
ABSORCIÓN DE FÁRMACOS
14
____________________________________________________________________________________________________________________________________________________________________________________ Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y Fitofarmacología Programa de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl UNIVERSIDAD DE CHILE FACULTAD DE MEDICINA PROGRAMA DE FARMACOLOGÍA MOLECULAR Y CLÍNICA LABORATORIO DE FARMACODINAMIA Y FITOFARMACOLOGÍA ABSORCIÓN DE FÁRMACOS POR Sandro E. Bustamante D., M.Sc. - 2005 -
Transcript of ABSORCIÓN DE FÁRMACOS

____________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
UNIVERSIDAD DE CHILEFACULTAD DE MEDICINA
PROGRAMA DE FARMACOLOGÍA MOLECULAR Y CLÍNICALABORATORIO DE FARMACODINAMIA Y FITOFARMACOLOGÍA
ABSORCIÓN DE FÁRMACOS
POR
Sandro E. Bustamante D., M.Sc.
- 2005 -

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
ABSORCIÓN DE FÁRMACOSMECANISMOS DE TRANSPORTE A TRAVÉS DE MEMBRANAS
BIODISPONIBILIDAD
a absorción es un proceso farmaco-cinético que comprende el ingresode la molécula de fármaco al
organismo, desde su sitio deadministración inicial hasta alcanzar lacirculación sistémica. Por ende, es muyrelevante comprender los mecanismos detransporte de las moléculas de fármacosa través de las membranas biológicas,las que constituyen barreras a su paso a
través de las células de los distintostejidos.
Existen varios factores quecondicionan la absorción como tambiéncircunstancias que pueden alterar esteproceso. La vía de administración, a suvez, impone determinadas característicasespeciales que determinan cambios en elproceso de absorción.
MECANISMOS DE TRANSPORTE A TRAVÉS DE MEMBRANAS
A excepción de la víaintravenosa, donde por definición noexiste el proceso de absorción, cualquiervía de administración de fármacos sitúa alas moléculas de fármaco en un lugarinicial en el cual dichas moléculas debenentrar en solución con los líquidos delmedio en el que se encuentran. Cualquierdesplazamiento de las moléculas defármaco dentro del organismo deberárealizarse a través de las membranasbiológicas, que no son otra cosa que lasmembranas plasmáticas de las célulasque componen los diferentes tejidos.
La membrana plasmática detodas las células, está constituída por unabicapa de lípidos y proteínas. Loslípidos son moléculas anfipáticas, con suporción polar o hidrófila orientada haciael exterior de la membrana y su porciónapolar o hidrófoba, se orienta hacia elinterior de la bicapa lipídica. De modoque el paso de una molécula a través deuna membrana celular debe enfrentar dosmedios polares separados por uno apolar,lo que termodinámicamente representa
barreras de energía que se oponen a sucruce.
Revisaremos tres tipos detransporte a través de membranas:transporte pasivo, transporte activo yfiltración.
1. TRANSPORTE PASIVO
El transporte pasivo secaracteriza porque la fuerza impulsoradel proceso para vencer la barrera deenergía que impone la bicapa lipídica alas moléculas de fármaco, se obtiene dela formación de una gradienteelectroquímica (electro por la gradientede moléculas que presentan cargaeléctrica neta y química por la gradientede concentración de las moléculas).Termodinámicamente, la gradientefavorece la dicipación de su energíaalmacenada en la dirección y sentido queva de mayor a menor concentraciónmolecular y de carga eléctrica de lasmoléculas del fármaco (figura 1). Por
L

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
tanto, es un proceso queenergéticamente, no representa gasto
alguno para la ecomomía celular.
Figura 1. Representación gráfica de la disipación de la energía libre almacenada en elgradiente electroquímico, la cual actúa como fuerza motríz del transporte pasivo a través de labicapa lipídica. –(z), magnitud de la gradiente eléctrica si la molécula presenta carga eléctrica.(d∇∇/dX), variación de la gradiente de concentración química de la molécula.
1.1. Difusión simple. La difusión simplees la forma más sencilla de transportepasivo. La mayoría de los fármacos, porsu peso molecular bajo a mediano,pueden cruzar la bicapa lipídicavenciendo la barrera de energía de ésta afavor del gradiente electroquímico.
