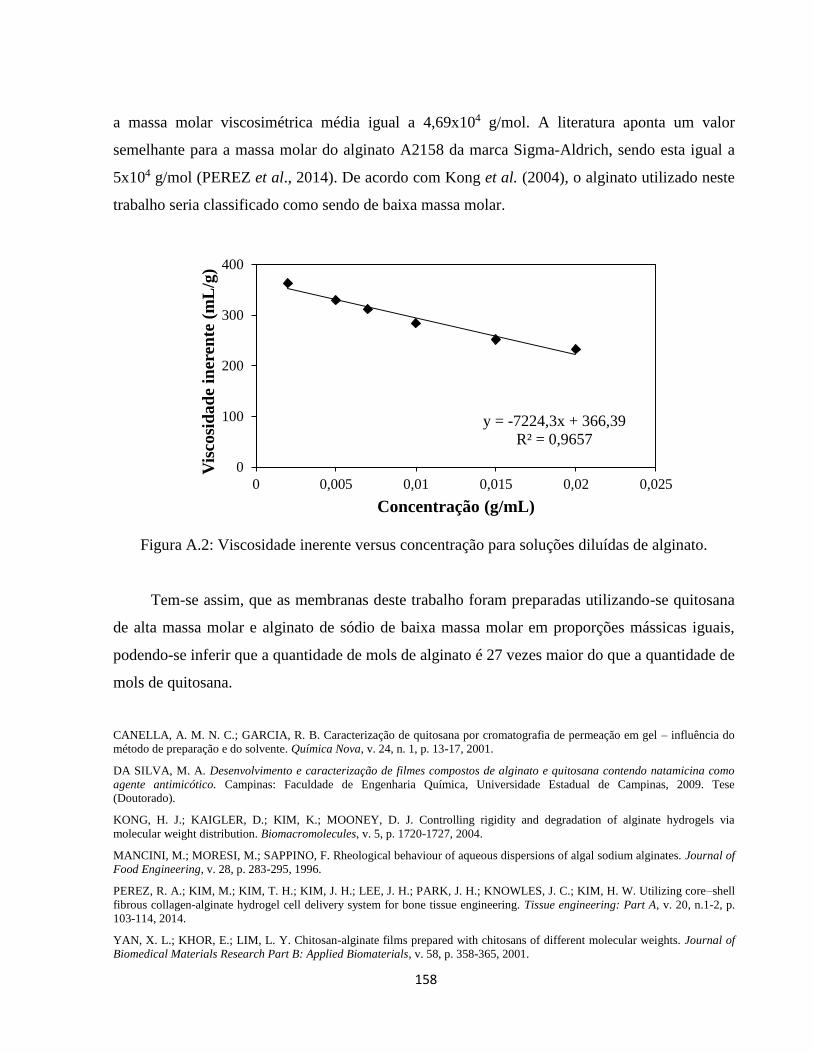
158
a massa molar viscosimétrica média igual a 4,69x104 g/mol. A literatura aponta um valor
semelhante para a massa molar do alginato A2158 da marca Sigma-Aldrich, sendo esta igual a
5x104 g/mol (PEREZ et al., 2014). De acordo com Kong et al. (2004), o alginato utilizado neste
trabalho seria classificado como sendo de baixa massa molar.
Figura A.2: Viscosidade inerente versus concentração para soluções diluídas de alginato.
Tem-se assim, que as membranas deste trabalho foram preparadas utilizando-se quitosana
de alta massa molar e alginato de sódio de baixa massa molar em proporções mássicas iguais,
podendo-se inferir que a quantidade de mols de alginato é 27 vezes maior do que a quantidade de
mols de quitosana.
CANELLA, A. M. N. C.; GARCIA, R. B. Caracterização de quitosana por cromatografia de permeação em gel – influência do
método de preparação e do solvente. Química Nova, v. 24, n. 1, p. 13-17, 2001.
DA SILVA, M. A. Desenvolvimento e caracterização de filmes compostos de alginato e quitosana contendo natamicina como
agente antimicótico. Campinas: Faculdade de Engenharia Química, Universidade Estadual de Campinas, 2009. Tese
(Doutorado).
KONG, H. J.; KAIGLER, D.; KIM, K.; MOONEY, D. J. Controlling rigidity and degradation of alginate hydrogels via
molecular weight distribution. Biomacromolecules, v. 5, p. 1720-1727, 2004.
MANCINI, M.; MORESI, M.; SAPPINO, F. Rheological behaviour of aqueous dispersions of algal sodium alginates. Journal of
Food Engineering, v. 28, p. 283-295, 1996.
PEREZ, R. A.; KIM, M.; KIM, T. H.; KIM, J. H.; LEE, J. H.; PARK, J. H.; KNOWLES, J. C.; KIM, H. W. Utilizing core–shell
fibrous collagen-alginate hydrogel cell delivery system for bone tissue engineering. Tissue engineering: Part A, v. 20, n.1-2, p.
103-114, 2014.
YAN, X. L.; KHOR, E.; LIM, L. Y. Chitosan-alginate films prepared with chitosans of different molecular weights. Journal of
Biomedical Materials Research Part B: Applied Biomaterials, v. 58, p. 358-365, 2001.
y = -7224,3x + 366,39
R² = 0,9657
0
100
200
300
400
0 0,005 0,01 0,015 0,02 0,025
Vis
cosi
dad
e in
eren
te (
mL
/g)
Concentração (g/mL)