Otra característica importanteque debe presentar una molécula paracruzar por difusión simple es suliposolubilidad, mientras mayor ésta sea,aumentará la velocidad de difusión. Laliposolubilidad de una molécula estáexpresada por su coeficiente de partición(o distribución) lípido/agua. Todas estascaracterísticas fisicoquímicas de lasmoléculas están incluídas en la ley deFick, que predice la velocidad de
difusión (ds/dt) de una molécula alcruzar una bicapa lipídica:
ds/dt = (A D ββ [[Ce-Ci]]) 1/∆∆X
donde A es el área de la bicapa sobre lacual ocurrirá la difusión y ∆∆X ladistancia que debe recorrer a través de labicapa la molécula que difunde. Lascaracterísticas fisicoquímicas propias dela molécula están representadas en D, laconstante de difusión de la molécula ypor ββ, el coeficiente de particiónlípido/agua. La fuerza electromotríz delproceso aparece en la expresión [[Ce-Ci]],es decir, la gradiente electroquímica.
La ausencia de carga eléctrica dela molécula es un factor muy importante,ya que una molécula apolar difunde con

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
mayor facilidad que otra de igualescaracterísticas, pero polar. Sin embargo,el grado de polaridad de las moléculas defármacos no es un valor absoluto, sinoque depende del pH del medio en el cualse encuentra la molécula.
La mayoría de los fármacos sonelectrolitos débiles, tanto ácidos comobases y, en el medio acuoso delorganismo, éstos se encuentranparcialmente ionizados.
k1AH A- + H+
k2
La ecuación anterior representa elequilibrio ácido-base de un ácido débil,dónde k1/k2 es la constante deasociación, Ka. El grado de ionizacióndependerá de la Ka de la molécula y dela concentración de H+ del medio
Ka - [[H+]] = {{[[A-]] / [[AH]]}}
Si aplicamos el logarítmo decimal aambos lados de la ecuación, ésta quedaexpresada como
Log Ka – Log [[H+]] = Log {{[[A-]] / [[AH]]}}
y reemplazando por la función p, ó –Log(argumento) y reordenando los términos:
pH = pKa + Log {{[[A-]] / [[AH]]}}
que es la conocida ecuación deHenderson-Hasselbach (para un ácidodébil). La forma no ionizada de lamolécula (AH, en el ejemplo del ácidodébil) difundirá libremente a través de labicapa lipídica en función de suliposolubilidad. Por el contrario, laforma ionizada (A- en nuestro ejemplo),
estará termodinámicamente dificultadapara cruzar la membrana celular debido ala presencia de carga eléctrica queinteractúa con los dipolos del agua.
Luego podemos concluir que siuna membrana semipermeable, como lamembrana celular, separa doscompartimientos (extra e intracelular,por ejemplo) en los que se encuentrandisueltas las moléculas del fármaco, sólopodrán difundir a su través la forma noionizada hasta que se logre el estado deequilibrio en el que las concentracionesde la forma no ionizada a ambos lados dela membrana se igualen.
En el caso de un fármaco ácidodébil, su absorción será favorecida enun ambiente ácido. Ahora bien, si los doscompartimientos separados por lamembrana tienen diferente pH, laconcentración del fármaco será tambiéndiferente una vez alcanzado el equilibrio.La concentración será mayor en el ladoen que el pH induzca mayor ionizaciónde la molécula. Este fenómeno se conocecomo atrapamiento de fármaco.
¿Y qué ocurrirá si el fármaco esuna base débil?
k1BH+ B + H+
k2
¿Cómo resulta la ecuación deHenderson-Hasselbach? ¿Bajo quécondiciones de pH aumenta la forma noionizada? ¿Qué valores de pHfavorecerían la absorción de un fármacobase débil?
Finalmente, debido a que elfármaco es retirado constantemente de sulugar de administración por la sangre que

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
lo distribuye en el organismo, nunca sealcanza el estado de equilibrio comoocurre in vitro, manteniéndosepermanentemente el gradienteelectroquímico entre el sitio deadministración y la sangre, lo quepermite que el proceso de absorción secomplete.
1.2 Difusión facilitada. Las proteínas demembrana que se encuentran en lassuperficies extra e intracelular de lamembrana cumplen variadas funciones.Una de ellas, las denominadas proteínastransportadoras, son capaces detransportar ciertas sustancias de un ladoa otro de la membrana plasmática. No seconoce con exactitud cómo ocurre elproceso; hay evidencia que sugiere que
algunas proteínas transportadoras semueven libremente dentro de la faselipídica de la membrana. Son capaces deligar, en sitios específicos, una sustanciaen uno de los lados de la memebrana y,usando la energía de la gradienteelectroquímica, pueden traslocar a lasustancia ligada al otro lado de lamembrana, donde es liberada.
Muchos aminoácidos sontransporatdos de esta manera (figura 2).Algunos fármacos poseen estructurasquímicas que les permiten reconocer yunirse al sitio de ligamen de la proteínatransportadora y ser translocados alinterior celular, usando la energíaalmacenada en su gradienteelectroquímico.
Figura 2. Esquema de difusión facilitada de aminoácidos (AA) y azúcar (Az) por medio deproteínas transportadoras al interior celular. La fuerza motríz para el proceso, proviene de laenergía almacenada en la gradiente de concentración de AA o de Az.

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
La existencia de sitios de uniónen la proteína transportadora, quenormalmente son usados por lassustancias endógenas, hacen que elsistema sea saturable cuando se ocupanen su totalidad los sitios de unión deltransportador. Por la misma razón, existela posibilidad de inhibición competitiva
del transporte por la presencia de lassustancias endógenas, o bien, pormoléculas afines por el mismotransportador. La figura 3 muestra lacomparación entre un sistema detransporte por difusión simple y otro pordifusión facilitada.
Figura 3. Gráfica comparativa entre el flujo y concentración del sustrato, transportado pordifusión simple (línea contínua) y por difusión facilitada (linea discontinua). Al ocupar elsustrato todos los sitios de unión del transportador, el transporte se satura, cosa que no ocurreen la difusión simple, donde un aumento de la concentración de sustrato induce un mayor flujo.
2. TRANSPORTE ACTIVO
Los sistemas de transporte activo,a menudo llamados bombas, utilizan laenergía libre almacenada en los enlacesde alta energía del fosfato del ATP comofuente de energía para producir elmovimiento de la proteína transportadoray la traslocación de la sustanciatransportada a través de la membrana, yaque la traslocación tiene lugar contra lagradiente electroquímica. Existen dostipos de transporte activo: primario ysecundario.
2.1 Transporte activo primario. Usa laenergía del ATP directamente en elproceso de transporte mediante la acciónde una ATP-asa que rompe el enlace delgrupo fosfato terminal del ATP. Lafigura 4 (parte superior) ilustra esteproceso. La bomba de Na+/K+ esresponsable de establecer y mantener lasgradientes de concentraciones de ionessodio y potasio que existen a través de lamembrana de las células.
Otros ejemplos de transporteactivo primario son la Ca2+-ATPasa de lamembrana celular externa (responsable

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
de transportar calcio fuera de la célula) y,la Ca2+-ATPasa del retículosarcoplásmico de las células musculares(transporta calcio desde el citoplasma alretículo sarcoplásmico).
2.2 Transporte activo secundario. Usala energía de una gradiente deconcentración previamente establecidapor un proceso de transporte activoprimario (figura 4, parte inferior). Por
tanto, el transporte activo secundario usaindirectamente la energía derivada de lahidrólisis del ATP. El intercambio deNa+/Ca2+ a través de la membranacelular es un ejemplo, como lo es eltransporte de azúcares y aminoácidos.Los fármacos pueden utilizar estesistema de transporte activo al sercapaces de unirse al sitio de ligamen deltransportador.
Figura 4. La bomba Na+/K+-ATPasa (parte superior) hidroliza el ATP a ADP y fosfatoinorgánico (Pi), liberando la energía libre del enlace fosfato terminal. La energía liberada seusa para cambiar la conformación de la proteína transportadora, resultando en el transporte de 3Na+ desde el interior al exterior celular y 2 K+ en el sentido opuesto. La energía almacenada enla gradiente de sodio es usada en varios procesos de transporte activo secundario (parte inferiorde la figura). Los aminoácidos (AA), cloruros y azúcares, como algunos fármacos, sontransportados al interior celular vía transporte activo secundario.