159
APÊNDICE II – Determinação do grau de desacetilação da quitosana
O grau de desacetilação da quitosana, definido como a porcentagem de unidades de D-
glicosamina em relação ao número de monômeros totais de D-glicosamina e N-
acetilglicosamina, foi determinado por titulação ácido-base segundo o procedimento descrito por
da Silva (2009) e Jiang et al. (2003). Cerca de 0,2 gramas de quitosana foram dissolvidos em
25 mL de HCl a 0,1 N, completando-se o volume para 100 mL com água destilada. A solução
resultante foi titulada com NaOH a 0,1 N sob agitação lenta até atingir pH 2,0. A partir deste
ponto, a solução titulante foi adicionada em intervalos de 0,5 mL, aguardando-se um tempo
mínimo de 25 segundos para que houvesse o equilíbrio da reação, obtendo-se um valor estável
de pH. A titulação foi interrompida quando o pH era igual a 6,0. Os dados de volume de solução
titulante e pH foram utilizados para o cálculo de f(x) de acordo com a Equação A.7:
OHH
N
VVxf
B
0)( (A.7)
onde V0 é o volume da solução de quitosana antes do início da titulação (mL), V é o volume de
solução de NaOH adicionada (mL), NB é a concentração real de NaOH (N), pH10H e
14pH10OH .
Uma curva de titulação foi obtida plotando-se f(x) versus o volume de NaOH, V,
correspondente. A partir do gráfico, obteve-se o volume de NaOH no ponto de extremidade, Ve,
quando 0xf )( .
O grau de desacetilação da quitosana (%GD) foi calculado por meio da Equação A.8:
100
204
161WGD
% (A.8)
onde 1000VNVN eBAA / , NA é a concentração de HCl (N), VA é o volume de HCl (mL),
NB é a concentração de NaOH (N), Ve é o volume do NaOH no ponto de extremidade (mL) e W é

160
a massa de amostra (g).
Os experimentos foram realizados em triplicata e as soluções de HCl e NaOH foram
padronizadas antes da realização das medidas.
Destaca-se que os métodos de determinação do grau de desacetilação da quitosana por
titulação são pouco onerosos e mais robustos do que outras técnicas como a espectroscopia no
UV-VIS e o FTIR. Os resultados obtidos por técnicas de titulação de quitosana apresentam um
desvio pequeno em relação à técnica de 1H-RMN, considerada como uma das mais precisas para
este tipo de objetivo, de cerca de 3,53% (CZECHOWSKA-BISKUP et al., 2012).
De acordo com esta metodologia, o grau de desacetilação da quitosana empregada neste
trabalho é de 95,24 ± 0,72 %.
Para haver interação completa entre os biopolímeros, a quantidade de mols de alginato
presente deve ser maior do que a quantidade de mols de quitosana, uma vez que a cadeia de
quitosana é maior em comparação com a de alginato. Calculou-se qual seria, em teoria, a relação
molar ideal entre a quitosana e o alginato utilizados neste trabalho de modo que todos os grupos
amino da quitosana e carboxila do alginato interagissem entre si. Para estes cálculos, levou-se
em conta o grau de desacetilação da quitosana medido por titulação, igual a 95,24 ± 0,72%, o
qual tem influência sobre a quantidade de grupos amino presentes na molécula de quitosana.
Levaram-se também em consideração as massas molares viscosimétricas mostradas
anteriormente. Assim, calculou-se que, numa situação ideal, deve haver 28 mols de alginato para
cada mol de quitosana, razão esta muito próxima à razão mássica entre os biopolímeros de 1:1.
CZECHOWSKA-BISKUP, R.; JAROSIŃSKA, D.; ROKITA, B.; ULAŃSKI, P.; ROSIAK, J. M. Determination of degree of
deacetylation of chitosan - comparison of methods. Progress on chemistry and application of chitin and its derivatives, v. XVII,
p. 5 -20, 2012.
DA SILVA, M. A. Desenvolvimento e caracterização de filmes compostos de alginato e quitosana contendo natamicina como
agente antimicótico. Campinas: Faculdade de Engenharia Química, Universidade Estadual de Campinas, 2009. Tese
(Doutorado).
JIANG, X.; CHEN, L.; ZHONG, W. A new linear potentiometric titration method for the determination of deacetylation degree
of chitosan. Carbohydrate Polymers, v. 54, p. 457–463, 2003.

161
APÊNDICE III – Determinação da razão M/G do alginato
A razão M/G do alginato de sódio foi determinada por 1H-RMN, que é a principal técnica
utilizada para esta finalidade (TORRES et al., 2007). Inicialmente foi realizado um pré-
tratamento das amostras de acordo com Davis et al. (2004), com o objetivo de despolimerizar o
alginato e reduzir a viscosidade da solução a um nível adequado para a aquisição de espectros
1H-RMN de alta resolução.
Para a despolimerização do alginato, 30 mg de amostra foram colocados em 90 mL de água
deionizada. A suspensão foi dissolvida ao longo de 24 h e o pH foi ajustado para 5,6 com HCl a
0,1M. A solução foi colocada em um banho de água a 95°C por uma hora. O pH foi então re-
ajustado para 3,8 e a solução retornou ao banho a 95°C por 45 minutos. Por fim, a solução foi
neutralizada com NaOH a 0,1M e liofilizada a -60°C sob vácuo por 24 horas (liofilizador Liobras
L101).
As amostras liofilizadas foram preparadas para a análise de 1H-RMN dissolvendo-se
30 mg em 0,7 mL de água deuterada. Utilizou-se como padrão 2,2-dimetil-2-silapentano-5-
sulfonato de sódio (DSS). As amostras foram analisadas em espectrômetro de ressonância
magnética nuclear Bruker Advance 500 MHz à temperatura de 80°C em triplicatas.
A fração de ácido gulurônico (FG) foi calculada de acordo com a Equação A.9
(KLEINÜBING, 2009; LARSEN et al., 2003):
CB
AG
II
IF
(A.9)
onde IA é a intensidade do pico em aproximadamente 5,06 ppm, IB é a intensidade do pico em
aproximadamente 4,4 ppm e IC é a intensidade do pico em aproximadamente 4,7 ppm.
A partir do valor da fração de ácido gulurônico (FG), calculou-se a razão M/G de acordo
com a Equação A.10 (KLEINÜBING, 2009; LARSEN et al., 2003; TORRES et al., 2007):
G
G
F
F1
G
M )( (A.10)
A quantidade relativa entre os blocos M e G e a sua conformação macromolecular
determinam as propriedades físicas e a afinidade do alginato por metais divalentes (HAUG et al.,