Al igual que en el transporte por difusiónfacilitada, en los sistemas de transporteactivo, por utilizar proteínastransportadoras con un número finito de
sitios de unión a ligando, poseeráncinética saturable (figura 3) y seránsuceptibles de inhibición competitivacon el sustrato endógeno.

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
El transporte activo puede serinhibido, además, por sustancias ocondiciones que (a) bloqueen laproducción de energía celular, comoinhibidores metabólicos (-CN), atmósferaanaerobia, cambios en la temperatura,etc., (b) sustancias que bloqueen a laproteína transportadora, como lauabaína, que bloquea la bombaNa+/K+-ATPasa y, (c) por inhibición dela síntesis de la proteína transportadora.
3. FILTRACIÓN
La filtración es un proceso detransporte a través de las hendidurasintercelulares, por ende, difieresustancialmente de los sistemas detransporte previante descritos. Depreferencia ocurre en la pared de loscapilares sanguíneos, pasando lasmoléculas del fármaco a través de losespacios existentes entre las células,junto a las moléculas de agua en la cualestá disuelto, desplazandose entre losintersticios celulares.
Este mecanismo de transporteintercelular coexiste con los otrosmecanismos de transporte que ocurrenen la membrana de las células.
La velocidad de filtración (esdecir, el flujo de las moléculas defármaco) depende de varios factores.
(a) Del tamaño de la molécula defármaco y del espacio intercelular.La fracción de fármaco unida aproteínas plasmáticas no puede pasara través de estos espacios. Además,la filtración observada de un tejido aotro varía considerablemente, debidoa las características histológicas de lared capilar que posea.
(b) De la concentración del fármaco enel líquido que se está filtrando, yaque una mayor gradiente deconcentración representa una mayorfuerza motríz para el proceso.
(c) De las presiones hidroestática ycoloidosmótica que se ejercen sobrela dosolución (diferentesdependiendo del territorio capilar) yque actúan como fuerza motríz. En laparte arterial del territorio capilar esmayor la influencia de la presiónhidroestática, en tanto que en la partevenosa, predomina la presióncoloidosmótica.
PROCESO DE ABSORCIÓN
Como ya mencionamos, elproceso de absorción de fármacoscomprende el paso del fármaco desde ellugar de administración hasta lacirculación sistémica. Existen múltiplesfactores que no solo condicionan elproceso de absorción, sino que tambiénfactores que pueden alterar el normaldesarrollo del proceso.
FACTORES DETERMINANTES EN LA TASADE ABSORCIÓN DE FÁRMACOS
La tasa de absorción de unfármaco depende de los siguientesparámetros: características fisico-químicas de la molécula de fármaco,forma farmacéutica, lugar de absorcióny elminación presistémica del fármaco.

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
1. Características fisicoquímicas de lamolécula. El peso molecular otamaño de la molécula es muyrelevante, en particular para eltransporte por difusión simple. Otrofactor determinante en el tipo detransporte es su carácter ácido obase y el valor de su pKa por cuantodeterminarán el grado de ionizaciónmolecular. El coeficiente departición lípido/agua, β, determinasu liposolubilidad. Estos factorescondicionan el mecanismo deabsorción (tipo de transporte) y lavelocidad del proceso.
2. Forma farmacéutica. Para queocurra el proceso de absorción, elfármaco debe estar en solución en loslíquidos tisulares. Por esta razón lasdiferentes formulacionesfarmacéuticas condicionan lavelocidad de disgregación ydisolución. Las principales formasfarmacéuticas son solución,suspención, polvo, cápsulas ycomprimidos. La presencia de losexcipientes y los aditivos, que juntoal principio activo conforman almedicamento, también tienen unainfluencia significativa en lacapacidad de disgregación ydisolución de la preparaciónfarmacéutica, condicionando lavelocidad de absorción.