162
1967), influenciando o tipo de gel formado pelo polímero quando na presença destes metais.
Géis quebradiços são obtidos a partir de alginatos com baixa razão M/G, enquanto géis elásticos
são obtidos a partir de alginatos com alta razão M/G (PENMAN e SANDERSON, 1972).
Calculou-se por meio das Equações A.9 e A.10, que a razão M/G do alginato utilizado é de
1,86±0,03, o que o classifica como um alginato alta razão M/G.
A literatura aponta um valor próximo para a razão M/G do alginato A2158 da marca
Sigma-Aldrich, sendo esta igual a 1,67 (PEREZ et al., 2014). A diferença entre este valor e o
encontrado seria provavelmente devida à diferença entre lotes.
DAVIS, T. A.; RAMIREZ, M.; MUCCI, A.; LARSEN, B. Extraction, isolation and cadmium binding of alginate from
Sargassum spp. Journal of Applied Phycology, v. 16, p. 275–284, 2004.
HAUG, A.; MYKLESTAD, S.; LARSEN, B.; SMIDSRØD, O. Correlation between chemical structure and physical properties
of alginates. Acta Chemica Scandinavica, v. 21, p. 768-778, 1967.
KLEINÜBING, S. J. Bioadsorção competitiva dos íons níquel e cobre em alginato e alga marinha Sargassum Filipendula.
Campinas: Faculdade de Engenharia Química, Universidade Estadual de Campinas, 2009. Tese (Doutorado).
LARSEN, B.; SALEM, D. M. S. A.; SALLAM, M. A. E.; MISHRIKEY, M. M.; BELTAGY, A. I. Characterization of the
alginates from algae harvested at the Egyptian Red Sea coast. Carbohydrate Research, v. 338, p. 2325-2336, 2003.
PENMAN, A.; SANDERSON, G. R. A method for the determination of uronic acid sequence in alginates. Carbohydrate
Research, v. 25, p. 273-282, 1972.
PEREZ, R. A.; KIM, M.; KIM, T. H.; KIM, J. H.; LEE, J. H.; PARK, J. H.; KNOWLES, J. C.; KIM, H. W. Utilizing core–shell
fibrous collagen-alginate hydrogel cell delivery system for bone tissue engineering. Tissue engineering: Part A, v. 20, n.1-2, p.
103-114, 2014.
TORRES, M. R.; SOUSA, A. P. A.; SILVA FILHO, E. A. T.; MELO, D. F.; FEITOSA, J. P. A.; DE PAULA, R. C. M.; LIMA,
M. G. S. Extraction and physicochemical characterization of Sargassum vulgare alginate from Brazil. Carbohydrate Research, v.
342, p. 2067–2074, 2007.

163
APÊNDICE IV – Estabelecimento da estratégia mais apropriada de quantificação do
PHMB incorporado
Diferentes abordagens foram utilizadas para a caracterização das membranas quanto à
quantidade de PHMB retida, sendo estas a análise direta da absorbância do PHMB em meio
líquido, a medida da coloração residual após a formação de um complexo de níquel com nioxima
e a análise por cromatografia líquida de alta eficiência (HPLC), conforme descrito nos subitens
que se seguem. Foi analisada também a potencial interferência dos componentes da membrana
na quantificação do agente antimicrobiano.
a) Espectrofotometria no UV-Visível
O espectro de absorção de luz do PHMB foi avaliado no intervalo de 200 a 500 nm e,
conhecendo-se a importância de determinar a interferência de eventuais componentes que
poderiam ser liberados das membranas devido à sua degradação durante a quantificação do
PHMB, foram feitas também varreduras de absorção no UV-VIS de soluções de tais
componentes nas concentrações explicitadas na Tabela A.1 (inclusas também as descrições das
soluções em branco utilizado nas análises).
Tabela A.1: Soluções dos constituintes das membranas utilizadas no levantamento dos perfis de
absorção de luz no espectro UV-VIS.
Composto Solução Branco
PHMB 20 µg/mL Água
Quitosana 0,5% em ácido acético 1,0% Ácido acético 1,0%
Alginato 0,25% em água Água
NaOH 0,25M em água Água
Pluronic F68 0,5% em água Água
Ácido acético 1,0% em água Água
CaCl2 1,0% em água Água
A análise por espectrofotometria no UV-VIS direta do PHMB em água e PBS indicou que,

164
para ambos os solventes, os comprimentos de onda de 235 ou 236 nm seriam os mais indicados
para a análise deste composto, dependendo do equipamento utilizado para as medidas.
Na Figura A.3 são mostrados os resultados das análises de varredura no UV-VIS dos
principais componentes das membranas preparadas neste trabalho. Por meio da análise dos
espectros obtidos, nota-se que os compostos mais propensos a causar interferências na
quantificação do PHMB, por apresentarem absorbância significativa a 236 nm são a quitosana, o
alginato e o ácido acético. Este último está presente em quantidades menores do que os
polímeros, pois grande parte do ácido acético utilizado na produção das membranas evapora ou é
removida durante as etapas de lavagem das membranas.
Um método muito utilizado para extrair e avaliar a cinética de liberação de agente ativos
contidos em dispositivos biomédicos é a análise de absorbância do meio líquido no qual o
material se encontra imerso. O tampão PBS é bastante utilizado para esta finalidade por possuir
pH igual a 7,4, simulando condições fisiológicas. Sendo assim, a varredura do meio de liberação
(tampão PBS) no qual as membranas ficaram imersas por 24 horas a 37°C e 100 rpm também foi
realizada principalmente porque, após o período de contato com as membranas, este líquido
apresentou-se turvo, provavelmente devido à degradação parcial do material.
A Figura A.4 mostra os espectros de absorbância do meio de liberação de membranas tipo
P2% preparadas na presença de PHMB a 10 mg/g. Nota-se que o pico característico do PHMB
(236 nm) é evidente neste gráfico, porém, a membrana que não contém PHMB também
apresenta absorbância no comprimento de onda característico do agente antimicrobiano. Sendo
assim, verificou-se que a presença de interferentes precisa ser computada nos cálculos da
concentração de PHMB no meio de liberação, utilizando-se como referência a membrana sem
PHMB. Verificou-se também que, para formulações contendo quantidades de PHMB menores
do que 10 mg/g, não é possível quantificar o antimicrobiano por UV-VIS, uma vez que os
valores de absorbância dos meios de liberação das membranas com e sem PHMB são
estatisticamente semelhantes.
b) Colorimetria
Alternativamente à análise por absorção direta de luz, a concentração do PHMB foi
também avaliada pelo método colorimétrico proposto por Rowhani e Lagalante (2007), o qual

165
consiste na reação entre íons níquel não reagidos com o PHMB e Nioxima (1,2-
ciclohexanodionadioxima).
Figura A.3: Espectros de absorbância no UV-VIS dos componentes das membranas (quitosana,
alginato, ácido acético, CaCl2, NaOH, Pluronic F68 e PHMB). A linha vertical em cada espectro
indica o comprimento de onda de 236 nm.
200 300 400 500
Ab
sorb
ân
cia
Comprimento de onda (nm)
Quitosana
236 200 300 400 500
Ab
sorb
ân
cia
Comprimento de onda (nm)
Alginato
236
200 300 400 500
Ab
sorb
ân
cia
Comprimento de onda (nm)
Ácido acético
236 200 300 400 500
Ab
sorb
ân
cia
Comprimento de onda (nm)
CaCl2
236
200 300 400 500
Ab
sorb
ân
cia
Comprimento de onda (nm)
NaOH
236 200 300 400 500
Ab
sorb
ân
cia
Comprimento de onda (nm)
Pluronic F68
236
200 300 400 500
Ab
sorb
ân
cia
Comprimento de onda (nm)
236
PHMB
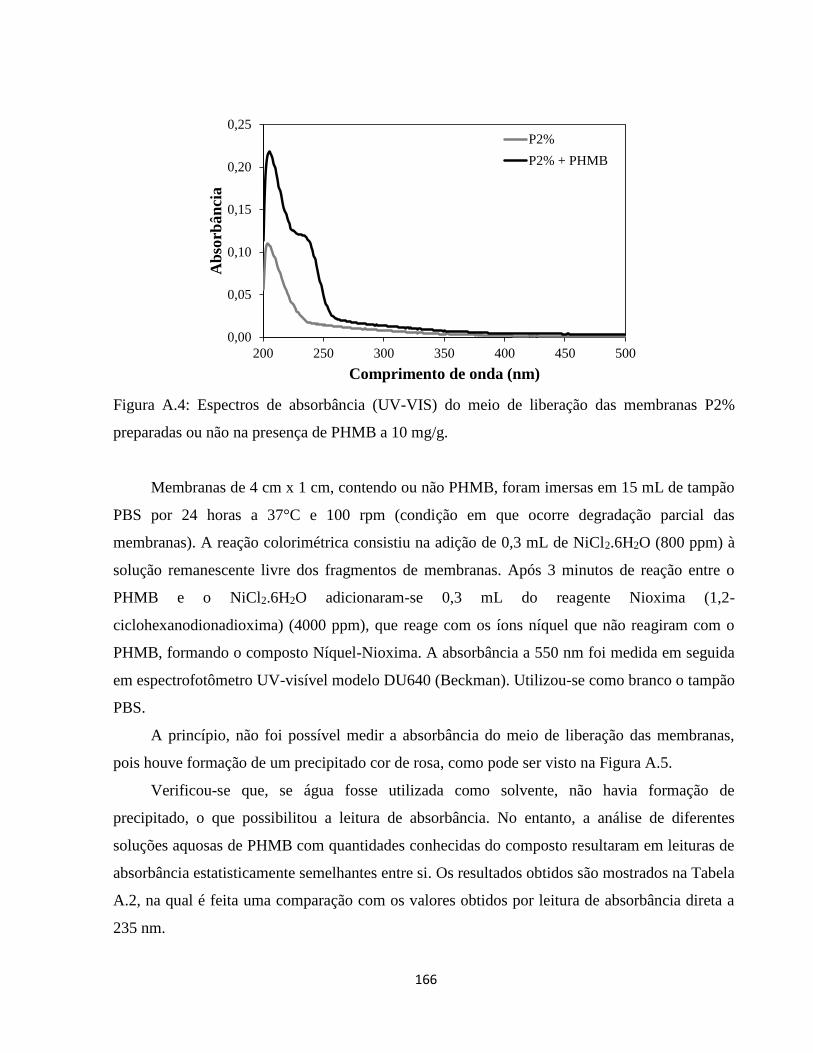
166
Figura A.4: Espectros de absorbância (UV-VIS) do meio de liberação das membranas P2%
preparadas ou não na presença de PHMB a 10 mg/g.
Membranas de 4 cm x 1 cm, contendo ou não PHMB, foram imersas em 15 mL de tampão
PBS por 24 horas a 37°C e 100 rpm (condição em que ocorre degradação parcial das
membranas). A reação colorimétrica consistiu na adição de 0,3 mL de NiCl2.6H2O (800 ppm) à
solução remanescente livre dos fragmentos de membranas. Após 3 minutos de reação entre o
PHMB e o NiCl2.6H2O adicionaram-se 0,3 mL do reagente Nioxima (1,2-
ciclohexanodionadioxima) (4000 ppm), que reage com os íons níquel que não reagiram com o
PHMB, formando o composto Níquel-Nioxima. A absorbância a 550 nm foi medida em seguida
em espectrofotômetro UV-visível modelo DU640 (Beckman). Utilizou-se como branco o tampão
PBS.
A princípio, não foi possível medir a absorbância do meio de liberação das membranas,
pois houve formação de um precipitado cor de rosa, como pode ser visto na Figura A.5.
Verificou-se que, se água fosse utilizada como solvente, não havia formação de
precipitado, o que possibilitou a leitura de absorbância. No entanto, a análise de diferentes
soluções aquosas de PHMB com quantidades conhecidas do composto resultaram em leituras de
absorbância estatisticamente semelhantes entre si. Os resultados obtidos são mostrados na Tabela
A.2, na qual é feita uma comparação com os valores obtidos por leitura de absorbância direta a
235 nm.
0,00
0,05
0,10
0,15
0,20
0,25
200 250 300 350 400 450 500
Ab
sorb
ân
cia
Comprimento de onda (nm)
P2%
P2% + PHMB