3. Lugar de absorción. La velocidadde absorción será mayor mientrasmayor sea el área, el tiempo deexposición y la irrigación del lugarde absorción. Recordemos que lairrigación mantiene el gradiente deconcentración del fármaco. Si el pH
del medio favorece la forma noionizada del fármaco se facilitará elproceso de absorción.
4. Eliminación presistémica. Laadministración de fármaco porcualquier vía, excepto la iv, puedeimpedir la llegada de todo el fármacoadministrado a la circulaciónsistémica. Pricipalmente el paso delfármaco desde su sitio deadministración al hígado por mediodel sistema vena porta antes dealcanzar la circulación sistémica ylos tejidos, en los que debe ejercer suacción farmacológica, se conocecomo efecto de primer paso. Elprimer paso del fármaco por elhígado puede significar un primerproceso de metabolización por partede las enzimas de las célulashepáticas.
El pulmón es otro órgano queproduce eliminación presistémica. Otrasformas de eliminación del fármacoadministrado es a través de las hecesantes de ser completamente absorvido,inactivación por el pH del medio o porenzimas digestivas y, la metabolizacióndel fármaco por las células del sistemagástrico o las bactéreas intestinales.
FACTORES QUE ALTERAN LA ABSOR-CIÓN DE FÁRMACOS
Existen tres factores principalescapaces de alterar significativamente elnormal proceso de absorción defármacos: fisiológicos, patológicos yyatrogénicos.

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
1. Factores fisiológicos. Son variadosdependiendo de la vía deadministración. Entre éstos podemosmencionar la presencia de alimentos,que disminuye el tiempo deexposición y área de absorción. Elembarazo altera la absorción debidoa cambios en la motilidad intestinal,cambios en el pH y alteraciones delflujo sanguíneo. La edad condicionacambios en la motilidad intestinal yalteraciones del pH, dependiendo siestamos en presencia de unprematuro, un adulto o un anciano.
2. Factores patológicos. En el caso dela administración de fármacos porvía oral, son muy importates elvómito y la diarrea, por disminuir eltiempo de permanencia del fármacoen el tracto gastrointestinal. En la vía
intramuscular los factores másimportantes que alteran el proceso deabsorción son la alteración del flujosanguíneo, estados de shock y lainsuficiencia cardíaca.
3. Factores yatrogénicos. Soninteracciones entre fármacos quealteran el proceso de absorción, yasea en forma directa por la formaciónde precipitados entre ellos queimpiden la normal absorción delfármaco, o bien interaccionesindirectas, al modificar el pH delmedio, alterar la motilidad intestinalo el flujo sanguíneo.
NIVELES PLASMÁTICOSPARÁMETROS FARMACOCINÉTICOS Y BIODISPONIBILIDAD
Curva de Niveles Plasmáticos
La curva de niveles plasmáticosrepresenta la evolución temporal de laconcentración en el plasma del fármacoadministrado. Usualmente, se determinaadministrando una dosis determinada delfármaco en estudio a un paciente (o a unvoluntario sano) y tomando muestrassanguíneas cada cierto intervalo detiempo. La muestra sanguínea esanalizada por HPLC para detectar lapresencia de las moléculas del fármaco ycuantificar su concentración, usualmenteen µg de fármaco por ml de plásma. Elanálisis de la curva de nivel plasmático,característico del fármaco y de la vía de