167
Figura A.5: Aspecto do meio de liberação das membranas P2% após a reação colorimétrica de
formação do complexo níquel-nioxima.
Tabela A.2: Absorbâncias médias de soluções aquosas com diferentes concentrações de PHMB
antes e depois da reação colorimétrica de formação do complexo níquel-nioxima.
Concentração de
PHMB (µg/mL)
ABS 235 nm
antes da reação
ABS 550 nm
antes da reação
ABS 550 nm depois da
reação
4 0,205 ± 0,003 a 0,000 ± 0,000 a 0,253 ± 0,003 a
10 0,537 ± 0,026 b 0,000 ± 0,000 a 0,208 ± 0,033 a
14 0,766 ± 0,010 c 0,001 ± 0,000 a 0,193 ± 0,030 a
20 1,122 ± 0,021 d 0,001 ± 0,000 a 0,183 ± 0,014 a
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de
Tukey, p<0,05).
A análise exploratória por colorimetria do PHMB liberado das membranas por extração
com água também resultou em dados pouco conclusivos (Tabela A.3), atingindo-se altas
absorbâncias, mas sem relação de proporcionalidade entre as concentrações. Concluiu-se que o
NiCl2.6H2O, empregado como primeiro reagente do método colorimétrico, pode estar reagindo
não somente com o PHMB, mas também com a quitosana, devido à sua capacidade de quelatar
diversos íons metálicos (DIVAKARAN et al., 2012), diminuindo a seletividade de detecção do
método. Como o PHMB está presente em pequenas quantidades, e a quitosana está presente em
Branco Sem
PHMB
[PHMB]
1 mg/g
[PHMB]
10 mg/g

168
grandes quantidades, se a membrana se desestabiliza, mesmo que parcialmente, liberando este
polissacarídeo, esta reação pode ter interferido fortemente nos resultados, inviabilizando o uso
confiável desta técnica.
Tabela A.3: Valores de absorbância a 550 nm das soluções de liberação das membranas
preparadas na ausência e na presença de PHMB a 10 mg/g (testes realizados em água).
[PHMB] (mg/g) Absorbância
0 0,408 ± 0,010 a
10 0,429 ± 0,011 a
Mesma letra na mesma coluna indica que não há diferença significativa entre os
valores médios (Teste de Tukey, p<0,05).
c) Cromatografia líquida de alta eficiência (HPLC)
Os ensaios foram realizados de acordo com o método proposto por Arora et al. (2009).
Este método foi escolhido dentre outras opções de quantificação do PHMB por HPLC devido a
sua elevada sensibilidade de detecção (2 µg/mL). Outros métodos considerados nesta escolha
foram os propostos por Yiping et al. (2005), com limite de detecção de 50 µg/mL, e pelo
fabricante do PHMB (ARCH CHEMICALS, 2007), com limite de detecção de 7,8 µg/mL.
Utilizou-se equipamento da marca Waters provido de bomba (Binary HPLC pump 1525) e
acoplado a um detector de absorbância (Dual λ absorbance detector 2487), utilizando-se uma
coluna Symmetry C18 de 4,6 mm x 50 mm com diâmetro de partícula de 5 μm da marca Waters.
Os dados foram armazenados e analisados por meio do software Breeze®. A fase móvel,
bombeada à vazão de 1 mL/min, consistiu de uma mistura de uma solução de acetonitrila a
1% (v/v) com uma solução aquosa de acetato de amônio a 20 mM (16:84 v/v), sendo o pH
ajustado para 4,0 com ácido acético glacial. O volume de amostra injetado no equipamento foi
de 20 μL e a temperatura foi mantida em 30°C durante as análises. De acordo com Arora et al.
(2009), o tempo de retenção do PHMB é de 5,883 min e, conforme já mencionado, o limite de
detecção do método é de 2 μg/mL.
De acordo com o método proposto por Arora et al. (2009), esperava-se observar o pico
característico do PHMB em cerca de 6 minutos, desta forma, as análises tiveram duração mínima
de 10 minutos.

169
Contudo, não foi possível reproduzir os dados da literatura. Os resultados obtidos não
permitiram identificar a presença de PHMB, não sendo observado nenhum pico no tempo de
retenção esperado.
Assim, dentre as técnicas utilizadas neste trabalho para a quantificação do PHMB, aquela
que se mostrou mais sensível e seletiva foi a análise por espectrofotometria direta em 235 ou
236 nm, a qual foi empregada nos ensaios de liberação e cálculo da eficiência de incorporação.
ARCH CHEMICALS. Determination of PHMB in 20% aqueous solution by HPLC. 2007. Documento confidencial recebido via
comunicação pessoal em 2012.
ARORA, A.; ALI, A.; ZZAMAN, M. T. Simple and sensitive method for determination of polyhexanide in multipurpose
solution. Electronic Journal of Biomedicine, v. 2, p. 30-37, 2009.
DIVAKARAN, R.; PAUL, A. A. J; ANOOP, K. K; KURIAKOSE, A. V. J; RAJESH, R. Adsorption of nickel(II) and
chromium(VI) ions by chitin and chitosan from aqueous solutions containing both ions. International Journal of Science and
Technology Education Research, v. 1, p. 43-50, 2012.
ROWHANI, T.; LAGALANTE, A. F. A colorimetric assay for the determination of polyhexamethylene biguanide in pool and
spa water using nickel–nioxime. Talanta, v. 71, p. 964-970, 2007.
YIPING, C.; XIAOJING, D.; SHAN, Z.; PENGYAN, L. Rapid determination of polyhexamethylenebiguanide in compound
chemical disinfectants by reversed-phase high performance liquid chromatography. Chemistry Magazine, v. 7, n. 8, p. 55, 2005.

170
APÊNDICE V – Verificação da estabilidade do PHMB nas condições de preparação das
membranas
Visando determinar se ocorrem alterações no comportamento de absorbância do PHMB no
UV-VIS nas condições de preparação das membranas, foram realizados preliminarmente ensaios
de simulação de produção e secagem das membranas na ausência de polímeros, contendo ácido
acético, NaOH e CaCl2 nas mesmas proporções em que são empregados na produção das
membranas. Dentre estes compostos, o ácido acético seria o mais propenso a prejudicar a
incorporação de PHMB, uma vez que há relatos de que a diminuição do pH pode provocar a
hidrólise imediata do antimicrobiano, particularmente na presença de HCl de 0,1 a 1 N (LUCAS,
2012).
O PHMB foi adicionado às soluções em quantidades equivalentes para o preparo de
membranas com 1% m/m (10 mg/g) e 10% m/m (100 mg/g) do antimicrobiano. Os espectros de
varredura no UV-VIS das soluções iniciais foram obtidos e, ao final da secagem em placas de
poliestireno, o sólido restante foi novamente diluído no mesmo volume de água, obtendo-se o
espectro de varredura no UV-VIS ao final da secagem.
As análises de absorbância a 235 nm antes e após a secagem indicaram que houveram
pequenas diminuições de absorbância (de 3,6% para a concentração inicial de PHMB de 10 mg/g
e de 1,3% para a concentração inicial de PHMB de 100 mg/g). Estes valores não são
significativos e são comparáveis à diminuição de absorbância em água, que é de 4% após
24 horas a 25°C. Desta forma, pode-se concluir que a absorbância no UV-VIS do PHMB é
estável durante a produção e secagem das membranas, podendo indicar que o antimicrobiano não
sofre degradação devido às alterações de pH.
LUCAS, A. D. Environmental fate of polyhexamethylene biguanide. Bulletin of Environmental Contamination and Toxicology,
v. 88, p. 322-325, 2012.