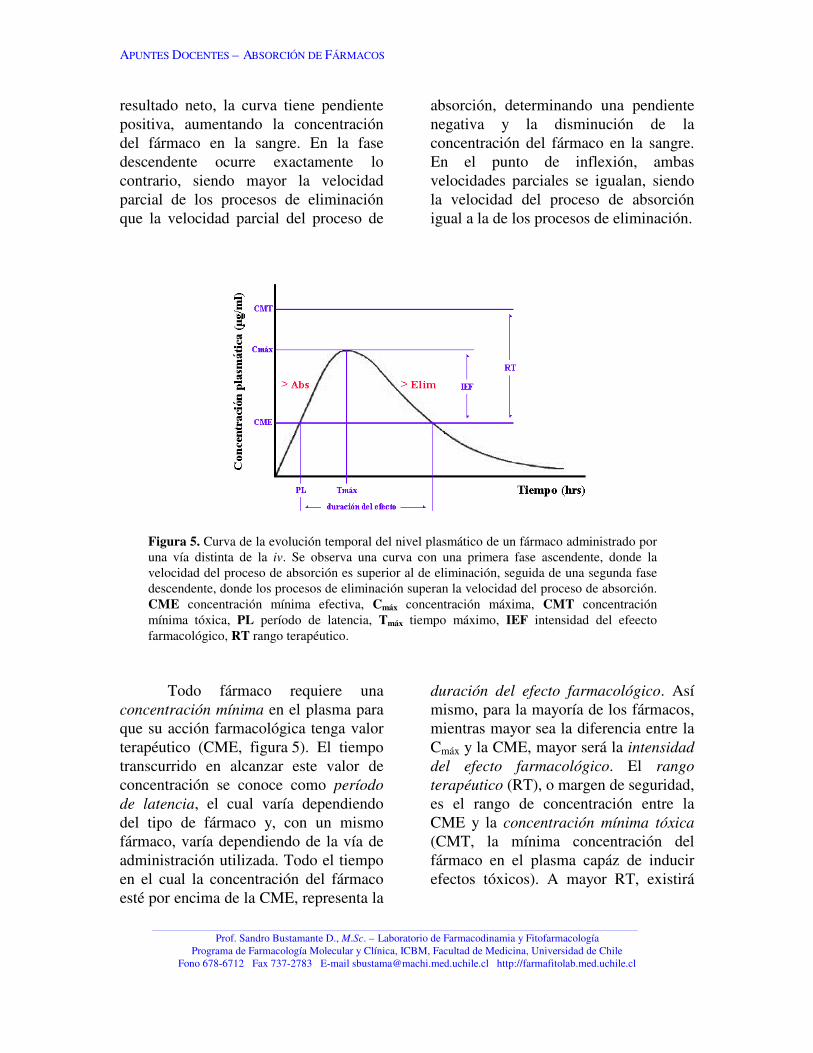
administración, permite determinar unaserie de parámetros farmacocinéticos,como se aprecia en la figura 5.
Es importante hacer notar quedurante el desarrollo temporal de lacurva de niveles plasmáticos, ocurrensimultáneamente los procesos deabsorción y eliminación. La faseascendente y descendente representan lavelocidad resultante de la sumación devolocidades parciales de los dosprocesos que antagonizan el nivelplasmático del fármaco. Así, en la faseascendente de la curva, la velocidadparcial del proceso de absorción esmayor que la velocidad parcial de losprocesos de eliminación y, como
APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
resultado neto, la curva tiene pendientepositiva, aumentando la concentracióndel fármaco en la sangre. En la fasedescendente ocurre exactamente locontrario, siendo mayor la velocidadparcial de los procesos de eliminaciónque la velocidad parcial del proceso de
absorción, determinando una pendientenegativa y la disminución de laconcentración del fármaco en la sangre.En el punto de inflexión, ambasvelocidades parciales se igualan, siendola velocidad del proceso de absorciónigual a la de los procesos de eliminación.
Figura 5. Curva de la evolución temporal del nivel plasmático de un fármaco administrado poruna vía distinta de la iv. Se observa una curva con una primera fase ascendente, donde lavelocidad del proceso de absorción es superior al de eliminación, seguida de una segunda fasedescendente, donde los procesos de eliminación superan la velocidad del proceso de absorción.CME concentración mínima efectiva, Cmáx concentración máxima, CMT concentraciónmínima tóxica, PL período de latencia, Tmáx tiempo máximo, IEF intensidad del efeectofarmacológico, RT rango terapéutico.
Todo fármaco requiere unaconcentración mínima en el plasma paraque su acción farmacológica tenga valorterapéutico (CME, figura 5). El tiempotranscurrido en alcanzar este valor deconcentración se conoce como períodode latencia, el cual varía dependiendodel tipo de fármaco y, con un mismofármaco, varía dependiendo de la vía deadministración utilizada. Todo el tiempoen el cual la concentración del fármacoesté por encima de la CME, representa la
duración del efecto farmacológico. Asímismo, para la mayoría de los fármacos,mientras mayor sea la diferencia entre laCmáx y la CME, mayor será la intensidaddel efecto farmacológico. El rangoterapéutico (RT), o margen de seguridad,es el rango de concentración entre laCME y la concentración mínima tóxica(CMT, la mínima concentración delfármaco en el plasma capáz de inducirefectos tóxicos). A mayor RT, existirá

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
mayor seguridad en el rango dedosificación del fármaco.