171
APÊNDICE VI – Análise do efeito de potenciais solventes para a solubilização das
membranas
Devido à capacidade de interação do PHMB com os polissacarídeos constituintes das
membranas por interações eletrostáticas entre os grupos biguanida do antimicrobiano e os grupos
–COO- do alginato, por ligações de hidrogênio entre os grupos biguanida e os grupos –OH dos
polissacarídeos e por interações hidrofóbicas entre os grupos hexametileno do PHMB e –CH dos
polissacarídeos, a simples extração do composto incorporado pelo uso de um tampão ou de outra
solução poderia não ser suficiente para a recuperação do PHMB visando sua quantificação, de
forma que se buscou opções para a dissolução das membranas.
De acordo com Yan et al. (2001) o PEC de quitosana-alginato apresenta-se insolúvel em
muitos solventes (água em faixa ampla de pH, etanol, metanol, dimetil sulfóxido, dimetil
formamida, LiCl-dimetil acetamida e ácido trifluoroacético), e mesmo após imersão por dois
dias em água régia (mistura de ácido nítrico e ácido clorídrico na proporção de 1:3 v/v), segundo
Bueno (2010), não ocorre solubilização efetiva do material.
Como um dos mecanismos de estabilização estrutural das membranas é a reticulação de
carboxilas provenientes do alginato com íons cálcio, sua exposição a soluções contendo EDTA,
um composto capaz de quelatar cálcio, poderia desestabilizá-las (HE et al., 2008; D’AYALA et
al., 2008).
Assim, a solubilização de membranas porosas de quitosana-alginato preparadas na
presença de 2% (m/m) de Pluronic F68 foi testada em uma solução contendo EDTA a 0,1M e
K2HPO4 a 0,1M, de acordo com o procedimento proposto por He et al. (2008) para a
solubilização de scaffolds de quitosana-alginato. Amostras de 32 mg de membrana cortadas em
fragmentos de cerca de 0,2 cm x 0,2 cm foram adicionadas a 15 mL da solução, sendo o ensaio
realizado em duplicata. Os frascos contendo as membranas e a solução de EDTA foram
mantidos em estufa a 37oC e 100 rpm por 24 horas e, em seguida, sonicados por 10 minutos e
centrifugados por 10 minutos a 5000 rpm, analisando-se a fase líquida.
Após a tentativa de solubilização, notou-se apenas uma leve fragmentação das amostras
devido provavelmente à ação quelante do EDTA, que causou a remoção de alguns íons cálcio.
Acredita-se que a solubilização do material preparado por He et al. (2008) foi possível pois o
mesmo era constituído pelo PEC de quitosana-alginato 1:1 (m/m) liofilizado e reticulado por

172
imersão em solução de CaCl2 a 2% (m/m) por apenas 2 minutos, apresentando estrutura mais
porosa e frágil do que as membranas do presente trabalho. Concluiu-se, assim, que novos ensaios
deveriam ser estabelecidos e realizados na busca de resolver este problema específico, como os
mostrados no Apêndice VII a seguir.
BUENO, C. Z. Influência do tipo de estratégia de reforço das propriedades mecânicas nas características de membranas de
quitosana e alginato projetadas para o recobrimento de lesões de pele. Campinas: Faculdade de Engenharia Química,
Universidade Estadual de Campinas, 2010. Dissertação (Mestrado).
D’AYALA, G. G; MALINCONICO, M.; LAURIENZO, P. Marine derived polysaccharides for biomedical applications:
Chemical modification approaches. Molecules, v. 13, p. 2069-2106, 2008.
HE, B.; LEUNG, M.; ZHANG, M. Optimizing creation and degradation of chitosan-alginate scaffolds for in vitro cell culture.
Journal of Undergraduate Research in Bioengineering, p. 31-35, 2008.
YAN, X. L.; KHOR, E.; LIM, L. Y. Chitosan-alginate films prepared with chitosans of different molecular weights. Journal of
Biomedical Materials Research Part B: Applied Biomaterials, v. 58, p. 358-365, 2001.

173
APÊNDICE VII – Ensaios de extração e quantificação de PHMB em soluções básicas
a) Análise do comportamento do PHMB em soluções básicas (testes de possíveis condições
de extração)
Como tentativa de extração e posterior quantificação do PHMB contido nas membranas de
quitosana-alginato, optou-se pela utilização de soluções básicas de NaOH. Sabe-se que, em altos
valores de pH, pode-se atingir uma situação na qual os grupos biguanida do PHMB e amino da
quitosana encontram-se desprotonados e os grupos carboxila do alginato apresentam carga
negativa. Desta forma, a interação entre a quitosana e o alginato diminuiria, facilitando a extração
do PHMB, o qual, por possuir carga neutra em altos valores de pH, não interagiria com os
polissacarídeos via interações eletrostáticas. O valor de pH neste caso, necessita ser alto o
suficiente para garantir que esteja acima dos valores de pKa do PHMB, que é igual a 10,96
(O’MALLEY et al., 2006), e dos polissacarídeos, que são de aproximadamente 6,3 a 7,3 para a
quitosana (KARAKEÇILI et al., 2007; IL’INA e VARLAMOV, 2005) e de 3,38 a 3,65 para o
alginato (LI et al., 2009; SÆTHER et al., 2008).
A utilização de baixos valores de pH, o que proporcionaria uma situação na qual o PHMB e
a quitosana apresentassem carga positiva e o alginato apresentasse carga neutra, também seria
interessante para a extração do PHMB. Nestas condições, além de a interação entre a quitosana e
o alginato ser reduzida devido à carga positiva da quitosana e neutra do alginato, o PHMB com
carga positiva seria repelido pelas cargas positivas da quitosana. No entanto, há relatos de que o
PHMB é imediatamente hidrolisado em soluções de HCl de 0,1 a 1N, enquanto que em soluções
de NaOH a 1N, o PHMB se degrada somente após 30 dias (LUCAS, 2012). Sendo assim, devido
à maior estabilidade do PHMB em soluções de NaOH em comparação ao HCl, optou-se pela
extração PHMB em meio básico, em detrimento da extração em condições ácidas.
Verificou-se em ensaios preliminares que, em soluções de NaOH, o comprimento de onda
de máxima absorbância do PHMB (λmáx) praticamente se manteve em relação ao λmáx em água,
que ocorre em 235 nm. Em solução de NaOH a 0,05M, o PHMB apresentou λmáx em 234 nm. Já
em solução de NaOH a 0,1M, o PHMB apresentou λmáx em 233 nm. Deslocamentos
significativos do λmáx de biguanidas em soluções básicas concentradas, devido à desprotonação,
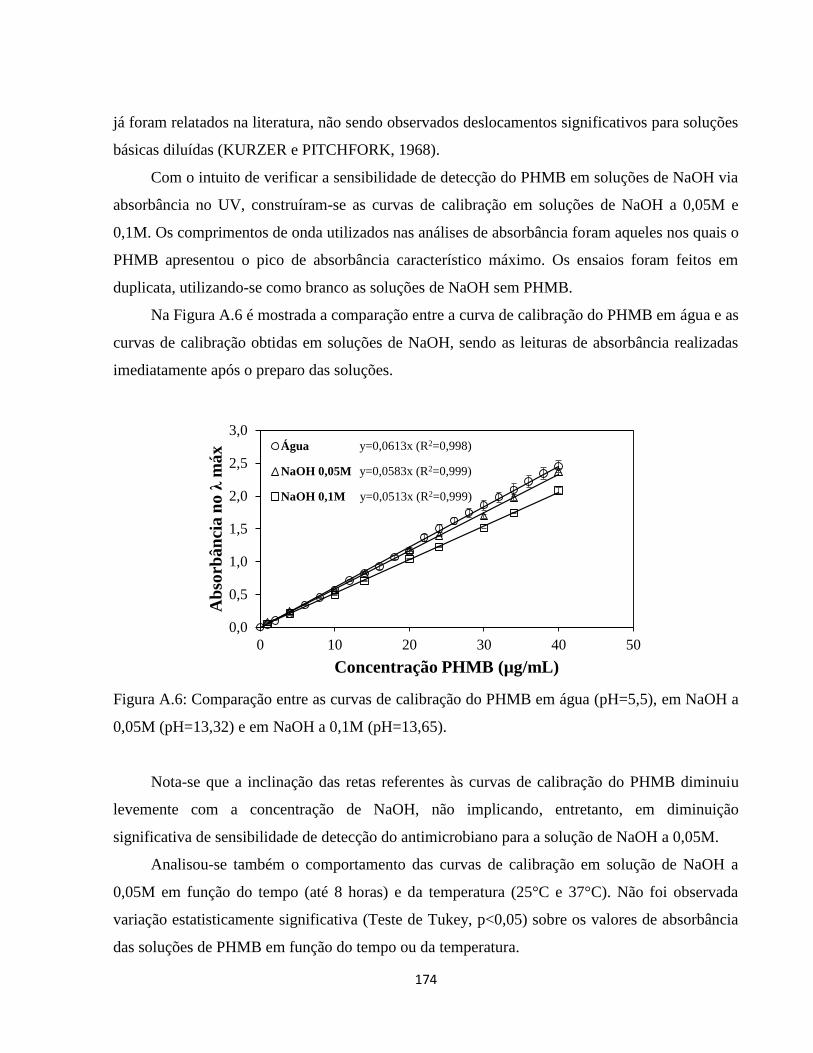
174
já foram relatados na literatura, não sendo observados deslocamentos significativos para soluções
básicas diluídas (KURZER e PITCHFORK, 1968).
Com o intuito de verificar a sensibilidade de detecção do PHMB em soluções de NaOH via
absorbância no UV, construíram-se as curvas de calibração em soluções de NaOH a 0,05M e
0,1M. Os comprimentos de onda utilizados nas análises de absorbância foram aqueles nos quais o
PHMB apresentou o pico de absorbância característico máximo. Os ensaios foram feitos em
duplicata, utilizando-se como branco as soluções de NaOH sem PHMB.
Na Figura A.6 é mostrada a comparação entre a curva de calibração do PHMB em água e as
curvas de calibração obtidas em soluções de NaOH, sendo as leituras de absorbância realizadas
imediatamente após o preparo das soluções.
Figura A.6: Comparação entre as curvas de calibração do PHMB em água (pH=5,5), em NaOH a
0,05M (pH=13,32) e em NaOH a 0,1M (pH=13,65).
Nota-se que a inclinação das retas referentes às curvas de calibração do PHMB diminuiu
levemente com a concentração de NaOH, não implicando, entretanto, em diminuição
significativa de sensibilidade de detecção do antimicrobiano para a solução de NaOH a 0,05M.
Analisou-se também o comportamento das curvas de calibração em solução de NaOH a
0,05M em função do tempo (até 8 horas) e da temperatura (25°C e 37°C). Não foi observada
variação estatisticamente significativa (Teste de Tukey, p<0,05) sobre os valores de absorbância
das soluções de PHMB em função do tempo ou da temperatura.
0,0
0,5
1,0
1,5
2,0
2,5
3,0
0 10 20 30 40 50
Ab
sorb
ân
cia n
o λ
má
x
Concentração PHMB (µg/mL)
Água
NaOH 0,05M
NaOH 0,1M
y=0,0613x (R2=0,998)
y=0,0513x (R2=0,999)
y=0,0583x (R2=0,999)