La administración de fármacospor vía intravenosa, por definición,carece del proceso de absorción (¿PodríaUd. dibujar la curva de nivelesplasmáticos iv sobre la curva de lafigura 5?). Así, la dosis administrada atiempo cero representa la Cmáx. Pero,para un mismo fármaco, ¿la Cmáx
alcanzada por iv es equivalente a laalcanzada por otra vía, digamos oral?
Aquí entra en juego un nuevoconcepto, la biodisponibilidad.
Biodisponibilidad
La biodisponibilidad indica lacantidad y forma en que un fármacollega a la circulación sistémica y, portanto, está disponible para acceder a lostejidos y producir su efectofarmacológico.
La biodisponibilidad dependegrandemente del proceso de absorción yde las variables que lo condicionan, de lavía de administración y del efecto deeliminación presistémica, en particular elefecto de primer paso hepático.
La vía intravenosa tiene unabiodisponibilidad del 100%, misma quees significativamente inferior por otravía. Para cuantificar la biodisponibilidadpor vo, por ejemplo, recurrimos a lacurva de niveles plasmáticos.
Ya que mientras más veloz sea elproceso de absorción mayor será la Cmáx
y aumentará la biodisponibilidad.Expresado matemáticamente no es otracosa que la derivada en el rango en queésta es mayor que cero, es decir:
d(Cp) / dt
Dado que la cantidad de fármacobiodisponible está representado por elárea bajo la curva, matemáticamenterepresenta la sumatoria de todas lasvariaciones infinitesimales de la Cp en eltiempo:
∫ d(Cp) / dt, entre t=0 y t=∞
Vemos entonces, que labiodisponibilidad está afectada por lavelocidad de absorción y se puedecuantificar mediante el área bajo la curvade niveles plasmáticos. Existen variadosprocedimientos para cuantificar el área yla velocidad a partir de la curva deniveles plasmáticos sin tener que recurrira las expresiones matemáticas que lasdescriben.
No obstante, es de mayor interésel definir una herramienta que nospermita comparar las biodisponibilidadesentre distintas vías de administración conla iv, la cual es máxima.
Se define la fracción deabsorción (f) como la razón entre labiodisponibilidad de la vía en estudio(oral en nuestro ejemplo) y labiodisponibilidad iv. El resultadosiempre será un número cuyo valor estéentre 0 y 1. Generalmente, se multiplicapor 100 para expresarlo en porcentaje:
f = [[ (ABC vo) / (ABC iv)]] 100
donde ABC representa el Área Bajo laCurva de la vía en cuestión.