175
b) Validação do método de extração de PHMB com solução básica
Estes ensaios consistiram em utilizar o método de spiking para verificar a validade do
método de extração de PHMB com solução básica de NaOH a 0,05M, com base na recuperação
de uma massa conhecida de antimicrobiano adicionada à amostra.
Os ensaios foram realizados com membranas de quitosana-alginato preparadas na presença
de 2% de Pluronic F68 e na ausência de PHMB. As membranas foram cortadas em corpos de
prova de 4 cm x 1 cm e armazenadas em dessecador por 24 horas para eliminação da umidade
residual. Após este período, mediu-se a massa inicial dos corpos de prova, os quais foram então
imersos em 15 mL de NaOH a 0,05M contendo uma quantidade inicial conhecida de PHMB
(cerca de 10 µg/mL) a 25°C e a 37°C a 100 rpm por 8 horas. A absorbância das soluções no λmáx
foi medida no início e no final do experimento, calculando-se então as quantidades de PHMB na
solução por meio da curva de calibração. Amostras controle sem PHMB foram usadas para
descontar as possíveis interferências dos polissacarídeos nas medidas de absorbância. Os ensaios
foram realizados em triplicata.
Os resultados obtidos nos testes de spiking, relativos à diferença entre as quantidades de
PHMB medidas no fim e no início do experimento são mostrados na Tabela A.4.
Tabela A.4: Diferença entre as quantidades de PHMB inicial e final utilizadas nos testes de
spiking para verificação da aplicabilidade dos testes de extração com solução de NaOH a 0,05M.
Condição [PHMB]inicial (µg/mg) [PHMB]final (µg/mg)
NaOH 0,05M 25°C 5,3 ± 0,3 a 5,6 ± 0,3 a
NaOH 0,05M 37°C 6,9 ± 0,4 a 6,6 ± 0,4 a
Mesma letra na mesma linha indica que não há diferença significativa entre os valores médios (Teste
De Tukey, p<0,05).
Como verificado pelo teste de Tukey (p<0,05), não foi observada variação significativa
entre as quantidades de PHMB presentes no início e no fim do experimento para as soluções de
NaOH a 0,05M a 25 e 37°C. Desta forma, os resultados obtidos nos testes de spiking indicam que
o método de extração de PHMB com soluções básicas de NaOH é aplicável.

176
c) Extração de PHMB contido nas membranas porosas de quitosana-alginato com solução
de NaOH a 0,05M
Os ensaios foram realizados com membranas de quitosana-alginato preparadas na presença
de 2% de Pluronic F68, contendo PHMB adicionado à mistura polimérica na proporção de
10 mg/g. As membranas foram cortadas em corpos de prova de 4 cm x 1 cm e imersas em 15 mL
de NaOH a 0,05M a 25°C ou 37°C a 100 rpm por até 8 horas. A absorbância das soluções no λmáx
foi medida ao final do experimento. Os ensaios foram realizados em triplicata utilizando-se como
controle amostras sem PHMB.
Os resultados obtidos são mostrados na Tabela A.5 e indicam que as quantidades de PHMB
extraídas nas duas condições empregadas foram muito menores do que a quantidade de PHMB
presente nas membranas, de 8,33 ± 0,12 mg/g, a qual foi calculada computando-se as perdas de
PHMB durante as etapas de reticulação secundária e lavagens com água.
Tabela A.5: Quantidade de PHMB extraído das membranas com solução de NaOH a 0,05M em
diferentes condições de temperatura.
Condição de extração [PHMB] extraído (mg/g) Perda de massa (%)
NaOH 0,05M a 25°C 0,41 ± 0,13 a 32,9 ± 3,2 a
NaOH 0,05M a 37°C 0,65 ± 0,03 a 31,3 ± 0,1 a
Mesma letra na mesma coluna indica que não há diferença significativa entre os valores médios (Teste de
Tukey, p<0,05).
Verificou-se também que o mesmo tipo de ensaio realizado em PBS a 37°C e 100 rpm
resultou na extração de 1,16 ± 0,01 mg de PHMB por grama de membrana, quantidade também
inferior àquela adicionada, porém superior às quantidades extraídas em soluções básicas. A perda
de massa das membranas em PBS, por outro lado, se mostrou estatisticamente similar à perda de
massa em soluções básicas, sendo igual a 30,6 ± 1,3 %.
Desta forma, apesar de os testes de spiking terem indicado a aplicabilidade da extração de
PHMB com solução de NaOH a 0,05M, esta solução não foi eficaz para a extração do
antimicrobiano total presente nas membranas.
As diferenças significativas observadas em PBS e NaOH a 0,05M podem ser devidas à
ação quelante do PBS sobre os íons cálcio utilizados para a reticulação das membranas, formando

177
sais de fosfato de cálcio, como a hidroxiapatita (PASPARAKIS e BOUROPOULOS, 2006) e
também à troca iônica entre os íons sódio presentes no PBS e o cálcio presente nas membranas,
fenômenos que podem ter sido mais pronunciados do que o efeito da solução de NaOH a 0,05M,
a qual agiria de forma a reduzir as interações eletrostáticas entre a quitosana e o alginato.
IL’INA, A. V.; VARLAMOV V. P. Chitosan-based polyelectrolyte complexes: a review. Applied Biochemistry and
Microbiology, v. 41, n. 1, p. 5-11, 2005.
KARAKEÇILI, A.G.; SATRIANO, C.; GÜMÜŞDERELIOĞLU, M.; MARLETTA, G. Surface characteristics of ionically
crosslinked chitosan membranes. Journal of Applied Polymer Science, v. 106, p. 3884-3888, 2007.
KURZER, F.; PITCHFORK, E.D. The chemistry of biguanides. Springer, 1968. Cap.: Fortschritte der Chemischen Forschung
v.10/3, p.375-472.
LI, X.; XIE, H.; LIN, J.; XIE, W.; MAA, X. Characterization and biodegradation of chitosan–alginate polyelectrolyte complexes.
Polymer Degradation and Stability, v. 94, p. 1–6, 2009.
LUCAS, A.D. Environmental fate of polyhexamethylene biguanide. Bulletin of Environmental Contamination and Toxicology,
v.88, p.322-325, 2012.
O’MALLEY, L. P.; COLLINS, A. N.; WHITE, G. F. Biodegradability of end-groups of the biocide polyhexamethylene biguanide
(PHMB) assessed using model compounds. Journal of Industrial Microbiology and Biotechnology, v. 33, p. 677–684, 2006.
PASPARAKIS, G.; BOUROPOULOS, N. Swelling studies and in vitro release of verapamil from calcium alginate and calcium
alginate chitosan beads. International Journal of Pharmaceutics, v. 323, p. 34–42, 2006.
SÆTHER, H.V.; HOLME, H.K.; MAURSTAD, G.; SMIDSRØD, O.; STOKKE, B.T. Polyelectrolyte complex formation using
alginate and chitosan. Carbohydrate Polymers, v. 74, p. 813–821, 2008.


