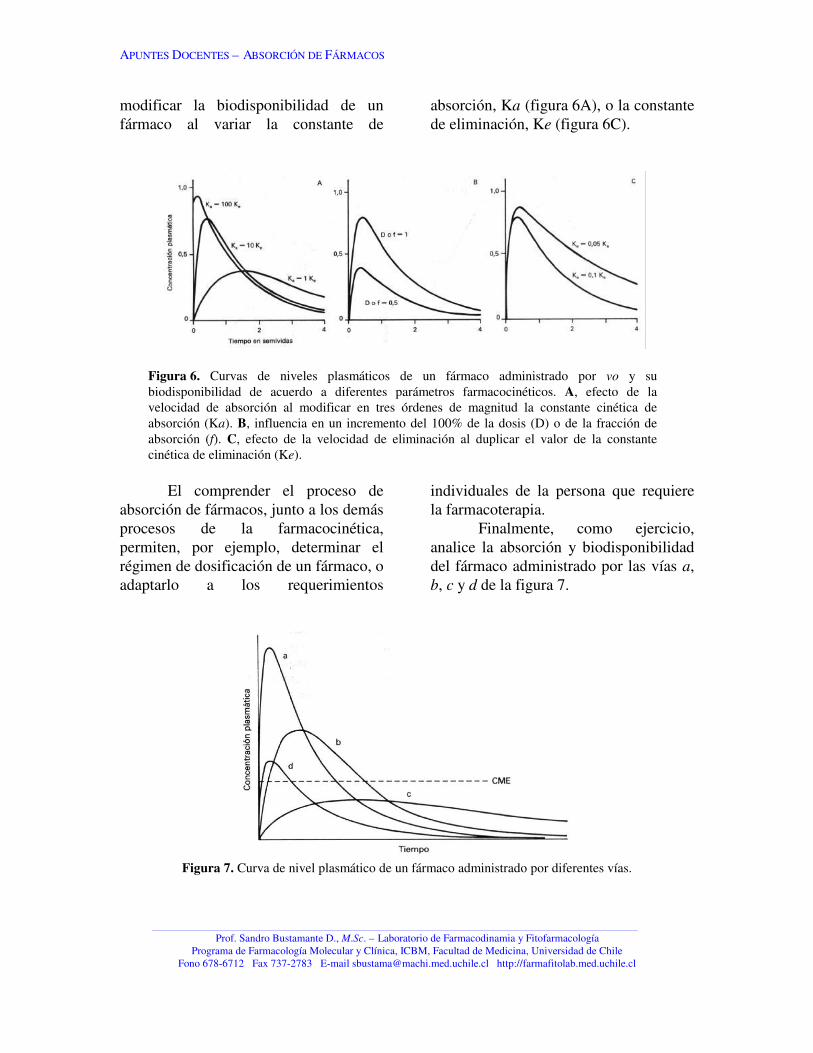
La figura 6B muestra el efectodel cambio de la f en la biodisponibilidadde un fármaco administrado por vo.Observe cómo otros parámetros pueden
APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
modificar la biodisponibilidad de unfármaco al variar la constante de
absorción, Ka (figura 6A), o la constantede eliminación, Ke (figura 6C).
Figura 6. Curvas de niveles plasmáticos de un fármaco administrado por vo y subiodisponibilidad de acuerdo a diferentes parámetros farmacocinéticos. A, efecto de lavelocidad de absorción al modificar en tres órdenes de magnitud la constante cinética deabsorción (Ka). B, influencia en un incremento del 100% de la dosis (D) o de la fracción deabsorción (f). C, efecto de la velocidad de eliminación al duplicar el valor de la constantecinética de eliminación (Ke).
El comprender el proceso deabsorción de fármacos, junto a los demásprocesos de la farmacocinética,permiten, por ejemplo, determinar elrégimen de dosificación de un fármaco, oadaptarlo a los requerimientos
individuales de la persona que requierela farmacoterapia.
Finalmente, como ejercicio,analice la absorción y biodisponibilidaddel fármaco administrado por las vías a,b, c y d de la figura 7.
Figura 7. Curva de nivel plasmático de un fármaco administrado por diferentes vías.

APUNTES DOCENTES – ABSORCIÓN DE FÁRMACOS
___________________________________________________________________________________________________________________________________________________________________________________
Prof. Sandro Bustamante D., M.Sc. – Laboratorio de Farmacodinamia y FitofarmacologíaPrograma de Farmacología Molecular y Clínica, ICBM, Facultad de Medicina, Universidad de Chile
Fono 678-6712 Fax 737-2783 E-mail [email protected] http://farmafitolab.med.uchile.cl
BIBLIOGRAFÍA
Biofísica y Fisiología Celular. Latorre,López-Barneo, Bezanilla y Llinás.Primera Edición. Editorial Universidadde Sevilla. España, 1996.
Las Bases Farmacológicas de laTerapéutica. J. Hardman, L. Limbird, P.Molinoff, R. Ruddon y A. GoodmanGilman. Novena Edición, Vol I. EditorialMc Graw-Hill-Interamericana,Traducción al Español. México, 1996.
Farmacología Humana. Flórez.Segunda Edición. Editorial Masson-Salvat. España, 1994.
Velázquez, Farmacología. A. Velasco,P. Lorenzo, J. Cerrano y F. Andrés-Trelles. Decimosexta Edición. EditorialMc Graw-Hill-Interamericana. Madrid,1993.


















